

Abstract
Transcription factors (TFs) are key regulatory proteins that control the transcriptional rate of cells by binding short DNA sequences called transcription factor binding sites (TFBS) or motifs. Identifying and characterizing TFBS is fundamental to understanding the regulatory mechanisms governing the transcriptional state of cells. During the last decades, several experimental methods have been developed to recover DNA sequences containing TFBS. In parallel, computational methods have been proposed to discover and identify TFBS motifs based on these DNA sequences. This is one of the most widely investigated problems in bioinformatics and is referred to as the motif discovery problem. In this manuscript, we review classical and novel experimental and computational methods developed to discover and characterize TFBS motifs in DNA sequences, highlighting their advantages and drawbacks. We also discuss open challenges and future perspectives that could fill the remaining gaps in the field.
Keywords: transcription factors, transcription factors motif discovery, motif discovery algorithms, motif models
INTRODUCTION
Transcription factors (TFs) are fundamental proteins regulating the transcriptional states, differentiation and developmental patterns of cells [1–3]. TFs exert their function by binding short and specific DNA sequences (~6–20 nt long [4]), called transcription factor binding sites (TFBS), recognized by their binding domains. TFBS are often located in gene promoters [5], distal regulatory elements, such as enhancers, silencers or insulators, and even within coding regions [6–8]. TFBS often correspond to recurring DNA sequence patterns, which are often referred to as motifs, and these patterns can differ by a few nucleotides. Importantly, TF function is critically linked to the motif sequences it can bind [9, 10]. Therefore, the identification of such regulatory motifs provides fundamental insights into the complex mechanisms governing gene expression.
Several experimental assays have been developed to determine the binding site sequences of TFs in living cells or organisms (in vivo), or in test tubes using synthetic or purified components (in vitro) [11] (Figure 1). Early methods, like electrophoretic mobility shift assay (EMSA) [12] or footprinting [13], generally analyze a relatively small number of target sequences to find TFBS. As a result, they return small datasets of bound sequences. In vitro and in vivo high-throughput protocols such as PBM, SELEX or ChIP methods [14–16] facilitated the analysis of most target sites for factors of interest. As a result, large datasets of bound sequences have been generated, presenting an unprecedented opportunity to study and determine the TF binding landscapes. Experimental assays can recover the sequences bound by TFs along with their relative or absolute binding affinity. However, such datasets can incorrectly report unbound sequences as binding sites. In addition, the assays usually capture extra nucleotides in target sites, reducing data resolution and making manual analysis challenging.
Figure 1.

Experimental and computational methods to discover TFBS and popular models to represent binding site motifs. Protein binding microarray (PBM), HT-SELEX and ChIP-seq have become the most popular assays to determine TF binding preferences and identify their target sites (TFBS) in recent years. Computational motif discovery methods can be grouped into five classes, based on the algorithms employed to discover TFBS: enumerative, alignment-based, probabilistic graphical model-based, SVM-based and DNN-based methods. TFBS sequences prioritized by motif discovery algorithms are encoded in computational models representing the binding preferences of the investigated TFs.
Motif discovery algorithms provide a computational framework to analyze these large datasets generated by experimental assays, discovering the sequences potentially bound by TFs and predicting their affinities [17–21]. Given a sequence dataset, these algorithms typically recover sets of short and similar sequence elements. The prioritized sequence elements are later used to construct a motif model, summarizing the diverse binding site configurations observed among the prioritized sequences, and encoding their recurrent patterns and similarities (Figure 1).
Several methods and models have been proposed to discover and represent TFBS motifs. Position weight matrices (PWMs) [22] are the most popular models. PWMs are simple yet powerful and interpretable models, encoding the probability of observing a given nucleotide in each TFBS position. However, PWMs have some limitations, like the assumption of independence among the binding site positions. Therefore, several alternative motif models have been proposed [23–25], as described below. The derived motif models can be employed in many downstream analyses, like searching potential binding site occurrences in regulatory genomic sequences, predicting the sets of genes regulated by the investigated TFs or assessing how genetic variants could affect their binding landscape (Section 5).
In this paper, we review the state-of-the-art of motif discovery, describing the classical and recent experimental and computational methods to discover and represent TFBS motifs in DNA sequences. We discuss the novelties brought to the field by each algorithm and model, highlighting advantages and drawbacks of motif discovery methods and motif models, and how researchers addressed their limitations over the years. We begin by introducing in Section 2 popular experimental technologies to identify TF target sequences (Table 1). In Section 3, we review the computational methods to discover TFBS motifs in the datasets recovered from experimental assays (Supplementary Table 1) and popular motif models to represent TFBS. In Section 4, we discuss widely used TF-related databases. In Section 5, we present common downstream analyses employing motif models. We conclude by discussing in Section 6 the open challenges and potential future research directions for the development of novel motif discovery algorithms.
Table 1.
In vivo and in vitro experimental assays to identify and validate transcription factor binding sites
| Experimental assay | Description | Output | De novo motif discovery capability | Type | Identification of genomic binding locations | Throughput |
|---|---|---|---|---|---|---|
| Competition EMSA | Bound DNA sequences are identified by observing changes in the electrophoretic migration of DNA sequences through non-denatured polyacrylamide gel | Bound DNA sequences | No. Used to validate known binding sites | In vitro | No | Low |
| DNase footprinting | Pools of DNA sequences are incubated with the TF of interest; then, the DNA is degraded using DNase I. The unbound fragments are cut in all positions, while the bound DNA is protected by the TF | Bound DNA sequences | No. Used to validate known binding sites | In vitro | No | Low |
| Protein binding microarrays | Arrays of ~40 000 spots with short, immobilized DNA sequences are incubated with a tagged TF, and then washed to remove weakly bound proteins. The bound sequences are identified through fluorescence-based detection | Continuous values describing fluorescence intensity on each array spot | Yes. Limited to short motifs (~12 bp) | In vitro | No | High |
| HT-SELEX | The TF is added to a pool of randomized DNA fragments. The bound sequences are selected and constitute the starting pool for the next experimental round. The procedure is repeated for several rounds. Sequencing is employed to recover the sequence of the bound DNA fragments | DNA sequences | Yes | In vitro | No | High |
| ChIP-based technologies | TF–DNA complexes are cross-linked with formaldehyde and immunoprecipitated employing TF-specific antibodies. The bound sequences are then prioritized employing qPCR microarrays (ChIP-on-Chip) or through sequencing (ChIP-seq). ChIP-exo integrates exonuclease treatment to enhance sequence resolution | Genomic binding location coordinates | Yes. Limited by the inability to distinguish direct and indirect binding | In vivo | Yes | Low |
Generally, EMSA and DNase footprinting are used to validate known TFBS, while currently PBMs, HT-SELEX and ChIP-based methods are preferred to discover novel binding sites. ChIP-based assays are the only methods that recover the TF genomic binding locations. The throughput column refers to the number of samples that can be processed in parallel by each method (high: hundreds of samples; low:a few samples).
EXPERIMENTAL METHODS TO DISCOVER TRANSCRIPTION FACTOR BINDING SITES
During the last decades, several techniques have been introduced to experimentally identify and assess TF binding sites and binding preferences [11] (Figure 1 and Table 1).
Early studies on TF binding focused their analysis on gene promoters [22] and employed in vitro methods, such as electrophoretic mobility shift assay (EMSA) [12] or DNase footprinting [26]. EMSA exploits non-denatured polyacrylamide gel properties to separate bound and unbound DNA sequences. DNase footprinting combines EMSA with DNase I cleavage, identifying uncut regions (footprints) due to the protection of the bound TF. Generally, these assays produce datasets of a few hundred of bound sequences, exploring a limited spectrum of TFs binding landscape. Moreover, EMSA and DNase footprinting may be subject to technical constraints that could lead to inaccuracies in the reported sequences and binding preferences [11].
The introduction of NGS technologies revolutionized the study of TFBS identification by encouraging researchers to develop methods that exploit the power of massively parallel sequencing (Figure 1). These methods have two major advantages: (i) they do not require any prior knowledge on the binding site sequence [11, 27] and (ii) produce datasets of thousands of bound sequences allowing a better characterization of TF binding preferences [28].
Protein binding microarrays (PBMs) [14, 29] recover short TFBS sequences (~10 bp) and measure TF binding preferences in vitro. In PBMs, a tagged TF is released on a glass slide containing thousands of spots filled with short, immobilized DNA sequences. The tagged TFs are then incubated with fluorescent antibodies against the tag and subsequently washed to remove weakly bound factors. The fluorescence and DNA sequence enrichment are then used to quantify the TF–DNA binding strength and capture the bound sequences. Generally, the recovered sequences do not contain nucleotides flanking the investigated binding sites, producing high-resolution datasets. However, since the number of possible sequences grows as a function of the target length, PBMs can assess only a limited number of target sequences [11, 27]. PBM analysis is usually constrained to binding sites ~10–12 bp long.
HT-SELEX [11, 15] is a widely used in vitro method, coupling SELEX with high-throughput sequencing. A TF is released on a pool of randomized DNA sequences to allow the factor to select its target sites. The resulting TF–DNA complexes are separated from unbound sequences using affinity capture, and subsequently amplified through polymerase chain reaction (PCR) and sequenced. The resulting DNA library is enriched in binding sites for the studied TF and is used as the starting pool for another SELEX run [11, 15]. SELEX does not require any prior knowledge on the target sites of the investigated factor [30]. Since SELEX reaction is typically performed in liquid phase and consequently does not suffer from physical constraints, the sequence space covered by HT-SELEX is often larger than that of PBMs. Moreover, by coupling sequencing with DNA barcode indexing, HT-SELEX allows to analyze hundreds of TFs in parallel. HT-SELEX produces datasets of thousands of high-resolution bound sequences, which include only a few nucleotides flanking the binding sites. However, since the starting DNA library is constituted by randomized sequences, HT-SELEX cannot recover the genomic binding locations for the investigated factor.
The introduction of chromatin immunoprecipitation (ChIP) technologies [16] radically changed the study of TFBS binding, enabling the genome-wide identification of regions bound by TFs in vivo. In ChIP, the TF–DNA complexes are cross-linked using formaldehyde. The DNA is then fragmented in ~100–1000 bp long fragments and subsequently immunoprecipitated with antibodies specific for the investigated TF. To recover the bound sequences, the cross-links are reverted. Then, the resulting fragments are amplified through microarray hybridization (ChIP-on-Chip [16, 31]) or sequencing (ChIP-seq [32, 33]). To locate the binding regions, the recovered DNA fragments are mapped onto the genome. After ChIP-seq reads mapping, peak calling algorithms [34–36] are employed to predict the genomic binding locations for the investigated factor. Peak calling algorithms identify the genomic regions showing greater enrichment in mapped DNA probes with respect to a control experiment and mark those regions as binding locations, or peaks [37]. ChIP methods produce large datasets of thousands of genomic regions, whose length ranges from few hundreds to thousands of nucleotides, from which we can identify the likely TFBS for the investigated factor. Although ChIP technologies, and particularly ChIP-seq, are currently considered the current ‘golden standard’, they have some limitations. (i) ChIP can detect indirect binding, identifying other TFBS not belonging to the investigated factor [38]. (ii) ChIP-seq peaks may be false positives, recovered because of poor antibody quality [39]. (iii) ChIP-seq returns low-resolution datasets, whose sequences include several nucleotides flanking the target TFBS. ChIP-exo [40] addresses the latter issue, employing a lambda exonuclease to trim ChIP sequences, removing some of the nucleotides flanking the target sites.
Alternatively, since most TFs bind their target sequences in open chromatin regions, experimental assays targeting open chromatin like ATAC-seq or DNase-seq [41, 42] can be employed to recover in vivo genomic locations likely to contain TFBS. ATAC-seq and DNase-seq are generally employed when the factors binding the target regions are not known.
In summary, the current high-throughput in vivo and in vitro assays generate datasets of thousands of sequences potentially containing several possible binding configurations of TFBS, thereby enabling better characterizations of TFs binding landscapes.
COMPUTATIONAL METHODS AND MODELS TO DISCOVER AND REPRESENT TRANSCRIPTION FACTOR BINDING SITES
The TFBS motif discovery problem can be formalized as follows. Given a set of positive DNA sequences 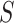 , obtained from an experimental assay targeting a certain TF, and a set of negative sequences
, obtained from an experimental assay targeting a certain TF, and a set of negative sequences 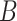 , the goal is to find one or more recurrent, short and similar subsequences in
, the goal is to find one or more recurrent, short and similar subsequences in 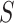 that maximize the discriminatory power between
that maximize the discriminatory power between 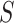 and
and 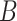 . Such subsequences are called patterns or motifs and are likely bound by the investigated TF. The negative set
. Such subsequences are called patterns or motifs and are likely bound by the investigated TF. The negative set 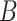 can contain randomly generated or selected genomic sequences, with similar nucleotide content and length of those in
can contain randomly generated or selected genomic sequences, with similar nucleotide content and length of those in 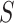 . The retrieved patterns are used to construct and train a computational model
. The retrieved patterns are used to construct and train a computational model 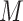 (motif model), representing the discovered motif. These models can then be used to identify new potential binding sites, given a new set of sequences, and to predict the strength of the TF–DNA binding. Motif discovery can be considered a classification or a regression problem, depending on the type of data used to train
(motif model), representing the discovered motif. These models can then be used to identify new potential binding sites, given a new set of sequences, and to predict the strength of the TF–DNA binding. Motif discovery can be considered a classification or a regression problem, depending on the type of data used to train 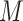 . The datasets derived by experimental assays like ChIP-seq or HT-SELEX provide hundreds or thousands of sequences containing binding sites. In this setting, motif discovery becomes a classification problem. In fact, the goal is to discriminate between bound and unbound sites in the input sequences and train the motif model with the identified binding sites. The datasets produced by other experimental technologies like PBMs provide the relative binding strength for large sets of sequences of equal length. Therefore, rather than discriminating between bound and unbound sequences, in this setting
. The datasets derived by experimental assays like ChIP-seq or HT-SELEX provide hundreds or thousands of sequences containing binding sites. In this setting, motif discovery becomes a classification problem. In fact, the goal is to discriminate between bound and unbound sites in the input sequences and train the motif model with the identified binding sites. The datasets produced by other experimental technologies like PBMs provide the relative binding strength for large sets of sequences of equal length. Therefore, rather than discriminating between bound and unbound sequences, in this setting 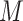 learns the relative binding affinities associated to each target site in the input dataset, transforming motif discovery into a regression problem. In both settings, the final goal is to derive a computational model
learns the relative binding affinities associated to each target site in the input dataset, transforming motif discovery into a regression problem. In both settings, the final goal is to derive a computational model 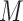 , describing the recovered TFBS and capable of predicting new binding events, along with their affinity, in sequences not used during model training. Motif discovery algorithms can be classified in enumerative, alignment-based, probabilistic graphical models, support vector machine (SVM)-based and deep neural network-based methods (Figure 1 and Supplementary Table 1).
, describing the recovered TFBS and capable of predicting new binding events, along with their affinity, in sequences not used during model training. Motif discovery algorithms can be classified in enumerative, alignment-based, probabilistic graphical models, support vector machine (SVM)-based and deep neural network-based methods (Figure 1 and Supplementary Table 1).
Other approaches to discover TFBS motifs in genomic sequences use phylogenetic footprinting [43, 44]. The core principle of phylogenetic footprinting is that functional elements, such as TFBS, are more likely to be conserved across evolutionarily related species, while non-functional elements are more susceptible to mutations. Although phylogenetic footprinting was one of the first techniques proposed for identifying TFBS, it is still widely used to examine TFBS conservation across different organisms [45–47]. In a recent study [48], the authors proposed a novel method that utilizes phylogenetic footprinting to discover TFBS.
Before describing the algorithms, we briefly review the models to describe TFBS motifs.
The most common models to represent TFBS are consensus sequences [49], PWMs [22, 50], high-order PWMs [23, 51], SVM-based [24] and deep neural network-based [25] models.
Consensus sequences summarize the discovered TFBS by denoting the most frequently observed nucleotide at each motif position in a prioritized sequence set. Although TFBS have conserved positions not tolerant to mutations [52], other binding site locations admit alternative nucleotides. Degenerate consensus accommodates ambiguous motif positions employing IUPAC symbols. However, consensus sequences cannot encode the contribution to TF–DNA binding of each nucleotide at each motif position.
PWMs address this limitation, providing an additive model with the contribution of each motif position to the binding site. PWMs construct an ungapped alignment between motif candidate sequences and count the frequency of each nucleotide at each position. The statistical significance of PWMs is often measured employing relative entropy (RE) [53]. RE quantifies the difference between computed nucleotide frequencies and those obtained from aligning random sequences. PWMs are visualized as logos [54], where the height of each nucleotide is proportional to its RE. Despite their wide success, PWMs still assume independence between motif positions.
Probabilistic graphical models address this limitation by modeling dependency between motif nucleotides. These models include high-order PWMs like dinucleotide weight matrices (DWMs), Bayesian networks (BNs), Markov models (MMs) or hidden Markov models (HMMs) [23, 51, 55, 56]. DWMs and high-order PWMs are often visualized as logos with 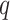 -mers replacing the single nucleotides, where
-mers replacing the single nucleotides, where 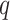 is the dependency order between neighboring nucleotides. Importantly, probabilistic graphical models can account for variable spacing between half-sites of two box motifs. However, the number of model’s parameters and its complexity grow exponentially with
is the dependency order between neighboring nucleotides. Importantly, probabilistic graphical models can account for variable spacing between half-sites of two box motifs. However, the number of model’s parameters and its complexity grow exponentially with 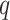 , often resulting in the model overfitting the input dataset.
, often resulting in the model overfitting the input dataset.
SVM-based models train a SVM kernel learning the binding site structure from the input sequence dataset. TFBS are represented by either a list of 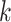 -mers with associated weights or support vectors used to discriminate between bound and unbound sequences, depending on the employed kernel [57]. In the former case, the weights reflect the
-mers with associated weights or support vectors used to discriminate between bound and unbound sequences, depending on the employed kernel [57]. In the former case, the weights reflect the 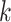 -mer contribution to the motif sequence. SVM-based models can account for variable spacing between the half-sites of two box motifs, like probabilistic graphical models. Importantly,
-mer contribution to the motif sequence. SVM-based models can account for variable spacing between the half-sites of two box motifs, like probabilistic graphical models. Importantly, 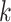 -mers indirectly capture
-mers indirectly capture 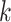 -th order dependencies between neighboring nucleotides. However, simple SVM-based models are limited to consider short
-th order dependencies between neighboring nucleotides. However, simple SVM-based models are limited to consider short 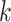 (~10 bp) and cannot represent longer motifs. Gapped
(~10 bp) and cannot represent longer motifs. Gapped 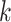 -mers [58] addressed this limitation, handling longer TFBS and sequence degeneration in non-informative motif positions. To visualize the discovered motifs, SVM-based models are often reduced to PWMs computed aligning the informative
-mers [58] addressed this limitation, handling longer TFBS and sequence degeneration in non-informative motif positions. To visualize the discovered motifs, SVM-based models are often reduced to PWMs computed aligning the informative 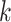 -mers.
-mers.
Deep neural network (DNN)-based models integrate the diverse, complex and hierarchical patterns governing TF–DNA binding events in input nucleotide sequences. Although DNN-based models are accurate and powerful, their ‘black box’ nature is a major limitation [59]. Many frameworks visualize the discovered motifs as PWMs, computed aligning the sequences activating the convolutional kernels of the DNN [60]. However, DNNs often learn distributed representations where multiple neurons cooperate to describe single patterns. Therefore, motifs learned by single kernels and the resulting PWMs are often redundant with each other. DeepLIFT [61] proposed a method to assign importance scores to the kernels. Comparing the activation of each neuron to a reference value, DeepLIFT selects which kernels contribute most to the TFBS definition, reducing motif redundancy. TF-MoDISco [62] extended this idea by clustering and aggregating the discovered motifs, using the importance scores assigned to the kernels. However, computing interpretable models without losing some information learned by the DNN is still an open challenge.
Enumerative methods
Enumerative motif discovery algorithms (Figure 1) assume that motifs are overrepresented patterns in the input dataset 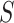 , with respect to a set of background genomic sequences
, with respect to a set of background genomic sequences 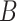 . Enumerative algorithms may assume that the motif length
. Enumerative algorithms may assume that the motif length 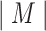 is known a priori. Given
is known a priori. Given 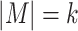 , the general idea is to collect the approximate occurrences of all potential
, the general idea is to collect the approximate occurrences of all potential 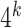
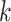 -mers in the sequences of
-mers in the sequences of 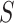 and assess if the difference between the number of matches found in
and assess if the difference between the number of matches found in 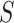 and
and 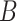 or the expected number of matches from a background model is statistically significant. Then, a PWM is obtained building an ungapped alignment from the statistically significant
or the expected number of matches from a background model is statistically significant. Then, a PWM is obtained building an ungapped alignment from the statistically significant 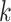 -mers. Searching the approximate occurrences of all
-mers. Searching the approximate occurrences of all 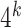
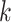 -mers quickly becomes impractical, even for small
-mers quickly becomes impractical, even for small 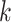 . Early proposals introduced the usage of heuristics to reduce the search space, for example, searching only patterns occurring at least once in each sequence
. Early proposals introduced the usage of heuristics to reduce the search space, for example, searching only patterns occurring at least once in each sequence 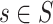 [63] or restricting mismatching locations to specific motif positions [64]. However, mismatches can occur at any motif position. Weeder [65, 66] and SMILE [67] proposed using suffix trees (STs) [68] to efficiently explore the entire motif search space. They leverage the indexing capabilities of STs to perform approximate pattern matching, without restrictions on mismatching positions. This enabled achieving high accuracy in motif discovery, while reducing computational costs. To determine the statistical significance of motif candidates, SMILE and Weeder compare the motifs frequencies in
[63] or restricting mismatching locations to specific motif positions [64]. However, mismatches can occur at any motif position. Weeder [65, 66] and SMILE [67] proposed using suffix trees (STs) [68] to efficiently explore the entire motif search space. They leverage the indexing capabilities of STs to perform approximate pattern matching, without restrictions on mismatching positions. This enabled achieving high accuracy in motif discovery, while reducing computational costs. To determine the statistical significance of motif candidates, SMILE and Weeder compare the motifs frequencies in 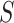 with those in a set of random genomic sequences or the promoters of the same organism, respectively (Supplementary File Section 1). However, these approaches can be computationally intensive and are not scalable on the large datasets generated by PBMs, HT-SELEX or ChIP assays [69]. Therefore, more efficient approaches specifically tailored to work on large datasets were proposed. MDscan [70] and Amadeus [71] use word enumeration to discover motif candidates in sequence datasets (Supplementary File Section 1). MDscan employs ChIP peaks shape to identify non-redundant patterns abundant in the most enriched sequences and uses a third-order Markov background model to assess motif statistical significance. Amadeus evaluates all
with those in a set of random genomic sequences or the promoters of the same organism, respectively (Supplementary File Section 1). However, these approaches can be computationally intensive and are not scalable on the large datasets generated by PBMs, HT-SELEX or ChIP assays [69]. Therefore, more efficient approaches specifically tailored to work on large datasets were proposed. MDscan [70] and Amadeus [71] use word enumeration to discover motif candidates in sequence datasets (Supplementary File Section 1). MDscan employs ChIP peaks shape to identify non-redundant patterns abundant in the most enriched sequences and uses a third-order Markov background model to assess motif statistical significance. Amadeus evaluates all 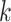 -mers in
-mers in 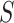 and groups similar patterns in list. Each list is grouped into motifs, statistically evaluated using a hypergeometric test. However, word enumeration can be still computationally demanding. To address this challenge, DREME [72] proposed using regular expressions to count approximate frequencies of motifs in
and groups similar patterns in list. Each list is grouped into motifs, statistically evaluated using a hypergeometric test. However, word enumeration can be still computationally demanding. To address this challenge, DREME [72] proposed using regular expressions to count approximate frequencies of motifs in 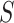 and
and 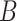 . To evaluate the motifs’ statistical significance, DREME employs Fisher’s exact test, comparing the number of sequences in
. To evaluate the motifs’ statistical significance, DREME employs Fisher’s exact test, comparing the number of sequences in 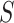 and
and 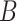 in which the motifs occur. However, regular expressions can be computationally expensive when analyzing large
in which the motifs occur. However, regular expressions can be computationally expensive when analyzing large 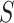 , and may detect false positives or miss motifs. Trawler, HOMER and STREME [73–75] reintroduced STs, proposing different optimizations to make the methods scalable on large datasets (Supplementary File Section 1). Trawler and HOMER optimized the statistical assessment step using
, and may detect false positives or miss motifs. Trawler, HOMER and STREME [73–75] reintroduced STs, proposing different optimizations to make the methods scalable on large datasets (Supplementary File Section 1). Trawler and HOMER optimized the statistical assessment step using 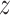 -scores derived from the normal approximation to the binomial distribution and the hypergeometric distribution, respectively. Instead of improving the statistical assessment, STREME reduces the motif search space by first identifying overrepresented seed words of different lengths on the ST. Then, STREME counts the number of approximate matches of the most significant words on the ST. By identifying seeds of different lengths, STREME discover motifs of different lengths in one single tree visit.
-scores derived from the normal approximation to the binomial distribution and the hypergeometric distribution, respectively. Instead of improving the statistical assessment, STREME reduces the motif search space by first identifying overrepresented seed words of different lengths on the ST. Then, STREME counts the number of approximate matches of the most significant words on the ST. By identifying seeds of different lengths, STREME discover motifs of different lengths in one single tree visit.
Alignment-based methods
Alignment-based motif discovery algorithms compute alignment profiles to describe motifs binding preferences (Figure 1), avoiding exhaustive 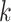 -mer enumeration. This approach involves constructing an alignment by selecting motif candidate sequences from the input dataset
-mer enumeration. This approach involves constructing an alignment by selecting motif candidate sequences from the input dataset 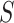 and evaluating the resulting profile using various measures, like nucleotide conservation, information content or profile statistical significance. Motif statistical significance is determined by computing the probability of obtaining the same alignment from either a background dataset
and evaluating the resulting profile using various measures, like nucleotide conservation, information content or profile statistical significance. Motif statistical significance is determined by computing the probability of obtaining the same alignment from either a background dataset 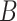 or random sequences. Alignment-based motif discovery algorithms typically assume that the motif length
or random sequences. Alignment-based motif discovery algorithms typically assume that the motif length 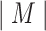 is known a priori. For alignment-based algorithms, motif discovery can be formalized as a combinatorial problem. Given
is known a priori. For alignment-based algorithms, motif discovery can be formalized as a combinatorial problem. Given 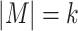 , the goal is to find the best alignment profile by combining
, the goal is to find the best alignment profile by combining 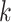 -mers from
-mers from 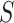 , according to a scoring criterion. The best alignments are then used to generate the corresponding PWMs. Most alignment-based algorithms assume that each sequence in
, according to a scoring criterion. The best alignments are then used to generate the corresponding PWMs. Most alignment-based algorithms assume that each sequence in 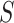 contains zero or one binding site. Therefore, there exist
contains zero or one binding site. Therefore, there exist 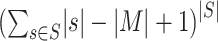 possible profiles, built by combining
possible profiles, built by combining  -mers in all possible ways. Since enumerating all possible solutions is computationally impractical even for small datasets, alignment-based algorithms employ heuristics, such as greedy [76], expectation–maximization (EM) [77], stochastic (e.g. Gibbs sampling) [78] or genetic algorithms [79] (Supplementary File Section 2). CONSENSUS [76] proposed a greedy approach to construct alignment profiles incrementally. It solves the problem initially on two sequences and progressively solves it by adding the remaining sequences
-mers in all possible ways. Since enumerating all possible solutions is computationally impractical even for small datasets, alignment-based algorithms employ heuristics, such as greedy [76], expectation–maximization (EM) [77], stochastic (e.g. Gibbs sampling) [78] or genetic algorithms [79] (Supplementary File Section 2). CONSENSUS [76] proposed a greedy approach to construct alignment profiles incrementally. It solves the problem initially on two sequences and progressively solves it by adding the remaining sequences  one by one. CONSENSUS stores the best partial alignments hoping to find the highest-scoring profiles. However, if motifs are not conserved, CONSENSUS may potentially discard the highest-scoring solutions. The MEME algorithm [77, 80, 81] proposed a different strategy based on EM. It iteratively refines an initial profile by substituting some
one by one. CONSENSUS stores the best partial alignments hoping to find the highest-scoring profiles. However, if motifs are not conserved, CONSENSUS may potentially discard the highest-scoring solutions. The MEME algorithm [77, 80, 81] proposed a different strategy based on EM. It iteratively refines an initial profile by substituting some  -mers in the profile, with others more likely to produce better solutions. MEME evaluates the fit of each
-mers in the profile, with others more likely to produce better solutions. MEME evaluates the fit of each 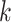 -mer in
-mer in 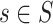 to the current profile, rather than a background model. MEME identifies motifs occurring more than once in each sequence and computes their statistical significance, and the method does not rely on TFBS conservation. However, the algorithm may converge prematurely to local maxima and convergence heavily depends on the algorithm starting conditions. In contrast to MEME, Gibbs sampling [82] employs a stochastic approach to add
to the current profile, rather than a background model. MEME identifies motifs occurring more than once in each sequence and computes their statistical significance, and the method does not rely on TFBS conservation. However, the algorithm may converge prematurely to local maxima and convergence heavily depends on the algorithm starting conditions. In contrast to MEME, Gibbs sampling [82] employs a stochastic approach to add 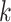 -mers to the alignment instead of a deterministic one based on the profile fit. Gibbs sampling replaces
-mers to the alignment instead of a deterministic one based on the profile fit. Gibbs sampling replaces 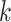 -mers in the profile with others selected with probability proportional to its likelihood score (Supplementary File Section 2). The algorithm’s stochastic nature reduces its likelihood to converge to local maxima, but it may require multiple runs to achieve reliable results. However, several methods using Gibbs sampling and its extensions have been proposed [83–89] (Supplementary File Section 2). Genetic algorithms are an alternative approach overcoming the limitations of EM and stochastic methods. GADEM [90] combined EM local search with genetic algorithms to refine profiles, avoid convergence to local maxima and overcome Gibbs sampling stochastic nature. However, due to their computational complexity, genetic algorithms are computationally demanding when analyzing thousands of sequences. Using alignment profiles, the solution space grows exponentially with the size of
-mers in the profile with others selected with probability proportional to its likelihood score (Supplementary File Section 2). The algorithm’s stochastic nature reduces its likelihood to converge to local maxima, but it may require multiple runs to achieve reliable results. However, several methods using Gibbs sampling and its extensions have been proposed [83–89] (Supplementary File Section 2). Genetic algorithms are an alternative approach overcoming the limitations of EM and stochastic methods. GADEM [90] combined EM local search with genetic algorithms to refine profiles, avoid convergence to local maxima and overcome Gibbs sampling stochastic nature. However, due to their computational complexity, genetic algorithms are computationally demanding when analyzing thousands of sequences. Using alignment profiles, the solution space grows exponentially with the size of 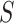 and even with employing heuristics analyzing thousands of sequences is computationally impractical [21]. Therefore, researchers focused on developing algorithms specifically tailored to analyze the large datasets produced by high-throughput assays (Supplementary File Section 2). MEME-ChIP [91] and STEME [92] improved the MEME algorithm to analyze ChIP datasets. While MEME-ChIP focuses the analysis on a random subset of sequences, STEME speeds up EM steps indexing the sequences in a suffix tree. However, using random subsets of
and even with employing heuristics analyzing thousands of sequences is computationally impractical [21]. Therefore, researchers focused on developing algorithms specifically tailored to analyze the large datasets produced by high-throughput assays (Supplementary File Section 2). MEME-ChIP [91] and STEME [92] improved the MEME algorithm to analyze ChIP datasets. While MEME-ChIP focuses the analysis on a random subset of sequences, STEME speeds up EM steps indexing the sequences in a suffix tree. However, using random subsets of 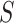 may cause missing critical motif instances and constructing ST from thousands of sequences may be computationally demanding. ChIPMunk [93] proposed a greedy profile optimization like EM developed to discover motifs in large ChIP-seq datasets, while accounting for ChIP peaks shape. XXmotif [94] and ProSampler [95] proposed methods combining enumerative motif discovery with iterative and stochastic profile refinement, respectively.
may cause missing critical motif instances and constructing ST from thousands of sequences may be computationally demanding. ChIPMunk [93] proposed a greedy profile optimization like EM developed to discover motifs in large ChIP-seq datasets, while accounting for ChIP peaks shape. XXmotif [94] and ProSampler [95] proposed methods combining enumerative motif discovery with iterative and stochastic profile refinement, respectively.
Probabilistic graphical model-based methods
The inclusion of dependencies between nucleotides in TFBS has been subject of debate [96–98]. Some studies have shown that dependencies exist between neighboring and non-neighboring nucleotides in TFBS [99, 100]. Enumerative and alignment-based algorithms represent motifs as PWMs, which do not account for dependencies between the binding site positions. PWMs can be extended to account for the frequency of di- or trinucleotides (high-order PWMs), like DWMs [23]. Dimont [101] and diChIPMunk [102] proposed extensions to alignment-based methods to discover and represent motifs as DWMs (Supplementary File Section 3). However, these methods capture dependencies only between neighboring nucleotides. Probabilistic graphical models (Figure 1) such as BNs, MMs or HMMs provide powerful frameworks for capturing dependencies between TFBS nucleotides. In [55], the authors proposed using BNs trained via EM to model TFBS. The proposed approach captures dependencies between neighboring and non-neighboring positions but assumes the same order of dependence throughout the entire motif. Similarly, in [103], the authors introduced VOBN models. VOBNs use BNs accounting for variable orders of dependencies between positions. However, training BNs is not computationally scalable when analyzing thousands of sequences and these models are prone to overfitting when trained on hundreds of sequences. MMs and HMMs provide more efficient and scalable frameworks than BNs to include dependencies between motif positions. Therefore, researchers focused on developing algorithms using these models to learn dependencies in large sequence datasets produced by NGS assays (Supplementary File Section 3). TFFMs [104] and Discrover [105] proposed HMM-based models learning the dinucleotide dependencies between neighboring motif positions in large sequence datasets. In addition, TFFMs learn the properties of the sequences flanking the TFBS. MMs can be extended to capture different orders of dependencies between neighboring nucleotides, as demonstrated in [106], where the authors proposed a method to discover CTCF [107] motifs using variable-order MMs. Similarly, MMs can also be extended to capture dependencies between non-neighboring nucleotides as proposed in Slim [108]. However, MMs and HMMs typically only capture low-order dependencies. BaMMotif [56, 109] proposed a motif discovery algorithm employing a Bayesian approach to efficiently train Markov models up to fifth-order dependencies on thousands of sequences.
SVM-based methods
SVMs [110] have been successfully applied to different problems in computational biology [111], including TFBS motif discovery (Figure 1). This is achieved by decomposing bound (foreground dataset 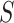 ) and unbound sequences (background dataset
) and unbound sequences (background dataset 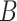 ) in
) in 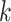 -mers and using their frequencies as features to train a sequence similarity kernel [111]. Generally, to each
-mers and using their frequencies as features to train a sequence similarity kernel [111]. Generally, to each 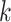 -mer is assigned a weight proportional to its contribution to the definition of the positive or negative training sets, or to its likelihood of being a motif candidate. While earlier methods [112–114] were designed for protein sequence homology, recent SVM-based algorithms have been developed to discover TFBS motifs. Furthermore, SVMs can efficiently analyze datasets of thousands of sequences. Kmer-SVM [115, 116] proposed a method to discover TFBS motifs in sequence datasets, using the spectrum kernel [112]. Kmer-SVM counts the exact matches for all contiguous
-mer is assigned a weight proportional to its contribution to the definition of the positive or negative training sets, or to its likelihood of being a motif candidate. While earlier methods [112–114] were designed for protein sequence homology, recent SVM-based algorithms have been developed to discover TFBS motifs. Furthermore, SVMs can efficiently analyze datasets of thousands of sequences. Kmer-SVM [115, 116] proposed a method to discover TFBS motifs in sequence datasets, using the spectrum kernel [112]. Kmer-SVM counts the exact matches for all contiguous 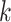 -mers in
-mers in 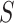 and
and 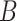 , building the
, building the 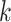 -mers feature space (Supplementary File Section 4). The mismatch and wildcard kernels [114, 117] were introduced to count
-mers feature space (Supplementary File Section 4). The mismatch and wildcard kernels [114, 117] were introduced to count 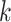 -mer frequencies while allowing a fixed number of mismatching positions for each
-mer frequencies while allowing a fixed number of mismatching positions for each 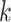 -mer. This approach was later extended to allow for less restrictive
-mer. This approach was later extended to allow for less restrictive 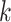 -mer frequency estimation, offering flexibility in the motif structure without affecting scalability on large datasets. Agius and coworkers [118] extended the concept of mismatch kernels by developing the di-mismatch kernel. The di-mismatch kernel is a first-order Markov mismatch kernel based on the dinucleotide alphabet, which handles sequence variability and accounts for dependencies between neighboring nucleotides (Supplementary File Section 4). To maintain scalability on large datasets small
-mer frequency estimation, offering flexibility in the motif structure without affecting scalability on large datasets. Agius and coworkers [118] extended the concept of mismatch kernels by developing the di-mismatch kernel. The di-mismatch kernel is a first-order Markov mismatch kernel based on the dinucleotide alphabet, which handles sequence variability and accounts for dependencies between neighboring nucleotides (Supplementary File Section 4). To maintain scalability on large datasets small 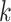 (
(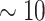 ) is used, discovering short motifs. However, TFBS lengths range between
) is used, discovering short motifs. However, TFBS lengths range between 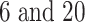 bp, making it challenging to fully characterize longer motifs with short
bp, making it challenging to fully characterize longer motifs with short 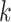 -mers. In addition, increasing
-mers. In addition, increasing 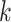 often results in sparse feature vectors overfitting the training dataset. Gapped
often results in sparse feature vectors overfitting the training dataset. Gapped 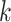 -mers [58] proposed to represent longer motifs as
-mers [58] proposed to represent longer motifs as 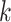 -mers with gaps in non-informative or degenerate TFBS positions, accounting for motif variability in sequence and length. Gkm-SVM [119, 120] extends kmer-SVM to train SVM kernels employing gapped
-mers with gaps in non-informative or degenerate TFBS positions, accounting for motif variability in sequence and length. Gkm-SVM [119, 120] extends kmer-SVM to train SVM kernels employing gapped 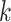 -mers as features. The algorithm considers larger
-mers as features. The algorithm considers larger 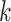 preventing model overfitting and reducing the method’s dependency on parameters’ choice. LS-GKM [121] optimizes the algorithm for scalable SVM training with gapped
preventing model overfitting and reducing the method’s dependency on parameters’ choice. LS-GKM [121] optimizes the algorithm for scalable SVM training with gapped 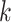 -mers on large-scale sequence datasets. LS-GKM also provides other kernels for SVM training (Supplementary File Section 4).
-mers on large-scale sequence datasets. LS-GKM also provides other kernels for SVM training (Supplementary File Section 4).
DNN-based methods
DNNs have become increasingly popular in computational biology [122–130] due to their ability to learn complex patterns [131] from large omics datasets [132]. Convolutional neural networks (CNNs) [133], originally developed for image classification [133–135], have been successfully applied to analyze in vivo TF–DNA interactions [136–139] (Figure 1). CNNs apply non-linear transformation to input data, learning and representing complex patterns in a high-dimensional space [140]. This simplifies classification tasks and enables accurate prediction of TFBS in genomic sequences. CNNs represent genomic sequences as 1D or 2D images with four associated channels (A, C, G, T) [139]. Therefore, classifying TFBS in genomic sequences becomes a two-class image classification problem. Typically, CNN architectures designed for motif discovery and classification consist of one or more sets of four layers: the convolutional layer, the max-pooling layer, the fully connected NN layer and the output layer [139] (Supplementary File Section 5). Deepbind [136] and Basset [138] proposed two CNN architectures to discover motifs in different datasets, such as ChIP-seq, HT-SELEX, PBM and DNase-seq (Supplementary File Section 5). The discovered motifs in DeepBind and Basset are visualized as PWMs. The PWMs are computed by aligning and grouping the sequences that activate the convolutional layer. While DeepBind and Basset have demonstrated promising results in predicting TFBS, their performance may be limited by the quality of training data and the significant computational resources and time required for model training. These limitations have led to the development of novel methods, such as BPNet [62], which address some of these issues by incorporating additional features in the model and using more efficient training processes. BPNet proposed a dilated CNN architecture, allowing the model to learn and integrate diverse complex features without sacrificing the spatial and base resolution of the input data (Supplementary File Section 5). However, TF–DNA interactions involve not only the direct binding between TF and DNA but also the interactions between multiple binding subregions (long-term interactions) and the nucleotides with high-order structures of TFs (short-term interactions). Long short-term memory networks (LSTMs) [141] and bi-directional LSTMs (BLSTMs) can efficiently capture long-term and short-term dependencies of sequential signals. LSTMs and BLSTMs are well suited for modeling TF–DNA interactions as genomic sequences can be viewed as sequential signals with long-term and short-term dependencies (Supplementary File Section 5). DeeperBind [142] introduced a hybrid CNN–LSTM architecture removing the pooling layer to maintain the positional information of potential motif instances. Similarly, DanQ [143] proposed a hybrid CNN–BLSTM architecture to capture the positional dynamics of genomic sequences for TFBS motif discovery. The BLSTM replaces the fully connected NN. Factornet [144] extended the DanQ approach by incorporating additional features in the model and using a Siamese BLSTM architecture to improve model training.
TF DATABASES
With the recent advancement in experimental technologies, a vast amount of TF-related data have been generated and stored in databases (Table 2). The ENCODE project [145] provides multiple data on functional elements in the human genome collected across different tissues and cell types. ENCODE stores TF-related genomic data such as ChIP-seq targeting several TFs and DNase-seq. Similarly, Cistrome [146] and GTRD [147] provide TF-related genomic data from different organisms and across different species, cell types and tissues, respectively. Furthermore, GTRD stores large collections of curated ChIP-seq, ChIP-exo and ChIP-nexus datasets. HOCOMOCO [148, 149] and JASPAR [150, 151] provide large collections of curated, experimentally derived and computationally predicted TFBS motifs for several TFs from different species. They store PWMs and DWMs obtained by analyzing ChIP-seq and SELEX datasets. In addition, HOCOMOCO models were generated integrating sequence datasets with evolutionary conservation and DNA shape. Similarly, Cis-BP [152] stores experimentally derived and computationally predicted PWMs, obtained integrating multiple sources, including published literature, other databases and experimental datasets. TRANSFAC [153, 154] collects experimentally validated and manually curated PWMs for various TFs from different eukaryotic organisms, and includes data on TF-associated proteins, DNA binding domains and, regulatory elements. FactorBook [155] provides computationally predicted PWMs generated analyzing ENCODE data and includes TF expression data across tissues and cell types. Unibind [156] collects experimentally validated and curated PWMs from different organisms, providing information on structural properties and conformation of TF–DNA complexes and their genomic binding locations across different cell types and tissues. UniPROBE [157] stores curated PWMs for several eukaryotic TFs, generated analyzing PBM datasets. HTRIdb [158] stores data on TF–target genes interactions in human, collected from published literature and other databases, in different cell types, experimental methods and disease state, also providing functional annotations for the target genes. TFcancer [159] collects TF–gene interactions across 33 cancer types, providing tools to identify TF expression alterations and their roles in biological processes and signaling pathways in cancer.
Table 2.
Transcription factor-related databases
| Type | Name | Reference | Data type | Model organisms | TFs |
|---|---|---|---|---|---|
| Sequence database | ENCODE | [145] | ChIP-seq DNase-seq ATAC-seq |
Caenorhabditis elegans Drosophila melanogaster Homo sapiens Mus musculus |
>1500 |
| Cistrome | [146] | ChIP-seq, DNase-seq |
H. sapiens M. musculus |
1773 (ChIP-seq) | |
| GTRD | [147] | ChIP-seq ChIP-exo, ChIP-nexus DNase-seq |
Arabidopsis thaliana C. elegans Danio rerio D. melanogaster H. sapiens M. musculus Rattus norvegicus Saccharomyces cerevisiae Schizosaccharomyces pombe |
3988 (ChIP-seq) 1708 (ChIP-exo + ChIP-nexus) |
|
| Motif models database | HOCOMOCO | [148, 149] | PWMs DWMs |
H. sapiens M. musculus |
680 (human) 453 (mouse) |
| JASPAR | [150, 151] | PWMs DWMs |
53 species | >1500 | |
| Cis-BP | [152] | PWMs |
A. thaliana C. elegans D. rerio D. melanogaster H. sapiens M. musculus Neurospora crassa R. norvegicus S. cerevisiae Xenopus tropicalis |
>5000 | |
| TRANSFAC | [153, 154] | PWMs | >300 species | >10 000 | |
| FactorBook | [155] | PWMs |
H. sapiens M. musculus |
881 (human) 49 (mouse) |
|
| Unibind | [156] | PWMs |
A. thaliana C. elegans D. rerio D. melanogaster H. sapiens M. musculus R. norvegicus S. cerevisiae S. pombe |
841 | |
| UniPROBE | [157] | PWMs |
C. elegans Cryptosporidium parvum H. sapiens M. musculus Plasmodium falciparum S. cerevisiae Vibrio harveyi |
726 | |
| TF–target gene interaction database | HTRIdb | [158] | TF–gene interaction networks | H. sapiens | 284 |
| TF–disease association database | TFcancer | [159] | TF–cancer associations | H. sapiens | 364 |
The table presents a summary of the TF-related databases discussed in Section 4. For each database, the table reports the database main purpose (Type), the available type of data (Data type), the model organisms for which data are provided (Model organisms), the number of TFs (TFs) and the database website (Website).
DOWNSTREAM ANALYSES
The discovered motifs can be employed in several downstream analyses: motif comparison, motif scanning, motif enrichment analysis and assessing genetic variants effects on TF–DNA binding affinity. Motif comparison measures the similarity between the discovered motifs and annotated TFBS. Motif comparison allows for linking known TFs to the newly discovered motifs [160] and inferring the relationship between the input sequences and function of the annotated TF [152]. For this task, several tools have been developed such as Tomtom, STAMP, MACRO-APE or MoSBAT [160–163]. These tools search annotated database for motifs matching the input consensus sequence or inferred motif matrix. Moreover, motif comparison tools have been developed to interpret and annotate the potential motifs encoded in the convolutional filters of a CNN model. Motif scanning scans sets of genomic regions searching for potential occurrences of the input motif. The goal is to recover sets of potential binding locations for the investigated factor. Given a motif model (e.g. a PWM) and a set of sequences, motif scanning algorithms assign a score to each sequence using the input model. A common challenge is to determine a reliable cutoff on the scores assigned to the sequences to discriminate between true and false binding events [57]. Several motif scanning tools are currently available such as MOODS, FIMO or PWMscan [164–166]. The HOMER suite [74] also provides a motif scanning functionality. Recently, MOODS was extended to search instances of motifs modeled as high-order PWMs [51]. GRAFIMO [167] extended classical motif scanning to panels of thousands of genomes encoded in genome graphs [168], considering individual genetic variants and haplotypes while searching for potential motif occurrences. Motif enrichment analysis (MEA) searches for over- and underrepresented motifs in gene regulatory regions. Analyzing the TFBS enrichment in regulatory regions governing sets of genes, researchers can link the investigated TFs to their function within the cell environment. MEA consists of two steps: (i) scanning regulatory regions for motif occurrences and (ii) statistical testing of motif enrichment. TFs whose motifs are significantly overrepresented (enriched) in the scanned regulatory regions are marked as transcriptional regulators for the target gene set. There are many MEA tools available to the community, such as Clover, Pscan, AME or oPOSSUM-3 [169–172]. HOMER [74] provides a functionality to perform MEA. Haystack [173] proposed an integrated MEA strategy, investigating motif enrichment in cell-type-specific regions and incorporating gene expression data to assess the transcriptional activity of the studied factors and their impact on the regulated genes.
Genetic variants have been shown to impact TF–DNA binding events [174–176], including variants associated with common diseases in regulatory elements [177], potentially altering the transcriptional state of the cell [178]. As a result, there has been a growing interest in developing tools to predict the impact of variants on TFBS (Table 3). TRAP [179] and CATO [10] use PWMs to predict the impact of variants on TFBS by comparing the binding affinity scores of reference and alternative sequences. TRAP repeats the procedure on a collection of TFBS, reporting the motif showing the largest score change. CATO, instead, provides a ranked list of disrupted motifs, obtained using a logistic model trained with the information content difference between reference and alternative sequences, TF occupancy and phylogenetic conservation. However, these methods are not scalable when analyzing thousands of single nucleotide polymorphisms (SNPs). atSNP [180] proposed a scalable strategy to assess the impact of thousands of SNPs on TFBS by computing the statistical significance of the computed affinity scores, in addition to the difference between the reference and alternative sequence binding scores using PWMs. GRAFIMO [167] extended the scalability to millions of SNPs by scanning collections of PWMs on genome graphs, while accounting for haplotypes. MotifRaptor [181] integrates chromatin accessibility, gene expression and GWAS summary statistics, to predict and annotate functional effects for large non-coding variant datasets, using PWMs. DeltaSVM [182] and GkmExplain [183] use SVM-based motif models to assess variant impact. DeltaSVM scans DNA positions overlapping each SNP in the input dataset using a pretrained list of 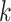 -mers with associated weights and computing the difference between the reference and alternative sequence scores. However, it assesses the impact of individual variants, not accounting for relationships between variants. GkmExplain overcomes this limitation by considering the impact of variants not in individual positions, but on sequence features, like entire
-mers with associated weights and computing the difference between the reference and alternative sequence scores. However, it assesses the impact of individual variants, not accounting for relationships between variants. GkmExplain overcomes this limitation by considering the impact of variants not in individual positions, but on sequence features, like entire  -mers. DeepBind [136] and DeepSEA [137] employ DNN-based models to predict variant impact on TFBS. DeepBind uses mutation maps to assess variant effect on binding affinities by considering the importance of each motif position within the model. DeepSEA uses in silico saturated mutagenesis to predict the impact of individual variants on the whole sequence context and features like TFBS. Similarly, Basset [138] employs in silico saturated mutagenesis by learning critical nucleotides governing chromatin accessibility. Basset assigns importance scores to each position in the input sequences and attempts to map the variants’ impact to the TFBS in the input sequences. Basenji [127] extends Basset’s workflow by providing functional annotations to SNPs affecting sequence features like TFBS and returning potential changes in gene expression patterns. However, Basenji is limited to predict SNP effects on distal regulatory elements within a 20 kb range. Enformer [184] overcomes this limitation by employing transformer architectures to extend the range up to 200 kb, providing more comprehensive and accurate functional effects of variants on sequence elements and gene expression.
-mers. DeepBind [136] and DeepSEA [137] employ DNN-based models to predict variant impact on TFBS. DeepBind uses mutation maps to assess variant effect on binding affinities by considering the importance of each motif position within the model. DeepSEA uses in silico saturated mutagenesis to predict the impact of individual variants on the whole sequence context and features like TFBS. Similarly, Basset [138] employs in silico saturated mutagenesis by learning critical nucleotides governing chromatin accessibility. Basset assigns importance scores to each position in the input sequences and attempts to map the variants’ impact to the TFBS in the input sequences. Basenji [127] extends Basset’s workflow by providing functional annotations to SNPs affecting sequence features like TFBS and returning potential changes in gene expression patterns. However, Basenji is limited to predict SNP effects on distal regulatory elements within a 20 kb range. Enformer [184] overcomes this limitation by employing transformer architectures to extend the range up to 200 kb, providing more comprehensive and accurate functional effects of variants on sequence elements and gene expression.
Table 3.
Software to assess genetic variants impact on transcription factor binding sites
| Motif model | Software | Reference | Original input data type | Output | Year | Availability |
|---|---|---|---|---|---|---|
| PWM | TRAP | [179] | ChIP-seq | Allele-specific score | 2011 | http://trap.molgen.mpg.de/cgi-bin/home.cgi |
| CATO score | [10] | DHS sites | Ranked list of TFBS affected by SNPs | 2015 | Available under request to the authors | |
| atSNP | [180] | Sequences overlapping input SNPs | Allele-specific score | 2015 | https://github.com/keleslab/atSNP | |
| GRAFIMO | [167] | ChIP-seq | Allele-specific score | 2021 |
https://github.com/pinellolab/GRAFIMO https://github.com/InfOmics/GRAFIMO |
|
| MotifRaptor | [181] | DNase-seq | Allele-specific score | 2021 | https://github.com/pinellolab/MotifRaptor | |
| SVM based | DeltaSVM | [182] | DNase-seq | Allele-specific score | 2015 | https://www.beerlab.org/deltasvm/ |
| GkmExplain | [183] | DNase-seq | SNP impact on whole TFBS | 2019 | https://github.com/kundajelab/gkmexplain | |
| DNN based | DeepBind | [136] | ChIP-seq, HT-SELEX | Single SNP impact | 2015 | http://tools.genes.toronto.edu/deepbind |
| DeepSEA | [137] | ATAC-seq, DNase-seq | Single SNP impact | 2015 | http://deepsea.princeton.edu | |
| Basset | [138] | ChIP-seq, DNase-seq | Single SNP impact | 2016 | https://github.com/davek44/Basset | |
| Basenji | [127] | ChIP-seq, DNase-seq | Single SNP functional impact | 2018 | https://github.com/calico/basenji | |
| Enformer | [184] | DNA sequences | SNP functional impact | 2021 | https://github.com/deepmind/deepmind-research/tree/master/enformer |
The table provides an overview of the tools for predicting the impact of variants on TFBS as discussed in Section 5. For each tool, the table report the employed TFBS model (Motif model), the original data type used to test each method in their original publication (Original input data type), the output type (Output), the year (Year), the associated publication (Reference) and their code or website (Availability).
DISCUSSION
Discovering TFBS motifs in DNA sequences has been extensively studied over the past few decades. This paper reviewed various algorithms and computational models for discovering and representing motifs. However, there are still several open issues and potential research directions that need to be addressed in this field.
The choice between simple and complex motif discovery algorithms is often debated. While classical enumerative and alignment-based methods have been shown to have comparable performance in scalability and accuracy to complex methods [185], they also offer user-friendly interfaces and generally do not require any computational expertise. In addition, they can be applied to any sequence dataset and do not require any additional information beyond the sequences themselves. Moreover, these tools have a strong user community and continue to be widely used in the field. MEME [77, 80, 81], HOMER [74] and the newer STREME [75] are particularly popular and continue to be well maintained by their developers, ensuring continued usability and relevance. In contrast, probabilistic graphical model-based algorithms often struggle with scalability when analyzing thousands of sequences due to the complexities involved in model training involving dependencies. SVM-based methods have demonstrated high scalability and accuracy in discovering TFBS motifs across various sequence datasets, as well as predicting PBM binding affinities. One major advantage of SVM-based algorithms is that they can learn features of the entire sequence context, contributing to their success in motif discovery. However, the performance of SVM-based methods heavily depends on the quality of the background dataset, which needs to be carefully designed based on different sequence characteristics, such as GC and repeat content. DNN-based motif discovery algorithms have been shown to be highly accurate compared to other methods. However, their complexity often requires expertise in the field and fine parameter tuning. DNN-based methods also hold the potential to integrate diverse genomic data sources for discovering TFBS (as discussed in Section 3.5). Although DNN-based methods are scalable in terms of dataset size, they often require significant computational resources and dedicated hardware components (e.g., GPUs) to train effective motif models. Nevertheless, DNN-based methods are rapidly gaining popularity within the community. The debate about which is the best method is ongoing. While several papers have benchmarked the performance of motif discovery algorithms on various datasets [69, 186], these benchmarks often focus on a small number of similar methods or homogenous datasets. A comprehensive benchmark that considers a wide range of datasets in terms of size and composition, as well as methods from different algorithm classes, is needed. Such a benchmark would offer crucial insights into which methods perform best with specific input data.
The need and effectiveness of motif models capturing dependencies between positions within a TFBS has been extensively discussed during the last decades [185, 187–189]. Although probabilistic models are expected to perform better, many studies showed that simpler models like PWMs perform as well as these models on both in vitro PBM and in vivo ChIP-seq data for most TFs [98, 185]. In [185], the authors suggested that these results could be explained by the degeneracy observed in eukaryotic TFBS. In fact, many TFs bind sequences showing variations with respect to the motif consensus, even though with less affinity. Since PWMs accommodate variations to the motif consensus, they can capture a wider range of target sites, including those weakly bound. However, this advantage comes at the cost of an increased susceptibility to noise, potentially recovering several false positives. By encoding dependencies between TFBS positions, probabilistic graphical models are expected to provide more robust models. However, since these models learn several parameters, they can easily overfit the training data if not trained on appropriate datasets. SVM-based motif models have been shown to perform generally better than PWMs when predicting potential TFBS [119]. However, these models are often reduced to PWMs for visualization and interpretation purposes, losing most of the learned information. Recent studies observed that DNN-based models better capture the sequence specificities underlying TF–DNA interactions, returning better predictions with respect to other models [190]. However, to visualize and interpret the discovered motifs, the DNN models are generally reduced to PWMs computed with the sequences activating the convolutional kernels. Therefore, complex motif models provide powerful frameworks sacrificing interpretability, while simpler models are more susceptible to noise but easily interpretable. The trade-off between model accuracy and interpretability is still an open challenge in the context of motif discovery.
Recently, many consortia like the ENCODE Project [145] and Roadmap Epigenomics Project [191] collected huge amounts of TF-related data, like ChIP-seq experiments performed on dozens of factors in different organisms, tissues and cell types. These datasets are often used as ground truth to evaluate the performance of motif discovery algorithms. However, ChIP-seq is susceptible to different sources of noise that could bias performance evaluation. The growth of HT-SELEX or ChIP-exo datasets available in public databases would address this limitation since they provide cleaner data (Section 2).
It is known that regulatory elements in multicellular organisms act in a cell-type-specific manner [192, 193]. TFBS show cell-type-specific patterns and configurations [194]. Moreover, cell-type-specific and individual-specific genetic variants can impact TFBS [195, 196]. However, capturing and modeling cell-type-specific TF–DNA interactions remain a key problem. Although NGS-based experimental assays like ChIP-seq enabled genome-wide TFBS analyses in vivo, they capture the binding landscape in a single cell type and rely on the availability of antibodies targeting the investigated factor. Since TFs are major drivers of chromatin accessibility [197], TFBS can be discovered on ATAC-seq or DNase-seq data by running motif discovery algorithms on the reported open chromatin sequences. Importantly, ATAC-seq data are easier to obtain than ChIP-seq even when targeting new cell types and unknown factors. Recently, Virtual ChIP-seq [198] proposed a method to predict TF binding in new cell types using only chromatin accessibility and transcriptomic data.
Often the motifs discovered in a certain cell type poorly generalize to other cell types [199] and the motifs discovered in DNase-seq or ATAC-seq datasets may not be sufficiently well calibrated to provide reliable predictions on the impact of genetic variants on TFBS [200]. DNN-based motif discovery algorithms potentially integrate data recovered from different cell types. However, the development of motif discovery algorithms and models explicitly integrating and representing different cell-type-specific motifs remains an open challenge. The recent introduction of experimental assays analyzing epigenetic marks at single-cell resolution provides new and powerful data to improve our knowledge on the mechanisms regulating individual cell environments [201].
Single-cell ChIP-seq (scChIP-seq) extends traditional ChIP assays to investigate the binding landscapes of DNA-binding proteins like TFs at single-cell resolution [202, 203]. The growing availability of such data would help to better learn cell-type-specific TF–DNA binding dynamics and epigenetic mechanisms. However, to our knowledge, only a few studies successfully performed scChIP-seq due to its challenging execution [201]. Single-cell ATAC-seq (scATAC-seq) identifies open chromatin regions at individual single-cell resolution [204]. scATAC-seq is not limited by technical constraints and the number of available datasets is rapidly growing. Interestingly, some tools performing different motif analyses on scATAC-seq, such as motif discovery [205] and MEA [206, 207], are already available. We expect that in a few years the availability of single-cell epigenetic data will increase, enabling the development of more motif discovery algorithms and models designed to analyze and describe TFBS data at single-cell resolution. This would provide more reliable datasets to train motif models and assess their predictive performances.
Key Points
The development of algorithms to discover transcription factor binding sites is one of the most studied problems in computational biology.
The introduction of in vitro and in vivo high-throughput experimental assays revolutionized transcription factor binding site discovery, returning large datasets of potential target sites that provide unprecedented opportunities to study and characterize the binding landscapes of transcription factors.
Several different algorithms to discover transcription factor binding site motifs along with different computational models to represent the discovered binding sites have been proposed over the last two decades; however, each proposed method and model show advantages and drawbacks.
The motif models derived by motif discovery algorithms are employed in several different downstream analysis, like motif comparison, motif scanning, motif enrichment and assessing the effects of genetic variants on transcription factor binding site affinity.
Supplementary Material
Acknowledgements
We would like to thank Vincenzo Bonnici, University of Parma, Parma, Italy, and Zain Patel, Molecular Pathology Unit Massachusetts General Hospital, Charlestown, MA, USA, for the feedbacks and fruitful discussions that helped improve this paper.
Manuel Tognon is a PhD student at the University of Verona. His research interests cover the development of novel computational methods to investigate the impact of genetic variants on epigenetic elements and CRISPR off-targets.
Rosalba Giugno is an associate professor at the University of Verona. Her laboratory, InfOmics, is devoted to developing computational methods for multi-omics data analysis, personal genomes analysis and network analysis.
Luca Pinello is an associate professor at Massachusetts General Hospital and Harvard Medical School. His laboratory is focused on developing computational methods to understand and characterize gene regulation and development.
Contributor Information
Manuel Tognon, Computer Science Department, University of Verona, Verona, Italy; Molecular Pathology Unit, Center for Computational and Integrative Biology and Center for Cancer Research, Massachusetts General Hospital, Charlestown, Massachusetts, United States of America; Broad Institute of MIT and Harvard, Cambridge, Massachusetts, United States of America.
Rosalba Giugno, Computer Science Department, University of Verona, Verona, Italy.
Luca Pinello, Molecular Pathology Unit, Center for Computational and Integrative Biology and Center for Cancer Research, Massachusetts General Hospital, Charlestown, Massachusetts, United States of America; Broad Institute of MIT and Harvard, Cambridge, Massachusetts, United States of America; Department of Pathology, Harvard Medical School, Boston, Massachusetts, United States of America.
Funding
R.G. is partially supported by the European Union’s Horizon 2020 research and innovation programme under grant agreement 814978 and JPcofuND2 Personalized Medicine for Neurodegenerative Diseases project JPND2019-466-037. L.P. is partially supported by the National Human Genome Research Institute (NHGRI) Genomic Innovator Award (R35HG010717).
Data Availability
No new data were generated or analysed in support of this research.
References
- 1. Lambert SA, Jolma A, Campitelli LF, et al. The human transcription factors. Cell 2018;172:650–65. [DOI] [PubMed] [Google Scholar]
- 2. Reimold AM, Iwakoshi NN, Manis J, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001;412:300–7. [DOI] [PubMed] [Google Scholar]
- 3. Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell 2013;152:1237–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stewart AJ, Hannenhalli S, Plotkin JB. Why transcription factor binding sites are ten nucleotides long. Genetics 2012;192:973–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Whitfield TW, Wang J, Collins PJ, et al. Functional analysis of transcription factor binding sites in human promoters. Genome Biol 2012;13:R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gotea V, Visel A, Westlund JM, et al. Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res 2010;20:565–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lemon B. Orchestrated response: a symphony of transcription factors for gene control. Genes Dev 2000;14:2551–69. [DOI] [PubMed] [Google Scholar]
- 8. Nolis IK, McKay DJ, Mantouvalou E, et al. Transcription factors mediate long-range enhancer-promoter interactions. Proc Natl Acad Sci 2009;106:20222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mendenhall EM, Williamson KE, Reyon D, et al. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol 2013;31:1133–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maurano MT, Haugen E, Sandstrom R, et al. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat Genet 2015;47:1393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jolma A, Taipale J. Methods for analysis of transcription factor DNA-binding specificity in vitro. Subcell Biochem 2011;52:155–73. [DOI] [PubMed] [Google Scholar]
- 12. Garner MM, Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res 1981;9:3047–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hampshire AJ, Rusling DA, Broughton-Head VJ, et al. Footprinting: a method for determining the sequence selectivity, affinity and kinetics of DNA-binding ligands. Methods 2007;42:128–40. [DOI] [PubMed] [Google Scholar]
- 14. Berger MF, Philippakis AA, Qureshi AM, et al. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat Biotechnol 2006;24:1429–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jolma A, Kivioja T, Toivonen J, et al. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res 2010;20:861–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Collas P, Dahl JA. Chop it, ChIP it, check it: the current status of chromatin immunoprecipitation. Front Biosci 2008;13:929–43. [DOI] [PubMed] [Google Scholar]
- 17. Pavesi G. In silico representation and discovery of transcription factor binding sites. Brief Bioinform 2004;5:217–36. [DOI] [PubMed] [Google Scholar]
- 18. Tompa M, Li N, Bailey TL, et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol 2005;23:137–44. [DOI] [PubMed] [Google Scholar]
- 19. D’haeseleer P. How does DNA sequence motif discovery work? Nat Biotechnol 2006;24:959–61. [DOI] [PubMed] [Google Scholar]
- 20. Das MK, Dai H-K. A survey of DNA motif finding algorithms. BMC Bioinformatics 2007;8(Suppl 7):S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zambelli F, Pesole G, Pavesi G. Motif discovery and transcription factor binding sites before and after the next-generation sequencing era. Brief Bioinform 2013;14:225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stormo GD. DNA binding sites: representation and discovery. Bioinformatics 2000;16:16–23. [DOI] [PubMed] [Google Scholar]
- 23. Siddharthan R. Dinucleotide weight matrices for predicting transcription factor binding sites: generalizing the position weight matrix. PloS One 2010;5:e9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gorkin DU, Lee D, Reed X, et al. Integration of ChIP-seq and machine learning reveals enhancers and a predictive regulatory sequence vocabulary in melanocytes. Genome Res 2012;22:2290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He Y, Shen Z, Zhang Q, et al. A survey on deep learning in DNA/RNA motif mining. Brief Bioinform 2021;22:bbaa229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Galas DJ, Schmitz A. DNAase footprinting a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res 1978;5:3157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zia A, Moses AM. Towards a theoretical understanding of false positives in DNA motif finding. BMC Bioinformatics 2012;13:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stormo GD, Zhao Y. Determining the specificity of protein–DNA interactions. Nat Rev Genet 2010;11:751–60. [DOI] [PubMed] [Google Scholar]
- 29. Berger MF, Bulyk ML. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat Protoc 2009;4:393–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jolma A, Yan J, Whitington T, et al. DNA-binding specificities of human transcription factors. Cell 2013;152:327–39. [DOI] [PubMed] [Google Scholar]
- 31. Pillai S, Chellappan SP. ChIP on chip and ChIP-Seq assays: genome-wide analysis of transcription factor binding and histone modifications. Methods Mol Biol 2015;1288:447–72. [DOI] [PubMed] [Google Scholar]
- 32. Johnson DS, Mortazavi A, Myers RM, et al. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007;316:1497–502. [DOI] [PubMed] [Google Scholar]
- 33. Mardis ER. ChIP-seq: welcome to the new frontier. Nat Methods 2007;4:613–4. [DOI] [PubMed] [Google Scholar]
- 34. Thomas R, Thomas S, Holloway AK, et al. Features that define the best ChIP-seq peak calling algorithms. Brief Bioinform 2017;18:441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guo Y, Mahony S, Gifford DK. High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLoS Comput Biol 2012;8:e1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat Methods 2009;6:S22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Worsley Hunt R, Wasserman WW. Non-targeted transcription factors motif are a systemic component of ChIP-seq datasets. Genome Biol 2014;15:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pickrell JK, Gaffney DJ, Gilad Y, et al. False positive peaks in ChIP-seq and other sequencing-based functional assays caused by unannotated high copy number regions. Bioinformatics 2011;27:2144–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rhee HS, Pugh BF. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 2011;147:1408–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buenrostro JD, Giresi PG, Zaba LC, et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 2013;10:1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. John S, Sabo PJ, Thurman RE, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet 2011;43(3):264–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McCue LA, Thompson W, Carmack CS, et al. Phylogenetic footprinting of transcription factor binding sites in probacterial genomes. Nucleic Acids Res 2001;29(3):774–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Blanchette M, Tompa M. Discovery of regulatory elements by a computational method for phulogenetic footprinting. Genome Res 2002;12(5):739–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Balazadeh S, Kwasniewski M, Caldana C, et al. ORS1, an H2O2-responsive NAC transcription factor, controls senescence in Arabidopsis thaliana. Mol Plant 2011;4(2):346–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu F, Park MR, Kitazumi A, et al. Cis-regulatory signatures of orthologous stress-associated bZIP transcription factors from rice, sorghum and Arabidopsis based on phylogenetic footprints. BMC Genomics 2012;13(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Katara P, Grover A, Sharma V. Phylogenetic footprinting: a boost for microbial regulatory genomics. Protoplasma 2012;249(4):901–7. [DOI] [PubMed] [Google Scholar]
- 48. Glenwinkel L, Wu D, Minevich G, et al. TargetOrtho: a phylogenetic footprinting tool to identify transcription factor targets. Genetics 2014;197(1):61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Day WH, McMorris FR. Critical comparison of consensus methods for molecular sequences. Nucleic Acids Res 1992;20:1093–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stormo GD. Modeling the specificity of protein-DNA interactions. Quant Biol 2013;1:115–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Korhonen JH, Palin K, Taipale J, et al. Fast motif matching revisited: high-order PWMs. SNPs and indels Bioinformatics 2017;33:514–21. [DOI] [PubMed] [Google Scholar]
- 52. Li S, Ovcharenko I. Human enhancers are fragile and prone to deactivating mutations. Mol Biol Evol 2015;32:2161–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stormo GD. Information content and free energy in DNA–protein interactions. J Theor Biol 1998;195:135–7. [DOI] [PubMed] [Google Scholar]
- 54. Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 1990;18:6097–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barash Y, Elidan G, Friedman N, et al. Modeling dependencies in protein-DNA binding sites. Proceedings of the seventh annual international conference on Research in computational molecular biology. 2003;1:28–37. [Google Scholar]
- 56. Siebert M, Söding J. Bayesian Markov models consistently outperform PWMs at predicting motifs in nucleotide sequences. Nucleic Acids Res 2016;44:6055–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Boeva V. Analysis of genomic sequence motifs for deciphering transcription factor binding and transcriptional regulation in eukaryotic cells. Front Genet 2016;7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ghandi M, Mohammad-Noori M, Beer MA. Robust k-mer frequency estimation using gapped k-mers. J Math Biol 2014;69:469–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Park S, Koh Y, Jeon H, et al. Enhancing the interpretability of transcription factor binding site prediction using attention mechanism. Sci Rep 2020;10:13413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koo PK, Ploenzke M. Deep learning for inferring transcription factor binding sites. Curr Opin Syst Biol 2020;19:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shrikumar A, Greenside P, Kundaje A. Learning important features through propagating activation differences. International conference on machine learning 2017;70:3145–53. [Google Scholar]
- 62. Avsec Ž, Weilert M, Shrikumar A, et al. Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat Genet 2021;53:354–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li M, Ma B, Wang L. Finding similar regions in many strings. Proceedings of the thirty-first annual ACM symposium on Theory of computing. 1999;1:473–82. [Google Scholar]
- 64. Califano A. SPLASH: structural pattern localization analysis by sequential histograms. Bioinformatics 2000;16:341–57. [DOI] [PubMed] [Google Scholar]
- 65. Pavesi G, Mauri G, Pesole G. An algorithm for finding signals of unknown length in DNA sequences. Bioinformatics 2001;17(Suppl 1):S207–14. [DOI] [PubMed] [Google Scholar]
- 66. Pavesi G, Mereghetti P, Mauri G, et al. Weeder Web: discovery of transcription factor binding sites in a set of sequences from co-regulated genes. Nucleic Acids Res 2004;32:W199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Marsan L, Sagot MF. Algorithms for extracting structured motifs using a suffix tree with an application to promoter and regulatory site consensus identification. J Comput Biol 2000;7:345–62. [DOI] [PubMed] [Google Scholar]
- 68. Weiner P. Linear pattern matching algorithms. 14th Annual Symposium on Switching and Automata Theory (swat 1973). USA: IEEE, 1973;1:1–11. [Google Scholar]
- 69. Liu B, Yang J, Li Y, et al. An algorithmic perspective of de novo cis-regulatory motif finding based on ChIP-seq data. Brief Bioinform 2018;19:1069–81. [DOI] [PubMed] [Google Scholar]
- 70. Liu XS, Brutlag DL, Liu JS. An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat Biotechnol 2002;20:835–9. [DOI] [PubMed] [Google Scholar]
- 71. Linhart C, Halperin Y, Shamir R. Transcription factor and microRNA motif discovery: the Amadeus platform and a compendium of metazoan target sets. Genome Res 2008;18:1180–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 2011;27:1653–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ettwiller L, Paten B, Ramialison M, et al. Trawler: de novo regulatory motif discovery pipeline for chromatin immunoprecipitation. Nat Methods 2007;4:563–5. [DOI] [PubMed] [Google Scholar]
- 74. Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38:576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bailey TL. STREME: accurate and versatile sequence motif discovery. Bioinformatics 2021;37:2834–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hertz GZ, Stormo GD. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 1999;15:563–77. [DOI] [PubMed] [Google Scholar]
- 77. Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 1994;2:28–36. [PubMed] [Google Scholar]
- 78. Lawrence CE, Altschul SF, Boguski MS, et al. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science 1993;262:208–14. [DOI] [PubMed] [Google Scholar]
- 79. Lee NK, Li X, Wang D. A comprehensive survey on genetic algorithms for DNA motif prediction. Inform Sci 2018;466:25–43. [Google Scholar]
- 80. Bailey TL, Elkan C. The value of prior knowledge in discovering motifs with MEME. Proc Int Conf Intell Syst Mol Biol 1995;3:21–9. [PubMed] [Google Scholar]
- 81. Bailey TL, Williams N, Misleh C, et al. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 2006;34:W369–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lawrence CE, Reilly AA. An expectation maximization (EM) algorithm for the identification and characterization of common sites in unaligned biopolymer sequences. Proteins 1990;7:41–51. [DOI] [PubMed] [Google Scholar]
- 83. Neuwald AF, Liu JS, Lawrence CE. Gibbs motif sampling: detection of bacterial outer membrane protein repeats. Protein Sci 1995;4:1618–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hughes JD, Estep PW, Tavazoie S, et al. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol 2000;296:1205–14. [DOI] [PubMed] [Google Scholar]
- 85. Workman CT, Stormo GD. ANN-Spec: a method for discovering transcription factor binding sites with improved specificity. Pac Symp Biocomput 2000;467–78. [DOI] [PubMed] [Google Scholar]
- 86. Liu X, Brutlag DL, Liu JS. BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac Symp Biocomput 2001;6:127–38. [PubMed] [Google Scholar]
- 87. Thijs G, Lescot M, Marchal K, et al. A higher-order background model improves the detection of promoter regulatory elements by Gibbs sampling. Bioinformatics 2001;17:1113–22. [DOI] [PubMed] [Google Scholar]
- 88. Frith MC, Hansen U, Spouge JL, et al. Finding functional sequence elements by multiple local alignment. Nucleic Acids Res 2004;32:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Frith MC, Saunders NFW, Kobe B, et al. Discovering sequence motifs with arbitrary insertions and deletions. PLoS Comput Biol 2008;4:e1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Li L. GADEM: a genetic algorithm guided formation of spaced dyads coupled with an EM algorithm for motif discovery. J Comput Biol 2009;16:317–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 2011;27:1696–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Reid JE, Wernisch L. STEME: efficient EM to find motifs in large data sets. Nucleic Acids Res 2011;39:e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kulakovskiy IV, Boeva VA, Favorov AV, et al. Deep and wide digging for binding motifs in ChIP-Seq data. Bioinformatics 2010;26:2622–3. [DOI] [PubMed] [Google Scholar]
- 94. Hartmann H, Guthöhrlein EW, Siebert M, et al. P-value-based regulatory motif discovery using positional weight matrices. Genome Res 2013;23:181–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Li Y, Ni P, Zhang S, et al. ProSampler: an ultrafast and accurate motif finder in large ChIP-seq datasets for combinatory motif discovery. Bioinformatics 2019;35:4632–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tomovic A, Oakeley EJ. Position dependencies in transcription factor binding sites. Bioinformatics 2007;23:933–41. [DOI] [PubMed] [Google Scholar]
- 97. Morris Q, Bulyk ML, Hughes TR. Jury remains out on simple models of transcription factor specificity. Nat Biotechnol 2011;29:483–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhao Y, Stormo GD. Quantitative analysis demonstrates most transcription factors require only simple models of specificity. Nat Biotechnol 2011;29:480–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Rohs R, Jin X, West SM, et al. Origins of specificity in protein-DNA recognition. Annu Rev Biochem 2010;79:233–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Slattery M, Zhou T, Yang L, et al. Absence of a simple code: how transcription factors read the genome. Trends Biochem Sci 2014;39:381–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Grau J, Posch S, Grosse I, et al. A general approach for discriminative de novo motif discovery from high-throughput data. Nucleic Acids Res 2013;41:e197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kulakovskiy I, Levitsky V, Oshchepkov D, et al. From binding motifs in ChIP-Seq data to improved models of transcription factor binding sites. J Bioinform Comput Biol 2013;11:1340004. [DOI] [PubMed] [Google Scholar]
- 103. Ben-Gal I, Shani A, Gohr A, et al. Identification of transcription factor binding sites with variable-order Bayesian networks. Bioinformatics 2005;21:2657–66. [DOI] [PubMed] [Google Scholar]
- 104. Mathelier A, Wasserman WW. The next generation of transcription factor binding site prediction. PLoS Comput Biol 2013;9:e1003214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Maaskola J, Rajewsky N. Binding site discovery from nucleic acid sequences by discriminative learning of hidden Markov models. Nucleic Acids Res 2014;42:12995–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Eggeling R, Gohr A, Keilwagen J, et al. On the value of intra-motif dependencies of human insulator protein CTCF. PloS One 2014;9:e85629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Bell AC, West AG, Felsenfeld G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 1999;98:387–96. [DOI] [PubMed] [Google Scholar]
- 108. Keilwagen J, Grau J. Varying levels of complexity in transcription factor binding motifs. Nucleic Acids Res 2015;43:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ge W, Meier M, Roth C, et al. Bayesian Markov models improve the prediction of binding motifs beyond first order. NAR Genom Bioinform 2021;3:lqab026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Boser BE, Guyon IM, Vapnik VN. A training algorithm for optimal margin classifiers. In: Haussler D. (ed.) Proceedings of the Fifth Annual Workshop on Computational Learning Theory - COLT ‘92. Pittsburgh, Pennsylvania, USA: Association for Computing Machinery, 1992; 1:144–52. [Google Scholar]
- 111. Ben-Hur A, Ong CS, Sonnenburg S, et al. Support vector machines and kernels for computational biology. PLoS Comput Biol 2008;4:e1000173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Leslie C, Eskin E, Noble WS. The spectrum kernel: a string kernel for SVM protein classification. Pac Symp Biocomput 2002;564–75. [PubMed] [Google Scholar]
- 113. Leslie CS, Eskin E, Cohen A, et al. Mismatch string kernels for discriminative protein classification. Bioinformatics 2004;20:467–76. [DOI] [PubMed] [Google Scholar]
- 114. Kuang R, Ie E, Wang K, et al. Profile-based string kernels for remote homology detection and motif extraction. Journal of bioinformatics and computational biology 2005;3:527–50. [DOI] [PubMed] [Google Scholar]
- 115. Lee D, Karchin R, Beer MA. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res 2011;21:2167–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Fletez-Brant C, Lee D, McCallion AS, et al. kmer-SVM: a web server for identifying predictive regulatory sequence features in genomic data sets. Nucleic Acids Res 2013;41:W544–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Leslie C, Kuang R. Fast kernels for inexact string matching. Learning Theory and Kernel Machines 2003;2777:114–28. [Google Scholar]
- 118. Agius P, Arvey A, Chang W, et al. High resolution models of transcription factor-DNA affinities improve in vitro and in vivo binding predictions. PLoS Comput Biol 2010;6:e1000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ghandi M, Lee D, Mohammad-Noori M, et al. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput Biol 2014;10:e1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ghandi M, Mohammad-Noori M, Ghareghani N, et al. gkmSVM: an R package for gapped-kmer SVM. Bioinformatics 2016;32:2205–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lee D. LS-GKM: a new gkm-SVM for large-scale datasets. Bioinformatics 2016;32:2196–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Talukder A, Barham C, Li X, et al. Interpretation of deep learning in genomics and epigenomics. Brief Bioinform 2021;22:bbaa177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zeng W, Wang Y, Jiang R. Integrating distal and proximal information to predict gene expression via a densely connected convolutional neural network. Bioinformatics 2020;36:496–503. [DOI] [PubMed] [Google Scholar]
- 124. Singh R, Lanchantin J, Robins G, et al. DeepChrome: deep-learning for predicting gene expression from histone modifications. Bioinformatics 2016;32:i639–48. [DOI] [PubMed] [Google Scholar]
- 125. Singh S, Yang Y, Póczos B, et al. Predicting enhancer-promoter interaction from genomic sequence with deep neural networks. Quant Biol 2019;7:122–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zeng W, Wu M, Jiang R. Prediction of enhancer-promoter interactions via natural language processing. BMC Genomics 2018;19:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kelley DR, Reshef YA, Bileschi M, et al. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res 2018;28:739–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Li W, Wong WH, Jiang R. DeepTACT: predicting 3D chromatin contacts via bootstrapping deep learning. Nucleic Acids Res 2019;47:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Yin Q, Wu M, Liu Q, et al. DeepHistone: a deep learning approach to predicting histone modifications. BMC Genomics 2019;20:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Manzanarez-Ozuna E, Flores D-L, Gutiérrez-López E, et al. Model based on GA and DNN for prediction of mRNA-Smad7 expression regulated by miRNAs in breast cancer. Theor Biol Med Model 2018;15:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Park Y, Kellis M. Deep learning for regulatory genomics. Nat Biotechnol 2015;33:825–6. [DOI] [PubMed] [Google Scholar]
- 132. Zhang Z, Zhao Y, Liao X, et al. Deep learning in omics: a survey and guideline. Brief Funct Genomics 2019;18:41–57. [DOI] [PubMed] [Google Scholar]
- 133. LeCun Y, Bengio Y, Hinton G. Deep learning. Nature 2015;521:436–44. [DOI] [PubMed] [Google Scholar]
- 134. Sainath TN, Mohamed A-R, Kingsbury B, et al. Deep convolutional neural networks for LVCSR. 2013 IEEE International Conference on Acoustics, Speech and Signal Processing. Vancouver, BC, Canada: IEEE, 2013; 8609–13.
- 135. Vu H, Cheng E, Wilkinson R, et al. On the use of convolutional neural networks for graphical model-based human pose estimation. 2017 International Conference on Recent Advances in Signal Processing, Telecommunications & Computing (SigTelCom). Da Nang, Vietnam: IEEE, 2017; 88–93.
- 136. Alipanahi B, Delong A, Weirauch MT, et al. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol 2015;33:831–8. [DOI] [PubMed] [Google Scholar]
- 137. Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods 2015;12:931–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kelley DR, Snoek J, Rinn JL. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res 2016;26:990–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zeng H, Edwards MD, Liu G, et al. Convolutional neural network architectures for predicting DNA-protein binding. Bioinformatics 2016;32:i121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Bengio Y, Courville A, Vincent P. Representation learning: a review and new perspectives. IEEE Trans Pattern Anal Mach Intell 2013;35:1798–828. [DOI] [PubMed] [Google Scholar]
- 141. Hochreiter S, Schmidhuber J. Long short-term memory. Neural Comput 1997;9:1735–80. [DOI] [PubMed] [Google Scholar]
- 142. Hassanzadeh HR, Wang MD. DeeperBind: enhancing prediction of sequence specificities of DNA binding proteins. Proceedings 2016;2016:178–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Quang D, Xie X. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res 2016;44:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Quang D, Xie X. FactorNet: a deep learning framework for predicting cell type specific transcription factor binding from nucleotide-resolution sequential data. Methods 2019;166:40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zheng R, Wan C, Mei S, et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res 2019;47(D1):D729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kolmykov S, Yevshin I, Kulyashov M, et al. GTRD: an integrated view of transcription regulation. Nucleic Acids Res 2021;49(D1):D104–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Kulakovskiy IV, Medvedeva YA, Schaefer U, et al. HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res 2013;41:D195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Kulakovskiy IV, Vorontsov IE, Yevshin IS, et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res 2018;46:D252–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Sandelin A, Alkema W, Engström P, et al. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res 2004;32:D91–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Fornes O, Castro-Mondragon JA, Khan A, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 2020;48:D87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Weirauch MT, Yang A, Albu M, et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 2014;158(6):1431–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wingender E. TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res 1996;24:238–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Wingender E, Chen X, Hehl R, et al. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res 2000;28:316–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Pratt HE, Andrews GR, Phalke N, et al. Factorbook: an updated catalog of transcription factor motifs and candidate regulatory motif sites. Nucleic Acids Res 2022;50(D1):D141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Puig RR, Boddie P, Khan A, et al. UniBind: maps of high-confidence direct TF-DNA interactions across nine species. BMC Genomics 2021;22(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Newburger DE, Bulyk ML. UniPROBE: an online database of protein binding microarray data on protein–DNA interactions. Nucleic Acids Res 2009;37.suppl_1:D77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Bovolenta L, Acencio M, Lemke N. HTRIdb: an open-access database for experimentally verified human transcriptional regulation interactions. BMC Genomics 2012;13:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Huang Q, Tan Z, Li Y, et al. Tfcancer: a manually curated database of transcription factors associated with human cancers. Bioinformatics 2021;37(22):4288–90. [DOI] [PubMed] [Google Scholar]
- 160. Gupta S, Stamatoyannopoulos JA, Bailey TL, et al. Quantifying similarity between motifs. Genome Biol 2007;8:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Mahony S, Benos PV. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res 2007;35:W253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Vorontsov IE, Kulakovskiy IV, Makeev VJ. Jaccard index based similarity measure to compare transcription factor binding site models. Algorithms Mol Biol 2013;8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Lambert SA, Albu M, Hughes TR, et al. Motif comparison based on similarity of binding affinity profiles. Bioinformatics 2016;32:3504–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Korhonen J, Martinmäki P, Pizzi C, et al. MOODS: fast search for position weight matrix matches in DNA sequences. Bioinformatics 2009;25:3181–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics 2011;27:1017–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Ambrosini G, Groux R, Bucher P. PWMScan: a fast tool for scanning entire genomes with a position-specific weight matrix. Bioinformatics 2018;34:2483–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Tognon M, Bonnici V, Garrison E, et al. GRAFIMO: variant and haplotype aware motif scanning on pangenome graphs. PLoS Comput Biol 2021;17:e1009444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Paten B, Novak AM, Eizenga JM, et al. Genome graphs and the evolution of genome inference. Genome Res 2017;27:665–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Frith MC, Fu Y, Yu L, et al. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res 2004;32:1372–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Zambelli F, Pesole G, Pavesi G. Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res 2009;37:W247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. McLeay RC, Bailey TL. Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 2010;11:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Kwon AT, Arenillas DJ, Hunt RW, et al. oPOSSUM-3: advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 Genes|Genomes|Genetics 2012;2:987–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Pinello L, Farouni R, Yuan G-C. Haystack: systematic analysis of the variation of epigenetic states and cell-type specific regulatory elements. Bioinformatics 2018;34:1930–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. De Gobbi M, Viprakasit V, Hughes JR, et al. A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter. Science 2006;312:1215–7. [DOI] [PubMed] [Google Scholar]
- 175. Wienert B, Funnell APW, Norton LJ, et al. Editing the genome to introduce a beneficial naturally occurring mutation associated with increased fetal globin. Nat Commun 2015;6:7085. [DOI] [PubMed] [Google Scholar]
- 176. Weinhold N, Jacobsen A, Schultz N, et al. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat Genet 2014;46:1160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012;337:1190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Deplancke B, Alpern D, Gardeux V. The genetics of transcription factor DNA binding variation. Cell 2016;166:538–54. [DOI] [PubMed] [Google Scholar]
- 179. Thomas-Chollier M, Hufton A, Heinig M, et al. Transcription factor binding predictions using TRAP for the analysis of ChIP-seq data and regulatory SNPs. Nat Protoc 2011;6:1860–9. [DOI] [PubMed] [Google Scholar]
- 180. Zuo C, Shin S, Keleş S. atSNP: transcription factor binding affinity testing for regulatory SNP detection. Bioinformatics 2015;31:3353–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Yao Q, Ferragina P, Reshef Y, et al. Motif-Raptor: a cell type-specific and transcription factor centric approach for post-GWAS prioritization of causal regulators. Bioinformatics 2021;37:2103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Lee D, Gorkin DU, Baker M, et al. A method to predict the impact of regulatory variants from DNA sequence. Nat Genet 2015;47:955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Shrikumar A, Prakash E, Kundaje A. GkmExplain: fast and accurate interpretation of nonlinear gapped k-mer SVMs. Bioinformatics 2019;35:i173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Avsec Ž, Agarwal V, Visentin D, et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat Methods 2021;18(10):1196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Weirauch MT, Cote A, Norel R, et al. Evaluation of methods for modeling transcription factor sequence specificity. Nat Biotechnol 2013;31:126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Castellana S, Biagini T, Parca L, et al. A comparative benchmark of classic DNA motif discovery tools on synthetic data. Brief Bioinform 2021;22(6):bbab303. [DOI] [PubMed] [Google Scholar]
- 187. Bulyk ML, Johnson PLF, Church GM. Nucleotides of transcription factor binding sites exert interdependent effects on the binding affinities of transcription factors. Nucleic Acids Res 2002;30:1255–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Benos PV. Additivity in protein-DNA interactions: how good an approximation is it? Nucleic Acids Res 2002;30:4442–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Siggers T, Gordân R. Protein-DNA binding: complexities and multi-protein codes. Nucleic Acids Res 2014;42:2099–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Trabelsi A, Chaabane M, Ben-Hur A. Comprehensive evaluation of deep learning architectures for prediction of DNA/RNA sequence binding specificities. Bioinformatics 2019;35:i269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Bernstein BE, Stamatoyannopoulos JA, Costello JF, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol 2010;28:1045–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Jenuwein T, Allis CD. Translating the histone code. Science 2001;293:1074–80. [DOI] [PubMed] [Google Scholar]
- 193. Arvey A, Agius P, Noble WS, et al. Sequence and chromatin determinants of cell-type-specific transcription factor binding. Genome Res 2012;22:1723–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Gertz J, Savic D, Varley KE, et al. Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol Cell 2013;52:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Kasowski M, Grubert F, Heffelfinger C, et al. Variation in transcription factor binding among humans. N Biotechnol 2010;27:S81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Yan J, Qiu Y, Ribeiro Dos Santos AM, et al. Systematic analysis of binding of transcription factors to noncoding variants. Nature 2021;591:147–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Thurman RE, Rynes E, Humbert R, et al. The accessible chromatin landscape of the human genome. Nature 2012;489:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Karimzadeh M, Hoffman MM. Virtual ChIP-seq: predicting transcription factor binding by learning from the transcriptome. Genome Biol 2022;23:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Bailey TL, Noble WS. Searching for statistically significant regulatory modules. Bioinformatics 2003;19 Suppl 2:ii16–25. [DOI] [PubMed] [Google Scholar]
- 200. Moyerbrailean GA, Kalita CA, Harvey CT, et al. Which genetics variants in DNase-Seq footprints are more likely to alter binding? PLoS Genet 2016;12:e100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Clark SJ, Lee HJ, Smallwood SA, et al. Single-cell epigenomics: powerful new methods for understanding gene regulation and cell identity. Genome Biol 2016;17:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Rotem A, Ram O, Shoresh N, et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol 2015;33:1165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Grosselin K, Durand A, Marsolier J, et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet 2019;51:1060–6. [DOI] [PubMed] [Google Scholar]
- 204. Buenrostro JD, Corces MR, Lareau CA, et al. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell 2018;173:1535–1548.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Fu L, Zhang L, Dollinger E, et al. Predicting transcription factor binding in single cells through deep learning. Sci Adv 2020;6:eaba9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Stuart T, Srivastava A, Madad S, et al. Single-cell chromatin state analysis with Signac. Nat Methods 2021;18:1333–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Yuan H, Kelley DR. scBasset: sequence-based modeling of single cell ATAC-seq using convolutional neural networks. bioRxiv 2021. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new data were generated or analysed in support of this research.