

Abstract
Background and Aims:
Tobacco smoking is a risk factor for impaired brain function, but its causal effect on white matter brain aging remains unclear. This study aimed to measure the causal effect of tobacco smoking on white matter brain aging.
Design:
Mendelian randomization (MR) analysis using two non-overlapping data sets (with and without neuroimaging data) from UK Biobank (UKB). The group exposed to smoking and control group consisted of current smokers and never smokers, respectively. Our main method was generalized weighted linear regression with other methods also included as sensitivity analysis.
Setting:
United Kingdom.
Participants:
The study cohort included 23 624 subjects [10 665 males and 12 959 females with a mean age of 54.18 years, 95% confidence interval (CI) = 54.08, 54.28].
Measurements:
Genetic variants were selected as instrumental variables under the MR analysis assumptions: (1) associated with the exposure; (2) influenced outcome only via exposure; and (3) not associated with confounders. The exposure smoking status (current versus never smokers) was measured by questionnaires at the initial visit (2006–10). The other exposure, cigarettes per day (CPD), measured the average number of cigarettes smoked per day for current tobacco users over the life-time. The outcome was the ‘brain age gap’ (BAG), the difference between predicted brain age and chronological age, computed by training machine learning model on a non-overlapping set of never smokers.
Findings:
The estimated BAG had a mean of 0.10 (95% CI = 0.06, 0.14) years. The MR analysis showed evidence of positive causal effect of smoking behaviors on BAG: the effect of smoking is 0.21 (in years, 95% CI = 6.5 × 10−3, 0.41; P-value = 0.04), and the effect of CPD is 0.16 year/cigarette (UKB: 95% CI = 0.06, 0.26; P-value = 1.3 × 10−3; GSCAN: 95% CI = 0.02, 0.31; P-value = 0.03). The sensitivity analyses showed consistent results.
Conclusions:
There appears to be a significant causal effect of smoking on the brain age gap, which suggests that smoking prevention can be an effective intervention for accelerated brain aging and the age-related decline in cognitive function.
Keywords: Brain aging, causal inference, cigarette per day, Mendelian randomization, smoking behaviors, smoking status, white matter fractional anisotropy
INTRODUCTION
Smoking behaviors are among the most studied modifiable risk factors related to accelerated brain atrophy and cognitive impairment [1–4]. This discovery has led to prosperous studies of the adverse impact of smoking behaviors on neuroimaging [1, 3, 5–8]. Substantial research has found that chronic smoking strongly impacts neurocognition and brain neurobiology [9–14]. For example, previous studies have reported that accelerated brain aging is associated with the production of oxidative stress and alteration of synaptic protein expressions from smoking behaviors [15, 16]. Also, the atherosclerotic processes of smoking had potentially harmful effects on brain aging [11, 17]. The microstructure of the human brain is constantly changing over the life-span with normal aging, reflecting brain shrinks, memory decline and increases in the development of severe chronic diseases such as vasculature diseases [18]. Therefore, it is critical to recognize the causal effect of smoking on brain aging and thus quantitatively understand the potential benefits of smoking intervention in reducing the acceleration of brain aging.
‘Brain age’ is a metric for brain aging status related to cognitive functions [19–21], calculated based on structural or functional neuroimaging data using machine learning (ML) algorithms [22–24]. In the current research, we adopt a commonly used chronological age-adjusted brain age metric, brain age gap (BAG), as the outcome variable that quantifies the difference between brain and chronological age [22–25]. BAG is a scalar metric that avoids the challenges of multiplicity, and dependence adjustment as in multivariate imaging models thus often yields more robust and interpretable results [22, 23, 25]. The recent literature showed that applying advanced ML methods to neuroimaging data can provide a reliable estimate for BAG with small predictive biases [25–34].
Various factors can influence BAG, including genetic, environmental, biological and behavioral factors [19, 35, 36]. Among these, smoking behavior is a well-studied factor that was adversely related to accelerated brain age in many previous studies [1, 37], and its causal effect on neuroimaging features was jointly examined with other risk factors, including diastolic blood pressure, systolic blood pressure, pulse pressure, low-density lipoprotein, high-density lipoprotein, triglycerides, total cholesterol, body mass index, Type 2 diabetes and alcoholic drinks consumed per week [38]. However, it is challenging to establish the causal relationship solely between smoking behavior and BAG in traditional observational studies because smoking often co-occurs with other health risk factors also linking to abnormal brain aging, including alcohol use, mental disorders (e.g. schizophrenia) and chronic diseases (e.g. hypertension, dyslipidemia and cardiovascular disease) [1, 39–42]. To address this challenge, we estimated the causal effect of smoking by Mendelian randomization (MR) analysis. Several addiction and neuropsychiatric research studies have employed MR analysis to investigate the causal effects of smoking behaviors on stroke, amyotrophic lateral sclerosis and psychiatric and behavioral disorders [14, 43–46].
The current study aimed to investigate the causal effects of smoking behaviors [i.e. smoking status (SS) and cigarette per day (CPD)] on BAG using the UK Biobank (UKB) cohort. We hypothesized that smoking behaviors, such as being a smoker or smoking a larger number of cigarettes per day, would significantly cause impairment in brain microstructures reflected by an increase in BAG among the participants in UKB. The estimated causal effect can reveal the overall effect of smoking and its biological consequences (e.g. cardiovascular condition worsening) on brain aging and thus unfolds the potential health benefits of the intervention and prevention for tobacco smoking.
METHODS
UKB cohort and variables
Our data were extracted from the UKB, a large-scale biomedical database consisting of phenotype and genotype, and imaging details of approximately 500 000 participants aged 40–69 years [47]. The initial measurement was conducted in 2006–10, and the repeat of baseline assessment started in 2013 [48]. We used the initial baseline measurement of smoking behavioral data in 2006–10 when conducting our current research to avoid poor data quality due to common loss to follow-up problem [48]. The neuroimaging data we used were collected since 2014 [48]. We performed quality control and restricted our analyses to participants with European ethnic backgrounds who had data available. To generate an accurate BAG estimation for normal brain aging, we excluded participants with health conditions (e.g. stroke, brain injury, brain cancer and psychiatric illnesses) to reduce the distortion of the fractional anisotropy (FA) measurements due to these diseases [49]. The study cohort included 23 624 subjects with neuroimaging data [10 665 males and 12 959 females with a mean age of 54.18 years, 95% confidence interval (CI) = 54.08, 54.28]. Supporting information, Fig. S1 shows a flow diagram of the number of participants for different analytical step. In each step we included participants with complete data, and thus no missing data presented in the analysis. As the primary research question and analysis plan were not pre-registered on a publicly available platform, the results of this study are considered exploratory.
Smoking behavior
This study focused up on two smoking behaviors (phenotypes): SS and CPD, following the commonly used definitions in the literature [50, 51]. SS was a binary variable classifying participants as current smokers and non-smokers who had never smoked according to the UKB data field 2016 (SS). The CPD was a quantitative variable representing the average number of cigarettes smoked per day for tobacco users, characterizing the level of nicotine craving in current smokers. We defined the CPD (ranges from 0 to 60) of current smokers based on three UKB data fields (IDs: 2887, 3456 and 6183) following the commonly used procedure in smoking genome-wide association studies (GWAS) [50, 51]. For those subjects who smoked fewer than one CPD, the CPD values were rounded down and recoded to 0; for those who smoked more than 60 CPD, the CPD values were truncated recoded to 60.
Neuroimaging data
We used diffusion magnetic resonance imaging (dMRI) data for the 23 624 participants for whom genotype and SS/CPD data were also available (see Supporting information, Fig. S1). The UKB acquired and processed these imaging data following its imaging protocol and pipeline [52]. White matter microstructure integrity was measured by FA derived based on the pre-processed dMRI, and then the per-tract mean values of 40 FA tracts were calculated [52]. The locations and names of the 40 FA tracts are provided in Supporting information, Data S1.
Genotype data
We used genotype data from the UKB cohort sequenced through two genotyping chip types, Affymetrix UK BiLEVE Axiom and UKBB Axiom® arrays. The data involved more than 90 million single-nucleotide variants (SNVs) among all participants [53]. More details regarding the quality control of genotype data are available in Supporting information, Appendix S1.
Analysis overview
Our analysis consisted of two steps (see Figure 1). The first step was to establish the function to compute the outcome variable, adjusted BAG, based on FA data and chronological age using machine learning techniques in a training set with only non-smokers (Figure 1a). In this step, we ensured the accuracy of the estimation and locked the optimal predictive model through an internal fivefold cross-validation (CV) within the training set. This predictive model was eventually applied to calculate the adjusted BAG of participants in another non-overlapping set consisting of both non-smokers and smokers (i.e. testing set) (see Figure 1b). In the second step, we conducted a two-sample MR analysis to investigate the causal effects of smoking behaviors on adjusted BAG [54]. The first sample consisted of UKB participants () with only smoking behavioral data (exposure) but no brain imaging data (outcome: adjusted BAG). The second sample included a non-overlapping set of UKB participants () who had neuroimaging data and the outcome data for adjusted BAG.
FIGURE 1.
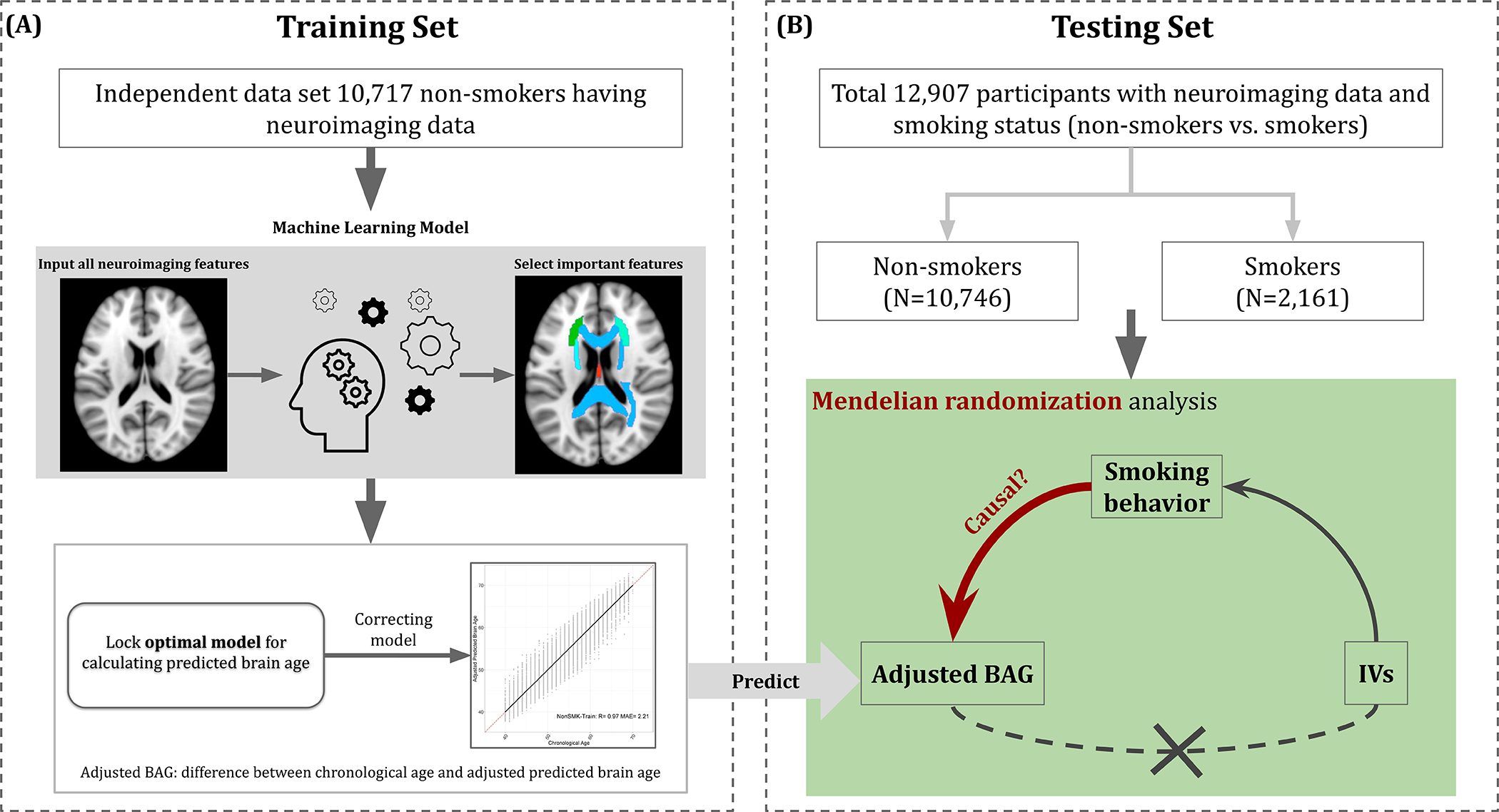
Overview of analysis procedures. (a) Within the non-overlapping training set, (i) the optimal predictive model for estimating predicted brain age was obtained using machine learning model; (ii) the predictive model was corrected to reduce the estimation bias and further used to estimate the adjusted predictive brain age; and (iii) the outcome variable [i.e. adjusted brain age gap (BAG)] was calculated by subtracting the chronological age by the adjusted predictive brain age. (b) In the testing set, the adjusted BAG was estimated based on the corrected predictive model in (a), and the causal effect of smoking behavior (e.g., smoking status) on adjusted BAG was evaluated through Mendelian randomization analysis. Smoking status was a binary trait consisting of nonsmokers and smokers
Adjusted BAG
The goal of the ML model was to compute a scalar brain age metric based on 40 FA measures. The UKB non-smokers with neuroimaging data were randomly split into training and testing sets by a 1:1 ratio, as shown in Supporting information, Fig. S1. The training set included 10 717 non-smokers for building an unbiased brain age estimating function for the general population [28]. The testing set consisted of 10 746 non-smokers and 2161 smokers serving as one of the samples used in the two-sample MR analysis. Among the smokers in the testing set, 1597 of them had CPD data available. Descriptive statistics were provided for training and testing sets separately, including mean and standard error for continuous variables and frequency and percentage for categorical variables.
Within the training set, we implemented ML analysis through an internal fivefold CV to achieve the optimal predictive performance using random forest (RF) regression [55, 56]. Based on the performance of the fivefold CV, we tuned the parameters of RF and then determined the set of FA measures via recursive feature selection according to their variable importance (i.e. Gini importance) based on accuracy criteria, including the coefficients of correlation (R) and mean absolute error [MAE (year)] [57, 58].
Given the optimal predictive model, we estimated the predicted brain age and obtained BAG by subtracting chronological age by the predicted brain age . This estimated BAG can be systematically biased because the prediction tends to overestimate brain age at low chronological age and underestimate brain age at high chronological age (see Supporting information, Fig. S6) [33, 34, 37]. Therefore, we regressed the BAG on chronological age to adjust the bias following a common bias adjustment procedure [33, 59–61]. We then obtained the adjusted predictive brain age and the adjusted BAG . A positive adjusted BAG implies accelerated brain aging, while a negative adjusted BAG implies decelerated brain aging [19]. We further inspected the data between current smokers (SMK) and non-smokers (non-SMK), summarizing results of distribution comparison and effect size of group differences (i.e. Cohen’s d) in box-plot and forest plot. Hereafter, we conducted the rest of the analyses to evaluate the potential causal effects of smoking phenotypes (i.e. SS and CPD) on in this study. In addition, we explored the association between the adjusted BAG and cognitive function measured from our previous study [62] at the 0.05 significance level. The cognitive function was represented by the intelligence g-factor estimated via factor analysis based on cognitive traits related to four domains in the UKB cohort: processing speed, perceptual reasoning, executive function and fluid intelligence [62].
Estimating causal effect by MR analysis
We evaluated the causal effects of smoking phenotypes (i.e. SS and CPD) on by performing a two-sample MR analysis [63], and a P-value of 0.05 indicates a significant causal relationship. To implement the analysis (see details in Supporting information, Fig. S1 and Appendix S1), we selected IVs for MR analysis based on smoking behavior GWAS using the first sample in the UKB after linkage disequilibrium pruning and clumping (removing genetic variants with within a 50-kb window). We performed IV–outcome association analysis using the second sample in the UKB, which is not non-overlapped with the first sample. The UKB participants in our analysis were family unrelated with European ethnic background. We selected genetic variants with genetic-exposure P-value <5 × 10−8, genetic-outcome P-value adjusted by the Benjamini–Hochberg false discovery rate method (BHp) > 0.05 and genetic-confounder BHP > 0.05 as the IVs for MR analysis. We provide the details of the IVs in Supporting information, Data S5.
Given the IVs selected, we implemented the two-sample MR analysis using the R package MendelianRandomization (version 0.5.1) [64]. We used the MR method with generalized weighted model (gen-IVW) by Burgess et al. [65] (see details in Supporting information, Data S5). Cochran’s Q-test, which provides evidence for heterogeneity in the causal effects between SNPs, was conducted alongside the MR analysis. We further carried out sensitivity analyses to examine the robustness of causal effects of SS and CPD on . The MR methods used in sensitivity analyses are MR-weighted-median [66], MR-PRESSO [67], MR-MIX [68] and contamination mixture method (MRconmix) [69]. MR-weighted-median provides consistent estimates when there are some invalid IVs using a weighted median estimator; however, it requires at least 50% of the weight derived from valid IVs [66]. MR-PRESSO removes IVs that tested to be outliers in the MR analysis, therefore generating MR estimates with less variability (tighter confidence intervals) [67]. Its standard deviation is restricted to be smaller than or equal to the standard deviation from MR-IVW [67]. This method also provides a global test to assess whether the IVs used in the MR model have significant horizontal pleiotropy [67]. MR-Mix is another method that uses a mixture model to incorporate those IVs having horizontal pleiotropy [68]. MRconmix provides robust analysis when invalid IVs present by constructing a likelihood function according to variant-specific causal estimates, assuming different normal distributions for valid and invalid IVs [69]. Additionally, we performed the leave-one-out analysis besides the sensitivity analysis to assess whether an IV was driving the causal relationship between the exposure and the outcome [70]. We also performed MR-Egger and evaluated the reliability of MR-Egger results via its I2 statistics [measurement of the ‘NO Measurement Error’ (NOME) assumption]. Moreover, we performed MR analysis to examine the other possible causal direction using BAG as the exposure and smoking behaviors (SS and CPD) as the outcome (see Appendix S2 for BAG GWAS and IV selection). In addition to UKB cohort, we performed the IV selection using summary statistics of CPD from the GSCAN (no GSCAN summary statistics available for SS) [39] and also performed the MR analysis using these IVs for CPD.
RESULTS
We summarized the participants’ demographic characteristics and health conditions in training and testing sets separately for non-smokers and smokers (Table 1; see Supporting information, Fig. S2 for their distributions). The training and the testing sets had balanced demographic factors such as age, body mass index (BMI) and sex. As shown in Table 1, the mean BMI, mean age and proportions of categorical characteristics were similar between smokers and non-smokers, showing that there is no systematic difference in the distribution of sample characteristics between different groups. Also, we explored the demographic factors and found that these factors were distributed similarly between participants included and excluded from our MR study (see Table 1). We provided frequency of additional health conditions between training and testing sets (Supporting information, Fig. S1) and the prevalence of different self-reported health conditions in Supporting information, Data S2. Most conditions had a low prevalence within the testing set.
Table 1.
Demographic characteristics of participants in training and testing data sets
| Training Set | Testing Set | |||
|---|---|---|---|---|
| Variable | Level | NonSMK (N=10,717) | NonSMK (N=10,746) | SMK (N=2,161) |
| BMI (kg/m2) | Mean (SE) | 26.30 (4.08×10−2) | 26.26 (4.02×10−2) | 26.48 (8.70×10−2) |
| Age (years) | Mean (SE) | 54.30 (7.15×10−2) | 54.28 (7.12×10−2) | 53.08 (1.57×10−2) |
| Townsend deprivation index at recruitment | Mean (SE) | −2.13 (2.57) | −2.18 (2.54) | −0.90 (3.19) |
| Age completed full time education (years) | Mean (SE) | 17.28 (2.34) | 17.27 (2.35) | 16.94 (2.35) |
| Average total household income before tax (n, %) | Less than 18,000 | 930 (8.68) | 899 (8.37) | 145 (6.71) |
| 18,000 to 30,999 | 1023 (9.55) | 1038 (9.66) | 314 (14.53) | |
| 31,000 to 51,999 | 2012 (18.77) | 2017 (18.77) | 455 (21.06) | |
| 52,000 to 100,000 | 2883 (26.9) | 3042 (28.31) | 635 (29.38) | |
| Greater than 100,000 | 3012 (28.1) | 2872 (26.73) | 495 (22.91) | |
| Do not know/ Prefer not to answer | 838 (7.82) | 803 (7.47) | 105 (4.86) | |
| Sex (n, %) | Female (%) | 6009 (56.07) | 5978 (55.63) | 972 (44.98) |
| Male (%) | 4708 (43.93) | 4768 (44.37) | 1189 (55.02) | |
| Hypertension (n, %) | Yes (%) | 1213 (11.32) | 1252 (11.65) | 259 (11.98) |
| No (%) | 9504 (88.68) | 9494 (88.35) | 1902 (88.01) | |
| Diabetes (n, %) | Yes (%) | 33 (3.08×10−3) | 43 (4.00×10−3) | 12 (5.55×10−3) |
| No (%) | 10684 (0.99) | 10703 (0.99) | 2149 (0.99) | |
| Stroke (n, %) | Yes (%) | 93 (8.68×10−3) | 86 (8.00×10−3) | 25 (1.16×10−3) |
| No (%) | 10660 (0.99) | 10624 (0.99) | 2136 (0.99) | |
SE = standard error; n = frequency; % = percentage within each column; NonSMK = non-smoker; SMK = smoker.
Model construction for calculating BAG
We constructed the ML model to calculate BAG from the training set using RF, locked the optimal model and applied it to the testing set. The optimal ML model chosen included 16 FA measures (see Supporting information, Data S1 and Fig. S3). The optimal model achieved excellent prediction performance in both training and testing sets: and for training set (see Figure 1a); and for non-smokers and and for smokers in the testing set, respectively (see Figure 2a). had a significant association with SS [ 95% confidence interval ; ] and CPD (; at the 0.05 significance level, as shown in Table 2. Such differences between smokers and non-smokers were consistent in all age categories (i.e. 40–49, 50–59 and 60–69 years) (see Figure 2b for the P-values). On average, mean of nonsmokers was 0.66 , 1.03 (0.77, 1.28) and 1.10 (0.81, 1.38) years younger than the of smokers in aged 40–49, 50–59 and 60–69 categories, respectively (see Cohen’s in Figure 2c for the effect size). Also, was significantly associated with cognitive function (; , data not shown), given the cognitive function represented by the intelligence calculated in our previous work [62].
FIGURE 2.
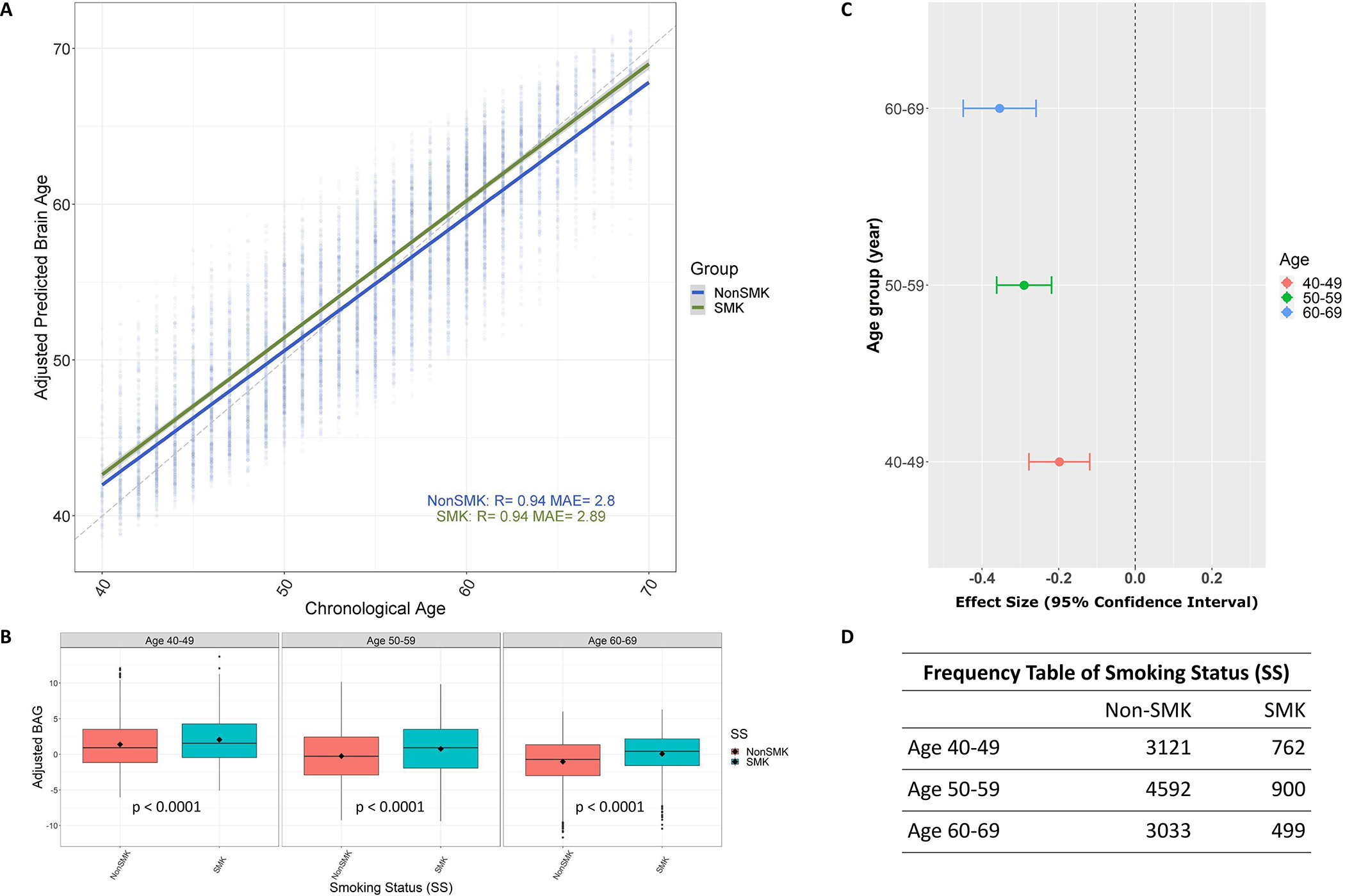
Adjusted brain age gap (BAG) and its relationships with smoking status in the testing set. (a) The relationship between the adjusted predicted brain age and chronological age in different smoking statuses (SS) (R = coefficients of correlation; MAE = mean absolute error). (b) The distribution of adjusted BAG between different SS groups in separate chronological age categories, where P indicates the P-value corresponding to the comparison between nonsmokers (non-SMK) and smokers (SMK) in each category. (c) The effect size with the 95% confidence interval for testing the difference between non-SMK and SMK within each age category based on (b). (d) The number of participants in each age group corresponding to comparisons between non-SMK and SMK in (b)
Table 2.
Results of Mendelian randomization and association analyses in the study sample.
| Association | Mendelian randomization | |||
|---|---|---|---|---|
| (95% CI) | p-value | (95% CI) | p-value | |
| SS | 0.88 ± 0.07 (0.74, 1.02) | 2.81× 10−36 | 0.21 ± 0.10 (6.5×10−3, 0.41) | 0.04 |
| CPD | 0.05 ± 0.01 (0.03, 0.07) | 5.49×10−6 | 0.16 ± 0.05 (0.06, 0.26) | 1.30×10−3 |
SE = standard error; CI = confidence interval; SS = smoking status; CPD = cigarette per day.
Causal effects of smoking behaviors on brain aging
We selected 248 and 54 genetic variants as IVs in the MR models for SS and CPD, respectively, following the criteria in Equations (1)–(3) in Supporting information, Data S5. These IVs were well aligned with findings in existing studies, such as the gene cluster CHRNA5–CHRNA3–CHRNB4 linked with CPD [71–73]. We further explored the gene information of these IVs via an SNP annotation portal, the functional annotation of variant on-line resource (FAVOR) (http://favor.genohub.org/, accessed 12 July 2022) (see Supporting information, Data S3) [74].
We performed MR analyses to test the estimated causal effect of the exposures using the selected IVs given as the outcome. As shown in Table 2 and Figure 3b, the primary MR method [i.e. gen-IVW (UKB)] showed a significant causal effect of SS on (). Also, had a significant causal effect on (UKB: ; GSCAN: ; ), indicating that the causal effect of CPD were robust across cohorts [see gen-IVW (GSCAN) in Figure 3b]. The results showed that smokers were 0.21 years older in brain age than comparable non-smokers at the same chronological age, and having an extra cigarette per day increased brain age by an additional 0.16 years.
FIGURE 3.
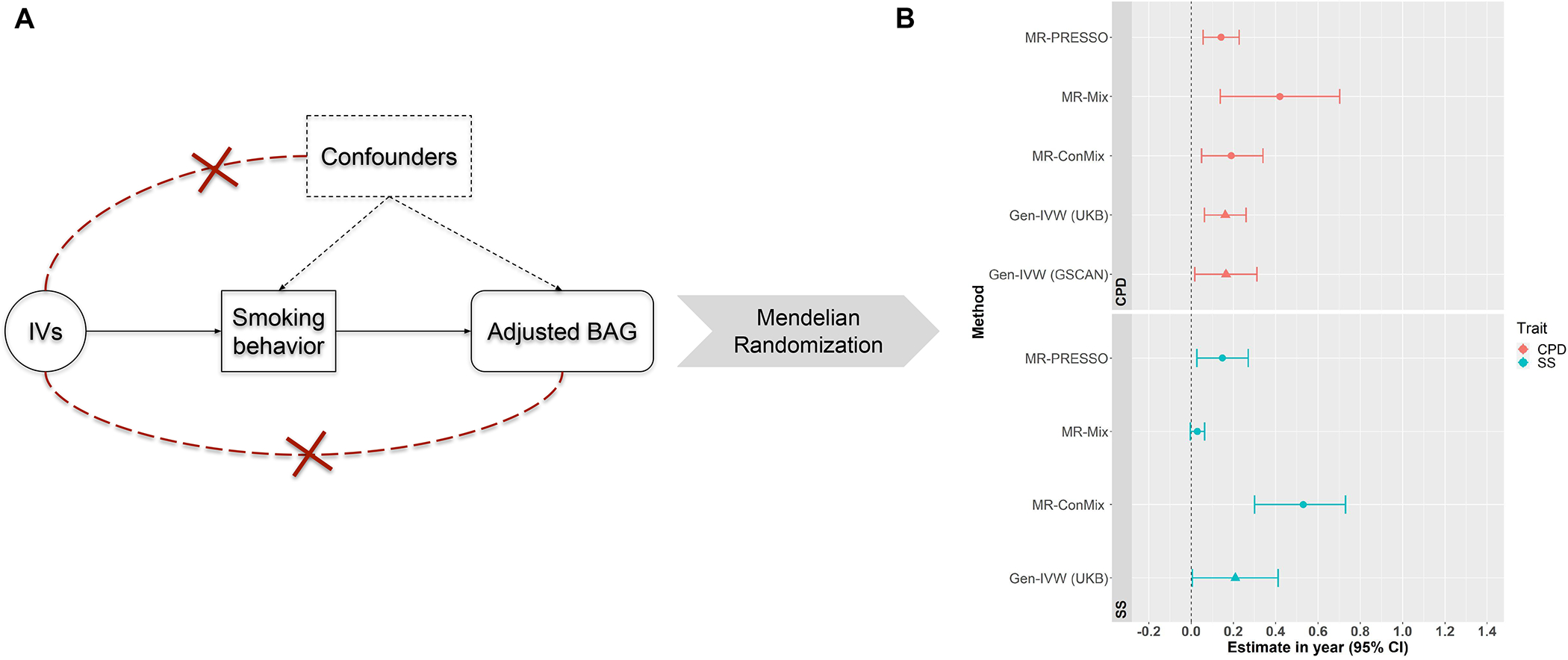
Mendelian randomization and the results. (a) The three fundamental instrumental variable (IV) assumptions in the Mendelian randomization (MR) analysis: (i) IVs are significantly associated with the exposure [i.e. smoking statuses (SS) or cigarettes per day (CPD)]; (ii) the exposure is not significantly associated with confounders of the exposure–outcome association; and (iii) IVs can affect the outcome variable only through the exposure. (b) The causal effect estimate with a 95% confidence interval using different MR methods for smoking traits SS and CPD separately, based on the IVs selected following the three IV assumptions. Gen-IVW (marked with a triangle) is the primary MR method (i.e. weighted generalized linear regression) and the other methods (marked with a dot) are the MR methods used in the sensitivity analysis. BAG = brain age gap
Moreover, sensitivity analyses showed robust causal effects of SS and CPD separately on (see Figure 3b). According to the MR-PRESSO’s global test, one of the methods in sensitivity analyses, the selected IVs did not show significant horizontal pleiotropic effects on both SS and CPD (). Cochran’s Q-test suggested no evidence of weak or pleiotropic effects of IVs in MR analyses for CPD (), but there were weak effects of IVs for SS (). The leave-one-out analysis indicated that no single IV was driving the causal relationship (see Supporting information, Figs S4 and S5 and a complete list of causal estimates in Supporting information, Data S4). The statistics suggested that MR-Egger had sufficiently low reliability for CPD . For SS, also suggested violation of the NOME assumption in MR-Egger. As MR-Egger is highly sensitive to the violation of the assumptions [75], we decided to focus upon results using other MR methods. Overall, most of the MR analyses showed evidence of significant causal effects of SS and CPD on BAG (see Figure 3b). We found no sufficient evidence of causal effect of BAG on smoking behaviors (SS:; CPD:). We further performed MR analysis using the instruments for CPD in non-smokers and found insignificant effect (P-value of gen-IVW = 0.11).
DISCUSSION
Our study investigated the potential causal effects of smoking behaviors on the brain aging process using the imaging-genetics data from the UKB cohort. Our findings confirmed the causal effect of smoking on accelerated neural degeneration during aging interpreted by age year. Also, the results revealed that the overall brain white matter deterioration was due to aging accelerated by smoking behaviors. The accelerated neural decline has been associated with multiple clinical symptoms of dementia, Alzheimer’s disease and vasculature diseases [15, 18]. Findings have important implications for clinical interventions, highlighting how reductions in smoking may have important neurological health benefits.
We identified significant causal effects of SS and CPD on BAG based on MR analysis, consistent with the strong associations reported in previous work [17, 62, 76, 77]. For example, the pack-year was reported as the significant risk factor associated with BAG, given an increase of 0.36 months of adjusted BAG associated with one pack-year [17]. Similarly, we observed that a person who was a current smoker or had a larger number of cigarettes per day at the time of data collection would have a higher BAG value than a person who was a non-smoker or had fewer cigarettes in the UKB cohort. In addition, our study revealed that smoking led to a significant increase in brain aging and consequently raised the risk of age-related diseases and symptoms (e.g. memory and cognitive function decline) which may, in part, explain the previous findings of abnormal brain aging and cognitive functions. Smoking can lead to multiple complications (e.g. encouraging acute cardiovascular events and increasing insulin resistance) [78–80]. Subsequently, these smoking-related biological conditions can lead to accelerated brain aging [38, 81–83]. Numerically, the estimated causal effects of smoking behaviors on brain aging are distinct from the association effects. The former provides a more accurate estimate of the potential health benefits by preventing smoking.
Moreover, the IVs used in our analysis coincided with previously reported smoking-behavior-related genes (see Supporting information, Data S1 for the complete list). For example, in chromosome 9 (CHR9), genes FAM163B and GAPVD1 had significant associations with CPD and smoking initiation, respectively [39, 51]. Genes REV3L (CHR6), CHRNB3 (CHR8), AS3MT (CHR10), NCAM1 (CHR11), CHRNA3–CHRNA5–CHRNB4 (CHR15) and CYP2T1P (CHR19) had association with smoking initiation, smoking cessation, nicotine dependence and smoking history [7, 39, 71, 72, 84–94]. These existing studies supported the validity of IVs used in our MR analysis and reliability of the MR results. In addition, the sensitivity analyses reassured that there was no evidence for significant horizontal pleiotropy corresponding to these IVs.
Results must be interpreted in the context of study limitations. First, the UKB sample was biased towards certain sampling characteristics: (1) being older, being female and living in less socioeconomically deprived areas compared to samples in other study cohorts; (2) the participants in UKB tended more likely to have obesity, smoke cigarettes, drink alcohol on a daily basis and have fewer self-reported health conditions compared to the general population [95]. Secondly, our current work was restricted to one ethnic background aged 40–69 years. Some other factors may potentially affect our MR findings, such as population structure, family and assortative mating [96]. To control these potential effects, we adjusted the effect of population structure by adding principal components to GWAS analysis and removed participants that were family related; the effect of assortative mating was not accounted because our data had limited information to assess this effect. Exposure to secondhand smoke can be a potential confounder of the study, the impact of which will be investigated in future studies as the data become available in UKB. We encourage future studies to examine the causal relationship using different cohorts and validate the generalization of the results in a broader range of age and ethnic backgrounds.
In conclusion, our study provided a thorough analysis of causal inference for smoking behaviors on BAG and revealed robust and consistent causal effects in a large study cohort. These results reinforced the evaluation of the impact coming from smoking and assisted in guiding future studies. This study provided a greater understanding of the causal effects of smoking behaviors on brain aging and cognitive disorders, connecting previous findings of neuroimaging and cognitive function [62, 76, 77]. Our results have important implications in how behavior may also improve neurological health status.
Supplementary Material
ACKNOWLEDGEMENTS
This research was carried out using the UK Biobank Database (https://www.ukbiobank.ac.uk/). This work was supported by the National Institute on Drug Abuse of the National Institutes of Health under Award Number 1DP1DA04896801 and the University of Maryland MPower Brain Health and Human Performance seed grant. Additional support for computer cluster was provided by the National Institutes of Health R01 grants EB008432 and EB008281.
Footnotes
DECLARATION OF INTERESTS
None.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
REFERENCES
- 1.Ning K, Zhao L, Matloff W, Sun F, Toga AW. Association of relative brain age with tobacco smoking, alcohol consumption, and genetic variants. Sci Rep. 2020;10:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duriez Q, Crivello F, Mazoyer B. Sex-related and tissue-specific effects of tobacco smoking on brain atrophy: assessment in a large longitudinal cohort of healthy elderly. Front Aging Neurosci. 2014;6:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Durazzo TC, Insel PS, Weiner MW, Alzheimer’s Disease Neuroimaging Initiative. Greater regional brain atrophy rate in healthy elderly subjects with a history of cigarette smoking. Alzheimers Dement. 2012;8:513–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallinat J, Meisenzahl E, Jacobsen LK, Kalus P, Bierbrauer J, Kienast T, et al. Smoking and structural brain deficits: a volumetric MR investigation. Eur J Neurosci. 2006;24:1744–50. [DOI] [PubMed] [Google Scholar]
- 5.Wassenaar TM, Yaffe K, van der Werf YD, Sexton CE. Associations between modifiable risk factors and white matter of the aging brain: insights from diffusion tensor imaging studies. Neurobiol Aging. 2019;80:56–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karama S, Ducharme S, Corley J, Chouinard-Decorte F, Starr JM, Wardlaw JM, et al. Cigarette smoking and thinning of the brain’s cortex. Mol Psychiatry. 2015;20:778–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao L, Matloff W, Ning K, Kim H, Dinov ID, Toga AW. Age-related differences in brain morphology and the modifiers in middle-aged and older adults. Cereb Cortex. 2019;29:4169–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitsel N, Reynolds CA, Buchholz EJ, Pahlen S, Pearce RC, Hatton SN, et al. Long-term associations of cigarette smoking in early mid-life with predicted brain aging from mid-to late life. Addiction. 2022;117:1049–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Durazzo TC, Meyerhoff DJ, Nixon SJ. Chronic cigarette smoking: implications for neurocognition and brain neurobiology. Int J Environ Res Public Health. 2010;7:3760–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang C, Xu X, Qian W, Shen Z, Zhang M. Altered human brain anatomy in chronic smokers: a review of magnetic resonance imaging studies. Neurol Sci. 2015;36:497–504. [DOI] [PubMed] [Google Scholar]
- 11.Swan GE, Lessov-Schlaggar CN. The effects of tobacco smoke and nicotine on cognition and the brain. Neuropsychol Rev. 2007;17:259–73. [DOI] [PubMed] [Google Scholar]
- 12.Traylor M, Persyn E, Tomppo L, Klasson S, Abedi V, Bakker MK, et al. Genetic basis of lacunar stroke: a pooled analysis of individual patient data and genome-wide association studies. Lancet Neurol. 2021;20:351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Firth J, Solmi M, Wootton RE, Vancampfort D, Schuch FB, Hoare E, et al. A meta-review of ‘lifestyle psychiatry’: the role of exercise, smoking, diet and sleep in the prevention and treatment of mental disorders. World Psychiatry. 2020;19:360–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Logtenberg E, Overbeek MF, Pasman JA, Abdellaoui A, Luijten M, van Holst RJ, et al. Investigating the causal nature of the relationship of subcortical brain volume with smoking and alcohol use. Br J Psychiatry. 2022;221:377–85. [DOI] [PubMed] [Google Scholar]
- 15.Ho YS, Yang X, Yeung SC, Chiu K, Lau CF, Tsang AWT, et al. Cigarette smoking accelerated brain aging and induced pre-Alzheimer-like neuropathology in rats. PLOS ONE. 2012;7:e36752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durazzo TC, Mattsson N, Weiner MW, Korecka M, Trojanowski JQ, Shaw LM, et al. History of cigarette smoking in cognitively-normal elders is associated with elevated cerebrospinal fluid biomarkers of oxidative stress. Drug Alcohol Depend. 2014;142:262–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bittner N, Jockwitz C, Franke K, Gaser C, Moebus S, Bayen UJ, et al. When your brain looks older than expected: combined lifestyle risk and BrainAGE. Brain Struct Funct. 2021;226:621–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peters R. Ageing and the brain. Postgrad Med J. 2006;82:84–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franke K, Gaser C. Ten years of brainage as a neuroimaging biomarker of brain aging: what insights have we gained? Front Neurol. 2019;10:789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Truelove-Hill M, Erus G, Bashyam V, Varol E, Sako C, Gur RC, et al. A multidimensional neural maturation index reveals reproducible developmental patterns in children and adolescents. J Neurosci. 2020;40:1265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erus G, Battapady H, Satterthwaite TD, Hakonarson H, Gur RE, Davatzikos C, et al. Imaging patterns of brain development and their relationship to cognition. Cereb Cortex. 2015;25:1676–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cole JH, Franke K. Predicting age using neuroimaging: innovative brain ageing biomarkers. Trends Neurosci. 2017;40:681–90. [DOI] [PubMed] [Google Scholar]
- 23.Cole JH, Marioni RE, Harris SE, Deary IJ. Brain age and other bodily ‘ages’: implications for neuropsychiatry. Mol Psychiatry. 2019;24:266–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franke K, Ziegler G, Klöppel S, Gaser C, Alzheimer’s Disease Neuroimaging Initiative. Estimating the age of healthy subjects from T1-weighted MRI scans using kernel methods: exploring the influence of various parameters. Neuroimage. 2010;50:883–92. [DOI] [PubMed] [Google Scholar]
- 25.Cole JH, Poudel RP, Tsagkrasoulis D, Caan MW, Steves C, Spector TD, et al. Predicting brain age with deep learning from raw imaging data results in a reliable and heritable biomarker. Neuroimage. 2017;163:115–24. [DOI] [PubMed] [Google Scholar]
- 26.Franke K, Gaser C. Longitudinal changes in individual BrainAGE in healthy aging, mild cognitive impairment, and Alzheimer’s disease. GeroPsych. 2012;25:235–45. [Google Scholar]
- 27.Koutsouleris N, Davatzikos C, Borgwardt S, Gaser C, Bottlender R, Frodl T, et al. Accelerated brain aging in schizophrenia and beyond: a neuroanatomical marker of psychiatric disorders. Schizophr Bull. 2014;40:1140–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Kochunov P, Sampath H, Hatch KS, Ryan MC, Xue F, et al. White matter brain aging in relationship to schizophrenia and its cognitive deficit. Schizophr Res. 2021;230:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han LK, Dinga R, Hahn T, Ching CR, Eyler LT, Aftanas L, et al. Brain aging in major depressive disorder: results from the ENIGMA major depressive disorder working group. Mol Psychiatry. 2021;26:5124–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liem F, Varoquaux G, Kynast J, Beyer F, Kharabian Masouleh S, et al. Predicting brain-age from multimodal imaging data captures cognitive impairment. Neuroimage. 2017;148:179–88. [DOI] [PubMed] [Google Scholar]
- 31.Gaser C, Franke K, Klöppel S, Koutsouleris N, Sauer H, Alzheimer’s Disease Neuroimaging Initiative. BrainAGE in mild cognitive impaired patients: predicting the conversion to Alzheimer’s disease. PLOS ONE. 2013;8:e67346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cole JH, Leech R, Sharp DJ, Alzheimer’s Disease Neuroimaging Initiative. Prediction of brain age suggests accelerated atrophy after traumatic brain injury. Ann Neurol. 2015;77:571–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith SM, Vidaurre D, Alfaro-Almagro F, Nichols TE, Miller KL. Estimation of brain age delta from brain imaging. Neuroimage. 2019;200:528–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butler ER, Chen A, Ramadan R, Le TT, Ruparel K, Moore TM, et al. Pitfalls in brain age analyses. Hum Brain Mapp. 2021;42:4092–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bocklandt S, Lin W, Sehl ME, Sánchez FJ, Sinsheimer JS, Horvath S, et al. Epigenetic predictor of age. PLOS ONE. 2011;6:e14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rando TA, Chang HY. Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell. 2012;148:46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Linli Z, Feng J, Zhao W, Guo S. Associations between smoking and accelerated brain ageing. Prog Neuropsychopharmacol Biol Psychiatry. 2022;113:110471. [DOI] [PubMed] [Google Scholar]
- 38.Taylor-Bateman V, Gill D, Georgakis MK, Malik R, Munroe P, Traylor M. Cardiovascular risk factors and MRI markers of cerebral small vessel disease: a Mendelian randomization study. Neurology. 2022;98:e343–51. [DOI] [PubMed] [Google Scholar]
- 39.23andMe Research Team, HUNT All-In Psychiatry, Liu M, Jiang Y, Wedow R, Li Y, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51:237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vancampfort D, Probst M, Scheewe T, de Herdt A, Sweers K, Knapen J, et al. Relationships between physical fitness, physical activity, smoking and metabolic and mental health parameters in people with schizophrenia. Psychiatry Res. 2013;207:25–32. [DOI] [PubMed] [Google Scholar]
- 41.van Kesteren CFMG, Gremmels H, de Witte LD, Hol EM, van Gool AR, Falkai PG, et al. Immune involvement in the pathogenesis of schizophrenia: a meta-analysis on postmortem brain studies. Transl Psychiatry. 2017;7:e1075–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ringen PA, Engh JA, Birkenaes AB, Dieset I, Andreassen OA. Increased mortality in schizophrenia due to cardiovascular disease–a non-systematic review of epidemiology, possible causes, and interventions. Front Psychol. 2014;5:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larsson SC, Burgess S, Michaëlsson K. Smoking and stroke: a Mendelian randomization study. Ann Neurol. 2019;86:468–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhan Y, Fang F. Smoking and amyotrophic lateral sclerosis: a Mendelian randomization study. Ann Neurol. 2019;85:482–4. [DOI] [PubMed] [Google Scholar]
- 45.Yuan S, Yao H, Larsson SC. Associations of cigarette smoking with psychiatric disorders: evidence from a two-sample Mendelian randomization study. Sci Rep. 2020;10:13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Treur JL, Munafò MR, Logtenberg E, Wiers RW, Verweij KJ. Using Mendelian randomization analysis to better understand the relationship between mental health and substance use: a systematic review. Psychol Med. 2021;51:1593–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLOS Med. 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conroy M, Sellors J, Effingham M, Littlejohns TJ, Boultwood C, Gillions L, et al. The advantages of UK Biobank’s open-access strategy for health research. J Intern Med. 2019;286:389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pfefferbaum A, Sullivan EV, Hedehus M, Lim KO, Adalsteinsson E, Moseley M. Age-related decline in brain white matter anisotropy measured with spatially corrected echo-planar diffusion tensor imaging. Magn Reson Med. 2000;44:259–68. [DOI] [PubMed] [Google Scholar]
- 50.Ye Z, Mo C, Liu S, Hatch KS, Gao S, Ma Y, et al. White matter integrity and nicotine dependence: evaluating vertical and horizontal pleiotropy. Front Neurosci. 2021;15:738037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Erzurumluoglu AM, Liu M, Jackson VE, Barnes DR, Datta G, Melbourne CA, et al. Meta-analysis of up to 622,409 individuals identifies 40 novel smoking behaviour associated genetic loci. Mol Psychiatry. 2020;25:2392–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alfaro-Almagro F, Jenkinson M, Bangerter NK, Andersson JL, Griffanti L, Douaud G, et al. Image processing and quality control for the first 10,000 brain imaging datasets from UK biobank. Neuroimage. 2018;166:400–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burgess S, Davey Smith G, Davies NM, Dudbridge F, Gill D, Glymour MM, et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res. 2019;4:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Breiman L Random forests. Mach Learn. 2001;45:5–32. [Google Scholar]
- 56.Geurts P, Ernst D, Wehenkel L. Extremely randomized trees. Mach Learn. 2006;63:3–42. [Google Scholar]
- 57.Willmott CJ, Matsuura K. Advantages of the mean absolute error (MAE) over the root mean square error (RMSE) in assessing average model performance. Climate Res. 2005;30:79–82. [Google Scholar]
- 58.De Leeuw J Models and methods for the analysis of correlation coefficients. J Econom. 1983;22:113–37. [Google Scholar]
- 59.Beheshti I, Nugent S, Potvin O, Duchesne S. Bias-adjustment in neuroimaging-based brain age frameworks: a robust scheme. Neuro-Image Clin. 2019;24:102063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chung Y, Addington J, Bearden CE, Cadenhead K, Cornblatt B, Mathalon DH, et al. Use of machine learning to determine deviance in neuroanatomical maturity associated with future psychosis in youths at clinically high risk. JAMA Psychiatry. 2018;75:960–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liang H, Zhang F, Niu X. Investigating systematic bias in brain age estimation with application to post-traumatic stress disorders. Hum Brain Mapp. 2019;40:3143–52. 10.1002/hbm.24588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mo C, Ye Z, Ke H, Lu T, Canida T, Liu S, et al. A new Mendelian randomization method to estimate causal effects of multivariable brain imaging exposures. Pac Symp Biocomput. 2022;27:73–84. [PMC free article] [PubMed] [Google Scholar]
- 63.Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat Med. 2017;36:1783–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46:1734–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med. 2016;35:1880–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Verbanck M, Chia YC, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qi G, Chatterjee N. Mendelian randomization analysis using mixture models for robust and efficient estimation of causal effects. Nat Commun. 2019;10:1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burgess S, Foley CN, Allara E, Staley JR, Howson JM. A robust and efficient method for Mendelian randomization with hundreds of genetic variants. Nat Commun. 2020;11:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiol Camb Mass. 2017;28:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berrettini WH, Doyle GA. The CHRNA5–A3–B4 gene cluster in nicotine addiction. Mol Psychiatry. 2012;17:856–66. [DOI] [PubMed] [Google Scholar]
- 72.Saccone NL, Wang JC, Breslau N, Johnson EO, Hatsukami D, Saccone SF, et al. The CHRNA5-CHRNA3-CHRNB4 nicotinic receptor subunit gene cluster affects risk for nicotine dependence in African-Americans and in European-Americans. Cancer Res. 2009;69:6848–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ducci F, Kaakinen M, Pouta A, Hartikainen AL, Veijola J, Isohanni M, et al. TTC12-ANKK1-DRD2 and CHRNA5-CHRNA3-CHRNB4 influence different pathways leading to smoking behavior from adolescence to mid-adulthood. Biol Psychiatry. 2011;69:650–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li X, Li Z, Zhou H, Gaynor SM, Liu Y, Chen H, et al. Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nat Genet. 2020;52:969–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burgess S, Bowden J, Dudbridge F, Thompson SG. Robust instrumental variable methods using multiple candidate instruments with application to Mendelian randomization. arXiv 160603729; 2016. [Google Scholar]
- 76.Elliott ML, Belsky DW, Knodt AR, Ireland D, Melzer TR, Poulton R, et al. Brain-age in midlife is associated with accelerated biological aging and cognitive decline in a longitudinal birth cohort. Mol Psychiatry. 2021;126:3829–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kirova AM, Bays RB, Lagalwar S. Working memory and executive function decline across normal aging, mild cognitive impairment, and Alzheimer’s disease. Biomed Res Int. 2015;2015:748212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zein CO, Unalp A, Colvin R, Liu YC, McCullough AJ, Network NSCR. Smoking and severity of hepatic fibrosis in nonalcoholic fatty liver disease. J Hepatol. 2011;54:753–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ohkuma T, Iwase M, Fujii H, Kaizu S, Ide H, Jodai T, et al. Dose- and time-dependent association of smoking and its cessation with glycemic control and insulin resistance in male patients with type 2 diabetes mellitus: the Fukuoka diabetes registry. PLOS ONE. 2015;10:e0122023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Benowitz NL, Burbank AD. Cardiovascular toxicity of nicotine: implications for electronic cigarette use. Trends Cardiovasc Med. 2016;26:515–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pase MP, Himali JJ, Mitchell GF, Beiser A, Maillard P, Tsao C, et al. Association of aortic stiffness with cognition and brain aging in young and middle-aged adults: the Framingham third generation cohort study. Hypertension. 2016;67:513–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ferreira LS, Fernandes CS, Vieira MN, De Felice FG. Insulin resistance in Alzheimer’s disease. Front Neurosci. 2018;12:830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Willette AA, Johnson SC, Birdsill AC, Sager MA, Christian B, Baker LD, et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimers Dement. 2015;11:504–510.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bidwell LC, McGeary JE, Gray JC, Palmer RHC, Knopik VS, MacKillop J. NCAM1-TTC12-ANKK1-DRD2 variants and smoking motives as intermediate phenotypes for nicotine dependence. Psychopharmacology. 2015;232:1177–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Icick R, Forget B, Cloëz-Tayarani I, Pons S, Maskos U, Besson M. Genetic susceptibility to nicotine addiction: Advances and shortcomings in our understanding of the CHRNA5/A3/B4 gene cluster contribution. Neuropharmacology. 2020;177:108234. [DOI] [PubMed] [Google Scholar]
- 86.Hancock DB, Wang JC, Gaddis NC, Levy JL, Saccone NL, Stitzel JA, et al. A multiancestry study identifies novel genetic associations with CHRNA5 methylation in human brain and risk of nicotine dependence. Hum Mol Genet. 2015;24:5940–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Minicã CC, Mbarek H, Pool R, Dolan CV, Boomsma DI, Vink JM. Pathways to smoking behaviours: biological insights from the tobacco and genetics consortium meta-analysis. Mol Psychiatry. 2017;22:82–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chuang YH, Paul KC, Sinsheimer JS, Bronstein JM, Bordelon YM, Ritz B. Genetic variants in nicotinic receptors and smoking cessation in Parkinson’s disease. Parkinsonism Relat Disord. 2019;62:57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wen L, Yang Z, Cui W, Li MD. Crucial roles of the CHRNB3–CHRNA6 gene cluster on chromosome 8 in nicotine dependence: update and subjects for future research. Transl Psychiatry. 2016;6:e843–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brazel DM, Jiang Y, Hughey JM, Turcot V, Zhan X, Gong J, et al. Exome chip meta-analysis fine maps causal variants and elucidates the genetic architecture of rare coding variants in smoking and alcohol use. Biol Psychiatry. 2019;85:946–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee CR, North KE, Bray MS, Fornage M, Seubert JM, Newman JW, et al. Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: the atherosclerosis risk in communities (ARIC) study. Hum Mol Genet. 2006;15:1640–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim W, Prokopenko D, Sakornsakolpat P, Hobbs BD, Lutz SM, Hokanson JE, et al. Genome-wide gene-by-smoking interaction study of chronic obstructive pulmonary disease. Am J Epidemiol. 2021;190:875–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Patel YM, Park SL, Han Y, Wilkens LR, Bickeböller H, Rosenberger A, et al. Novel association of genetic markers affecting CYP2A6 activity and lung cancer risk. Cancer Res. 2016;76:5768–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Matoba N, Akiyama M, Ishigaki K, Kanai M, Takahashi A, Momozawa Y, et al. GWAS of smoking behaviour in 165,436 Japanese people reveals seven new loci and shared genetic architecture. Nat Hum Behav. 2019;3:471–7. [DOI] [PubMed] [Google Scholar]
- 95.Fry A, Littlejohns TJ, Sudlow C, Doherty N, Adamska L, Sprosen T, et al. Comparison of sociodemographic and health-related characteristics of UK biobank participants with those of the general population. Am J Epidemiol. 2017;186:1026–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brumpton B, Sanderson E, Heilbron K, Hartwig FP, Harrison S, Vie GÅ, et al. Avoiding dynastic, assortative mating, and population stratification biases in Mendelian randomization through withinfamily analyses. Nat Commun. 2020;11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.