

Key Points
Question
How does risk prediction based on proteomics data compare with clinical risk factors and polygenic risk scores?
Findings
In a retrospective analysis using measurements of thousands of plasma proteins in primary- and secondary-event populations, a protein risk score, developed using more than 4900 plasma protein levels to predict major atherosclerotic cardiovascular disease events, stratified risk as well as a risk factor score and was slightly better than polygenic risk scores for coronary artery disease and stroke. The protein score modestly improved discriminative accuracy (measured by C index) and risk classification (measured by category-free net reclassification improvement and integrated discrimination improvement) when added to a model using clinical risk factors.
Meaning
A protein risk score, derived from large-scale proteomics, yielded a modest improvement in discrimination when added to models based on clinical risk factors.
Abstract
Importance
Whether protein risk scores derived from a single plasma sample could be useful for risk assessment for atherosclerotic cardiovascular disease (ASCVD), in conjunction with clinical risk factors and polygenic risk scores, is uncertain.
Objective
To develop protein risk scores for ASCVD risk prediction and compare them to clinical risk factors and polygenic risk scores in primary and secondary event populations.
Design, Setting, and Participants
The primary analysis was a retrospective study of primary events among 13 540 individuals in Iceland (aged 40-75 years) with proteomics data and no history of major ASCVD events at recruitment (study duration, August 23, 2000 until October 26, 2006; follow-up through 2018). We also analyzed a secondary event population from a randomized, double-blind lipid-lowering clinical trial (2013-2016), consisting of individuals with stable ASCVD receiving statin therapy and for whom proteomic data were available for 6791 individuals.
Exposures
Protein risk scores (based on 4963 plasma protein levels and developed in a training set in the primary event population); polygenic risk scores for coronary artery disease and stroke; and clinical risk factors that included age, sex, statin use, hypertension treatment, type 2 diabetes, body mass index, and smoking status at the time of plasma sampling.
Main Outcomes and Measures
Outcomes were composites of myocardial infarction, stroke, and coronary heart disease death or cardiovascular death. Performance was evaluated using Cox survival models and measures of discrimination and reclassification that accounted for the competing risk of non-ASCVD death.
Results
In the primary event population test set (4018 individuals [59.0% women]; 465 events; median follow-up, 15.8 years), the protein risk score had a hazard ratio (HR) of 1.93 per SD (95% CI, 1.75 to 2.13). Addition of protein risk score and polygenic risk scores significantly increased the C index when added to a clinical risk factor model (C index change, 0.022 [95% CI, 0.007 to 0.038]). Addition of the protein risk score alone to a clinical risk factor model also led to a significantly increased C index (difference, 0.014 [95% CI, 0.002 to 0.028]). Among White individuals in the secondary event population (6307 participants; 432 events; median follow-up, 2.2 years), the protein risk score had an HR of 1.62 per SD (95% CI, 1.48 to 1.79) and significantly increased C index when added to a clinical risk factor model (C index change, 0.026 [95% CI, 0.011 to 0.042]). The protein risk score was significantly associated with major adverse cardiovascular events among individuals of African and Asian ancestries in the secondary event population.
Conclusions and Relevance
A protein risk score was significantly associated with ASCVD events in primary and secondary event populations. When added to clinical risk factors, the protein risk score and polygenic risk score both provided statistically significant but modest improvement in discrimination.
This study of 13 540 individuals in Iceland (aged 40-75 years) evaluated the utility of protein risk scores for prediction of atherosclerotic cardiovascular disease events compared to risk prediction using polygenic risk scores in addition to risk scores based on clinical risk factors
Introduction
Atherosclerotic cardiovascular disease (ASCVD) is largely preventable through management of known risk factors, but risk assessment needs to be improved.1,2,3,4,5
Sequence variants have been combined into polygenic risk scores for the prediction of coronary artery disease,6,7,8,9,10,11 and there is evidence that polygenic risk scores complement conventional risk prediction in White populations for whom large data sets of genotyped individuals are available.8,9,10 However, the clinical utility of polygenic risk scores for ASCVD risk assessment in the general population has yet to be demonstrated in prospective studies.
Proteins mediate phenotypic effects of genomic sequence diversity and many environmental factors. They also reflect ongoing biological processes. As such, protein levels can supplement information regarding health status12 and disease risk. The recent emergence of technologies for large-scale measurements of protein levels13,14 has allowed for the identification of new protein biomarkers predictive of ASCVD events.15,16,17,18,19,20
The objective of this study was to evaluate the utility of protein risk scores for prediction of ASCVD events compared with risk prediction using polygenic risk scores in addition to risk scores based on clinical risk factors.
Methods
A detailed description of methods and populations is provided in eAppendix 1 in Supplement 1.
Study Cohorts
The primary event population was a subset of a population-based cohort of Icelanders21 with protein levels measured in plasma (Table 1, Figure 1). The study start date was the day of plasma sampling, which was conducted in the years 2000-2006 (eFigure 1 in Supplement 1). The end point considered was the first major ASCVD event, composed of first myocardial infarction, first fatal or nonfatal ischemic or hemorrhagic stroke, or coronary heart disease death. Events were ascertained based on International Classification of Diseases, Ninth Revision, Clinical Modification and the International Statistical Classification of Diseases and Related Health Problems, Tenth Revision, Clinical Modification diagnosis codes from the Icelandic health care system and International Classification of Diseases, Eighth Revision diagnosis codes for the FinnGen study (eAppendix 1 in Supplement 1). The population consisted of participants aged 40 to 75 years who had not experienced major ASCVD events or undergone a percutaneous coronary intervention or coronary artery bypass grafting. Participants were followed-up through 2018.
Table 1. Baseline Characteristics of Primary and Secondary Event Populations.
| No. (%)a | ||||
|---|---|---|---|---|
| Primary event population (Iceland) | Secondary event population | |||
| Derivation set (n = 9522) | Test set (n = 4018) | With proteomics (n = 6307)b | With proteomics and genotyping (n = 3887)b,c | |
| Age at plasma sampling, mean (SD), y | 57.2 (10.3) | 57.2 (10.1) | 62.8 (8.9) | 63.0 (8.8) |
| Women | 5720 (60.1) | 2370 (59.0) | 1537 (24.4) | 968 (24.9) |
| Men | 3802 (39.9) | 1648 (41.0) | 4770 (75.6) | 2919 (75.1) |
| Body mass index, mean (SD)d | 26.9 (4.5) | 26.9 (4.6) | 30 (5.1) | 30 (5.1) |
| Current smoker | 1465 (15.4) | 610 (15.2) | 1798 (30.1)e | 1140 (30.9)e |
| Type 2 diabetes | 299 (3.1) | 139 (3.5) | 2098 (33.3) | 1263 (32.5) |
| Hypertension | 3230 (33.9)f | 1282 (31.9)f | 5121 (85.7)e | 3139 (85.0)e |
| Statin medication | 527 (5.5) | 236 (5.9) | 6307 (100.0) | 3887 (100.0) |
| Prior coronary artery disease | 211 (2.2) | 96 (2.4) | 5473 (86.8) | 3380 (87.0) |
| Prior peripheral arterial disease | 931 (14.8) | 609 (15.7) | ||
| Prior myocardical infarction | 5151 (81.7) | 3167 (81.5) | ||
| Prior stroke | 1105 (17.5) | 658 (16.9) | ||
| Length of follow-up, median (IQR), y | 15.8 (13.4 to 16.8) | 15.8 (13.6 to 16.8) | 2.2 (1.8 to 2.5) | 2.3 (1.9 to 2.6) |
| Events (first atherosclerotic cardiovascular disease event or major adverse cardiovascular event)g | 1042 (10.9) | 465 (11.6) | 432 (6.8) | 271 (7) |
| Nonfatal myocardial infarction | 581 (6.1) | 239 (5.9) | 290 (4.6) | 183 (4.7) |
| Coronary heart disease death | 110 (1.2) | 55 (1.4) | ||
| Cardiovascular death | 49 (0.8) | 26 (0.7) | ||
| Stroke | 347 (3.6) | 170 (4.2) | 89 (1.4) | 61 (1.6) |
| Ischemic stroke | 282 (3.0) | 129 (3.2) | ||
| Hemorrhagic stroke | 64 (0.7) | 41 (1.0) | ||
| Multiple event typesh | 4 (<0.1) | 1 (<0.1) | 4 (0.1) | 1 (<0.1) |
| Years until event, median (IQR) | 8.5 (4.9 to 12.0) | 8.6 (4.4 to 12.0) | 1.1 (0.6 to 1.7) | 1.2 (0.6 to 1.7) |
| Death from other causes | 1705 (17.9) | 678 (16.9) | 3 (0.05) | 2 (0.05) |
| Years until death from other causes, median (IQR) | 9.0 (4.5 to 13.0) | 9.6 (4.7 to 13.4) | ||
Numeric values are reported as No. (%) unless otherwise indicated.
The secondary event population in this table provided data regarding White participants. More information regarding participants of African and Asian ancestries in the secondary event population is provided in eAppendix 1 in Supplement 1, eFigure 14 in Supplement 1, and in eTable 11 in Supplement 2.
Not all individuals in the secondary event population had both genotype and proteomics data available. This set includes White participants with both proteomics and genotyping.
Calculated as weight in kilograms divided by height in meters squared.
Some percentages do not calculate because of missing values (<6%).
In the primary event population, information regarding use of hypertension medication was used to infer whether individuals had hypertension.
In the primary event population, the event considered was the first hard atherosclerotic cardiovascular disease event. In the secondary event population most participants previously had a first atherosclerotic cardiovascular disease event, and the event considered was the next major adverse cardiovascular event.
Indicates individuals who had a concurrent stroke and myocardial infarction diagnosis. Numeric values for the secondary event population indicate those who died of myocardial infarction or stroke or who had concurrent stroke and myocardial infarction diagnoses.
Figure 1. Overview of Study of Derivation of Protein Risk Score, Polygenic Risk Scores, and the Comparative Analysis.
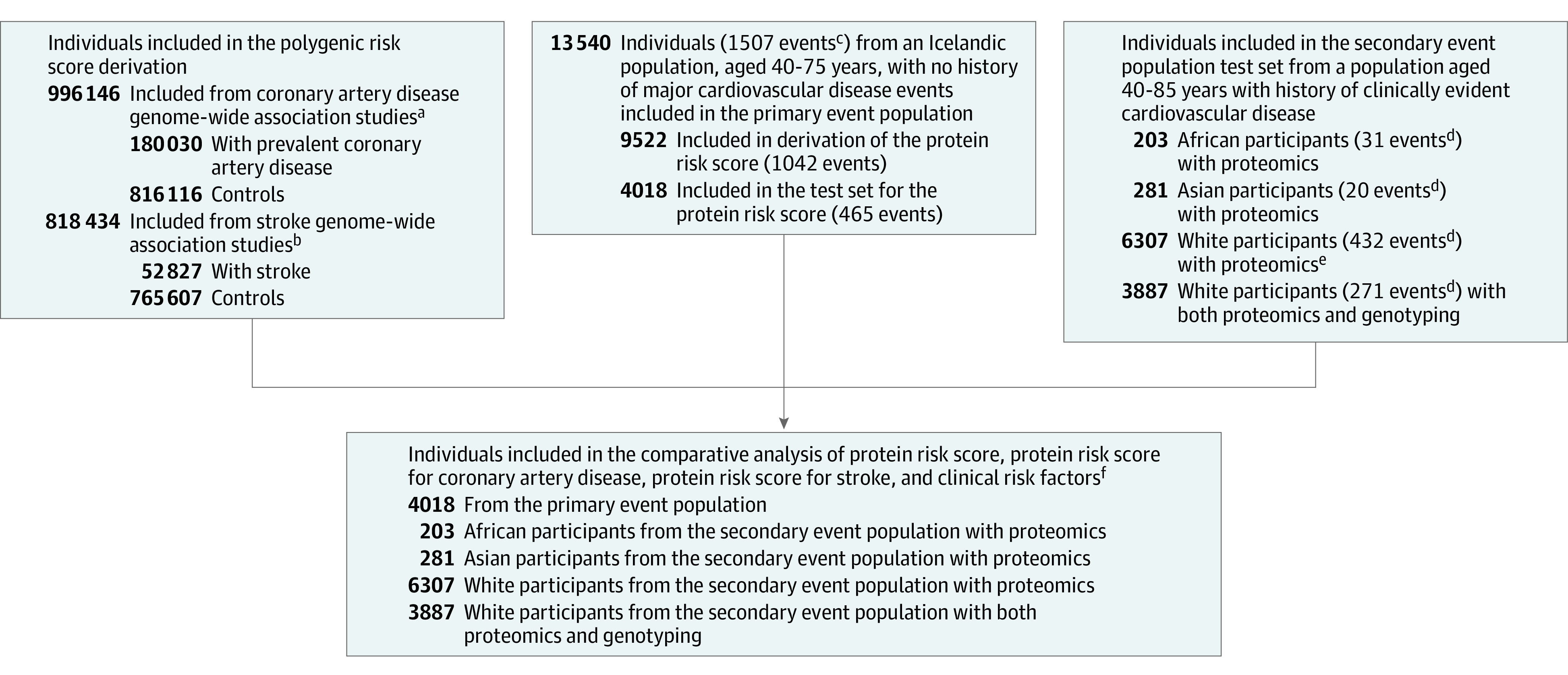
The diagram gives an overview of the populations used and how the study is divided into derivation and testing data sets. Some boxes do not show total individuals because subcategories are not mutually exclusive.
aPopulation sources: UK Biobank, FinnGen, Copenhagen Hospital Biobank (part of the Genetics of Cardiovascular Disease Study), Danish Blood Donor Study, and CARDIoGRAMplusC4D.
bPopulation sources: UK Biobank, Finngen, and Megastroke.
cComposite end point of first myocardial infarction, first stroke, or coronary heart disease death.
dIndicates the composite end point of myocardial infarction, stroke, or cardiovascular disease death.
eThe counts for the proteomics set include those with both proteomics and genotyping (ie, the sets are not mutually exclusive).
fTest data for this analysis were separate from derivation of polygenic and protein risk scores.
The secondary event population consisted of participants with established ASCVD in the placebo group of the FOURIER clinical trial,22,23 conducted in 2013 through 2016. The primary analysis was composed of White participants (6307 with protein measurements; 6583 who were genotyped; 3887 with protein measurements as well as genotyping). The end point considered in the FOURIER trial was first recurrent major adverse cardiovascular event (a composite of myocardial infarction, stroke, or cardiovascular death). The total number of composite events among the 6307 White participants with protein measurements at baseline was 432 with median follow-up of 2.2 years (Table 1, Figure 1). A secondary analysis involved participants with protein measurements who were of Asian (281 participants; 20 events) and African (203 participants; 31 events) ancestries.
Proteomics Measurements
In both the secondary and primary event populations, 4963 protein levels were measured using the SomaScan13 v4 platform, with 1 thawing cycle for the majority of the samples during preparation. For the primary event population, the participants were nonfasting at plasma collection, and all samples were analyzed between January 2019 and June 2019. For the secondary event population, the participants fasted for at least 9 hours prior to plasma collection, and all samples were analyzed between August 2020 and January 2021.
Clinical Risk Factors and Clinical Risk Factor Models
For analysis in the primary event population, selection of clinical risk factors took into account the 2013 American College of Cardiology/American Heart Association Pooled Cohort Equations3 and data availability near the time of plasma sampling. In addition to age and sex, other factors collected were information on statin use, hypertension treatment, type 2 diabetes, body mass index (calculated as weight in kilograms divided by height in meters squared), and smoking status at the time of plasma sampling (Table 1; eTable 1 in Supplement 2). Cholesterol and blood pressure measurements at the time of plasma sampling were not available for the majority of participants in the primary event population, and information regarding statin use and hypertension treatment were used instead.
For comparison in the secondary event population, we mainly considered a refitted version of the updated Second Manifestations of Arterial disease (SMART2) score.24 The score includes age, age squared, sex, use of antithrombotic medication, history of cerebrovascular disease, peripheral artery disease, coronary artery disease, C-reactive protein levels, non–high-density lipoprotein levels, systolic blood pressure, estimated glomerular filtration rate, estimated glomerular filtration rate squared, years since first ASCVD diagnosis, years since first ASCVD diagnosis squared, current smoking status, and diabetes. Information on medical history for abdominal aortic aneurysms, which is part of the SMART2 score, was not available for a large proportion of the secondary event population and was therefore omitted. We refitted the SMART2 score to our data by estimating weights using a Cox proportional hazards model and including individuals from the secondary event population who were separate from the test set (5403 individuals [413 events] in the placebo group of the FOURIER trial who did not have proteomics data. The weights for the refitted SMART2 score refitted to our data are included in eTable 2 in Supplement 2.
Protein Risk Score Development
For deriving the protein risk score, we used a derivation set (ie, training set) consisting of a random 70% subset of the primary event population (9522 participants; 1042 events) reserving the remaining 30% (4018 participants; 465 events) for testing the score. The score was constructed using all the 4963 protein levels, age, and sex in a Cox proportional hazard model25 in which participants who died from causes other than ASCVD were censored. Lasso penalty26 was used for model selection, and cross-validation was used to select penalization strength to determine the number of variables included in the model (eFigure 2 in Supplement 1). The whole derivation set and the penalty from the cross-validation were then used in the lasso objective function to obtain the coefficients in the protein risk score. The protein risk score was then calculated for all individuals in both populations with available protein measurements as the linear part of the Cox model, excluding age and sex, using the protein score coefficients obtained using the derivation set (eTable 3 in Supplement 2).
Polygenic Risk Score Development
Polygenic risk scores were generated using methods previously described.27 Additionally, these scores were developed using effect estimates from coronary artery disease and stroke genome-wide association studies for populations of White individuals that were separate from the primary and secondary event populations (ie, populations that were entirely external to the sets in which they were validated). Weights for the polygenic risk score were based on effect estimates in a combination of study cohorts for prevalent coronary artery disease (180 030 participants with prevalent coronary artery disease; 816 116 control participants) and ischemic stroke (52 827 ischemic stroke participants; 765 607 control participants). Detailed description of the development of polygenic risk scores is given in eAppendix 1 in Supplement 1.
Statistical Analysis
For evaluation of prediction performance in the primary event population, coefficients for clinical risk factors, polygenic risk scores, and protein risk score were fitted in the training set for each model (the weights in the polygenic risk scores and protein risk score formulas were not refitted). For survival analyses, hazard ratios (HRs), CIs, and significance levels were calculated using Cox proportional hazards models; participants who died from causes other than ASCVD were censored. The HR for a score (eg, protein risk score or polygenic risk score) was reported in 1–standard deviation (SD) increase in the score. To estimate the discrimination ability of models, we used the Wolbers C index28 to account for the competing risk of non-ASCVD death. Bootstrapping was used to estimate the CIs of C indices and their differences between models. In the case of nested models, change in model deviance based on log partial likelihood was used to statistically test the contribution of a predictor. Associations of variables with the protein risk score were calculated using linear regression with age and sex as covariates.
Model calibration was assessed by comparing observed vs predicted event rates based on Cox modeling. To evaluate the improvement in prediction performance gained by adding a variable to a baseline predictor, the net reclassification improvement using 7.5% and 20% predicted 10-year risk thresholds,29 the integrated discrimination improvement,30 and the category-free net reclassification improvement31 were used. Bootstrapping was used to estimate CIs.
R software was used for all statistical analysis, hypothesis tests were 2-sided, P values less than .05 were considered statistically significant, and Bonferroni correction was used to account for multiple testing. Sensitivity analysis was performed for the primary event population by separating at the median age.
Results
The primary event population consisted of 13 540 genotyped Icelanders (8090 [59.7%] women) with meedian follow-up of 15.8 years. A major ASCVD event was experienced by 1507 individuals with a median time to event of 8.5 years (Table 1, Figure 1, eFigure 1 in Supplement 1, and eTable 1 in Supplement 2).
The protein risk score formula obtained from the derivation set was composed of 70 proteins (eFigure 3 in Supplement 1, eTable 3 in Supplement 2). When considering a Cox model with protein risk score, age, and sex in the primary event population test set (4018 participants [465 events]; eFigures 3 and 4 in Supplement 1), the protein risk score was statistically significantly associated with primary ASCVD events (HR, 1.93 per SD [95% CI, 1.75 to 2.13]; P = 1.5e-39; Table 2). The C index for the model was 0.737 compared with a C index of 0.719 for a basic model with only age and sex (Table 3).
Table 2. Cox Survival Analysis of Protein and Polygenic Risk Scores for White Participants in the Primary Event and Secondary Event Populations.
| Exposure | Covariates | Hazard ratio per SD (95% CI)a | |
|---|---|---|---|
| Primary event populationb | Secondary event populationc | ||
| Protein risk score models | |||
| No. of participants/events | 4018/465 | 6307/432 | |
| Protein risk score | Age + sex | 1.93 (1.75 to 2.13) | 1.62 (1.48 to 1.79) |
| Protein risk score | Clinical risk factorsb,c | 1.83 (1.64 to 2.04) | 1.35 (1.22 to 1.50) |
| Polygenic risk score models | |||
| No. of participants/events | 4018/465 | 6583/489 | |
| Polygenic risk score for coronary artery disease | Age + sex | 1.39 (1.26 to 1.52) | 1.08 (0.99 to 1.18) |
| Polygenic risk score for stroke | Age + sex | 1.21 (1.10 to 1.32) | 1.14 (1.04 to 1.24) |
| Both polygenic risk scores | Age + sex | ||
| Polygenic risk score for coronary artery disease | 1.36 (1.24 to 1.50) | 1.06 (0.96 to 1.16) | |
| Polygenic risk score for stroke | 1.16 (1.06 to 1.27) | 1.12 (1.03 to 1.23) | |
| Protein risk score + polygenic risk score models | |||
| No. of participants/events | 4018/465 | 3887/271 | |
| Protein risk score + polygenic risk scores | Age + sex | ||
| Protein risk score | 1.87 (1.69 to 2.07) | 1.56 (1.38-1.76) | |
| Polygenic risk score for coronary artery disease | 1.27 (1.15 to 1.39) | 1.04 (0.92-1.18) | |
| Polygenic risk score for stroke | 1.16 (1.06 to 1.28) | 1.14 (1.01-1.29) | |
| Protein risk score + polygenic risk scores | Clinical risk factorsb,c | ||
| Protein risk score | 1.79 (1.60 to 2.00) | 1.28 (1.12 to 1.46) | |
| Polygenic risk score for coronary artery disease | 1.25 (1.14 to 1.38) | 1.02 (0.90 to 1.15) | |
| Polygenic risk score for stroke | 1.16 (1.06 to 1.28) | 1.13 (1.00 to 1.28) | |
Abbreviation: SMART, secondary manifestations of arterial disease.
The hazard ratio is given in a standard deviation unit of the variable in the test set used. In all instances, the 95% CI for the protein risk score indicates that it is a statistically significant variable in the Cox model (significance levels are provided in eTables 5, 11, and 17 in Supplement 2).
In the primary event population, the clinical risk factors are age, sex, smoking, body mass index (calculated as weight in kilograms divided by height in meters squared), type 2 diabetes, hypertension treatment, and statin use.
The clinical risk factors correspond to the refitted SMART2 risk score using a training set in the secondary event population.
Table 3. Concordance and Reclassification Indices.
| Model | Wolbers C index (95%CI)a | Comparison to reference models | |
|---|---|---|---|
| Wolbers C index increase (95% CI)a,b | 10-y Risk integrated discrimination improvement (95% CI) | ||
| Primary event population: No. of participants/events, 4018/465 | |||
| Age + sex | 0.719 (0.698 to 0.741) | ||
| Reference model: age + sex | |||
| Age + sex + polygenic risk scores | 0.737 (0.716 to 0.759) | 0.018 (0.009 to 0.028) | 0.014 (0.008 to 0.020) |
| Age + sex + protein risk score | 0.747 (0.727 to 0.768) | 0.028 (0.013 to 0.045) | 0.056 (0.037 to 0.074) |
| Age + sex + protein risk score + polygenic risk scores | 0.756 (0.737 to 0.777) | 0.038 (0.021 to 0.056) | 0.066 (0.046 to 0.084) |
| Clinical risk factorsc | 0.736 (0.713 to 0.759) | 0.017 (0.007 to 0.027) | 0.015 (0.008 to 0.021) |
| Reference model: age + sex + polygenic risk scores | |||
| Age + sex + protein risk score + polygenic risk scores | 0.756 (0.737 to 0.777) | 0.019 (0.005 to 0.034) | 0.052 (0.034 to 0.069) |
| Reference model: age + sex + protein risk score | |||
| Age + sex + protein risk score + polygenic risk scores | 0.756 (0.737 to 0.777) | 0.009 (0.003 to 0.015) | 0.010 (0.005 to 0.015) |
| Reference model: clinical risk factors | |||
| Age + sex + protein risk score | 0.747 (0.727 to 0.768) | 0.011 (−0.002 to 0.025) | 0.041 (0.023 to 0.057) |
| Clinical risk factors + polygenic risk scores | 0.749 (0.728 to 0.770) | 0.013 (0.005 to 0.021) | 0.009 (0.004 to 0.015) |
| Clinical risk factors + protein risk score | 0.750 (0.730 to 0.771) | 0.014 (0.002 to 0.028) | 0.045 (0.028 to 0.062) |
| Clinical risk factors + protein risk score + polygenic risk scores | 0.758 (0.738 to 0.778) | 0.022 (0.007 to 0.038) | 0.053 (0.034 to 0.071) |
| Secondary event population | |||
| No. of participants/events, 6307/432 | 2-y Risk integrated discrimination improvement (95% CI) | ||
| Age + sex | 0.570 (0.541 to 0.602) | ||
| Reference model: age + sex | |||
| Age + sex + protein risk score | 0.667 (0.634 to 0.702) | 0.096 (0.062 to 0.128) | 0.015 (0.011 to 0.019) |
| Refitted SMART2 scored | 0.651 (0.617 to 0.689) | 0.081 (0.042 to 0.119) | 0.017 (0.013 to 0.022) |
| Reference model: refitted SMART2 scored | |||
| Age + sex + protein risk score | 0.667 (0.634 to 0.702) | 0.015 (−0.014 to 0.045) | −0.002 (−0.007 to 0.002) |
| Refitted SMART2 scored + protein risk score | 0.678 (0.646 to 0.710) | 0.026 (0.011 to 0.042) | 0.004 (0.002 to 0.007) |
Abbreviation: SMART, secondary manifestations of arterial disease.
This table shows Wolbers C indices that account for the competing risk of non–athersclerotic cardiovascular disease death (Harrell C indices are shown in eTable 4 in Supplement 2).
C index increase corresponds to difference relative to the reference model.
In the primary event population, the clinical risk factors are as follows: age, sex, smoking, body mass index (calculated as weight in kilograms divided by height in meters squared), type 2 diabetes, hypertension treatment, and statin use.
Indicates a refitted (to our data) SMART2 risk score using a training set in the secondary event population.
The polygenic risk score for coronary artery disease and the polygenic risk score for stroke were statistically significantly associated with major ASCVD events in the primary event population test data, both when considered separately in univariate models and in a joint model (Table 2). In the test set, the 2 polygenic risk scores were correlated (ρ = 0.16 [95% CI, 0.13 to 0.19]), while the protein risk score did not correlate with the polygenic risk score for stroke (ρ = 0.02 [95% CI, −0.01 to 0.05]) and was only weakly correlated with the polygenic risk score for coronary artery disease (ρ = 0.08 [95% CI, 0.05 to 0.11]) (eFigure 3 and eFigure 5 in Supplement 1). When the protein risk score was added to the model with the 2 polygenic risk scores, all 3 variables were statistically significant, and the joint estimates were slightly attenuated compared with the univariate models (Table 2). This joint model had a marginally higher C index than the model with the protein risk score, age, and sex (difference, 0.009 [95% CI, 0.003 to 0.015]). Adding the protein risk score to a model including the polygenic risk scores increased the C index more (difference, 0.019 [95% CI, 0.005 to 0.034]) (Table 3).
Comparing models with and without the protein risk score, the HRs for the clinical risk factors (age, sex, statin use, hypertension treatment, type 2 diabetes, body mass index, and smoking status) were all attenuated with the inclusion of the protein risk score (eTable 5 in Supplement 2). The effect of the protein risk score was less affected by the inclusion of these clinical risk factors in the models, decreasing from an HR of 1.87 to 1.79 per SD (95% CI, 1.60 to 2.00) (Table 2). All of the clinical risk factors correlated with the protein risk score (eTable 6 in Supplement 2). Discrimination increased when adding the protein risk score to the clinical risk factors, with C index increasing from 0.736 to 0.750 (difference, 0.014 [95% CI, 0.002 to 0.028]; Table 3) (for concordance indices at different time points, see eTable 7 in Supplement 2 and eFigure 6 in Supplement 1). The addition of the polygenic risk scores to the model with the protein risk score and the clinical risk factors increased the C index (from 0.750 to 0.758; difference,0.008 [95% CI, 0.001 to 0.014]).
Overall, the models appeared to be well calibrated (eFigure 7 in Supplement 1), with the difference between predicted and observed risk being less than 5% in all deciles. Inclusion of the protein risk score, with or without polygenic risk scores, in addition to the clinical risk factors did not result in significant net reclassification improvement of classification into low- and intermediate-risk groups using a predicted 7.5% 10-year risk threshold in which the overall proportion of individuals reclassified was 15.1% with the addition of the protein risk score and 16.0% when the protein risk score and polygenic risk scores were added (net reclassification improvement for the protein risk score, 0.04 [95% CI, −0.009 to 0.094]; net reclassification improvement for the protein risk score and polygenic risk scores, 0.053 [95% CI, −0.006 to 0.110]) (Figure 2). When considering a high-risk threshold of 20% 10-year predicted risk, including the protein risk score and polygenic risk scores in addition to clinical risk factors resulted in a net reclassification improvement of 0.084 (95% CI, 0.034 to 0.134) with an overall proportion of 8.1% of individuals being reclassified (eFigure 8 in Supplement 2). The metrics integrated discrimination improvement, and category-free net reclassification improvement showed significant improvement when the protein risk score, the polygenic risk scores, or both were added to a model with the clinical risk factors (Table 3; eTable 8 in Supplement 2). We observed that individuals who died of causes other than ASCVD within 10 years tended to have higher predicted risk than those who survived 10 years without ASCVD for all models (eFigure 9 in Supplement 1).
Figure 2. Reclassification in the Primary Event Population.
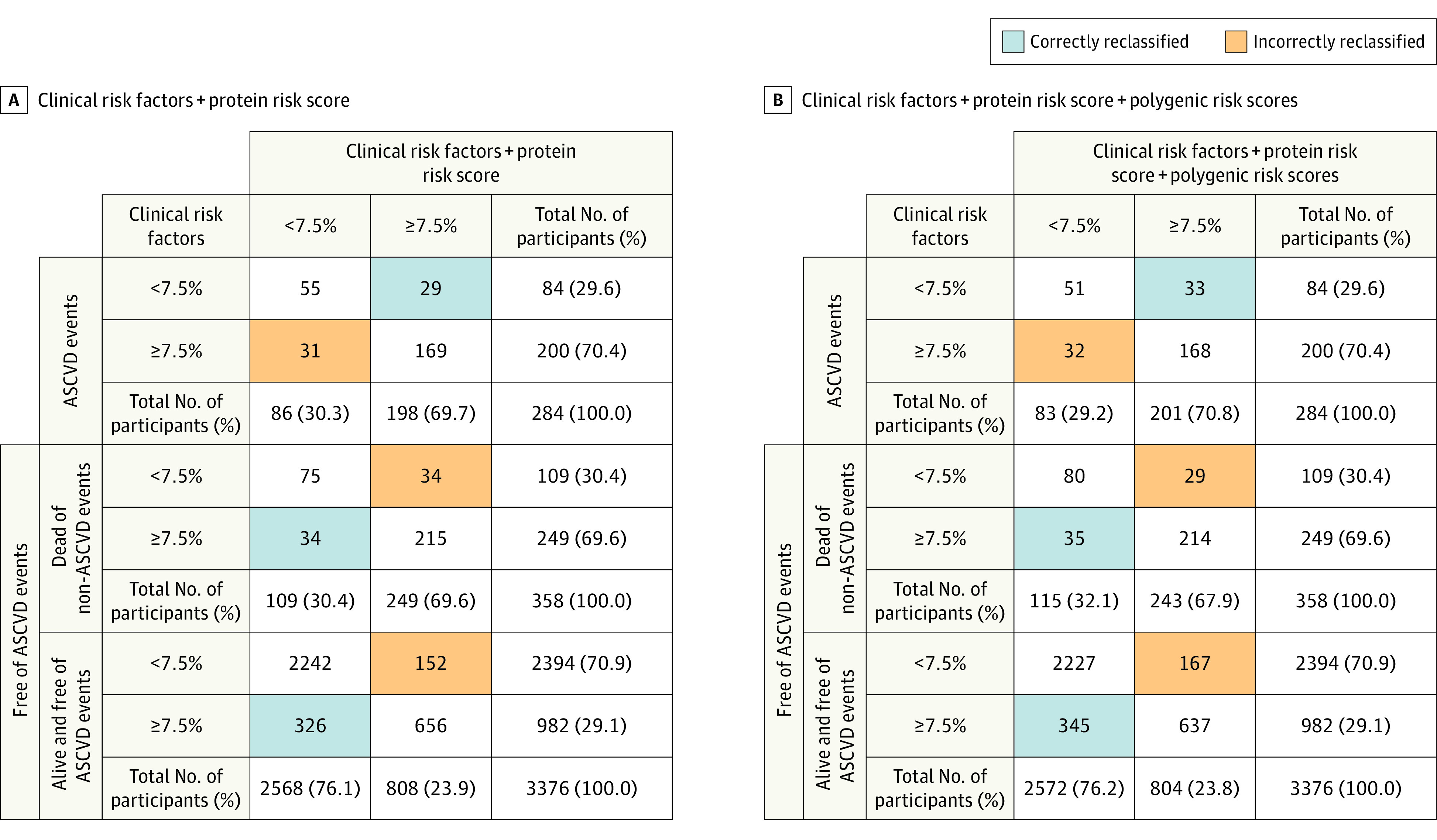
The figure shows reclassification results when protein risk score and polygenic risk scores are added on top of clinical risk factors (age, sex, statin use, hypertension treatment, type 2 diabetes, body mass index [calculated as weight in kilograms divided by height in meters squared], and smoking status at the time of plasma collection) in the primary event population test set (4018 participants; 465 events; 284 events within 10 years). The results are shown for predicted 10-year risk using a 7.5% risk threshold to separate between low and intermediate risk. Three groups are considered: (i) those with an atherosclerotic cardiovascular disease (ASCVD) event within 10 years; (ii) those who die of causes other than ASCVD within 10 years; and (iii) those who survive 10 years without an ASCVD event.
Panel A shows a reclassification table for the addition of protein risk score on top of the clinical risk factors. The total categorical net reclassification improvement is 0.040 (95% CI, −0.009 to 0.094) and 0.044 (95% CI, −0.013 to 0.096) when excluding those who die from non-ASCVD causes. Net reclassification improvement for ASCVD events is −0.007 (95% CI, −0.056 to 0.046), 0.047 (95% CI, 0.035 to 0.059) for ASCVD nonevents (groups [ii] and [iii]), and 0.052 (95% CI, 0.039 to 0.064) for those who survive 10 years without ASCVD event (group [iii]).
Panel B shows a reclassification table when both protein risk score and polygenic risk scores are added on top of the clinical risk factors. The total net reclassification improvement is 0.053 (95% CI, −0.006 to 0.110) and 0.056 (95% CI, −0.001 to 0.114) when excluding those who die from non-ASCVD causes. Net reclassification improvement for ASCVD events is 0.004 (95% CI, −0.054 to 0.058), 0.049 (95% CI, 0.037 to 0.061) for ASCVD nonevents (groups [ii] and [iii]), and 0.053 (95% CI, 0.039 to 0.066) for those who survive 10 years without ASCVD event (group [iii]).
The protein risk score was evaluated in a sensitivity analysis in both younger and older age groups. After adjusting for the clinical risk factors and polygenic risk scores, the protein risk score was statistically significantly associated with the composite end point when restricting to individuals younger than the median age of 56.6 years (P = 4.5e-12) and when considering only those older than the median age (P = 2.4e-16) (eFigure 10 in Supplement 1 and eTable 9 in Supplement 2).
In a secondary analysis of White individuals in the secondary event population (6307 participants [432 events]; median follow-up, 2.2 years; median time to event, 1.1 years), the protein risk score was statistically significantly associated with the major adverse cardiovascular event end point, with an HR of 1.62 per SD (95% CI, 1.48 to 1.79) after adjusting for age and sex. For the analysis of the protein risk score in conjunction with clinical risk factors, we used an adapted SMART2 score in which coefficients for risk factors were refitted in a subset of the secondary event population separate from the test set (eTable 2 in Supplement 2). Alone, the adapted SMART2 score refitted to our data was statistically significantly associated with an HR of 1.59 per SD (95% CI, 1.46 to 1.73). When assessing discrimination based on C indices, we did not observe a significant difference between the adapted SMART2 score (C index, 0.651) and a model with only the protein risk score, age, and sex (C index, 0.667; difference, 0.015 [95% CI, −0.014 to 0.045]).
In a joint model with the adapted SMART2 score, the protein risk score was statistically significantly associated with an adjusted HR of 1.35 per SD (95% CI, 1.22 to 1.50). The addition of the protein risk score to a model with the adapted SMART2 score resulted in a statistically significant change in C index from 0.651 to 0.678 (difference, 0.026 [95% CI, 0.011 to 0.042]) (Table 3). Similar results were observed when looking at the area under the curve at different time points (eFigure 11 in Supplement 1).
Calibration was assessed by comparing expected and actual 2-year event rates for the protein risk score and the adapted SMART2 score both separately and together in a single model. Overall, these 3 models appeared to be well calibrated, with the difference between predicted and observed risk being less than 5% in all deciles (eFigure 12 in Supplement 1). Results on reclassification metrics are provided in Table 3 and in eTable 8 in Supplement 2.
Among the genotyped individuals in the secondary event population (6583 participants; 489 events), the polygenic risk score for stroke was statistically significantly associated with the major adverse cardiovascular event end point (HR, 1.14 per SD [95% CI, 1.04 to 1.24]) while the polygenic risk score for coronary artery disease was not (HR, 1.08 per SD [95% CI, 0.99 to 1.18]) (Table 2). Among White individuals with both genotype and proteomics data in the secondary event population (3887 participants; 271 events), the protein risk score correlated neither with polygenic risk score for stroke (ρ = 0.00 [95% CI, −0.03 to 0.03]) nor polygenic risk score for coronary artery disease (ρ = 0.03 [95% CI, −0.01 to 0.06]; eFigure 13 in Supplement 1). The HR of the protein risk score in the secondary event population data set was not statistically significantly altered when the 2 polygenic risk scores were added to the model (Table 2). The resulting change in C index was not statistically significant (difference, 0.007 [95%CI, −0.006 to 0.019]), while adding the protein risk score to a model including the polygenic risk scores increased the C index from 0.561 to 0.655 (difference, 0.094 [95% CI, 0.027 to 0.149]). When both the protein risk score and the polygenic risk scores were added to the adapted SMART2 score model, the C index increased from 0.668 to 0.684 (difference, 0.016 [95% CI, 0.000 to 0.031]) (eTable 10 in Supplement 2).
We tested the association of the protein score in individuals who self-reported as having Asian and African ancestry in the secondary event population. There were considerably fewer participants from these groups than participants who self-reported as White (281 Asian ancestry [20 events]; 203 African ancestry [31 events]). The protein risk score was statistically significantly associated with a major adverse cardiovascular event in both groups when considered alone with age and sex adjustment (HR, 1.82 per SD [P = .008] for participants with Asian ancestry; HR, 1.82 per SD [P = .001] for participants with African ancestry). When performing a meta-analysis that included all 3 populations, the combined protein risk score effect was an HR of 1.64 (95% CI, 1.50 to 1.79), and no heterogeneity in effect was observed (P value for heterogeneity = .74). The protein risk score remained significant in the Asian ancestry and African ancestry populations when added to models including clinical risk factors through the SMART2 score (eFigure 14 in Supplement 1 and eTable 11 in Supplement 2).
Results and considerations related to interpretation of the protein risk score formula and methodology for its construction are provided in eAppendix 1 in Supplement 1 along with further analysis involving clinical risk factors in the secondary event population (eAppendix 1, eAppendix 2, and eFigures 15-18 in Supplement 1 and eTables 12-17 in Supplement 2).
Discussion
In this study, we have developed a protein risk score derived from circulating proteins in a large primary ASCVD event cohort. When added to a model that included clinical risk factors and treatment-related variables and also polygenic risk scores for coronary artery disease and stroke, the protein risk score modestly improved risk prediction for first and recurrent ASCVD events.
The protein risk score correlates with clinical risk factors for ASCVD and remains significantly associated with ASCVD after accounting for them. This is supported by the HR conveyed by the protein risk score, along with statistically significant improvement in discrimination that was observed in the primary and the secondary event populations. When considering reclassification between low- and intermediate-risk groups in the primary event population, based on a predicted 10-year risk threshold of 7.5%, approximately 16% of the individuals were reclassified when adding both protein risk score and polygenic risk scores to a model with clinical risk factors. Among those individuals who subsequently experienced an ASCVD event, we did not observe a statistically significant improvement in reclassification. We therefore cannot conclude that the protein risk score, with or without including polygenic risk scores, has clinical utility for screening programs based on a 7.5% risk threshold. However, we observed statistically significant reclassification at higher risks, based on predicted 10-year risk threshold of 20%, with 8.1% of the individuals reclassified when adding the protein risk score and polygenic risk scores. This indicates that the protein risk score and polygenic risk scores may not be able to improve prediction when added to clinical risk factors over all risk groups. When considering competing risks, the protein risk score seemed to also capture risk of dying from causes other than ASCVD (this was also the case for the model based on clinical risk factors alone).
In light of their good performance in previous studies,32 we considered nonlinear protein risk score models (ie, gradient-boosted trees using XGBoost33) and did not observe improvement in performance over the linear protein risk score used in the analysis. Based on this and alternative methods we considered for constructing linear protein risk scores, we believe that the general conclusions of this study are not limited to the particular method chosen to construct the protein risk score used in the analysis (see eAppendix 1 in Supplement 1).
The populations in this study are mainly composed of White participants. The protein risk score developed in the primary event population in Iceland was transferable to a group of individuals in the secondary event population who self-reported as White as well as Asian and African. In contrast, it has been demonstrated that polygenic risk scores do not transfer well between different racial groups and are limited by the lack of genome-wide association studies data in people who are not White.34,35 However, the contrast between predictions based on protein and genetic scores is notable and underlines the robustness of protein-based predictors. When comparing the protein risk score alone with clinical risk models, the protein risk score had a statistically significant greater C index and 10-year integrated discrimination improvement for the primary event population. This observation suggests that a meaningful risk assessment can be made based on a single plasma sample using a similar proteomics-based predictive model.
Limitations
The cohorts in this study are mainly White participants. It has been demonstrated that the polygenic risk score for coronary artery disease developed across several racial groups shows weaker prediction performance among individuals of African ancestry.35 This, along with this study’s limited sample size for individuals who are not White, makes it difficult to show what polygenic risk score–related findings hold for people who are not White.
Another limitation is that the study used a test set for primary event prediction based on the same population from which the protein risk score was derived. This could lead to overestimation of prediction performance in the primary event population. We note that the coefficients for the clinical risk factors in the primary event population were fitted in the derivation set to put the risk models on an equal footing in the sense that they were both being adapted to the population. This limitation does not apply to the testing in the secondary event population since the proteins and coefficients in the protein risk score were derived in the primary event population.
We also note that the analysis in the population-based primary event cohort was limited by availability of risk factor data at the time of plasma sampling. Since cholesterol and blood pressure measurements were not available for the majority of the participants, information regarding hypertension treatment and statin use was used instead.
Finally, proteomics technology is currently rapidly evolving, with increases in the number of targeted proteins. The protocols are still evolving with innovations involving automated sample preparation and the use of next-generation sequencing for high throughput.36 The parameters of protein risk scores might be sensitive to the method of measurement. There is more than 1 proteomics platform, and transferring a protein score formula derived using one platform to another will need to be addressed.
Conclusions
When protein risk scores and polygenic risk scores are added to a model with clinical risk factors, either individually or jointly, they provide modest improvement in discrimination. Further research is needed to determine whether the protein risk score and polygenic risk scores are clinically useful for screening purposes.
eAppendix. Methods
eAppendix 2. Results
eAppendix 3. References
eFigure 1. Age at Plasma Collection and Follow-Up in the Icelandic Population
eFigure 2. Cross-Validation for Selecting Penalization Strength for Fitting Protein ASCVD Risk Prediction Models
eFigure 3. Protein Risk Score Weights and Relationship With Polygenic Risk Scores and Age in the Primary Event Test Set
eFigure 4. Protein Risk Scores With Respect to Age and Sex in Test Set for Primary Event Population
eFigure 5. Polygenic Risk Scores for CAD and Stroke With Respect to Age and Sex in Primary Event Population
eFigure 6. AUCs and ROC Curves with Different Handling of Censoring and Competing Risk in the Primary Event Population
eFigure 7. Calibration in the Primary Event Population
eFigure 8. Reclassification in the Primary Event Population
eFigure 9. Predicted 10-Year Risk by Different Models in Three Separate Groups in the Primary Event Population
eFigure 10. Cumulative Rate of Cardiovascular Events in Primary Event Population
eFigure 11. AUCs and ROC Curves With Different Handling of Censoring in the Secondary Event Population
eFigure 12. Calibration in the Secondary Event Population
eFigure 13. Polygenic Risk Scores for CAD and Stroke and Protein Risk Score in the Secondary Event Population
eFigure 14. Cumulative Rate of Cardiovascular Events in Secondary Event Population
eFigure 15. Cumulative Event Rate of Cardiovascular Events Stratified by Protein Risk Scores in the Primary Event Population
eFigure 16. Prediction of Age and Sex Using Proteomics Data in the Primary Event Population
eFigure 17. Comparison of Models to Calculate Protein Risk Scores
eFigure 18. Cumulative Event Rate of Cardiovascular Events for Several Clinical Risk Scores in the Secondary Primary Event Population
eAppendix 4. List of eTables
eTable 1. Inclusion Criteria and Missing Data and Imputation in the Icelandic Proteomics Population
eTable 2. Weights in ASCVD Risk Scores Based on Clinical Risk Factors in the Secondary Event Population
eTable 3. Probes and Weights for Protein Risk Score Trained Including Age and Sex Covariates
eTable 4. Harrell's C-Indices in Primary and Secondary Event Populations
eTable 5. Cox Proportional Hazard Joint Survival Analysis for Clinical Risk Factors, PRSs and ProtRS in the Primary Event Population Test Set (N = 4,018, N events = 465)
eTable 6. Association of Clinical Risk Factors With ProtRS and the ASCVD Event in the Primary Event Population Test Set (N = 4018, N events = 465)
eTable 7. Prediction Performance in the Primary Event Population for Events Within 2-, 5-, or 10-Years
eTable 8. Category-Free Net Reclassification Index in Primary and Secondary Event Populations
eTable 9. Risk Estimates for Models With Protein Risk Score, Clinical Risk Factors, and Polygenic Risk Scores for CAD and Stroke in the Test Set of the Primary Event Population
eTable 10. C-Indices and Reclassification in the Secondary Event Population With Protein and Genotype Data Available
eTable 11. Cox Survival Analysis of the ProtRS in Secondary Event Populations of European, Asian, and African Ancestries
eTable 12. Cox Survival Analysis of Protein Risk Scores Trained With and Without Including NT-proBNP and MMP-12 in Primary and Secondary Event Populations
eTable 13. Association and Prediction Performance of Protein Risk Scores Trained Using Small Sets of Proteins Selected With Stability Selection
eTable 14. Protein Risk Scores Trained in Secondary Event Population or Primary Event Population Tested in Secondary Event Population Test Subset (N = 1863, N events = 127)
eTable 15. Correlations of Lab Measurements and Medical History at Baseline With ProtRS in Secondary Event Population
eTable 16. Association of Lab Measurements and Medical History Parameters, and Protein Risk Score with MACE Endpoint in the Secondary Event Population
eTable 17. Cox Survival Analysis of Protein Risk Scores, PRSs, and Clinical Risk Factor Scores in the Secondary Event Population
Data Sharing Statement
References
- 1.Wilson PW, D’Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97(18):1837-1847. doi: 10.1161/01.CIR.97.18.1837 [DOI] [PubMed] [Google Scholar]
- 2.Conroy RM, Pyörälä K, Fitzgerald AP, et al. ; SCORE Project Group . Estimation of ten-year risk of fatal cardiovascular disease in Europe: the SCORE project. Eur Heart J. 2003;24(11):987-1003. doi: 10.1016/S0195-668X(03)00114-3 [DOI] [PubMed] [Google Scholar]
- 3.Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. ; American College of Cardiology/American Heart Association Task Force on Practice Guidelines . 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25)(suppl 2):S49-S73. doi: 10.1161/01.cir.0000437741.48606.98 [DOI] [PubMed] [Google Scholar]
- 4.Collins GS, Altman DG. Predicting the 10 year risk of cardiovascular disease in the United Kingdom: independent and external validation of an updated version of QRISK2. BMJ. 2012;344:e4181. doi: 10.1136/bmj.e4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mach F, Baigent C, Catapano AL, et al. ; ESC Scientific Document Group . 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111-188. doi: 10.1093/eurheartj/ehz455 [DOI] [PubMed] [Google Scholar]
- 6.Ripatti S, Tikkanen E, Orho-Melander M, et al. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet. 2010;376(9750):1393-1400. doi: 10.1016/S0140-6736(10)61267-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inouye M, Abraham G, Nelson CP, et al. ; UK Biobank CardioMetabolic Consortium CHD Working Group . Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention. J Am Coll Cardiol. 2018;72(16):1883-1893. doi: 10.1016/j.jacc.2018.07.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knowles JW, Ashley EA. Cardiovascular disease: the rise of the genetic risk score. PLoS Med. 2018;15(3):e1002546. doi: 10.1371/journal.pmed.1002546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elliott J, Bodinier B, Bond TA, et al. Predictive accuracy of a polygenic risk score-enhanced prediction model vs a clinical risk score for coronary artery disease. JAMA. 2020;323(7):636-645. doi: 10.1001/jama.2019.22241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosley JD, Gupta DK, Tan J, et al. Predictive accuracy of a polygenic risk score compared with a clinical risk score for incident coronary heart disease. JAMA. 2020;323(7):627-635. doi: 10.1001/jama.2019.21782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nikpay M, Goel A, Won HH, et al. A comprehensive 1,000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47(10):1121-1130. doi: 10.1038/ng.3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eiriksdottir T, Ardal S, Jonsson BA, et al. Predicting the probability of death using proteomics. Commun Biol. 2021;4(1):758. doi: 10.1038/s42003-021-02289-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rohloff JC, Gelinas AD, Jarvis TC, et al. Nucleic acid ligands with protein-like side chains: modified aptamers and their use as diagnostic and therapeutic agents. Mol Ther Nucleic Acids. 2014;3(10):e201. doi: 10.1038/mtna.2014.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Assarsson E, Lundberg M, Holmquist G, et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One. 2014;9(4):e95192. doi: 10.1371/journal.pone.0095192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams SA, Kivimaki M, Langenberg C, et al. Plasma protein patterns as comprehensive indicators of health. Nat Med. 2019;25(12):1851-1857. doi: 10.1038/s41591-019-0665-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho JE, Lyass A, Courchesne P, et al. Protein biomarkers of cardiovascular disease and mortality in the community. J Am Heart Assoc. 2018;7(14):e008108. doi: 10.1161/JAHA.117.008108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganz P, Heidecker B, Hveem K, et al. Development and validation of a protein-based risk score for cardiovascular outcomes among patients with stable coronary heart disease. JAMA. 2016;315(23):2532-2541. doi: 10.1001/jama.2016.5951 [DOI] [PubMed] [Google Scholar]
- 18.Hoogeveen RM, Pereira JPB, Nurmohamed NS, et al. Improved cardiovascular risk prediction using targeted plasma proteomics in primary prevention. Eur Heart J. 2020;41(41):3998-4007. doi: 10.1093/eurheartj/ehaa648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nurmohamed NS, Belo Pereira JP, Hoogeveen RM, et al. Targeted proteomics improves cardiovascular risk prediction in secondary prevention. Eur Heart J. 2022;43(16):1569-1577 doi: 10.1093/eurheartj/ehac055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams SA, Ostroff R, Hinterberg MA, et al. A proteomic surrogate for cardiovascular outcomes that is sensitive to multiple mechanisms of change in risk. Sci Transl Med. 2022;14(639):eabj9625. doi: 10.1126/scitranslmed.abj9625 [DOI] [PubMed] [Google Scholar]
- 21.Ferkingstad E, Sulem P, Atlason BA, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet. 2021;53(12):1712-1721. doi: 10.1038/s41588-021-00978-w [DOI] [PubMed] [Google Scholar]
- 22.Sabatine MS, Giugliano RP, Keech A, et al. Rationale and design of the Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk trial. Am Heart J. 2016;173:94-101. doi: 10.1016/j.ahj.2015.11.015 [DOI] [PubMed] [Google Scholar]
- 23.Sabatine MS, Giugliano RP, Keech AC, et al. ; FOURIER Steering Committee and Investigators . Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713-1722. doi: 10.1056/NEJMoa1615664 [DOI] [PubMed] [Google Scholar]
- 24.Hageman SHJ, McKay AJ, Ueda P, et al. Estimation of recurrent atherosclerotic cardiovascular event risk in patients with established cardiovascular disease: the updated SMART2 algorithm. Eur Heart J. 2022;43(18):1715-1727. doi: 10.1093/eurheartj/ehac056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cox DR. Regression models and life-tables. J R Stat Soc B. 1972;34(2):187-202. doi: 10.1111/j.2517-6161.1972.tb00899.x [DOI] [Google Scholar]
- 26.Tibshirani R. The lasso method for variable selection in the Cox model. Stat Med. 1997;16(4):385-395. doi: [DOI] [PubMed] [Google Scholar]
- 27.Kong A, Frigge ML, Thorleifsson G, et al. Selection against variants in the genome associated with educational attainment. Proc Natl Acad Sci U S A. 2017;114(5):E727-E732. doi: 10.1073/pnas.1612113114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolbers M, Blanche P, Koller MT, Witteman JCM, Gerds TA. Concordance for prognostic models with competing risks. Biostatistics. 2014;15(3):526-539. doi: 10.1093/biostatistics/kxt059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lloyd-Jones DM, Braun LT, Ndumele CE, et al. Use of risk assessment tools to guide decision-making in the primary prevention of atherosclerotic cardiovascular disease: a special report from the American Heart Association and American College of Cardiology. Circulation. 2019;139(25):e1162-e1177. doi: 10.1161/CIR.0000000000000638 [DOI] [PubMed] [Google Scholar]
- 30.Pencina MJ, D’Agostino RB Sr, D’Agostino RB Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27(2):157-172. doi: 10.1002/sim.2929 [DOI] [PubMed] [Google Scholar]
- 31.Pencina MJ, D’Agostino RB Sr, Steyerberg EW. Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. Stat Med. 2011;30(1):11-21. doi: 10.1002/sim.4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rousset A, Dellamonica D, Menuet R, et al. Can machine learning bring cardiovascular risk assessment to the next level? a methodological study using FOURIER trial data. Eur Heart J. 2021;3(1):38-48. doi: 10.1093/ehjdh/ztab093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen T, Guestrin C. XGBoost: a scalable tree boosting system. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. KDD ’16. Association for Computing Machinery; 2016:785-794. doi: 10.1145/2939672.2939785 [DOI] [Google Scholar]
- 34.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51(4):584-591. doi: 10.1038/s41588-019-0379-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fahed AC, Aragam KG, Hindy G, et al. Transethnic transferability of a genome-wide polygenic score for coronary artery disease. Circ Genom Precis Med. 2021;14(1):e003092. doi: 10.1161/CIRCGEN.120.003092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wik L, Nordberg N, Broberg J, et al. Proximity extension assay in combination with next-generation sequencing for high-throughput proteome-wide analysis. Mol Cell Proteomics. 2021;20:100168. doi: 10.1016/j.mcpro.2021.100168 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eAppendix. Methods
eAppendix 2. Results
eAppendix 3. References
eFigure 1. Age at Plasma Collection and Follow-Up in the Icelandic Population
eFigure 2. Cross-Validation for Selecting Penalization Strength for Fitting Protein ASCVD Risk Prediction Models
eFigure 3. Protein Risk Score Weights and Relationship With Polygenic Risk Scores and Age in the Primary Event Test Set
eFigure 4. Protein Risk Scores With Respect to Age and Sex in Test Set for Primary Event Population
eFigure 5. Polygenic Risk Scores for CAD and Stroke With Respect to Age and Sex in Primary Event Population
eFigure 6. AUCs and ROC Curves with Different Handling of Censoring and Competing Risk in the Primary Event Population
eFigure 7. Calibration in the Primary Event Population
eFigure 8. Reclassification in the Primary Event Population
eFigure 9. Predicted 10-Year Risk by Different Models in Three Separate Groups in the Primary Event Population
eFigure 10. Cumulative Rate of Cardiovascular Events in Primary Event Population
eFigure 11. AUCs and ROC Curves With Different Handling of Censoring in the Secondary Event Population
eFigure 12. Calibration in the Secondary Event Population
eFigure 13. Polygenic Risk Scores for CAD and Stroke and Protein Risk Score in the Secondary Event Population
eFigure 14. Cumulative Rate of Cardiovascular Events in Secondary Event Population
eFigure 15. Cumulative Event Rate of Cardiovascular Events Stratified by Protein Risk Scores in the Primary Event Population
eFigure 16. Prediction of Age and Sex Using Proteomics Data in the Primary Event Population
eFigure 17. Comparison of Models to Calculate Protein Risk Scores
eFigure 18. Cumulative Event Rate of Cardiovascular Events for Several Clinical Risk Scores in the Secondary Primary Event Population
eAppendix 4. List of eTables
eTable 1. Inclusion Criteria and Missing Data and Imputation in the Icelandic Proteomics Population
eTable 2. Weights in ASCVD Risk Scores Based on Clinical Risk Factors in the Secondary Event Population
eTable 3. Probes and Weights for Protein Risk Score Trained Including Age and Sex Covariates
eTable 4. Harrell's C-Indices in Primary and Secondary Event Populations
eTable 5. Cox Proportional Hazard Joint Survival Analysis for Clinical Risk Factors, PRSs and ProtRS in the Primary Event Population Test Set (N = 4,018, N events = 465)
eTable 6. Association of Clinical Risk Factors With ProtRS and the ASCVD Event in the Primary Event Population Test Set (N = 4018, N events = 465)
eTable 7. Prediction Performance in the Primary Event Population for Events Within 2-, 5-, or 10-Years
eTable 8. Category-Free Net Reclassification Index in Primary and Secondary Event Populations
eTable 9. Risk Estimates for Models With Protein Risk Score, Clinical Risk Factors, and Polygenic Risk Scores for CAD and Stroke in the Test Set of the Primary Event Population
eTable 10. C-Indices and Reclassification in the Secondary Event Population With Protein and Genotype Data Available
eTable 11. Cox Survival Analysis of the ProtRS in Secondary Event Populations of European, Asian, and African Ancestries
eTable 12. Cox Survival Analysis of Protein Risk Scores Trained With and Without Including NT-proBNP and MMP-12 in Primary and Secondary Event Populations
eTable 13. Association and Prediction Performance of Protein Risk Scores Trained Using Small Sets of Proteins Selected With Stability Selection
eTable 14. Protein Risk Scores Trained in Secondary Event Population or Primary Event Population Tested in Secondary Event Population Test Subset (N = 1863, N events = 127)
eTable 15. Correlations of Lab Measurements and Medical History at Baseline With ProtRS in Secondary Event Population
eTable 16. Association of Lab Measurements and Medical History Parameters, and Protein Risk Score with MACE Endpoint in the Secondary Event Population
eTable 17. Cox Survival Analysis of Protein Risk Scores, PRSs, and Clinical Risk Factor Scores in the Secondary Event Population
Data Sharing Statement