

Abstract
Tuberculosis (TB) remains a significant public health concern in the 21st century, especially due to drug resistance, coinfection with diseases like immunodeficiency syndrome (AIDS) and coronavirus disease 2019, and the lengthy and costly treatment protocols. In this review, we summarize the pathogenesis of TB infection, therapeutic targets, and corresponding modulators, including first‐line medications, current clinical trial drugs and molecules in preclinical assessment. Understanding the mechanisms of Mycobacterium tuberculosis (Mtb) infection and important biological targets can lead to innovative treatments. While most antitubercular agents target pathogen‐related processes, host‐directed therapy (HDT) modalities addressing immune defense, survival mechanisms, and immunopathology also hold promise. Mtb’s adaptation to the human host involves manipulating host cellular mechanisms, and HDT aims to disrupt this manipulation to enhance treatment effectiveness. Our review provides valuable insights for future anti‐TB drug development efforts.
Keywords: biological targets, host‐directed therapy, inhibitors, modulaters, Mycobacterium tuberculosis, pathogenesis
In this review, we summarize the pathogenesis of TB infection, therapeutic targets, and corresponding modulators, including first‐line medications, current clinical trial drugs and molecules in preclinical assessment. Our review provides valuable insights for future antituberculosis drug development efforts.
1. INTRODUCTION
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), remains a significant global health problem, despite advances in diagnosis and treatment. Approximately 10 million new cases of TB are diagnosed each year, with over a million deaths attributed to the disease. 1 TB is particularly prevalent in developing countries, where coinfection with human immunodeficiency virus (HIV) and poor living conditions are common. 2 Additionally, the emergence of drug‐resistant strains of Mtb poses a significant challenge to global TB control efforts. 3 The current standard regimen for drug‐sensitive TB involves a combination of drugs, including isoniazid (INH), rifampicin (RIF), pyrazinamide (PZA), and ethambutol (EMB). However, treatment can be lengthy and costly, with patients often requiring 6 months of therapy. 4 Moreover, the emergence of multidrug‐resistant TB (MDR‐TB) and extensively drug‐resistant TB (XDR‐TB) has further complicated the treatment landscape (Figure 1).
FIGURE 1.
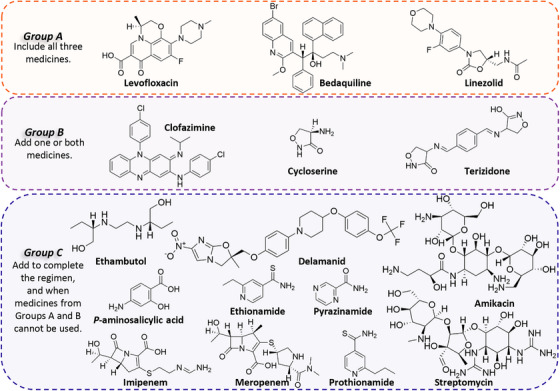
Grouping of medicines recommended for use in longer multidrug‐resistant TB (MDR‐TB) regimens by the WHO. 2 Group A drugs are strongly recommended for inclusion in all longer MDR‐TB regimens containing fluoroquinolones, bedaquiline, and linezolid. Group B drugs were recommended to add one or two to the regimen to improve outcomes. Group C drugs were recommended as a secondary option to Group A and B drugs.
While progress has been made in the development of new TB drugs, there remains a significant need for novel treatment options. Several drug candidates are presently in diverse phases of clinical development (Figure 2), comprising reoptimized adaptations of current TB drugs and some involving original mechanisms of action. Additionally, several new targets for drug development have been identified. 4 In the context of the coronavirus disease 2019 (COVID‐19) pandemic, the impact of TB on global health has become even more severe. Individuals who have recovered from COVID‐19 have been found to have a higher risk of developing TB, likely due to the negative impact of COVID‐19 on the immune system. 5 Therefore, new TB treatment drugs remain an urgent research priority.
FIGURE 2.
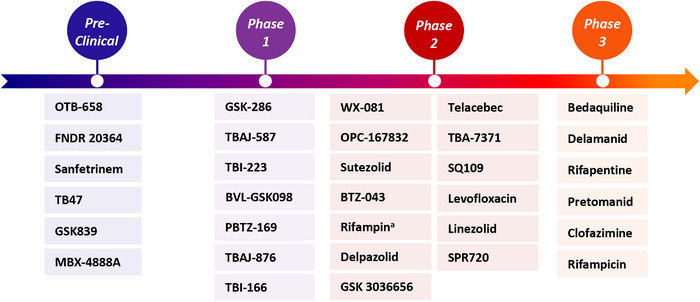
Current global clinical pipeline of new tuberculosis drugs based on information provided by the Working Group for New TB Drugs (WGND). 156 aTrial of high‐dose Rifampin in patients with TB.
This review aims to identify potential targets that show promise for the development of new therapeutic approaches to treat TB, based on an understanding of the pathogenesis of the disease. The ability of Mtb to survive in the microenvironment of the human host is one of the greatest challenges faced when developing new drugs. We first elucidate the pathogenesis of Mtb, which encompasses the phases of invasion, proliferation, latency, and revival of the pathogen. By understanding the mechanisms, we can identify new drug targets and advance the development of more effective TB treatments. In the pathogenic process, Mtb acts as a pathogen causing TB, so targets within the bacterium and drugs that directly kill Mtb are of great interest. We reviewed all kinds of targets and drugs targeting Mtb. Simultaneously, Mtb provokes an immunoreaction in its host, while skillfully tampering with vital cellular processes, leading to protracted treatment and unfavorable prognosis. This review will summarize the latest research identifying high‐value targets in host‐targeted therapies and describe the related modulaters.
2. PATHOGENESIS OF Mtb
Mtb complex (MTBC) has persistently accompanied our species, anatomically modern humans, during our evolutionary journey and widespread dispersion across the globe throughout the past 70 millennia. Mtb initiates its life cycle upon invasion of the respiratory tract and lungs (Figure 3), as the microorganism is classically believed to thrive only within living organisms. 6 , 7 This initial interaction, commonly referred to as primary infection, marks the onset of contact between the pathogen and the respiratory system. There has been a contention that the coevolution between Mtb and its human host does not follow the conventional trajectory of an evolutionary arms‐race. Instead, it is suggested to be characterized by mechanisms of manipulation. 7 Following exposure to multiple host cell receptors (including toll‐like receptors [TLRs], C‐type lectin receptors [CLRs], dendritic cells [DCs], mannose receptors [MRs], and NOD‐like receptors [NLRs]) and internalization by alveolar macrophages and DCs, Mtb undergoes a phase of intracellular replication. Subsequently, infected cells spread to lymph nodes while attempting to destroy the bacteria with various proteolytic enzymes and cytokines (e.g., tumor necrosis factor alpha [TNF‐α] and interferon gamma [IFN‐γ]), perpetuating spread of the pathogen throughout the host's lung parenchyma upon arrival. 8 Activation of macrophages results in recruitment of additional innate immune cells, thereby fostering inflammatory responses that contribute to host defense against the pathogen. Neutrophils demonstrate a heightened intensity of phagocytosis compared to macrophages, coupled with elevated levels of reactive oxygen species (ROS)‐mediated oxidative burst. 9 Upon recruitment of lymphocytes to the site of infection, a cascade of cell‐mediated immune responses is initiated, leading to the arrival of additional immune cells aimed at localizing bacterial colonization and inhibiting their proliferation. 10 , 11 During the initial stages, a characteristic delay is observed in the T‐cell response, which aids the pathogen in establishing a persistent infection. 10 At this point, complete eradication of Mtb is feasible if the host immune capacity remains intact. 12 In most cases, however, the immune response is not sufficient to eradicate Mtb. Monocytes accumulate in the vicinity of infected macrophages, precipitating the formation of solid granulomas, a pathological feature characteristic of TB. Additionally, Mtb can traverse the mucosa upon infection of alveolar epithelial cells and stimulation of cellular necrosis, further exacerbating pathogenesis. 13 , 14 Hence, the gathered data suggest that local immune responses play a pivotal role in mitigating progression of TB disease. 15
FIGURE 3.
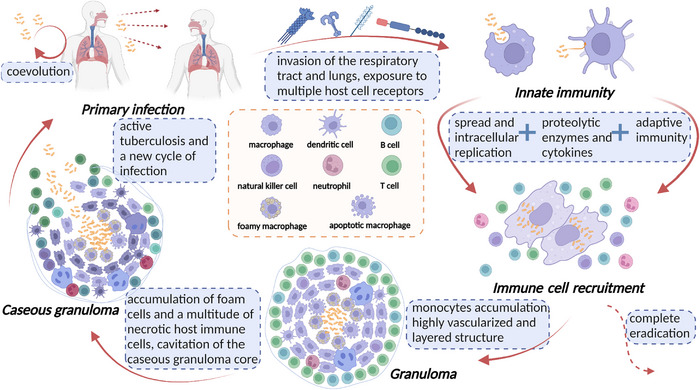
Pathophysiology of pulmonary TB. Upon entering the respiratory tract and lungs of the host, Mtb incites an innate immune response and is engulfed by pivotal immune cells such as macrophages and dendritic cells. Subsequently, Mtb replicates within these cells as more immune cells are recruited to the site of infection. Whilst it is possible for the host to completely eliminate Mtb at this stage, the formation of solid granulomas is often prompted. These granulomas are composed of foam cells derived from macrophages, as well as a multitude of necrotic immune cells, culminating in caseous granulomas that eventually rupture and release bacteria, giving way to the subsequent development of active TB. Ultimately, these Mtb bacteria are released as infectious aerosol droplets, reinstating a new cycle of infection. The elements in the figure were drawn using BioRender online tool (https://biorender.com).
Despite being formed with the intent of containing the spread of bacteria, granulomas can also serve as a refuge for bacterial populations, thus evading further recognition and removal by the host immune system, resulting in a clinically defined state of latent TB infection (LTBI). 12 , 16 Ensuring screening, diagnosis, and treatment of LTBI is critical in facilitating the global decline in TB incidence and ultimately achieving TB elimination. During early development, the granuloma is highly vascularized (via vascular endothelial growth factor (VEGF)) and the vessels have extensive lymphocytic cuffs. As the granuloma progresses, macrophages undergo differentiation into various morphotypes, resulting in the formation of a layered structure with a layer of lymphocytes aggregated outside a fibrous cuff surrounding a macrophage‐rich layer. In this scenario, patients harbouring granulomas are asymptomatic and noninfectious. 17 , 18 Studies have revealed that Mtb harnesses the production of mycolic acid (MA) to instigate differentiation of macrophages into foam cells. The core of the granuloma can give rise to a caseous granuloma, characterised by the accumulation of foam cells and a multitude of necrotic host immune cells. 19 During late‐stage TB, cavitation of the caseous granuloma core may ensue, resulting in the release of bacteria and consequent progression to active TB disease. 20 Consequently, the reactivation of LTBI, and ensuing progression to symptomatic disease, can enable transmission of the bacteria to a new host, perpetuating a new cycle of infection. Moreover, Mtb can disseminate through the bloodstream 21 and lymphatic endothelial cells, 22 disseminating beyond infected lungs and leading to the development of extrapulmonary TB (EPTB). EPTB can manifest in nearly any part of the body, akin to lymph nodes, pleura, genitourinary system, bones and joints, and other organs. 23
Pursuing an in‐depth comprehension of the pathogenesis of TB may unlock novel therapeutic avenues. Despite having been identified and isolated over a century ago, Mtb has continued to inflict protracted distress and fatalities worldwide. Moreover, the latest data from the World Health Organization (WHO) regarding drug‐resistant TB are alarming, with approximately 450,000 new cases of RIF‐resistant TB reported in 2021. 1 The emergence of drug‐resistant strains of TB correlates with the epidemic of HIV, and early incidence of drug‐resistant TB epidemics were witnessed primarily among HIV–Mtb coinfected patients. 24 , 25 Furthermore, the detrimental effects of the COVID‐19 pandemic have impeded the identification and management of TB cases, undermining the gains made in combatting TB in recent years. 5 , 26 Therefore, it is imperative to recognize the urgency of discovering effective therapies for TB.
Several studies have explored the potential use of Bacillus Calmette‐Guérin (BCG) as prophylaxis for TB; however, results indicate that BCG exerts suboptimal effects on immune memory. 27 , 28 , 29 Nonetheless, promising breakthroughs in the analysis of pathogen–host interactions and evolutionary investigations of Mtb offer prospective avenues for identifying pathogenicity and virulence factors that could catalyze the development of novel therapies for TB. 30
3. DRUG TARGETS AND INHIBITORS TARGETING Mtb
Invasion of Mtb causes TB and its ability to replicate and maintain using host cellular mechanisms makes it a major target for killing by drugs. The publication of the whole genome sequencing of Mtb has advanced our understanding of the molecular biology of this bacterium, making it easier to identify specific targets. In fact, the most potent drugs targeting Mtb primarily aim at eradicating the pathogen currently. 31 Unquestionably, novel drug discovery and development efforts concentrate predominantly on direct Mtb killing. This section reviews the drug targets and inhibitors targeting Mtb, which are classified according to the different actions associated with bacterial survival and are discussed in different subsections.
3.1. Cell wall synthesis and assembly
Mtb, the causative agent of TB, possesses an atypical cellular envelope comprising primarily lipids and carbohydrates. This envelope is characterized by the complex mycolyl‐arabinogalactan‐peptidoglycan (mAGP) and phosphatidyl‐myo‐inositol‐based lipoglycans. The mAGP complex is constructed from several critical components, including peptidoglycan (PG), arabinogalactan (AG), MA units, and the indispensable lipoarabinomannan (LAM). 32 , 33 It is estimated that the cellular envelope synthesis and assembly pathway of Mtb harbors a minimum of 60 prospective enzymatic targets. 4 , 34 , 35 The targets described in this section involve the synthesis of MA, AG, LAM, PG, and the transport of MA.
3.1.1. MA biosynthesis
MA, a crucial component of Mtb’s cellular envelope, is synthesized predominantly through the fatty acid synthesis (FAS) pathway. It is noteworthy that unlike mammalian FAS‐I synthase, which utilizes a multidomain protein, FAS in Mtb is carried out by a combination of FAS‐I synthase and several FAS‐II monofunctional enzymes. The FAS‐II type system is responsible for the extension of acyl‐CoA (C16:0 to C18:0), the products of de novo synthesis by FAS‐I. 36 This unique contrast renders the FAS pathway a viable therapeutic target for pharmacological agents aimed at combating Mtb infection.
FAS‐II pathway
InhA, an enoyl‐acyl carrier protein (ACP) reductase, facilitates the reduction of long‐chain trans‐2‐enoyl‐ACP by forming covalent adducts between nicotinamide cofactors and enoyl‐CoA substrates in the FAS‐II pathway. 37 The role of several first‐line anti‐TB drugs such as INH, ethionamide (ETH, Group C), and prothionamide (PTH, Group C) 38 , 39 (Table 1) demonstrates that InhA is an important therapeutic target for the treatment of Mtb infection.
TABLE 1.
Biological targets and inhibitors targeting Mtb.
| Mechanism of action | Target | Typical compound | Drug stage for TB | |
|---|---|---|---|---|
| Cell wall synthesis and sssembly | MA a biosynthesis | Enoyl‐acyl carrier protein reductase (InhA) | Isoniazid 38 | Approved |
| Ethionamide 39 | Approved | |||
| β‐Ketoacyl synthase (KasA) | Thiolactomycin 66 , 67 , 68 , 69 | Biological test | ||
| Fatty acid degradation protein D32 (FadD32) | Quinoline‐2‐carboxamide 78 | Biological test | ||
| Polyketide synthase 13 (Pks13) | TAM16 79 | Biological test | ||
| Mycolic acid methyltransferase 4 (MmaA4) | SADAE 88 | Biological test | ||
| Cyclopropane mycolic acid synthase (CMAS) | / | In silico docking | ||
| AG biosynthesis | N‐acetylglucosamine‐1‐phosphate transferase (WecA) | CPZEN‐45 97 | Preclinical | |
| LAM biosynthesis | Arabinosyl transferase C (EmbC) | Amikacin 106 | Approved | |
| Decaprenylphosphoryl‐β‐D‐ribose‐2′‐epimerase (DprE1) | PBTZ‐169 110 | Phase I | ||
| OPC‐167832 111 | Phase II | |||
| TBA‐7371 112 | Phase II | |||
| BTZ‐043 113 | Phase II | |||
| PG biosynthesis | Alanine racemase (Alr) | Cycloserine 130 , 131 | Approved | |
| D‐alanyl‐D‐alanine ligase (Ddl) | ||||
| L,D‐transpeptidase type 2 (LdtMt2) | Meropenem 138 | Approved | ||
| Phospho‐N‐acetylmuramoyl‐pentapeptide transferase (MurX) | SQ641 148 , 149 | Preclinical | ||
| MA transporter | Mamalian membrane protein large 3 (MmpL3) | SQ109 155 | Phase II | |
| Lipid metabolism | Aspartate decarboxylase (PanD) | Pyrazinamide 171 , 172 | Approved | |
| Protein synthesis and breakdown | Ribosome | TBI‐223 191 | Phase I | |
| Leucyl‐tRNA synthase (LeuRS) | GSK656 194 | Phase II | ||
| Caseinolytic protease P (ClpP) | EZ120 200 | Preclinical | ||
| Proteasome | Phenylimidazole 210 | Preclinical | ||
| Proteasome accessory factor A (PafA) | ST1926 213 | Biological test | ||
| Amino acid synthesis and metabolism | Ser/Thr protein kinases (protein kinase G, PknG) | AX20017 220 | Biological test | |
| NU‐6027 224 | Biological test | |||
| Shikimate pathway (3‐dehydroquinate synthase, DHQS) | IMB‐T130 228 | Biological test | ||
| Tryptophan synthase (TrpAB) | BRD4592 233 | Preclinical | ||
| Nucleotide synthesis | Type I topoisomerase (topoI) | m‐AMSA 241 | Biological test | |
| Type II topoisomerase (DNA gyrase) | Levofloxacin 244 | Phase II | ||
| SPR720 246 | Phase II | |||
| RNA polymerase (RNAP) | Rifampicin 252 | Approved | ||
| transcriptional repressor of ethA (EthR) | BVL‐GSK098 257 , 258 | Phase 1 | ||
| Energy metabolism | Type II NADH dehydrogenase (NDH‐2) | Clofazimine 263 | Phase III | |
| TBI‐166 264 | Phase I | |||
| β Subunit of cytochrome bc 1 complex (QcrB) | Telacebec 268 | Phase II | ||
| ATP synthase | Bedaquiline 276 | Phase III | ||
| TBAJ‐876 280 | Phase I | |||
| TBAJ‐587 280 | Phase I | |||
| WX‐081 281 | Phase II | |||
| Virulence | Two‐component system | PhoPR | Ethoxzolamide 294 | Biological test |
| Artemisinin 299 | Biological test | |||
| DosRST | HC102A 299 | Biological test | ||
| HC103A 299 | Biological test | |||
| ESX‐1 secretion system | ESAT‐6 secretion system‐1 (ESX‐1) | BBH7 306 | Biological test | |
| BTP15 306 | Biological test | |||
| Folic acid and mycobactin biosynthesis | Dihydrofolate reductase (DHFR) | P‐aminosalicylic acid 308 | Approved | |
| AF‐353 311 | Biological test | |||
| Metal uptake | Iron‐dependent regulator (IdeR) | / | In silico docking | |
| Cholesterol metabolism | Adenylyl cyclase (AC) | GSK2556286 316 | Phase I | |
MA, mycolic acid; AG, arabinogalactan; LAM, lipoarabinomannan; PG, peptidoglycan.
Various studies have demonstrated that INH, ETH, and PTH exert their inhibitory effect on InhA by forming an INH/ETH/PTH‐NAD+ adduct, requiring prior activation. 40 , 41 , 42 , 43 Mtb possesses two enzymes—KatG, a catalase‐peroxidase, 44 and EthA, a monooxygenase 45 —which catalyze the generation of free radical inhibitors that bind to NADH to form binary complexes. 38 Recent research has revealed that during the catalytic cycle of InhA, 37 NADH cofactors and octenoyl‐CoA substrates form covalent adducts, including a C2‐ene adduct, which offers insight into the function of key amino acid residues at the active site of InhA.
The emergence of drug resistance due to mutations of activating enzymes is a formidable challenge in the case of INH, ETH, and PTH. As a result, researchers have shifted their focus to identifying compounds that can directly bind to InhA, spurred by the discovery of targets for the broad‐spectrum fungicide triclosan. 46 , 47 , 48 Promising direct inhibitors of InhA have been identified, such as diphenyl ethers, 47 , 49 , 50 pyrrolidine carboxamides, 51 arylamides, 52 indole‐5‐Amides, 53 pyridones, 54 imidazopiperidines, 55 thiadiazoles, 56 diazaborines, 57 and benzimidazoles. 58 Most InhA inhibitors occupy the substrate binding sites. However, pyridomycin, 59 produced by Streptomyces pyridomyceticus 60 , 61 or Dactylosporangium fulvum, 62 represented the first direct competitive inhibitor of NADH binding with specific activity against mycobacteria. The crystal structures of InhA, either WT (PDB: 4BII) or the INH‐resistant S94A mutant (PDB: 4BGE), 63 bound to pyridomycin were determined and confirmed that pyridomycin occupied simultaneously the NADH and the substrate binding sites. 64
Similar to InhA, β‐ketoacyl synthase KasA also plays a pivotal role in the elongation of long‐chain fatty acids by facilitating the initial step of the FAS‐II pathway. 65 Thiolactomycin is a thiolactone natural product that inhibits all three annotated Mtb β‐ketoacyl synthases, including KasA, in various functional assays. 66 , 67 , 68 , 69 DG167, an indazole sulfonamide, was first identified as an antitubercular through a high‐throughput screening (HTS) campaign. 70 , 71 The X‐ray crystallography has been employed to determine the molecular structure of this compound while it was bound to KasA. 72 , 73 The in vivo efficacy of transposed indazole sulfonamide derivatives based on DG167 has shown substantial improvement in an acute infection model of Mtb in mice. 74
Fatty acid degradation protein D32/Polyketide synthase 13 crosstalk
Fatty acid degradation protein D32 (FadD32) acts as a fatty acyl‐AMP ligase, transferring the resulting acyl‐adenylate to specific thioester acceptors. 75 Polyketide synthase 13 (Pks13) is a module encoding several enzymatic and transport functions to the viability and virulence of Mtb. 76 Once produced by the FadD32 enzyme, the resulting acyl‐AMPs are specifically transferred to the ketosynthase domain of Pks13 after binding to the phosphopantetheinyl moiety of its N‐terminal ACP domain (N‐ACP(Pks13)). Together, FadD32 and N‐ACP(Pks13) constitute the initiation module of the mycolic condensation system. 77 Recent research reports that several quinoline‐2‐carboxamides effectively reduce the mycobacterial burden in mouse lungs by inhibiting FadD32 activity upon oral administration. 78 TAM16, a benzofuran derivative, has also been identified as a promising inhibitor of Pks13. 79 Additionally, coumarin 80 and chromen‐4‐one derivatives 81 have likewise demonstrated Pks13 inhibition, although a continued focus on optimizing in vivo therapeutic efficacy is warranted.
Delamanid (DLM, Group C) and pretomanid (Pa) are prodrugs that need to be activated by the deazaflavin F420‐dependent nitroreductase enzyme. 82 , 83 DLM and Pa, two promising nitroimidazole candidates, exert their anti‐Mtb effects by selectively inhibiting the biosynthesis of specific MA, such as methoxy‐ and keto‐MA. 84 By contrast, INH inhibits the synthesis of all MA classes—methoxy‐, keto‐, and α‐MA. 85 The precise enzyme targets of nitroimidazoles in cell wall biosynthesis have not yet been identified, though current research supports their multitargeted nature. Further, Pa has been shown to act as a direct nitric oxide (NO) donor, offering insight into its mechanisms for Mtb killing under hypoxic, nonreplicating conditions. 86 This is an encouraging prospect for the treatment of on LTBI. Spontaneous drug‐resistant mutants of Mtb were found to carry mutations in MA methyltransferases, MmaA4 and MmaA2. 87 Crystallographic studies identify the interaction of S‐adenosyl‐N‐decyl‐aminoethyl (SADAE) with MmaA4 and provide ideas for inhibitor design. 88 MAs cyclopropanation contributes to virulence, antibiotic resistance, and intracellular survival and is catalyzed by enzymes of the cyclopropane MA synthase (CMAS) family. 89 , 90 MA cyclopropane synthase PcaA (also known as UmaA2), which is essential for the nucleation morphology of Mtb 91 and is expressed at high levels during Mtb dormancy, 92 appears to be a potential target for dormant mycobacteria. In addition to PcaA, the cyclopropane synthases CmaA1 and CmaA2 are also involved in the cyclopropanation of MAs. 93 , 94 Several skeletons have been obtained by in silico docking, 92 , 95 , 96 and subsequent biochemical validation and optimization are expected. These targets have received limited research attention and hold promising potential for further investigation and development.
3.1.2. AG biosynthesis
N‐acetylglucosamine‐1‐phosphate transferase
N‐acetylglucosamine‐1‐phosphate transferase (WecA) responsible for initiating AG biosynthesis in Mtb, has been identified as a potential target for the caprazamycin derivative CPZEN‐45. 97 This preclinical drug candidate has shown promise as an inhalation treatment option for TB. 98 , 99 Studies have demonstrated that transcriptional silencing of the gene encoding WecA has a bactericidal effect on Mtb both in vitro and in vivo. 100 The discovery of novel WecA inhibitors has been supported by medium‐ to HTS methods targeting WecA. Such screening methods have contributed to the identification of potential inhibitors of WecA, which can serve as lead compounds for the development of new drugs to treat TB. 101
3.1.3. LAM biosynthesis
Arabinosyl transferase C
Arabinosyl transferases EmbA, EmbB, and EmbC are critical components of the mycobacterial cell wall biosynthesis pathway. While EmbA and EmbB are known to interact and form a heterodimeric complex, EmbC functions as a homodimeric enzyme. 102 Specifically, EmbA and EmbB are involved in the formation of the terminal hexaarabinofuranoside motif in AG, 103 while EmbC is responsible for chain lengthening of LAM. 104 Recent data from crystallography and overexpression studies suggest that EMB (Group C) competes with substrates for binding to the EmbB and EmbC subunits. 102 , 105 Notably, a high‐throughput virtual screening of the United States Food and Drug Administration (US FDA) library has identified two additional EmbC inhibitors, terlipressin and amikacin 106 (Group C).
Decaprenylphosphoryl‐β‐d‐ribose‐2′‐epimerase
Decaprenylphosphoryl‐β‐D‐ribose‐2′‐epimerase (DprE) is a heterodimeric diastereoselective enzyme containing DprE1 and DprE2. 107 DprE1 catalyzes the two‐step epimerization of decaprenyl‐phospho‐ribose to decaprenyl‐phospho‐arabinose, the precursor for AG and LAM synthesis. 108 DprE1 was originally discovered as a major target of benzothiazinones, which have demonstrated potent antimicrobial activity against Mtb. 109 Currently, several DprE1 inhibitors are in various stages of clinical development, as of February 2023. These include PBTZ‐169 110 (macozinone, phase I), OPC‐167832 111 (phase II), TBA‐7371 112 (phase II), and BTZ‐043 113 (phase II). Remarkably, both BTZ‐043 and PBTZ‐169 are covalent inhibitors of DprE1. The nitro group of the benzothiazinone scaffold is reduced to form its nitroso derivative, which binds to the Cys387 residue in DprE1, resulting in irreversible enzyme inhibition. 114 , 115 In contrast, OPC‐167832 is a carbostyril derivative and TBA‐7371 is a 1,4‐azaindole, both of which are noncovalent DprE1 inhibitors. Recent research have identified a variety of new DprE1 inhibitors, including benzothiazinones containing 2‐benzyl‐2,7‐diazaspiro[3.5]nonane, 116 benzothiopyranones, 117 morpholino‐pyrimidines, 118 hydantoins, 119 thiophene‐arylamides, 120 4‐aminoquinolone piperidine amides, 121 2‐carboxyquinoxalines, 122 N‐alkyl‐5‐hydroxypyrimidinone carboxamides, 123 selamectin, 124 with promising antimycobacterial activity. 125
3.1.4. PG biosynthesis
d‐Alanyl‐d‐alanine dipeptide synthesis pathway
Alanine racemase (Alr) is a crucial pyridoxal 5′‐phosphate‐dependent amino acid racemase enzyme that facilitates the conversion of l‐alanine to d‐alanine, which is utilized by bacterial cell walls for PG biosynthesis. 126 d‐Alanyl‐d‐alanine ligase (Ddl) is a multistructural domain protein that is dependent on adenosine triphosphate (ATP) and is involved in the biosynthesis of PG precursors. Ddl catalyzes the ligation of two d‐alanine molecules into one d‐alanyl‐d‐alanine dipeptide. 127 The inhibition of both Alr and Ddl enzymes in Mtb can lead to a significant weakening of the cell wall, making these enzymes crucial targets for intervention. 128 Terizidone (Group B), a compound comprising of two cycloserine (CS) moieties linked to a terephthalaldehyde molecule, undergoes in vivo hydrolysis to CS and exhibits activity against these enzymes. 129 CS (Group B), a second‐line medication utilized in the management of TB and MDR‐TB, is effective in inhibiting the synthesis of PG by concurrently targeting the Alr and Ddl enzymes. 130 , 131 However, CS has limited clinical utility owing to its nonspecific nature. Other Alr inhibitors, including alanine phosphonates and thiadiazolidinones that are currently underutilized in clinical practice, similarly lack specificity due to their effects on multiple other phosphate‐dependent enzymes. 126 Although several Ddl inhibitors have been demonstrated to be effective at the molecular and cellular level, they require further development for clinical application. 127 , 132
l,d‐Transpeptidase type 2
The presence of β‐lactamase, BlaC, in Mtb has long impeded the development of β‐lactam anti‐TB antibiotics. 133 However, the combination of carbapenem antibiotics and β‐lactamase inhibitors has recently been revisited as a strategy in the fight against TB. Studies have shown that meropenem‐clavulanate (Group C) is highly effective against extensively XDR‐TB. 134 , 135 LdtMt2 is a critical enzyme involved in cell wall synthesis, virulence and amoxicillin tolerance of Mtb. 136 , 137 Complexation of l,d‐transpeptidase type 2 (LdtMt2) and meropenem demonstrates that inactivation of LdtMt2 may be the main mechanism of meropenem‐clavulanate effectiveness against Mtb. 138 Hybrid quantum mechanics/molecular mechanics offer a potential avenue to obtain active molecules that inhibit LdtMt2 139. Biapenem, a carbapenem that boasts superior stability, has been evaluated against RIF‐resistant Mtb. 140 , 141 Recent investigations have implicated both reversible reactions and nonhydrolytic off‐loading reactions from the cysteine transpeptidase LdtMt2 in the effectiveness of meropenem. 142 These findings provide a direction for future optimization of next‐generation anti‐TB carbapenems. The development of carbapenem antibiotics is a remarkable accomplishment, and it is essential to proceed quickly with in vitro and in vivo experimental validations to establish effective treatment regimens for XDR‐TB. Recently, a low‐molecular‐weight organoselenium compound ebselen was shown to inhibit LdtMt2, suggesting that cysteine‐reactive reagents may act as potential LdtMt2 inhibitors. 143
Phospho‐N‐acetylmuramoyl‐pentapeptide transferase
Phospho‐N‐acetylmuramoyl‐pentapeptide transferase (MurX), also called translocase I, converts UDP‐MurNAc‐pentapeptide into prenyl‐MurNAc‐pentapeptide (lipid I) in PG biosynthesis. 144 MurX has been identified as the target for five families of nucleoside antibacterial natural products, which include the tunicamycins, mureidomycins, liposidomycins, muraymycins, capuramycins, and sansanmycins. 145 , 146 , 147 The capuramycin analogue SQ641 has been shown to be effective in killing Mtb by inhibiting MurX. 148 , 149 However, to accommodate higher drug loads, SQ641 requires a phospholipid‐based nanoemulsion formulation. 150 This formulation allows for increased drug delivery and improved efficacy in treating TB. The chemical properties of lipid I renders MurX enzyme assays impractical for screening and lacks reproducibility of the enzyme assays. 151 However, in vitro experiments have shown that a water‐soluble analogue of lipid I can be used as a substitute to quantify the inhibitory activity of library molecules against MurX. 152 This approach has allowed researchers to identify potential new inhibitors of MurX that could be further developed for use in TB treatment.
3.1.5. MA transporter
Mamalian membrane protein large 3
Trehalose monomycolates (TMMs), crucial components in the robust barrier against Mtb, are transferred from the cytoplasm to the periplasm via the Mamalian membrane protein large 3 (MmpL3). Further, TMMs act as the essential building blocks for the synthesis of MAs, an indispensable feature of the protective envelope of Mtb, rendering the latter vulnerable to the effects of TMM depletion. 153 The discovery of knockdown mutants has illuminated the critical role of MmpL3 in both the replication and activity of Mtb, thereby enhancing our comprehension of its multifaceted pathogenesis. 154 Notably, recent advances have led to the discovery of several structurally unique MmpL3 inhibitors, including the 1,2‐ ethylenediamine SQ109, which has successfully advanced to clinical Phase 2 trials. 155 , 156 Other inhibitors, such as the adamantyl urea, the 1,5‐diarylpyrrole, the tetrahydropyrazolopyrimidine, and the indolecarboxamide, have demonstrated significant potential and warrant further investigation. 157 , 158 , 159 , 160
3.2. Lipid metabolism
Complex lipids of Mtb demonstrate a striking ability to function as critical effector molecules that engage in dynamic interactions with the host. By modulating host metabolism and inciting a robust immune response, these lipids play a paramount role in shaping both the mycobacterium's own physiology and that of the host cells. This section reviews the discovery process of targeting aspartate decarboxylase (PanD).
3.2.1. Aspartate decarboxylase
PanD is a key enzyme implicated in the biosynthesis of β‐alanine, a critical precursor molecule for pantothenate and CoA biosynthesis, derived from l‐asparate. 161 Pantothenate, also known as vitamin B5, serves as a crucial building block for CoA production, which is primarily involved in the synthesis of mycolic MA. 162 Notably, PanD is conspicuously absent in mammalian systems, thereby indicating the therapeutic potential of PanD inhibitors. 163 PanD, along with pantothenate synthase, has piqued the interest of researchers in the development of PZA, a Group C anti‐TB drug. 164 PZA functions as a prodrug that is enzymatically activated by nicotinamidase or pyrazinamidase (PZAse), encoded by the pncA gene in Mtb, thereby promoting its conversion into the pharmacologically active pyrazinoic acid (POA). 165 PZA's discovery was the result of a therapeutic test on Mtb‐infected mice. 166 Although PZA exhibited limited anti‐TB activity in vitro in the standard neutral pH broth medium, the exact mechanism remained an enigma. 167 , 168 Nevertheless, years of research have resulted in significant breakthroughs in understanding PZA's mode of action. Genome sequencing data have identified mutations in the panD gene responsible for PanD production in PZA‐resistant strains, independent of mutations in pncA and rpsA, which were previously presumed as targets for PZA. 169 , 170 Here, it was discovered that while PZA was ineffective, the active metabolite POA competes with PanD, thereby disrupting the CoA biosynthetic pathway. 171 , 172 This novel insight may help clarify why POA can effectively target Mtb under nonreplicating conditions, leading to a reduction of the treatment regimen by three months. 169
The other enzymes of the pantothenate synthetase pathway, ketopantoate hydroxymethyltransferase PanB, pantothenate synthetase PanC, and ketopantoate reductase PanE, are also worthy targets for development. 161 , 173 , 174 Research on pantothenate synthetase inhibition is currently focused on two main approaches. The first approach involves the synthesis of nonreactive analogues of the reaction intermediate. 175 , 176 The second approach involves identifying hits for pantothenate synthetase inhibition through HTS, followed by structure‐based validation to determine their efficacy and safety as potential drug candidates. 177 These methods contribute pantoyl adenylate analogues, 175 nafronyl oxalate, 178 actinomycin D, 179 3‐phenyl‐4,5,6,7‐tetrahydro‐1H‐pyrazolo[4,3‐c]pyridine derivatives, 180 and 5‐tert‐butyl‐N‐pyrazol‐4‐yl‐4,5,6,7‐tetrahydrobenzo[d]isoxazole‐3‐carboxamide derivatives 181 as pantothenate synthetase inhibitors. In addition to the two approaches previously mentioned, there are also groups focused on fragment growing, 177 virtual screening, 182 and molecular hybridization 183 , 184 aimed at providing compounds with higher whole‐cell activity. These approaches can lead to the development of novel compounds with improved efficacy and safety for the treatment of TB.
3.3. Protein synthesis and breakdown
Protein synthesis represents an indispensable process vital for the survival and replication of all living organisms, taking place within the ribosomes of cells. 185 This section describes the Mtb ribosome and the leucyl‐tRNA synthase (LeuRS) involved in protein synthesis, as well as the caseinolytic protease P (ClpP) system and the proteasome system responsible for the degradation of intracellular damaged proteins.
3.3.1. Ribosome
In the instance of Mtb, the ribosomal machinery forms a large, functional 70S ribosome consisting of a 2.7 MDa complex. 186 Notably, the 50S large subunit contains 37 ribosomal proteins, as well as 23S and 5S rRNAs, whereas the smaller 30S subunit is comprised of 21 ribosomal proteins and 16S rRNA. 187 , 188 In particular, Mycobacterial‐specific protein Y successfully binds to the 30S subunit, inducing ribosomal hibernation and mediating resistance to aminoglycoside antibiotics. 189 This phenomenon is implicated in the development of nonreplicating Mtb, 190 highlighting the potential of ribosomal targeting to impede protein synthesis. Aminoglycosides, oxazolidinones (including TBI‐223, 191 currently in phase I, sutezolid, 192 and delpazolid, 193 presently in phase II) represent classes of drugs effective for targeting ribosomes.
3.3.2. Leucyl‐tRNA synthase
LeuRS, belonging to the class I aminoacyl‐tRNA synthase subgroup, represents a critical player in intracellular transport. Specifically, GSK656 (phase II), a benzoxazole compound, has shown considerable potential as a specific inhibitor of LeuRS through its targeting of the catalytic site of hydrolysis of incorrectly ligated aminoacylated tRNA. 194 Unlike the initial lead compounds, GSK656 exhibits enhanced selectivity over human cytoplasmic LeuRS while effectively inhibiting protein synthesis in intact human cells. 195 Upon computer screening, N‐benzylidene‐N’‐thiazol‐2‐yl‐hydrazines 196 and 5‐Phenylamino‐2H‐[1,2,4] triazin‐3‐ones 197 were successfully synthesized as promising inhibitors of MtbLeuRS. However, further elucidation of the structure–activity relationships between these molecules and their targets is necessary to gain a deeper understanding of the inhibitory mechanisms and potential structural optimization of these molecules.
3.3.3. Caseinolytic protease P
ClpP represents an ATP‐dependent, unfolding peptidase protein vital in preserving cellular homeostasis by degrading damaged and misfolded proteins. The joint expression of Mtb’s two clpP genes (ClpP1 and ClpP2) generates an active structure capable of hydrolyzing oligopeptides. However, aside from ClpP, ClpX, or ClpC1 is also necessary for the efficient hydrolysis of large, globular proteins. 198 Candidate compounds can selectively bind to either the catalytic active center or the chaperone‐binding site of ClpP, while also partially influencing ClpC1 and ultimately affecting ClpP activity. 199
β‐lactone derivatives, such as EZ120, represent promising inhibitors of ClpP, although further optimization is necessary to enhance their efficacy. 200 The Bortezomib analog Pyr‐FL‐CMK displays MtbClpP selective inhibitory activity. 201 Acyldepsipeptide antibiotics target the ATP‐binding site to curb the activity of ClpP, 202 thereby stunting Mtb growth. In the same way, pyrrole derivatives are also targeted‐ClpP1P2 regulators. 203 Lassomycin, a cyclic peptide synthesized by ribosomes, as well as ecumicin, a macrocyclic tridecapeptide, eradicate Mtb by targeting the ClpC1 ATPase complex. 204 , 205 Additionally, cyclomarin A serves to excessively activate ClpC1, thereby interfering with the usual function of ClpP, 206 eventually resulting in the development of novel strategies for regulating ClpP activity.
3.3.4. Proteasome
Mtb has a proteasomal degradation system that is responsible for the prompt degradation of the majority of damaged proteins. In this system, the prokaryotic ubiquitin‐like protein (Pup) plays the role of ubiquitin in the degradation process. 207 Mtb proteasomes consists of 1 α and 1 β subunit, encoded by genes prcA and prcB, respectively. 208 Peptidyl boronates, macrocyclic peptides, and phenylimidazole derivatives were reported as inhibitors of the proteasome. 208 , 209 , 210 Previous studies have shown that deamidase of Pup (Dop) deamidates the C‐terminal glutamine of Pup to glutamate, preparing it for ligation to target proteins by proteasome accessory factor A (PafA). 211 PafA can efficiently move Pup from one proteasome substrate, inositol 1‐phosphate synthetase, to two different proteins, malonyl‐CoA:ACP transacylase (FabD) and lonely guy (LOG). 212 We recently developed a mutant of MtbPafA, purified active PafA on a large scale, and conducted HTS to identify two promising PafA inhibitors, ST1926 and bithionol. 213 In addition, the computational approach also yields potential inhibitors of the proteasome. 214 , 215
3.4. Amino acid synthesis and metabolism
The levels of various amino acids vary throughout distinct stages of infection and disease progression, reflecting the dynamic nature of infection. 216 Amino acid synthesis and metabolic processes represent crucial determinants in Mtb survival and pathogenesis, underscoring their significance in combatting TB. This section describes the Ser/Thr protein kinases (STPKs) that regulate glutamate metabolism, the shikimate pathway involved in aromatic amino acid synthesis and the tryptophan synthase (TrpAB) involved in L‐tryptophan synthesis.
3.4.1. Ser/Thr protein kinases
STPKs are phosphorylating enzymes that play essential roles in regulating various cellular functions. Within Mtb, 11 STPKs have been identified, of which PknA, B, and G (protein kinases A, B, and G) are indispensable for survival. Studies have highlighted the limited effectiveness of PknA and PknB inhibitors in targeting TB. 107 Our recent work reports a series of antitubercular compounds based on ceritinib derivatives LPX‐16j, 217 of which 5a has good efficacy and safety profile. The differential scanning fluorescence, isothermal titration calorimetry and molecular docking assays suggest that PknB may be one of the targets of 5a. 218 Notably, PknG represents a vital kinase regulating glutamate metabolism. 219 The inhibition of PknG kinase activity was first reported in vitro with the use of AX20017, a pioneering tetrahydrobenzothiophene compound that was specifically designed for this purpose. 220 The inhibition of PknG activity has also been observed with nitro‐fatty acids, 221 sclerotiorin, 222 and steroidal lactones, 223 which have all been reported as effective inhibitors of this kinase. NU‐6027, a dual inhibitor of PknG and PknD, has emerged as a potent inhibitor of Mtb growth in macrophages and mouse tissues. 224 In recent studies, it has been suggested that PknG may additionally block autophagic flux by inhibiting phagosome maturation. 225
3.4.2. Shikimate pathway
The shikimate pathway comprises seven critical enzymatic steps, which are fundamental in Mtb and are connected with the synthesis of vital aromatic molecules 226 (e.g., tryptophan 227 ). Among these, inhibitors 3‐dehydroquinate synthase (DHQS), shikimate dehydrogenase, and shikimate kinase have been specifically targeted and have demonstrated noteworthy activity against Mtb. One of the important targets of multitarget compound IMB‐T130 is DHQS, which can effectively inhibit Mtb. 228 IMB‐SD62, a lead triazolothiadiazole, and its derivatives were identified as inhibitors of shikimate dehydrogenase with antitubercular activity. 229 , 230 , 231 Shikimate kinase inhibitor Compound 5631296, which was acquired through a comprehensive screening process, has demonstrated a remarkably low toxicity to HepG2 cells. Furthermore, it has exhibited synergistic activity when combined with RIF, resulting in the effective eradication of Mtb. 232
3.4.3. Tryptophan synthase
The indispensability of TrpAB for the sustenance of Mtb within macrophages and circumvention of host immune milieu renders it a highly auspicious therapeutic target. 227 , 233 In bacteria, fungi, and plants, the TrpAB bifunctional enzyme catalyzes the ultimate two steps of tryptophan biosynthesis and modulates pyridoxal 5′‐phosphate as an indispensable cofactor. 234 , 235 , 236 TrpA converts indole‐3‐glycerol phosphate into glyceraldehyde‐3‐phosphate and indole. TrpB catalyzes PLP‐dependent β‐replacement reaction in which indole displaces the hydroxyl group of l‐Ser to produce l‐Trp. 237 , 238 An allosteric, mixed‐type inhibitor BRD4592 inhibits enzyme subunits and shows in vitro antitubercular efficacy. 233 The same group also reported GSK1 and GSK2, which were found to target TrpAB in 2017 by Abrahams et al., 239 both bind to TrpAB very similarly to BRD4592. 238
3.5. Nucleotide synthesis
Mtb must execute conserved DNA replication to transmit genetic information, a highly regulated process that represents a rich source of potential drug targets. This section describes the topoisomerase, the RNA polymerase (RNAP), and the transcriptional repressor of ethA (EthR).
3.5.1. Topoisomerase
The genome of Mtb encodes a solitary type I topoisomerase (topoI) and a single type II topoisomerase (gyrase), comprising gyrA (Rv0006) and gyrB (Rv0005). 133 Biochemical studies utilizing monoclonal antibodies and oligonucleotides have specifically demonstrated the site‐specificity of MtbtopoI. 240 Various compounds have demonstrated inhibitory activity against MtbtopoI, including m‐AMSA, 241 polyamines, 242 imipramine, and norclomipramine, 243 but appear limited in their cytostatic abilities. In comparison, DNA gyrase has emerged as a promising drug target for anti‐TB drug development. Several fluoroquinolone derivatives (Group A) have exhibited substantial inhibitory potential against TB, and are currently undergoing evaluation for the treatment of MDR‐TB and XDR‐TB. 1 , 244 X‐ray crystallography has proven to be an instrumental tool in the concerted effort to comprehend the precise mechanism by which fluoroquinolones affect DNA gyrase and to develop novel inhibitors for this crucial enzyme. 245 In sharp contrast to fluoroquinolones—which chiefly target the N‐terminal domain of GyrA along with the C‐terminal domain of GyrB fused to GyrA—the newly developed phase II drug, SPR720, 246 selectively targets GyrB. This aminobenzimidazole is both structurally and mechanistically dissimilar to fluoroquinolones, thus significantly reducing the risk of cross‐resistance. Thiazolopyridine ureas, 247 thiazole‐aminopiperidine hybrid analogues, 248 substituted benzofurans, 249 and 4‐aminoquinolines 250 have shown promising results as GyrB inhibitors with anti‐TB activity. Undoubtedly, the triumph of fluoroquinolones and the existence of other potential ligand‐binding sites 251 in topoisomerase clearly suggest that the search for new topoisomerase inhibitors is a worthwhile scientific pursuit.
3.5.2. RNA polymerase
RNAP is an evolutionarily conserved enzyme that plays a vital role in both transcription initiation and RNA elongation, and is subject to diverse regulatory mechanisms mediated by multiple transcription factors. In the case of Mtb, the RNAP is comprised of a central core that consists of five subunits (α2ββ’ω), with the β subunit being susceptible to inhibition by RIF. 252 Several RIF analogues (e.g. rifamycin, rifalazil, and rifabutin) have been developed with the aim of enhancing the therapeutic efficacy of RIF through the same mechanistic pathway. Regrettably, a significant number of RIF‐resistant TB cases have emerged in clinical settings. Gene mutations that arise in rpoB are the primary culprits behind this phenomenon, rendering RNAP an unviable target for novel drug development. 253 , 254 Nevertheless, strategies aimed at targeting other critical transcriptional processes continue to be promising avenues for future investigations. 255
3.5.3. Transcriptional repressor of ethA
EthR is a repressor of ethA, a gene encoding a monooxygenase required for the activation of the prodrug ETH. Overexpression of ethR, which codes for the repressor EthR belonging to the TetR/CamR family of transcriptional regulators, has been found to induce potent inhibition of ethA. 256 As previously mentioned, various thiocarbamide‐containing drugs, including ETH, rely on the activity of the monooxygenase EthA for activation. A breakthrough inhibitor of EthR, BVL‐GSK098, was developed via a combination of molecular design, screening, and optimization. This compound demonstrated impressive synergy with ETH combination therapy, as evidenced by a mouse model of TB. 257 , 258 Notably, the molecular targeting of EthR presents a groundbreaking approach that may help reverse ETH‐induced resistance. Spiroisoxazoline analogues, 258 oxadiazole derivatives, 259 and N‐phenylphenoxyacetamides 260 have been discovered to possess EthR‐inhibitory and ETH‐enhancing properties. It is worth noting that first‐line drugs such as INH also require mycobacterial enzyme activation, making the development of transcriptional regulator‐targeting agents a pressing clinical need, as drug resistance often arises at this stage.
3.6. Energy metabolism
In recent years, significant attention has been devoted to Mtb’s energy metabolism—particularly, the oxidative phosphorylation pathway—with the aim of identifying novel strategies for pathogen control and drug discovery. Among these promising strategies are classes of antibacterial agents that target different elements of the oxidative phosphorylation pathway, which have shown significant efficacy in controlling dormant or latent mycobacterial infections. These novel therapeutic approaches hold tremendous potential for shortening the chemotherapy regimen for TB. In oxidative phosphorylation, the respiratory chain protein complexes facilitate the generation of a proton motive force (PMF) across a biomembrane, which is then harnessed by ATP synthase to produce ATP. 261 This process involves several key steps, including (a) the transfer of electrons from NADH via the type II NADH dehydrogenase (NDH‐2) into the electron transport chain and (b) the acceptance of electrons by oxygen via a supercomplex comprising the cytochrome bc 1 complex and the cytochrome aa 3‐type terminal oxidase. Additionally, a cytochrome bd‐type terminal oxidase can directly accept electrons from the menaquinone pool. 262
Clofazimine 263 (Group B), a phase III drug currently used to target NDH‐2 in leprosy, is also undergoing repurposing as a treatment for TB. Additionally, clinical trials (phase I) have been initiated for TBI‐166, 264 a riminophenazine analogue that may enhance the efficacy of clofazimine while reducing potential side effects. However, recent findings suggest that the activity of these drugs is not solely dependent on NDH‐2. 265 Other agents that inhibit NDH‐2 and are commonly employed for the treatment of psychiatric disorders—such as thioridazine 266 and other phenothiazines 267 —are currently being assessed as alternatives to conventional anti‐TB therapy. Telacebec (Q203), an imidazopyridine amide that targets the QcrB subunit of respiratory cytochrome bc 1 complex, disrupts ATP synthesis. Encouraging results from phase I clinical trials regarding safety and tolerability have led to the initiation of phase II clinical trials examining the efficacy of Telacebec against MDR‐TB and XDR‐TB strains. 268 , 269 Another drug, PZA, affects PMF 270 and is commonly used in combination with other respiratory chain inhibitors. Although quinazoline derivatives, 271 morpholino thiophenes, 272 arylvinylpiperazine amides, 273 heterobiaryl side chain analogues, 274 and imidazo[2,1‐b]thiazole derivatives 275 have shown promise as inhibitors of QcrB, their metabolic stability requires further optimization.
3.6.1. ATP synthase
ATP synthase is another crucial factor in Mtb’s energy metabolism and, as such, represents a critical target for drug development. Bedaquiline (BDQ) 276 , 277 , 278 (Group A), a diarylquinoline compound, exerts potent antimycobacterial activity by binding to the c and Ɛ subunits of F‐ATP synthase, leading to the blockade of its proton pumping function. BDQ has been granted approval by the US FDA as a crucial component of short‐term XDR‐TB treatment regimens (BPaL regimen). 279 This achievement confirms that ATP synthesis is a prime vulnerability in Mtb and that impairing the energy metabolism holds significant promise for shortening the duration of TB treatment. The successful initiation of phase I clinical trials for two dialkoxypyridine analogues 280 (TBAJ‐876 and TBAJ‐587), which exhibit higher potency, significantly reduced lipophilicity, and pose a lower risk of cardiotoxicity, is a notable breakthrough following the clinical triumph of BDQ. Sudapyridine (WX‐081), a novel compound displaying similar efficacy to BDQ in the TB mouse model, boasts superior pharmacokinetic and toxicological profiles when compared with BDQ. WX‐081 is currently undergoing investigations in phase 2 clinical trials involving patients. 281 In addition, squaramides 282 and pyrazolopyrimidines 283 are being investigated in preclinical studies.
3.7. Virulence
Mtb is an opportunistic slow‐growing intracellular organism whose multifaceted virulence mechanisms support the establishment of infection, persistence and reactivation. 284 Consequently, efforts to develop Mtb virulence inhibitors are gaining increasing attention as a potential avenue for advancing TB control programs. This section describes the two‐component system (TCS) and the ESX‐1 secretion system of Mtb.
3.7.1. Two‐component system
The TCS, a key pathogen–host signaling pathway constituted by two proteins responsible for transducing environmental cues into physiological responses, has emerged as a potent target for TB therapy. The canonical two‐component signaling pathway is comprised of a sensor kinase (SK) that detects specific environmental cues, and a cognate response regulator (RR) that mobilizes the necessary biological response in return. 285 Several TCSs, including PhoPR, 286 DosRST, 287 , 288 PdtaRS, 289 and MtrAB, 290 have demonstrated significant contributions to in vivo virulence and are, therefore, particularly attractive targets for future TB drug development.
PhoPR
PhoPR is recognized as a central regulator of pathogenic traits in MTBC strains, influencing the secretion of the virulence factor ESAT‐6, biosynthesis of acyltrehalose‐based lipids, and modulation of antigen export. 291 Studies have revealed that low pH causes PhoPR phosphorylation, which, in turn, triggers the activation of the cytosolic redox sensor WhiB3. 292 Given its role in Mtb virulence, PhoPR represents a compelling target candidate for TB therapy. 293 Ethoxzolamide, a drug commonly used to manage duodenal ulcers, has been shown to inhibit PhoPR and significantly reduce Mtb load in both infected macrophages and mice. 294
DosRST
DosRS was initially discovered to play a critical role in the survival and virulence of Mycobacterial spp. under hypoxic conditions. 295 , 296 Another SK, DosT, also contributes to sensing hypoxia and NO, alongside DosRS. 297 Mtb exploits DosRST to establish and maintain nonreplicating persistence in response to hypoxia, NO, acid stress, or starvation. 298 A recent whole‐cell phenotypic HTS campaign identified three inhibitors of DosRST, including artemisinin, HC102A, and HC103A. Artemisinin functions by disrupting heme‐based SKs DosS and DosT via the oxidation of ferrous heme, and subsequent heme‐artemisinin adduct formation. In contrast, HC102A and HC103A do not regulate DosS/T heme, but have been found to inhibit SK autophosphorylation. 299 , 300
3.7.2. ESX‐1 secretion system
ESAT‐6 secretion system‐1 (ESX‐1) is a sophisticated type VII secretion system which is encoded by the RD1 locus. 301 Its primary function is to facilitate the secretion of a variety of substrates such as ESAT‐6, EsxA, and EspB, among others, with the ultimate goal of inducing macrophage lysis. 302 , 303 , 304 In addition, recent studies have proven that PhoPR inhibitors are highly effective in regulating ESX‐1 due to the fact that PhoPR serves as an essential mediator in activating ESX‐1 secretion. 305 Leveraging the EsxA‐dependent cytolytic activity of Mtb, HTS has yielded two promising compounds. Notably, BBH7 and BTP15 not only significantly reduce intracellular bacterial load, but also promote phagolysosomal fusion in Mtb‐infected THP‐1 macrophages. 306 More recently, HTS has produced new lead compound 3,5‐dinitrobenzamide. 307
3.8. Others
3.8.1. Folic acid and mycobactin biosynthesis
Dihydrofolate reductase
At the core of the folate pathway lies the pivotal role of dihydrofolate reductase (DHFR), responsible for catalyzing the transformation of dihydrofolate (DHF) into tetrahydrofolate (THF) using NADPH as an electron donor. Notably, existing DHFR inhibitors have shown limited efficacy against MtbDHFR or are only weakly effective in inhibiting Mtb. However, P‐aminosalicylic acid (PAS), classified as a Group C drug, serves a dual role as both a substrate and prodrug within the folate pathway, with DHFR serving as one of its key targets. 308 , 309 Further analysis of the mechanism of PAS’ antifolate action has highlighted the potential benefits of utilizing compounds that can target multiple targets within the same pathway, thereby simplifying treatment regimens. 310 Our team acquired the DHFR inhibitor AF‐353 through virtual screening and confirmed its selectivity between MtbDHFR and hDHFR. 311 In addition, we has predicted that ceritinib, a classical antilung cancer drug and its derivatives, may hold significant promise in combatting Mtb by serving as an effective DHFR inhibitor. 217
3.8.2. Metal uptake
Iron‐dependent regulator
In response to the stress imposed by the host, Mtb employs iron chelators known as siderophores, notably the mycobactin, to acquire iron. Meanwhile, Mtb has developed highly sophisticated intracellular iron sensing mechanisms, which are tightly regulated by the Fur (ferric uptake regulator) or DtxR (diphtheria toxin regulator) families. 312 Among these, the mycobacterial iron‐dependent regulator (IdeR), a crucial metal binding transcriptional regulator of the DtxR family, plays a key role in maintaining mycobacterial iron homeostasis and facilitating virulence. 313 Through virtual screening, numerous IdeR inhibitors, including acid alizarin violet N derivatives 314 and short peptides, 315 have been identified, though further analysis pertaining to structure–activity relationships is necessary to identify even more potent candidates.
3.8.3. Cholesterol metabolism
Adenylyl cyclase
GSK2556286 (GSK‐286, phase I) is a pyrimidine‐2,4‐dione derivative that was uncovered from a HTS library of Mtb‐infected macrophages. 316 Intriguingly, several in‐depth analyses of the Mtb survival cycle have revealed that cholesterol metabolism plays a pivotal role in facilitating Mtb’s survival within macrophages. 317 In fact, in the absence of cholesterol utilization, Mtb is unable to establish an effective infection in macrophages and cannot effectively elicit pathogenesis. 318 Notably, the MtbAC possesses a specific ATP pocket that is distinct from its mammalian counterpart, thus playing a key role in converting NTPs into respective 3′,5′‐cyclic nucleoside monophosphates. 319 Through direct adenylyl cyclase (AC) activation, GSK‐286 induces the generation of high levels of 3′,5′‐cyclic AMP (cAMP), thereby disrupting the Mtb cAMP signaling network. 320 Furthermore, the agonist V‐58 is known to operate via a similar mechanism, thereby modulating cAMP signaling and inhibiting cholesterol metabolism by Mtb. 321 It is worth noting that cAMP signaling is multifaceted in terms of its impact on Mtb pathogenesis, with studies indicating that it can regulate TNF‐α by macrophages. 322
Figure 4 provides a comprehensive summary of the main targets of anti‐TB compounds targeting pathogens. Indirect effects of GSK‐286 and V‐58 on host macrophages get us thinking. Notably, the interaction between pathogens and their host is a crucial factor in determining bacterial pathogenicity and virulence. It is true that all drugs in the current clinical pipeline target the pathogen directly. In fact, by targeting cell wall synthesis and assembly, protein synthesis, nucleotide synthesis, energy metabolism, folic acid, and cholesterol metabolic pathways, existing drugs have already shown great promise in achieving high clinical outcomes. 323 Current trends in drug development involve bi‐directional screening for both cellular and target‐based activity, with a focus on multitarget candidates. Finding new targets and their inhibitors in Mtb remains a promising strategy for combating drug resistance and developing potent lead molecules, though it is important to acknowledge the potential drawbacks such as the lengthy investment required for development and their limitations in treating both active and latent TB. Due to these challenges, an increasing number of studies have recently devoted their efforts towards host‐directed therapies for TB treatment.
FIGURE 4.
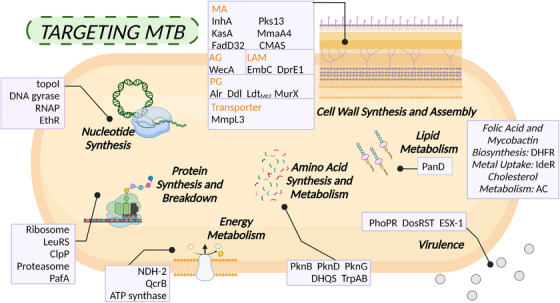
Overview of antituberculosis targets aimed at Mtb. Disruption of crucial pathways in Mtb, such as cell wall synthesis and assembly, protein synthesis and breakdown, and energy metabolism, has been regarded as a potent strategy for combating tuberculosis. Recently, there has been a growing interest in interventions focused on lipid metabolism, amino acid synthesis and metabolism, nucleotide synthesis and virulence. The elements in the figure were drawn using BioRender online tool (https://biorender.com).
4. HOST‐DIRECTED THERAPY
The infection process of Mtb is highly dependent on host cells and requires the utilization of multiple strategies to persist within infected cells. Although host‐directed therapy (HDT) is often viewed as an adjunctive regimen, recent clinical studies have shown that it can lead to rapid anti‐TB effects and improved prognosis. 324 The complex immune events that occur during Mtb infection and pathogenesis offer numerous opportunities for HDT, and ongoing discoveries pertaining to the involved pathways and molecular participants continue to expand the list of potential molecules that can serve as anti‐TB treatments. Currently, TB HDT strategies are focused on three main objectives: (a) enhancing host immune defense, (b) interfering with the use of host mechanism, and (c) limiting immunopathology.
4.1. Enhancing host immune defense
Innate immune responses include cells and mechanisms that are either constantly present or are activated within minutes to hours following an infection to suppress the replication and spread of the invading Mtb. Multiple immune cells are endowed with a repertoire of pattern recognition receptors, including TLRs, NLRs, and CLRs, each of which has been implicated in the recognition and internalization of Mtb. 325 , 326 These innate mechanisms act as a first line of defense against pathogenic microorganisms and are essential in shaping the subsequent adaptive immune response. 327 Phagocytic DCs present Mtb antigen to T lymphocytes and promote activation and differentiation of naïve CD4 T cells and naïve CD8 T cells. 328 , 329 This process necessitates the presentation of antigen in the context of major histocompatibility complex (MHC), costimulatory molecules, and the necessary cytokines. 11
The immunity‐centered HDT approach emphasizes the development of IFN‐α and IFN‐γ supplementation regimens, which, when utilized in conjunction with antimycobacterial therapy, may potentially influence the progression of pulmonary TB. 330 , 331 , 332 Nonetheless, we are predominantly concerned with regulating the abundance of them to influence the immune process. Several methods have been devised to enhance the endogenous IFN response, depending mainly on the activation of TLRs, but their use in the treatment of TB has not been reported. 333 , 334 Metformin (MET), the most widely administered diabetes drug, has been proposed as a candidate adjunctive HDT for TB. 335 In humans, MET exhibits a multitude of effects, such as the production of TNF‐α, IFN‐γ, and interleukin 1β (IL‐1β), augmented phagocytosis activity, and increased production of ROS. 336 Myeloid‐derived suppressor cells (MDSCs) are increasingly recognized as a critical driver of TB pathogenesis and represent an immunosuppressive cell population. 337 In fact, elevated levels of MDSCs have been observed in both blood and sputum of patients with active TB in studies, 338 and have also been induced in healthy individuals after exposure to Mtb. 339 These cells play a detrimental role in diminishing protective T‐cell responses and may contribute to the inability of hosts to eradicate the infection, which subsequently leads to the development of TB disease. 340 Tasquinimod, 341 an experimental quinoline‐3‐carboxamide, has demonstrated tremendous promise in inhibiting tumor growth in murine cancer models and has recently been shown to deplete MDSCs and reduce the relative bacterial burden in the lung and spleen of murine models of TB 342 (Table 2).
TABLE 2.
Targets, drug candidates, and actions of host‐directed therapeutics for tuberculosis.
| Effect of HDT | Target | Typical compound | Mechanism of action | Drug stage for TB | |
|---|---|---|---|---|---|
| Enhancing host immune defense | Myeloid‐derived suppressor cells (MDSCs) | Tasquinimod 341 | Antagonizing MDSCs to activate T cells | Biological test | |
| Tyrosine kinase (TK) | Imatinib 348 | Inhibition of TK to activate phagolysosomal acidification | Phase II | ||
| Ca2+‐adenosine monophosphate‐activated protein kinase (AMPK) | GABA 349 | Inhibition of Ca2+‐AMPK to enhance phagosomal maturation | Biological test | ||
| Indoleamine 2,3‐dioxygenase (IDO) | D‐1MT 355 | Inhibition of IDO to increase CD4 T cells | Biological test | ||
| Heme oxygenase‐1 (HO‐1) | SnPPIX 356 | Inhibition of HO‐1 to increase iNOS and NO production | Biological test | ||
| Histone deacetylases (HDACs) | Trichostatin A 362 | Inhibition of HDAC6 to modify epigenetics | Biological test | ||
| Interfering with the use of host mechanism | Regulation of autophagy | Mammalian target of rapamycin (mTOR)/AMPK | Rapamycin 375 | Regulation of mTOR/AMPK to enhance autophagy | Biological test |
| Extracellular regulated protein kinase ERK1/2 | Pasakbumin A 393 | Activating ERK1/2 to enhance autophagy | Biological test | ||
| Epidermal growth factor receptor (EGFR) | Gefitinib 399 | Inhibition of STAT3 to enhance autophagy | Biological test | ||
| Tyrosine kinase (TK) | Ibrutinib 401 | Inhibition of BTK/Akt/mTOR to enhance autophagy | Biological test | ||
| Transcription factor EB (TFEB) | Statins 403 , 404 , 405 | Activating AMPK/mTORC1/TFEB to enhance autophagy | Biological test | ||
| Sirtuin 1/3 (SIRT1/3) | Resveratrol 406 | Activation of SIRT1/3 to enhance autophagy | Biological test | ||
| Toll‐like receptor 7 (TLR7) | Imiquimod 409 | Agonism of TLR7 to enhance autophagy | Biological test | ||
| Intervention of granuloma formation | Vascular endothelial growth factor (VEGF) | Pazopanib 415 | Inhibition of VEGF to impede granuloma angiogenesis | Biological test | |
| Angiopoietin‐2 (ANG‐2) | AKB‐9778 418 | Inhibition of ANG‐2/TIE2/VE‐PTP to reduce infection‐induced vascular permeability | Biological test | ||
| Regulation of cell death | Myeloid cell leukemia sequence‐1 (Mcl‐1) | Sabutoclax 443 | Inhibition of Mcl‐1 to activate apoptosis | Biological test | |
| Complex I | Metformin 396 | Inhibition of complex I to reduce necrosis | Phase II | ||
| Ferroptosis | Ferrostatin‐1 446 | Inhibition of ferroptosis to reduce necrosis | Biological test | ||
| Mitogen‐activated protein kinase (MAPK) | Corticosteroids 447 | Inhibition of p38 MAPK to reduce necrosis | Phase II | ||
| Domain‐like receptor protein 3 (NLRP3) | Baicalin 453 | Inhibition of PERK/TXNIP/NLRP3 to reduce pyroptosis | Biological test | ||
| Limiting immunopathology | Matrix metalloproteinase (MMP) | Doxycycline 466 | Inhibition of MMP to reduce cavitary pathology | Phase III | |
| 5‐Lipoxygenase (5‐LOX) | Zileuton 477 | Inhibition of 5‐LOX to regulate lipid metabolism | Biological test | ||
| Cyclooxygenase‐2 (COX‐2) | NSAIDs 481 | Inhibition of COX‐2 to reduce inflammatory response | Phase II | ||
| Glucocorticoid receptor (GR) | Corticosteroids 484 | Binding to GR to reduce inflammatory response | Phase II | ||
| Poly(ADP‐ribose) polymerase 1 (PARP1), | PJ‐34 487 | Inhibition of PARP1 to reduce inflammatory response | Biological test | ||
| Phosphodiesterase‐4 (PDE‐4) | Dovramilast 488 | Inhibition of PDE‐4 to reduce inflammatory response | Phase II | ||
Notably, studies have shown that enhancing phagocytosis can effectively limit the intracellular growth of Mtb. 343 , 344 Phagocytosis serves as the fundamental link between the innate and adaptive branches of the immune system. 345 In addition to effectively isolating and eliminating pathogens, the phagocytic process also plays a critical role in triggering the activation of adaptive immune responses. Following the ingestion of pathogens, phagosomes must undergo a sequence of dynamic transformations involving both the membrane and internal components, ultimately allowing for their maturation and subsequent fusion with lysosomes. 346 Pathogens frequently resort to various cunning strategies to evade capture, including evading detection, disrupting signaling pathways, or disabling the machinery that drives the phagocytic process. 347 Imatinib, a chemical inhibitor of tyrosine kinase (TK), has been found to promote Mtb killing through the activation of cathepsin D and phagolysosomal acidification. 348 Gamma‐aminobutyric acid, an inhibitory neurotransmitter, is known to regulate the Ca2+‐AMPK (adenosine monophosphate‐activated protein kinase) pathway, thereby enhancing phagosomal maturation. 349
In the context of adaptive immune responses, antigen‐presenting cells (APCs) play a pivotal role in phagocytosing antigens and attaching them to MHC class I or II molecules, thereby presenting the antigens to T cells to initiate adaptive T cell responses. Facilitating the proper activation of APCs is an effective strategy to help hosts recognize Mtb. G1‐4A, a polysaccharide derived from Tinospora cordifolia and a reported TLR inhibitor, has shown promising results in improving host defense against Mtb. In fact, G1‐4A has been found to increase NO and proinflammatory cytokine secretion (such as TNF‐α, IL‐β, IL‐6, IL‐12, IFN‐γ) through upregulating MHC‐II, thus leading to reduced intracellular survival of Mtb. 350 Additionally, vitamin D, which is required for TLR production, has been recognized as a key molecule in host defense against TB. 351 However, more research is necessary to fully elucidate the role of vitamin D in the prevention and treatment of TB. 352 In fact, Mtb devotes considerable energy to directing the induction of the cellular response to infection. 353 Mtb induces the expression of indoleamine 2,3‐dioxygenase (IDO), which degrades tryptophan and attenuates T cell and NK cell proliferation to suppress immunity. 354 The specific inhibitor of IDO activity, D‐1MT, has been shown to improve clinical outcomes by increasing the entry of CD4 T cells into granulomas. 355 Heme oxygenase‐1 (HO‐1), an antioxidant enzyme, is induced by Mtb to be expressed in the lung. The inhibition of HO‐1 activity with tin protoporphyrin XI (SnPPIX) was found to enhance iNOS expression and NO production by Mtb‐infected macrophages following activation by IFN‐γ produced by T lymphocytes, consequently allowing for a more efficient control of bacterial replication by host cells. 356
The epigenetic changes elicited by Mtb infection play a pivotal role in circumventing the immune response of the host and thereby inducing bacterial persistence and dissemination. 357 Epigenetic modifications used by Mtb to evade host immune responses include histone acetylation, 358 noncoding RNA expression 359 , 360 and DNA methylation. 361 Targeting epigenetics works have been undertaken for the management of TB. Studies have shown that Trichostatin A impedes host histone deacetylases (HDACs) and, as a result, augments both in vitro and in vivo antimycobacterial efficacy in human macrophages. 362 Tubastatin A, an inhibitor of HDAC6, fortifies the immune response and curbs the growth of mycobacteria in an Mtb‐infected mouse model. 363 Valproic acid and suberoylanilide hydroxamic acid (vorinostat), both inhibitors of HDACs, have supplementary potential to INH and RIF regimens. 364 Also as an adjunct to standard TB treatment, the combination of 4‐phenyl butyrate (a nonspecific HDACs inhibitor) with vitamin D3 administered orally has shown beneficial effects on clinical recovery. 365 , 366 DNA methylation inhibitors, such as 5‐azacytidine, belong to another class of compounds that target host epigenetics. 367 Bristol‐Myers Squibb has previously submitted a phase Ib/IIa open label, nonrandomized clinical trial to investigate whether the use of injectable azacitidine affects DNA methylation levels and immune signaling pathways during the treatment of pulmonary TB. Unfortunately this study has now been withdrawn. 368 Developing methods to utilize small‐molecule drugs to influence the course of immune events is a promising avenue of research, especially given the fact that most of events are still being explored.
4.2. Interfering with the use of host mechanism
Mtb is an intracellular parasitic bacterium that relies on host cell mechanisms in order to proliferate and persist. Its replication and persistence is determined by a multitude of cellular processes, including autophagy, granuloma formation, and the specific type of cell death that occurs within infected cells (such as apoptosis, necrosis, and pyroptosis). 369 The majority of novel HDT strategies operate by disrupting these processes, thereby inhibiting the survival potential of Mtb.
4.2.1. Regulation of autophagy
Autophagy is a key mediator responsible for the degradation of damaged macromolecules and organelles. Autophagy is regulated primarily by the mammalian target of rapamycin (mTOR) complex 1 and the AMPK. 370 , 371
mTOR/AMPK
The 5′‐adenosine AMPK plays a crucial role in maintaining cellular material and energy homeostasis via phosphorylation. 372 The mammalian target of mTOR is a serine/threonine kinase that operates through two distinct complexes (mTORC1 and mTORC2) and regulates cellular metabolism in response to environmental cues. 373 Autophagic signaling is typically divided into mTOR‐independent and mTOR‐dependent pathways, with the latter serving as a negative regulatory pathway through which Mtb inhibits host autophagy. 374 Inhibition of mTOR has therefore emerged as a viable strategy for counteracting the low autophagic state observed in Mtb infections. One classical example of an mTOR inhibitor is rapamycin, which is capable of inducing in situ autophagy in lung macrophages and has been found to effectively alleviate Mtb burden when utilized in conjunction with INH or RIF via inhalation. This effect is mediated by the phosphorylation of S6 kinase. 375 It is worth noting that the use of rapamycin is currently limited by its potential for immunosuppression. 376 In contrast, the antiprotozoal drug nitazoxanide has been found to strongly stimulate autophagy while inhibiting mTOR signaling. 377 Additionally, the antidepressant amoxapine has demonstrated the ability to induce autophagy and protect macrophages during infection. 378 The anticonvulsant drugs carbamazepine and valproic acid, meanwhile, can induce mTOR‐independent autophagy through AMPK activation, a unique characteristic that sets them apart from other drugs. 379 MET, the antidiabetic drug, has been reported to significantly reduce intracellular Mtb growth in an AMPK‐dependent manner. 380
TNF‐α
The TNF‐α signaling pathway has proved amenable for therapy of autoimmune and other chronic inflammatory noninfectious diseases. 381 , 382 Multiple cells synthesize this cytokine in response to mycobacterial infection to induce a phagocytosis program, 383 , 384 , 385 , 386 , 387 and the regulatory process involves IFN‐γ, 384 enzymes, 388 and lipid mediators. 389 TNF‐deficient mice infected with Mtb exhibit delayed chemokine induction and immune cell recruitment. 390 Maintaining its normal level is essential to activate autophagy. 391 TNF‐α is also associated with granuloma biogenesis and integrity, driving the formation of durable solid granulomas. 392 Since none of the approved TNF modulators are small‐molecule drugs, they are not discussed in this review. Pasakbumin A, which is extracted from Eurycoma longifolia Jack, effectively inhibits intracellular Mtb killing by inducing both autophagy and TNF‐α production through the extracellular regulated protein kinase ERK1/2‐ and nuclear factor NF‐κB‐mediated signaling pathways in Mtb‐infected cells. 393 It is noteworthy to mention that the studies have reported a higher incidence of active TB among patients receiving TNF‐neutralizing therapy. 394 , 395 Notwithstanding its beneficial effects on granuloma formation, overexpression of TNF‐α can promote inflammation while contributing to the hyperactivation of infected macrophages in the granuloma and provoke programmed necrosis 396 , 397 (as detailed below).
Protein kinase
Kinases are central to mammalian signaling pathways. The screening process for identifying compounds that suppress the proliferation of Mtb in macrophages yielded two promising candidates, namely imatinib, which was previously mentioned, and gefitinib, an inhibitor of the epidermal growth factor receptor (EGFR). 398 The therapeutic application of gefitinib in Mtb‐infected macrophages has been demonstrated to effectively restrain the STAT3 signaling pathway, a transcription factor that has been found to impede effective immune responses in vivo. 399 Additionally, gefitinib treatment was observed to stimulate the expression of genes associated with lysosomal biogenesis and function, resulting in an increased production of functional lysosomes with enhanced autophagy. 400 According to recent research, the TK inhibitor, ibrutinib, which is commonly used in the treatment of chronic lymphocytic leukemia, has been identified to have efficacy against Mtb infection by inhibiting the BTK/Akt/mTOR signaling pathway and inducing autophagy. This treatment has demonstrated a significant reduction in bacterial load in Mtb‐infected mice models. 401
In addition to the aforementioned drugs, there is growing interest in the development of modulators that regulate autophagy through alternative pathways. Autophagy‐related proteins are regulated by the transcription factor EB (TFEB). 402 Recent studies have demonstrated that statins can induce autophagy by altering cellular AMP:ATP ratios in Mtb‐infected macrophages and activating the AMPK/mTORC1/TFEB axis. 403 , 404 , 405 Meanwhile, the activation of host sirtuin 1 (SIRT1) has also been shown to reduce intracellular growth of drug‐susceptible and drug‐resistant strains of Mtb, inducing phagosome‐lysosome fusion and autophagy through a SIRT1‐dependent mechanism. 406 In recent studies, it has been demonstrated that the sirtuin 3 (SIRT3), coordinates mitochondrial function and autophagy activation to facilitate anti‐Mtb responses via peroxisome proliferator activated receptor alpha (PPARA). 407 Natural activators such as resveratrol 406 and honokiol 407 activate SIRT1 and SIRT3, respectively, exerting their inhibitory effects on intracellular growth of Mtb. Additionally, the use of 4‐phenyl butyrate in combination with vitamin D shows potential in counteracting the inhibition of human antimicrobial peptide LL‐37 expression by bacilli, thereby promoting autophagy. 408 This finding suggests that regulation of antimicrobial peptides could serve as an approach to promote autophagy. 408 Imiquimod (IMQ), a TLR7 agonist, has been shown to act as a radiosensitizer for melanoma by inducing autophagy. 409 Recent studies indicate that IMQ can stimulate TLR7 and activate autophagy by increasing the production of ROS through the p38‐ or MEK/ERK1/2‐mediated signaling pathways during the early phase. 410 Further research has outlined the two E3‐Ubiquitin (E3‐Ub) ligases, PRKN and SMURF1, that specifically target phagophores by tagging poly‐Ub, 411 highlighting the efficacy of targeting ubiquitin machinery in regulating autophagy.
4.2.2. Intervention of granuloma formation
Pathogenic mycobacteria have been shown to induce the formation of complex cellular aggregates known as granulomas, which are a hallmark of TB. These granulomas are composed of various immune cells, including macrophages, DCs, and T cells, which surround and attempt to contain the mycobacteria through the formation of a barrier. However, the bacteria can persist within the granuloma, which can lead to recurrent or chronic infections. If uncontrolled, the formation of granulomas can lead to the development of active TB in human patients. 412
Angiogenesis obstruction
In tumors, the hypoxic environment induces VEGF expression and stimulates angiogenesis. 413 Significantly high levels of serum VEGF were also found in patients with active TB. 414 In 2015, Oehlers et al. reported that intercepting VEGF pathway signaling by pazopanib (a VEGF receptor inhibitor 415 ) or SU5416 (a prototypical TK receptor 416 ) inhibits granuloma‐associated angiogenesis, reduces Mtb burden and limits the spread. 417 Subsequent studies have demonstrated that angiopoietin‐2 (ANG‐2) expression is prominently upregulated within granulomas. Specifically, pharmacological inhibition of vascular endothelial‐protein tyrosine phosphatase 418 (AKB‐9785, a structurally similar and equivalent inhibitor to AKB‐9778, patented from Aerpio Therapeutics 419 , 420 ) interrupts the ANG‐2/TIE2/VE‐PTP axis and severs the stabilizing effects of this pathway, culminating in vascular permeability alterations that effectively limit the growth of Mtb. 421
Metabolism blockage
Previous studies have reviewed the strategies by which Mtb influences host‐pathogen interactions by regulating metabolism. 422 The metabolic alterations culminating due to stressors present in the microenvironment of granuloma—namely a paucity of nutrients, hypoxia, and acidic pH—hold great significance. The metabolic transition from oxidative phosphorylation to aerobic glycolysis of cells in the presence of sufficient oxygen, 423 a process known as the Warburg effect, 424 is present in tumors 425 and has parallels within TB granulomas. 426 As a consequence, regulation of glycolysis, glucose transport and glucose homeostasis are important strategies to limit granuloma metabolism. Treatment to limit aerobic glycolysis can limit granuloma as well as Mtb replication. Mtb‐infected macrophages in granuloma undergo metabolic changes, including upregulation of hypoxia‐inducible factor‐1α (HIF‐1α) and using glutamine as an important carbon and nitrogen source. 427 , 428 Insufficient cytoplasmic aspartate delivery leads to cell death when TCA circulating carbon is reduced after glutamine inhibition. 429 Foamy macrophages, a granuloma‐specific cell population characterized by its high lipid content, constitute a long‐term reservoir for Mtb in the human host. 430 Lipid bodies serve as both a source of nutrients and a secure niche for the bacterium. 431 Formation of Mtb‐dependent lipid bodies is mediated through the G protein‐coupled receptor GPR109A. Inhibition of the GPR109A leads to a reduced bacterial load and the reduction in alveolar macrophage lipid bodies in vivo, which are associated with granuloma caseation. 431 The significance of glycolysis and lipid metabolism in the advancement of Mtb infection highlights the potential for HDT. In addition, metabolic alterations within granulomas can lead to the accumulation of advanced glycation end products (AGEs). These AGEs are known to affect macrophage death and activation, potentially playing a role in the progression of TB. 432
4.2.3. Regulation of cell death
Cell death can be classified into two main categories based on molecular mechanisms, morphological characteristics, and signal dependency: programmed cell death (PCD) and nonprogrammed cell death (n‐PCD). 433 The interaction between Mtb and the host in relation to cell death is complex, with Mtb initially inhibiting host cell apoptosis to ensure replication niches, and subsequently inducing necroptosis and pyroptosis after successful replication to release and infect new cells. 434
Apoptosis activation
Numerous studies 435 , 436 have demonstrated that Mtb has the ability to regulate proapoptotic proteins to inhibit the intrinsic pathway of host cell apoptosis, and this escape ability is thought to be a critical virulence factor. 437 Currently, much work is being done to regulate antiapoptotic genes and controlling apoptotic factors with the aim of enhancing host cell apoptosis. 438 Peroxisome proliferator‐activated receptor gamma (PPARγ), a member of the nuclear receptor superfamily, is a transcriptional factor that governs inflammation. 439 This protein exhibits elevated expression within activated alveolar macrophages and macrophage‐derived foam cells, each of which plays a significant role in the pathogenesis of TB. 440 , 441 Mtb and its cell wall mannose‐capped lipoprotein mannan activate PPARγ and stimulate MRs to enhance their intracellular survival. 442 To prevent apoptosis, Mtb differentially regulates the Bcl‐2 (B‐cell lymphoma protein 2) family members Bax (proapoptotic) and Mcl‐1 (prosurvival) expression through PPARγ. 443 Therapeutics aimed at Mcl‐1, alongside other Bcl‐2 prosurvival proteins, is currently under development as potential cancer treatments. 444 The Mcl‐1 inhibitors sabutoclax, TW‐37, A‐1210477, and MIM‐1 have been shown to significantly decrease Mtb survival within human macrophages via the activation of apoptosis. 443 These promising results indicate the feasibility of Mcl‐1 and other antiapoptotic proteins as viable targets for HDT.
Necrosis suppression
Conversely, another idea that is being explored is the reversing of cell necrosis. The necrosis of infected macrophages represents a crucial event in the pathogenesis of TB as it results in the release of mycobacteria into the extracellular environment, which is permissive for the growth and spread of the pathogen. Excess TNF has been shown to trigger programmed necrosis. 397 Recent research has elucidated the mechanism by which excess TNF induces mitochondrial ROS (mROS) production in TB. It has been found that excess TNF in macrophages infected with mycobacterium triggers an increase in mROS production through the reverse electron transport process, which occurs through complex I. MET may inhibit complex I and thus prevent TNF‐induced mROS and macrophage necrosis. 396 Moreover, excess TNF‐α following infection has been found to upregulate mROS and induce the formation of the mitochondrial permeability transition pore complex, subsequently triggering necrosis via cyclophilin D (CypD). The inhibitory peptide alisporivir of CypD has been found to synergistically inhibit Mycobacterium zebrafish infection when used in combination with the ceramide inhibitor desipramine. 445 Interestingly, it has been found that ceramide is induced to be produced by TNF‐α. 445 Studies have demonstrated a significant correlation between Mtb‐induced host cell necrosis and ferroptosis, with decreased lung bacterial load observed in mice with acute TB infection following ferroptosis inhibitor, Ferrostatin‐1 treatment. 446 Furthermore, corticosteroids have been found to inhibit necrosis by targeting mitochondrial membrane stability through the inhibition of p38 mitogen‐activated protein kinase (MAPK) in addition to their known actions through glucocorticoid receptors (GRs). 447
Pyroptosis suppression
Pyroptosis is a proinflammatory PCD pathway distinct from other forms of cell death, mediated by pyroptosomes, dependent on N‐terminal fragment of the caspase‐1‐cleaved gasdermin D (GSDMD) and causes plasma membrane rupture. 448 , 449 Two known mechanisms for initiating pyroptosis are through (a) damage to the plasma membrane of host cell by the ESX‐1 system, which activates the NLRP3 (domain‐like receptor protein 3) inflammasome, 450 and (b) interaction between EST12, a protein that induces cell pyroptosis, and active C kinase 1, leading to activation of the macrophage NLRP3. 451 Ultimately, both mechanisms result in the activation of the inflammasome‐caspase‐1‐GSDMD pyroptosis‐IL‐1β immune pathway. Also, upon internalization in the phagosome, Mtb create phagosomal pores that allow for the passage of bacterial factors, ultimately activating the cGAS/STING pathway and resulting in an IFN‐γ response. 452 Research shows that baicalin reduced pyroptosis by inhibiting the PERK/TXNIP/NLRP3 axis. 453 Additionally, Tanshinone IIA inhibited upstream signals of NLRP3 inflammasome activation in Mtb‐infected macrophages. 454 Despite this understanding of the mechanisms behind pyroptosis induction in Mtb, there are currently no known modulators for the pyroptosis suppression for the clearance of Mtb.
4.3. Limiting immunopathology
The immunopathological response seen in TB, characterized by excessive inflammation, can present a significant challenge in managing the disease. 455 Research has confirmed that immune cells utilize distinct strategies to control Mtb invasion, with proinflammatory mechanisms playing a crucial role in slowing, sequestering, and ultimately eliminating the pathogen. The successful host response, however, relies on balancing the timing and expression levels of pro‐ and anti‐inflammatory responses. 456 Consequently, regulation of inflammation to balance protective and pathological elements of the immune response is an integral component of HDT.
Cavitary pathology is associated with host‐protease imbalance driven by Mtb. 457 Unbiased investigations conducted by various research groups, using a diversity of methodological approaches, have consistently identified matrix metalloproteinases (MMPs) as one of the most highly upregulated proteins in TB. 458 , 459 , 460 , 461 , 462 , 463 , 464 MMPs depend on zinc ions to carry out the hydrolysis of protein substrates, and are highly associated with TB cavity in patients. 465 The original ribosome inhibitor, doxycycline, has been found to decrease MMP activity in cellular models and suppress mycobacterial growth in vitro and in guinea pigs, as evidenced by clinical trials. 466 Marimastat, an antitumor drug specifically designed to inhibit MMP, was not ultimately approved for use due to musculoskeletal toxicity, recent studies have demonstrated its efficacy in reducing granuloma formation and Mtb bacterial load when combined with RIF or INH. 467 Other MMP inhibitors that have been reported to improve granuloma production are batimastat, prinomastat, Sb‐3ct, 467 and some specific antibodies. 468 However, the MMP‐7 inhibitor cipemastat increased the frequency of lung cavitation in mice with TB. 469 Additional research is necessary to comprehend the potential of MMP inhibitors as supplementary therapeutic interventions for pulmonary TB.
Host lipid metabolism plays an essential role in nodule–host interactions. 470 Lipid peroxidation, a process that causes oxidative damage to lipids in host cell membranes, has been found to induce tissue necrosis and facilitate the transmission of Mtb. 471 In vivo experiments have shown that lung necrosis in acutely Mtb‐infected mice is associated with reduced glutathione peroxidase‐4 (Gpx4) expression as well as increased lipid peroxidation. 446 N‐acetylcysteine restores the reduced form of glutathione and counteracts TB‐induced oxidative stress. 472 The addition of N‐acetylcysteine to the standard drug regimens for TB has been demonstrated to increase the levels of glutathione peroxidase in TB patients. 473 As a result of this finding, clinical trials assessing the efficacy of N‐acetylcysteine‐assisted treatment for TB patients are currently underway. 474 Host eicosanoids, lipid mediators of inflammation, have been shown to respond to the severity of TB and TB‐diabetes comorbidity, 475 , 476 and have a significant impact on the outcome of Mtb infection through their correlation with two major counter‐regulatory classes of inflammatory cytokines: IL‐1 and type I IFNs. 477 Currently, modulation of lipid metabolic pathways (5‐lipoxygenase, 5‐LOX modulators) has been reported for the treatment of TB infection. Zileuton, a 5‐LOX inhibitor originally developed for asthma, has been found to improve survival, reduce bacterial burden, and alleviate lung injury in Mtb‐infected mice. 477 In oncology studies, cyclooxygenase‐2 (COX) has been shown to be associated with MDSC activation, and COX‐2 inhibitors such as aspirin and ibuprofen have been utilized to test their efficacy in treating mouse models of infection, where they demonstrated an increase in bacterial clearance when combined with PZA. 478 , 479 However, recent studies have reported that COX‐2 inhibitor treatment significantly reduces Type‐1 helper (Th1) differentiation and downregulates IFN‐γ expression, which exacerbates Mtb aerosol infection. 480 As such, more experimental evidence is required to determine the appropriate role of nonsteroidal anti‐inflammatory drugs (NSAIDs) in adjuvant anti‐TB therapy. 481
Corticosteroid therapy probably improves neurological outcomes of, and decreases mortality due to, tuberculous meningitis of moderate severity. 482 Due to their potent anti‐inflammatory properties, these drugs are capable of suppressing the host's immune response, thereby reducing the potential for immunopathology. A recent study has demonstrated a differential expression of GR pathway‐regulated genes in patients with TB. 483 Promisingly, the corticosteroids dexamethasone and prednisolone have shown efficacy in reducing mortality from all forms of TB, including pulmonary TB. 484 A randomized, double‐blind, placebo‐controlled trial has demonstrated that the administration of prednisone is effective in reducing the need for hospitalization and therapeutic procedures, while also accelerating improvements in symptoms, performance, and quality of life. 485
Poly(ADP‐ribose) Polymerase 1 (PARP1), a member of the PARP family and a regulatory factor, is known to induce further activation of itself through the actions of inflammatory mediators. 486 In light of this, PARP1 inhibitors PJ‐34 have recently been proposed as an HDT approach to TB, due to their ability to reduce inflammation and alleviate lung disease. 456 , 487 The phosphodiesterase‐4 (PDE‐4) inhibitor Dovramilast (CC‐11050) has been found to effectively downregulate TNF‐α production in macrophages through the increase of intracellular cAMP, thereby reducing inflammation in rabbits with pulmonary TB during INH treatment. 488 When administered in combination with INH, roflumilast, another PDE‐4 inhibitor, was shown to effectively decrease Mtb bacterial load in mice. 489 Notably, the role of the Janus kinases (JAKs)/signal transducers and activators of transcription (STATs) pathway in the development of TB has received interest. The JAK/STAT pathway regulates multiple TB‐related cytokines, including IL‐27 490 and IFN‐γ, 491 affecting T cell subset differentiation, T cell activation and generation of memory. 492 LINC00870, an upregulated long, noncoding RNAs in Mtb‐infected peripheral blood mononuclear cells (PBMCs), promoted p‐STAT5 and p‐JAK2 protein expression, thus activating JAK/STAT signaling in PBMCs. 493 Blocking STAT7 or IL‐10 signaling led to a diminished bacterial load in the lungs of infected mice, yet without a significant alteration in their inflammatory response. 494
Figure 5 provides a comprehensive summary of the primary cellular processes and prospective targets implicated in HDT. The administration of HDT medications to enhance TB outcomes represents a propitious approach, particularly for MDR‐ and XDR‐TB. Despite this fact, the available clinical data regarding HDT are grossly insufficient. For the nascent HDT, forthcoming research must prioritize drug repositioning alongside validating efficacy. Due to the intricacy of the diverse cellular processes in the host and the inconsistency in response to the identical medication, HDT necessitates comprehensive analysis across multiple models to reveal the implications of novel treatment opportunities.
FIGURE 5.
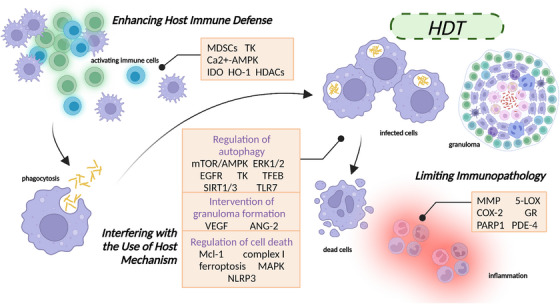
Overview of the host‐directed therapies (HDT) addressed. At the level of the host, targeting important processes such as immune defense, the use of host mechanism by Mtb, and inflammation regulation are invigorated to address and overcome drug resistance. The elements in the figure were drawn using BioRender online tool (https://biorender.com).
5. DISCUSSION AND PERSPECTIVE
Although TB is a malady that can generally be treated and even eradicated, the process often entails a long and financially demanding regimen. This underscores the urgent need for the discovery and development of novel therapeutic agents against TB. It is disappointing that the occurrence of drug‐resistant genes and multidrug‐resistant strains of Mtb is distressingly common. This not only complicates the treatment and management of TB but also poses a significant threat to global health. 495 Consequently, any new therapeutic strategy must prioritize the circumvention of any crosstalk or interaction with first‐line drugs whilst simultaneously minimizing the risk of inducing resistance to existing therapies. Achieving this goal necessitates not only exhaustive characterization of current resistance mechanisms and the establishment of effective strategies to circumvent any potential cross‐resistance, but also the identification and validation of novel biological targets, and action mechanisms and potent lead compounds with novel scaffold to guide subsequent preclinical research and development initiatives.
Thanks to advances in pathogenomics, structure–function analysis, and systems biology, the endeavor of identifying targets in infectious agents has been accelerated and refined. Historically, disruption of crucial cellular pathways in Mtb, such as cell wall synthesis and assembly, protein synthesis, and energy metabolism, has been regarded as a potent strategy for combating TB. However, current research has shifted its focus to newly uncovered targets within Mtb, such as DNA gyrase, ATP synthase, DprE1, and MmpL3, which offer a promising alternative approach to subvert existing drug resistance mechanisms and have yielded remarkable outcomes. Structural determination of enzyme–ligand complexes lays the foundation for identifying key binding sites and designing high‐affinity therapeutics. Nonetheless, these drug candidates remain challenged in their ability to sufficiently engage both active and latent forms of Mtb, thus leaving behind a vicious cycle of recurrence.
As known, Mtb has coevolved with humans and has developed a remarkable degree of adaptation to the human host. Meanwhile, the latent form of the disease evades host immunity and retains the ability to cause disease upon reactivation. HDT seeks to achieve enhanced therapeutic outcomes and prognoses by judiciously manipulating host cellular processes. In healthy individuals, the foremost response upon exposure to Mtb is immunization: a cascade that begins with uptake of the pathogen by innate immune cells, which trigger the activation of adaptive immunity. Approaches aimed at optimizing this sequence represent an efficacious strategy for harnessing HDT. Interventions aimed at augmenting autophagy, dampening granuloma formation, and regulating host cell death to interfere with pathogen replication and retention have emerged as promising avenues for influencing the outcome of TB treatment. Active infection with Mtb elicits a potent immune response that often results in host‐mediated tissue damage. Combining other treatments with modulation of the inflammatory response to decrease tissue damage and cavitation can enhance the effectiveness of TB treatment.
In the context of comorbidities such as HIV infection or diabetes, HDT offers the opportunity to tailor TB management to each patient's unique clinical circumstances. Such patient‐specific tailoring of TB management cannot be readily achieved with antibiotics alone. Furthermore, the administration of combination HDT has consistently yielded improved therapeutic efficacy and abbreviated treatment durations across a diverse array of clinical trials. Additional benefits include the modulation of immunopathology and the facilitation of immune memory development. However, the toxicity potential of HDT must still be vigilantly monitored and actively assessed, and the combination strategies and underlying mechanisms of action must be subjected to rigorous scrutiny across multiple experimental models.
Undoubtedly, delving more deeply into the pathogenesis of TB will unveil additional targets for therapeutic intervention. These newly identified targets demand urgent attention and intensive investment to identify and optimize suitable drug candidates. For instance, target‐based virtual library HTS can be a potent and efficient method for expeditiously identifying leading compound candidates while substantially mitigating the time and cost of drug development. Drug repositioning strategies likewise hold great promise, as numerous US FDA‐approved drugs are currently under active investigation for their potential utility in the management of TB. It is clear that the successful control of TB represents a shared goal of both governments and civic society, and we must remain unwavering in their efforts to attain this objective.
AUTHOR CONTRIBUTIONS
Y. L. and J. Y. conceived the study and wrote the paper. L. Z. revised the figures. Y. L. and W. Q. revised the article. All authors read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to declare.
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
This study was funded by National Natural Science Foundation of China (No. 81973368, 82173861, and 81473253), Open Project of State Key Laboratory of Respiratory Disease (No. SKLRD‐OP‐202113)
Yang J, Zhang L, Qiao W, Luo Y. Mycobacterium tuberculosis: Pathogenesis and therapeutic targets. MedComm. 2023;4:e353. 10.1002/mco2.353
Contributor Information
Wenliang Qiao, Email: qwl_sum@126.com.
Youfu Luo, Email: luo_youfu@scu.edu.cn.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Bagcchi S. WHO's Global Tuberculosis Report 2022. The Lancet Microbe. 2023;4(1):e20. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. On the Road to Ending TB: Highlights from the 30 Highest TB Burden Countries.
- 3. Żukowska L, Zygała‐Pytlos D, Struś K, et al. An overview of tuberculosis outbreaks reported in the years 2011–2020. BMC Infect Dis. 2023;23(1):253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fernandes GFS, Thompson AM, Castagnolo D, Denny WA, Dos Santos JL. Tuberculosis drug discovery: challenges and new horizons. J Med Chem. 2022;65(11):7489‐7531. [DOI] [PubMed] [Google Scholar]
- 5. McQuaid CF, Vassall A, Cohen T, Fiekert K, White RG. The impact of COVID‐19 on TB: a review of the data. Int J Tuberc Lung Dis. 2021;25(6):436‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Comas I, Coscolla M, Luo T, et al. Out‐of‐Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45(10):1176‐1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gagneux S. Host‐pathogen coevolution in human tuberculosis. Philos Trans R Soc Lond B Biol Sci. 2012;367(1590):850‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leung AN. Pulmonary tuberculosis: the essentials. Radiology. 1999;210(2):307‐322. [DOI] [PubMed] [Google Scholar]
- 9. Hilda JN, Das S, Tripathy SP, Hanna LE. Role of neutrophils in tuberculosis: a bird's eye view. Innate Immun. 2020;26(4):240‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Winslow GM, Cooper A, Reiley W, Chatterjee M, Woodland DL. Early T‐cell responses in tuberculosis immunity. Immunol Rev. 2008;225:284‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cooper AM. Cell‐mediated immune responses in tuberculosis. Annu Rev Immunol. 2009;27:393‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schluger NW. The pathogenesis of tuberculosis: the first one hundred (and twenty‐three) years. Am J Respir Cell Mol Biol. 2005;32(4):251‐256. [DOI] [PubMed] [Google Scholar]
- 13. Bermudez LE, Goodman J. Mycobacterium tuberculosis invades and replicates within type II alveolar cells. Infect Immun. 1996;64(4):1400‐1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Danelishvili L, McGarvey J, Li YJ, Bermudez LE. Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell Microbiol. 2003;5(9):649‐660. [DOI] [PubMed] [Google Scholar]
- 15. Torrelles JB, Schlesinger LS. Integrating lung physiology, immunology, and tuberculosis. Trends Microbiol. 2017;25(8):688‐697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luies L, du Preez I. The echo of pulmonary tuberculosis: mechanisms of clinical symptoms and other disease‐induced systemic complications. Clin Microbiol Rev. 2020;33(4):e00036‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chee CBE, Reves R, Zhang Y, Belknap R. Latent tuberculosis infection: opportunities and challenges. Respirology. 2018;23(10):893‐900. [DOI] [PubMed] [Google Scholar]
- 18. Cohen A, Mathiasen VD, Schön T, Wejse C. The global prevalence of latent tuberculosis: a systematic review and meta‐analysis. Eur Respir J. 2019;54(3):1900655. [DOI] [PubMed] [Google Scholar]
- 19. Huszár S, Chibale K, Singh V. The quest for the holy grail: new antitubercular chemical entities, targets and strategies. Drug Discov Today. 2020;25(4):772‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grosset J. Mycobacterium tuberculosis in the extracellular compartment: an underestimated adversary. Antimicrob Agents Chemother. 2003;47(3):833‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smith DT, Abernathy RS, Smith GB, Bondurant S. The apical localization of reinfection pulmonary tuberculosis. I. The stream flow theory. Am Rev Tuberc. 1954;70(4):547‐556. [DOI] [PubMed] [Google Scholar]
- 22. Lerner TR, Queval CJ, Lai RP, et al. Mycobacterium tuberculosis cords within lymphatic endothelial cells to evade host immunity. JCI Insight. 2020;5(10):e136937. 136937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heye T, Stoijkovic M, Kauczor HU, Junghanss T, Hosch W. Extrapulmonary tuberculosis: radiological imaging of an almost forgotten transformation artist. Rofo. 2011;183(11):1019‐1029. [DOI] [PubMed] [Google Scholar]
- 24. Ritacco V, Di Lonardo M, Reniero A, et al. Nosocomial spread of human immunodeficiency virus‐related multidrug‐resistant tuberculosis in Buenos Aires. J Infect Dis. 1997;176(3):637‐642. [DOI] [PubMed] [Google Scholar]
- 25. Wells CD, Cegielski JP, Nelson LJ, et al. HIV infection and multidrug‐resistant tuberculosis: the perfect storm. J Infect Dis. 2007;196(1):S86‐107. Suppl. [DOI] [PubMed] [Google Scholar]
- 26. Alemu A, Bitew ZW, Seid G, et al. Tuberculosis in individuals who recovered from COVID‐19: a systematic review of case reports. PLoS ONE. 2022;17(11):e0277807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fatima S, Kumari A, Das G, Dwivedi VP. Tuberculosis vaccine: a journey from BCG to present. Life Sci. 2020;252:117594. [DOI] [PubMed] [Google Scholar]
- 28. Darrah PA, Zeppa JJ, Maiello P, et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature. 2020;577(7788):95‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Negi K, Bhaskar A, Dwivedi VP. Progressive host‐directed strategies to potentiate BCG vaccination against tuberculosis. Front Immunol. 2022;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gagneux S. Ecology and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol. 2018;16(4):202‐213. [DOI] [PubMed] [Google Scholar]
- 31. Peloquin CA, Davies GR. The treatment of tuberculosis. Clin Pharmacol Ther. 2021;110(6):1455‐1466. [DOI] [PubMed] [Google Scholar]
- 32. Jankute M, Cox JAG, Harrison J, Besra GS. Assembly of the mycobacterial cell wall. Annu Rev Microbiol. 2015;69:405‐423. [DOI] [PubMed] [Google Scholar]
- 33. Vergne I, Gilleron M, Nigou J. Manipulation of the endocytic pathway and phagocyte functions by Mycobacterium tuberculosis lipoarabinomannan. Front Cell Infect Microbiol. 2015;4:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abrahams KA, Besra GS. Mycobacterial cell wall biosynthesis: a multifaceted antibiotic target. Parasitology. 2018;145(2):116‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jankute M, Grover S, Rana AK, Besra GS. Arabinogalactan and lipoarabinomannan biosynthesis: structure, biogenesis and their potential as drug targets. Future Microbiol. 2012;7(1):129‐147. [DOI] [PubMed] [Google Scholar]
- 36. PaweŁczyk J, Kremer L. The molecular genetics of mycolic acid biosynthesis. Microbiol Spectr. 2014;2(4). 2.4.24. [DOI] [PubMed] [Google Scholar]
- 37. Vögeli B, Rosenthal RG, Stoffel GMM, et al. InhA, the enoyl‐thioester reductase from Mycobacterium tuberculosis forms a covalent adduct during catalysis. J Biol Chem. 2018;293(44):17200‐17207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johnsson K, King DS, Schultz PG. Studies on the mechanism of action of isoniazid and ethionamide in the chemotherapy of tuberculosis. J Am Chem Soc. 1995;117(17):5009‐5010. [Google Scholar]
- 39. Wang F, Langley R, Gulten G, et al. Mechanism of thioamide drug action against tuberculosis and leprosy. J Exp Med. 2007;204(1):73‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vilchèze C, Weisbrod TR, Chen B, et al. Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrob Agents Chemother. 2005;49(2):708‐720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. DeBarber AE, Mdluli K, Bosman M, Bekker LG, Barry CE. Ethionamide activation and sensitivity in multidrug‐resistant Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2000;97(17):9677‐9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rawat R, Whitty A, Tonge PJ. The isoniazid‐NAD adduct is a slow, tight‐binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc Natl Acad Sci USA. 2003;100(24):13881‐13886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vale N, Gomes P, Santos HA. Metabolism of the antituberculosis drug ethionamide. Curr Drug Metab. 2013;14(1):151‐158. [PubMed] [Google Scholar]
- 44. Zhang Y, Heym B, Allen B, Young D, Cole S. The catalase‐peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature. 1992;358(6387):591‐593. [DOI] [PubMed] [Google Scholar]
- 45. Vilchèze C, Jacobs WR. Resistance to isoniazid and ethionamide in mycobacterium tuberculosis: genes, mutations, and causalities. Microbiol Spectr. 2014;2(4). MGM2‐0014‐2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Roujeinikova A, Sedelnikova S, de Boer GJ, et al. Inhibitor binding studies on enoyl reductase reveal conformational changes related to substrate recognition. J Biol Chem. 1999;274(43):30811‐30817. [DOI] [PubMed] [Google Scholar]
- 47. Freundlich JS, Wang F, Vilchèze C, et al. Triclosan derivatives: towards potent inhibitors of drug‐sensitive and drug‐resistant Mycobacterium tuberculosis. ChemMedChem. 2009;4(2):241‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Parikh S, Xiao G, Tonge P. Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid. Biochemistry. 2000;39(26):7645‐7650. [DOI] [PubMed] [Google Scholar]
- 49. Wang H, Liu L, Lu Y, et al. Radiolabelling and positron emission tomography of PT70, a time‐dependent inhibitor of InhA, the Mycobacterium tuberculosis enoyl‐ACP reductase. Bioorg Med Chem Lett. 2015;25(21):4782‐4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kini SG, Bhat AR, Bryant B, Williamson JS, Dayan FE. Synthesis, antitubercular activity and docking study of novel cyclic azole substituted diphenyl ether derivatives. Eur J Med Chem. 2009;44(2):492‐500. [DOI] [PubMed] [Google Scholar]
- 51. He X, Alian A, Stroud R, Ortiz de Montellano PR. Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis. J Med Chem. 2006;49(21):6308‐6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guardia A, Gulten G, Fernandez R, et al. N‐Benzyl‐4‐((heteroaryl)methyl)benzamides: a new class of direct NADH‐dependent 2‐trans Enoyl‐Acyl Carrier Protein Reductase (InhA) inhibitors with antitubercular activity. ChemMedChem. 2016;11(7):687‐701. [DOI] [PubMed] [Google Scholar]
- 53. Kuo MR, Morbidoni HR, Alland D, et al. Targeting tuberculosis and malaria through inhibition of Enoyl reductase: compound activity and structural data. J Biol Chem. 2003;278(23):20851‐20859. [DOI] [PubMed] [Google Scholar]
- 54. Manjunatha UH, Rao SPS, Kondreddi RR, et al. Direct inhibitors of InhA are active against Mycobacterium tuberculosis. Sci Transl Med. 2015;7(269):269ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wall MD, Oshin M, Chung GAC, et al. Evaluation of N‐(phenylmethyl)‐4‐[5‐(phenylmethyl)‐4,5,6,7‐tetrahydro‐1H‐imidazo[4,5‐c]pyridin‐4‐yl]benzamide inhibitors of Mycobacterium tuberculosis growth. Bioorg Med Chem Lett. 2007;17(10):2740‐2744. [DOI] [PubMed] [Google Scholar]
- 56. Šink R, Sosič I, Živec M, et al. Design, synthesis, and evaluation of new thiadiazole‐based direct inhibitors of enoyl acyl carrier protein reductase (InhA) for the treatment of tuberculosis. J Med Chem. 2015;58(2):613‐624. [DOI] [PubMed] [Google Scholar]
- 57. Xia Y, Zhou Y, Carter DS, et al. Discovery of a cofactor‐independent inhibitor of Mycobacterium tuberculosis InhA. Life Sci Alliance. 2018;1(3):e201800025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kamsri P, Hanwarinroj C, Phusi N, et al. Discovery of new and potent InhA inhibitors as antituberculosis agents: structure‐based virtual screening validated by biological assays and X‐ray crystallography. J Chem Inf Model. 2020;60(1):226‐234. [DOI] [PubMed] [Google Scholar]
- 59. Hartkoorn RC, Sala C, Neres J, et al. Towards a new tuberculosis drug: pyridomycin—nature's isoniazid. EMBO Mol Med. 2012;4(10):1032‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Maeda K, Kosaka H, Okami Y, Umezawa H. A new antibiotic, pyridomycin. J Antibiot (Tokyo). 1953;6(3):140. [PubMed] [Google Scholar]
- 61. Yagishita K. Studies on the pyridomycin production by Streptomyces albidofuscus. I. On pyridomycin production of a lactose‐utilizing mutant. J Antibiot (Tokyo). 1954;7(5):143‐148. [PubMed] [Google Scholar]
- 62. Shomura T, Amano S, Yoshida J, Kojima M. Dactylosporangium fulvum sp. nov. Int J Syst Evol Microbiol. 1986;36(2):166‐169. [Google Scholar]
- 63. Dodge GJ, Patel A, Jaremko KL, McCammon JA, Smith JL, Burkart MD. Structural and dynamical rationale for fatty acid unsaturation in Escherichia coli. Proc Natl Acad Sci USA. 2019;116(14):6775‐6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hartkoorn RC, Pojer F, Read JA, et al. Pyridomycin bridges the NADH‐ and substrate‐binding pockets of the enoyl reductase InhA. Nat Chem Biol. 2014;10(2):96‐98. [DOI] [PubMed] [Google Scholar]
- 65. DeJesus MA, Gerrick ER, Xu W, et al. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio. 2017;8(1):e02133‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schaeffer ML, Agnihotri G, Volker C, Kallender H, Brennan PJ, Lonsdale JT. Purification and biochemical characterization of the Mycobacterium tuberculosis beta‐ketoacyl‐acyl carrier protein synthases KasA and KasB. J Biol Chem. 2001;276(50):47029‐47037. [DOI] [PubMed] [Google Scholar]
- 67. Kim P, Zhang YM, Shenoy G, et al. Structure‐activity relationships at the 5‐position of thiolactomycin: an intact (5R)‐isoprene unit is required for activity against the condensing enzymes from Mycobacterium tuberculosis and Escherichia coli. J Med Chem. 2006;49(1):159‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Machutta CA, Bommineni GR, Luckner SR, et al. Slow onset inhibition of bacterial β‐ketoacyl‐acyl carrier protein synthases by thiolactomycin. J Biol Chem. 2010;285(9):6161‐6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kremer L, Douglas JD, Baulard AR, et al. Thiolactomycin and related analogues as novel anti‐mycobacterial agents targeting KasA and KasB condensing enzymes in Mycobacterium tuberculosis. J Biol Chem. 2000;275(22):16857‐16864. [DOI] [PubMed] [Google Scholar]
- 70. Ballell L, Bates RH, Young RJ, et al. Fueling open‐source drug discovery: 177 small‐molecule leads against tuberculosis. ChemMedChem. 2013;8(2):313‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rebollo‐Lopez MJ, Lelièvre J, Alvarez‐Gomez D, et al. Release of 50 new, drug‐like compounds and their computational target predictions for open source anti‐tubercular drug discovery. PLoS One. 2015;10(12):e0142293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Abrahams KA, Chung CW, Ghidelli‐Disse S, et al. Identification of KasA as the cellular target of an anti‐tubercular scaffold. Nat Commun. 2016;7:12581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kumar P, Capodagli GC, Awasthi D, et al. Synergistic lethality of a binary inhibitor of Mycobacterium tuberculosis KasA. mBio. 2018;9(6):e02101‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Inoyama D, Awasthi D, Capodagli GC, et al. A preclinical candidate targeting Mycobacterium tuberculosis KasA. Cell Chem Biol. 2020;27(5):560‐570.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Léger M, Gavalda S, Guillet V, et al. The dual function of the Mycobacterium tuberculosis FadD32 required for mycolic acid biosynthesis. Chem Biol. 2009;16(5):510‐519. [DOI] [PubMed] [Google Scholar]
- 76. Kim SK, Dickinson MS, Finer‐Moore J, et al. Structure and dynamics of the essential endogenous mycobacterial polyketide synthase Pks13. Nat Struct Mol Biol. 2023;30(3):296‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gavalda S, Léger M, van der Rest B, et al. The Pks13/FadD32 crosstalk for the biosynthesis of mycolic acids in Mycobacterium tuberculosis. J Biol Chem. 2009;284(29):19255‐19264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fang C, Lee KK, Nietupski R, et al. Discovery of heterocyclic replacements for the coumarin core of anti‐tubercular FadD32 inhibitors. Bioorg Med Chem Lett. 2018;28(22):3529‐3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Aggarwal A, Parai MK, Shetty N, et al. Development of a novel lead that targets M. tuberculosis polyketide synthase 13. Cell. 2017;170(2):249‐259.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhang W, Lun S, Wang SH, et al. Identification of novel coumestan derivatives as polyketide synthase 13 inhibitors against Mycobacterium tuberculosis. J Med Chem. 2018;61(3):791‐803. [DOI] [PubMed] [Google Scholar]
- 81. Zhao W, Wang B, Liu Y, et al. Design, synthesis, and biological evaluation of novel 4H‐chromen‐4‐one derivatives as antituberculosis agents against multidrug‐resistant tuberculosis. Eur J Med Chem. 2020;189:112075. [DOI] [PubMed] [Google Scholar]
- 82. Stover CK, Warrener P, VanDevanter DR, et al. A small‐molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405(6789):962‐966. [DOI] [PubMed] [Google Scholar]
- 83. Ryan NJ, Lo JH. Delamanid: first global approval. Drugs. 2014;74(9):1041‐1045. [DOI] [PubMed] [Google Scholar]
- 84. M M, H H, T T, et al. OPC‐67683, a nitro‐dihydro‐imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS medicine. 2006;3(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wellington S, Hung DT. The expanding diversity of Mycobacterium tuberculosis drug targets. ACS Infect Dis. 2018;4(5):696‐714. [DOI] [PubMed] [Google Scholar]
- 86. R S, U M, Hi B, et al. PA‐824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science (New York, NY). 2008;322(5906). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Grzegorzewicz AE, Korduláková J, Jones V, et al. A common mechanism of inhibition of the Mycobacterium tuberculosis mycolic acid biosynthetic pathway by isoxyl and thiacetazone. J Biol Chem. 2012;287(46):38434‐38441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vaubourgeix J, Bardou F, Boissier F, et al. S‐Adenosyl‐N‐decyl‐aminoethyl, a potent bisubstrate inhibitor of Mycobacterium tuberculosis mycolic acid methyltransferases. J Biol Chem. 2009;284(29):19321‐19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Glickman MS, Cox JS, Jacobs WR. A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol Cell. 2000;5(4):717‐727. [DOI] [PubMed] [Google Scholar]
- 90. Corrales RM, Molle V, Leiba J, Mourey L, de Chastellier C, Kremer L. Phosphorylation of mycobacterial PcaA inhibits mycolic acid cyclopropanation: consequences for intracellular survival and for phagosome maturation block. J Biol Chem. 2012;287(31):26187‐26199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Huang CC, Smith CV, Glickman MS, Jacobs WR, Sacchettini JC. Crystal structures of mycolic acid cyclopropane synthases from Mycobacterium tuberculosis. J Biol Chem. 2002;277(13):11559‐11569. [DOI] [PubMed] [Google Scholar]
- 92. Verma J, Subbarao N. Designing novel inhibitors against cyclopropane mycolic acid synthase 3 (PcaA): targeting dormant state of Mycobacterium tuberculosis. J Biomol Struct Dyn. 2021;39(17):6339‐6354. [DOI] [PubMed] [Google Scholar]
- 93. Yuan Y, Lee RE, Besra GS, Belisle JT, Barry CE. Identification of a gene involved in the biosynthesis of cyclopropanated mycolic acids in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 1995;92(14):6630‐6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Glickman MS, Cahill SM, Jacobs WR. The Mycobacterium tuberculosis cmaA2 gene encodes a mycolic acid trans‐cyclopropane synthetase. J Biol Chem. 2001;276(3):2228‐2233. [DOI] [PubMed] [Google Scholar]
- 95. Choudhury C, Priyakumar UD, Sastry GN. Dynamics based pharmacophore models for screening potential inhibitors of mycobacterial cyclopropane synthase. J Chem Inf Model. 2015;55(4):848‐860. [DOI] [PubMed] [Google Scholar]
- 96. Taira J, Ito T, Nakatani H, et al. In silico structure‐based drug screening of novel antimycobacterial pharmacophores by DOCK‐GOLD tandem screening. Int J Mycobacteriol. 2017;6(2):142‐148. [DOI] [PubMed] [Google Scholar]
- 97. Ishizaki Y, Hayashi C, Inoue K, et al. Inhibition of the first step in synthesis of the mycobacterial cell wall core, catalyzed by the GlcNAc‐1‐phosphate transferase WecA, by the novel caprazamycin derivative CPZEN‐45. J Biol Chem. 2013;288(42):30309‐30319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Young EF, Durham PG, Perkowski EF, Malik S, Hickey AJ, Braunstein M. Efficacy of inhaled CPZEN‐45 in treating tuberculosis in the guinea pig. Tuberculosis. 2022;135:102207. [DOI] [PubMed] [Google Scholar]
- 99. Salomon JJ, Galeron P, Schulte N, et al. Biopharmaceutical in vitro characterization of CPZEN‐45, a drug candidate for inhalation therapy of tuberculosis. Ther Deliv. 2013;4(8):915‐923. [DOI] [PubMed] [Google Scholar]
- 100. Huszár S, Singh V, Polčicová A, et al. N‐acetylglucosamine‐1‐phosphate transferase, WecA, as a validated drug target in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2017;61(11):e01310‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Mitachi K, Siricilla S, Yang D, et al. Fluorescence‐based assay for polyprenyl phosphate‐GlcNAc‐1‐phosphate transferase (WecA) and identification of novel antimycobacterial WecA inhibitors. Anal Biochem. 2016;512:78‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhang L, Zhao Y, Gao Y, et al. Structures of cell wall arabinosyltransferases with the anti‐tuberculosis drug ethambutol. Science. 2020;368(6496):1211‐1219. [DOI] [PubMed] [Google Scholar]
- 103. Escuyer VE, Lety MA, Torrelles JB, et al. The role of the embA and embB gene products in the biosynthesis of the terminal hexaarabinofuranosyl motif of Mycobacterium smegmatis arabinogalactan. J Biol Chem. 2001;276(52):48854‐48862. [DOI] [PubMed] [Google Scholar]
- 104. Berg S, Kaur D, Jackson M, Brennan PJ. The glycosyltransferases of Mycobacterium tuberculosis—roles in the synthesis of arabinogalactan, lipoarabinomannan, and other glycoconjugates. Glycobiology. 2007;17(6):35‐56R. [DOI] [PubMed] [Google Scholar]
- 105. Goude R, Amin AG, Chatterjee D, Parish T. The arabinosyltransferase EmbC is inhibited by ethambutol in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2009;53(10):4138‐4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Singh A, Somvanshi P, Grover A. Drug repurposing against arabinosyl transferase (EmbC) of Mycobacterium tuberculosis: essential dynamics and free energy minima based binding mechanics analysis. Gene. 2019;693:114‐126. [DOI] [PubMed] [Google Scholar]
- 107. Mi J, Gong W, Wu X. Advances in key drug target identification and new drug development for tuberculosis. BioMed Res Int. 2022;2022:5099312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wolucka BA. Biosynthesis of D‐arabinose in mycobacteria—a novel bacterial pathway with implications for antimycobacterial therapy. FEBS J. 2008;275(11):2691‐2711. [DOI] [PubMed] [Google Scholar]
- 109. Brecik M, Centárová I, Mukherjee R, et al. DprE1 is a vulnerable tuberculosis drug target due to its cell wall localization. ACS Chem Biol. 2015;10(7):1631‐1636. [DOI] [PubMed] [Google Scholar]
- 110. Mariandyshev AO, Khokhlov AL, Smerdin SV, et al. The main results of clinical trials of the efficacy, safety and pharmacokinetics of the perspective anti‐tuberculosis drug makozinone (PBTZ169). Ter Arkh. 2020;92(3):61‐72. [DOI] [PubMed] [Google Scholar]
- 111. Hariguchi N, Chen X, Hayashi Y, et al. OPC‐167832, a novel carbostyril derivative with potent antituberculosis activity as a DprE1 inhibitor. Antimicrob Agents Chemother. 2020;64(6):e02020‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Finger V, Kufa M, Soukup O, Castagnolo D, Roh J, Korabecny J. Pyrimidine derivatives with antitubercular activity. Eur J Med Chem. 2023;246:114946. [DOI] [PubMed] [Google Scholar]
- 113. Eckhardt E, Li Y, Mamerow S, et al. Pharmacokinetics and efficacy of the benzothiazinone BTZ‐043 against tuberculous mycobacteria inside granulomas in the guinea pig model. Antimicrob Agents Chemother. 2023;67(4):e0143822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Trefzer C, Rengifo‐Gonzalez M, Hinner MJ, et al. Benzothiazinones: prodrugs that covalently modify the decaprenylphosphoryl‐β‐D‐ribose 2’‐epimerase DprE1 of Mycobacterium tuberculosis. J Am Chem Soc. 2010;132(39):13663‐13665. [DOI] [PubMed] [Google Scholar]
- 115. Tiwari R, Moraski GC, Krchňák V, et al. Thiolates chemically induce redox activation of BTZ043 and related potent nitroaromatic anti‐tuberculosis agents. J Am Chem Soc. 2013;135(9):3539‐3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wang A, Ma C, Chai Y, et al. Identification of benzothiazinones containing 2‐benzyl‐2,7‐diazaspiro[3.5]nonane moieties as new antitubercular agents. Eur J Med Chem. 2020;200:112409. [DOI] [PubMed] [Google Scholar]
- 117. Li P, Wang B, Zhang X, et al. Identification of novel benzothiopyranone compounds against Mycobacterium tuberculosis through scaffold morphing from benzothiazinones. Eur J Med Chem. 2018;160:157‐170. [DOI] [PubMed] [Google Scholar]
- 118. Borthwick JA, Alemparte C, Wall I, et al. Mycobacterium tuberculosis decaprenylphosphoryl‐β‐d‐ribose oxidase inhibitors: expeditious reconstruction of suboptimal hits into a series with potent in vivo activity. J Med Chem. 2020;63(5):2557‐2576. [DOI] [PubMed] [Google Scholar]
- 119. Balabon O, Pitta E, Rogacki MK, et al. Optimization of hydantoins as potent antimycobacterial decaprenylphosphoryl‐β‐d‐ribose oxidase (DprE1) inhibitors. J Med Chem. 2020;63(10):5367‐5386. [DOI] [PubMed] [Google Scholar]
- 120. Wang P, Batt SM, Wang B, et al. Discovery of novel thiophene‐arylamide derivatives as DprE1 inhibitors with potent antimycobacterial activities. J Med Chem. 2021;64(9):6241‐6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Naik M, Humnabadkar V, Tantry SJ, et al. 4‐aminoquinolone piperidine amides: noncovalent inhibitors of DprE1 with long residence time and potent antimycobacterial activity. J Med Chem. 2014;57(12):5419‐5434. [DOI] [PubMed] [Google Scholar]
- 122. Neres J, Hartkoorn RC, Chiarelli LR, et al. 2‐Carboxyquinoxalines kill mycobacterium tuberculosis through noncovalent inhibition of DprE1. ACS Chem Biol. 2015;10(3):705‐714. [DOI] [PubMed] [Google Scholar]
- 123. Oh S, Park Y, Engelhart CA, et al. Discovery and structure‐activity‐relationship study of N‐alkyl‐5‐hydroxypyrimidinone carboxamides as novel antitubercular agents targeting decaprenylphosphoryl‐β‐d‐ribose 2’‐oxidase. J Med Chem. 2018;61(22):9952‐9965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Ezquerra‐Aznárez JM, Degiacomi G, Gašparovič H, et al. The veterinary anti‐parasitic selamectin is a novel inhibitor of the Mycobacterium tuberculosis DprE1 enzyme. Int J Mol Sci. 2022;23(2):771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Amado PSM, Woodley C, Cristiano MLS, O'Neill PM. Recent advances of DprE1 inhibitors against mycobacterium tuberculosis: computational analysis of physicochemical and ADMET properties. ACS Omega. 2022;7(45):40659‐40681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Azam MA, Jayaram U. Inhibitors of alanine racemase enzyme: a review. J Enzyme Inhib Med Chem. 2016;31(4):517‐526. [DOI] [PubMed] [Google Scholar]
- 127. Qin Y, Xu L, Teng Y, Wang Y, Ma P. Discovery of novel antibacterial agents: recent developments in D‐alanyl‐D‐alanine ligase inhibitors. Chem Biol Drug Des. 2021;98(3):305‐322. [DOI] [PubMed] [Google Scholar]
- 128. Chen Y, Xu Y, Yang S, Li S, Ding W, Zhang W. Deficiency of D‐alanyl‐D‐alanine ligase A attenuated cell division and greatly altered the proteome of Mycobacterium smegmatis. Microbiologyopen. 2019;8(9):e00819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zítková L, Tousek J. Pharmacokinetics of cycloserine and terizidone. A comparative study. Chemotherapy. 1974;20(1):18‐28. [DOI] [PubMed] [Google Scholar]
- 130. Walsh CT. Enzymes in the D‐alanine branch of bacterial cell wall peptidoglycan assembly. J Biol Chem. 1989;264(5):2393‐2396. [PubMed] [Google Scholar]
- 131. Lambert MP, Neuhaus FC. Mechanism of D‐cycloserine action: alanine racemase from Escherichia coli W. J Bacteriol. 1972;110(3):978‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Meng J, Gao P, Wang X, Guan Y, Liu Y, Xiao C. Digging deeper to save the old anti‐tuberculosis target: d‐Alanine‐D‐Alanine ligase with a novel inhibitor, IMB‐0283. Front Microbiol. 2019;10:3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393(6685):537‐544. [DOI] [PubMed] [Google Scholar]
- 134. Hugonnet JE, Tremblay LW, Boshoff HI, Barry CE, Blanchard JS. Meropenem‐clavulanate is effective against extensively drug‐resistant Mycobacterium tuberculosis. Science. 2009;323(5918):1215‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Payen MC, De Wit S, Martin C, et al. Clinical use of the meropenem‐clavulanate combination for extensively drug‐resistant tuberculosis. Int J Tuberc Lung Dis. 2012;16(4):558‐560. [DOI] [PubMed] [Google Scholar]
- 136. Gupta R, Lavollay M, Mainardi JL, Arthur M, Bishai WR, Lamichhane G. The Mycobacterium tuberculosis protein LdtMt2 is a nonclassical transpeptidase required for virulence and resistance to amoxicillin. Nat Med. 2010;16(4):466‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Cordillot M, Dubée V, Triboulet S, et al. In vitro cross‐linking of Mycobacterium tuberculosis peptidoglycan by L,D‐transpeptidases and inactivation of these enzymes by carbapenems. Antimicrob Agents Chemother. 2013;57(12):5940‐5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Li WJ, Li DF, Hu YL, Zhang XE, Bi LJ, Wang DC. Crystal structure of L,D‐transpeptidase LdtMt2 in complex with meropenem reveals the mechanism of carbapenem against Mycobacterium tuberculosis. Cell Res. 2013;23(5):728‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Silva JRA, Govender T, Maguire GEM, et al. Simulating the inhibition reaction of Mycobacterium tuberculosisL,D‐transpeptidase 2 by carbapenems. Chem Commun. 2015;51(63):12560‐12562. [DOI] [PubMed] [Google Scholar]
- 140. Kaushik A, Makkar N, Pandey P, Parrish N, Singh U, Lamichhane G. Carbapenems and rifampin exhibit synergy against Mycobacterium tuberculosis and Mycobacterium abscessus. Antimicrob Agents Chemother. 2015;59(10):6561‐6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kaushik A, Ammerman NC, Tasneen R, et al. In vitro and in vivo activity of biapenem against drug‐susceptible and rifampicin‐resistant Mycobacterium tuberculosis. J Antimicrob Chemother. 2017;72(8):2320‐2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Zandi TA, Townsend CA. Competing off‐loading mechanisms of meropenem from an l,d‐transpeptidase reduce antibiotic effectiveness. Proc Natl Acad Sci USA. 2021;118(27):e2008610118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. de Munnik M, Lohans CT, Lang PA, et al. Targeting the Mycobacterium tuberculosis transpeptidase LdtMt2 with cysteine‐reactive inhibitors including ebselen. Chem Commun (Camb). 2019;55(69):10214‐10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Ikeda M, Wachi M, Jung HK, Ishino F, Matsuhashi M. The Escherichia coli mraY gene encoding UDP‐N‐acetylmuramoyl‐pentapeptide: undecaprenyl‐phosphate phospho‐N‐acetylmuramoyl‐pentapeptide transferase. J Bacteriol. 1991;173(3):1021‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Bugg TDH, Lloyd AJ, Roper DI. Phospho‐MurNAc‐pentapeptide translocase (MraY) as a target for antibacterial agents and antibacterial proteins. Infect Disord Drug Targets. 2006;6(2):85‐106. [DOI] [PubMed] [Google Scholar]
- 146. Kimura KI. Liposidomycin, the first reported nucleoside antibiotic inhibitor of peptidoglycan biosynthesis translocase I: the discovery of liposidomycin and related compounds with a perspective on their application to new antibiotics. J Antibiot (Tokyo). 2019;72(12):877‐889. [DOI] [PubMed] [Google Scholar]
- 147. Tran W, Kusay AS, Hawkins PME, et al. Synthetic sansanmycin analogues as potent Mycobacterium tuberculosis translocase I inhibitors. J Med Chem. 2021;64(23):17326‐17345. [DOI] [PubMed] [Google Scholar]
- 148. Reddy VM, Einck L, Nacy CA. In vitro antimycobacterial activities of capuramycin analogues. Antimicrob Agents Chemother. 2008;52(2):719‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Dubuisson T, Bogatcheva E, Krishnan MY, et al. In vitro antimicrobial activities of capuramycin analogues against non‐tuberculous mycobacteria. J Antimicrob Chemother. 2010;65(12):2590‐2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Nikonenko B, Reddy VM, Bogatcheva E, Protopopova M, Einck L, Nacy CA. Therapeutic efficacy of SQ641‐NE against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2014;58(1):587‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Kurosu M, Mahapatra S, Narayanasamy P, Crick DC. Chemoenzymatic synthesis of Park's nucleotide: toward the development of high‐throughput screening for MraY inhibitors. Tetrahedron Lett. 2007;48(5):799‐803. [Google Scholar]
- 152. Siricilla S, Mitachi K, Skorupinska‐Tudek K, Swiezewska E, Kurosu M. Biosynthesis of a water‐soluble lipid I analogue and a convenient assay for translocase I. Anal Biochem. 2014;461:36‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Varela C, Rittmann D, Singh A, et al. MmpL genes are associated with mycolic acid metabolism in mycobacteria and corynebacteria. Chem Biol. 2012;19(4):498‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Li W, Obregón‐Henao A, Wallach JB, et al. Therapeutic potential of the Mycobacterium tuberculosis mycolic acid transporter, MmpL3. Antimicrob Agents Chemother. 2016;60(9):5198‐5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Li W, Stevens CM, Pandya AN, et al. Direct inhibition of MmpL3 by novel antitubercular compounds. ACS Infect Dis. 2019;5(6):1001‐1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pipeline | Working Group for New TB Drugs.
- 157. Grzegorzewicz AE, Pham H, Gundi VAKB, et al. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat Chem Biol. 2012;8(4):334‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Remuiñán MJ, Pérez‐Herrán E, Rullás J, et al. Tetrahydropyrazolo[1,5‐a]pyrimidine‐3‐carboxamide and N‐benzyl‐6′,7′‐dihydrospiro[piperidine‐4,4′‐thieno[3,2‐c]pyran] analogues with bactericidal efficacy against Mycobacterium tuberculosis targeting MmpL3. PLOS ONE. 2013;8(4):e60933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Deidda D, Lampis G, Fioravanti R, et al. Bactericidal activities of the pyrrole derivative BM212 against multidrug‐resistant and intramacrophagic Mycobacterium tuberculosis strains. Antimicrob Agents Chemother. 1998;42(11):3035‐3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Rao SPS, Lakshminarayana SB, Kondreddi RR, et al. Indolcarboxamide is a preclinical candidate for treating multidrug‐resistant tuberculosis. Sci Transl Med. 2013;5(214):214ra168. [DOI] [PubMed] [Google Scholar]
- 161. Suresh A, Srinivasarao S, Khetmalis YM, Nizalapur S, Sankaranarayanan M, Sekhar KVGC. Inhibitors of pantothenate synthetase of Mycobacterium tuberculosis—a medicinal chemist perspective. RSC Adv. 2020;10(61):37098‐37115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Björkelid C, Bergfors T, Raichurkar AKV, et al. Structural and biochemical characterization of compounds inhibiting Mycobacterium tuberculosis pantothenate kinase. J Biol Chem. 2013;288(25):18260‐18270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Devi PB, Jogula S, Reddy AP, et al. Design of Novel Mycobacterium tuberculosis pantothenate synthetase inhibitors: virtual screening, synthesis and in vitro biological activities. Mol Inform. 2015;34(2‐3):147‐159. [DOI] [PubMed] [Google Scholar]
- 164. Shi W, Chen J, Feng J, et al. Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerg Microbes Infect. 2014;3(8):e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Scorpio A, Zhang Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat Med. 1996;2(6):662‐667. [DOI] [PubMed] [Google Scholar]
- 166. Malone L, Schurr A, Lindh H, McKenzie D, Kiser JS, Williams JH. The effect of pyrazinamide (aldinamide) on experimental tuberculosis in mice. Am Rev Tuberc. 1952;65(5):511‐518. [PubMed] [Google Scholar]
- 167. Zhang Y, Permar S, Sun Z. Conditions that may affect the results of susceptibility testing of Mycobacterium tuberculosis to pyrazinamide. J Med Microbiol. 2002;51(1):42‐49. [DOI] [PubMed] [Google Scholar]
- 168. Tarshis MS. Lack of significant in vitro sensitivity of Mycobacterium tuberculosis to pyrazinamide on three different solid media. Am Rev Tuberc. 1953;67(3):391‐395. [DOI] [PubMed] [Google Scholar]
- 169. Dillon NA, Peterson ND, Feaga HA, Keiler KC, Baughn AD. Anti‐tubercular activity of pyrazinamide is independent of trans‐translation and RpsA. Sci Rep. 2017;7(1):6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Zhang Y, Shi W, Zhang W, Mitchison D. Mechanisms of pyrazinamide action and resistance. Microbiol Spectr. 2014;2(4). MGM2‐0023‐2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Gopal P, Nartey W, Ragunathan P, et al. Pyrazinoic acid inhibits mycobacterial coenzyme A biosynthesis by binding to aspartate decarboxylase PanD. ACS Infect Dis. 2017;3(11):807‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Sun Q, Li X, Perez LM, Shi W, Zhang Y, Sacchettini JC. The molecular basis of pyrazinamide activity on Mycobacterium tuberculosis PanD. Nat Commun. 2020;11(1):339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Zheng R, Blanchard JS. Steady‐state and pre‐steady‐state kinetic analysis of Mycobacterium tuberculosis pantothenate synthetase. Biochemistry. 2001;40(43):12904‐12912. [DOI] [PubMed] [Google Scholar]
- 174. Merkel WK, Nichols BP. Characterization and sequence of the Escherichia coli panBCD gene cluster. FEMS Microbiol Lett. 1996;143(2‐3):247‐252. [DOI] [PubMed] [Google Scholar]
- 175. Ciulli A, Scott DE, Ando M, et al. Inhibition of Mycobacterium tuberculosis pantothenate synthetase by analogues of the reaction intermediate. Chembiochem. 2008;9(16):2606‐2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Xu Z, Yin W, Martinelli LK, et al. Reaction intermediate analogues as bisubstrate inhibitors of pantothenate synthetase. Bioorg Med Chem. 2014;22(5):1726‐1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Hung AW, Silvestre HL, Wen S, Ciulli A, Blundell TL, Abell C. Application of fragment growing and fragment linking to the discovery of inhibitors of Mycobacterium tuberculosis pantothenate synthetase. Angew Chem Int Ed Engl. 2009;48(45):8452‐8456. [DOI] [PubMed] [Google Scholar]
- 178. White EL, Southworth K, Ross L, et al. A novel inhibitor of Mycobacterium tuberculosis pantothenate synthetase. J Biomol Screen. 2007;12(1):100‐105. [DOI] [PubMed] [Google Scholar]
- 179. Yang Y, Gao P, Liu Y, et al. A discovery of novel Mycobacterium tuberculosis pantothenate synthetase inhibitors based on the molecular mechanism of actinomycin D inhibition. Bioorg Med Chem Lett. 2011;21(13):3943‐3946. [DOI] [PubMed] [Google Scholar]
- 180. Samala G, Devi PB, Nallangi R, Yogeeswari P, Sriram D. Development of 3‐phenyl‐4,5,6,7‐tetrahydro‐1H‐pyrazolo[4,3‐c]pyridine derivatives as novel Mycobacterium tuberculosis pantothenate synthetase inhibitors. Eur J Med Chem. 2013;69:356‐364. [DOI] [PubMed] [Google Scholar]
- 181. Velaparthi S, Brunsteiner M, Uddin R, Wan B, Franzblau SG, Petukhov PA. 5‐tert‐butyl‐N‐pyrazol‐4‐yl‐4,5,6,7‐tetrahydrobenzo[d]isoxazole‐3‐carboxamide derivatives as novel potent inhibitors of Mycobacterium tuberculosis pantothenate synthetase: initiating a quest for new antitubercular drugs. J Med Chem. 2008;51(7):1999‐2002. [DOI] [PubMed] [Google Scholar]
- 182. Samala G, Nallangi R, Devi PB, et al. Identification and development of 2‐methylimidazo[1,2‐a]pyridine‐3‐carboxamides as Mycobacterium tuberculosis pantothenate synthetase inhibitors. Bioorg Med Chem. 2014;22(15):4223‐4232. [DOI] [PubMed] [Google Scholar]
- 183. Samala G, Devi PB, Nallangi R, et al. Development of novel tetrahydrothieno[2,3‐c]pyridine‐3‐carboxamide based Mycobacterium tuberculosis pantothenate synthetase inhibitors: molecular hybridization from known antimycobacterial leads. Bioorg Med Chem. 2014;22(6):1938‐1947. [DOI] [PubMed] [Google Scholar]
- 184. Hassan NW, Saudi MN, Abdel‐Ghany YS, et al. Novel pyrazine based anti‐tubercular agents: design, synthesis, biological evaluation and in silico studies. Bioorg Chem. 2020;96:103610. [DOI] [PubMed] [Google Scholar]
- 185. Green R, Noller HF. Ribosomes and translation. Annu Rev Biochem. 1997;66:679‐716. [DOI] [PubMed] [Google Scholar]
- 186. Selmer M, Dunham CM. Structure of the 70S ribosome complexed with mRNA and tRNA. Science (New York, NY). 2006;313(5795):1935‐1942. [DOI] [PubMed] [Google Scholar]
- 187. Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science (New York, NY). 2000;289(5481):905‐920. [DOI] [PubMed] [Google Scholar]
- 188. Wimberly BT, Brodersen DE. Structure of the 30S ribosomal subunit. Nature. 2000;407(6802):327‐339. [DOI] [PubMed] [Google Scholar]
- 189. Li Y, Sharma MR, Koripella RK, et al. Zinc depletion induces ribosome hibernation in mycobacteria. Proc Natl Acad Sci USA. 2018;115(32):8191‐8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Li Y, Sharma MR, Koripella RK, Banavali NK, Agrawal RK, Ojha AK. Ribosome hibernation: a new molecular framework for targeting nonreplicating persisters of mycobacteria. Microbiology. 2021;167(2):001035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Global Alliance for TB Drug Development. A Phase 1, Partially‐Blinded, Placebo‐Controlled, Randomized, Single Ascending Dose (SAD) with a Food Effect Cohort Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of TBI‐223 in Healthy Adult Participants. clinicaltrials.gov; 2021.
- 192. Wallis RS, Jakubiec WM, Kumar V, et al. Pharmacokinetics and whole‐blood bactericidal activity against Mycobacterium tuberculosis of single doses of PNU‐100480 in healthy volunteers. J Infect Dis. 2010;202(5):745‐751. [DOI] [PubMed] [Google Scholar]
- 193. Jeong JW, Jung SJ, Lee HH, et al. In vitro and in vivo activities of LCB01‐0371, a new oxazolidinone. Antimicrob Agents Chemother. 2010;54(12):5359‐5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Shetye GS, Franzblau SG, Cho S. New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Transl Res. 2020;220:68‐97. [DOI] [PubMed] [Google Scholar]
- 195. Li X, Hernandez V, Rock FL, et al. Discovery of a potent and specific M. tuberculosis Leucyl‐tRNA synthetase inhibitor: (S)‐3‐(Aminomethyl)‐4‐chloro‐7‐(2‐hydroxyethoxy)benzo[c][1,2]oxaborol‐1(3H)‐ol (GSK656). J Med Chem. 2017;60(19):8011‐8026. [DOI] [PubMed] [Google Scholar]
- 196. Gudzera OI, Golub AG, Bdzhola VG, et al. Discovery of potent anti‐tuberculosis agents targeting leucyl‐tRNA synthetase. Bioorg Med Chem. 2016;24(5):1023‐1031. [DOI] [PubMed] [Google Scholar]
- 197. Gudzera OI, Golub AG, Bdzhola VG, et al. Identification of Mycobacterium tuberculosis leucyl‐tRNA synthetase (LeuRS) inhibitors among the derivatives of 5‐phenylamino‐2H‐[1,2,4]triazin‐3‐one. J Enzyme Inhib Med Chem. 2016;31(sup2):201‐207. [DOI] [PubMed] [Google Scholar]
- 198. Akopian T, Kandror O, Raju RM, UnniKrishnan M, Rubin EJ, Goldberg AL. The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J. 2012;31(6):1529‐1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Xu X, Zhang L, Yang T, Qiu Z, Bai L, Luo Y. Targeting caseinolytic protease P and its AAA1 chaperone for tuberculosis treatment. Drug Discov Today. 2023;28(3):103508. [DOI] [PubMed] [Google Scholar]
- 200. Lehmann J, Cheng TY, Aggarwal A, et al. An antibacterial β‐lactone kills Mycobacterium tuberculosis by infiltrating mycolic acid biosynthesis. Angew Chem Int Ed Engl. 2018;57(1):348‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Moreira W, Santhanakrishnan S, Dymock BW, Dick T. Bortezomib warhead‐switch confers dual activity against mycobacterial caseinolytic protease and proteasome and selectivity against human proteasome. Front Microbiol. 2017;8:746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Akopian T, Kandror O, Tsu C, et al. Cleavage specificity of Mycobacterium tuberculosis ClpP1P2 protease and identification of novel peptide substrates and boronate inhibitors with anti‐bacterial activity. J Biol Chem. 2015;290(17):11008‐11020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Liu P, Yang Y, Ju Y, et al. Design, synthesis and biological evaluation of novel pyrrole derivatives as potential ClpP1P2 inhibitor against Mycobacterium tuberculosis. Bioorg Chem. 2018;80:422‐432. [DOI] [PubMed] [Google Scholar]
- 204. Gavrish E, Sit CS, Cao S, et al. Lassomycin, a ribosomally synthesized cyclic peptide, kills mycobacterium tuberculosis by targeting the ATP‐dependent protease ClpC1P1P2. Chem Biol. 2014;21(4):509‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Gao W, Kim JY, Anderson JR, et al. The cyclic peptide ecumicin targeting ClpC1 is active against Mycobacterium tuberculosis in vivo. Antimicrob Agents Chemother. 2015;59(2):880‐889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Taylor G, Frommherz Y, Katikaridis P, et al. Antibacterial peptide CyclomarinA creates toxicity by deregulating the Mycobacterium tuberculosis ClpC1‐ClpP1P2 protease. J Biol Chem. 2022;298(8):102202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Burns KE, Darwin KH. Pupylation: a signal for proteasomal degradation in Mycobacterium tuberculosis. Subcell Biochem. 2010;54:149‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Hu G, Lin G, Wang M, et al. Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol Microbiol. 2006;59(5):1417‐1428. [DOI] [PubMed] [Google Scholar]
- 209. Zhang H, Hsu HC, Kahne SC, et al. Macrocyclic peptides that selectively inhibit the Mycobacterium tuberculosis proteasome. J Med Chem. 2021;64(9):6262‐6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Zhan W, Hsu HC, Morgan T, et al. Selective phenylimidazole‐based inhibitors of the Mycobacterium tuberculosis proteasome. J Med Chem. 2019;62(20):9246‐9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Burns KE, Cerda‐Maira FA, Wang T, Li H, Bishai WR, Darwin KH. “Depupylation” of prokaryotic ubiquitin‐like protein from mycobacterial proteasome substrates. Mol Cell. 2010;39(5):821‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Shang L, Li G, Lin Q, et al. Crystal structure of the cytokinin‐producing enzyme “lonely guy” (LOG) from Mycobacterium tuberculosis. Biochemical and biophysical research communications. 2022;598:113‐118. [DOI] [PubMed] [Google Scholar]
- 213. Li C, Liu S, Dong B, et al. Discovery and mechanistic study of Mycobacterium tuberculosis PafA inhibitors. J Med Chem. 2022;65(16):11058‐11065. [DOI] [PubMed] [Google Scholar]
- 214. Almeleebia TM, Shahrani MA, Alshahrani MY, et al. Identification of new Mycobacterium tuberculosis proteasome inhibitors using a knowledge‐based computational screening approach. Molecules. 2021;26(8):2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Tyagi R, Srivastava M, Jain P, et al. Development of potential proteasome inhibitors against Mycobacterium tuberculosis. J Biomol Struct Dyn. 2022;40(5):2189‐2203. [DOI] [PubMed] [Google Scholar]
- 216. Amalia F, Syamsunarno MRAA, Triatin RD, Fatimah SN, Chaidir L, Achmad TH. The role of amino acids in tuberculosis infection: a literature review. Metabolites. 2022;12(10):933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Liu P, Yang Y, Tang Y, et al. Design and synthesis of novel pyrimidine derivatives as potent antitubercular agents. Eur J Med Chem. 2019;163:169‐182. [DOI] [PubMed] [Google Scholar]
- 218. Li C, Tian X, Huang Z, et al. Structure‐activity relationship of novel pyrimidine derivatives with potent inhibitory activities against Mycobacterium tuberculosis. J Med Chem. 2023;66(4):2699‐2716. [DOI] [PubMed] [Google Scholar]
- 219. Bhattacharyya N, Nkumama IN, Newland‐Smith Z, et al. An aspartate‐specific solute‐binding protein regulates protein kinase G activity to control glutamate metabolism in mycobacteria. mBio. 2018;9(4):e00931‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Walburger A, Koul A, Ferrari G, et al. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science. 2004;304(5678):1800‐1804. [DOI] [PubMed] [Google Scholar]
- 221. Gil M, Graña M, Schopfer FJ, et al. Inhibition of Mycobacterium tuberculosis PknG by non‐catalytic rubredoxin domain specific modification: reaction of an electrophilic nitro‐fatty acid with the Fe‐S center. Free Radic Biol Med. 2013;65:150‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Chen D, Ma S, He L, Yuan P, She Z, Lu Y. Sclerotiorin inhibits protein kinase G from Mycobacterium tuberculosis and impairs mycobacterial growth in macrophages. Tuberculosis (Edinb). 2017;103:37‐43. [DOI] [PubMed] [Google Scholar]
- 223. Santhi N, Aishwarya S. Insights from the molecular docking of withanolide derivatives to the target protein PknG from Mycobacterium tuberculosis. Bioinformation. 2011;7(1):1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Kidwai S, Bouzeyen R, Chakraborti S, et al. NU‐6027 inhibits growth of Mycobacterium tuberculosis by targeting protein kinase D and protein kinase G. Antimicrob Agents Chemother. 2019;63(9):e00996‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Ge P, Lei Z, Yu Y, et al. tuberculosis PknG manipulates host autophagy flux to promote pathogen intracellular survival. Autophagy. 2022;18(3):576‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Nunes JES, Duque MA, de Freitas TF, et al. Mycobacterium tuberculosis shikimate pathway enzymes as targets for the rational design of anti‐tuberculosis drugs. Molecules. 2020;25(6):1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Zhang YJ, Reddy MC, Ioerger TR, et al. Tryptophan biosynthesis protects mycobacteria from CD4 T cell‐mediated killing. Cell. 2013;155(6):1296‐1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Zhu N, Wang X, Li D, et al. IMB‐T130 targets 3‐dehydroquinate synthase and inhibits Mycobacterium tuberculosis. Sci Rep. 2018;8(1):17439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Deng Q, Meng J, Liu Y, Guan Y, Xiao C. IMB‐SD62, a triazolothiadiazoles derivative with promising action against tuberculosis. Tuberculosis (Edinb). 2018;112:37‐44. [DOI] [PubMed] [Google Scholar]
- 230. Li Z, Bai X, Deng Q, et al. Preliminary SAR and biological evaluation of antitubercular triazolothiadiazine derivatives against drug‐susceptible and drug‐resistant Mtb strains. Bioorg Med Chem. 2017;25(1):213‐220. [DOI] [PubMed] [Google Scholar]
- 231. Li Z, Liu Y, Bai X, et al. SAR studies on 1,2,4‐triazolo[3,4‐b][1,3,4]thiadiazoles as inhibitors of Mtb shikimate dehydrogenase for the development of novel antitubercular agents. RSC Adv. 2015;5(118):97089‐97101. [Google Scholar]
- 232. Rajput VS, Mehra R, Kumar S, Nargotra A, Singh PP, Khan IA. Screening of antitubercular compound library identifies novel shikimate kinase inhibitors of Mycobacterium tuberculosis. Appl Microbiol Biotechnol. 2016;100(12):5415‐5426. [DOI] [PubMed] [Google Scholar]
- 233. Wellington S, Nag PP, Michalska K, et al. A small‐molecule allosteric inhibitor of Mycobacterium tuberculosis tryptophan synthase. Nat Chem Biol. 2017;13(9):943‐950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Blumenstein L, Domratcheva T, Niks D, et al. BetaQ114N and betaT110V mutations reveal a critically important role of the substrate alpha‐carboxylate site in the reaction specificity of tryptophan synthase. Biochemistry. 2007;46(49):14100‐14116. [DOI] [PubMed] [Google Scholar]
- 235. Dunn MF, Niks D, Ngo H, Barends TRM, Schlichting I. Tryptophan synthase: the workings of a channeling nanomachine. Trends Biochem Sci. 2008;33(6):254‐264. [DOI] [PubMed] [Google Scholar]
- 236. Dunn MF. Allosteric regulation of substrate channeling and catalysis in the tryptophan synthase bienzyme complex. Arch Biochem Biophys. 2012;519(2):154‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Schneider TR, Gerhardt E, Lee M, Liang PH, Anderson KS, Schlichting I. Loop closure and intersubunit communication in tryptophan synthase. Biochemistry. 1998;37(16):5394‐5406. [DOI] [PubMed] [Google Scholar]
- 238. Michalska K, Chang C, Maltseva NI, et al. Allosteric inhibitors of Mycobacterium tuberculosis tryptophan synthase. Protein Sci. 2020;29(3):779‐788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Abrahams KA, Cox JAG, Fütterer K, et al. Inhibiting mycobacterial tryptophan synthase by targeting the inter‐subunit interface. Sci Rep. 2017;7:9430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Godbole AA, Leelaram MN, Bhat AG, Jain P, Nagaraja V. Characterization of DNA topoisomerase I from Mycobacterium tuberculosis: dNA cleavage and religation properties and inhibition of its activity. Arch Biochem Biophy. 2012;528(2):197‐203. [DOI] [PubMed] [Google Scholar]
- 241. Godbole AA, Ahmed W, Bhat RS, Bradley EK, Ekins S, Nagaraja V. Inhibition of Mycobacterium tuberculosis topoisomerase I by m‐AMSA, a eukaryotic type II topoisomerase poison. Biochem Biophys Res Commun. 2014;446(4):916‐920. [DOI] [PubMed] [Google Scholar]
- 242. Sandhaus S, Annamalai T, Welmaker G, et al. Small‐molecule inhibitors targeting topoisomerase i as novel antituberculosis agents. Antimicrob Agents Chemother. 2016;60(7):4028‐4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Godbole AA, Ahmed W, Bhat RS, Bradley EK, Ekins S, Nagaraja V. Targeting Mycobacterium tuberculosis topoisomerase I by small‐molecule inhibitors. Antimicrob Agents Chemother. 2015;59(3):1549‐1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Asif M, Siddiqui AA, Husain A. Quinolone derivatives as antitubercular drugs. Med Chem Res. 2013;22(3):1029‐1042. [Google Scholar]
- 245. Blower TR, Williamson BH, Kerns RJ, Berger JM. Crystal structure and stability of gyrase‐fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2016;113(7):1706‐1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Talley AK, Thurston A, Moore G, et al. First‐in‐human evaluation of the safety, tolerability, and pharmacokinetics of SPR720, a novel oral bacterial DNA Gyrase (GyrB) inhibitor for mycobacterial infections. Antimicrob Agents Chemother;65(11):e01208‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Kale MG, Raichurkar A, SH P, et al. Thiazolopyridine ureas as novel antitubercular agents acting through inhibition of DNA Gyrase B. J Med Chem. 2013;56(21):8834‐8848. [DOI] [PubMed] [Google Scholar]
- 248. Jeankumar VU, Renuka J, Santosh P, et al. Thiazole‐aminopiperidine hybrid analogues: design and synthesis of novel Mycobacterium tuberculosis GyrB inhibitors. Eur J Med Chem. 2013;70:143‐153. [DOI] [PubMed] [Google Scholar]
- 249. Renuka J, Reddy KI, Srihari K, et al. Design, synthesis, biological evaluation of substituted benzofurans as DNA gyraseB inhibitors of Mycobacterium tuberculosis. Bioorg Med Chem. 2014;22(17):4924‐4934. [DOI] [PubMed] [Google Scholar]
- 250. Medapi B, Suryadevara P, Renuka J, Sridevi JP, Yogeeswari P, Sriram D. 4‐Aminoquinoline derivatives as novel Mycobacterium tuberculosis GyrB inhibitors: structural optimization, synthesis and biological evaluation. Eur J Med Chem. 2015;103:1‐16. [DOI] [PubMed] [Google Scholar]
- 251. Nagaraja V, Godbole AA, Henderson SR, Maxwell A. DNA topoisomerase I and DNA gyrase as targets for TB therapy. Drug Discov Today. 2017;22(3):510‐518. [DOI] [PubMed] [Google Scholar]
- 252. Campbell EA, Korzheva N, Mustaev A, et al. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell. 2001;104(6):901‐912. [DOI] [PubMed] [Google Scholar]
- 253. Koch A, Mizrahi V, Warner DF. The impact of drug resistance on Mycobacterium tuberculosis physiology: what can we learn from rifampicin? Emerg Microbes Infect. 2014;3(3):e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Zaw MT, Emran NA, Lin Z. Mutations inside rifampicin‐resistance determining region of rpoB gene associated with rifampicin‐resistance in Mycobacterium tuberculosis. J Infect Public Health. 2018;11(5):605‐610. [DOI] [PubMed] [Google Scholar]
- 255. Stephanie F, Tambunan USF, Siahaan TJM. tuberculosis transcription machinery: a review on the mycobacterial RNA polymerase and drug discovery efforts. Life. 2022;12(11):1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Engohang‐Ndong J, Baillat D, Aumercier M, et al. EthR, a repressor of the TetR/CamR family implicated in ethionamide resistance in mycobacteria, octamerizes cooperatively on its operator. Mol Microbiol. 2004;51(1):175‐188. [DOI] [PubMed] [Google Scholar]
- 257. Willand N, Dirié B, Carette X, et al. Synthetic EthR inhibitors boost antituberculous activity of ethionamide. Nat Med. 2009;15(5):537‐544. [DOI] [PubMed] [Google Scholar]
- 258. Blondiaux N, Moune M, Desroses M, et al. Reversion of antibiotic resistance in Mycobacterium tuberculosis by spiroisoxazoline SMARt‐420. Science. 2017;355(6330):1206‐1211. [DOI] [PubMed] [Google Scholar]
- 259. Villemagne B, Machelart A, Tran NC, et al. Fragment‐based optimized EthR inhibitors with in vivo ethionamide boosting activity. ACS Infect Dis. 2020;6(3):366‐378. [DOI] [PubMed] [Google Scholar]
- 260. Flipo M, Willand N, Lecat‐Guillet N, et al. Discovery of novel N‐phenylphenoxyacetamide derivatives as EthR inhibitors and ethionamide boosters by combining high‐throughput screening and synthesis. J Med Chem. 2012;55(14):6391‐6402. [DOI] [PubMed] [Google Scholar]
- 261. Black PA, Warren RM, Louw GE, van Helden PD, Victor TC, Kana BD. Energy metabolism and drug efflux in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2014;58(5):2491‐2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Bald D, Villellas C, Lu P, Koul A. Targeting energy metabolism in Mycobacterium tuberculosis, a new paradigm in antimycobacterial drug discovery. mBio. 2017;8(2):e00272‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Diacon AH, Dawson R, von Groote‐Bidlingmaier F, et al. Bactericidal activity of pyrazinamide and clofazimine alone and in combinations with pretomanid and bedaquiline. Am J Respir Crit Care Med. 2015;191(8):943‐953. [DOI] [PubMed] [Google Scholar]
- 264. Xu J, Wang B, Fu L, et al. In vitro and in vivo activities of the riminophenazine TBI‐166 against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2019;63(5):e02155‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Beites T, O'Brien K, Tiwari D, et al. Plasticity of the Mycobacterium tuberculosis respiratory chain and its impact on tuberculosis drug development. Nat Commun. 2019;10(1):4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Kristiansen JE, Dastidar SG, Palchoudhuri S, et al. Phenothiazines as a solution for multidrug resistant tuberculosis: from the origin to present. Int Microbiol. 2015;18(1):1‐12. [DOI] [PubMed] [Google Scholar]
- 267. Salie S, Labuschagné A, Walters A, et al. In vitro and in vivo toxicity evaluation of non‐neuroleptic phenothiazines, antitubercular drug candidates. Regul Toxicol Pharmacol. 2019;109:104508. [DOI] [PubMed] [Google Scholar]
- 268.Qurient Co., Ltd. An Open‐Label Randomized Study to Evaluate the Early Bactericidal Activity, Safety, Tolerability, and Pharmacokinetics of Multiple Oral Doses of Telacebec (Q203) in Treatment‐Naïve Patients With Newly Diagnosed Drug‐Sensitive Sputum Smear‐Positive Pulmonary Tuberculosis. clinicaltrials.gov; 2019.
- 269. Pethe K, Bifani P, Jang J, et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med. 2013;19(9):1157‐1160. [DOI] [PubMed] [Google Scholar]
- 270. Zhang Y, Wade MM, Scorpio A, Zhang H, Sun Z. Mode of action of pyrazinamide: disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J Antimicrob Chemother. 2003;52(5):790‐795. [DOI] [PubMed] [Google Scholar]
- 271. Lupien A, Foo CSY, Savina S, et al. New 2‐Ethylthio‐4‐methylaminoquinazoline derivatives inhibiting two subunits of cytochrome bc1 in Mycobacterium tuberculosis. PLoS Pathog. 2020;16(1):e1008270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Cleghorn LAT, Ray PC, Odingo J, et al. Identification of morpholino thiophenes as novel Mycobacterium tuberculosis inhibitors, targeting QcrB. J Med Chem. 2018;61(15):6592‐6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Foo CS, Lupien A, Kienle M, et al. Arylvinylpiperazine amides, a new class of potent inhibitors targeting QcrB of Mycobacterium tuberculosis. mBio. 2018;9(5):e01276‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Hu X, Wan B, Liu Y, et al. Identification of pyrazolo[1,5‐a]pyridine‐3‐carboxamide diaryl derivatives as drug resistant antituberculosis agents. ACS Med Chem Lett. 2019;10(3):295‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Moraski GC, Seeger N, Miller PA, et al. Arrival of Imidazo[2,1‐b]thiazole‐5‐carboxamides: potent anti‐tuberculosis agents that target QcrB. ACS Infect Dis. 2016;2(6):393‐398. [DOI] [PubMed] [Google Scholar]
- 276. Kakkar AK, Dahiya N. Bedaquiline for the treatment of resistant tuberculosis: promises and pitfalls. Tuberculosis (Edinb). 2014;94(4):357‐362. [DOI] [PubMed] [Google Scholar]
- 277. Preiss L, Langer JD, Yildiz Ö, et al. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti‐TB drug bedaquiline. Sci Adv. 2015;1(4):e1500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Kundu S, Biukovic G, Grüber G, Dick T. Bedaquiline targets the ε subunit of mycobacterial F‐ATP synthase. Antimicrob Agents Chemother. 2016;60(11):6977‐6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Migliori GB, Tiberi S, Zumla A, et al. MDR/XDR‐TB management of patients and contacts: challenges facing the new decade. The 2020 clinical update by the Global Tuberculosis Network. Int J Infect Dis. 2020;92S:S15‐S25. [DOI] [PubMed] [Google Scholar]
- 280. Sutherland HS, Tong AST, Choi PJ, et al. 3,5‐Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Bioorg Med Chem. 2019;27(7):1292‐1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Yao R, Wang B, Fu L, et al. Sudapyridine (WX‐081), a novel compound against Mycobacterium tuberculosis. Microbiol Spectr;10(1):e02477‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Tantry SJ, Markad SD, Shinde V, et al. Discovery of Imidazo[1,2‐a]pyridine ethers and squaramides as selective and potent inhibitors of mycobacterial Adenosine Triphosphate (ATP) synthesis. J Med Chem. 2017;60(4):1379‐1399. [DOI] [PubMed] [Google Scholar]
- 283. Tantry SJ, Shinde V, Balakrishnan G, et al. Scaffold morphing leading to evolution of 2,4‐diaminoquinolines and aminopyrazolopyrimidines as inhibitors of the ATP synthesis pathway. Med Chem Commun. 2016;7(5):1022‐1032. [Google Scholar]
- 284. Sundararajan S, Muniyan R. Latent tuberculosis: interaction of virulence factors in Mycobacterium tuberculosis. Mol Biol Rep. 2021;48(8):6181‐6196. [DOI] [PubMed] [Google Scholar]
- 285. Capra EJ, Laub MT. The evolution of two‐component signal transduction systems. Annu Rev Microbiol. 2012;66:325‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Walters SB, Dubnau E, Kolesnikova I, Laval F, Daffe M, Smith I. The Mycobacterium tuberculosis PhoPR two‐component system regulates genes essential for virulence and complex lipid biosynthesis. Mol Microbiol. 2006;60(2):312‐330. [DOI] [PubMed] [Google Scholar]
- 287. Converse PJ, Karakousis PC, Klinkenberg LG, et al. Role of the dosR‐dosS two‐component regulatory system in Mycobacterium tuberculosis virulence in three animal models. Infect Immun. 2009;77(3):1230‐1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Mehra S, Foreman TW, Didier PJ, et al. The DosR regulon modulates adaptive immunity and is essential for Mycobacterium tuberculosis persistence. Am J Respir Crit Care Med. 2015;191(10):1185‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Buglino JA, Sankhe GD, Lazar N, Bean JM, Glickman MS. Integrated sensing of host stresses by inhibition of a cytoplasmic two‐component system controls M. tuberculosis acute lung infection. Elife. 2021;10:e65351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Banerjee SK, Lata S, Sharma AK, et al. The sensor kinase MtrB of Mycobacterium tuberculosis regulates hypoxic survival and establishment of infection. J Biol Chem. 2019;294(52):19862‐19876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Broset E, Martín C, Gonzalo‐Asensio J. Evolutionary landscape of the Mycobacterium tuberculosis complex from the viewpoint of PhoPR: implications for virulence regulation and application to vaccine development. mBio. 2015;6(5):e01289‐01215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Feng L, Chen S, Hu Y. PhoPR positively regulates whiB3 expression in response to low pH in pathogenic mycobacteria. J Bacteriol. 2018;200(8):e00766‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Ryndak M, Wang S, Smith I. PhoP, a key player in Mycobacterium tuberculosis virulence. Trends Microbiol. 2008;16(11):528‐534. [DOI] [PubMed] [Google Scholar]
- 294. Johnson BK, Colvin CJ, Needle DB, Mba Medie F, Champion PAD, Abramovitch RB. The carbonic anhydrase inhibitor ethoxzolamide inhibits the Mycobacterium tuberculosis PhoPR regulon and Esx‐1 secretion and attenuates virulence. Antimicrob Agents Chemother. 2015;59(8):4436‐4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Dasgupta N, Kapur V, Singh KK, et al. Characterization of a two‐component system, devR‐devS, of Mycobacterium tuberculosis. Tuber Lung Dis. 2000;80(3):141‐159. [DOI] [PubMed] [Google Scholar]
- 296. Malhotra V, Sharma D, Ramanathan VD, et al. Disruption of response regulator gene, devR, leads to attenuation in virulence of Mycobacterium tuberculosis. FEMS Microbiol Lett. 2004;231(2):237‐245. [DOI] [PubMed] [Google Scholar]
- 297. Roberts DM, Liao RP, Wisedchaisri G, Hol WGJ, Sherman DR. Two sensor kinases contribute to the hypoxic response of Mycobacterium tuberculosis. J Biol Chem. 2004;279(22):23082‐23087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Zheng H, Abramovitch RB. Inhibiting DosRST as a new approach to tuberculosis therapy. Future Med Chem. 2020;12(5):457‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Zheng H, Colvin CJ, Johnson BK, et al. Inhibitors of Mycobacterium tuberculosis DosRST signaling and persistence. Nat Chem Biol. 2017;13(2):218‐225. [DOI] [PubMed] [Google Scholar]
- 300. Zheng H, Williams JT, Aleiwi B, Ellsworth E, Abramovitch RB. Inhibiting Mycobacterium tuberculosis DosRST signaling by targeting response regulator DNA binding and sensor kinase heme. ACS Chem Biol. 2020;15(1):52‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Abdallah AM, Gey van Pittius NC, Champion PAD, et al. Type VII secretion–mycobacteria show the way. Nat Rev Microbiol. 2007;5(11):883‐891. [DOI] [PubMed] [Google Scholar]
- 302. Houben D, Demangel C, van Ingen J, et al. ESX‐1‐mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol. 2012;14(8):1287‐1298. [DOI] [PubMed] [Google Scholar]
- 303. Wong KW, Jacobs WR Jr. Critical role for NLRP3 in necrotic death triggered by Mycobacterium tuberculosis. Cell Microbiol. 2011;13(9):1371‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. De Leon J, Jiang G, Ma Y, Rubin E, Fortune S, Sun J. Mycobacterium tuberculosis ESAT‐6 exhibits a unique membrane‐interacting activity that is not found in its ortholog from non‐pathogenic Mycobacterium smegmatis. J Biol Chem. 2012;287(53):44184‐44191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Frigui W, Bottai D, Majlessi L, et al. Control of M. tuberculosis ESAT‐6 secretion and specific T cell recognition by PhoP. PLOS Pathogens. 2008;4(2):e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Rybniker J, Chen JM, Sala C, et al. Anticytolytic screen identifies inhibitors of mycobacterial virulence protein secretion. Cell Host Microbe. 2014;16(4):538‐548. [DOI] [PubMed] [Google Scholar]
- 307. Jia P, Zhang Y, Xu J, et al. IMB‐BZ as an inhibitor targeting ESX‐1 secretion system to control mycobacterial infection. J Infect Dis. 2022;225(4):608‐616. [DOI] [PubMed] [Google Scholar]
- 308. Chakraborty S, Gruber T, Barry CE, Boshoff HI, Rhee KY. Para‐aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science. 2013;339(6115):88‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Zheng J, Rubin EJ, Bifani P, et al. para‐Aminosalicylic acid is a prodrug targeting dihydrofolate reductase in Mycobacterium tuberculosis. J Biol Chem. 2013;288(32):23447‐23456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Hajian B, Scocchera E, Shoen C, et al. Drugging the folate pathway in Mycobacterium tuberculosis: the role of multi‐targeting agents. Cell Chem Biol. 2019;26(6):781‐791.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. He J, Li C, Hu W, et al. Identification of selective mtbDHFR inhibitors by virtual screening and experimental approaches. Chem Biol Drug Des. 2022;100(6):1005‐1016. [DOI] [PubMed] [Google Scholar]
- 312. Waldron KJ, Robinson NJ. How do bacterial cells ensure that metalloproteins get the correct metal? Nat Rev Microbiol. 2009;7(1):25‐35. [DOI] [PubMed] [Google Scholar]
- 313. Pandey R, Rodriguez GM. IdeR is required for iron homeostasis and virulence in Mycobacterium tuberculosis. Mol Microbiol. 2014;91(1):98‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Rohilla A, Khare G, Tyagi AK. Virtual Screening, pharmacophore development and structure based similarity search to identify inhibitors against IdeR, a transcription factor of Mycobacterium tuberculosis. Sci Rep. 2017;7:4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Salimizand H, Jamehdar SA, Nik LB, Sadeghian H. Design of peptides interfering with iron‐dependent regulator (IdeR) and evaluation of Mycobacterium tuberculosis growth inhibition. Iran J Basic Med Sci. 2017;20(6):722‐728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Nuermberger EL, Martínez‐Martínez MS, Sanz O, et al. GSK2556286 is a novel antitubercular drug candidate effective in vivo with the potential to shorten tuberculosis treatment. Antimicrob Agents Chemother. 2022;66(6):e0013222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. VanderVen BC, Fahey RJ, Lee W, et al. Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium's metabolism is constrained by the intracellular environment. PLoS Pathog. 2015;11(2):e1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Rengarajan J, Bloom BR, Rubin EJ. Genome‐wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci USA. 2005;102(23):8327‐8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Shenoy AR, Visweswariah SS. Mycobacterial adenylyl cyclases: biochemical diversity and structural plasticity. FEBS Lett. 2006;580(14):3344‐3352. [DOI] [PubMed] [Google Scholar]
- 320. Brown KL, Wilburn KM, Montague CR, et al. Cyclic AMP‐mediated inhibition of cholesterol catabolism in Mycobacterium tuberculosis by the novel drug candidate GSK2556286. Antimicrob Agents Chemother. 2023;67(1):e0129422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Johnson RM, Bai G, DeMott CM, et al. Chemical activation of adenylyl cyclase Rv1625c inhibits growth of Mycobacterium tuberculosis on cholesterol and modulates intramacrophage signaling. Mol Microbiol. 2017;105(2):294‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Wall EA, Zavzavadjian JR, Chang MS, et al. Suppression of LPS‐induced TNF‐α production in macrophages by cAMP is mediated by PKA‐AKAP95‐p105. Sci Signal. 2009;2(75):ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Tiberi S, du Plessis N, Walzl G, et al. Tuberculosis: progress and advances in development of new drugs, treatment regimens, and host‐directed therapies. Lancet Infect Dis. 2018;18(7):e183‐e198. [DOI] [PubMed] [Google Scholar]
- 324. Palucci I, Delogu G. Host directed therapies for tuberculosis: futures strategies for an ancient disease. Chemotherapy. 2018;63(3):172‐180. [DOI] [PubMed] [Google Scholar]
- 325. Killick KE, Ní Cheallaigh C, O'Farrelly C, Hokamp K, MacHugh DE, Harris J. Receptor‐mediated recognition of mycobacterial pathogens. Cell Microbiol. 2013;15(9):1484‐1495. [DOI] [PubMed] [Google Scholar]
- 326. Liu CH, Liu H, Ge B. Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell Mol Immunol. 2017;14(12):963‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Lerner TR, Borel S, Gutierrez MG. The innate immune response in human tuberculosis. Cell Microbiol. 2015;17(9):1277‐1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Orme IM. The kinetics of emergence and loss of mediator T lymphocytes acquired in response to infection with Mycobacterium tuberculosis. J Immunol. 1987;138(1):293‐298. [PubMed] [Google Scholar]
- 329. Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T‐cell immunity. Infect Immun. 2002;70(8):4501‐4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Giosué S, Casarini M, Alemanno L, et al. Effects of aerosolized interferon‐alpha in patients with pulmonary tuberculosis. Am J Respir Crit Care Med. 1998;158(4):1156‐1162. [DOI] [PubMed] [Google Scholar]
- 331. Giosuè S, Casarini M, Ameglio F, et al. Aerosolized interferon‐alpha treatment in patients with multi‐drug‐resistant pulmonary tuberculosis. Eur Cytokine Netw. 2000;11(1):99‐104. [PubMed] [Google Scholar]
- 332. Raad I, Hachem R, Leeds N, Sawaya R, Salem Z, Atweh S. Use of adjunctive treatment with interferon‐gamma in an immunocompromised patient who had refractory multidrug‐resistant tuberculosis of the brain. Clin Infect Dis. 1996;22(3):572‐574. [DOI] [PubMed] [Google Scholar]
- 333. Bilu D, Sauder DN. Imiquimod: modes of action. Br J Dermatol. 2003;149(66):5‐8. Suppl. [DOI] [PubMed] [Google Scholar]
- 334. Wang Y, Zhang S, Li H, et al. Small‐molecule modulators of toll‐like receptors. Acc Chem Res. 2020;53(5):1046‐1055. [DOI] [PubMed] [Google Scholar]
- 335.MD HK. A Prospective, Randomized Open‐Label Phase II Study of the Safety and Tolerability of Metformin in Combination With Standard Antimicrobial Treatment of Pulmonary Tuberculosis in People With TB and Co‐Infected With HIV. clinicaltrials.gov; 2023.
- 336. Lachmandas E, Eckold C, Böhme J, et al. Metformin alters human host responses to Mycobacterium tuberculosis in healthy subjects. J Infect Dis. 2019;220(1):139‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Dorhoi A, Kotzé LA, Berzofsky JA, et al. Therapies for tuberculosis and AIDS: myeloid‐derived suppressor cells in focus. J Clin Invest. 2020;130(6):2789‐2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Knaul JK, Jörg S, Oberbeck‐Mueller D, et al. Lung‐residing myeloid‐derived suppressors display dual functionality in murine pulmonary tuberculosis. Am J Respir Crit Care Med. 2014;190(9):1053‐1066. [DOI] [PubMed] [Google Scholar]
- 339. du Plessis N, Loebenberg L, Kriel M, et al. Increased frequency of myeloid‐derived suppressor cells during active tuberculosis and after recent mycobacterium tuberculosis infection suppresses T‐cell function. Am J Respir Crit Care Med. 2013;188(6):724‐732. [DOI] [PubMed] [Google Scholar]
- 340. Zhang MN, Yuan YL, Ao SH. Advances in the study of myeloid‐derived suppressor cells in infectious lung diseases. Front Immunol. 2023;14:1125737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Shen L, Pili R. Tasquinimod targets suppressive myeloid cells in the tumor microenvironment. Oncoimmunology. 2019;8(10):e1072672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Gupta S, Krug S, Pokkali S, et al. Pharmacologic exhaustion of suppressor cells with tasquinimod enhances bacterial clearance during tuberculosis. Am J Respir Crit Care Med. 2019;199(3):386‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119(6):753‐766. [DOI] [PubMed] [Google Scholar]
- 344. Adikesavalu H, Gopalaswamy R, Kumar A, Ranganathan UD, Shanmugam S. Autophagy induction as a host‐directed therapeutic strategy against Mycobacterium tuberculosis infection. Medicina (Kaunas). 2021;57(6):522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Flannagan RS, Cosío G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7(5):355‐366. [DOI] [PubMed] [Google Scholar]
- 346. Vergne I, Chua J, Singh SB, Deretic V. Cell biology of mycobacterium tuberculosis phagosome. Annu Rev Cell Dev Biol. 2004;20:367‐394. [DOI] [PubMed] [Google Scholar]
- 347. Sarantis H, Grinstein S. Subversion of phagocytosis for pathogen survival. Cell Host Microbe. 2012;12(4):419‐431. [DOI] [PubMed] [Google Scholar]
- 348. Bruns H, Stegelmann F, Fabri M, et al. Abelson tyrosine kinase controls phagosomal acidification required for killing of Mycobacterium tuberculosis in human macrophages. J Immunol. 2012;189(8):4069‐4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Kim JK, Kim YS, Lee HM, et al. GABAergic signaling linked to autophagy enhances host protection against intracellular bacterial infections. Nat Commun. 2018;9(1):4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Gupta PK, Chakraborty P, Kumar S, et al. G1‐4A, a polysaccharide from Tinospora cordifolia inhibits the survival of Mycobacterium tuberculosis by modulating host immune responses in TLR4 dependent manner. PLoS One. 2016;11(5):e0154725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Liu PT, Stenger S, Li H, et al. Toll‐like receptor triggering of a vitamin D‐mediated human antimicrobial response. Science. 2006;311(5768):1770‐1773. [DOI] [PubMed] [Google Scholar]
- 352. Papagni R, Pellegrino C, Di Gennaro F, et al. Impact of vitamin D in prophylaxis and treatment in tuberculosis patients. Int J Mol Sci. 2022;23(7):3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Mayer‐Barber KD, Barber DL. Innate and adaptive cellular immune responses to mycobacterium tuberculosis infection. Cold Spring Harb Perspect Med. 2015;5(12):a018424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Mbongue JC, Nicholas DA, Torrez TW, Kim NS, Firek AF, Langridge WHR. The role of indoleamine 2, 3‐dioxygenase in immune suppression and autoimmunity. Vaccines (Basel). 2015;3(3):703‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Gautam US, Foreman TW, Bucsan AN, et al. In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune‐mediated control of Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2018;115(1):E62‐E71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Costa DL, Amaral EP, Namasivayam S, et al. Heme oxygenase‐1 inhibition promotes IFNγ‐ and NOS2‐mediated control of Mycobacterium tuberculosis infection. Mucosal Immunol. 2021;14(1):253‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Marimani M, Ahmad A, Duse A. The role of epigenetics, bacterial and host factors in progression of Mycobacterium tuberculosis infection. Tuberculosis. 2018;113:200‐214. [DOI] [PubMed] [Google Scholar]
- 358. Ghorpade DS, Leyland R, Kurowska‐Stolarska M, Patil SA, Balaji KN. MicroRNA‐155 is required for Mycobacterium bovis BCG‐mediated apoptosis of macrophages. Mol Cell Biol. 2012;32(12):2239‐2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Pennini ME, Liu Y, Yang J, Croniger CM, Boom WH, Harding CV. CCAAT/enhancer‐binding protein beta and delta binding to CIITA promoters is associated with the inhibition of CIITA expression in response to Mycobacterium tuberculosis 19‐kDa lipoprotein. J Immunol. 2007;179(10):6910‐6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Ma F, Xu S, Liu X, et al. The microRNA miR‐29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon‐γ. Nat Immunol. 2011;12(9):861‐869. [DOI] [PubMed] [Google Scholar]
- 361. Zheng L, Leung ETY, Wong HK, et al. Unraveling methylation changes of host macrophages in Mycobacterium tuberculosis infection. Tuberculosis (Edinb). 2016;98:139‐148. [DOI] [PubMed] [Google Scholar]
- 362. Moreira JD, Koch BEV, van Veen S, et al. Functional inhibition of Host Histone Deacetylases (HDACs) enhances in vitro and in vivo anti‐mycobacterial activity in human macrophages and in zebrafish. Front Immunol. 2020;11:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Wang X, Tang X, Zhou Z, Huang Q. Histone deacetylase 6 inhibitor enhances resistance to Mycobacterium tuberculosis infection through innate and adaptive immunity in mice. Pathog Dis. 2018;76(6). [DOI] [PubMed] [Google Scholar]
- 364. Rao M, Valentini D, Zumla A, Maeurer M. Evaluation of the efficacy of valproic acid and suberoylanilide hydroxamic acid (vorinostat) in enhancing the effects of first‐line tuberculosis drugs against intracellular Mycobacterium tuberculosis. Int J Infect Dis. 2018;69:78‐84. [DOI] [PubMed] [Google Scholar]
- 365. Mily A, Rekha RS, Kamal SMM, et al. Significant effects of oral phenylbutyrate and vitamin D3 adjunctive therapy in pulmonary tuberculosis: a randomized controlled trial. PLoS One. 2015;10(9):e0138340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Rekha RS, Mily A, Sultana T, et al. Immune responses in the treatment of drug‐sensitive pulmonary tuberculosis with phenylbutyrate and vitamin D3 as host directed therapy. BMC Infect Dis. 2018;18(1):303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Kaminskas E, Farrell AT, Wang YC, Sridhara R, Pazdur R. FDA drug approval summary: azacitidine (5‐azacytidine, Vidaza) for injectable suspension. Oncologist. 2005;10(3):176‐182. [DOI] [PubMed] [Google Scholar]
- 368. Dinardo A, Phase 1b/2a Safety and Immunogenicity of the DNMT Inhibitor Azacitidine During Anti‐Tuberculosis Therapy. clinicaltrials.gov; 2023.
- 369. Kimmey JM, Huynh JP, Weiss LA, et al. Unique role for ATG5 in neutrophil‐mediated immunopathology during M. tuberculosis infection. Nature. 2015;528(7583):565‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self‐digestion. Nature. 2008;451(7182):1069‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Kim J, Guan KL. Amino acid signaling in TOR activation. Annu Rev Biochem. 2011;80:1001‐1032. [DOI] [PubMed] [Google Scholar]
- 372. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19(2):121‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Sarkar S. Regulation of autophagy by mTOR‐dependent and mTOR‐independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans. 2013;41(5):1103‐1130. [DOI] [PubMed] [Google Scholar]
- 375. Gupta A, Sharma D, Meena J, et al. Preparation and preclinical evaluation of inhalable particles containing rapamycin and anti‐tuberculosis agents for induction of autophagy. Pharm Res. 2016;33(8):1899‐1912. [DOI] [PubMed] [Google Scholar]
- 376. Turnquist HR, Fischer RT, Thomson AW. Pharmacological modification of dendritic cells to promote their tolerogenicity in transplantation. Methods Mol Biol. 2010;595:135‐148. [DOI] [PubMed] [Google Scholar]
- 377. Lam KKY, Zheng X, Forestieri R, et al. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog. 2012;8(5):e1002691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Wang J, Sha J, Strong E, Chopra AK, Lee S. FDA‐approved amoxapine effectively promotes macrophage control of mycobacteria by inducing autophagy. Microbiol Spectr. 2022;10(5):e0250922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Schiebler M, Brown K, Hegyi K, et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR‐independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol Med. 2015;7(2):127‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Singhal A, Jie L, Kumar P, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med. 2014;6(263):263ra159. [DOI] [PubMed] [Google Scholar]
- 381. Sfikakis PP. The first decade of biologic TNF antagonists in clinical practice: lessons learned, unresolved issues and future directions. Curr Dir Autoimmun. 2010;11:180‐210. [DOI] [PubMed] [Google Scholar]
- 382. Jang DI, Lee AH, Shin HY, et al. The role of Tumor Necrosis Factor Alpha (TNF‐α) in autoimmune disease and current TNF‐α inhibitors in therapeutics. Int J Mol Sci. 2021;22(5):2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Barnes PF, Fong SJ, Brennan PJ, Twomey PE, Mazumder A, Modlin RL. Local production of tumor necrosis factor and IFN‐gamma in tuberculous pleuritis. J Immunol. 1990;145(1):149‐154. [PubMed] [Google Scholar]
- 384. Valone SE, Rich EA, Wallis RS, Ellner JJ. Expression of tumor necrosis factor in vitro by human mononuclear phagocytes stimulated with whole Mycobacterium bovis BCG and mycobacterial antigens. Infect Immun. 1988;56(12):3313‐3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Henderson RA, Watkins SC, Flynn JL. Activation of human dendritic cells following infection with Mycobacterium tuberculosis. J Immunol. 1997;159(2):635‐643. [PubMed] [Google Scholar]
- 386. Barnes PF, Lu S, Abrams JS, Wang E, Yamamura M, Modlin RL. Cytokine production at the site of disease in human tuberculosis. Infect Immun. 1993;61(8):3482‐3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. O'Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MPR. The immune response in tuberculosis. Annu Rev Immunol. 2013;31:475‐527. [DOI] [PubMed] [Google Scholar]
- 388. Spencer CT, Abate G, Sakala IG, et al. Granzyme A produced by γ(9)δ(2) T cells induces human macrophages to inhibit growth of an intracellular pathogen. PLoS Pathog. 2013;9(1):e1003119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Tobin DM, Roca FJ, Oh SF, et al. Host genotype‐specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. 2012;148(3):434‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Roach DR, Bean AGD, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol. 2002;168(9):4620‐4627. [DOI] [PubMed] [Google Scholar]
- 391. Gautam US, Mehra S, Ahsan MH, Alvarez X, Niu T, Kaushal D. Role of TNF in the altered interaction of dormant Mycobacterium tuberculosis with host macrophages. PLoS One. 2014;9(4):e95220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Dorhoi A, Kaufmann SHE. Tumor necrosis factor alpha in mycobacterial infection. Semin Immunol. 2014;26(3):203‐209. [DOI] [PubMed] [Google Scholar]
- 393. Lee HJ, Ko HJ, Kim SH, Jung YJ. Pasakbumin A controls the growth of Mycobacterium tuberculosis by enhancing the autophagy and production of antibacterial mediators in mouse macrophages. PLoS One. 2019;14(3):e0199799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha‐neutralizing agent. N Engl J Med. 2001;345(15):1098‐1104. [DOI] [PubMed] [Google Scholar]
- 395. Xie X, Li F, Chen JW, Wang J. Risk of tuberculosis infection in anti‐TNF‐α biological therapy: from bench to bedside. J Microbiol Immunol Infect. 2014;47(4):268‐274. [DOI] [PubMed] [Google Scholar]
- 396. Roca FJ, Whitworth LJ, Prag HA, Murphy MP, Ramakrishnan L. Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science. 2022;376(6600):eabh2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Roca FJ, Whitworth LJ, Redmond S, Jones AA, Ramakrishnan L. TNF induces pathogenic programmed macrophage necrosis in tuberculosis through a mitochondrial‐lysosomal‐endoplasmic reticulum circuit. Cell. 2019;178(6):1344‐1361.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Stanley SA, Barczak AK, Silvis MR, et al. Identification of host‐targeted small molecules that restrict intracellular Mycobacterium tuberculosis growth. PLoS Pathog. 2014;10(2):e1003946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Queval CJ, Song OR, Deboosère N, et al. STAT3 represses nitric oxide synthesis in human macrophages upon Mycobacterium tuberculosis infection. Sci Rep. 2016;6:29297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Sogi KM, Lien KA, Johnson JR, Krogan NJ, Stanley SA. The tyrosine kinase inhibitor gefitinib restricts Mycobacterium tuberculosis growth through increased lysosomal biogenesis and modulation of cytokine signaling. ACS Infect Dis. 2017;3(8):564‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401. Hu Y, Wen Z, Liu S, et al. Ibrutinib suppresses intracellular mycobacterium tuberculosis growth by inducing macrophage autophagy. J Infect. 2020;80(6):e19‐e26. [DOI] [PubMed] [Google Scholar]
- 402. Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129(13):2475‐2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Parihar SP, Guler R, Khutlang R, et al. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis. 2014;209(5):754‐763. [DOI] [PubMed] [Google Scholar]
- 404. Lobato LS, Rosa PS, Ferreira J, da S, et al. Statins increase rifampin mycobactericidal effect. Antimicrob Agents Chemother. 2014;58(10):5766‐5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Bruiners N, Dutta NK, Guerrini V, et al. The anti‐tubercular activity of simvastatin is mediated by cholesterol‐driven autophagy via the AMPK‐mTORC1‐TFEB axis. J Lipid Res. 2020;61(12):1617‐1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Cheng CY, Gutierrez NM, Marzuki MB, et al. Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci Immunol. 2017;2(9):eaaj1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Kim TS, Jin YB, Kim YS, et al. SIRT3 promotes antimycobacterial defenses by coordinating mitochondrial and autophagic functions. Autophagy. 2019;15(8):1356‐1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Rekha RS, Rao Muvva SSVJ, Wan M, et al. Phenylbutyrate induces LL‐37‐dependent autophagy and intracellular killing of Mycobacterium tuberculosis in human macrophages. Autophagy. 2015;11(9):1688‐1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Cho JH, Lee HJ, Ko HJ, et al. The TLR7 agonist imiquimod induces anti‐cancer effects via autophagic cell death and enhances anti‐tumoral and systemic immunity during radiotherapy for melanoma. Oncotarget. 2017;8(15):24932‐24948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Lee HJ, Kang SJ, Woo Y, Hahn TW, Ko HJ, Jung YJ. TLR7 stimulation with imiquimod induces selective autophagy and controls Mycobacterium tuberculosis growth in mouse macrophages. Front Microbiol. 2020;11:1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Shariq M, Quadir N, Alam A, et al. The exploitation of host autophagy and ubiquitin machinery by Mycobacterium tuberculosis in shaping immune responses and host defense during infection. Autophagy. 2023;19(1):3‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Pagán AJ, Ramakrishnan L. Immunity and immunopathology in the tuberculous granuloma. Cold Spring Harb Perspect Med. 2014;5(9):a018499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Forsythe JA, Jiang BH, Iyer NV, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia‐inducible factor 1. Mol Cell Biol. 1996;16(9):4604‐4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Matsuyama W, Hashiguchi T, Matsumuro K, et al. Increased serum level of vascular endothelial growth factor in pulmonary tuberculosis. Am J Respir Crit Care Med. 2000;162(3):1120‐1122. Pt 1. [DOI] [PubMed] [Google Scholar]
- 415. Podar K, Tonon G, Sattler M, et al. The small‐molecule VEGF receptor inhibitor pazopanib (GW786034B) targets both tumor and endothelial cells in multiple myeloma. Proc Natl Acad Sci USA. 2006;103(51):19478‐19483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Fong TA, Shawver LK, Sun L, et al. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk‐1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999;59(1):99‐106. [PubMed] [Google Scholar]
- 417. Oehlers SH, Cronan MR, Scott NR, et al. Interception of host angiogenic signalling limits mycobacterial growth. Nature. 2015;517(7536):612‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Bartz RR, McCord T, Suliman H, Orr T, Piantadosi CA, Kontos C, AKB‐9785, a VE‐PTP inhibitor, decreases inflammation and alters the angiopoietin/Tie2 system dynamic response in a S. aureus sepsis model. In: C67. Acute Lung Injury. American Thoracic Society International Conference Abstracts. American Thoracic Society; Am J Respir Crit Care Med. 2016;193:A5751‐A5751. [Google Scholar]
- 419. Shalwitz R, Peters KG, Methods for treating vascular leak syndrome. US Patent US20110268694A1. December 25, 2014.
- 420. Shirakura K, Baluk P, Nottebaum AF, et al. Shear stress control of vascular leaks and atheromas through Tie2 activation by VE‐PTP sequestration. EMBO Mol Med. 2023;15(4):e16128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Oehlers SH, Cronan MR, Beerman RW, et al. Infection‐induced vascular permeability aids mycobacterial growth. J Infect Dis. 2017;215(5):813‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Warner DF. Mycobacterium tuberculosis metabolism. Cold Spring Harb Perspect Med. 2014;5(4):a021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. MacFarlane M, Robinson GL, Cain K. Glucose–a sweet way to die: metabolic switching modulates tumor cell death. Cell Cycle. 2012;11(21):3919‐3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424. Menchikov LG, Shestov AA, Popov AV. Warburg effect revisited: embodiment of classical biochemistry and organic chemistry. Current state and prospects. Biochemistry (Mosc). 2023;88(1):S1‐S20. Suppl. [DOI] [PubMed] [Google Scholar]
- 425. Wang T, Liu G, Wang R. The intercellular metabolic interplay between tumor and immune cells. Front Immunol. 2014;5:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Shi L, Eugenin EA, Subbian S. Immunometabolism in tuberculosis. Front Immunol. 2016;7:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL‐1β through HIF‐1α. Nature. 2013;496(7444):238‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Jiang Q, Qiu Y, Kurland IJ, et al. Glutamine is required for M1‐like polarization of macrophages in response to Mycobacterium tuberculosis infection. mBio. 2022;13(4):e0127422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Alkan HF, Walter KE, Luengo A, et al. Cytosolic aspartate availability determines cell survival when glutamine is limiting. Cell Metab. 2018;28(5):706‐720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Peyron P, Vaubourgeix J, Poquet Y, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient‐rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008;4(11):e1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Singh V, Jamwal S, Jain R, Verma P, Gokhale R, Rao KVS. Mycobacterium tuberculosis‐driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe. 2012;12(5):669‐681. [DOI] [PubMed] [Google Scholar]
- 432. Rachman H, Kim N, Ulrichs T, et al. Critical role of methylglyoxal and AGE in mycobacteria‐induced macrophage apoptosis and activation. PLoS One. 2006;1(1):e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19(1):107‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Briken V, Miller JL. Living on the edge: inhibition of host cell apoptosis by Mycobacterium tuberculosis. Future Microbiol. 2008;3(4):415‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Spira A, Carroll JD, Liu G, et al. Apoptosis genes in human alveolar macrophages infected with virulent or attenuated Mycobacterium tuberculosis: a pivotal role for tumor necrosis factor. Am J Respir Cell Mol Biol. 2003;29(5):545‐551. [DOI] [PubMed] [Google Scholar]
- 436. Maiti D, Bhattacharyya A, Basu J. Lipoarabinomannan from Mycobacterium tuberculosis promotes macrophage survival by phosphorylating Bad through a phosphatidylinositol 3‐kinase/Akt pathway. J Biol Chem. 2001;276(1):329‐333. [DOI] [PubMed] [Google Scholar]
- 437. Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. 2000;164(4):2016‐2020. [DOI] [PubMed] [Google Scholar]
- 438. Mohareer K, Asalla S, Banerjee S. Cell death at the cross roads of host‐pathogen interaction in Mycobacterium tuberculosis infection. Tuberculosis. 2018;113:99‐121. [DOI] [PubMed] [Google Scholar]
- 439. Moraes LA, Piqueras L, Bishop‐Bailey D. Peroxisome proliferator‐activated receptors and inflammation. Pharmacol Ther. 2006;110(3):371‐385. [DOI] [PubMed] [Google Scholar]
- 440. Ricote M, Huang J, Fajas L, et al. Expression of the peroxisome proliferator‐activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci USA. 1998;95(13):7614‐7619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441. Rigamonti E, Fontaine C, Lefebvre B, et al. Induction of CXCR2 receptor by peroxisome proliferator‐activated receptor gamma in human macrophages. Arterioscler Thromb Vasc Biol. 2008;28(5):932‐939. [DOI] [PubMed] [Google Scholar]
- 442. Rajaram MVS, Brooks MN, Morris JD, Torrelles JB, Azad AK, Schlesinger LS. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator‐activated receptor γ linking mannose receptor recognition to regulation of immune responses. J Immunol. 2010;185(2):929‐942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443. Arnett E, Weaver AM, Woodyard KC, et al. PPARγ is critical for Mycobacterium tuberculosis induction of Mcl‐1 and limitation of human macrophage apoptosis. PLoS Pathog. 2018;14(6):e1007100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Besbes S, Mirshahi M, Pocard M, Billard C. New dimension in therapeutic targeting of BCL‐2 family proteins. Oncotarget. 2015;6(15):12862‐12871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153(3):521‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Amaral EP, Costa DL, Namasivayam S, et al. A major role for ferroptosis in Mycobacterium tuberculosis‐induced cell death and tissue necrosis. J Exp Med. 2019;216(3):556‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. Gräb J, Suárez I, van Gumpel E, et al. Corticosteroids inhibit Mycobacterium tuberculosis‐induced necrotic host cell death by abrogating mitochondrial membrane permeability transition. Nat Commun. 2019;10:688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. Maltez VI, Miao EA. Reassessing the evolutionary importance of inflammasomes. J Immunol. 2016;196(3):956‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450. Beckwith KS, Beckwith MS, Ullmann S, et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat Commun. 2020;11:2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. Qu Z, Zhou J, Zhou Y, et al. Mycobacterial EST12 activates a RACK1–NLRP3–gasdermin D pyroptosis–IL‐1β immune pathway. Sci Adv. 2020;6(43):eaba4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452. Wassermann R, Gulen MF, Sala C, et al. Mycobacterium tuberculosis differentially activates cGAS‐ and inflammasome‐dependent intracellular immune responses through ESX‐1. Cell Host Microbe. 2015;17(6):799‐810. [DOI] [PubMed] [Google Scholar]
- 453. Fu Y, Shen J, Li Y, et al. Inhibition of the PERK/TXNIP/NLRP3 axis by Baicalin reduces NLRP3 inflammasome‐mediated pyroptosis in macrophages infected with Mycobacterium tuberculosis. Mediators Inflamm. 2021;2021:1805147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454. Li Y, Fu Y, Sun J, et al. Tanshinone IIA alleviates NLRP3 inflammasome‐mediated pyroptosis in Mycobacterium tuberculosis‐(H37Ra‐) infected macrophages by inhibiting endoplasmic reticulum stress. J Ethnopharmacol. 2022;282:114595. [DOI] [PubMed] [Google Scholar]
- 455. Tezera LB, Mansour S, Elkington P. Reconsidering the optimal immune response to Mycobacterium tuberculosis. Am J Respir Crit Care Med. 2020;201(4):407‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456. Krug S, Parveen S, Bishai WR. Host‐directed therapies: modulating inflammation to treat tuberculosis. Front Immunol. 2021;12:660916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457. Kübler A, Luna B, Larsson C, et al. Mycobacterium tuberculosis dysregulates MMP/TIMP balance to drive rapid cavitation and unrestrained bacterial proliferation. J Pathol. 2015;235(3):431‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458. Reichmann MT, Tezera LB, Vallejo AF, et al. Integrated transcriptomic analysis of human tuberculosis granulomas and a biomimetic model identifies therapeutic targets. J Clin Invest. 2021;131(15):e148136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459. Pollara G, Turner CT, Rosenheim J, et al. Exaggerated IL‐17A activity in human in vivo recall responses discriminates active tuberculosis from latent infection and cured disease. Sci Transl Med. 2021;13(592):eabg7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460. Elkington PTG, Nuttall RK, Boyle JJ, et al. Mycobacterium tuberculosis, but not vaccine BCG, specifically upregulates matrix metalloproteinase‐1. Am J Respir Crit Care Med. 2005;172(12):1596‐1604. [DOI] [PubMed] [Google Scholar]
- 461. Elkington P, Shiomi T, Breen R, et al. MMP‐1 drives immunopathology in human tuberculosis and transgenic mice. J Clin Invest. 2011;121(5):1827‐1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462. Tadokera R, Meintjes GA, Wilkinson KA, et al. Matrix metalloproteinases and tissue damage in HIV‐tuberculosis immune reconstitution inflammatory syndrome. Eur J Immunol. 2014;44(1):127‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463. Elkington PT, Green JA, Emerson JE, et al. Synergistic up‐regulation of epithelial cell matrix metalloproteinase‐9 secretion in tuberculosis. Am J Respir Cell Mol Biol. 2007;37(4):431‐437. [DOI] [PubMed] [Google Scholar]
- 464. Sabir N, Hussain T, Mangi MH, Zhao D, Zhou X. Matrix metalloproteinases: expression, regulation and role in the immunopathology of tuberculosis. Cell Prolif. 2019;52(4):e12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465. Elkington PTG, Friedland JS. Matrix metalloproteinases in destructive pulmonary pathology. Thorax. 2006;61(3):259‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466. Walker NF, Clark SO, Oni T, et al. Doxycycline and HIV infection suppress tuberculosis‐induced matrix metalloproteinases. Am J Respir Crit Care Med. 2012;185(9):989‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467. Xu Y, Wang L, Zimmerman MD, et al. Matrix metalloproteinase inhibitors enhance the efficacy of frontline drugs against Mycobacterium tuberculosis. PLoS Pathog. 2018;14(4):e1006974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468. Ordonez AA, Pokkali S, Kim S, et al. Adjunct antibody administration with standard treatment reduces relapse rates in a murine tuberculosis model of necrotic granulomas. PLoS One. 2018;13(5):e0197474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469. Ordonez AA, Pokkali S, Sanchez‐Bautista J, et al. Matrix metalloproteinase inhibition in a murine model of cavitary tuberculosis paradoxically worsens pathology. J Infect Dis. 2019;219(4):633‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470. Gago G, Diacovich L, Gramajo H. Lipid metabolism and its implication in mycobacteria‐host interaction. Curr Opin Microbiol. 2018;41:36‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471. Conrad M, Kagan VE, Bayir H, et al. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018;32(9‐10):602‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472. Vilchèze C, Jacobs WR. The promises and limitations of N‐acetylcysteine as a potentiator of first‐line and second‐line tuberculosis drugs. Antimicrob Agents Chemother. 2023;65(5):e01703‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473. Mahakalkar SM, Nagrale D, Gaur S, Urade C, Murhar B, Turankar A. N‐acetylcysteine as an add‐on to directly observed therapy short‐I therapy in fresh pulmonary tuberculosis patients: a randomized, placebo‐controlled, double‐blinded study. Perspect Clin Res. 2017;8(3):132‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.The Aurum Institute NPC. A Prospective Randomized Controlled Trial of Adjunctive N‐Acetylcysteine (NAC) in Adult Patients with Pulmonary Tuberculosis: A Sub‐Study of TB Sequel. clinicaltrials.gov; 2021.
- 475.Pavan Kumar N, Moideen K, Nancy A, et al. Plasma eicosanoid levels in tuberculosis and tuberculosis‐diabetes co‐morbidity are associated with lung pathology and bacterial burden. Front Cell Infect Microbiol. 2019;9:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476. Vinhaes CL, Oliveira‐de‐Souza D, Silveira‐Mattos PS, et al. Changes in inflammatory protein and lipid mediator profiles persist after antitubercular treatment of pulmonary and extrapulmonary tuberculosis: a prospective cohort study. Cytokine. 2019;123:154759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477. Mayer‐Barber KD, Andrade BB, Oland SD, et al. Host‐directed therapy of tuberculosis based on interleukin‐1 and type I interferon crosstalk. Nature. 2014;511(7507):99‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid‐derived suppressor cells. Blood. 2011;118(20):5498‐5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479. Byrne ST, Denkin SM, Zhang Y. Aspirin and ibuprofen enhance pyrazinamide treatment of murine tuberculosis. J Antimicrob Chemother. 2007;59(2):313‐316. [DOI] [PubMed] [Google Scholar]
- 480. Mortensen R, Clemmensen HS, Woodworth JS, et al. Cyclooxygenase inhibitors impair CD4 T cell immunity and exacerbate Mycobacterium tuberculosis infection in aerosol‐challenged mice. Commun Biol. 2019;2:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481. Daniel C, Bhakta S. Immunobiology of tubercle bacilli and prospects of immunomodulatory drugs to tackle tuberculosis (TB) and other non‐tubercular mycobacterial infections. Immunobiology. 2022;227(3):152224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482. Dooley DP, Carpenter JL, Rademacher S. Adjunctive corticosteroid therapy for tuberculosis: a critical reappraisal of the literature. Clin Infect Dis. 1997;25(4):872‐887. [DOI] [PubMed] [Google Scholar]
- 483. Gallucci G, Díaz A, Fernandez RDV, et al. Differential expression of genes regulated by the glucocorticoid receptor pathway in patients with pulmonary tuberculosis. Life Sci. 2022;301:120614. [DOI] [PubMed] [Google Scholar]
- 484. Critchley JA, Young F, Orton L, Garner P. Corticosteroids for prevention of mortality in people with tuberculosis: a systematic review and meta‐analysis. Lancet Infect Dis. 2013;13(3):223‐237. [DOI] [PubMed] [Google Scholar]
- 485. Meintjes G, Wilkinson RJ, Morroni C, et al. Randomized placebo‐controlled trial of prednisone for paradoxical tuberculosis‐associated immune reconstitution inflammatory syndrome. AIDS. 2010;24(15):2381‐2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486. Ba X, Garg NJ. Signaling mechanism of poly(ADP‐ribose) polymerase‐1 (PARP‐1) in inflammatory diseases. Am J Pathol. 2011;178(3):946‐955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487. Liaudet L, Pacher P, Mabley JG, et al. Activation of poly(ADP‐Ribose) polymerase‐1 is a central mechanism of lipopolysaccharide‐induced acute lung inflammation. Am J Respir Crit Care Med. 2002;165(3):372‐377. [DOI] [PubMed] [Google Scholar]
- 488. Subbian S, Koo MS, Tsenova L, et al. Pharmacologic inhibition of host phosphodiesterase‐4 improves isoniazid‐mediated clearance of Mycobacterium tuberculosis. Front Immunol. 2016;7:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489. Maiga MC, Ahidjo BA, Maiga M, Bishai WR. Roflumilast, a type 4 phosphodiesterase inhibitor, shows promising adjunctive, host‐directed therapeutic activity in a mouse model of tuberculosis. Antimicrob Agents Chemother. 2015;59(12):7888‐7890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490. Abdalla AE, Li Q, Xie L, Xie J. Biology of IL‐27 and its role in the host immunity against Mycobacterium tuberculosis. Int J Biol Sci. 2015;11(2):168‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491. Sabri A, Grant AV, Cosker K, et al. Association study of genes controlling IL‐12‐dependent IFN‐γ immunity: sTAT4 alleles increase risk of pulmonary tuberculosis in Morocco. J Infect Dis. 2014;210(4):611‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492. Rottenberg ME, Carow B. SOCS3 and STAT3, major controllers of the outcome of infection with Mycobacterium tuberculosis. Semin Immunol. 2014;26(6):518‐532. [DOI] [PubMed] [Google Scholar]
- 493. Jianfang W, Hui W, Le K. LINC00870 regulates Th1/Th2 via the JAK/STAT pathway in peripheral blood mononuclear cells infected with Mycobacterium tuberculosis. Int Immunopharmacol. 2022;102:107188. [DOI] [PubMed] [Google Scholar]
- 494. Gao Y, Basile JI, Classon C, et al. STAT3 expression by myeloid cells is detrimental for the T‐ cell‐mediated control of infection with Mycobacterium tuberculosis. PLoS Pathog. 2018;14(1):e1006809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495. Rossini NO, Dias MVB. Mutations and insights into the molecular mechanisms of resistance of Mycobacterium tuberculosis to first‐line. Genet Mol Biol. 2023;46:e20220261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.