

Abstract
Background
Evaluating the impact of amino acid variants has been a critical challenge for studying protein function and interpreting genomic data. High-throughput experimental methods like deep mutational scanning (DMS) can measure the effect of large numbers of variants in a target protein, but because DMS studies have not been performed on all proteins, researchers also model DMS data computationally to estimate variant impacts by predictors.
Results
In this study, we extended a linear regression-based predictor to explore whether incorporating data from alanine scanning (AS), a widely used low-throughput mutagenesis method, would improve prediction results. To evaluate our model, we collected 146 AS datasets, mapping to 54 DMS datasets across 22 distinct proteins.
Conclusions
We show that improved model performance depends on the compatibility of the DMS and AS assays, and the scale of improvement is closely related to the correlation between DMS and AS results.
Keywords: deep mutational scanning, alanine scanning, machine learning, predictor
Introduction
Deep mutational scanning (DMS) is a functional genomics method that can experimentally measure the impact of many thousands of protein variants by combining high-throughput sequencing with a functional assay [1]. In a typical DMS, a complementary DNA library of genetic variants of a target gene is generated, containing all possible single amino acid substitutions. This variant library is then expressed in a functional assay system where the DMS variants can be selected based on their properties. The change in variant frequency in the pre- and postselection populations is determined by high-throughput sequencing, which is then used to calculate a multiplexed functional score that captures the variant's impact [2–4]. The versatility of DMS assays makes it possible to measure variant impact on a wide range of protein properties, including protein binding affinity [5, 6], protein abundance [7–9], enzyme activity [10, 11], and cell survival [12–14]. So far, hundreds of DMS studies covering tens of thousands of nucleotides have been published [15], and experiments targeting over a hundred additional genes are under way according to MaveRegistry [16].
Computational studies have used DMS data to build predictive models of variant impact. These predictors use supervised or semi-supervised learning models trained on experimental DMS data and various protein features to make predictions [17–23]. Envision is one such method that used protein structural, physicochemical, and evolutionary features to predict variant effect scores and was trained on DMS data from 8 proteins using gradient boosting [17]. Another method, DeMaSk, predicted DMS scores by combining 2 evolutionary features (protein positional conservation and variant homologous frequency) with a DMS substitution matrix and was trained on data from 17 proteins using a linear model [19]. Deep learning algorithms have also been applied to build protein fitness predictors [18, 20], which are usually based only on variant sequences. These variant effect predictors can also be benchmarked using DMS experimental results and assist in the interpretation of experimental data [20, 24, 25].
Low-throughput mutagenesis experiments that measure tens of variants at a time have also been used extensively to study diverse protein properties, including substrate binding affinity [26, 27], protein stability [28, 29], and protein-specific activities [30, 31]. Alanine scanning (AS) is a widely used low-throughput mutagenesis method [32, 33], and AS data are available for many proteins. In this method, each targeted protein residue is substituted with alanine, and the impacts of these variants are measured by a functional assay [34]. AS experiments are typically used to identify functional hot spots or critical residues in the target protein [35, 36] and have been used as a source of independent validation for DMS studies [31, 37–39].
In this study, we explore whether a predictive model can be improved by incorporating low-throughput mutagenesis data (Fig. 1). We find that AS data can increase prediction accuracy and that the improvement is related to the similarity of the functional assays and the correlation of DMS and AS results.
Figure 1:

Workflow for model training and testing. DMS and AS datasets are collected from online resources and are normalized. DMS and AS datasets targeting the same protein are then matched, filtered, and merged. Two predictors are constructed and tested: the first uses DMS data, AS data, and other protein features, and the second uses only DMS data and the same other protein features.
Methods
DMS data collection
DMS data were downloaded from MaveDB [40, 41], which were then filtered and curated. DMS experiments targeting antibody and virus proteins were removed because of their potentially unique functionality. We retrieved the UniProt accession ID of target proteins by searching the protein names or sequences in UniProt [42], and proteins lacking available UniProt ID were also excluded. Datasets that are computationally processed or their wild-type-like and nonsense-like scores (see Normalization) cannot be identified were also filtered out (Supplementary Table S1). All missense variants with only a single amino acid substitution were curated from the DMS studies for our analysis. A total of 130 DMS experiments from 53 studies [5, 6, 9–14, 24, 31, 37–39, 43–80] were collected for our analysis.
Collection of AS data and other features
The following process was used to search for candidate AS studies. Papers were identified by searching on PubMed and Google Scholar for the “alanine scan” or “alanine scanning” together with the name of candidate proteins. While searching in Google Scholar, we included the protein's UniProt ID rather than molecule name as the search term to reduce false positives. Appropriate AS data were collected from the search results. Western blot results were transformed to values by ImageJ if it was the only experimental data available in the study. A total 146 AS experiments were collected from 45 distinct studies [26–28, 30, 31, 81–86, 70, 87–119].
Protein features of Shannon entropy and the logarithm of variant amino acid frequency were downloaded from the DeMaSk online toolkit [19]. The substitution score matrix feature was calculated from the mean of training DMS scores for each of the 380 possible amino acid substitutions before each iteration of cross-validation.
Normalization
DMS and AS datasets were normalized to a common scale using the following approach adapted from previous studies [17, 120]. Let D denote a protein study measuring scores 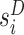 for a single variant i,
for a single variant i, 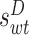 denote the scores for wild-type, and
denote the scores for wild-type, and 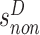 represent the score for nonsense-like variants. The normalized scores
represent the score for nonsense-like variants. The normalized scores 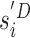 are given by
are given by
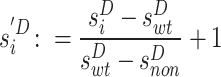 |
Wild-type scores were directly identified from the paper or the median score of synonymous variants. For DMS data, since not all DMS studies report the score of nonsense variants, we defined the nonsense-like scores as the median DMS scores for the 1% missense variants with the strongest loss of function for each dataset. For AS data, nonsense-like scores were defined according to the paper or by using the extreme values (Supplementary Table S1).
AS data filtering and matching
AS data subsets were filtered/matched according to either assay compatibility or score correlation. For assay compatibility filtering, we first categorized each DMS or AS assay by the protein property or function using the following assay types: binding affinity, enzyme activity, protein abundance, cell survival, pathogen infection, drug response, ability to perform a novel function, or other protein-specific activities (e.g., transcription activity for transcription factors) (Supplementary Table S1). The DMS/AS assay pairs were then classified into 3 levels of compatibility based on these categories (Supplementary Fig. S2). For each DMS dataset, we first tried to use only AS data with high assay compatibility for further modeling, removing AS data of medium and low assay compatibility. We then also tried to model with AS data of both high and medium assay compatibility.
For score correlation matching, Spearman's correlation (ρ) is calculated between alanine substitution scores in each pair of AS and DMS data. To avoid influence from the size of AS datasets, we estimated the ρ value with the empirical copula, which is related to the standard estimator by a factor of (n – 1)/(n + 1) [121, 122]:
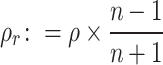 |
where 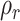 is the regularized correlation coefficient, and n is the number of alanine substitutions used for correlation calculation. For each DMS dataset, the AS result with the highest
is the regularized correlation coefficient, and n is the number of alanine substitutions used for correlation calculation. For each DMS dataset, the AS result with the highest 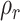 was picked for modeling.
was picked for modeling.
AS data preprocessing
AS data were preprocessed prior to modeling. For variants without available (filtered/matched) AS data, their AS scores were imputed with the mean value of all available AS scores across all studies. Then the AS data were encoded by the wild-type and variant amino acid type with one-hot encoding. For each variant, the AS feature is expanded with 2 one-hot vectors. Each of the vectors has 19 zeros and 1 nonzero value that was the AS score, with the location of the nonzero value indicating the wild-type or variant amino acid type.
Training and evaluation of DMS score predictor
To build the predictors, we performed linear regression using the function sklearn.linear_model.LinearRegression from scikit-learn [123]. Training and validation data were separated with leave-one-protein-out cross-validation. In this process, data from 1 protein were withheld for subsequent validation, and the rest were used for training. This process was iterated over all proteins in the data. Variants were inversely weighted during the training process by the number of measurements available, thus compensating for some regions having greater coverage with DMS and AS assays. Predictors were trained on protein features, DMS data, and (optionally) AS data using 4 different filtering or matching strategies: (i) all DMS/AS data, (ii) compatibility-filtered DMS/AS data, (iii) correlation-matched DMS/AS data, and (iv) a control, constructed using DMS data only.
In the evaluation process, let V be protein variants assayed by both DMS study D and AS study A. Variant scores are predicted by the previously mentioned predictors either using AS data (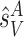 ) or not (
) or not (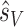 ). Spearman's correlation (ρ) was calculated between the DMS scores
). Spearman's correlation (ρ) was calculated between the DMS scores 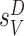 and each set of predicted scores. The difference of ρ was used to evaluate the performance change (
and each set of predicted scores. The difference of ρ was used to evaluate the performance change (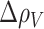 ).
).
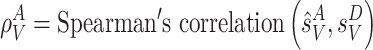 |
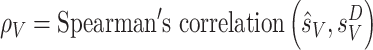 |
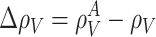 |
To evaluate, we iterated over variants from each pair of DMS/AS studies. Results were dropped for variants V with only 1 protein residue available during analysis and visualization. Model performance was compared using the following statistical tests. Results in Fig. 5 and Supplementary Fig. S5 were tested with Welch's test, and results in Supplementary Figs. S4 and S6 were tested with paired t-tests. The P values were jointly corrected using the Holm–Šidák method. The 95% confidence interval of median values was calculated by Gaussian-based asymptotic approximation [124].
Figure 5:

Performance of variant impact prediction is improved using AS data with high assay compatibility. The change in prediction ρ achieved by including the AS data feature for each DMS and AS data pair is shown as box plots. A higher value represents higher prediction accuracy achieved for using AS data. Different approaches to filtering/matching the data are shown on the x-axis: “All AS data” used all available data; “Compatibility filtered” used only data of high assay compatibility; “Correlation matched” used only data with the highest regularized correlation for each DMS dataset. Results for data pairs with only 1 residue are not shown. P values were calculated using Welch's test and jointly corrected using Holm–Šidák (Methods), *P < 0.05. Notches show the 95% confidence interval around the median, and whiskers show the full value range.
Prediction with other variant effect predictors
For PROVEAN [125] and SIFT [126], prediction results on target variants were directly downloaded from the precalculated database for PROVEAN. For PolyPhen-2 [127] and GEMME [128], variant scores were computed through their online toolkits, using the default settings. ESM-1v [129] was set up locally and run according to its examples and documentations. EVE [130] results were collected from their precalculated database and a benchmarking study [131].
Results
Overview of DMS and AS data
To build the predictive model, 130 DMS datasets were collected from MaveDB [40, 41] (Supplementary Table S1). We searched the literature and found 146 AS datasets targeting the same proteins as 54 of the DMS datasets. In total, we obtained both DMS and AS data for 22 different proteins: 17 human proteins, 3 yeast proteins, and 2 bacterial proteins. Most DMS experiments were highly complete, with a mean coverage of 95.0% of all possible single amino acid substitutions assayed in the target region, comprising 373,219 total protein variant measurements. AS data were only available on a small number of protein residues (Fig. 2), and we were able to curate 1,480 alanine substitution scores from the 146 studies. Variant scores from collected DMS and AS studies were linearly normalized to a common scale (see Methods) to make them comparable across datasets (Supplementary Fig. S1).
Figure 2:

DMS data generally cover more protein residues than AS data. Each bar shows the number of residues assayed by DMS studies on given target proteins. Color indicates the number of AS studies available for the DMS-tested residues.
The correlation of DMS and AS scores is related to assay compatibility
To evaluate the similarity of AS and DMS scores, we calculated Spearman's correlation (ρ) between the AS scores and DMS scores for the same alanine substitutions. Since each protein may have results from several AS and DMS experiments, we calculated ρ between each possible pair. The median ρ over DMS and AS data (DMS/AS) pairs was 0.2, indicating that the experimental scores were poorly correlated overall (Fig. 3).
Figure 3:

Correlation between DMS and AS data shows substantial variation. We calculated Spearman's ρ between alanine substitution scores in each pair of AS and DMS data. The results for pairs with fewer than 3 alanine substitutions are not shown. The red dashed line shows the median ρ.
We then considered if differences between AS and DMS assay designs might contribute to this low agreement between scores. To explore this, we developed a decision tree (Supplementary Fig. S2) to classify whether DMS/AS pairs had low, medium, or high assay compatibility, which we defined as a similarity measurement of the functional assays performed. For example, the DMS assay measuring the binding affinity of a cell surface protein, CXCR4, to its natural ligand [43] has high compatibility with the AS experiment also measuring this ligand binding but has low compatibility with the study on CXCR4’s ability to facilitate virus infection [81]. A full assay compatibility table can be found in Supplementary Table S1 with the compatibility classifications and justification for each pair. We then compared DMS and AS score correlation for each compatibility class and found that score correlations were closely related to assay compatibility. Data from low-compatibility assays had a median correlation of 0.15, rising to 0.19 for medium-compatibility assays and 0.40 for high-compatibility assays (Fig. 4). This trend of increased correlation for high-compatibility assay pairs holds across secondary structures (Supplementary Table S4). This link between assay compatibility and score correlation indicates that our decision tree approach was able to capture the similarity between assay systems.
Figure 4:

DMS and AS data pairs with high assay compatibility show a higher score correlation. Each box shows the Spearman's ρ between DMS and AS data pairs for each level of assay compatibility or overall. The correlation coefficients were calculated between alanine substitution scores in each pair of AS and DMS datasets. Results for pairs with fewer than 3 alanine substitutions were removed. P values calculated using Welch's test and corrected using Holm–Šidák, *P < 0.05; notches show 95% confidence interval around median, and whiskers show the full value range.
Compatible AS data improve DMS score prediction accuracy
To test if incorporating AS data into DMS score models would improve prediction accuracy, we decided to build a new model based on DeMaSk [19]. We chose DeMaSk because it showed better performance compared to similar methods and was straightforward to modify. The published DeMaSk model predicts DMS scores using protein positional conservation, variant homologous frequency, and substitution score matrix, and we incorporated AS data as an additional feature. Our new predictor was modeled with all 130 DMS we collected, and we applied a leave-one-protein-out cross-validation approach to training and testing, avoiding information leakage for variants of the same protein target [17]. Prediction performance was evaluated using the Spearman's correlation (ρ) between the experimentally derived DMS scores and the predicted scores for each pair of DMS and AS studies. The performance of our DMS/AS model was compared with a model trained only on DMS data, equivalent to retrained DeMaSk (Supplementary Fig. S3), by calculating the change of prediction ρ (see Methods).
We trained our model with either all or a subset of AS data we collected (Fig. 5, Supplementary Table S5). We first integrated all 146 AS data collected for training and evaluation but observed only a modest improvement of prediction ρ (Fig. 5, left box, and Supplementary Fig. S4). We then retrained and evaluated our model on filtered AS data with only high-compatibility assays and observed a median increase in prediction Spearman's ρ of 0.1 compared to the results with no AS data (Fig. 5, middle box, and Supplementary Fig. S4). However, training with both high- and medium-compatibility pairs reduced the performance improvement (Supplementary Fig. S5). These results indicate that medium- and low-compatibility pairs might provide inconsistent training data, degrading model performance. We also evaluated the impact of including high-compatibility AS data in an alternative model based on Envison [17, 132] and found similar results (Supplementary Fig. S6 and Supplementary Information). To differentiate between high assay compatibility and high DMS/AS score correlation, we trained the model using the most highly correlated AS result for each DMS dataset (see Methods). Although the upper quartile was high, the median performance change of this predictor was lower than the high assay compatibility model, suggesting that matching with the highest score correlation alone is insufficient (Fig. 5, right box). However, when applying a stricter threshold, the correlation matched models still show limited improvement (Supplementary Fig. S7). Additionally, to ensure the models performance is not biased by pseudo-replication of multiple datasets, we averaged DMS and AS scores that were part of the same study and type of assay and saw similar results (Supplementary Fig. S8).
Our compatibility-filtered predictor shows improved prediction accuracy for these regions compared to not only the baseline model but other widely used predictors as well (Supplementary Fig. S9). To further explore the higher performance of this compatibility-filtered predictor, we examined the relationship between prediction ρ change and score correlation for each high-compatibility DMS/AS pair (Fig. 6). For most pairs, prediction performance was improved by using AS data, and the scale of improvement was also related to the score correlation. This relationship could also be observed for multiple DMS/AS pairs from an individual protein, such as CXCR4 and CCR5. We saw the same trend in the predictor trained with all DMS/AS pairs but noted that the performance even of highly correlated pairs was worse, likely due to the influence of low-compatibility training data on the model (Supplementary Fig. S10).
Figure 6:

Prediction performance change is related to DMS and AS score correlation. Each dot represents a filtered DMS/AS data pair of high assay compatibility. The vertical axis shows the change of prediction ρ by using AS data (larger means higher performance achieved by using AS data). The horizontal axis shows the DMS/AS score correlation for all variants on the matched residues rather than just alanine substitutions. The colors and shapes of the dots correspond to the target protein, and size indicates the number of variants in each data pair. Results for data pairs with only 1 residue are not shown.
We also explored the consequences of the sparsity of AS data on our model in 3 ways: (i) by training only with variants that have AS data available (Supplementary Fig. S11), (ii) by using a boosting approach that focuses only on residues with AS data (Supplementary Fig. S12 and Supplementary Information), and (iii) by using complete alanine substitution information from DMS as the AS feature (Supplementary Fig. S13 and Supplementary Information). The first approach gave lower absolute prediction performance, presumably because the model was underfitted due to the small number of variants. The last 2 approaches performed very similarly to the primary model constructed using high-compatibility DMS/AS data and simple mean score imputation.
To test the influence of amino acids on our predictor, we grouped the prediction results by either wild-type or variant amino acid and calculated the prediction improvement when AS data were included (Fig. 7). We found that 14 of 19 wild-type amino acids performed better with the addition of AS data, with cysteine showing the largest improvement and performing worst in the model lacking AS data. Eighteen of 20 variant amino acids benefited from the inclusion of AS data, with marginal performance decrease on lysine and aspartic acid (|Δρ| < 0.01) (Fig. 7). We also noticed that variants to alanine are not most improved, but we observed an overall trend showing higher improvement for amino acids that are physiochemically similar to alanine (Supplementary Fig. S15).
Figure 7:

Model performance is generally improved for each wild-type and variant amino acid. Prediction Spearman's ρ when using (y-axis) or not using (x-axis) AS data on each wild-type (A) or variant (B) amino acid is shown in the scatterplots. The results are colored according to the property of each amino acid type. Alanine (A) result is not applicable in the first figure since alanine scanning data are always missing when the wild-type is alanine itself. Absolute count for each amino acid can be found in Supplementary Fig. S14. Neg., negatively; Pos., positively.
Discussion
In this study, we integrated AS data into DMS score prediction, leading to modest improvements in the accuracy of variant score prediction. We also explored the impact of the diversity of protein properties measured by DMS and AS. Filtering DMS and AS data based on our manual classification of assay type compatibility led to improved prediction performance.
A potential shortcoming of our current approach is that AS data were available for only a small proportion of the DMS data. Although most recent DMS studies can analyze variants of the whole protein, most AS experiments only cover a handful of residues in the target protein, leaving missing AS scores for the vast majority of residues. We explored this here and found that alternative methods for addressing the sparsity of AS data did not improve or degrade performance, but we anticipate further improved prediction accuracy if the low completeness and unevenness of AS data are appropriately handled before modeling.
In this study, we identified the importance of DMS/AS assay compatibility as a crucial factor for improving prediction accuracy. An issue with using this concept is that it further shrinks already sparse data. It also fails to take advantage of the fact that even for low-compatible assays, some fundamental information like protein abundance can still be mutually captured. Instead of hard filtering, proper implementation of this underlying information may facilitate variant impact prediction in the future. Nonetheless, filtering on assay compatibility still leads to performance improvement. We also briefly explored whether the consistency of DMS and AS scores can be considered more directly by matching the best-correlated AS data for each DMS dataset. Consistency is partially driven by assay compatibility but also reflects other features of the data, such as bias and noise.
The concepts of compatibility and data quality are also relevant to training any DMS-based predictors. DMS assays have been developed to measure variant impacts to distinct protein properties, and a variant can behave similarly to wild-type when measured by one assay yet show altered protein properties in other assay results, which are frequently found in regions with specific biochemical functions [25, 133–137]. With more experimental assays to be applied, the diverse measurements may impede the progress of future DMS-based predictors unless this assay effect is properly addressed, for example, by building assay-specific predictors. Measurement error is another source of DMS data heterogeneity that potentially affects the model performance. In our current study, DMS scores of protein variants are weighted equally while training. Adjustable weighting can be applied in future studies to adapt the distinct experimental error between individual variants and datasets, reducing the influence of low-confident data.
In summary, we conclude that the careful inclusion of low-throughput mutagenesis data improves the prediction of DMS scores, and the approaches described here can potentially be applied to other prediction methods.
Availability of Supporting Source Code and Requirements
Project name: DMS_with_Alanine_scan
Project homepage: https://github.com/PapenfussLab/DMS_with_Alanine_scan
Operating system: Platform independent
Programming language: Python
Other requirements: Python 3.10 or higher
License: MIT license
RRID: SCR_023949
Supplementary Material
Joseph Ng -- 3/21/2023 Reviewed
Joseph Ng -- 7/12/2023 Reviewed
Leopold Parts -- 4/7/2023 Reviewed
Leopold Parts -- 7/21/2023 Reviewed
Contributor Information
Yunfan Fu, The Walter and Eliza Hall Institute of Medical Research, Bioinformatics Division, 1G Royal Pde, Parkville, Victoria 3052, Australia; The University of Melbourne, Department of Medical Biology, Parkville, Victoria 3010, Australia.
Justin Bedő, The Walter and Eliza Hall Institute of Medical Research, Bioinformatics Division, 1G Royal Pde, Parkville, Victoria 3052, Australia; The University of Melbourne, Department of Medical Biology, Parkville, Victoria 3010, Australia.
Anthony T Papenfuss, The Walter and Eliza Hall Institute of Medical Research, Bioinformatics Division, 1G Royal Pde, Parkville, Victoria 3052, Australia; The University of Melbourne, Department of Medical Biology, Parkville, Victoria 3010, Australia; Peter MacCallum Cancer Centre, Melbourne, Victoria 3000, Australia.
Alan F Rubin, The Walter and Eliza Hall Institute of Medical Research, Bioinformatics Division, 1G Royal Pde, Parkville, Victoria 3052, Australia; The University of Melbourne, Department of Medical Biology, Parkville, Victoria 3010, Australia.
Data Availability
A copy of the data analysis code and a full set of data files required to reproduce this work are openly available in the GigaScience repository, GigaDB, under the record described in [138]. MaveDB accession numbers, UniProt accession numbers, and other metadata describing the matched DMS-AS datasets are listed in Supplementary Table S1 (see supporting information).
Additional Files
Supplementary Table S1. All candidate DMS and alanine scanning data with detailed dataset information.
Supplementary Table S2. Normalized DMS dataset with protein property features.
Supplementary Table S3. Normalized alanine scanning dataset.
Supplementary Table S4. DMS/AS correlation on each secondary structural region.
Supplementary Table S5. Amount of data with AS scores available.
Supplementary Fig. S1. DMS and AS score distribution. The figure shows the kernel estimated density of normalized AS scores and DMS scores for variants with or without available AS data.
Supplementary Fig. S2. Decision tree for classifying DMS and AS assay compatibility. The similarity of DMS and AS assays is compared (Methods) and the DMS/AS assay pairs are classified using 3 levels of compatibility (low, medium, high). The leaf-node text and color show the classified assay compatibility. The number indicates the count of assay pairs for each compatibility level.
Supplementary Fig. S3. Comparison between published and reimplemented predictors. The plot shows leave-one-protein-out cross-validation performance on predictors built from the published DeMaSk code or our code. The predictors were trained and evaluated on DMS data either provided by the DeMaSk study or curated by our own. The “DeMaSk data & code” result is similar to the published result. For the “Our data & DeMaSk code” result, we used our own data and published code, which shows a median performance around 0.35. This is probably because many more DMS results are included in our data. The similarity of results achieved using “Our data & code” demonstrates the correctness of our reimplementation. (Whiskers show the full value range.)
Supplementary Fig. S4. Performance comparison between predictors with or without AS data. The Spearman's ρ between DMS scores and predicted scores for each DMS and AS data pair are shown as box plots. Different approaches to filtering the data are shown on the x-axis: “All AS data” used all available data; “Compatibility filtered” used only data of high assay compatibility; “Correlation matched” used only data with the highest regularized correlation for each DMS dataset. The figure does not include data without available AS scores. This means that the different results are not directly comparable since they are computed for different subsets of DMS/AS data pairs (for example, “All AS data” contains all DMS/AS data pairs, but “Compatibility filtered” contains only data pairs of high assay compatibility). Control results are shown as green boxes for predictions on the same residues without AS data as a feature. The underlying ρ for each data pair in the control results is the same, but the boxes are shifted due to data filtering. Results for data pairs with only 1 residue are not shown. P values were calculated using paired t-test and jointly corrected using Holm–Šidák (Methods), *P < 0.05. Notches show the 95% confidence interval around the median, and whiskers show the full value range.
Supplementary Fig. S5. The change in prediction performance for using data of different assay compatibility levels. The change of prediction Spearman's ρ for each DMS and AS data pair is shown as box plots. A higher value represents higher prediction accuracy achieved for using AS data. Different data-filtering methods are shown on the x-axis. Results for data pairs with only 1 residue are not shown. P values were calculated using Welch's test and jointly corrected using Holm–Šidák (Methods), *P < 0.05. Notches show the 95% confidence interval around the median, and whiskers show the full value range.
Supplementary Fig. S6. Prediction performance is improved while incorporating high-compatibility AS data into the Envision model. The Spearman's ρ between experiment DMS scores and predicted scores for each DMS/AS assay pair with high compatibility are shown as box plots. The x-axis shows the predictor used, either Envision or DeMaSk. Control results are shown as green boxes for predictions on the same residues without AS data as a feature. Results for data pairs with only 1 residue are not shown. P values were calculated using paired t-test and jointly corrected using Holm–Šidák (Methods), *P < 0.05. Notches show the 95% confidence interval around the median, and whiskers show the full value range.
Supplementary Fig. S7. Performance improvement on thresholded correlation matching. The change of prediction ρ for each DMS and AS data pair is shown as box plots. Different approaches to filtering/matching the data are shown on the x-axis: “All AS data,” “Compatibility filtered,” and “Correlation matched” are the same results as previously discussed; while doing correlation matching, a further thresholding (0, 0.25, or 0.5) on the regularized DMS/AS correlation values (ρr) was applied. Notches show the 95% confidence interval around the median, and whiskers show the full value range.
Supplementary Fig. S8. Performance improvement on averaged DMS/AS testing data. This figure shows model performance when we averaged variant scores for DMS or AS data that are (i) published in the same paper, (ii) targeting the same protein region, and (iii) measured by the same type of assays (Supplementary Table S1). The change of prediction ρ for each averaged DMS and AS data pair is shown. A higher value represents higher prediction accuracy achieved when using AS data. Different approaches to filtering/matching the data are shown on the x-axis: “All AS data” used all available data; “Compatibility filtered” used only data of high assay compatibility; “Correlation matched” used only data with the highest regularized correlation for each DMS dataset. Results for data pairs with only 1 residue are not shown. Notches show the 95% confidence interval around the median, and whiskers show the full value range.
Supplementary Fig. S9. Model performance on various variant effect predictors. The Spearman's ρ between DMS scores and predicted scores from different variant effect predictors for each DMS and AS pair are shown as box plots. Results are evaluated on different sets of variant data shown on the x-axis: “All AS data” used all available data; “Compatibility filtered” used only data of high assay compatibility; “Correlation matched” used only AS data with the highest regularized correlation for each DMS dataset. The figure does not include residues without available AS scores. Results for data pairs with only 1 residue are not shown. Notches show the 95% confidence interval around the median, and whiskers show the full value range.
Supplementary Fig. S10. Prediction performance change for using all AS data. Each dot represents a DMS/AS data pair. The vertical axis shows the change of prediction ρ by using AS data (larger means higher performance achieved by using AS data). The horizontal axis shows the DMS/AS score correlation for all variants on the matched residues rather than just alanine substitutions. The colors and shapes of the dots correspond to the target protein, and size indicates the number of variants in each data pair. Results for data pairs with only 1 residue are not shown.
Supplementary Fig. S11. Model performance for training with AS data-available residues. The predictors were trained only on variants that have AS data available. Panel A shows the performance visualized by prediction Spearman's ρ for DMS scores and predicted scores for each DMS and AS data pair. Different approaches to filtering the data are shown on the x-axis: “All AS data” used all available data; “Compatibility filtered” used only data of high assay compatibility; “Correlation matched” used only AS data with the highest regularized correlation for each DMS dataset. Control results are shown as green boxes for predictions on the same residues without AS data as a feature. Panel B shows change of prediction ρ for each DMS and AS data pair. A higher value indicates higher prediction accuracy achieved when using AS data. Different approaches to filtering the data are also shown on the x-axis as described. Notches show the 95% confidence interval around the median, and whiskers show the full value range.
Supplementary Fig. S12. Boosting setup shows similar performance as the main result. Each dot represents a filtered DMS/AS data pair of high assay compatibility. The vertical and horizontal axes show the prediction Spearman's ρ for either modeled with boosting or the 1-step (main result) setup. The colors and shapes of the dots correspond to the target protein, and size indicates the number of variants in each data pair.
Supplementary Fig. S13. Training with DMS scores of alanine substitutions shows similar performance as the main result. The vertical and horizontal axes show the prediction Spearman's ρ for predictors either trained with DMS score of alanine substitutions (DMS-Ala) or AS data of high assay compatibility (main result), yet all evaluated on high-compatibility AS data. The colors and shapes of the dots correspond to the target protein, and size indicates the number of variants in each data pair.
Supplementary Fig. S14. Count of variant entries for each wild-type or variant amino acid of high assay compatibility data. Neg., negatively; Pos., positively.
Supplementary Fig. S15. Relationship between amino acid similarity and model performance. For each amino acid, its similarity to alanine was computed by their DMS score correlation or using BLOSUM scores as shown on the x-axis. The performance improvement (Δρ) for each wild-type (left) or variant (right) amino acid while using AS data was computed as previously mentioned (Fig. 7), with their Spearman's correlation against the similarity measurements shown in the figure. The label for each amino acid is colored by the amino acid physicochemical property. Neg., negatively; Pos., positively.
Supplementary Information. Alternative baseline predictor and approaches to incoporate AS data.
Abbreviations
AS: alanine scanning; DMS: deep mutational scanning.
Competing Interests
The authors declare that they have no competing interests.
Funding
Y.F. is supported by the Melbourne Research Scholarship. A.T.P. was supported by an Australian National Health and Medical Research Council (NHMRC) Senior Research Fellowship (1116955). J.B., A.F.R., and A.T.P. were supported by the Lorenzo and Pamela Galli Medical Research Trust. J.B. and A.T.P. were supported by the Stafford Fox Medical Research Foundation. A.F.R. was supported by the National Human Genome Research Institute of the National Institutes of Health under award numbers RM1HG010461 and UM1HG011969. The research benefited from support from the Victorian State Government Operational Infrastructure Support and Australian Government NHMRC Independent Research Institute Infrastructure Support. This project received grant funding from the Australian Government.
Authors’ Contributions
Y.F. developed the software and wrote the initial draft of the manuscript. A.F.R. conceived the study. J.B., A.F.R., and A.T.P. oversaw the project. All authors reviewed, contributed to, and approved the manuscript.
References
- 1. Fowler DM, Fields S. Deep mutational scanning: a new style of protein science. Nat Methods. 2014;11:801–7.. 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Findlay GM. Linking genome variants to disease: scalable approaches to test the functional impact of human mutations. Hum Mol Genet. 2021;30:R187–97.. 10.1093/hmg/ddab219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Geck RC, Boyle G, Amorosi CJ, et al. Measuring pharmacogene variant function at scale using multiplexed assays. Annu Rev Pharmacol Toxicol. 2022;62:531–50.. 10.1146/annurev-pharmtox-032221-085807. [DOI] [PubMed] [Google Scholar]
- 4. Weile J, Roth FP. Multiplexed assays of variant effects contribute to a growing genotype–phenotype atlas. Hum Genet. 2018;137:665–78.. 10.1007/s00439-018-1916-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Diss G, Lehner B. The genetic landscape of a physical interaction. eLife. 2018;7:e32472. 10.7554/eLife.32472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fowler DM, Araya CL, Fleishman SJ, et al. High-resolution mapping of protein sequence-function relationships. Nat Methods. 2010;7:741–6.. 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Amorosi CJ, Chiasson MA, McDonald MG, et al. Massively parallel characterization of CYP2C9 variant enzyme activity and abundance. Am Hum Genet. 2021;108:1735–51.. 10.1016/j.ajhg.2021.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Faure AJ, Domingo J, Schmiedel JM, et al. Mapping the energetic and allosteric landscapes of protein binding domains. Nature. 2022;604:175–83.. 10.1038/s41586-022-04586-4. [DOI] [PubMed] [Google Scholar]
- 9. Matreyek KA, Starita LM, Stephany JJ, et al. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat Genet. 2018;50:874–82.. 10.1038/s41588-018-0122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mighell TL, Evans-Dutson S, O'Roak BJ. A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotype relationships. Am Hum Genet. 2018;102:943–55.. 10.1016/j.ajhg.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stiffler MA, Hekstra DR, Ranganathan R. Evolvability as a function of purifying selection in TEM-1 β-lactamase. Cell. 2015;160:882–92.. 10.1016/j.cell.2015.01.035. [DOI] [PubMed] [Google Scholar]
- 12. Ahler E, Register AC, Chakraborty S, et al. A combined approach reveals a regulatory mechanism coupling Src's kinase activity, localization, and phosphotransferase-independent functions. Mol Cell. 2019;74:393–408.. e20. 10.1016/j.molcel.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Giacomelli AO, Yang X, Lintner RE, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet. 2018;50:1381–7.. 10.1038/s41588-018-0204-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roscoe BP, Thayer KM, Zeldovich KB, et al. Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J Mol Biol. 2013;425:1363–77.. 10.1016/j.jmb.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tabet D, Parikh V, Mali P, et al. Scalable functional assays for the interpretation of human genetic variation. Annu Rev Genet. 2022;56:441–65.. 10.1146/annurev-genet-072920-032107. [DOI] [PubMed] [Google Scholar]
- 16. Kuang D, Weile J, Kishore N, et al. MaveRegistry: a collaboration platform for multiplexed assays of variant effect. Bioinformatics. 2021;37:3382–3.. 10.1093/bioinformatics/btab215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gray VE, Hause RJ, Luebeck J, et al. Quantitative missense variant effect prediction using large-scale mutagenesis data. Cell Syst. 2018;6:116–24..e3. 10.1016/j.cels.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alley EC, Khimulya G, Biswas S, et al. Unified rational protein engineering with sequence-based deep representation learning. Nat Methods. 2019;16:1315–22.. 10.1038/s41592-019-0598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Munro D, Singh M. DeMaSk: a deep mutational scanning substitution matrix and its use for variant impact prediction. Bioinformatics. 2021;36:5322–9.. 10.1093/bioinformatics/btaa1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Biswas S, Khimulya G, Alley EC, et al. Low-N protein engineering with data-efficient deep learning. Nat Methods. 2021;18:389–96.. 10.1038/s41592-021-01100-y. [DOI] [PubMed] [Google Scholar]
- 21. Høie MH, Cagiada M, Beck Frederiksen AH, et al. Predicting and interpreting large-scale mutagenesis data using analyses of protein stability and conservation. Cell Rep. 2022;38:110207. 10.1016/j.celrep.2021.110207. [DOI] [PubMed] [Google Scholar]
- 22. Wu Y, Li R, Sun S, et al. Improved pathogenicity prediction for rare human missense variants. Am Hum Genet. 2021;108:1891–906.. 10.1016/j.ajhg.2021.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hsu C, Nisonoff H, Fannjiang C, et al. Learning protein fitness models from evolutionary and assay-labeled data. Nat Biotechnol. 2022;40:1114–22.. 10.1038/s41587-021-01146-5. [DOI] [PubMed] [Google Scholar]
- 24. Findlay GM, Daza RM, Martin B, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562:217–22.. 10.1038/s41586-018-0461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cagiada M, Bottaro S, Lindemose S, et al. Discovering functionally important sites in proteins. Nat Commun. 2023;14:4175. 10.1038/s41467-023-39909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Block C, Janknecht R, Herrmann C, et al. Quantitative structure-activity analysis correlating ras/raf interaction in vitro to raf activation in vivo. Nat Struct Mol Biol. 1996;3:244–51.. 10.1038/nsb0396-244. [DOI] [PubMed] [Google Scholar]
- 27. Sloan DJ, Hellinga HW. Dissection of the protein G B1 domain binding site for human IgG Fc fragment. Protein Sci. 1999;8:1643–8.. 10.1110/ps.8.8.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fleming KG, Engelman DM. Specificity in transmembrane helix–helix interactions can define a hierarchy of stability for sequence variants. Proc Natl Acad Sci USA. 2001;98:14340–4.. 10.1073/pnas.251367498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shibata Y, White JF, Serrano-Vega MJ, et al. Thermostabilization of the neurotensin receptor NTS1. J Mol Biol. 2009;390:262–77.. 10.1016/j.jmb.2009.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brzovic PS, Heikaus CC, Kisselev L, et al. The acidic transcription activator Gcn4 binds the mediator subunit Gal11/Med15 using a simple protein interface forming a fuzzy complex. Mol Cell. 2011;44:942–53.. 10.1016/j.molcel.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gajula KS, Huwe PJ, Mo CY, et al. High-throughput mutagenesis reveals functional determinants for DNA targeting by activation-induced deaminase. Nucleic Acids Res. 2014;42:9964–75.. 10.1093/nar/gku689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kortemme T, Kim DE, Baker D. Computational alanine scanning of protein-protein interfaces. Sci STKE. 2004;2004:pl2. 10.1126/stke.2192004pl2. [DOI] [PubMed] [Google Scholar]
- 33. Morrison KL, Weiss GA. Combinatorial alanine-scanning. Curr Opin Chem Biol. 2001;5:302–7.. 10.1016/S1367-5931(00)00206-4. [DOI] [PubMed] [Google Scholar]
- 34. Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–5.. 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 35. DeLano WL. Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol. 2002;12:14–20.. 10.1016/S0959-440X(02)00283-X. [DOI] [PubMed] [Google Scholar]
- 36. Eustache S, Leprince J, Tufféry P. Progress with peptide scanning to study structure-activity relationships: the implications for drug discovery. Expert Opin Drug Discov. 2016;11:771–84.. 10.1080/17460441.2016.1201058. [DOI] [PubMed] [Google Scholar]
- 37. Olson CA, Wu NC, Sun R. A comprehensive biophysical description of pairwise epistasis throughout an entire protein domain. Curr Biol. 2014;24:2643–51.. 10.1016/j.cub.2014.09.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Staller MV, Holehouse AS, Swain-Lenz D, et al. A high-throughput mutational scan of an intrinsically disordered acidic transcriptional activation domain. Cell Syst. 2018;6:444–55..e6. 10.1016/j.cels.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gray VE, Sitko K, Kameni FZN, et al. Elucidating the molecular determinants of aβ aggregation with deep mutational scanning. G3 (Bethesda). 2019;9:3683–9.. 10.1534/g3.119.400535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Esposito D, Weile J, Shendure J et al. MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol. 2019;20:223. 10.1186/s13059-019-1845-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rubin AF, Min JK, Rollins NJ, et al. MaveDB v2: a curated community database with over three million variant effects from multiplexed functional assays. Biorxiv. 10.1101/2021.11.29.470445. [DOI] [Google Scholar]
- 42. The UniProt Consortium, Bateman A, Martin M-J, et al. UniProt: the Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–9.. 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Heredia JD, Park J, Brubaker RJ, et al. Mapping interaction sites on human chemokine receptors by deep mutational scanning. J Immunol. 2018;200:3825–39.. 10.4049/jimmunol.1800343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Andrews B, Fields S. Distinct patterns of mutational sensitivity for λ resistance and maltodextrin transport in Escherichia coli LamB. Microb Genom. 2020;6:e000364. 10.1099/mgen.0.000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bandaru P, Shah NH, Bhattacharyya M, et al. Deconstruction of the Ras switching cycle through saturation mutagenesis. eLife. 2017;6:e27810. 10.7554/eLife.27810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bolognesi B, Faure AJ, Seuma M, et al. The mutational landscape of a prion-like domain. Nat Commun. 2019;10:4162. 10.1038/s41467-019-12101-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bridgford JL, Lee SM, Lee CMM, et al. Novel drivers and modifiers of MPL-dependent oncogenic transformation identified by deep mutational scanning. Blood. 2020;135:287–92.. 10.1182/blood.2019002561. [DOI] [PubMed] [Google Scholar]
- 48. Chan KK, Dorosky D, Sharma P, et al. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science. 2020;369:1261–5.. 10.1126/science.abc0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chiasson MA, Rollins NJ, Stephany JJ, et al. Multiplexed measurement of variant abundance and activity reveals VKOR topology, active site and human variant impact. eLife. 2020;9:e58026. 10.7554/eLife.58026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Elazar A, Weinstein J, Biran I, et al. Mutational scanning reveals the determinants of protein insertion and association energetics in the plasma membrane. eLife. 2016;5:e12125. 10.7554/eLife.12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Firnberg E, Labonte JW, Gray JJ, et al. A comprehensive, high-resolution map of a gene's fitness landscape. Mol Biol Evol. 2014;31:1581–92.. 10.1093/molbev/msu081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hietpas RT, Jensen JD, Bolon DNA. Experimental illumination of a fitness landscape. Proc Natl Acad Sci USA. 2011;108:7896–901.. 10.1073/pnas.1016024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hietpas RT, Bank C, Jensen JD et al. Shifting fitness landscapes in response to altered environments. Evolution. 2013;67:3512–22.. 10.1111/evo.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jiang L, Mishra P, Hietpas RT, et al. Latent effects of Hsp90 mutants revealed at reduced expression levels. PLoS Genet. 2013;9:e1003600. 10.1371/journal.pgen.1003600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jiang RJ. Exhaustive Mapping of Missense Variation in Coronary Heart Disease-related Genes. [Thesis]. University of Toronto. [Google Scholar]
- 56. Keskin A, Akdoğan E, Dunn CD. Evidence for amino acid snorkeling from a high-resolution, in vivo analysis of Fis1 tail-anchor insertion at the mitochondrial outer membrane. Genetics. 2017;205:691–705.. 10.1534/genetics.116.196428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kitzman JO, Starita LM, Lo RS, et al. Massively parallel single-amino-acid mutagenesis. Nat Methods. 2015;12:203–6.. 10.1038/nmeth.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kotler E, Shani O, Goldfeld G, et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol Cell. 2018;71:178–90.. e8. 10.1016/j.molcel.2018.06.012. [DOI] [PubMed] [Google Scholar]
- 59. Kowalsky CA, Whitehead TA. Determination of binding affinity upon mutation for type I dockerin–cohesin complexes from Clostridium thermocellum and Clostridium cellulolyticum using deep sequencing. Proteins. 2016;84:1914–28.. 10.1002/prot.25175. [DOI] [PubMed] [Google Scholar]
- 60. McLaughlin RN Jr, Poelwijk FJ, Raman A, et al. The spatial architecture of protein function and adaptation. Nature. 2012;491:138–42.. 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Melamed D, Young DL, Gamble CE, et al. Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)-binding protein. RNA. 2013;19:1537–51.. 10.1261/rna.040709.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mishra P, Flynn JM, Starr TN, et al. Systematic mutant analyses elucidate general and client-specific aspects of Hsp90 function. Cell Rep. 2016;15:588–98.. 10.1016/j.celrep.2016.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nedrud D, Coyote-Maestas W, Schmidt D. A large-scale survey of pairwise epistasis reveals a mechanism for evolutionary expansion and specialization of PDZ domains. Proteins. 2021;89:899–914.. 10.1002/prot.26067. [DOI] [PubMed] [Google Scholar]
- 64. Newberry RW, Arhar T, Costello J, et al. Robust sequence determinants of α-synuclein toxicity in yeast implicate membrane binding. ACS Chem Biol. 2020;15:2137–53.. 10.1021/acschembio.0c00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Newberry RW, Leong JT, Chow ED, et al. Deep mutational scanning reveals the structural basis for α-synuclein activity. Nat Chem Biol. 2020;16:653–9.. 10.1038/s41589-020-0480-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Roscoe BP, Bolon DNA. Systematic exploration of ubiquitin sequence, E1 activation efficiency, and experimental fitness in yeast. J Mol Biol. 2014;426:2854–70.. 10.1016/j.jmb.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sarkisyan KS, Bolotin DA, Meer MV, et al. Local fitness landscape of the green fluorescent protein. Nature. 2016;533:397–401.. 10.1038/nature17995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Silverstein RA, Sun S, Verby M, et al. A systematic genotype-phenotype map for missense variants in the human intellectual disability-associated gene GDI1. Biorxiv. 10.1101/2021.10.06.463360. [DOI] [Google Scholar]
- 69. Starita LM, Pruneda JN, Lo RS, et al. Activity-enhancing mutations in an E3 ubiquitin ligase identified by high-throughput mutagenesis. Proc Natl Acad Sci USA. 2013;110:E1263–72.. 10.1073/pnas.1303309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Starita LM, Young DL, Islam M, et al. Massively parallel functional analysis of BRCA1 RING domain variants. Genetics. 2015;200:413–22.. 10.1534/genetics.115.175802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Starita LM, Islam MM, Banerjee T, et al. A multiplex homology-directed DNA repair assay reveals the impact of more than 1,000 BRCA1 missense substitution variants on protein function. Am Hum Genet. 2018;103:498–508.. 10.1016/j.ajhg.2018.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Suiter CC, Moriyama T, Matreyek KA, et al. Massively parallel variant characterization identifies NUDT15 alleles associated with thiopurine toxicity. Proc Natl Acad Sci USA. 2020;117:5394–401.. 10.1073/pnas.1915680117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sun S, Weile J, Verby M, et al. A proactive genotype-to-patient-phenotype map for cystathionine beta-synthase. Genome Med. 2020;12:13. 10.1186/s13073-020-0711-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Thompson S, Zhang Y, Ingle C, et al. Altered expression of a quality control protease in E. coli reshapes the in vivo mutational landscape of a model enzyme. eLife. 2020;9:e53476. 10.7554/eLife.53476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Trenker R, Wu X, Nguyen JV, et al. Human and viral membrane–associated E3 ubiquitin ligases MARCH1 and MIR2 recognize different features of CD86 to downregulate surface expression. J Biol Chem. 2021;297:100900. 10.1016/j.jbc.2021.100900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Weile J, Sun S, Cote AG, et al. A framework for exhaustively mapping functional missense variants. Mol Syst Biol. 2017;13:957. 10.15252/msb.20177908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Weile J, Kishore N, Sun S, et al. Shifting landscapes of human MTHFR missense-variant effects. Am Hum Genet. 2021;108:1283–300. 10.1016/j.ajhg.2021.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wrenbeck EE, Bedewitz MA, Klesmith JR, et al. An automated data-driven pipeline for improving heterologous enzyme expression. ACS Synth Biol. 2019;8:474–81.. 10.1021/acssynbio.8b00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhang L, Sarangi V, Moon I et al. CYP2C9 and CYP2C19: deep mutational scanning and functional characterization of genomic missense variants. Clin Transl Sci. 2020;13:727–42.. 10.1111/cts.12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zinkus-Boltz J, DeValk C, Dickinson BC. A phage-assisted continuous selection approach for deep mutational scanning of protein–protein interactions. ACS Chem Biol. 2019;14:2757–67.. 10.1021/acschembio.9b00669. [DOI] [PubMed] [Google Scholar]
- 81. Tian S, Choi W-T, Liu D, et al. Distinct functional sites for human immunodeficiency virus type 1 and stromal cell-derived factor 1α on CXCR4 transmembrane helical domains. J Virol. 2005;79:12667–73.. 10.1128/JVI.79.20.12667-12673.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chabot DJ, Zhang P-F, Quinnan GV, et al. Mutagenesis of CXCR4 identifies important domains for human immunodeficiency virus type 1×4 isolate envelope-mediated membrane fusion and virus entry and reveals cryptic coreceptor activity for R5 isolates. J Virol. 1999;73:6598–609.. 10.1128/JVI.73.8.6598-6609.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Han DP, Penn-Nicholson A, Cho MW. Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology. 2006;350:15–25.. 10.1016/j.virol.2006.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fujita–Yoshigaki J, Shirouzu M, Ito Y, et al. A constitutive effector region on the C-terminal side of switch I of the ras protein. J Biol Chem. 1995;270:4661–7.. 10.1074/jbc.270.9.4661. [DOI] [PubMed] [Google Scholar]
- 85. Hidalgo P, Ansari AZ, Schmidt P, et al. Recruitment of the transcriptional machinery through GAL11P: structure and interactions of the GAL4 dimerization domain. Genes Dev. 2001;15:1007–20.. 10.1101/gad.873901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Rodríguez-Escudero I, Oliver MD, Andrés-Pons A, et al. A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes. Hum Mol Genet. 2011;20:4132–42.. 10.1093/hmg/ddr337. [DOI] [PubMed] [Google Scholar]
- 87. Bernier-Villamor V, Sampson DA, Matunis MJ, et al. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP. Cell. 2002;108:345–56.. 10.1016/S0092-8674(02)00630-X. [DOI] [PubMed] [Google Scholar]
- 88. Blanpain C, Doranz BJ, Vakili J, et al. Multiple charged and aromatic residues in CCR5 amino-terminal domain are involved in high affinity binding of both chemokines and HIV-1 env protein. J Biol Chem. 1999;274:34719–27.. 10.1074/jbc.274.49.34719. [DOI] [PubMed] [Google Scholar]
- 89. Brzovic PS, Keeffe JR, Nishikawa H, et al. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc Natl Acad Sci USA. 2003;100:5646–51.. 10.1073/pnas.0836054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chen S, Wu J, Zhong S, et al. iASPP mediates p53 selectivity through a modular mechanism fine-tuning DNA recognition. Proc Natl Acad Sci USA. 2019;116:17470–9.. 10.1073/pnas.1909393116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chupreta S, Holmstrom S, Subramanian L, et al. A small conserved surface in SUMO is the critical structural determinant of its transcriptional inhibitory properties. Mol Cell Biol. 2005;25:4272–82.. 10.1128/MCB.25.10.4272-4282.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cobb JA, Roberts DM. Structural requirements for N-trimethylation of lysine 115 of Calmodulin. J Biol Chem. 2000;275:18969–75. 10.1074/jbc.M002332200. [DOI] [PubMed] [Google Scholar]
- 93. Coyne RS, McDonald HB, Edgemon K, et al. Functional characterization of BRCA1 sequence variants using a yeast small colony phenotype assay. Cancer Biol Ther. 2004;3:453–7.. 10.4161/cbt.3.5.809. [DOI] [PubMed] [Google Scholar]
- 94. Denker K, Orlik F, Schiffler B, et al. Site-directed mutagenesis of the greasy slide aromatic residues within the LamB (Maltoporin) channel of Escherichia coli: effect on ion and maltopentaose transport. J Mol Biol. 2005;352:534–50.. 10.1016/j.jmb.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 95. Dragic T, Trkola A, Lin SW, et al. Amino-terminal substitutions in the CCR5 coreceptor impair gp120 binding and Human Immunodeficiency virus type 1 entry. J Virol. 1998;72:279–85.. 10.1128/JVI.72.1.279-285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Dragic T, Trkola A, Thompson DAD, et al. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc Natl Acad Sci USA. 2000;97:5639–44.. 10.1073/pnas.090576697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ecsédi P, Gógl G, Hóf H, et al. Structure determination of the transactivation domain of p53 in complex with S100A4 using Annexin A2 as a crystallization chaperone. Structure. 2020;28:943–53.. e4. 10.1016/j.str.2020.05.001. [DOI] [PubMed] [Google Scholar]
- 98. Kopecká J, Krijt J, Raková K, et al. Restoring assembly and activity of cystathionine β-synthase mutants by ligands and chemical chaperones. J Inher Metab Dis. 2011;34:39–48.. 10.1007/s10545-010-9087-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kožich V, Sokolová J, Klatovská V, et al. Cystathionine β-synthase mutations: effect of mutation topology on folding and activity. Hum Mutat. 2010;31:809–19.. 10.1002/humu.21273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kruger WD, Wang L, Jhee KH, et al. Cystathionine β-synthase deficiency in Georgia (USA): correlation of clinical and biochemical phenotype with genotype. Hum Mutat. 2003;22:434–41.. 10.1002/humu.10290. [DOI] [PubMed] [Google Scholar]
- 101. Lee SY, Pullen L, Virgil DJ, et al. Alanine scan of core positions in ubiquitin reveals links between dynamics, stability, and function. J Mol Biol. 2014;426:1377–89.. 10.1016/j.jmb.2013.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Li W, Zhang C, Sui J, et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24:1634–43.. 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lin G, Baribaud F, Romano J, et al. Identification of gp120 binding sites on CXCR4 by using CD4-independent human immunodeficiency virus type 2 env proteins. J Virol. 2003;77:931–42.. 10.1128/JVI.77.2.931-942.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mascle XH, Lussier-Price M, Cappadocia L, et al. Identification of a non-covalent ternary complex formed by PIAS1, SUMO1, and UBC9 proteins involved in transcriptional regulation. J Biol Chem. 2013;288:36312–27.. 10.1074/jbc.M113.486845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Matthews EE, Thévenin D, Rogers JM, et al. Thrombopoietin receptor activation: transmembrane helix dimerization, rotation, and allosteric modulation. FASEB J. 2011;25:2234–44.. 10.1096/fj.10-178673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Mayfield JA, Davies MW, Dimster-Denk D, et al. Surrogate genetics and metabolic profiling for characterization of Human disease alleles. Genetics. 2012;190:1309–23.. 10.1534/genetics.111.137471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Navenot J-M, Wang Z, Trent JO, et al. Molecular anatomy of CCR5 engagement by physiologic and viral chemokines and HIV-1 envelope glycoproteins: differences in primary structural requirements for RANTES, MIP-1α, and vMIP-II binding. J Mol Biol. 2001;313:1181–93.. 10.1006/jmbi.2001.5086. [DOI] [PubMed] [Google Scholar]
- 108. Peng L, Damschroder MM, Cook KE, et al. Molecular basis for the antagonistic activity of an anti-CXCR4 antibody. mAbs. 2016;8:163–75.. 10.1080/19420862.2015.1113359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Peterson BR, Sun LJ, Verdine GL. A critical arginine residue mediates cooperativity in the contact interface between transcription factors NFAT and AP-1. Proc Natl Acad Sci USA. 1996;93:13671–6.. 10.1073/pnas.93.24.13671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Rabut GEE, Konner JA, Kajumo F, et al. Alanine substitutions of polar and nonpolar residues in the amino-terminal domain of CCR5 differently impair entry of macrophage- and dualtropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72:3464–8.. 10.1128/JVI.72.4.3464-3468.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ransburgh DJR, Chiba N, Ishioka C, et al. Identification of breast tumor mutations in BRCA1 that abolish its function in homologous DNA recombination. Cancer Res. 2010;70:988–95.. 10.1158/0008-5472.CAN-09-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tan Y, Tong P, Wang J, et al. The membrane-proximal region of C–C chemokine receptor type 5 participates in the infection of HIV-1. Front Immunol. 2017;8:478. 10.3389/fimmu.2017.00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Towler WI, Zhang J, Ransburgh DJR, et al. Analysis of BRCA1 variants in double-strand break repair by homologous recombination and single-strand annealing. Hum Mutat. 2013;34:439–45.. 10.1002/humu.22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Trent JO, Wang Z, Murray JL, et al. Lipid bilayer simulations of CXCR4 with inverse agonists and weak partial agonists. J Biol Chem. 2003;278:47136–44.. 10.1074/jbc.M307850200. [DOI] [PubMed] [Google Scholar]
- 115. Van Gelder P, Dumas F, Bartoldus I, et al. Sugar transport through maltoporin of Escherichia coli : role of the greasy slide. J Bacteriol. 2002;184:2994–9.. 10.1128/JB.184.11.2994-2999.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. VanBerkum MF, Means AR. Three amino acid substitutions in domain I of calmodulin prevent the activation of chicken smooth muscle myosin light chain kinase. J Biol Chem. 1991;266:21488–95.. 10.1016/S0021-9258(18)54665-2. [DOI] [PubMed] [Google Scholar]
- 117. Wei Q, Wang L, Wang Q, et al. Testing computational prediction of missense mutation phenotypes: functional characterization of 204 mutations of human cystathionine beta synthase. Proteins. 2010;78:2058–74.. 10.1002/prot.22722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Williams AD, Shivaprasad S, Wetzel R. Alanine scanning mutagenesis of aβ(1-40) amyloid fibril stability. J Mol Biol. 2006;357:1283–94.. 10.1016/j.jmb.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 119. Zhang J, Rao E, Dioszegi M, et al. The second extracellular loop of CCR5 contains the dominant epitopes for highly potent anti-human immunodeficiency virus monoclonal antibodies. Antimicrob Agents Chemother. 2007;51:1386–97.. 10.1128/AAC.01302-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Gray VE, Hause RJ, Fowler DM. Analysis of large-scale mutagenesis data to assess the impact of single amino acid substitutions. Genetics. 2017;207:53–61.. 10.1534/genetics.117.300064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Nelsen RB. An Introduction to Copulas. 2nd ed. New York: Springer; 2006. [Google Scholar]
- 122. Bedő J, Ong CS. Multivariate Spearman's rho for aggregating ranks using copulas. J Mach Learn Res. 2016;17:1–30.. arXiv. 10.48550/ARXIV.1410.4391. [DOI] [Google Scholar]
- 123. Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit-learn: machine learning in Python. J Mach Learn Res. 2011;12:2825–30. [Google Scholar]
- 124. Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9:90–5.. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 125. Choi Y, Sims GE, Murphy S, et al. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Vaser R, Adusumalli S, Leng SN, et al. SIFT missense predictions for genomes. Nat Protoc. 2016;11:1–9.. 10.1038/nprot.2015.123. [DOI] [PubMed] [Google Scholar]
- 127. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.. 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Laine E, Karami Y, Carbone A. GEMME: a simple and fast global epistatic model predicting mutational effects. Mol Biol Evol. 2019;36:2604–19.. 10.1093/molbev/msz179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Meier J, Rao R, Verkuil R, et al. Language models enable zero-shot prediction of the effects of mutations on protein function. Biorxiv. 10.1101/2021.07.09.450648. [DOI] [Google Scholar]
- 130. Frazer J, Notin P, Dias M, et al. Disease variant prediction with deep generative models of evolutionary data. Nature. 2021;599:91–5.. 10.1038/s41586-021-04043-8. [DOI] [PubMed] [Google Scholar]
- 131. Livesey BJ, Marsh JA. Updated benchmarking of variant effect predictors using deep mutational scanning. Mol Syst Biol. 2023;19:e11474. 10.15252/msb.202211474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. González J, Dai Z, Hennig P, et al. Batch bayesian optimization via local penalization. arXiv. 10.48550/arXiv.1505.08052. [DOI] [Google Scholar]
- 133. Cagiada M, Johansson KE, Valanciute A, et al. Understanding the origins of loss of protein function by analyzing the effects of thousands of variants on activity and abundance. Mol Biol Evol. 2021;38:3235–46.. 10.1093/molbev/msab095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Jepsen MM, Fowler DM, Hartmann-Petersen R, et al. Classifying disease-associated variants using measures of protein activity and stability. In: Pey AL, editor. Protein Homeostasis Diseases. Academic Press; Cambridge:2020; pp. 91–107. [Google Scholar]
- 135. Matreyek KA, Stephany JJ, Ahler E, et al. Integrating thousands of PTEN variant activity and abundance measurements reveals variant subgroups and new dominant negatives in cancers. Genome Med. 2021;13:165. 10.1186/s13073-021-00984-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Mighell TL, Thacker S, Fombonne E, et al. An integrated deep-mutational-scanning approach provides clinical insights on PTEN genotype-phenotype relationships. Am Hum Genet. 2020;106:818–29.. 10.1016/j.ajhg.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Nielsen SV, Hartmann-Petersen R, Stein A, et al. Multiplexed assays reveal effects of missense variants in MSH2 and cancer predisposition. PLoS Genet. 2021;17:e1009496. 10.1371/journal.pgen.1009496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Fu Y, Bedő J, Papenfuss AT, et al. Supporting data for “Integrating Deep Mutational Scanning and Low-Throughput Mutagenesis Data to Predict the Impact of Amino Acid Variants.”. GigaScience Database. 2023. 10.5524/102429. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Fu Y, Bedő J, Papenfuss AT, et al. Supporting data for “Integrating Deep Mutational Scanning and Low-Throughput Mutagenesis Data to Predict the Impact of Amino Acid Variants.”. GigaScience Database. 2023. 10.5524/102429. [DOI] [PMC free article] [PubMed]
Supplementary Materials
Joseph Ng -- 3/21/2023 Reviewed
Joseph Ng -- 7/12/2023 Reviewed
Leopold Parts -- 4/7/2023 Reviewed
Leopold Parts -- 7/21/2023 Reviewed
Data Availability Statement
A copy of the data analysis code and a full set of data files required to reproduce this work are openly available in the GigaScience repository, GigaDB, under the record described in [138]. MaveDB accession numbers, UniProt accession numbers, and other metadata describing the matched DMS-AS datasets are listed in Supplementary Table S1 (see supporting information).