

Abstract
The presence and role of microbes in human cancers has come full circle in the last century. Tumors are no longer considered aseptic, but implications for cancer biology and oncology remain underappreciated. Opportunities to identify and build translational diagnostics, prognostics, and therapeutics that exploit cancer’s second genome—the metagenome—are manifold, but require careful consideration of microbial experimental idiosyncrasies that are distinct from host-centric methods. Furthermore, the discoveries of intracellular and intra-metastatic cancer bacteria necessitate fundamental changes in describing clonal evolution and selection, reflecting bidirectional interactions with non-human residents. Reconsidering cancer clonality as a multispecies process similarly holds key implications for understanding metastasis and prognosing therapeutic resistance while providing rational guidance for the next generation of bacterial cancer therapies. Guided by these new findings and challenges, this Review describes opportunities to exploit cancer’s metagenome in oncology and proposes an evolutionary framework as a first step towards modeling multispecies cancer clonality.
Keywords: cancer microbiome, clonal evolution, diagnostics, therapeutic modulation, prognostics
INTRODUCTION
A rich history exists between microbes and cancer. As early as 1550 BCE, Egyptian writings suggested a crude tumor therapy through incision and application of a poultice, inciting an infection.[1–3] Nearly three millennia later, Saint Peregrine Laziosi (c. 1265–1345) documented spontaneous regression of a septic leg sarcoma large enough to pierce skin.[2] Although these accounts predated modern germ theory, they associated acute infections with cancer retrogression, independently re-discovered between 1868–1893 by Wilhelm Busch, Friedrich Fehleisen, and William Coley.[4–6]
These physicians linked spontaneous tumor regressions with Streptococcus pyogenes and erysipelas. However, only Coley treated patients with live bacteria, and, later, heat-killed Streptococcus and Serratia toxins. These experiments revealed >10-year disease-free survival in 60 of 210 patients across multiple cancer types—efficacy only matched by modern immunotherapy.[7] Nonetheless, unknown mechanisms and side effects made ‘Coley’s toxins’ unpalatable to oncology, especially compared to radiotherapy and chemotherapy.[8,9] Another century passed before recognition of Coley’s approach as the first intentional immunotherapy, and its prediction of a causal relationship between immunotherapy efficacy and endogenous or exogenously-administered microbiomes.[10–16]
Viruses also catalyzed our understanding of cancer and its genetic material. Peyton Rous’s 1911 discovery of his eponymous, transmissible, oncogenic, RNA virus galvanized investigation of cancer’s viral origins, linking Epstein-Barr, human papilloma (HPV), hepatitis, and Merkel cell polyomavirus to carcinogenesis.[17–19] Although decades of research concluded that viruses cause a minority of cancers, the pursuit of oncogenic viruses led to the definition of and search for ‘oncogenes’ that transform benign tissue into malignant tissue.[19] One key oncogene was src, a protein kinase identified in transforming-only strains of Rous’s Sarcoma Virus (RSV), also found by Michael Bishop and Harold Varmus in cells of non-infected, phylogenetically-divergent birds.[20] Their data suggested a non-viral, cellular origin of src: hosts normally contain oncogenes, and transforming strains of RSV had acquired one. This discovery earned the 1989 Nobel Prize in Medicine.[19,20] Realizing oncogenes were internal to cancer motivated characterization of all possible oncogenes in the human cancer genome by sequencing the normal human genome as a reference.[21] Modern cancer genomics thus stems from tumor virology.
RSV’s story and its hijacking of src showed how genetic information could transfer between tumors, microbes, and their hosts over evolutionary time. Successive passaging of RSV later enabled researchers to evolve the chicken-specific virus to induce tumors in various animals (e.g., ducks, pigeons, rats, rabbits, mice), presumably activating similar kinase-related oncogenic pathways.[19,22,23] This process represented early examples of intentional transfection and directed evolution, whereby recipient cells received genetic cargo that changed cellular fitness. Decades later, bacterial capability to transfect human or microbial genetic material[24–28] to cells—including cancer cells[29]—with subsequent protein expression would be demonstrated and coined “bactofection.”[30] Bactofection was pursed as an alternative to conventional gene therapy or vaccination, but received little attention.[27,30,31]
Since Bishop and Varmus’s discovery, the last 30 years of cancer research primarily focused on characterizing all major coding, noncoding, structural, and copy number aberrations in the cancer genome.[32–36] Substantial study of the tumor microenvironment (TME) elucidated impacts of heterogeneous tumor architecture, spatial organization, and multifaceted cellular agents (e.g., immune and stromal cells) on cancer evolution, clonality, antitumor immunity, and metastasis.[37–39] Further work revealed similarities between microbial and cancer evolution. For example, ubiquitous plasmid-like, extrachromosomal DNA (ecDNA) segments and their unequal segregation during cancer cell division is analogous to unequal segregation of high-copy plasmids during bacterial replication.[40–44] Hybrid viral-human sequences on ecDNA segments in HPV-infected cancers even contribute to immune evasion and carcinogenesis.[45,46] Nonetheless, most cancer ‘omic’ studies portrayed tumors as sterile, and microbial constituents as unrelated to tumor evolution or clinical care.
The last five years have revealed metabolically-active, immunoreactive, intracellular, cancer type-specific communities of bacteria and viruses living within tumor tissues, several of which modulate cancer therapies.[47–60] These microbes may move during metastasis and facilitate leaving and/or seeding of metastatic cancer cells.[53,54,61–63] Critically, intratumoral and gut microbes can create chemo-, radio-, and hormonal therapeutic resistance without cancer genome changes.[47,64,65] Conversely, microbiota may render other chemo-, radio-, hormonal, and immunotherapies possible and/or effective without any cancer cell intervention.[12–14,64,66–68] Trace amounts of cancer type-specific bacterial DNA have been identified in cancer patient blood, suggesting a novel class of microbial cancer diagnostics.[58,69] Most, if not all, human cancers lack sterility, and their microbes are clinically relevant, provoking a third revision of the classic cancer ‘hallmarks’ to include “polymorphic microbiomes” of the gut and tumors.[70]
Towards building a microbially-conscious framework of cancer, we posit cancer-bearing humans as meta-organisms colonized by numerous, diverse microbial constituents (Box 1).[71,72] We propose the clinical utility of microbial information as cancer diagnostics, prognostics, and therapeutics and consider (intracellular) microbes as live, mobile agents within tumors that encounter selection pressures alongside/within cancer cells. Finally, we hypothesize that fundamental ecological rules governing microbial activity and spatial placement (e.g., redox, chemotactic, oxygen gradients)[73] outside tumors also govern them inside tumors. This Review details the study of cancer’s “second genome,” its clinical use, and its impact on cancer clonal evolution.
BOX 1— Quantifying the cancer microbiome.
Broadly speaking, the human body microbiota include ~4×103 species accounting for ~4×1013 total microorganisms, with ~97% of those cells comprising colonic bacteria and ~2–3% comprising extra-colonic bacteria while archaea and eukarya—including fungi—comprise smaller populations around ~0.1–1% of the total microbial abundance.[71,77] Human virus and phage abundance estimates remain undercharacterized but likely have even greater diversity than bacteria.[174] The human gut microbiome contains the largest bodily microbial biomass by far—roughly 0.2 kilograms[71,175]—with substantial effects on host antitumor immunity.[3] Biomass estimates of other body sites or tissues remain unknown.
Intratumoral microbiome diversity estimates with stringent decontamination controls (~1:2 control to sample ratio) suggest that at least 500 distinct bacterial species inhabit tumors.[57] Intratumoral microbiome abundance estimates have been inferred using shotgun read quantification and quantitative polymerase chain reaction (qPCR) of 16S rRNA.[57,58] Microbial profiling of all whole genome and transcriptome studies from The Cancer Genome Atlas (TCGA) revealed an average of 2.5% of total sequencing reads to be microbial and an average of 0.9% of total reads that were resolvable at the genus-level (both estimates were based on raw, non-decontaminated data).[58] To quantitate intratumoral bacterial abundance, Nejman et al. performed qPCR of the V6 region of 16S rRNA among DNA extraction and paraffin controls and several cancer types (Table 1). Bootstrapping these qPCR data reveal a heterogeneous average number of bacteria per cancer type, ranging from ~13 to ~70 per 40 nanograms (ng) of DNA, among seven major human cancers (Table 1), although we note that abundances varied over three orders of magnitude between patients within the same cancer type.[57] The pan-cancer average was 34.19 bacteria per 40 ng of DNA (95% CI: [24.04, 46.56]; Table 1), or, as will be calculated below, approximately 0.68% bacterial composition (i.e., a ratio of 1 bacteria for every ~147 cancer cells).
The discrepancy between the shotgun metagenomic read abundance and qPCR (V6 16S rRNA amplicon) bacterial abundance may be due to several factors: (1) 16S rRNA, though abundant in bacteria, does not account for total microbial DNA or RNA, nor does it account for non-bacterial nucleic acids (e.g., viruses, archaea) measured by the shotgun read approach; (2) technical amplification biases of the V6 region may under-represent certain bacteria, with a recent in silico study showing ~55% of bacteria being inadequately covered by V6-V8 primers;[176] and (3) the qPCR data were normalized to non-template contamination controls[57] whereas the shotgun estimates[58] did not take into account contamination, which may comprise as much as 90–95% of raw microbial data. To proceed with calculations, however, we used the qPCR data from Nejman et al. as a conservative approach that accounted for contamination using hundreds of experimental controls.
To translate qPCR values from Nejman and colleagues to percent tumor composition, it is necessary to first estimate the number of tumor cells per 40 ng of DNA. One way to estimate this for haploid cells is as follows:
To translate from haploid cell to tumor cell, an estimate of ploidy is needed, which can be derived from the most recent Pan-Cancer Analysis of Whole Genomes (PCAWG) dataset.[32] The mean estimated ploidy in PCAWG across all human cancers is 2.36 and ranges from a low of 1.28 to a high of 6.22. If we assume average cancer ploidy, the average DNA mass per cancer cell is thus:
Similarly, the range of DNA masses per cancer cell based on ploidy would be 4.48 pg to 21.77 pg. For simplicity, one can round the average mass value to 8 pg/cancer cell. Assuming that the DNA mass of microbes is negligible compared to that of the host, since its genome is roughly 103-fold smaller and there are fewer of them expected, then the estimated percent composition is as follows:
This estimate, however, assumes 100% tumor purity. Fortunately, PCAWG estimated tumor purity across the same samples, showing an average tumor purity of 63.8%.[32] Instead of 5000 cancer cells per 40 ng of DNA, assuming 8 pg per cancer cell, average tumor purity suggests 3190 cancer cells with the remaining cells comprising the TME. While this does not change the percent bacterial composition of the tumor, it does change the ratio of bacteria to cancer cells to approximately ~1:100 or ~1% (i.e. 34.19 bacteria:3190 cancer cells; Table 1). Using the 95% confidence interval bounds of the pan-cancer bootstrapped average number of bacteria per tumor (Table 1) gives a range of 0.75% to 1.46% bacterial.
In the case of high tumor ploidy and low tumor purity, it may become important to weigh the contributions between tumor (aneuploid) and stroma (diploid) to the number of cells within 40 ng of DNA. This may be done as follows, for example using a tumor ploidy of 6.0 and 20% purity:
whereas a tumor of 100% purity at a ploidy of 6.0 would provide an average tumor bacterial composition of 1.86%. More generally, cases with high ploidy and high purity will maximize this percentage value, in addition to when there is more observed bacteria.
To compare these bacterial abundances to intratumor immune cell populations, which are usually reported as densities of immune cell counts per square millimeter, it is necessary to first estimate the total number of cells per square millimeter in a tumor. While a handful of density estimates exist in the literature, such as a mean of 5,558 cells (std. dev. 1,980) per mm2 in metastatic melanoma,[177] it can be inferred directly from circle packing theory.[178] Specifically, given the average diameter of cells in a tissue, then the number of possible cells within the 1 mm2 square can be calculated. In one way, this can be interpreted as a conservative estimate since cells are often compressed and non-circular in real tissues; conversely, it may overestimate cell density in regions with dense blood or lymphatic vessels. The typical diameter of lymphocytes approximates 6–7 μm in diameter[179] while the diameter of cancer cells vary by type and are approximately ~20 μm in diameter across many cancer cell lines.[180] Using average cell diameters of 12 μm, 15 μm, and 18 μm, circle packing theory predicts the following total cell abundances per 1 mm2: 8213 cells, 5208 cells, and 3589 cells.
Then, using the previously calculated average pan-cancer tumor bacterial composition of 0.68% (assuming tumor homogeneity), the estimated number of bacteria inferred as the following: 56, 35, 24 bacteria/mm2 (assuming 12 μm, 15 μm, and 18 μm average diameter cells, respectively). Notably, these bacterial abundance estimates are similar to the proportion of PD1+ cells identified in a recent pan-cancer imaging dataset (~22 PD1+ cells/mm2) and roughly one-tenth of CD8+ T-cell density (~385 cells/mm2).[181] Overall, the values reflected in this analysis may vary from tumor to tumor, depending on the assumptions made above—tumor ploidy, purity, homogeneity—but the analysis provides a rough approximation and analogy of intratumor bacterial abundances to immune cell abundances.
To summarize, these calculations estimate an average pan-cancer bacterial composition of ~0.68% with two- and three-dimensional estimates of ~35 bacteria/mm2 (assuming 5200 cells/mm2), or approximately 6×105 to 6×106 bacteria per palpable 1 cm3 tumor (assuming 108-109 cells/cm3).[182] Again, we note that these estimates can vary between patients by three orders of magnitude and require further examination in additional cohorts.
Notwithstanding the above calculations, Nejman and colleagues noted that many and different proportions of samples per cancer type had qPCR abundances that were lower than the 99th-percentile of their DNA extraction controls, which comprised their cutoff for background contamination.[57] Specifically, just 14.3% of melanoma samples, 16.8% of lung cancer samples, 24.6% of ovarian cancer samples, 44.4% of glioblastoma samples, 68.2% of pancreatic cancer samples, 66.7% of bone cancer samples, and 62.7% of breast cancer samples exceeded this conservative threshold.[57] However, because large inter-patient variability was also observed within cancer types in their study, these values may change and become more accurate with broader cohorts.
CANCER MICROBIOME DIAGNOSTICS AND PROGNOSTICS
The concept of “strength in numbers” applies to cancer diagnostics, especially for low-biomass material. Liquid biopsies in cancer detect minute quantities of analytes (e.g., DNA, RNA, proteins) shed from the tumor to diagnose cancer presence and/or type.[74] The low-biomass, limited unique number, and limits of analyte detection usually restricts application of liquid biopsies to later-stage tumors, several cubic centimeters and above.[74,75] Adding analytes or modifications, even if rare, increases overall test sensitivity .[76] Cristiano et al. demonstrated this principle using Monte Carlo simulations of liquid biopsies, showing that a test examining DNA modifications comprising ≤0.001% of total plasma material could still have near-perfect sensitivity if at least 256 alterations were interrogated.[76]
These cancer genomics conclusions suggest the diversity of the intratumoral microbiome (≥500 unique bacterial species)[57] and gut microbiome (~4×103 bacterial species)[77] substantiate microbiome-focused cancer diagnostics, even if all individual microbes are rare or lowly abundant. Two alternative ways of phrasing this idea are: (i) high microbial diversity provides “many shots on goal” for a single diagnosis and (ii) interrogating the microbiome is like employing an ensemble of many weak learners that collectively provide strong prediction performance—the conceptual basis of boosting in machine learning. For diagnostic purposes, detected microbes need not be causally carcinogenic but only correlated with cancer presence, absence, and/or growth. These microbial-informed diagnostics and prognostics could dramatically improve clinical cancer care (Figure 1).
FIGURE 1.
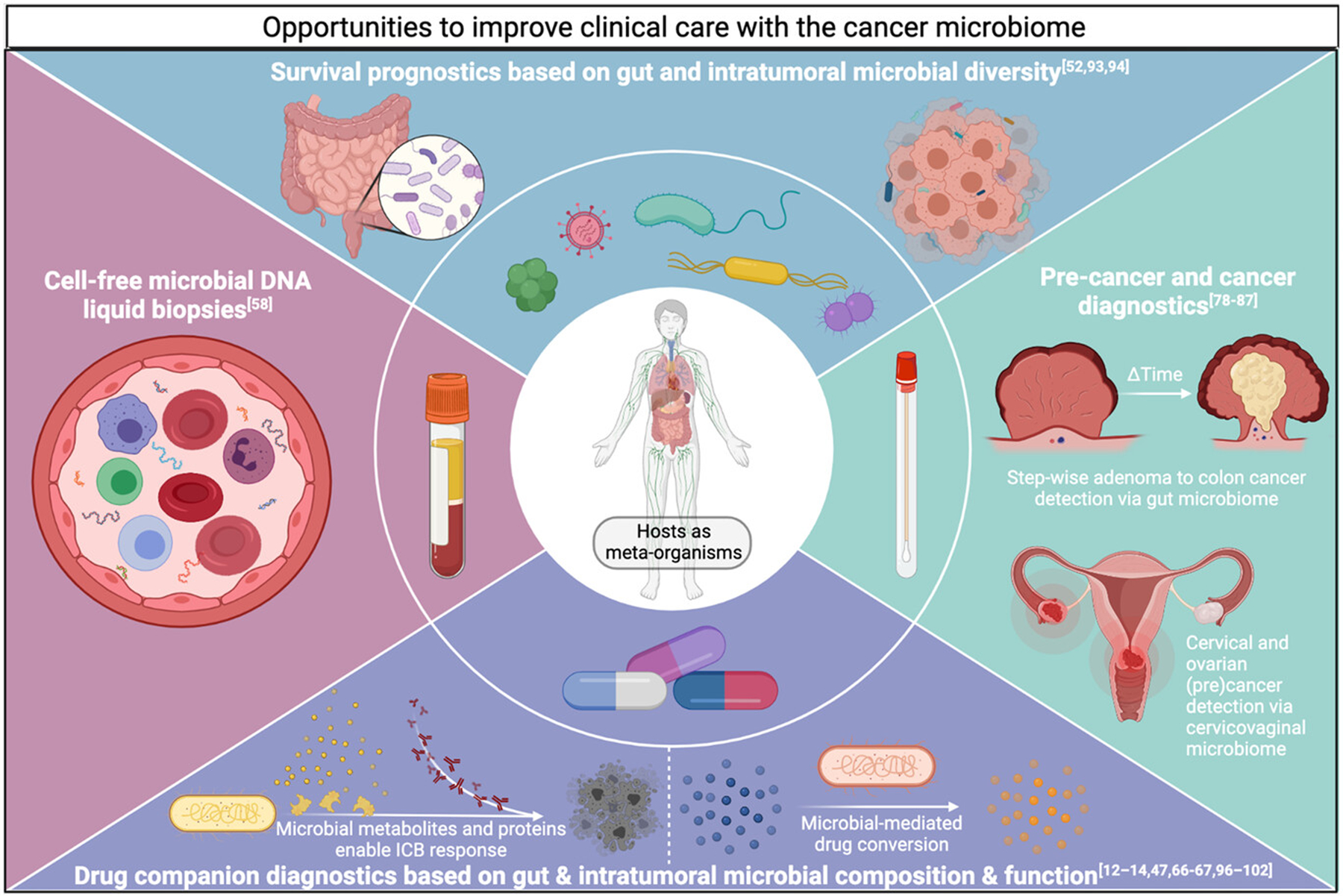
Illustration of opportunities to enhance clinical cancer diagnostics and prognostics using the cancer microbiome. Relevant references are listed in the title of each quadrant.
Pre-cancer and cancer microbiome diagnostics
Pre-cancer diagnostics identify lesions of high malignant potential, typically cervical and colorectal cancer (CRC) precursors. Focusing on gut microbiota, metagenomic studies identified distinct fecal microbial compositions between colonic adenoma-bearing human patients and healthy individuals, often with increases in Proteobacteria relative abundance.[78–81] Yachida et al. characterized shotgun metagenomic and metabolomic shifts in patients’ guts with no disease, polypoid adenomas, and stage 0 to stage IV CRCs, revealing distinct stage-wise microbial and metabolic compositions sufficient to build fecal stage-specific classifiers.[78] Vaginal microbiome studies also revealed distinguishable microbial compositions and functions between healthy humans, patients with cervical intraepithelial neoplasia or cervical cancer, and modifying effects of HPV or HIV status.[82–84] In a longitudinal trial, women with high-risk HPV infection and abundant vaginal Lactobacillus were more likely to clear the infection by their second clinical visit (average 1.5 years later); conversely, patients with abundant vaginal Gardnerella upon presentation were more likely to show disease progression by the second visit.[82] These studies suggest the opportunity for minimally-invasive, swab-based stool and vaginal microbiome diagnostics that detect precursor cancer lesions and/or forecast cancer conversion risk.
Pre-cancerous syndromes are also pertinent for microbiome diagnostics, such as genetically-driven familial adenomatous polyposis (FAP), pre-leukaemic myeloproliferation (PMP), and BRCA1 status, by auguring subsequent carcinogenesis in ways not fully predicted by host genomics. Comparing gut microbiota from clinical patients with and without FAP, Dejea et al. elucidated that FAP encourages biofilm formation comprising genotoxic strains of Escherichia coli and Bacteroides fragilis with greater expression of their colibactin and B. fragilis toxins, increasing IL-17-dependent inflammation, DNA damage, and cancer conversion.[85] Meisel and colleagues demonstrated in preclinical mouse models that gut microbial translocation to systemic circulation with resultant IL-6 production drives conversion from predisposing Tet2 germline mutations to PMP.[86] Nené et al. reported significant cervicovaginal microbiome changes (absence of Lactobacillus species) among BRCA1-positive, non-cancer-carrying clinical patients—changes shared among women with ovarian cancer, suggesting that germline mutations affect microbial composition and may show continuity with subsequent cancer conversion.[87] Collectively, these studies argue that pre-cancerous syndromes modify and interact with microbiota, highlighting an opportunity to develop diagnostics and interventions that reduce cancer conversion rates.
For solid tumor and blood microbiome diagnostics, Nejman et al. and Poore et al. provide the most comprehensive analyses to date.[57,58] Specifically, Nejman and colleagues combined imaging, cultivation, qPCR, and multi-region 16S rRNA sequencing to characterize intratumoral bacteria among breast, bone, pancreas, brain, ovarian, lung, melanoma, and colon cancers. Inclusion of 811 experimental contamination controls (i.e., DNA extraction controls, no-template PCR controls, paraffin controls) for 1010 tumor and 516 normal samples enabled stringent decontamination that removed 94.5% of detected bacterial species, leaving 528 confident species-level calls. Statistical analyses of decontaminated tumor microbiomes exhibited cancer type-specific differences. Fluorescent and electron microscopic imaging data also revealed the intracellular localization—both in cancer and immune cells—of many intratumoral bacteria, and further experiments showing D-alanine metabolism (a xenobiotic) in breast tumors suggested these bacteria were metabolically active.
Using a distinct approach, Poore and colleagues mined all whole genome and transcriptome sequencing data in TCGA (n=18,116 samples; 14,007 primary tumors) and employed shotgun metagenomic strategies to derive ~2000 genus-level calls across bacteria, viruses, and archaea (59,974 total genomes searched).[58] In silico decontamination based on sample DNA or RNA concentrations[88] removed up to 92.3% of microbial information, but machine learning performance distinguishing between cancer types and tumor from adjacent normal tissue remained strong. Based on historical and epidemiological data associating bacteremias with subsequent CRC diagnosis,[69,89] they showed that blood-derived microbes in TCGA could distinguish CRC from other cancer types, and that blood-derived microbiomes discriminated more broadly among 19 other cancer types, including restricting the analyses to stage 1–2 tumors or tumors without any canonical mutations on two commercial cell-free tumor DNA (ctDNA) panels. Applying this approach to 100 plasma samples from three cancer types (lung, prostate, melanoma) and 69 HIV-negative, non-cancer controls suggested that cell-free microbial DNA (cf-mbDNA) could distinguish healthy individuals from patients with cancer, and distinguish cancer types, suggesting novel microbial-based, minimally-invasive cancer diagnostics.[58]
The blood and plasma analyses reported by Poore and colleagues identified novel taxonomic compositions not previously appreciated to play a role in cancer biology, despite stringent in silico decontamination.[58] Although these compositions must be confirmed in further cohorts and using metagenome assemblies, preceding efforts reported substantially new, previously uncharacterized, and compositionally divergent microbes in non-cancer-associated human plasma.[90] In particular, Kowarsky and colleagues performed metagenome assembly on cell-free DNA from 1351 sequenced blood samples (188 human subjects) in four longitudinally-sampled cohorts (heart, lung, bone marrow transplants and during pregnancy), identifying 7190 contigs larger than 1 kilobases.[90] 3761 (52.3%) of these contigs had little or no homology to any contemporaneous multi-domain (i.e., bacterial, viral, fungal, eukaryotic pathogens) microbial databases, suggesting that cell-free DNA from hundreds of previously uncharacterized bacteria and viruses existed in human plasma, several of which were validated by orthogonal analyses.[90] Future characterization of novel, cell-free microbial diversity in patients with cancer may thus further improve performance of microbiome-informed, plasma cancer diagnostics, which have thus far relied on mapping non-human reads against curated databases of microbial genomes. Additionally, although cf-mbDNA’s origins remain unknown, possible sources include oral, gut, and intratumoral microbiomes.[53,61,62,91,92] We speculate that the cf-mbDNA test’s strength derives from the number of microbial biomarkers assayed rather than the amount of microbial DNA in plasma, as shown in fragmentomic-based liquid biopsies.[76]
Prognostics and companion diagnostics
The impact of gut and intratumoral microbiomes on local and systemic immune tone and host metabolites makes them versatile prognostics and companion diagnostics.[3] In the clinical setting of hematopoietic stem cell transplantation, higher gut microbiome diversity prognoses better survival, whereas lower gut diversity correlates with higher risks of transplant-related and graft-versus-host disease deaths (adjusted hazard ratios in two cohorts of 0.71 and 0.49).[93] Gut microbial diversity is also a prognostic indicator of overall survival in patients with cervical cancer in the context of chemoradiation (cisplatin plus brachytherapy) independent of body mass index.[94] Intratumoral microbial diversities and their impact on survival may be cancer type-specific: high intratumoral microbial diversity predicts long-term survival (median 10.1 years) in pancreatic cancer patients,[52] but high diversity in stomach adenocarcinoma tumors predicts worse patient survival.[95] Colorectal cancer stages reflect successive microbial changes in the fecal microbiome,[78,81] and early versus late stage lung cancer in clinical patients can be distinguished through lower airway microbiota compositions.[51] Intratumoral microbiomes in patients can also distinguish stage I from stage IV tumors in multiple gastrointestinal cancers (stomach, colon, rectal) and renal cell cancer.[58]
Therapeutically, numerous studies in preclinical and clinical cohorts link efficacy of anti-CTLA-4 and anti-PD-(L)1 immune checkpoint blockade (ICB) to gut microbiome composition and function,[12–14,67,96–100] and the intratumoral microbiome in human melanoma samples.[49,57] Efficacies and toxicities of cyclophosphamide,[66,101] gemcitabine,[47] and platinum-based[67,102] chemotherapy depend on the composition and metabolic capacity of gut and intratumoral microbiota.[103] Bacterial enzymes can degrade specific chemotherapy compounds into non-functional byproducts,[47] diminishing drug responses in colonized patients. In mouse models of HER2-positive breast cancer, antibiotic administration impairs trastuzumab efficacy and clinical patients with less diverse gut microbiomes are less likely to respond to trastuzumab;[68] moreover, fecal microbiota transplants from patient responders and non-responders into recipient mice recapitulate differential response to trastuzumab in patients, implicating gut microbiota as critical for HER2-targeted therapeutic responses.[68]
Gut microbiota also affect hormonal therapies. Administration of abiraterone acetate (AA) in the setting of castrate-resistant prostate cancer promoted outgrowth of Akkermansia muciniphila and appeared to aid overall AA therapeutic efficacy.[104] However, androgen deprivation therapy also increases gut-residing Ruminococcus species containing enzyme(s) (currently uncharacterized) that catalyze pregnenolone conversion to the sex hormone precursor dehydroepiandrosterone (DHEA) and testosterone, enhancing progression to castration-resistant prostate cancer; notably, CYP17A1-selective abiraterone administration inhibited androgen synthesis in cultivated bacteria, and incubation of R. gnavus with pregnenolone up-regulated 22 genes, several sharing sequence homology to CYP17, although the precise enzymes are unknown.[65] Thus, longitudinal profiling of implicated gut microbes may indicate failing androgen deprivation therapy while substantiating their removal. Estrogen-receptor-positive breast cancer may be affected by microbial hormone metabolism,[105,106] possibly including intratumoral microbes. Altogether, gut and intratumoral microbiomes affect virtually every domain of cancer therapy, supporting their clinical utility as prognostics and companion diagnostics.
Challenges for cancer microbiome diagnostics and prognostics
Low-biomass microbial sampling creates analysis challenges requiring care and decontamination.[107,108] While less impactful in gut microbiome studies or large-scale meta-analyses, external (e.g., environmental) and internal (e.g., well-to-well) contamination can skew small-to-moderate scale profiling of the cancer microbiome.[107,108] Standardized experimental contamination controls (Figure 2) alongside in silico decontamination methods[88,108] can enable more robust and reproducible results, and therefore improve microbiome-augmented cancer diagnostics and prognostics. Few cancer genomics studies implement contamination controls, but adding them would allow broad utilization of “cancer-specific” data for microbial analyses, although this may be addressable by integrating thousands of samples.
FIGURE 2.
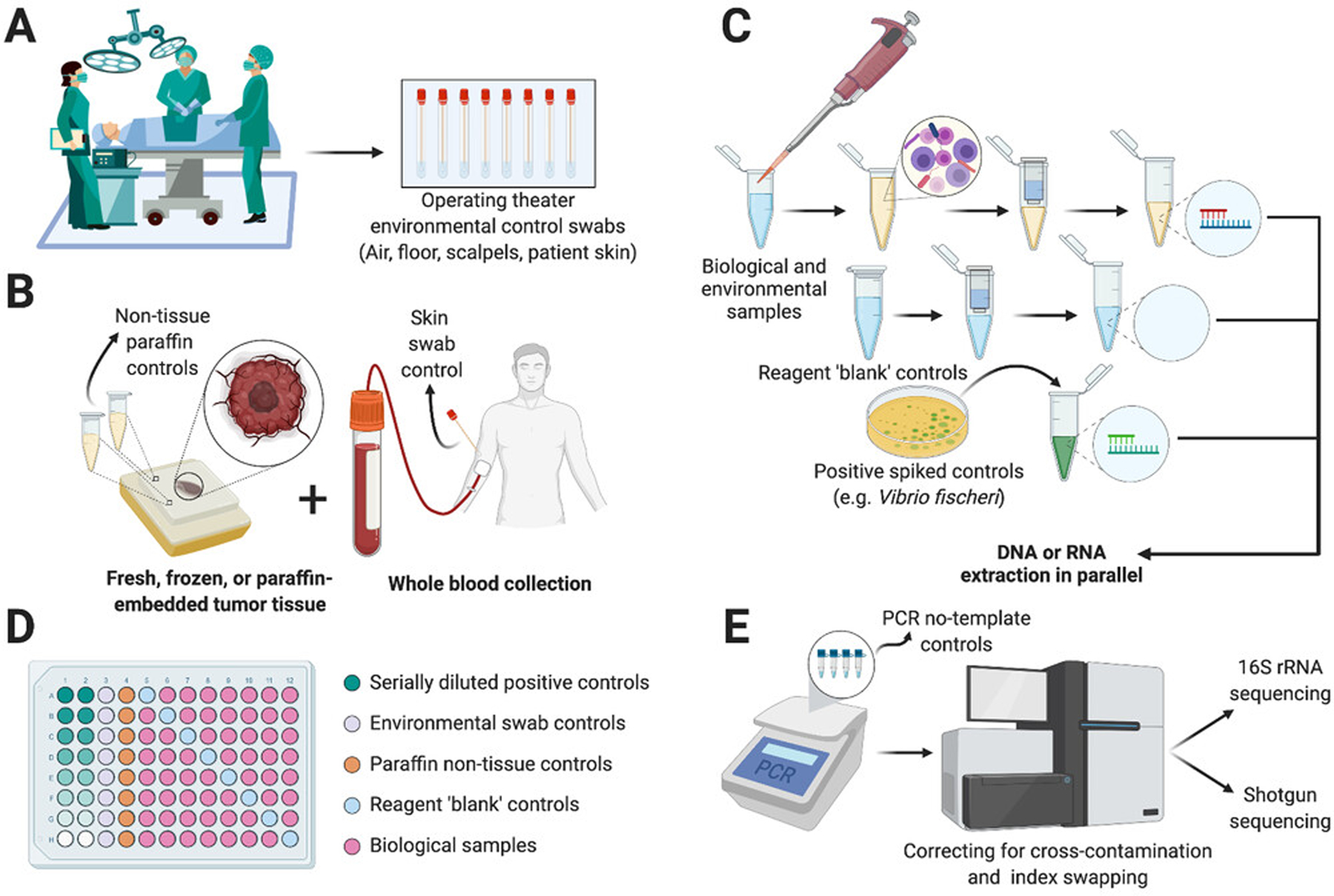
Extracting and analyzing low-biomass microbiomes requires special care to control external and internal contamination.[88,107,108] (A) Collection of environmental controls ideally begins in the operating room to account for non-patient environmental sources. (B) Post-operative tissues, if paraffin embedded, can have non-tissue paraffin controls taken to ensure the embedding process is not contaminated. Whole blood should ideally be collected with a skin swab to account for peri-needle contamination. (C) Negative reagent-only ‘blank’ controls and positive titrated controls should be processed simultaneously alongside nucleic acid extraction from biological and environmental samples. (D) Plating strategies should be considered to reduce cross-contamination; controls may include up to 40% of total samples. (E) Amplification steps may include PCR no-template controls and sequencing may include correction for cross-contamination or index swapping, although the latter remains challenging.
Other challenges with microbiome studies include (i) the degree of variation between sample and bioinformatics processing choices;[109] (ii) fundamental differences in data properties and statistics with relative abundances compared to host ‘omic’ data;[110–112] and (iii) geographical and ethnic compositional differences, particularly when assaying gut microbiota.[113–115] One or more of these factors have, for example, resulted in three major microbiome studies[12–14] concluding that different gut microbes predict anti-PD(L)1 immunotherapy response—irreconcilable despite analyses that equally reprocessed the data or instead examined microbial functions.[98] Large meta-analyses can surmount some of these problems, with two studies identifying conserved gut microbial signatures predictive of colorectal cancer across diverse cohorts and geographies.[116,117]
Strategies for assaying low-biomass microbes
Alternative strategies involving host-depletion and microbial enrichment methods may circumvent low-biomass challenges. Three categories of host depletion strategies exist: pre-extraction, post-extraction, and during sequencing.[118,119] Pre-extraction methods use differential lysis plus enzymatic or chemical (e.g., propidium monoazide) cell-free DNA removal.[119,120] Post-extraction methods remove nucleic acids by targeting species-specific chemical moieties (e.g., methylation marks).[121,122] Nanopore sequencing methods enable dynamic rejection of human-aligning reads,[118,123,124] and combining strategies can optimize host depletion.[118]
Microbial enrichment strategies often use post-extraction amplicon sequencing but with limited taxonomic resolution and amplification biases.[125] qPCR[126] and microarrays[127] are “post-extraction” but limited by genomic region-specificity, fragment size, and practical multiplexing. Stitching multiple 16S rRNA amplicons bioinformatically[128] improves taxonomic resolution and biases; this method identified intratumoral bacterial species.[57] High fidelity, long-read sequencing of full-length 16S with single-base precision can provide species or strain-level resolution.[125,129] Pre-extraction methods (e.g., microfluidics) selectively isolate bacteria[130] but present challenges in solid tissues. Selective microbial sequencing can improve low-biomass read depths.[123] Although challenges remain for commercial host depletion and microbial enrichment strategies,[119,131] advancements would tremendously aid low-biomass, intratumoral and blood-based cancer microbiome characterization.
CANCER CLONALITY AS A MULTISPECIES PROCESS
Redefining conventional cancer clonal evolution and selection
Cancer cells evolve through space and time. Although clonal evolution has traditionally centered on genetic alterations,[37,132,133] non-genetic alterations (e.g., epimutations) also contribute.[134–136] Single-cell multi-omics and longitudinal studies offer more inclusive, multi-analyte views of intratumor heterogeneity and clonal evolution.[137,138] Recognizing the role of multi-omics in functional clonal diversity advocates for broader definitions beyond cancer genomics.[38]
Research demonstrating effects of extracellular and intracellular microbes on cancer cells’ genomes,[85,139] transcriptomes,[48,50,51,140] proteomes,[49] and metabolomes[47,65] strongly justify microbial inclusion in multi-omic clonal evolution models (Figure 3). Additional microbial functions that enable or abolish chemo-, radio-, and/or immunotherapy efficacy without any cancer genomic changes further substantiate their inclusion.[3,11,141] Intracellular localization of metabolically active, immunogenic cancer microbes that shape cancer immunoediting—evolutionary processes and selection pressures previously privileged to cancer clonal selection—also provides justification.[49,57,63] Identification of hybrid microbial-human reads involved in carcinogenesis on plasmid-like ecDNA segments intimately links cancer and microbial fitness.[45,46] Microbial mechanisms that modify immunosurveillance also impact when and where tumors grow and/or metastasize.[49,53,54,61,62,142] As distinct agents from cancer cells with separate genetic material that can face discordant selection pressure(s) from the cancer genome (e.g., antibiotic therapy for bacteria, targeted kinase therapies for cancer cells), cancer microbes cannot merely be added as another “-ome.” Studies examining cancer microbiota roles have not yet seriously considered their clonality or impacts on cancer cell clonality. Thus, there is a theoretical gap between the cancer microbiota and clonal evolution modeling that should be bridged.
FIGURE 3.
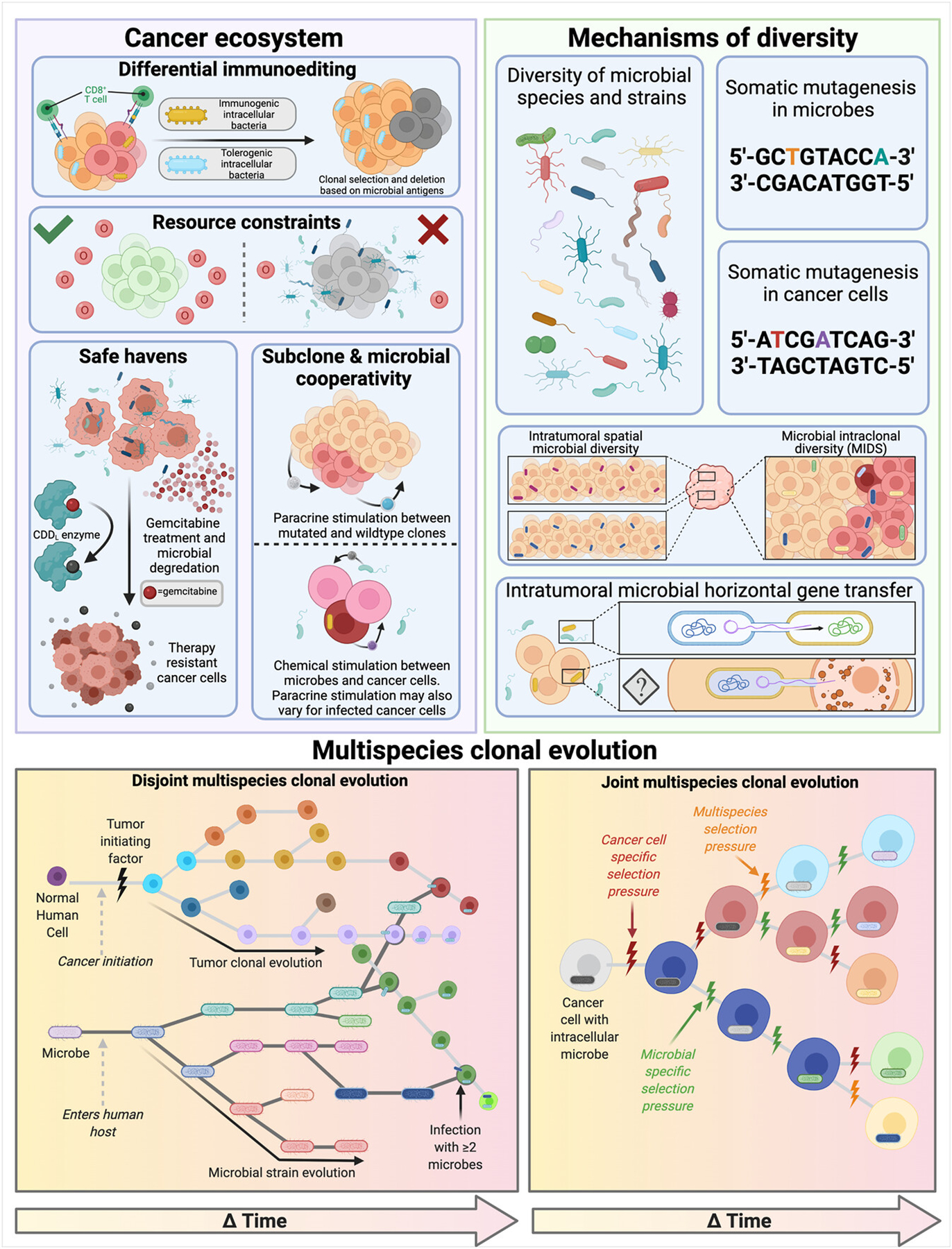
Impacts of intratumoral microbes on cancer evolution and arguments for multispecies clonal evolution. Effects are summarized into three major categories: modulation of ecosystem effects, mechanisms of clonal diversity, and example disjoint and joint phylogenetic clonal evolution. Upper figure subpanels depict supported and hypothesized microbial counterparts to the host-specific clonal selection modes, mechanisms, and ecosystems originally described by McGranahan and Swanton.[37]
Key evidence advocating for multispecies cancer clonality
Decoupling of therapeutic efficacy from human genetic changes
The genetic model of clonal evolution explains mutagenic-induced relapses. For example, epidermal growth factor receptor (EGFR) mutations induce resistance to EGFR inhibitors while creating favorable selection pressures for mutated cells over non-mutated counterparts.[143,144] Consequently, EGFR-mutated cells outcompete their neighbors and clonally expand.
However, this model has its limitations. For instance, isocitrate dehydrogenase (IDH1/IDH2)-mutated acute myeloid leukemia patients treated with IDH1/2 inhibitors can show complete and sustainable responses to treatment without eliminating mutated cells.[145–147] The same is observed in chronic myelomonocytic leukemia responsive to hypomethylating agents—no decrease in the mutational load and no specific selection events explain secondary escape.[148] Moreover, despite clear cancer cell burden reductions, thereby generating a selective bottleneck, relapse can occur without genetic selection. For example, in childhood B-cell precursor acute lymphoblastic leukemia, Turati et al. demonstrated how vincristine and dexamethasone drastically reduced the leukemic burden with no change in clonal composition.[149] Conversely, a transcriptional bottleneck was observed in single-cell RNA-Seq, with reduced transcriptomic intratumor heterogeneity. A similar resistant transcriptomic profile was found in the leukemic cells before treatment, suggesting positive selection of these rare pre-existing resistant cells rather than induction of that phenotype under treatment exposure. These resistant cells comprised a subfraction of low cycling cells and have been associated with distinct metabolic programming.[149,150] Several hypotheses concerning non-genetic resistance to therapies are debated.[151]
Genetic clonal evolution also fails to account for microbial-mediated treatment efficacy or failure of (i) cyclophosphamide,[66,101] gemcitabine,[47] and platinum-based[67,102] chemotherapy; (ii) anti-CTLA-4 and anti-PD-(L)1 ICB efficacy;[12–14,96–100] (iii) androgen deprivation therapy in prostate cancer;[65] and (iv) trastuzumab in HER-2-positive breast cancer.[68] Notably, some of these examples rely on microbial genetic content (e.g., cytidine deaminase long (CDDL) isoforms degrading gemcitabine),[47] which may be shared through conventional horizontal gene transfer and may also be intracellular. Similarly, clonal selection may entirely occur on CDDL-containing microbes by providing growth advantages to microbes that can metabolize the concentrated carbon source, and cancer cell survival may be tied to CDDL+-microbe proximity. Yet, cancer genome-centric evolutionary models miss all these effects and fail to accurately forecast evolutionary changes.
Impact of intracellular bacteria on cancer cell properties and fitness
Immunohistochemistry, immunofluorescence, and electron microscopy data document intracellular localizations of intratumoral bacteria.[49,57,61,63] Bacteria inside cancer cells modify their properties—transcriptional states,[63] proteomes,[49] and metabolic repertoires[47,57]—in ways tied to clonal evolution. Extracellular bacteria also modulate these properties and cause cancer cell genomic mutations.[85,139,152] Key affected clonal aspects comprise cancer cell metabolism, immunoediting, clonal expansion and metastasis, and mutagenesis.
First, intracellular microbes change host cell metabolism, including degradation of exogenous chemotherapy[47] and xenobiotic D-alanine.[57] Geller et al. identified microbial gemcitabine resistance through incidental Mycoplasma contamination of cell cultures, which caused drug resistance.[47] Isolating the responsible enzyme and its drug-degrading isoform (CDDL), and bioinformatic searches, revealed >300 CDDL+ species, 98.4% within Gammaproteobacteria. Imaging, sequencing, and cultivation from gemcitabine-associated pancreatic cancer patient biopsies revealed CDDL+ bacteria that conferred gemcitabine resistance in co-cultures with cancer cell lines.[47]
Second, intratumoral microbes modulate the immune response. Fusobacterium nucleatum inhibits natural killer cell (NK)-dependent tumor killing through Fap2 interaction with TIGIT, constituting a bacterium-dependent mechanism of tumor-immune evasion.[153] Pancreatic cancer bacteria induce innate and adaptive immune suppression, including via selective Toll-like receptor ligation leading to T-cell anergy.[48] Conversely, a metastatic melanoma study elucidated immunogenic, MHC I and II-bound bacterial peptides presented on cancer and immune cells that putatively shape cancer immunoediting, positting gut-tumor antigenic overlap.[49] Thus, uneven partitioning of microbes among cancer cells could result in differential elimination or maintenance, enriching the traditional “3Es” of elimination, equilibrium, and escape[154] and documenting how cancer cell fitness is decoupled from the cell’s own genome.
Third, intratumoral microbes can favor metastases. Bullman et al. found Fusobacterium persistence in colorectal cancers through successive mouse xenografts and similar bacterial compositions in matched primary-metastasis (colorectal-liver) patient samples.[61] Metronidazole treatment reduced tumor growth, implying greater fitness from Fusobacterium colonization.[61] Bertocchi et al. then showed that colorectal bacteria stepwise enter tumor tissue, modify the gut vascular barrier, migrate to the liver, and foster formation of a premetastatic niche favoring metachronous metastasis.[62] Parhi et al. noted earlier metastasis of Fusobacterium-seeded breast cancers.[53] Hence, intratumoral bacteria enhance metastasis.
Fourth, microbes cause genotoxin-mediated mutagenesis.[85,139] pks+ E. coli generates mutational signatures in head and neck, colorectal, and urinary tract cancers.[139] Gut-residing Proteobacteria produce cytolethal distending toxin (CDT) that induces single- and double-stranded DNA breaks.[152] Collectively, these mechanisms shape cancer cell properties and fitness.
Implications and hypotheses if cancer clonality is multispecies
Imaging data portray intracellular bacteria as heterogeneously distributed among cancer cells and tumor regions,[57,61] suggesting differential fitness at the single cell level that may not correspond with mutational status. This challenges the definition of cancer clones as private lineages of mutated cells stemming from common ancestors, and violates modeling assumptions whereby clonal lineages comprise homogeneous cell populations. Although no two cancer cells are equal in every respect, the primary assertion of clonality is that intraclonal individual differences are negligible.[155] Moreover, if intracellular bacteria differentially alter phenotypes, behaviors, and fitness of spatially-adjacent cancer cells, then they create major intraclonal heterogeneity, which we define as “microbial intraclonal diversity” (MIDS). MIDS challenges clonal lineage homogeneity and motivates revising clonal boundaries, most simplistically through further subsetting (e.g., KRAS-mutated, Fusobacterium-infected cells versus KRAS-mutated, uninfected cells) or more accurately through revised modeling approaches that account for discordant microbe-cancer selection pressures. MIDS also includes mimicry between microbial and cancer antigens.[156,157] Should genetic cargo be shared between intracellular bacteria and host cells, as biotechnology already shows is possible[24] and cancer virology affirms,[45,46] MIDS must account for DNA and RNA from multiple species.
Beyond challenging clonal boundaries, intracellular bacteria may require evolutionary tree revision, particularly if future evidence demonstrates heritability (i.e., at time of division during mitosis or infection post-division) of intracellular microbes. Typical clonal evolution models depict evolutionary trees with one trunk and several branches, assuming vertically transmitted traits from mother to daughter cells. If future research affirms horizontal/lateral gene transfers between intracellular bacteria and host cancer cells, multiple tree trunks and branch connections would be required. A similar debate has taken place in evolutionary biology, challenging the traditional Darwinian “tree of life.”[158–160] Clonal evolution may be better articulated as “reticulated evolution,” wherein horizontal/lateral transfers change the fitness, function, and/or phenotype of host cancer cells.
Considerations for cancer microbiome therapeutics under multispecies clonality
Multispecies cancer clonality offers new therapeutic strategies that neither human nor microbial clonality proposes. For instance, Byndloss et al. demonstrated an interplay between fastidious anaerobic gut bacteria and butyrate-mediated, PPAR-γ-dependent host signaling that maintained low oxygen levels in the gut and prevented outgrowth of facultative pathogens.[161] Conversely, antibiotics increased gut oxygen concentration and pathogen outgrowth.[161] Analogously, there may be opportunities to target host processes that facilitate microbial homeostasis to mitigate microbial-mediated carcinogenesis in favor of blunted antibiotics. Butler et al. provide another example whereby administration of a bacterial protease depleted cellular MYC in colon and bladder cancers.[162] Similarly, identification of anticancer microbial enzymes or metabolites may provide effective host-modulating cancer therapies or improve efficacy of existing therapies—a strategy several groups have already taken with immunotherapy.[99,100]
EVOLUTIONARY MODELING OF THE CANCER MICROBIOME
Example of Helicobacter pylori
Helicobacter pylori is a well-studied example of microbial effects on the TME,[163] and has adapted to thrive in the majority of humans long enough to trace migration events.[164] H. pylori has co-evolved protective and pathogenic roles: protective against gastric cardia and esophageal adenocarcinoma[165,166] and pathogenic in noncardiac gastric cancer.[163] Most H. pylori-positive patients carry multiple strains, including at least one strain unique to their body alongside more common strains like VacA, CagA, and BabA.[164] This extreme genetic diversity stems from slipped-strand mispairing in multiple genes and H. pylori’s lack of typical DNA repair genes.[164] High strain diversity across individual human hosts also enhances H. pylori’s population-wide resilience, expanding the strain with highest fitness in each setting. Collectively, high diversity and concomitant mutagenesis of H. pylori, combined with human immune selection pressures and pathological impacts on noncardiac gastric carcinogenesis, help portray an exemplary “big picture” of multispecies cancer evolution. Building on this idea, we describe how existing clonal evolution modeling may take intratumoral microbes into account.
Common constraints of the tumor microenvironment
The TME contains intracellular and extracellular microbes that affect cancer fitness and comprise independent clonal agents. The TME contexture applies simultaneous, shared selective pressures and environmental constraints on co-located cancer cells and microbes. Shared resources necessitate cooperative use and/or competition, which may further limit their abundance. For instance, oxygen availability drives spatial organization and metabolic capacities of cancer cells[167] and similarly affects microbes in environmental contexts and model systems (e.g., Winogradsky columns).[168–170] Common selection pressures may drive common evolutionary solutions, such as metabolic symbiosis between cancer cells[171] or between microbes positioned along an oxygen gradient.[170] pH gradients are tied to oxygen and common in tumors,[167] and shape microbial compositions in environmental contexts.[172] Hence, multispecies evolutionary models should include joint environmental constraints.
Anderson and colleagues presented compatible multiscale mathematical models of cancer growth including cellular biophysical properties and TME factors.[173–175] Their models determined that aggressive cancer clones establish under the harshest TME conditions (e.g., hypoxia, heterogenous extracellular matrix) but tumor invasiveness was blunted under milder TME conditions.[173,174] Hence, chemotherapy that creates a harsh microenvironmental condition may worsen long-term cancer invasiveness. Incorporating microbes into similar multiscale model equations—their reliance and impacts on TME chemical gradients, cancer metabolism, and therapy—could inform TME conditions that improve multispecies clonal dynamics.
Microbes affect clonal fitness
Current fitness models account for factors including probabilities of cell division and cell death alongside inferred mutation rates and human cancer driver genes. Intracellular microbes may also need to be included, particularly their mutational, division, and death rates. In circumstances of microbial enzymatic degradation of TME metabolites or drugs,[47] transcriptional rates and enzymatic efficiencies may comprise important variables. In genotoxin-mediated mutagenesis (e.g, pks+ E. coli),[139] the base-pair motif and rate of mutations in cancer cells may be instructive to add, since cancer cells in the presence of pks+ E. coli may have higher mutation rates.
Likelihood of clone development, treatment resistance, and fitness are all major parts of clonal evolution models and related to extracellular and intracellular microbes, but microbes have not been typically considered within models of cancer evolution. In common population genetics models of clonal evolution, including Wright-Fisher diffusion type models[176] and Moran type models,[177] clonal fitness may be considered as a function of time-dependent fluctuations in microbial abundances or presence/absence of particular species. Branching-process stochastic models of tumor growth that parameterize evolution in terms of proliferation and mutation rates[178–180] may also benefit from considering microbial colonization rates and species-specific transcriptional or mutational effects when found to have a significant effect on cancer cell evolution. Furthermore, cancer microbes individually (and perhaps jointly) undergo somatic clonal evolution, as with H. pylori, and similar modeling techniques quantify evolutionary dynamics in microbial populations (e.g., emergence of community-level heredity).[181] Phylogenetic tree reconstructions of clonal evolution[182] may need to include multispecies lineages, but specialized methods are likely necessary. Finally, existing software can construct genome-scale metabolic models (GEMs), including host-microbe interactions,[183] to quantify microbial production/consumption of metabolites and determine impacts on host metabolism.[184,185] GEMs recently characterized bacterial passengers in colorectal cancer (CRC) based on metabolite availability[186] and predicted CRC therapeutic drug targets.[187] To summarize, we created a table listing suggested strategies for adapting existing models to study multispecies clonal interactions and evolution while incorporating cancer cell and microbial information (Table 2).
TABLE 2.
Microbial integration into mathematical models of cancer evolution. Overview of each model’s characteristics and references provided with modeling examples, as well as suggested ways that both microbes and cancer cells could be incorporated into models if not yet commonly applied in this context. Hybrid models that include aspects of more than one model type are also utilized in practice. CRC: colorectal cancer.
| Model Type | Overview of Model | Examples of Potential Incorporation of Host-Microbe Cancer Interactions |
References |
|---|---|---|---|
| Genome-scale metabolic model (GEM) | • Models metabolic interactions between microbiome and host • Analyzes genotype-phenotype relationships by connecting metabolic genes with their corresponding metabolic pathways • Can be used to analyze the Warburg effect in cancer cells • GEM algorithms (e.g., CASINO) allow several microbial species (≥5 species) to be modeled |
• Current uses include modeling host-microbe interactions (e.g., quantifying how microbiota interactions impact host physiology, including applications in CRC) • Potential to explicitly incorporate metabolite release by microbes and consumption (e.g., glucose) by cancer cells in an evolutionary model |
[169–173,198–202] |
| Generalized Lotka-Volterra (gLV) model | • Predicts the population dynamics of cancer cells or microbes and incorporates growth rates, interaction strength or competition between groups, and environmental changes • Classical predator-prey LV model defined as two competing populations that affect one another’s growth, but can be generalized to an arbitrary number of coexisting populations |
• Quantify competition between cancer cells using relative abundance of microbes • Microbes and cancer cells could both be considered as distinct populations competing amongst each other over shared resources or sustaining a mutualistic relationship in the model with defined interaction strengths |
[170,203,204] |
| Agent-based model | • Define ‘agents’ as individuals or members of the microenvironment with specific properties and actions on a structured grid or 3D space • Can have stochastic and deterministic components with spatial constraints • Define environmental rules such as chemotaxis, and the presence of factors in space, such as signalling proteins like VEGF • Define agent-agent interaction rules |
• Create microbe as one agent type in the microenvironment and cancer cell as another agent type • Allow clonal evolution of cancer cells and separate evolution of microbes in equations • Create biophysical rules accounting for spatial movement of microbes (e.g., quorum sensing) and effect of microbes on evolutionary rates, such as proliferation or death of cancer cells during drug delivery |
[205–208] |
| Wright-Fisher type model | • Population size remains constant over time (can be extended to growing populations) • Models consider finite number of population species/k-alleles among either microbes (e.g., bacteria, viruses) or cancer cells • To create the next non-overlapping generation, alleles are randomly sampled with replacement • Allele frequency in the new generation is the combination of random sampling of population and the fitness of alleles • Captures genetic drift and natural selection if included |
• Microbial species could undergo distinct Wright-Fisher evolutionary dynamics that are independent of, or, in turn, affect cancer cell evolution • Effects of microbes present could also be interwoven into cancer cell fitness evolving under Wright-Fisher dynamics • Fitness parameter of certain cancer cell genotypes may depend on metabolites, proteins, and antigens from intracellular bacteria, which in certain cases may drive differential immunoediting between cancer cell-bearing bacteria |
[162,209–211] |
| Moran-type model | • Two or more species considered in a population of either microbes or cancer cells • Asexual reproduction, overlapping generations • Simultaneous birth and death events occur • As in the Wright-Fisher model, can be formulated as a diffusion approximation |
• Similar to the Wright-Fisher type model, microbial species could be considered distinct population genotypes undergoing evolutionary dynamics, or the effects of microbes could be interwoven into the fitness of cancer cells • Fitness parameter of certain cancer cell genotypes may depend on metabolites, proteins, and antigens from intracellular bacteria, which in certain cases may drive differential immunoediting between cancer cell-bearing bacteria |
[163,212–214] |
| Birth-death stochastic process | • Continuous time Markov model (branching process) where ‘birth’ or ‘death’ events can change the state/population size • A ‘birth’ increases the state by one, a ‘death’ decreases the state by one • Allows for multiple cell types (e.g., with/without driver mutations), fluctuations in total population size, stochastic extinction of cells, and mutation to other types • Can also be used to model eco-evolutionary dynamics of microbial communities |
• Define properties of stochastic events such as survival for human cancer cells with: ◦ Probability of birth, death, and/or mutation affected by products of microbes in the microenvironment ◦ Probability of birth, death, and/or mutation dependent on a function of the fluctuating populations of intracellular microbes present • Define stochastic events in terms of both human cancer cells and microbial populations |
[167,215,216] |
| Evolutionary game theory model | • Includes density-dependent fitness with cell-cell interactions • Models cooperation (e.g., between tumor and stromal cells, or between bacteria) • Fitness landscapes in non-cancer models have been central to understanding microbial evolution such as E. coli |
• Include microbes as a type of “player” in the modeled ecosystem alongside tumor cells for limited chemicals and nutrients (e.g., oxygen, sugars) • “Public good” produced by tumor cells, such as lactate, included in a game as competing resources with microbial populations |
[217–222] |
CONCLUSIONS
Rigorous studies provide extensive evidence for the existence and functionality of cancer-associated gut and intratumoral microbes while echoing ancient historical narratives of microbial-mediated recovery. Drawing from cancer genomics, the high diversity of the cancer microbiome substantiates its strong predictive power, even with rare or low-abundance microbes. Cancer microbiota can distinguish healthy, pre-cancer, and cancerous tissues across multiple cancer and sample types, although most diagnostics remain unvalidated in large, multi-national, prospective cohorts. Cancer microbiota also demonstrate stage-specific differences that may enable simultaneous identification and prognostication of tumors. Nonetheless, contamination challenges in low-biomass settings and analytic idiosyncrasies of microbiomic data have hitherto complicated routine clinical application of microbial-informed cancer diagnostics or prognostics. Recent and future advances in host depletion and microbial enrichment strategies may help mitigate low-biomass characterization challenges.
Numerous microbial mechanisms affect the cancer genome, transcriptome, proteome, and metabolome, advocating for their inclusion in cancer evolution models. Extracellular and intracellular microbes affect most cancer medication classes’ efficacies, sometimes without any cancer cell(s) interventions. Negatively, it is not possible to accurately model cancer-drug dynamics, clonality, or fitness without accounting for microbes. Serious consideration of multispecies clonality is complex because microbes carry distinct genetic cargo with discordant selection pressures from the cancer genome. Flexible evolutionary models treating intratumoral microbes as independent, albeit rule-abiding, agents within the TME may be appropriate. Multispecies clonality also informs treatments: modifying cancer pathways may be more effective to restore healthy microbial ecologies than targeted antimicrobials, and vice versa. Multispecies treatment strategies may benefit from target selectivity, because targeting microbial genes or proteins generally carries fewer side effects than targeting human counterparts. Altogether, understanding cancer’s metagenome carries key ramifications for cancer care and clonal evolution for the benefit of patients worldwide.
TABLE 1.
Abundance estimates of intratumoral bacteria among seven major human cancers profiled by Nejman et al. (Ravid Straussman, personal communication).[57] One thousand iteration-bootstraps of the mean and median approximated the average number of bacteria per 40 nanograms of DNA on a per-cancer and pan-cancer basis. Conversions and assumptions of tumor ploidy, purity, and homogeneity are detailed in Box 1. Area density estimates assume 5200 total cells/mm2 and volume density estimates assume 109 total cells/cm3. In the two far-right columns, “med.” denotes the propagated “median” bootstrapped value and “avg.” denotes the propagated “average” bootstrapped value.
| Cancer type in Nejman et al. 2020[57] | qPCR sample size (n) | Absolute range (bacteria/40ng) (min, max) | Bootstrapped estimate of median bacteria per 40 ng DNA (median, 95% CI; 1000 iterations) | Bootstrapped estimate of average bacteria per 40 ng DNA (mean, 95% CI; 1000 iterations) | Area density estimate (bacteria/mm2) | Volume density estimate (bacteria/cm3) |
|---|---|---|---|---|---|---|
| Melanoma | 200 | (0.85, 3023) | 6.72 (5.99, 7.77) | 31.69 (9.71, 71.20) | ~7 med., ~33 avg. | ~1.3×106 med., ~6.3×106 avg. |
| Lung | 274 | (1.2, 3663) | 5.83 (5.13, 6.48) | 22.50 (7.90, 50.35) | ~6 med., ~23 avg. | ~1.2×106 med., ~4.5×106 avg. |
| Ovarian | 57 | (1.84, 73.2) | 9.88 (7.59, 12.37) | 12.72 (10.25, 16.00) | ~10 med., ~13 avg. | ~2.0×106 med., ~2.5×106 avg. |
| GBM | 37 | (3.41, 77.4) | 10.06 (6.62, 15.85) | 15.55 (10.89, 20.85) | ~10 med., ~16 avg. | ~2.0×106 med., ~3.1×106 avg. |
| Pancreatic | 66 | (3.82, 2147) | 17.17 (11.72, 22.36) | 70.43 (26.19, 147.78) | ~18 med., ~73 avg. | ~3.4×106 med., ~14×106 avg. |
| Bone | 30 | (1.62, 76.4) | 15.53 (12.78, 18.53) | 19.33 (13.97, 25.51) | ~16 med., ~20 avg. | ~3.1×106 med., ~3.9×106 avg. |
| Breast | 352 | (0.765, 1723) | 14.82 (13.35, 16.39) | 44.63 (31.41, 59.83) | ~15 med., ~46 avg. | ~3.0×106 med., ~8.9×106 avg. |
| Pan-cancer | 1016 | (0.765, 3663) | 9.15 (8.70, 9.60) | 34.19 (24.04, 46.56) | ~10 med., ~35 avg. | ~1.8×106 med., ~6.8×106 avg. |
ACKNOWLEDGEMENTS
We thank C. Sepich-Poore for providing critical review and feedback on the manuscript and figures. Figures were created with BioRender.com. The authors also wish to acknowledge the patients and their families who have helped contribute towards a better understanding of this field.
FUNDING SOURCES
G.D.S-P. is supported by a fellowship from the US National Institutes of Health, National Cancer Institute (F30 CA243480). T.P. has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme - grant agreement n° 637647 – IDEM (P.I.: T. Pradeu). L.L. is funded by the Cancéropôle Île-de-France (n°2021-1-EMERG-54-CNRS DR 5–1) and her team is supported by the Ligue National contre le Cancer (P.I.: F. Porteu). R.K. is funded in part by grants from the National Cancer Institute within the National Institutes of Health (R01 CA255206, U24 CA248454). R.K. and C.G. are funded by Mapping host-microbe-metabolite interactions in 3D to find diet-derived enhancers of immunity (NIH 1DP1AT010885); Advancing our Understanding of Cancer and the Human Microbiome with QIIME 2 (NIH U24CA248454) and NIH Director’s Pioneer Award (NIH DP1AT010885). C.G. and K.C. are funded by Moores Cancer Center Support Grant NIH (NCI P30 CA023100).
Abbreviations:
- cf-mbDNA
cell-free microbial DNA
- ctDNA
cell-free tumor DNA
- CDDL
cytidine deaminase long isoform
- CRC
colorectal cancer
- ecDNA
extrachromosomal DNA
- FAP
familial adenomatous polyposis
- GEM
genome-scale metabolic model
- HPV
human papilloma
- ICB
immune checkpoint blockade
- MIDS
microbial intraclonal diversity
- PCAWG
Pan-Cancer Analysis of Whole Genomes
- PMP
pre-leukaemic myeloproliferation
- qPCR
quantitative polymerase chain reaction
- RSV
Rous’s Sarcoma Virus
- TCGA
The Cancer Genome Atlas
- TME
tumor microenvironment
Footnotes
CONFLICTS OF INTEREST
G.D.S.-P. and R.K. are inventors on a US patent application (PCT/US2019/059647) submitted by The Regents of the University of California and licensed by Micronoma, covering methods of diagnosing and treating cancer using microbial biomarkers in blood and cancer tissues. G.D.S.-P. and R.K. are founders of and report stock interest in Micronoma. G.D.S.-P. has filed several additional US patent applications on cancer microbiome diagnostics that are owned by The Regents of the University of California. R.K. additionally is a member of the scientific advisory board for GenCirq, holds an equity interest in GenCirq, and can receive reimbursements for expenses up to US $5,000 per year.
REFERENCES
- 1.Ebbell B (1937). The Papyrus Ebers: the greatest Egyptian medical document Levin & Munksgaard. [Google Scholar]
- 2.Hoption Cann SA, van Netten JP, & van Netten C (2003). Dr William Coley and tumour regression: a place in history or in the future. Postgraduate Medical Journal, 79(938), 672–680. [PMC free article] [PubMed] [Google Scholar]
- 3.Sepich-Poore GD, Zitvogel L, Straussman R, Hasty J, Wargo JA, & Knight R (2021). The microbiome and human cancer. Science, 371(6536). 10.1126/science.abc4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busch W (1868). Aus der Sitzung der medicinischen Section vom 13 November 1867. Berl Klin Wochenschr, 5, 137. [Google Scholar]
- 5.Fehleisen F (1882). Ueber die Züchtung der Erysipelkokken auf künstlichem Nährboden und ihre Übertragbarkeit auf den Menschen. Deutsche Medizinische Wochenschrift , 8(31), 553–554. [Google Scholar]
- 6.Coley WB (1893). The treatment of malignant tumors by repeated inoculations of erysipelas: With a report of ten original cases. 1. The American Journal of the Medical Sciences, 105(6), 487. [PubMed] [Google Scholar]
- 7.Starnes CO (1992). Coley’s toxins in perspective. Nature, 357(6373), 11–12. [DOI] [PubMed] [Google Scholar]
- 8.Hobohm U (2001). Fever and cancer in perspective. Cancer Immunology, Immunotherapy: CII, 50(8), 391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Unproven Methods of Cancer Treatment. (1961). CA: A Cancer Journal for Clinicians, 11(5), 191–193. [DOI] [PubMed] [Google Scholar]
- 10.Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, & Wargo JA (2018). The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell, 33(4), 570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zitvogel L, Ma Y, Raoult D, Kroemer G, & Gajewski TF (2018). The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science, 359(6382), 1366–1370. [DOI] [PubMed] [Google Scholar]
- 12.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, Cogdill AP, Zhao L, Hudgens CW, Hutchinson DS, Manzo T, Petaccia de Macedo M, Cotechini T, Kumar T, Chen WS, … Wargo JA (2018). Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science, 359(6371), 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, Fidelle M, Flament C, Poirier-Colame V, Opolon P, Klein C, Iribarren K, Mondragón L, Jacquelot N, Qu B, … Zitvogel L (2018). Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science, 359(6371), 91–97. [DOI] [PubMed] [Google Scholar]
- 14.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre M-L, Luke JJ, & Gajewski TF (2018). The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science, 359(6371), 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin J-M, Morrison RM, Deblasio RN, Menna C, Ding Q, Pagliano O, Zidi B, Zhang S, Badger JH, Vetizou M, Cole AM, Fernandes MR, Prescott S, Costa RGF, Balaji AK, … Zarour HM (2021). Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science, 371(6529), 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, Adler K, Dick-Necula D, Raskin S, Bloch N, Rotin D, Anafi L, Avivi C, Melnichenko J, Steinberg-Silman Y, Mamtani R, Harati H, Asher N, Shapira-Frommer R, … Boursi B (2020). Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 10.1126/science.abb5920 [DOI] [PubMed]
- 17.White MK, Pagano JS, & Khalili K (2014). Viruses and human cancers: a long road of discovery of molecular paradigms. Clinical Microbiology Reviews, 27(3), 463–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rous P (1911). A SARCOMA OF THE FOWL TRANSMISSIBLE BY AN AGENT SEPARABLE FROM THE TUMOR CELLS. The Journal of Experimental Medicine, 13(4), 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin GS (2004). The road to Src. Oncogene, 23(48), 7910–7917. [DOI] [PubMed] [Google Scholar]
- 20.Stehelin D, Varmus HE, Bishop JM, & Vogt PK (1976). DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature, 260(5547), 170–173. [DOI] [PubMed] [Google Scholar]
- 21.Dulbecco R (1986). A turning point in cancer research: sequencing the human genome. Science, 231(4742), 1055–1056. [DOI] [PubMed] [Google Scholar]
- 22.Simons PJ, & Dougherty RM (1963). ANTIGENIC CHARACTERISTICS OF THREE VARIANTS OF ROUS SARCOMA VIRUS. Journal of the National Cancer Institute, 31, 1275–1283. [PubMed] [Google Scholar]
- 23.Svet-Moldavsky GJ (1957). Development of multiple cysts and of haemorrhagic affections of internal organs in albino rats treated during the embryonic or new-born period with Rous sarcoma virus. Nature, 180(4597), 1299–1300. [DOI] [PubMed] [Google Scholar]
- 24.Mulligan RC, & Berg P (1980). Expression of a bacterial gene in mammalian cells. Science, 209(4463), 1422–1427. [DOI] [PubMed] [Google Scholar]
- 25.Grillot-Courvalin C, Goussard S, Huetz F, Ojcius DM, & Courvalin P (1998). Functional gene transfer from intracellular bacteria to mammalian cells. Nature Biotechnology, 16(9), 862–866. [DOI] [PubMed] [Google Scholar]
- 26.Laner A, Goussard S, Ramalho AS, Schwarz T, Amaral MD, Courvalin P, Schindelhauer D, & Grillot-Courvalin C (2005). Bacterial transfer of large functional genomic DNA into human cells. Gene Therapy, 12(21), 1559–1572. [DOI] [PubMed] [Google Scholar]
- 27.Darji A, Guzmán CA, Gerstel B, Wachholz P, Timmis KN, Wehland J, Chakraborty T, & Weiss S (1997). Oral somatic transgene vaccination using attenuated S. typhimurium. Cell, 91(6), 765–775. [DOI] [PubMed] [Google Scholar]
- 28.Sizemore DR, Branstrom AA, & Sadoff JC (1995). Attenuated Shigella as a DNA delivery vehicle for DNA-mediated immunization. Science, 270(5234), 299–302. [DOI] [PubMed] [Google Scholar]
- 29.Johnson SA, Ormsby MJ, McIntosh A, Tait SWG, Blyth K, & Wall DM (2019). Increasing the bactofection capacity of a mammalian expression vector by removal of the f1 ori. Cancer Gene Therapy, 26(7–8), 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pálffy R, Gardlík R, Hodosy J, Behuliak M, Resko P, Radvánský J, & Celec P (2006). Bacteria in gene therapy: bactofection versus alternative gene therapy. Gene Therapy, 13(2), 101–105. [DOI] [PubMed] [Google Scholar]
- 31.Baban CK, Cronin M, O’Hanlon D, O’Sullivan GC, & Tangney M (2010). Bacteria as vectors for gene therapy of cancer. Bioengineered Bugs, 1(6), 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. (2020). Pan-cancer analysis of whole genomes. Nature, 578(7793), 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, Khurana E, Waszak S, Korbel JO, Haber JE, Imielinski M, PCAWG Structural Variation Working Group, Weischenfeldt J, Beroukhim R, Campbell PJ, & PCAWG Consortium. (2020). Patterns of somatic structural variation in human cancer genomes. Nature, 578(7793), 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.PCAWG Transcriptome Core Group, Calabrese C, Davidson NR, Demircioğlu D, Fonseca NA, He Y, Kahles A, Lehmann K-V, Liu F, Shiraishi Y, Soulette CM, Urban L, Greger L, Li S, Liu D, Perry MD, Xiang Q, Zhang F, Zhang J, … PCAWG Consortium. (2020). Genomic basis for RNA alterations in cancer. Nature, 578(7793), 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rheinbay E, Nielsen MM, Abascal F, Wala JA, Shapira O, Tiao G, Hornshøj H, Hess JM, Juul RI, Lin Z, Feuerbach L, Sabarinathan R, Madsen T, Kim J, Mularoni L, Shuai S, Lanzós A, Herrmann C, Maruvka YE, … PCAWG Consortium. (2020). Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature, 578(7793), 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cortés-Ciriano I, Lee JJ-K, Xi R, Jain D, Jung YL, Yang L, Gordenin D, Klimczak LJ, Zhang C-Z, Pellman DS, PCAWG Structural Variation Working Group, Park, P. J., & PCAWG Consortium. (2020). Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nature Genetics, 52(3), 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGranahan N, & Swanton C (2017). Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell, 168(4), 613–628. [DOI] [PubMed] [Google Scholar]
- 38.Black JRM, & McGranahan N (2021). Genetic and non-genetic clonal diversity in cancer evolution. Nature Reviews. Cancer, 21(6), 379–392. [DOI] [PubMed] [Google Scholar]
- 39.Hanahan D, & Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell, 144(5), 646–674. [DOI] [PubMed] [Google Scholar]
- 40.Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, Kornblum HI, Taylor MD, Kaushal S, Cavenee WK, Wechsler-Reya R, Furnari FB, Vandenberg SR, Rao PN, Wahl GM, … Mischel PS (2017). Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature, 543(7643), 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, Luebeck J, Rajkumar U, Diao Y, Li B, Zhang W, Jameson N, Corces MR, Granja JM, Chen X, Coruh C, Abnousi A, Houston J, Ye Z, … Mischel PS (2019). Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature, 575(7784), 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim H, Nguyen N-P, Turner K, Wu S, Gujar AD, Luebeck J, Liu J, Deshpande V, Rajkumar U, Namburi S, & Others. (2020). Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nature Genetics, 52(9), 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verhaak RGW, Bafna V, & Mischel PS (2019). Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nature Reviews. Cancer, 19(5), 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Münch K, Münch R, Biedendieck R, Jahn D, & Müller J (2019). Evolutionary model for the unequal segregation of high copy plasmids. PLoS Computational Biology, 15(3), e1006724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deshpande V, Luebeck J, Nguyen N-PD, Bakhtiari M, Turner KM, Schwab R, Carter H, Mischel PS, & Bafna V (2019). Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nature Communications, 10(1), 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pang J, Nguyen N-P, Luebeck J, Ball L, Finegersh A, Ren S, Nakagawa T, Flagg M, Sadat S, Mischel PS, Xu G, Fisch K, Guo T, Cahill G, Panuganti B, Bafna V, & Califano J (2021). Extrachromosomal DNA in HPV mediated oropharyngeal cancer drives diverse oncogene transcription. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 10.1158/1078-0432.CCR-21-2484 [DOI] [PMC free article] [PubMed]
- 47.Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, Gavert N, Zwang Y, Cooper ZA, Shee K, Thaiss CA, Reuben A, Livny J, Avraham R, Frederick DT, Ligorio M, Chatman K, Johnston SE, Mosher CM, … Straussman R (2017). Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science, 357(6356), 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, Mohan N, Aykut B, Usyk M, Torres LE, Werba G, Zhang K, Guo Y, Li Q, Akkad N, Lall S, Wadowski B, Gutierrez J, Kochen Rossi JA, … Miller G (2018). The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discovery, 8(4), 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kalaora S, Nagler A, Nejman D, Alon M, Barbolin C, Barnea E, Ketelaars SLC, Cheng K, Vervier K, Shental N, Bussi Y, Rotkopf R, Levy R, Benedek G, Trabish S, Dadosh T, Levin-Zaidman S, Geller LT, Wang K, … Samuels Y (2021). Identification of bacteria-derived HLA-bound peptides in melanoma. Nature, 592(7852), 138–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, Ameh S, Sandel D, Liang XS, Mazzilli S, Whary MT, Meyerson M, Germain R, Blainey PC, Fox JG, & Jacks T (2019). Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell, 176(5), 998–1013.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsay J-CJ, Wu BG, Sulaiman I, Gershner K, Schluger R, Li Y, Yie T-A, Meyn P, Olsen E, Perez L, Franca B, Carpenito J, Iizumi T, El-Ashmawy M, Badri M, Morton JT, Shen N, He L, Michaud G, … Segal LN (2021). Lower Airway Dysbiosis Affects Lung Cancer Progression. Cancer Discovery, 11(2), 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, Quesada P, Sahin I, Chandra V, San Lucas A, Scheet P, Xu H, Hanash SM, Feng L, Burks JK, Do K-A, Peterson CB, Nejman D, Tzeng C-WD, … McAllister F (2019). Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell, 178(4), 795–806.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parhi L, Alon-Maimon T, Sol A, Nejman D, Shhadeh A, Fainsod-Levi T, Yajuk O, Isaacson B, Abed J, Maalouf N, Nissan A, Sandbank J, Yehuda-Shnaidman E, Ponath F, Vogel J, Mandelboim O, Granot Z, Straussman R, & Bachrach G (2020). Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nature Communications, 11(1), 3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Le Noci V, Guglielmetti S, Arioli S, Camisaschi C, Bianchi F, Sommariva M, Storti C, Triulzi T, Castelli C, Balsari A, Tagliabue E, & Sfondrini L (2018). Modulation of Pulmonary Microbiota by Antibiotic or Probiotic Aerosol Therapy: A Strategy to Promote Immunosurveillance against Lung Metastases. Cell Reports, 24(13), 3528–3538. [DOI] [PubMed] [Google Scholar]
- 55.Shi Y, Zheng W, Yang K, Harris KG, Ni K, Xue L, Lin W, Chang EB, Weichselbaum RR, & Fu Y-X (2020). Intratumoral accumulation of gut microbiota facilitates CD47-based immunotherapy via STING signaling. The Journal of Experimental Medicine, 217(5). 10.1084/jem.20192282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mima K, Sukawa Y, Nishihara R, Qian ZR, Yamauchi M, Inamura K, Kim SA, Masuda A, Nowak JA, Nosho K, Kostic AD, Giannakis M, Watanabe H, Bullman S, Milner DA, Harris CC, Giovannucci E, Garraway LA, Freeman GJ, … Ogino S (2015). Fusobacterium nucleatum and T Cells in Colorectal Carcinoma. JAMA Oncology, 1(5), 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, Rotter-Maskowitz A, Weiser R, Mallel G, Gigi E, Meltser A, Douglas GM, Kamer I, Gopalakrishnan V, Dadosh T, Levin-Zaidman S, Avnet S, Atlan T, Cooper ZA, … Straussman R (2020). The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science, 368(6494), 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, Kosciolek T, Janssen S, Metcalf J, Song SJ, Kanbar J, Miller-Montgomery S, Heaton R, Mckay R, Patel SP, Swafford AD, & Knight R (2020). Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature, 579(7800), 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Wieland A, Patel MR, Cardenas MA, Eberhardt CS, Hudson WH, Obeng RC, Griffith CC, Wang X, Chen ZG, Kissick HT, Saba NF, & Ahmed R (2021). Defining HPV-specific B cell responses in patients with head and neck cancer. Nature, 597(7875), 274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zapatka M, Borozan I, Brewer DS, Iskar M, Grundhoff A, Alawi M, Desai N, Sültmann H, Moch H, PCAWG Pathogens, Cooper CS, Eils R, Ferretti V, Lichter P, & PCAWG Consortium. (2020). The landscape of viral associations in human cancers. Nature Genetics, 52(3), 320–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, Neuberg D, Huang K, Guevara F, Nelson T, Chipashvili O, Hagan T, Walker M, Ramachandran A, Diosdado B, Serna G, Mulet N, Landolfi S, Ramon Y Cajal S, … Meyerson M (2017). Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science, 358(6369), 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bertocchi A, Carloni S, Ravenda PS, Bertalot G, Spadoni I, Lo Cascio A, Gandini S, Lizier M, Braga D, Asnicar F, Segata N, Klaver C, Brescia P, Rossi E, Anselmo A, Guglietta S, Maroli A, Spaggiari P, Tarazona N, … Rescigno M (2021). Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell, 39(5), 708–724.e11. [DOI] [PubMed] [Google Scholar]
- 63.Casasanta MA, Yoo CC, Udayasuryan B, Sanders BE, Umaña A, Zhang Y, Peng H, Duncan AJ, Wang Y, Li L, Verbridge SS, & Slade DJ (2020). Fusobacterium nucleatum host-cell binding and invasion induces IL-8 and CXCL1 secretion that drives colorectal cancer cell migration. In Science Signaling (Vol. 13, Issue 641, p. eaba9157). 10.1126/scisignal.aba9157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shiao SL, Kershaw KM, Limon JJ, You S, Yoon J, Ko EY, Guarnerio J, Potdar AA, McGovern DPB, Bose S, Dar TB, Noe P, Lee J, Kubota Y, Maymi VI, Davis MJ, Henson RM, Choi RY, Yang W, … Underhill DM (2021). Commensal bacteria and fungi differentially regulate tumor responses to radiation therapy. Cancer Cell, 39(9), 1202–1213.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pernigoni N, Zagato E, Calcinotto A, Troiani M, Mestre RP, Calì B, Attanasio G, Troisi J, Minini M, Mosole S, Revandkar A, Pasquini E, Elia AR, Bossi D, Rinaldi A, Rescigno P, Flohr P, Hunt J, Neeb A, … Alimonti A (2021). Commensal bacteria promote endocrine resistance in prostate cancer through androgen biosynthesis. Science, 374(6564), 216–224. [DOI] [PubMed] [Google Scholar]
- 66.Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, Schlitzer A, Ginhoux F, Apetoh L, Chachaty E, Woerther P-L, Eberl G, Bérard M, Ecobichon C, Clermont D, … Zitvogel L (2013). The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science, 342(6161), 971–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, Dai R-M, Kiu H, Cardone M, Naik S, Patri AK, Wang E, Marincola FM, Frank KM, Belkaid Y, … Goldszmid RS (2013). Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science, 342(6161), 967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Di Modica M, Gargari G, Regondi V, Bonizzi A, Arioli S, Belmonte B, De Cecco L, Fasano E, Bianchi F, Bertolotti A, Tripodo C, Villani L, Corsi F, Guglielmetti S, Balsari A, Triulzi T, & Tagliabue E (2021). Gut Microbiota Condition the Therapeutic Efficacy of Trastuzumab in HER2-Positive Breast Cancer. Cancer Research, 81(8), 2195–2206. [DOI] [PubMed] [Google Scholar]
- 69.Kwong TNY, Wang X, Nakatsu G, Chow TC, Tipoe T, Dai RZW, Tsoi KKK, Wong MCS, Tse G, Chan MTV, Chan FKL, Ng SC, Wu JCY, Wu WKK, Yu J, Sung JJY, & Wong SH (2018). Association Between Bacteremia From Specific Microbes and Subsequent Diagnosis of Colorectal Cancer. Gastroenterology, 155(2), 383–390.e8. [DOI] [PubMed] [Google Scholar]
- 70.Hanahan D (2022). Hallmarks of Cancer: New Dimensions. Cancer Discovery, 12(1), 31–46. [DOI] [PubMed] [Google Scholar]
- 71.Sender R, Fuchs S, & Milo R (2016). Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biology, 14(8), e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Belkaid Y, & Naik S (2013). Compartmentalized and systemic control of tissue immunity by commensals. Nature Immunology, 14(7), 646–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ramírez-Flandes S, González B, & Ulloa O (2019). Redox traits characterize the organization of global microbial communities. Proceedings of the National Academy of Sciences of the United States of America, 116(9), 3630–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Avanzini S, Kurtz DM, Chabon JJ, Moding EJ, Hori SS, Gambhir SS, Alizadeh AA, Diehn M, & Reiter JG (2020). A mathematical model of ctDNA shedding predicts tumor detection size. Science Advances, 6(50). 10.1126/sciadv.abc4308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, Le Quesne J, Moore DA, Veeriah S, Rosenthal R, Marafioti T, Kirkizlar E, Watkins TBK, McGranahan N, Ward S, Martinson L, Riley J, Fraioli F, Al Bakir M, … Swanton C (2017). Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature, 545(7655), 446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cristiano S, Leal A, Phallen J, Fiksel J, Adleff V, Bruhm DC, Jensen SØ, Medina JE, Hruban C, White JR, Palsgrove DN, Niknafs N, Anagnostou V, Forde P, Naidoo J, Marrone K, Brahmer J, Woodward BD, Husain H, … Velculescu VE (2019). Genome-wide cell-free DNA fragmentation in patients with cancer. Nature, 570(7761), 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Almeida A, Nayfach S, Boland M, Strozzi F, Beracochea M, Shi ZJ, Pollard KS, Sakharova E, Parks DH, Hugenholtz P, Segata N, Kyrpides NC, & Finn RD (2021). A unified catalog of 204,938 reference genomes from the human gut microbiome. Nature Biotechnology, 39(1), 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, Watanabe H, Masuda K, Nishimoto Y, Kubo M, Hosoda F, Rokutan H, Matsumoto M, Takamaru H, Yamada M, Matsuda T, Iwasaki M, Yamaji T, Yachida T, … Yamada T (2019). Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nature Medicine, 25(6), 968–976. [DOI] [PubMed] [Google Scholar]
- 79.Dadkhah E, Sikaroodi M, Korman L, Hardi R, Baybick J, Hanzel D, Kuehn G, Kuehn T, & Gillevet PM (2019). Gut microbiome identifies risk for colorectal polyps. BMJ Open Gastroenterology, 6(1), e000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peters BA, Dominianni C, Shapiro JA, Church TR, Wu J, Miller G, Yuen E, Freiman H, Lustbader I, Salik J, Friedlander C, Hayes RB, & Ahn J (2016). The gut microbiota in conventional and serrated precursors of colorectal cancer. Microbiome, 4(1), 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kordahi MC, Stanaway IB, Avril M, Chac D, Blanc M-P, Ross B, Diener C, Jain S, McCleary P, Parker A, Friedman V, Huang J, Burke W, Gibbons SM, Willis AD, Darveau RP, Grady WM, Ko CW, & DePaolo RW (2021). Genomic and functional characterization of a mucosal symbiont involved in early-stage colorectal cancer. Cell Host & Microbe, 29(10), 1589–1598.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Usyk M, Zolnik CP, Castle PE, Porras C, Herrero R, Gradissimo A, Gonzalez P, Safaeian M, Schiffman M, Burk RD, & Costa Rica HPV Vaccine Trial (CVT) Group. (2020). Cervicovaginal microbiome and natural history of HPV in a longitudinal study. PLoS Pathogens, 16(3), e1008376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tango CN, Seo S-S, Kwon M, Lee D-O, Chang HK, & Kim MK (2020). Taxonomic and Functional Differences in Cervical Microbiome Associated with Cervical Cancer Development. Scientific Reports, 10(1), 9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klein C, Gonzalez D, Samwel K, Kahesa C, Mwaiselage J, Aluthge N, Fernando S, West JT, Wood C, & Angeletti PC (2019). Relationship between the Cervical Microbiome, HIV Status, and Precancerous Lesions. mBio, 10(1). 10.1128/mBio.02785-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, Wu X, DeStefano Shields CE, Hechenbleikner EM, Huso DL, Anders RA, Giardiello FM, Wick EC, Wang H, Wu S, Pardoll DM, Housseau F, & Sears CL (2018). Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science, 359(6375), 592–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, Pierre JF, Ernest JD, Galipeau HJ, Thuille N, Bouziat R, Buscarlet M, Ringus DL, Wang Y, Li Y, Dinh V, Kim SM, McDonald BD, Zurenski MA, … Jabri B (2018). Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature, 557(7706), 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nené NR, Reisel D, Leimbach A, Franchi D, Jones A, Evans I, Knapp S, Ryan A, Ghazali S, Timms JF, Paprotka T, Bjørge L, Zikan M, Cibula D, Colombo N, & Widschwendter M (2019). Association between the cervicovaginal microbiome, BRCA1 mutation status, and risk of ovarian cancer: a case-control study. The Lancet Oncology, 20(8), 1171–1182. [DOI] [PubMed] [Google Scholar]
- 88.Davis NM, Proctor DM, Holmes SP, Relman DA, & Callahan BJ (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome, 6(1), 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Klein RS, Recco RA, Catalano MT, Edberg SC, Casey JI, & Steigbigel NH (1977). Association of Streptococcus bovis with carcinoma of the colon. The New England Journal of Medicine, 297(15), 800–802. [DOI] [PubMed] [Google Scholar]
- 90.Kowarsky M, Camunas-Soler J, Kertesz M, De Vlaminck I, Koh W, Pan W, Martin L, Neff NF, Okamoto J, Wong RJ, Kharbanda S, El-Sayed Y, Blumenfeld Y, Stevenson DK, Shaw GM, Wolfe ND, & Quake SR (2017). Numerous uncharacterized and highly divergent microbes which colonize humans are revealed by circulating cell-free DNA. Proceedings of the National Academy of Sciences of the United States of America, 114(36), 9623–9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Abed J, Emgård JEM, Zamir G, Faroja M, Almogy G, Grenov A, Sol A, Naor R, Pikarsky E, Atlan KA, Mellul A, Chaushu S, Manson AL, Earl AM, Ou N, Brennan CA, Garrett WS, & Bachrach G (2016). Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host & Microbe, 20(2), 215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Abed J, Maalouf N, Manson AL, Earl AM, Parhi L, Emgård JEM, Klutstein M, Tayeb S, Almogy G, Atlan KA, Chaushu S, Israeli E, Mandelboim O, Garrett WS, & Bachrach G (2020). Colon Cancer-Associated Fusobacterium nucleatum May Originate From the Oral Cavity and Reach Colon Tumors via the Circulatory System. Frontiers in Cellular and Infection Microbiology, 10, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Peled JU, Gomes ALC, Devlin SM, Littmann ER, Taur Y, Sung AD, Weber D, Hashimoto D, Slingerland AE, Slingerland JB, Maloy M, Clurman AG, Stein-Thoeringer CK, Markey KA, Docampo MD, Burgos da Silva M, Khan N, Gessner A, Messina JA, … van den Brink MRM (2020). Microbiota as Predictor of Mortality in Allogeneic Hematopoietic-Cell Transplantation. The New England Journal of Medicine, 382(9), 822–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sims TT, El Alam MB, Karpinets TV, Dorta-Estremera S, Hegde VL, Nookala S, Yoshida-Court K, Wu X, Biegert GWG, Delgado Medrano AY, Solley T, Ahmed-Kaddar M, Chapman BV, Sastry KJ, Mezzari MP, Petrosino JF, Lin LL, Ramondetta L, Jhingran A, … Klopp AH (2021). Gut microbiome diversity is an independent predictor of survival in cervical cancer patients receiving chemoradiation. Communications Biology, 4(1), 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rodriguez RM, Menor M, Hernandez BY, Deng Y, & Khadka VS (2021). Bacterial Diversity Correlates with Overall Survival in Cancers of the Head and Neck, Liver, and Stomach. Molecules , 26(18). 10.3390/molecules26185659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CPM, Poirier-Colame V, Roux A, Becharef S, Formenti S, Golden E, Cording S, Eberl G, Schlitzer A, Ginhoux F, … Zitvogel L (2015). Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science, 350(6264), 1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre M-L, Chang EB, & Gajewski TF (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science, 350(6264), 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Limeta A, Ji B, Levin M, Gatto F, & Nielsen J (2020). Meta-analysis of the gut microbiota in predicting response to cancer immunotherapy in metastatic melanoma. JCI Insight, 5(23). 10.1172/jci.insight.140940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, Paik S, Stagg J, Groves RA, Gallo M, Lewis IA, Geuking MB, & McCoy KD (2020). Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science, 369(6510), 1481–1489. [DOI] [PubMed] [Google Scholar]
- 100.Griffin ME, Espinosa J, Becker JL, Luo J-D, Carroll TS, Jha JK, Fanger GR, & Hang HC (2021). Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science, 373(6558), 1040–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Daillère R, Vétizou M, Waldschmitt N, Yamazaki T, Isnard C, Poirier-Colame V, Duong CPM, Flament C, Lepage P, Roberti MP, Routy B, Jacquelot N, Apetoh L, Becharef S, Rusakiewicz S, Langella P, Sokol H, Kroemer G, Enot D, … Zitvogel L (2016). Enterococcus hirae and Barnesiella intestinihominis Facilitate Cyclophosphamide-Induced Therapeutic Immunomodulatory Effects. Immunity, 45(4), 931–943. [DOI] [PubMed] [Google Scholar]
- 102.Lam KC, Araya RE, Huang A, Chen Q, Di Modica M, Rodrigues RR, Lopès A, Johnson SB, Schwarz B, Bohrnsen E, Cogdill AP, Bosio CM, Wargo JA, Lee MP, & Goldszmid RS (2021). Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell, 184(21), 5338–5356.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alexander JL, Wilson ID, Teare J, Marchesi JR, Nicholson JK, & Kinross JM (2017). Gut microbiota modulation of chemotherapy efficacy and toxicity. Nature Reviews. Gastroenterology & Hepatology, 14(6), 356–365. [DOI] [PubMed] [Google Scholar]
- 104.Daisley BA, Chanyi RM, Abdur-Rashid K, Al KF, Gibbons S, Chmiel JA, Wilcox H, Reid G, Anderson A, Dewar M, Nair SM, Chin J, & Burton JP (2020). Abiraterone acetate preferentially enriches for the gut commensal Akkermansia muciniphila in castrate-resistant prostate cancer patients. Nature Communications, 11(1), 4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kwa M, Plottel CS, Blaser MJ, & Adams S (2016). The Intestinal Microbiome and Estrogen Receptor-Positive Female Breast Cancer. Journal of the National Cancer Institute, 108(8). 10.1093/jnci/djw029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Komorowski AS, & Pezo RC (2020). Untapped “-omics”: the microbial metagenome, estrobolome, and their influence on the development of breast cancer and response to treatment. Breast Cancer Research and Treatment, 179(2), 287–300. [DOI] [PubMed] [Google Scholar]
- 107.Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, & Weyrich LS (2019). Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends in Microbiology, 27(2), 105–117. [DOI] [PubMed] [Google Scholar]
- 108.Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, & Walker AW (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biology, 12(1), 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sinha R, Abu-Ali G, Vogtmann E, Fodor AA, Ren B, Amir A, Schwager E, Crabtree J, Ma S, Microbiome Quality Control Project Consortium, Abnet, Knight R, White O, & Huttenhower C (2017). Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nature Biotechnology, 35(11), 1077–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Morton JT, Marotz C, Washburne A, Silverman J, Zaramela LS, Edlund A, Zengler K, & Knight R (2019). Establishing microbial composition measurement standards with reference frames. Nature Communications, 10(1), 2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lin H, & Peddada SD (2020). Analysis of compositions of microbiomes with bias correction. Nature Communications, 11(1), 3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lin H, & Peddada SD (2020). Analysis of microbial compositions: a review of normalization and differential abundance analysis. NPJ Biofilms and Microbiomes, 6(1), 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, … Gordon JI (2012). Human gut microbiome viewed across age and geography. Nature, 486(7402), 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.He Y, Wu W, Zheng H-M, Li P, McDonald D, Sheng H-F, Chen M-X, Chen Z-H, Ji G-Y, Zheng Z-D-X, Mujagond P, Chen X-J, Rong Z-H, Chen P, Lyu L-Y, Wang X, Wu C-B, Yu N, Xu Y-J, … Zhou H-W (2018). Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nature Medicine, 24(10), 1532–1535. [DOI] [PubMed] [Google Scholar]
- 115.Deschasaux M, Bouter KE, Prodan A, Levin E, Groen AK, Herrema H, Tremaroli V, Bakker GJ, Attaye I, Pinto-Sietsma S-J, van Raalte DH, Snijder MB, Nicolaou M, Peters R, Zwinderman AH, Bäckhed F, & Nieuwdorp M (2018). Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nature Medicine, 24(10), 1526–1531. [DOI] [PubMed] [Google Scholar]
- 116.Wirbel J, Pyl PT, Kartal E, Zych K, Kashani A, Milanese A, Fleck JS, Voigt AY, Palleja A, Ponnudurai R, Sunagawa S, Coelho LP, Schrotz-King P, Vogtmann E, Habermann N, Niméus E, Thomas AM, Manghi P, Gandini S, … Zeller G (2019). Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nature Medicine, 25(4), 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, Beghini F, Manara S, Karcher N, Pozzi C, Gandini S, Serrano D, Tarallo S, Francavilla A, Gallo G, Trompetto M, Ferrero G, Mizutani S, Shiroma H, … Segata N (2019). Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nature Medicine, 25(4), 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Charalampous T, Kay GL, Richardson H, Aydin A, Baldan R, Jeanes C, Rae D, Grundy S, Turner DJ, Wain J, Leggett RM, Livermore DM, & O’Grady J (2019). Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nature Biotechnology, 37(7), 783–792. [DOI] [PubMed] [Google Scholar]
- 119.Marotz CA, Sanders JG, Zuniga C, Zaramela LS, Knight R, & Zengler K (2018). Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome, 6(1), 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Horz H-P, Scheer S, Huenger F, Vianna ME, & Conrads G (2008). Selective isolation of bacterial DNA from human clinical specimens. Journal of Microbiological Methods, 72(1), 98–102. [DOI] [PubMed] [Google Scholar]
- 121.Feehery GR, Yigit E, Oyola SO, Langhorst BW, Schmidt VT, Stewart FJ, Dimalanta ET, Amaral-Zettler LA, Davis T, Quail MA, & Pradhan S (2013). A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PloS One, 8(10), e76096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gu W, Crawford ED, O’Donovan BD, Wilson MR, Chow ED, Retallack H, & DeRisi JL (2016). Depletion of Abundant Sequences by Hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biology, 17, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Payne A, Holmes N, Clarke T, Munro R, Debebe BJ, & Loose M (2021). Readfish enables targeted nanopore sequencing of gigabase-sized genomes. Nature Biotechnology, 39(4), 442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Edwards HS, Krishnakumar R, Sinha A, Bird SW, Patel KD, & Bartsch MS (2019). Real-Time Selective Sequencing with RUBRIC: Read Until with Basecall and Reference-Informed Criteria. Scientific Reports, 9(1), 11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Johnson JS, Spakowicz DJ, Hong B-Y, Petersen LM, Demkowicz P, Chen L, Leopold SR, Hanson BM, Agresta HO, Gerstein M, Sodergren E, & Weinstock GM (2019). Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nature Communications, 10(1), 5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kralik P, & Ricchi M (2017). A Basic Guide to Real Time PCR in Microbial Diagnostics: Definitions, Parameters, and Everything. Frontiers in Microbiology, 8, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Miller MB, & Tang Y-W (2009). Basic concepts of microarrays and potential applications in clinical microbiology. Clinical Microbiology Reviews, 22(4), 611–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fuks G, Elgart M, Amir A, Zeisel A, Turnbaugh PJ, Soen Y, & Shental N (2018). Combining 16S rRNA gene variable regions enables high-resolution microbial community profiling. Microbiome, 6(1), 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Karst SM, Ziels RM, Kirkegaard RH, Sørensen EA, McDonald D, Zhu Q, Knight R, & Albertsen M (2021). High-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or PacBio sequencing. Nature Methods, 18(2), 165–169. [DOI] [PubMed] [Google Scholar]
- 130.Zelenin S, Hansson J, Ardabili S, Ramachandraiah H, Brismar H, & Russom A (2015). Microfluidic-based isolation of bacteria from whole blood for sepsis diagnostics. Biotechnology Letters, 37(4), 825–830. [DOI] [PubMed] [Google Scholar]
- 131.Couto N, Schuele L, Raangs EC, Machado MP, Mendes CI, Jesus TF, Chlebowicz M, Rosema S, Ramirez M, Carriço JA, Autenrieth IB, Friedrich AW, Peter S, & Rossen JW (2018). Critical steps in clinical shotgun metagenomics for the concomitant detection and typing of microbial pathogens. Scientific Reports, 8(1), 13767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nowell PC (1976). The clonal evolution of tumor cell populations. Science, 194(4260), 23–28. [DOI] [PubMed] [Google Scholar]
- 133.Greaves M, & Maley CC (2012). Clonal evolution in cancer. Nature https://www.nature.com/articles/nature10762 [DOI] [PMC free article] [PubMed]
- 134.Mazor T, Pankov A, Johnson BE, Hong C, Hamilton EG, Bell RJA, Smirnov IV, Reis GF, Phillips JJ, Barnes MJ, Idbaih A, Alentorn A, Kloezeman JJ, Lamfers MLM, Bollen AW, Taylor BS, Molinaro AM, Olshen AB, Chang SM, … Costello JF (2015). DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell, 28(3), 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li S, Garrett-Bakelman FE, Chung SS, Sanders MA, Hricik T, Rapaport F, Patel J, Dillon R, Vijay P, Brown AL, Perl AE, Cannon J, Bullinger L, Luger S, Becker M, Lewis ID, To LB, Delwel R, Löwenberg B, … Mason CE (2016). Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nature Medicine, 22(7), 792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vaz M, Hwang SY, Kagiampakis I, Phallen J, Patil A, O’Hagan HM, Murphy L, Zahnow CA, Gabrielson E, Velculescu VE, Easwaran HP, & Baylin SB (2017). Chronic Cigarette Smoke-Induced Epigenomic Changes Precede Sensitization of Bronchial Epithelial Cells to Single-Step Transformation by KRAS Mutations. Cancer Cell, 32(3), 360–376.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nam AS, Chaligne R, & Landau DA (2021). Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nature Reviews. Genetics, 22(1), 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gutierrez C, Al’Khafaji AM, Brenner E, Johnson KE, Gohil SH, Lin Z, Knisbacher BA, Durrett RE, Li S, Parvin S, Biran A, Zhang W, Rassenti L, Kipps TJ, Livak KJ, Neuberg D, Letai A, Getz G, Wu CJ, & Brock A (2021). Multifunctional barcoding with ClonMapper enables high-resolution study of clonal dynamics during tumor evolution and treatment. Nature Cancer, 2(7), 758–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J, Gurjao C, Manders F, Dalmasso G, Stege PB, Paganelli FL, Geurts MH, Beumer J, Mizutani T, Miao Y, van der Linden R, van der Elst S , Genomics England Research Consortium, Garcia KC, … Clevers H (2020). Mutational signature in colorectal cancer caused by genotoxic pks E. coli. Nature, 580(7802), 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, Qian Y, Kryczek I, Sun D, Nagarsheth N, Chen Y, Chen H, Hong J, Zou W, & Fang J-Y (2017). Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell, 170(3), 548–563.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Garrett WS (2015). Cancer and the microbiota. Science, 348(6230), 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V, Ritz T, Longerich T, Theriot CM, McCulloch JA, Roy S, Yuan W, Thovarai V, Sen SK, Ruchirawat M, … Greten TF (2018). Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science, 360(6391). 10.1126/science.aan5931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Goss G, Tsai C-M, Shepherd FA, Bazhenova L, Lee JS, Chang G-C, Crino L, Satouchi M, Chu Q, Hida T, Han J-Y, Juan O, Dunphy F, Nishio M, Kang J-H, Majem M, Mann H, Cantarini M, Ghiorghiu S, & Mitsudomi T (2016). Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): a multicentre, open-label, single-arm, phase 2 study. The Lancet Oncology, 17(12), 1643–1652. [DOI] [PubMed] [Google Scholar]
- 144.Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, Barrett JC, Jänne PA, & Oxnard GR (2015). Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nature Medicine, 21(6), 560–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, Swords R, Collins RH, Mannis GN, Pollyea DA, Donnellan W, Fathi AT, Pigneux A, Erba HP, Prince GT, Stein AS, Uy GL, Foran JM, Traer E, … Kantarjian HM (2018). Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. The New England Journal of Medicine, 378(25), 2386–2398. [DOI] [PubMed] [Google Scholar]
- 146.Quek L, David MD, Kennedy A, Metzner M, Amatangelo M, Shih A, Stoilova B, Quivoron C, Heiblig M, Willekens C, Saada V, Alsafadi S, Vijayabaskar MS, Peniket A, Bernard OA, Agresta S, Yen K, MacBeth K, Stein E, … Vyas P (2018). Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nature Medicine, 24(8), 1167–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Roboz GJ, DiNardo CD, Stein EM, de Botton S, Mims AS, Prince GT, Altman JK, Arellano ML, Donnellan W, Erba HP, Mannis GN, Pollyea DA, Stein AS, Uy GL, Watts JM, Fathi AT, Kantarjian HM, Tallman MS, Choe S, … Stone RM (2020). Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood, 135(7), 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Merlevede J, Droin N, Qin T, Meldi K, Yoshida K, Morabito M, Chautard E, Auboeuf D, Fenaux P, Braun T, Itzykson R, de Botton S, Quesnel B, Commes T, Jourdan E, Vainchenker W, Bernard O, Pata-Merci N, Solier S, … Solary E (2016). Mutation allele burden remains unchanged in chronic myelomonocytic leukaemia responding to hypomethylating agents. Nature Communications, 7, 10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Turati VA, Guerra-Assunção JA, Potter NE, Gupta R, Ecker S, Daneviciute A, Tarabichi M, Webster AP, Ding C, May G, James C, Brown J, Conde L, Russell LJ, Ancliff P, Inglott S, Cazzaniga G, Biondi A, Hall GW, … Enver T (2021). Chemotherapy induces canalization of cell state in childhood B-cell precursor acute lymphoblastic leukemia. Nature Cancer, 2(8), 835–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dobson SM, García-Prat L, Vanner RJ, Wintersinger J, Waanders E, Gu Z, McLeod J, Gan OI, Grandal I, Payne-Turner D, Edmonson MN, Ma X, Fan Y, Voisin V, Chan-Seng-Yue M, Xie SZ, Hosseini M, Abelson S, Gupta P, … Dick JE (2020). Relapse-Fated Latent Diagnosis Subclones in Acute B Lineage Leukemia Are Drug Tolerant and Possess Distinct Metabolic Programs. Cancer Discovery, 10(4), 568–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Marine J-C, Dawson S-J, & Dawson MA (2020). Non-genetic mechanisms of therapeutic resistance in cancer. Nature Reviews. Cancer, 20(12), 743–756. [DOI] [PubMed] [Google Scholar]
- 152.Barrett M, Hand CK, Shanahan F, Murphy T, & O’Toole PW (2020). Mutagenesis by Microbe: the Role of the Microbiota in Shaping the Cancer Genome. Trends in Cancer Research, 6(4), 277–287. [DOI] [PubMed] [Google Scholar]
- 153.Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, Enk J, Bar-On Y, Stanietsky-Kaynan N, Coppenhagen-Glazer S, Shussman N, Almogy G, Cuapio A, Hofer E, Mevorach D, Tabib A, Ortenberg R, Markel G, Miklić K, … Mandelboim O (2015). Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity, 42(2), 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dunn GP, Bruce AT, Ikeda H, Old LJ, & Schreiber RD (2002). Cancer immunoediting: from immunosurveillance to tumor escape. Nature Immunology, 3(11), 991–998. [DOI] [PubMed] [Google Scholar]
- 155.Jourdain A, Duchmann M, Willekens C, Solary É, Perié L, & Laplane L (2021). Évolution clonale: Qu’est-ce qu’un clone. La Biologie Au Défi de L’histoire; Chapitre, 10, 167–188. [Google Scholar]
- 156.Fluckiger A, Daillère R, Sassi M, Sixt BS, Liu P, Loos F, Richard C, Rabu C, Alou MT, Goubet A-G, Lemaitre F, Ferrere G, Derosa L, Duong CPM, Messaoudene M, Gagné A, Joubert P, De Sordi L, Debarbieux L, … Zitvogel L (2020). Cross-reactivity between tumor MHC class I–restricted antigens and an enterococcal bacteriophage. Science https://science.sciencemag.org/content/369/6506/936.abstract [DOI] [PubMed]
- 157.Bessell CA, Isser A, Havel JJ, Lee S, Bell DR, Hickey JW, Chaisawangwong W, Glick Bieler J, Srivastava R, Kuo F, Purohit T, Zhou R, Chan TA, & Schneck JP (2020). Commensal bacteria stimulate antitumor responses via T cell cross-reactivity. JCI Insight, 5(8). 10.1172/jci.insight.135597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Doolittle WF (1999). Phylogenetic classification and the universal tree. Science, 284(5423), 2124–2129. [DOI] [PubMed] [Google Scholar]
- 159.Doolittle WF, & Bapteste E (2007). Pattern pluralism and the Tree of Life hypothesis. Proceedings of the National Academy of Sciences of the United States of America, 104(7), 2043–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.O’Malley MA, & Koonin EV (2011). How stands the Tree of Life a century and a half after The Origin? Biology Direct, 6(1), 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Byndloss MX, Olsan EE, Rivera-Chávez F, Tiffany CR, Cevallos SA, Lokken KL, Torres TP, Byndloss AJ, Faber F, Gao Y, Litvak Y, Lopez CA, Xu G, Napoli E, Giulivi C, Tsolis RM, Revzin A, Lebrilla CB, & Bäumler AJ (2017). Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science, 357(6351), 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Butler DSC, Cafaro C, Putze J, Wan MLY, Tran TH, Ambite I, Ahmadi S, Kjellström S, Welinder C, Chao SM, Dobrindt U, & Svanborg C (2021). A bacterial protease depletes c-MYC and increases survival in mouse models of bladder and colon cancer. Nature Biotechnology, 39(6), 754–764. [DOI] [PubMed] [Google Scholar]
- 163.Kusters JG, van Vliet AHM, & Kuipers EJ (2006). Pathogenesis of Helicobacter pylori infection. Clinical Microbiology Reviews, 19(3), 449–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Suerbaum S, & Josenhans C (2007). Helicobacter pylori evolution and phenotypic diversification in a changing host. In Nature Reviews Microbiology (Vol. 5, Issue 6, pp. 441–452). 10.1038/nrmicro1658 [DOI] [PubMed] [Google Scholar]
- 165.Anandasabapathy S, Jhamb J, Davila M, Wei C, Morris J, & Bresalier R (2007). Clinical and endoscopic factors predict higher pathologic grades of Barrett dysplasia. Cancer, 109(4), 668–674. [DOI] [PubMed] [Google Scholar]
- 166.Islami F, & Kamangar F (2008). Helicobacter pylori and Esophageal Cancer Risk: A Meta-analysis. In Cancer Prevention Research (Vol. 1, Issue 5, pp. 329–338). 10.1158/1940-6207.capr-08-0109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Petrova V, Annicchiarico-Petruzzelli M, Melino G, & Amelio I (2018). The hypoxic tumour microenvironment. Oncogenesis, 7(1), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rundell EA, Banta LM, Ward DV, Watts CD, Birren B, & Esteban DJ (2014). 16S rRNA gene survey of microbial communities in Winogradsky columns. PloS One, 9(8), e104134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hoshino T, Doi H, Uramoto G-I, Wörmer L, Adhikari RR, Xiao N, Morono Y, D’Hondt S, Hinrichs K-U, & Inagaki F (2020). Global diversity of microbial communities in marine sediment. Proceedings of the National Academy of Sciences of the United States of America, 117(44), 27587–27597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fenchel T, & Finlay B (2008). Oxygen and the spatial structure of microbial communities. Biological Reviews of the Cambridge Philosophical Society, 83(4), 553–569. [DOI] [PubMed] [Google Scholar]
- 171.Semenza GL (2008). Tumor metabolism: cancer cells give and take lactate [Review of Tumor metabolism: cancer cells give and take lactate]. The Journal of Clinical Investigation, 118(12), 3835–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bahram M, Hildebrand F, Forslund SK, Anderson JL, Soudzilovskaia NA, Bodegom PM, Bengtsson-Palme J, Anslan S, Coelho LP, Harend H, Huerta-Cepas J, Medema MH, Maltz MR, Mundra S, Olsson PA, Pent M, Põlme S, Sunagawa S, Ryberg M, … Bork P (2018). Structure and function of the global topsoil microbiome. Nature, 560(7717), 233–237. [DOI] [PubMed] [Google Scholar]
- 173.Anderson ARA, Weaver AM, Cummings PT, & Quaranta V (2006). Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell, 127(5), 905–915. [DOI] [PubMed] [Google Scholar]
- 174.Damaghi M, West J, Robertson-Tessi M, Xu L, Ferrall-Fairbanks MC, Stewart PA, Persi E, Fridley BL, Altrock PM, Gatenby RA, Sims PA, Anderson ARA, & Gillies RJ (2021). The harsh microenvironment in early breast cancer selects for a Warburg phenotype. Proceedings of the National Academy of Sciences of the United States of America, 118(3). 10.1073/pnas.2011342118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Frankenstein Z, Basanta D, Franco OE, Gao Y, Javier RA, Strand DW, Lee M, Hayward SW, Ayala G, & Anderson ARA (2020). Stromal reactivity differentially drives tumour cell evolution and prostate cancer progression. Nature Ecology & Evolution, 4(6), 870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Salehi S, Kabeer F, Ceglia N, Andronescu M, Williams MJ, Campbell KR, Masud T, Wang B, Biele J, Brimhall J, Gee D, Lee H, Ting J, Zhang AW, Tran H, O’Flanagan C, Dorri F, Rusk N, de Algara TR, … Shah SP (2021). Clonal fitness inferred from time-series modelling of single-cell cancer genomes. Nature, 595(7868), 585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Temko D, Cheng Y-K, Polyak K, & Michor F (2017). Mathematical Modeling Links Pregnancy-Associated Changes and Breast Cancer Risk. Cancer Research, 77(11), 2800–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Williams MJ, Werner B, Heide T, Curtis C, Barnes CP, Sottoriva A, & Graham TA (2018). Quantification of subclonal selection in cancer from bulk sequencing data. Nature Genetics, 50(6), 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Avanzini S, & Antal T (2019). Cancer recurrence times from a branching process model. PLoS Computational Biology, 15(11), e1007423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Curtius K, Dewanji A, Hazelton WD, Rubenstein JH, & Luebeck GE (2021). Optimal Timing for Cancer Screening and Adaptive Surveillance Using Mathematical Modeling. Cancer Research, 81(4), 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Doulcier G, Lambert A, De Monte S, & Rainey PB (2020). Eco-evolutionary dynamics of nested Darwinian populations and the emergence of community-level heredity. eLife, 9. 10.7554/eLife.53433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Schwartz R, & Schäffer AA (2017). The evolution of tumour phylogenetics: principles and practice. Nature Reviews. Genetics, 18(4), 213–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gu C, Kim GB, Kim WJ, Kim HU, & Lee SY (2019). Current status and applications of genome-scale metabolic models. In Genome Biology (Vol. 20, Issue 1). 10.1186/s13059-019-1730-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kumar M, Ji B, Zengler K, & Nielsen J (2019). Modelling approaches for studying the microbiome. Nature Microbiology, 4(8), 1253–1267. [DOI] [PubMed] [Google Scholar]
- 185.Shoaie S, Ghaffari P, Kovatcheva-Datchary P, Mardinoglu A, Sen P, Pujos-Guillot E, de Wouters T, Juste C, Rizkalla S, Chilloux J, Hoyles L, Nicholson JK, MICRO-Obes Consortium, Dore J, Dumas ME, Clement K, Bäckhed F, & Nielsen J (2015). Quantifying Diet-Induced Metabolic Changes of the Human Gut Microbiome. Cell Metabolism, 22(2), 320–331. [DOI] [PubMed] [Google Scholar]
- 186.Garza DR, Taddese R, Wirbel J, Zeller G, Boleij A, Huynen MA, & Dutilh BE (2020). Metabolic models predict bacterial passengers in colorectal cancer. Cancer & Metabolism, 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bhalla P, Rengaswamy R, Karunagaran D, Suraishkumar GK, & Sahoo S (2022). Metabolic modeling of host-microbe interactions for therapeutics in colorectal cancer. NPJ Systems Biology and Applications, 8(1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Dion MB, Oechslin F, & Moineau S (2020). Phage diversity, genomics and phylogeny. Nature Reviews. Microbiology, 18(3), 125–138. [DOI] [PubMed] [Google Scholar]
- 189.Sender R, Fuchs S, & Milo R (2016). Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell, 164(3), 337–340. [DOI] [PubMed] [Google Scholar]
- 190.Abellan-Schneyder I, Matchado MS, Reitmeier S, Sommer A, Sewald Z, Baumbach J, List M, & Neuhaus K (2021). Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing. mSphere, 6(1). 10.1128/mSphere.01202-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, Patterson JW, & Slingluff CL Jr. (2012). Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Research, 72(5), 1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Stephenson K (2005). Introduction to Circle Packing: The Theory of Discrete Analytic Functions Cambridge University Press. [Google Scholar]
- 193.Downey GP, Doherty DE, Schwab B 3rd, Elson EL, Henson PM, & Worthen GS (1990). Retention of leukocytes in capillaries: role of cell size and deformability. Journal of Applied Physiology, 69(5), 1767–1778. [DOI] [PubMed] [Google Scholar]
- 194.Shashni B, Ariyasu S, Takeda R, Suzuki T, Shiina S, Akimoto K, Maeda T, Aikawa N, Abe R, Osaki T, Itoh N, & Aoki S (2018). Size-Based Differentiation of Cancer and Normal Cells by a Particle Size Analyzer Assisted by a Cell-Recognition PC Software. Biological & Pharmaceutical Bulletin, 41(4), 487–503. [DOI] [PubMed] [Google Scholar]
- 195.Kather JN, Suarez-Carmona M, Charoentong P, Weis C-A, Hirsch D, Bankhead P, Horning M, Ferber D, Kel I, Herpel E, Schott S, Zörnig I, Utikal J, Marx A, Gaiser T, Brenner H, Chang-Claude J, Hoffmeister M, Jäger D, & Halama N (2018). Topography of cancer-associated immune cells in human solid tumors. eLife, 7. 10.7554/eLife.36967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Monte UD, & Del Monte U (2009). Does the cell number 109still really fit one gram of tumor tissue? In Cell Cycle (Vol. 8, Issue 3, pp. 505–506). 10.4161/cc.8.3.7608 [DOI] [PubMed] [Google Scholar]
- 197.Ankrah NYD, Bernstein DB, Biggs M, Carey M, Engevik M, García-Jiménez B, Lakshmanan M, Pacheco AR, Sulheim S, & Medlock GL (2021). Enhancing Microbiome Research through Genome-Scale Metabolic Modeling. mSystems, 6(6), e0059921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zorrilla F, Buric F, Patil KR, & Zelezniak A (2021). metaGEM: reconstruction of genome scale metabolic models directly from metagenomes. Nucleic Acids Research, 49(21), e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.van der Ark KCH, van Heck RGA, Martins Dos Santos VAP, Belzer C, & de Vos WM (2017). More than just a gut feeling: constraint-based genome-scale metabolic models for predicting functions of human intestinal microbes. Microbiome, 5(1), 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Nilsson A, & Nielsen J (2017). Genome scale metabolic modeling of cancer. Metabolic Engineering, 43(Pt B), 103–112. [DOI] [PubMed] [Google Scholar]
- 201.Shlomi T, Benyamini T, Gottlieb E, Sharan R, & Ruppin E (2011). Genome-scale metabolic modeling elucidates the role of proliferative adaptation in causing the Warburg effect. PLoS Computational Biology, 7(3), e1002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Faust K, Bauchinger F, Laroche B, de Buyl S, Lahti L, Washburne AD, Gonze D, & Widder S (2018). Signatures of ecological processes in microbial community time series. Microbiome, 6(1), 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bucci V, & Xavier JB (2014). Towards predictive models of the human gut microbiome. Journal of Molecular Biology, 426(23), 3907–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Robertson-Tessi M, Gillies RJ, Gatenby RA, & Anderson ARA (2015). Impact of metabolic heterogeneity on tumor growth, invasion, and treatment outcomes. Cancer Research, 75(8), 1567–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ghaffarizadeh A, Heiland R, Friedman SH, Mumenthaler SM, & Macklin P (2018). PhysiCell: An open source physics-based cell simulator for 3-D multicellular systems. PLoS Computational Biology, 14(2), e1005991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Din MO, Danino T, Prindle A, Skalak M, Selimkhanov J, Allen K, Julio E, Atolia E, Tsimring LS, Bhatia SN, & Hasty J (2016). Synchronized cycles of bacterial lysis for in vivo delivery. Nature, 536(7614), 81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Mina P, di Bernardo M, Savery NJ, & Tsaneva-Atanasova K (2013). Modelling emergence of oscillations in communicating bacteria: a structured approach from one to many cells. Journal of the Royal Society, Interface / the Royal Society, 10(78), 20120612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Beerenwinkel N, Antal T, Dingli D, Traulsen A, Kinzler KW, Velculescu VE, Vogelstein B, & Nowak MA (2007). Genetic progression and the waiting time to cancer. PLoS Computational Biology, 3(11), e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Park S-C, & Krug J (2007). Clonal interference in large populations. Proceedings of the National Academy of Sciences of the United States of America, 104(46), 18135–18140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Favero M, Hult H, & Koski T (2021). A dual process for the coupled Wright–Fisher diffusion. In Journal of Mathematical Biology (Vol. 82, Issues 1–2). 10.1007/s00285-021-01555-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tomasetti C, Vogelstein B, & Parmigiani G (2013). Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. In Proceedings of the National Academy of Sciences (Vol. 110, Issue 6, pp. 1999–2004). 10.1073/pnas.1221068110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kaveh K, McAvoy A, Chatterjee K, & Nowak MA (2020). The Moran process on 2-chromatic graphs. PLoS Computational Biology, 16(11), e1008402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Måløy M, Måløy F, Lahoz-Beltrá R, Carlos Nuño J, & Bru A (2019). An extended Moran process that captures the struggle for fitness. Mathematical Biosciences, 308, 81–104. [DOI] [PubMed] [Google Scholar]
- 214.Curtius K, Hazelton WD, Jeon J, & Georg Luebeck E (2015). A Multiscale Model Evaluates Screening for Neoplasia in Barrett’s Esophagus. In PLOS Computational Biology (Vol. 11, Issue 5, p. e1004272). 10.1371/journal.pcbi.1004272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Reiter JG, Makohon-Moore AP, Gerold JM, Bozic I, Chatterjee K, Iacobuzio-Donahue CA, Vogelstein B, & Nowak MA (2017). Reconstructing metastatic seeding patterns of human cancers. Nature Communications, 8, 14114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Archetti M, Ferraro DA, & Christofori G (2015). Heterogeneity for IGF-II production maintained by public goods dynamics in neuroendocrine pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America, 112(6), 1833–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Basanta D, Scott JG, Fishman MN, Ayala G, Hayward SW, & Anderson ARA (2012). Investigating prostate cancer tumour-stroma interactions: clinical and biological insights from an evolutionary game. British Journal of Cancer, 106(1), 174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Nichol D, Rutter J, Bryant C, Hujer AM, Lek S, Adams MD, Jeavons P, Anderson ARA, Bonomo RA, & Scott JG (2019). Antibiotic collateral sensitivity is contingent on the repeatability of evolution. Nature Communications, 10(1), 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kaznatcheev A, Vander Velde R, Scott JG, & Basanta D (2017). Cancer treatment scheduling and dynamic heterogeneity in social dilemmas of tumour acidity and vasculature. British Journal of Cancer, 116(6), 785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lambert G, Vyawahare S, & Austin RH (2014). Bacteria and game theory: the rise and fall of cooperation in spatially heterogeneous environments. Interface Focus, 4(4), 20140029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zomorrodi AR, & Segrè D (2017). Genome-driven evolutionary game theory helps understand the rise of metabolic interdependencies in microbial communities. Nature Communications, 8(1), 1563. [DOI] [PMC free article] [PubMed] [Google Scholar]