

Abstract
Gut microbiome disruptions may lead to adverse effects on wildlife fitness and viability, thus maintaining host microbiota biodiversity needs to become an integral part of wildlife conservation. The highly-endangered callitrichid golden lion tamarin (GLT-Leontopithecus rosalia) is a rare conservation success, but allochthonous callitrichid marmosets (Callithrix) serve as principle ecological GLT threats. However, incorporation of microbiome approaches to GLT conservation is impeded by limited gut microbiome studies of Brazilian primates. Here, we carried out analysis of gut metagenomic pools from 114 individuals of wild and captive GLTs and marmosets. More specifically, we analyzed the bacterial component of ultra filtered samples originally collected as part of a virome profiling study. The major findings of this study are consistent with previous studies in showing that Bifidobacterium, a bacterial species important for the metabolism of tree gums consumed by callitrichids, is an important component of the callitrichid gut microbiome - although GTLs and marmosets were enriched for different species of Bifidobacterium. Additionally, the composition of GLT and marmoset gut microbiota is sensitive to host environmental factors. Overall, our data expand baseline gut microbiome data for callitrichids to allow for the development of new tools to improve their management and conservation.
Subject terms: Microbiome, Zoology
Introduction
The gut microbiome provides the host with a number of essential metabolic, immune, and physiological functions1,2, and various factors influence gut microbiome community structure. Host phylogeny shapes gut microbiome composition through vertical transmission of microbiota and host-microbe interactions related to immune genes3,4. Diet, on the other hand, individualizes the gut microbiome profiles of a given host by enriching the gut with microbiota related to a host’s feeding strategy5,6. In wild mammals, the microbiome functional landscape is significantly associated with host diet, life span, body mass, and social structure7. Importantly, the physiological and functional benefits of host-associated microbial communities are susceptible to disruptions by several anthropogenic factors like deforestation, land-use change, urbanization, and captivity8–10.
As gut microbiome disruptions may lead to adverse effects on wildlife fitness and viability, there is a growing awareness that maintaining the biodiversity of host-associated microbiota may need to become an integral part of wildlife management and conservation practices2,9. The recovery of the highly endangered golden-lion tamarin (Leontopithecus rosalia, GLT) within its native Brazilian Atlantic Forest biome, one of the world’s 25 biodiversity hot spots11, is a rare conservation success story12. Although conservation of the species continues through strategic plans coordinated by the Brazilian organization Associação Mico-Leão-Dourado (AMLD)13, the species remains threaten by anthropogenic activities related to land use and urbanization that lead to habitat degradation and fragmentation14,15. Furthermore, the presence of anthropogenic marmoset hybrids, which arose after human introductions of allochthonous marmoset species (common marmosets Callithrix jacchus and black-tufted marmosets C. penicillata) into forest fragments located within the native GLT range16,17, represent additional ecological and health threats to GLT recovery18. Incorporation of microbiome approaches for future GLT conservation efforts is currently impeded by a restricted understanding of the gut microbiome of most Brazilian primates. With the exception of a number 16S rRNA and ITS microbial profiling studies (e.g.19–22), microbiome studies of Brazilian primates are limited.
Prior microbiome work has shown that the Callitrichidae family, which includes GLTs and marmosets, possesses the highest average primate gut microbiome abundance of Bifidobacterium (> 30%) as well as significant host co-evolutionary signal with Bifidobacterium21. This bacterial genus is a natural commensal that functionally supports carbohydrate metabolism within their hosts21,23 and is involved in other crucial host biological processes (see24). GLTs and marmosets both exploit viscous plant exudates composed of polysaccharides as part of their dietary intake, albeit to a different degree. A number of marmoset species are considered obligate exudivores25, and some species exhibit high morphological specialization for exudivory26,27. In wild common marmosets, exudate consumption provides key nutrients (calcium, protein) for balancing their diet, especially during the dry season. Exudates vary in complex carbohydrate content (38–81%) and can contain more calcium than insects, more protein than other plant sources, and even high water content28. As frugivore-insectivores, GLTs eat exudates opportunistically29,30, but exudates overall are considered an essential part of the callitrichid diet31. In fact, Bifidobacterium seems to play a key functional role in the wild marmoset gut microbiome19, and captivity reduces gut Bifidobacterium abundance in marmosets as well as other primate hosts19,21. It is indeed thought that such changes in host gut microbiome composition between the wild and captivity affect the health and viability of captive marmosets19.
To further expand understanding of the callitrichid gut microbiome, especially that of GLTs, we took advantage of opportunistically-available data from ultra-filtered metagenomic sequencing short-read pooled libraries (metagenomic pools hereafter) from feces and anal swabs of captive and wild GLTs and marmosets. The biological samples were originally collected as part of a virome profiling study, and were therefore enriched for viral-like particles (VPLs) through ultra-filtration and nuclease treatment. Nonetheless, the presence of non-viral genetic material is common in such genomic libraries regardless of enrichment protocol32,33. Although the non-viral genetic material is usually considered a contaminant in virome studies33, from the microbiome perspective, such material may still contain valuable taxonomic and functional bacterial information.
Here, we carried out metagenomic analysis of callitrichid VPL pools using both microbial community and metagenome-assembled genome (MAG) approaches from a total of 114 individuals of wild and captive GLTs, captive black-tufted marmosets (C. penicillata) and wild C. jacchus x C. penicillata hybrids. We hypothesize that the microbiome component of VPL-enriched genomic libraries represents only the most abundant bacterial taxa present within the sampled host material. Further, we hypothesize that host gut microbiome metagenomic pools from both GLTs and marmosets show strong enrichment for Bifidobacterium, but that this bacterial taxon is more abundant in the gut of wild callitrichids than that of captive callitrichids. As it has been previously shown that Bifidobacterium species are specific for different host species and taxa21, we predict that the gut microbiome profiles of Leontopithecus and Callithrix metagenomic pools will respectively possess distinct Bifidobacterium species. We do expect the functional profile of the bacterial component of our analyzed host gut microbiome metagenomic pools to be biased towards carbohydrate function in both GLTs and marmosets.
Methods
Sample collection
We studied six readily accessible groups of wild golden lion tamarins (Leontopithecus rosalia; GLT; n = 71) and four groups of wild marmosets (Callithrix sp.; n = 9) that were habituated to regular human contact and consistently monitored by AMLD in Silva Jardim and Rio Bonito municipalities in Rio de Janeiro state, Brazil (Fig. 1). Previous genetic analysis of marmoset populations from this region indicate that these populations comprise an anthropogenic hybrid swarm descended from introduced populations of C. jacchus and C. penicillata17,34. Callitrichid groups were sampled in several small fragments of the Atlantic Forest at the São João river basin: Afetiva (Afe; GLT = 12 and marmoset = 4), Igarapé (Igar; GLT = 11 and marmoset = 2), Iguape (Igua; GLT = 5 and marmoset = 3), Nova Esperança (NEs; GLT = 16), Santa Helena 1 (StH; GLT = 13) and Rio Vermelho (RV; GLT = 14). Wild marmosets and tamarins were immobilized for routine veterinary check-ups with injection of ketamine (approximately 10–15 mg/kg) into the intramuscular region of the inner thigh. Then, fecal samples were collected in 15 mL tubes and this volume was mixed in a proportion of 1:1 with RNAlater, followed by vigorous homogenization. Tubes were kept at ambient temperature in the field and sent to the Laboratory of Viral Diversity and Disease (LDDV), in the Department of Genetics of the Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, Brazil, to freeze at − 80 C until processing. Field information collected for each sampled animal included all the following: ID number, group, specie, age, sex, weight and clinical conditions.
Figure 1.

Map showing the sample collection sites with host environment (C/circle = captive and W/triangle = wild) and host species information (Leontopithecus rosalia shown as teal and Callithrix sp. shown as purple). The map inset represents the sampling localities of Silva Jardim and Rio Bonito, Rio de Janeiro state (RJ), Brazil. The Callithrix sampling locality of Brasília, Federal District is also shown on the main map as “DF.”
We also included in the study a captive GLT group (n = 3) housed at Centro de Primatologia do Rio de Janeiro (CPRJ; Primate Center of Rio de Janeiro) located in Guapimirim, Rio de Janeiro, Brazil. Finally, we sampled six marmosets groups of C. penicillata (n = 39; 4–6 animals by group) housed at Centro de Primatologia da Universiade de Brasília (CP/UnB; Primate Center of the Brasilia University), Brasília, Federal District, Brazil. Animals were kept in enclosures surrounded by natural vegetation and were maintained in couples or in groups according to the rules of IBAMA (Brazilian Institute of Environment and Renewable Natural Resources). The marmosets from CP/UnB were anesthetized also for routine veterinary check-ups and anal swab samples were collected using sterile cotton swabs with plastic shafts. Swab samples were placed in 1.5 mL centrifuge tubes with 500 L of PBS and later excised close to the cotton tip using flame-sterilized scissors. Tubes were kept at − 20 C until processing. For the GLTs from CPRJ, only fecal samples were collected using a similar method as specified for wild GLTs. General information was collect for all sampled animals as: ID number, enclosure, specie, age, sex, weight and clinical conditions. Although our sampling included both anal swabs and fecal samples, several previous studies indicate that anal swabs are reliable proxies for fecal samples35–38, thus we treated both sample types as equivalent.
General sampling information is summarized in Fig. 1 and Table 1. Host taxon identification at the genus level, Callithrix and Leontopithecus, followed previously published phenotype descriptions34 and official registered identification by primate captive facilities and the AMLD. Hosts were also classified by their environment as wild (captured as free-range individuals) or captive (maintained in captivity). All samples were collected following the national guidelines and provisions of CONCEA (National Council for Animal Experimentation Control, Brazil), which included animal welfare standard operating procedures. This project was approved by the Ethics Committee on the Use of Animals (CEUA) of UFRJ (reference number 037/14). This study is reported in accordance to ARRIVE guidelines (https://arriveguidelines.org/resources/questionnaire). All methods were carried out in accordance with relevant international guidelines and regulations.
Table 1.
Information summary on golden lion tamarin (GLT) and marmoset gut metagenomic pools with host taxon, sampling location, sampling coordinates, number of individual hosts included in each pool, and host environment. For sampling locations, the following abbreviations are used: CP/UnB = Centro de Primatologia de Brasília / University of Brasília. For host environment, the following abbreviations are used: W = Wild, C = Captive. For host location the following abbreviations are used: RJ = Rio de Janeiro, DF = Federal District.
| Host species | Host common name | Location (ID) | Pool ID | Sample approximate collection geographic coordinates | Sample type | Pool N | Host environment |
|---|---|---|---|---|---|---|---|
| Leontopithecus rosalia | GLT | Afetiva (Afe), Silva Jardim, RJ | MLDAfe | 22 3758.84 S–42 281.47 W | Feces | 4 | W |
| Leontopithecus rosalia | GLT | Afetiva (Afe), Silva Jardim, RJ | LrAfef | 22 3758.84 S–42 281.47 W | Feces | 8 | W |
| Leontopithecus rosalia | GLT | Igarapé (Igar), Silva Jardim, RJ | MLDIgar | 22 3027.41 S–42 1834.66 W | Feces | 3 | W |
| Leontopithecus rosalia | GLT | Igarapé (Igar), Silva Jardim, RJ | LrIgarf | 22 3027.41 S–42 1834.66 W | Feces | 8 | W |
| Leontopithecus rosalia | GLT | Iguape (Igua), Silva Jardim, RJ | MLDIgua | 22 3020.74 S–42 1924.27 W | Feces | 5 | W |
| Leontopithecus rosalia | GLT | Rio Vermelho (RV), Rio Bonito, RJ | MLDRV | 22 433.26 S–42 3253.37 W | Feces | 9 | W |
| Leontopithecus rosalia | GLT | Rio Vermelho (RV), Rio Bonito, RJ | LrRVf | 22 433.26 S–42 3253.37 W | Feces | 5 | W |
| Leontopithecus rosalia | GLT | Nova Esperança (NEs), Silva Jardim, RJ | LrNEs1f | 22 386.54 S–42 2547.38 W | Feces | 8 | W |
| Leontopithecus rosalia | GLT | Nova Esperança (NEs), Silva Jardim, RJ | LrNEs2f | 22 386.54 S–42 2547.38 W | Feces | 8 | W |
| Leontopithecus rosalia | GLT | Santa Helena (StH) 1, Silva Jardim, RJ | LrStH1f | 22 3137.33 S–42 2052.51 W | Feces | 7 | W |
| Leontopithecus rosalia | GLT | Santa Helena (StH) 1, Silva Jardim, RJ | LrStH2f | 22 3137.33 S–42 2052.51 W | Feces | 6 | W |
| Leontopithecus rosalia | GLT | CPRJ, Guapimirim, RJ | PoolRosalia | 22 2917.458 S–42 5448.432 W | Feces | 3 | C |
| Callithrix sp. | Marmoset | Afetiva (Afe), Silva Jardim, RJ | SAfe | 22 3758.84 S–42 281.47 W | Feces | 4 | W |
| Callithrix sp. | Marmoset | Igarapé (Igar), Silva Jardim, RJ | SIgar | 22 3027.41 S–42 1834.66 W | Feces | 2 | W |
| Callithrix sp. | Marmoset | Iguape (Igua), Silva Jardim, RJ | SIgua | 22 3020.74 S–42 1924.27 W | Feces | 3 | W |
| Callithrix sp. | Marmoset | CP/UnB, DF | P1 | 15 56 54.624 S–47 56 2.575 W | Anal Swab | 5 | C |
| Callithrix sp. | Marmoset | CP/UnB, DF | P2 | 15 56 54.624 S–47 562.575 W | Anal Swab | 5 | C |
| Callithrix sp. | Marmoset | CP/UnB, DF | P3 | 15 5654.624 S–47 56 2.575 W | Anal Swab | 5 | C |
| Callithrix sp. | Marmoset | CP/UnB, DF | P4 | 15 56 54.624 S–4756 2.575 W | Anal Swab | 4 | C |
| Callithrix sp. | Marmoset | CP/UnB, DF | P7 | 15 56 54.624 S–47 56 2.575 W | Anal Swab | 6 | C |
| Callithrix sp. | Marmoset | CP/UnB, DF | P8 | 15 56 54.624 S–47 56 2.575 W | Anal Swab | 6 | C |
Sample processing and sequencing
The molecular protocol was conducted as follows: after thawing and vigorous homogenization for 1 min, 1 mL of fecal sample was disrupted by Lysing Matrix E extraction beads (MPbio) and clarified by centrifugation at 6000g for 10 min at 4 C. Between 100–250 L of fecal supernatants or PBS direct from swab samples was mixed with samples from animals of the same location, resulting in 18 pools (GLT = 9 and Marmoset = 9). Sampling pools are summarized in Table 1. The pooled samples were filtered through an Ultra-free-MC HV 0.45 sterile filter (Millipore, UFC30HV0S). The filtrates were submitted to sucrose density gradient ultracentrifuge separation at 35,000g for 90 min at 4 C and degradation of unprotected nucleic acid by nuclease digestion at 37 C for 60 min. Remaining nucleic acid (DNA and RNA) were then isolated using QIAamp® MinElute® Virus Spin kit (Qiagen), followed the manufacturer’s instructions, with the following modifications: (i) Carrier RNA was omitted from the AL Buffer; (ii) the protease was resuspended in AVE Buffer, instead of Protease Resuspension Buffer; (iii) the washing step with AW1 was suppressed; (iv) the final elution was performed in 20 of ultra-pure water. Thereafter, a RT-PCR reaction was performed with the SuperScript® III First-Strand Synthesis System (Invitrogen) for first-strand cDNA synthesis from RNA, using random primers, while preserving DNA. The second-strand cDNA synthesis was performed using a DNA Polymerase I Large (Klenow 3–5 exo) Fragment (New England Biolabs®). All those reactions were conducted according to the manufacturer’s instructions. Total DNA (DNA and cDNA) quantification was performed using the High Sensitivity dsDNA Assay kit in a Qubit 2.0 Fluorometer (Thermo Fisher Scientific). The libraries were constructed using the Nextera XT - DNA Library Preparation Kit (Illumina), purified with the Agencourt AMPure XP -PCR Purification (Beckman Coulter) kit and quantified using both High Sensitivity DNA Kits from Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and 2100 Bioanalyzer (Agilent Technologies). The sequencing was conducted by applying 2 pM of each library in the MiSeq Illumina platform using the MiSeq V2 300-cycle kit (Illumina) in paired-end mode 2 150 bp with dual barcode for each pooled sample.
Bioinformatics and statistical analysis
Metagenomic community profiling
Initially, analyses for quality filtering, taxonomic profiling, and functional profiling of callitrichid gut microbiomes was carried out with the bioBakery metagomic environment39. We first used KneadData v0.7.10 (https://huttenhower.sph.harvard.edu/kneaddata/) with default settings for quality control of raw pair-ended fastq files from individual pooled metagenomic sequencing libraries, which included removal of potential host reads and trimming of low quality regions from the reads. Kneaddata used Trimmomatic v0.3940 for trimming and Bowtie2 v 2.4.241 for removal of contaminating host reads. For reference host genomes, we combined four publicly available Neotropical primate genomes from GenBank (Saimiri boliviensis boliviensis - GCA_000235385.1; Callithrix jacchus - GCA_002754865.1; Aotus nancymaae - GCA_000952055.2; Cebus capucinus imitator - GCA_001604975.1) into a single fasta file, which was then turned into a reference database for use by Bowtie2 and thus promoted a deep cleaning of the host reads, especially in the case of the GLT that does not have a published genome. We installed the CHOCOPhlAn_201901 database for use with MetaPhlAn v3.0.7. Then we profiled the composition of microbial community composition of our pooled metagenomic shotgun sequencing data with MetaPhlAn using the following command “metaphlan *.kneaddata_paired_1.fastq, *.kneaddata_paired_2.fastq –bowtie2out *.bowtie2.counts.bz2 –input_type fastq -o *.profiled_metagenome.counts.txt -t rel_ab_w_read_stats.” The “-t rel_ab_w_read_stats” option was used to profile each pooled metagenome in terms of relative abundances and estimate the number of reads for each identified bacterial clade. Resulting MetaPhlAn count tables of bacterial species from individual metagenomic pools were merged into one large table (Supplementary Table S1) with R v4.2.242. However, at this point, metagenomic pools LrRVf, LrIgarf, LrNEs1f, and MLDRV were excluded as they did not contain sequencing reads identified as bacteria during the MetaPhlAn analysis. Code for the above analyses is available at https://github.com/Callithrix-omics/callitrichidae_microbiome/blob/main/Metagenomic_Community_Profiling/biobakery.sh.
The merged MetaPhlAn table was then read into R for alpha and beta diversity analysis with phyloseq 1.34.043 and vegan 2.5-744. Input files to create the phyloseq object are accessible at https://github.com/Callithrix-omics/callitrichidae_microbiome/blob/main/additional_files/phyloseq_OTU.tsv and https://github.com/Callithrix-omics/callitrichidae_microbiome/blob/main/additional_files/phyloseq_taxaonomy.tsv. We first normalized bacterial read counts in each gut metagenomic pool using median bacterial abundance values across the entire data set. The resulting normalized counts by host taxon are listed in Supplementary Table S2 and by host environment in Supplementary Table S3. We first created a phyloseq object which was then turned into a vegan compatible data object. We calculated the Shannon diversity index with vegan to measure the gut microbiome alpha diversity of callitrichid metagenomic pools while classifying hosts by genus and environment, respectively. To better understand the effects of both host environment and host genus on callitrichid gut microbiome alpha diversity, we fitted a two-way ANOVA model. Shannon index measures were used as model response variables, and host environment and host genus as independent variables. Levene’s test indicated homogeneous variances in the two independent variables (F(3,14) = 1.10, p = 0.38). No interaction between the independent variables was used, as we assumed host genus was independent of host environment. A diagnostic residuals Q-Q plot was used to check the data for normality. The code for the analyses described in this paragraph is available specifically at https://github.com/Callithrix-omics/callitrichidae_microbiome/blob/main/Metagenomic_Community_Profiling/R_MLD_marmoset_microbiome_vegan_phyloseq.rmd.
Phyloseq was also used to estimate gut microbiome bacterial species abundance for callitrichid metagenomic pools and construct a bacterial abundance plot. To test for significance in differential bacterial taxa abundance according to host environment and host genus, respectively, we used LEfSe45 at species level for bacterial taxa. The merged table of MetaPhlAn bacterial species counts within each pooled sample library, was loaded into the LEfSe submodule within MicrobiomeAnalyst46. The analysis was carried out with the default settings of a FDR-adjusted p-value cutoff set to 0.1 and the log LDA cut-off at 2.0. Prior to carrying out the LEfSe analysis, data were normalized in MicrobiomeAnalyst with settings of the ‘Low count filter’ set to ‘Mean abundance value’ and the option under ‘Low variance filter’ set to 10% based on the interquantile range. Next, at the data normalization step, data were scaled by ‘total sum scaling,’ and we did not apply any data transformations.
To explore beta diversity of the callitrichid gut microbiome in R, using the same phyloseq object as above, we calculated the Bray–Curtis dissimilarity indices for each host, and then used the indices to make a Principle Coordinates Analysis (PCoA) plot in vegan. We superimposed both host environmental and host taxon information onto the PCoA plot. To understand the effects that host environment and host taxon had on marmoset gut microbiome Bray–Curtis dissimilarity indices, we used adonis2 function in the phyloseq package43. We fitted PERMANOVA47 models which included the marginal effects of host environment and host taxon as independent variables and Bray–Curtis dissimilarity indices as the dependent variables. The PERMANOVA models were run with the adnois2 function. PERMANOVA post-hoc tests of Bray–Curtis dissimilarity indices were carried out as pairwise adonis tests with the adonis.pair function from the the EcolUtils48 R package. The test was run for 1000 permutations and p-values were corrected by the false discovery rate. The code for the analyses described in this paragraph is available at https://github.com/Callithrix-omics/callitrichidae_microbiome/blob/main/Metagenomic_Community_Profiling/R_MLD_marmoset_microbiome_vegan_phyloseq.rmd.
Metagenomic community functional profiling
In order to identify functional gut microbiome pathways among our pooled samples we used HUMAnN3 v3.0.0.alpha.449. Since HUMAnN does not utilize pair-ended fastqs, we first merged all such pairs of Kneaddata-filtered fastqs into a single file that served as input to HUMAnN. We ran the program under default settings with the ChocoPhlAn pangenome database (part of the bioBakery environment) to profile callitrichid functional gut microbiome Metacyc50 metabolic pathway abundance. Analyses ran for individual sample pools were merged with the HUMAnN humann_renorm_tabl command and normalized from reads per kilobase (RPK) units to counts per million (CPM). After filtering out unidentified functional pathways among sample metagenomic pools, we conducted a multivariate analysis to search for significant differential abundance between gut microbiome function pathways among our sample pools and host genus and environment with MaAsLin 2.051. Names of resulting HUMAnN3 pathways and recoded numbering of these pathways is available at https://github.com/Callithrix-omics/callitrichidae_microbiome/blob/main/additional_files/bacterioma_pathways_coded_Humann_Maasalin.tsv. The input for MaAsLin 2.0 is provide at https://github.com/Callithrix-omics/callitrichidae_microbiome/blob/main/additional_files/bacterioma_pathways.cleaned.transpose.dummy.tsv. We ran this program in R by fitting a fixed effects model (“expr Host Genus + Host Environment”). For functional pathways with differential abundance in the callitrichid gut, we identified their higher order ‘superclass’ in the Metacyc database (https://metacyc.org). Finally, for significantly differentially abundant gut microbiome functional pathways in our sample pools, we calculated contributions of bacterial species to each pathway with the Humann3 humann_barplot command. Each given pathway was considered as the focal feature within the humann_barplot command. Code for the HUMAnN analyses described above is available at https://github.com/Callithrix-omics/callitrichidae_microbiome/blob/main/Metagenomic_Community_Profiling/biobakery.sh. Code for the MaAsLin analysis is provided at https://github.com/Callithrix-omics/callitrichidae_microbiome/blob/main/Metagenomic_Functional_Profiling/R_MLD_marmoset_microbiome_maasalin.rmd.
Recovery and characterization of MAGs
All steps of MAG recovery were performed through a co-assembly approach with the KBase platform52, using the the same set of raw pair-ended fastq files as used above with the bioBakery metagomic environment. Gut metagenomic pools LrRVf, LrIgarf, LrNEs1f, and MLDRV were excluded from MAG analyses, as they did not contain sequencing reads identified as bacteria during community profiling analysis. Prior to uploading these reads into a Kbase narrative, they were pre-processed for quality in fastp 0.23.453 with default settings for removing low quality reads, removing adaptors, trimming of low quality base calls, and read de-duplication. For co-assembly, we then merged all fastqs together from wild L. rosalia sample pools, captive Callithrix sample pools, and wild Callithrix sample pools, respectively. Only reads from the single sample pool from captive L. rosalia were not merged with any additional samples. Merged filtered paired-end reads were uploaded using the KBase app Upload File to Staging from Web v1.0.12 and Import FASTQ/SRA File as Reads from Staging Area52, respectively. Bins from each merged set of sample pools were recovered by first assembling metagenome contigs using metaSPades v.3.15.354 and MEGAHIT v1.2.935. As ultimately full MAG analysis was carried out only for data from captive marmoset hosts (see below), we continued with analysis of metaSPADEs contigs as they provided better results than MEGAHIT for captive marmoset gut microbiome pools in terms of N50 and the longest length contig (Supplementary Fig. S1 and Supplementary Fig. S2). We then conducted binning of contigs with CONCOCT v1.155 and MAXBIN2 v2.2.456. Resulting bins from CONCOCT and MAXBIN2 from each co-assembly were optimized using the app DAS Tool v1.1.257 and optimized bins were quality-checked using CheckM v1.0.18. After CheckM bin quality checks, only the captive Callithrix optimized co-assembly bins showed completeness above 30% and marker lineage beyond “root.” Thus, from this point we only focused on the 4 recovered captive Callithrix optimized co-assembly bins. Although only a single bin (bin.002) out of these four met the minimum Metagenome-Assembled Genome (MIMAG) standards of completeness (> 90%) and contamination (< 5%) for high-quality drafts of MAGS52, we included all four due to their relevance for exploratory aims of this study. The four bins were functionally annotated by the Annotate and Distill Assemblies app with DRAM v.0.1.2 (Distilled and Refined Annotation of Metabolism)58. Taxonomic classification of MAGs was done with the GTDB-Tk29 v1.76.059 taxonomic classification tool.
Ethics
Tissues were collected under the approval of the Brazilian Environmental Ministry (SISBIO protocols 17409 and 35931). Biological tissue sampling complied with all institutional, national, and international guidelines.
Results
Community profiling of callitrichid metagenomic pools
L.rosalia and Callithrix sp. metagenomic pool gut microbiome alpha and beta-diversity
Shannon index box plots of callitrichid gut metagenomic pool alpha diversity are shown in Fig. 2. Considering host environment, alpha diversity values were higher in wild hosts than in captive hosts (Fig. 2A). For host genus, gut microbiome alpha diversity was higher for Callithrix hosts than for Leontopithecus hosts (Fig. 2B). A two-way ANOVA model (Shannon Index Genus+Environment) was fit to test for differences in host gut microbiome Shannon diversity indices when considering both host environment and host genus. Callitrichid gut microbiome alpha diversity differences due to host genus were significant (ANOVA, F(1) = 8.861, p-value = 0.01), but those due to host environment were not (ANOVA, F(1) = 3.6, p-value = 0.08).
Figure 2.

Boxplots of callitrichid gut microbiome Shannon indices for sample pools by host taxon (A) and host environment (B). Panel (C) shows a PCoA plot for host pool gut microbiome beta-diversity measured by the Bray–Curtis dissimilarity index.
A PCoA plot of host metagenomic pool Bray–Curtis dissimilarity index measures with superimposition of host environment and host taxon is shown in Fig. 2C. This plot shows gut microbiome dissimilarity in the callitrichid gut microbiome when considering host genus as well as host environment. We fit a PERMANOVA model with independent variable margins, to explain the effects of host environment and host taxon on callitrichid gut microbiome Bray–Curtis indices. The effects of both host environment (PERMANOVA, F(1) = 1.46, p = 0.001) and host genus (PERMANOVA, F(2) = 1.51, p = 0.012) were significant for metagenomic pool callitrichid gut microbiome beta diversity.
L.rosalia and Callithrix sp. metagenomic gut microbiome composition
The relative abundances of gut microbiome bacteria among callitrichid hosts are shown in Fig. 3A and absolute and relative abundance counts are also shown by host taxon in Supplementary Table S2 and by host environment in Supplementary Table S3. Bifidobacterium is more abundant in the gut microbiome of Atlantic Forest hosts than that of captive hosts. For example, Bifidobacterium callitrichos is more abundant in the former (see label “9” in bars for Safe and SIgua in Fig. 3A) than in the latter (see label “9” in bars for P1, P3, and P4 in Fig. 3A). Among GLT hosts, we see Bifidobacterium biavatii being the most abundant Bifidobacterium species in the gut of wild hosts, but Bifidobacterium is absent from the single captive gut microbiome GLT pool (Fig. 3A). Bifidobacterium tissieri is also in the gut microbiome of wild GLT hosts (abundance label “15” in LrNes2f and MLDAfe in Fig. 3A). For Enterococcus, the bacterial species most abundant in the gut of captive marmoset hosts were Enterococcus faecalis, Enterococcus facium, and Enterococcus hirae (see abundance labels “26–28” in bars representing P1–P4, and P7 in Fig. 3A). Only a single species of Enterococcus, Enterococcus italicus, was identified in a single gut microbiome pool of wild Callithrix hosts (bar for SIgua Fig. 3A). Then, Serratia marcescens was unique to gut microbiome pools of captive Callithrix hosts (see abundance label “51” in P3 and P4 bars in Fig. 3A), and Megamonas funiformis occurred only in the single captive GLT gut microbiome pool (see abundance labels “40–31” for PoolRosalia bar in Fig. 3A).
Figure 3.

Gut metagenomic pools from callitrichids are enriched for Bifidobacterium species. (A) Relative levels of bacterial species abundance by host environment (W = Wild, C = Captive) and taxon classification of pooled samples. Each bacterial species represented in the legend on the right hand side is represented by a color and a number. Bacterial abundances for a specific bacterial species are labeled by the corresponding color and number in bars representing each individual gut metagenonic pool. Bacterial species with relatively ultra low abundances were blacked out in the bar representing gut metagenomic pool P7. (B). LefSe analysis of gut microbiome bacterial species abundance by host taxon of sampled pools. (C) LefSe analysis of gut microbiome bacterial species taxa abundance by host taxon of sampled pools.
Results of LefSe differential abundance testing of callitrichid gut microbiome metagenomic pools show that gut microbiome pools from marmosets were enriched for B. callitrichos and three other bacterial species (Fig. 3B). GLTs were enriched for B. biavatii (Fig. 3B). Then LefSe analysis indicated that enriched gut microbiome bacterial species included Enterococcus hirae and Enterococcus faecalis, and Leuconostoc citreium among captive pool samples, and B. biavatii was among the enriched bacterial species in Atlantic Forest sample pools (Fig. 3C).
Gut microbiome functional pathways of Callithrix and Leontopithecus metagenomic pools
From the 1700+ metabolic pathways found in our data set, fitting a MaAsLin fixed effects model identified 38 Metacyc functional pathways with significant differential abundance among our gut metagenomic sample pools. We removed a total of five Metacyc pathways which were either attributed to non-bacterial organisms like plants or mammals or not identifiable within the Metacyc database. A heatmap of the finalized set of enriched pathways is shown in Fig. 4, along with the higher order functional Metacyc ‘superclass’ of each pathway. Enrichment information for each pathway within a specific host classification is given in Supplementary Table S4. With one exception, all these pathways were significantly enriched in captive callitrichids (Fig. 4). These pathways fell under Metacyc superclasses related to metabolite biosynthesis, degradation, and fermentation. More specifically, pathways included Entner–Doudoroff Glycolysis pathway and carbohydrate degradation. A single pathway was enriched in GLTs (PWY-5100) which was related to pyruvate fermentation.
Figure 4.

Callitrichid microbiome functional pathways are enriched for metabolite biosynthesis, degradation, and fermentation. Heatmap of MaAsLin analysis of significant associations Metacyc functional pathway superclasses and host environment and genus classifications of callitrichid sample pools, respectively. Level of association between each functional pathway and host category is indicated by the legend on the left side.
Bacterial species associations with callitrichid gut microbiome functional pathways are shown in Fig. 5 and Supplementary Fig. S3. We see multiple associations of the same recurrent set of gut bacterial species across a wide range of Metacyc pathways. These bacterial species include Serratia marcescens, Escherichia coli, Ewingella americana, and Pseudomonas helleri, with most pathways being enriched for captivity. One exception is shown for Pathway PWY-5100, which is associated with pyruvate fermentation to acetate and lactate II (Supplementary Fig. S3). This pathway shows that B. callitrichos carries out this function in wild Callithrix host.
Figure 5.
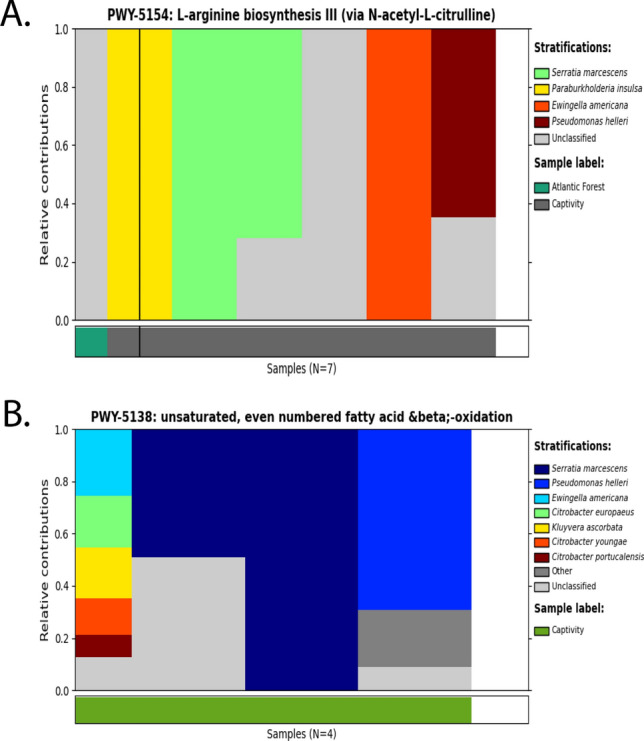
Relative abundance plots of gut microbiome bacterial contributions to differentially abundant functional Metacyc pathways. Each separate plot within the figure represents an individual pathway, each bar represents an individual sample pool, and stratification within each bar represent bacterial species.
MAG recovery from callitrichid metagenomic pools
MetaSPades (MEGAHIT results are shown in Supplementary Figs. S1 and S2) generated the following contig results from callitrichid gut metagenomic pools: (1) 637 co-assembled contigs (65,079 bp for the largest contig) with N50 of 2932 bp (base pairs) and L50 of 183 bp from wild GLTs; (2) 72 co-assembled contigs (92,239 bp for the largest contig) with N50 of 12,354 bp and L50 of 10 from wild marmosets; (3) 4146 co-assembled contigs (79,182 bp for the largest contig) with N50 of 4421 bp and L50 of 1008 from captive marmosets; and (4) 11 contigs (6058 for the largest contig) with N50 of 2949 bp and L50 of 4 from captive GLTs. MAXBIN2 produced 4 bins from captive marmoset metagenomic pools and 2 bins from wild GLT metagenomic pools, but failed to produce bins for the remaining data. CONCOCT produced 37 bins from captive marmoset metagenomic pools, 26 bins from wild GLT metagenomic pools, 8 bins from wild marmoset metagenomic pools, and 4 bins from the captive GLT metagenomic pool. The DAS Tool produced a total of 4 captive marmoset bins, but failed to produce optimized bins for captive GLTs, wild GLTs, and wild marmosets. Marker lineage beyond “root” during CheckM bin quality checks were only determined for the the 4 captive Callithrix optimized co-assembly bins (detailed information on these bins is available in Supplementary Table S5). Further MAG taxonomic and functional classification were only carried out for these 4 bins.
Taxonomic classification of MAGs from callitrichid metagenomic pools
Taxonomic classification of bins from captive marmoset hosts (Supplementary Table S5) placed two bins within the Gammaproteobacteria bacterial class, 1 bin in the the Actinomycetales bacterial class, and 1 bin into the Actinomycetia. All bins were classified down to the genus level (Microbacterium, Serratia, Pseudomonas, and Leuconostoc). One bin was classified at the species level as Serratia marcescens, which was the bin with the highest level of completeness and lowest level of contamination among these 4 bins (Supplementary Table S5).
Functional classification of MAGs from callitrichid metagenomic pools
Based on DRAM annotations, genes related to glycolysis (Embden–Meyerhof pathway), pentose phosphate pathway (pentose phosphate cycle), citrate (TCA or Krebs cycle), glyoxylate, reductive pentose phosphate (Calvin cycle), reductive citrate (Arnon–Buchanan cycle), dicarboxylate–hydroxybutyratecycles, and the reductive acetyl-CoA pathway (Wood–Ljungdahl pathway) were found in the bin classified as Serratia marcescens (Fig. 6). The same set of pathways except the Wood–Ljungdahl pathway were also represented, but to a lesser degree of completeness, in the bins classified as Microbacterium and Pseudomonas (Fig. 6). Carbohydrate-active enzymes (CAZymes) genes were mostly absent from bins except for the bin classified as Leuconostoc, for which CAZymes for amorphous cellulose, xyloglucan, and arabinan were present (Fig. 7). Serratia, Microbacterium, and Pseudomonas bins possessed genes involved in several short chain fatty acids (SCFAs) and alcohol conversion modules. SCFA modules in these bins included those for pyruvate to acetyl CoAv1 conversion and lactate L. Supplementary Table S6 shows gene annotations for the four bins that represent recovered captive Callithrix gut microbiome MAGs. Supplementary Table S7 gives gene counts of functional modules across a wide variety of metabolisms based on DRAM annotations.
Figure 6.

DRAM annotations of MAGs (metagenome-assembled genomes) recovered from four bins (bin.001–bin.004) of captive Callithrix gut metagenomic pools. The colors in the heatmap represent the completeness of relevant pathways and electron transport chain complexes (ETC) in each MAG. The heatmap was as part of DRAM output and shows modules present in at least one MAG.
Figure 7.

Presence/absence of relevant metabolic functions based on DRAM annotations of MAGs (metagenome-assembled genomes) recovered from four bins (bin.001–bin.004) of captive Callithrix gut metagenomic pools. The colours in the heatmap represent the presence or absence of relevant metabolic functions in each MAG. The heatmap was a part of DRAM output and shows modules present in at least one MAG. Abbreviations used in the figure are as follows: CAZy carbohydrate-active enzymes, metab. metabolism, red. reductases, SCFA short-chain fatty acids.
Discussion
The aim of this study was to expand our compositional and functional knowledge of the callitrichid gut microbiome. Thereby, we analyzed the bacterial component of VLP-filtered metagenomic pools that were opportunistically available from GLTs and marmosets. Although such data are not conventionally utilized for microbiome studies, our utilization of these data was largely motivated by (1) the importance that a healthy gut microbiome may play in the conservation of endangered animals; (2) the threatened conservation status of GLTs; (3) the potential health impact of hybrid marmosets introduced to the GLT geographic distribution; and (4) the relatively small number of studies conducted on Brazilian primate gut microbiomes. Metatranscriptomic data represent transcribed bacteria, and seem to be dominated by a small set of bacterial genera60. VLP-filtered metagenomic pools, which are specifically processed to enrich for VLPs and ideally remove all other biological material present in a sample, are even likely more biased in their representation of a few select bacterial taxa. These limitations are important to keep in mind while interpreting the results obtained in this present study.
Despite these limitations and when specifically considering results from community profiling approaches, we observed a number of parallel patterns between our findings and previously available callitrichid metagenomic studies. First, we observe significant effects of host taxon on callitrichid gut microbiome alpha and beta diversity, which may be related to relative differences for exudivory specialization between callitrichid taxa26,27,29,30. A similar pattern was observed for gut microbiome diversity across and within several Callithrix species and hybrids19, which differ in their level of specialization for exudivory. In wild lemur species, alpha diversity of gut microbiome composition also was significantly influenced by host taxon61. Further microbiomes, metagenomes, and metabolomes have been found to be species-specific in lemurs and attuned to host dietary specializations and associated gastrointestinal morphology61. Significant differences in GLT and marmoset gut microbiome diversity are also likely related to differences in dietary specialization between hosts. For callitrichids, a similar systematic study of taxa adapted to different dietary strategies as well as differing levels of exudivory specialization is necessary. Undertaking such work, especially in wild populations, will allow us to better understand how host phylogeny influences gut microbiome diversity.
In general, gut microbiome studies show that there is a significant difference in gut microbiome diversity between captive and wild hosts (marmosets19, kiwis62, Tasmanian devil63, mice64, primates22, raptors65, rhinos66, woodrats67). For our callitrichid data set, we found a significant effect of host environment on gut microbiome beta diversity but not alpha diversity. Cluster analysis of our data in fact shows almost no overlap of beta-diversity measures between captive and wild hosts, independent of host taxon. We do note that the effect of this host variable on gut alpha diversity was nearly significant, and it is likely that a larger sample size for our data would have produced a significant effect of host environment on callitrichid gut microbiome alpha diversity. Several previous studies agree that dietary differences between host captive and wild environments are one of the main factors driving some of these gut microbiome changes22,62–70.
In terms of bacterial taxa abundance in our metagenomic pool data set, we hypothesized that GLT and Callithrix gut microbiomes are enriched for Bifidobacterium, a bacterial genus important for host carbohydrate metabolism21,71,72. Our hypothesis is based on evidence that Neotropical primates gut microbiomes are significantly enriched for this bacterial genus relative to Old World primates73. Further Callithrix and Leontopithecus are the two primate genera with the highest average abundance of Bifidobacterium in the primate gut microbiome21. Based on community profiling, we found gut microbiome metagenomic pools of wild GLTs and Callithrix sp. to be significantly enriched for Bifidobacterium relative to their captive counterparts, a pattern which has been previously observed in the gut microbiome of wild and captive Callithrix19. Several other captive C. jacchus studies have also shown that Bifidobacterium plays an important compositional and functional role, particularly for carbohydrate mechanism, in Callithrix21,74. The results from our study extend this observation to also include the gut microbiome of GLTs. As our results provide further evidence of the importance of Bifidobacterium in the callitrichid gut, our current work and several other studies support to the idea that an important evolutionary relationship exists between the Callitrichidae family and Bifidobacterium21.
The types of Bifidobacterium species of that inhabit the gut microbiome of Neotropical primates seem to vary and depend on the taxon of the host73. While one recent study of the gut microbiome of wild and captive marmosets19 could only determine that bacteria from the Bifidobacterium genus were present within the gut microbiome of sampled hosts, captive studies have found that B. callitrichos and B. myosotis are common within the Callithrix gut microbiome21,72–74. We also show that Callithrix gut microbiome metagenomic pools were specifically enriched for B. callitrichos and B. myosotis. Strains of B. callitrichos previously found in the gut of captive C. jacchus were thought to possess genes that contributed to galactose, arabinose, and trehalose metabolic pathways74. Further, different B. callitrichos strains with significant genomic differences were found within the same marmoset host, results that suggest that different Bifidobacterium strains support various roles for carbohydrate metabolism within individual hosts74. Bifidobacterium myosotis is a relatively recently recognized species of Bifidobacterium that was identified in the feces of a baby C. jacchus72. Our data also indicated that gut metagenomic pools from Atlantic Forest hosts and GLTs were enriched for B. biavatii. This bacterial species has been isolated from the red-handed tamarin Saguinus midas, but interestingly, B. biavatii is common among primate taxa in general21.
By applying metagenomic community profiling approaches, we observed significant enrichment and high relative abundance of other bacterial species besides Bifidobacterium in callitrichid gut metagenomic pools. Metagenomic pools from hosts that were both Callithrix and captive were significantly enriched for Leuconostoc citreum, Enterococcus facelis, and Enterococcus hirae. Leuconostoc citreum is considered a lactic acid bacterium, a bacterial type known to be involved in sugar fermentation and that can colonize the gut75. Some lactic acid bacteria are able to metabolize oligosaccharides76,77. Leuconostoc citreum produces SCFAs78, a function also observed in Bifidobacterium, which may guard against the proliferation of pathogenic bacteria in the gut and decrease chronic inflammation79,80. It is plausible that within our sample of captive Callithrix, lactic acid bacteria may provide some of the same functional and protective properties in the gut as provided by Bifidobacterium to the gut of wild marmoset hosts. Enterococcus is present in the gastrointestinal tract of human and non-human animals, usually serving as commensals that participate in metabolism of carbohydrates and other nutrients but can turn into opportunistic pathogens in other environments81. Enterococcus facelis has been found in the enclosures of captive C. jacchus, which was recognized as a potential but not active ‘veterinary risk’82. Serratia marcescens, which was found in two captive Callithrix gut metagenomic pools, has been found to be injurious to intestinal epithelial cells in humans83. Finally, the only host category to possess Megamonas funiformis, another potentially pathogenic bacterial species, was found in the captive GLT gut microbiome pool. Other gut microbial community studies have associated Megamonas with obesity, inflammation, and prediabetes (see the discussion in84).
Relative to our community profiling approach, co-assembly MAG recovery approaches provided much sparser results for callitrichid gut metagenomic pools. For example, we were only able to classify bacterial species for co-assembled bins recovered from gut metagenomic pools of captive Callithrix hosts. Given that our data set was initially enriched for VLPs, the resulting filtered data most likely do not possesses a sufficient number of sequencing reads from the bacterial component to produce high quality MAGs. This point is evident from the low number of co-assembled contigs obtained from the majority of callitrichid gut metagenomic pools. However, some of the bacterial taxa classified through MAG recovery in bins from the gut of captive marmoset hosts (Enterococcus, Pseudomonas, and Serratia marcescens) overlapped with taxa also identified by community profiling approaches. However, except for the Serratia marcescens bin, all other bins did not meet MIMAG standards of completeness (> 90%) and contamination (< 5%) for high-quality MAG drafts52.
Functional analysis of callitrichid gut microbiome metagenomic pools via community profiling showed enrichment of 38 Metacyc pathways related to biosynthesis, degradation, and fermentation of metabolites. It is important to keep in mind that our functional results only give a partial look into the microbiome of sampled GLTs and marmosets. Functional pathways related to carbohydrate metabolism included those for pyruvate fermentation, carbohydrate degradation, and sugar alcohol degradation. In terms of host environment, functional pathways were enriched in captive but not wild callitrichids. For host genus, a single functional pathways was enriched in GLTs. Given that our data set only represents a select group of bacteria from callitrichid guts, these results likely show a limited representation of functional pathway enrichment of different classes of callitrichid hosts. However, we do see some parallel patterns in results with that of Malukiewicz et al.19 as to which bacterial species are preforming which functional roles within the gut of callitrichids. The latter study showed that whereas Bifidobacterium plays an important role in carbohydrate metabolism in wild Callithrix, other bacterial species carry out these functions in captive Callithrix. Our data indicate that pathways related to carbohydrate metabolism as well as other functions is carried out in captive callitrichids by Serratia marcescens, Escherichia coli Pseudomonas helleri, and Ewingella americana.
Despite the lack of high quality MAG recovery for our data set, functional annotation was possible for some of the recovered MAGs. For example, functional annotation of genes in the Serratia marcescens bin pointed to a role of the bacterial species in pyruvate fermentation in captive Callithrix hosts. Interestingly, functional annotation via community profiling pointed to pyruvate fermentation in the gut microbiome being carried out by Bifidobacterium callithricos in a metagenomic pool from one of the wild Callithrix hosts. These results point to the likelihood that metabolic functions usually carried out by Bifidobacterium in the gut of wild marmoset hosts are carried out by potentially pathogenic bacteria in the gut of captive marmoset hots.
The major findings of this study are consistent with previous studies in showing that Bifidobacterium is an important component of the callitrichid gut microbiome, and that the composition of GLT and marmoset gut microbiota is sensitive to host environmental factors. It will be, nonetheless, important for future studies to further confirm, replicate, as well as build upon our findings due to some of the inherent limitations of our opportunistic data set. A major goal of integrating the study of microbiomes into conservation research is determining what indicates a “healthy baseline” microbiome for a given host taxon9. This task requires the determination of reliable microbial indicators that consider the specific conservation needs of the host9. Given that urbanization and land use are considered major threats to GLT conservation, we recommend that future studies focus on the composition and functional aspects of the GLT gut microbiome among forest fragments which vary in terms of factors such as size, level of degradation, dietary intake, access to nutritional resources and proximity to urbanized areas. Ultimately, such data should facilitate the not so simple tasks of distinguishing between the causes and effects of community changes, and determination of whether these changes are functionally consequential for the host9.
For gut microbiome studies of marmosets, with the exception of this study and that of19, other studies are highly biased towards C. jacchus, such that future studies should strive to expand sampling to other marmoset species. Hybridization is also an extremely common occurrence in marmosets, which should be further explored in relation to the marmoset microbiome. This data set was highly biased toward C. jacchus x C. penicillata hybrids, expanding sampling of other types of free-ranging and captive marmoset hybrids is necessary to move marmoset microbiome studies forward. Overall, such information will expand baseline gut microbiome data available for wild and non-wild callitrichids to allow for the development of new tools to improve their management, welfare, and conservation. For new research into the callitrichid microbiome, we especially recommend utilizing shotgun whole metagenomic and/or transcriptomic approaches (which naturally do not intentionally filter out bacteria) in lieu of 16s rRNA approaches that have been previously utilized to significantly increase the inferential power to characterized the functional as well as taxonomic aspects of the callitrichid gut microbiome85,86.
Supplementary Information
Acknowledgements
We thank all the workers of the Centro de Primatologia de Brasília, Universidade de Brasília (Distrito Federal, Brazil), Centro de Primatologia do Rio de Janeiro (Rio de Janeiro, Brazil) and Associação Mico-Leão-Dourado (Rio de Janeiro, Brazil) for assistance with sample collection. We are also grateful to Carolina Furtado from Instituto Nacional de Câncer José de Alencar (Rio de Janeiro, Brazil) for assistance with high throughput sequencing and Matheus Cosentino from UFRJ for making the maps used in Fig. 1. This work was supported in part by the Conselho Nacional de Desenvolvimento Científico e Tecnológico/CNPq (grant 313005/2020-6) and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro/FAPERJ (E26/211.040/2019 , E-26/211.355/2021 and E-26/201.193/2022) for funding this study. This work was also supported by Associação Mico-Leão-Dourado.
Author contributions
J.M. carried out bioinformatics and statistical analysis and wrote the majority of the manuscript. MD carried out the study conceptualization and design, data generation, manuscript writing, reviewing and editing. A.F.A.S. provided reagents for metagenomic laboratory protocols, acquired financial support for the project, and provided manuscript feedback during writing. A.D.G. was involved with study conceptualization and manuscript writing. C.R.R.M. acquired financial support for the project, provided access to biological samples, and was involved with manuscript writing. R.A.C. provided access to computational infrastructure and was involved with manuscript writing. C.A.D., S.B.M., M.C.H.T., A.R.S., and A.P. provided access to biological samples.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Data availability
Supplementary material for this work is available at https://doi.org/10.5281/zenodo.8271171. The data set supporting the conclusions of this article is available in the NCBI SRA repository under Bioproject PRJNA847605 (SAMN28946310–SAMN28946330). The authors declare that they have no competing interests. Supplementary material for this work is available at https://doi.org/10.5281/zenodo.8271171.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Joanna Malukiewicz and Mirela D’arc.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-023-42059-4.
References
- 1.Huttenhower C, et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.West AG, et al. The microbiome in threatened species conservation. Biol. Conserv. 2019;229:85–98. doi: 10.1016/j.biocon.2018.11.016. [DOI] [Google Scholar]
- 3.McKnite AM, et al. Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS One. 2012;7:e39191. doi: 10.1371/journal.pone.0039191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kohl KD. Ecological and evolutionary mechanisms underlying patterns of phylosymbiosis in host-associated microbial communities. Philos. Trans. R. Soc. B Biol. Sci. 2020;375:20190251. doi: 10.1098/rstb.2019.0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley RE, et al. Evolution of mammals and their gut microbes. Science. 2008;320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Youngblut ND, et al. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 2019 doi: 10.1038/s41467-019-10191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levin D, et al. Diversity and functional landscapes in the microbiota of animals in the wild. Science. 2021 doi: 10.1126/science.abb5352. [DOI] [PubMed] [Google Scholar]
- 8.Amato KR, et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013;7:1344–1353. doi: 10.1038/ismej.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trevelline BK, Fontaine SS, Hartup BK, Kohl KD. Conservation biology needs a microbial renaissance: A call for the consideration of host-associated microbiota in wildlife management practices. Proc. R. Soc. B Biol. Sci. 2019;286:20182448. doi: 10.1098/rspb.2018.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barelli C, et al. Habitat fragmentation is associated to gut microbiota diversity of an endangered primate: implications for conservation. Sci. Rep. 2015 doi: 10.1038/srep14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Myers N, Mittermeier RA, Mittermeier CG, da Fonseca GAB, Kent J. Biodiversity hotspots for conservation priorities. Nature. 2000;403:853–858. doi: 10.1038/35002501. [DOI] [PubMed] [Google Scholar]
- 12.Russo G. Biodiversity: Biodiversity’s bright spot. Nature. 2009;462:266–269. doi: 10.1038/462266a. [DOI] [PubMed] [Google Scholar]
- 13.Ruiz-Miranda CR, et al. Estimating population sizes to evaluate progress in conservation of endangered golden lion tamarins (Leontopithecus rosalia) PLoS One. 2019;14:e0216664. doi: 10.1371/journal.pone.0216664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruiz-Miranda, C. et al.Leontopithecus rosalia (amended version of 2019 assessment). The IUCN red list of threatened species 2021: e.t11506a192327291. Tech. Rep. (2021). 10.2305/IUCN.UK.2021-1.RLTS.T11506A192327291.en. Accessed 11 Feb 2022.
- 15.Carvalho FA, Braga JMA, Nascimento MT. Tree structure and diversity of lowland Atlantic forest fragments: Comparison of disturbed and undisturbed remnants. J. For. Res. 2015;27:605–609. doi: 10.1007/s11676-015-0165-2. [DOI] [Google Scholar]
- 16.Miranda C, Affonso A, Martins A, Beck B. Distribuição do sagüi (Callithrix jacchus) nas áreas de ocorrência do mico leão dourado no estado de Rio de Janeiro. Neotrop. Primates. 2000;8:98–101. [Google Scholar]
- 17.Malukiewicz J, et al. Natural and anthropogenic hybridization in two species of eastern Brazilian marmosets (Callithrix jacchus and C. penicillata) PLoS One. 2015;10:e0127268. doi: 10.1371/journal.pone.0127268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.dos Santos Sales, I., Ruiz-Miranda, C. R. & de Paula Santos, C. Helminths found in marmosets (Callithrix penicillata and Callithrix jacchus) introduced to the region of occurrence of golden lion tamarins (Leontopithecus rosalia) in Brazil. Vet. Parasitol.171, 123–129. 10.1016/j.vetpar.2010.02.044 (2010). [DOI] [PubMed]
- 19.Malukiewicz J, et al. The gut microbiome of exudivorous marmosets in the wild and captivity. Sci. Rep. 2022 doi: 10.1038/s41598-022-08797-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grassotti TT, et al. Fecal bacterial communities of wild black capuchin monkeys (Sapajus nigritus) from the Atlantic forest biome in southern Brazil are divergent from those of other non-human primates. Curr. Res. Microb. Sci. 2021;2:100048. doi: 10.1016/j.crmicr.2021.100048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lugli GA, et al. Evolutionary development and co-phylogeny of primate-associated Bifidobacteria. Environ. Microbiol. 2020;22:3375–3393. doi: 10.1111/1462-2920.15108. [DOI] [PubMed] [Google Scholar]
- 22.Clayton JB, et al. Captivity humanizes the primate microbiome. Proc. Natl. Acad. Sci. 2016;113:10376–10381. doi: 10.1073/pnas.1521835113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pokusaeva K, Fitzgerald GF, van Sinderen D. Carbohydrate metabolism in Bifidobacteria. Genes Nutr. 2011;6:285–306. doi: 10.1007/s12263-010-0206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milani C, et al. Unveiling bifidobacterial biogeography across the mammalian branch of the tree of life. ISME J. 2017;11:2834–2847. doi: 10.1038/ismej.2017.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cabana F, Dierenfeld ES, Wirdateti DG, Nekaris KAI. Exploiting a readily available but hard to digest resource: A review of exudativorous mammals identified thus far and how they cope in captivity. Integr. Zool. 2018;13:94–111. doi: 10.1111/1749-4877.12264. [DOI] [PubMed] [Google Scholar]
- 26.Rylands AB, de Faria DS. Habitats, Feeding Ecology, and Home Range Size in the Genus Callithrix. Oxford University Press; 2011. pp. 262–271. [Google Scholar]
- 27.Malukiewicz J, et al. An introduction to the Callithrix genus and overview of recent advances in marmoset research. ILAR J. 2021 doi: 10.1093/ilar/ilab027. [DOI] [PubMed] [Google Scholar]
- 28.Francisco TM, et al. Vegetable exudates as food for Callithrix spp. (Callitrichidae): Exploratory patterns. PLoS One. 2014;9:e112321. doi: 10.1371/journal.pone.0112321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rylands AB. The Ecology of the Lion Tamarins Leontopithecus: Some Intrageneric Differences and Comparisons with Other Callitrichids. Oxford University Press; 2011. pp. 297–313. [Google Scholar]
- 30.Coimbra-Filho A. Animais predados ou rejeitados pelo sauí-piranga, Leontopithecus rosalia (Linnaeus, 1766) na sua área de ocorrência primitiva (Callitrichidae, primates) Rev. Brasil. Biol. 1981;41:717–731. [Google Scholar]
- 31.Ferrari S. Ecological Differentiation in the Callitrichidae. Oxford University Press; 2011. pp. 316–326. [Google Scholar]
- 32.Roux S, Krupovic M, Debroas D, Forterre P, Enault F. Assessment of viral community functional potential from viral metagenomes may be hampered by contamination with cellular sequences. Open Biol. 2013;3:130160. doi: 10.1098/rsob.130160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zolfo M, et al. Detecting contamination in viromes using ViromeQC. Nat. Biotechnol. 2019;37:1408–1412. doi: 10.1038/s41587-019-0334-5. [DOI] [PubMed] [Google Scholar]
- 34.Malukiewicz J, et al. Hybridization effects and genetic diversity of the common and black-tufted marmoset (Callithrix jacchus and Callithrix penicillata) mitochondrial control region. Am. J. Phys. Anthropol. 2014;155:522–536. doi: 10.1002/ajpa.22605. [DOI] [PubMed] [Google Scholar]
- 35.Bassis CM, et al. Comparison of stool versus rectal swab samples and storage conditions on bacterial community profiles. BMC Microbiol. 2017 doi: 10.1186/s12866-017-0983-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reyman M, van Houten MA, Arp K, Sanders EAM, Bogaert D. Rectal swabs are a reliable proxy for faecal samples in infant gut microbiota research based on 16s-rRNA sequencing. Sci. Rep. 2019 doi: 10.1038/s41598-019-52549-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choudhury R, Middelkoop A, Bolhuis JE, Kleerebezem M. Legitimate and reliable determination of the age-related intestinal microbiome in young piglets rectal swabs and fecal samples provide comparable insights. Front. Microbiol. 2019 doi: 10.3389/fmicb.2019.01886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Short MI, et al. Comparison of rectal swab, glove tip, and participant-collected stool techniques for gut microbiome sampling. BMC Microbiol. 2021 doi: 10.1186/s12866-020-02080-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McIver LJ, et al. bioBakery: A metaomic analysis environment. Bioinformatics. 2017;34:1235–1237. doi: 10.1093/bioinformatics/btx754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2020).
- 43.McMurdie PJ, Holmes S. phyloseq: An r package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oksanen, J. et al.vegan: Community Ecology Package. R package version 2.5-7 (2020).
- 45.Segata N, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chong J, Liu P, Zhou G, Xia J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020;15:799–821. doi: 10.1038/s41596-019-0264-1. [DOI] [PubMed] [Google Scholar]
- 47.Anderson, M. J. Permutational multivariate analysis of variance (PERMANOVA). 10.1002/9781118445112.stat07841 (2017).
- 48.Salazar, G. EcolUtils: Utilities for community ecology analysis. R package version 0.1 (2021).
- 49.Beghini F, et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife. 2021;10:1. doi: 10.7554/elife.65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caspi R, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2015;44:D471–D480. doi: 10.1093/nar/gkv1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mallick H, et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 2021 doi: 10.1101/2021.01.20.427420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chivian D, et al. Metagenome-assembled genome extraction and analysis from microbiomes using KBase. Nat. Protoc. 2022;18:208–238. doi: 10.1038/s41596-022-00747-x. [DOI] [PubMed] [Google Scholar]
- 53.Chen S, Zhou Y, Chen Y, Gu J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017;27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alneberg J, et al. Binning metagenomic contigs by coverage and composition. Nat. Methods. 2014;11:1144–1146. doi: 10.1038/nmeth.3103. [DOI] [PubMed] [Google Scholar]
- 56.Wu Y-W, Simmons BA, Singer SW. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2015;32:605–607. doi: 10.1093/bioinformatics/btv638. [DOI] [PubMed] [Google Scholar]
- 57.Sieber CMK, et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018;3:836–843. doi: 10.1038/s41564-018-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shaffer M, et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 2020;48:8883–8900. doi: 10.1093/nar/gkaa621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-tk: A toolkit to classify genomes with the genome taxonomy database. Bioinformatics. 2019;36:1925–1927. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mühr LSA, Dillner J, Ure AE, Sundström K, Hultin E. Comparison of DNA and RNA sequencing of total nucleic acids from human cervix for metagenomics. Sci. Rep. 2021;11:1. doi: 10.1038/s41598-021-98452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Greene LK, et al. A role for gut microbiota in host niche differentiation. ISME J. 2020;14:1675–1687. doi: 10.1038/s41396-020-0640-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Juan PAS, Castro I, Dhami MK. Captivity reduces diversity and shifts composition of the brown kiwi microbiome. Anim. Microbiome. 2021 doi: 10.1186/s42523-021-00109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chong R, et al. Looking like the locals—Gut microbiome changes post-release in an endangered species. Anim. Microbiome. 2019 doi: 10.1186/s42523-019-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosshart SP, et al. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell. 2017;171:1015–1028.e13. doi: 10.1016/j.cell.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oliveira BCM, Murray M, Tseng F, Widmer G. The fecal microbiota of wild and captive raptors. Anim. Microbiome. 2020 doi: 10.1186/s42523-020-00035-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gibson KM, et al. Gut microbiome differences between wild and captive black rhinoceros—Implications for rhino health. Sci. Rep. 2019;9:1. doi: 10.1038/s41598-019-43875-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kohl KD, Skopec MM, Dearing MD. Captivity results in disparate loss of gut microbial diversity in closely related hosts. Conserv. Physiol. 2014;2:cou009. doi: 10.1093/conphys/cou009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McKenzie VJ, et al. The effects of captivity on the mammalian gut microbiome. Integr. Comp. Biol. 2017;57:690–704. doi: 10.1093/icb/icx090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Houtz JL, Sanders JG, Denice A, Moeller AH. Predictable and host-species specific humanization of the gut microbiota in captive primates. Mol. Ecol. 2021 doi: 10.1111/mec.15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Greene LK, et al. Gut microbiota of frugo-folivorous sifakas across environments. Anim. Microbiome. 2021 doi: 10.1186/s42523-021-00093-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu L, et al. Captive common marmosets (Callithrix jacchus) are colonized throughout their lives by a community of Bifidobacterium species with species-specific genomic content that can support adaptation to distinct metabolic niches. mBio. 2021 doi: 10.1128/mbio.01153-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Michelini S, et al. Bifidobacterium myosotis sp. nov., Bifidobacterium tissieri sp. nov. and Bifidobacterium hapali sp. nov., isolated from faeces of baby common marmosets (Callithrix jacchus) Int. J. Syst. Evol. Microbiol. 2016;66:255–265. doi: 10.1099/ijsem.0.000708. [DOI] [PubMed] [Google Scholar]
- 73.Modrackova N, et al. The bifidobacterial distribution in the microbiome of captive primates reflects parvorder and feed specialization of the host. Sci. Rep. 2021 doi: 10.1038/s41598-021-94824-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Albert K, Rani A, Sela DA. The comparative genomics of Bifidobacterium callitrichos reflects dietary carbohydrate utilization within the common marmoset gut. Microb. Genom. 2018;4:1. doi: 10.1099/mgen.0.000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pessione E. Lactic acid bacteria contribution to gut microbiota complexity: Lights and shadows. Front. Cell. Infect. Microbiol. 2012 doi: 10.3389/fcimb.2012.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Endo A, Nakamura S, Konishi K, Nakagawa J, Tochio T. Variations in prebiotic oligosaccharide fermentation by intestinal lactic acid bacteria. In.t J. Food Sci. Nutr. 2016;67:125–132. doi: 10.3109/09637486.2016.1147019. [DOI] [PubMed] [Google Scholar]
- 77.Gänzle M, Follador R. Metabolism of oligosaccharides and starch in Lactobacilli: A review. Front. Microbiol. 2012;3:1. doi: 10.3389/fmicb.2012.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Silva MS, et al. Probiotic properties of Weissella cibaria and Leuconostoc citreum isolated from Tejuino—a typical Mexican beverage. LWT. 2017;86:227–232. doi: 10.1016/j.lwt.2017.08.009. [DOI] [Google Scholar]
- 79.DeGruttola AK, Low D, Mizoguchi A, Mizoguchi E. Current understanding of dysbiosis in disease in human and animal models. Inflamm. Bowel Dis. 2016;22:1137–1150. doi: 10.1097/mib.0000000000000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grosse CSJ, Christophersen CT, Devine A, Lawrance IC. The role of a plant-based diet in the pathogenesis, etiology and management of the inflammatory bowel diseases. Expert Rev. Gastroenterol. Hepatol. 2020;14:137–145. doi: 10.1080/17474124.2020.1733413. [DOI] [PubMed] [Google Scholar]
- 81.Krawczyk B, Wityk P, Gałęcka M, Michalik M. The many faces of Enterococcus spp.—commensal, probiotic and opportunistic pathogen. Microorganisms. 2021;9:1900. doi: 10.3390/microorganisms9091900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bakker J, Ouwerling B, Heidt P, Kondova I, Langermans J. Advantages and risks of husbandry and housing changes to improve animal well being in a breeding colony of common marmosets (Callithrix jacchus) J. Am. Assoc. Lab. Anim. Sci. 2015;54:273–279. [PMC free article] [PubMed] [Google Scholar]
- 83.Ochieng JB, et al. Serratia marcescens is injurious to intestinal epithelial cells. Gut Microbes. 2014;5:729–736. doi: 10.4161/19490976.2014.972223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sheng S, et al. Gut microbiome is associated with metabolic syndrome accompanied by elevated gamma-glutamyl transpeptidase in men. Front. Cell. Infect. Microbiol. 2022 doi: 10.3389/fcimb.2022.946757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Durazzi F, et al. Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Sci. Rep. 2021 doi: 10.1038/s41598-021-82726-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ranjan R, Rani A, Metwally A, McGee HS, Perkins DL. Analysis of the microbiome: Advantages of whole genome shotgun versus 16s amplicon sequencing. Biochem. Biophys. Res. Commun. 2016;469:967–977. doi: 10.1016/j.bbrc.2015.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supplementary material for this work is available at https://doi.org/10.5281/zenodo.8271171. The data set supporting the conclusions of this article is available in the NCBI SRA repository under Bioproject PRJNA847605 (SAMN28946310–SAMN28946330). The authors declare that they have no competing interests. Supplementary material for this work is available at https://doi.org/10.5281/zenodo.8271171.