

Abstract
HLA allelic variation has been well studied and documented in many parts of the world. However, African populations have been relatively under-represented in studies of HLA variation. We have characterized HLA variation from 489 individuals belonging to 13 ethnically diverse populations from rural communities from the African countries of Botswana, Cameroon, Ethiopia, and Tanzania, known to practice traditional subsistence lifestyles using next generation sequencing (Illumina) and long-reads from Oxford Nanopore Technologies. We identified 342 distinct alleles among the 11 HLA targeted genes: HLA-A, -B, -C, -DRB1, -DRB3, -DRB4, -DRB5, -DQA1, -DQB1, -DPA1 and -DPB1, with 140 of those alleles containing novel sequences that were submitted to the IPD-IMGT/HLA database. Sixteen of the 140 alleles contained novel content within the exonic regions of the genes, while 110 alleles contained novel intronic variants. Four alleles were found to be recombinants of already described HLA alleles and 10 alleles extended the sequence content of already described alleles. All 140 alleles include complete allelic sequence from the 5’ UTR to the 3’ UTR that are inclusive of all exons and introns. This report characterizes the HLA allelic variation from these individuals and describes the novel allelic variation present within these specific African populations.
Keywords: HLA, genetic variation, novel alleles, Illumina, Oxford Nanopore Technologies
Introduction:
It has been hypothesized that the human populations may have millions of uncharacterized and unreported HLA alleles; admittedly rare alleles but nevertheless unreported1,2. In this report we have characterized HLA alleles from 13 different populations across Africa. The significant finding is that among 489 subjects we identified 130 unique alleles with new sequences not previously described. It suggests that poorly characterized populations may include large numbers of new HLA alleles. HLA genes determine the antigen-specific immune responses and, therefore, any disease with an immune-related component may be influenced by these HLA genes/proteins. Considering the increasing globalization of our world today, issues related to infectious diseases, vaccinations and responses to the vaccines or any diseases with an immunological basis, will be inherently related to HLA polymorphisms. Therefore, it becomes imperative to develop a more comprehensive understanding of the genetic variation of HLA genes in many world populations. In this report we describe new HLA class I alleles with either single nucleotide polymorphisms (SNPs) or already known alleles with incomplete sequences that we now have characterized for the entire sequence between the 5’ and the 3’ untranslated regions (UTR). Regarding the new HLA class II alleles, they are more numerous and have a higher degree of variation, whereby SNPs are the main type of variation observed across the length of the genes, however, insertions, and deletions are also found. Additionally, the majority of novel variants observed in HLA class II alleles were found to occur in intron 1, in part due to the extended length of intron 1 in these genes. We conclude that the scientific and medical communities need to be aware of this potentially enormous variation of HLA genes and systematically undertake the study and characterization of the many understudied world populations.
Materials & Methods:
Samples:
Whole blood samples were collected from 504 volunteers, coming from 13 ethnically diverse African populations in Botswana, Cameroon, Ethiopia and Tanzania. Before sample collection permits were received from the Ministry of Health and National Ethics Committee in Cameroon; Commission for Science and Technology (COSTECH), National Institute for Medical Research (NIMR) in Dar es Salaam, Tanzania; the University of Addis Ababa and the Federal Democratic Republic of Ethiopia Ministry of Science and Technology National Health Research Ethics Review Committee; the University of Botswana and the Ministry of Health in Gaborone, Botswana. We obtained Informed consent from all research participants. In addition, appropriate IRB approval was obtained from the University of Pennsylvania (IRB Protocol #807981). All individuals were sampled from rural communities, known to practice traditional subsistence lifestyles (hunter-gathering, agriculture or pastoralism). These populations (listed in Table 1) have highly diverse genetic ancestry and live in diverse environments with differing exposure to pathogens3–5. Individuals included in this study have unknown consanguinity.
Table 1.
Country and population information for the 504 individuals who were HLA genotyped. Population size derived from Ethnologue 25th Edition19, unless otherwise noted. †Population size within the country studied.
| Country | Population | Number of Individuals included in study | Approximate Population Size |
|---|---|---|---|
| Botswana | Herero | 40 | 18,700† |
| Botswana | Ju|’hoansi | 34 | 5,000† |
| Botswana | !Xoo | 42 | 2,000† |
| Cameroon | Bagyeli | 11 | 4,250† |
| Cameroon | Baka | 24 | 40,000† |
| Cameroon | Mbororo Fulani | 83 | 85,00019 - 100,00020 |
| Cameroon | Tikari | 26 | 110,000 |
| Ethiopia | Amhara | 41 | 19,880,00021 |
| Ethiopia | Dizi | 39 | 34,70021 |
| Ethiopia | Mursi | 30 | 7,48021 |
| Ethiopia | Chabu | 39 | 40019 – 1,50022 |
| Tanzania | Hadza | 53 | 1,200 |
| Tanzania | Sandawe | 42 | 60,000 |
| Total | 504 |
Sample Preparation and Sequencing:
Genomic DNA was isolated from white blood cells with Puregene DNA extraction kits (Qiagen, Germany) for these 504 volunteers of African ancestry. All 11 HLA class I and class II genes (HLA-A, -B, -C, -DRB1, -DRB3, -DRB4, -DRB5, -DQA1, -DQB1, -DPA1 and -DPB1) for the aforementioned 504 individuals were sequenced on an Illumina MiSeq platform (San Diego, CA) on multiple sequencing runs using targeted amplicon-based NGS with Omixon Holotype HLA™ V2 kits (Budapest, Hungary)6. Fastq files were analyzed with Omixon Twin (version 3.1.3) and GenDx NGSengine (Utrecht, Netherlands, version 2.13) using IPD-IMGT/HLA database7 version 3.38. The resulting genotype and quality metrics from both software programs using the MiSeq data were compared to produce a high confidence final genotype through an in-house software program (HLAInspector) that has been validated for use in the Immunogenetics Laboratory of Children’s Hospital of Philadelphia, a CLIA and ASHI accredited clinical laboratory, using clinical protocols with appropriate quality controls and standards.
Consensus Sequence Generation:
Consensus sequences, extending from the 5′ UTR to the 3′ UTR of the respective HLA gene, were then extracted from Twin and GenDx for every successfully sequenced allele and compared with each other. Differences due to errors in Twin or GenDx, as well as differences in low complexity regions, were investigated and resolved manually. After this review and manual editing, the finalized consensus sequences were compared and grouped if they were identical. Each unique consensus sequence was annotated with the fully characterized alleles reported in the IPD-IMGT/HLA database version 3.38. Consensus sequences that either perfectly matched a known allele or did not match a known allele but had been observed as phased, one or more times, in the dataset were considered complete.
Additional work was performed on selected loci using Oxford Nanopore Technologies (ONT) (Oxford, UK) to complete the consensus sequences for alleles that had either exonic or intronic novel variants that were not phased elsewhere in the entire genotyping dataset of these populations. Briefly, these loci were amplified using the Omixon Holotype HLA V2 kits, with libraries prepared using the ONT HLA_1D Native barcoding kit for genomic DNA with EXP-NBD104, EXP-NBD114, SQD-LSK109, according to the manufacturer’s protocol version HLA V1, NBE_9065_v109_revH_23May2018, beginning at the end repair step. The final barcoded pool was loaded on an ONT SpotON SQK-LSK109 flow cell and sequenced on the MinION platform. During analysis, basecalling and demultiplexing of ONT reads was performed using Guppy (ONT), version 3.2.2 initially and version 3.4.3 later. Correction of the ONT reads with Illumina reads was performed with FMLRC8 version 0.1.2, using default settings to produce long hybrid reads that were then used to create an intermediate consensus sequence with MAFFT9 version 7.394, which served as a reference allele in subsequent alignment steps. A total of 1,000 corrected ONT reads were aligned to the intermediate consensus sequence using minimap210 version 2.17. Variants were called and corrected reads were split into separate alleles before a final consensus sequence was generated for each allele. Alternatively, if a locus was homozygous, then a single consensus sequence was generated. A final allele-specific consensus sequence was derived from manual inspection and resolution of any differences found between the consensus sequences generated from the original Illumina data and the corrected ONT data.
All complete consensus sequences, generated using either only Illumina data or Illumina and ONT data, were compared again and grouped if they were identical. Each group represented a unique allele sequence, observed at least once in our dataset, and was annotated with the fully characterized alleles in the most recent IPD-IMGT/HLA database version (3.46, October 2021). Next, the alleles that were found to be either new or extending the sequence of known alleles were reviewed according to the IPD-IMGT/HLA database criteria for submission and a total of 140 HLA alleles were finally submitted to GenBank and to the IPD-IMGT/HLA database in November/December 2021. If any of these alleles had been observed only once in our dataset, it was verified with a second round of amplification and sequencing on the Illumina MiSeq platform before being submitted.
IPD-IMGT/HLA database version 3.48 (April 2022) was used for 1) all comparisons between the novel HLA alleles described herein and the closest reference allele found in the database, and 2) the determination of the prevalence of the variant(s) found in each novel HLA allele.
Results:
Out of the 504 DNA samples initially received, 15 samples failed to amplify and were removed early from further analysis. Four hundred eighty-nine (489) samples were amplified, prepared, and sequenced at all 11 HLA genes (HLA-A, -B, -C, -DRB1, -DRB3, -DRB4, -DRB5, -DQA1, -DQB1, -DPA1, and -DPB1) on the Illumina MiSeq platform (NGS). A total of 4,524 loci were genotyped among the 11 targeted HLA genes (Table 2).
Table 2.
Number of samples genotyped at each HLA locus.
| HLA class I | Number of samples genotyped | HLA class II | Number of samples genotyped |
|---|---|---|---|
| HLA-A | 489 | HLA-DRB1 | 489 |
| HLA-B | 489 | HLA-DRB3 | 305 |
| HLA-C | 489 | HLA-DRB4 | 192 |
| HLA-DRB5 | 125 | ||
| HLA-DPB1 | 488 | ||
| HLA-DPA1 | 489 | ||
| HLA-DQB1 | 481 | ||
| HLA-DQA1 | 488 |
Consensus sequences were generated using only Illumina data for the alleles present within 4,253 loci (94%), while 271 loci (6%) had consensus sequences completed using both Illumina and ONT data, for a total of 4,524 loci with completed consensus sequences. We were unable to generate final consensus sequences for 84 loci, which were due to various reasons, including sample or data insufficiency impacting both Illumina and ONT results, and failure to generate consensus sequences in low complexity and/or homopolymer regions.
Next, all finalized consensus sequences, generated with either technology, were compared and grouped if they were identical. A total of 743 unique allele consensus sequences were identified and each unique allele sequence was compared to the fully characterized alleles in the most recent IPD-IMGT/HLA database version (3.46, 2021–10). Of the 743 unique alleles, 401 (54%) perfectly matched fully characterized alleles already reported in the IPD-IMGT/HLA database (A: 57, B: 71, C: 56, DRB1: 24, DRB4: 4, DRB5: 3, DQA1: 46, DQB1: 39, DPA1: 40, DPB1: 53). The remaining 342 unique allele sequences (46%) were either novel alleles or extended/completed the sequence of known, already reported alleles (exonic, intronic and/or UTRs). Supplemental Table 1 describes the 743 consensus sequences identified in this study and the populations from which they derive.
Of the aforementioned 342 unique sequences, we found 177 allele sequences in genes that were partially characterized (DRB1: 49, DRB3: 22, DRB4: 10, DRB5: 2, DPB1: 94); however, since they did not cover the entire length of the gene, from exon 1 to the final exon of the gene, they were not considered further. The remaining 165 allele sequences were observed in genes whereby we were able to characterize the full gene sequence and were considered complete (A: 13, B: 5, C: 12, DQA1: 53, DQB1: 30, DPA1: 52), of which 140 allele sequences met the IPD-IMGT/HLA database criteria for submission and were submitted to the IPD-IMGT/HLA database in November/December 2021 for official HLA allele names. Forty-five of the sequences submitted had been observed only once in our dataset and were re-sequenced successfully on the Illumina MiSeq platform before submission. Of the 25 complete allele sequences not submitted, 22 (C: 1, DQA1: 12, DQB1: 6, DPA1: 3) were observed only once in our dataset and could not be sequenced again due to lack of genetic material, while 3 were DQA1 alleles with weak evidence of only homopolymer length differences against their closest known alleles.
The 140 HLA allele sequences submitted to the IPD-IMGT/HLA database were either new or extended the sequence (exonic, intronic and/or UTRs’s sequence) of known, already reported alleles (A: 13, B: 5, C: 11, DQA1: 38, DQB1: 24, DPA1: 49). A full description of the 140 alleles can be found in Supplemental Table 2. In summary, the 140 alleles are grouped as follows:
15 allele sequences were found to be novel exonic variants of known alleles (A: 1, B: 2, C: 1, DPA1: 5, DQA1: 3, DQB1: 3), with 11 of these novel variants representing non-synonymous exonic mutations leading to new proteins (A: 1, B: 2, C: 1, DPA1: 3, DQA1: 2, DQB1: 2) and 4 novel variants representing synonymous (silent) exonic mutations (DPA1: 2, DQA1: 1, DQB1: 1). Out of these 15 novel exonic variants, 10 included only exonic differences when compared to their closest known alleles, whereas 5 included other differences as well (intronic/UTR differences or sequence extension).
1 allele sequence was found to represent a novel null DQB1 allele.
110 allele sequences were found to be novel intronic variants of known alleles (A: 7, B: 2, C: 9, DPA1: 38, DQA1: 34, DQB1: 20).
4 allele sequences were found to be novel hybrid (recombinant) alleles between known alleles (DPA1: 3, DQA1: 1).
10 allele sequences were found to extend the sequence of known alleles (A: 5, B: 1, C:1, DPA1: 3), and now provide complete allelic sequence from the 5’ UTR to the 3’ UTR, including all exons and introns.
All the above-mentioned HLA allele sequences that were submitted were approved and official names were assigned to them by the World Health Organization (WHO) Nomenclature Committee For Factors of the HLA System in January-March 2022 following the policy stipulated in the most recent Nomenclature Report11,12.
Figure 1 indicates the region where the novelties occur in each of the genes characterized at the full length. For HLA class I alleles, we only observed single nucleotide polymorphisms or sequence extensions for already known alleles that are now characterized for the entire sequence between the 5’ UTR and the 3’ UTR including all exons and introns. Figure 2 panels A – C show the precise location of all these novel variants and the regions that were completed for each allele.
Figure 1.
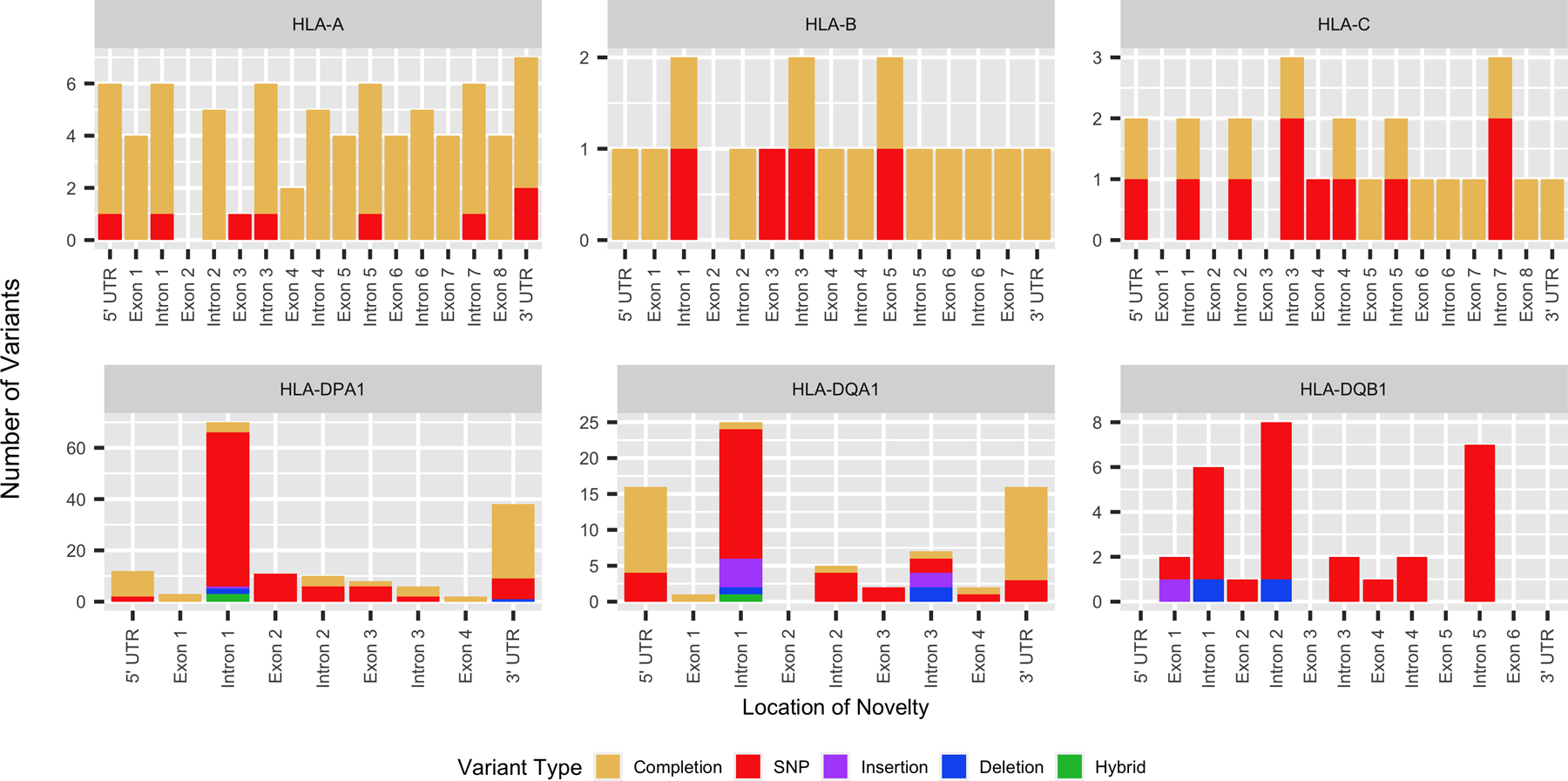
General location of novel contributions within the 140 newly described alleles. The color indicates the type of variation observed (yellow= completion of alleles sequence, red = single nucleotide polymorphism (SNP), purple = insertion, blue = deletion, green = hybrid allele).
Figure 2.
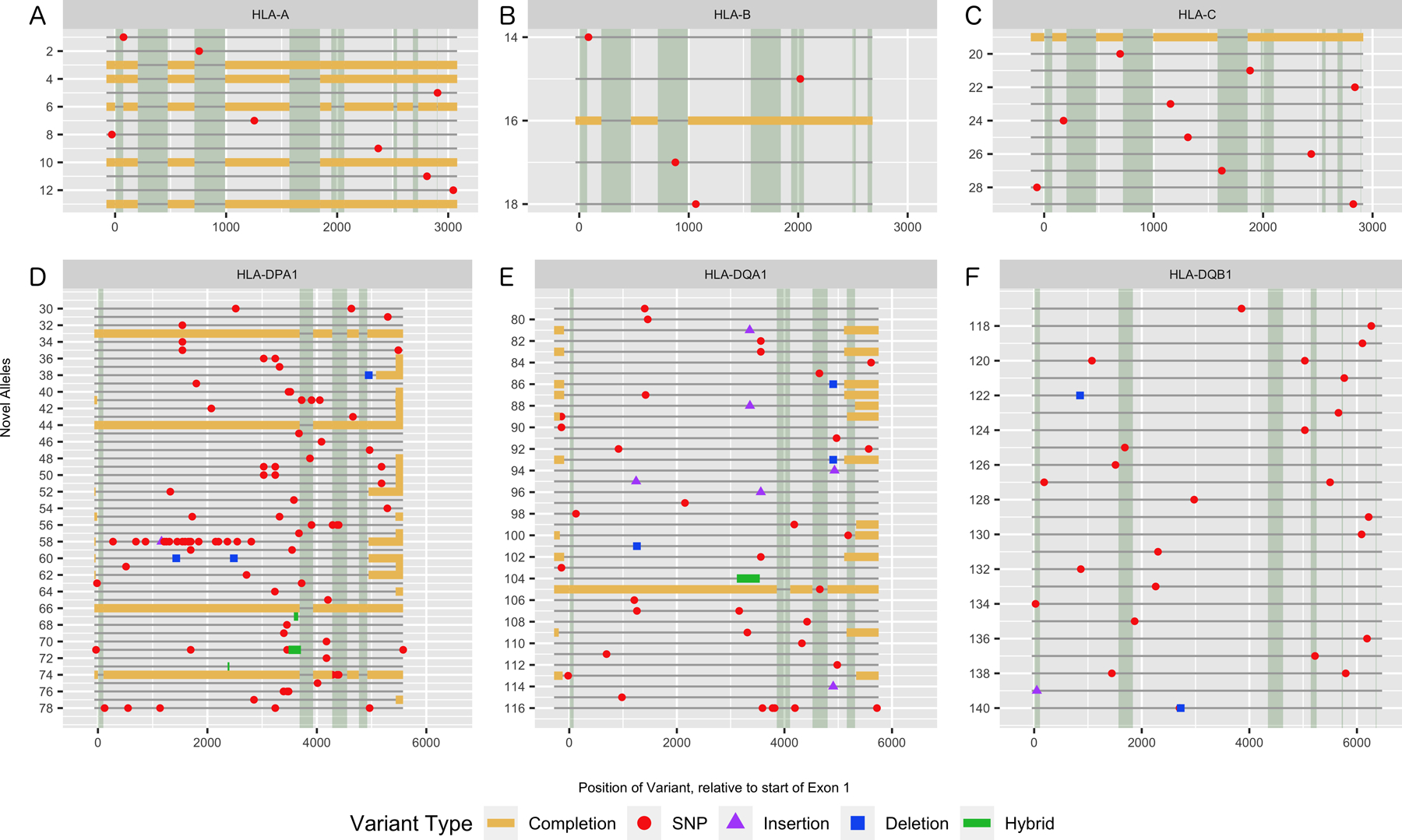
Precise mapping of novel variants and extended sequences per allele, by HLA gene. Novel alleles are numbered consecutively as shown on the y-axis, separated by locus and correspond to the allele numbering used in Supplemental Table 1. The position of the variant is relative to the start of exon 1. Exonic regions have the background shaded a darker gray. For hybrid alleles, the region where the recombination is believed to occur is indicated in green. A) HLA-A B) HLA-B C) HLA-C D) HLA-DPA1 E) HLA-DQA1 F) HLA-DQB1.
As shown in Figure 1, the novelties observed in HLA class II alleles are more extensive and varied. SNPs are the main type of variation observed across the length of the HLA class II genes, however, unlike HLA class I genes, insertions and deletions are also found. All but one of the insertions and deletions are found within the intronic regions of HLA class II genes. The single exonic insertion occurs in exon 1 of DQB1 resulting in a null allele (described in detail below). Additionally, the majority of novel variants observed in HLA class II alleles were found to occur in intron 1, in part due to the extended length of intron 1 in these genes, which accounts for approximately 36.7% of DPA1, 58.1% of DQA1 and 20.6% of DQB1 gene content. The exact distribution of novel variants in these HLA class II genes can be found outlined in Figure 2 panels D – F.
Following are the descriptions of exonic, null and hybrid novel HLA alleles (n=20) that were identified in the current study.
Novel HLA-A*43 allele, A*43:03
A*43:03 is a new HLA-A allele that most closely resembles A*43:01, differing at a single nucleotide position in the antigen recognition site (α2 domain), whereby the novel allele shows a guanine (G) instead of a cytosine (C) in the second position of codon 105 in exon 3 (Figure 2A, HLA-A, Novel Allele #2). The non-synonymous substitution (CCG → CGG) results in the replacement of a proline (P) with an arginine (R) residue in the mature protein. This nucleotide substitution and resulting amino acid have also been observed in two other HLA alleles: A*01:153 and A*26:212.
A*43:03 was separately observed in seven individuals, all coming from Botswana (populations: Ju|’hoansi and !Xoo). Five individuals shared the same haplotype which is likely to carry the new allele: A*43:03 ~ B*07:06:01 ~ C*07:02:01 ~ DRB1*13:02:01 ~ DRB3*03:01:01 ~ DQA1*01:02:01 ~ DQB1*06:04:01 ~ DPA1*02:09 ~ DPB1*01:01:01.
Novel HLA-B*41 allele, B*41:75
B*41:75 is a new HLA-B allele that most closely resembles B*41:01:01:01, differing at a single nucleotide position in the transmembrane domain of the α chain, whereby the novel allele shows a cytosine (C) instead of a guanine (G) in the first position of codon 303 in exon 5 (Figure 2B, HLA-B, Novel Allele #15). The non-synonymous substitution (GTC → CTC) results in the replacement of a valine (V) with a leucine (L) residue in the mature protein. No other HLA-B alleles possess this same nucleotide substitution in codon 303 or possess a leucine at this position in the HLA-B protein.
B*41:75 was observed in one individual from the Dizi population in Ethiopia. The complete HLA typing of this individual was A*02:05:01, A*02:05:01, B*15:31, B*41:75, C*04:07:01, C*07:01:01, DRB1*07:01:01, DRB1*11:01:02, DRB3*02:02:01, DRB4*01:03:01, DQA1*02:01:01, DQA1*05:05:01, DQB1*03:01:01, DQB1*03:03:02, DPA1*01:03:01, DPA1*01:03:01, DPB1*02:01:02G, DPB1*03:01:01G.
Novel HLA-B*58 allele, B*58:135
B*58:135 is a new HLA-B allele that most closely resembles B*58:01:01:01, differing at a single nucleotide position in the antigen recognition site (α2 domain), whereby the novel allele shows a guanine (G) instead of a cytosine (C) in the first position of codon 145 in exon 3 (Figure 2B, HLA-B, Novel Allele #17). The non-synonymous substitution (CGC → GGC) results in the replacement of an arginine (R) with a glycine (G) residue in the mature protein. Three additional HLA-B alleles also share this same nucleotide substitution in codon 145 and the resulting amino acid: B*27:60, B*51:84 and B*57:50.
B*58:135 was observed in one individual from Cameroon (Mbororo Fulani individual). The complete HLA typing of this individual was A*30:02:01, A*33:01:01, B*58:135, B*78:01:01, C*07:18:01, C*16:01:01, DRB1*08:04:01, DRB1*13:01:01, DRB3*01:01:02, DQA1*01:03:01, DQA1*05:05:01, DQB1*03:01:04, DQB1*06:04:01, DPA1*01:03:01, DPA1*01:03:01, DPB1*02:01:19, DPB1*04:02:01.
Novel HLA-C*16 allele, C*16:193
C*16:193 is a new HLA-C allele that most closely resembles C*16:01:01:01, differing at a single nucleotide position in the α3 extracellular domain, whereby the novel allele shows an adenine (A) instead of a cytosine (C) in the third position of codon 196 in exon 4 (Figure 2C, HLA-C, Novel Allele #27). The non-synonymous substitution (GAC → GAA) results in the replacement of an aspartic acid (D) with a glutamic acid (E) residue in the mature protein. This particular nucleotide substitution and resulting amino acid are not observed in any other HLA-C allele.
C*16:193 was observed in one individual from Cameroon (Tikari population). The complete HLA typing of this individual was A*24:02:01, A*29:02:01, B*27:05:02, B*78:01:01, C*02:02:02, C*16:193, DRB1*04:05:01, DRB1*13:01:01, DRB3*01:01:02, DRB4*01:03:01, DQA1*01:03:01, DQA1*03:03:01, DQB1*02:02:01, DQB1*06:04:01, DPA1*01:03:01, DPA1*01:03:01, DPB1*04:02:01, DPB1*04:02:01.
Novel HLA-DPA1*01 allele, DPA1*01:62
DPA1*01:62 is a new HLA-DPA1 allele that most closely resembles DPA1*01:58, differing at two single nucleotide positions in the antigen recognition site (α1 domain) (Figure 2D, HLA-DPA1, Novel Allele #41. The first mismatch occurs in the third position of codon 15 in exon 2, whereby the novel allele shows an adenine (A) instead of a guanine (G). Codon 15 typically encodes for a threonine (T) residue and the synonymous substitution (ACG → ACA) does not result in any amino acid replacement in the mature protein; it is a silent mutation. This nucleotide substitution is also observed in DPA1*01:03:32 (described below) but is not observed in any other HLA-DPA1 allele. The second mismatch occurs in the first position of codon 76 in exon 2, whereby the novel allele shows a thymine (T) instead of a cytosine (C). The non-synonymous substitution (CGT → TGT) results in the replacement of an arginine (R) with a cysteine (C) residue in the mature protein. The substitution in codon 76 is also observed in DPA1*01:15, DPA1*02:47, DPA1*03:08 (described below), and all DPA1*02:09:01 alleles. Furthermore, compared to DPA1*01:58, the novel allele shows a nucleotide mismatch in intron 2 [guanine (G) → adenine (A)]. Additionally, the DPA1*01:58 had truncated 5’ and 3’ UTR sequences as compared to other DPA1 alleles in the IPD-IMGT/HLA database, and we have contributed an additional 53 nucleotides to the 5’ UTR and 141 nucleotides to the 3’ UTR sequences.
DPA1*01:62 was separately observed in two individuals, both coming from separate populations of Cameroon: Bagyeli and Baka. These individuals also shared a DPB1*04:02:01G allele.
Novel HLA-DPA1*02 allele, DPA1*02:55
DPA1*02:55 is a new HLA-DPA1 allele that most closely resembles DPA1*02:01:01:06, differing at a single nucleotide position in the antigen recognition site (α1 domain), whereby the novel allele shows a cytosine (C) instead of a thymine (T) in the second position of codon 66 in exon 2 (Figure 2D, HLA-DPB1, Novel Allele #48). The non-synonymous substitution (TTG → TCG) results in the replacement of a leucine (L) with a serine (S) residue in the mature protein. This particular nucleotide substitution and resulting amino acid are also shared with DPA1*01:12 and all HLA-DPA1*03 alleles except DPA1*03:02. Furthermore, compared to DPA1*02:01:01:06, the novel allele shows an extended 3’UTR sequence by 136 additional nucleotides downstream of the existing 3’end.
DPA1*02:55 was separately observed in seven individuals all coming from Ethiopia (Dizi and Mursi populations). All individuals shared the same haplotype which is likely to carry the new allele: B*57:02:01 ~ C*18:02:01 ~ DRB1*15:03:01 ~ DRB5*01:01:01 ~ DQA1*01:02:01 ~ DQB1*06:02:01 ~ DPA1*02:55 ~ DPB1*13:01:01G. Also, five of the seven individuals shared an A*03:01:01 allele and the other two individuals shared an A*02:05:01 allele.
Novel HLA-DPA1*03 allele, DPA1*03:08
DPA1*03:08 is a new HLA-DPA1 allele that most closely resembles DPA1*03:07:02, differing at four single nucleotide positions (Figure 2D, HLA-DPA1, Novel Allele #56). The first novelty occurs in the antigen recognition site (α1 domain), in the first position of codon 76 in exon 2, whereby the novel allele possesses a thymine (T) instead of a cytosine (C). The non-synonymous substitution (CGT → TGT) results in the replacement of an arginine (R) with a cysteine (C) residue in the mature protein. This substitution is also shared by DPA1*01:15, DPA1*01:62 (described above), DPA1*02:47 and all DPA1*02:09:01 alleles. The remaining three novel variants are synonymous substitutions occuring in the α2 extracellular domain proximal to the membrane: in the third position of codon 90 in exon 3, whereby the novel allele shows a cytosine (C) instead of a guanine (G), ACG → ACC, both encoding a threonine (T) residue; in the third position of codon 118 in exon 3, whereby the novel allele shows cytosine (C) instead of a thymine (T), AAT → AAC, both encoding an asparagine (N) residue; in the third position of codon 127 in exon 3, whereby the novel allele shows adenine (A) instead of guanine (G), CCG → CCA, both encoding a proline (P) residue. All of these silent substitutions in exon 3 are common within the HLA-DPA1 gene. Furthermore, compared to DPA1*03:07:02 sequence which includes only exons 2–4, the novel allele sequence is complete including exon 1, introns 1–3 and partial UTRs as well.
DPA1*03:08 was observed in one individual of the Herero population in Botswana. The complete HLA typing of this individual was A*02:14, A*23:17, B*44:03:01, B*47:01:01, C*02:10:01, C*07:18, DRB1*07:01:01, DRB1*13:03:01, DRB3*01:01:02, DRB4*01:03:01N, DQA1*02:01:01, DQA1*05:05:01, DQB1*02:02:01, DQB1*03:01:01, DPA1*03:01:01, DPA1*03:08, DPB1*40:01:01, DPB1*55:01:01.
Novel HLA-DPA1*01 allele, DPA1*01:03:32
DPA1*01:03:32 is a new HLA-DPA1 allele that most closely resembles DPA1*01:03:01:02, differing at a single nucleotide position in the antigen recognition site (α1 domain), whereby the novel allele shows an adenine (A) instead of a guanine (G) in the third position of codon 15 in exon 2 (Figure 2D, HLA-DPA1, #63). Codon 15 typically encodes for a threonine (T) residue and the synonymous substitution (ACG → ACA) does not result in any amino acid replacement in the mature protein. This nucleotide substitution at codon 15 is only shared with DPA1*01:62, as described above. Furthermore, compared to DPA1*01:03:01:02, the novel allele shows a nucleotide mismatch in 5’UTR [adenine (A) → guanine (G)] as well.
DPA1*01:03:32 was separately observed in three individuals of the Baka population in Cameroon. All individuals shared a DPB1*04:02:01G allele, and two individuals shared the same set of HLA class II alleles which is likely to carry the new allele: DRB1*15:03:01 ~ DRB5*01:01:01 ~ DQA1*01:02:01 ~ DQB1*06:02:01 ~ DPA1*01:03:32 ~ DPB1*04:02:01G.
Novel HLA-DPA1*01 allele, DPA1*01:33:02
DPA1*01:33:02 is a new HLA-DPA1 allele that most closely resembles DPA1*01:33, differing at three single nucleotide positions in the extracellular domain proximal to the membrane, all of which are synonymous substitutions: in the third position of codon 90 in exon 3, whereby the novel allele has a guanine (G) instead of a cytosine (C), ACC → ACG, both encoding a threonine (T) residue; in the third position of codon 118 in exon 3, whereby the novel allele has thymine (T) instead of a cytosine (C), AAC → AAT, both encoding an asparagine (N) residue; in the third position of codon 127 in exon 3, whereby the novel allele shows a guanine (G) instead of an adenine (A), CCA → CCG, both encoding a proline residue (Figure 2D, HLA-DPA1, Novel Allele #74). All three substitutions are common within HLA-DPA1*02 alleles. Furthermore, compared to the DPA1*01:33 sequence, which includes only exons 1–4, the novel allele sequence is complete including introns 1–3 and partial UTRs as well.
DPA1*01:33:02 was separately observed in two individuals of the Sandawe population from Tanzania. These individuals shared the same haplotype which is likely to carry the new allele: A*29:01:01 ~ B*53:01:01 ~ C*06:02:01 ~ DRB1*04:01:01 ~ DRB4*01:01:01 ~ DQA1*03:03:01. They also shared the following genotypes: DQB1*03:02:01, DQB1*06:04:01; DPA1*01:33:02, DPA1*03:01:01; DPB1*04:01:01G, DPB1*04:02:01G.
Novel HLA-DQA1*03 allele, DQA1*03:03:07
DQA1*03:03:07 is a new HLA-DQA1 allele that most closely resembles DQA1*03:03:01:01, differing at a single nucleotide position in the extracellular domain proximal to the membrane, whereby the novel allele shows a guanine (G) instead of a cytosine (C) in the third position of codon 131 in exon 3 (Figure 2E, HLA-DQA1, Novel Allele #85). Codon 131 typically encodes for a valine (V) residue and the synonymous substitution (GTC → GTG) does not result in any amino acid replacement in the mature protein. It is the only instance of this nucleotide substitution within the HLA-DQA1 alleles.
DQA1*03:03:07 was observed in one individual in the Amhara population from Ethiopia. The complete HLA typing of this individual was A*01:01:01, A*01:03:01, B*15:17:01, B*41:01:01, C*07:01:01, C*07:01:02, DRB1*01:02:01, DRB1*04:05:01, DRB4*01:03:01, DQA1*01:01:02, DQA1*03:03:07, DQB1*03:02:01, DQB1*05:01:01, DPA1*01:03:01, DPA1*03:01:01, DPB1*03:01:01, DPB1*55:01:01.
Novel HLA-DQA1*02 allele, DQA1*02:25
DQA1*02:25 is a new HLA-DQA1 allele that most closely resembles DQA1*02:01:01:02, differing at a single nucleotide position in the transmembrane domain or the cytoplasmic tail of the α chain, whereby the novel allele shows a thymine (T) instead of a cytosine (C) in the second position of codon 190 in exon 4 (Figure 2E, HLA-DQA1, Novel Allele #100). The non-synonymous substitution (TCA → TTA) results in the replacement of a serine (S) with a leucine (L) residue in the mature protein. This nucleotide substitution and resulting amino acid are also shared with the DQA1*01:51:01 alleles. Furthermore, compared to DQA1*02:01:01:02, the novel allele shows extended 5’UTR and 3’UTR sequences by 107 and 264 additional nucleotides upstream and downstream of the existing 5’end and 3’end respectively.
DQA1*02:25 was observed in one individual of the Dizi population from Ethiopia. The complete HLA typing of this individual was A*02:01:01, A*26:12, B*15:220, B*57:03:01, C*04:01:01, C*18:02, DRB1*07:01:01, DRB1*07:01:01, DRB4*01:03:01N, DRB4*01:03:01N, DQA1*02:01:01, DQA1*02:25, DQB1*03:03:02, DQB1*03:03:02, DPA1*02:01:01, DPA1*03:01:01, DPB1*13:01:01G, DPB1*49:01:01.
Novel HLA-DQA1*01 allele, DQA1*01:63:01:01
DQA1*01:63:01:01 is a new HLA-DQA1 allele that most closely resembles DQA1*01:08, differing at a single nucleotide position in the extracellular domain proximal to the membrane, whereby the novel allele shows a guanine (G) instead of a cytosine (C) in the second position of codon 134 in exon 3 (Figure 2E, HLA-DQA1, Novel Allele #105). The non-synonymous substitution (GCT → GGT) results in the replacement of an alanine (A) with a glycine (G) residue in the mature protein. Having a glycine at codon 134, represented by the codon GGT, is shared among all HLA-DQA1 alleles except DQA1*01:08. Furthermore, compared to DQA1*01:08 sequence which includes only exons 2 and 3, the novel allele sequence is complete including exons 1 and 4, introns 1–3 and partial UTRs as well.
DQA1*01:63:01:01 was separately observed in two individuals from the Mursi population within Ethiopia. These individuals shared the same HLA class I genes, which are likely part of the haplotype carrying the new allele: A*74:03 ~ B*44:03:01 ~ C*07:01:01 However, they had an identical genotype for HLA class II alleles and the novelty allele could not be directly attributed to a particular haplotype: DRB1*03:01:01G, DRB1*11:01:02, DRB3*02:02:01, DQA1*01:63:01, DQA1*05:01:01; DQB1*02:01:01, DQB1*05:01:01.
Novel HLA-DQB1*02 allele, DQB1*02:02:17
DQB1*02:02:17 is a new HLA-DQB1 allele that most closely resembles DQB1*02:02:01:02, differing at a single nucleotide position in the antigen recognition site (β1 domain), whereby the novel allele shows an adenine (A) instead of a guanine (G) in the third position of codon 44 in exon 2 (Figure 2F, HLA-DQB1, Novel Allele #125). Codon 44 typically encodes for a valine (V) residue and the synonymous substitution (GTG → GTA) does not result in any amino acid replacement in the mature protein. This substitution is only observed in three other HLA-DQB1 alleles: DQB1*05:01:25, DQB1*06:02:03 and DQB1*06:03:30.
DQB1*02:02:17 was observed in one individual of the Herero population from Botswana. The complete HLA typing of this individual was A*02:02:01, A*02:14, B*18:03, B*18:03, C*04:01:01, C*07:02:01, DRB1*01:02:01, DRB1*07:01:01, DRB4*01:01:01, DQA1*01:01:02, DQA1*03:03:01, DQB1*02:02:17, DQB1*05:01:01, DPA1*01:03:01, DPA1*01:03:01, DPB1*04:01:01, DPB1*104:01:01.
At the time the initial analysis was performed, the novel allele was identical to the allele named as DQB1*02:01:14, which was characterized only at exon 2 in the respective IPD-IMGT/HLA database version. However, upon completion of its sequence, the novel allele turned out to encode the same amino acid sequence as DQB1*02:02 and thus, it was officially assigned the name DQB1*02:02:17 by the WHO Nomenclature Committee For Factors of the HLA System in January 2022. DQB1*02:01:14 has currently been deleted from the IPD-IMGT/HLA database, with its sequence having been extended and renamed as DQB1*02:02:17.
Novel HLA-DQB1*02 allele, DQB1*02:198
DQB1*02:198 is a new HLA-DQB1 allele that most closely resembles DQB1*02:01:01:01, differing at a single nucleotide position in the leader peptide of the β chain, whereby the novel allele shows an adenine (A) instead of a thymine (T) in the second position of codon −24 in exon 1 (Figure 2F, HLA-DQB1, Novel Allele #134). The non-synonymous substitution (ATC → AAC) results in the replacement of an isoleucine (I) with an asparagine (N) residue in the mature protein. This nucleotide substitution and resulting amino acid are not found in any other HLA-DQB1 allele.
DQB1*02:198 was separately observed in two individuals, both coming from the Chabu population of Ethiopia. These individuals shared the same haplotype which is likely to carry the new allele: A*74:03 ~ B*73:01 ~ C*15:05:01 ~ DRB1*04:05:01 ~ DRB4*01:03:01 ~ DQA1*03:04 ~ DQB1*02:198 ~ DPA1*01:03:01 ~ DPB1*03:01:01G.
Novel HLA-DQB1*04 allele, DQB1*04:52
DQB1*04:52 is a new HLA-DQB1 allele that most closely resembles DQB1*04:02:01:08, differing at a single nucleotide position in the transmembrane domain of the β chain, whereby the novel allele shows an cytosine (C) instead of a guanine (G) in the second position of codon 216 in exon 4 (Figure 2F, HLA-DQB1, Novel Allele #137). The non-synonymous substitution (GGC → GCC) results in the replacement of a glycine (G) with an alanine (A) residue in the mature protein and is not observed in any other HLA-DQB1 allele.
DQB1*04:52 was observed in one individual of the !Xoo population from Botswana. The complete HLA typing of this individual was A*30:02:01, A*34:02:01, B*08:01:01, B*44:03:01, C*04:01:01, C*07:01:01, DRB1*03:01:01, DRB1*08:04:01, DRB3*02:02:01, DQA1*04:03N, DQA1*05:01:01, DQB1*02:01:01, DQB1*04:52, DPA1*02:02:02, DPA1*04:02, DPB1*04:02:01G, DPB1*04:02:01G.
Novel HLA-DQB1 null allele, DQB1*06:422N
DQB1*06:422N is a new HLA-DQB1 null allele that most closely resembles DQB1*06:02:01:04, differing by an insertion of four nucleotides in the leader peptide of the β chain, between codons −15 and −14 in exon 1, which typically encode a valine (V) and threonine residue (T) respectively (Figure 2F, HLA-DQB1, Novel Allele #139). The four-base insertion of a thymine (T), guanine (G), thymine (T), and cytosine (C), sequentially (TGTC), causes a frame shift leading to a premature stop codon (TGA) approximately 40 nucleotides downstream within exon 1 (Figure 3) and has not been observed in any other HLA-DQB1 allele.
Figure 3.
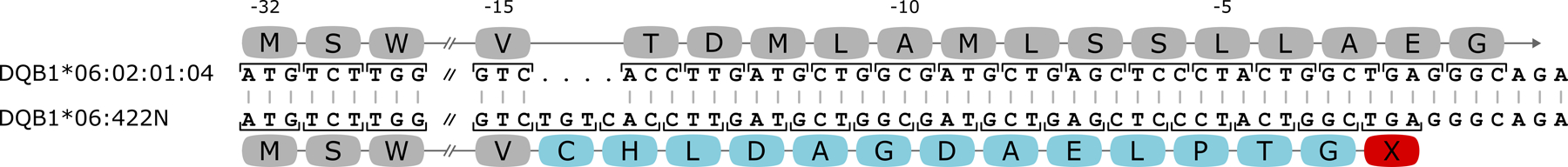
Comparison of DQB1*06:422N to the closest reference allele DQB1*06:02:01:04. The nucleotide sequence and resulting amino acids (blocks) are shown for both alleles starting from the beginning of exon 1. Codons −29 through −16 are skipped as they are identical between the alleles. Amino acids that are different for DQB1*06:422N due to the four nucleotide insertion are colored in blue, with the premature stop codon shown in red with an ‘X’. The numbering indicates the amino acid with respect to the start of the mature protein.
DQB1*06:422N was observed in one individual from the Dizi population of Ethiopia. The complete HLA typing of this individual was A*68:02:01, A*68:02:01, B*15:10:01, B*81:02:02, C*03:04:02, C*07:01:01, DRB1*08:04:01, DRB1*11:01:02, DRB3*02:02:01, DQA1*01:02:01, DQA1*04:01:02, DQB1*04:02:13, DQB1*06:422N, DPA1*02:02:02, DPA1*04:02, DPB1*05:01:01G, DPB1*04:02:01G.
Novel HLA-DPA1 hybrid allele, DPA1*02:01:01:25
DPA1*02:01:01:25 is a new HLA-DPA1 allele that appears to be a hybrid between DPA1*01:03:01:02 (5’UTR through the split point) and DPA1*02:01:01:09 (split point through 3’UTR) (Figure 2D, HLA-DPA1, Novel Allele #67). The split occurs near the end of intron 1, approximately 100bp upstream of exon 2, within a 78bp region between positions 3580 and 3658 using complete genomic alignment.
DPA1*02:01:01:25 was separately observed in two individuals of the Mursi population from Ethiopia. These individuals shared the following alleles in addition to the new hybrid allele: A*02:01:01, B*58:01:01, DPA1*02:01:01, and DPB1*03:01:01G.
Novel HLA-DPA1 hybrid allele, DPA1*02:02:02:14
DPA1*02:02:02:14 is a new HLA-DPA1 allele that appears to be a hybrid between DPA1*02:01:01:02 (5’UTR through the split point) and DPA1*02:02:02:09 (split point through 3’UTR) (Figure 2D, HLA-DPA1, Novel Allele #71). The split occurs near the end of intron 1, approximately 200bp upstream of exon 2, within a 225bp region between positions 3483 and 3708 using complete genomic alignment. However, the first part of the novel allele sequence that most closely resembles DPA1*02:01:01:02 still shows one 5’UTR and two intronic nucleotide mismatches against it, whereas the second part of the novel allele sequence that most closely resembles DPA1*02:02:02:09 still shows a 3’UTR nucleotide mismatch against it.
DPA1*02:02:02:14 was separately observed in three individuals of the !Xoo population from Botswana. Two individuals shared the same set of alleles: B*15:10:01, C*03:04:02, DQA1*01:02:01, DQB1*06:02:01, DPA1*02:02:02, and DPB1*01:01:01.
Novel HLA-DPA1 hybrid allele, DPA1*02:01:01:26
DPA1*02:01:01:26 is a new HLA-DPA1 allele that appears to be a hybrid between DPA1*01:03:01:02 (5’UTR through the split point) and DPA1*02:01:01:06 (split point through 3’UTR) (Figure 2D, HLA-DPA1, Novel Allele #73). The split occurs around the middle of intron 1, within a 32bp region between positions 2369 and 2401 using complete genomic alignment.
DPA1*02:01:01:26 was separately observed in three individuals of the Sandawe population from Tanzania. All individuals shared the same haplotype which is likely to carry the new hybrid allele: A*34:02:01 ~ B*53:01:01 ~ C*06:02:01 ~ DRB1*04:01:01 ~ DRB4*01:01:01 ~ DQA1*03:03:01 ~ DQB1*03:02:01 ~ DPA1*02:01:01 ~ DPB1*30:01:01.
Novel HLA-DQA1 hybrid allele, DQA1*01:01:02:03
DQA1*01:01:02:03 is a new HLA-DQA1 allele that appears to be a hybrid between DQA1*01:02:01:03 (5’UTR through the split point) and DQA1*01:01:02:01 (split point through 3’UTR) (Figure 2E, HLA-DQA1, Novel Allele #104). The split occurs towards the end of intron 1, approximately 700bp upstream of exon 2, within a 426bp region between positions 3115 and 3541 using complete genomic alignment.
DQA1*01:01:02:03 was separately observed in four individuals of the Mursi population from Ethiopia. All individuals shared the same haplotype which is likely to carry the new hybrid allele: B*35:01:01 ~ DRB1*11:01:02 ~ DRB3*02:02:01 ~ DQA1*01:01:02 ~ DQB1*05:01:01 ~ DPA1*03:01:01 ~ DPB1*49:01:01. Also, three individuals shared a C*16:01:01 allele.
Discussion:
The genomic characterization of the 11 classical HLA genes of 489 African participants collected from four countries and thirteen ethnic groups from within these countries, revealed the presence of 140 alleles with new sequence information, whereby 130 alleles have novel sequences and 10 alleles that have been updated to include additional sequence information, as they had been described but incompletely characterized. It should be noted that the characterization of HLA class II sequences does not include the reporting of new alleles from HLA-DPB1, DRB1, DRB3, DRB4 or DRB5 because of the absence of exon 1 and intron 1 sequences from the amplicons of these genes. The majority (110) of the new alleles were novel intronic variants of known alleles. Regarding HLA class I genes, all new alleles were genomic variations involving SNPs. No new polymorphisms were detected in exon 2 of any of the HLA class I genes A, B or C (Figure 1) and only one new polymorphism was detected in exon 3 of each of the respective HLA-A and -B genes. The distribution of these new alleles within each of these African populations is reported, and the genomic variations, primarily SNPs, identified are compared to other alleles within the IPD-IMGT/HLA database (Figure 1). Some appear to be unique, but others have already been described in other alleles. Expectedly most of the new information is in regions not previously extensively characterized (Figure 1). This may suggest that most of the polymorphisms associated with the peptide binding domains may have already been described worldwide. It will be interesting to see whether new populations, characterized within Africa or elsewhere, demonstrate patterns similar to the observations made in this study.
In contrast, the newly described HLA class II genes were generated by various types of genomic variations besides SNPs, including insertions, deletions and hybrid formations through recombination. The differences we identified in the HLA class II genes are found throughout almost all regions of these genes, including exonic, intronic and UTRs. The characterization of the HLA genes has been driven by laboratories focusing on A, B, C, DRB1 and DQB1 for transplantation purposes, and to a lesser extent on DPA1 and DQA1. However, more recently the DPA1 and DQA1 genes are being included in the compatibility assessment and it is expected that more new alleles will be described for these HLA class II genes in the future.
Furthermore, even though we have not submitted the novel alleles from HLA-DPB1, we have observed 10 new alleles with exonic differences that are found throughout exons 2–5 of the HLA-DPB1 gene, eight of which alter the mature protein. A similar situation has been observed with the DRB genes, but there will be another report on these findings after the amplicons generated for these genes include both exon 1 and intron 1 sequences.
Regarding the hybrid new alleles, we have not reported any patterns of sequences as being observed in any other HLA alleles in the IPD-IMGT/HLA database, because it is not clear what part of the hybrid sequence we should use for comparisons. Even though the name assigned by the Nomenclature Committee is suggestive of which allele of the two comprises the majority of the hybrid sequence, we felt uncertain of the rational to use the one or the other part as the sequence for comparisons within the IPD-IMGT/HLA database. All hybrid alleles of this report have been formed with breaking points within intron 1.
It should be noted that the sequences of many of the novel alleles described in this report have been generated and phased using the Illumina (MiSeq) sequencing platform. However, a number of alleles that had either exonic or intronic novelties and could not be phased using only the Illumina data given the fragmented DNA necessary for sequencing on the platform, and were not observed elsewhere in the dataset, were subsequently sequenced using Oxford Nanopore Technologies (ONT) to generate complete consensus sequences. The combination of the two technologies added confidence to the new allele call. The strength of the ONT platform is the ability to sequence very long pieces of DNA (>10 kb), such that the entire amplified region for the gene of interest is sequenced as a single read, guaranteeing phase. However, the ONT kits used for sequencing in this project have the limitation of not accurately sequencing through low complexity regions, including homopolymers. There are three homopolymer regions, all occurring in intron 1 of HLA-DQA1, that were particularly challenging: 1) a poly-A region ranging between 7 and 16 consecutive adenine bases, 2) a second poly-A region ranging from 7 to 23 consecutive adenine bases, and 3) a poly-T region ranging from 7 to 14 consecutive thymine bases. Given these complexities, we were unable to rely on ONT alone to determine the consensus sequence and had to use the higher quality Illumina data to resolve these regions. New updates to the ONT sequencing platforms offer higher quality sequencing reads that in future projects may allow sole use of the ONT platform to determine full-length allele sequences.
Even though the polymorphisms in the intronic regions of the HLA genes are not known for functional significance, it has been reported that these non-coding regions harbor miRNAs13,14 and not infrequently include SNPs associated with diseases15. More specifically, miR-6891 is encoded from intron 4 of HLA-B and, based on a computational analysis related to the presence of miRNAs in the MHC and the HLAs, like DRA and DRB5, include identifiable miRNAs of unknown significance and functionality13. Clearly the functional role of intronic sequences of the HLA genes is a domain that requires focused attention. Recent reports reveal that information content in the intronic sequences is comparable to exonic sequences16.
The HLA polymorphisms in individual populations and their evolutionary relationships will be analyzed in the context of their geographical locations in another report. Their connections to other known alleles will be identified and possible environmental pressures that have contributed to the formation of these new alleles will be assessed and discussed further (manuscript in preparation).
Finally, it is expected that thorough characterization of the HLA genes of the African populations may also contribute and influence other health related domains like transplantation, vaccine development, autoimmunity or even cancer development and the therapeutic approaches devised to address these problems. Examples of the above can be the identification of better matched donors for patients with sickle cell disease, a very frequent disease in Africa, whereby stem cell transplantation can be a viable therapeutic option, the possible differential response of vaccines directed to infectious diseases (HIV, SARS-COV2) that may depend on HLA allele distribution in different populations in Africa17,18, or the HLA dependent efficacy of cancer-related therapeutics whereby knowledge of HLA frequencies in a particular population are relevant.
Supplementary Material
Supplemental Table 1. Complete listing of all 743 consensus sequences derived from the genotyping of the 489 African participants. The alleles are separated into the genes that were characterized for the full length of the gene (HLA-A, B, C, DPA1, DQA1 and DQB1) and genes that were partially characterized (HLA-DPB1, DRB1, DRB3, DRB4 and DRB5). The alleles are listed with their internal consensus sequence index, along with the allele name and the number of times each allele was observed in each population and a total count of the total number of times each allele was observed in the study. For alleles that were matched a known allele (black text), all alleles that exactly matched the consensus sequence were reported (as of IPD-IMGT/HLA version 3.46). The 140 alleles that were submitted to the WHO nomenclature committee for official names and or sequence updates have blue text. Any novel alleles that were not submitted to the WHO have allele names with red text, with a note for novel alleles in fully characterized genes describing the reason the allele was not submitted. Any novel allele from the genes that were partial characterized were not considered for submission due to their incomplete nature.
Supplemental Table 2. Description of the 140 novel or completed alleles. Here we list the detailed information for all the alleles submitted for naming or sequence extension and include the GenBank and IPD-IMGT/HLA submission numbers, the official allele name, the closest reference allele, the type of variant(s) found, the location of such variants, the total number of times the allele was observed in the study, and the country and populations for which the allele was found.
Acknowledgments
This research was supported by the following funding sources: NIH Grant T32 DK07314, the Penn Training Grant in Diabetes, Endocrine and Metabolic Diseases to Eric Mbunwe, American Diabetes Association Pathway award 1–19-VSN-02 and NIH grants 1R01DK104339, 1R35GM134957, and R01AR076241 to Sarah A. Tishkoff, NIH grant R01AR070873 and institutional funds from the Children’s Hospital of Philadelphia to Dimitri S. Monos and Office of Naval Research Grant (N00014–18-1–2045) to Martin Maiers
Footnotes
Ethics Approval Number:
University of Pennsylvania IRB Protocol #807981
Conflicts of Interest
DSM is Chair of the Scientific Advisory Board of Omixon and owns options in Omixon. DSM, JLD and DF receive royalties from Omixon. No other authors have any conflicts of interest.
Data Availability Statement:
The data that supports the findings of this study are available in the supplementary material of this article
References:
- 1.Klitz W, Hedrick P, Louis EJ. New reservoirs of HLA alleles: pools of rare variants enhance immune defense. Trends Genet 2012;28(10):480–486. doi: 10.1016/j.tig.2012.06.007 [DOI] [PubMed] [Google Scholar]
- 2.Robinson J, Guethlein LA, Cereb N, et al. Distinguishing functional polymorphism from random variation in the sequences of >10,000 HLA-A, -B and -C alleles. PLOS Genetics 2017;13(6):e1006862. doi: 10.1371/journal.pgen.1006862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tishkoff SA, Reed FA, Friedlaender FR, et al. The genetic structure and history of Africans and African Americans. Science 2009;324(5930):1035–1044. doi: 10.1126/science.1172257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheinfeldt LB, Soi S, Lambert C, et al. Genomic evidence for shared common ancestry of East African hunting-gathering populations and insights into local adaptation. Proc Natl Acad Sci U S A 2019;116(10):4166–4175. doi: 10.1073/pnas.1817678116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan S, Kelly DE, Beltrame MH, et al. African evolutionary history inferred from whole genome sequence data of 44 indigenous African populations. Genome Biol 2019;20(1):82. doi: 10.1186/s13059-019-1679-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Margolis DJ, Mitra N, Duke JL, et al. Human leukocyte antigen class-I variation is associated with atopic dermatitis: A case-control study. Hum Immunol 2021;82(8):593–599. doi: 10.1016/j.humimm.2021.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE. IPD-IMGT/HLA Database. Nucleic Acids Res 2020;48(D1):D948–D955. doi: 10.1093/nar/gkz950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang JR, Holt J, McMillan L, Jones CD. FMLRC: Hybrid long read error correction using an FM-index. BMC Bioinformatics 2018;19(1):50. doi: 10.1186/s12859-018-2051-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013;30(4):772–780. doi: 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018;34(18):3094–3100. doi: 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marsh SGE. Nomenclature for factors of the HLA system, update October, November and December 2021. HLA 2022;99(3):231–278. doi: 10.1111/tan.14538 [DOI] [PubMed] [Google Scholar]
- 12.Marsh SGE. Nomenclature for factors of the HLA system, update January, February, and March 2022. HLA 2022;99(6):674–701. doi: 10.1111/tan.14642 [DOI] [PubMed] [Google Scholar]
- 13.Clark PM, Chitnis N, Shieh M, Kamoun M, Johnson FB, Monos D. Novel and Haplotype Specific MicroRNAs Encoded by the Major Histocompatibility Complex. Sci Rep 2018;8(1):3832. doi: 10.1038/s41598-018-19427-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ladewig E, Okamura K, Flynt AS, Westholm JO, Lai EC. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome Res 2012;22(9):1634–1645. doi: 10.1101/gr.133553.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper DN. Functional intronic polymorphisms: Buried treasure awaiting discovery within our genes. Hum Genomics 2010;4(5):284–288. doi: 10.1186/1479-7364-4-5-284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karakatsanis LP, Pavlos EG, Tsoulouhas G, et al. Spatial constrains and information content of sub-genomic regions of the human genome. iScience 2021;24(2):102048. doi: 10.1016/j.isci.2021.102048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sriwanthana B, Mori M, Tanaka M, et al. The Effect of HLA Polymorphisms on the Recognition of Gag Epitopes in HIV-1 CRF01_AE Infection. PLoS One 2012;7(7):e41696. doi: 10.1371/journal.pone.0041696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crocchiolo R, Gallina AM, Pani A, et al. Polymorphism of the HLA system and weak antibody response to BNT162b2 mRNA vaccine. HLA 2022;99(3):183–191. doi: 10.1111/tan.14546 [DOI] [PubMed] [Google Scholar]
- 19.Eberhard DM, Simons GF, Fennig CD, eds. Ethnologue: Languages of the World 25th ed. SIL International; 2022. http://www.ethnologue.com [Google Scholar]
- 20.Boesen E Gleaming Like The Sun: Aesthetic Values in Wodaabe Material Culture. Africa 2008;78(4):582–602. doi: 10.3366/E0001972008000454 [DOI] [Google Scholar]
- 21.Population Census Commission. Population and Housing Census 2007 - National Statistical Published online December 2008. Accessed January 30, 2023. http://www.statsethiopia.gov.et/wp-content/uploads/2019/06/Population-and-Housing-Census-2007-National_Statistical.pdf
- 22.Survival International. Chabu hunter-gatherers in Ethiopia killed by settlers Published October 4, 2014. Accessed January 19, 2023. https://www.survivalinternational.org/news/10461
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Complete listing of all 743 consensus sequences derived from the genotyping of the 489 African participants. The alleles are separated into the genes that were characterized for the full length of the gene (HLA-A, B, C, DPA1, DQA1 and DQB1) and genes that were partially characterized (HLA-DPB1, DRB1, DRB3, DRB4 and DRB5). The alleles are listed with their internal consensus sequence index, along with the allele name and the number of times each allele was observed in each population and a total count of the total number of times each allele was observed in the study. For alleles that were matched a known allele (black text), all alleles that exactly matched the consensus sequence were reported (as of IPD-IMGT/HLA version 3.46). The 140 alleles that were submitted to the WHO nomenclature committee for official names and or sequence updates have blue text. Any novel alleles that were not submitted to the WHO have allele names with red text, with a note for novel alleles in fully characterized genes describing the reason the allele was not submitted. Any novel allele from the genes that were partial characterized were not considered for submission due to their incomplete nature.
Supplemental Table 2. Description of the 140 novel or completed alleles. Here we list the detailed information for all the alleles submitted for naming or sequence extension and include the GenBank and IPD-IMGT/HLA submission numbers, the official allele name, the closest reference allele, the type of variant(s) found, the location of such variants, the total number of times the allele was observed in the study, and the country and populations for which the allele was found.
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article