

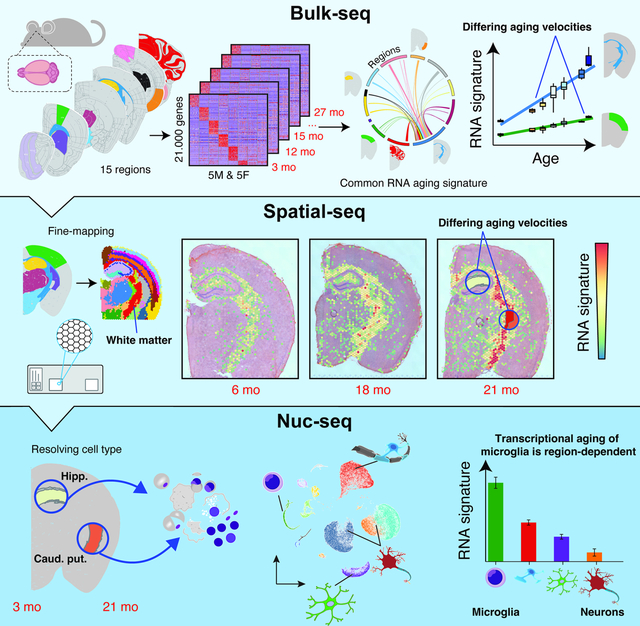
Summary:
Aging is the key risk factor for cognitive decline, yet the molecular changes underlying brain aging remain poorly understood. Here, we conducted spatiotemporal RNA-seq of the mouse brain, profiling 1,076 samples from 15 regions across 7 ages and 2 rejuvenation interventions. Our analysis identified a brain-wide gene signature of aging in glial cells, which exhibited spatially defined changes in magnitude. By integrating spatial and single-nucleus transcriptomics, we found that glia aging was particularly accelerated in white matter compared to cortical regions, while specialized neuronal populations showed region-specific expression changes. Rejuvenation interventions, including young plasma injection and dietary restriction, exhibited distinct effects on gene expression in specific brain regions. Furthermore, we discovered differential gene expression patterns associated with three human neurodegenerative diseases, highlighting the importance of regional aging as a potential modulator of disease. Our findings identify molecular foci of brain aging, providing a foundation to target age-related cognitive decline.
In Brief:
A spatiotemporal transcriptome map of the aging mouse brain identifies region-specific acceleration of glial aging, particularly in white matter, distinctive regional responses to rejuvenation interventions, and regional age-associated expression patterns of genes tied to human neurodegenerative diseases.
Graphical Abstract
Introduction
Aging is the predominant risk factor for cognitive dysfunction 1,2 and several neurodegenerative disorders, including Alzheimer’s disease (AD) and Parkinson’s disease (PD) 3–5. It remains unclear though, how aging contributes to the development of these distinct diseases of the brain, given their differences in pathological hallmarks, time of onset, and, notably, the regions affected4. A quantitative understanding of the dynamics of aging across the brain may provide new insight into the relationship between aging and neurodegeneration. Interestingly, neuroimaging studies using structural and functional magnetic resonance imaging (MRI) data indicate that aging impacts the brain in a region-specific manner 6,7. However, these structural manifestations provide limited insight into the underlying molecular alterations occurring during brain aging. In contrast, changes in gene expression can be a readout of cellular deterioration and molecular processes accompanying aging, permitting quantitative comparisons of aging rates between tissues 8 and cell types 9. Previous studies have profiled age-related gene expression changes in human brain tissue, yet these microarray-based experiments capture a limited set of transcripts and cover usually one to four regions 10,11 or quantify the transcriptome at low temporal resolution 12,13. Expression profiling during human brain aging is particularly challenging since it can take hours to days before postmortem tissue is stabilized 13–15. Alternatively, expression profiling in model organisms like M. musculus enables quantitative data with minimal confounding factors, but comprehensive studies covering more than a few regions and at high temporal resolution 16–19 do not - to our knowledge - yet exist. In consequence, this limitation also complicates the dissection of molecular mechanisms which mediate the effects of experimental disease models or interventions targeting the aging process, such as dietary restriction or young plasma injection, which delay molecular and cognitive phenotypes of brain aging 20.
Results
Spatiotemporal quantification of age-related gene expression across the mouse brain
Seeking a molecular understanding of the spatiotemporal changes of the mammalian brain, we punched out 15 regions from each hemisphere of coronal brain sections of 59 mice (Figure 1A; n = 3–6 males per age; aged 3, 12, 15, 18, 21, 26 and 28 months; n = 5 females per age; aged 3, 12, 15, 18 and 21 months; all C57BL/6JN strain): Three cortical regions (motor area, visual area and entorhinal cortex; Mot.cor., Vis.cor and Ent.cor, respectively), anterior (dorsal) and posterior (ventral) hippocampus (Hipp.ant and Hipp.post., respectively), hypothalamus (Hypoth.), thalamus, caudate putamen (part of the striatum; Caud.put.), pons, medulla, cerebellum (Cereb.) and the olfactory bulb (Olf.bulb). We further isolated three regions that were enriched with the corpus callosum (Corp.cal.), choroid plexus (Chor.plx.) and the neurogenic subventricular zone (SVZ), (Methods S1, section 1). We obtained a total of 1,770 samples (885 samples from each hemisphere). Regions from the left hemisphere were stored, while right hemisphere regions were processed through a custom-built bulk RNA-seq (bulk-seq) pipeline (Figure 1B, STAR Methods). We achieved robust tissue sampling with high RNA quality while minimizing perfusion artifacts (Methods S1, section 1; median RIN of 9.45).
Figure 1. Brain regions exhibit distinct transcriptional patterns of aging.
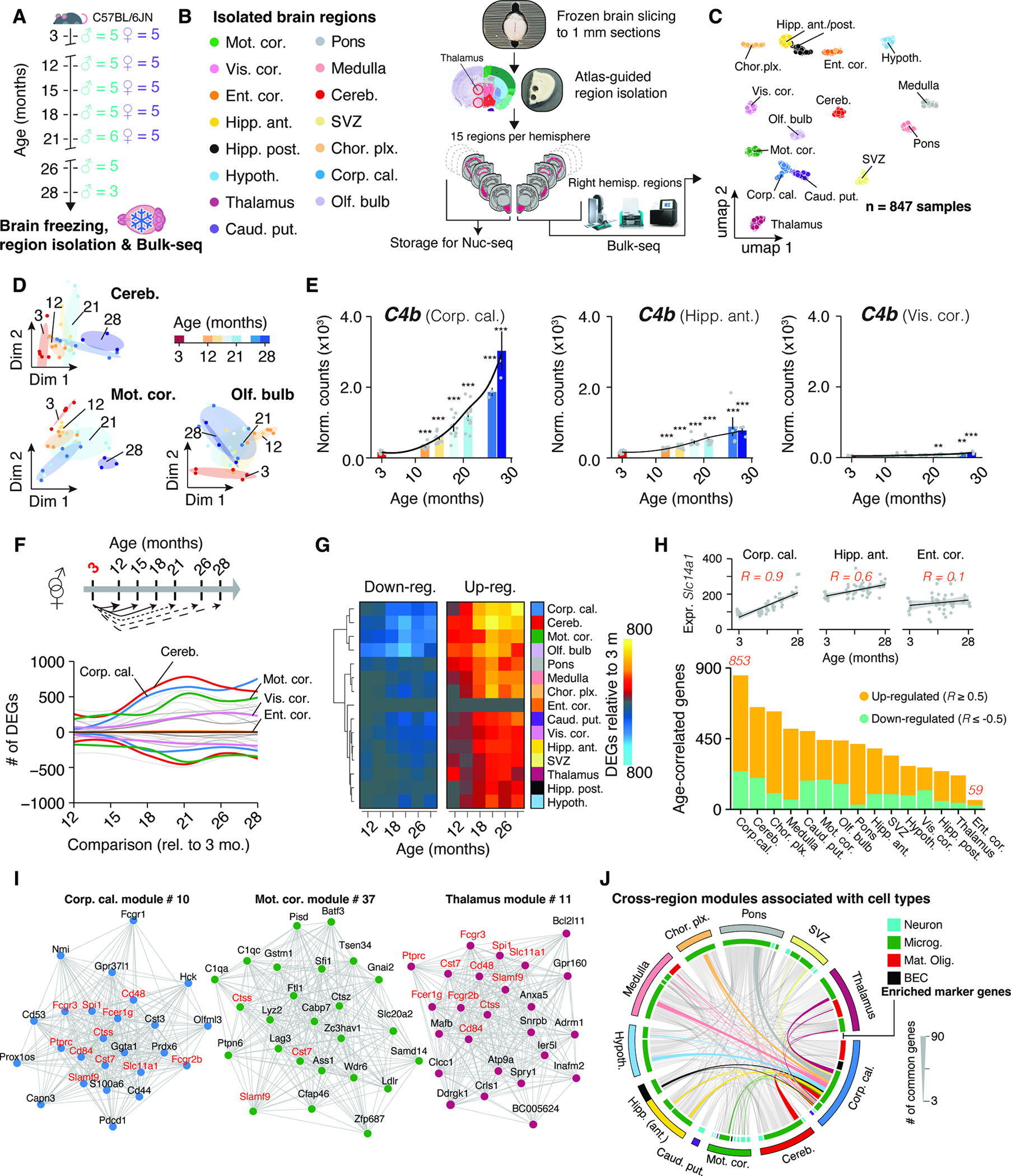
(A) Cohort overview. Whole brains were collected from male (n = 3–5, 3–28 months) and female (n = 5, 3–21 months) C57BL/6JN mice. (B) Study outline. 15 brain regions were isolated and analyzed using Bulk-seq. (C) UMAP representation of brain region transcriptomes. (D) Diffusion maps of region transcriptomes from selected areas. (E) C4b expression in selected regions. Black lines indicate smoothed gene expression. Differential expression compared to 3 months group is indicated. Mean ± s.e.m. Two-sided Wald test. (F) Smoothed line plot displaying DEGs for pairwise comparisons. Positive (negative) values represent up-regulated (down-regulated) genes. DEGs that reached significance in ≥ 2 pairwise comparisons were included. (G) Heat map of data in (F). (H) Number of age-correlated genes, colored by regulation. (I) Networks of the most connected genes (‘eigengenes’) in selected regions. (J) Chord diagram of genes shared in age-associated modules across regions. Modules and associated genes are listed in Table S1.
Post-quality control, we obtained 847 single-region transcriptomes. Visualization in uniform manifold approximation and projection (UMAP) space separated samples by region (Figure 1C), but not sex or age, concurring with deterministic hierarchical clustering (Methods S1, section 2). However, samples segregated transcriptionally by age within individual regions, highlighting the necessity for precise isolation of brain tissue to resolve the subtle effect of aging on expression (Figure 1D).
To assess if isolated regions capture a given brain structure’s transcriptome, we analyzed region-enriched genes (‘marker genes’; Table S1) in a publicly-available mouse brain spatial transcriptomics dataset22–24, creating ‘signatures’ 25 for each region. Signature scores were distinctly elevated in areas corresponding to the anatomical structures annotated in the Allen brain reference atlas 26 (Methods S1, section 3). Further, a significant decline with age in a signature score for activated neural stem cells (aNSCs, based on single-cell data 27) was observed in the SVZ region, indicating a loss of aNSCs with age, that is in agreement with diminished neurogenic capacity in aged mice 27 (Methods S1, section 3). These findings affirm the quality of our tissue isolation and bulk-seq workflow and demonstrate the robust capture of region’s transcriptome across animals. The data can be interactively explored at https://twc-stanford.shinyapps.io/spatiotemporal_brain_map/.
Region identity is linked to expression dynamics during aging
RNA-seq permits quantitative comparisons of aging rates 8,19 based on gene expression shifts. For instance, we found substantial region-dependence in the magnitude and timing of C4b expression (Figure 1E), a complement component and major schizophrenia risk factor 28 that is up-regulated in aged mice 29 and models of neurodegeneration 30. Notably, recent single-cell sequencing and spatial imaging studies revealed that the composition of major cell types remains almost constant throughout the aging mouse brain between 3 and 21 months 31,32, thus the expression dynamics observed in bulk are unlikely to be driven primarily by shifts in cell type abundance. In cases of stable cell populations and substantial replicate numbers, bulk RNA-seq is particularly suitable to investigate subtle, yet robust expression changes by taking advantage of well-established, replicate-sensitive statistical approaches 33 that currently do not exist for single-cell data 34. Thus, we could use our temporally resolved data to probe the per-region impact of aging on gene expression, as this could help to identify structures with selective vulnerability.
We performed pairwise differential expression between 3 months group and every subsequent age group to determine when differentially expressed genes arise (DEGs; referring from heron to genes that change with age). We treated sex as a covariate given the lack of significant interaction between sex and age, and similar expression changes during aging in each sex (Figure S1A–E). A gene had to pass the statistical cutoff in at least two comparisons to be classified as a DEG (Figure 1E–G). The general trend across regions indicated an increase of DEGs over time plateauing around 21 months (Figure 1F,G), though individual regions exhibited profoundly differing trajectories of DEG accumulation (Figure 1F and Table S1). For instance, the visual cortex showed a steady increase of DEGs until late age, while the motor cortex already exhibited significant perturbation at 12 months, with little additional increase until a jump at 21 months (Figure 1F,G). In contrast, entorhinal cortex’ transcriptome appeared largely refractory to the effects of age (Figure 1F,G). This agrees with human MRI 7 and microarray 35 studies demonstrating that the entorhinal cortex displays only mild alterations during cognitive aging, despite exhibiting the first amyloid deposition in AD patients 36. Together, these results reveal the striking region- and time-dependent nature of expression shifts during brain aging, thus necessitating region-resolved quantification and analysis. Notably, regions with the most profound and earliest shifts in gene expression were the white matter-rich caudate putamen, cerebellum, and corpus callosum, the latter showing a tenfold increase in the number of DEGs between 12 and 18 months.
To validate these results with independent analyses, we first probed all genes for correlation with age (STAR Methods; Table S1), thus taking all age groups into account (Figure 1H). Regions differed in the number of age-correlated genes, confirming that the effect size of age depends on the region, and the corpus callosum and cerebellum were the most impacted, while the entorhinal cortex remained largely unaffected (Figure 1H). As a second validation, we performed weighted gene co-expression network analysis (WGCNA) 37 for each region (STAR Methods; Table S1), clustering genes into modules. We filtered for modules exhibiting significant association with age and found that the number of modules differed between regions. Agreeing with the above results, we found seven or more modules in the corpus callosum and cerebellum, whereas we detected no age-related modules in the entorhinal cortex (Table S1). We compiled cell type- and pathway-enrichment for each age-related module into reports for each region as resource (https://twc-stanford.shinyapps.io/spatiotemporal_brain_map/). Interestingly, we discovered in 10 regions at least one module with increased expression over time that was enriched for microglia- and inflammation-related genes (Figure 1I,J). Consistent with these findings, we found a small, common set of DEGs, including neuroinflammatory markers Fcgr2b, Ctss, Cst7 38 in modules across regions, suggesting the presence of a minimal group of co-regulated genes changing throughout the brain. In summary, we found the results of three independent analyses (pairwise tests, age-correlation and WGCNA) congruent, demonstrating that the observed effects of aging on the transcriptome are region-dependent.
A minimal gene set forms a common fingerprint of brain aging
While the vast majority of DEGs appeared to change only in three or less regions, we found 82 genes that were differentially regulated in 10 or more regions (Figure 2A,B; Table S1). These were strongly enriched for up-regulated genes with immune-modulatory functions (Table S1), including MHC I-mediated antigen presentation, interferon-response, and complement cascade, as well as regulators of microglia activity (Figure 2C) including Cd22 39, Trem2 and Tyrobp 40. Of the only 7 down-regulated genes in this set, we found protein homeostasis genes Dnajb1, Hsph1 and Ahsa1, as well as collagen synthesis gene P4ha1 3 (Figure 2B). We combined these 82 genes into a common RNA aging signature to calculate their expression as a single ‘common aging score’ (CAS; STAR Methods) for each mouse and region. While the CAS expectedly showed significant increases in every region (Figure 2D, Figure S2A), the shape and amplitude of the trajectories varied profoundly. We employed linear models to approximate these trajectories, using the slope of the linear fit as a metric to comparatively assess the ‘CAS velocity’ across regions (Figure 2D,E). Of note, the CAS at baseline (i.e. the offset of the linear fit) did not predict a region’s CAS velocity (Figure 2F). Our analysis revealed a gradient of velocities, with cortical areas ranking last, at one-third of the velocity of the corpus callosum, the ‘fastest’ region (Figure 2G,H,I). Other white matter-rich areas, including the caudate putamen, also exhibited high velocities, while hippocampus, thalamus and hypothalamus ranked below average. The median CAS across all regions associated with the animals’ chronological age (Figure S2B). Yet, the regions’ differing velocities resulted in increased per-animal variance, indicating that the transcriptional state of this gene set becomes profoundly desynchronized across the brain. Importantly, we found no association between the CAS velocity and the regions’ cell composition at young age as quantified in a brain-wide in-situ single-cell dataset 41 (Figure 2J, Methods S2, section 1). This suggests that the heterogeneous CAS velocities are unlikely to result from cell proportions differing across regions.
Figure 2. Common gene signature identifies regions with accelerated aging.
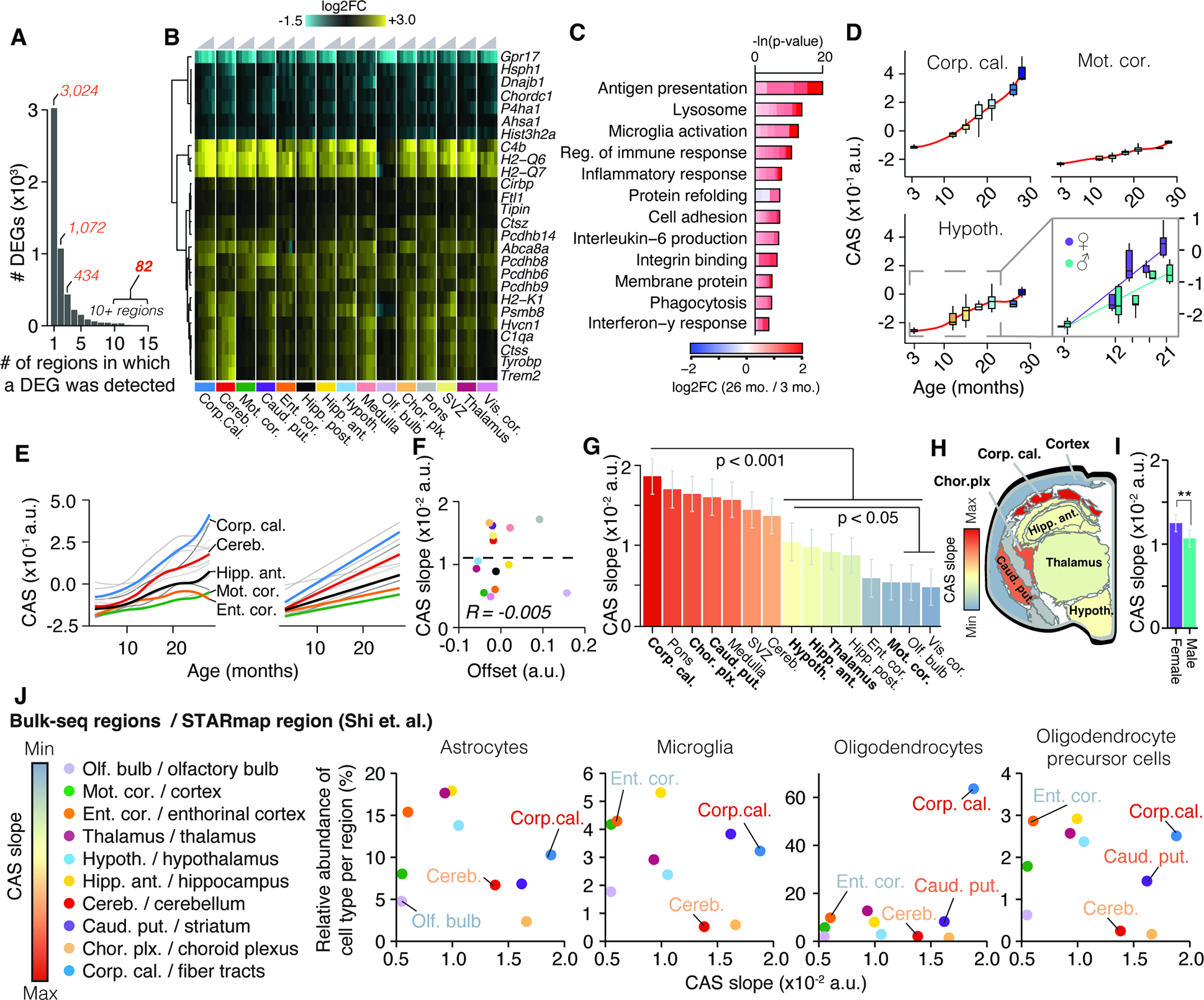
(A) Bar graph indicating the number of regions in which a DEG was detected (Table S1). (B) Region-wise expression changes for genes with shifts in 10 of 15 collected regions.(C) Representative GO analysis of 82 genes forming the CAS. Lengths of bars represent negative ln-transformed Padj using Fisher’s exact test. Colors indicate gene-wise log2 fold-changes in the corpus callosum. Table S1 contains full results list. (D) CAS trajectories selected regions. Insert indicates trajectories for male and females in the hypothalamus. (E) CAS trajectories of all regions approximated via LOESS and linear regression (F) Offset and slope comparison for linear models. (G) Slope of linear regressions in (D), colored by slope. Mean ± 95% confidence intervals. Two-sided Tukey’s HSD test. Bolded regions are highlighted in the following panel. (H) Mouse brain cross-section, with regions colored by CAS linear slopes.. (I) Slope of linear regression across all brain regions, colored by sex. Mean ± 95% confidence intervals. Two-sided Tukey’s HSD test. The highest (least significant) Pval is indicated. (J) Correlation of the abundance of major glia cell types (as measured in 41) with the regions’ respective CAS slopes. Significance tested through spearman correlation and linear regression.
When we examined the CAS trajectories for the interval between 3 and 21 months, we observed a moderate but significant CAS acceleration in females (Figure 2I, S3A,B), with the hypothalamus exhibited the most pronounced acceleration (Figure S3B). While overall age-related expression changes were well-correlated between both sexes (Figure S3D; P value for Fisher’s Exact test < 2.2×10−16), genes related to lipid metabolism, stress response and unfolded protein response, including CAS genes Rbm3 and Cirbp 42, were stronger regulated in males (Figure S3D,E). In contrast, females exhibited a more accentuated regulation of neuroinflammatory markers (including Gfap) and antigen-presentation genes (Figure S3F), as well as several CAS genes related to immune response. In line with this, we found Cish, a known regulator of T cell immune response increasing in females 43,44, being the only gene exhibiting significant, opposite regulation in both sexes. Critically, the female-specific regulation of pro-inflammatory genes was not observable in other regions with similar CAS slopes (Figure S3F). These findings are in line with human studies reporting more pronounced expression of immune-related genes in the hippocampus and cortex of aged women 35,45. Of note, adenomas in the hypothalamus-adjacent pituitary gland can develop at high frequency in female C57BL/6J mice older than 20 months of age46. Since we did not record adenomas in our study, it is possible that this phenomenon could contribute to the accelerated aging patterns observed here. Our data could advance the understanding of several sexual-dimorphisms observed in the brain, including the higher age-specific risk of dementia among women 47, given the hypothalamus’ critical role in regulating reproduction, development and metabolism 48.
Fiber tracts are foci of accelerated brain aging
Bulk-seq data may obscure shifts in sub-structures within regions, like a specific cortical layer. To verify our CAS analysis, we sought a method that could simultaneously examine multiple regions at high-resolution. To this end, we performed spatial transcriptomics (10X Visium) of the brain, isolating coronal sections from an independent cohort of male mice aged 6, 18, and 21 months (Figure 3A). Using a clustering-based we annotated the regional identity of Visium spots (Figure S4A,B; Table S1), identifying them as belonging to the hippocampus, cortex, thalamus, hypothalamus, striatum, choroid plexus and white matter fiber tracts (Figure 3B and S4C–F). Our data demonstrated robust capture of the same regions across age groups and individuals (Figure S4G–L), permitting a comparison of DEGs found in bulk-seq with Visium data (Table S1). We confirmed a more pronounced regulation of DEGs in the white matter cluster (equivalent to the dissected corpus callosum region) compared to the cortex cluster (equivalent to the motor cortex region), This includes several of the 82 CAS genes (Figure 3C, Table S1) such as Trem2 (Figure 3D). Calculating CAS for each Visium spot identified a clear, spatially-defined increase in the score along the white matter tracts, encompassing the corpus callosum and other fiber tract sub-structures (Figure 3E,F). In the cortex, however, we observed only a modest increase in CAS. Generally, CAS velocities determined by bulk-seq and spatial transcriptomics were well-correlated (Figure 3G), confirming vastly differing aging velocities between proximal regions in-situ.
Figure 3. Spatially-resolved CAS detects accelerated aging in white matter tracts.
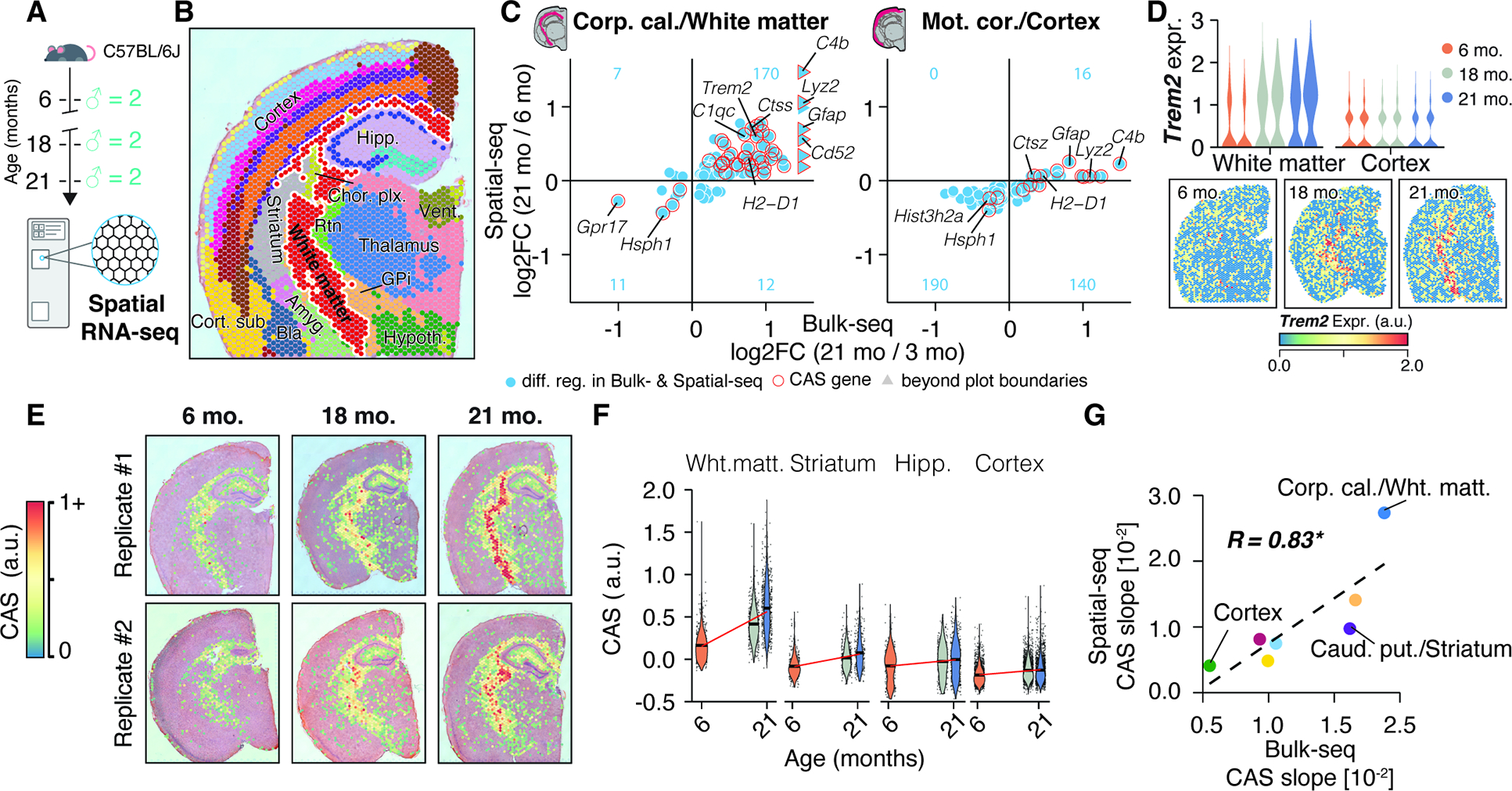
(A) 10X Visium experiment overview. Brain tissue was collected from an independent male C57BL/6J mouse cohort (n = 2 mice; 6, 18 and 21 months). (B) Spatial transcriptome data, colored by cluster-based annotation. Labels represent region-level annotation. Labels represent region-level annotation according to Figure S4. Complete data description and abbreviations are in Figure S4. (C) Comparison of Bulk-seq and Visium differential expression results in selected regions. DEGs found in both datasets are shown, with CAS genes highlighted.The number of overlapping DEGs in each quadrant is indicated in blue. (D) Spatially-resolved expression of Trem2 across age. Violin plots represent expression in white matter- and cortex-associated spots, split by replicates. (E) Spatial representation of CAS. Spots with values ≥ 0 are shown. (F) CAS across spatial clusters of selected regions. Red line indicates linear regression fit. (G) Comparison of CAS slopes for linear models in Bulk-seq and Visium data, colored by region. Corpus callosum, caudate putamen and motor cortex regions were chosen to represent white matter, striatum and cortex, respectively.
Heterogeneous velocity of CAS is encoded by glial transcripts
We aimed to quantify the activity of CAS genes at the single-cell level to pinpoint the cell type(s) influencing the heterogeneous expression dynamics across brain regions. We chose the anterior hippocampus given its intermediate CAS velocity (Figure 2G), utilizing frozen punches from the left hemispheres of the bulk-seq cohort (Figure 4A). Single-nuclei sequencing (nuc-seq) yielded all major cell types, with no age- or sex-related shifts in cell composition. Microglia exhibited the highest baseline CAS, which aligns with many CAS genes being known immune-response genes (Figure 2B,C). Although CAS displayed a significant increase in all cell types (Figure 4B), microglia exhibited the most accentuated increase (Figure 4C), followed by mature oligodendrocytes, brain endothelial cells (BECs), astrocytes and oligodendrocyte progenitor cells (OPCs).
Figure 4. Aging in glia and endothelial cells is the major contributor to CAS increase.
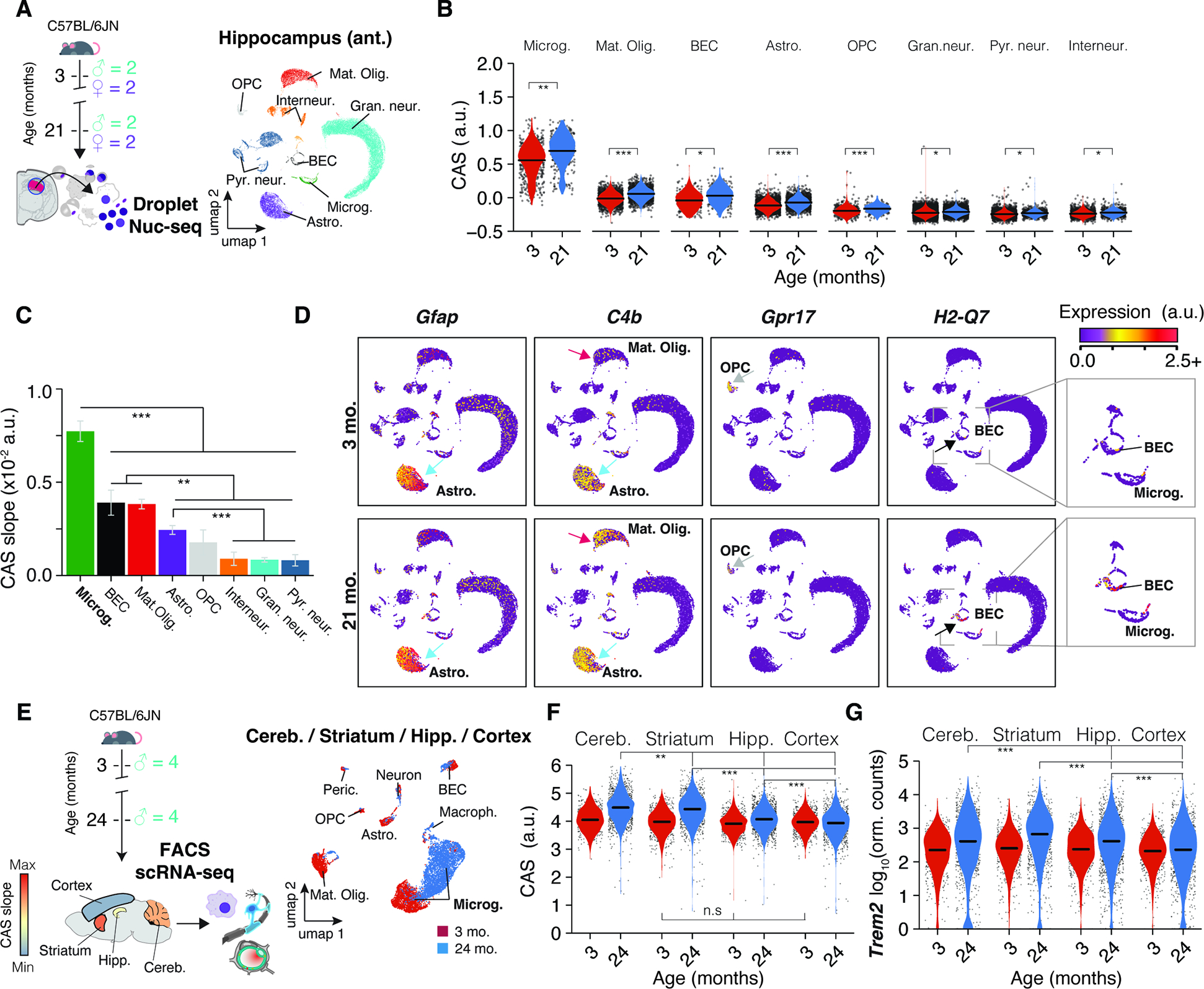
(A) Nuc-seq experiment overview. Nuc-seq of anterior hippocampus from same mice used for bulk RNA-seq (n = 4; 3, 21 months). UMAP of nuclei populations (n = 36,339). (B) CAS across hippocampal cell types. P values from two-tailed t-test on per-replicate median of CAS. (C) CAS slope of linear regressions in (B). Two-sided Tukey’s HSD test. The highest (least significant) Pval is indicated. (D) Expression of CAS genes Gfap, C4b, Gpr17, H2-Q7. Additional details in Methods S2, section 2 and 3. (E) Meta-analysis of scRNA-seq data from 19 of microglia from different brain regions. UMAP of all cell populations (n = 6,373). (F,G) CAS and Trem2 expression across microglia from different brain regions. (MAST, Benjamini–Hochberg correction; false discovery rate (FDR).<.0.05 and logFC.>.0.2).
Upon closer examination of the 82 genes, it was evident that the CAS could mirror aging dynamics in several cell types beyond microglia through cell type-specific or selective gene expression shifts (Figure 4D), including Gfap (Astrocytes), C4b (Astrocytes and mature oligodendrocytes; Methods S2, section 2), Gpr17 (OPCs) and H2-Q7 (BECs; Methods S2, section 3). Notably, aging could trigger the expression of genes undetected at young age. For instance, C4b, mostly detected in young astrocytes, became detectable and increased foremost with age in mature oligodendrocytes (Methods S2, section 2). Similarly, expression of H2-Q7 only became detectable in BECs with old age (Methods S2, section 3). We validated our findings in an independent dataset of publicly available scRNA-seq data from young and old mice’ SVZ 27. Though generated using a different cohort and method, the CAS increase was most pronounced in microglia, consistent with our data (Methods S2, section 4). CAS also significantly increased in aNSCs, but a small cell count at 28 months (under 50 per animal) complicated robust CAS calculations. Thus, differences in CAS velocity between regions predominantly reflect age effects in non-neuronal cells, with microglia having the strongest contribution.
Transcriptional aging of microglia is region-dependent
We aimed to discern varying CAS dynamics between microglia from regions with fast or slow CAS velocity. For this, we analyzed Smartseq2 scRNA-seq data from the Tabula Muris consortium (Figure 4E), which contains comparable numbers of microglia from freshly-isolated cerebellum (high velocity), striatum (equivalates to caudate putamen, high velocity),, hippocampus (medium velocity) and cortex (low velocity). Smartseq2, due to its efficient per-cell transcript capture rate, is particularly apt for examining subtle aging-related transcriptional effects. 49–51.In agreement with our bulk-seq results, the CAS increased in microglia across regions, though with greater magnitude in the cerebellum and striatum, followed by the hippocampus and cortex, respectively (Figure 4F). These shifts were consistent across biological replicates (Methods S2, section 5), and was also reflected on the level of individual CAS genes, like Trem2 (Figure 4G). Notably, there was no detectable CAS difference among microglia at young age across the striatum, hippocampus and cortex, while the cerebellum-derived microglia exhibited a slightly higher baseline CAS. Consistent with our data, aged mice’s microglia isolated from white matter exhibited significantly elevated CAS compared to those derived from gray matter 51 (Methods S2, section 5). Further, we meta-analyzed a well-powered bulk microarray dataset of microglia isolated from cerebellum, striatum, hippocampus and cortex 52, of mice aged 4, 12 and 22 months. We identified more differentially expressed genes with age in the cerebellum and striatum, together with a more pronounced up-regulation of CAS genes (Methods S2, section 5), particularly in the period of 12 to 22 months.
In conclusion, CAS velocities observed in bulk-seq and Visium data partly represent microglia that exhibit region-specific aging rates.
Neuronal transcripts encode region-specific expression patterns
Since CAS genes represent only 1.5% of all DEGs (Figure 2A; Table S1), we hypothesized that the remainder could represent region-specific expression shifts. We first compared age-related DEGs across mouse organs to construct organ-specific aging signatures (Methods S3, section 1). The identification of specific signatures in functionally distinct organs led us to investigate whether individual brain regions display a similar degree of specificity during aging. The number of region-specific DEGs varied significantly (Figure 5A), which we utilized to build aging signatures for each region (Figure 5B,C, Methods S3, section 2). As exemplified for the caudate putamen, we found that most region-specific signatures generally increased with age predominantly in the region they were based on (Figure 5B–D and Methods S3, section 2). Thus, dozens to hundreds of genes in the brain are regulated in a region-specific or -selective manner, revealing highly compartmentalized effects of aging within a single organ.
Figure 5. Neuronal transcripts encode region-specific expression shifts. (A) UpSet plot showing regional specificity of DEGs.
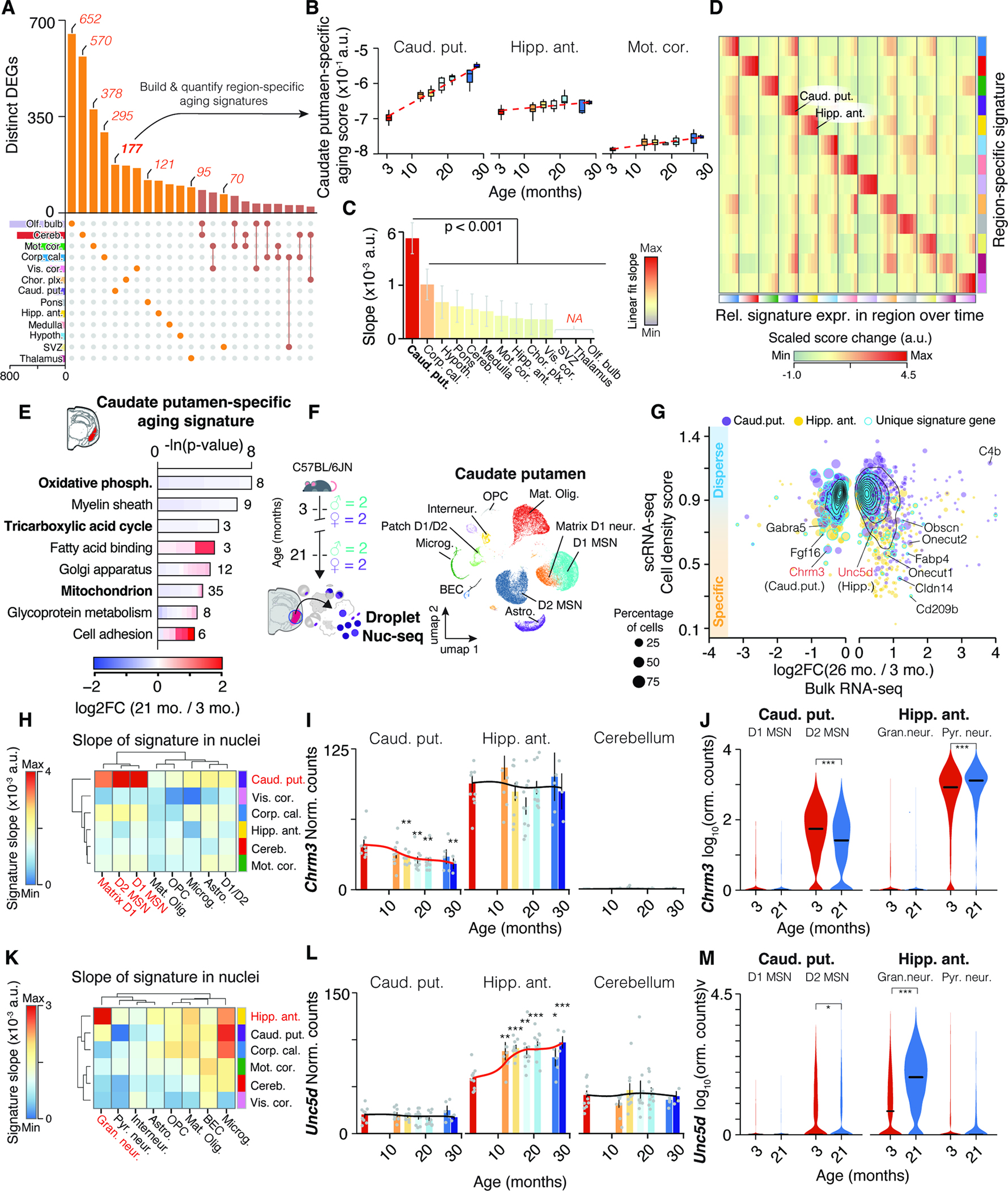
Unique gene sets were used to construct region-specific aging signatures. (B) Trajectories of caudate putamen-specific aging score in selected regions. (C) Slope of linear regressions in (B), colored by slope. Mean ± 95% confidence intervals. (D) Score changes for region-specific signatures relative to 3 months. Statistical analysis in Methods S3, section 2. (E) Representative GO enrichment for 177 DEGs unique to caudate putamen. Table S1 contains full results list. (F) Nuc-seq experiment overview of left-hemisphere regions from same mice used for bulk RNA-seq (n = 4; 3, 21 months). (G) Single-nuclei dispersion scores vs. log2-transformed expression ratios for different regions. Region-specific score genes are highlighted. (H) Slope of cell type-wise changes with age for caudate putamen-specific signature. D1 and D2 Medium spiny neuron populations (MSN) are highlighted. (I,J) Bulk and cell type-wise and expression changes for Chrm3. (K) Slope of cell type-wise changes with age for hippocampus-specific signature. (L-M) Bulk and cell type-wise and expression changes for Unc5d. Additional details in Methods S3, section 2.
Signature genes appeared to be functionally connected, as exemplified by the caudate putamen-specific signature which was enriched for down-regulated mitochondrial processes and up-regulated cell adhesion and lipid binding functions (Figure 5E and Table S1). We analyzed nuc-seq data from the left hemisphere punches of the anterior hippocampus (Figure 4A) and caudate putamen (Figure 5F), where we captured non-neuronal cell types as well as striatum-specific D1- and D2-type medium spiny neurons (D1 and D2 MSNs, respectively). Mapping signature genes like Fgf16, S100a10 and Fabp4 (Methods S3, section 2) to distinct cell populations (Figure 5G, STAR Methods and Table S1) suggested that bulk tissue can indeed capture the expression dynamics of specific cell subsets. We calculated several region-specific signature scores for each cell type at young and old age. We found a distinct increase of the caudate putamen-specific signature in D1 and D2 MSNs which was not seen with signatures from other regions (Figure 5H J, Methods S3, section 2). In comparison, dentate gyrus granule cells of the hippocampus exhibited a distinct increase of the hippocampus-specific signature (Figure 5K), and we found respective regulation of signature genes such as axon-guidance receptor Unc5d 53 and transcription factor Onecut1 (Figure 5L,M, Methods S3, section 3). Notably, granule neurons are highly abundant in the cerebellum 54 yet the hippocampus-specific signature, as well as expression of Unc5d or Onecut1, exhibited no age-related change in the bulk data of the cerebellum. Our approach could thus identify aging signatures of a given cell type occurring selectively in a specific region.
Finally, we explored whether the biological processes associated with signature genes could indicate differential transcriptional activity across whole pathways or organelles. We observed significant down-regulation of mitochondria-related genes in the caudate putamen, including electron transport chain subunits, which could indicate impaired mitochondrial function (Figure 5E). We identified in this region a global, gradual down-regulation of all genes coding for mitochondria-related proteins (Methods S3, section 3), and a corresponding mitochondrial signature dropped significantly in aged D2 MSNs, mature oligodendrocytes, and astrocytes. This was not detected in cell types from other regions (Methods S3, section 2). This specific down-regulation of mitochondrial processes in aged striatum could help to explain the selective vulnerability to mitochondrial toxins and stresses in the striatum of old animals 55,56.
In conclusion, we discovered extensive region-specific transcriptional signatures of aging, largely encoded by expression shifts in distinct neuronal subpopulations reflecting a region’s specialization.
Rejuvenating interventions act on distinct regions and cell types affected during normal aging
Given the substantial region-specific expression changes during normal aging, we wondered if interventions known to stave off age-related pathologies may also act regionally. We performed region-resolved bulk-seq on brains of 19-month-old mice following either four weeks of acute dietary restriction (aDR), a well established nutritional intervention 57,58, or recurring injections of young mouse plasma (YMP) 59, a paradigm to administer circulatory factors found at young age. Both aDR and YMP have previously been shown to exert molecular, structural, and cognitive improvements even at this late age 58,59. Understanding the trajectories of region-specific transcriptional shifts in response to ‘rejuvenating’ interventions, may help to decipher the mechanism mediating their effects.
For the dietary intervention, 19-month-old female mice were either treated with four weeks of aDR or ad libitum feeding (AL; n = 4–5 female C57BL/6JN; Figure 6A). The 25% aDR paradigm (i.e. food reduction to 75% of the AL group) resulted in the expected metabolic shifts, marked by weight loss (Figure S5A,B) and induction of well-recognized expression changes in the liver, albeit to a milder degree than those observed in studies employing chronic DR over years57 (Figure S5C–E). For the plasma intervention, we profiled brains of 19-month-old male mice receiving recurring injections of either YMP or PBS (n = 3–4 male C57BL/6JN; Figure S5F). Critically, the resulting 229 single-region transcriptomes clustered well with the bulk-seq data from the aging cohort (Figure S5G), suggesting a robust sampling of regions across experimental cohorts.
Figure 6. Young plasma injection and acute dietary restriction trigger distinct spatial gene expression changes in the aged brain.
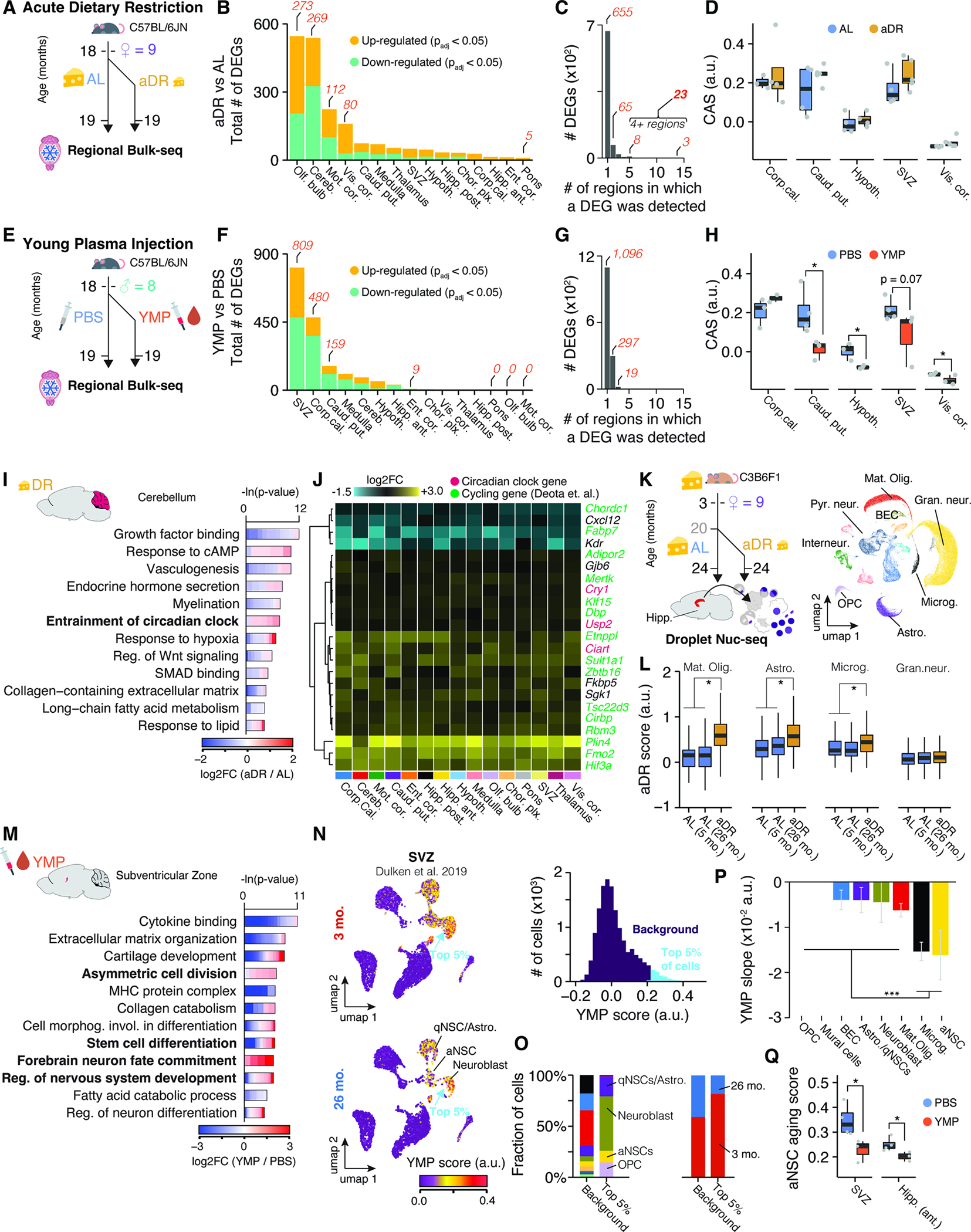
(A) Experiment overview. Aged female mice (n = 4–5) either underwent acute dietary restriction (aDR) for five weeks or continued with ad libitum (AL) feeding before brain collection and bulk-seq analysis on 15 regions. (B) The number of DEGs, split by region and regulation. (C) Bar graph showing the regions where a particular DEG was detected. Refer to Table S1 for the list of DEGs (D) CAS shifts in response to aDR across selected regions. One-tailed t-test. (E) Experiment overview. Aged male mice (n – 3–4) were injected with either young mouse plasma (YMP) or PBS over four weeks. (F-H) Similar to (B-D) for YMP experiments. (I) Representative GO analysis of DEGs with shifts in cerebellum due to aDR. Table S1 contains full results list. (J) Region-wise expression changes in aDR for 24 genes with shifts in at least four of the collected regions. (K) Experiment overview. Nuc-seq of whole hippocampus from female C3B6F1 mice undergoing AL-to-aDR dietary switch at 20 months. UMAP representation of all nuclei is depicted (n=69,253) (L) Boxplot representation of common aDR scores in four cell types. two-tailed t-test on per-replicate median. (M) Similar to (I) but for YMP-induced DEGs in SVZ. (N) UMAP representation of single-cell SVZ data with scores for YMP signature. Cells are colored with scores for YMP signature (representing DEGs up-regulated in response to YMP). Histogram of score distribution is depicted on the right-hand side. Signature genes can be found in Table S1. (O) Composition of cell types and age groups in cells showing the highest YMP scores. (P) YMP score slope of linear regressions against age, colored by cell type. Mean ± 95% confidence intervals. (Q) Boxplot representation of scores for aNSC aging in SVZ and hippocampus in YMP- or PBS-injected mice. Two-tailed t-test on per-replicate median of score
Remarkably, aDR and YMP exerted distinct expression changes across the brain (Figure 6B–D, F–H). aDR was primarily marked by differential regulation in the olfactory bulb, cerebellum and cortical areas, as well as fewer expression changes across all regions (Figure 6A–C). DEGs under aDR exhibited little overlap with DEGs occurring during aging (Figure S5H–J) and the CAS remained unaffected across the brain (Figure 6D, S5K). In several regions, particularly the cerebellum, we found a strong functional enrichment for aDR-induced genes related to regulation of the circadian clock. A set of 23 genes, including three direct members of the circadian clock (Cry1, Usp2, and Ciart) and other genes with cycling expression60, was differentially regulated in at least four regions (Figure 6C,J). We utilized these 23 genes to construct an aDR signature, which was robustly and evenly induced across all brain regions examined (Figure S6A,B). To map out the cell types driving the aDR signature, we performed nuc-seq on whole, frozen hippocampus tissue of 24-month-old female mice that had been fed AL or subjected to 40% aDR since 20 months of age (Figure 6K; n = 3–4 female C3B6F1; cohort previously described in 57,61). The signature was specifically up-regulated under aDR in mature oligodendrocytes, astrocytes, microglia and OPCs, but unaffected in any neuronal subpopulation (Figure 6L, S6C–E). Thus, aDR induces a brain-wide transcriptional program acting on the same cell types affected by the CAS, albeit through molecular pathways orthogonal to those changing during aging.
In contrast, YMP caused region-selective expression shifts, affecting the SVZ in particular. (Figure 6E–H). Here, we observed profound up-regulation of pathways related to stem cell differentiation and neuronal maturation (Figure 6M). We mapped a signature representing all up-regulated genes under YMP to single-cell data of the SVZ 27, where it demarcated neuroblasts, quiescent, and activated neural stem cells (aNSCs), which were primarily found in young mice (Figure 6N,O). SVZ cells from aged mice decreased the YMP signature, and, conversely, age-related DEGs found in aNSCs were down-regulated in YMP-treated mice (Figure 6P,Q). Thus, YMP injection reactivates an expression pattern in the neurogenic lineage that becomes down-regulated with age. In addition to effects on the SVZ, YMP caused significant down-regulation of genes like C4b, B2m, Trem2 or Gfap in selected regions, and led to a significant CAS reduction in caudate putamen, hypothalamus, SVZ and several cortical areas (Figure S5I,L–M).
In summary, we uncovered that the rejuvenating interventions aDR and YMP act in profoundly different, region-specific manners. While aDR instigates a reprogramming of genes related to the circadian clock across all glia, YMP causes a selective reversal of age-related expression signatures, particularly in the neurogenic lineage of the SVZ.
Aging results in region-specific expression changes of genes associated with human diseases
Region-specific gene expression of a gene could influence not just the aging of the brain, but also susceptibility to disease and selective vulnerability of regional cell populations. This is due to differences in basal gene expression levels across the young adult brain and trajectories of age-related changes. This is demonstrated with the example of the C4b gene, a key genetic risk factor for schizophrenia 28. The motor cortex and hippocampus, for instance, show up to a 3-fold difference in basal expression of C4b at three months of age, with age-related increases of up to 10-fold observed from the least affected visual cortex to the corpus callosum (Methods S2, section 2). If such region-specific differences and aging trajectories exist in human brains, it could likely impact the pathogenesis and clinical manifestations of a disease.
To explore this concept in the context of neurodegenerative diseases such as AD or PD, we analyzed the expression of genes linked to autosomal dominant forms of disease or genes linked to the risk of developing sporadic forms of disease. We ranked regions based on the highly variable expression of either Apoe, Trem2, Plcg2 or Scna (α-synuclein) at 3 months (Figure 7A–D). Crucially, the expression distribution across the brain for each gene was substantially rearranged in older animals due to region-specific differential regulation. To systematically assess the regulation of disease risk-linked genes, we assembled lists of Genome-wide association studies (GWAS) genes for AD or PD, and investigated whether they were significantly enriched among age-related DEGs of a given region (Table S1) 62,63. Each disease-associated gene set exhibited a different enrichment pattern (Figure 7E–G) and a varying number of associated genes (Figure 7H–I). AD-related genes, including Apoe, Ms4a6d and Plcg2 64, were part of DEGs that were co-regulated in a small cluster of three regions: the choroid plexus, corpus callosum, and pons (Figure 7E). Conversely, PD-related genes, like the neuroprotective gene Ip6k2 65, were distributed across several regions with limited overlap (Figure 7F). Of note, the substantia nigra, a major region where PD typically manifests, was not quantified in our study.
Figure 7. Interplay of region and age determines expression of disease variant homologues.
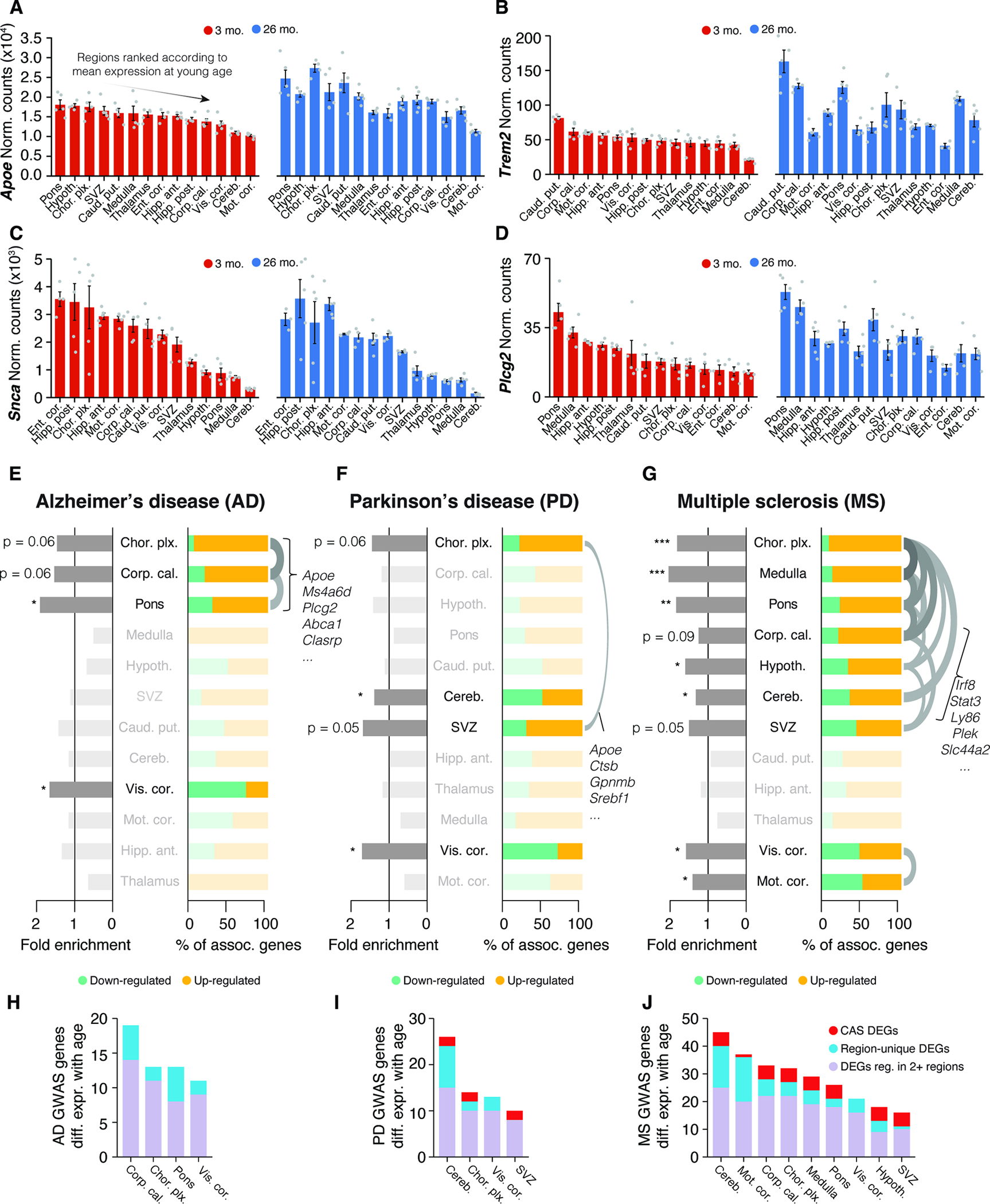
(A-D) Bulk expression for (A) Apoe, (B) Trem2, (C) Scna (α-synuclein) and (D) Plcg2 at 3 and 26 months of age, represented with only male samples. Regions are arranged by descending order of mean expression at young age. Mean ± s.e.m. (E-G) Enrichment analysis of region-resolved DEGs for human GWAS variants for AD, PD, and MS, with associated genes listed in Table S1. Fold enrichment and relative composition of disease-associated DEGs with respect to their regulation are indicated. One-sided Fisher’s exact test with hypergeometric distribution. Order of regions results from hierarchical clustering on a pairwise Jacquard Distance matrix. Overlaps with a Jaccard index ≥ 0.25 are indicated with an arc. One-sided Fisher’s exact test. (H-J) Number of DEGs per region that are homologues of human GWAS variant for AD, PD, and MS. Colors group the genes into CAS DEGs, region-specific DEGs, or other (DEG in 2 or more but fewer than 10 regions).
We also analyzed GWAS genes for multiple sclerosis (MS) due to observed age-related effects in white matter-rich regions. These genes had significant associations with DEGs from nine different regions that fell into two clusters, indicating two disparate subsets. One cluster consisted of regions including the corpus callosum and cerebellum, that up-regulated a shared set of inflammation-related genes such as Stat3, Ly86 and Irf8, all part of the CAS (Figure 7G). This hints at similarities between the pathophysiology of inflammation and demyelination associated with MS and the accelerated aging observed in white matter-rich areas. The visual and motor cortex formed a second cluster, exhibiting even numbers of up- and down-regulated MS genes. This supports evidence of transcriptional shifts (e.g. of Cbln2) in cortical areas that can occur far away from the actual lesions 66 and highlights the need to broadly study regional patterns of gene expression to understand the role of MS-associated genes. To better contextualize the biological relevance of the observed expression changes, we further compared the up-regulation of GWAS homologues with their baseline expression across regions (Figure S7B,C; STAR Methods), which confirmed that age-related differential expression in specific regions led to a significant redistribution of where in the brain a given GWAS homologue was predominantly expressed (Figure S7C–E).
Our data demonstrate that genetic risk factors linked to three major neurodegenerative diseases are affected by age in a region-selective manner. While we cannot predict whether the directionality of the regulation itself has a biological consequence, this region-specific differential regulation of such genes could be an additional factor modulating disease risk.
Discussion
The advent of single-cell technologies and cell dissociation methods have enabled the exploration of an ever increasing number of cell populations in the brain 67, allowing for cell type-specific characterization of gene expression during aging 9. The interplay between cell type and regional niche during aging is, however, yet to be more deeply understood. Our results underscore the importance of region identity in modulating gene expression dynamics in the context of aging and neurodegeneration. Future studies should probe if these diverse expression patterns lead to shifts in the proteome or bring about functional changes in neuronal activity and plasticity. Given comparable observations that astrocytes exhibit stronger age-related expression changes in striatum and cerebellum as compared to cortical areas 32,68,69, it is likely that other glial cells contribute to the heterogeneous CAS increase at the bulk level. Further exploration of the CAS in other non-neuronal cell types could clarify whether microglia actively drive the regional expression dynamics described here, or if they simply respond to cues from other cell types in the region.
Our data reveal that certain brain regions are selectively vulnerable to aging, with white matter fiber tracts exhibiting a particular sensitivity. These areas, dense with myelinated axons and myelinating cells, form the basis of neurotransmission across brain regions 70. The strong activation of immune- and inflammation-related genes, and differential expression of remyelination regulators like Gpr17 71,72 suggests that the homeostasis of this region is compromised at old age potentially affecting myelin sheath integrity and impairing axonal signal transmission as an early event in brain aging. In line with this, rejuvenation oligodendrogenesis in aged mice via FGF17 improves long-term memory consolidation, demonstrating a causal role of compromised myelin on cognition73.
We found evidence that aDR induces brain-wide reprogramming of genes associated with circadian rhythmicity, independent of feeding time (C57BL/6JN were fed in the afternoon and C3B6F1 fed in the morning 57). This aligns with recent findings that the lifespan-extending effects of DR are dependent on a shift in circadian rhythm74–76. Future work should investigate how altered circadian rhythmicity impacts cell function and why only glia, not neurons, are affected. Conversely, YMP appeared to directly reverse age-related expression shifts in regions near the ventricles, which are highly permissive for peripheral plasma proteins77. We provide evidence that YMP specifically reverses aging signatures of the neurogenic lineage, indicating restoration of adult neurogenesis that should be assessed specifically with cell cycling tracing assays78.
Our findings strongly support the notion that the impacts of aging on brain function are region-specific. This may explain the regional vulnerability across different diseases and the varied manifestations of neurodegeneration at the individual level. We demonstrate that key genetic risk genes are differentially expressed in a region-specific manner, potentially locally amplifying or attenuating their impact on disease pathways. Importantly, our findings also suggest that aging may drive dysfunction in brain regions that are not affected by classical pathological hallmarks. The translation of these findings to humans may serve as a new brain cartography leading to novel treatment strategies and interventions.
Limitations of this study
Because region-specific and age-related changes in gene expression may be distinct for each species, the conclusions drawn here from mouse data may not be translatable in their entirety to humans. The analysis of our single-nuclei RNA-seq data computationally pools animals of the same age and cell type, and thus was not analyzed in a replicate-sensitive manner, and the presence of pseudo-replication effects cannot be excluded. We also combined sexes for most of our analyses, potentially masking subtle sex-specific gene expression differences. Relatedly, limitations in mouse availability for this study resulted in the two oldest ages being profiled only in male mice. We suggest further interrogation of potential sex differences of murine brain aging late in life. Lastly, determining the exact distribution of major cell types remains a challenge in the field, preventing us from fully eliminating the possibility that baseline differences in abundance play some role in detected aging effects.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Requests for resources and reagents should be directed to the lead contact, Tony Wyss-Coray (twc@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and Code Availability
The sequencing data have been deposited at Gene Expression Omnibus repository and are publicly available as of the date of publication. Publicly available datasets were obtained from the following repositories: BioProject, Gene Expression Omnibus and 10X Genomics’ public resources. DOIs are listed in the key resources table.
All original code has been deposited at https://github.com/OliInTheValley/SpatioTemporal_Analysis and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2,2,2-Tribromoethanol (Avertin) | Sigma Aldrich | Cat# T48402-100G |
| UltraPure™ 0.5M EDTA, pH 8.0 | Thermo Fisher Scientific | Cat# 15575020 |
| PBS, pH 7.4 | Thermo Fisher Scientific | Cat# 10010049 |
| Slide-A-Lyzer™ Dialysis Cassettes, 10K MWCO, 3 mL | Thermo Fisher Scientific | Cat# 66380 |
| VWR brand, razor blades, 0.009” | VWR | Cat# 55411-050 |
| Miltex® Disposable Biopsy Punches with Plunger System 1.5mm | AliMed | Cat# 98PUN6-2 |
| Miltex® Disposable Biopsy Punches with Plunger System 2mm | AliMed | Cat# 98PUN6-3 |
| 0.1% sodium dodecyl sulfate | Teknova | Cat# S0180 |
| EZ Prep lysis buffer | Sigma Aldrich | Cat# NUC-101 |
| UltraPure™ BSA | Thermo Fisher Scientific | Cat# AM2618 |
| Hoechst 33342, 10 mg/mL Solution in Water | Thermo Fisher Scientific | Cat# H3570 |
| Recombinant RNase Inhibitor | Takara | Cat# 2313B |
| Critical Commercial Assays | ||
| RNeasy 96 Kit | Qiagen | Cat# 74181 |
| Qubit™ 1X dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q33231 |
| Quant-iT™ dsDNA Assay Kit, high sensitivity | Thermo Fisher Scientific | Cat# Q33120 |
| Illumina Tagment DNA Enzyme and Buffer large Kit | Illumina | Cat# 20034198 |
| Agencourt AMPure XP, 60 mL | Beckman Coulter | Cat# A63881 |
| SMARTScribe™ Reverse Transcriptase | Takara | Cat# 639538 |
| KAPA HiFi HotStart ReadyMix PCR Kit | Roche | Cat# 07958935001 |
| Visium Spatial Tissue Optimization Reagents Kit | 10X Genomics | Cat# 1000193 |
| Visium Spatial Gene Expression Reagent Kit | 10X Genomics | Cat# 1000184 |
| Chromium Single Cell 3’ GEM & Gel Bead Kit v3.1 | 10X Genomics | Cat# 1000121 |
| Chromium Single Cell 3’ Library Construction Kit | 10X Genomics | Cat# 1000121 |
| Deposited Data | ||
| scRNA-seq of SVZ | Dulken et al.,27 | Bioproject:PRJNA450425 |
| 10X Sagittal Visium Dataset Anterior | https://www.10xgenomics.com/resources/datasets/mouse-brain-serial-section-1-sagittal-anterior-1-standard-1-0-0 | 10xgenomics.com: V1_Mouse_Brain_Sagittal_Anterior |
| 10X Sagittal Visium Dataset Posterior | https://www.10xgenomics.com/resources/datasets/mouse-brain-serial-section-1-sagittal-posterior-1-standard-1-0-0 | 10xgenomics.com: V1_Mouse_Brain_Sagittal_Posterior |
| 10X Coronal Visium Dataset | https://www.10xgenomics.com/resources/datasets/mouse-brain-section-coronal-1-standard-1-0-0 | 10xgenomics.com: V1_Adult_Mouse_Brain |
| SS2 scRNA-seq of grey/white matter microglia | Safaiyan et al.,51 | GEO:GSE166548 |
| Bulk-RNA microarray of microglia from various regions | Grabert et al.,52 | GEO:GSE62420 |
| SS2 scRNA-seq of microglia from various regions | Tabula Muris Consortium et al.,19 | GEO:GSE109774 |
| Bulk-seq of brain regions (aging) | This paper | GEO:GSE212336 |
| Nuc-seq of the mouse hippocampus and caudate putamen at young and old age | This paper | GEO:GSE212576 |
| Spatial transcriptomics of the mouse brain across three age groups | This paper | GEO:GSE212903 |
| Bulk-seq of brain regions (rejuvenation) | This paper | GEO:GSE227689 |
| Nuc-seq of the mouse hippocampus under dietary restriction | This paper | GEO:GSE227515 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6JN | NIA | N/A |
| C57BL/6J | Jackson Laboratory | 000664 |
| C3B6F1 | Max-Planck-Institute for Biology of Ageing | N/A |
| Oligonucleotides | ||
| SS2 Oligo-dT30VN: AAGCAGTGGTATCAACGCAGAGTACTTT TTTTTTTTTTTTTTTTTTTTTTTTTTTTVN | Integrated DNA Technologies | N/A |
| Software and Algorithms | ||
| Custom analysis software | This paper | https://github.com/OliInTheValley/SpatioTemporal_Analysis |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animal husbandry and organ collection
For Bulk-seq and nuc-seq, male and female C57BL/6JN mice were shipped from the National Institute on Aging colony at Charles River. 5–6 male and 5 female mice were used for each 3, 12, 15, 18, and 21 months group, while only 5 and 3 male mice were used for the 26 and 28 months groups, respectively. For the 10X Visium experiments, aged C57BL/6J mice (000664, Jackson Laboratory) were shipped from Jackson Laboratory. 2 male mice/age were used for the 6, 18, and 21 months groups. All mice of the aging and aDR cohorts were housed in cages of 2–3 mice at the Stanford ChEM-H animal facility under a 12 h/12 h light/dark cycle at 67–73 °F and provided with food and water ad libitum. Mice were housed in the ChEM-H animal facility for one month before euthanasia, except for mice older than 18 months, which were housed at the ChEM-H animal facility beginning at 18 months. Takedown of the bulk- and nuc-seq cohort was conducted between 10:00am-12:00pm over four days. Takedown of mice for 10X Visium was conducted between 10:00am-10:15am on a single day. Age groups and sexes were rotated through over the duration of the takedowns to average out the impact of takedown time and day. After anaesthetization with 2.5% v/v Avertin, ~700ul blood was drawn via cardiac puncture before transcardial perfusion with 20 ml cold PBS. The brain was immediately removed and snap-frozen by submerging for 60 seconds in liquid nitrogen-cooled isopentane. Brains were stored at −80°C until further processing. All animal care and procedures complied with the Animal Welfare Act and were in accordance with institutional guidelines and approved by the institutional administrative panel of laboratory animal care at Stanford University.
For the aDR study with C57BL/6JN mice, 18-months-old mice were randomly assigned to AL or aDR. aDR treatment was initiated by transferring mice from AL to 10% aDR for 7 days. After that, aDR was increased to 25%. aDR animals were fed once per day between 3–5 p.m., and all animals were checked daily for their well-being and any deaths. For the first 16 days, weights were checked daily. Mice were euthanized at the ages of 19 months. All mice were euthanized in the morning within a period of 6 hours prior to the regular feeding time of the DR mice.
The aDR study with C3B6F1 mice was performed in accordance with the recommendations and guidelines of the Federation of the European Laboratory Animal Science Association (FELASA), with all protocols approved by the Landesamt für Natur, Umwelt und Verbraucherschutz, Nordrhein-Westfalen, Germany (84–02.04.2015.A437). Female F1 hybrid mice (C3B6F1) were generated in-house by crossing C3H/HeOuJ females with C57BL/6NCrl males (strain codes 626 and 027, respectively, Charles River Laboratories). Lifespans of chronic DR and AL C3B6F1 mice were previously published 61. Pups were weaned at 3–4 weeks of age and were randomly assigned to cages upon weaning. Animals were housed in groups of 5 females in individually ventilated cages under specific-pathogen-free conditions with constant temperature (21.°C), 50–60% humidity and a 12 h/12 h light/dark cycle. For environmental enrichment, mice had constant access to nesting material and chew sticks. All mice received commercially available rodent chow (ssniff R/M-Low phytoestrogen, ssniff Spezialdiäten, Germany) and were provided with filtered water ad libitum. aDR animals received 60% of the food amount consumed by AL animals. aDR treatment was initiated at 20 months of age by directly transferring mice from AL to 40% DR. aDR animals were fed once per day, and all animals were checked daily for their well-being and any deaths. Mice were euthanized at the ages of 24 months. All mice were euthanized in the morning within a period of 3 hours prior to the regular feeding time of the DR mice. Mice were euthanized by cervical dislocation, and tissues were rapidly collected and snap-frozen in liquid nitrogen. The cohort of mice treated with YMP or PBS were housed at the Palo Alto VA animal facility under a 12 h/12 h light/dark cycle at 68–73.°F under 40–60% humidity. All experiments were performed in accordance with institutional guidelines approved by the VA Palo Alto Committee on Animal Research. Euthanasia and organ collection was conducted in the same way as the aging cohorts.
METHOD DETAILS
Processing and administration of plasma
Young Mouse Plasma (YMP) was collected following the protocol described by59,79. Briefly, C57Bl/6J male mice aged 2 months were group-housed and anesthetized with 2.5% v/v Avertin. Approximately 700 μl of blood was drawn via cardiac puncture prior to transcardial perfusion. Blood was collected using 15 μl of 250 mM EDTA (Thermo Fisher Scientific, 15575020) and centrifuged at 4°C for 15 minutes at 1,000g to obtain plasma. The plasma from 20–25 mice was pooled together and dialyzed in 1X PBS using cassettes (Slide-A-Lyzer Dialysis Cassettes, 3.5 kDa molecular weight cut-off, 3–12 ml) before being frozen at −80°C.
For plasma transfer experiments, male C57BL/6JN mice aged 18 months were injected retro-orbitally with 150 μl of YMP per injection. Prior to injection, mice were habituated by being placed on the procedure table in their cage. Injections were administered every 3–4 days, alternating between the left and right eye to allow for recovery. Mice were rested for four days before tissue collection.
Brain region dissection
Dissociating the mouse brain at scale poses several challenges, as the tissue consists of a multitude of biologically distinct structures that require careful, time-consuming separation to avoid cross-region contamination - all while avoiding tissue degradation and loss of RNA quality. We systematically assessed several isolation, dissection, and freezing strategies, most of which yielded low-quality RNA or were not scalable to the intended set of samples and regions. We found success in perfusing the animal before isolating and freezing the whole brain in under 5 minutes, thus rapidly stabilizing the tissue and RNA. Region isolation via slicing and atlas-guided tissue punching was subsequently conducted at sub −0°C temperatures (Methods S1, section 1). In detail, brain regions were dissected from frozen mouse brains through a modification of a previously developed protocol 80. Frozen brains were sliced into 1mm thick coronal slices at −20°C using a metal brain matrix and .22mm razor blades (Ted Pella, 15045; VWR, 55411–050) and were then placed on dry ice and covered to prevent condensation. One slice at a time was placed on a metal block cooled on wet ice and 1.5mm and 2mm diameter regions of interest were dissected quickly via disposable biopsy punches (Alimed, 98PUN6–2, 98PUN6–3) from the left and right hemispheres guided by visual landmarks and the Allen Mouse Brain Atlas. The same biopsy punch was used for identical regions between left and right hemispheres, but replaced between regions and mice. 15 regions were collected: three cortical regions (motor cortex, visual cortex and entorhinal cortex), anterior (dorsal) and posterior (ventral) hippocampus, hypothalamus, thalamus, caudate putamen (part of the striatum), pons, medulla, cerebellum and the olfactory bulb, corpus callosum, choroid plexus and the subventricular zone. The following regions required overlapping punches and were thus sequentially collected: (1) motor cortex, (2) caudate putamen, (3) subventricular zone, (4) corpus callosum (Methods S1, section 1).
Regions were stored at −80°C until further processing.
Bulk-seq preparation and sequencing
We isolated RNA from the right hemisphere brain regions described above using the RNeasy 96 kit (Qiagen, 74181) and a TissueLyser II (Qiagen, 85300), according to RNeasy 96 Handbook protocol “Purification of Total RNA from Animal Tissues using Spin Technology” without the optional on-plate DNase digestion. Quality control of RNA was conducted using a Bioanalyzer (Agilent) at the Stanford Protein and Nucleic Acid Facility for three randomly selected samples per brain region.
cDNA and library syntheses were performed in house using the Smart-seq2 protocol as previously described 8,79 with the following modifications: Extracted RNA (2 ul at a concentration of 25 ng/ul) was reverse-transcribed and the resulting cDNA amplified using 10 cycles. After bead clean-up using 0.7x ratio with AMPure beads (Thermo Fisher, A63881), cDNA concentration was measured using the Quant-iT dsDNA HS kit (Thermo Fisher, Q33120) and normalized to 0.4 ng/ul as input for library prep. 0.8 ul of each normalized sample was mixed with 2.4 ul of tagmentation mix containing Tn5 Tagmentation enzyme (20034198, Illumina) and then incubated at 55°C for 12 minutes. The reaction was stopped by burying the plate in ice for 2 minutes followed by quenching with 0.8 ul 0.1% sodium dodecyl sulfate (Teknova, S0180). 1.6 ul indexing primer (IDT) was added and amplified using 12 cycles. Libraries were pooled and purified using two purification rounds with a ratio of 0.8x and 0.7x AMPure beads. Library quantity and quality was assessed using a Bioanalyzer (Agilent) and Qubit dsDNA HS kit. Pipetting steps were performed using the liquid-handling robots Dragonfly or Mosquito HV (SPT Labtech) using 384 well-plates and PCR reactions were carried out on a 384-plate Thermal Cycler (BioRad). Illumina sequencing of the resulting libraries was performed by Novogene (https://en.novogene.com/) on an Illumina NovaSeq S4 (Illumina). Base calling, demultiplexing, and generation of FastQ files were conducted by Novogene.
10X Visium preparation and sequencing
Frozen brains (n = 2 males per age; aged 6, 18 and 21 months; C57BL/6J strain) were embedded in OCT for cryosectioning at 16 micron thickness (app. Bregma −1.655mm; Allen brain reference atlas coronal section 71). Reactions were carried out with the Visum Spatial Gene Expression (GEX) and Tissue Optimization (TO) Slide & Reagent Kits according to the manufacturer’s protocol with recommended reagents (10X Genomics, 1000193 and 1000184). Sections were placed on designated capture areas of slides for TO and GEX and stored at −80°C until further processing. TO and GEX slides were fixed with methanol and stained with hematoxylin and eosin (H&E) for visualization of tissue morphology on a AxioImager Widefield Fluorescence Microscope (Zeiss) at 10-fold magnification. To determine the optimal permeabilization time, TO slides were incubated with permeabilization enzyme for various timeframes followed by incubation with reverse transcriptase (RT) and fluorescently labeled nucleotides (FLNs) and enzymatic tissue removal. After visualizing cDNA signal via fluorescence microscopy, we selected 20 minutes as the optimal permeabilization time. GEX slides were incubated with permeabilization enzyme for 20 minutes followed by incubation with RT. cDNA was then transferred into tubes and amplified for 15 cycles using a Thermal Cycler (BioRad). Library construction steps were performed according to the manufacturer’s protocol and included cDNA fragmentation, end repair and A-tailing, adaptor ligation, and sample indexing and amplification. Quality control of the constructed library was conducted via Bioanalyzer (Agilent). Illumina sequencing of the resulting libraries was performed by Novogene (https://en.novogene.com/) on an Illumina NovaSeq S4 (Illumina). Base calling, demultiplexing, and generation of FastQ files were conducted by Novogene.
Nuc-seq preparation and sequencing
Single-nuclei preparation (n = 2 males and females per age and region; aged 3 and 21 months; all C57BL/6JN strain) and sequencing was performed as previously described29 with the following modifications: Nuclei from left hemisphere brain region punches were isolated with EZ Prep lysis buffer (Sigma, NUC-101) on ice. Single-nuclei isolation from the whole hippocampus of C3B6F1 mice (aDR) was performed similarly with the exception that tissues were not pooled. Samples were placed into 2 ml cold EZ lysis buffer in a 2 ml glass dounce tissue grinder (Sigma, D8938) and homogenized by hand 25 times with pestle A followed by 25 times with pestle B while incorporating a 180-degree twist. Tissue homogenate was transferred to a fresh 15 ml tube on ice. The tissue grinder was rinsed with 2 ml fresh lysis buffer and transferred to the tube holding the homogenate for a total volume of 4 ml. Samples were incubated on ice for 5 minutes. Nuclei were centrifuged at 500 × g for 5 minutes at 4°C, supernatant removed and pellet resuspended with 4 ml EZ lysis buffer, and incubated on ice for 5 minutes. Centrifugation at 500 × g for 5 minutes at 4°C was repeated. After removing supernatant, the pellet was resuspended with 4 ml chilled PBS and filtered through a 35-um cell strainer into a 5 ml round bottom FACS tube (Corning, 352235). Following centrifugation at 300 × g for 10 minutes at 4°C with break 3, supernatant was gently poured out leaving behind the nuclei pellet. Pellet was resuspended in 400 ul PBS containing 1% BSA (Thermo Fisher, BP9700100), 0.2 ul Hoechst dye (Thermo Fisher, H3570), and 2 ul recombinant RNase inhibitor (Takara, 2313B). Isolated nuclei were sorted on a MA900 Multi-Application Cell Sorter (Sony Biotechnology). 25,000 single nuclei per sample were collected into 1.5 ml DNA lo-bind tubes (Eppendorf, 022431021) containing 1 ml buffer mix with PBS, UltraPure BSA (Thermo Fisher, AM2618), and RNase inhibitor (Takara, 2313B). One male and one female sample from the same time point and region were pooled at this stage by FACS collecting into the same sample tube (thus yielding 50,000 nuclei per tube). Collected nuclei were centrifuged at 400 × g for 5 minutes at 4°C with break 2. Supernatant was removed leaving 40 ul suspended nuclei. Nuclei were counted using a hemocytometer (Sigma, Z359629–1EA) and assessed for concentration and quality.
Reagents of the Chromium Single Cell 3’ GEM & Gel Bead Kit v3.1 (10X Genomics, 1000121) were thawed and prepared according to the manufacturer’s protocol. Nuclei and master mix solution was adjusted to target 10,000 nuclei per sample and loaded on a standard Chromium Controller (10X Genomics, 1000204) according to manufacturer protocols. We applied 11 PCR cycles to generate cDNA. Library construction was conducted using Chromium Single Cell 3’ Library Construction Kit v3 (10X Genomics, 1000121). All reaction and quality control steps were carried out according to the manufacturer’s protocol and with recommended reagents, consumables, and instruments. We chose 11 PCR cycles for library generation. Quality control of cDNA and libraries was conducted using a Bioanalyzer (Agilent) at the Stanford Protein and Nucleic Acid Facility. Illumina sequencing of the resulting libraries was performed by Novogene (https://en.novogene.com/) on an Illumina NovaSeq S4 (Illumina). Base calling, demultiplexing, and generation of FastQ files were conducted by Novogene.
QUANTIFICATION AND STATISTICAL ANALYSIS
Bulk-seq quantification, quality control
Raw sequence reads were trimmed to remove adaptor contamination and poor-quality reads using Trim Galore! (v.0.4.4, parameters: --paired --length 20 --phred33 --q 30). Trimmed sequences were aligned using STAR (v.2.5.3, default parameters). Multi-mapped reads were filtered. Read quality control and counting were performed using SeqMonk v.1.48.0 and RStudio v.3.6. Data visualization and analysis were performed using custom Rstudio scripts and the following Bioconductor packages: Deseq2 33, topGO, destiny and org.Mm.eg.db. Finally, we excluded pseudogenes and predicted genes from the count matrix to focus predominantly on well-annotated, protein-coding genes. In total, all of the following analyses were performed on the same set of 21,076 genes.
To assess the quality of our dataset, the count matrix was analyzed using Seurat’s built-in, default dimensionality reduction workflow 81 (Normalization: ‘LogNormalize’; Variable feature discovery: selection.method=‘vst’, features=2000). Umaps were calculated using Seurat’s built-in functions, based on the first 40 principle components (PC) dimensions (Figure 1C; Methods S1, section 2). A shared-nearest-neighbors graph was constructed using the first 40 PC dimensions before clustering samples using Seurat’s built-in FindClusters function with a resolution of 0.8 to identify samples that would not cluster with their region of origin.
We corroborated the Seurat-based quality assessment by loading and normalizing the count matrix using DEseq2 before conducting the built-in variance stabilizing transformation 33. We then performed hierarchical sample-to-sample clustering using Ward’s clustering algorithm across all 21,076 genes (Methods S1, section 2). To detect whether samples within a given tissue would show profound clustering by age, we finally calculated diffusion maps using the R package destiny with default parameters (Figure 1D).
For bar graph visualization of gene expression (e.g. Figure 1E), we used DEseq2-normalized counts after calculating factors and dispersion estimates across all regions using the factor design ~age + region. Trajectories were smoothed via triangular moving average across the interval between 3 and 28 months. This quantification and smoothing was solely used for visualization and was not the basis for any statistical testing in this study.
Bulk-seq differential expression
To identify significant differential expression changes with age, we used the raw count matrix as recommended for the DEseq2 standard analysis pipeline. Factors and dispersion estimates were calculated for each region separately. We conducted differential expression analysis comparing samples from 3 months to each consecutive time point, using sex as covariate. This is consistent with previously published differential expression analyses performed across whole organs in mice8. P values were adjusted for multiple testing, and genes with an adjusted P value of less than 0.05 were determined to be statistically significant. Finally, we required a gene to reach statistical significance (after multiple testing correction) in at least 2 pairwise comparisons (e.g. 3 months vs 18 months and 3 months vs 21 months) to be called a differentially expressed gene (DEG). We chose this criterion to retain only genes with robust differential expression patterns across age groups. We recognize that this tends to select against genes that are differentially expressed very late in life (i.e. 3 months vs 28 months).
To demonstrate the validity of using sex as a model covariate in the differential gene expression analysis, we performed gene-wise likelihood-ratio tests (LRT analysis, as implemented in DESeq2 7). This assesses the goodness of fit between a ‘complete’ model formula ( expr. ~ age + sex + age:sex interaction) and the model formula implemented in our study (expr. ~ age + sex). This analysis was run across ages 3 to 21 months, due to the lack of female samples for ages 26 and 28 months. If aging trajectories would be reasonably similar between sexes, then the LRT would indicate a significantly better goodness of fit for the complete model in only very few genes, if any (i.e. the interaction term improves the fit). In addition, we repeated the differential expression analysis for the age groups 3 to 21 months, for which we had data from both sexes. In addition, we repeated the differential expression analysis across age groups only in female or male samples, respectively. For the males, we further excluded samples of age groups older than 21 months. To demonstrate that expression shifts in both sexes were overall strongly associated, we extracted for each age comparison relative to 3 months (i.e. 12 vs 3 months, 15 vs 3 months, etc.) the genes passing the significance threshold of padj < 0.05 in at least one of the sexes. We plotted the respective log2FoldChanges for each sex and counted the number of genes in each of the resulting four quadrants. This was used as a 2-by-2 Fisher Matrix and tested for significant association using one-sided Fisher’s exact test. We exemplified this with the motor cortex and the comparison of 18 vs 3 months (Figure S1D). The resulting p-values (adjusted for multiple testing) were visualized on top of a heat map that is colored according to the fraction of genes following the same trend in regulation (upper right and lower left quadrant) relative to all the plotted genes (all four quadrants combined). We found consistently high, statistically significant overlaps in regulation during aging between both sexes (Figure S1E).
Differential expression for the rejuvenation interventions was performed as described for the aging cohorts, except that no additional filter of 2 pairwise comparisons was employed.
DEG Gene Ontology functional enrichment
Unless stated otherwise, we performed functional enrichment analysis for DEGs using the Biocunductor package topGO as described in detail before 8,57. Unless stated otherwise, the set of expressed genes (defined as passing the independent filtering criterion of DEseq2 33) was used as background for all functional enrichment analyses involving expression data. Top-ranked, representative Gene Ontology (GO) terms were selected and visualized using the CellPlot package. The full-length GO terms were shortened to fit into the figure format.
Bulk-seq GWAS gene enrichment and expression distribution analysis
We analyzed if DEGs of a given region would be enriched for disease-associated genes using a previously assembled list of GWAS hits for several neurodegenerative diseases 63. The analysis was focused on Alzheimer’s disease and Parkinson’s disease (both age-related forms of dementia) and multiple sclerosis as we had observed several white matter-related effects in our dataset. We refer to these as ‘disease-associated genes’. Disease-associated genes that were expressed in a given region (defined as passing the independent filtering criterion of DESeq2 33) were analyzed. To determine if disease-associated genes were enriched among the DEGs of a given region, we used a one-sided hypergeometric test with expressed genes as background. Resulting P values were corrected for multiple testing. We chose the anterior hippocampus region as representative for the hippocampus, and further excluded the entorhinal cortex (too few DEGs) and olfactory bulb. For each disease, we plotted the enrichment and the relative composition of disease-associated DEGs with respect to their regulation (i.e., up- or down-regulated) using the CellPlot package. We clustered regions for using a pairwise Jacquard Distance matrix, so that regions with overlapping diseases-associated DEGs will cluster together. Gene overlaps with a Jaccard index ≥ 0.25 were indicated with an arc.
Additionally, we performed a systematic analysis of expression shifts that could affect a given GWAS homologue’s distribution across the brain. To this end, we focused on sets of GWAS-DEGs in each region and ranked their mean expression at young (3 months) and very old (26 months) age, respectively. For instance, Irf8, one of the GWAS-DEGs associated with MS, became differentially regulated in multiple regions, including the corpus callosum (Figure S7B). However, the actual rank of the top Irf8-expressing regions (caudate putamen, SVZ and corpus callosum) stayed relatively constant. In contrast, the expression of Nlrc5, a gene from the same set, was only the 8th-highest in the corpus callosum at young age, but became the highest across all tissues with age (Figure S7B). Here, the differential regulation with age led to a significant redistribution of where the gene is predominantly expressed. Expanding the analysis to all GWAS-DEGs in a given tissue/disease set with least 15 genes to focus on (to ensure statistical power), we tested if there would be a systematic shift in rank-based expression using paired two-sided Wilcoxon rank-sum tests.
Bulk-seq correlation of gene expression with age
For each region separately, we probed the expression of each gene (using DEseq2-normalized counts) for positive or negative correlation with age using Spearman’s method and tested for significant association. P values were adjusted for multiple testing using the Benjamini-Hochberg method. Genes with Spearman’s rho ≥ 0.5 or ≤ −0.5, respectively, and padj ≤ 0.05 were called as significantly age-correlated in a given region. The total number of age-correlated genes was used to evaluate the impact of aging on a given region.
The sex-specific age-correlations in the hypothalamus were performed similarly, by subsetting the dataset to the ages 3 to 21 months (for which data of both sexes was available). Correlation with age was then calculated for each gene based on the male or female samples only. Criterion for age-correlated genes remained the same.
Weighted gene co-expression network analysis (WGCNA)
Network analysis was performed with the Weighted Gene Correlation Network Analysis (WGCNA) 82 package to identify significant modules that were associated with a specific aging group and brain region. Modules were independently detected in each brain region. For each brain region the soft-thresholding (ß value) was set based on scale-free topology (R2>0.8) to construct a correlation adjacency matrix. ß values 18, 10, 9, 8, 12, 4, 4, 5, 7, 9, 24, 14, 4 and 13 were used for the corpus callosum, cerebellum, motor cortex, entorhinal cortex, anterior hippocampus, posterior hippocampus, hypothalamus, medulla, olfactory bulb, choroid plexus, pons, SVZ, thalamus and visual cortex respectively. The ‘blockwiseModules’ function was used to construct the network. Biweight midcorrelation (‘bicor’) was used to compute the correlation between each pair of genes. Network analysis was performed with the “signed” network. The “deepSplit” argument value was 2 and a minimum cluster size was 25. (blockwiseModules parameters: datExpr=(datExpr), maxBlockSize=22000, networkType=“signed”, corType=“bicor”, power=ß, saveTOMFileBase=(file=‘TOM_signed’), minModuleSize=25, deepSplit=2, saveTOMs=TRUE). The average linkage hierarchical clustering of the topological overlap dissimilarity matrix (1-TOM) was used to generate the network dendrogram. The hybrid dynamic tree-cutting method was used to define modules. Modules were summarized by their first principal component (ME, module eigengene) and modules with eigengene correlations >0.9 were merged.
Module-aging group associations were evaluated using a linear model within each brain region. Significance values were corrected for multiple testing using Benjamini-Hochberg method. Results from module-eigengene association tests are shown in Table S1. Genes within each module were prioritized based on their module membership (kME), defined as correlation to the module eigengene. The top ‘hub’ genes for several of the modules are shown in supplementary Table S1. Cell type enrichment analyses were performed using several mouse derived cell type specific expression datasets 83–85. Enrichment was performed for cell type specific marker genes using Fisher’s exact test, followed by Benjamini-Hochberg-correction for multiple testing. The WGCNA results were assembled in summarizing figures that can be browsed through our interactive shiny app website (https://twc-stanford.shinyapps.io/spatiotemporal_brain_map/).
Estimating the variance of the data depending on metadata
To estimate the variance in the data depending on age, tissue or gender we made use of principal variance component analysis (PVCA) as implemented in the Bioconductor Package pvca and described in detail in 8. PVCA combines the strength of principal component analysis and variance components analysis (VCA). Originally it was applied to quantify batch effects in microarray data. In our case, however, we do not provide experimental batches but rather groups of meta data as input
Gene signature generation and score calculation
Gene signatures are used in this study to quantify the expression of a gene set, thus representing the aggregated expression of multiple genes in a given transcriptome (e.g. a regional bulk-seq transcriptome or a single-nuclei transcriptome). The resulting value is defined as a score. Throughout the manuscript we generated signatures and quantified scores using the VISION (v.3.0) package as detailed in the original study 25. Notably, VISION z-normalizes signature scores with random gene signatures to account for global sample-level metrics (such as total number of counts/UMIs, which can be affected by age 86). While VISION was originally intended for the analysis of signatures in single-cell data we found its analysis workflow applicable for bulk, spatial and single-cell/-nuclei datasets. We note that due to differences in baseline expression across regions or cell types as well as the z-normalization mentioned above, VISION scores can be negative. However, our analyses are focused - unless stated otherwise - on the relative score changes (i.e. increase or decrease relative to 3 months) occurring with age in a given region or cell type.
Bulk-seq marker genes and score calculation
Seurat’s FindAllMarkers function was run using the ‘DESeq2’ test with parameters and Bonferroni correction for multiple testing to identify region-specific marker genes (P value of less than 0.05; Methods S1, section 3). For each region, we constructed unsigned signatures 25 based on a given region’s significant marker genes. For each signature, we calculated scores across a publicly available spatial transcriptome dataset from 10X Genomics (https://www.10xgenomics.com/resources) and compared the patterns to structural annotations in Allen Mouse Brain Atlas.
Bulk-seq Common Aging Score (CAS) calculation and CAS velocity comparison
We ranked genes on the basis of their regulation across regions, to summarize in how many regions a given gene would be called as a DEG (i.e. reach statistical significance in at least two comparisons between samples from 3 months and any following age group). We included only the anterior hippocampus region in the selection of cross-region DEGs to prevent a potential bias towards aging effects in the hippocampus. This led to the identification of 82 genes that were marked as DEG in at least 10 out of 14 regions (15 regions minus the posterior hippocampus region). We constructed a signed gene signature25 based on 75 up- and 7 down-regulated DEGs. We used the signature to calculate CAS for each single-region transcriptome. To quantify a region’s score increase over time (aging velocity), we constructed a linear model with the design: score ~ age + region + age:region (score explained by a two factor model including interaction term) using the linear model function in R. We used the lstrends function of the lsmeans package87 that utilizes least-square means to estimate and compare the slopes of fitted lines for each region. We subsequently used Tukey’s range test across all possible region-to-region comparisons to assess which regions exhibited statistically significant (P value < 0.05) slope differences. In addition, we repeated the analysis resolved for sex-specific effects across the 3, 12, 15, 18 and 21 months groups (for which we had both male and female samples). We assessed if there was a differential aging velocity between sexes across all regions (Figure 2I), for which analyzed a linear model with the design: score ~ age + sex + age:sex + region. We further performed the same analysis iteratively for each region individually (Figure S1D) using a model with the design: score ~ age + sex + age:sex. We corrected the resulting P values for each region-wise analysis using the Benjamini-Hochberg method.
Comparing CAS velocity with STARmap single-cell composition
We obtained meta data from Shi et al. 41 via the Single Cell Portal where the authors had quantified spatial distribution of major cell types across the entire mouse brain using their previously published, imaging-based STARmap method88. This atlas contains data on 422,766 single cells of 27 major cell types quantified across 73 brain structures and several sagittal sections. We aggregated the 73 reported brain structures into 10 regions that we considered meaningful equivalents of regions profiled in our Bulk-seq study (e.g. data from CA1, CA2, CA3, dentate gyrus etc. were grouped together into a ‘hippocampus’ region). We calculated for each of the 10 regions the relative abundance of each of the 27 cell types and then correlated these with the regions’ respective CAS slopes. If, for example, the relative abundance of microglia across brain regions at young age was responsible for the observed differences in CAS slopes, then we would assume a significant correlation between those two metrics across the analyzed brain regions. We did not find any significant relationship (as tested with spearman correlation and linear regression) between relative cell abundance and CAS increase over time for any of the investigated cell types (Figure 2J, Methods S2, section 1).
Microarray analysis of microglia
We obtained annotated and pre-processed microarray data from 52 (GSE62420) using limma89, GEOquery90 and GEO’s online GEO2R tool (https://www.ncbi.nlm.nih.gov/geo/info/geo2r.html). Pairwise-differential testing between age groups of the same region was performed using empirical bayes moderation as implemented in limma with multiple testing correction.
Organ-specific aging signature identification and velocity comparison
To explore the feasibility of detecting gene expression patterns with organ-specific regulation during aging, we re-analyzed a previously published bulk RNA-seq dataset of 17 mouse tissues profiled across ten age groups (n = 4 males; aged 1, 3, 6, 9, 12, 15, 18, 21, 24 and 27 months; n = 2 females; aged 1, 3, 6, 9, 12, 15, 18 and 21 months)8. The dataset comprised the following organs: bone, brain, brown adipose tissue (BAT), gonadal adipose tissue (GAT), heart, kidney, limb muscle (muscle), liver, lung, bone marrow (marrow), mesenteric adipose tissue (MAT), pancreas, skin, small intestine (intestine), spleen, subcutaneous adipose tissue (SCAT), and white blood cells (WBC). We obtained pre-processed data as described in the original study and performed differential expression analysis accordingly8. We identified age-related DEGs in the same manner as described for the bulk-seq data: we used the raw count matrix as recommended for the DEseq2 standard analysis pipeline. Factors and dispersion estimates were calculated for each tissue separately. We conducted differential expression analysis comparing samples from 3-months-old mice to each consecutive time point, using age and sex as covariates. P values were adjusted for multiple testing, and genes with an adjusted P value of less than 0.05 were determined to be statistically significant. Finally, we required a gene to reach statistical significance (after multiple testing correction) in at least 2 pairwise comparisons (e.g. 3-months-old vs 12 months-old and 3-months-old vs 21 months-old) to be called a differentially expressed gene (DEG). We analyzed age groups that would be comparable to the age groups profiled in our study (3, 12, 15, 18, 21, 24 and 27 months). We ranked genes on the basis of their regulation across organs, to summarize in how many organs a given gene would be called as a DEG (i.e. reach statistical significance in at least two comparisons between samples from 3-months-old mice and any following age group). DEGs that were only detected in a single organ were assembled into signed, organ-specific aging signatures using VISION 25, comparable to the CAS (Methods S3, section 1). For organs that exhibited fewer than 25 unique DEGs we did not construct a signature. For each organ-specific signature, we performed the following analysis: We first tested for each organ separately, if the respective signature would show a significant correlation with age using linear models with the design: score ~ age. Organs that showed no significant (P val < 0.05, t-test) association with the age were excluded. Next, we constructed a linear model with the design: score ~ age + organ + age:organ (organ-specific score explained by a two factor model including interaction term) using the linear model function in R. We used the lstrends function of the lsmeans package87 that utilizes least-square means to estimate and compare the slopes of fitted lines for each organ. We subsequently used Tukey’s range test across all possible organ-to-organ comparisons to assess which organs exhibited statistically significant (P value < 0.05) slope differences. Notably, we asked if the organ where the signature was identified (the ‘reference’ organ) would show a significantly higher slope compared to all other organs. The summarized results are displayed in the heatmap in Methods S3, section 1.
Bulk-seq region-specific aging signature identification and velocity comparison
We ranked genes on the basis of their regulation across regions, to summarize in how many regions a given gene would be called as a DEG (i.e. reach statistical significance in at least two comparisons between samples from 3-months-old mice and any following age group). DEGs that were only detected in a single region were assembled into signed, region-specific aging signatures using VISION 25, comparable to the CAS (Methods S3, section 2). We excluded the posterior hippocampus region in the selection of region-specific DEGs. Further, there were less than 20 unique DEGs found for the entorhinal cortex, which we considered too small to construct a signature. For each region-specific signature, we performed the following analysis: We first tested for each region separately, if the respective signature would show a significant correlation with age using linear models with the design: score ~ age. Regions that showed no significant (P val < 0.05, t-test) association with age were excluded. Next, we constructed a linear model with the design: score ~ age + region + age:region (region-specific score explained by a two factor model including interaction term) using the linear model function in R. We used the lstrends function of the lsmeans package87 that utilizes least-square means to estimate and compare the slopes of fitted lines for each region. We subsequently used Tukey’s range test across all possible region-to-region comparisons to assess which regions exhibited statistically significant (P value < 0.05) slope differences. Notably, we asked if the region where the signature was identified (the ‘reference region’) would show a significantly higher slope compared to all other regions. The summarized results are displayed in the heatmap in (Methods S3, section 2).
10X Visium mapping, embedding, clustering and region identification
Space Ranger analysis pipelines were utilized to align image and FASTQ files, detect tissue and fiducial frames, count barcodes/UMIs. Throughout the manuscript we refer to the barcoded areas from a given dataset as ‘spots’. Spots with less than 5 UMIs were removed as well as all spots at the outline of the tissue as these can be affected by RNA diffusion. We integrated all six sample-wise datasets (two from 6 months, two from 18 months and two from 21 months), using Seurat’s built-in SCTransform and integration workflow81, with 2000 genes set as integration features. Integrated datasets were then used as input for spot embedding and clustering. A shared-nearest-neighbors graph was constructed using the first 30 PC dimensions before clustering spots using Seurat’s built-in FindClusters function with a resolution of 0.8 and default parameters. Umaps and tSNEs were calculated using Seurat’s built-in functions, based on the first 30 PC dimensions. Count data was subsequently normalized and scaled using SCTransform across all spots to allow for visualization of expression values and differential gene expression analysis.
We chose a data-driven approach to group spots and map them to anatomical structures of the brain (Figure S4A): Transcriptional clustering yielded 29 clusters and we used Seurat’s FindAllMarkers function (parameters: min.pct=0.1, thresh.use=0.1 assay=‘SCT’, only.pos=TRUE) to identify cluster markers. We compared the expression of marker genes to in-situ hybridization (ISH) data from the Allen Mouse Brain Atlas26 and visual landmarks from the H&E microscopy images (e.g. cornu Ammonis and dentate gyrus of the hippocampus; cell-sparse structure of the white matter fiber tracts). To enable comparisons with the regions isolated for Bulk-seq, we additionally grouped annotated clusters into meaningful region-level sets, guided by their anatomical location and the hierarchical ordering of structures in the Allen Mouse Brain Atlas. Ontology and nomenclature of clusters is indicated in Figure S4B.
10X Visium differential expression analysis and comparison with Bulk-seq data
Given comparable representation of clusters across all samples and age groups, we considered differential expression analysis across age groups feasible. We analyzed differential expression in the white matter and cortex cluster as we considered them comparable to the corpus callosum (high CAS velocity) and motor cortex (low CAS velocity) region from the bulk-seq dataset. Differential gene expression of genes comparing 10X Visium data from 6 months to 21 months was done using the ‘DESeq2’ algorithm implemented in Seurat on Spatial count data. Seurat natural log(fold change).>.0.2 (absolute value), adjusted P value (Benjamini-Hochberg correction).<.0.05, and expression in greater than 10% of spots in both comparison groups were required to consider a gene differentially expressed. To test for a potential association between gene-expression changes measured in 10X Visium and bulk-seq data, we considered only genes that changed significantly in both datasets. For both regions, we confirmed significant overlap between the DEGs found in Bulk-seq and 10X Visium dataset (Fisher’s exact test, P Value < 0.05). Next, we plotted log2 fold expression changes during aging as measured via Visium versus log2 fold expression changes on the bulk-seq level. The distribution of genes among the four resulting quadrants was tested for directionality using Fisher’s exact test.
Visium CAS calculation
Calculation of CAS and CAS velocities for Visium data was carried out in a similar manner as described above for bulk-seq data: CAS for each Visium spot were calculated and score increase over time was calculated for region clusters with equivalent Bulk-seq regions: White matter (compared to corpus callosum), cortex (compared to motor cortex), striatum (compared to caudate putamen), hippocampus, hypothalamus, choroid plexus and thalamus. To quantify a region’s score increase over time (aging velocity), we constructed a linear model with the design: score ~ age + region + age:region and carried out slope estimation and differential analysis as described above. We acknowledge that this analysis does not account for biological replicates but treats each spot belonging to the same region as replicate. We therefore visualized CAS in Visium for each replicate, to demonstrate that age-related changes in CAS supersede the intra-replicate CAS differences.
Nuc-seq mapping, embedding, clustering, sample demultiplexing and cell type identification
Cell Ranger (v.6.1.2) analysis pipelines were utilized to align reads to mm10 reference genome and count barcodes/UMIs. To account for unspliced nuclear transcripts, reads mapping to pre-mRNA were counted. Throughout the manuscript we use nuclei and ‘cells’ synonymously. Outliers with a high ratio of mitochondrial (more than 5%, fewer than 400 features) relative to endogenous RNAs and homotypic doublets (more than 6,000 features in hippocampus; more than 7,000 features in caudate putamen) were removed in Seurat81. We integrated all sample-wise datasets, using Seurat’s built-in SCTransform and integration workflow81, with 500 genes set as integration features. Integrated datasets were then used as input for cell embedding and clustering. A shared-nearest-neighbors graph was constructed using the first 12 PC dimensions before clustering spots using Seurat’s built-in FindClusters function with a resolution of 0.4 and default parameters. Umaps and tSNEs were calculated using Seurat’s built-in functions, based on the first 12 PC dimensions. A given nuc-seq sample represented nuclei from a male and female animal (of the same age) that were pooled in equal numbers during the nuclei isolation steps. To demultiplex a sample by sex, we calculated the ratio of counts belonging to female- (Xist, Tsix) and male-specific (Ddx3y, Eif2s3y, Uty, Kdm5d) genes, and identified nuclei with a log2 cutoff of 1 and −1 as female- and male-derived nuclei, respectively. Ambiguous nuclei, which had reads from both female and male nuclei, were removed from the analysis. Count data was subsequently normalized and scaled to allow for visualization of expression values and differential gene expression analysis. Seurat’s FindAllMarkers function (parameters: min.pct=0.15, thresh.use=0.15 assay=‘SCT’) was run to identify cluster markers. Clusters were annotated based on marker genes. Finally, nuclei were manually inspected using known cell type-specific marker genes and nuclei expressing more than one cell type-specific marker were defined as doublets and removed91.
Publicly available scRNA-seq data embedding
We re-analyzed two previously published single-cell RNA-seq datasets: (1) Droplet-based scRNA-seq of freshly dissected SVZ at young and old age (n = 3 males per age; aged 3 and 28 months; all C57BL/6JN strain)27; (2) Smart-seq2-based 49 scRNA-seq of freshly isolated cells from the myeloid and non-myeloid fraction of the striatum, cerebellum, hippocampus and cortex at young and old age (n = 4 males per age; aged 3 and 24 months; all C57BL/6JN strain) 19. For the SVZ data, we obtained and analyzed pre-processed count matrices. For visualization purposes, we integrated all sample-wise datasets, using Seurat’s built-in SCTransform and integration workflow81, with 2000 genes set as integration features. Integrated datasets were then used as input for cell embedding. Umaps and tSNEs were calculated using Seurat’s built-in functions, based on the first 12 PC dimensions. Cell annotations were transferred from the original study. Count data was subsequently normalized and scaled to allow for visualization of expression values. For the second dataset, we obtained and analyzed pre-processed count matrices. We followed previous analyses on the same dataset73. For visualization purposes, we integrated all sample-wise datasets, using Seurat’s built-in SCTransform workflow81. Integrated datasets were then used as input for cell embedding. Umaps and tSNEs were calculated using Seurat’s built-in functions, based on the first 12 PC dimensions. Cell annotations were transferred from the original study. Count data was subsequently normalized and scaled to allow for visualization of expression values.
Differential expression in scRNA- and Nuc-seq data
Differential gene expression of genes comparing young and old samples was done using the MAST92 algorithm, which implements a two-part hurdle model. Random effects accounting for individual brain samples were not included in the mixed model. Seurat natural log(fold change).>.0.2 (absolute value), adjusted P value (Benjamini-Hochberg correction).<.0.05, and expression in greater than 10% of cells in both comparison young and old samples. We acknowledge that due to the omission of a dedicated replication-sensitive analysis, an impact of pseudo-replication effects on differential expression results cannot be excluded.
Signature calculations in scRNA- and Nuc-seq data
Calculation of CAS and CAS velocities for scRNA- and Nuc-seq data was carried out in a similar manner as described above for 10X Visium data: CAS for each cell were calculated and CAS increase over time was calculated for cell types. To quantify a cell type’s score increase over time (aging velocity), we constructed a linear model with the design: score ~ age + cell type + age:cell type and carried out slope estimation and differential analysis as described above. We acknowledge that this analysis does not account for biological replicates but treats each cell belonging to the same cell type as replicate. To account for this, we further calculated the cell type-median CAS for each biological replicate and tested for differential CAS regulation with two-tailed t-test on per-replicate median of CAS. For the comparison of CAS increase with age across microglia from different brain regions (Figure 4A–C), we used two-sided Wilcoxon rank-sum tests to test for CAS differences between microglia from the same age group.
For region-specific signatures, we performed a per-cell type slope quantifications as detailed for the CAS and clustered the resulting slope estimates using hierarchical clustering (Figure 5H,K).
Single-nuclei dispersion score
We employed a previously published strategy to quantify if a given DEG detected in bulk data would be expressed in a specific cell type8. For each gene in each brain region, we selected cells expressing the gene (log-CPM expression > 0). Next, we assigned to the cells the log-CPM expression values of the gene as weights. Based on these, we calculated the weighted center of the cells in the single-cell landscape defined by the UMAP embeddings. We defined the ‘single-cell dispersion’ of the gene as the weighted mean distance of the cells from their weighted center. Finally, we introduced region specific factors to account for differences between brain region specific embeddings. Per region, we set pseudo log-CPM count 1 to all cells and calculated the dispersion of them. We normalized the dispersion scores by these region-specific factors.
ADDITIONAL RESOURCES
We created an web application app to provide interactive access to the processed bulk-seq and Visium data. The app can be accessed via: https://twc-stanford.shinyapps.io/spatiotemporal_brain_map/.
Supplementary Material
Methods S1 Bulk-seq method details and quality control, related to Figure 1 and STAR Methods.
Overview of collected brain regions, performance values for RNA isolation and quality control analyses confirming isolation of the correct anatomical structures.
Methods S2 Cell type- and replicate-resolved statistical analysis of CAS genes and validation in independent datasets, related to Figures 2–4 and STAR Methods
Replicate-resolved expression analysis of CAS genes and CAS scores across single-cell and bulk transcriptomes from this study and independent datasets.
Methods S3 Method details for identification and statistical analysis for region-specific expression signatures, related to Figure 5 and STAR Methods
Overview of method for identification of region-specific aging signatures and statistical analysis results.
Figure S1 Validation of differential gene expression analysis, related to Figure 1
(A) Number of differentially expressed genes in 2+ age comparisons (12 mo., 15 mo., 18 mo., 21 mo., 26 mo. and 28 mo.) relative to 3 mo. Split by region, colored by up- and down-regulation. padj ≥ 0.5 in 2+ comparisons. (B) Number of differentially expressed genes in 2+ age comparisons (12 mo., 15 mo., 18 mo., 21 mo.) relative to 3 mo. Split by region, colored by up- and down-regulation. padj ≥ 0.5 in 2+ comparisons. (C) Total number of genes exhibiting a significant interaction between sex and age split by region. padj < 0.05 LRT33. (D) Scatterplot representation of genes that are differentially regulated at 18 months of age in motor cortex of female or male samples, respectively. Plotted are genes that pass statistical testing in at least on of the sexes. The number of genes in each quadrant is indicated in blue. (E) Heatmap representing results of association analyses of DEGs in males and females. Indicated are fraction of DEGs that were co-regulated in males and females for pairwise comparisons, referenced to data at 3 months. Fractions were calculated as sum of genes following the same trend in both sexes relative to all genes passing the significance threshold in at least one sex. Results of one-sided Fisher’s exact test for association of gene regulation in both sexes are indicated as well. One-sided Fisher’s exact test, adjusted for multiple testing, *** p.<.0.001, ** p.<.0.01, * p.<.0.05.
Figure S2 A common RNA aging signature quantifies the region-specific pace and magnitude of transcriptional shifts in the brain, related to Figure 2
(A) CAS trajectories of all regions, colored by age. Red lines indicate averaged-smoothed values. (B) CAS for each sample grouped by animal. Each boxplot represents distribution of CAS across the 15 regions as measured by bulk RNA-seq. Animals are ranked by median score and colored by chronological age. Distribution of chronological age groups is indicated in the background.
Figure S3 Hypothalamus aging is accelerated in females, related to Figure 2
(A) CAS trajectories of all regions for the interval of 3 to 21 months, colored by sex. (B) Sex-specific slope of linear regressions in (A), colored by sex. Data are mean ± 95% confidence intervals. Pair-wise, two-sided Tukey’s HSD test. For each region, we calculated a separate model limited to data from the respective tissue only. *** p.<.0.001, ** p.<.0.01, * p.<.0.05. The highest (least significant) Pval is indicated. (C) Scatterplot representation of genes that significantly correlate with age (Spearman’s rho ≥ 0.5) in males, females or both. The number of overlapping DEGs in each quadrant is indicated in blue. (D) Representative GO analysis of genes with significant age-correlation in males or females. Lengths of bars represent negative ln-transformed Padj using two-sided Fisher’s exact test. Colors indicate gene-wise age-correlation coefficient. (E) Bulk expression of genes with age-correlation in males, split by male and female samples. Lines indicate linear fit of gene expression resolved by sex. Data are mean ± s.e.m. (F) same as (E) for genes with significant age-correlation in females. Expression in hypothalamus, cerebellum and corpus callosum are shown to demonstrate the specificity of sexual dimorphisms in the hypothalamus.
Figue S4 Robust capture of spatial transcriptomes across age, related to Figure 3
(A) 10X Visium processing and analysis overview. Whole brains were frozen prior to OCT embedding and cryo-sectioning. Coronal sections were placed on a 10X Visium Spatial Gene Expression slide, followed by H&E staining and spatial reverse-transcription reaction. Single-spot transcriptomes were integrated, clustered with default settings and visualized as UMAP. Clustered spatial spot transcriptomes were mapped to their original location. To annotate the clusters, their marker genes (Table S1) were visualized, compared to the Allen Brain Atlas26. (B) Complete data description and abbreviations of ontology and nomenclature for spatial transcriptome data. Regional-level annotated manually, and cluster-level determined by Seurat clustering. (C,D) Representative spatial transcriptome data (6 months replicate #2), colored by cluster-level annotation and represented as (C) UMAP and (D) spatial transcriptome. (E,F) Representative spatial transcriptome data (6 months replicate #2), colored by region-level annotation and represented as (E) UMAP and (F) spatial transcriptome. (G,H,I) Cluster-level annotation across replicates and datasets represented as (G) UMAP and (H) spatial transcriptome. (I) Fraction of spots corresponding to each cluster. (J,K,L) Region-level annotation across replicates and datasets represented as (J) UMAP and (K) spatial transcriptome. (L) Fraction of spots corresponding to each region.
Figure S5 Mapping the shared and distinct expression patterns of dietary restriction and young mouse plasma injection, related to Figure 6
(A) Body weight change for AL and aDR-fed animals (n = 9). Each animal’s individual trajectory is depicted with gray points. Per-group averages are indicated by trajectories colored in black. Start of the complete 25% aDR phase is indicated. (B) Body weights for each animal at the beginning and end of the aDR phase. P values calculated with two-tailed, paired t-test. Bonferroni correction; padj.<.0.05. *** p.<.0.001, ** p.<.0.01, * p.<.0.05. (C) PCA of Bulk-seq data of liver tissue from the aDR and AL-fed mice. (D) Bar graph indicating the number of detected DEGs in liver tissue of aDR-fed mice. (E) Venn diagrams depicting the overlap of DEGs in liver of C57BL/6JN and chronically DR-fed B6D2F1 mice (data from 57). (F) Injection paradigm for YMP-treated animals. Injections were conducted retro-orbitally every 3–4 days. Injections were alternated between left and right eye. (G) UMAP representation of co-integrated brain region transcriptomes from rejuvenation cohorts and aging time course (n = 131 total samples from DR/AL fed mice; n = 98 samples from YMP/PBS mice; n = 847 samples from aging cohort; from Figure 1), based on the first 40 principal components. Cells are colored by experiment (left) or region (right). (H,I), Barplots showing the proportion of genes that are differentially expressed age which are reversed by (H) aDR or (I) YMP-injection. Region, and whether the genes increase or decrease with age. (J) Gfap and Cst7 expression in caudate putamen. Differential expression relative to control group is indicated. Data are mean ± s.e.m. Two-sided Wald test, adjusted for multiple testing. *** p.<.0.001, ** p.<.0.01, * p.<.0.05. (K) Boxplot representation of CAS across all regions. P values calculated with two-tailed t-test on per-replicate median of score. *** p.<.0.001, ** p.<.0.01, * p.<.0.05. (L) same as (J) for YMP-treated mice. (M) same as (K) for YMP experiments.
Figure S6 Acute DR causes brain-wide expression shifts in glia, related to Figure 6
(A) CAS shifts in response to aDR across all regions. P values calculated with two-tailed t-test. Bonferroni correction; padj.<.0.05. *** p.<.0.001, ** p.<.0.01, * p.<.0.05. (B) same as (A) for YMP experiments. (C,D) Expression of (C) Adipor2 and (D) Hif3a mapped onto aDR hippocampus Nuc-seq data. Quantification and statistical analysis is plotted on the right as violin plots. Points indicate nuclei-wise expression levels, and the violin indicates average distribution of expression split by age. (MAST, Benjamini–Hochberg correction; false discovery rate (FDR).<.0.05 and logFC.>.0.2 to be significant). *** p.<.0.001, ** p.<.0.01, * p.<.0.05. (E) Boxplot representation of common aDR score across all cell types. P values calculated with two-tailed t-test on per-replicate median of score. *** p.<.0.001, ** p.<.0.01, * p.<.0.05.
Figure S7 Expression of disease variant homologues across the brain is shifted as a result of age-related expression changes, related to Figure 7
(A) Trem2 expression across all bulk regions, colored by age. Red lines indicate averaged-smoothed gene expression. Data are mean ± s.e.m. (B) Analysis outline to probe brain-wide expression of GWAS genes at young and old age. Enrichment analysis of region-resolved DEGs for human GWAS variants for multiple sclerosis from Figure 7 is depicted. The expression of corpus callosum DEGs associated with MS GWAS hits were ranked across brain regions at young (3 months) and old (26 months) of age. Expression of two exemplary genes, Irf8 (top) and Nlrc5 (bottom) across bulk regions at 3 mo. and 26 mo is depicted. The rank of mean expression for selected regions is highlighted. (C) Box plots displaying ranked expression changes from 3 mo. to 26 mo. of up-regulated GWAS-DEGS for multiple sclerosis across regions, filtered for regions with at least 15 genes to focus on trends with specific statistical power. Irf8 (green) and Nlrc5 (teal) rank changes in the corpus callosum are highlighted. Significance determined by paired, pairwise Wilcoxon test between regions. *** padj.<.0.001, ** padj.<.0.01, * padj.<.0.05. (D) Box plot displaying ranked expression changes from 3 mo. to 26 mo. of up-regulated GWAS-DEGS for Alzheimer’s in the corpus callosum. Ms4a6d (teal) highlighted. Significance determined by paired, pairwise Wilcoxon test between regions. *** padj.<.0.001, ** padj.<.0.01, * padj.<.0.05. (E) Ms4a6d expression across bulk regions at 3 mo. and 26 mo. The rank of expression for selected regions is highlighted.
Table S1. Complete gene lists for signatures and functional enrichment analysis, related to all figures
Each tab contains complete gene lists and annotations for gene signatures created in this study, and GO results lists for functional enrichment analysis.
Highlights for CELL-D-22–02981 “Atlas of the aging mouse brain reveals white matter as vulnerable foci“.
Brain-wide gene signature of aging in glial cells, with spatially defined changes
Glial cell aging is accelerated in white matter
Rejuvenation interventions have region-specific effects on gene expression
Genes implicated in neurodegenerative diseases show regional aging patterns
Acknowledgments:
We thank all members of the Wyss-Coray laboratory for feedback and support, and H. Zhang, D. Berdnik, K. Dickey and D. Channappa for laboratory management. We further thank G. Wang for microscopy assistance in acquiring images for Visium. We thank S. Buschbaum, L. Drews, J. Fröhlich, O. Hendrich, R. Hoppe and A. Pahl for preparing the murine tissues from the C3B6F1 cohort. We thank A. Bastian for help with Visium experiments. The research leading to these results received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007–2013) / ERC grant agreement n° 268739 (L.P.), the Max Planck Society (L.P.), the Schaller-Nikolich Foundation (A.Ke.), the Wu Tsai Neurosciences Institute and Fondation Bertarelli (T.W.-C.), the Simons Foundation (T.W.-C.), the Cure Alzheimer’s Fund (T.W.-C.), a National Institute of Aging grant R01-AG072255 (T.W.-C.), the Milky Way Research Foundation (T.W.-C.), the American Heart Association-Allen Initiative in Brain Health and Cognitive Impairment (T.W.-C.), The Phil and Penny Knight Initiative for Brain Resilience (T.W.-C.), Michael J. Fox Foundation for Parkinson’s Research grants 125491594 (A.Ke and F.K.) and MJFF-021418 (T.W.-C., A.Ke. and F.K.).
Footnotes
Declaration of interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kalache A, and Gatti A (2003). Active ageing: a policy framework. Adv. Gerontol. 11, 7–18. [PubMed] [Google Scholar]
- 2.Partridge L, Deelen J, and Slagboom PE (2018). Facing up to the global challenges of ageing. Nature 561, 45–56. [DOI] [PubMed] [Google Scholar]
- 3.López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, and Bohr VA (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. [DOI] [PubMed] [Google Scholar]
- 5.Niccoli T, Partridge L, and Isaacs AM (2017). Ageing as a risk factor for ALS/FTD. Hum. Mol. Genet. 26, R105–R113. [DOI] [PubMed] [Google Scholar]
- 6.Pomponio R, Erus G, Habes M, Doshi J, Srinivasan D, Mamourian E, Bashyam V, Nasrallah IM, Satterthwaite TD, Fan Y, et al. (2020). Harmonization of large MRI datasets for the analysis of brain imaging patterns throughout the lifespan. Neuroimage 208, 116450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng X, Guo J, Sigmon HC, Sloan RP, Brickman AM, Provenzano FA, Small SA, and Alzheimer’s Disease Neuroimaging Initiative (2020). Brain regions vulnerable and resistant to aging without Alzheimer’s disease. PLoS One 15, e0234255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaum N, Lehallier B, Hahn O, Pálovics R, Hosseinzadeh S, Lee SE, Sit R, Lee DP, Losada PM, Zardeneta ME, et al. (2020). Ageing hallmarks exhibit organ-specific temporal signatures. Nature 583, 596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buckley MT, Sun E, George BM, Liu L, Schaum N, Xu L, Reyes JM, Goodell MA, Weissman IL, Wyss-Coray T, et al. (2022). Cell type-specific aging clocks to quantify aging and rejuvenation in regenerative regions of the brain. bioRxiv, 2022.01.10.475747. 10.1101/2022.01.10.475747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, Colantuoni EA, Elkahloun AG, Herman MM, Weinberger DR, et al. (2011). Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478, 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ham S, and Lee S-JV (2020). Advances in transcriptome analysis of human brain aging. Exp. Mol. Med. 52, 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AMM, Pletikos M, Meyer KA, Sedmak G, et al. (2011). Spatio-temporal transcriptome of the human brain. Nature 478, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soreq L, UK Brain Expression Consortium, North American Brain Expression Consortium, Rose J., Soreq E., Hardy J., Trabzuni D., Cookson MR., Smith C., Ryten M., et al. (2017). Major Shifts in Glial Regional Identity Are a Transcriptional Hallmark of Human Brain Aging. Cell Rep. 18, 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trabzuni D, Ryten M, Walker R, Smith C, Imran S, Ramasamy A, Weale ME, and Hardy J (2011). Quality control parameters on a large dataset of regionally dissected human control brains for whole genome expression studies. J. Neurochem. 119, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, et al. (2015). Human genomics. The human transcriptome across tissues and individuals. Science 348, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee CK, Weindruch R, and Prolla TA (2000). Gene-expression profile of the ageing brain in mice. Nat. Genet. 25, 294–297. [DOI] [PubMed] [Google Scholar]
- 17.Zahn JM, Poosala S, Owen AB, Ingram DK, Lustig A, Carter A, Weeraratna AT, Taub DD, Gorospe M, Mazan-Mamczarz K, et al. (2007). AGEMAP: a gene expression database for aging in mice. PLoS Genet. 3, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hargis KE, and Blalock EM (2017). Transcriptional signatures of brain aging and Alzheimer’s disease: What are our rodent models telling us? Behav. Brain Res. 322, 311–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tabula Muris Consortium (2020). A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bieri G, Schroer AB, and Villeda SA (2023). Blood-to-brain communication in aging and rejuvenation. Nat. Neurosci. 26, 379–393. [DOI] [PubMed] [Google Scholar]
- 21.Mueller O RNA Integrity Number (RIN) – Standardization of RNA Quality Control. [Google Scholar]
- 22.https://www.10xgenomics.com/resources/datasets/mouse-brain-serial-section-1-sagittal-anterior-1-standard-1-0-0.
- 23.https://www.10xgenomics.com/resources/datasets/mouse-brain-serial-section-1-sagittal-posterior-1-standard-1-0-0.
- 24.https://www.10xgenomics.com/resources/datasets/mouse-brain-section-coronal-1-standard-1-0-0.
- 25.DeTomaso D, Jones MG, Subramaniam M, Ashuach T, Ye CJ, and Yosef N (2019). Functional interpretation of single cell similarity maps. Nat. Commun. 10, 4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- 27.Dulken BW, Buckley MT, Navarro Negredo P, Saligrama N, Cayrol R, Leeman DS, George BM, Boutet SC, Hebestreit K, Pluvinage JV, et al. (2019). Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature 571, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, et al. (2016). Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hahn O, Fehlmann T, Zhang H, Munson CN, Vest RT, Borcherding A, Liu S, Villarosa C, Drmanac S, Drmanac R, et al. (2020). CoolMPS for robust sequencing of single-nuclear RNAs captured by droplet-based method. Nucleic Acids Res. 10.1093/nar/gkaa1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, et al. (2020). Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 26, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ximerakis M, Lipnick SL, Innes BT, Simmons SK, Adiconis X, Dionne D, Mayweather BA, Nguyen L, Niziolek Z, Ozek C, et al. (2019). Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 22, 1696–1708. [DOI] [PubMed] [Google Scholar]
- 32.Allen WE, Blosser TR, Sullivan ZA, Dulac C, and Zhuang X (2022). Molecular and spatial signatures of mouse brain aging at single-cell resolution. Cell. 10.1016/j.cell.2022.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lähnemann D, Köster J, Szczurek E, McCarthy DJ, Hicks SC, Robinson MD, Vallejos CA, Campbell KR, Beerenwinkel N, Mahfouz A, et al. (2020). Eleven grand challenges in single-cell data science. Genome Biol. 21, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berchtold NC, Cribbs DH, Coleman PD, Rogers J, Head E, Kim R, Beach T, Miller C, Troncoso J, Trojanowski JQ, et al. (2008). Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc. Natl. Acad. Sci. U. S. A. 105, 15605–15610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Hoesen GW, Hyman BT, and Damasio AR (1991). Entorhinal cortex pathology in Alzheimer’s disease. Hippocampus 1, 1–8. [DOI] [PubMed] [Google Scholar]
- 37.Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen W-T, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, Qian X, Laláková J, Kühnemund M, Voytyuk I, et al. (2020). Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 182, 976–991.e19. [DOI] [PubMed] [Google Scholar]
- 39.Pluvinage JV, Haney MS, Smith BAH, Sun J, Iram T, Bonanno L, Li L, Lee DP, Morgens DW, Yang AC, et al. (2019). CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 568, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yeh FL, Hansen DV, and Sheng M (2017). TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med. 23, 512–533. [DOI] [PubMed] [Google Scholar]
- 41.Shi H, He Y, Zhou Y, Huang J, Wang B, Tang Z, Tan P, Wu M, Lin Z, Ren J, et al. (2022). Spatial Atlas of the Mouse Central Nervous System at Molecular Resolution. bioRxiv, 2022.06.20.496914. 10.1101/2022.06.20.496914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu X, Bührer C, and Wellmann S (2016). Cold-inducible proteins CIRP and RBM3, a unique couple with activities far beyond the cold. Cell. Mol. Life Sci. 73, 3839–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palmer DC, Guittard GC, Franco Z, Crompton JG, Eil RL, Patel SJ, Ji Y, Van Panhuys N, Klebanoff CA, Sukumar M, et al. (2015). Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J. Exp. Med. 212, 2095–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, Stannard K, Zhang J-G, Teh C, Firth M, et al. (2016). CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 17, 816–824. [DOI] [PubMed] [Google Scholar]
- 45.Yuan Y, Chen Y-PP, Boyd-Kirkup J, Khaitovich P, and Somel M (2012). Accelerated aging-related transcriptome changes in the female prefrontal cortex. Aging Cell 11, 894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schechter JE, Felicio LS, Nelson JF, and Finch CE (1981). Pituitary tumorigenesis in aging female C57BL/6J mice: a light and electron microscopic study. Anat. Rec. 199, 423–432. [DOI] [PubMed] [Google Scholar]
- 47.Corrada MM, Brookmeyer R, Berlau D, Paganini-Hill A, and Kawas CH (2008). Prevalence of dementia after age 90: results from the 90+ study. Neurology 71, 337–343. [DOI] [PubMed] [Google Scholar]
- 48.Mozhui K, Lu L, Armstrong WE, and Williams RW (2012). Sex-specific modulation of gene expression networks in murine hypothalamus. Front. Neurosci. 6, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, and Sandberg R (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098. [DOI] [PubMed] [Google Scholar]
- 50.Wang X, He Y, Zhang Q, Ren X, and Zhang Z (2021). Direct Comparative Analyses of 10X Genomics Chromium and Smart-seq2. Genomics Proteomics Bioinformatics 19, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Safaiyan S, Besson-Girard S, Kaya T, Cantuti-Castelvetri L, Liu L, Ji H, Schifferer M, Gouna G, Usifo F, Kannaiyan N, et al. (2021). White matter aging drives microglial diversity. Neuron 109, 1100–1117.e10. [DOI] [PubMed] [Google Scholar]
- 52.Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, and McColl BW (2016). Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 19, 504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamagishi S, Hampel F, Hata K, Del Toro D, Schwark M, Kvachnina E, Bastmeyer M, Yamashita T, Tarabykin V, Klein R, et al. (2011). FLRT2 and FLRT3 act as repulsive guidance cues for Unc5-positive neurons. EMBO J. 30, 2920–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Consalez GG, Goldowitz D, Casoni F, and Hawkes R (2020). Origins, Development, and Compartmentation of the Granule Cells of the Cerebellum. Front. Neural Circuits 14, 611841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brouillet E, Jenkins BG, Hyman BT, Ferrante RJ, Kowall NW, Srivastava R, Roy DS, Rosen BR, and Beal MF (1993). Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J. Neurochem. 60, 356–359. [DOI] [PubMed] [Google Scholar]
- 56.Patki G, Che Y, and Lau Y-S (2009). Mitochondrial dysfunction in the striatum of aged chronic mouse model of Parkinson’s disease. Front. Aging Neurosci. 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hahn O, Drews LF, Nguyen A, Tatsuta T, Gkioni L, Hendrich O, Zhang Q, Langer T, Pletcher S, Wakelam MJO, et al. (2019). A nutritional memory effect counteracts benefits of dietary restriction in old mice. Nat Metab 1, 1059–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wahl D, Solon-Biet SM, Wang Q-P, Wali JA, Pulpitel T, Clark X, Raubenheimer D, Senior AM, Sinclair DA, Cooney GJ, et al. (2018). Comparing the Effects of Low-Protein and High-Carbohydrate Diets and Caloric Restriction on Brain Aging in Mice. Cell Rep. 25, 2234–2243.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, Smith LK, Bieri G, Lin K, Berdnik D, et al. (2014). Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 20, 659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deota S, Lin T, Chaix A, Williams A, Le H, Calligaro H, Ramasamy R, Huang L, and Panda S (2023). Diurnal transcriptome landscape of a multi-tissue response to time-restricted feeding in mammals. Cell Metab. 35, 150–165.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaeser SA, Lehallier B, Thinggaard M, Häsler LM, Apel A, Bergmann C, Berdnik D, Jeune B, Christensen K, Grönke S, et al. (2021). A neuronal blood marker is associated with mortality in old age. Nature Aging 1, 218–225. [DOI] [PubMed] [Google Scholar]
- 62.Yang AC, Kern F, Losada PM, Agam MR, Maat CA, Schmartz GP, Fehlmann T, Stein JA, Schaum N, Lee DP, et al. (2021). Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 595, 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang AC, Vest RT, Kern F, Lee DP, Agam M, Maat CA, Losada PM, Chen MB, Schaum N, Khoury N, et al. (2022). A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 603, 885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reiman EM, Webster JA, Myers AJ, Hardy J, Dunckley T, Zismann VL, Joshipura KD, Pearson JV, Hu-Lince D, Huentelman MJ, et al. (2007). GAB2 alleles modify Alzheimer’s risk in APOE epsilon4 carriers. Neuron 54, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nagpal L, Kornberg MD, and Snyder SH (2022). Inositol hexakisphosphate kinase-2 non-catalytically regulates mitophagy by attenuating PINK1 signaling. Proc. Natl. Acad. Sci. U. S. A. 119, e2121946119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kihara Y, Zhu Y, Jonnalagadda D, Romanow W, Palmer C, Siddoway B, Rivera R, Dutta R, Trapp BD, and Chun J (2022). Single-Nucleus RNA-seq of Normal-Appearing Brain Regions in Relapsing-Remitting vs. Secondary Progressive Multiple Sclerosis: Implications for the Efficacy of Fingolimod. Front. Cell. Neurosci. 16, 918041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.BRAIN Initiative Cell Census Network (BICCN) (2021). A multimodal cell census and atlas of the mammalian primary motor cortex. Nature 598, 86–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boisvert MM, Erikson GA, Shokhirev MN, and Allen NJ (2018). The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep. 22, 269–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, and Barres BA (2018). Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. U. S. A. 115, E1896–E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu H, Yang Y, Xia Y, Zhu W, Leak RK, Wei Z, Wang J, and Hu X (2017). Aging of cerebral white matter. Ageing Res. Rev. 34, 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dziedzic A, Miller E, Saluk-Bijak J, and Bijak M (2020). The GPR17 Receptor-A Promising Goal for Therapy and a Potential Marker of the Neurodegenerative Process in Multiple Sclerosis. Int. J. Mol. Sci. 21. 10.3390/ijms21051852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rivera AD, Pieropan F, Chacon-De-La-Rocha I, Lecca D, Abbracchio MP, Azim K, and Butt AM (2021). Functional genomic analyses highlight a shift in Gpr17-regulated cellular processes in oligodendrocyte progenitor cells and underlying myelin dysregulation in the aged mouse cerebrum. Aging Cell 20, e13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Iram T, Kern F, Kaur A, Myneni S, Morningstar AR, Shin H, Garcia MA, Yerra L, Palovics R, Yang AC, et al. (2022). Young CSF restores oligodendrogenesis and memory in aged mice via Fgf17. Nature 605, 509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Acosta-Rodríguez VA, de Groot MHM, Rijo-Ferreira F, Green CB, and Takahashi JS (2017). Mice under Caloric Restriction Self-Impose a Temporal Restriction of Food Intake as Revealed by an Automated Feeder System. Cell Metab. 26, 267–277.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Di Francesco A, Di Germanio C, Bernier M, and de Cabo R (2018). A time to fast. Science 362, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Acosta-Rodríguez V, Rijo-Ferreira F, Izumo M, Xu P, Wight-Carter M, Green CB, and Takahashi JS (2022). Circadian alignment of early onset caloric restriction promotes longevity in male C57BL/6J mice. Science 376, 1192–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang AC, Stevens MY, Chen MB, Lee DP, Stähli D, Gate D, Contrepois K, Chen W, Iram T, Zhang L, et al. (2020). Physiological blood–brain transport is impaired with age by a shift in transcytosis. Nature 583, 425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, et al. (2011). The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.De Miguel Z, Khoury N, Betley MJ, Lehallier B, Willoughby D, Olsson N, Yang AC, Hahn O, Lu N, Vest RT, et al. (2021). Exercise plasma boosts memory and dampens brain inflammation via clusterin. Nature 600, 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wager-Miller J, Murphy Green M, Shafique H, and Mackie K (2020). Collection of Frozen Rodent Brain Regions for Downstream Analyses. J. Vis. Exp. 10.3791/60474. [DOI] [PubMed] [Google Scholar]
- 81.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ferreira M, Francisco S, Soares AR, Nobre A, Pinheiro M, Reis A, Neto S, Rodrigues AJ, Sousa N, Moura G, et al. (2021). Integration of segmented regression analysis with weighted gene correlation network analysis identifies genes whose expression is remodeled throughout physiological aging in mouse tissues. Aging 13, 18150–18190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goldmann T, Wieghofer P, Jordão MJC, Prutek F, Hagemeyer N, Frenzel K, Amann L, Staszewski O, Kierdorf K, Krueger M, et al. (2016). Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 17, 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, Marques S, Munguba H, He L, Betsholtz C, et al. (2015). Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- 85.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MSB, Li G, et al. (2016). Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pálovics R, Keller A, Schaum N, Tan W, Fehlmann T, Borja M, Kern F, Bonanno L, Calcuttawala K, Webber J, et al. (2022). Molecular hallmarks of heterochronic parabiosis at single-cell resolution. Nature 603, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lenth RV (2016). Least-Squares Means: The R Package lsmeans. J. Stat. Softw. 69, 1–33. [Google Scholar]
- 88.Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, Evans K, Liu C, Ramakrishnan C, Liu J, et al. (2018). Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361. 10.1126/science.aat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Davis S, and Meltzer PS (2007). GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 23, 1846–1847. [DOI] [PubMed] [Google Scholar]
- 91.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods S1 Bulk-seq method details and quality control, related to Figure 1 and STAR Methods.
Overview of collected brain regions, performance values for RNA isolation and quality control analyses confirming isolation of the correct anatomical structures.
Methods S2 Cell type- and replicate-resolved statistical analysis of CAS genes and validation in independent datasets, related to Figures 2–4 and STAR Methods
Replicate-resolved expression analysis of CAS genes and CAS scores across single-cell and bulk transcriptomes from this study and independent datasets.
Methods S3 Method details for identification and statistical analysis for region-specific expression signatures, related to Figure 5 and STAR Methods
Overview of method for identification of region-specific aging signatures and statistical analysis results.
Figure S1 Validation of differential gene expression analysis, related to Figure 1
(A) Number of differentially expressed genes in 2+ age comparisons (12 mo., 15 mo., 18 mo., 21 mo., 26 mo. and 28 mo.) relative to 3 mo. Split by region, colored by up- and down-regulation. padj ≥ 0.5 in 2+ comparisons. (B) Number of differentially expressed genes in 2+ age comparisons (12 mo., 15 mo., 18 mo., 21 mo.) relative to 3 mo. Split by region, colored by up- and down-regulation. padj ≥ 0.5 in 2+ comparisons. (C) Total number of genes exhibiting a significant interaction between sex and age split by region. padj < 0.05 LRT33. (D) Scatterplot representation of genes that are differentially regulated at 18 months of age in motor cortex of female or male samples, respectively. Plotted are genes that pass statistical testing in at least on of the sexes. The number of genes in each quadrant is indicated in blue. (E) Heatmap representing results of association analyses of DEGs in males and females. Indicated are fraction of DEGs that were co-regulated in males and females for pairwise comparisons, referenced to data at 3 months. Fractions were calculated as sum of genes following the same trend in both sexes relative to all genes passing the significance threshold in at least one sex. Results of one-sided Fisher’s exact test for association of gene regulation in both sexes are indicated as well. One-sided Fisher’s exact test, adjusted for multiple testing, *** p.<.0.001, ** p.<.0.01, * p.<.0.05.
Figure S2 A common RNA aging signature quantifies the region-specific pace and magnitude of transcriptional shifts in the brain, related to Figure 2
(A) CAS trajectories of all regions, colored by age. Red lines indicate averaged-smoothed values. (B) CAS for each sample grouped by animal. Each boxplot represents distribution of CAS across the 15 regions as measured by bulk RNA-seq. Animals are ranked by median score and colored by chronological age. Distribution of chronological age groups is indicated in the background.
Figure S3 Hypothalamus aging is accelerated in females, related to Figure 2
(A) CAS trajectories of all regions for the interval of 3 to 21 months, colored by sex. (B) Sex-specific slope of linear regressions in (A), colored by sex. Data are mean ± 95% confidence intervals. Pair-wise, two-sided Tukey’s HSD test. For each region, we calculated a separate model limited to data from the respective tissue only. *** p.<.0.001, ** p.<.0.01, * p.<.0.05. The highest (least significant) Pval is indicated. (C) Scatterplot representation of genes that significantly correlate with age (Spearman’s rho ≥ 0.5) in males, females or both. The number of overlapping DEGs in each quadrant is indicated in blue. (D) Representative GO analysis of genes with significant age-correlation in males or females. Lengths of bars represent negative ln-transformed Padj using two-sided Fisher’s exact test. Colors indicate gene-wise age-correlation coefficient. (E) Bulk expression of genes with age-correlation in males, split by male and female samples. Lines indicate linear fit of gene expression resolved by sex. Data are mean ± s.e.m. (F) same as (E) for genes with significant age-correlation in females. Expression in hypothalamus, cerebellum and corpus callosum are shown to demonstrate the specificity of sexual dimorphisms in the hypothalamus.
Figue S4 Robust capture of spatial transcriptomes across age, related to Figure 3
(A) 10X Visium processing and analysis overview. Whole brains were frozen prior to OCT embedding and cryo-sectioning. Coronal sections were placed on a 10X Visium Spatial Gene Expression slide, followed by H&E staining and spatial reverse-transcription reaction. Single-spot transcriptomes were integrated, clustered with default settings and visualized as UMAP. Clustered spatial spot transcriptomes were mapped to their original location. To annotate the clusters, their marker genes (Table S1) were visualized, compared to the Allen Brain Atlas26. (B) Complete data description and abbreviations of ontology and nomenclature for spatial transcriptome data. Regional-level annotated manually, and cluster-level determined by Seurat clustering. (C,D) Representative spatial transcriptome data (6 months replicate #2), colored by cluster-level annotation and represented as (C) UMAP and (D) spatial transcriptome. (E,F) Representative spatial transcriptome data (6 months replicate #2), colored by region-level annotation and represented as (E) UMAP and (F) spatial transcriptome. (G,H,I) Cluster-level annotation across replicates and datasets represented as (G) UMAP and (H) spatial transcriptome. (I) Fraction of spots corresponding to each cluster. (J,K,L) Region-level annotation across replicates and datasets represented as (J) UMAP and (K) spatial transcriptome. (L) Fraction of spots corresponding to each region.
Figure S5 Mapping the shared and distinct expression patterns of dietary restriction and young mouse plasma injection, related to Figure 6
(A) Body weight change for AL and aDR-fed animals (n = 9). Each animal’s individual trajectory is depicted with gray points. Per-group averages are indicated by trajectories colored in black. Start of the complete 25% aDR phase is indicated. (B) Body weights for each animal at the beginning and end of the aDR phase. P values calculated with two-tailed, paired t-test. Bonferroni correction; padj.<.0.05. *** p.<.0.001, ** p.<.0.01, * p.<.0.05. (C) PCA of Bulk-seq data of liver tissue from the aDR and AL-fed mice. (D) Bar graph indicating the number of detected DEGs in liver tissue of aDR-fed mice. (E) Venn diagrams depicting the overlap of DEGs in liver of C57BL/6JN and chronically DR-fed B6D2F1 mice (data from 57). (F) Injection paradigm for YMP-treated animals. Injections were conducted retro-orbitally every 3–4 days. Injections were alternated between left and right eye. (G) UMAP representation of co-integrated brain region transcriptomes from rejuvenation cohorts and aging time course (n = 131 total samples from DR/AL fed mice; n = 98 samples from YMP/PBS mice; n = 847 samples from aging cohort; from Figure 1), based on the first 40 principal components. Cells are colored by experiment (left) or region (right). (H,I), Barplots showing the proportion of genes that are differentially expressed age which are reversed by (H) aDR or (I) YMP-injection. Region, and whether the genes increase or decrease with age. (J) Gfap and Cst7 expression in caudate putamen. Differential expression relative to control group is indicated. Data are mean ± s.e.m. Two-sided Wald test, adjusted for multiple testing. *** p.<.0.001, ** p.<.0.01, * p.<.0.05. (K) Boxplot representation of CAS across all regions. P values calculated with two-tailed t-test on per-replicate median of score. *** p.<.0.001, ** p.<.0.01, * p.<.0.05. (L) same as (J) for YMP-treated mice. (M) same as (K) for YMP experiments.
Figure S6 Acute DR causes brain-wide expression shifts in glia, related to Figure 6
(A) CAS shifts in response to aDR across all regions. P values calculated with two-tailed t-test. Bonferroni correction; padj.<.0.05. *** p.<.0.001, ** p.<.0.01, * p.<.0.05. (B) same as (A) for YMP experiments. (C,D) Expression of (C) Adipor2 and (D) Hif3a mapped onto aDR hippocampus Nuc-seq data. Quantification and statistical analysis is plotted on the right as violin plots. Points indicate nuclei-wise expression levels, and the violin indicates average distribution of expression split by age. (MAST, Benjamini–Hochberg correction; false discovery rate (FDR).<.0.05 and logFC.>.0.2 to be significant). *** p.<.0.001, ** p.<.0.01, * p.<.0.05. (E) Boxplot representation of common aDR score across all cell types. P values calculated with two-tailed t-test on per-replicate median of score. *** p.<.0.001, ** p.<.0.01, * p.<.0.05.
Figure S7 Expression of disease variant homologues across the brain is shifted as a result of age-related expression changes, related to Figure 7
(A) Trem2 expression across all bulk regions, colored by age. Red lines indicate averaged-smoothed gene expression. Data are mean ± s.e.m. (B) Analysis outline to probe brain-wide expression of GWAS genes at young and old age. Enrichment analysis of region-resolved DEGs for human GWAS variants for multiple sclerosis from Figure 7 is depicted. The expression of corpus callosum DEGs associated with MS GWAS hits were ranked across brain regions at young (3 months) and old (26 months) of age. Expression of two exemplary genes, Irf8 (top) and Nlrc5 (bottom) across bulk regions at 3 mo. and 26 mo is depicted. The rank of mean expression for selected regions is highlighted. (C) Box plots displaying ranked expression changes from 3 mo. to 26 mo. of up-regulated GWAS-DEGS for multiple sclerosis across regions, filtered for regions with at least 15 genes to focus on trends with specific statistical power. Irf8 (green) and Nlrc5 (teal) rank changes in the corpus callosum are highlighted. Significance determined by paired, pairwise Wilcoxon test between regions. *** padj.<.0.001, ** padj.<.0.01, * padj.<.0.05. (D) Box plot displaying ranked expression changes from 3 mo. to 26 mo. of up-regulated GWAS-DEGS for Alzheimer’s in the corpus callosum. Ms4a6d (teal) highlighted. Significance determined by paired, pairwise Wilcoxon test between regions. *** padj.<.0.001, ** padj.<.0.01, * padj.<.0.05. (E) Ms4a6d expression across bulk regions at 3 mo. and 26 mo. The rank of expression for selected regions is highlighted.
Table S1. Complete gene lists for signatures and functional enrichment analysis, related to all figures
Each tab contains complete gene lists and annotations for gene signatures created in this study, and GO results lists for functional enrichment analysis.
Data Availability Statement
The sequencing data have been deposited at Gene Expression Omnibus repository and are publicly available as of the date of publication. Publicly available datasets were obtained from the following repositories: BioProject, Gene Expression Omnibus and 10X Genomics’ public resources. DOIs are listed in the key resources table.
All original code has been deposited at https://github.com/OliInTheValley/SpatioTemporal_Analysis and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2,2,2-Tribromoethanol (Avertin) | Sigma Aldrich | Cat# T48402-100G |
| UltraPure™ 0.5M EDTA, pH 8.0 | Thermo Fisher Scientific | Cat# 15575020 |
| PBS, pH 7.4 | Thermo Fisher Scientific | Cat# 10010049 |
| Slide-A-Lyzer™ Dialysis Cassettes, 10K MWCO, 3 mL | Thermo Fisher Scientific | Cat# 66380 |
| VWR brand, razor blades, 0.009” | VWR | Cat# 55411-050 |
| Miltex® Disposable Biopsy Punches with Plunger System 1.5mm | AliMed | Cat# 98PUN6-2 |
| Miltex® Disposable Biopsy Punches with Plunger System 2mm | AliMed | Cat# 98PUN6-3 |
| 0.1% sodium dodecyl sulfate | Teknova | Cat# S0180 |
| EZ Prep lysis buffer | Sigma Aldrich | Cat# NUC-101 |
| UltraPure™ BSA | Thermo Fisher Scientific | Cat# AM2618 |
| Hoechst 33342, 10 mg/mL Solution in Water | Thermo Fisher Scientific | Cat# H3570 |
| Recombinant RNase Inhibitor | Takara | Cat# 2313B |
| Critical Commercial Assays | ||
| RNeasy 96 Kit | Qiagen | Cat# 74181 |
| Qubit™ 1X dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q33231 |
| Quant-iT™ dsDNA Assay Kit, high sensitivity | Thermo Fisher Scientific | Cat# Q33120 |
| Illumina Tagment DNA Enzyme and Buffer large Kit | Illumina | Cat# 20034198 |
| Agencourt AMPure XP, 60 mL | Beckman Coulter | Cat# A63881 |
| SMARTScribe™ Reverse Transcriptase | Takara | Cat# 639538 |
| KAPA HiFi HotStart ReadyMix PCR Kit | Roche | Cat# 07958935001 |
| Visium Spatial Tissue Optimization Reagents Kit | 10X Genomics | Cat# 1000193 |
| Visium Spatial Gene Expression Reagent Kit | 10X Genomics | Cat# 1000184 |
| Chromium Single Cell 3’ GEM & Gel Bead Kit v3.1 | 10X Genomics | Cat# 1000121 |
| Chromium Single Cell 3’ Library Construction Kit | 10X Genomics | Cat# 1000121 |
| Deposited Data | ||
| scRNA-seq of SVZ | Dulken et al.,27 | Bioproject:PRJNA450425 |
| 10X Sagittal Visium Dataset Anterior | https://www.10xgenomics.com/resources/datasets/mouse-brain-serial-section-1-sagittal-anterior-1-standard-1-0-0 | 10xgenomics.com: V1_Mouse_Brain_Sagittal_Anterior |
| 10X Sagittal Visium Dataset Posterior | https://www.10xgenomics.com/resources/datasets/mouse-brain-serial-section-1-sagittal-posterior-1-standard-1-0-0 | 10xgenomics.com: V1_Mouse_Brain_Sagittal_Posterior |
| 10X Coronal Visium Dataset | https://www.10xgenomics.com/resources/datasets/mouse-brain-section-coronal-1-standard-1-0-0 | 10xgenomics.com: V1_Adult_Mouse_Brain |
| SS2 scRNA-seq of grey/white matter microglia | Safaiyan et al.,51 | GEO:GSE166548 |
| Bulk-RNA microarray of microglia from various regions | Grabert et al.,52 | GEO:GSE62420 |
| SS2 scRNA-seq of microglia from various regions | Tabula Muris Consortium et al.,19 | GEO:GSE109774 |
| Bulk-seq of brain regions (aging) | This paper | GEO:GSE212336 |
| Nuc-seq of the mouse hippocampus and caudate putamen at young and old age | This paper | GEO:GSE212576 |
| Spatial transcriptomics of the mouse brain across three age groups | This paper | GEO:GSE212903 |
| Bulk-seq of brain regions (rejuvenation) | This paper | GEO:GSE227689 |
| Nuc-seq of the mouse hippocampus under dietary restriction | This paper | GEO:GSE227515 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6JN | NIA | N/A |
| C57BL/6J | Jackson Laboratory | 000664 |
| C3B6F1 | Max-Planck-Institute for Biology of Ageing | N/A |
| Oligonucleotides | ||
| SS2 Oligo-dT30VN: AAGCAGTGGTATCAACGCAGAGTACTTT TTTTTTTTTTTTTTTTTTTTTTTTTTTTVN | Integrated DNA Technologies | N/A |
| Software and Algorithms | ||
| Custom analysis software | This paper | https://github.com/OliInTheValley/SpatioTemporal_Analysis |