

Abstract
Pirfenidone and nintedanib are only anti-pulmonary fibrosis (PF) drugs approved by the FDA. However, they are not target specific, and unable to modify the disease status. Therefore, it is still desirable to discover more effective agents against PF. Vimentin (VIM) plays key roles in tissue regeneration and wound healing, but its molecular mechanism remains unknown. In this work, we demonstrated that atractylodinol (ATD) significantly inhibits TGF-β1-induced epithelial-mesenchymal transition and fibroblast-to-myofibroblast transition in vitro. ATD also reduces bleomycin-induced lung injury and fibrosis in mice models. Mechanistically, ATD inhibited TGF-β receptor I recycling by binding to VIM (KD = 454 nM) and inducing the formation of filamentous aggregates. In conclusion, we proved that ATD (derived from Atractylodes lancea) modified PF by targeting VIM and inhibiting the TGF-β/Smad signaling pathway. Therefore, VIM is a druggable target and ATD is a proper drug candidate against PF. We prove a novel VIM function that TGF-β receptor I recycling. These findings paved the way to develop new targeted therapeutics against PF.
Keywords: pulmonary fibrosis, vimentin, TGF-β receptor, atractylodinol, TGF-β/Smad
Graphical abstract
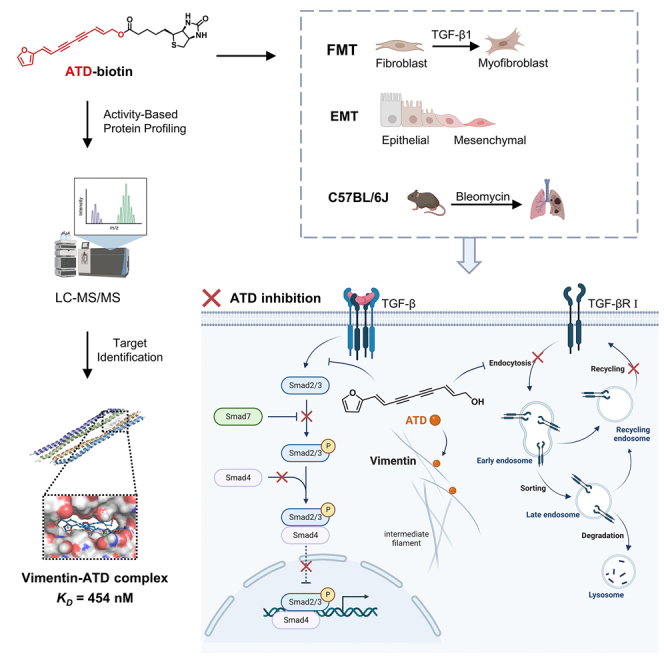
Gu, Xu, and colleagues found that vimentin (VIM) facilitates TGF-βRI recycling and thereby contributes to pulmonary fibrosis (PF) progression. Targeting VIM can be a therapeutic strategy against PF. Atractylodinol (ATD) modifies PF disease status by binding VIM to inhibit TGF-βRI recycling. Thus, ATD is a potent therapeutic agent against PF.
Introduction
Pulmonary fibrosis (PF) is the result of excessive deposition of the extracellular matrix due to dysregulated tissue repair. The incidence of PF is increasing worldwide. Idiopathic pulmonary fibrosis (IPF) of unknown etiology affects about 3 million people worldwide.1 The incidence PF ranges from 0.09 to 1.30 per 10,000 persons.2 PF is one of the most severe and frequently reported COVID-19 sequelae.3 Since the beginning of the COVID-19 outbreak, more than 628 million people have been infected worldwide, of whom 622 million patients have recovered.4 According to a meta-analysis in 2022, approximately 44.9% of COVID-19 survivors had PF.5 PF has become a global medical burden. Recently, the FDA approved pirfenidone (PFD) and nintedanib as only anti-PF drugs, but they are not target-specific drugs, and can only alleviate the symptoms. Dysfunctions of the liver and kidney are common consequences of COVID-19 disease. Because PFD and nintedanib side effects also include liver and kidney toxicities, COVID-19 patients with PF are temporarily withheld antifibrotic therapy pending resolution of liver and kidney dysfunction.3 Twenty-eight small molecular agents are currently in anti-PF clinical trials. Among them, 5 agents were terminated due to side effects; 8, 12, and 1 agents are under phase I, II, and III trials, respectively.6,7 Hence, disease-modifying anti-PF drugs are demanded.
Known anti-PF drug targets are the transforming growth factor beta (TGF-β), receptor tyrosine kinases (RTKs), connective tissue growth factor (CTGF), and WNT/β-catenin.8 Agents against these targets are still under clinical trials. Because the pathogenesis of PF is complicated, the target or lead identification is sluggish.
Vimentin (VIM) belongs to the type III intermediate filament protein family. The known functions of VIM are to regulate cell mobility, mechanosensing, transduction, signaling pathways, and inflammation.9 Therefore, VIM was proposed as a target against virus infection (including COVID-19), cancer, and inflammatory diseases, although a VIM regulator is still not clinically available yet.10,11,12 Withaferin-A (WFA) does inhibit tumor growth through covalently binding to VIM, but remains in lab studies due to toxicities.13 It was reported that VIM-knockout mice can still grow well and reproduce normally.14 Therefore, VIM can be a druggable target. Studies indicate that VIM is a key regulator of fibrosis.15 However, the molecular mechanisms of VIM are under investigation.
In this study, atractylodinol (ATD) (derived from Atractylodes lancea) is found to be a safe agent against PF by screening 98 compounds of an in-house library. ATD was reported as an antimicrobial or anti-respiratory virus agent and a potent bore reproduction inhibitor.16,17 To our best knowledge, this is the first time that ATD has been reported as an anti-PF agent. Therefore, we conducted evaluations of ATD’s safety and the mechanism of actions in vitro and in vivo and identified its target. This work proves that ATD is a safe and promising therapeutic agent against PF.
Results
ATD as an anti-PF agent
A fibroblast-to-myofibroblast transition (FMT) model was developed with transforming growth factor β1 (TGF-β1)-induced human fetal lung fibroblasts (HFL1) cells.18 With the FMT assay, a collagen staining with picrosirius red is used to determine accumulated collagen in HFL1 cells after a compound has been incubated with the cells for 48 h.
To identify new anti-PF leads, a screening campaign against collagen deposition in FMT on an in-house natural product library was conducted in our lab. The library contains 98 compounds, which were derived from Atractylodes lancea,19 Atractylodes chinensis,20 Hypericum japonicum,21 Ajuga decumbens,22 and Siegesbeckia pubescens23 (traditional Chinese medicines). Twenty-four hits were determined (collagen deposition inhibition >50%). Among them, ATD (the structure is depicted in Figure 1A) derived from Atractylodes lancea significantly inhibited collagen deposition at 99.67% inhibition with a concentration of 10 μM (Figure S1A). ATD was incubated with HFL1 cells at concentrations from 0.003 to 300 μM for 48 and 72 h. As shown in Figure S1B, no ATD-induced toxicity was observed at 30 μM or lower. Since the active concentration window for anti-PF ranges between 0.01 and 1 μM, ATD is a safe and potent PF inhibitor in vitro.
Figure 1.

ATD blocks EMT
(A) ATD chemical structure. (B) The protein expression of E-cadherin and VIM in A549 cells was examined by western blotting analysis. Quantification of (C) E-cadherin and (D) VIM expression using ImageJ. Each data point was repeated 6 times (n = 6). (E) Young’s modulus represents cell stiffness. (F) Rq represents the root mean-square roughness of cells. (G) Ra represents the average roughness of cells. (H) Representative images of the ultrastructural characterization of A549 cells include the deflection image size of 10 × 10 μm2 and 3D images. (I) Typical force curves of A549 cells. (J) Typical force curves of A549 cells were stimulated by 5 ng/mL TGF-β1 for 48 h. (K) Typical force curves of A549 cells were treated with 1,000 nM ATD and 5 ng/mL TGF-β1 for 48 h. The data are presented as the mean ± SEM. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the control group; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with the TGF-β1 group. ns, no statistical difference.
ATD blocks the EMT
EMT is important for PF pathogenesis.24 Therefore, we use the EMT model to investigate if ATD inhibits fibrogenesis in A549 cell lines, in which human alveolar epithelial cells were stimulated by TGF-β1.25 E-Cadherin (a biomarker for epithelial cells) was decreased and VIM (a biomarker for mesenchymal cells) was increased in TGF-β1-induced A549 cells (Figures 1B–1D). When ATD or PFD were incubated with the A549 cells, E-cadherin was significantly increased and VIM was significantly decreased.
Interconversion between epithelial and mesenchymal phenotypes is a re-organization of their cytoskeletal systems.26 EMT-associated cytoskeletal restructuring changes cellular biomechanics.27 A549 cells were imaged with atomic force microscopy (AFM) to assess the cell phenotype changes, while TGF-β1 stimulations were applied. The results are depicted in Figures 1E–1G. The interquartile range represents the central tendency of data. The cell stiffness was significantly increased after TGF-β1 stimulation. As shown in Figure 1E, Young’s modulus increases from 1.46 ± 0.03 to 2.40 ± 0.05 kPa. Rq and Ra values increase from 14.55 ± 1.39 to 31.30 ± 2.62 nm and from 11.72 ± 1.22 to 23.68 ± 1.81 nm, respectively, after being stimulated by TGF-β1 (Figures 1F and 1G). The images (10 × 10 μm2) shown in Figure 1H demonstrate the detailed nano-structure and roughness of cellular surfaces in A549 cells, which became more ruffled after being stimulated by TGF-β1. The slope of the force curve after ATD treatment was relatively smoother than that corresponding to the curve of the TGF-β1-stimulated cell (Figures 1I–1K). This notable distinction suggests a stiffer cellular structure upon exposure to TGF-β1.
Thus, we have proved that TGF-β1 does induce PF formation in A549 cells; ATD can significantly alleviate the PF formation through blocking EMT (Young’s modulus, Rq, and Ra were decreased to 1.40 ± 0.03 kPa, 15.08 ± 0.69 nm, and 11.77 ± 0.54 nm, respectively).
The EMT process acquires migratory characteristics resulting in PF during organ healing. We also conducted A549 cell scratch closure experiments, which indicated that the scratch closure rate significantly increased from 36.81% to 74.84% after TGF-β1 stimulation. After ATD treatment, the scratch closure rate significantly decreased in a dose-dependent manner (Figures S1C and S1D).
Taken together, these experiments demonstrated that ATD can reverse the occurrence of TGF-β1-induced EMT. In addition, ATD prevents TGF-β1-induced A549 fibrosis and is a potent anti-PF agent.
ATD blocks the FMT
Excessive proliferation of myofibroblasts is critical to PF, and FMT is an important source of myofibroblasts. Experiments were accomplished to investigate if ATD inhibits PF by blocking FMT. HFL1 cells were incubated with ATD and TGF-β1, then stained by picrosirius red. Results show that collagen was increased by TGF-β1 stimulation, and ATD dose dependently reduces collagen accumulation (Figure 2A).
Figure 2.

ATD blocks FMT
(A) Picrosirius red staining of HFL1 cells was treated with various concentrations of ATD (10, 100, and 1,000 nM) in the presence or absence of TGF-β1 (5 ng/mL) stimulation for 48 h. Scale bars, 400 μm. (B–D) Quantitative RT-PCR assays were performed to determine mRNA expression of genes related to extracellular matrix (FN1 and COL1A1) and fibrotic marker (ACTA2) in HFL1 cells (n = 6). (E) The expression levels of fibrosis biomarkers. Quantification of (F) fibronectin, (G) collagen I, (H) collagen III, and (I) α-SMA expression using ImageJ. Each data point was repeated 6 times (n = 6). PFD-treated cells were the positive control group. The data are presented as the mean ± SEM. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the control group; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with the TGF-β1 group. ns, no statistical difference.
Within FMT, myofibroblast cells could attain apoptosis resistance during differentiation.7,28 ATD increased total apoptosis from 7.00% to 15.03% in myofibroblasts (Figures S2A and S2C), suggesting that ATD ameliorates PF. Compared with PFD, ATD is more active in promoting TGF-β1-induced apoptosis in HFL1 cells.
TGF-β1-induced cell-cycle prolongation into the G2-M phase limits the potential of cells to repair and regenerate, exacerbating chronic fibrosis.29,30 We prove that ATD-induced HFL1 cells return to G0-G1 and resume cell growth and division to alleviate PF (Figures S2B and S2D). In contrast, PFD did not demonstrate activity at a concentration of 10 μM. TGF-β1 stimulation results in increased HFL1 cell proliferation. With EdU assays, we observed that ATD inhibited TGF-β1-induced proliferation in HFL1 cells at concentrations from 10 to 1,000 nM (Figures S2E and S2F) and PFD had similar behavior.
The fibronectin (FN), collagen I, and α-SMA mRNA and protein levels increased after TGF-β1 induction (Figures 2B–2I). ATD significantly reduced the mRNA and protein expression of α-SMA (a fibroblast marker) (Figures 2D, 2E, 2I, S3A, and S3B). ATD suppressed extracellular matrix (ECM) (such as FN, collagen I, and collagen III) deposition induced by TGF-β1 in a concentration-dependent manner (Figures 2E–2H, S3C, S3D, S5A, and S5B). Meanwhile, ATD decreases the mRNA expression of FN1 and COL1A1 (Figures 2B and 2C). Overall, the findings imply that ATD can ameliorate PF by blocking FMT.
Safety evaluation of ATD in C57BL/6J mice
To measure the short-term safety of ATD, we used an up-and-down method to detect the oral acute toxicity of ATD. Acute toxicity experiments result in an oral median lethal dose (LD50) of 3,129 mg/kg (Table S1). For efficacy experiments, we use concentrations below 1/300 of the LD50. Hence, ATD is safe for experimental dosage in mice according to the Globally Harmonized System (GHS) for the Classification and Labeling of Chemicals.
To measure safety during ATD administration, we performed biochemical index detection on the serum of mice administered with ATD for 21 days. There was no significant change in organ coefficients of the heart, liver, kidney, and spleen after ATD administration (Table S2). In addition, all serum biochemistry remained in the normal range (Table S3), indicating normal liver, kidney, heart, and overall metabolic functions. However, creatinine (CREA) was significantly increased in the PFD group, indicating that PFD can cause nephrotoxicity. These results indicate that ATD is stable and safe in vivo, and a potent agent for PF treatment.
ATD ameliorates PF induced by bleomycin in vivo
PF is a scarring disease of the lungs that eventually leads to respiratory failure. In animal experiments, PFD was used as a positive control.31 We next examined the anti-fibrotic effect of ATD in vivo (Figure 3A).
Figure 3.

ATD ameliorates PF induced by bleomycin in vivo
(A) Protocol for bleomycin to induce PF in C57BL/6J mice. The PF model mice underwent a single intratracheal instillation of bleomycin (2 mg/kg) at day 0 and were treated with CMC-Na (control group), ATD, or PFD at 1 day and were euthanized at day 21 (n = 14 mice per group). (B) Survival curve. (C) Change in body weight measured over time. Representative images of micro-CT (D), H&E staining (E), and Masson staining (F). Scale bars, 100 μm. (G) H&E staining score for each group (n = 6). (H) Ashcroft score for each group (n = 6). (I) Western blot for collagen I, α-SMA, E-cadherin, VIM, and PCNA protein in mouse lung tissue. (J) The content of hydroxyproline in lung tissue (n = 6). (K) Quantification of collagen I, α-SMA, E-cadherin, VIM, and PCNA expression using ImageJ (n = 6). The data are presented as the mean ± SEM. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the control group; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with the bleomycin group. ns, no statistical difference.
Acute toxicity experiments result in ATD having an oral LD50 of 3129 mg/kg (Table S1). Hence, ATD is safe for experimental dosage in mice according to the GHS for the Classification and Labeling of Chemicals. For efficacy experiments, we use concentrations below 1/300 of the LD50. The C57BL/6J mouse successfully induced PF by a single intratracheal instillation of bleomycin (BLM). Compared with the control group, the BLM group decreased overall survival (Figure 3B), body weight (Figure 3C), lung coefficient (Table S2), and lung function, including increased enhance pause (Penh) (Figure S6D) and end-expiratory pause (EEP) (Figure S6E). ATD (2.5, 5, and 10 mg/kg) or PFD (300 mg/kg) treatments significantly improved overall survival, alleviated weight loss, and reversed BLM-caused pulmonary dysfunction.
Microcomputer tomography (micro-CT) experiments showed that the area and fibrotic foci (white patches) were decreased after ATD or PFD treatment (Figure 3D). Hematoxylin and eosin (H&E) and Masson staining assays indicated that ATD reduced the inflammatory cell infiltration and the thickening of alveolar septa in a dose-dependent manner (Figures 3D–3F). Compared with PFD, ATD showed a better therapeutic effect in reducing inflammatory cells and the degree of fibrosis. As shown in Figures S7A and S7B, the secretion level associated with fibrosis (TGF-β1 and vascular endothelial growth factor A [VEGFA]) and inflammation (tumor necrosis factor alpha [TNF-α], interleukin-6 [IL-6], and chemokine ligand 1 [CXCL1]) in serum and bronchoalveolar lavage fluid (BALF) were significantly inhibited by ATD administration.
Hydroxyproline is the principal component of collagen. Our experiments show that ATD treatment reduced hydroxyproline in lung tissue (Figure 3J). As shown in Figures 3I and 3K, western blotting results indicate that ATD significantly reduced collagen I expression. In addition, compared with the BLM group, the epithelial cell marker (E-cadherin) was significantly increased, and the mesenchymal cell marker (VIM) and the myofibroblast marker (α-SMA) were significantly decreased after ATD administration. Proliferating cell nuclear antigen (PCNA) is an essential DNA replication and repair protein that increases significantly in proliferating cells. ATD inhibited the expression of PCNA (Figures 3I and 3K). Collectively, these results suggest that ATD can inhibit BLM-induced EMT and FMT processes in vivo.
ATD inhibits the TGF-β/Smad signaling pathway
To explore the molecular mechanisms of ATD, we conducted mRNA transcript sequencing to compare the differential genes between the ATD group and the TGF-β1 group (Figure S4). Compared with the HFL1 group, 316 genes were differentially downregulated and 143 genes were upregulated after TGF-β1 stimulation for 48 h (log2 (fold change) >5 or <1/5). After ATD treatment, 282 genes were differentially upregulated and 138 genes were differentially downregulated (Figure S4B). The heatmap demonstrated that ATD treatment reversed the TGF-β1-induced changes in fibroblast gene expression (Figure S4C). Among them, the COL1A1 and ACTA2 genes were significantly changed, and they were mainly affected by the TGF-β/Smad signaling pathway. We conclude that ATD ameliorates PF through this pathway.
TGF-β1 induces fibrosis by activating canonical Smad-dependent and Smad-independent pathways.32 As shown in Figures 4A–4C, TGF-β1 treatment for 48 h selectively activated TGF-βRI (TGF-β receptor type Ι) in HFL1 cells. TGF-βRII (TGF-β receptor type II) is not activated by TGF-β1. Western blotting analyses indicate that TGF-β1 treatment for 30 min significantly increased Smad2 phosphorylation (Ser255 and Ser465/Ser467) (Figures 4D–4F) and Smad3 phosphorylation (Ser204 and Ser423/Ser425) (Figures 4H–4J). Smad2 phosphorylation (Ser465/Ser467) is also confirmed by immunofluorescent staining experiments (Figure 4G). ATD decreased the phosphorylation of Smad2 and Smad3 by suppressing TGF-βRI expression in a concentration-dependent manner.
Figure 4.

ATD ameliorates PF through the TGF-β/Smad signaling pathway
(A) The protein expression of TGF-βRI and TGF-βRII in HFL1 cells was examined by western blotting analysis. Quantification of (B) TGF-βRI and (C) TGF-βRII expression using ImageJ. Each data point was repeated 6 times (n = 6). (D) The phosphorylation level of p-Smad2Ser255 and p-Smad2Ser465/467 was analyzed by western blotting analysis. Quantification of (E) p-Smad2Ser255 and (F) p-Smad2Ser465/467 expression using ImageJ (n = 6). (G) The subcellular localization of p-Smad2Ser465/467 was observed by immunofluorescence microscopy. Scale bar, 30 μm. (H) The phosphorylation level of p-Smad3Ser204 and p-Smad3Ser423/425 was analyzed by western blotting analysis. Quantification of (I) p-Smad3Ser204 and (J) p-Smad3Ser423/425 expression using ImageJ (n = 6). (K) The protein expression of Smad4 and Smad7 in HFL1 cells was examined by western blotting analysis. Quantification of (L) Smad4 and (M) Smad7 expression using ImageJ (n = 6). The data are presented as the mean ± SEM. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the control group; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with the TGF-β1 group. ns, no statistical difference.
In the TGF-β/Smad signaling pathway, Smad7 inhibits TGF-βRI activation of Smad2 and Smad3. Smad4 forms a heterotrimeric complex with activated Smad2 and Smad3 and translocates to the nucleus. However, Smad7 is not affected by ATD (Figures 4K and 4M), while Smad4 expression is significantly suppressed by ATD only at 1,000 nM (Figures 4K and 4L). Overall, our finding indicates that ATD inhibits the TGF-β/Smad signaling pathway.
TGF-β1 may also affect the Smad pathway through the mitogen-activated protein kinase pathways. TGF-β-induced activation of ERK1/2 and JNK1/2 pathways can lead to Smad phosphorylation and regulate Smad activation.33,34,35 ATD treatment significantly decreased the phosphorylation of ERK1/2 and JNK1/2 in TGF-β1-induced HFL1 cells (Figures S5E–S5H). However, ATD did not affect the phosphorylation of p38 with or without TGF-β1 (Figures S5C and S5D).
Thus, we conclude that ATD modifies PF status by inhibiting TGF-β/Smad-dependent and Smad-independent (ERK1/2 and JNK1/2) signaling pathways.
ATD inhibits the TGF-β/Smad signaling pathway through binding VIM
Our mRNA transcript sequencing results also indicated that the differential genes after ATD treatment were mainly enriched in ECM-receptor interaction and focal adhesion pathways by KEGG analysis (Figure S4A). Focal adhesions are dynamic protein complexes through which the cytoskeleton of a cell connects to the ECM.36 The cytoskeleton regulates focal contact size and helps stabilize cell-matrix adhesions in endothelial cells.37 We recognize that ATD can inhibit ECM deposition through the cytoskeleton, a new pathway for improving PF.
To identify the target of ATD, we synthesized an ATD-biotin chemical probe (the structure is depicted in Figure 5A). The probe was incubated with HFL1 cells. Western blotting assays indicated that the ATD-biotin is still active (Figures S8B–S8E). ATD-biotin was used in biotin-streptavidin pull-down experiments. A distinct protein band was identified between molecular weights 50 and 70 kDa (Figure S8F). The band was cleaved and confirmed that ATD was bound to VIM with mass spectroscopy (Table S4). The VIM cytoskeleton regulates focal contact size and helps stabilize cell-matrix adhesions in endothelial cells.
Figure 5.

ATD targets the intermediate filament protein VIM
(A) ATD-biotin chemical structures. (B) Purified His-VIM was incubated with the indicated concentrations of ATD for 1 h followed by incubation with ATD-biotin (10 μM) for 1 h at 37°C. ATD-biotin bound to purified VIM was detected after SDS-PAGE with streptavidin-HRP. (C) Ligand-observed 1D 1H-NMR of the peaks of 2 mM ATD (red) becomes wider (green) in the presence of 20 μM VIM. Positive and negative signals identify VIM binding and non-interacting molecules, respectively. (D) Surface plasmon resonance sensorgrams for the binding of ATD to VIM. (E) The detectable remaining amount of VIM after ATD treatment varies with the temperature used for heat treatment of HFL1 cells. (F) CETSA melt curve quantified by western blotting (n = 3). (G) VIM was polymerized in the presence of 5 mM MgCl2, 100 mM KCl, and 170 mM NaCl by incubation at 37°C for 1 h. The protein was fixed with 0.5% glutaraldehyde, stained with uranyl acetate, and observed by transmission electron microscopy. Polymerization of tetrameric VIM in the presence of 20 μM ATD produces extensive filamentous aggregates. (H) HFL1 cells treated with DMSO or 1 μM ATD for 24 h were stained for VIM with anti-VIM antibody (green) and co-stained with F-actin (red). The presence of numerous cytoplasmic particulate granules that co-stain for VIM and disrupted F-actin in ATD-treated cells compared with control. Scale bar, 30 μm. (I) Cell lysates treated with or without ATD samples were crosslinked with 0.005% glutaraldehyde, and the VIM forms were detected by western blotting (n = 3). The data are presented as the mean ± SEM.
The full-length VIM was specifically pulled down by ATD-biotin. The binding of ATD-biotin and VIM can be reduced by ATD competition in vitro, confirming the ATD-VIM binding (Figure 5B). The NMR experiments were performed to prove the ATD-VIM binding. As shown in Figure 5C, the feature peaks (red) of pure ATD become wider (green), meaning that ATD binds to VIM. The binding affinity of ATD-VIM was measured with surface plasmon resonance, and the equilibrium dissociation constant (KD) was 454 nM (Figure 5D). Consistently, the circular dichroism (CD) spectrum results obtained a characteristic signal of α-helical structure, featuring two broad negative bands at 222 and 208 nm and one positive band at 192 nm. This further proved that ATD altered VIM structure, indicating the interaction of ATD and VIM (Figure S8G).
In HFL1 cells, the thermal stability of VIM bound to ATD was investigated using a cellular thermal shift assay. The melting curve experiments in the absence (control) and presence of ATD are shown in Figures 5E and 5F. The apparent Tagg value of the control group (the temperature at which 50% of the protein aggregates) is 48.88°C ± 0.96°C, and with ATD the Tagg value was 55.56°C ± 2.02°C. The binding of ATD and VIM stabilizes VIM conformations. Overall, we have proved that ATD binds to VIM, the intermediate filament protein.
VIM proteins are assembled to form intermediate filaments, with which a network of cytoplasmic filaments is generated. The network extends from the cell nucleus to the cell edge. VIM exists in cells as soluble tetramers and insoluble filaments, and the two forms are dynamically exchanged.38 By covalently binding to VIM, WFA induces VIM to form filamentous aggregates, thereby perturbing the cytoskeletal structure.39 We hypothesized that ATD could interfere with the VIM intermediate filaments by binding to VIM. Therefore, we assayed the effect of ATD on VIM formation of intermediate filaments in vivo and in vitro. In the filament polymerization assays in vitro, electron microscopy data indicate that ATD promotes the filament assembly while inducing the formation of filamentous aggregates (amorphous condensed structures) (Figure 5G). Immunofluorescence staining experiments with VIM also confirmed this observation in vivo, with 1 μM ATD-treated HFL1 cells showing condensation of VIM filaments around the perinuclear region and the presence of abundant VIM-stained granules in the cytoplasm. The results also indicate that ATD disturbs the structure of VIM filaments (Figure 5H). ATD increased high-molecular-weight forms of VIM (soluble tetramers or possibly unit-length filaments) through glutaraldehyde crosslinking found in the presence or absence of TGF-β1 (Figure 5I). This further suggests that ATD stabilizes VIM in a high-molecular-weight form, disturbing the homeostasis of VIM assembly in vivo.
Molecular dynamics (MD) simulations resulted in a stable binding complex with three ATD and one VIM tetramer (Figure S9C). As shown in Figure S9D, the three ATD molecules bind to a pocket formed by four VIM monomers consisting of two head-to-tail α helix dimers. ATD binds with Arg321 (chain B), Arg320 (chain B), and Glu277 (chain C) by forming hydrogen bonds and π-H interactions between the furan-ring of ATD and Gln285 (chain C). Hydrophobic contacts or π-stacking between ATD and nonpolar amino acids also provided a certain affinity. Thus, ATD molecules stabilize VIM tetramers. All the experimental data demonstrate that ATD targets VIM and disrupts the VIM filament structure against PF disease.
ATD blocks the TGF-βRI recycle by targeting VIM
To further confirm that ATD alleviates PF through its interaction with VIM, we have overexpressed VIM-Flag and knocked down VIM, respectively, in a fibroblast-to-myofibroblast transformation model of HFL1 cells. We performed transient overexpression experiments with vector encoding VIM-Flag (Figures 6A and 6B). We observed that VIM overexpression increased the protein expression of PF biomarkers (FN, collagen I, and α-SMA) in HFL1 cells. TGF-β1-induced fibrosis was further exacerbated after VIM overexpression (Figures 6C–6F). This is also confirmed by picrosirius red staining experiments (Figure 6G), where overexpression of VIM prevented ATD from inhibiting TGF-β1-induced collagen deposition in HFL1 cells (Figures 6C–6G).
Figure 6.

Overexpression of VIM attenuates the protective effect of ATD on PF
(A) qPCR analysis of VIM expression in VIM-overexpressed HFL1 cells (n = 3). (B) The expression of VIM and flag in VIM-overexpressed HFL1 cells was examined by western blot analysis (n = 3). Quantification of (C) fibronectin, (D) collagen I, and (E) α-SMA expression using ImageJ (n = 3). (F) The protein expression was examined by western blot analysis in VIM-overexpressed HFL1 cells (n = 3). (G) Picrosirius red staining of VIM-overexpressed HFL1 cells treated with or without TGF-β1 (5 ng/mL) and visualized. Scale bar, 400 μm. The data are presented as the mean ± SEM. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the VIM-Flag group; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with the pCMV6 group; Δp < 0.05, ΔΔp < 0.01, ΔΔΔp < 0.001 compared with TGF-β1 group. ns, no statistical difference.
In the BLM-induced mouse PF model, immunohistochemistry showed that the expression of VIM in the lung tissue of the BLM group was significantly increased (Figure S7D). ATD (at dosages 2.5, 5, and 10 mg/kg) significantly suppressed the expression of VIM in a dose-dependent manner. This is consistent with the change in the expression of α-SMA, indicating that the expression of VIM is related to fibrosis level (Figures S7C and S7D). Together, we can conclude that VIM has a pro-fibrotic effect and that ATD’s anti-PF activity is related to the interaction with VIM.
To further study VIM functions, we used four different short hairpin RNAs (shRNA) to knock down VIM. shVIM-1 and shVIM-2 were able to significantly knockdown VIM in HFL1 cells for subsequent research (Figures 7A and 7B). Experimental results demonstrated that the collagen I and α-SMA were reduced after VIM knockdown (Figures 7C–7G). Therefore, VIM is essential to PF.
Figure 7.

VIM knockdown prevents PF
(A) qPCR analysis of VIM expression in HFL1 cells (n = 3). (B) Western blot analysis of VIM expression in VIM-knockdown HFL1 cells (n = 3). Quantification of (C) VIM, (D) collagen I, and (E) α-SMA expression using ImageJ (n = 3). (F) Western blot analysis of protein expression treated with or without TGF-β1 (5 ng/mL) in VIM-knockdown HFL1 cells (n = 3). (G) Picrosirius red staining of VIM-knockdown HFL1 cells treated with or without TGF-β1 (5 ng/mL) and visualized. Scale bar, 400 μm. The data are presented as the mean ± SEM. #P < 0.05, ##P < 0.01, ###P < 0.001 compared with the vector group. ns, no statistical difference.
TGF-β signaling is a canonical pathway in fibrosis, TGF-βRI, a receptor of TGF-β, is endocytosed and recycled to the cell surface, and intracellular trafficking of TGF-βRI is required for TGF-β signaling.40,41 Nestin (the intermediate filament protein) knockdown inhibits the recycling of TGF-βRI to the cell surface.42 ATD-VIM binding will alleviate PF symptoms by inhibiting TGF-βRI recycling. The effect of ATD on TGF-βRI cycling can be assayed by biotinylation of cell surface proteins. The experimental data indicated that TGF-βRI endocytosis decreased from 87.61% to 49.53% after ATD administration (Figures 8A, 8B, and 8E). Only 11.33% of internalized TGF-βRI was recycled compared with 80.48% in the control group (Figures 8C, 8D, and 8F).
Figure 8.

ATD inhibits the recycling of TGF-βRI to the cell surface
(A) The serum-starved HFL1 cells were first treated with DMSO or ATD (1 μM) and chloroquine (Chlq) (100 μM) for 4 h and then performed the following experiments. The biotinylated protein was pulled down with streptavidin agarose, and quantification of the plasma membrane TGF-βRI was performed by immunoblotting. (B) Biotinylated serum-starved HFL1 cells were placed at 37°C for 30 min to allow for TGF-βRI endocytosis. The biotinylated protein was pulled down with streptavidin agarose, and quantification of the endocytosed TGF-βRI was performed by immunoblotting. (C) Then, the biotinylated HFL1 cells were placed at 37°C for 30 min to allow for TGF-βRI recycling. The quantification of biotinylated TGF-βRI included the internalized and recycled TGF-βRI that were subjected to streptavidin agarose pull-down and analyzed by immunoblotting. (D) The internalized TGF-βRI were subjected to streptavidin agarose pull-down and analyzed by immunoblotting. (E) Quantification of the percentage of internalized TGF-βRI. TGF-βRI internalization rate = total internalized TGF-βRI at 30 min/plasma membrane TGF-βRI × 100%. (F) Quantification of the percentage of recycled TGF-βRI. TGF-βRI recycling rate = (internalized and recycled TGF-βRI at 60 min – internalized TGF-βRI at 60 min)/total internalized TGF-βRI at 30 min × 100%). Three separate experiments were performed. The data are presented as the mean ± SEM. ##P < 0.01, compared with control.
Taken together, the above data suggest that the binding of ATD and VIM inhibits TGF-βRI recycling and blocks the TGF-β/Smad signaling pathway, thereby ameliorating PF.
Discussion
ATD is a safe and potent anti-PF agent derived from Atractylodes lancea (Chinese medicine). We have proved that ATD blocks EMT and FMT in vivo and in vitro by inhibiting the TGF-β/Smad signaling pathway. Interestingly, we found that ATD could also inhibit collagen deposition in HFL1 cells in the absence of TGF-β1 stimulation, suggesting that ATD might regulate collagen deposition through a pathway distinct from the TGF-β/Smad pathway. This observation warrants further in-depth studies.
Specifically, the target of ATD is VIM. Recently, it was reported that VIM was highly expressed during fibrosis development in multiple organs.15,43,44,45 VIM intermediate filaments are increased in myofibroblasts in pulmonary fibrotic lesions of IPF patients. Therefore, VIM is associated with enhanced myofibroblast invasiveness and disease progression.46,47 A recent report also suggests that VIM can be a drug target for lung fibrogenesis-related diseases.12
In our in vivo models, we observed that VIM was increased in BLM-induced mice. This resulted in collagen deposition and PF, similar to observations in previous studies.15,43,44,45 Our data indicated that overexpressed VIM upregulated the protein levels of α-SMA, FN, and collagen I. VIM knockdown experiments exhibited significant attenuation of PF by alleviating collagen accumulation. Further exploration of the mechanism of action of targeting VIM in PF will be of interest.
VIM maintains the structural integrity of the cells, but also plays important roles in various intracellular signaling systems,48 such as TGF-β149 and ERK signaling.50 The cytoskeleton composed of VIM determines the mechanical integrity and wound-healing ability of cells.51,52,53 We demonstrated that ATD treatment promotes the collapse of VIM architecture in HFL1 cells and in vitro. The known VIM inhibitors, such as WFA39 and FiVe1,53 could also depress VIM, while ATD stabilizes VIM neither changing VIM expression nor affecting cell viability. Our MD simulations helped us to discover a novel ATD-binding pocket in a VIM tetramer. Therefore, ATD is a valuable chemical probe to discover VIM properties.
The TGF-β/Smad family signaling pathway is an important pathogenic mechanism in PF.54 Many studies have shown that the endocytosis, intracellular trafficking, and recycling of TGF-β receptors regulate TGF-β/Smad signaling.42,55,56 With the chemical probe ATD-biotin, we reveal a new VIM function that promoted TGF-βRI recycling to the cell membrane, thereby contributing to the progression of PF. ATD stabilizes VIM multimers and promotes the formation of filamentous aggregates, thereby inhibiting TGF-βRI recycling. ATD reduces the recycling of TGF-βRI to the cell surface, thereby attenuating the signaling response induced by TGF-β factors (a linear signaling pathway from type II to type I receptor kinase to Smad2/3 activation). At the same time, ATD inhibits the expression of Smad4. ATD reduces the complex formed by activated Smad2/3 and Smad4. Furthermore, ATD inhibits the translocation of the complex into the nucleus. This process reduces the transcription of downstream target genes, including α-SMA and collagen I. Thus, ATD reduces myofibroblast activation and ECM deposition. PF status is alleviated.
Thus, VIM is identified as the target of ATD. And ATD is a safe and potent drug candidate against PF in C57BL/6J mice model. This finding paves a new road to developing new therapeutics against PF.
Materials and methods
Cell culture and cytotoxicity assay
ATD (purity >98%) was isolated as per the previously described method.19 PFD was purchased from TargetMol (Boston, USA). The HFL1 and A549 cells were purchased from Procell Life Science & Technology (Wuhan, China). The Cells were cultured in F-12K medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Lonsera, Shanghai, China) and 1% penicillin-streptomycin (Thermo Fisher Scientific) in a humidified 5% CO2 incubator at 37°C. According to the manufacturer’s protocol, the cell cytotoxicity was measured using the cell counting kit-8 (CCK8) (Dojindo, Kyushu, Japan). The cells were seeded in 96-well plates at a density of 2 × 103 cells per well. After incubating for 24 h, the cells were treated with a medium containing various concentrations of ATD (0–300 μM) for 48 or 72 h. Following incubation, each well was incubated at 37°C for 2 h with 10 μL of CCK8 solution. At the end of the incubation, the absorbance was measured at 450 nm on a full-function microplate reader (BioTek).
Cell wound healing assay
The EMT model constructed by TGF-β-induced A549 cells has the characteristic of migration. A549 cells were seeded in 6-well plates at a density of 1 × 105 cells per well. After incubating for 24 h, the confluent monolayer of cells was wounded gently by scraping a narrow 200 μL tip across the diameter of the well. Cells were incubated in serum-free F-12K medium with 5 ng/mL TGF-β1 (Peprotech, Rochy Hill) and various concentrations of ATD (0, 10, 100, and 1,000 nM). After incubation for 48 h, cells were visualized with a microscope under 100× magnification. ImageJ software was used to calculate the scratch area. Scratch closure (%) = (scratch area at 0 h – scratch area at 48 h)/(scratch area at 0 h) × 100%.
AFM imaging
In the EMT model constructed by TGF-β1-induced A549 cells, the cytoskeleton reorganized and changed the cell biomechanics. A549 cells were seeded in 60-mm2 culture disks and incubated with 5 ng/mL TGF-β1 in the presence or absence of 1,000 nM ATD for 48 h. Then, cells were fixed in 4% paraformaldehyde for 30 min. Ultrastructural assays were performed using a Dimension FastScan AFM system (Bruker BioSpin, Switzerland) and high-quality etched silicon probes (RTESPA-150, radius 8–12 nm; Bruker BioSpin). For cell imaging, a triangular cantilever with a normal spring constant of 5 N/m was employed.
Cell mechanical properties by AFM
The elasticity of A549 cells was also detected in contact mode. Measurements were conducted above the nucleus region of the cell to avoid the influence of the underlying substrate. One hundred force-distance curves were obtained from 10 different cells in each group at a ramp rate of 0.5 Hz. Force curves were obtained at the same loading rate and performed using silicon nitride probes (MLCT-O10, 0.07 N/m; Bruker BioSpin). The Hertz model was used to compute Young’s modulus according to the following formula:
where F is loading force, is Poisson ratio, E is Young’s modulus, and R is the radius of the curvature of the AFM tip.
Picrosirius red for collagen staining
Since myofibroblasts localize at sites undergoing active ECM deposition and display elevated collagen synthetic capacity, myofibroblasts are considered to play a major role in the pathology of PF. HFL1 cells were seeded in 96-well plates at a density of 2 × 104 cells per well. After incubating for 24 h, the cells were treated with a medium containing 5 ng/mL TGF-β1, various concentrations of ATD (0, 10, 100, and 1,000 nM), and 10 μM PFD for 48 h. Then, cells were immediately fixed in 4% neutral paraformaldehyde for 30 min. Cells were washed twice with PBS and then incubated with a 0.1% picrosirius red solution dissolved in aqueous saturated picric acid for 4 h. The cells were washed twice with acidified water (0.5% hydrogen chloride), dehydrated, and visualized under the microscope cell-imaging system (EVOS FL Auto, Life Technologies). For the quantitative determinations of the accumulated collagen, the stained cells were destained with 0.1 M NaOH (100 μL per well) for 10 min. The absorbance was measured at 540 nm on a full-function microplate reader.
EdU cell proliferation assay
Cell proliferation was assessed using the BeyoClick EdU-594 kit (Beyotime, Shanghai, China) according to the manufacturer’s protocol. In brief, the HFL1 cells were treated with a medium containing various concentrations of ATD (0, 10, 100, and 1,000 nM) or 10 μM PFD in the presence or absence of 5 ng/mL TGF-β1 stimulation for 48 h. EdU-594 was added to medium resulting in a final concentration of 10 μM for 2 h. The click reaction solution includes 430 μL click reaction buffer, 20 μL CuSO4 solution, 1 μL azide 594, and 50 μL click additive solution (sodium ascorbate). Then, cells were fixed in 4% paraformaldehyde for 15 min, and permeabilized with 0.3% Triton X-100 for 15 min. The click reaction solution includes 430 μL click reaction buffer, 20 μL CuSO4 solution, 1 μL azide 594, and 50 μL click additive solution (sodium ascorbate). Cells were incubated with the click reaction solution in the dark at room temperature for 30 min, then washed three times with 3% BSA/PBS. Cells were incubated with Hoechst 33342 (diluted 1:100) in the dark at room temperature for 10 min. After washing, digital images were recorded using an FV3000 laser scanning confocal microscope (Olympus) with 100× magnification.
Cell-cycle and apoptosis analysis with flow cytometry
HFL1 cells were seeded in 6-well plates and treated with various concentrations of ATD (0, 10, 100, and 1,000 nM) or 10 μM PFD in the presence or absence of 5 ng/mL TGF-β1 stimulation for 48 h. After incubation, cells were harvested and fixed with 70% ethanol for 4 h at −20°C. The cells were washed with PBS three times, then incubated with ribonuclease A (RNase A) and propidium iodide (PI) for 30 min in the dark. The cell-cycle analysis was analyzed by using CytoFLEX (Beckman Coulter) after cells passed 300-mesh sieves. The apoptosis assay was performed using the Annexin V-Alexa Fluor 488/PI apoptosis detection kit according to the manufacturer’s instructions (Yeasen, Shanghai, China).
Immunofluorescence assay
HFL1 cells were fixed in 4% paraformaldehyde for 10 min, then permeabilized with 0.1% Triton X-100 (Sangon Biotech, Shanghai, China, no. 9002-93-1) for 10 min and blocked with goat serum (BOSTER, Wuhan, China, no. AR0009) for 30 min. After that, cells were incubated with primary antibodies for FN, collagen I, α-SMA, or VIM (diluted 1:100) for 24 h at 4°C and secondary antibodies for 1 h at room temperature. Nuclei were stained with Hoechst 33342 (diluted 1:100) for 10 min at room temperature. The fluorescence images were captured using an FV3000 laser scanning confocal microscope (Olympus, Japan).
Real-time qPCR
The total RNA was extracted from differentiated tissues using RNAiso Plus according to the manufacturer’s instructions. The real-time reaction was carried out using 1 μg of total RNA. The RNA was reverse transcribed into cDNA using a ReverTra Ace qPCR RT Master Mix (Toyobo, Katata Otsu, Japan). The gene expression levels were measured with qPCR using a StepOnePlus Real-Time PCR instrument (Life Technologies) and SYBR Green Real-Time PCR Master Mix (Toyobo). The primers used are shown in Table S5. The relative gene expression quantification was normalized to the β-actin mRNA expression using the 2−ΔΔCt analysis method.
RNA sequencing library preparation and data analyses
HFL1 cells were treated with a medium incubated with (1 μM)/without ATD in the presence or absence of 5 ng/mL TGF-β1 stimulation for 48 h. Total RNA was isolated from HFL1 cells by TRIzol (Sangon Biotech). RNA sequencing libraries were prepared with the NEBNextUltraTM RNA Library Prep Kit for Illumina. RNA integrity was evaluated with a 1.0% agarose gel. DNA concentration was checked by using Qubit 2.0 Fluorometer. Thereafter, the quality and quantity of RNA were assessed using a NanoPhotometer spectrophotometer (Implen, CA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, CA). The cDNA libraries were sequenced by DIATRE Biotechnology (Shanghai, China) using an Illumina Nova 6000 instrument (Illumina, San Diego, CA). FastQC initially processed raw sequence reads for quality control, and then adapter sequences and poor-quality reads were removed. Quality-filtered reads were then mapped to the reference genome using STAR, and only uniquely mapped reads were kept. Sam files were converted to Bam format using Samtools. Cufflinks software was used to compute gene expression levels and differences. R packages such as DEseq2 and edgeR were further used to perform data analyses.
Animal experiments
All animal studies were conducted by the laboratory animal center of Sun Yat-sen University under the recommendations of the Guide for the Care and Use of Laboratory Animals (NIH Publication no. 85-23, revised 1996). The C57BL/6J mice were purchased from Guangdong Medical Laboratory Animal Center (SPF grade, certification no. 44007200087781) and housed in standard isolator cages and maintained at a room temperature of 23°C ± 1°C and humidity level of 50%–65% in an environmentally controlled room with a 12-h:12-h light/dark cycle.
Acute toxicity in mice
An up-and-down method for acute toxicity test was approved by the Research Ethics Committee of Sun Yat-sen University (animal ethics approval no. SYSU-IACUC-2021-000503). According to previous literature results, ATD is a low-toxic substance. The dosage was calculated by AOT425StatPgm. Therefore, the estimated maximum experimental dosage was 5,000 mg/kg. The sequential dosages were 175, 550, 1,750, and 5,000 mg/kg. ATD solutions were prepared in 0.5% sodium carboxymethyl cellulose (CMC-Na) and stored at 4°C before use. C57BL/6J mice (8 male, 8 female, 7 weeks old, 25 ± 3 g) were used in the study. In the experiment, each mouse was weighed and fasted for 4 h with drinking water freely before administration. After administration of ATD, the mice fasted for 1 h with drinking water freely. The first dosage of 175 mg/kg was given to the first mouse. Symptoms of poisoning were recorded within 48 h. If it survived, 550 mg/kg was given as the second dosage. The experimental sequence was followed until the standard stopping rules appeared. For the main test, the criteria for stopping the test were as follows: five reversals occurring in any six subsequent animals administered.57 When the experiment was stopped, all the surviving mice were humanely killed and necropsied after a 14-day observation. The pathological changes in organs were observed and recorded. The LD50 for acute toxicity of ATD was calculated by AOT425StatPgm.
Animal models and experimental design
This study was approved by the Research Ethics Committee of Sun Yat-sen University (animal ethics approval no. SYSU-IACUC-2021-000170). The PF model was induced through a single intratracheal instillation of BLM (2 mg/kg, Macklin, Shanghai, China) in male mice. Control animals received an equal volume of intratracheal sterile saline. To explore the dose-response effects of ATD in PF therapy, we selected 2.5, 5, and 10 mg/kg as therapeutic doses in BLM-induced mice. After a 3-day observation period, the mice were randomly divided into six groups (n = 12, mice per group): (1) saline + CMC-Na, vehicle control group, (2) BLM + CMC-Na, BLM control group, (3) BLM + ATD (2.5 mg/ kg/day), low dose of ATD-treated group, (4) BLM + ATD (5 mg/kg/day), medium dose of ATD-treated group, (5) BLM + ATD (10 mg/kg/day), high dose of ATD-treated group, and (6) BLM + PFD (300 mg/kg/day), positive group. On day 1 after BLM induction, mice in ATD groups and the PFD group were intragastrically administered ATD or PFD dissolved in 0.5% CMC-Na while mice in the vehicle group and BLM group were intragastrically administered an equal volume 0.5% CMC-Na for 21 days. Body weight was measured every 3 days.
Pulmonary function assay
At the end of the trial, the respiratory parameters of mice were measured by whole-body plethysmography (EMKA pulmonary system, USA) in conscious unrestrained subjects. The respiratory parameters include Penh, EEP, end-inspiratory pause, minute volume, relaxation time, and peak inspiratory flow. Finally, the system software automatically recorded and displayed the pulmonary function parameters.
Micro-CT of the Lung
Three mice in each group were anesthetized with 0.5% pentobarbital and fixed in supine position. The degree of PF in mice of each group was evaluated using a Latheta LCT-200 (Hitachi Aloka Medical, Japan).
Serum biochemical assays
The levels of alanine transaminase, aspartate transaminase, alkaline phosphatase, cholesterol, CREA, glucose, lactate dehydrogenase, blood urea nitrogen, creatine kinase, total bile acids, and triglyceride in serum were determined using certain commercial assay kits (Guangzhou Donglin Biotechnology, China) using an automatic biochemical analyzer (3100, Hitachi, Japan). Fasting adiponectin and insulin were quantified in plasma samples. The secretion levels of TGF-β1, VEGFA, TNF-α, IL-6, and CXCL1 in the BALF and serum were assessed by corresponding commercially available Elisa kits (Cloud-Clone Crop).
Morphological and histology analysis
Afterward, the mice were sacrificed, and their whole lungs were quickly removed and weighed. The organ coefficient was calculated using the following formula: organ coefficient = (organ weight (g))⁄(body weight (g)) ×100%. The left lung tissues were fixed immediately in 4% paraformaldehyde for 48 h, embedded in a paraffin block, and cut into 5 μm sections. The sections were stained with H&E and Masson trichrome staining to assess histopathological changes in the lungs. H&E staining and Masson staining were used to semi-quantitatively analyze the degree of alveolitis and PF.58,59
Immunohistochemical analysis
Paraffin-embedded lung slices were deparaffinized and rehydrated, and antigen retrieval was performed under high pressure and temperature in 0.01 M sodium citrate buffer (pH 6.0). To quench the endogenous peroxidase activity, the slices were then incubated with 1% hydrogen peroxide in methanol for 10 min at room temperature. Then slices were incubated with corresponding primary antibodies. After washing, horseradish peroxidase (HRP)-conjugated secondary antibodies specific to the species of the primary antibodies were used.
Measurement of hydroxyproline assay
The content of hydroxyproline (HYP) reflects the metabolism of collagen and its regulation. In this study, HYP content in the lung tissues was measured by the alkali hydrolysis method.60 First, 20 mg of the fresh right lung tissue was cut and weighed accurately, and then put into a test tube and 2 mL hydrolysate was added. Next, the test tube was placed in a boiling water bath for 2 h for the hydrolysis reaction. The lysate was centrifuged at 16,000 × g and 25°C for 20 min. After the lysate was cooled to room temperature, the pH value of the lysate was adjusted to 6.0–8.0. Distilled water was added to the supernatant to a volume of 4 mL. The following procedures complied with the instructions of the HYP assay kit instructions for the HYP assay kit (Solarbio, China, no. BC0255). Finally, the absorbance of each sample at 560 nm was detected and the HYP content calculated using the following formula:
Western blot analysis
After washing with cold PBS, cells were lysed in a radioimmunoprecipitation (RIPA) solution (Beyotime) with a protease inhibitor cocktail. Total protein was centrifuged at 12,000 × g and 4°C for 15 min. The supernatant was collected, and the total protein concentration was detected using the Pierce bicinchoninic acid protein assay kit (BCA, Thermo Fisher Scientific). The total protein was diluted in SDS-PAGE protein loading buffer (Yeasen Biotechnology, Shanghai, China, no. 20315ES05) and denatured at 100°C for 10 min. An equal quantity of protein was loaded and separated on 10% SDS-PAGE gels. After electrophoresis, proteins were transferred to polyvinylidene difluoride membranes (Millipore, Burlington, MA). The membranes were blocked with 5% fat-free milk for 1 h at room temperature and incubated with primary antibodies overnight at 4°C, washed in Tris-buffered saline with 0.1% Tween 20 three times, and the membranes were incubated with corresponding HRP-conjugated secondary antibodies at room temperature for 1.5 h. The signal was visualized with an ECL detection system and analyzed by densitometric scanning. The band intensity was quantified using the ImageJ software (Bio-Rad).
ATD target identification
HFL1 cells were exposed to 20 μM of ATD-biotin (Figures S10–S13) or the addition of either biotin for 2 h at 37°C. HFL1 cells were lysed in RIPA. The streptavidin agarose (Sigma-Aldrich, no. S1638) was then washed three times in the wash buffer (PBS supplemented with 0.05% Tween 20 and 0.1% BSA). The lysates were preincubated with streptavidin agarose overnight at 4°C. Individual fractions were separated via SDS-PAGE and stained with Coomassie blue. A relevant gel slice was excised and VIM identity was identified by liquid chromatography-tandem mass spectrometry.
Protein expression and purification
Human VIM with an N-terminal 6× His-tag is recombinantly expressed in Escherichia coli and purified from inclusion bodies. The VIM cDNA-coding region was synthesized and cloned into the BamHI and EcoRI sites of pET28a. Constructs were transformed into BL21(DE3) E. coli competent cells by heat shock and induced with 1 mM isopropyl β-D-thiogalactopyranoside at OD600 of 0.7. Cells were pelleted by centrifugation at 5,000 × g and 30 min, resuspended in 50 mL 20 mM phosphate buffer (PB, pH 7.5), 500 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF), 80 μL deoxyribonuclease I (50 mg/mL), and 80 μL RNase A (10 mg/mL), and lysed using sonication for 45 min (3 s on, 3 s off), then centrifuged at 1,200 × g and 20 min, with repeated sonication and centrifugation. The lysate was resuspended in 50 mL 20 mM PB (pH 7.5), 500 mM NaCl, and 2 M urea for 1 h, followed by centrifugation. The inclusion bodies were dissolved in 50 mL 20 mM PB (pH 7.5), 500 mM NaCl, and 8 M urea. Cellular debris was removed by centrifugation at 12,000 × g for 20 min. The fusion product was purified on a pre-equilibrated BeaverBeads IDA-Nickel (Beaver, China, no. 70501) and eluted with 20 mM PB (pH 7.5), 500 mM NaCl, 5–500 mM imidazole, and 8 M urea. Purified VIM appears at 100–500 mM imidazole. Vimentin renaturation was initiated by a 30-min stepwise dialysis (8, 6, 4, 2, 1, and 0 M urea in 2 mM PB [pH 7.5]) at 8°C to reduce urea concentration. Finally, tetramer formation was induced by additional overnight dialysis against 2 mM PB at pH 7.5 and 8°C.
NMR experiments
WaterLOGSY experiments were conducted using the pulse sequence as described by Dalvit et al.61 All spectra were recorded at a 1H frequency of 600 MHz using a Bruker Avance IIIT 600HD (Bruker BioSpin). All experiments used 5-mm diameter NMR tubes with a sample volume of 500 μL. Solutions were buffered using 20 mM PB (pH 7.5) dissolved in 50% D2O.
SPR analysis
The binding affinity of ATD to VIM was measured using Biacore 8K (GE Healthcare) with a CM5 sensor chip (GE Healthcare). Solutions of compounds were prepared with a running buffer by serial dilutions from stock solutions. The samples were then injected at a flow rate of 30 μL/min for 120 s of the association phase, followed by 100 s of the disassociation phase at 25°C. Data were analyzed using Biacore Insight Evaluation software.
CD spectroscopy
CD spectra of VIM (5 μM) and VIM ATD (5 μM) complexes were recorded on the Chirascan Operation and Good Practice (Applied Photophysics, UK) over the spectral range 260−190 nm.
Pull-down assay
Binding assays were performed by adding 1 μg of VIM protein to 100 μL of 20 mM Tris buffer (pH 7.4). The proteins and 10 μM biotin and 10 μM ATD-biotin were incubated for 1 h at 37°C with ATD as a competitor. The samples were diluted in an SDS-PAGE protein loading buffer and denatured at 100°C for 10 min. The samples were separated by SDS-PAGE and immunoblotted for anti-biotin.
Collection of VIM subunits
HFL1 cells were exposed to 20 μM of ATD for 24 h at 37°C. Cell lysates were centrifuged at 12,000 × g and 15 min, and supernatants were collected. Vimentin subunits were crosslinked with 0.005% glutaraldehyde and detected by western blotting.
Cellular thermal shift assay
HFL1 cells were treated with ATD or DMSO for 1 h. Cells were washed with PBS, trypsinized, and resuspended in PBS containing PMSF in each respective tube. The cell suspension was allocated into 10 different 0.2 mL PCR tubes with 100 μL of volume in each tube. Cells were heated at 40°C–67°C for 3 min using a thermal cycler (Bio-Rad) and removed at room temperature for 3 min. Then, cell suspensions were immediately snap-frozen in liquid nitrogen. Cells were frozen-thawed twice using liquid nitrogen and a thermal circulator. Cells lysates were centrifuged at 12,000 × g for 20 min at 4°C. Soluble proteins were detected by western blotting.
Transmission electron microscopy of VIM filaments
VIM (0.5 mg/mL) was mixed with ATD (20 μM) or DMSO in filament polymerization buffer (170 mM NaCl, 100 mM KCl, 5 mM MgCl2 final concentration) and incubated for 30 min at 37°C. Protein was immediately fixed in 0.5% glutaraldehyde, stained with uranyl acetate, and applied to copper grids for EM staining (University of Kentucky Core Microscopy and Imaging Facilities). Over 100 grids for each treatment were viewed at 80 kV on a JEM-1400 electron microscope (JEOL, Japan) and 25 representative images were collected. The entire experiment was then repeated.
MD simulation
MOE (The Molecular Operating Environment, Chemical Computing Group) version 2019.0102 was employed to perform the docking. The structure of dimer-VIM was predicted with AlphaFold2 (Alphafold2-multimer). Then we developed the three-dimensional model of tetrameric VIM with MOE modeling based on the dimer-VIM structure. The potential binding sites were predicted by MOE. The docking workflow followed the “induced fit” protocol, which allows the side chains of the receptor pocket to move according to the ligand conformations, with a constraint on their positions. All docked poses of ATD were ranked by London dG scoring first, and then a force field refinement was carried out on the top 10 poses followed by a rescoring of GBVI/WSA dG. Then, the system was minimized and equilibrated, and 100-ns long production MD simulations were performed with AMBER 2018. To prevent the α helix transforming, position restraints (5 kcal/mol/Ų) were used on atoms of main chains. During the MD simulations, the equations of motion were integrated with a 2-fs time step in the NPT ensemble. The SHAKE algorithm was applied to all hydrogen atoms. The van der Waals cutoff was set to 8 Å. The temperature was maintained at 310 K, employing the Nosé-Hoover thermostat method with a relaxation time of 1 ps. Long-range electrostatic forces were taken into account using the particle-mesh Ewald approach. Data were collected every 10 ps during the MD runs. Molecular visualization of the system and MD trajectory analysis were carried out with the PYTRAJ software. The last stable 50 ns dynamic trajectory was selected for cluster analysis, and the main constructions were extracted and displayed for graphical representation with PyMOL.
Transfection studies
For loss of VIM function, VIM shRNA was synthesized in pLKO.1-TRC-copGFP-2A-PURO by Tsingke (Beijing, China). Scramble shRNA served as a control. Details on the plasmids are provided in Table S6. The shRNA transfections were performed using Lipofectamine 3000 Transfection Reagent (Invitrogen) according to the manufacturer’s instructions. For VIM overexpression, full-length VIM was cloned into the pCMV6-Entry vector. The pCMV6-Entry served as the empty control vector. Cells were transfected for 24 h and subsequently treated with vehicle or 1,000 nM ATD in the presence or absence of 5 ng/mL TGF-β1 stimulation for 48 h.
Biotinylation assay for TGF-βRI recycling
Biotinylation assays for TGF-βRI recycling refer to previously published methods.42,62 Serum-starved HFL1 cells were first treated with DMSO or ATD (1 μM) and chloroquine (100 μM) for 4 h before the following experiments. To quantitate plasma membrane TGF-βRI, cells were placed on ice, washed twice with ice-cold PBS, incubated with 0.2 mM biotin in PBS at 4°C for 30 min, and washed twice with 0.1 M glycine. Biotinylated proteins were pulled down with streptavidin agarose (Sigma-Aldrich, S1638) and immunoblotted for TGF-βRI. To quantitate endogenous TGF-βRI endocytosis, cells were labeled with cleavable biotin. Cells were resuspended in culture medium and incubated at 37°C to allow TGF-βRI endocytosis. After 30 min, the cells were then returned to 4°C and washed once with ice-cold PBS to stop membrane trafficking. To strip the remaining biotin from the cell surface, cells were treated twice with stripping buffer (50 mM glutathione, 75 mM NaCl, 10 mM EDTA, 1% BSA, 0.075 N NaOH) at 4°C for 15 min. Cell lysates were then subjected to streptavidin agarose pull-down and immunoblotted for TGF-βRI. To quantitate TGF-βRI recycling at 37°C, cells were labeled with cleavable biotin and incubated for 30 or 60 min at 37°C. Cells were treated without stripping buffer and harvested at the end of incubation or subjected to two additional washes at 4°C for 15 min with stripping buffer to ensure the complete de-biotinylated of recycled TGF-βRI. Cell lysates were subjected to streptavidin agarose pull-down and analyzed by immunoblotting. The recycling rate of TGF-βRI after 60 min of incubation was calculated by the following formula: TGF-βRI recycling rate = (internalized and recycled TGF-βRI at 60 min – internalized TGF-βRI at 60 min)/total internalized TGF-βRI at 30 min × 100%.
Antibodies for western blotting
Antibodies used for western blotting and immunofluorescence assays are listed in Table S7.
Statistical analysis
The data are represented as the mean ± SEM. Statistical analysis was performed using the GraphPad Prism 6.0 software (San Diego, CA). The significant differences between two groups were statistically analyzed using an unpaired Student’s t test. The significant differences between various groups were statistically analyzed using the one-way analysis of variance followed by a post hoc test (least significant difference). All differences were considered statistically significant at p < 0.05. The numbers of technical replicates or biological replicates in each group were stated in the figure legends.
Acknowledgments
This research was supported by the Fund of State Key Laboratory of Phytochemistry and Plant Resources in West China (E2654291z1) and the Administration of Traditional Chinese Medicine of Guangdong Province, China (20231069), and the Key Area Research and Development Program of Guangdong Province (no. 2020B1111110003).
Author contributions
M.H., Q.G., and J.X. conceived and planned the experiments. M.H. carried out the experiments and data analyses. Z.G. assisted with the animal experiments. Z.Z. assisted with the molecular dynamics simulation. H.A. supported the synthesis of the ATD-biotin. X.P. and Q.G. extracted and purified the natural products studied in this work. H.Z. supported guidance on the expression of protein. M.H. wrote the initial manuscript. M.H., Q.G., and J.X. finalized the revised manuscript. All authors provided critical feedback and helped shape the research, analysis, and manuscript. All authors agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of interests
Q.G., J.X., and M.H. are filing a patent application based on this work.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.08.017.
Contributor Information
Jun Xu, Email: xujun9@mail.sysu.edu.cn.
Qiong Gu, Email: guqiong@mail.sysu.edu.cn.
Supplemental information
Data and code availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplemental information.
References
- 1.Martinez F.J., Collard H.R., Pardo A., Raghu G., Richeldi L., Selman M., Swigris J.J., Taniguchi H., Wells A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers. 2017;3 doi: 10.1038/nrdp.2017.74. [DOI] [PubMed] [Google Scholar]
- 2.Maher T.M., Bendstrup E., Dron L., Langley J., Smith G., Khalid J.M., Patel H., Kreuter M. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir. Res. 2021;22:197. doi: 10.1186/s12931-021-01791-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.George P.M., Wells A.U., Jenkins R.G. Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. Lancet Respir. Med. 2020;8:807–815. doi: 10.1016/S2213-2600(20)30225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.WHO Coronavirus (COVID-19) Dashboard. 2022. https://covid19.who.int/
- 5.Hama Amin B.J., Kakamad F.H., Ahmed G.S., Ahmed S.F., Abdulla B.A., mohammed S.H., Mikael T.M., Salih R.Q., Ali R.k., Salh A.M., Hussein D.A. Post COVID-19 pulmonary fibrosis; a meta-analysis study. Ann. Med. Surg. 2022;77 doi: 10.1016/j.amsu.2022.103590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mora A.L., Rojas M., Pardo A., Selman M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017;16:810. doi: 10.1038/nrd.2017.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao M., Wang L., Wang M., Zhou S., Lu Y., Cui H., Racanelli A.C., Zhang L., Ye T., Ding B., et al. Targeting fibrosis: mechanisms and clinical trials. Signal Transduct. Tar. 2022;7:206. doi: 10.1038/s41392-022-01070-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henderson N.C., Rieder F., Wynn T.A. Fibrosis: from mechanisms to medicines. Nature. 2020;587:555–566. doi: 10.1038/s41586-020-2938-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ridge K.M., Eriksson J.E., Pekny M., Goldman R.D. Roles of vimentin in health and disease. Genes Dev. 2022;36:391–407. doi: 10.1101/gad.349358.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z., Wu J., Zhou J., Yuan B., Chen J., Wu W., Mo L., Qu Z., Zhou F., Dong Y., et al. A vimentin-targeting oral compound with host-directed antiviral and anti-inflammatory actions addresses multiple features of COVID-19 and related diseases. mBio. 2021;12:e02542211. doi: 10.1128/mBio.02542-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strouhalova K., Přechová M., Gandalovičová A., Brábek J., Gregor M., Rosel D. Vimentin intermediate filaments as potential target for cancer treatment. Cancers. 2020;12:184. doi: 10.3390/cancers12010184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Z., Paulin D., Lacolley P., Coletti D., Agbulut O. Vimentin as a target for the treatment of COVID-19. BMJ Open Respir. Res. 2020;7 doi: 10.1136/bmjresp-2020-000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vanden Berghe W., Sabbe L., Kaileh M., Haegeman G., Heyninck K. Molecular insight in the multifunctional activities of withaferin A. Biochem. Pharmacol. 2012;84:1282–1291. doi: 10.1016/j.bcp.2012.08.027. [DOI] [PubMed] [Google Scholar]
- 14.Colucci-Guyon E., Portier M.M., Dunia I., Paulin D., Pournin S., Babinet C. Mice lacking vimentin develop and reproduce without an obvious phenotype. Cell. 1994;79:679–694. doi: 10.1016/0092-8674(94)90553-3. [DOI] [PubMed] [Google Scholar]
- 15.Li F.J., Surolia R., Li H., Wang Z., Liu G., Kulkarni T., Massicano A.V.F., Mobley J.A., Mondal S., de Andrade J.A., et al. Citrullinated vimentin mediates development and progression of lung fibrosis. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.aba2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans A.B., Dong P., Loyd H., Zhang J., Kraus G.A., Carpenter S. Identification and characterization of small molecule inhibitors of porcine reproductive and respiratory syndrome virus. Antivir. Res. 2017;146:28–35. doi: 10.1016/j.antiviral.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Xu L., Zhou Y., Xu J., Xu X., Lu G., Lv Q., Wei L., Deng X., Shen X., Feng H., Wang J. Anti-inflammatory, antioxidant and anti-virulence roles of atractylodin in attenuating Listeria monocytogenes infection. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.977051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Q., Liu J., Hu Y., Pan T., Xu Y., Yu J., Xiong W., Zhou Q., Wang Y. Local administration of liposomal-based Srpx2 gene therapy reverses pulmonary fibrosis by blockading fibroblast-to-myofibroblast transition. Theranostics. 2021;11:7110–7125. doi: 10.7150/thno.61085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun Z., Zhang Y., Peng X., Huang S., Zhou H., Xu J., Gu Q. Diverse sesquiterpenoids and polyacetylenes from Atractylodes lancea and their anti-osteoclastogenesis activity. J. Nat. Prod. 2022;85:866–877. doi: 10.1021/acs.jnatprod.1c00997. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y., Peng X., Sun Z., Hu C., Zhou H., Xu J., Gu Q. Diverse polyacetylenes from Atractylodes chinensis and their anti-osteoclastogenesis activity. Fitoterapia. 2022;161 doi: 10.1016/j.fitote.2022.105233. [DOI] [PubMed] [Google Scholar]
- 21.Peng X., Tan Q., Zhou H., Xu J., Gu Q. Discovery of phloroglucinols from Hypericum japonicum as ferroptosis inhibitors. Fitoterapia. 2021;153 doi: 10.1016/j.fitote.2021.104984. [DOI] [PubMed] [Google Scholar]
- 22.Peng X., Tan Q., Wu L., Wu D., Xu J., Zhou H., Gu Q. Ferroptosis inhibitory aromatic abietane diterpenoids from Ajuga decumbens and structural revision of two 3,4-epoxy group-containing abietanes. J. Nat. Prod. 2022;85:1808–1815. doi: 10.1021/acs.jnatprod.2c00352. [DOI] [PubMed] [Google Scholar]
- 23.Sun Z., Zhang Y., Zhou H., Xu J., Gu Q. Diverse diterpenoids and sesquiterpenoids from Siegesbeckia pubescens and their activity against RANKL-induced osteoclastogenesis. Bioorg. Chem. 2021;107 doi: 10.1016/j.bioorg.2020.104537. [DOI] [PubMed] [Google Scholar]
- 24.King T.E., Pardo A., Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z., Qu J., Zheng C., Zhang P., Zhou W., Cui W., Mo X., Li L., Xu L., Gao J. Nrf2 antioxidant pathway suppresses Numb-mediated epithelial–mesenchymal transition during pulmonary fibrosis. Cell Death Dis. 2018;9:83. doi: 10.1038/s41419-017-0198-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Júnior C., Narciso M., Marhuenda E., Almendros I., Farré R., Navajas D., Otero J., Gavara N. Baseline stiffness modulates the non-linear response to stretch of the extracellular matrix in pulmonary fibrosis. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms222312928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato K., Logsdon N.J., Shin Y.J., Palumbo S., Knox A., Irish J.D., Rounseville S.P., Rummel S.R., Mohamed M., Ahmad K., et al. Impaired myofibroblast dedifferentiation contributes to nonresolving fibrosis in aging. Am. J. Respir. Cell Mol. Biol. 2020;62:633–644. doi: 10.1165/rcmb.2019-0092OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lovisa S., LeBleu V.S., Tampe B., Sugimoto H., Vadnagara K., Carstens J.L., Wu C.C., Hagos Y., Burckhardt B.C., Pentcheva-Hoang T., et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015;21:998–1009. doi: 10.1038/nm.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lv X., Liu C., Liu S., Li Y., Wang W., Li K., Hua F., Cui B., Zhang X., Yu J., et al. The cell cycle inhibitor P21 promotes the development of pulmonary fibrosis by suppressing lung alveolar regeneration. Acta Pharm. Sin. B. 2022;12:735–746. doi: 10.1016/j.apsb.2021.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cottin V., Maher T. Long-term clinical and real-world experience with pirfenidone in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2015;24:58–64. doi: 10.1183/09059180.00011514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Derynck R., Zhang Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 33.de Caestecker M.P., Parks W.T., Frank C.J., Castagnino P., Bottaro D.P., Roberts A.B., Lechleider R.J. Smad2 transduces common signals from receptor serine-threonine and tyrosine kinases. Genes Dev. 1998;12:1587–1592. doi: 10.1101/gad.12.11.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Funaba M., Zimmerman C.M., Mathews L.S. Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J. Biol. Chem. 2002;277:41361–41368. doi: 10.1074/jbc.M204597200. [DOI] [PubMed] [Google Scholar]
- 35.Engel M.E., McDonnell M.A., Law B.K., Moses H.L. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J. Biol. Chem. 1999;274:37413–37420. doi: 10.1074/jbc.274.52.37413. [DOI] [PubMed] [Google Scholar]
- 36.Zaidel-Bar R., Cohen M., Addadi L., Geiger B. Hierarchical assembly of cell-matrix adhesion complexes. Biochem. Soc. Trans. 2004;32:416–420. doi: 10.1042/BST0320416. [DOI] [PubMed] [Google Scholar]
- 37.Tsuruta D., Jones J.C.R. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J. Cell Sci. 2003;116:4977–4984. doi: 10.1242/jcs.00823. [DOI] [PubMed] [Google Scholar]
- 38.Soellner P., Quinlan R.A., Franke W.W. Identification of a distinct soluble subunit of an intermediate filament protein: tetrameric vimentin from living cells. Proc. Natl. Acad. Sci. USA. 1985;82:7929–7933. doi: 10.1073/pnas.82.23.7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bargagna-Mohan P., Hamza A., Kim Y.E., Khuan Abby Ho Y., Mor-Vaknin N., Wendschlag N., Liu J., Evans R.M., Markovitz D.M., Zhan C.G., et al. The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem. Biol. 2007;14:623–634. doi: 10.1016/j.chembiol.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di Guglielmo G.M., Le Roy C., Goodfellow A.F., Wrana J.L. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat. Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y.-G. Endocytic regulation of TGF-β signaling. Cell Res. 2009;19:58–70. doi: 10.1038/cr.2008.315. [DOI] [PubMed] [Google Scholar]
- 42.Wang J., Lai X., Yao S., Chen H., Cai J., Luo Y., Wang Y., Qiu Y., Huang Y., Wei X., et al. Nestin promotes pulmonary fibrosis via facilitating recycling of TGF-β receptor I. Eur. Respir. J. 2022;59 doi: 10.1183/13993003.03721-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang D., Zhuang R., Guo Z., Gao M., Huang L., You L., Zhang P., Li J., Su X., Wu H., et al. Desmin- and vimentin-mediated hepatic stellate cell-targeting radiotracer (99m)Tc-GlcNAc-PEI for liver fibrosis imaging with SPECT. Theranostics. 2018;8:1340–1349. doi: 10.7150/thno.22806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao Y.H., Lv L.L., Zhang X., Hu H., Ding L.H., Yin D., Zhang Y.Z., Ni H.F., Chen P.S., Liu B.C. Urinary vimentin mRNA as a potential novel biomarker of renal fibrosis. Am. J. Physiol. Ren. Physiol. 2015;309:F514–F522. doi: 10.1152/ajprenal.00449.2014. [DOI] [PubMed] [Google Scholar]
- 45.Surolia R., Antony V.B. Pathophysiological role of vimentin intermediate filaments in lung diseases. Front. Cell Dev. Biol. 2022;10 doi: 10.3389/fcell.2022.872759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Surolia R., Li F.J., Wang Z., Li H., Dsouza K., Thomas V., Mirov S., Pérez-Sala D., Athar M., Thannickal V.J., Antony V.B. Vimentin intermediate filament assembly regulates fibroblast invasion in fibrogenic lung injury. JCI insight. 2019;4 doi: 10.1172/jci.insight.123253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li F.J., Surolia R., Li H., Wang Z., Kulkarni T., Liu G., de Andrade J.A., Kass D.J., Thannickal V.J., Duncan S.R., Antony V.B. Autoimmunity to vimentin is associated with outcomes of patients with idiopathic pulmonary fibrosis. J. Immunol. 2017;199:1596–1605. doi: 10.4049/jimmunol.1700473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ivaska J., Pallari H.M., Nevo J., Eriksson J.E. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell Res. 2007;313:2050–2062. doi: 10.1016/j.yexcr.2007.03.040. [DOI] [PubMed] [Google Scholar]
- 49.Eckes B., Colucci-Guyon E., Smola H., Nodder S., Babinet C., Krieg T., Martin P. Impaired wound healing in embryonic and adult mice lacking vimentin. J. Cell Sci. 2000;113:2455–2462. doi: 10.1242/jcs.113.13.2455. [DOI] [PubMed] [Google Scholar]
- 50.Kidd M.E., Shumaker D.K., Ridge K.M. The role of vimentin intermediate filaments in the progression of lung cancer. Am. J. Respir. Cell Mol. Biol. 2014;50:1–6. doi: 10.1165/rcmb.2013-0314TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nunes Vicente F., Lelek M., Tinevez J.Y., Tran Q.D., Pehau-Arnaudet G., Zimmer C., Etienne-Manneville S., Giannone G., Leduc C. Molecular organization and mechanics of single vimentin filaments revealed by super-resolution imaging. Sci. Adv. 2022;8 doi: 10.1126/sciadv.abm2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schepers A.V., Lorenz C., Nietmann P., Janshoff A., Klumpp S., Köster S. Multiscale mechanics and temporal evolution of vimentin intermediate filament networks. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2102026118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bollong M.J., Pietilä M., Pearson A.D., Sarkar T.R., Ahmad I., Soundararajan R., Lyssiotis C.A., Mani S.A., Schultz P.G., Lairson L.L. A vimentin binding small molecule leads to mitotic disruption in mesenchymal cancers. Proc. Natl. Acad. Sci. USA. 2017;114:E9903–E9912. doi: 10.1073/pnas.1716009114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu H.H., Chen D.Q., Wang Y.N., Feng Y.L., Cao G., Vaziri N.D., Zhao Y.Y. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 2018;292:76–83. doi: 10.1016/j.cbi.2018.07.008. [DOI] [PubMed] [Google Scholar]
- 55.Grant B.D., Donaldson J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mitchell H., Choudhury A., Pagano R.E., Leof E.B. Ligand-dependent and -independent transforming growth factor-beta receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Mol. Biol. Cell. 2004;15:4166–4178. doi: 10.1091/mbc.E04-03-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y.Y., Huang Y.F., Liang J., Zhou H. Improved up-and-down procedure for acute toxicity measurement with reliable LD50 verified by typical toxic alkaloids and modified Karber method. BMC Pharmacol. Toxicol. 2022;23:3. doi: 10.1186/s40360-021-00541-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Szapiel S.V., Elson N.A., Fulmer J.D., Hunninghake G.W., Crystal R.G. Bleomycin-induced interstitial pulmonary disease in the nude, athymic mouse. Am. Rev. Respir. Dis. 1979;120:893–899. doi: 10.1164/arrd.1979.120.4.893. [DOI] [PubMed] [Google Scholar]
- 59.Hübner R.H., Gitter W., El Mokhtari N.E., Mathiak M., Both M., Bolte H., Freitag-Wolf S., Bewig B. Standardized quantification of pulmonary fibrosis in histological samples. Biotechniques. 2008;44:514. doi: 10.2144/000112729. [DOI] [PubMed] [Google Scholar]
- 60.da Silva C.M.L., Spinelli E., Rodrigues S.V. Fast and sensitive collagen quantification by alkaline hydrolysis/hydroxyproline assay. Food Chem. 2015;173:619–623. doi: 10.1016/j.foodchem.2014.10.073. [DOI] [PubMed] [Google Scholar]
- 61.Dalvit C., Fogliatto G., Stewart A., Veronesi M., Stockman B. WaterLOGSY as a method for primary NMR screening: practical aspects and range of applicability. J. Biomol. NMR. 2001;21:349–359. doi: 10.1023/a:1013302231549. [DOI] [PubMed] [Google Scholar]
- 62.Liu Y., Tao Y.M., Woo R.S., Xiong W.C., Mei L. Stimulated ErbB4 internalization is necessary for neuregulin signaling in neurons. Biochem. Biophys. Res. Commun. 2007;354:505–510. doi: 10.1016/j.bbrc.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplemental information.