

Abstract
NK cells are major effector cells involved in the elimination of early tumors and prevent metastasis. They often have an impaired function in patients with cancer. Preclinical studies have demonstrated NK cell activation as the adjunctive effect of invariant NKT (iNKT) cells. Activation of iNKT cells after administration of the glycolipid ligand α-galactosylceramide, loaded with CD1d-expressing human PBMC-derived APCs (APC/Gal), is an attractive cancer therapy to optimize the use of NK cells. However, the subsets of NK cells that are activated following iNKT cell activation as well as the period of NK cell activation remain unclear. In this study, we report that the granzyme B–expressing NK cell response in postoperative lung cancer patients was enhanced 49 d after administration of APC/Gal in a phase II study. We found maximum IFN-γ production on day 49 in 13 out of 27 APC/Gal-treated patients. On day 49, 14 out of 27 patients (51.9%) had higher IFN-γ production by iNKT cells (>6-fold higher than the baseline level). This increment significantly correlated with granzyme B–expressing NK cells. Although IFN-γ production was lower in patients in the nontreated group, we detected maximum IFN-γ production 12 mo after the resection of lung cancer (9 out of 29 patients [31%]). These findings suggest that elimination of cancer cells leads to increased NK cell function, which can be further enhanced by APC/Gal therapy.
Introduction
Lung cancer is the leading cause of cancer incidence (11.6% of all cases) and mortality (18.4% of total cancer deaths) according to a recent status report on worldwide cancer (1). Particularly, non–small cell lung cancer (NSCLC) represents 80% of all lung cancer cases, and more than half of the patients with early-stage (stage I or II) cancers are generally cured by standard therapy, that is, surgery and additional platinum-based chemotherapy or radiation (stage I, 70–90%, stage II, 40–70%). However, some patients experience disease recurrence within the first 5 y of surgery (2). To prevent this recurrence, several adjuvant therapies following the completion of first-line therapy have been trialed. Standard chemotherapy frequently causes unfavorable complications. Therefore, additional radiation therapy and/or chemotherapy are used. Recently, immunotherapy is emerging as a new effective treatment for lung cancer (3, 4). Particularly, anti–PD-1 Ab therapy has beneficial effects in patients with NSCLC; however, its efficacy is still suboptimal. This highlights the need for novel immunotherapies with an alternate mechanism and low toxicity.
The NK cell frequency is associated with a favorable prognosis in many cancers (5–7). Tumor cells and associated stromal cells have shown immunosuppression of NK cells by secreting TGF-β, IL-10, and PGE2. NK cells from patients with NSCLC are lower in number and have impaired IFN-γ production and lower cytotoxicity (8, 9). The CD56brightCD16dim subset is enriched in NSCLC tumor samples and exhibits increased cytokine release but low cytotoxicity (10). It further produces proangiogenic factors, such as VEGF, PlGF, and IL-8, in tumor-bearing hosts promoting tumor growth and metastases (11). Therefore, the restoration of NK cell responses is a desirable therapeutic concept targeted against malignancy. The efficacy of NK cell therapies is currently being explored, for example, NK cell therapies including autologous and allogeneic NK cells, CAR-NK cells, and combination with immune checkpoint blockade therapies (7). To activate NK cells systemically, we have focused on invariant NKT (iNKT) cell activation.
iNKT cells in humans express a Vα24-Jα18 rearranged TCRα chain associated with a Vβ11 TCRβ chain (12, 13). iNKT cells are capable of producing a large number of cytokines, such as IFN-γ, TNF-α, IL-2, and IL-4, when stimulated by a ligand such as α-galactosylceramide (α-GalCer) (14, 15). Because the CD1d molecule resembles MHC class I molecules and has high homology across species (16, 17), human CD1d+ cells loaded with α-GalCer stimulate murine iNKT cells. Trans-species iNKT activation between humans and mice has been shown to be possible (18). Generally, iNKT cells can exert potent NK cell–mediated antitumor activity. At this point, IL-2 and IFN-γ production by iNKT cells is known as an important factor for NK cell activation (19). In fact, the antitumor effect of NK cells was enhanced by IL-2 and IFN-γ released from α-GalCer–stimulated iNKT cells in mice (20). In humans, we have demonstrated that after the activation of iNKT cells in vitro, NK cell cytotoxicity against tumor cells was enhanced (21). Thus, we primarily focused on the NK cell response in this study.
For iNKT cell activation, immunotherapy using α-GalCer–pulsed dendritic cells (DCs) (DC/Gal) showed tumor eradication (22, 23). Several clinical trials using monocyte-derived DC/Gal have also confirmed iNKT cell expansion, cytokine production, and tolerability of this therapy (24–28). Whole-cultured PBMCs were also used for pulsing α-GalCer (i.e., α-GalCer loaded with CD1d-expressing human PBMC-derived APCs [APC/Gal]) (29), instead of DCs, in which whole-cultured PBMCs cultured with α-GalCer were verified to expand and activate iNKT cells. Using this approach, several clinical studies in patients with advanced NSCLC in phase I and II clinical trials were conducted (29–32). Patients with advanced NSCLC who completed the standard treatment were enrolled, and the median survival time was relatively prolonged without severe adverse events (31, 33). These studies have provided evidence of tolerability as well as restoration of iNKT cell function following APC/Gal therapy. In this study, we evaluated the iNKT and NK cell functional response in APC/Gal-treated or nontreated groups involving patients with postoperative early-stage NSCLC and low tumor burden. Particularly, we evaluated the NK cell response following iNKT activation.
Materials and Methods
Patient eligibility criteria
The inclusion criteria were as follows: 1) patients with histopathological confirmation of NSCLC; 2) hilar and mediastinal lymph node were deserted, which were, together with the lobe, selectively resected; 3) complete resection of NSCLC (complete resection is defined as the tumor being completely removed macroscopically at the time of surgery and the pathological absence of tumor cells in the resection line); 4) pathological stages IIA, IIB, and IIIA (the Union Internationale Contre le Cancer and American Joint Committee on Cancer staging system, 7th Ed.) (34); 5) patients between 20 and 75 y of age; 6) no tumor recurrence observed; 7) patients with a performance status of the Eastern Cooperative Oncology Group score of 0 or 1; 8) combined adjuvant chemotherapy (three to four cycles) of cisplatin (total ≥200 mg/m2) and vinorelbine (total 100 mg/m2 or more), and final treatment was performed: at 4 wk after administration and within 16 wk, 9) the function of major organs (e.g., bone marrow, liver, kidney) was sufficiently maintained in clinical examination within 4 wk before enrollment, and with the following criteria: leukocyte count ≥3000/ml, thrombotic count ≥75,000/μl, hemoglobin ≥9.0 g/dl, serum creatine ≤1.5 mg, total bilirubin ≤2.0 mg/dl, aspartate aminotransferase (glutamic oxaloacetic transaminase) and alanine aminotransferase (glutamic pyruvic transaminase) ≤100 U/l, and oxygen saturation (room air) ≥93%; 10) the number of iNKT cells was ≥10 cells/ml in peripheral blood; and 11) patients who gave written consent after a sufficient explanation of the study purpose and procedures before enrollment in this study.
Clinical protocol and study design
The study was conducted in National Hospital Organization (NHO) Nagoya Medical Center (Aichi, Japan) and NHO Kyushu Cancer Center (Fukuoka, Japan), according to the standards of Good Clinical Practice for Trials on Medicinal Products in Japan. The protocol and related documents were reviewed and approved by the central certified committee for regenerative medicine of NHO (35). This trial was registered at the University Hospital Medical Information Network Center as UMIN000010386 and jRCTc040190130. The primary endpoint was recurrence-free survival, and the secondary endpoints were an iNKT cell–specific immune response, the frequency of toxic effects and safety, and overall survival. The study was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all patients before screening evaluation to determine eligibility.
Preparation of APC/Gal
All procedures were performed according to good manufacturing practice standards. Eligible patients in the APC/Gal therapy group underwent peripheral blood leukapheresis (Spectra; COBE), and PBMCs were separated by density gradient centrifugation (OptiPrep; Axis-Shield). Thereafter, whole PBMCs were cultured with GM-CSF and IL-2, as previously described (31). Briefly, PBMCs were washed three times and resuspended in AIM-V (Invitrogen) with 800 U/ml human GM-CSF (Genentech) and 100 Japan reference units/ml recombinant human IL-2 (Immunace; Shionogi). The whole cultured cells were pulsed with 100 ng/ml of a specific ligand, α-GalCer (KRN7000, good manufacturing practice grade), on the day before administration. After 7 or 14 d of cultivation, the cells were harvested, washed three times, and resuspended in 100 ml of 2.5% albumin in saline (APC1/Gal, APC2/Gal, APC3/Gal, and APC4/Gal were generated). APC/Gal (1 × 109 cells/m2/injection) was i.v. administered four times.
Human samples and preparation
Peripheral blood samples from 56 patients with NSCLC who were enrolled in this phase II study were obtained from 15 NHOs. PBMCs were washed twice with PBS (Nacalai Tesque, Kyoto, Japan) and stored in liquid nitrogen until use. All experiments were performed with authorization from the Institutional Review Board for Human Research at the NHO and RIKEN Center for Integrative Medical Sciences. Written informed consent was obtained from all healthy donors and patients according to the Declaration of Helsinki. HLA-A24+ donors were selected using flow cytometry.
Flow cytometry analysis
The surface phenotypes of PBMCs and APC/Gal were determined by a FACSCanto II, LSRFortessa X-20, or FACSCalibur (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (Tree Star, Ashland, OR). The following mAbs were from BioLegend (San Diego, CA): anti-human mAbs; FITC-conjugated anti-CD3, anti-CD8, anti-CD45RA, and lineage mixture; Alexa Fluor 488–conjugated anti-CD45; allophycocyanin-conjugated anti-CD3, anti-CD141, and anti-HLA-DR; Alexa Fluor 647–conjugated anti-CD14; allophycocyanin-Cy7–conjugated anti-CD16; PE-conjugated anti-CD1d, anti-CD8, anti-CD33, and anti-CD56; PE-Cy7–conjugated anti-CD3, anti-CD11c, anti-CD226, anti-NKG2D, and anti-DNAM-1; PerCP-conjugated anti–HLA-DR; PerCP-Cy5.5–conjugated anti-CD1c and anti-CD4; and Pacific Blue–conjugated anti-CD4, anti-CD11b, anti-CD16, anti-CD19, anti–HLA-DR, and anti–granzyme B. Anti-human Vα24-FITC and anti–Vβ11-PE mAbs were purchased from Beckman Coulter (Fullerton, CA). FITC-conjugated anti-CD8 and anti-CD15, PE-conjugated anti-CD123, Alexa Fluor 700–conjugated anti-CD11c, and BUV395-conjugated anti-CD3 Abs were purchased from BD Biosciences (San Jose, CA). PE-conjugated anti-Foxp3 Ab was purchased from eBioscience (San Diego, CA). A Fixable Aqua or Violet Dead Cell Stain Kit (Thermo Fisher Scientific) was used to eliminate dead cells. Intracellular staining for granzyme B and Foxp3 was performed using a BD intracellular cytokine staining kit (BD Biosciences) and eBioscience Foxp3 staining buffer kit (Thermo Fisher Scientific), respectively. Gating strategies for iNKT cells and NK cells are shown in Supplemental Fig. 1.
Mice
Specific pathogen-free 6- to 8-wk-old C57BL/6 mice were purchased from Charles River Laboratories (Kanagawa, Japan). All mice were maintained under specific pathogen-free conditions and studied in compliance with RIKEN institutional guidelines.
α-GalCer presentation by APC/Gal
In addition to the evidence that murine iNKT cells recognize α-GalCer on human CD1d and proliferate (18), an approach to evaluate the iNKT cell–stimulating function of human APCs including DC/Gal was established previously (36). Using this method, APC/Gal (1 × 106 cells/mouse) was injected to mice i.v., and spleen cells were harvested 4 d after immunization. The frequencies of iNKT cells in the spleen were analyzed using flow cytometry.
CMV-specific T cell response
PBMCs from patients treated with APC/Gal were cultured in the presence of CMVpp65 matrix protein-derived CMV peptide (QYDPVAALF aa 341–349). Seven days later, CMV-specific CD8+ T cells were analyzed using HLA-A*2402-CMVpp65 tetramer staining.
ELISPOT assay
For detecting IFN-γ–secreting cells, 96-well filtration plates (Millipore) were coated overnight at 4°C with anti-human IFN-γ (BD Biosciences), according to the manufacturer’s instructions. Cells were then resuspended (5 × 105/well) and stimulated with α-GalCer (100 ng/ml) for 16 h in complete medium (RPMI 1640 [Sigma-Aldrich] containing 10% FBS [Sigma-Aldrich], 10 mM HEPES [Nacalai Tesque], 50 U/ml penicillin [Life Technologies], 50 μg/ml streptomycin [Life Technologies, Grand Island, NY], and 55 μM 2-ME [Life Technologies]). PMA (1 μg/ml) plus ionomycin (1 μM) was used as a positive control. After cultivation, the plates were washed and incubated with biotinylated anti–IFN-γ. The color development was performed using 3,3′-diaminobenzidine substrate solution (Falma, Tokyo, Japan). Spot-forming cells (SFCs) were quantified by microscopy.
Quantitative PCR assay for NK cells
For gene-specific reverse transcription and quantitative PCR (qPCR), oligonucleotide primer pairs and corresponding hydrolysis probes (UPL Probes) were designed at the Universal ProbeLibrary Assay Design Center on the Roche Diagnostics Web site. Primers used were as follows: IFNG forward, 5′-GGCATTTTGAAGAATTGGAAAG-3′, IFNG reverse, 5′-TTTGGATGCTCTGGTCATCTT-3′; GZMB forward, 5′-AGATGCAACCAATCCTGCTT-3′, GZMB reverse, 5′-CATGTCCCCCGATGATCT-3′; GAPDH forward, 5′-AGCCACATCGCTCAGACAC-3′, GAPDH reverse, 5′-GCCCAATACGACCAAATCC-3′. Probes as follows: probe no. 21 (Roche Diagnostics, 04686942001) for IFNG, probe no. 18 (Roche Diagnostics, 04686918001) for GZMB, and probe no. 60 (Roche Diagnostics, 04688589001) for GAPDH. To obtain cDNA, NK cells were sorted using FACSAria, and every 100 cells were directly lysed in 10 µl of CellsDirect reaction buffer (Thermo Fisher Scientific) containing reverse transcriptase, Taq DNA polymerase, and a set of 200 nM primers. The lysates were then subjected to RT-PCR (30 min at 50°C followed by 2 min at 94°C) and subsequent pre-PCR (18 cycles of 30 s at 94°C and 4 min at 60°C) on a thermal cycler.
For qPCR, the reaction mixture contained a diluted aliquot of the above cDNA as a template, 500 nM of the same but single primer pair used for pre-PCR, 250 nM UPL probe, and FastStart universal probe master (ROX; Roche). The qPCR consisted of an initial activation step of 10 min at 95°C, followed by 40 cycles of 10 s at 95°C and 30 s at 60°C. qPCR was performed on a StepOne Plus (Thermo Fisher Scientific), and the average of duplicated results was analyzed using the comparative cycle threshold method with GAPDH expression as a reference.
Genetic analysis of APC/Gal-treated patients
To elucidate genomic variants associated with the interindividual variability of the clinical outcomes and immune responses in 27 APC/Gal-treated patients, we conducted a genome-wide association study (GWAS), for which 659,184 variants were genotyped using the Illumina Infinium Asian Screening Array-24 kit (version 1, Illumina, San Diego, CA). Quality control of subjects was performed using the following exclusion criteria: 1) sample call rate <0.98, 2) closely related individuals with a genome-wide identity-by-descent score (PI_HAT) ≥0.175 identified using the PLINK version 1.9.0 software, and 3) outliers from Asian clusters estimated by principal component analysis with 1000 Genomes Project samples using the EIGENSOFT software (version 7.2.1). Variants satisfying the following criteria were also excluded: 1) single-nucleotide polymorphism (SNP) call rate <0.99, 2) minor allele frequency <0.01, and (3) Hardy–Weinberg equilibrium p value ≤1.0 × 10−6. Whole-genome imputation was performed on the reference genome (hg19) to maximize genetic coverage. Haplotype phasing of the GWAS data was performed using the SHAPEIT2 software, and genotype dosages were imputed using the IMPUTE2 software with the Japanese reference panel, which integrates whole-genome sequence data of 1000 Genomes Project phase 3.5. After applying the probability threshold of 0.9 to obtain high-quality genotype calls, the number of imputed variants in the patients was 13,821,119. To reduce false-positive results after statistical analysis, variants satisfying the following criteria were excluded: minimum minor allele count <5, missingness <0.01, and Hardy–Weinberg equilibrium p value ≤1 × 10−6. Finally, we obtained 3,111,052 variants for autosomal chromosomes. The GWASs of overall survival and recurrence-free survival were performed using log-rank test in survival, an R package for survival analysis. We also conducted GWASs using a linear mixed model in the PLINK software between the imputed genotypes and the fold change of IFN-γ production or that of granzyme B–expressing NK cells before and 49 d after the APC/Gal treatment. All genetic analyses were performed by the dominant model. We set the genome-wide association significance threshold to a p value < 5.0 × 10−8. This study was reviewed and approved by Central Review Board of NHO.
Statistical analysis
All analyses for clinical outcomes were conducted on the full analysis set. Recurrence-free survival and overall survival within 2 y were evaluated as the primary and secondary endpoints, respectively. The survival probabilities were estimated using the Kaplan–Meier method and compared between treatment groups by log-rank tests. Hazard ratios (APC/Gal-treated/nontreated) for recurrence and death were also evaluated using Cox proportional hazard regression analyses. Statistical analyses were performed using R software version 3.6.3 (R Foundation for Statistical Computing) and StatMate (version 5.01). Differences were analyzed using a two-tailed Pearson’s test of correlation coefficients, χ2 and Fisher’s exact tests, or the ANOVA Tukey–Kramer method. The significance threshold was set to a two-sided α value of 0.05.
Results
Patient characteristics
A scheme of the study design is shown in (Fig. 1. Patients received a total of four i.v. injections of APC/Gal. Before recruitment, all patients were assessed by clinical history, physical examination, and laboratory data, including complete blood cell count and blood chemistry. Physical examination and laboratory data assessments were conducted at least once a week in the APC/Gal therapy group, and at least once a month in the nontreated control group. Adverse events and changes in laboratory values were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 3.0). For immune monitoring, PBMC samples were obtained from patients twice before APC/Gal administration and 10 times after APC/Gal administration (Fig. 1C), and PBMCs were obtained from blood samples of patients in the nontreated control group, as indicated in (Fig. 1D. A total of 57 patients, from June 2013 to September 2018, fulfilled the eligibility criteria of this study and were enrolled. The profile of the 56 patients is summarized in Table I. Twenty-seven patients were treated using APC/Gal therapy, and 26 of them completed the trial. The nontreated group (control) included 29 patients (Fig. 1A).
FIGURE 1.
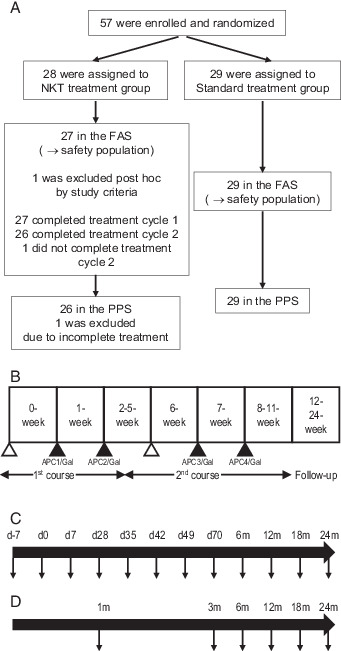
Design of an open-label study randomized trial using α-GalCer–pulsed APCs.
(A) Postoperative patients with NSCLC separated into two groups (APC/Gal therapy or nonimmunotherapy) in this open-label randomized trial. (B) Study design of α-GalCer–pulsed APC (APC/Gal) administration. The timing of apheresis (△) and APC/Gal (▲) administration is shown. The first administration was defined as day 0. The patients received two courses and two injections of APC/Gal per course. (C and D) Schemes of the PBMC sampling points (↓) of the APC/Gal therapy group (C) or nonimmunotherapy group (D).
Table I.
Profile of patients who completed phase II of the APC/Gal therapy study
| APC/Gal-Treated Group (n = 27) | Nontreated Group (n = 29) | |
|---|---|---|
| Age, y | 56 (32–69) | 62 (40–74) |
| Age group | ||
| <65 y | 18 (67%) | 13 (45%) |
| ≥65 y | 9 (33%) | 16 (55%) |
| Sex | ||
| Male | 19 (70%) | 20 (69%) |
| Female | 8 (30%) | 9 (31%) |
| ECOG performance status | ||
| 0 | 22 (81%) | 24 (83%) |
| 1 | 5 (19%) | 5 (17%) |
| Histology | ||
| Squamous | 6 (22%) | 5 (17%) |
| Nonsquamous | 21 (78%) | 24 (83%) |
| Stage | ||
| IIA | 9 (33%) | 11 (38%) |
| IIB | 6 (22%) | 5 (17%) |
| IIIA | 12 (44%) | 13 (45%) |
| Regional lymph node stage | ||
| N0 | 6 (22%) | 6 (21%) |
| N1 | 11 (41%) | 11 (38%) |
| N2 | 10 (37%) | 12 (41%) |
| Type of surgery | ||
| Lobectomy | 27 (100%) | 28 (97%) |
| Pneumonectomy | 0 (0%) | 1 (3%) |
| EGFR mutation status | ||
| Yes | 7 (26%) | 10 (34%) |
| No | 18 (68%) | 15 (52%) |
| Unknown | 2 (7%) | 4 (14%) |
Data were acquired at the initial phase before treatment. ECOG, Eastern Cooperative Oncology Group.
Characterization of APCs
The surface phenotype of APCs prepared for each administration was analyzed using flow cytometry (Fig. 2). Representative profiles of one patient (case 038) are shown in (Fig. 2A. The percentage of HLA-DR+, CD11c+, CD86+, and CD3+ cells was determined by the overtone subtraction test using the population comparison platform in the FlowJo software package, and the results are summarized in Supplemental Table I. As reported previously (31), >60% of APCs were HLA-DR+, 20–70% were CD11c+ cells, and 50–80% were CD86+ cells. Multiparameter flow cytometry analysis was performed to characterize APCs, and representative profiles (case 038) are shown in (Fig. 2B. Both CD3+ T cells and CD3− cells expressed various but considerable levels of HLA-DR, CD11c, and CD86, similar to results in previous studies (31). The results of the multiparameter flow cytometry analysis of APCs are summarized in Supplemental Table II. A substantial number of CD3+ T cells and CD3− cells in APCs express HLA-DR, CD11c, and CD86. We also confirmed CD1d expression on APCs (Fig. 2C). To estimate iNKT ligand–presenting activity of the human APC/Gal, we previously administered them to mice (36). Along with this assay, APC/Gal was injected into C57BL/6 mice. Four days after APC/Gal administration, iNKT cell frequency in the spleen was analyzed. By this approach, we checked the quality of APC/Gal for the four APC/Gal administrations. Each manipulated APC/Gal was defined as APC1/Gal, APC2/Gal, APC3/Gal, and APC4/Gal (Fig. 2D). The average iNKT cell proliferation in vivo in APC/Gal-injected mice was higher than that in naive mice, suggesting that the quality of APC/Gal is apparently significant for iNKT cell proliferation in vivo. However, there was no statistical difference among APC1 (n = 27), APC2 (n = 27), APC3 (n = 25), APC4 (n = 26), and control (n = 27) groups (Fig. 2D). Thus, we verified that the average APC/Gal at each injection time point was consistent and sufficient to activate iNKT cells.
FIGURE 2.

Flow cytometry analysis and functional analysis of APC/Gal.
(A) The expression levels of HLA-DR and CD86 on APC/Gal (APC1, APC2, APC3, and APC4) at the time of administration were assessed by flow cytometry analysis. Gray-filled histogram indicates background staining with an isotype control. Representative data of APC/Gal (numbers 1–4 for case 038) are shown. (B) Representative flow cytometry profiles of the expression of HLA-DR, CD11c, and CD86 on CD3+ and CD3− cells in APC/Gal. The percentage of cells detected is represented in each quadrant. (C) CD1d expression in APC/Gal. Histograms results show CD1d expression for 006 DC1. Data are expressed as the mean ± SEM of APC/Gal for eight APC/Gal samples from seven patients. (D) Evaluation of iNKT ligand–presenting capacity of APC/Gal. APC/Gal from each patient was injected into C57BL/6 mice. The frequency of murine iNKT cells in the spleen was analyzed 4 d later. Data are expressed as the mean ± SEM of the NKT cell response to APC/Gal of all patients. The control was naive mice. Difference was estimated using the ANOVA Tukey–Kramer method.
Immunological monitoring in the APC/Gal-treated or nontreated group
Immunological assays were performed in the study before and after treatment with APC/Gal. The frequency of peripheral blood Vα24+Vβ11+ iNKT cells and CD3−CD56+ NK cells was measured using flow cytometry analysis (Supplemental Fig. 1). Eight patients (cases 002, 014, 024, 034, 035, 038, 045, and 046) showed a dramatic increase (≥2-fold) in the circulating iNKT cell number after the first (014, 024, and 038), second (002 and 034), third (046), and fourth (035 and 045) APC/Gal administration (Fig. 3). The number of circulating iNKT cells in 7 patients out of the remaining 20 cases moderately increased (<1.5- to <2-fold) following APC/Gal administration. High baseline iNKT cell numbers did not always lead to the augmentation of peripheral blood iNKT cell frequency after APC/Gal injection (Table II, cases 020 and 049). Alternatively, a dramatic increase (≥2-fold) in the circulating NK cell number after the second (case 051) and fourth (cases 004, 009, 018, and 035) administration could be detected in five patients. Noticeable immune responses in iNKT and NK cells after APC/Gal administration were detected in the APC/Gal administration group, although each patient showed various activities in both iNKT and NK cells (Table II). We also monitored other immune cells, such as CD4 T cells, CD8 T cells, DCs, and regulatory T cells; however, there was no significant change during APC/Gal treatment in those cell populations (Table III). In addition, to assess whether the Ag-specific T cell response was driven by the bystander effect of iNKT cells, we compared them before and after APC/Gal therapy. As shown in Table III, we did not find an enhancement in the CMV-specific T cell response.
FIGURE 3.

Number of circulating iNKT and NK cells during the course of APC/Gal treatment.
The percentages of peripheral blood Vα24+Vβ11+ iNKT cells and CD56+CD3− NK cells during the course of the trial were monitored by flow cytometry analysis. The absolute number of iNKT and NK cells in 1 million PBMCs was calculated. Six cases whose iNKT cell number expanded after APC/Gal injection are shown. Apheresis (△) and APC/Gal (▲) are shown.
Table II.
Immune monitoring and clinical responses of patients completing APC/Gal therapy
| Case No. | Baseline NKT (%) | NKT (day max)a | Baseline NK (%) | NK (day max)a | ELISPOT (day max)b | Clinical Response | Follow (y) | Outcome (last point) |
|---|---|---|---|---|---|---|---|---|
| 002 | 0.0077 | 2.2 (d7, d28) | 7.0 | 0.9 (d7) | 110.0 (d70) | RF | 7.0 | Alive |
| 043 | 0.00496 | 1.4 (d49) | 6.1 | 1.1 (d35) | 56.5 (d49) | RF | 3.0 | Alive |
| 009 | 0.03 | 0.1 (d42) | 3.6 | 3.2 (d70) | 42.3 (d49) | R | 2.0 | Dead |
| 018 | 0.0183 | 0.5 (d35) | 3.7 | 2.8 (d70) | 28.3 (d49) | RF | 5.0 | Alive |
| 014 | 0.00131 | 2.6 (d7) | 11.7 | 1.1 (d35) | 26.0 (d49) | R | 3.7 | Alive |
| 045 | 0.00316 | 2.1 (d70) | 1.9 | 1.6 (d7) | 17.1 (d49) | R | 2.9 | Alive |
| 035 | 0.00424 | 2.3 (d70) | 4.2 | 5.1 (d70) | 14 (d28) | R | 3.6 | Alive |
| 038 | 0.00314 | 2.2 (d7) | 12.0 | 1.8 (d28) | 12.0 (d35) | R | 3.3 | Alive |
| 004 | 0.00297 | 1.6 (d7) | 7.2 | 3.7 (d70) | 10.7 (d49) | R | 6.3 | Alive |
| 006 | 0.023 | 0.3 (d7) | 2.3 | 1.2 (d70) | 9.9 (d49) | RF | 5.8 | Alive |
| 030 | 0.00365 | 1.6 (d42) | 14.1 | 1.0 (d7) | 9.0 (d28, d70) | RF | 3.8 | Alive |
| 034 | 0.00697 | 6.6 (d42) | 5.3 | 1.8 (d35) | 14 (d28) | RF | 3.5 | Alive |
| 016 | 0.0071 | 1.7 (d7) | 5.9 | 1.2 (d35) | 8 (d49) | R | 5.1 | Alive |
| 024 | 0.000697 | 6.5 (d7) | 9.0 | 1.6 (d70) | 7.7 (d28) | RF | 4.6 | Alive |
| 047 | 0.019 | 0.4 (d35) | 3.6 | 1.3 (d70) | 7.4 (d49) | RF | 2.9 | Alive |
| 026 | 0.00654 | 0.7 (d28) | 5.7 | 1.9 (d35) | 6.4 (d49) | R | 3.5 | Dead |
| 054 | 0.00267 | 1.1 (d28) | 16.5 | 1.2 (d35) | 5.8 (d49) | R | 0.9 | Alive |
| 046 | 0.00753 | 2.1 (d49) | 9.4 | 0.9 (d70) | 5.6 (d49) | RF | 2.8 | Alive |
| 022 | 0.014 | 1.6 (d7) | 2.2 | 1.1 (d35) | 4.7 (d7) | RF | 4.8 | Alive |
| 040 | 0.016 | 1.9 (d70) | 15.5 | 0.8 (d70) | 3.3 (d35) | RF | 3.0 | Alive |
| 013 | 0.00334 | 1.5 (d70) | 7.4 | 1.2 (d35) | 3.1 (d49) | RF | 5.0 | Alive |
| 051 | 0.018 | 0.7 (d7) | 1.8 | 2.5 (d49) | 2.8 (d7) | R | 2.8 | Alive |
| 055 | 0.015 | 0.7 (d49) | 11.8 | 0.8 (d28) | 2.3 (d49) | RF | 2.0 | Alive |
| 049 | 0.16 | 1.4 (d7) | 6.4 | 1.9 (d35) | 1.5 (d28) | RF | 2.8 | Alive |
| 041 | 0.011 | 0.4 (d7) | 9.9 | 1.4 (d7) | 1.2 (d28) | R | 3.2 | Alive |
| 020 | 1.6 | 0.5 (d35) | 3.8 | 1.9 (d28) | 0.8 (d42) | R | 4.8 | Alive |
| 039 | 0.012 | 1.6 (d28) | 8.9 | 0.9 (d35) | 0.6 (d28, d35, d49) | RF | 3.0 | Alive |
The line in the table shows the boundary of 2.0 × the increase rate of ELISPOT (day max). d, day; max, maximum; R, recurrence progressive; RF, recurrence-free.
Maximal fold increase to baseline in NKT cell or NK cell number during treatment. The time point is depicted in parentheses.
ELISPOT maximal fold increase to baseline in IFN-γ–producing cell number. The time point is depicted in parentheses.
Table III.
Summary of T cell responses of patients completing APC/Gal therapy
| Baseline | Day 49 | Day 70 | |
|---|---|---|---|
| CD4 Ta | 16.3 ± 1.3 | 17.4 ± 1.1 | 17.2 ± 1.2 |
| Tn CD4 Tb | 51.3 ± 3.2 | 49.3 ± 3.3 | 50.9 ± 3.0 |
| TEM/TE CD4 Tb | 24.5 ± 2.3 | 26.7 ± 2.8 | 24.5 ± 2.5 |
| TCM CD4 Tb | 20.4 ± 1.5 | 20.7 ± 1.4 | 20.5 ± 1.4 |
| Regulatory T cellsb | 0.7 ± 0.2 | 0.6 ± 0.1 | 0.6 ± 0.2 |
| CD8 Ta | 8.6 ± 0.9 | 9.6 ± 1.0 | 10.5 ± 1.1 |
| Tn CD8 Tc | 30.0 ± 4.2 | 30.5 ± 4.6 | 25.8 ± 4.0 |
| TEM/TE CD8 Tc | 26.6 ± 2.9 | 29.6 ± 3.4 | 30.1 ± 3.5 |
| TCM CD8 Tc | 3.7 ± 0.6 | 3.8 ± 0.7 | 3.3 ± 0.4 |
| CMV-reactive CD8 Tc | 0.3 ± 0.1 | n.d. | 0.2 ± 0.1 |
| cDCd | 63.7 ± 3.0 | 59.0 ± 3.7 | 61.7 ± 3.2 |
| pDCd | 18.1 ± 1.6 | 18.2 ± 2.0 | 21.0 ± 2.0 |
| MDSCa | 3.5 ± 0.5 | 2.8 ± 0.3 | 2.3 ± 0.2 |
CMV-specific CD8+ T cells were detected by HLA-A2+ or HLA-A24+ tetramer. n.d., not done; cDC, conventional DC; pDC, plasmacytoid DC; TCM, central memory T (CD45RA−CCR7+); TEM/EF, effector memory/effector T (CD45RA−CCR7−); Tn, naive T (CD45RA+CCR7+).
Percentage in leukocytes.
Percentage in CD4 T cells.
Percentage in CD8 T cells.
Percentage in lineage−HLA-DR+ leukocytes.
Next, as a functional analysis of circulating iNKT and NK cells, we assessed IFN-γ–producing iNKT cells using an ELISPOT assay of all APC/Gal-treated and nontreated patients. The assay was used to assess the number of IFN-γ–producing cells in PBMCs after restimulation with α-GalCer in vitro (Fig. 4). Previous reports showed maximum IFN-γ production by iNKT cells as a potential marker of advanced-stage NSCLC (31, 33). In 23 APC/Gal-treated patients in this study, the number of cells with IFN-γ production increased >2-fold after the administration of APC/Gal (Table II). In the remaining four patients, a minimal alteration (<2.0) in IFN-γ–producing capacity was observed. We also found that the maximum IFN-γ production was on day 49 in 13 of the APC/Gal-treated patients (Fig. 5A, Table IV). As the NK cells are major effector cells following the activation of Vα24+NKT cells, we evaluated the NK cell response following APC/Gal therapy. Granzyme B–expressing NK cells (by qPCR) were observed in patients with a >6-fold increase in IFN-γ production after the administration of APC/Gal (Fig. 5B, Table IV). Thus, higher innate activation of iNKT cells can elicit granzyme B–expressing NK cells. Regarding the nontreated group, the number of IFN-γ–producing cells increased >2-fold in 9 patients, whereas the remaining 18 patients showed a <2-fold increase in IFN-γ production (Table V). Surprisingly, we detected the maximum IFN-γ production after 12 mo in the nontreated group (Fig. 5C, left). Most of the nontreated patients after the surgery also showed an immunological peak at 12 mo (Fig. 5C, right). Resection of tumor cells enhanced the immune response of Vα24+NKT cells, whereas the expression of granzyme B in NK cells did not increase in the nontreated group (Fig. 5D, Supplemental Table III). We compared granzyme B–expressing NK cells at the protein level on day 49 (treated group) and at 12 mo (nontreated group), which were the time points with the highest response. We verified the result of granzyme B expression by RNA analysis using real-time PCR and that of the protein level using flow cytometric analysis in the treated (Fig. 5Ei, ii) and nontreated groups (Fig. 5Fi, ii). Finally, we demonstrated the correlation between IFN-γ spots using the ELISPOT assay and granzyme B expression in NK cells at the protein level in the treated group, but not in the nontreated group (Fig. 5). Furthermore, in the nontreated group, IFN-γ expression of NK cells was higher at 12 mo in the group that did not relapse compared with the group that relapsed (Fig. 5G, right), suggesting that removal of lung cancer leads to a recovery of the NK cell responses. On the contrary, there was no difference in IFN-γ expression of NK cells at 12 mo in both the recurrent and nonrecurrent APC/Gal-treated groups. Because we already found a series of effector functions of iNKT and NK cells, we next focused on NKG2D and DNAM-1 expression of iNKT and NK cells in terms of effector and coreceptor usage molecules. NKG2D expression of iNKT cells was significantly higher after treatment than before treatment, whereas DNAM-1 expression of iNKT cells was not (Supplemental Fig. 1C). In contrast, there was no difference in NKG2D and DNAM-1 expression of NK cells between APC/Gal-treated and nontreated groups (Supplemental Fig. 1D).
FIGURE 4.

Detection of α-GalCer–reactive IFN-γ–producing cells by the ELISPOT assay.
Cryopreserved PBMCs were thawed and cultured for 16 h in the presence of α-GalCer. IFN-γ–producing cells were analyzed by an IFN-γ ELISPOT assay. The mean IFN-γ spot-forming cell (SFC) number for duplicate cultures in nine representative cases is shown. Apheresis (△) and APC/Gal (▲) are shown.
FIGURE 5.

Relationship between α-GalCer–reactive IFN-γ production and NK cell activation.
(A–D) Results of APC/Gal-treated (A and B) and nontreated groups (C and D) are shown. (A) Apheresis (△) and APC/Gal (▲) are shown. (A, left and C, left) Time points of the maximum score of IFN-γ SFCs in patients treated with or without APC/Gal therapy. (A, right and C, right) The number of patients with the maximum IFN-γ SFC score is shown. Difference was estimated using a χ2 test. (B) Analysis of correlation between fold of IFN-γ SFCs (x-axis) and fold of granzyme B expression of NK cells (by qPCR) (y-axis) after third APC/Gal administration (day 49). The patients were divided into two groups (day 49/prestatus SFCs) using the threshold of a 6-fold increase in scores (<6-fold, >6-fold)). (D) Same as (B), but analysis of correlation between IFN-γ SFCs (x-axis) and granzyme B expression of NK cells (y-axis) after enrollment (12 mo). The patients were divided into two groups (12 mo/prestatus SFCs) using the threshold of a 6-fold increase in scores (<6-fold, >6-fold). (E and F) Granzyme B expression of NK cells was assessed at the protein level using flow cytometric analysis. Solid lines in the histograms indicate granzyme B, and gray indicates the isotype. (Ei) Granzyme B expression of NK cells in the treated group detected using flow cytometric analysis. (Eii) Correlation between the amount of granzyme B protein and IFN-γ ELISPOT at day 49 in the treated group. (Fi) Granzyme B expression of NK cells in the nontreated group detected using flow cytometric analysis. (Fii) Correlation between granzyme B protein level and IFN-γ ELISPOT for patients with ELISPOT >6 at 12 mo in the nontreated group (n = 4). Statistical analyses were performed using StatMate software, and correlation coefficients were determined by Pearson’s correlation tests. (G) IFN-γ expression of NK cells was assessed using real-time PCR analysis. IFN-γ expression of NK cells (12 mo/prestatus) in APC/Gal-treated and nontreated groups was evaluated in recurrent or nonrecurrent patients. Differences were analyzed using a two-tailed Pearson’s test of correlation, χ2 and Fisher’s exact tests, or the ANOVA Tukey–Kramer method. The significance threshold was set at a two-sided α value of 0.05. Statistical analyses were performed using StatMate (version 5.01).
Table IV.
NKT and NK cell responses of patients at day 49 after APC/Gal therapy
| Case No. | Baseline NKT (%) | NKT (day 49)a | Baseline NK (%) | NK (day 49)a | ELISPOT (day 49)b | NK GzmB (day 49) |
|---|---|---|---|---|---|---|
| 043 | 0.00496 | 1.4 | 6.1 | 5.9 | 56.5 | 6.5 |
| 002 | 0.0077 | 1.2 | 7.0 | 4.4 | 44.0 | 0.9 |
| 009 | 0.03 | 0.0 | 3.6 | 6.9 | 42.3 | 0.8 |
| 018 | 0.0183 | 0.2 | 3.7 | 5.9 | 28.3 | 1.8 |
| 014 | 0.00131 | 1.2 | 11.7 | 5.4 | 26.0 | 1.1 |
| 045 | 0.00316 | 0.4 | 1.9 | 1.6 | 17.1 | 1.7 |
| 004 | 0.00297 | 0.9 | 7.2 | 10.6 | 10.7 | 0.4 |
| 006 | 0.023 | 0.1 | 2.3 | 2.5 | 9.9 | 0.7 |
| 035 | 0.00424 | 0.3 | 4.2 | 9.6 | 8.5 | 0.0 |
| 016 | 0.0071 | 0.4 | 5.9 | 4.7 | 8.0 | 0.9 |
| 047 | 0.019 | 0.1 | 3.6 | 3.7 | 7.4 | 0.7 |
| 034 | 0.00697 | 1.6 | 5.3 | 7.4 | 7.3 | 0.0 |
| 024 | 0.000697 | 3.4 | 9.0 | 7.9 | 6.6 | 1.6 |
| 026 | 0.00654 | 0.1 | 5.7 | 4.8 | 6.4 | 1.9 |
| 054 | 0.00267 | 0.2 | 16.5 | 18.9 | 5.8 | 1.4 |
| 046 | 0.00753 | 2.1 | 9.4 | 2.1 | 5.6 | 7.7 |
| 030 | 0.00365 | 0.8 | 14.1 | 9.3 | 5.3 | 1.4 |
| 022 | 0.014 | 1.0 | 2.2 | 2.2 | 3.4 | 1.5 |
| 013 | 0.00334 | 0.9 | 7.4 | 7.6 | 3.1 | 0.8 |
| 038 | 0.00314 | 1.1 | 12.0 | 7.9 | 3.0 | 2.4 |
| 040 | 0.016 | 0.8 | 15.5 | 11.0 | 2.5 | 2.6 |
| 055 | 0.015 | 0.7 | 11.8 | 4.6 | 2.3 | 2.0 |
| 049 | 0.16 | 0.4 | 6.4 | 5.4 | 0.7 | 1.4 |
| 039 | 0.012 | 0.8 | 8.9 | 2.7 | 0.6 | 0.7 |
| 020 | 1.6 | 0.1 | 3.8 | 4.1 | 0.4 | 0.5 |
| 041 | 0.011 | 0.3 | 9.9 | 10.5 | 0.3 | 1.1 |
| 051 | 0.018 | 0.1 | 1.8 | 4.5 | 0.0 | 1.8 |
The line in the table shows the boundary of 6.0 × the increase rate of ELISPOT on day 49. GzmB, granzyme B.
Fold increase of day 49 to baseline in NKT cell or NK cell number during treatment.
ELISPOT fold increase of day 49 to baseline in IFN-γ–producing cell number.
Table V.
Immune monitoring and clinical responses of patients in the nontreated group
| Case No. | Baseline NKT (%) | NKT (3 mo)a | Baseline NK (%) | NK (3 mo)a | ELISPOT (3 mo)b | Clinical Response | Follow (y) | Outcome (last point) |
|---|---|---|---|---|---|---|---|---|
| 010 | 0.011 | 0.9 | 8.3 | 0.7 | 11.5 | RF | 5.0 | Alive |
| 056 | 0.01 | 0.9 | 48.9 | 0.9 | 8.5 | R | 0.3 | Dead |
| 021 | 0.00912 | 0.7 | 5.9 | 1.0 | 6.0 | RF | 0.5 | Alive |
| 029 | 0.00407 | 2.7 | 17.1 | 0.8 | 6.0 | R | 1.5 | Dead |
| 025 | 0.00403 | 1.2 | 5.1 | 2.2 | 4.8 | RF | 0.2 | Alive |
| 012 | 0.00589 | 0.1 | 6.7 | 0.8 | 4.5 | R | 0.5 | Dead |
| 028 | 0.011 | 0.9 | 11.9 | 1.1 | 3.6 | R | 0.3 | Dead |
| 007 | 0.01 | 0.7 | 15.9 | 1.2 | 2.5 | RF | 5.3 | Alive |
| 019 | 0.00482 | 0.5 | 9.1 | 1.9 | 2.0 | RF | 4.5 | Alive |
| 027 | 0.000949 | 0.3 | 9.6 | 0.8 | 1.8 | R | 2.9 | Alive |
| 032 | 0.081 | 0.9 | 2.9 | 0.8 | 1.2 | R | 3.4 | Dead |
| 011 | 0.00456 | 1.5 | 15.8 | 1.1 | 1.0 | RF | 5.4 | Alive |
| 057 | 0.01 | 1.4 | 8.4 | 0.6 | 1.0 | RF | 2.0 | Alive |
| 042 | 0.018 | 1.2 | 14.5 | 1.0 | 0.9 | RF | 3.0 | Alive |
| 031 | 0.00815 | 1.7 | 9.7 | 1.3 | 0.9 | RF | 3.7 | Alive |
| 017 | 0.00209 | 0.7 | 18.0 | 0.8 | 0.8 | RF | 3.8 | Alive |
| 037 | 0.0062 | 0.3 | 6.6 | 0.8 | 0.8 | R | 0.9 | Alive |
| 008 | 0.013 | 1.4 | 7.1 | 1.4 | 0.5 | RF | 5.5 | Alive |
| 015 | 0.00596 | 0.3 | 9.2 | 1.0 | 0.5 | RF | 5.1 | Alive |
| 033 | 0.11 | 0.0 | 4.7 | 1.6 | 0.5 | R | 0.6 | Dead |
| 023 | 0.00149 | 1.7 | 31.2 | 0.7 | 0.4 | RF | 4.3 | Alive |
| 044 | 0.015 | 0.8 | 8.5 | 1.6 | 0.3 | R | 1.0 | Alive |
| 036 | 0.00257 | 0.6 | 9.9 | 1.0 | 0.0 | RF | 3.0 | Alive |
| 005 | 0.009 | 0.8 | 4.8 | 1.0 | 0 | RF | 6.1 | Alive |
| 048 | 0.000345 | 2.5 | 2.4 | 3.6 | 0.0 | R | 2.6 | Alive |
| 053 | 0.00756 | 3.0 | 8.9 | 0.9 | 0.0 | R | 2.0 | Alive |
| 058 | 0.00142 | 3.5 | 8.4 | 0.6 | 0.0 | RF | 2.0 | Alive |
| 001 | n.d. | n.d. | n.d. | n.d. | n.d. | R | 4.1 | Alive |
| 052 | n.d. | n.d. | n.d. | n.d. | n.d. | RF | 2.5 | Alive |
The line in the table shows the boundary of 2.0 × the increase rate of ELISPOT at 3 mo. n.d., not determined; R, recurrence; RF, recurrence-free.
Fold increase of 3 mo to baseline in NKT cell or NK cell number during treatment.
ELISPOT fold increase of 3 mo to baseline in IFN-γ–producing cell number.
Clinical outcome in the APC/Gal-treated patients
A total of 10 and 7 recurrences and one and three deaths occurred in the APC/Gal-treated (n = 27) and nontreated groups (n = 29), respectively. The 2-y survival rate was analyzed for the full analysis set population. At 2 y, recurrence-free survival or overall survival between the two groups did not significantly differ (Fig. 6); the recurrence-free survival at 2 y was 63.0% (95% confidence interval [CI], 47.1–84.1) in the APC/Gal-treated group and 74.3% (95% CI, 59.5–92.7) in the nontreated group (p value in the log-rank test was 0.43). The 2-y survival rate was 96.2% (95% CI, 89.0–100.0) in the APC/Gal-treated group and 89.0% (95% CI, 78.0–100.0) in the nontreated group (Fig. 6). The p values of 0.43 and 0.31, respectively, indicated that the APC/Gal therapy arm was not considered as a treatment worth extending to a phase III trial.
FIGURE 6.
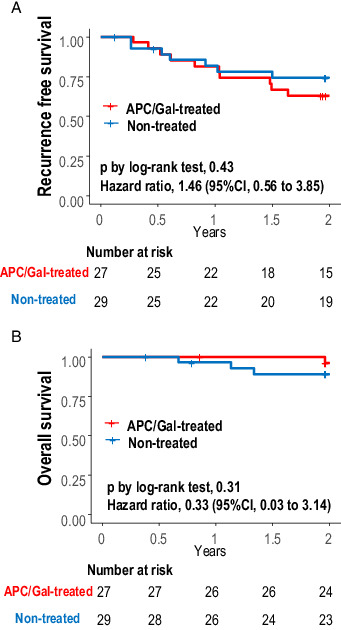
Kaplan–Meier plot of recurrence-free survival and overall survival.
(A and B) Kaplan–Meier estimate of recurrence-free survival (A) and overall survival (B) with comparison between APC/Gal-treated and nontreated control groups. The survival probabilities were estimated using the Kaplan–Meier method and compared between treatment groups by log-rank tests. Hazard ratios (APC/Gal-treated/nontreated) for recurrence and death were also evaluated using Cox proportional hazard regression analyses. Statistical analyses were performed using the R software, version 3.6.3 (R Foundation for Statistical Computing).
Adverse events in APC/Gal-treated patients
dverse events are summarized in Table VI. Systemic scleroderma was a serious adverse event that occurred during the course of this cell therapy. However, the causal relationship of it was unclear, but was subsequently ruled out as “causal relationship undeniable” based on the opinion of the Cell Therapy Effectiveness and Safety Evaluation Committee. Other adverse events occurred in 11 cases in four patients, but all these cases were less than grade 2, that is, increased laboratory test values and fever. Two out of 11 cases had a causal relationship that could be ruled out, and 8 cases were judged to have an undeniable causal relationship. One case of pulmonary inflammation was grade 1.
Table VI.
Summary of adverse events in APC/Gal therapy study
| Adverse Events | APC/Gal-Treated Group | Nontreated Group |
|---|---|---|
| Any grade | 5 (19%) | 0 (0%) |
| Grade ≤2 | 4 (15%) | 0 (0%) |
| Serious | 1 (4%) | 0 (0%) |
| Grade 5 | 0 (0%) | 0 (0%) |
GWASs of functional analysis with iNKT cells and clinical outcomes
When the IFN-γ–producing iNKT cells (ELISPOT assay) and the expression of granzyme B in the 27 APC/Gal-treated patients were assessed, no SNPs achieved genome-wide significance (Fig. 7A, 7B). Similarly negative results were seen when overall survival was examined. These results implied that immunological responses in this study were not derived from individual differences at birth, but were the result induced by immunotherapy. In contrast, when focusing on recurrence-free survival, several SNPs were found to have genome-wide significance (Fig. 7C, 7D). The peak signal (p = 5.63 × 10−10) was for SNP rs2870863 located at an intron of PTPRR, the gene that encodes protein tyrosine phosphatase receptor type R (Table VII).
FIGURE 7.
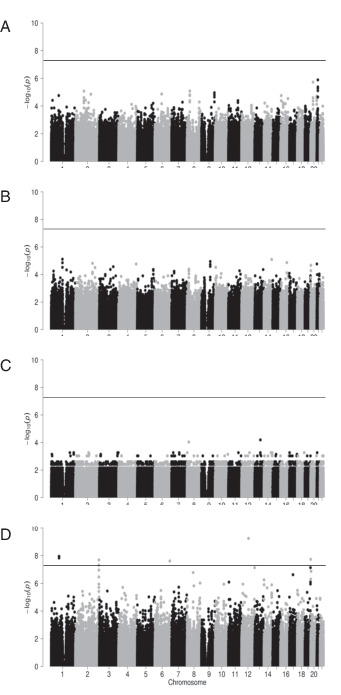
Manhattan plots of GWAS for the clinical outcomes and immune responses in 27 APC/Gal-treated patients.
(A–D) Manhattan plots for IFN-γ-production (A), expression of granzyme B–expressing NK cells (B), overall survival (C), and recurrence-free survival (D). The x-axis indicates chromosomal positions, and the y-axis shows −log10 p values calculated using an appropriate statistical analysis for each phenotype. Each dot represents a variant. The horizontal line indicates the genome-wide significance threshold (p < 5.0 × 10−8).
Table VII.
Summary of significant SNPs in the GWAS for recurrence-free survival
| Index | SNP ID | Chromosome | Position (build hg19) | RA | NRA | p Value | RA Freq. | Gene |
|---|---|---|---|---|---|---|---|---|
| 1 | rs12041399 | 1 | 81,151,125 | T | G | 1.11E−08 | 0.111 | —a |
| 2 | rs841671 | 1 | 81,156,849 | C | T | 1.46E−08 | 0.120 | — |
| 3 | rs76311403 | 1 | 81,157,874 | G | A | 1.46E−08 | 0.120 | — |
| 4 | rs78032611 | 1 | 81,158,081 | T | G | 1.46E−08 | 0.120 | — |
| 5 | rs13392396 | 2 | 239,318,016 | C | T | 4.86E−08 | 0.148 | — |
| 6 | rs112295092 | 2 | 239,319,148 | GAGTT | G | 4.86E−08 | 0.148 | — |
| 7 | rs61237304 | 2 | 239,319,762 | C | T | 2.04E−08 | 0.135 | — |
| 8 | rs13389337 | 2 | 239,319,900 | T | G | 4.86E−08 | 0.148 | — |
| 9 | rs13415491 | 2 | 239,319,901 | G | C | 4.86E−08 | 0.148 | — |
| 10 | rs13389451 | 2 | 239,320,012 | A | G | 4.86E−08 | 0.148 | — |
| 11 | rs10084236 | 2 | 239,320,550 | A | G | 4.86E−08 | 0.148 | — |
| 12 | rs10084472 | 2 | 239,320,570 | G | A | 4.86E−08 | 0.148 | — |
| 13 | rs7596847 | 2 | 239,321,118 | A | T | 4.86E−08 | 0.148 | — |
| 14 | rs11884498 | 2 | 239,321,618 | G | A | 4.86E−08 | 0.148 | — |
| 15 | rs6752291 | 2 | 239,322,058 | C | T | 4.86E−08 | 0.148 | — |
| 16 | rs10084421 | 2 | 239,322,265 | A | C | 4.86E−08 | 0.148 | — |
| 17 | rs10084217 | 2 | 239,322,436 | C | T | 4.86E−08 | 0.148 | — |
| 18 | rs10084192 | 2 | 239,322,485 | G | A | 4.86E−08 | 0.148 | — |
| 19 | rs9287628 | 2 | 239,322,744 | A | G | 4.86E−08 | 0.148 | — |
| 20 | rs7563974 | 2 | 239,323,764 | A | G | 4.86E−08 | 0.148 | — |
| 21 | rs11887955 | 2 | 239,324,429 | G | A | 4.86E−08 | 0.148 | — |
| 22 | rs11889150 | 2 | 239,324,542 | G | A | 4.86E−08 | 0.148 | — |
| 23 | rs10677780 | 2 | 239,327,673 | ACT | A | 4.86E−08 | 0.148 | — |
| 24 | rs58986127 | 2 | 239,331,582 | C | T | 4.86E−08 | 0.148 | — |
| 25 | rs10194338 | 2 | 239,339,251 | T | C | 4.86E−08 | 0.148 | ASB1 |
| 26 | rs9630961 | 2 | 239,341,772 | G | C | 4.86E−08 | 0.148 | ASB1 |
| 27 | rs10203153 | 2 | 239,347,083 | T | C | 4.86E−08 | 0.148 | ASB1 |
| 28 | rs6747427 | 2 | 239,350,536 | C | T | 4.86E−08 | 0.148 | ASB1 |
| 29 | rs6748424 | 2 | 239,351,532 | C | A | 4.86E−08 | 0.148 | ASB1 |
| 30 | rs2278768 | 2 | 239,355,561 | G | A | 4.86E−08 | 0.148 | ASB1 |
| 31 | rs311328 | 6 | 153,137,443 | T | C | 2.42E−08 | 0.423 | — |
| 32 | rs311326 | 6 | 153,137,621 | A | G | 2.42E−08 | 0.423 | — |
| 33 | rs2870863 | 12 | 71,177,098 | C | T | 5.63E−10 | 0.130 | PTPRR |
| 34 | rs6140884 | 20 | 968,892 | T | C | 1.83E−08 | 0.100 | RSPO4 |
NRA, non-risk allele; RA, risk allele; RA Freq., risk allele frequency.
The SNP is not located in genes.
Discussion
In the current study, APC/Gal was administered to patients with early-stage NSCLC who underwent complete excision of the lung tumor and subsequently received adjuvant chemotherapy. We evaluated the immune responses in APC/Gal-treated patients in the early stage as well as that in untreated patients. We evaluated the variety of iNKT and NK cell responses by comparing immunological data to previous data using the same protocol and analysis. Our immunological analysis revealed that 15 out of 27 patients had a dramatic increase in the circulating iNKT cell frequency after the APC/Gal therapy. With regard to a direct and rapid functional iNKT cell analysis, we previously established an in vitro assay for ligand-specific IFN-γ–producing iNKT cells, involving culture for 16 h in the presence of α-GalCer (23, 37). We found that the number of iNKT cells with IFN-γ production increased >2-fold after immunotherapy in 23 out of 27 patients (85.1%), indicative of a high number of responders. Surprisingly, the number of cells with IFN-γ production increased >6-fold after immunotherapy in 16 out of 27 patients (59.1%). Thus, the iNKT cell response was much more enhanced in the patients with early-stage NSCLC as compared with a previous report in which 10 out of 17 APC/Gal-treated patients with advanced-stage NSCLC (58.8%) showed a >2-fold increase in the number of iNKT cells with IFN-γ production after therapy (31). In addition, the peak iNKT cell response was detected on day 49, that is, 7 d after the third APC/Gal therapy in most patients. In contrast to the iNKT cell response, the frequency of T cells, myeloid-derived suppressor cells (MDSCs), and regulatory T cells did not change following immunotherapy. We previously observed some memory iNKT cells after activation in murine models (38, 39). Robust iNKT cell expansion occurred 1 wk after the third administration of APC/Gal, indicating enhanced memory iNKT cells. Further studies are required to elucidate this observation. Furthermore, several studies have demonstrated that iNKT cell activation is followed by augmentation of NK cells. When focusing on the iNKT/NK cell axis, we surprisingly found an adjunctive effect of granzyme B–expressing NK cells following iNKT cell activation in patients with lung cancer. This finding implies a close association between iNKT activation (i.e., IFN-γ production) and NK cytolytic activity (granzyme B).
We also evaluated the clinical response. A previous report demonstrated that ligand-specific IFN-γ–producing SFCs, as representatives of iNKT cell function, are correlated with clinical relevance (31). Although we demonstrated the heightened and durable activation of innate immunity, we could not see any significant differences in clinical responses between the APC/Gal-treated and nontreated groups (Fig. 6). The difference between our study and the previous report may be due to disease status, that is, the tumor burden. Generally, tumor-bearing patients in an advanced stage have an impaired immunological status (40). As tumors progress and increase in burden, tumor cells potentially acquire heterogeneity or clonal cancer evolution. During progression, some tumor cells may express ligands of NK cell activating receptors (e.g., NKG2D-L or DNAM-1), or lose the HLA (41, 42). This could have caused differences in the sensitivity of tumors toward innate immunity. In the current study, the innate immune response was indeed augmented by APC/Gal in early-stage patients with NSCLC. In this study, a GWAS was performed to confirm the influence of genetic factors on peripheral blood NKT cell–specific immune responses and clinical outcomes in the 27 APC/Gal-treated patients. Despite the immunological response, the absence of significant SNPs in IFN-γ–producing iNKT cells and granzyme B expression of NK cells indicates that there may be no associated genetic factors (Fig. 7). These results indicated that immunotherapy using CD1d+cells loaded with Gal can be applicable for any patients without any genomic influences. Next, when assessing recurrence-free survival, several significant SNPs were found, indicating the possibility of accounting for interindividual variability in the efficacy of administration of APC/Gal. Some SNPs with genome-wide significance in recurrence-free survival would be potential biomarkers for DC/Gal therapy. However, further studies are required for confirmation.
Intriguingly, patients in the nontreated, control group had altered immune responses. This is the first finding of the iNKT cell response after the resection of cancer in NSCLC patients. We found that the peak of the iNKT cell response was detected 12 mo after the initiation of the trial, implying that iNKT cell responses recover 12 mo after the resection of lung cancer in patients without immunotherapy. iNKT cells may be affected by the tumor burden in patients with cancer (21, 43). We also detected the elevation of IFN-γ in NK cells at 12 mo in nonrecurrent patients. Nevertheless, we could not find a correlation between iNKT and NK cell responses in our observation (Fig. 5D), suggesting that NK cell recovery is independent of iNKT cells. Other immune cells, such as T cells, regulatory T cells, and MDSCs did not change between the baseline and 12 mo. These findings suggest that tumor cells might suppress iNKT cells, whereas iNKT cells could be enhanced by APC/Gal immunotherapy. In a study of colon carcinoma that monitored the subset distribution of immune cells both before and after operation (44), CD8 T cells significantly increased 4 mo after surgery; however, the restoration of CD8 T cell response following resection of colon cancer in that study is in contrast to our findings. Thus, the restoration of cancer immunity may be dependent on the cancer type or progression stages, and further investigation is required to reveal the exact mechanism of this phenomenon.
In this study we demonstrate that elimination of lung cancer enhances innate immunity. In addition, we found the enhancement of cytolytic NK cells (i.e., granzyme B–expressing NK cells) by APC/Gal therapy. As future therapies, there are some candidates. This treatment may be effective when combined with other therapies, such as with molecular-targeted drugs (e.g., anti-EGFR Ab or anti–PD-1 Ab) or irradiation. Otherwise, other types of therapeutic vaccines generating Ag-specific T cells in addition to innate immunity, such as the artificial adjuvant vector cells recently established by us (36), would be another candidate. Such a combined treatment or a treatment with new modality may enhance the immunotherapeutic effect in the early stages of NSCLCs.
Supplementary Material
Acknowledgements
We thank all coworkers from all units of national hospital organizations as well as all patients who participated in this trial and their families. We also thank the following investigators: Dr. Toshiki Saito (Clinical Research Center, National Hospital Organization Headquarters, Nagoya Medical Center), Drs. Tatsuro Okamoto and Tatsunori Wada (Kyushu Cancer Center), Dr. Hidenori Ibata (Mie Central Medical Center), Dr. Tatsuo Kato (Nagara Medical Center), Dr. Koji Takami (Osaka National Hospital), Dr. Motohiro Yamashita (Shikoku Cancer Center), Dr. Tadashi Maeda (Yamaguchi-Ube Medical Center), Dr. Sadanori Takeo (National Hospital Organization Kyushu Medical Center), Dr. Hitoshi Ueda (National Hospital Organization Fukuoka National Hospital), Dr. Kan Okabayashi (Fukuoka-Higashi Medical Center), Dr. Seiji Nagashima (Nagasaki Medical Center), Dr. Katsumi Nakatomi (Ureshino Medical Center), Dr. Hidenori Koso (Oita Medical Center), Dr. Seiichi Fukuyama (Beppu Medical Center), and Dr. Kentaro Yoshimoto (Minamikyushu National Hospital) for skillful assistance in making this trial run successfully, including the cell therapy staff, and for the support they provided to the patients under their care. We also thank the Department of Clinical Research Planning and Management, National Hospital Organization Nagoya Medical Center for special contributions to operating this clinical trial, Miyuki Namatame (Laboratory of Cell Therapy, Clinical Research Center, National Hospital Organization Nagoya Medical Center) for cell preparation, and Dr. Yusuke Sato and An Sanpei (RIKEN, Center for Integrative Medical Sciences Laboratory of Immunotherapy) for providing technical assistance in the analysis.
Footnotes
This work was supported by the National Hospital Organization collaborative clinical research grant.
This clinical trial was registered in the University Hospital Medical Information Network Clinical Trials Registry under number UMIN000010386 and in the Japan Registry for Clinical Trials under number jRCTc040190130.
The online version of this article contains supplemental material.
- APC/Gal
- α-GalCer loaded with CD1d-expressing human PBMC-derived APCs
- CI
- confidence interval
- DC
- dendritic cell
- DC/Gal
- α-GalCer–pulsed DCs
- α-GalCer
- α-galactosylceramide
- GWAS
- genome-wide association study
- iNKT
- invariant NKT
- MDSC
- myeloid-derived suppressor cell
- NHO
- National Hospital Organization
- NSCLC
- non–small cell lung cancer
- qPCR
- quantitative PCR
- SFC
- spot-forming cell
- SNP
- single-nucleotide polymorphism
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R. L., Torre L. A., Jemal A.. 2018. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. [Published erratum appears in 2020 CA Cancer J. Clin. 70: 313.] CA Cancer J. Clin. 68: 394–424. [DOI] [PubMed] [Google Scholar]
- 2.Uramoto H., Tanaka F.. 2014. Recurrence after surgery in patients with NSCLC. Transl. Lung Cancer Res. 3: 242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ortega-Franco A., Calvo V., Franco F., Provencio M., Califano R.. 2020. Integrating immune checkpoint inhibitors and targeted therapies in the treatment of early stage non-small cell lung cancer: a narrative review. Transl. Lung Cancer Res. 9: 2656–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou F., Qiao M., Zhou C.. 2021. The cutting-edge progress of immune-checkpoint blockade in lung cancer. Cell. Mol. Immunol. 18: 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nersesian S., Schwartz S. L., Grantham S. R., MacLean L. K., Lee S. N., Pugh-Toole M., Boudreau J. E.. 2021. NK cell infiltration is associated with improved overall survival in solid cancers: a systematic review and meta-analysis. Transl. Oncol. 14: 100930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang S., Liu W., Hu B., Wang P., Lv X., Chen S., Shao Z.. 2020. Prognostic significance of tumor-infiltrating natural killer cells in solid tumors: a systematic review and meta-analysis. Front. Immunol. 11: 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russell É., Conroy M. J., Barr M. P.. 2022. Harnessing natural killer cells in non-small cell lung cancer. Cells 11: 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Platonova S., Cherfils-Vicini J., Damotte D., Crozet L., Vieillard V., Validire P., André P., Dieu-Nosjean M. C., Alifano M., Régnard J. F., et al. 2011. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 71: 5412–5422. [DOI] [PubMed] [Google Scholar]
- 9.Cong J., Wei H.. 2019. Natural killer cells in the lungs. Front. Immunol. 10: 1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamilton G., Plangger A.. 2021. The impact of NK cell-based therapeutics for the treatment of lung cancer for biologics: targets and therapy. Biologics 15: 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruno A., Focaccetti C., Pagani A., Imperatori A. S., Spagnoletti M., Rotolo N., Cantelmo A. R., Franzi F., Capella C., Ferlazzo G., et al. 2013. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia 15: 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porcelli S., Yockey C. E., Brenner M. B., Balk S. P.. 1993. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J. Exp. Med. 178: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dellabona P., Padovan E., Casorati G., Brockhaus M., Lanzavecchia A.. 1994. An invariant V alpha 24-J alpha Q/V beta 11 T cell receptor is expressed in all individuals by clonally expanded CD4-8- T cells. J. Exp. Med. 180: 1171–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers P. R., Matsumoto A., Naidenko O., Kronenberg M., Mikayama T., Kato S.. 2004. Expansion of human Vα24+ NKT cells by repeated stimulation with KRN7000. J. Immunol. Methods 285: 197–214. [DOI] [PubMed] [Google Scholar]
- 15.Coquet J. M., Chakravarti S., Kyparissoudis K., McNab F. W., Pitt L. A., McKenzie B. S., Berzins S. P., Smyth M. J., Godfrey D. I.. 2008. Diverse cytokine production by NKT cell subsets and identification of an IL-17-producing CD4−NK1.1− NKT cell population. Proc. Natl. Acad. Sci. USA 105: 11287–11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brossay L., Kronenberg M.. 1999. Highly conserved antigen-presenting function of CD1d molecules. Immunogenetics 50: 146–151. [DOI] [PubMed] [Google Scholar]
- 17.Godfrey D. I., Pellicci D. G., Patel O., Kjer-Nielsen L., McCluskey J., Rossjohn J.. 2010. Antigen recognition by CD1d-restricted NKT T cell receptors. Semin. Immunol. 22: 61–67. [DOI] [PubMed] [Google Scholar]
- 18.Shimizu K., Hidaka M., Kadowaki N., Makita N., Konishi N., Fujimoto K., Uchiyama T., Kawano F., Taniguchi M., Fujii S.. 2006. Evaluation of the function of human invariant NKT cells from cancer patients using α-galactosylceramide-loaded murine dendritic cells. J. Immunol. 177: 3484–3492. [DOI] [PubMed] [Google Scholar]
- 19.Smyth M. J., Crowe N. Y., Pellicci D. G., Kyparissoudis K., Kelly J. M., Takeda K., Yagita H., Godfrey D. I.. 2002. Sequential production of interferon-γ by NK1.1+ T cells and natural killer cells is essential for the antimetastatic effect of α-galactosylceramide. Blood 99: 1259–1266. [DOI] [PubMed] [Google Scholar]
- 20.Fujii S., Shimizu K., Smith C., Bonifaz L., Steinman R. M.. 2003. Activation of natural killer T cells by α-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J. Exp. Med. 198: 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iyoda T., Yamasaki S., Hidaka M., Kawano F., Abe Y., Suzuki K., Kadowaki N., Shimizu K., Fujii S. I.. 2018. Amelioration of NK cell function driven by Vα24+ invariant NKT cell activation in multiple myeloma. Clin. Immunol. 187: 76–84. [DOI] [PubMed] [Google Scholar]
- 22.Toura I., Kawano T., Akutsu Y., Nakayama T., Ochiai T., Taniguchi M.. 1999. Cutting edge: inhibition of experimental tumor metastasis by dendritic cells pulsed with alpha-galactosylceramide. J. Immunol. 163: 2387–2391. [PubMed] [Google Scholar]
- 23.Fujii S., Shimizu K., Kronenberg M., Steinman R. M.. 2002. Prolonged interferon-γ-producing NKT response induced with α-galactosylceramide-loaded dendritic cells. Nat. Immunol. 3: 867–874. [DOI] [PubMed] [Google Scholar]
- 24.Nieda M., Okai M., Tazbirkova A., Lin H., Yamaura A., Ide K., Abraham R., Juji T., Macfarlane D. J., Nicol A. J.. 2004. Therapeutic activation of Vα24+Vβ11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood 103: 383–389. [DOI] [PubMed] [Google Scholar]
- 25.Chang D. H., Osman K., Connolly J., Kukreja A., Krasovsky J., Pack M., Hutchinson A., Geller M., Liu N., Annable R., et al. 2005. Sustained expansion of NKT cells and antigen-specific T cells after injection of α-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J. Exp. Med. 201: 1503–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richter J., Neparidze N., Zhang L., Nair S., Monesmith T., Sundaram R., Miesowicz F., Dhodapkar K. M., Dhodapkar M. V.. 2013. Clinical regressions and broad immune activation following combination therapy targeting human NKT cells in myeloma. Blood 121: 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Exley M. A., Friedlander P., Alatrakchi N., Vriend L., Yue S., Sasada T., Zeng W., Mizukami Y., Clark J., Nemer D., et al. 2017. Adoptive transfer of invariant NKT cells as immunotherapy for advanced melanoma: a phase I clinical trial. Clin. Cancer Res. 23: 3510–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Godfrey D. I., Le Nours J., Andrews D. M., Uldrich A. P., Rossjohn J.. 2018. Unconventional T cell targets for cancer immunotherapy. Immunity 48: 453–473. [DOI] [PubMed] [Google Scholar]
- 29.Ishikawa A., Motohashi S., Ishikawa E., Fuchida H., Higashino K., Otsuji M., Iizasa T., Nakayama T., Taniguchi M., Fujisawa T.. 2005. A phase I study of α-galactosylceramide (KRN7000)-pulsed dendritic cells in patients with advanced and recurrent non-small cell lung cancer. Clin. Cancer Res. 11: 1910–1917. [DOI] [PubMed] [Google Scholar]
- 30.Motohashi S., Ishikawa A., Ishikawa E., Otsuji M., Iizasa T., Hanaoka H., Shimizu N., Horiguchi S., Okamoto Y., Fujii S., et al. 2006. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin. Cancer Res. 12: 6079–6086. [DOI] [PubMed] [Google Scholar]
- 31.Motohashi S., Nagato K., Kunii N., Yamamoto H., Yamasaki K., Okita K., Hanaoka H., Shimizu N., Suzuki M., Yoshino I., et al. 2009. A phase I-II study of α-galactosylceramide-pulsed IL-2/GM-CSF-cultured peripheral blood mononuclear cells in patients with advanced and recurrent non-small cell lung cancer. J. Immunol. 182: 2492–2501. [DOI] [PubMed] [Google Scholar]
- 32.Yamasaki K., Horiguchi S., Kurosaki M., Kunii N., Nagato K., Hanaoka H., Shimizu N., Ueno N., Yamamoto S., Taniguchi M., et al. 2011. Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin. Immunol. 138: 255–265. [DOI] [PubMed] [Google Scholar]
- 33.Toyoda T., Kamata T., Tanaka K., Ihara F., Takami M., Suzuki H., Nakajima T., Ikeuchi T., Kawasaki Y., Hanaoka H., et al. 2020. Phase II study of α-galactosylceramide-pulsed antigen-presenting cells in patients with advanced or recurrent non-small cell lung cancer. J. Immunother. Cancer 8: e000316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Detterbeck F. C., Boffa D. J., Tanoue L. T.. 2009. The new lung cancer staging system. Chest 136: 260–271. [DOI] [PubMed] [Google Scholar]
- 35.Saka H., Kitagawa C., Ichinose Y., Takenoyama M., Ibata H., Kato T., Takami K., Yamashita M., Maeda T., Takeo S., et al. 2017. A randomized phase II study to assess the effect of adjuvant immunotherapy using α-GalCer-pulsed dendritic cells in the patients with completely resected stage II-IIIA non-small cell lung cancer: study protocol for a randomized controlled trial. Trials 18: 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujii S., Kawamata T., Shimizu K., Nakabayashi J., Yamasaki S., Iyoda T., Shinga J., Nkazato H., Sanpei A., Kawamura M., et al. 2022. Reinvigoration of innate and adaptive immunity via therapeutic cellular vaccine for patients with AML. Mol. Ther. Oncolytics DOI: 10.1016/j.omto.2022.09.001. [DOI] [Google Scholar]
- 37.Fujii S., Shimizu K., Steinman R. M., Dhodapkar M. V.. 2003. Detection and activation of human Vα24+ natural killer T cells using α-galactosyl ceramide-pulsed dendritic cells. J. Immunol. Methods 272: 147–159. [DOI] [PubMed] [Google Scholar]
- 38.Shimizu K., Sato Y., Shinga J., Watanabe T., Endo T., Asakura M., Yamasaki S., Kawahara K., Kinjo Y., Kitamura H., et al. 2014. KLRG+ invariant natural killer T cells are long-lived effectors. Proc. Natl. Acad. Sci. USA 111: 12474–12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimizu K., Sato Y., Kawamura M., Nakazato H., Watanabe T., Ohara O., Fujii S. I.. 2019. Eomes transcription factor is required for the development and differentiation of invariant NKT cells. Commun. Biol. 2: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hiam-Galvez K. J., Allen B. M., Spitzer M. H.. 2021. Systemic immunity in cancer. Nat. Rev. Cancer 21: 345–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGranahan N., Rosenthal R., Hiley C. T., Rowan A. J., Watkins T. B. K., Wilson G. A., Birkbak N. J., Veeriah S., Van Loo P., Herrero J., Swanton C.; TRACERx Consortium . 2017. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 171: 1259–1271.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones A. B., Rocco A., Lamb L. S., Friedman G. K., Hjelmeland A. B.. 2022. Regulation of NKG2D stress ligands and its relevance in cancer progression. Cancers (Basel) 14: 2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Konishi J., Yamazaki K., Yokouchi H., Shinagawa N., Iwabuchi K., Nishimura M.. 2004. The characteristics of human NKT cells in lung cancer—CD1d independent cytotoxicity against lung cancer cells by NKT cells and decreased human NKT cell response in lung cancer patients. Hum. Immunol. 65: 1377–1388. [DOI] [PubMed] [Google Scholar]
- 44.Krijgsman D., De Vries N. L., Andersen M. N., Skovbo A., Tollenaar R. A. E. M., Bastiaannet E., Kuppen P. J. K., Hokland M.. 2020. The effects of tumor resection and adjuvant therapy on the peripheral blood immune cell profile in patients with colon carcinoma. Cancer Immunol. Immunother. 69: 2009–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.