

Abstract
Background & aims:
Aberrant DNA methylation is frequent in colorectal cancer (CRC), but underlying mechanisms and pathological consequences are poorly understood.
Methods:
We disrupted active DNA demethylation genes Tet1 and/or Tdg from ApcMin mice, and characterized the methylome and transcriptome of colonic adenomas. Data were compared to human colonic adenocarcinomas (COAD) in TCGA.
Results:
There were increased numbers of small intestinal adenomas in ApcMin mice expressing the TdgN151A allele, whereas Tet1-deficient and Tet1/TdgN151A-double heterozygous ApcMin colonic adenomas were larger with features of erosion and invasion. We detected reduction in global DNA hypomethylation in colonic adenomas from Tet1- and Tdg-mutant ApcMin mice, and hypermethylation of CpG islands in Tet1-mutant ApcMin adenomas. Upregulation of inflammatory, immune and interferon response genes was present in Tet1- and Tdg-mutant colonic adenomas compared to control ApcMin adenomas. This upregulation was also seen in murine colonic organoids and human CRC lines infected with lentiviruses expressing TET1 or TDG shRNA. A 127-gene inflammatory signature separated COAD into four groups, closely aligned with their microsatellite or chromosomal instability, and characterized by different levels of DNA methylation and DNMT1 expression that anti-correlated with TET1 expression. Tumors with the CpG island methylator phenotype (CIMP) had concerted high DNMT1/low TET1 expression. TET1 or TDG knockdown in CRC lines enhanced killing by NK cells.
Conclusions:
Our findings reveal a novel epigenetic regulation, linked to the type of genomic instability, by which TET1-TDG-mediated DNA demethylation decreases methylation levels and inflammatory/interferon/immune responses. CIMP in CRC is triggered by an imbalance of methylating activities over demethylating activities. These mice represent a model of CIMP CRC.
Keywords: DNA methylation, DNA demethylation, CpG Island Methylator Phenotype, inflammatory response, interferon response
Introduction
Colorectal cancer (CRC) is the second leading cause of cancer death in Western countries. Every year, approximately 140,000 new cases are diagnosed in U.S.A. and approximately 50,000 deaths occur1. Successful control of this disease, via appropriate preventive, interventional and therapeutic strategies, will depend on the precise characterization of its molecular basis. CRC, like many other epithelial malignancies, is characterized by multi-step carcinogenesis, in which accumulation of (epi)genetic alterations in critical genes affect rate-limiting steps of cell proliferation, differentiation and death2–4. Common genetic alterations in CRC initiation and progression include inactivating mutations of the tumor suppressor genes APC, SMAD2–4 and TP53, and activating mutations of the oncogene KRAS. In addition, epigenetic changes during CRC progression involve DNA methylation, chromatin modification and remodeling, and microRNA expression2, 4, 5.
Although several discrete alterations in DNA methylation patterns have been identified in CRC their causes are poorly defined. These include loss of imprinting marks distinguishing the paternal and maternal alleles; genome-wide DNA hypomethylation, particularly evident at satellite sequences; and DNA hypermethylation and silencing of CpG-rich loci and promoters (i.e., the CpG island methylator phenotype (CIMP)6–8. While some hypermethylation events are linked to aging9, it is clear that even after correcting for age, consistent hypermethylation of clusters of CpG islands is found in a subset of tumors, in which CIMP represents a powerful pathogenetic mechanisms because it can achieve inactivation of multiple tumor suppressor genes at once7, 10. Pan-analyses of The Cancer Genome Atlas (TCGA) have identified CIMP in multiple cancer types11. Whereas CIMP in glioma (G-CIMP) is associated with gain-of-function mutations in IDH1 that lead to inactivation of dioxygenases, suggesting a generalized impact on the epigenome12, the molecular basis of CIMP in CRC is unknown13.
Ten-Eleven Translocation (TET) family dioxygenases play a crucial role in active DNA demethylation by oxidizing 5-methylcytosine to 5-hydroxymethylcytosine (5hmC), and converting 5hmC to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC)14–16. Low levels of 5hmC have been reported in CRC, suggesting an involvement of TET enzymes in CRC tumorigenesis17, 18. In past work, we showed that TET1 binds preferentially to hypomethylated CpG islands and maintains their hypomethylated status by removing and preventing the spreading of aberrant DNA methylation from nearby loci19. In addition, we and others identified the base excision repair enzyme Thymine DNA Glycosylase (TDG) as a critical effector of DNA demethylation downstream of TET enzymes, mediating the removal of 5fC, 5caC and 5-hydroxymethyluracil (5hmU), a deamination product of 5hmC20–25. We also found that the expression of TDG is frequently reduced in CRC cell lines, and that TDG acts as a tumor suppressor in the pathogenesis of a subset of intestinal tumors26. Importantly, inactivation of TDG by knock-out in developing murine embryos22, 24 and by knockdown in melanoma cell lines27 is associated with increased DNA methylation.
Based on this work, we hypothesized that CIMP in CRC may originate from an imbalance of methylating activities over demethylating activities that normally protect CpG islands from methylation7, 28. To functionally study the role of TET1 and TDG in epigenome modulation in CRC formation, we crossed mice bearing the Tet1− knock-out allele29 and/or the dominant-negative, glycosylase-dead TdgN151A allele22 with ApcMin (Multiple Intestinal Neoplasia) mice, which are predisposed to colorectal neoplasia30. Analysis of the resulting tumor phenotypes revealed complex roles for TET1 and TDG in modulating methylation patterns/CIMP and gene expression, and revealed a tumor suppressive role associated with inhibition of inflammatory, interferon and immune response pathways. Analysis of the colon adenocarcinoma (COAD) dataset in TCGA confirmed the association between low DNA methylation levels and low inflammatory, interferon and immune response, which may have important diagnostic and (immuno)therapeutic implications. To our knowledge, this is the first study in which TET1 and TDG have been studied and compared in the context of cancer disposition and inflammation.
Material and Methods
Experimental animals
The Institutional Animal Care and Use Committee of the Fox Chase Cancer Center approved animal protocols and mouse handling procedures. Tet1+/− (from The Jackson Laboratory)29 and ApcMin mice31 were mated with TdgN151A/+ mice22. The resulting F1 Tet1+/−ApcMin/+ and Tet1+/−TdgN151A/+ mice were crossed to generate 92 F2 mice: Tet1−/−Tdg+/+ApcMin/+ (n=8); Tet1+/−TdgN151A/+ApcMin/+ (n=22); Tet1+/+TdgN151A/+ApcMin/+ (n=33); and Tet1+/+Tdg+/+ApcMin/+ (n=29)(Suppl. Fig. 1). All the strains used in this study have a C57BL/6J genetic background.
Histopathology and tumor load analyses
Animals were monitored weekly for rectal prolapse and/or bleeding and sacrificed at 150 days. At the time of sacrifice, small intestine, cecum, colorectum and other organs were collected and inspected. Small intestinal and colorectal adenomas were counted, evaluated for hemorrhagic features and measured with a ruler. Proximal, middle, and distal small intestine and colorectum were embedded in paraffin, and sections were stained with hematoxylin/eosin26 for histopathological evaluation by a pathologist (RN) with expertise in gastrointestinal cancer.
Immunohistochemistry
Colonic adenoma sections were fixed for 24hr in 10% buffered formalin and embedded in paraffin. Sections were deparaffinized, rehydrated, and antigen retrieval was performed in citrate buffer (pH 6.0) for 1hr at 95°C. Sections were incubated with 3%H2O2 for 10min and blocked with 5% goat serum in 1%BSA in PBS for 30min. Sections were incubated overnight at 4°C with rat anti-mouse F4/80 and anti CD45R/B220 antibodies (BD-Pharmingen), and rabbit anti-mouse CD3 antibody (Dako/Aligent). After incubation with biotinylated goat anti-rat and anti-rabbit secondary antibodies, staining was visualized using streptavidin–horseradish peroxidase (BD-Pharmingen) and chromogen diaminobenzidine (Sigma) for 10min.
DNA methylation analysis by DREAM
Genomic DNA was extracted from normal colonic mucosa (n=6 from 6 C57BL/6J mice) and colonic adenomas (n=12, from three mice each of the four genotypes), using Gentra Puregene Tissue Kit (Qiagen). Adenomas had similar size (4–6mm) and lacked hemorrhagic features. DNA methylation was determined with Digital restriction enzyme analysis of methylation (DREAM)32. Sequencing reads were mapped to SmaI/XmaI restriction sites in mouse genome mm10, and methylation ratios were calculated as a proportion of methylated counts to the sum of unmethylated and methylated counts32. Differentially methylated CpG sites had minimal methylation differences of 5% with false discovery rate (FDR)< 0.05.
Whole Genome Methylome by Enzymatic Methyl-Seq (EM-Seq)
Genomic DNA from duplicate colonic adenomas (n=8, from two mice each of the four genotypes) was processed for library preparation using NEBNext Enzymatic-Methyl-seq Kit (New England Biolabs). Libraries were pair-end sequenced (150 cycles) on NextSeq550 (Illumina). EM-seq reads were trimmed and aligned to reference mouse genome (mm10). Duplicate reads were removed and methylation ratios were calculated per CpG.
Analysis of DNA Methylation by Bisulfite Modification Sequencing
Genomic DNA (250–500ng) was modified by sodium bisulfite using EZ DNA Methylation-Lightning kit (Zymo Research). Amplicons were designed using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/); primer sequences are available upon request. PCR products were purified with PCR Purification Kit (QIAGEN), subcloned into pGEM-T-Easy-I (Promega), and sequenced (n=12–19). Methylation levels were analyzed by QUMA (http://quma.cdb.riken.jp/).
RNA sequencing
Total RNA was isolated from colonic adenomas from all genotype groups in triplicate using TRIzol (Invitrogen); adenomas had similar size (4–6mm) and lacked hemorrhagic features. Sequencing libraries were constructed using TruSeq RNA Sample pre-kit-V2 (Illumina), and single-read sequenced (75 cycles) on HiSeq2500 (Illumina). Reads were aligned to mouse mm10 genome using Tophat233. Cufflinks algorithm34 was implemented to assemble transcripts and estimate their abundance. Cuffdiff35 was used to statistically assess expression changes; genes with FDR<0.05 and fold change ≥2 were considered differentially expressed. Analysis of differentially expressed genes among genotypes was conducted with SuperExactTest package in R36. Gene ontology was done with Ingenuity Pathway Analysis (http://www.qiagen.com).
Preparation and lentiviral transduction of colonic organoids
AP and AKP organoids, provided by Grivennikov lab, were obtained37 from colons of tamoxifen-treated CDX2::ER-T2-Cre Apcflox/floxp53flox/flox and CDX2::ER-T2-Cre Apcflox/floxp53flox/floxKras LSL-G12D mice. After dissociation into single cells with TrypLE (Invitrogen), 1×105 cells were mixed with polybrene (8μg/ml) and lentiviral control pLKO or C8 lentivirus27 expressing shRNA against Tdg. The mixture was transferred to Matrigel (BD Sciences)-coated well at 37°C for 24hr. After puromycin selection (4 and 5μg/ml for AP and APK, respectively), organoids were expanded in Matrigel-DMEM F12 (supplemented with antibiotics, 10mm Hepes, L-Glut-Max, N2, B27 and EGF 50μg/ml) for RNA isolation.
Bioinformatic analysis
The Cancer Genome Atlas (TCGA) COAD (colon adenocarcinoma) and READ (rectal adenocarcinoma) datasets38 were downloaded from NCI Genomic Data Commons (https://gdc.cancer.gov/). cBioPortal (https://www.cbioportal.org/) and Oncomine (http://www.oncomine.org) were used to explore the alterations and expression levels, respectively, of TET1–3 and TDG in TCGA COAD and READ. Hierarchical clustering of TCGA COAD mRNA expression data from 278 tumor samples was performed using the heatmap.2 function from the R gplots package; rows were centered and scaled, and the distance metric was Euclidean with complete linkage. Boxplots of gene expression used Z-scores from cBioPortal39; and p-values for expression differences among groups are from Kruskal-Wallis tests. Processed TCGA data to analyze counts of copy number variants (CNVs) in C2 and C4 tumors were obtained from UCSC Xena Functional Genomics Explorer; CNVs were calculated for each chromosome and compared between tumor groups (C2/C4) using Poisson regression, including chromosome as a random effect; exponent of Poisson regression coefficient estimates ratio of mean chromosome aberration rates between groups.
Quantitative Real-Time PCR
RNA was reverse transcribed to cDNA using SuperScript-IV-VILO (ThermoFisher); qPCR was performed using SYBR Green (ThermoFisher). Primer sequences are available upon request.
Killing by Natural Killer Cells
Killing of HT29 by NK-92 cells was monitored on xCELLigence platform (ACEA Biosciences), measuring electrical impedance of adherent cells; impedance decrease over time reflects cytotoxicity. HT29 cells transfected with scramble siRNA or siRNA against TET1 or TDG (Horizon Discovery) were plated in triplicate at 4,000 cells/well on xCELLigence plates. The next day, NK-92 cells were added at 50,000 cells/well. Impedance was recorded at 15-minute intervals for additional 24hr.
Statistical analysis
Comparison of adenoma size/number between different genotypes was by binomial test of proportions, whereas comparison of fraction of hemorrhagic adenomas was by Mann-Whitney test; all tests were two-sided and used a Type-I Error of 5% to determine statistical significance; computations were done with R language40. Differences of hypo- and hypermethylated CpG sites in volcano plots between adenomas of various genotypes and normal colonic mucosa were analyzed using two-sided one sample test of proportions whereby comparison involving Tet1+/+Tdg+/+ApcMin/+ adenomas vs. normal colonic mucosa was used as reference; a Type-I Error of 5% was used to determine statistical significance.
Data availability
Datasets are in GEO repository at accession numbers GSE179313 and GSE179526.
Results
Tet1 and Tdg act as intestinal tumor suppressors in the context of the ApcMin mutation
To investigate the biological significance of the TET-TDG demethylation axis (Suppl. Fig. 1A) in intestinal tumorigenesis, we crossed Tet1 knockout mice29 and/or mice bearing the dominant-negative, glycosylase-dead TdgN151A allele22 with ApcMin mice, predisposed to intestinal adenomas30, 31; we employed the ApcMin Fox Chase Cancer Center variant characterized by high incidence of colonic adenomas31. We studied mice with the following 4 genotypes (Suppl. Fig. 1B): Tet1−/−Tdg+/+ (Tet1 knockout) ApcMin/+ (hereinafter referred to as Te1-Te1-Am mice); Tet1+/−TdgN151A/+ (double heterozygotes) ApcMin/+ (Te1-Td-Am mice); Tet1+/+TdgN151A/+ (Tdg heterozygotes) ApcMin/+ (Td-Am mice), and Tet1+/+Tdg+/+ (“wild type”) ApcMin/+ (control Am mice). We note that, as the TdgN151A mutation is embryonically lethal in homozygosity22, we were unable to include these mice in our analysis. In addition, live-birth Tet1−/−TdgN151A/+ApcMin/+ mice were never obtained, due to embryonic lethality.
We first investigated whether Tet1 and /or Tdg defects modify tumor multiplicity or size in the ApcMin background. For tumor multiplicity, we found that Td-Am mice showed a predisposition to develop a large number of adenomas (>30) in the small intestine, a phenotype that was not observed in Te1-Te1-Am, Te1-Td-Am or control Am mice; in contrast, Te1-Td-Am and Te1-Te1-Am mice tended to have <10 small intestinal adenomas (less than control Am mice), but more colonic adenomas than Am mice (Fig. 1A; Suppl. Fig. 2A). For tumor size, Td-Am mice tended to develop small (<2mm) adenomas of the small intestine (Fig. 1B, left); in contrast, both Te1-Te1-Am and Te1-Td-Am mice exhibited a tendency to develop large colonic adenomas (>3mm) compared with control Am mice (Fig. 1B; Suppl. Fig. 2B). Histopathological analysis revealed more frequent surface erosion in adenomas from Te1-Te1-Am, Te1-Td-Am and Td-Am mice, in comparison with adenomas from control Am mice (Fig. 1C–D).
Figure 1 -. Tet1 and Tdg inactivation modifies tumor multiplicity or size in the ApcMin background.
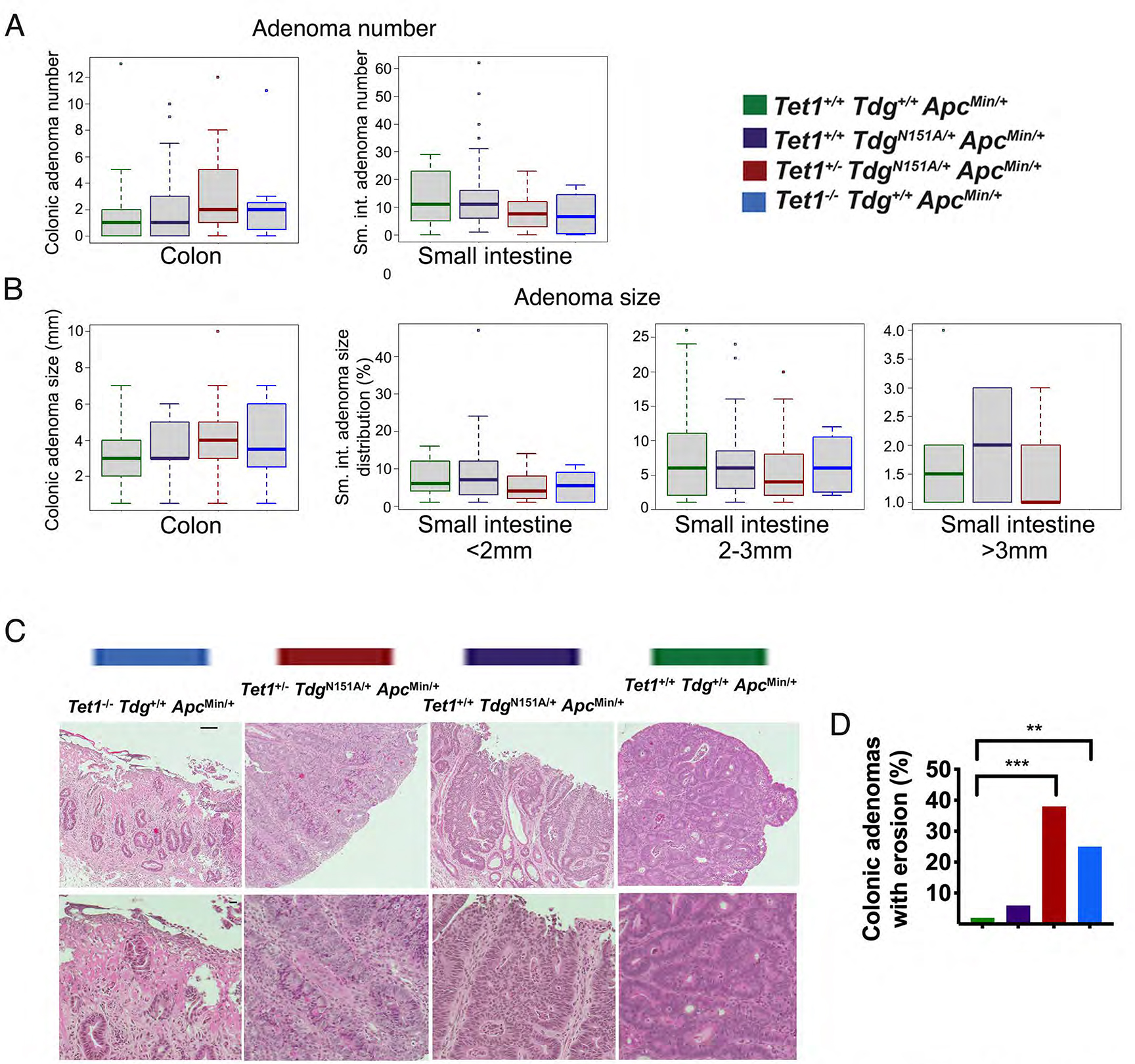
Box plots representations of number (A) and size distribution (B) of colonic and small intestinal adenomas in Tet1−/−Tdg+/+ApcMin/+ mice; Tet1+/−TdgN151A/+ApcMin/+ mice; Tet1+/+TdgN151A/+ApcMin/+ mice, and control Tet1+/+Tdg+/+ApcMin/+ mice. Small intestinal adenomas were binned in: <2mm, 2–3mm and >3mm. (C) Histopathological analysis of adenomas of different genotypes stained by hematoxylin-eosin and visualized at 10x (top row; scale bar=50 microns) and 20x magnification (bottom row; scale bar=10 microns). (D) Frequency of surface erosion in adenomas of different genotypes. **p-value <0.01, ***p-value <0.001.
A statistically significant increase of congested, hemorrhagic adenomas, limited to the colon, was observed in Te1-Te1-Am (p-value=0.0044) and Te1-Td-Am (p-value=0.0008) mice, compared with control Am mice, in which they were rarely detected (Fig. 2A). In addition, histopathological analysis of hemorrhagic adenomas from Te1-Te1-Am and Te1-Td-Am mice revealed features of invasion compared with control Am mice (Fig. 2B). Infiltration of macrophages (F4/80 staining) in the stroma surrounding adenomatous crypts was seen in Te1-Te1-Am, Te1-Td-Am and Td-Am mice, but not control Am mice, whereas there was no difference in T-cell infiltration (CD3 staining) among the four genotypes (Suppl. Fig. 3A,B). No B-cell infiltration (CD45R/B220 staining) was noted (not shown). Gene expression (see below) deconvolution confirmed these infiltration results (Suppl. Fig. 3C). For all genotypes, tumors were only observed in the digestive tract, and no significant differences in survival among the four genotypes (i.e., no spontaneous deaths) were detected before planned euthanasia at 150–160 days. Te1-Te1, Te1-Td and Td mice (lacking ApcMin mutation) did not manifest tumors when aged up to 1 year.
Figure 2 -. Hemorrhagic colonic adenomas in Tet1+/−TdgN151A/+ApcMin/+ mice and Tet1−/−Tdg+/+ApcMin/+ mice.
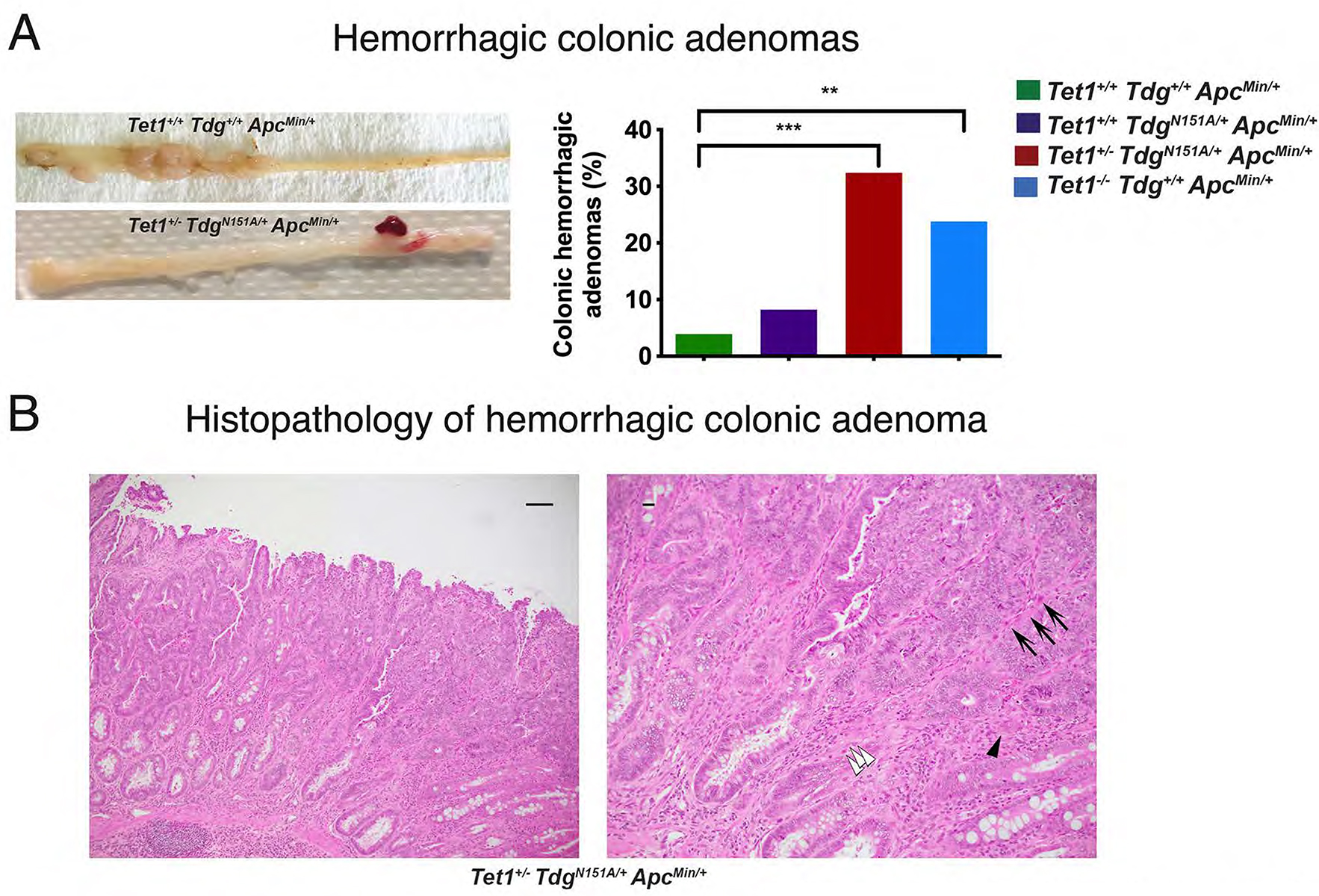
(A) Gross morphology and percent of hemorrhagic colonic adenomas in the four genotype-groups. **p-value=0.0044, ***p-value=0.0008. (B) Representative pictures of hematoxylin-eosin-stained sections of a hemorrhagic colonic adenoma developed in Tet1+/−TdgN151A/+ApcMin/+ mouse at 10x (left; scale bar=50 microns) and 20x magnification (right; scale bar=10 microns), showing evidence of malignant transformation, evidenced by overall disorganized architecture, irregular, ragged and invading glands. Frame in left panel corresponds to enlarged right panel. White arrowheads mark cancer cells invading the stroma; black arrowhead points to a budding gland; arrows point to capillaries responsible for the characteristic hemorrhagic macroscopic appearance.
These observations indicate that both Tdg and Tet1 act as tumor suppressor genes in the intestine in the context of the ApcMin background, affecting tumor size (Tet1) or multiplicity (Tdg). In addition, Tet1 homozygous mutations or double heterozygous Tet1/Tdg mutations enhance intestinal tumorigenesis in the ApcMin background, by causing hemorrhagic adenomas with features of invasion, indicating an incipient conversion to malignancy.
Tet1 and Tdg mutations affect the methylome of colonic adenomas in the ApcMin background
To determine whether comparable epigenomic changes are associated with Tet1 and Tdg defects in the context of the ApcMin/+ background, we conducted a genome-wide DNA methylation analysis of adenomas by Digital restriction enzyme analysis of methylation (DREAM)32, which interrogates approximately 25K CpG sites in the murine genome, including both CpG islands (CGI) and non-CpG islands (NCGI), and provides high-coverage of target CpG sites41. DREAM analysis was conducted on three adenomas from mice of each of the four genotypes, and on six samples of normal colonic mucosa from wild type C57BL/6J mice. Analyzed adenomas from each genotype were chosen based on similar size (4–6mm) and lack of hemorrhagic features.
We first focused on methylation levels at NCGIs. Compared to normal colonic mucosa, a global hypomethylation at NCGIs was detected in control Am adenomas (Fig. 3A) and is reminiscent of the genome-wide hypomethylation reported in human colonic adenomas as one of the earliest manifestations of multistep tumorigenesis6, 42. Remarkably, this global hypomethylation was progressively decreased in Td-Am, Te1-Td-Am and Te1-Te1-Am adenomas (Fig. 3A,C), which indicates a shift towards increased methylation levels in the absence of a functional TET-TDG axis.
Figure 3 -. Loss of global DNA hypomethylation in Tet1- and Tdg-mutant ApcMin adenomas.
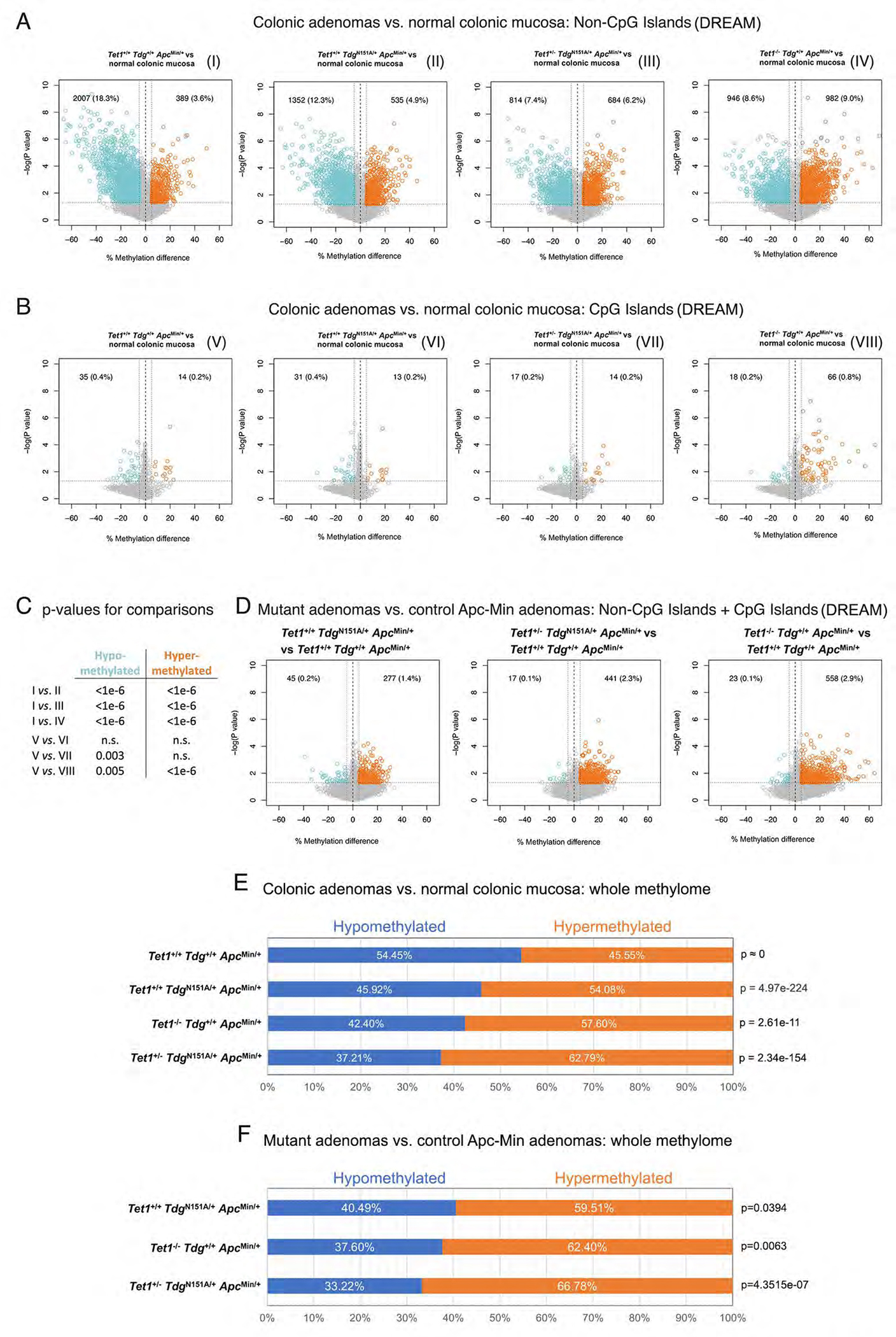
Volcano plots showing differences in DNA methylation at (A) non-CpG island (NCGI) and (B) CpG island sites (CGI) in each comparison of Tet1−/−Tdg+/+ApcMin/+ adenomas, Tet1+/−TdgN151A/+ApcMin/+ adenomas, Tet1+/+TdgN151A/+ApcMin/+ adenomas and control Tet1+/+Tdg+/+ApcMin/+ adenomas vs. normal colonic mucosa. CpG sites showing average methylation changes ≥5% and p-value<0.05 are highlighted in blue (hypomethylated sites) and orange (hypermethylated sites); number and percent of hypo-/hypermethylated CpG sites over total number of CpG sites analyzed is shown in each Volcano plot. Volcano plots are labeled I through VIII. (C) Summary of p-values for differences in hypo- and hypermethylated CpG sites between the indicated volcano plots in A and B, labeled I through VIII; n.s.=non-significant. (D) Volcano plots showing the global (NCGI + CGI) difference in average DNA methylation in each comparison between Tet1−/−Tdg+/+ApcMin/+ adenomas, Tet1+/−TdgN151A/+ApcMin/+ adenomas, Tet1+/+TdgN151A/+ApcMin/+ adenomas vs. control Tet1+/+Tdg+/+ApcMin/+ adenomas. CpG sites showing average DNA methylation changes ≥5% and p-value<0.05 are highlighted in blue (hypomethylated sites) and orange (hypermethylated sites). (E) Percent hypo- and hyper-methylation in comparisons between normal murine colonic mucosa methylome (GSE57527)44 and methylome of Tet1−/−Tdg+/+ApcMin/+ adenomas, Tet1+/−TdgN151A/+ApcMin/+ adenomas, Tet1+/+TdgN151A/+ApcMin/+ adenomas and control Tet1+/+Tdg+/+ApcMin/+ adenomas. (F) Percent hypo- and hyper-methylation in each comparison between Tet1−/−Tdg+/+ApcMin/+ adenomas, Tet1+/−TdgN151A/+ApcMin/+ adenomas, Tet1+/+TdgN151A/+ApcMin/+ adenomas vs. control Tet1+/+Tdg+/+ApcMin/+ adenomas. Percent hypo-/hypermethylation is computed by identifying CpGs in the intersection between each methylome comparison; p-value was computed with Wilcoxon rank-sum test.
Previous DNA methylation analysis of ApcMin tumors showed little evidence of CGI hypermethylation43. However, Te1-Te1-A adenomas showed elevated methylation at CpG sites (Fig. 3B,C), which resembles the CIMP in humans. Microsatellite instability (MSI), usually associated with CIMP in humans, was not detected in Te1-Te1-Am adenomas (not shown).
An overall shift towards increased methylation was also detected when Te1-Te1-Am, Te1-Td-Am and Td-Am adenomas were compared to control Am adenomas (Fig. 3D and Suppl. Fig. 4).
Because a restriction enzyme-based method like DREAM could potentially introduce a bias, we employed whole-genome enzymatic methylation-sequencing (WGEM-seq) for duplicate adenoma samples of four genotypes, covering approximately 80% of CpG sites in the genome (Suppl. Table 1). Am adenomas demonstrated hypomethylation (54.45% of the CpG sites) in comparison to normal colonic mucosa44, but the fraction of hypomethylated sites was decreased in Td-Am (45.92%), Te1-Te1-Am (42.40%) and Te1-Td-Am (37.21%) adenomas (Fig. 3E). When we compared mutant adenomas to control Am adenomas, hypermethylation was detected in Td-Am (59.51%), Te1-Te1-Am (62.40%) and Te1-Td-Am (66.78%) adenomas (Fig. 3F), confirming DREAM analysis.
Thus, Tet1 and Tdg mutations shape the methylome of colonic adenomas in the ApcMin background: colonic adenomas from Te1-Te1-Am, Te1-Td-Am and Td-Am mice exhibit increased methylation at NCGIs in comparison to control Am adenomas, whereas only Te1-Te1-Am adenomas display evidence of increased methylation at CGI - a CIMP-like phenotype.
Tet1- and Tdg-mutant ApcMin colonic adenomas upregulate inflammatory, immune and interferon response genes relative to control ApcMin adenomas
To clarify the role of Tet1 and Tdg mutations in CRC tumorigenesis and evaluate the impact of the observed methylation changes on gene expression, we conducted RNA-seq of adenomas from Te1-Te1-Am, Te1-Td-Am, Td-Am and control Am mice, identifying 88, 74 and 123 differentially expressed genes, respectively (Fig. 4A). By SuperExactTest, the commonality of differentially expressed genes among genotypes was statistically significant when calculated against the background (null hypothesis) of 25,000 genes in the mouse genome (Fig. 4B). Gene ontology analysis of each group of differentially expressed genes revealed an enrichment of common processes of inflammatory response, inflammatory disease, and immunological disease (Fig. 4C). A direct comparison of these 224 genes to interferon beta-stimulated genes45 revealed that the differentially expressed genes between Tet1 and Tdg mutant adenomas and the ApcMin/+ control adenomas are also enriched for interferon response genes (Fig. 4D).
Figure 4 -. Tet1- and/or Tdg-mutant ApcMin adenomas exhibit upregulation of inflammatory, interferon and immune response pathways.
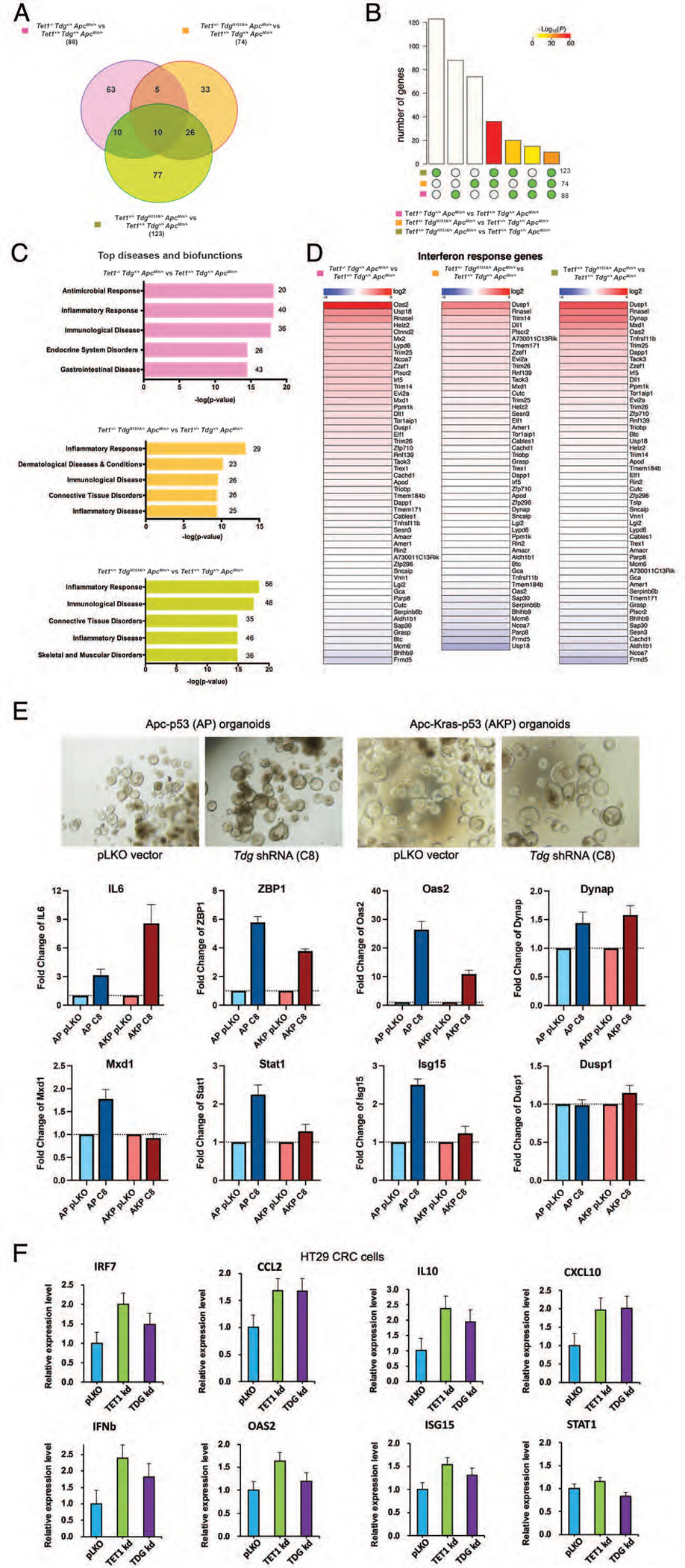
(A) Venn diagrams showing the number of common and differentially expressed genes between Tet1−/−Tdg+/+ApcMin/+ adenomas, Tet1+/−TdgN151A/+ApcMin/+ adenomas, Tet1+/+TdgN151A/+ApcMin/+ adenomas vs. control Tet1+/+Tdg+/+ApcMin/+ adenomas. (B) Bar chart illustrating all possible intersections, by SuperExactTest, among genes differentially expressed (y-axis) in Tet1−/−Tdg+/+ApcMin/+ adenomas, Tet1+/−TdgN151A/+ApcMin/+ adenomas, Tet1+/+TdgN151A/+ApcMin/+ adenomas vs. control Tet1+/+Tdg+/+ApcMin/+ adenomas confirms statistically significant involvement of the same genes in Tet1 and Tdg mutant adenomas. The matrix of solid and empty circles at the bottom illustrates “presence” (solid green) or “absence” (empty) of data sets in each intersection; numbers to the right of the matrix are set sizes; colored bars represent intersection sizes with color intensity showing p-value significance. (C) Ingenuity pathway analysis of differentially expressed genes in Tet1−/−Tdg+/+ApcMin/+ adenomas, Tet1+/−TdgN151A/+ApcMin/+ adenomas, Tet1+/+TdgN151A/+ApcMin/+ adenomas vs. control Tet1+/+Tdg+/+ApcMin/+ adenomas. The most highly enriched gene ontology categories (top diseases and biofunctions) are shown. (D) Differentially expressed genes between Tet1−/−Tdg+/+ApcMin/+ adenomas, Tet1+/−TdgN151A/+ApcMin/+ adenomas, Tet1+/+TdgN151A/+ApcMin/+ adenomas vs. control Tet1+/+Tdg+/+ApcMin/+ adenomas are compared with a list of genes induced by interferon-beta45. (E) qRT-PCR showing induction of interferon and inflammatory response genes following Tdg knockdown (C8) versus lentivirus control (pLKO) infection of colonic organoids bearing Apc/p53 mutation (AP) or Apc/Kras/p53 mutation (AKP). Morphology of organoids (phase-contrast) is shown. (F) qRT-PCR showing induction of interferon and inflammatory response genes in HT29 CRC cells following TET1 or TDG knockdown versus lentivirus control (pLKO) infection.
To determine whether upregulation of inflammatory and interferon response is, at least initially, cell autonomous, i.e., occurring in epithelial cells, we studied double mutant Apc-p53 (AP) and triple mutant Apc-Kras-p53 (AKP) murine colonic organoids. Infection of these organoids with a lentivirus expressing shRNA against Tdg led to increased expression of key inflammatory and interferon response genes, including Il6, Zbp1, Oas2, in both organoid models, although the response was somewhat blunted in AKP organoids (Fig. 4E). Upregulation of inflammatory and interferon response genes, including Irf7, Ccl2, IL10, IFNB and OAS2, was also seen in human CRC HT29 cells infected with lentiviruses expressing shRNA against TET1 and TDG (Fig. 4F).
Taking advantage of WGEM-seq data, we explored the connection between changes in methylation and changes in gene expression. The vast majority of differentially expressed genes do not change their methylation status in mutant adenomas compared to control adenomas (Fig. 5B), regardless of whether they contain a CGI-promoter (like Dusp1) or not (like Cxcr2)(Fig. 5C), which suggest an indirect mode of regulation. However, two genes upregulated in Te1-Te1-Am adenomas only, Hspa1a (Fig. 5C) and Hspa1b (not shown), showed increased gene body methylation, likely responsible for increased expression.
Figure 5 -. Tet1/Tdg-related inflammatory signature and methylation changes in differentially expressed genes.
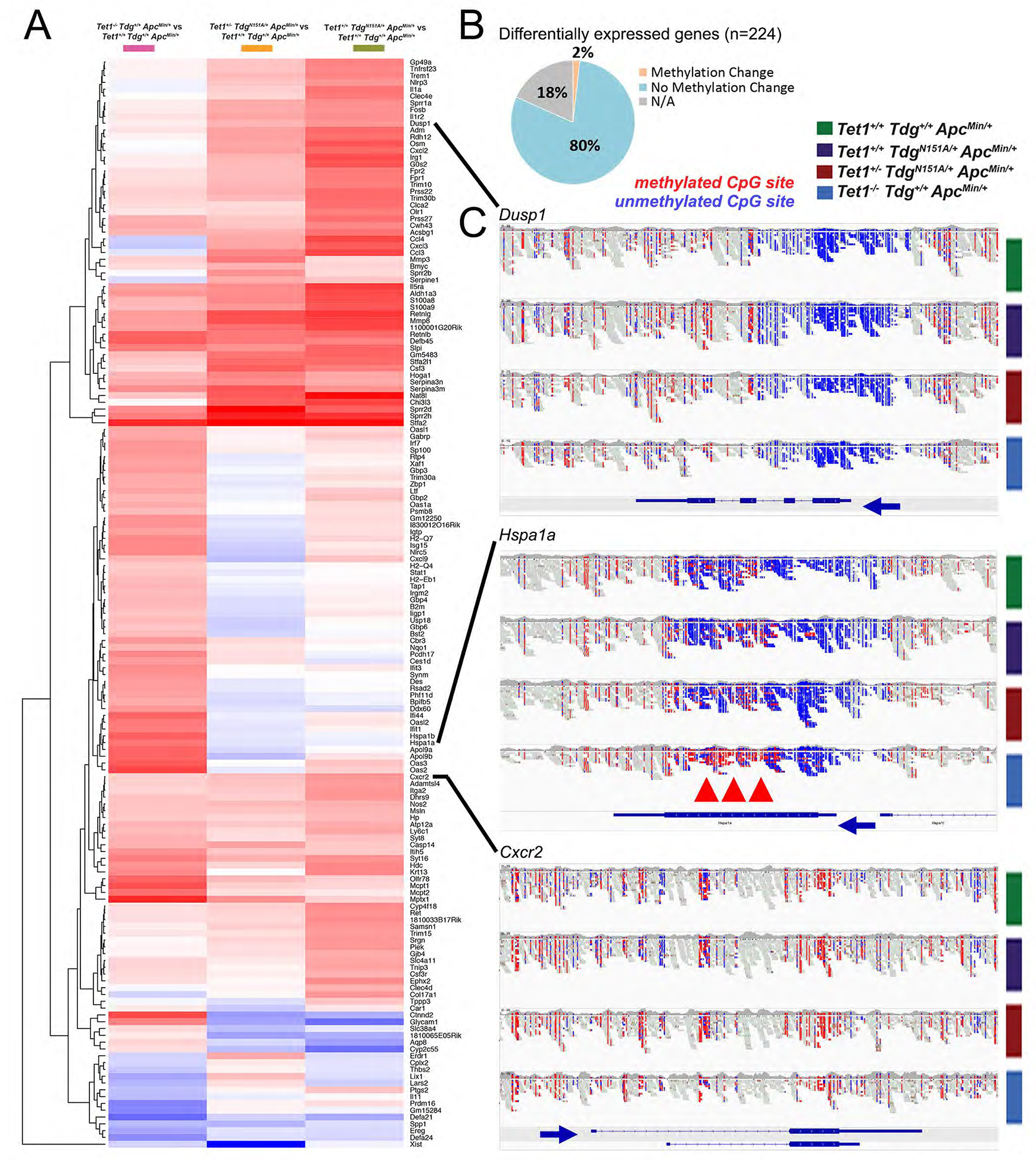
(A) Hierarchical clustering of 160 genes differentially expressed between Tet1−/−Tdg+/+ApcMin/+ adenomas, Tet1+/−TdgN151A/+ApcMin/+ adenomas, Tet1+/+TdgN151A/+ApcMin/+ adenomas vs. control Tet1+/+Tdg+/+ApcMin/+ adenomas. (B) Summary of changes in methylation of differentially expressed genes in comparisons between Tet1−/−Tdg+/+ApcMin/+ adenomas, Tet1+/−TdgN151A/+ApcMin/+ adenomas, Tet1+/+TdgN151A/+ApcMin/+ adenomas vs. control Tet1+/+Tdg+/+ApcMin/+ adenomas. NA: for 18% of genes no methylation data were available in WGEM-seq dataset. (C) Genome browser views of WGEM-seq reads covering select differentially expressed genes. Blue: unmethylated CpG site; red: methylated CpG site. Blue arrows indicate direction of transcription. Red arrowheads mark gene body methylation of Hspa1a.
Thus, in comparison to control ApcMin adenomas, adenomas from the three Tet1 and Tdg mutant genotypes exhibited a similar transcriptional profile characterized by enrichment, and prevalently upregulation, of genes belonging to the inflammatory, immune and interferon response. Because this upregulation occurs in organoids and human CRC cells, it is, by definition, cell autonomous. In general (with two notable exceptions), the mechanism of upregulation does not appear to be directly linked to methylation changes in CGI/promoter or gene body.
The TET1/TDG-related inflammatory signature is linked to genomic instability in human CRC
By conducting unsupervised hierarchical cluster analysis, we identified 160 genes, highly divergent between Tet1 or Tdg mutant adenomas and control ApcMin/+ adenomas (Fig. 5A), composing an inflammatory signature that included mostly upregulated genes. To assess the significance of this inflammatory signature, we conducted an analysis of the colon adenocarcinoma (COAD) TCGA. We first converted the murine 160-gene signature to a human 127-gene signature (Suppl. Table 2) by discarding genes that did not have a human counterpart, then conducted an unsupervised cluster analysis and looked for association with clinico-pathological parameters annotated in TCGA. The human 127-gene signature separates COAD samples into four clusters (Clusters C1–4), that do not correspond (adjusted Rand index=0.142) to the Consensus Molecular Subtypes46, but remarkably align strongly with the genomic instability profile, i.e., with either their chromosomal instability (CIN) status or MSI status (p<0.0001)(Fig. 6A). Whereas Cluster C1 has mixed MSI-CIN status and separates early from the other groups, Clusters C2 and C4 are CIN with low and high expression of the inflammatory signature; Cluster C3 corresponds mostly to MSI cases that are characterized by CIMP. We next overlayed on these four clusters the overall DNA methylation levels, that are known to be established mostly by maintenance DNA methyltransferase 1 (DNMT1). Cluster C2 (low inflammatory score, cold) exhibited high TET1/low DNMT1 and low methylation; Cluster C4 (high inflammatory score, hot) exhibited low TET1/high DNMT1 and high methylation; Cluster C3 has the highest levels of DNMT1/methylation/inflammatory score, and the lowest TET1 expression (Fig. 6B). TDG expression did not vary significantly across the four clusters, with the caveat that we are not measuring its activity. Thus, a TET1/TDG-derived inflammatory signatures separates COAD cases into four groups all characterized by anti-correlation between DNMT1/methylation levels and TET1 levels. Further, in Clusters C2 and C4, characterized by CIN, there is anti-correlation between expression levels of inflammatory genes and TET1 levels.
Figure 6 – The TET1/TDG-related inflammatory signature separates human colon adenocarcinoma cases according to their genomic instability features; concerted high DNMT1 expression and low TET1 expression in human CIMP-H colonic adenocarcinoma.
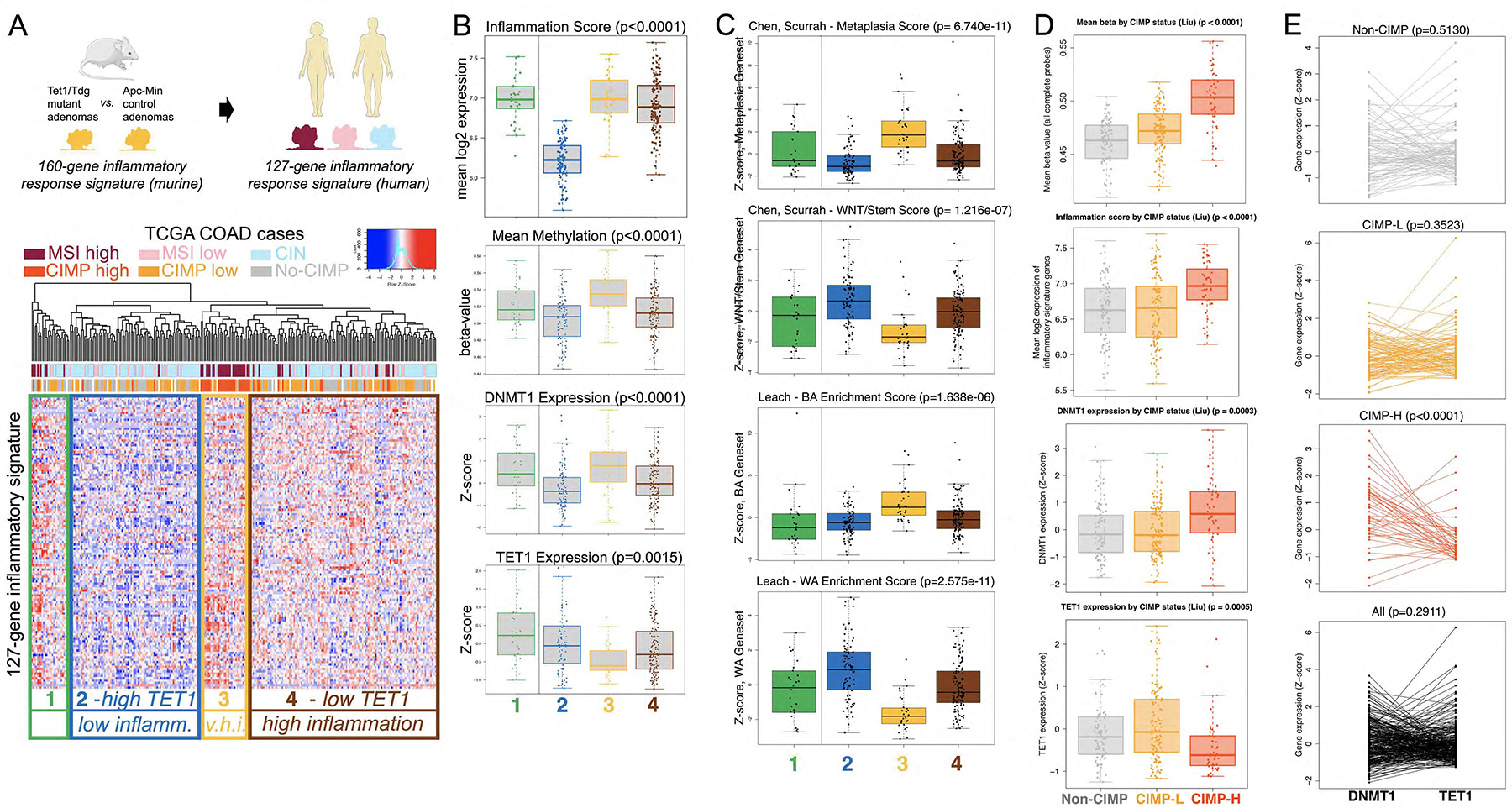
(A) The 127-gene human TET1/TDG-related inflammatory signature, derived from the 160-gene murine Tet1/Tdg-inflammatory signature, identifies 4 clusters in TCGA COAD (colon adenocarcinoma) differing in microsatellite instability (MSI) or chromosomal instability (CIN) status. V.h.i.=very high inflammation. (B) Box plot representation of inflammation score (expression levels of 127-gene signature), mean methylation, DNMT1 and TET1 expression levels in the 4 clusters. (C) Box plot representation of expression (Z-score) of Metaplasia and WNT/Stem signatures from Chen, Scurrah et al.48, and BA (Braf-Alk5) and WA (Wnt activation) signatures from Leach et al.49. (D) Box plot representation of mean methylation (β-value), inflammation score (expression levels of 127-gene signature), DNMT1 and TET1 expression levels in TCGA COAD cases, separated according to CpG Island Methylator Phenotype (no-CIMP, CIMP-L, and CIMP-H, in gray, orange and red, respectively, as in panel A). (E) Relationship between DNMT1 and TET1 expression levels in TCGA COAD cases (mRNA expression Z-scores from cBioPortal; from top to bottom: no-CIMP, CIMP-L, CIMP-H and all cases); DNMT1 expression was higher than TET1 expression in CIMP-H samples (36/46 cases, 78.3%, p-value< 0.0001 for paired Wilcoxon test).
Recently, the metaplasia/BA (Braf-Alk5) and WNT/WA (Wnt activation) gene signatures have been found to characterize the serrated and tubulovillous CRC pathways, respectively47, 48. There is no overlap between the 127-gene set and any of these signatures (Suppl. Table 4). However, the metaplasia and BA z-scores are highest in C3, whereas the WNT and WA signatures are highest in C2 and second highest in C4 (Fig. 6C).
Taken together, these observations suggest that the (epi)genetic program set forth by TET1-TDG suppresses inflammation in human colonic adenocarcinomas; and subversion of this program is strongly linked to, and helps stratify, the intrinsic features of genomic instability (MSI vs. CIN). Specifically, whereas inactivation of the WNT pathway characterizes, to different extent in C2 and C4, the tubulovillous/CIN pathway, impairment of active demethylation and increased DNA methylation drive tumors towards the serrated/CIMP pathway.
CIMP in human CRC is characterized by a concerted imbalance of DNMT1 and TET1 expression
The observation that highest levels of DNMT1/methylation/inflammatory score and lowest TET1 expression were found in Cluster C3, corresponding mostly to MSI cases characterized by CIMP, prompted us to directly evaluate these parameters according to CIMP status. Human COAD cases were divided in three groups: non-CIMP, low frequency (CIMP-L) and high frequency (CIMP-H) of DNA hypermethylation49.
Highest methylation (β-value), inflammation score and DNMT1 expression were found in the CIMP-H group, along with lowest TET1 expression (Fig. 6D), further supporting our original hypothesis that CIMP in CRC may originate from an imbalance of methylating activities over demethylating activities. In order to corroborate these results, we determined, within each tumor sample, how DNMT1 levels related to TET1 levels. There was no statistically significant pattern for the relationship between DNMT1 and TET1 levels in all COAD, non-CIMP and CIMP-L tumors, whereas DNMT1 expression was higher than TET1 expression in most CIMP-H samples (36/46, 78.3%, p-value<0.0001 for paired Wilcoxon test) (Fig. 6E). These results support the notion that CIMP-H in CRC is characterized by a concerted imbalance of methylating (DNMT1) activities over demethylating (TET1) activities.
Knockdown of TET1 and TDG enhances killing of CRC cells by Natural Killer cells
The link between blockade of active DNA demethylation and induction of inflammatory and interferon response has potentially important therapeutic implications. Because activation of the interferon response is implicated in cancer cell killing by natural killer (NK) cells50, we reasoned that activation of this response may enhance NK killing of CRC cells. We studied HT29 cells, as they are responsive to TET1 and TDG blockade (Fig. 4F). Indeed, TET1 or TDG knockdown in HT29 cells accelerate their killing by NK92 cells (Fig. 7).
Figure 7 – Knockdown of TET1 or TDG sensitizes HT29 cells to killing by Natural Killer cells.
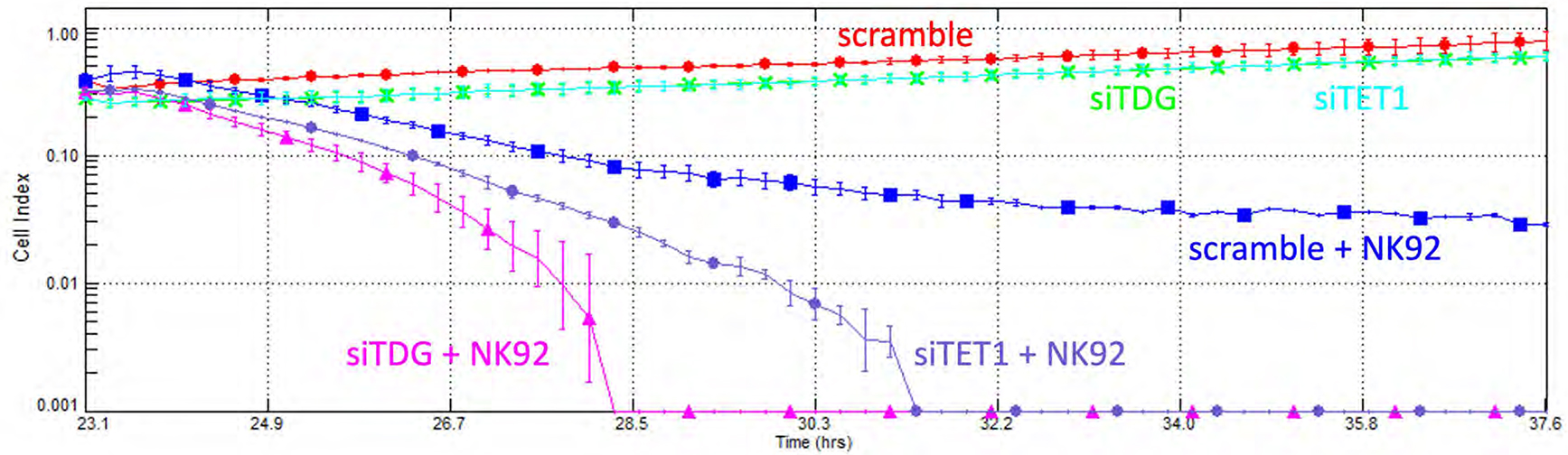
Cell killing analysis with xCELLigence system: HT29 cells were transfected with scramble siRNA or siRNA against TET1 or TDG, and the next day incubated or not with NK92 cells, and monitored in real-time for loss of adherent cells.
Discussion
In this article, we show that both Tet1 and Tdg act as tumor suppressors in intestinal tumorigenesis associated with ApcMin mutation, by affecting the number (Td-Am mice) or the size (Te1-Te1-Am and Te1-Td-Am mice) of colonic adenomas. In addition, a fraction of colonic adenomas from Te1-Te1-Am and Te1-Td-Am mice show congestive/hemorrhagic appearance, corresponding to morphological features of invasion and incipient malignancy, suggesting a role in tumor progression.
This tumor suppressive function of Tet1 and Tdg is likely linked to their role in active DNA demethylation, as their mutations had similar impact on epigenome of murine adenomas. However, only Tet1 inactivation was associated with CIMP-like phenotype; this may reflect a specific function of Tet1 compared to Tdg or alternatively may be due to the relative strength of the mutation (stronger Tet1 homozygous inactivation vs. weaker Tdg heterozygous knock-in) or the fact that, being upstream, the TET1 phenotype takes priority over the TDG phenotype.
The data support our hypothesis that an imbalance between methylating and demethylating activities causes CIMP in CRC, as Cluster C3 MSI cases, which largely align with CIMP cases in TCGA COAD38, have the highest levels of DNA methylation and DNMT1 expression and lowest levels of TET1 expression (Fig. 6). This hypothesis is further supported by the detection of concerted high DNMT1 expression and low TET1 expression specifically in CIMP-H cases, but not in no-CIMP or CIMP-L colonic adenocarcinomas (Fig. 6). Downregulation of TET1/2 in BRAFV600E-mutated CRC has been described51, but, to our knowledge, this concerted high DNMT1 expression / low TET1 expression has not been reported before and offers a clear and testable model for future mechanistic studies aimed at clarifying molecular basis of CIMP in CRC.
Analysis of gene expression revealed transcriptional activation of the inflammatory, interferon and immune response pathways in Tet1- and Tdg-mutant colonic adenomas, murine colonic organoids and HT29 cells. Further, CIN Clusters C2 and C4 with low and high levels of expression of the inflammatory gene expression signature exhibited high and low TET1 levels, respectively (Fig. 6). Thus, in an animal model of intestinal tumorigenesis, murine colonic organoids, human CRC cells and COAD dataset, TET1 and TDG suppress inflammatory, interferon and immune response pathways. Upregulation of the immune/inflammatory mRNAs in isolated epithelial cells (organoids and HT29)(Fig. 4E–F) suggests that suppression of these pathways by TET1 and TDG is largely cell-autonomous, at least initially, and later in tumor progression, via the secretion of inflammatory chemokines and cytokines, may lead to the recruitment of immune and stromal cells, particularly of the myelomonocytic and T-cell lineage (Suppl. Fig. 3). Mechanistically, upregulation of immune/inflammatory mRNAs in Tet1-Tdg mutant adenomas does not appear to be directly linked to methylation changes in CGI-promoter or gene body in most cases (Fig. 5B), suggesting that this transcriptional output could be an indirect response to epigenetic stress/damage/imbalance caused by impairment of active DNA demethylation. However, increased gene body methylation due to Tet1 deficiency might be the relevant mechanism in some cases, as for Hspa1a (Fig. 5C) and Hspa1b, suggesting that TET-mediated dynamic regulation of gene body (hydroxy)methylation has complex effects on transcription.
An important and unexpected observation in this study is that the TET1/TDG inflammatory signature separates TCGA COAD tumors based on the features of their genomic instability (MSI vs. CIN) status. It is thought that high tumor mutation burden (TMB), and consequent generation of neo-antigens, is responsible for immune infiltration and inflammation in MSI tumors52, most of which are CIMP. However, because the Te1-Te1-Am adenomas exhibit CIMP, but not MSI, our results suggest that the imbalance of methylation/demethylation in CIMP-H tumors may be a contributing factor to the proinflammatory features, which are then exacerbated by the MSI-H status. As recent clinical observations indicate that response to immune checkpoint blockade (ICB) may depend not on an absolute threshold of TMB, but rather on the causal context of high TMB53, future evaluation of epigenetic milieu, combined with high TMB, may help better stratify patients in ICB-responders and ICB-non-responders. The TET1/TDG inflammatory signature also separates TCGA COAD in CIN-cold (C2) and CIN-hot (C4), which is consistent with recent studies highlighting a role of TET enzymes and 5hmC in genome integrity, DNA damage response and aneuploidy suppression54–57.
Taken together, the findings reported here suggest an important role of active DNA demethylation mediated by TET-TDG in both epigenome and genome modulation during CRC tumorigenesis: specifically, the TET-TDG axis enforces methylation patterns and transcriptional programs that not only prevent activation of immune, interferon and inflammatory response pathways, but also impinge on the features of genomic instability. In fact, even in the presence of the strong ApcMin mutation driving tumorigenesis towards the tubulovillous adenomatous trajectory, inactivation of TET1-TDG propels tumors towards the serrated/CIMP/inflammatory pathway.
In the future, knowledge acquired through the analysis of active DNA demethylation mediated by TET-TDG might suggest innovative cancer prevention or therapeutic strategies for inflammatory bowel disease, colitis-associated cancer, and likely sporadic CRC as well. Mechanistic exploration of the processes involved in colonic inflammation and immune response regulated by TET1 and TDG has the potential to disclose novel targets to prevent transition to malignancy, implement early diagnosis and/or improve the efficacy of cancer immunotherapy.Our experiments with NK cells (Fig. 7) are in line with this contention.
Supplementary Material
Supplementary Figure 1 - (A) Schematic of the main pathway of DNA demethylation. TET enzymes (TET1–3) progressively oxidize 5mC to 5hmC, 5fC, and 5caC. TDG excises 5fC and 5caC, and downstream BER enzymes yield cytosine. DNMTs convert cytosine into 5mC. (B) Schematic of the mating strategy employed to generate mice with the four genotypes of interest.
Supplementary Figure 2 - Tet1 and Tdg inactivation modifies tumor multiplicity or size in the ApcMin background. Graphical representation of the total number (A) and size distribution (B) of small intestinal (left panels) and colonic (right panels) adenomas in Tet1−/−Tdg+/+ (Tet1 knockout) ApcMin/+ mice; Tet1+/−TdgN151A/+ (double heterozygotes) ApcMin/+ mice; Tet1+/+TdgN151A/+ (Tdg heterozygotes) ApcMin/+ mice, and control Tet1+/+Tdg+/+ (“wild type”) ApcMin/+ mice. * p-value <0.05, ** p-value <0.01.
Supplementary Figure 3 - Immunohistochemistry of adenomas of different genotypes stained with anti-F4/80 antibody (A) or anti-CD3 antibody (B); the scale bar is 100 microns. (C) Deconvolution of differential gene expression between control ApcMin/+ adenomas and Tet1 and Tdg mutant adenomas (see Fig. 4) using GEDIT (https://webtools.mcdb.ucla.edu/) confirms the prevalent involvement of myelomonocytic and T-cell lineages.
Supplementary Figure 4 - (A) Pie chart showing the percentage of hypermethylation and hypomethylation at CpG sites by genomic feature/location (promoter, exonic, intronic and intergenic) for comparison between Tet1−/−Tdg+/+ (Tet1 knockout) ApcMin/+ adenomas, Tet1+/−TdgN151A/+ (double heterozygous) ApcMin/+ adenomas, Tet1+/+TdgN151A/+ (Tdg heterozygous) ApcMin/+ adenomas vs. control Tet1+/+Tdg+/+ (“wild type”) ApcMin/+ adenomas. (B) Examples of DNA methylation level validation by bisulfite sequencing of a CGI (Semaf5b) and a NCGI (Calb2) locus differentially methylated between Tet1−/−Tdg+/+ (Tet1 knockout) ApcMin/+ colonic adenomas and Tet1+/+Tdg+/+ (“wild type”) ApcMin/+ colonic adenomas.
Supplementary Figure 5 - (A) cBioPortal Oncoprint showing alterations of TET1–3 and TDG in various CRC data sets. Each column denotes an individual tumor and each row represents a gene. The total frequency of alterations per gene is noted on the left. Colors and symbols refer to type of genomic alteration and study of origin, as indicated. (B) cBioPortal Oncoprint showing alterations and mRNA expression changes of TET1–3 and TDG levels in TCGA COAD and READ data sets. Each column denotes an individual tumor and each row represents a gene. The total frequency of alterations and changes in expression levels is noted on the left. Colors and symbols refer to type of genomic alteration or change in expression and study of origin, as indicated.
Supplementary Figure 6 - (A) Expression levels of TET1–3 and TDG mRNA (z-score relative to all samples, log RNA Seq V2 RSEM) in CRC samples in TCGA COAD (n=439) and READ (n=155) data sets. (B) TET1 expression by lower/upper quartile of TDG expression, and (C) TDG expression by lower/upper quartile of TET1 expression in TCGA COAD and READ data sets, showing that lower TET1 expression corresponds to lower TDG levels and vice versa, as indicated.
Supplementary Figure 7 - Cluster analysis of TCGA COAD (colon adenocarcinoma) cases with the 127-gene human TET1/TDG-related inflammatory signature, as in Fig. 6A, to mark clusters C1-C4 (A), with the signature unique to Te1-Te1-Am adenomas which identified the MSI/CIMP cases (yellow) (B), with the signature unique to Te1-Td-Am (double het) adenomas (C), and with the signature unique to Td-Am adenomas (D).
Supplementary Figure 8 - Cluster analysis of TCGA COAD (colon adenocarcinoma) cases with a 44-gene signature derived from the 127-gene inflammation set by linear discriminant analysis (LDA) followed by a leave-one-out cross validation; clusters C1-C4 are identified with a success rate of approximately 75%.
Supplementary Figure 9 - (A) Hypoxia score and (B) Female/Male ratio in TCGA COAD (colon adenocarcinoma) cases in clusters CIN-cold (C2) and CIN-hot (C4).
Acknowledgments
We thank Drs. E. Golemis, R. Moore and C. Sapienza for critical reading of the manuscript, and L. Cathay for secretarial assistance. We are grateful to the anonymous Reviewers for constructive criticism and suggestions. We thank the following core services at Fox Chase Cancer Center: Genomics, Cell Culture, Cell Sorting, Biological Imaging, High-Throughput Screening, Biostatistics and Bioinformatics, Transgenic Mouse and Laboratory Animal Facilities. We thank John and Robin Spurlino for their generous support of this research. AB would like to dedicate this article to the memory of his friend Antonio, who left us at the young age of 35 after a strenuous battle against MSI colorectal cancer.
Funding Sources
This study was supported by NIH grants CA78412 (to AB), CA191956 (to AB and TJY), CA218133 (to SG), R35GM144131 (to JRW) and NIH/NCI Cancer Center Support grant P30 CA006927 (to Fox Chase Cancer Center), and an appropriation from the Commonwealth of Pennsylvania, to the Fox Chase Cancer Center. Work related to this study in the Bellacosa laboratory is also supported by Melanoma Research Alliance grant 693670, American Lung Association grant 817618, and DOD LCRP grant W81XWH-21-1-0648. RT was supported in part by a William J. Avery Endowed Postdoctoral Fellowship.
Footnotes
Conflicts of interest
J.R.W. has served or is serving as a consultant or advisor for Qsonica, Salarius Pharmaceuticals, Daiichi Sankyo, Inc., Vyne Therapeutics and Lily Asia Ventures. J.R.W also receives funding for research from Salarius Pharmaceuticals and Oryzon Genomics. No relevant conflicts of interest exist for the other authors.
DNA methylation data:
To review GEO accession GSE179313:
Go to https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE179313
Enter token mjaragikttwxhyz into the box
RNA sequencing data:
To review GEO accession GSE179526:
Go to https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE179526
Enter token mjsbkuuglhcrdez into the box
References
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61:759–767. [DOI] [PubMed] [Google Scholar]
- 3.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004;4:143–53. [DOI] [PubMed] [Google Scholar]
- 4.Bellacosa A Genetic hits and mutation rate in colorectal tumorigenesis: versatility of Knudson’s theory and implications for cancer prevention. Genes Chromosomes Cancer 2003;38:382–388. [DOI] [PubMed] [Google Scholar]
- 5.Grady WM, Yu M, Markowitz SD. Epigenetic Alterations in the Gastrointestinal Tract: Current and Emerging Use for Biomarkers of Cancer. Gastroenterology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goelz SE, Vogelstein B, Hamilton SR, et al. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science 1985;228:187–90. [DOI] [PubMed] [Google Scholar]
- 7.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer 2004;4:988–93. [DOI] [PubMed] [Google Scholar]
- 8.Goel A, Boland CR. Epigenetics of Colorectal Cancer. Gastroenterology 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Issa JP. Aging and epigenetic drift: a vicious cycle. J Clin Invest 2014;124:24–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 2011;11:726–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes LA, Melotte V, de Schrijver J, et al. The CpG island methylator phenotype: what’s in a name? Cancer Res 2013;73:5858–68. [DOI] [PubMed] [Google Scholar]
- 12.Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010;17:510–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hughes LA, Khalid-de Bakker CA, Smits KM, et al. The CpG island methylator phenotype in colorectal cancer: progress and problems. Biochim Biophys Acta 2012;1825:77–85. [DOI] [PubMed] [Google Scholar]
- 14.Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 2013;14:341–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009;324:930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009;324:929–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neri F, Dettori D, Incarnato D, et al. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene 2015;34:4168–76. [DOI] [PubMed] [Google Scholar]
- 18.Mariani CJ, Madzo J, Moen EL, et al. Alterations of 5-hydroxymethylcytosine in human cancers. Cancers (Basel) 2013;5:786–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin C, Lu Y, Jelinek J, et al. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Research 2014;42:6956–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuermann D, Weber AR, Schar P. Active DNA demethylation by DNA repair: Facts and uncertainties. DNA Repair (Amst) 2016;44:92–102. [DOI] [PubMed] [Google Scholar]
- 21.Bellacosa A, Drohat AC. Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair (Amst) 2015;32:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cortellino S, Xu J, Sannai M, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011;146:67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dalton SR, Bellacosa A. DNA demethylation by TDG. Epigenomics 2012;4:459–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cortazar D, Kunz C, Selfridge J, et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 2011;470:419–423. [DOI] [PubMed] [Google Scholar]
- 25.Weber AR, Krawczyk C, Robertson AB, et al. Biochemical reconstitution of TET1-TDG-BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat Commun 2016;7:10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu J, Cortellino S, Tricarico R, et al. Thymine DNA Glycosylase (TDG) is involved in the pathogenesis of intestinal tumors with reduced APC expression. Oncotarget 2017;8:89988–89997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mancuso P, Tricarico R, Bhattacharjee V, et al. Thymine DNA glycosylase as a novel target for melanoma. Oncogene 2019;38:3710–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prasad R, Yen TJ, Bellacosa A. Active DNA Demethylation – the Epigenetic Gatekeeper of Development, Immunity, and Cancer. Advanced Genetics;n/a:e10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dawlaty MM, Ganz K, Powell BE, et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 2011;9:166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990;247:322–324. [DOI] [PubMed] [Google Scholar]
- 31.Cooper HS, Chang WC, Coudry R, et al. Generation of a unique strain of multiple intestinal neoplasia (Apc(+/Min-FCCC)) mice with significantly increased numbers of colorectal adenomas. Mol Carcinog 2005;44:31–41. [DOI] [PubMed] [Google Scholar]
- 32.Jelinek J, Madzo J. DREAM: A Simple Method for DNA Methylation Profiling by High-throughput Sequencing. Methods Mol Biol 2016;1465:111–27. [DOI] [PubMed] [Google Scholar]
- 33.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trapnell C, Hendrickson DG, Sauvageau M, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 2013;31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang M, Zhao Y, Zhang B. Efficient Test and Visualization of Multi-Set Intersections. Sci Rep 2015;5:16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dmitrieva-Posocco O, Wong AC, Lundgren P, et al. β-Hydroxybutyrate suppresses colorectal cancer. Nature 2022;605:160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing, 2021. [Google Scholar]
- 41.Maegawa S, Lu Y, Tahara T, et al. Caloric restriction delays age-related methylation drift. Nat Commun 2017;8:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grimm C, Chavez L, Vilardell M, et al. DNA-methylome analysis of mouse intestinal adenoma identifies a tumour-specific signature that is partly conserved in human colon cancer. PLoS Genet 2013;9:e1003250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fini L, Piazzi G, Daoud Y, et al. Chemoprevention of intestinal polyps in ApcMin/+ mice fed with western or balanced diets by drinking annurca apple polyphenol extract. Cancer Prev Res 2011;4:907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abu-Remaileh M, Bender S, Raddatz G, et al. Chronic inflammation induces a novel epigenetic program that is conserved in intestinal adenomas and in colorectal cancer. Cancer Res 2015;75:2120–30. [DOI] [PubMed] [Google Scholar]
- 45.Basagoudanavar SH, Thapa RJ, Nogusa S, et al. Distinct roles for the NF-kappa B RelA subunit during antiviral innate immune responses. J Virol 2011;85:2599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21:1350–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen B, Scurrah CR, McKinley ET, et al. Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell 2021;184:6262–6280.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leach JDG, Vlahov N, Tsantoulis P, et al. Oncogenic BRAF, unrestrained by TGFβ-receptor signalling, drives right-sided colonic tumorigenesis. Nat Commun 2021;12:3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y, Sethi NS, Hinoue T, et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018;33:721–735.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lam AR, Bert NL, Ho SS, et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res 2014;74:2193–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Noreen F, Küng T, Tornillo L, et al. DNA methylation instability by BRAF-mediated TET silencing and lifestyle-exposure divides colon cancer pathways. Clin Epigenetics 2019;11:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rousseau B, Foote MB, Maron SB, et al. The Spectrum of Benefit from Checkpoint Blockade in Hypermutated Tumors. N Engl J Med 2021;384:1168–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.An J, González-Avalos E, Chawla A, et al. Acute loss of TET function results in aggressive myeloid cancer in mice. Nat Commun 2015;6:10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang D, Wei S, Chen F, et al. TET3-mediated DNA oxidation promotes ATR-dependent DNA damage response. EMBO Rep 2017;18:781–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kafer GR, Li X, Horii T, et al. 5-Hydroxymethylcytosine Marks Sites of DNA Damage and Promotes Genome Stability. Cell Rep 2016;14:1283–1292. [DOI] [PubMed] [Google Scholar]
- 57.López-Moyado IF, Tsagaratou A, Yuita H, et al. Paradoxical association of TET loss of function with genome-wide DNA hypomethylation. Proc Natl Acad Sci U S A 2019;116:16933–16942. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 - (A) Schematic of the main pathway of DNA demethylation. TET enzymes (TET1–3) progressively oxidize 5mC to 5hmC, 5fC, and 5caC. TDG excises 5fC and 5caC, and downstream BER enzymes yield cytosine. DNMTs convert cytosine into 5mC. (B) Schematic of the mating strategy employed to generate mice with the four genotypes of interest.
Supplementary Figure 2 - Tet1 and Tdg inactivation modifies tumor multiplicity or size in the ApcMin background. Graphical representation of the total number (A) and size distribution (B) of small intestinal (left panels) and colonic (right panels) adenomas in Tet1−/−Tdg+/+ (Tet1 knockout) ApcMin/+ mice; Tet1+/−TdgN151A/+ (double heterozygotes) ApcMin/+ mice; Tet1+/+TdgN151A/+ (Tdg heterozygotes) ApcMin/+ mice, and control Tet1+/+Tdg+/+ (“wild type”) ApcMin/+ mice. * p-value <0.05, ** p-value <0.01.
Supplementary Figure 3 - Immunohistochemistry of adenomas of different genotypes stained with anti-F4/80 antibody (A) or anti-CD3 antibody (B); the scale bar is 100 microns. (C) Deconvolution of differential gene expression between control ApcMin/+ adenomas and Tet1 and Tdg mutant adenomas (see Fig. 4) using GEDIT (https://webtools.mcdb.ucla.edu/) confirms the prevalent involvement of myelomonocytic and T-cell lineages.
Supplementary Figure 4 - (A) Pie chart showing the percentage of hypermethylation and hypomethylation at CpG sites by genomic feature/location (promoter, exonic, intronic and intergenic) for comparison between Tet1−/−Tdg+/+ (Tet1 knockout) ApcMin/+ adenomas, Tet1+/−TdgN151A/+ (double heterozygous) ApcMin/+ adenomas, Tet1+/+TdgN151A/+ (Tdg heterozygous) ApcMin/+ adenomas vs. control Tet1+/+Tdg+/+ (“wild type”) ApcMin/+ adenomas. (B) Examples of DNA methylation level validation by bisulfite sequencing of a CGI (Semaf5b) and a NCGI (Calb2) locus differentially methylated between Tet1−/−Tdg+/+ (Tet1 knockout) ApcMin/+ colonic adenomas and Tet1+/+Tdg+/+ (“wild type”) ApcMin/+ colonic adenomas.
Supplementary Figure 5 - (A) cBioPortal Oncoprint showing alterations of TET1–3 and TDG in various CRC data sets. Each column denotes an individual tumor and each row represents a gene. The total frequency of alterations per gene is noted on the left. Colors and symbols refer to type of genomic alteration and study of origin, as indicated. (B) cBioPortal Oncoprint showing alterations and mRNA expression changes of TET1–3 and TDG levels in TCGA COAD and READ data sets. Each column denotes an individual tumor and each row represents a gene. The total frequency of alterations and changes in expression levels is noted on the left. Colors and symbols refer to type of genomic alteration or change in expression and study of origin, as indicated.
Supplementary Figure 6 - (A) Expression levels of TET1–3 and TDG mRNA (z-score relative to all samples, log RNA Seq V2 RSEM) in CRC samples in TCGA COAD (n=439) and READ (n=155) data sets. (B) TET1 expression by lower/upper quartile of TDG expression, and (C) TDG expression by lower/upper quartile of TET1 expression in TCGA COAD and READ data sets, showing that lower TET1 expression corresponds to lower TDG levels and vice versa, as indicated.
Supplementary Figure 7 - Cluster analysis of TCGA COAD (colon adenocarcinoma) cases with the 127-gene human TET1/TDG-related inflammatory signature, as in Fig. 6A, to mark clusters C1-C4 (A), with the signature unique to Te1-Te1-Am adenomas which identified the MSI/CIMP cases (yellow) (B), with the signature unique to Te1-Td-Am (double het) adenomas (C), and with the signature unique to Td-Am adenomas (D).
Supplementary Figure 8 - Cluster analysis of TCGA COAD (colon adenocarcinoma) cases with a 44-gene signature derived from the 127-gene inflammation set by linear discriminant analysis (LDA) followed by a leave-one-out cross validation; clusters C1-C4 are identified with a success rate of approximately 75%.
Supplementary Figure 9 - (A) Hypoxia score and (B) Female/Male ratio in TCGA COAD (colon adenocarcinoma) cases in clusters CIN-cold (C2) and CIN-hot (C4).
Data Availability Statement
Datasets are in GEO repository at accession numbers GSE179313 and GSE179526.