

SUMMARY
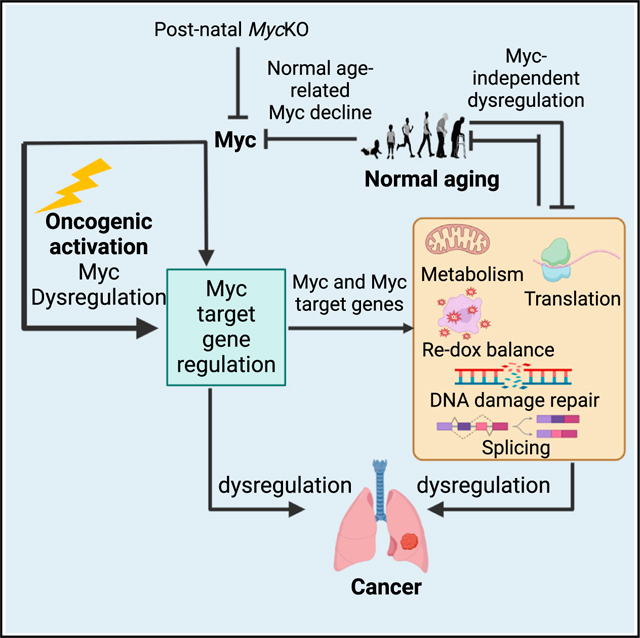
MYC proto-oncogene dysregulation alters metabolism, translation, and other functions in ways that support tumor induction and maintenance. Although Myc+/− mice are healthier and longer-lived than control mice, the long-term ramifications of more complete Myc loss remain unknown. We now describe the chronic consequences of body-wide Myc inactivation initiated postnatally. “MycKO” mice acquire numerous features of premature aging, including altered body composition and habitus, metabolic dysfunction, hepatic steatosis, and dysregulation of gene sets involved in functions that normally deteriorate with aging. Yet, MycKO mice have extended lifespans that correlate with a 3- to 4-fold lower lifetime cancer incidence. Aging tissues from normal mice and humans also downregulate Myc and gradually alter many of the same Myc target gene sets seen in MycKO mice. Normal aging and its associated cancer predisposition are thus highly linked via Myc.
In brief
Wang et al. show that the postnatal elimination of Myc causes premature aging and the deterioration of age-sensitive functions. Yet, these mice have extended lifespans and a reduced cancer incidence. Gradual Myc downregulation accompanies normal aging in many tissues. Thus, the strong relationship between aging and cancer can be severed by eliminating a single gene.
Graphical Abstract
INTRODUCTION
Precisely how the c-Myc oncoprotein (hereafter Myc) contributes to the pathogenesis of cancer has been well chronicled.1,2 Tumors often deregulate MYC, which encodes a basic helixloop-helix-leucine zipper (bHLH-ZIP) transcription factor that, upon dimerizing with its partner protein Max, binds to “E boxes” in its target genes’ promoters and enhances transcription.3–7 Negative regulation by Myc-Max is mediated by interaction with and suppression of positively acting transcription factors such as Miz1.6,8,9 Much Myc-mediated regulation involves context-dependent control over cell-cycle progression, metabolism, and translation.4,6,10–14 The magnitude of each of these reflects Myc protein levels, its accessibility to and affinity for E boxes, and their occupancy by competing factors.6,11 Secondary roles for Myc-mediated tumorigenesis include the promotion of angiogenesis and immune system evasion.15–18 Continuous Myc expression is usually needed to maintain high rates of proliferation.6,13,14,19–23
Less is known about Myc’s roles in normal development, since germline Myc inactivation in mice is embryonic lethal due to placental, hematopoietic, and vascular defects.24–27 In adult mice, body-wide Myc inhibition by the dominant interfering “Omomyc” causes reversible aplastic anemia, colonic epithelial hypoplasia, and the regression of lung neoplasms.21 However, neither the degree of Myc’s incapacitation nor its long-term consequences were described. Tissue-specific Myc inactivation has demonstrated differential dependencies, although the observation times were again limited.12,27–30 Thus, the long-term consequences of Myc inactivation remain unknown.
Relative to Myc+/+ mice, Myc+/− mice viable, are smaller, age more slowly, have longer lifespans, and develop fewer age-related pathologies.31 Their longevity may reflect a lower cancer incidence.32,33 Whether survival was influenced by slower aging rather than altered Myc levels is unclear, since aging is the strongest independent predictor of cancer development.31,32,34,35 These findings raise questions concerning Myc’s role in maintaining normal body homeostasis and imply that the consequences of Myc loss are incremental.27,31 The small number of transcript differences between Myc+/− and Myc+/+ mouse tissues also suggested that half-normal levels of Myc can exert normal or near-normal control over its target genes.11,31 Partial Myc expression might therefore forestall deleterious phenotypes in a dose-dependent manner.27
We describe here the body-wide, near-complete elimination of Myc initiated at weaning.25,27 Unlike Myc+/− mice, “MycKO” mice age prematurely yet live longer than wild-type (WT) mice while displaying a 3- to 4-fold lower lifetime cancer incidence. Transcriptional profiling in 3 tissues sensitive to Myc loss and/or aging shows widespread and early-onset dysregulation of genes involved in mitochondrial and ribosomal structure/function, oxidative stress, aging/senescence, DNA damage recognition/repair, and mRNA splicing, which is consistent with that in Myc−/− hepatocytes and murine embryonic fibroblasts (MEFs).12–14,23,31,36–38 The transcriptomic changes resemble and precede by several months those arising during normal aging, including changes in both Myc itself and its target genes. The link between aging and cancer is therefore genetically maintained by Myc.
RESULTS
Near total-body MycKO mice
B6.129S6-Myctm2Fwa/Mmjax mice13,29 (Figures S1A and S1B) were crossed with B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J mice, which express a ROSA26-driven Cre recombinase-estrogen receptor (CreER) transgene.39 Progeny strains with one or two copies of CreER were examined to determine how copy number influenced Myc excision efficiency initiated at weaning (ca. 4weeks). Because TaqMan-based assays (Figures S1A–S1D) performed 2 weeks after tamoxifen administration showed Myc locus excision efficiency in some cases to be dependent on CreER copy number (Figure S1E), subsequent studies were performed with mice carrying two copies of CreER (Figures S1E and S1F and Table S1). Tamoxifen-treated offspring of B6.129S6-Myctm2Fwa//Mmjax 3 C57BL/6 mice with intact Myc genes served as wild-type (WT} controls. Myc transcript levels correlated well with the degree of Myc deletion (Figure S1E and Table S1). Follow-up qPCR/qRT-PCR studies indicated Myc loss persistence beyond 30 months, although in some tissues it was incomplete and less prominent (Figure S1F and Table S1). Myc protein was reduced in certain tissues with a proliferative compartment (Figures S1G and S1H).
MycKO mice age prematurely, survive longer, and have a lower cancer incidence
Growth rates and body masses of WT and MycKO cohorts remained indistinguishable until ~10 months of age, when they diverged in both sexes. They then converged at 18–20 months, when body masses began their age-related decline (Figure 1A). MycKO mice showed earlier decreases in lean mass and increases in adiposity and fat:lean mass ratios that explained the otherwise identical weights of younger mice. Thus, the overall body habitus of younger MycKO mice prematurely assumed that of older WT mice.40 In females, the differences became less pronounced as WT and MycKO mice eventually acquired the same overall body composition. Although male MycKO mice showed the same tendencies, the differences from WT mice persisted throughout life.
Figure 1. Young MycKO mice display aging-related phenotypes.

(A) Weight and body composition of male and female WT and MycKO mice. Each point represents the mean of measurements performed on 10–20 animals performed over 2–3 days. Times during which differences existed between the two groups are indicated by gray shading.
(B) Premature alopecia in MycKO mice.
(C) Premature achromotrichia in MycKO mice.
(D) Appearance of representative WT and MycKO mice. See Video S1 for additional examples.
(E) Close-up images of fur from 20-month-old WT and MycKO mice showing the interspersion of dark and gray strands in the former cohort versus the greater uniformity of gray color among individual strands in the latter.
(F) Four-limb GripMeter testing performed on male and female animals. n = 9–13.
(G) Rotarod testing of WT and MycKO mice. n = 5–14.
(H) Treadmill running. Cohorts of WT and MycKO mice were allowed to maintain a continuous pace on an automated treadmill until becoming exhausted. n = 6–13.
(I) Diurnal activity of WT and MycKO mice of the indicated ages as measured in metabolic cages. n = 5–10 males and 5–10 females at each age. White and gray-shaded regions of the plots denote day and night, respectively. (A, F, G, and H) Unpaired t test; (B and C) log rank test; (I) ANOVA145; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars: standard error of the mean (SEM).
MycKO mice developed alopecia and achromotrichia as early as 3–4 months of age, which first appeared peri-orbitally and/or peri-nasally (Figures 1B–1D and Video S1). In WT mice, achromotrichia comprised alternating dark and light gray hairs, whereas in MycKO mice, all hairs were uniformly light gray, occurred in patches, and resembled those from melanocytespecific MycKO mice (Figure 1E).41,42 Some hair shafts also comprised alternating light-dark segments. Skin from alopecic areas showed epidermal thickening, hyperkeratinization, loss of surface invaginations, and reduced numbers of hair follicles and sebaceous glands (Figure S2A). Focal regions of perifollicular, senescence-associated b-galactosidase-positive cells were also noted (Figure S2B).
MycKO mice, particularly younger males, were generally weaker, less coordinated, and less active (Figures 1F–1H). However, the magnitude of these differences, when they were first detected, and their duration were age-, sex-, and test-type dependent. For example, reduced grip strength, first noted in 3-month-old male MycKO mice, did not persist beyond about 10 months (Figure 1F). This occurred in parallel with the premature muscle mass loss and its eventual equalization as WT mice aged (Figure 1A). Lessened ability to balance on a Rotarod apparatus was noted in MycKO mice of both sexes by 11 months and persisted in males (Figure 1G). Beginning at 13–16 months, male MycKO mice also showed less treadmill endurance (Figure 1H).43 Finally, diurnal ambulatory activity of MycKO mice was reduced in younger animals and decreased further by 20 months in MycKO females (Figure 1I). MycKO mice thus acquired age-related features and behaviors earlier than WT mice, although at different rates.44–47 These differences either persisted or converged as WT mice aged.
Bone marrow failure accompanies Myc loss in both the embryo and the adult,21,25,27 and MycKO mice showed mild-moderate anemia and leukopenia within 10–15 days of initiating tamoxifen treatment (Figures S3A and S3B). The accompanying bone marrow hypoplasia reflected that this was well tolerated (Figure S3C). The peripheral findings resolved within several weeks despite the long-term persistence of Myc loss (Figures S3A and S3B).21 However, the bone marrow of some MycKO mice remained hypoplastic and resembled that of middle-aged normal animals (Figure S3C). Inactivating Myc prior to weaning and/or attaining weights of 15–16 g was associated with severe and usually fatal pancytopenia. Thus, the bone marrow’s greatest Myc dependency declines during the first month of life.27
Myc loss is associated with transient flattening of the intestinal epithelium and abnormal crypts.21,28,48 We observed similar changes in the colons in young MycKO mice (ca. 2.5 months) that normalized by 5–6 months despite the persistence of Myc gene loss (Figure S3D).
Non-alcoholic fatty liver disease (NAFLD) increases in the context of age-related dyslipidemia, obesity, and insulin resistance.36,49 Neutral lipid accumulation also follows Myc (and Mycn) loss/inhibition in a variety of cells and tissues and in livers lacking other members of the “Extended Myc Network” such as ChREBP and/or Mlx.14,50–53 Yet, the contribution of aging to Myc-dependent NAFLD development is unknown.13,14,29 Consistent with this, the neutral lipid and triglyceride content of 5-month-old MycKO livers was higher than that of WT controls and rivaled that of even the oldest WT mice (Figure 2). These differences became less pronounced as the excess hepatic lipid in MycKO livers was eventually matched by WT mice, indicating that MycKO livers show acceleration of an otherwise normal age-related process.
Figure 2. MycKO mice prematurely develop NAFLD.

(A) Representative oil red O (ORO)-stained liver sections of WT and MycKO mice.
(B) Quantification of ORO-stained sections. At least 3 liver sections from 4 or 5 mice were scanned, quantified, and combined.
(C) Higher-power magnification of the sections from (A) showing a greater prominence of large lipid droplets in MycKO livers.
(D) Triglyceride content of WT and MycKO livers. (B and C) Unpaired t test, *p < 0.05, ***p < 0.001. Error bars: standard deviation (SD).
Despite aging prematurely, MycKO mice lived significantly longer than WT mice (Figure 3A). Postmortem necropsies showed that 58.1% of WT animals had tumors, with 64.3% of these resembling B cell lymphomas (Figure 3B).33,34 These were frequently of high grade, were associated with hepatosplenomegaly, and often displayed leukemic dissemination. In contrast, only 17.3% of MycKO mice had obvious tumors (p < 0.0001). The tumor spectra of the cohorts were similar, and in the few cases where mice had two or more tumors, they were histologically indistinguishable lymphomas (Figures 3C–3H). The 3.4-fold lower cancer incidence observed in MycKO mice is consistent with the Myc dependency of most can-cers.12–14,19,21,50,54 Thus, its most striking aspect was that it was dissociated from the premature aging phenotype.
Figure 3. MycKO mice have extended lifespans and lower cancer incidence.
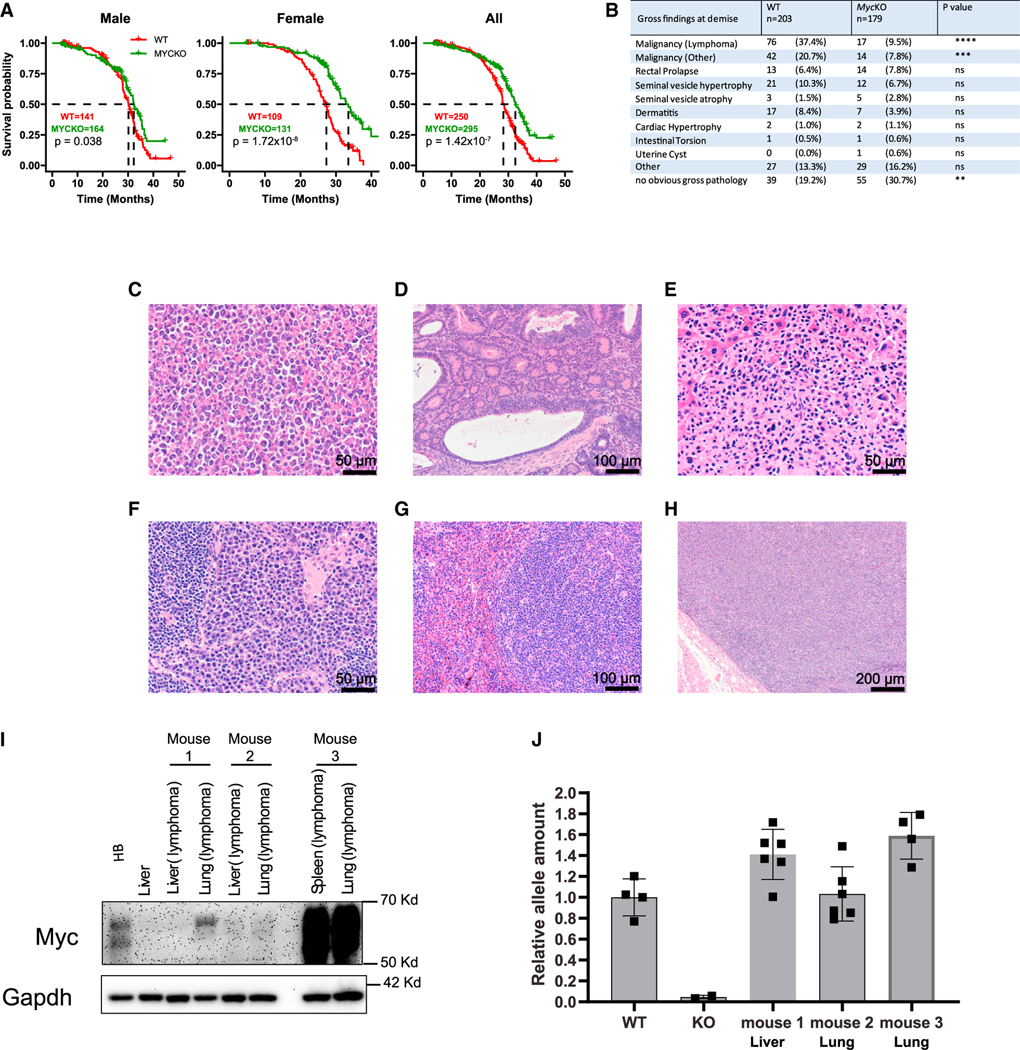
(A) Natural lifespans of WT and MycKO male, female, and all mice.
(B) Incidence of associated gross pathologies in WT and MycKO mice at the time of demise.
(C) High-grade lymphoma from a MycKO mouse forming a nodular mass adjacent to a loop of bowel.
(D) Well-differentiated MycKO colonic adenocarcinoma.
(E) High-grade MycKO lymphoma replacing normal liver parenchyma.
(F) Probable MycKO plasmacytoma.
(G) Splenic MycKO lymphoma.
(H) Lymphoma from the mouse in (G) effacing a lymph node adjacent to the pleural surface.
(I) Myc protein expression. Control tissues included normal liver and a hepatoblastoma.55 Lymphomas from three MycKO mice (#1 to #3) were sampled from the two indicated sites.
(J) Myc alleles in MycKO lymphomas (I). Myc copy number quantification was performed on several sections of each tumor (Figure S1). DNAs from WT and MycKO primary MEFs (n = 4 each) served as controls for two copies or zero copies, respectively, of an intact Myc allele.23 (A) Log rank test; (B) unpaired t test, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant; (J) error bars: SD.
Lymphomas involving 2 organs from 3 MycKO mice were examined for Myc protein. Control tissues included a WT normal liver with low to undetectable endogenous Myc levels and a hepatoblastoma (HB) with high levels.14,55 Myc levels in MycKO lymphomas ranged from undetectable-low to severalfold higher than in HB (Figure 3I). Intact and amplified Myc gene loci were detected even when they expressed little protein (Figure 3J). Thus, some tumors arising in MycKO mice originate in rare cells with intact Myc alleles that retain the ability to be amplified.
MycKO mice display metabolic and mitochondrial dysfunction
Age-related deterioration of mitochondrial structure and function affects organelle size, electron transport chain (ETC) activity, fatty acid β-oxidation (FAO), and redox balance, which are compounded by aging-related co-morbidities such as obesity, NAFLD, and insulin resistance.37,56–63 Conversely, mitochondrial dysfunction and its excessive production of reactive oxygen species (ROS) can accelerate aging.61,63,64 mtDNA and protein content and ETC function are reduced in individuals with type 2 diabetes, metabolic syndrome, and NAFLD.62,65 Myc’s roles in these processes include its maintenance of mitochondrial structure and function and the oxidation of glucose, glutamine, and fatty acids.66–72 Linked to this is ROS overproduction in response to the ETC dysfunction associated with both over and underexpression of Myc.69,71–74
High nocturnal respiratory exchange ratios (RERs) of the youngest WT mice indicated near-complete reliance on glucose as the primary energy source (Figure 4A). Normally, RERs >1 are seen in juvenile mice and following post-starvation re-feeding, where they signify high levels of both de novo fatty acid synthesis (FAS) and glucose utilization.75,76 The lower, adult-like nocturnal RERs of MycKO mice during this time indicated their disproportionate reliance on FAO and/or reduced FAS efficiency. A glycolysis → FAO switch following hepatocyte-specific loss of Myc may increase fatty acid uptake in excess of that needed for FAO, with the difference being stored as neutral lipid that causes NAFLD (Figure 2).13,14,23,29 The overreliance of MycKO mice on FAO suggested the loss of Myc-dependent glycolysis, reduced provision of pyruvate for mitochondrial ATP production, or pyruvate’s diversion into other pathways.67,71,77–79 Regardless of the cause(s), the RERs of younger MycKO mice resembled those of older animals. The differences persisted as mice aged but became more erratic, with the lower RERs of the latter now observed during the day.
Figure 4. Metabolic defects in MycKO mice are consistent with premature aging.

(A) Respiratory exchange ratios (RERs) calculated from the formula RER = VCO2/VO2.29 At 60 h, mice were fasted for 12 h and then provided with ad lib standard (re-feed) or high-fat diets (HFD) for consecutive 24 h periods. Each point is the mean of n = 11–13 mice/group ± 1 SE.
(B) Fasting glucose, lactate, and ketone levels.
(C) Hourly food and water intake (A).
(D) Glucose tolerance tests (GTTs) and serum insulin levels. Mice were fasted for 5 h and then administered a single i.p. bolus of glucose. n = 5.
(E) Oroboros respirometry results performed on mitochondria from the indicated WT and MycKO tissues. Pyruvate responses were determined following the addition of malate and ADP, whereas total complex I activity was determined following the subsequent addition to glutamate.12,13,23.
(F) Fifty-one serum acyl carnitine levels in 5 month-old WT and KO mice obtained after overnight fasting. n = 5 mice/group. Also see Figures S4 and S5.
(G) The same serum acyl carnitines were assessed in ~20-month-old WT and KO mice as described in (F). n = 5 mice/group. Boxes indicate significant intergroup differences. Also see Figures S4 and S5.
(H) Gene set enrichment analysis (GSEA) for liver transcripts involved in FAO from 20-month-old MycKO mice and additional negative enrichment in 5- and 20-month-old MycKO mice for genes comprising the BCAA catabolic pathway. Results were generated from RNA-seq data obtained from liver, adipose tissue, and skeletal muscle of each of the indicated cohorts, but were significant only in the liver as shown. (A and C) ANOVA,145 (B, D, E, F, and G) unpaired t test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars: SD.
WT and MycKO RERs converged during fasting, indicating that both groups responded similarly when demands for FAO were high. Re-feeding was again associated with low MycKO RERs, emphasizing the lifelong overreliance on FAO and/or suboptimal glucose utilization. MycKO mice also showed higher levels of serum ketones in the face of normal glucose and lactate levels (Figure 4B). Finally, younger MycKO mice episodically reduced water and food intake (Figure 4C and Data S1). While the latter might have forced a somewhat greater reliance on FAO in the youngest mice (Figure 4A), it seems unlikely that it fully explains the lower RERs, given that they persisted and were not associated with hypoglycemia (Figure 4B). Myc compromise thus promotes dysfunctional glycolysis and oxidative phosphorylation (Oxphos) and FAO dependency.6,51–53,69,71,72
Less efficient FAO accompanies aging and correlates with adiposity, NAFLD, ketosis, hyperglycemia, and insulin resistance.60,80 In contrast, glycolytic and Oxphos dysfunction following Myc inhibition is accompanied by increased FAO that maintains ATP levels.12,13,23,51,52,71,72,81 Lower RERs of young MycKO mice might reflect an integration of these opposing factors that favors FAO. The convergence of aging WT and MycKO RERs (Figure 4A) might also indicate a more rapid age-related decline in FAO in the latter group balanced by persistently high FAO needed to maintain dysfunctional mitochondria. Although baseline fasting glucose levels were similar in the two groups following a glucose challenge, younger MycKO mice displayed the exaggerated hyperglycemia and hyperinsulinemia that characterizes type 2 diabetes (Figures 4B and 4D). MycKO mice thus demonstrate defects in glucose metabolism consistent with their greater reliance on FAO. Eventual age-related metabolic compensation appears to be the result of other defects.
Liver, white adipose tissue, and skeletal muscle mitochondria from 5-month-old mice were assessed for complex I and II function.12–14 Complex I responses of MycKO liver and adipose tissue mitochondria were lower than those of WT mitochondria (Figure 4E). No differences were observed in succinate-driven complex II activities. In livers, where signals were sufficiently strong,82 MycKO palmitoyl CoA-driven oxygen consumption rates (OCRs) were again lower, thus suggesting a generalized complex I dysfunction.
Carnitine-long-chain fatty acid (LCFA) conjugation and the bidirectional transport of acyl carnitines across mitochondrial membranes are key steps in FAO.83 Complex I disorders are associated with elevated serum levels of 3-hydroxy-C14-carnitine (C14-OH), which reflects inefficient LCFA oxidation.83 Indeed, a mass spectrometry-based evaluation of 51 serum acylcarnitines in 5-month-old mice documented higher C14OH levels in the MycKO group (Figures 4F and S4A). This difference disappeared by 20 months of age and was replaced by 12 new changes, mostly involving the accumulation of even longer chain (C16 and C18) serum acylcarnitines, suggesting a progressive deterioration of the FAO pathway in aging MycKO mice akin to that of aging humans with type 2 diabetes (Figures 4G and S4B).84 The normalization of C14-OH in this older cohort likely reflected reduced C14 pools resulting from accumulated longer-chain precursors and their defective oxidation to shorter-chain acylcarnitines. Aging cohorts continued to show aberrant serum acylcarnitine profiles that were consistent with the observed NAFLD, low RERs, and insulin resistance of young MycKO mice (Figures 2, 4A, 4D, and S5). Twenty-month-old MycKO mice also accumulated C5-carnitine (Figure S4B), suggesting errors in mitochondrial branched-chain amino acid (BCAA) catabolism and implying a broadening of energy-generating defects in aging MycKO mice.85 Supporting this was a significant negative enrichment of FAO-related gene sets in 20-month-old MycKO livers and negative enrichment in 5- and 20-month-old MycKO mice for BCAA catabolic pathway-related gene sets (Figure 4H).
Blue native gel electrophoresis (BNGE) of ETC complexes and in situ enzymatic measurements showed no significant cohort- or age-related structural or functional differences (Figures 5A and 5B).55,72,86 However, tissue- and age-dependent differences between WT and MycKO mice were found among a subset of important and, in some cases, rate-limiting Myc-regulated glucose transporters and glycolytic enzymes (Figure 5C).14,71,78,87 The Glut1 glucose transporter, whose gene is a Myc target,78 and Glut487 were regulated oppositely in 5-month-old MycKO livers, whereas Glut2 did not change. Glut4 is expressed at low to undetectable levels in the liver, suggesting that it might be a negative Myc target.87 This also appeared true in MycKO skeletal muscle, where Glut4 is the major transporter.87 Also demonstrating differences in expression between WT and MycKO 5-month-old mice was the muscle-specific phosphofructokinase isoform PFK-M, which contrasted with no change in liver-specific PFK-L. Finally, pyruvate dehydrogenase (PDH) activity appeared to be increased in 5-month-old MycKO livers by virtue of its reduced level of phosphorylation (pPDH), whereas in skeletal muscle, this activity was decreased.79 Tissue-specific increases in PDH activity might represent responses to the impaired hepatic function of complex I that increases acetyl-coenzyme A availability and ETC activity (Figures 4E–4H).
Figure 5. ETC structure and function and glucose handling differ between WT and MycKO livers and skeletal muscle.

(A) BNGE profiles of liver and skeletal muscle ETC complexes I–IV, complex V, and supercomplexes (SCs) from mitochondria of 5- and 20-month-old mice.72 SCs comprise higher-order assemblies of complexes I, III, and IV.72
(B) In situ enzymatic activity of complexes I, III, IV, and V from (A).72
(C) Immunoblot analyses of proteins involved in glucose and pyruvate transport and metabolism from the tissues shown in (A) and (B).
(D) ROS production by WT and MycKO MEFs measured by the oxidation of CM-H2DCFDA.23 n = 6.
(E) Mitochondrial-specific ROS production measured by the superoxide-mediated oxidation of MitoSOX red. n = 6. Unpaired t test, ****p < 0.0001.
Twenty-month-old mice also showed tissue-, cohort-, and age-specific differences (Figure 5C). Lower Glut1 levels originally observed in MycKO livers and skeletal muscle persisted only in the latter tissue of older mice, whereas the initially higher levels of Glut4 expression in MycKO mice remained elevated in both sets of older tissues. Elevated PFK-M levels also persisted. Neither PDH nor pPDH levels changed in livers, whereas pPDH levels in MycKO skeletal muscle were reduced, as observed in 5-month-old livers.
Myc overexpression and underexpression both promote ROS.23,72,74 In the former case, hyperactive mitochondria with a normal ETC simply generate more ROS, whereas in the latter case, ETC dysfunction increases electron “leakage.”69,70,88 MycKO primary MEFs23 generated more ROS and superoxide, indicating that disproportionate amounts originated in mitochondria (Figures 5D and 5E). These findings and others (Figures 4A, 4E, 4F, 4G, S4, and S5) argue that younger MycKO mice acquire mitochondrial and ETC functional defects normally observed in aged WT mice.
RNA-seq differences between WT and MycKO tissues correlate with phenotypes
RNA sequencing (RNA-seq) was performed on liver, mesenteric white adipose tissue, and skeletal muscle from 5-month-old mice because they are Myc dependent for tissue homeostasis and/or because they undergo age-related changes (Figures 1A and 1B).31,32,36–38,89 We first verified the dysregulation of multiple Myc target genes in these MycKO tissues using gene set enrichment analysis (GSEA). The results were consistent with the previously documented inactivation of Myc (Figure S1E and Table S1), although enrichment patterns were tissue specific (Figures S6A–S6F).14
The paucity of gene expression differences between WT and Myc+/− tissues likely reflects low basal Myc levels in WT tissues and/or modest effects of Myc haploinsufficiency on high-affinity targets.11,19,31 To capture the greatest possible variation among WT and MycKO tissues, to avoid bias, and to identify functional categories, we performed GSEA. Seven particularly noteworthy categories were identified, with all having been previously identified in association with aging, senescence, or conditional Myc inactivation (Figures 6A and S7 and Data S1).23,56–58,90–97
Figure 6. Tissues from 5-month-old MycKO mice are enriched for aging- and senescence-associated transcripts.

(A) GSEA from tissues of 5-month-old WT and MycKO mice.146–148 clusterProfiler displays representative examples of the most recurrent and prominent of the gene sets within each category.149,150 Numbers to the right of each profile indicate its normalized enrichment score. Curves shown in gray and lacking enrichment scores indicate gene sets that were not significantly enriched. Values >0 along the abscissas indicate gene sets that were upregulated in MycKO tissues, whereas values <0 indicate gene sets that were downregulated. See Figure S7 for standard GSEA plots of these.
(B and C) GSEA and heatmap for transcripts that correlate with aging in most tissues and across species in livers and adipose tissue of 5-month-old WT and MycKO mice.
(D) Gene sets associated with types 1 and 2 diabetes selectively enriched in the indicated tissues of 5-month-old MycKO mice.
(E) Gene sets associated with cancer selectively enriched in the indicated tissues of 5-month-old MycKO mice.
(F) Examples of immunostaining for γ-H2AX in the indicated mice depicting double-stranded DNA breaks. Shown are merged micrographs: γ-H2AX immunostaining (red) and DAPI staining (blue).
The first category, “translation/ribosomal structure and function,” contained gene sets encoding ribosomal subunits and factors involved in translation and the synthesis/processing of rRNAs and tRNAs (Figures 6A and S7A). A second category, “mitochondrial structure and function,” encoded components of the mitochondrial membrane, the matrix, the ETC, and mitochondrial ribosomes (Figures 6A and S7B). We previously identified these categories in MEFs and hepatocytes lacking Myc and/or extended Myc network members.6,12–14,23
An “oxidative stress response” category (Figures 6A and S7C) encoded transcripts pertaining to the redox-responsive transcription factor NFE2L2/NRF2, the generation of superoxide, and the response to hydrogen peroxide previously documented in other MycKO cells and tissues.23,29,70,86
Also, strongly dysregulated in 5-month-old MycKO tissues were transcript categories associated with aging and senescence (Figures 6A, S7D, and S7E). A 79-member subset selected for its near-universal association with aging was also dysregulated in MycKO liver and adipose tissue in ways that, again, marked them as possessing an “older” transcriptional profile (Figures 6B and 6C and Data S1). Also identified were gene sets known to be enriched in tissues from individuals with types 1 and 2 diabetes and cancer (Figures 6D and 6E and Data S1). In MycKO tissues, the former sets were generally dysregulated in the directions seen in diabetic tissues, whereas in the latter, the directions of enrichment were opposite those seen in cancers and thus consistent with a low cancer risk (Figure 3B).
“DNA damage recognition and repair” comprised the sixth GSEA category in MycKO tissues (Figures 6A and S7F). This category pertains to recognition/repair of radiation-induced lesions, DNA breaks and other damage, and telomere/shelterin complexes and was dysregulated in MycKO MEFs, which show abnormal responses to DNA damage.23 Monogenic disorders involving these genes include Werner syndrome, Nijmegen break syndrome, and “telomeropathies,” such as dyskeratosis congenita and aplastic anemia, which are associated with premature aging and cancer.95,98–100 In most cases, genes in MycKO tissues were enriched in the same direction as occurs in these human conditions and in MycKO MEFs.23 MycKO livers also showed more double-stranded DNA breaks (Figure 6F). These findings indicate that Myc oversees interconnected pathways that participate in the recognition/repair of DNA damage and that are dysregulated in premature aging syndromes with high cancer susceptibility.
Enriched in MycKO livers were gene sets encoding spliceosome components, which orchestrate intron-exon junction recognition, lariat formation/removal, and exon-exon ligation (Figures 6A and S7G).101 However, we initially found no evidence increases in the frameshifts or indels that accumulate during aging and senescence as a result of aberrant splicing.90,91,102
Myc target gene dysregulation in MycKO mice occurs with normal aging
Knowing that ~10% of the transcript differences of Myc+/− mice originate from direct Myc targets,31 we compared RNA-seq profiles of similarly aged WT and MycKO cohorts. Focusing on previously enriched gene sets permitted two major observations. First, more differences existed between WT and MycKO liver and adipose tissues at 5 months than at ~20 months (Figure 7A and Data S1). This indicated that much of the dysregulation of young MycKO tissues eventually occurred in older WT tissues, thus equalizing the previous differences. Indeed, many gene sets that distinguished the livers and adipose tissues of younger and older WT mice were dysregulated following Myc loss. This Myc-dependent transcript fingerprint of young mice was consistent with their age-related features (Figures 1A, 2, and 4).32,36,76,94
Figure 7. Gene expression differences in young and old mouse tissues reflect declines in Myc and Myc target genes.

(A) Age- and Myc-dependent gene set enrichment differences among 5- and 20-month-old WT and MycKO tissues. n = 5. The total number of gene sets for which significant enrichment was observed is indicated beneath each category. Colored lines within each category represent a single gene set, the top 30 of which are shown for each category. Data S1 lists all relevant gene sets and others depicted here.
(B) Heatmap for the 79 transcripts shown in Figures 6B and 6C that correlate with aging in most tissues examined and across species.
(C) Heatmap of individual gene sets related to types 1 and 2 diabetes, including those depicted in Figure 6D from 5- and 20-month-old WT and MycKO mice.
(D) Heatmap for the expression of individual gene sets related to cancer, including those depicted in Figure 6E from 5- and 20-month-old WT and MycKO mice.
(E) Transcriptome-wide quantification of non-canonically spliced transcripts.149–152 Unpaired t test, *p < 0.05.
(F) Significant declines in Myc transcript levels in 12 of 90 single-cell populations derived from 23 individual young (1–3 months) and old (18–30 months) mousetissues.103 Results are expressed as q values based upon correlation coefficients that compared transcript levels across aging populations.
(G) Overrepresentation analysis of 58 Myc target gene sets analyzed using the above-cited single-cell RNA-seq data from young versus old mice.103 Gene sets for which significant dysregulation was observed in at least 40 of the 90 single-cell populations are shown, although 74 of the cell populations (82.2%) showed enriched representation of at least one gene set (Data S1). Down-reg. sets, downregulated in response to Myc overexpression; Up-reg. sets, upregulated in response to Myc overexpression; ND, sets comprising both positive and negative targets whose overall direction of response could not be determined. (H) Overlap between direct Myc target genes and those that undergo significant age-related changes in expression (q < 0.05).104,105 Gene expression differences were compared from 76 single-cell populations derived from 23 tissues from 1- to 3- and 18- to 30-month-old mice.103 (I) Myc transcript differences in young and old human tissues. Results are from the Broad Institute’s GTEx database.
(J) Enrichment of Myc target gene sets (see Figures S6A–S6C) in aging and senescent human tissues and cell lines. (E and I) Unpaired t test, *p < 0.05, **p < 0.01, ****p < 0.0001.
Muscle of young WT and MycKO mice again showed much of the same Myc-dependent gene set enrichment previously seen in livers and adipose tissue (Figure 7A). However, many of these, notably in the “aging” category, were enriched in directions opposite those seen in livers and adipose tissue. Also seen was a more pronounced gene set enrichment in WT and MycKO muscle at 20 months of age than in liver and adipose tissue and particularly for gene sets from the “translation/ribosomal structure and function” and “mitochondrial structure and function” categories. This suggested that GSEA differences in muscle were less equalized during aging. Indeed, the greater directional change in the enrichment of some gene sets in muscle, from down in young MycKO mice to up in old mice, appeared to result from aging-related declines in WT mice (Figure 7A).
The 79-member aging-associated transcript set (Figures 6B and 6C and Data S1) was re-examined in older livers and adipose tissues, where differences between WT and MycKO were again noted, although they were less pronounced than those of 5-month-old tissues (Figure 7B and Data S1). This again suggested that aging-related gene signatures associated with younger MycKO tissues appear in WT tissues by 20 months of age.
More comprehensive assessments of types 1 and 2 diabetes-associated gene sets than used previously (Figure 6D) were performed on young and old tissues. Tissue-specific dysregulation of these was again observed among tissues from young mice (Figure 7C and Data S1). This persisted in older mice, although the numbers and identities of the gene sets and the enrichment levels changed in tissue-specific ways (Figure 7C). Thus, the dysregulation of these gene sets was already quite extensive in young MycKO mice and remained so throughout life. Similar analyses of a larger number of gene sets associated with and/or deregulated in cancer also showed enrichment in MycKO tissues (Figures 6E and 7D and Data S1).
As already noted, the “RNA splicing” gene set category was enriched in young MycKO livers but was unassociated with any changes in non-canonical mRNA splicing. Although only 8 such gene sets remained enriched at 20 months of age (Table S2), significant increases in non-canonically spliced transcripts were now observed (Figure 7E). Thus, aberrantly spliced transcripts accumulated only in older livers and thus likely required additional age-dependent and Myc-independent functions as well as tissue context.
A previous comparative study of young and old mice103 showed significant age-related declines in Myc transcripts in 12 of 90 (13.3%) single-cell populations from 23 tissues (Figure 7F and Data S1). Thirty-five of 58 Myc target gene sets (60.3%) from the MSigDB database were also dysregulated in one or more single-cell populations of most of these tissues (Figure 7G and Data S1). Where the directionality of dysregulation could be determined, it usually correlated with the age-related declines in Myc levels. These results thus documented extensive age-related alterations of direct Myc target gene transcript collections that would not have been anticipated based solely on Myc expression changes.
The above single-cell RNA-seq data were used to search the ENCODE and ChEA databases.104,105 We found that 89.5% of genes whose expression changed significantly during normal aging were direct Myc targets, and 67.2% of chromatin immunoprecipitation sequencing (ChIP-seq)-confirmed direct Myc target genes from ENCODE and ChEA significantly altered their expression during aging (Figure 7H).
Myc expression also declines during the propagation of primary human fibroblasts, and the accompanying senescence can be prolonged or hastened by enforcing or inhibiting Myc, respectively.23,106 Upon querying the GTEx database, which contains RNA-seq results from numerous normal human tissues, we found age-related declines in Myc expression to be common, particularly in adipose tissue, sigmoid colon, and leukocytes (Figure 7I). Myc transcripts were also lower in older individuals’ fibroblasts, as noted previously in murine fibroblasts.106,107 Consistent with Myc’s role in maintaining the replication of most cell types, a previous study of >650 primary human fibroblast lines showed that those from older individuals become senescent sooner than those from younger individuals.22,23,27,51,108 Interrogating the above samples with the collection of direct Myc target gene sets from the MSigDB database (Figures S6A–S6C) confirmed that positively regulated Myc target gene sets were negatively enriched in older tissues, and negatively regulated gene sets were positively enriched (Figure 7J). Thus, in both mice and humans, normal aging and senescence are commonly associated with Myc downregulation and appropriate changes in its target genes. The deliberate Myc inactivation and the ensuing dysregulation of its target genes in young MycKO mice thus accelerated the changes that otherwise occur with normal aging.
DISCUSSION
By postponing Myc inactivation until weaning, we have avoided the factors that contribute most strongly to prenatal demise while allowing ourselves to assess the consequences of its loss on multiple, interdependent whole-body phenotypes.24,26,27 While differing from the method previously used to generate Myc+/− mice,31 our approach permitted a comparison of the 2 mouse strains over their lifetimes.
MycKO mice presented 2 disadvantages. First, Myc’s contributions to the substantial growth and development of the immediate postnatal period could not be determined (Figures S3A–S3C) (data not shown).21 Second, the prominence of MycKO phenotypes may be skewed in favor of tissues with the highest and most persistent Myc loss. The partial re-appearance of intact Myc alleles suggested that stem cell populations with incomplete Myc excision and proliferative advantages are responsible (Figure S1F and Table S1). Similarly, infrequent MycKO mouse tumors appeared to originate from a minority population of cells that retained Myc (Figures 3I and 3J). Myc’s excisional variability might reflect the degree to which tamoxifen penetrates different tissues, the efficiency of its activation, and differential accessibility of the Myc locus to CreER.109 Nonetheless, our approach provided a means to assess the life-long consequences of global Myc loss on health and fitness. Our studies also demonstrate that some pathologies and phenotypes observed with tissue-specific and/or prenatal Myc inactivation are replicated when inactivation is delayed.14,27,29,42 Certain phenotypes of Myc+/− mice that we did not observe, such as an overall reduced body size, are likely determined during embryogenesis.27,31
Myc inactivation in adults and juveniles causes bone marrow hypoplasia, peripheral cytopenias, colonic epithelial flattening, and villous atrophy (Figure S3).21,24–28,110 While the other findings resolved, bone marrow continued to resemble that of aged mice (Figure S3C). The normal weights of young MycKO mice and the absence of steatorrhea provided evidence that any malabsorption did not impair growth (Figure 1A and not shown). These observations point to Myc’s variable importance at different developmental stages and that deleterious consequences of its loss are often mitigated when inactivation is delayed and/or incomplete.6
Relatively young MycKO mice often displayed progressive age-related phenotypes. Appearing at different times and sometimes influenced by gender, they included increased fat:lean mass ratios, alopecia and achromotrichia, and reduced strength, endurance, and balance (Figure 1). Notable additional findings include NAFLD, glucose intolerance, and mitochondrial dysfunction, with a preferential reliance on FAO (Figures 1, 2, and 5D).46,56,61,62,64 Mitochondrial abnormalities and steatosis occur in mice with hepatocyte-specific Myc loss of relatively short duration, with the current findings confirming and extending these earlier ones by showing that the maximal hepatic lipid content accumulates earlier in MycKO mice (Figure 2).13,14,29 Age-related changes in skin (Figure S2) also recapitulate some of the milder consequences of melanocyte-specific Myc KO.42 Many of the abnormal phenotypes of MycKO mice are thus attributable to interactions between Myc inactivation and normal aging phenotypes (Figure S8).
While the extended lifespan of Myc+/− mice was originally ascribed to a lower cancer incidence, their relative youthfulness may also have contributed.31 Aging and cancer therefore remained temporally linked. Neoplasms are common in normal aging mice, and age is the strongest independent predictor of cancer development.32,34,35 This association is exaggerated in human and murine disorders of premature aging despite the chronological youthfulness of affected individuals.93,96,99,111,112 Highlighting this relationship is the critical contribution of Myc to cancer pathogenesis.2,6,13–15 The lower lifetime cancer incidence of MycKO mice (Figure 3B) indicates that its strict association with aging is maintained by a single gene, namely Myc. The reduced cancer incidence and increased longevity of MycKO mice are even more remarkable given that several of their associated co-morbidities are independent risk factors for cancer development and shortened lifespan.93,99,113–116
Given Myc’s link to cancer, the dissociation of aging and neoplasia in MycKO mice raised the question of how occasional tumors do arise (Figure 3B).11,13,14,54 The variable levels of their Myc expression suggested that at least some originated from a minority population of cells with intact Myc alleles (Figures 3I and 3J). Whether tumorigenesis in MycKO mice is reduced due to a lower initiation rate or a slower growth rate (Figure 3B) requires further investigation, since roles for Myc in both steps have been demonstrated.13,14,19,23,54 Aside from Myc’s loss, indirect mechanisms may contribute to the low cancer incidence in MycKO mice. These potentially include the suppression of Myc target genes by Max-Mxd family heterodimers or by cross-binding members of the Mlx Network.6,14,117 MycKO cells could have higher baseline rates of neoantigen generation resulting from DNA damage response/repair and mRNA splicing defects that could enhance immune surveillance (Figures 6A, 7A, 7E, S7F, and S7G).23,118–120 Slower MycKO tumor growth might allow for longer periods of neoantigen presentation and immune response maturation.12–14 However, the degree to which anti-tumor immunity is actually enhanced might be limited given the Myc dependence of T cell expansion.121
The relationships among Myc, aging, and cancer likely cannot be explained by any single mechanism, since many of the gene sets under Myc’s control functionally converge upon the “hallmarks” of both aging and cancer (Figures 6A, 7, and S8).37,61,92,94,97,122–125 Both normal aging and Myc loss generate ROS production due to progressive ETC decline and/or increased reliance on FAO (Figures 4A and S8).63,88,126 Excessive ROS and impaired ribosomal biogenesis/translation both accelerate aging.97 Nuclear and mtDNA damage, aberrant splicing, and senescence also increase in the face of aging and Myc loss.90,91,102,127–130 Genotoxic ROS also inhibit translation, thus highlighting how individual Myc- and/or age-linked functions crosstalk and influence one another (Figure S8).131
Do MycKO mice better mimic normal aging than other models, which are largely based upon rare monogenic disorders of DNA damage recognition/repair?132,133 Importantly, the aging of MycKO mice directly reproduced the dysregulation of Myc target genes that normally accompanies aging in mice and humans and that correlates with declines in Myc itself (Figures 6 and 7). These findings indicate that Myc inactivation in juvenile mice prematurely re-creates the dysregulation of its downstream target genes and the aging-like deterioration of their collective functions with similar molecular and phenotypic outcomes (Figures 7F–7J and S8).
The aging-related enrichment of Myc target genes involved more tissues than did the declines in Myc. There are at least 3 explanations for this finding. First, in some tissues, Myc paralogs might play a larger role in regulating these gene sets. Although none of the above cells or tissues expressed significant levels of Mycn, some expressed Mycl, which sometimes declined during aging when Myc itself did not. Myc target gene sets may therefore be preferentially responsive to Mycl in certain tissues. Second, some Myc targets may be selectively sensitive to one or more Mxd proteins.6,11 Finally, Mlx Network members might displace Myc-Max complexes in some tissues and modify Myc’s transcriptional impact.6,11
“Heterozygous advantage” applies to genes such as those encoding α and β globins and the cystic fibrosis transmembrane conductance regulator, where single mutant alleles protect against malaria and diarrheal diseases, respectively, whereas mutational homozygosity can be lethal.134–137 Myc hemizygosity’s association with a spectrum of health benefits versus the pathologies of MycKO mice is consistent with Myc being a somewhat different example of heterozygous advantage, despite its association only in the experimental context described here.11,25,27,31 Nonetheless, single-nucleotide polymorphisms far upstream of the Myc coding region can significantly affect its expression and correlate with cancer susceptibilities.138,139 Normal declines in Myc might thus have very different lifetime consequences depending upon its genetically predetermined initial levels. The heterozygous advantage of Myc might thus relate more to the genetic constraints upon its normal expression, which would become increasingly consequential as its levels decline with age and reach pathologic thresholds at different times.
Although inhibiting Myc to treat cancer has proved elusive, the finding that Myc+/− mice displayed increased longevity, a lower cancer incidence, and additional health dividends provides additional incentive to pursue this objective.31,140–142 However, the current work suggests that caution is warranted in the use of Myc inhibitors, particularly to extend longevity.143 Our work thus raises questions that will need to be confronted before such inhibitors can be employed clinically, particularly in children, where even short-term treatment with traditional chemotherapeutics can accelerate aging.144 Among these is the degree to which Myc inhibition unintentionally accelerates aging, whether certain age-associated phenotypes will be differentially manifested, and whether some phenotypes can be “rejuvenated” when Myc expression is restored. Another question is whether MycKO phenotypes will appear when Myc inactivation is implemented later in life. Finally, might young age be a contraindication when cancer therapy demands that Myc inhibition be both efficient and prolonged? More refined evaluation in appropriate experimental and clinical settings will likely be necessary before answers to such questions are forthcoming.
Limitations of the study
Among this study’s unanswered questions are how the pathways that are affected by Myc’s loss cooperate to promote premature aging and the nature of their tissue dependencies (Figure S8). Myc-dependent alterations in mitochondrial and ribosomal structure and function, energy metabolism, and genome integrity drive both normal and premature aging. In our model, Myc inactivation is also not 100% efficient, and whether more complete Myc elimination remains compatible with extended longevity and allows better cataloging of all potential phenotypes remains unknown. Residual Myc expression may mask additional phenotypes, as seen with Myc+/− mice. In addition, those described here may be incomplete and/or milder than what is potentially achievable. Some tissues that were not carefully examined may also possess overlooked abnormalities. Finally, it remains unclear how delaying Myc inactivation until later in life affects the age-related findings we have reported.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
All additional information and requests for resources, reagents, and methods should be directed to the lead contact, Edward V. Prochownik (procev@chp.edu).
Materials availability
All unique reagents generated in this study will be made available from the lead contact (E.V.P.) and may require a completed materials transfer agreement.
Data and code availability
All raw RNA-seq files have been deposited in the NCBI Gene Expression Omnibus154 and are accessible through GEO Series accession number GSE223676 database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE223676 ). Data underlying the display items in the manuscript, related to Figures 1, 2, 3, 4, 5, 6, 7, and S1–S7 are available as Data S1 – Source data. The original full-length western blots for Figures 3I, 5C, and S1G have been deposited in Mendeley Data (https://doi.org/10.17632/4t9xbmxszn.1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animal models
Animal work was conducted in compliance with the Public Health Service Policy on Humane Care and Use of Laboratory Animal Research (ILAR) Guide for Care and Use of Laboratory Animals. All experimental procedures, diets and tests were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Pittsburgh. All mice were housed in a specific pathogenfree facility, maintained under standard conditions at UPMC Children’s Hospital of Pittsburgh. The B6.129S6-Myctm2Fwa/Mmjax mouse strain, in which the second and third exons of the Myc gene are flanked by loxP sites, was originally obtained as a gift from I. Moreno de Alboran.13,14,29 These were crossed with the B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J strain, which expresses a Cre recombinase-estrogen receptor (CreER) fusion transgene under the control of the ROSA26 promoter.39. 2 MycLoxP/LoxP progeny strains were derived, containing one or 2 CreER transgene copies, which allowed for a determination of the efficiency of Myc excision in response to CreER dose. CreER activation and Myc excision were initiated at the time of weaning in mice that had attained a weight of 15 g or greater. Each mouse received 5 daily i.p. injections of freshly-prepared tamoxifen (75 mg/Kg) in corn oil. To ensure the complete metabolism and excretion of tamoxifen and to avoid any of its non-specific side effects, we allowed at least 8 wks before initiating any testing other than that specifically designed to confirm the extent of Myc exon 2 excision and full-length Myc transcript expression (Figure S1). As a further control for any long-term effects of tamoxifen treatment, control (WT) mice for all studies consisted of the offspring of matings between B6.129S6-Myctm2Fwa/Mmjax and wild-type C57BL/6 mice treated with tamoxifen in the manner described above. Equal numbers of males and female were used for all studies that were conducted during the entire lifetimes (Figure 3).
Myc excisional efficiency was determined using a quantitative TaqMan-based qPCR assay that compared the exon 2 : exon 1 ratio using tissues from the above mice and standard curves generated with known ratios of WT and MycKO DNAs as described previously (Figures S1A–S1E).13,14,29 Cre-ER transgene copy number was determined by a separate TaqMan-based assay using the primers listed in Figure S1D. 10 ng of total DNA was used in each TaqMan assay. 3 primer sets were designed to amplify regions to identify specifically unfloxed, floxed (WT) and MycKO alleles. All primers and probes (Figure S1D) were synthesized by IDT, Inc. (Coralville, IA). PCR reactions were performed on CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad, Inc.) using the following conditions: 95 °C for 5 min; 10 cycles at 95 °C for 20 s, and 65 °C~60 °C (decreasing by 0.5 °C per cycle) for 15 s, and 68°C for 10 s; 40 cycles at 95 °C for 15 s, and 60 °C for 1 min.
METHOD DETAILS
Derivation and propagation of primary murine embryo fibroblasts (MEFs)
Briefly, 10–12 e14 embryos from pregnant WT mothers were decapitated, eviscerated, rinsed in PBS, placed into sterile 0.25% trypsin-EDTA and incubated 1 hr at 37C as described previously.23,155 They were then finely minced and digested for an additional 1–2 h at 37C before transferring to fresh Dulbecco’s modified minimum essential medium (DMEM) containing 10% FBS, 100 mM glutamine and penicillin/streptomycin as previously described.69 After expanding for 3–4 days, these early passage cells were trypsinized and frozen at −80C to serve as subsequent stocks. These primary MEFs were designated as passage 1. To excise the floxed Myc alleles from the above cells, in vitro culturing was continued in fresh medium containing 500 nM 4-hydroxytamoxifen (4-OHT) (Sigma-Aldrich, St. Louis, MO), which was changed daily. On day 8 an aliquot of cells was harvested, DNA was isolated as described below and the ratio of WT and MycKO Myc alleles was calculated using the same approach as described above for individual mouse tissues. Under these conditions, Myc allele excision routinely exceeded 95%.23
Strength and endurance testing
Strength testing was performed using a Grip Strength Meter (Harvard Apparatus, Holliston, MA) according to the direction of the supplier. Rotarod testing (SPW Industrial, Laguna Hills, CA) was based on a modification of the standard operating procedure from Jackson Laboratories: https://www.jax.org/-/media/jaxweb/files/research-and-faculty/tools-and-resources/peripheral-neuropathy-resource/rotarod.pdf?la=en&hash=78228ECB294E38BC773843500CDE2E8C99A96316. Briefly, animals were initially placed on the slowly rotating rod (5 rpm) and maintained at this speed for 20 sec. The speed was then increased by 5 rpm increments each lasting 20 sec. The recorded numbers indicate the total time that each mouse was able to maintain its balance.
Treadmill performance
Treadmill performance was monitored with a Columbus Instruments Exer 3/6 apparatus (Columbus, OH). Groups of 6 mice at a time (3 WT and 3 MycKO) were evaluated according to a published protocol.156 Briefly, mice were allowed to run along a treadmill (elevated 10° from the horizontal) at a gradually increasing pace until reaching exhaustion, which was defined as the time at which they preferred to rest for >5 sec. upon an immobile metal shock plate at the bottom of the treadmill. The total distance run until reaching the point of exhaustion was recorded for each animal.
Metabolic cage profiling
These were performed essentially as described previously.29 Briefly, control and MycKO mice of the indicated ages were housed individually in metabolic cages (Columbus Instruments) and allowed to acclimate for 24 hr while being provided ad lib access to water and a standard mouse chow containing 5% fat (Picolab 5053; LabDiet, St. Louis, MO, USA). VO2 and VCO2 were recorded every 20 min over the subsequent 48 hr along with food intake and overall activity. At the conclusion of this observation period, mice were starved overnight (12 hr) and then provided with a standard diet for 24 hr followed by a high-fat diet (45%) for an additional 24 hr while again monitoring RERs. Data analyses were performed with a web-based analysis software package CalR (https://calrapp.org/cite.html).
Glucose tolerance tests and serum glucose, lactate, and ketone measurements
Mice were fasted for 5 hr. at which time whole blood glucose, lactate and ketone levels were obtained using meters and compatible strips according to the directions provided by the suppliers (Glucose AimStrip Plus, Germaine Laboratories, Inc. San Antonio, TX; Lactate Plus Analyzer, Sports Resource Group, Inc., Hawthorne NY; Keto-Mojo Ketone Meter, Keto-Check, Inc. Napa, CA). To perform glucose tolerance tests and to measure insulin levels, the above mice were injected with 2g of dextrose/kg body mass with blood glucose levels being subsequently measured at the indicated times. Serum insulin levels were measured using an Ultra Sensitive Mouse Insulin ELISA Kit according to the directions provided by the supplier (Crystal Chem, Elk Grove Village, IL).
ImageJ quantification of ORO staining
ORO- and hematoxylin-stained tissue sections were imaged on a Leica DFC7000T microscope with 5x and 40x magnification. Multiple overlapping images of each section were acquired for the full area. The images of each section were joined using the stitching plugin of the open source software FIJI.157–159 After subtracting background from each image, color de-convolution160 was performed in FIJI where the colors were specified in advance from ROIs respectively corresponding to unstained tissue, strongly stained tissue and the slide background. Quantification of Oil-Red-O positive staining was performed as described in ImageJ documentation (https://imagej.nih.gov/ij/docs/examples/stained-sections/index.html). Higher resolution images were acquired at 5x magnification(Figure 2C).
Nucleic acid isolation
DNAs and RNAs were isolated from mouse tissues using DNeasy and RNeasy kits, respectively according to the directions of the supplier (Qiagen, Inc. Germantown, MD). Exceptions to this were made in the case of adipose tissue and skeletal muscle for which we utilized a RNeasy Lipid Tissue extraction Kit and QIAzol Lysis Reagent (Qiagen, Inc., Germantown, MD), respectively. Total RNAs were reverse transcribed using a SuperScript IV First-Strand Synthesis System according to the directions of the supplier (Thermo Fisher Scientific, Pittsburgh, PA). To determine the degree of Myc transcript reduction in control and MycKO tissues, 2 separate TaqMan-based qRT-PCR assays were performed that compared the exon 2: exon 1 ratio signals in each WT and MycKO tissue (Figures S1D and S1E).
Blue native gel electrophoresis (BNGE), in situ enzymatic assays for ETC enzymatic function
Non-denaturing gel electrophoresis was performed largely as described previously.55,72 Briefly, purified mitochondria (approx. 1 mg of total protein), were lysed by the addition of digitonin and then incubated on ice for 20 min. Coomassie blue solution (5% Coomassie blue G250 in 750 mM 6-aminocaproic acid) was added and the suspension was then centrifuged at 14,000 × g for 20 min at 4°C. The supernatant was diluted in the supplier’s buffer, loaded onto a 3–12% Native PAGE Novex Bis-Tris gel (Life Technologies, Carlsbad, CA) and electrophoresed for 4 hr at 4C at 80 V. Gels were then stained with Bio-Safe Coomassie G250 (Bio-Rad, Hercules, CA) for 30 min and de-stained exhaustively in deionized water. Stained gels were scanned and the imaged using an AlphaEaseFC 2200 scanner and AlphaEaseFC software. Enzymatic assays for mitochondrial complexes and super-complexes were performed as previously described for Complex I (NADH ubiquinone oxidoreductase), Complex III (CIII) (decylubiquinol cytochrome c oxidoreductase), Complex IV (CIV) (cytochrome c oxidase) and Complex V (ATPase).72 Band intensities were measured and quantified using Image J software and normalized with their corresponding bands on the Coomassie stained blue native gel.
ROS assessment
CM-H2DCFDA and MitoSOX™ Red dyes were utilized to measure reactive oxygen species (ROS) levels (Molecular Probes, Eugene, OR, USA) according to the manufacturer’s protocol. This was achieved by exposing monolayer cultures of mouse embryonic fibroblasts (MEFs) maintained at a temperature of 37 °C. Quantifications were performed on 6 biological replicates comprising 20,000 cells/sample using a BD LSRII flow cytometer (Becton-Dickinson Biosciences, San Jose, CA, USA) and results were analyzed using FlowJo v10 software. This was done as described in Wang et al.23
β-galactosidase staining
Tissue sections were stained for β-galactosidase using a Senescence Detection Kit (ab65351) according to the directions of the supplier (Abcam, Inc., Waltham, MA).
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting
At the time of sacrifice, individual tissues were removed, and immediately placed on ice. They were then divided into small sections, snap-frozen in liquid nitrogen and maintained at −80C for long-term storage. To prepare samples for SDS page, tissue fragments were disrupted in PAGE buffer using a Bullet Blender as previously described.55,161 Protein concentration was quantified using the Bradford reagent (Bio-Rad, Inc., Hercules, CA). Electrophoresis, semi-dry blotting and protein detection was performed as previously described.55 Antibodies used for the detection of specific proteins were used largely according to the directions of the suppliers and are shown in Table S3.
Immunohistochemistry and immunohistofluorescence staining
All tissues were fixed in 10% formalin, paraffin embedded and cut into 4 μm thick sections for standard hematoxylin/eosin staining or immunostaining procedures as previously described (Wang 2022). Prior to staining for Myc, heat-induced antigen retrieval was performed using a citrate buffer (pH 6.0) for 30 minutes. Sections were incubated with a rabbit anti-Myc antibody (1:250; N262, SantaCruz) at 4C for 72 hours. A biotinylated secondary antibody was used to amplify the signal using an avidin–biotin substrate (Vector Laboratories, Inc., Newark, CA). Immunohistofluorescence staining for γH2AX was done as described in Wang et al23.
Transcriptional profiling
RNAs were purified from omental adipose tissue, liver and skeletal muscle as described above followed by DNAase digestion.13,14,23 RIN values were determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Foster City, CA) and only those with values of >8.5 were processed further. Sequencing libraries were generated with a NEBNext Ultra Directional RNA Library Prep kit according to the supplier’s directions (New England Biolabs, Beverly, MA). Sequencing was performed as previously described on a NovaSeq 600 instrument (Illumina, Inc., San Diego, CA) by Novagene, Inc. (Sacramento, CA).13,14,23 Original data were deposited in the NCBI Gene Expression database and are available through the Gene Expression Omnibus (GEO)154 under accession number GSE223676.
To identify differentially expressed transcripts, we utilized CLC Genomic Workbench version 21(Qiagen) and mapped raw reads to the GRCm38.p6 mouse reference genome. Functionally related and differentially expressed groups were identified using clusterProfiler (R package version 4.2)149,150 by first screening the MSigDB data bases (http://www.gseamsigdb.org/gsea/msigdb/as described previously.13,14 We also screened the Enrichr collection to identify additional groups of gene sets that were either absent from or underrepresented in MSigDB (http://amp.pharm.mssm.edu/Enrichr).146–148 Representative gene sets along with their normalized enrichment score (NES) and q values were displayed graphically using the Ridgeline plot application from Clusterprofiler (https://rdrr.io/bioc/enrichplot/man/ridgeplot.html).
To identify non-canonically spliced transcripts, we utilized the nf-core/rnaseq-3.4 analysis pipeline with the percentage of non-canonical splices being calculated from multi-qc of STAR section pct_noncanonical_splices = num_noncanonical_splices/total_ reads*100.151,152 Tabula Muris Consortium mosue single cell RNAseq data to evaluate the expression of Myc and Myc targets expression were obtained from https://figshare.com/ndownloader/files/27856758 and analyzed as described.103,162 Myc transcript levels in tissues obtained from young and old human tissues were downloaded from the GTEx Portal (GTEx Analysis V8 release: RNAseq gene TPMs by tissue) (https://gtexportal.org/home/datasets dbGaP: phs000424.v8.p2).163
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and statistical analysis were performed using R software v4.2.0164 (R Foundation for Statistical Computing, Vienna, Austria) and GraphPad Prism v9.00 (GraphPad Software Inc., USA). The ComplexHeatmap and ggplot2 packages were utilized for boxplot and heatmap visualizations, while the survminer package was used for survival curve plotting. The number of samples per group (n) for each experiment is indicated either in the figure legend or within the figure itself. A two-tailed, unpaired t-test was employed to assess significant differences between normally distributed populations, while a two-tailed Mann-Whitney exact test was used for non-normally distributed populations. A p-value below 0.05 was considered statistically significant. Significance is denoted as follows: * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001, and “ns” indicates not significant. Detailed statistical analysis information can also be found in each figure legend.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Glut 1 | Abcam | Cat#Ab115730; RRID:AB_10903230 |
| Glut 2 | Proteintech | Cat#20436–1-AP; RRID:AB_2750600 |
| Glut 4 | Cell signaling | Cat#2213; RRID:AB_823508 |
| GAPDH | Sigma | Cat#G8795; RRID:AB_1078991 |
| γH2A.X | Abcam | Cat#ab81299; RRID:AB_1640564 |
| PDH | Cell signaling | Cat#3205; RRID:AB_2162926 |
| c-Myc | Cell signaling | Cat#13987; RRID:AB_2631168 |
| c-Myc | Santa Cruz | Cat# sc-764; RRID:AB_631276 |
| p-PDH | Millipore | Cat# AP1062; RRID:AB_10616069 |
| PFK-L | Avivva sys bio | Cat# ARP45774_T100; RRID:AB_1294624 |
| PFK-M | R and D Systems | Cat# MAB7687; RRID:AB_2861389 |
| Rabbit IgG | Cell signaling | Cat# 7074; RRID:AB_2099233 |
| Mouse IgG | Cell signaling | Cat# 7076; RRID:AB_330924 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| ABC-HRP Kit | Vector Laboratories | Cat#PK-6101 |
| 0.25% trypsin-EDTA | Corning | Cat#25–053-CI |
| DMEM | Cytiva | Cat#SH30022.01 |
| Fetal Bovine Serum (FBS) | Biowest | Cat# S1620 |
| 4-hydroxytamoxifen (4-OHT) | Sigma-Aldrich | Cat#H7904 |
| Tamoxifen | Sigma-Aldrich | Cat#T10540–29-1 |
| D-(+)-Glucose solution | Sigma-Aldrich | Cat#G8769 |
| MitoSOX™ Red | Thermo Fisher Scientific | Cat#M36008 |
| CM-H2DCFDA | Thermo Fisher Scientific | Cat#D399 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Ultra Sensitive Mouse Insulin ELISA Kit | Crystal Chem | Cat#90080 |
| Senescence Detection Kit | Abcam | Cat#ab65351 |
|
| ||
| Deposited data | ||
|
| ||
| Raw data of RNAseq | This paper | GEO: GSE223676 |
| uncropped western blots deposited at Mendeley Data | This paper | https://doi.org/10.17632/4t9xbmxszn.1 |
| Tabula Muris Consortium mosue single cell RNAseq data | Tabula Muris Consortium | https://figshare.com/ndownloader/files/27856758 |
| RNAseq gene TPMs from young and old human tissues (GTEx Analysis V8 release: RNAseq gene TPMs by tissue) |
GTEx |
https://gtexportal.org/home/datasets (dbGaP: phs000424.v8.p2) |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| MEFs | In this paper | Wang et al23 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: B6.129S6-Myctm2Fwa/Mmjax | JACKSON LABORATORY | Strain # 032046-JAX; RRID:MMRRC_032046-JAX |
| Mouse: B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J | JACKSON LABORATORY | Strain # 008463; RRID:IMSR_JAX:008463 |
| Mouse: C57BL/6 | JACKSON LABORATORY | Strain # 000664; RRID:IMSR_JAX:000664 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers and probes for qRT-PCR | In this paper | Figure S1D |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ- Fiji | NIH | https://fiji.sc/#; RRID:SCR_002285 |
| FlowJo v10 | Becton-Dickinson Biosciences |
https://www.flowjo.com/solutions/flowjo/;RRID:SCR_008520 |
| R studio | Posit | https://posit.co/download/rstudio-desktop/; RRID:SCR_000432 |
| GraphPad Prism v9.0 | GraphPad Software Inc. | https://www.graphpad.com;RRID:SCR_002798 |
| Clusterprofiler | Wu et al149 |
https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html; RRID:SCR_016884 |
| ComplexHeatmap | Gu et al153 |
https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html; RRID:SCR_017270 |
| ggplot2 | CRAN |
https://ggplot2.tidyverse.org/index.html; RRID:SCR_014601 |
| survminer | CRAN |
https://cran.r-project.org/web/packages/survminer/index.html; RRID:SCR_021094 |
| CLC Genomic Workbench version 21 | QIAGEN | https://digitalinsights.qiagen.com/;RRID:SCR_011853 |
| nf-core/rnaseq-3.4 | Ewels, et al152 | https://github.com/nf-core/rnaseq |
|
| ||
| Other | ||
|
| ||
| DNeasy Blood & Tissue Kit | QIAGEN | Cat#69504 |
| RNeasy kit | QIAGEN | Cat#74004 |
| QIAzol Lysis Reagent | QIAGEN | Cat#79306 |
| Glucose AimStrip Plus | Germaine Laboratories | Cat#37321 |
| Lactate Plus Analyzer | Sports Resource Group | Model#SN0000101115 |
| Keto-Mojo Ketone Meter | Keto-Check | Model#TD-4279 |
| RNeasy Lipid Tissue Mini Kit | QIAGEN | Cat#74804 |
| SuperScript™ IV First-Strand Synthesis System | Thermo Fisher Scientific | Cat#18091050 |
| 3–12% Native PAGE Novex Bis-Tris gel | Thermo Fisher Scientific | Cat#BN1003BOX |
Highlights.
Postnatal body-wide deletion of the Myc gene in mice causes premature aging
“MycKO” mice dysregulate numerous genes involved in aging, senescence, and cancer
MycKO mice live longer and have a low lifetime cancer incidence
Normal aging in mice and humans is associated with Myc downregulation
ACKNOWLEDGMENTS
This work was supported by NIH grant RO1 CA174713, a Hyundai Hope on Wheels Scholar grant, Rally Foundation Independent Investigator grant 22IN42, and The UPMC Children’s Hospital of Pittsburgh Foundation (all to E.V.P). J.E.V. was supported by NIH grant DK RO1 109907. M.S.T. was supported by NIH grant P50 CA 210964. Analysis of RNA-seq data was partly supported by The University of Pittsburgh Center for Research Computing. Acylcarnitine profiling was performed in collaboration with the Rangos Research Center Metabolic Core LC/MS/MS services at the Department of Pediatrics, University of Pittsburgh Medical Center.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112830.
REFERENCES
- 1.Kalkat M, De Melo J, Hickman KA, Lourenco C, Redel C, Resetca D, Tamachi A, Tu WB, and Penn LZ (2017). MYC deregulation in primary human cancers. Genes 8, 151. 10.3390/genes8060151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nesbit CE, Tersak JM, and Prochownik EV (1999). MYC oncogenes and human neoplastic disease. Oncogene 18, 3004–3016. 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 3.Ge Z, Leighton JS, Wang Y, Peng X, Chen Z, Chen H, Sun Y, Yao F, Li J, Zhang H, et al. (2018). Integrated genomic analysis of the ubiquitin pathway across cancer types. Cell Rep. 23, 213–226.e3. 10.1016/j.celrep.2018.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, and Li F. (2006). The c-Myc target gene network. Semin. Cancer Biol 16, 253–264. 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 5.Kalkat M, Resetca D, Lourenco C, Chan PK, Wei Y, Shiah YJ,Vitkin N, Tong Y, Sunnerhagen M, Done SJ, et al. (2018). MYC protein interactome profiling reveals functionally distinct regions that cooperate to drive tumorigenesis. Mol. Cell 72, 836–848.e7. 10.1016/j.molcel.2018.09.031. [DOI] [PubMed] [Google Scholar]
- 6.Prochownik EV (2022). Regulation of normal and neoplastic proliferation and metabolism by the extended myc network. Cells 11. 10.3390/cells11243974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, and Young RA (2010). c-Myc regulates transcriptional pause release. Cell 141, 432–445. 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gartel AL, and Shchors K. (2003). Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp. Cell Res 283, 17–21. 10.1016/s0014-4827(02)00020-4. [DOI] [PubMed] [Google Scholar]
- 9.Herkert B, and Eilers M. (2010). Transcriptional repression: the darkside of myc. Genes Cancer 1, 580–586. 10.1177/1947601910379012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez-Roman N, Felton-Edkins ZA, Kenneth NS, Goodfellow SJ,Athineos D, Zhang J, Ramsbottom BA, Innes F, Kantidakis T, Kerr ER, et al. (2006). Activation by c-Myc of transcription by RNA polymerases I, II and III. Biochem. Soc. Symp 73, 141–154. 10.1042/bss0730141. [DOI] [PubMed] [Google Scholar]
- 11.Prochownik EV, and Wang H. (2022). Normal and neoplastic growth suppression by the extended myc network. Cells 11. 10.3390/cells11040747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H, Lu J, Edmunds LR, Kulkarni S, Dolezal J, Tao J, Ranganathan S, Jackson L, Fromherz M, Beer-Stolz D, et al. (2016). Coordinated activities of multiple myc-dependent and myc-independent biosynthetic pathways in hepatoblastoma. J. Biol. Chem 291, 26241–26251. 10.1074/jbc.M116.754218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Dolezal JM, Kulkarni S, Lu J, Mandel J, Jackson LE, Alencastro F, Duncan AW, and Prochownik EV (2018). Myc and ChREBP transcription factors cooperatively regulate normal and neoplastic hepatocyte proliferation in mice. J. Biol. Chem 293, 14740–14757. 10.1074/jbc.RA118.004099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Lu J, Alencastro F, Roberts A, Fiedor J, Carroll P, Eisenman RN, Ranganathan S, Torbenson M, Duncan AW, and Prochownik EV (2022). Coordinated cross-talk between the myc and mlx networks in liver regeneration and neoplasia. Cell. Mol. Gastroenterol. Hepatol 13, 1785–1804. 10.1016/j.jcmgh.2022.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gabay M, Li Y, and Felsher DW (2014). MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med 4, a014241. 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kress TR, Pellanda P, Pellegrinet L, Bianchi V, Nicoli P, Doni M,Recordati C, Bianchi S, Rotta L, Capra T, et al. (2016). Identification of MYC-dependent transcriptional programs in oncogene-addicted liver tumors. Cancer Res. 76, 3463–3472. 10.1158/00085472.CAN-16-0316. [DOI] [PubMed] [Google Scholar]
- 17.Soucek L, and Evan GI (2010). The ups and downs of Myc biology.Curr. Opin. Genet. Dev 20, 91–95. 10.1016/j.gde.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swaminathan S, Hansen AS, Heftdal LD, Dhanasekaran R, Deutzmann A, Fernandez WDM, Liefwalker DF, Horton C, Mosley A, Liebersbach M, et al. (2020). MYC functions as a switch for natural killer cell-mediated immune surveillance of lymphoid malignancies. Nat. Commun 11, 2860. 10.1038/s41467-020-16447-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dolezal JM, Wang H, Kulkarni S, Jackson L, Lu J, Ranganathan S, Goetzman ES, Bharathi SS, Beezhold K, Byersdorfer CA, and Prochownik EV (2017). Sequential adaptive changes in a c-Myc-driven model of hepatocellular carcinoma. J. Biol. Chem 292, 10068–10086. 10.1074/jbc.M117.782052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mateyak MK, Obaya AJ, Adachi S, and Sedivy JM (1997). Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. 8, 1039–1048. [PubMed] [Google Scholar]
- 21.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, and Evan GI (2008). Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683. 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Mannava S, Grachtchouk V, Zhuang D, Soengas MS,Gudkov AV, Prochownik EV, and Nikiforov MA (2008). c-Myc depletion inhibits proliferation of human tumor cells at various stages of the cell cycle. Oncogene 27, 1905–1915. 10.1038/sj.onc.1210823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, Stevens T, Lu J, Airik M, Airik R, and Prochownik EV(2022). Disruption of multiple overlapping functions following stepwise inactivation of the extended myc network. Cells 11. 10.3390/cells11244087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, and Cleveland JL (2002). c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 16, 2530–2543. 10.1101/gad.1024602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis AC, Wims M, Spotts GD, Hann SR, and Bradley A. (1993). Anull c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 7, 671–682. 10.1101/gad.7.4.671. [DOI] [PubMed] [Google Scholar]
- 26.Dubois NC, Adolphe C, Ehninger A, Wang RA, Robertson EJ,and Trumpp A. (2008). Placental rescue reveals a sole requirement for c-Myc in embryonic erythroblast survival and hematopoietic stem cell function. Development 135, 2455–2465. 10.1242/dev.022707. [DOI] [PubMed] [Google Scholar]
- 27.Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, Martin GR, and Bishop JM (2001). c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414, 768–773. 10.1038/414768a. [DOI] [PubMed] [Google Scholar]
- 28.Bettess MD, Dubois N, Murphy MJ, Dubey C, Roger C, Robine S, and Trumpp A. (2005). c-Myc is required for the formation of intestinal crypts but dispensable for homeostasis of the adult intestinal epithelium. Mol. Cell Biol 25, 7868–7878. 10.1128/MCB.25.17.7868-7878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edmunds LR, Otero PA, Sharma L, D’Souza S, Dolezal JM, David S, Lu J, Lamm L, Basantani M, Zhang P, et al. (2016). Abnormal lipid processing but normal long-term repopulation potential of myc−/− hepatocytes. Oncotarget 7, 30379–30395. 10.18632/oncotarget.8856 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosselot C, Kumar A, Lakshmipathi J, Zhang P, Lu G, Katz LS,Prochownik EV, Stewart AF, Lambertini L, Scott DK, and Garcia-Ocaña A. (2019). Myc is required for adaptive beta-cell replication in young mice but is not sufficient in one-year-old mice fed with a high-fat diet. Diabetes 68, 1934–1949. 10.2337/db18-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hofmann JW, Zhao X, De Cecco M, Peterson AL, Pagliaroli L,Manivannan J, Hubbard GB, Ikeno Y, Zhang Y, Feng B, et al. (2015). Reduced expression of MYC increases longevity and enhances healthspan. Cell 160, 477–488. 10.1016/j.cell.2014.12.016 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pettan-Brewer C, and M Treuting P. (2011). Practical pathology of aging mice. Pathobiol. Aging Age Relat. Dis 1, 7202. 10.3402/pba.v1i0.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ward JM (2006). Lymphomas and leukemias in mice. Exp. Toxicol. Pathol 57, 377–381. 10.1016/j.etp.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Snyder JM, Ward JM, and Treuting PM (2016). Cause-of-Death analysis in rodent aging studies. Vet. Pathol 53, 233–243. 10.1177/0300985815610391. [DOI] [PubMed] [Google Scholar]
- 35.White MC, Holman DM, Boehm JE, Peipins LA, Grossman M,and Henley SJ (2014). Age and cancer risk: a potentially modifiable relationship. Am. J. Prev. Med 46, S7–S15. 10.1016/j.amepre.2013.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Honma T, Yanaka M, Tsuduki T, and Ikeda I. (2011). Increased lipid accumulation in liver and white adipose tissue in aging in the SAMP10 mouse. J. Nutr. Sci. Vitaminol 57, 123–129. 10.3177/jnsv.57.123. [DOI] [PubMed] [Google Scholar]
- 37.Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J,Raghavakaimal S, and Nair KS (2005). Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 102, 5618–5623. 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uchitomi R, Hatazawa Y, Senoo N, Yoshioka K, Fujita M, Shimizu T, Miura S, Ono Y, and Kamei Y. (2019). Metabolomic analysis of skeletal muscle in aged mice. Sci. Rep 9, 10425. 10.1038/s41598-019-46929-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J,Lintault L, Newman J, Reczek EE, Weissleder R, and Jacks T. (2007). Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665. 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 40.Pappas LE, and Nagy TR (2019). The translation of age-related bodycomposition findings from rodents to humans. Eur. J. Clin. Nutr 73, 172–178. 10.1038/s41430-018-0324-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernandez-Flores A, Saeb-Lima M, and Cassarino DS (2019). Histopathology of aging of the hair follicle. J. Cutan. Pathol 46, 508–519. 10.1111/cup.13467. [DOI] [PubMed] [Google Scholar]
- 42.Pshenichnaya I, Schouwey K, Armaro M, Larue L, Knoepfler PS, Eisenman RN, Trumpp A, Delmas V, and Beermann F. (2012). Constitutive gray hair in mice induced by melanocyte-specific deletion of c-Myc. Pigment Cell Melanoma Res. 25, 312–325. 10.1111/j.1755-148X.2012.00998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goh J, and Ladiges W. (2015). Voluntary wheel running in mice.Curr. Protoc. Mol. Biol 5, 283–290. 10.1002/9780470942390.mo140295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dall’Ara E, Boudiffa M, Taylor C, Schug D, Fiegle E, Kennerley AJ, Damianou C, Tozer GM, Kiessling F, and Müller R. (2016). Longitudinal imaging of the ageing mouse. Mech. Ageing Dev 160, 93–116. 10.1016/j.mad.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 45.Howlett SE (2015). Assessment of frailty in animal models. Interdiscip. Top. Gerontol. Geriatr 41, 15–25. 10.1159/000381131. [DOI] [PubMed] [Google Scholar]
- 46.Ponti F, Santoro A, Mercatelli D, Gasperini C, Conte M, Martucci M, Sangiorgi L, Franceschi C, and Bazzocchi A. (2019). Aging and imaging assessment of body composition: from fat to facts. Front. Endocrinol 10, 861. 10.3389/fendo.2019.00861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitehead JC, Hildebrand BA, Sun M, Rockwood MR, Rose RA, Rockwood K, and Howlett SE (2014). A clinical frailty index in aging mice: comparisons with frailty index data in humans. J. Gerontol. A Biol. Sci. Med. Sci 69, 621–632. 10.1093/gerona/glt136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muncan V, Sansom OJ, Tertoolen L, Phesse TJ, Begthel H, Sancho E, Cole AM, Gregorieff A, de Alboran IM, Clevers H, and Clarke AR (2006). Rapid loss of intestinal crypts upon conditional deletion of the Wnt/Tcf-4 target gene c-Myc. Mol. Cell Biol 26, 8418–8426. 10.1128/MCB.00821-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bertolotti M, Lonardo A, Mussi C, Baldelli E, Pellegrini E, Ballestri S, Romagnoli D, and Loria P. (2014). Nonalcoholic fatty liver disease and aging: epidemiology to management. World J. Gastroenterol 20, 14185–14204. 10.3748/wjg.v20.i39.14185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Müller I, Larsson K, Frenzel A, Oliynyk G, Zirath H, Prochownik EV, Westwood NJ, and Henriksson MA (2014). Targeting of the MYCN protein with small molecule c-MYC inhibitors. PLoS One 9, e97285. 10.1371/journal.pone.0097285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang H, Sharma L, Lu J, Finch P, Fletcher S, and Prochownik EV(2015). Structurally diverse c-Myc inhibitors share a common mechanism of action involving ATP depletion. Oncotarget 6, 15857–15870. 10.18632/oncotarget.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang H, Teriete P, Hu A, Raveendra-Panickar D, Pendelton K, Lazo JS, Eiseman J, Holien T, Misund K, Oliynyk G, et al. (2015). Direct inhibition of c-Myc-Max heterodimers by celastrol and celastrol-inspired triterpenoids. Oncotarget 6, 32380–32395. 10.18632/oncotarget.6116 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zirath H, Frenzel A, Oliynyk G, Segerström L, Westermark UK, Larsson K, Munksgaard Persson M, Hultenby K, Lehtiö J, Einvik C, et al. (2013). MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc. Natl. Acad. Sci. USA 110, 10258–10263. 10.1073/pnas.1222404110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S,Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, et al. (2004). MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 431, 1112–1117. 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 55.Zhang W, Meyfeldt J, Wang H, Kulkarni S, Lu J, Mandel JA, Marburger B, Liu Y, Gorka JE, Ranganathan S, and Prochownik EV (2019). beta-Catenin mutations as determinants of hepatoblastoma phenotypes in mice. J. Biol. Chem 294, 17524–17542. 10.1074/jbc.RA119.009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bratic A, and Larsson NG (2013). The role of mitochondria in aging. J. Clin. Invest 123, 951–957. 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jang JY, Blum A, Liu J, and Finkel T. (2018). The role of mitochondriain aging. J. Clin. Invest 128, 3662–3670. 10.1172/JCI120842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kauppila TES, Kauppila JHK, and Larsson NG (2017). Mammalian mitochondria and aging: an update. Cell Metabol. 25, 57–71. 10.1016/j.cmet.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 59.Lenaz G, Bovina C, Castelluccio C, Fato R, Formiggini G, Genova ML, Marchetti M, Pich MM, Pallotti F, Parenti Castelli G, and Biagini G. (1997). Mitochondrial complex I defects in aging. Mol. Cell. Biochem 174, 329–333. [PubMed] [Google Scholar]
- 60.Lesnefsky EJ, Chen Q, and Hoppel CL (2016). Mitochondrial metabolism in aging heart. Circ. Res 118, 1593–1611. 10.1161/CIRCRESAHA.116.307505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Srivastava S. (2017). The mitochondrial basis of aging and age-related disorders. Genes 8, 398. 10.3390/genes8120398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pessayre D, Berson A, Fromenty B, and Mansouri A. (2001). Mitochondria in steatohepatitis. Semin. Liver Dis 21, 57–69. 10.1055/s-2001-12929 . [DOI] [PubMed] [Google Scholar]
- 63.Rosca MG, Vazquez EJ, Chen Q, Kerner J, Kern TS, and Hoppel CL (2012). Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes 61, 2074–2083. 10.2337/db11-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trifunovic A, and Larsson NG (2008). Mitochondrial dysfunction as acause of ageing. J. Intern. Med 263, 167–178. 10.1111/j.1365-2796.2007.01905.x. [DOI] [PubMed] [Google Scholar]
- 65.Fazzini F, Lamina C, Raftopoulou A, Koller A, Fuchsberger C, Pattaro C, Del Greco FM, Döttelmayer P, Fendt L, Fritz J, et al. (2021). Association of mitochondrial DNA copy number with metabolic syndrome and type 2 diabetes in 14 176 individuals. J. Intern. Med 290, 190–202. 10.1111/joim.13242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Camarda R, Williams J, and Goga A. (2017). In vivo reprogramming of cancer metabolism by MYC. Front. Cell Dev. Biol 5, 35. 10.3389/fcell.2017.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dang CV (2011). Therapeutic targeting of Myc-reprogrammed cancer cell metabolism. Cold Spring Harbor Symp. Quant. Biol 76, 369–374. 10.1101/sqb.2011.76.011296. [DOI] [PubMed] [Google Scholar]
- 68.Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Edmunds LR, Sharma L, Kang A, Lu J, Vockley J, Basu S, Uppala R, Goetzman ES, Beck ME, Scott D, and Prochownik EV (2015). c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate. J. Biol. Chem 290, 20100. 10.1074/jbc.A114.580662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Edmunds LR, Sharma L, Wang H, Kang A, d’Souza S, Lu J, McLaughlin M, Dolezal JM, Gao X, Weintraub ST, et al. (2015). c-Myc and AMPK control cellular energy levels by cooperatively regulating mitochondrial structure and function. PLoS One 10, e0134049. 10.1371/journal.pone.0134049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goetzman ES, and Prochownik EV (2018). The role for myc in coordinating glycolysis, oxidative phosphorylation, glutaminolysis, and fatty acid metabolism in normal and neoplastic tissues. Front. Endocrinol 9, 129. 10.3389/fendo.2018.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Graves JA, Wang Y, Sims-Lucas S, Cherok E, Rothermund K,Branca MF, Elster J, Beer-Stolz D, Van Houten B, Vockley J, and Prochownik EV (2012). Mitochondrial structure, function and dynamics are temporally controlled by c-Myc. PLoS One 7, e37699. 10.1371/journal.pone.0037699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prochownik EV, and Li Y. (2007). The ever expanding role for c-Myc inpromoting genomic instability. Cell Cycle 6, 1024–1029. 10.4161/cc.6.9.4161. [DOI] [PubMed] [Google Scholar]
- 74.Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, and Wahl GM (2002). c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol. Cell 9, 1031–1044. 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 75.Bruss MD, Khambatta CF, Ruby MA, Aggarwal I, and Hellerstein MK (2010). Calorie restriction increases fatty acid synthesis and whole body fat oxidation rates. Am. J. Physiol. Endocrinol. Metab 298, E108–E116. 10.1152/ajpendo.00524.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Houtkooper RH, Argmann C, Houten SM, Cantó C, Jeninga EH, Andreux PA, Thomas C, Doenlen R, Schoonjans K, and Auwerx J. (2011). The metabolic footprint of aging in mice. Sci. Rep 1, 134. 10.1038/srep00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dang CV (2010). Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res. 70, 859–862. 10.1158/0008-5472.CAN-09-3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y,Wonsey D, Lee LA, and Dang CV (2000). Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem 275, 21797–21800. 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- 79.Prochownik EV, and Wang H. (2021). The metabolic fates of pyruvatein normal and neoplastic cells. Cells 10. 10.3390/cells10040762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Toth MJ, and Tchernof A. (2000). Lipid metabolism in the elderly. Eur.J. Clin. Nutr 54 (Suppl 3), S121–S125. 10.1038/sj.ejcn.1601033. [DOI] [PubMed] [Google Scholar]
- 81.Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA,Kim JW, Yustein JT, Lee LA, and Dang CV (2005). Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell Biol 25, 6225–6234. 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Houten SM, Violante S, Ventura FV, and Wanders RJA (2016). The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders. Annu. Rev. Physiol 78, 23–44. 10.1146/annurev-physiol-021115-105045. [DOI] [PubMed] [Google Scholar]
- 83.El-Gharbawy A, and Vockley J. (2018). Inborn errors of metabolism with myopathy: defects of fatty acid oxidation and the carnitine shuttle system. Pediatr. Clin 65, 317–335. 10.1016/j.pcl.2017.11.006 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mihalik SJ, Goodpaster BH, Kelley DE, Chace DH, Vockley J,Toledo FGS, and DeLany JP (2010). Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity 18, 1695–1700. 10.1038/oby.2009.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gibson KM, Lee CF, and Hoffmann GF (1994). Screening for defects of branched-chain amino acid metabolism. Eur. J. Pediatr 153, S62–S67. 10.1007/BF02138780. [DOI] [PubMed] [Google Scholar]
- 86.Wang H, Lu J, Mandel JA, Zhang W, Schwalbe M, Gorka J, Liu Y, Marburger B, Wang J, Ranganathan S, and Prochownik EV (2021). Patient-derived mutant forms of NFE2L2/NRF2 drive aggressive murine hepatoblastomas. Cell. Mol. Gastroenterol. Hepatol 12, 199–228. 10.1016/j.jcmgh.2021.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chadt A, and Al-Hasani H. (2020). Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflugers Arch. 472, 1273–1298. 10.1007/s00424-020-02417-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kirkinezos IG, and Moraes CT (2001). Reactive oxygen species and mitochondrial diseases. Semin. Cell Dev. Biol 12, 449–457. 10.1006/scdb.2001.0282. [DOI] [PubMed] [Google Scholar]
- 89.Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, Khosla S, Jensen MD, and Kirkland JL (2010). Fat tissue, aging, and cellular senescence. Aging Cell 9, 667–684. 10.1111/j.1474-9726.2010.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bhadra M, Howell P, Dutta S, Heintz C, and Mair WB (2020). Alternative splicing in aging and longevity. Hum. Genet 139, 357–369. 10.1007/s00439-019-02094-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Deschênes M, and Chabot B. (2017). The emerging role of alternative splicing in senescence and aging. Aging Cell 16, 918–933. 10.1111/acel.12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gonskikh Y, and Polacek N. (2017). Alterations of the translation apparatus during aging and stress response. Mech. Ageing Dev 168, 30–36. 10.1016/j.mad.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 93.Knoch J, Kamenisch Y, Kubisch C, and Berneburg M. (2012). Rare hereditary diseases with defects in DNA-repair. Eur. J. Dermatol 22, 443–455. 10.1684/ejd.2012.1654. [DOI] [PubMed] [Google Scholar]
- 94.López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G. (2013). The hallmarks of aging. Cell 153, 1194–1217. 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.O’Driscoll M. (2012). Diseases associated with defective responses to DNA damage. Cold Spring Harbor Perspect. Biol 4, a012773. 10.1101/cshperspect.a012773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Park Y, and Gerson SL (2005). DNA repair defects in stem cell function and aging. Annu. Rev. Med 56, 495–508. 10.1146/annurev.med.56.082103.104546. [DOI] [PubMed] [Google Scholar]
- 97.Turi Z, Lacey M, Mistrik M, and Moudry P. (2019). Impaired ribosomebiogenesis: mechanisms and relevance to cancer and aging. Aging (Albany NY) 11, 2512–2540. 10.18632/aging.101922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Donate LE, and Blasco MA (2011). Telomeres in cancer and ageing. Philos. Trans. R. Soc. Lond. B Biol. Sci 366, 76–84. 10.1098/rstb.2010.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Opresko PL, and Shay JW (2017). Telomere-associated aging disorders. Ageing Res. Rev 33, 52–66. 10.1016/j.arr.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roake CM, and Artandi SE (2020). Regulation of human telomerase in homeostasis and disease. Nat. Rev. Mol. Cell Biol 21, 384–397. 10.1038/s41580-020-0234-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yan C, Wan R, and Shi Y. (2019). Molecular mechanisms of pre-mRNA splicing through structural biology of the spliceosome. Cold Spring Harbor Perspect. Biol 11, a032409. 10.1101/cshperspect.a032409 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Meshorer E, and Soreq H. (2002). Pre-mRNA splicing modulations in senescence. Aging Cell 1, 10–16. 10.1046/j.1474-9728.2002.00005.x. [DOI] [PubMed] [Google Scholar]
- 103.Tabula Muris Consortium (2020). A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595. 10.1038/s41586-020-2496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.ENCODE Project Consortium (2004). The ENCODE (ENCyclopedia ofDNA elements) project. Science 306, 636–640. 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 105.Keenan AB, Torre D, Lachmann A, Leong AK, Wojciechowicz ML, Utti V, Jagodnik KM, Kropiwnicki E, Wang Z, and Ma’ayan A. (2019). ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 47, W212–W224. 10.1093/nar/gkz446 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Benanti JA, Wang ML, Myers HE, Robinson KL, Grandori C, and Galloway DA (2007). Epigenetic down-regulation of ARF expression is a selection step in immortalization of human fibroblasts by c-Myc. Mol. Cancer Res 5, 1181–1189. 10.1158/1541-7786.MCR-06-0372. [DOI] [PubMed] [Google Scholar]
- 107.Dean R, Kim SS, and Delgado D. (1986). Expression of c-myc onco-gene in human fibroblasts during in vitro senescence. Biochem. Biophys. Res. Commun 135, 105–109. 10.1016/0006-291x(86)90948-4 . [DOI] [PubMed] [Google Scholar]
- 108.Smith JR, Venable S, Roberts TW, Metter EJ, Monticone R, andSchneider EL (2002). Relationship between in vivo age and in vitro aging: assessment of 669 cell cultures derived from members of the Baltimore Longitudinal Study of Aging. J. Gerontol. A Biol. Sci. Med. Sci 57, B239–B246. 10.1093/gerona/57.6.b239. [DOI] [PubMed] [Google Scholar]
- 109.Cronin-Fenton DP, Damkier P, and Lash TL (2014). Metabolism and transport of tamoxifen in relation to its effectiveness: new perspectives on an ongoing controversy. Future Oncol. 10, 107–122. 10.2217/fon.13.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bastide P, Darido C, Pannequin J, Kist R, Robine S, Marty-Double C, Bibeau F, Scherer G, Joubert D, Hollande F, et al. (2007). Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J. Cell Biol 178, 635–648. 10.1083/jcb.200704152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Blasco MA (2005). Telomeres and human disease: ageing, cancer and beyond. Nat. Rev. Genet 6, 611–622. 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- 112.Kalb R, Neveling K, Nanda I, Schindler D, and Hoehn H. (2006). Fanconi anemia: causes and consequences of genetic instability. Genome Dyn. 1, 218–242. 10.1159/000092510. [DOI] [PubMed] [Google Scholar]
- 113.Iwase T, Wang X, Shrimanker TV, Kolonin MG, and Ueno NT(2021). Body composition and breast cancer risk and treatment: mechanisms and impact. Breast Cancer Res. Treat 186, 273–283. 10.1007/s10549-020-06092-5. [DOI] [PubMed] [Google Scholar]
- 114.Kang C, LeRoith D, and Gallagher EJ (2018). Diabetes, obesity, and breast cancer. Endocrinology 159, 3801–3812. 10.1210/en.2018-00574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ling S, Brown K, Miksza JK, Howells L, Morrison A, Issa E, Yates T, Khunti K, Davies MJ, and Zaccardi F. (2020). Association of type 2 diabetes with cancer: a meta-analysis with bias analysis for unmeasured confounding in 151 cohorts comprising 32 million people. Diabetes Care 43, 2313–2322. 10.2337/dc20-0204. [DOI] [PubMed] [Google Scholar]
- 116.Renehan AG, Tyson M, Egger M, Heller RF, and Zwahlen M(2008). Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 371, 569–578. 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 117.Billin AN, and Ayer DE (2006). The Mlx network: evidence for a parallelMax-like transcriptional network that regulates energy metabolism. Curr. Top. Microbiol. Immunol 302, 255–278. 10.1007/3-540-32952-8_10. [DOI] [PubMed] [Google Scholar]
- 118.Germano G, Lamba S, Rospo G, Barault L, Magrì A, Maione F, Russo M, Crisafulli G, Bartolini A, Lerda G, et al. (2017). Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 552, 116–120. 10.1038/nature24673. [DOI] [PubMed] [Google Scholar]
- 119.Jiang T, Shi T, Zhang H, Hu J, Song Y, Wei J, Ren S, and Zhou C. (2019). Tumor neoantigens: from basic research to clinical applications. J. Hematol. Oncol 12, 93. 10.1186/s13045-019-0787-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.O’Donnell T, Christie EL, Ahuja A, Buros J, Aksoy BA, Bowtell DDL, Snyder A, and Hammerbacher J. (2018). Chemotherapy weakly contributes to predicted neoantigen expression in ovarian cancer. BMC Cancer 18, 87. 10.1186/s12885-017-3825-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D,McCormick LL, Fitzgerald P, Chi H, Munger J, and Green DR (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Anisimova AS, Alexandrov AI, Makarova NE, Gladyshev VN, and Dmitriev SE (2018). Protein synthesis and quality control in aging. Aging (Albany NY) 10, 4269–4288. 10.18632/aging.101721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 124.Molenaars M, Janssens GE, Williams EG, Jongejan A, Lan J, Rabot S, Joly F, Moerland PD, Schomakers BV, Lezzerini M, et al. (2020). A conserved mito-cytosolic translational balance links two longevity pathways. Cell Metabol. 31, 549–563.e7. 10.1016/j.cmet.2020.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stefanatos R, and Sanz A. (2011). Mitochondrial complex I: a central regulator of the aging process. Cell Cycle 10, 1528–1532. 10.4161/cc.10.10.15496. [DOI] [PubMed] [Google Scholar]
- 126.Balaban RS, Nemoto S, and Finkel T. (2005). Mitochondria, oxidants, and aging. Cell 120, 483–495. 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 127.Edgar D, Shabalina I, Camara Y, Wredenberg A, Calvaruso MA, Nijtmans L, Nedergaard J, Cannon B, Larsson NG, and Trifunovic A. (2009). Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metabol. 10, 131–138. 10.1016/j.cmet.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 128.Schumacher B, Pothof J, Vijg J, and Hoeijmakers JHJ (2021). The central role of DNA damage in the ageing process. Nature 592, 695–703. 10.1038/s41586-021-03307-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhuang D, Mannava S, Grachtchouk V, Tang WH, Patil S, Wawrzyniak JA, Berman AE, Giordano TJ, Prochownik EV, Soengas MS, and Nikiforov MA (2008). C-MYC overexpression is required for continuous suppression of oncogene-induced senescence in melanoma cells. Oncogene 27, 6623–6634. 10.1038/onc.2008.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Solvie D, Baluapuri A, Uhl L, Fleischhauer D, Endres T, Papadopoulos D, Aziba A, Gaballa A, Mikicic I, Isaakova E, et al. (2022). MYC multimers shield stalled replication forks from RNA polymerase. Nature 612, 148–155. 10.1038/s41586-022-05469-4. [DOI] [PubMed] [Google Scholar]
- 131.Ghosh A, and Shcherbik N. (2020). Effects of oxidative stress on protein translation: implications for cardiovascular diseases. Int. J. Mol. Sci 21, 2661. 10.3390/ijms21082661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Folgueras AR, Freitas-Rodríguez S, Velasco G, and López-Otín C. (2018). Mouse models to disentangle the hallmarks of human aging. Circ. Res 123, 905–924. 10.1161/CIRCRESAHA.118.312204. [DOI] [PubMed] [Google Scholar]
- 133.Kõks S, Dogan S, Tuna BG, González-Navarro H, Potter P, and Vandenbroucke RE (2016). Mouse models of ageing and their relevance to disease. Mech. Ageing Dev 160, 41–53. 10.1016/j.mad.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 134.Cuthbert AW, Halstead J, Ratcliff R, Colledge WH, and Evans MJ(1995). The genetic advantage hypothesis in cystic fibrosis heterozygotes: a murine study. J. Physiol 482, 449–454. 10.1113/jphysiol.1995.sp020531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Destro-Bisol G, D’Aloja E, Spedini G, Scatena R, Giardina B, and Pascali V. (1999). Brief communication: resistance to Falciparum malaria in alpha-thalassemia, oxidative stress, and hemoglobin oxidation. Am. J. Phys. Anthropol 109, 269–273. . [DOI] [PubMed] [Google Scholar]
- 136.Jones TR (1997). Quantitative aspects of the relationship between the sickle-cell gene and malaria. Parasitol. Today 13, 107–111. 10.1016/s0169-4758(96)10083-1. [DOI] [PubMed] [Google Scholar]
- 137.Hedrick PW (2012). What is the evidence for heterozygote advantage selection? Trends Ecol. Evol 27, 698–704. 10.1016/j.tree.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 138.Grisanzio C, and Freedman ML (2010). Chromosome 8q24-associated cancers and MYC. Genes Cancer 1, 555–559. 10.1177/1947601910381380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Matthews SM, Eshelman MA, Berg AS, Koltun WA, and Yochum GS (2019). The Crohn’s disease associated SNP rs6651252 impacts MYC gene expression in human colonic epithelial cells. PLoS One 14, e0212850. 10.1371/journal.pone.0212850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Beaulieu ME, Jauset T, Massó -Vallé s D, Martínez-Martín S, Rahl P, Maltais L, Zacarias-Fluck MF, Casacuberta-Serra S, Serrano Del Pozo E, Fiore C, et al. (2019). Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med 11, eaar5012. 10.1126/scitranslmed.aar5012 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B,Sagar V, Luan Y, Chalmers ZR, Unno K, et al. (2019). Small-Molecule MYC inhibitors suppress tumor growth and enhance immunotherapy. Cancer Cell 36, 483–497.e15. 10.1016/j.ccell.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang H, Chauhan J, Hu A, Pendleton K, Yap JL, Sabato PE, Jones JW, Perri M, Yu J, Cione E, et al. (2013). Disruption of Myc-Max heterodimerization with improved cell-penetrating analogs of the small molecule 10074-G5. Oncotarget 4, 936–947. 10.18632/oncotarget.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Prochownik EV, and Vogt PK (2010). Therapeutic targeting of myc. Genes Cancer 1, 650–659. 10.1177/1947601910377494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ness KK, Kirkland JL, Gramatges MM, Wang Z, Kundu M,McCastlain K, Li-Harms X, Zhang J, Tchkonia T, Pluijm SMF, and Armstrong GT (2018). Premature physiologic aging as a paradigm for understanding increased risk of adverse health across the lifespan of survivors of childhood cancer. J. Clin. Oncol 36, 2206–2215. 10.1200/JCO.2017.76.7467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mina AI, LeClair RA, LeClair KB, Cohen DE, Lantier L, and Banks AS (2018). CalR: a web-based analysis tool for indirect calorimetry experiments. Cell Metabol. 28, 656–666.e1. 10.1016/j.cmet.2018.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A. (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 14, 128. 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. (2021). Gene set knowledge discovery with Enrichr. Curr. Protoc 1, e90. 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). ClusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141. 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S,Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, Garcia MU, Di Tommaso P, and Nahnsen S. (2020). The nf-core frame-work for community-curated bioinformatics pipelines. Nat. Biotechnol 38, 276–278. 10.1038/s41587-020-0439-x. [DOI] [PubMed] [Google Scholar]
- 153.Gu Z, Eils R, and Schlesner M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 154.Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Xu J. (2005). Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr Protoc Mol Biol Chapter 28, mb2801s70, Unit 28 21. 10.1002/0471142727. [DOI] [PubMed] [Google Scholar]
- 156.Castro B, and Kuang S. (2017). Evaluation of muscle performance inmice by treadmill exhaustion test and whole-limb grip strength assay. Bio. Protoc 7, e2237. 10.21769/BioProtoc.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Preibisch S, Saalfeld S, and Tomancak P. (2009). Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25, 1463–1465. 10.1093/bioinformatics/btp184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ruifrok AC, and Johnston DA (2001). Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol 23, 291–299. [PubMed] [Google Scholar]
- 161.Jackson LE, Kulkarni S, Wang H, Lu J, Dolezal JM, Bharathi SS, Ranganathan S, Patel MS, Deshpande R, Alencastro F, et al. (2017). Genetic dissociation of glycolysis and the TCA cycle affects neither normal nor neoplastic proliferation. Cancer Res. 77, 5795–5807. 10.1158/0008-5472.CAN-17-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang MJ, Pisco AO, Darmanis S, and Zou J. (2021). Mouse aging cell atlas analysis reveals global and cell type-specific aging signatures. Elife 10, e62293. 10.7554/eLife.62293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.GTEx Consortium (2013). The genotype-tissue expression (GTEx) project. Nat. Genet 45, 580–585. 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.R Core Team (2022). R: A Language and Environment for Statistical Computing. https://www.R-project.org/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw RNA-seq files have been deposited in the NCBI Gene Expression Omnibus154 and are accessible through GEO Series accession number GSE223676 database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE223676 ). Data underlying the display items in the manuscript, related to Figures 1, 2, 3, 4, 5, 6, 7, and S1–S7 are available as Data S1 – Source data. The original full-length western blots for Figures 3I, 5C, and S1G have been deposited in Mendeley Data (https://doi.org/10.17632/4t9xbmxszn.1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.