

Abstract
During tumor progression, mechanical abnormalities in the tumor microenvironment (TME) trigger signaling pathways in cells that activate cellular programs, resulting in tumor growth and drug resistance. In this review, we describe mechanisms of action for anti-cancer therapies and mechanotransduction programs that regulate cellular processes, including cell proliferation, apoptosis, survival and phenotype switching. We discuss how the therapeutic response is impacted by the three main mechanical TME abnormalities: high extracellular matrix (ECM) composition and stiffness; interstitial fluid pressure (IFP); and elevated mechanical forces. We also review drugs that normalize these abnormalities or block mechanosensors and mechanotransduction pathways. Finally, we discuss current challenges and perspectives for the development of new strategies targeting mechanically induced drug resistance in the clinic.
Mechanical abnormalities in the TME
In many tumor types, such as breast, colon, and pancreatic cancer, and sarcomas, the TME (see Glossary) is characterized by a desmoplastic reaction, which is directly linked to ECM stiffness [1–3]. ECM stiffness is perhaps the only mechanical aspect of a tumor that can be assessed by clinicians and patients via palpation and can lead to clinical detection of many solid tumors (e.g., in breast or prostate cancer) [1]. As cancer cells proliferate rapidly within the stiff ECM of a solid tumor at the expense of the host tissue, a reciprocal compressive force is applied by the host tissue to resist tumor expansion [2,3]. This leads to accumulation of mechanical stress (i.e., force per unit area) within the tumor [2,4], which is compressive in the tumor interior (i.e., tends to reduce the size of an object). At the interface between the tumor and normal tissue, the stress is tensile (i.e., tends to increase the size of an object) [2,3]. At the cellular level, mechanical compression affects cell cycle progression, migration, invasion, and proliferation [4–8]. At the tissue level, increased compressive forces lead to blood and lymphatic vessel compaction that can impair the transport of nutrients, oxygen (resulting in hypoxia), therapeutic agents, and cells inside the tumor [3]. Hypoxic environments can further fuel tumor progression via induction of the cancer stem cell (CSC) phenotype, epithelial-to-mesenchymal transition (EMT), angiogenesis, inflammation, fibrosis, and immunosuppression [9]. The dense ECM can also act as a physical barrier to immune cell infiltration, contributing to the formation of an immune-excluded phenotype [10,11]. In response to a hypoxic environment, new blood vessels are formed to supply oxygen to the rapidly growing tumor. However, tumor-induced angiogenesis can lead to immature vessels, characterized by reduced pericyte coverage, detachment or loss of the basement membrane, and large openings between endothelial cells, causing vessel hyperpermeability (i.e., leakiness) and transport of large amounts of fluid from the vascular to the extracellular space of the tumor [12,13]. In turn, this results in uniformly elevated IFP, washing drugs out of the tumor [12,13].
Sensing of mechanical cues arising from these TME abnormalities activates mechanotransduction-triggered pathways, which regulate multiple cellular phenotypes (e.g., proliferation, invasion, and survival). Mechanotransduction is essential to tissue homeostasis; however, cancer cells co-opt these aberrant TME signals to activate downstream signaling pathways that lead to abnormal cellular responses, including cell cycle arrest, disruption of apoptosis, immune evasion, and improved DNA repair. Current strategies to overcome therapy resistance have had limited success, because they focus on cell-intrinsic pathways and genetic alterations without considering the impact of the ECM and TME mechanical cues that enable cancer cells to evade death. To this end, a better understanding of the mechanisms involved in mechanical cue sensing and how they regulate cellular programs that determine drug sensitivity will advance the development of novel therapies that target mechanically induced therapy resistance.
In this review, we describe the mechanisms of action and resistance of anti-cancer therapeutic strategies, with a focus on chemotherapy. In addition, we highlight how the biomechanical abnormalities of TME lead to drug resistance, illustrating the crosstalk with mechanical signaling pathways. We next focus on two different therapeutic approaches that either restore/normalize the TME or target components of mechanical signaling, and classify them based on whether they have been tested preclinically, whether they are currently in clinical trials, or are FDA approved. We also discuss opportunities and challenges in mechanoresistance, as well as implementing these mechanical-targeted strategies in the clinic.
Mechanisms of chemotherapy action and resistance
Chemotherapies have diverse mechanisms of action to induce cytotoxic and cytostatic effects on cancer cells. However, response to chemotherapy critically depends on intrinsic mechanisms of chemoresistance that upregulate DNA repair, drug efflux, prosurvival signaling, and cell cycle arrest. The mechanisms of action and resistance of the most common chemotherapies (platinum alkylating agents, topoisomerase inhibitors, mitotic inhibitors, and antimetabolites) that have also been linked with mechanical-induced signaling are presented below. For more details about these mechanisms, we refer readers to several comprehensive reviews [14,15].
Chemotherapeutics can be broadly classified into several categories with diverse mechanisms of action. Platinum alkylating agents, such as cisplatin and carboplatin, covalently bind to DNA to form inter- and intrastrand adducts that disrupt DNA repair, induce DNA damage, and activate proapoptotic genes [16]. Topoisomerase II inhibitors, such as doxorubicin, etoposide, and mitoxantrone, stabilize the topoisomerase II cleavage complex during DNA replication, resulting in DNA single- and double-strand breaks that induce apoptosis [17]. Paclitaxel and docetaxel are microtubule inhibitors that cause cell cycle arrest due to their disruption of microtubule formation and disassembly, which are necessary for the formation of mitotic asters and the kinetochore during cell division [18]. Antimetabolites, such as 5-fluorouracil (5-FU) and gemcitabine, incorporate into RNA or DNA instead of their normal nucleotide counterpart. 5-FU incorporates into DNA to inhibit DNA and RNA synthesis [19], whereas gemcitabine also induces DNA damage to prevent DNA replication [20].
Cancer cells can evade the effects of chemotherapy through several resistance mechanisms. Here, we discuss the preclinical and clinical evidence for mechanisms that are relevant to mechanoresistance. For example, heightened DNA repair capacity can correct chemotherapy-induced lesions to prevent cell death. Greater expression of genes associated with DNA repair signaling pathways is correlated with increased resistance to cisplatin in ovarian and lung cancer, and copy number amplification of these genes is associated with reduced overall survival for several cancers [21]. ATP-binding cassette (ABC) family multidrug efflux pumps sequester or remove drugs from the cell. P-glycoprotein (ABCB1) is the most prominent efflux pump [22] and is associated with poor response to many chemotherapies and overall survival in patients with ovarian cancer [23]. A greater ratio of proapoptotic proteins to antiapoptotic proteins triggers apoptosis [24]. The expression of these proteins is regulated by many pathways, such as PI3K/Akt and p38-MAPK signaling [24,25]. Increased Akt expression is associated with paclitaxel and cisplatin resistance in ovarian cancer cells and doxorubicin resistance in gastric cancer [26–28]. Additionally, greater expression of antiapoptotic proteins BCL2L1 and MCL1 is associated with worse survival outcomes in patients with ovarian cancer [29]. Cell cycle checkpoints regulate progression through the cell cycle. Cancer cell sensitivity to certain chemotherapies (e.g., platinum based and taxanes) are cell cycle specific; thus, arrested cells may be less sensitive to the effects of chemotherapy [30,31]. Patients with cisplatin-treated non-small-cell lung cancer expressing high p27Kip1, which prevents cell cycle progression from G1 to S phase, experienced poorer overall survival compared with patients with low p27Kip1 expression [32]. These pathways mediating chemoresistance can be activated in a cell-intrinsic manner (i.e., genetic alterations) or in a cell-extrinsic manner via environment-mediated chemoresistance. We discuss this intricate connection between therapeutic resistance and mechanical forces/mechanotransduction signaling mechanisms in the next section.
Mechanical signaling crosstalk with drug resistance mechanisms
Mechanical cues can regulate response to chemotherapy by activating chemoresistance programs that are associated with cell states, such as EMT and cancer stemness. Cells that have undergone EMT exhibit increased expression of drug efflux pump genes, resistance to apoptosis, anchorage-independent growth (anoikis), and stem cell characteristics [33]. Furthermore, CSCs exhibit chemoresistance due to their slow proliferation, high prosurvival programs via Akt/PKB signaling, greater drug efflux, and efficient DNA damage repair [34]. Here, we discuss the role of these cell states on chemoresistance and how mechanical abnormalities impact chemotherapy response (see Box 1 for mechanosensing and Figure 1 for shared chemotherapy with mechanotransduction signaling responses). Studies related to mechanically induced chemoresistance are summarized in Table 1. We also discuss several studies that provide evidence for the mechanisms of mechanically induced immunotherapy and targeted therapy resistance (see Box 2).
Box 1. Mechanisms of sensing and transmitting mechanical cues.
By forming a physical boundary between intracellular components and the extracellular environment, the cellular membrane is a key interface mediating response to mechanical stimuli. Cellular membranes contain a variety of mechanosensitive proteins that translate extracellular mechanical cues into intracellular signals (see Figure 1 in main text). Among the most well-characterized mechanosensitive proteins are integrins, found to be overexpressed in solid tumors, including breast, pancreatic, and lung carcinomas [134]. Upon activation, integrins recruit the adhesion plaque protein talin, creating mature focal adhesions [135]. Focal adhesions physically connect integrins to the cytoskeleton by their interaction with actin linker molecules, inducing the formation of actin stress fibers in the cytoplasm [135]. In addition, focal adhesions interact with signaling molecules, including focal adhesion kinase (FAK), which in turn presents a docking site for the nonreceptor tyrosine kinase Src [135]. Downstream, the FAK/Src complex activates the NF-κB, Hippo, MAPK, and PI3K signaling pathways, which can also interact with several receptor tyrosine kinase (RTK) and growth factor receptors (GFRs), including the epidermal (EGFR), insulin (IGFR), and transforming growth factor beta (TGFβR) receptors [136].
In addition to integrins, mechanically activated ion channels, including Piezo and transient receptor potential (TRP), and GPCRs are cell surface receptors of mechanical signals. Ion channels are nonselective cation channels responding with channel gating to mechanical stress [137] or indirectly by integrins or other scaffolding elements, such as ECM components [94]. Ion channels sense mechanical cues in the tissue microenvironment and cooperate with focal adhesions to regulate cellular responses. In breast cancer cells, upon application of mechanical stress, activated mechanosensitive ion channels were detected in 54% of cell membrane patches, leading to higher metastatic potential [138]. Similarly, GPCRs interact with nearby G proteins on the plasma membrane and cooperate with integrins and GFRs to regulate cytoskeletal rearrangements and cell shape changes [139]. Studies have shown that GPCRs are overexpressed in a variety of cancer types, contributing to cell proliferation in response to ligands or mechanical cues [140].
Mechanical sensing and mechanotransduction pathways are also necessary for other cellular components of the TME, including stromal and immune cells. The cellular responses mediated in these cell types can also affect tumor progression and resistance to therapy, and are extensively reviewed elsewhere [141,142].
Figure 1. Shared signaling responses induced by mechanical cues and mechanisms of chemoresistance in cancer cells.
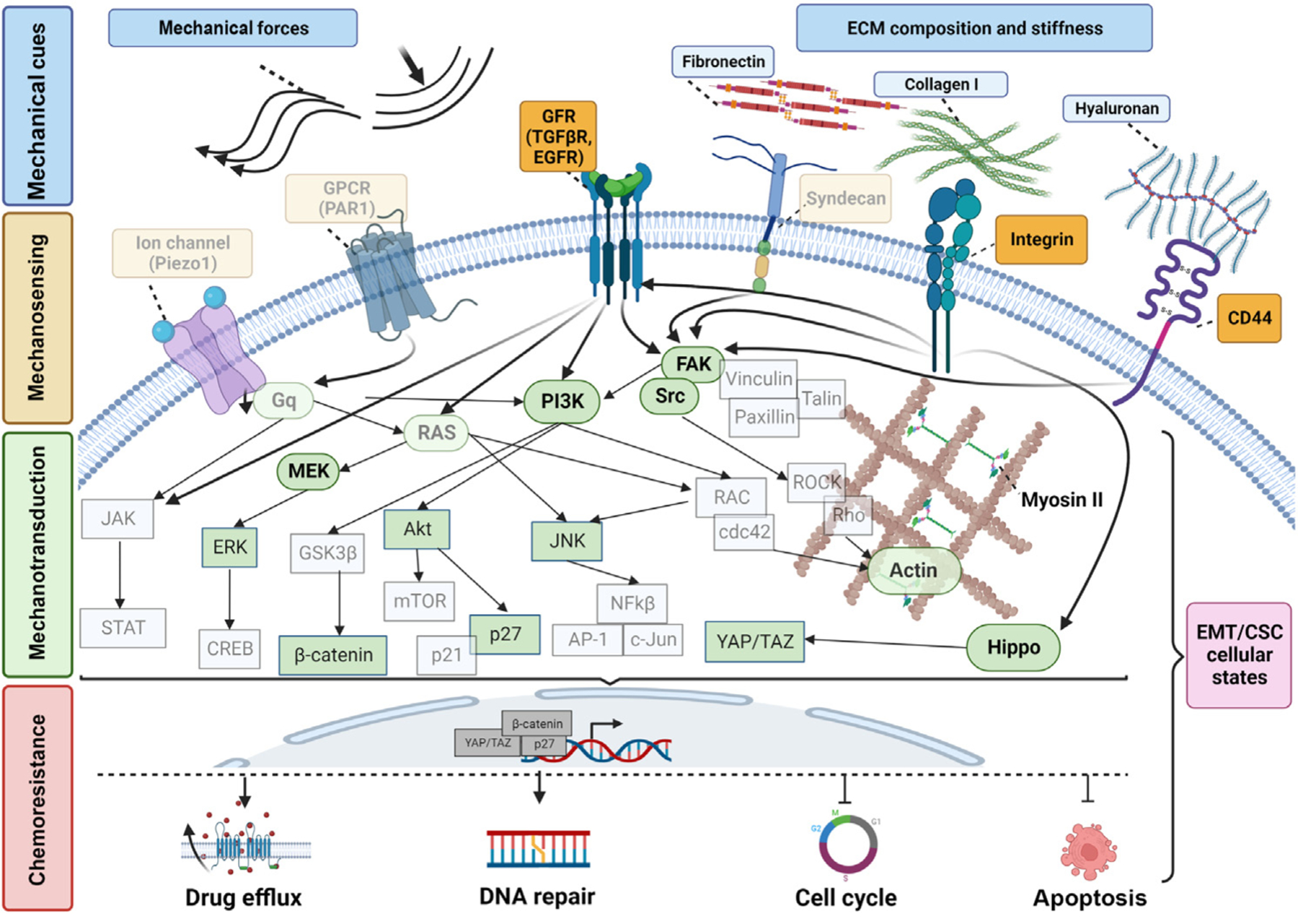
Mechanical cues that are present in the tumor microenvironment [TME; i.e., mechanical forces or extracellular matrix (ECM) stiffness] are sensed by: (i) membrane receptors, such as integrins; (ii) G protein-coupled receptors (GPCRs); and (iii) ion channels. At the same time, mechanosensors regulate the activity of GFRs, together leading to the activation of major signaling pathways, including RAS, PI3K, FAK, and Hippo. These pathways interact with each other to promote cytoskeleton rearrangement, which is mainly facilitated by the activation of ROCK/Rho, RAC/cdc42, and myosin II, translocation of transcription factors into the nucleus that directly regulate gene expression, and eventually regulation of epithelial-to-mesenchymal transition (EMT) and cancer stemness. Altogether, these mechanically driven responses lead to four main chemoresistance mechanisms: activation of (i) drug efflux and (ii) DNA repair, and alterations in (iii) cell cycle and (iv) apoptosis. Faded structures represent known mechanically regulated pathways in normal cells, while structures shown in full color represent those structures shown to be regulated by mechanical cues in cancer cells. Abbreviation: CSC, cancer stem cell. Created with BioRender.com.
Table 1.
Studies that identified mechanically induced chemoresistance mechanisms
| Experimental setup (simulated mechanical cue of TME) | Cancer type | Mechanotransduction mechanism | Chemoresistance mechanism | Resistance to | Refs |
|---|---|---|---|---|---|
| ECM composition and stiffness | |||||
| Fibronectin (FN)-coated substrate (ECM component, 2D) | Ovarian; breast | FN/PI3K/Akt2/survivin axis activation | Inhibition of apoptosisa | Docetaxel (microtubule inhibitor) | [37] |
| Myeloma | FN/β1-integrins/p27kip1 axis activation | Inhibition of DNA synthesis → cell cycle arrestb | Etoposide VP-16 (topoisomerase II inhibitor) | [38,39] | |
| Breast | FN/β1-integrins/PI3K/Akt axis activation | Inhibition of cytochrome c release/sustained expression of Bcl-2 proteina | Paclitaxel (microtubule inhibitor) | [40] | |
| Collagen I and FN substrate (ECM components, 2D) | Breast | Collagen I–integrin activation | Activation of integrin-induced ABC efflux transporter (CAM-DR)c | Cisplatin (platinum alkylating agent), doxorubicin, mitoxantrone (topoisomerase II inhibitors) | [46] |
| Collagen I substrate (ECM component, 2D) | Jurkat and HSB2 leukemic T cells | Collagen I–integrin/Erk activation | Upregulation of ABC drug transporters → drug effluxc | Doxorubicin (topoisomerase II inhibitor) | [48] |
| 3D collagen I matrix (ECM component) | Pancreatic | Increased membrane type 1 matrix metalloproteinase (MT1-MMP)/ERK1/2 phosphorylation → increased HMGA2 expression → chromatin remodeling | Activation of checkpoint kinase 1 (Chk1) and cell cycle regulationb | Gemcitabine (antimetabolite) | [145] |
| Ovarian | Activation of TGFβ and Wnt signaling, increased MMP, fibronectin, vimentin, N-cadherin, Slug, Snail, and integrin expression | Activation of TGFβ-, Wnt-induced EMTd → decreased growth ratese | Carboplatin (platinum alkylating agent), 5-FU (antimetabolite), paclitaxel (microtubule inhibitor) | [55] | |
| Increased stiffness of polyacrylamide gels (ECM stiffness) | Pancreatic | Increased vimentin and decreased E-cadherin expression; nuclear translocation of β-catenin, YAP and TAZ | Induction of EMTd,e | Paclitaxel (microtubule inhibitor) | [56] |
| Increased stiffness of methylcellulose matrix (ECM stiffness) | Pancreatic | Alterations in gene expression (drug transporter, collagen, fibronectin, and metabolic genes) | Increase in drug efflux, CAM-DR, alterations in cell metabolismc | Multiple drugs | [51] |
| Increased stiffness of polyacrylamide hydrogel (ECM stiffness) | Breast | Inactivation of Hippo pathway protein; Merlin/MST/LATS/ILK-induced activation of YAP | Increase in cell survivala, upregulation of ABCB1 expression → drug effluxc | Doxorubicin (topoisomerase II inhibitor) | [47] |
| HA-containing Matrigel matrix (ECM stiffness) | CSC of head and neck squamous cell carcinoma | HA-CD44v3 activation → increased miR-302 expression | Upregulation of prosurvival proteins (e.g., AP1) and CSC markers (e.g., Oct-4), cancer cell self-renewal, stemness, and clonal formatione | Cisplatin (platinum alkylating agent) | [69] |
| Increased stiffness of alginate beads (ECM stiffness) | Hepatocellular | Not identified | Alterations in expression of ABC drug transporters → drug effluxc; upregulation of antiapoptotic programa | 5-FU (antimetabolite), cisplatin (platinum alkylating agent), paclitaxel (microtubule inhibitor) | [52] |
| Increased stiffness of collagen I matrix (ECM stiffness) | Hepatocellular | Not identified | Alterations in expression of drug metabolism enzyme CYP3A4c,d | Doxorubicin (topoisomerase II inhibitor) | [146] |
| Colorectal | FAK/Wnt signaling activation | Induction of CSC → inhibition of cell growth | 5-FU-based chemotherapies (antimetabolite) | [66] | |
| Increased stiffness of alginate hydrogels (ECM stiffness) | Breast | Nuclear translocation of YAP, decreased E-cadherin expression | Induction of EMTd,e | Doxorubicin (topoisomerase II inhibitor) | [57] |
| Increased stiffness of polyacrylamide hydrogels (ECM stiffness) | Breast | Regulation through MAP4K4/6/7 kinases/RNF8-mediated ubiquitin signaling at DNA double-stranded breaks (DSBs) | Induction of DNA DSB repairf | Etoposide (topoisomerase II inhibitor), cisplatin (platinum alkylating agent) | [42] |
| Mechanical forces (compression/shear) | |||||
| Flow-induced shear stress using 3D tumoroid flow system (ECM stiffness, IFP-induced shear stress) | Breast | Upregulation of Snail/MMP14 and downregulation of E-cadherin/vimentin | EMTd,e; inhibition of cell viability/proliferation | Doxorubicin (topoisomerase II inhibitor) | [91] |
| Hydrostatic pressure generated using microfluidic platform (increased fluid pressure) | Breast | Not identified | Modulation of expression of drug transporter gene ABCC1 → drug effluxc | Doxorubicin (topoisomerase II inhibitor) | [88] |
| 3D-matrigel flow culture (increased flow-induced shear stress) | Ovarian | EGFR/MEK/ERK signaling activation | – | Carboplatin (platinum alkylating agent) | [48] |
| PDMS microfluidic channel (increased flow-induced shear stress) | Ovarian | miR-199a-3p/PI3K/Akt/ABCG2 drug transporter activation | Alterations in drug effluxc; EMTd,e; CSCd | Cisplatin (platinum alkylating agent); paclitaxel (microtubule inhibitor) | [89] |
| 3D hydrogel embedded in bioreactor that applies shear stress (increased flow-induced shear stress) | Breast | Upregulation of PLAU expression and increase in uPA activity | Increase in cell proliferation and invasionb | Paclitaxel (microtubule inhibitor) | [90] |
| Compression bioreactor system; 3D (compression-induced mechanical stress) | Ovarian | CDC42 upregulation | Upregulation of cdc42, Oct4 → EMT and CSCe; increase in ABC drug transporters expression → drug effluxc; inhibition of apoptotic cell deatha | Paclitaxel (microtubule inhibitor) and carboplatin (platinum alkylating agent) | [99] |
| Spheroids embedded in 3D agarose matrix (compression-induced mechanical stress) | Pancreatic | Inactivation of ERK | Decrease in cell proliferationb | Gemcitabine (antimetabolite) | [98] |
Apoptosis/cell survival.
Cell cycle.
Drug efflux.
The exact cellular process that leads to chemoresistance has not been specified.
EMT/CSC.
DNA repair.
Box 2. Mechanisms of targeted and immunotherapies.
Mechanism of action and resistance of targeted therapies
Targeted therapies present a personalized approach for treating cancer based on genetic alterations in individual patients. Prominent examples include agents targeting the EGFR, RAF/MEK, and AKT signaling pathways, which are aberrantly activated in cancer cells to drive proliferation and survival, and evade apoptosis. In addition, VEGF inhibitors represent another class of targeted therapies that are used to interfere with the formation of new blood vessels and limit tumor growth. Despite the initial benefits offered by targeted therapies, their effectiveness often diminishes over time due to new genetic alterations or reactivation of the target molecule, upstream or downstream signaling activation, epigenetic changes, by-pass signaling changes, and cancer cell–ECM interactions that enable cell survival under therapy (extensively reviewed in [143]).
Immunotherapy mechanisms of action and resistance
Distinct from chemotherapy, which impacts all cell types, immunotherapy harnesses the immune system to kill cancer cells. Immune checkpoint inhibitors (ICIs) represent one of the most successful immunotherapies in the clinic. Their mode of action involves inhibition of checkpoint molecules that act as ‘brakes’ on the immune system, with PD-1 being a widely targeted molecule. The PD-1 receptor, which is expressed on T cells, recognizes PD-L1 ligand on cancer cells and prevents T cell-mediated killing. By blocking PD-1/PD-L1 recognition, the cytotoxicity of T cells is restored, allowing them to recognize and attack cancer cells more effectively [11]. However, despite the significant advances in ICI as a cancer treatment, many patients do not respond to this approach. ICI resistance is usually attributed to the absence of tumor-infiltrating effector immune cells and TME properties [80].
Although cell-intrinsic mechanisms of drug resistance have been extensively studied, recent research highlights the role of extracellular cues, including mechanical forces in the TME, in promoting drug resistance [144]. Therefore, investigating the coupling between mechanical signaling and therapy resistance mechanisms is crucial to developing novel strategies that can enhance the effectiveness of targeted and immune-directed therapies.
ECM mechanisms of therapy resistance
Adhesion of cancer cells to ECM components, such as collagen and fibronectin, or their growth in a stiff matrix, drives resistance to chemotherapy [35]. This ECM-mediated chemoresistance mechanism is primarily based on activation of integrin signaling, which results in overexpression of prosurvival and antiapoptotic proteins, cell cycle arrest, modulation of drug efflux, and a phenotype switching of cancer cells (e.g., EMT or cancer stemness), which is referred to as cell adhesion-mediated drug resistance (CAM-DR) [36]. For example, ovarian and breast cancer cells exhibited docetaxel resistance through PI3K/Akt/survivin axis-induced inhibition of apoptosis [37], while, in myeloma cells, fibronectin-induced integrin activation led to cell cycle arrest and etoposide resistance [38,39]. In breast cancer cells, it has been also shown that fibronectin-induced integrin activation inhibited the release of cytochrome c and the expression of the antiapoptotic protein Bcl-2 through the PI3K/Akt signaling axis, resulting in inhibition of apoptosis triggered by microtubule-directed chemotherapeutic agents [40]. Another study demonstrated that gemcitabine resistance can be induced by activation of checkpoint kinase 1 (CHK1) and subsequent cell cycle regulation through the MMP/ERK1/2 signaling axis in pancreatic cancer cells growing in a 3D collagen matrix [41]. In breast cancer cells, increased stiffness of fibronectin-coated substrates was also shown to facilitate DNA repair of double-stranded DNA breaks, impairing the efficacy of several drugs, including etoposide and cisplatin. This mechanism was regulated by MAP4K4/6/7 kinase and subsequent ubiquitin phosphorylation, which recruits H2AX at DNA-damaged sites to activate DNA repair mechanisms [42]. Transcriptomic-based analysis in the clinic of patients with breast and gastric cancer also provides evidence for the role of ECM constituents, such as fibronectin and collagen type I, and poor treatment responses [43–45].
Recent studies showed that ECM stiffness has also a significant effect on the activity of ABC drug transporters that determine intracellular drug uptake [46]. When the ECM is stiff, ABC transporters are less active and less effective at removing drugs from cells. Conversely, when the ECM is more compliant or soft, ABC transporters are more active, which can enhance drug clearance and improve treatment outcomes [47]. Breast cancer cells growing on collagen I and fibronectin substrates exhibited resistance to cisplatin, doxorubicin, and mitoxantrone through elevated expression of ABC efflux transporters [46,47]. In a similar manner, Jurkat and HSB2 leukemic cells grown on collagen substrate acquired resistance to doxorubicin through integrin/Erk signaling activation and ABCC1 drug transporter upregulation [48]. Clinically, matrix stiffness has been measured directly through elastography or inferred through lysyl hydroxylase (LOX) expression, and is correlated with poorer pathological complete response and relapse-free survival [49,50]. Alterations in the expression of ABC drug transporter genes have also been observed in pancreatic [51] and hepatocellular carcinoma cells [52] grown on substrates of increased stiffness, which led to resistance to 5-FU, cisplatin, and paclitaxel. These findings highlight the importance of ABC transporters as key regulators of ECM-induced chemoresistance, and should be a focus of future research and therapeutic development efforts.
Cell adhesion to collagen type I and fibronectin upregulates the expression of mesenchymal markers, including Slug, Snail, and vimentin, and downregulates E-cadherin to promote EMT transition through the activation of Src and the ERK/MAPK pathway [53] or the focal adhesion kinase (FAK)/Src/β-catenin signaling axis [54]. In line with this, ovarian tumor spheroids embedded in type I collagen-rich ECM showed increased invasive abilities and EMT, which subsequently led to chemoresistance, although the exact mechanism was unclear [55]. In pancreatic cancer cells, increased stiffness upregulated EMT markers and promoted the nuclear localization of β-catenin, YAP, and TAZ mechanoresponsive transcriptional regulators, leading to paclitaxel resistance [56]. In breast cancer cells, dynamic ECM stiffening induced doxorubicin resistance through YAP nuclear translocation and an EMT state [57].
ECM signals, including transforming growth factor beta (TGFβ) release in the TME, may also induce a CSC phenotype by activation of EMT in cancer cells [58,59] and maintenance of cancer cell dormancy [60]. In colorectal and breast cancer, type I collagen promotes the expression of CSC markers [61–63], while in squamous cell carcinoma and breast cancer, it increases CSC enrichment through activation of FAK signaling [64] and Akt/mTOR/YAP signaling [65], respectively. In a similar manner, it was shown that cancer cells reversed their mesenchymal phenotype, reduced their proliferation rate, and acquired chemoresistance when co-cultured with hepatic stem cells in organoids embedded in dense collagen type I matrices [66]. Similar to collagen type I, hyaluronic acid (HA)-rich ECM also provides a favorable environment for the maintenance of CSCs. This is evidenced by studies showing that hyaluronan synthase (HAS2) is necessary for the metastatic ability of breast CSCs [67], HA accumulation increased CSC enrichment in breast tumor models through Twist- and TGFβ-snail signaling axis [68], and the interaction of HA with CD44 in head and neck squamous CSC promoted their self-renewal and cisplatin resistance [69]. Altogether, these studies support that ECM composition has an important role in CSC survival/growth, which, in turn, leads to chemoresistance and the increased metastatic potential of cancer cells.
In addition to chemoresistance, mechanical cues also affect the efficacy of targeted therapies. Indeed, patient-derived glioblastoma cells grown in stiff hydrogels comprising HA, showed reduced response to the FDA-approved epidermal growth factor receptor (EGFR) inhibitor erlotinib [70]. Furthermore, collagen-driven activation of integrin signaling in lung cancer cells also conferred resistance to multiple EGFR inhibitors [71]. Increased ECM stiffness in colorectal liver metastasis reduced the efficacy of the antiangiogenic agent bevacizumab [72], as well as the efficacy of the Raf inhibitor sorafenib in breast cancer cells through the β1-integrin/JNK signaling axis [73]. Finally, ECM stiffness induced resistance of breast cancer cells to the HER2 inhibitor lapatinib through YAP/TAZ transcriptional activity [74], and to BRAF inhibitors in melanoma cells [75].
A dense ECM constitutes a physical barrier to the diffusion of immune checkpoint therapies [α-programmed cell death protein 1 (α-PD-1)/programmed death-ligand 1 (PDL-1)] and T cell infiltration, which limit cancer cell cytotoxicity [76,77]. Trafficking of T cells through dense collagen fibers can result in nuclear damage, impaired motility, and eventually cell death [78]. Increased matrix stiffness has also been shown to upregulate PD-L1 expression in cancer cells that was linked with YAP/TAZ nuclear translocation [79]. Other cellular components of the immune system are also affected by increased matrix stiffness, including dendritic cells, natural killer cells, and macrophages, which exhibit chemotaxis toward cancer cells and determine the balance between an immunostimulatory versus immunosuppressive microenvironment [80]. For example, when lysyl oxidases were used to target ECM remodeling and stiffness, T cell migration improved with increased efficacy of anti-PD-1 blockade [81]. In line with this, restoring TME abnormalities using the mechanotherapeutic tranilast, combined with immune checkpoint inhibition, increased T cell infiltration in TME, and immunological memory in mice bearing immunotherapy-resistant breast cancer [82].
Force-induced therapy resistance
In solid tumors, ECM stiffness is mainly determined by ECM composition and organization, while mechanical/physical forces are exerted during tumor growth. At the tissue level, the compressed and abnormal tumor blood vessels along with the increased deposition of matrix components in the tumor ECM, hinder the delivery of therapeutic agents to the tumor interior [83,84]. At the same time, the elevated intratumoral IFP, which drops to normal values at the tumor periphery, induces outward fluid flow from the tumor interior to the surrounding tissue, which washes drugs out of the tumor [83,85,86]. At the cellular level, while the effect of ECM stiffness on therapy resistance is actively being investigated, studies focused on force-induced drug resistance remain limited. Here, we provide evidence for how mechanical forces compromise the effectiveness of anti-cancer therapies, illustrating the molecular mechanisms identified so far.
Elevated IFP-induced shear stress and therapy resistance
Within the TME, the accumulation of interstitial fluid due to blood vessel hyperpermeability and lymphatic dysfunction leads to IFP, which hinders drug delivery. High IFP values have been also measured in patients with cervical cancer, and are considered an independent poor prognostic factor for tumor recurrence after radiotherapy [87]. However, the direct effects of IFP on cancer cell chemosensitivity remain poorly understood. Breast cancer cells exposed to hydrostatic pressure exhibited poor response to doxorubicin, which was mediated by upregulation of the ABCC1 drug transporter and reduced intracellular doxorubicin concentration [88]. High IFP also generates shear forces on tumor cells via fluid flow. In ovarian cancer spheroids, shear stress increased the expression of EMT markers, ABCC1 drug transporters, and cancer stemness markers, with reduced sensitivity to cisplatin and paclitaxel [89]. Additionally, another study showed that adherent ovarian cancer cells exhibited a poor response to carboplatin following exposure to shear stress through activation of EGFR-driven MEK and ERK prosurvival signaling [48]. Consistent with these results, breast cancer cells exposed to fluid shear stress also exhibited increased motility and resistance to paclitaxel [90]. When shear stress was applied on breast cancer cells embedded in a 3D collagen matrix, it was shown to induce the expression of EMT markers and result in poor response to doxorubicin across multiple cell lines [91]. Importantly, transcriptomic analysis of patients with triple-negative breast cancer demonstrated that the expression of chemoresistance-related genes was correlated with a shear stress-induced gene expression profile [92].
Although little is known about IFP- and shear stress-induced targeted therapy resistance, it was shown that shear stress enhanced the resistance of sarcoma cells to insulin growth factor-1 receptor blockade by dalotuzumab [93]. IFP also impairs the delivery of immunotherapies to the tumor interior, and induces flow of tumor- and stromal cell-derived immunosuppressive exosomes toward the tumor boundary. These exosomes may recruit immunosuppressive immune cells to fuel tumor progression [94].
Compression-induced mechanical stress and therapy resistance
Mechanical forces result not only internally from TME structural remodeling, but also externally from the cancer–host tissue reactive boundary due to tumor expansion. These compressive mechanical cues have been linked with alterations in the proliferation rate, higher metastatic potential, and survival of cancer cells [4–8,95,96]. However, their role in chemoresistance remains elusive [97]. It has been shown that compressive stress results in mitotic arrest through inhibition of the bipolar spindle assembly, which eventually impairs proliferation [5]. Given that most common chemotherapeutic agents target proliferating cells, this compression-induced mechanism may limit therapeutic responses. Another study that combined mathematical modeling with experiments showed that high compressive stress generated during tumor spheroid growth in a confined agarose matrix led to decreased cell proliferation and response to gemcitabine, which targets actively proliferating cancer cells [98]. Mechanical compression applied in a 3D setting also resulted in ovarian cancer invasion and chemoresistance through upregulation of CDC42 expression; however, the underlying mechanotransduction mechanism is not yet defined [99]. Mechanical stresses were also shown to induce immunotherapy resistance through activation of the PI3K/Akt pathway in tumor cells, which blunts T cell-induced apoptosis, increases PD-L1 expression, and promotes the recruitment of immunosuppressive immune cells (e.g., Tregs) [61]. The effect of compression-induced mechanical stress on the efficacy of targeted therapies remains to be explored.
Strategies to target biomechanical abnormalities in the TME and mechanically induced signaling pathways
Mechanotransduction pathways and the mechanically remodeled TME can be therapeutically targeted by blocking mechanical signaling components and normalizing the TME (Figure 2, Key figure). In Table 2, we list drugs that have been tested in clinical trials in combination with the standard-of-care, as well as the outcome of the clinical trial.
Figure 2. Drugs that target mechanosensors, growth factor receptors, mechanotransduction pathways, or components of the extracellular matrix (ECM).
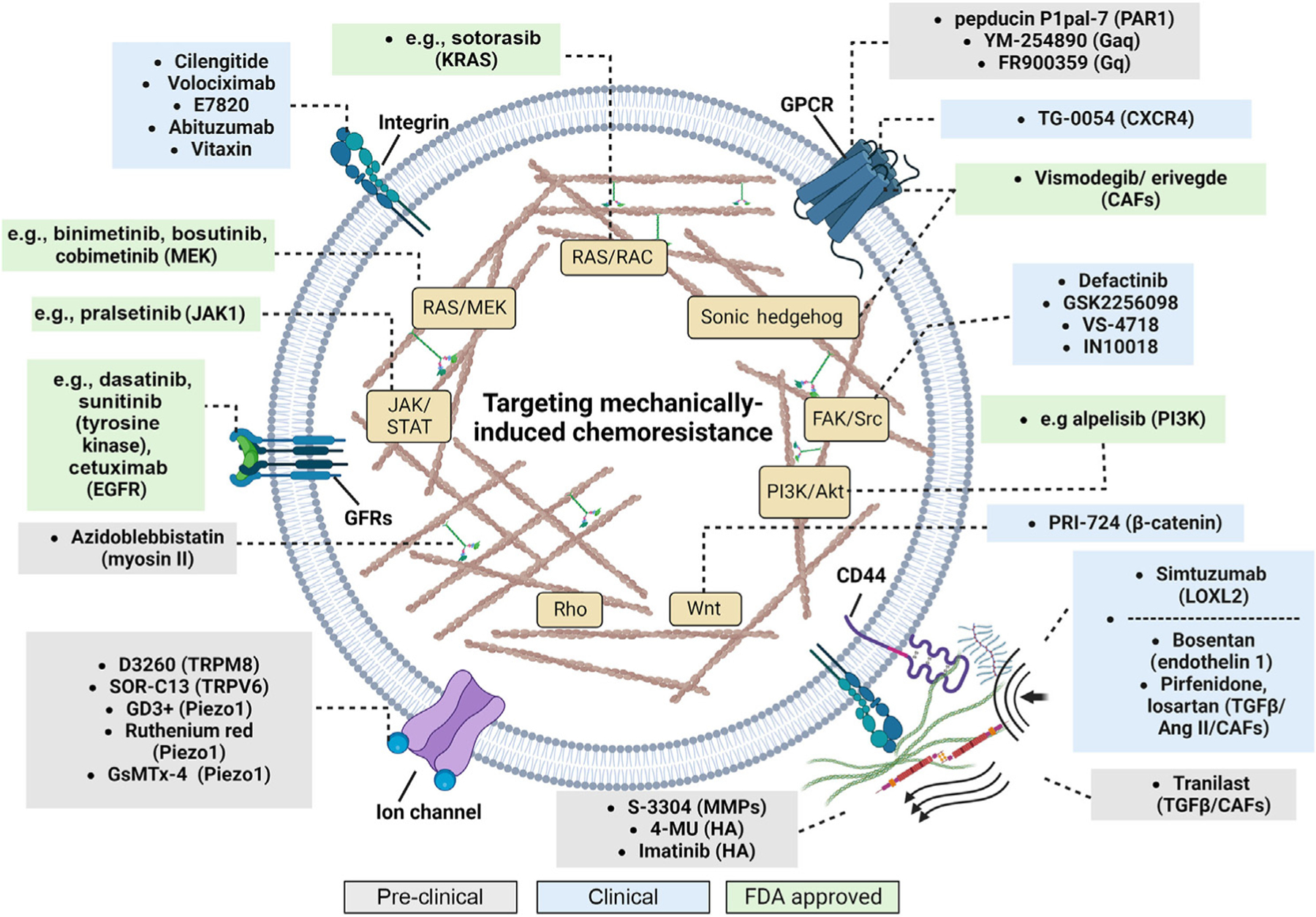
Grey boxes indicate drugs that are in preclinical studies; blue boxes include drugs that are being tested in clinical trials; and green boxes include drugs that are FDA approved for the treatment of patients with cancer. Names in parentheses correspond to the target of each drug. Abbreviations: CAFs, cancer-associated fibroblasts; GFRs, growth factor receptors; HA, hyaluronic acid; MMPs, matrix metalloproteinases. Created with BioRender.com.
Table 2.
Representative clinical trials of agents targeting the mechanical aspects of TME
| Target | Agent | Phase | Cancer type | Combined with | Clinicaltrial.gov ID |
|---|---|---|---|---|---|
| Mechanosensors/mechanotransduction | |||||
| Integrin | Cilengitide | 2 | Squamous cell carcinoma | 5-FU + cisplatin + cetuximab (EGFR | NCT00705016 a |
| inhibitor) (PFE) | |||||
| 1 | Breast | Paclitaxel | NCT01276496 b | ||
| 2 | Lung | Cisplatin, gemcitabine, cetuximab | NCT00842712 | ||
| 2 | Glioblastoma | Temozolomide | NCT00085254 | ||
| 3 | Glioblastoma | Temozolomide | NCT00689221 a | ||
| Volociximab (M200) | 1b | Lung | Carboplatin, paclitaxel, bevacizumab (anti-VEGF) | NCT00666692b, NCT00654758b | |
| 2 | Lung | Erlotinib | NCT00278187 b | ||
| 2 | Pancreatic | Gemcitabine | NCT00401570 b | ||
| 1/2 | Ovarian | Doxorubicin | NCT00635193 a | ||
| 2 | Melanoma | Dacarbazine | NCT00099970 b | ||
| E7820 | 1&2 | Colon, rectal | Irinotecan versus FOLFIRI | NCT01347645 b | |
| 2 | Colorectal | Cetuximab | NCT00309179 b | ||
| 1/2 | Colorectal | FOLFIRI versus bevacizumab | NCT01133990 b | ||
| Abituzumab (EMD 525797) | 1/2 | Colorectal | Cetuximab + irinotecan | NCT01008475 a | |
| 2 | Colorectal | Cetuximab + FOLFIRI | NCT03688230 a | ||
| ATN-161 | 1/2 | Glioma | Carboplatin | NCT00352313 b | |
| Vitaxin | 2 | Prostate | Docetaxel | NCT00072930 b | |
| 1/2 | Renal | Bevacizumab | NCT00684996 a | ||
| 2 | Melanoma | Dacarbazine | NCT00066196 c | ||
| FAK | Defactinib | 1 | Lung | VS-6766 (RAF/MEK inhibitor) | NCT03875820 d |
| 1/1b | Ovarian | Paclitaxel | NCT01778803 b | ||
| 1/2 | Ovarian | Paclitaxel, carboplatin | NCT03287271 d | ||
| 1 | Pancreatic | Gemcitabine + pembrolizumab (immunotherapy) | NCT02546531 b | ||
| GSK2256098 | 2 | Pancreatic | Trametinib (MEK inhibitor) | NCT02428270 b | |
| 2 | Meningiomas | Vismodegib (Hedgehog inhibitor), capivasertib (Akt inhibitor), abemaciclib (CDK inhibitor) | NCT02523014 d | ||
| VS-4718 | 1 | Pancreatic | Nab-paclitaxel + gemcitabine | NCT02651727 e | |
| IN10018 | 1 | Gastric | Docetaxel | NCT05327231 d | |
| 1 | Melanoma | Cobimetinib (MEK inhibitor) | NCT04109456 d | ||
| β-catenin | PRI-724 | 2 | Colorectal | Bevacizumab + oxaliplatin + leucovorin + fluorouracil | NCT02413853 e |
| 1 | Pancreatic | Gemcitabine | NCT01764477 b | ||
| Tyrosine kinases | Dasatinib (>300 trials) | 1/2 | Lung | Erlotinib | NCT00826449 b |
| 2 | Pancreatic | Mfolfox6 | NCT01652976 a | ||
| FDA approved | Myeloid leukemia | – | – | ||
| CXC4R (GPCR) | TG-0054 (burixafor) | 2 | Multiple myeloma, lymphoma | G-CSF | NCT02104427 b |
| TME normalization | |||||
| Stromal components | S-3304 (MMP) | 1/2 | Lung | Chemo-irradiation | NCT00078390 b |
| PEGPH20 (HA) (discontinued) | 1/2 | Pancreatic | Gemcitabine/nab-paclitaxel + dexamethasone + gemcitabine + enoxaparin | NCT01453153c, NCT01839487c | |
| 3 | Pancreatic | Gemcitabine + nab-paclitaxel | NCT02715804 a | ||
| 1 | Lung, gastric | Pembrolizumab | NCT02563548 b | ||
| Not applicable | Pancreatic | Cetuximab | NCT02241187 b | ||
| Not applicable | Pancreatic | Gemcitabine + nab-paclitaxel + rivaroxaban | NCT02921022 b | ||
| 1/2 | Pancreatic | mFOLFIRINOX | NCT01959139 a | ||
| 1/2 | Gastric/gastroesophageal/esophageal | Leucovorin, cisplatin, paclitaxel, oxaliplatin, fluorouracil, atezolizumab, cobimetinib, ramucirumab, BL-8040, linagliptin, tiragolumab | NCT03281369 d | ||
| Simtuzumab (LOXL2) | 2 | Pancreatic | Gemcitabine | NCT01472198 a | |
| Vismodegib (GPCR inhibitor) | 2 | Breast | Paclitaxel + epirubicin | NCT02694224 b | |
| 2 | Colorectal | mFOLFOX, FOLFIRI, bevacizumab | NCT00636610 a | ||
| 2 | Pancreatic | Gemcitabine | NCT01195415a, NCT01064622a | ||
| 2 | Ovarian, colorectal, basal cell carcinoma | FOLFIRI, FOLFOX, bevacizumab | NCT00959647 b | ||
| 2 | Pancreatic | Gemcitabine, nab-paclitaxel | NCT01088815 | ||
| 1 | Pancreatic | Gemcitabine, erlotinib | NCT00878163 b | ||
| 2 | Gastric | Oxaliplatin, leucovorin, fluorouracil | NCT00982592 a | ||
| 2 | Solid tumors | Trastuzumab, pertuzumab, erlotinib, vemurafenib, cobimetinib, alectinib, atezolizumab | NCT02091141 b | ||
| FDA approved | Basal cell | – | – | ||
| 1/2 | Skin basal | Pembrolizumab | NCT02690948 a | ||
| 2 | Lung | Cisplatin, cixutumumab, etoposide | NCT00887159 a | ||
| 2 | Meningiomas | GSK2256098, capivasertib, abemaciclib | NCT02523014 b | ||
| Saridegib (GPCR inhibitor) | 1/2 | Pancreatic | Gemcitabine | NCT01130142 a | |
| 1 | Head and neck squamous cell carcinoma | Cetuximab | NCT01255800 b | ||
| 1 | Pancreatic | FOLFIRINOX | NCT01383538 b | ||
| Pirfenidone (TGFβ inhibitor, CAFs) | 1 | Lung | Carboplatin + paclitaxel + pemetrexed | NCT03177291 b | |
| 1/2 | Lung | Atezolizumab | NCT04467723 d | ||
| e.g., Losartan (Ang II inhibitor, TGFβ inhibitor, CAFs) | 2 | Pancreatic | Nivolumab + FOLFIRINOX | NCT03563248 d | |
| 2 | Pancreatic | FOLFIRINOX | NCT01821729 c | ||
| 2 | Breast | Doxorubicin + camrelizumab | NCT05097248 d | ||
| 1 | Pancreatic | Gemcitabine | NCT01276613 e | ||
| 2 | Pancreatic | mFOLFIRINOX/gemcitabine + nab-paclitaxel | NCT04539808 d | ||
| 2 | Pancreatic | FOLFIRINOX 1 + 9-ING-41 (NF-κB inhibitor) | NCT05077800 d | ||
| 1 | Osteosarcoma | Sunitinib | NCT03900793 d | ||
| e.g., Bosentan (endothelin I inhibitor) | 1 | Pancreatic | Gemcitabine + nab-paclitaxel | NCT04158635 d | |
No change compared with standard treatment.
No results posted.
Improved progression-free survival and overall survival.
Recruiting.
Terminated/withdrawn.
Approaches to target components of mechanical sensing and mechanotransduction
Integrin inhibitors have been investigated in Phase 1 and 2 clinical trials with promising results [62,63,100]. However, the survival benefits of chemotherapeutics combined with mechanical pathway-focused inhibitors compared with single-agent chemotherapy have been modest, with only a few exceptions improving outcomes (e.g., cilengitide, vitaxin, and dasatinib).
Targeting mechanosensors presents an alternative approach to exploit aberrant activation of mechanotransduction pathways and restore chemosensitivity. To this end, expression of the transient receptor potential (TRP) and Piezo ion channels correlates with poor patient survival, making these mechanosensors promising therapeutic targets [101,102]. In breast cancer cells, TRPM2 was linked with doxorubicin and tamoxifen resistance [103], while pharmacological and genetic inhibition of TRPC5-mediated autophagy was shown to increase the cytotoxic effect of adriamycin [104]. Furthermore, inhibitors that disrupt the ionic homeostasis of TRP channels via altering the Ca2+ and Na+ influx can induce apoptosis and inhibit cell proliferation and migration [105]. Additionally, the metabolite of ginseng saponin, 20-GPPD, stimulated Ca2+ influx through TRPC channel activation, which resulted in apoptosis and G1-phase arrest in colon cancer cells [106]. Cell cycle arrest has also been shown to be induced in gastric cancer cells via TRPC6 blockade and decreased Ca2+ influx [107]. Clinical trials against mechanosensitive ion channels are in early stages. For example, the TRPM8 activator D3263 is being tested as a single agent in a Phase 1 dose escalation study (NCT00839631). Targeting TRPV6 represents another approach to suppress Ca2+-mediated cancer proliferation and metastasis [108,109]. A Phase 1 clinical trial of the TRPV6 inhibitor (NCT03784677) was recently designed to investigate tolerable doses and safety of this inhibitor in patients with advanced solid tumors. A detailed review of approaches targeting TRP channels can be found elsewhere [105].
Inhibition of piezo ion channels represents another mechanosensor-targeted approach that is in early clinical development. Currently, only a few inhibitors of Piezo1 are available, with low specificity, including Gd3+, ruthenium red, and GsMTx-4 [110,111]. The G protein-coupled receptor (GPCR) family of mechanosensors could also serve as potential targets of mechanically induced chemoresistance. In mice bearing breast and lung tumors, pepducin P1pal-7, which targets the interaction of PAR1 with G proteins, was shown to block tumor progression and metastasis [112]. More recently, doxycycline was shown to selectively inhibit PAR1 in several cancer cell lines, including breast and melanoma cancer cells [113]. Furthermore, YM-254890 and FR900359 represent promising and specific inhibitors of GPCR signaling. In melanoma, YM-254890 induced tumor killing when combined with MEK inhibitors in vitro and in vivo [114]. GPCR-targeting drugs have entered clinical trial testing, such as the CXCR4 inhibitor TG-0054 [115,116].
Approaches to normalize the TME
Agents that inhibit matrix metalloproteinase (MMP)-driven ECM degradation have been tested in clinical trials to block metastatic progression. In preclinical studies, 4-methylumbelliferone (4-MU) and imatinib were shown to reduce HA accumulation in pancreatic and prostate cancers in vivo [117,118]. The subsequent reduced HA-mediated CD44 activation led to decreased PI3K, Akt, and ERK signaling, which impaired migration and invasion [117,118]. PEGylated human recombinant PH20 hyaluronidase (PEGPH20) is another therapy that has shown improved responses to gemcitabine and doxorubicin in in vivo pancreatic tumor models [119]. However, in a Phase 3 clinical trial, administration of PEGPH20 combined with nab-paclitaxel plus gemcitabine failed to improve survival outcomes [120].
Inhibiting collagen remodeling represents another ECM-targeted therapeutic strategy. Specifically, ECM remodeling involves collagen fiber crosslinking, which is mediated by the LOX family of enzymes [121]. Preclinically, inhibition of lysyl oxidases in murine breast cancer models improved doxorubicin responses by reducing tissue stiffness [122]. However, in the clinic, combination of the LOX2 antibody, simtuzumab, with gemcitabine did not improve therapeutic outcomes in patients with pancreatic cancer [123].
Targeting cancer-associated fibroblast (CAF) activation represents another strategy to alleviate CAF-mediated ECM remodeling and reduce tumor stiffness. For example, vismodegib (or saridegib) and pirfenidone, which target Hedgehog and TGFβ pathway activation in CAFs, respectively, have been shown to improve the efficacy of chemotherapeutics and immunotherapies in breast and pancreatic tumor models in vivo [124,125]. Vismodegib has already been approved by the FDA for the treatment of basal cell carcinoma; however, further investigation is needed for the treatment of other solid tumors characterized by high CAF density, including breast and pancreatic cancer. Additionally, CAF-targeted approaches involve repurposing of several FDA-approved drugs, including the antihypertensives losartan and bosentan, the corticosteroid dexamethasone, and the antihistamines tranilast and ketotifen [82,126–128].
Therapeutics that directly modulate the mechanical TME are also known as mechanotherapeutics [129]. It was shown that administering mechanotherapeutics, such as pirfenidone, losartan, tranilast, and dexamethasone, reduces tumor stiffness and mechanical forces, and increases tumor perfusion and drug delivery in preclinical tumor models of pancreatic and breast cancer [125,126,130–132]. Importantly, bosentan is being tested in a Phase 1 clinical trial in patients with pancreatic cancer (NCT04158635). Losartan has already been studied successfully in a Phase 2 clinical trial showing that, when used in combination with radiation and chemotherapy, it converted 60% of unresectable, locally advanced pancreatic ductal adenocarcinoma to a resectable status, thereby making it a ‘potentially curable’ treatment [133]. Given that only a subset of patients with PDAC respond to this therapy, further studies are needed to develop predictive biomarkers and elucidate mechanisms of treatment response.
Concluding remarks and future directions
Emerging evidence suggests that mechanical cues present in the TME affect the ability of chemotherapeutic treatments to induce cancer cell death. Recent studies have shown that mechanically induced chemoresistance is mediated through integrin or receptor tyrosine kinase (RTK) activation of prosurvival pathways, including PI3K/Akt, FAK/Src, ERK/MEK, or Wnt, which also regulate the expression of drug transporters. However, many critical questions remain (see Outstanding questions). First, the responses of cancer cells to mechanical cues may depend on the genetic alterations driving tumorigenesis, including activating mutations of the Ras, EGFR, or PI3K signaling pathways. Responses to targeted therapies against aberrant activation of these pathways may be impacted by crosstalk with mechanotransduction pathways. To this end, it would be critical to characterize directly in patient samples both cancer cells and the TME before therapy for genetic heterogeneity and mechanical alterations compared with normal tissue. Continuous monitoring of ECM stiffness, vessel function, and matrix deformation using an ultrasound imaging-based approach could provide further insights into mechanical cues acting on tumor cells [82,132]. Integrating this mechanical characterization with multi-omic profiling would enable us to dissect how TME mechanical abnormalities impact signaling responses and drug sensitivity. Importantly, a large-scale analysis of signaling pathways in surviving cancer cells and other cells present in TME, including immune cells, will lead to rational target identification for developing therapeutic approaches that target mechanoresistance mechanisms and personalized treatments.
Outstanding questions.
How do genetic alterations that drive oncogenic signaling impact transduction of mechanical cues in cancer cells?
Do mechanosensors crosstalk with different molecular complexes involved in chemotherapy mechanisms of action (e.g., disruption, transcription, or DNA replication)?
Is there overlap in the mechanically induced resistance mechanisms between chemo-, targeted, and immunotherapies?
Which mechanosensors and downstream signaling nodes should be targeted to restore sensitivity to cancer therapy when an association with mechanical cues has been established in the clinic?
Is there a therapeutic window for mechanosensitive pathways with limited on-target toxicity in normal cells compared with cancer cells?
What are the effects of mechanical cues on immune and stromal cell types in the TME with respect to promoting resistance to chemotherapy, targeted therapy, and immunotherapy?
Importantly, out of the clinical trials shown in Table 2, only a subset showed therapeutic benefits. Analysis of mechanosensor expression, such as Piezo1 or GPCRs, in multiple tissues, is critical for successful therapeutic development and strategies to enable tumor-specific delivery. Genetic studies in Piezo1-knockout mice demonstrated a lethal phenotype in utero with impaired vascular development, highlighting the importance of careful consideration when targeting mechanosensors. Similarly, targeting integrins in patients with cancer has been studied extensively, but most have not shown significant results. This could be because of the central role of integrins in normal physiology and the lack of a therapeutic window. With the emergence of more therapeutic strategies that target biophysical cues to reverse drug resistance, there is a need for experimental platforms to comprehensively explore mechanically induced drug resistance in model systems and clinically relevant samples. Such platforms could use 3D tumor cell- or patient-derived organoids embedded in a supportive matrix or exposed to mechanical forces (both compressive and tensile) within mechanical bioreactors. Cancer-on-a-chip models that allow for precise control of mechanical stimuli and fluid flow could also be used to study mechanoresistance mechanisms and test novel therapeutic strategies. Computational models using transport modeling and gene regulatory networks also present a powerful approach to study the interplay between forces, mechanotransduction, and drug responses. Finally, machine learning-based tools promise to integrate large-scale imaging and multi-omics data sets in the clinic to stratify patients based on mechanically informed signatures. Advances in these areas will lead to the identification of novel, highly targeted therapies that intercept mechanically induced drug resistance and enhance the efficacy of available treatments for patients with cancer.
Highlights.
Mechanical abnormalities present within the tumor microenvironment, including compressive forces and matrix stiffness, affect cancer cells at the molecular, cellular, and tissue level to fuel tumor growth and metastasis.
Chemotherapy, targeted therapy, and immunotherapy mechanisms of action can crosstalk with mechanotransduction signaling mechanisms that can shape drug sensitivity and resistance.
Restoring mechanical abnormalities in the tumor microenvironment and targeting mechanosensitive pathways provide opportunities for designing rational combination therapies in drug-resistant solid tumors.
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (2019-CoG-863955 and 2022-PoC1–101069207 to T.S.), the Department of Bioengineering at the University of Pittsburgh (to I.K.Z.), NCI (R00 CA222554, to I.K.Z.), and NIH (T32 EB001026 to M.D.P.).
Glossary
- ATP-binding cassette (ABC) family multidrug efflux pumps:
family of membrane proteins that utilize energy from ATP hydrolysis to pump substances, including drugs, out of cells or into intracellular compartments
- Cancer-associated fibroblasts (CAFs):
type of fibroblast present in the microenvironment of cancer tissues
- Cancer stem cell (CSC):
small fraction of tumor cells that have high tumorigenic potential due to their self-renewing and tumor-initiating properties (stem cell characteristics).
- Desmoplasia:
condition in which CAFs produce excessive amounts of extracellular proteins, which form a dense matrix network around and within the tumor
- ECM stiffness:
material property that refers to the extent to which a material can resist deformation or deflection in response to an applied force. A relatively stiff matrix requires greater forces to deform compared with a more compliant matrix
- Epithelial-to-mesenchymal transition (EMT):
biological process in which epithelial cells undergo a phenotypic change and acquire mesenchymal characteristics, including loss of cell–cell adhesion and polarity, and acquisition of migratory and invasive properties.
- Extracellular matrix (ECM):
mixture of extracellular components, including collagen, fibronectin, and HA.
- G protein-coupled receptor (GPCR):
large family of cell surface receptors that trigger the activation of intracellular signaling proteins, called G proteins, and initiate downstream signaling cascades
- Interstitial fluid pressure (IFP):
result of increased fluid leakage from the vascular to the interstitial space due to the hyperpermeability of tumor vessels (new blood vessels formed: angiogenesis or abnormal tumor vasculature). This fluid leakage reduces blood flow and impairs drug delivery to the tumor
- Mechanical forces:
mechanical stresses and pressures exerted within TME due to tumor growth and interaction of tumor cells with the surrounding tissue. In the tumor interior, these forces are compressive in all directions, while, at the tumor periphery, forces are compressive in the radial direction and tensile at the circumferential direction
- Mechanotransduction:
process of translating extracellular mechanical cues into downstream intracellular signals that regulate cellular programs, cell states, and phenotypes.
- Transient receptor potential (TRP):
family of transmembrane proteins that form ion channels, allowing the flow of ions across cell membranes in response to external stimuli. Upon activation, they can initiate downstream signaling cascades to regulate cellular responses
- Tumor microenvironment (TME):
complex mixture of cellular and noncellular components surrounding a tumor. Cellular components include cancer, stromal, endothelial, and immune cells. Noncellular components include the ECM
Footnotes
Declaration of interests
The authors declare that there are no conflicts of interest.
References
- 1.Mahoney L and Csima A (1982) Efficiency of palpation in clinical detection of breast cancer. Can. Med. Assoc. J 127, 729–730 [PMC free article] [PubMed] [Google Scholar]
- 2.Kalli M and Stylianopoulos T (2018) Defining the role of solid stress and matrix stiffness in cancer cell proliferation and metastasis. Front. Oncol 8, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stylianopoulos T (2017) The solid mechanics of cancer and strategies for improved therapy. J. Biomech. Eng 139, 021004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delarue M et al. (2014) Compressive stress inhibits proliferation in tumor spheroids through a volume limitation. Biophys. J 107, 1821–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desmaison A et al. (2013) Mechanical stress impairs mitosis progression in multi–cellular tumor spheroids. PLoS ONE 8, e80447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalli M et al. (2021) Mechanical stress signaling in pancreatic cancer cells triggers p38 MAPK- and JNK-dependent cytoskeleton remodeling and promotes cell migration via Rac1/cdc42/myosin II. Mol. Cancer Res 20, 485–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalli M et al. (2019) Solid stress-induced migration is mediated by GDF15 through Akt pathway activation in pancreatic cancer cells. Sci. Rep 9, 978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalli M et al. (2018) Solid stress facilitates fibroblasts activation to promote pancreatic cancer cell migration. Ann. Biomed. Eng 46, 657–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jain RK (2014) Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 26, 605–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin JD et al. (2020) Improving cancer immunotherapy using nanomedicines: progress, opportunities and challenges. Nat. Rev. Clin. Oncol 17, 251–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen DS and Mellman I (2017) Elements of cancer immunity and the cancer–immune set point. Nature 541, 321–330 [DOI] [PubMed] [Google Scholar]
- 12.Jain RK (2005) Antiangiogenic therapy for cancer: current and emerging concepts. Oncology 19, 7–16 [PubMed] [Google Scholar]
- 13.Carmeliet P and Jain RK (2000) Angiogenesis in cancer and other diseases. Nature 407, 249–257 [DOI] [PubMed] [Google Scholar]
- 14.Tilsed CM et al. (2022) Cancer chemotherapy: insights into cellular and tumor microenvironmental mechanisms of action. Front. Oncol 12, 960317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng HC (2017) The molecular mechanisms of chemoresistance in cancers. Oncotarget 8, 59950–59964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Basourakos SP et al. (2017) Combination platinum-based and DNA damage response-targeting cancer therapy: evolution and future directions. Curr. Med. Chem 24, 1586–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nitiss JL (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 9, 338–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mosca L et al. (2021) Taxanes in cancer treatment: activity, chemoresistance and its overcoming. Drug Resist. Updat 54, 100742. [DOI] [PubMed] [Google Scholar]
- 19.Longley DB et al. (2003) 5-Fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 3, 330–338 [DOI] [PubMed] [Google Scholar]
- 20.Plunkett W et al. (1995) Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin. Oncol 22, 3–10 [PubMed] [Google Scholar]
- 21.Wu Z et al. (2020) Copy number amplification of DNA damage repair pathways potentiates therapeutic resistance in cancer. Theranostics 10, 3939–3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Binkhathlan Z and Lavasanifar A (2013) P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr. Cancer Drug Targets 13, 326–346 [DOI] [PubMed] [Google Scholar]
- 23.Gillet JP et al. (2012) Multidrug resistance-linked gene signature predicts overall survival of patients with primary ovarian serous carcinoma. Clin. Cancer Res 18, 3197–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan S et al. (2016) Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol 43, 723–737 [DOI] [PubMed] [Google Scholar]
- 25.Johnstone RW et al. (2002) Apoptosis: a link between cancer genetics and chemotherapy. Cell 108, 153–164 [DOI] [PubMed] [Google Scholar]
- 26.Hayakawa J et al. (2000) Inhibition of BAD phosphorylation either at serine 112 via extracellular signal-regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res 60, 5988–5994 [PubMed] [Google Scholar]
- 27.Yu H et al. (2008) Phosphoinositide 3-kinase/Akt pathway plays an important role in chemoresistance of gastric cancer cells against etoposide and doxorubicin induced cell death. Int. J. Cancer 122, 433–443 [DOI] [PubMed] [Google Scholar]
- 28.Page C et al. (2000) Overexpression of Akt/AKT can modulate chemotherapy-induced apoptosis. Anticancer Res 20, 407–416 [PubMed] [Google Scholar]
- 29.Stover EH et al. (2019) Pooled genomic screens identify anti-apoptotic genes as targetable mediators of chemotherapy resistance in ovarian cancer. Mol. Cancer Res 17, 2281–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donaldson KL et al. (1994) Cytotoxicity of the anticancer agents cisplatin and taxol during cell proliferation and the cell cycle. Int. J. Cancer 57, 847–855 [DOI] [PubMed] [Google Scholar]
- 31.Gascoigne KE and Taylor SS (2008) Cancer cells display profound intra-and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14, 111–122 [DOI] [PubMed] [Google Scholar]
- 32.Filipits M et al. (2007) Cell cycle regulators and outcome of adjuvant cisplatin-based chemotherapy in completely resected non-small-cell lung cancer: the International Adjuvant Lung Cancer Trial Biologic Program. J. Clin. Oncol 25, 2735–2740 [DOI] [PubMed] [Google Scholar]
- 33.Ahmed N et al. (2010) Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer. Curr. Cancer Drug Targets 10, 268–278 [DOI] [PubMed] [Google Scholar]
- 34.Visvader JE and Lindeman GF (2008) Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer 8, 755–768 [DOI] [PubMed] [Google Scholar]
- 35.Darvishi B et al. (2022) Matrix stiffening and acquired resistance to chemotherapy: concepts and clinical significance. Br. J. Cancer 126, 1253–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meads MB et al. (2009) Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat. Rev. Cancer 9, 665–674 [DOI] [PubMed] [Google Scholar]
- 37.Xing H et al. (2008) Activation of fibronectin/PI-3K/Akt2 leads to chemoresistance to docetaxel by regulating survivin protein expression in ovarian and breast cancer cells. Cancer Lett 261, 108–119 [DOI] [PubMed] [Google Scholar]
- 38.Hazlehurst LA et al. (2000) Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene 19, 4319–4327 [DOI] [PubMed] [Google Scholar]
- 39.Lwin T et al. (2007) Cell adhesion induces p27Kip1-associated cell-cycle arrest through down-regulation of the SCFSkp2 ubiquitin ligase pathway in mantle-cell and other non-Hodgkin B-cell lymphomas. Blood 110, 1631–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aoudjit F and Vuori K (2001) Integrin signaling inhibits paclitaxel-induced apoptosis in breast cancer cells. Oncogene 20, 4995–5004 [DOI] [PubMed] [Google Scholar]
- 41.Dangi-Garimella S et al. (2013) Three-dimensional collagen I promotes gemcitabine resistance in vitro in pancreatic cancer cells through HMGA2-dependent histone acetyltransferase expression. PLoS ONE 8, e64566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng M et al. (2020) Extracellular matrix stiffness determines DNA repair efficiency and cellular sensitivity to genotoxic agents. Sci. Adv 6, eabb2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Z et al. (2021) Extracellular matrix characterization in gastric cancer helps to predict prognosis and chemotherapy response. Front. Oncol 11, 753330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fatherree JP et al. (2022) Chemotherapy-induced collagen IV drives cancer cell motility through activation of Src and focal adhesion kinase. Cancer Res 82, 2031–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J et al. (2020) A novel model incorporating tumor stiffness, blood flow characteristics, and Ki-67 expression to predict responses after neoadjuvant chemotherapy in breast cancer. Front. Oncol 10, 603574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baltes F et al. (2020) β1-Integrin binding to collagen type 1 transmits breast cancer cells into chemoresistance by activating ABC efflux transporters. Biochim. Biophys. Acta Mol. Cell Res 1867, 118663. [DOI] [PubMed] [Google Scholar]
- 47.Qin X et al. (2020) Matrix stiffness modulates ILK-mediated YAP activation to control the drug resistance of breast cancer cells. Biochim. Biophys. Acta Mol. Basis Dis 1866, 165625. [DOI] [PubMed] [Google Scholar]
- 48.Nath S et al. (2020) Flow-induced shear stress confers resistance to carboplatin in an adherent three-dimensional model for ovarian cancer: a role for EGFR-targeted photoimmunotherapy informed by physical stress. J. Clin. Med 9, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saatci O et al. (2020) Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nat. Commun 11, 2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hayashi M et al. (2012) Evaluation of tumor stiffness by elastography is predictive for pathologic complete response to neoadjuvant chemotherapy in patients with breast cancer. Ann. Surg. Oncol 19, 3042–3049 [DOI] [PubMed] [Google Scholar]
- 51.Longati P et al. (2013) 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 13, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu C et al. (2015) Role of three-dimensional matrix stiffness in regulating the chemoresistance of hepatocellular carcinoma cells. Biotechnol. Appl. Biochem 62, 556–562 [DOI] [PubMed] [Google Scholar]
- 53.Park J and Schwarzbauer JE (2014) Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene 33, 1649–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koenig A et al. (2006) Collagen type I induces disruption of E-cadherin–mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res 66, 4662–4671 [DOI] [PubMed] [Google Scholar]
- 55.Liu M et al. (2018) Collagen-based three-dimensional culture microenvironment promotes epithelial to mesenchymal transition and drug resistance of human ovarian cancer in vitro. RSC Adv 8, 8910–8919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rice AJ et al. (2017) Matrix stiffness induces epithelial–mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 6, e352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Joyce MH et al. (2018) Phenotypic basis for matrix stiffness-dependent chemoresistance of breast cancer cells to doxorubicin. Front. Oncol 8, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chikamatsu K et al. (2013) Alteration of cancer stem cell-like phenotype by histone deacetylase inhibitors in squamous cell carcinoma of the head and neck. Cancer Sci 104, 1468–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katsuno Y et al. (2019) Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci. Signal 12, eaau8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brown JA et al. (2017) TGF-β-Induced quiescence mediates chemoresistance of tumor-propagating cells in squamous cell carcinoma. Cell Stem Cell 21, 650–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Donnell JS et al. (2018) PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol 48, 91–103 [DOI] [PubMed] [Google Scholar]
- 62.Bell-McGuinn KM et al. (2011) A phase II, single-arm study of the anti-α5β1 integrin antibody volociximab as monotherapy in patients with platinum-resistant advanced epithelial ovarian or primary peritoneal cancer. Gynecol. Oncol 121, 273–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haddad T et al. (2017) A phase I study of cilengitide and paclitaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol 79, 1221–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schober M and Fuchs E (2011) Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-β and integrin/focal adhesion kinase (FAK) signaling. Proc. Natl. Acad. Sci. U. S. A 108, 10544–10549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shea MP et al. (2018) High collagen density augments mTOR-dependent cancer stem cells in ERα+ mammary carcinomas, and increases mTOR-independent lung metastases. Cancer Lett 433, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dominijanni A et al. (2020) Manipulating the tumor microenvironment in tumor organoids induces phenotypic changes and chemoresistance. iScience 23, 101851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Okuda H et al. (2012) Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res 72, 537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chanmee T et al. (2014) Excessive hyaluronan production promotes acquisition of cancer stem cell signatures through the coordinated regulation of twist and the transforming growth factor β (TGF-β)-Snail signaling axis. J. Biol. Chem 289, 26038–26056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bourguignon LY et al. (2012) Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. J. Biol. Chem 287, 32800–32824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pedron S et al. (2019) Hyaluronic acid-functionalized gelatin hydrogels reveal extracellular matrix signals temper the efficacy of erlotinib against patient-derived glioblastoma specimens. Biomaterials 219, 119371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Y et al. (2019) Stromal extracellular matrix is a microenvironmental cue promoting resistance to EGFR tyrosine kinase inhibitors in lung cancer cells. Int. J. Biochem. Cell Biol 106, 96–106 [DOI] [PubMed] [Google Scholar]
- 72.Shen Y et al. (2020) Reduction of liver metastasis stiffness improves response to bevacizumab in metastatic colorectal cancer. Cancer Cell 37, 800–817 [DOI] [PubMed] [Google Scholar]
- 73.Nguyen TV et al. (2014) Sorafenib resistance and JNK signaling in carcinoma during extracellular matrix stiffening. Biomaterials 35, 5749–5759 [DOI] [PubMed] [Google Scholar]
- 74.Lin C et al. (2015) Microenvironment rigidity modulates responses to the HER2 receptor tyrosine kinase inhibitor lapatinib via YAP and TAZ transcription factors. Mol. Biol. Cell 26, 3946–3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Long JE et al. (2019) Therapeutic resistance and susceptibility is shaped by cooperative multi-compartment tumor adaptation. Cell Death Differ 26, 2416–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Salmon H et al. (2012) Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Invest 122, 899–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hartmann N et al. (2014) Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin. Cancer Res 20, 3422–3433 [DOI] [PubMed] [Google Scholar]
- 78.Moreau J et al. (2017) The emerging role of ECM crosslinking in T cell mobility as a hallmark of immunosenescence in humans. Ageing Res. Rev 35, 322–335 [DOI] [PubMed] [Google Scholar]
- 79.Miyazawa A et al. (2018) Regulation of PD-L1 expression by matrix stiffness in lung cancer cells. Biochem. Biophys. Res. Commun 495, 2344–2349 [DOI] [PubMed] [Google Scholar]
- 80.Zhang T et al. (2022) Targeting the tumor biophysical microenvironment to reduce resistance to immunotherapy. Adv. Drug Deliv. Rev 186, 114319. [DOI] [PubMed] [Google Scholar]
- 81.Nicolas-Boluda A et al. (2021) Tumor stiffening reversion through collagen crosslinking inhibition improves T cell migration and anti-PD-1 treatment. eLife 10, e58688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Panagi M et al. (2022) Polymeric micelles effectively reprogram the tumor microenvironment to potentiate nanoimmunotherapy in mouse breast cancer models. Nat. Commun 13, 7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jain RK et al. (2014) The role of mechanical forces in tumor growth and therapy. Annu. Rev. Biomed. Eng 16, 321–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Spill F et al. (2016) Impact of the physical microenvironment on tumor progression and metastasis. Curr. Opin. Biotechnol 40, 41–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jain RK and Stylianopoulos T (2010) Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol 7, 653–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stylianopoulos T et al. (2018) Reengineering the physical microenvironment of tumors to improve drug delivery and efficacy: from mathematical modeling to bench to bedside. Trends Cancer 4, 292–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Milosevic M et al. (2001) Interstitial fluid pressure predicts survival in patients with cervix cancer independent of clinical prognostic factors and tumor oxygen measurements. Cancer Res 61, 6400–6405 [PubMed] [Google Scholar]
- 88.Shang M et al. (2021) Microfluidic studies of hydrostatic pressure-enhanced doxorubicin resistance in human breast cancer cells. Lab Chip 21, 746–754 [DOI] [PubMed] [Google Scholar]
- 89.Ip CK et al. (2016) Stemness and chemoresistance in epithelial ovarian carcinoma cells under shear stress. Sci. Rep 6, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Novak CM et al. (2019) Fluid shear stress stimulates breast cancer cells to display invasive and chemoresistant phenotypes while upregulating PLAU in a 3D bioreactor. Biotechnol. Bioeng 116, 3084–3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Azimi T et al. (2020) Cancer cells grown in 3D under fluid flow exhibit an aggressive phenotype and reduced responsiveness to the anti-cancer treatment doxorubicin. Sci. Rep 10, 12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huo Y et al. (2022) Subpathway analysis of transcriptome profiles reveals new molecular mechanisms of acquired chemotherapy resistance in breast cancer. Cancers (Basel) 14, 4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Santoro M et al. (2015) Flow perfusion effects on three-dimensional culture and drug sensitivity of Ewing sarcoma. Proc. Natl. Acad. Sci. U. S. A 112, 10304–10309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang L and Yu D (2019) Exosomes in cancer development, metastasis, and immunity. Biochim. Biophys. Acta Rev. Cancer 1871, 455–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cheng G et al. (2009) Micro-environmental mechanical stress controls tumor spheroid size and morphology by suppressing proliferation and inducing apoptosis in cancer cells. PLoS ONE 44, e4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tse JM et al. (2012) Mechanical compression drives cancer cells toward invasive phenotype. Proc. Natl. Acad. Sci. U. S. A 109, 911–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chaudhuri PK et al. (2018) Mechanobiology of tumor growth. Chem. Rev 118, 6499–6515 [DOI] [PubMed] [Google Scholar]
- 98.Rizzuti IF et al. (2020) Mechanical control of cell proliferation increases resistance to chemotherapeutic agents. Phys. Rev. Lett 125, 128103. [DOI] [PubMed] [Google Scholar]
- 99.Novak CM et al. (2020) Compressive stimulation enhances ovarian cancer proliferation, invasion, chemoresistance, and mechanotransduction via CDC42 in a 3D bioreactor. Cancers (Basel) 12, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McNeel DG et al. (2005) Phase I trial of a monoclonal antibody specific for αvβ3 integrin (MEDI-522) in patients with advanced malignancies, including an assessment of effect on tumor perfusion. Clin. Cancer Res 11, 7851–7860 [DOI] [PubMed] [Google Scholar]
- 101.Cui C et al. (2017) Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 7, 3–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kischel P et al. (2019) Ion channels: new actors playing in chemotherapeutic resistance. Cancers 11, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Koh D et al. (2015) Enhanced cytotoxicity in triple-negative and estrogen receptor-positive breast adenocarcinoma cells due to inhibition of the transient receptor potential melastatin-2 channel. Oncol. Rep 34, 1589–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang P et al. (2017) TRPC5-induced autophagy promotes drug resistance in breast carcinoma via CaMKKβ/AMPKα/mTOR pathway. Sci. Rep 7, 3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gautier M et al. (2014) New insights into pharmacological tools to TR(i)P cancer up. Br. J. Pharmacol 171, 2582–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hwang JA et al. (2013) 20-O-β-d-glucopyranosyl-20(S)-protopanaxadiol, a metabolite of ginseng, inhibits colon cancer growth by targeting TRPC channel-mediated calcium influx. J. Nutr. Biochem 24, 1096–1104 [DOI] [PubMed] [Google Scholar]
- 107.Cai R et al. (2009) Blockade of TRPC6 channels induced G2/M phase arrest and suppressed growth in human gastric cancer cells. Int. J. Cancer 125, 2281–2287 [DOI] [PubMed] [Google Scholar]
- 108.Kim DY et al. (2021) RNA interference mediated suppression of TRPV6 inhibits the progression of prostate cancer in vitro by modulating cathepsin B and MMP9 expression. Investig. Clin. Urol 62, 447–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Song H et al. (2018) Expression and prognostic significance of TRPV6 in the development and progression of pancreatic cancer. Oncol. Rep 39, 1432–1440 [DOI] [PubMed] [Google Scholar]
- 110.Bowman CL et al. (2007) Mechanosensitive ion channels and the peptide inhibitor GsMTx-4: history, properties, mechanisms and pharmacology. Toxicon 49, 249–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Volkers L et al. (2015) Piezo channels: from structure to function. Pflugers Arch 467, 95–99 [DOI] [PubMed] [Google Scholar]
- 112.Cisowski J et al. (2011) Targeting protease-activated receptor-1 with cell-penetrating pepducins in lung cancer. Am. J. Pathol 179, 513–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhong W et al. (2017) Doxycycline directly targets PAR1 to suppress tumor progression. Oncotarget 8, 16829–16842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hitchman TD et al. (2021) Combined inhibition of Gαq and MEK enhances therapeutic efficacy in uveal melanoma. Clin. Cancer Res 27, 1476–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tsou LK et al. (2020) Comparative study between deep learning and QSAR classifications for TNBC inhibitors and novel GPCR agonist discovery. Sci. Rep 10, 16771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hauser AS et al. (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov 16, 829–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Skandalis SS et al. (2019) Hyaluronan–CD44 axis orchestrates cancer stem cell functions. Cell. Signal 63, 109377. [DOI] [PubMed] [Google Scholar]
- 118.Lompardía SL et al. (2017) 4-methylumbelliferone and imatinib combination enhances senescence induction in chronic myeloid leukemia cell lines. Investig. New Drugs 35, 1–10 [DOI] [PubMed] [Google Scholar]
- 119.Jacobetz MA et al. (2013) Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 62, 112–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Van Cutsem E et al. (2020) Randomized phase III trial of pegvorhyaluronidase alfa with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J. Clin. Oncol 38, 3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Siegel RC (1974) Biosynthesis of collagen crosslinks: increased activity of purified lysyl oxidase with reconstituted collagen fibrils. Proc. Natl. Acad. Sci. U. S. A 71, 4826–4830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rossow L et al. (2018) LOX-catalyzed collagen stabilization is a proximal cause for intrinsic resistance to chemotherapy. Oncogene 37, 4921–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Benson III AB et al. (2017) A phase II randomized, double-blind, placebo-controlled study of simtuzumab or placebo in combination with gemcitabine for the first-line treatment of pancreatic adenocarcinoma. Oncologist 22, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mpekris F et al. (2017) Sonic-hedgehog pathway inhibition normalizes desmoplastic tumor microenvironment to improve chemo-and nanotherapy. J. Control. Release 261, 105–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Polydorou C et al. (2017) Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget 8, 24506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Panagi M et al. (2020) TGF-beta inhibition combined with cytotoxic nanomedicine normalizes triple negative breast cancer microenvironment towards anti-tumor immunity. Theranostics 10, 1910–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chauhan VP and Jain RK (2013) Strategies for advancing cancer nanomedicine. Nat. Mater 12, 958–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mpekris F et al. (2021) Normalizing the microenvironment overcomes vessel compression and resistance to nanoimmunotherapy in breast cancer lung metastasis. Adv. Sci. (Weinh) 8, 2001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sheridan C (2019) Pancreatic cancer provides testbed for first mechanotherapeutics. Nat. Biotechnol 37, 829–831 [DOI] [PubMed] [Google Scholar]
- 130.Chauhan VP et al. (2013) Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun 4, 2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Martin JD et al. (2019) Dexamethasone increases cisplatin-loaded nanocarrier delivery and efficacy in metastatic breast cancer by normalizing the tumor microenvironment. ACS Nano 13, 6396–6408 [DOI] [PubMed] [Google Scholar]
- 132.Voutouri C et al. (2021) Endothelin inhibition potentiates cancer immunotherapy revealing mechanical biomarkers predictive of response. Adv. Ther 4, 2000289 [Google Scholar]
- 133.Murphy JE et al. (2019) Total neoadjuvant therapy with FOLFIRINOX in combination with losartan followed by chemoradiotherapy for locally advanced pancreatic cancer: a phase 2 clinical trial. JAMA Oncol 5, 1020–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kechagia JZ et al. (2019) Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol 20, 457–473 [DOI] [PubMed] [Google Scholar]
- 135.Wozniak MA et al. (2004) Focal adhesion regulation of cell behavior. Biochim. Biophys. Acta 1692, 103–119 [DOI] [PubMed] [Google Scholar]
- 136.Ivaska J and Heino J (2011) Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu. Rev. Cell Dev. Biol 27, 291–320 [DOI] [PubMed] [Google Scholar]
- 137.Petho Z et al. (2019) Mechanosensitive ion channels push cancer progression. Cell Calcium 80, 79–90 [DOI] [PubMed] [Google Scholar]
- 138.Li C et al. (2015) Piezo1 forms mechanosensitive ion channels in the human MCF-7 breast cancer cell line. Sci. Rep 5, 8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Heifetz A et al. (2015) GPCR structure, function, drug discovery and crystallography: report from Academia-Industry International Conference (UK Royal Society) Chicheley Hall, 1–2 September 2014. Naunyn Schmiedeberg’s Arch. Pharmacol 388, 883–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Insel PA et al. (2018) GPCRomics: GPCR expression in cancer cells and tumors identifies new, potential biomarkers and therapeutic targets. Front. Pharmacol 9, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang X et al. (2020) Unraveling the mechanobiology of immune cells. Curr. Opin. Biotechnol 66, 236–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Northcott JM et al. (2018) Feeling stress: the mechanics of cancer progression and aggression. Front. Cell Dev. Biol 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Labrie M et al. (2022) Therapy resistance: opportunities created by adaptive responses to targeted therapies in cancer. Nat. Rev. Cancer 22, 323–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Correia AL and Bissell MJ (2012) The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updat 15, 39–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dangi-Garimella S et al. (2011) Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP-mediated expression of HMGA2. Cancer Res 71, 1019–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ozkan A et al. (2021) Tumor microenvironment alters chemoresistance of hepatocellular carcinoma through CYP3A4 metabolic activity. Front. Oncol 11, 1956. [DOI] [PMC free article] [PubMed] [Google Scholar]