

Abstract
Mimics of protein secondary and tertiary structure offer rationally-designed inhibitors of biomolecular interactions. β-Sheet mimics have a storied history in bioorganic chemistry and are typically designed with synthetic or natural turn segments. We hypothesized that replacement of terminal inter-β-strand hydrogen bonds with hydrogen bond surrogates (HBS) may lead to conformationally-defined macrocyclic β-sheets without the requirement for natural or synthetic β-turns, thereby providing a minimal mimic of protein β-sheets. To access turn-less antiparallel β-sheet mimics, we developed a facile solid phase synthesis protocol. We surveyed a dataset of protein β-sheets for naturally observed interstrand side chain interactions. This bioinformatics survey highlighted an over-abundance of aromatic–aromatic, cation-π and ionic interactions in β-sheets. In correspondence with natural β-sheets, we find that minimal HBS mimics show robust β-sheet formation when specific amino acid residue pairings are incorporated. In isolated β-sheets, aromatic interactions endow superior conformational stability over ionic or cation-π interactions. Circular dichroism and NMR spectroscopies, along with high-resolution X-ray crystallography, support our design principles.
Keywords: β-sheets, protein mimics, protein-protein-interactions, macrocycles, constrained peptides
Grapical Abstract
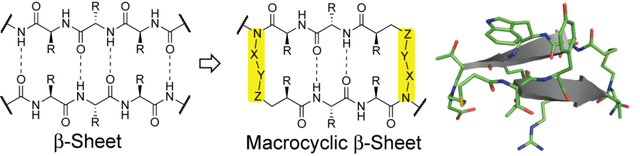
This manuscript describes synthesis of turn-less β-sheet mimics. Synthetic β-turns are important components of β-sheet mimics; however, the impact of the designed β-turns on β-sheet stabilization is often dependent on the strand sequence. We show that conformationally-defined macrocyclic β-sheet mimics can be accessed by replacing interstrand hydrogen bonds with covalent bonds.
Protein-protein interactions (PPIs) are hubs for cellular function, and misregulation of these interactions is implicated in diseases.[1] Several strategies have been employed to mimic protein epitopes to develop rationally designed inhibitors of PPIs as leads for drug discovery.[2] Classical approaches to controlling conformational stability of synthetic β-sheets/β-hairpins have yielded elegant examples of designed β-turns, covalent cross-strand interactions, and β-strand stabilizing auxiliaries.[3] β-turn mimics have built upon the turn inducing property of prolines or acyclic chains constrained by torsional strain; examples include D-Pro-L-Pro,[4] D-Pro-Gly,[3c] methylornithine,[3f] and dibenzofuran (Figure 1A).[5] These pioneering approaches for β-sheet stabilization provide a reproducible framework for rigidifying peptide conformation. However, the impact of β-turn units on β-sheet stabilization is often dependent on the strand sequence.[6] Given the potential biomedical importance of β-sheet mimics, several efforts to develop context-independent turn units are underway.[3f] Here we sought to determine if β-turn sequences, which are an intimate part of natural and synthetic β-sheets, can be entirely removed and replaced with a hydrogen bond surrogate (HBS) (Figure 1B).[7] We have extensively evaluated the potential of the hydrogen bond surrogate to stabilize α-helical domains. These previous efforts resulted in olefin, thioether and disulfide linkages that can mimic the ii+4 main-chain hydrogen bonds in helical sequences.[7d, 8] Judicious replacement of chosen hydrogen bonds with covalent bonds results in nucleation of the desired peptide conformation.[9]
Figure 1.
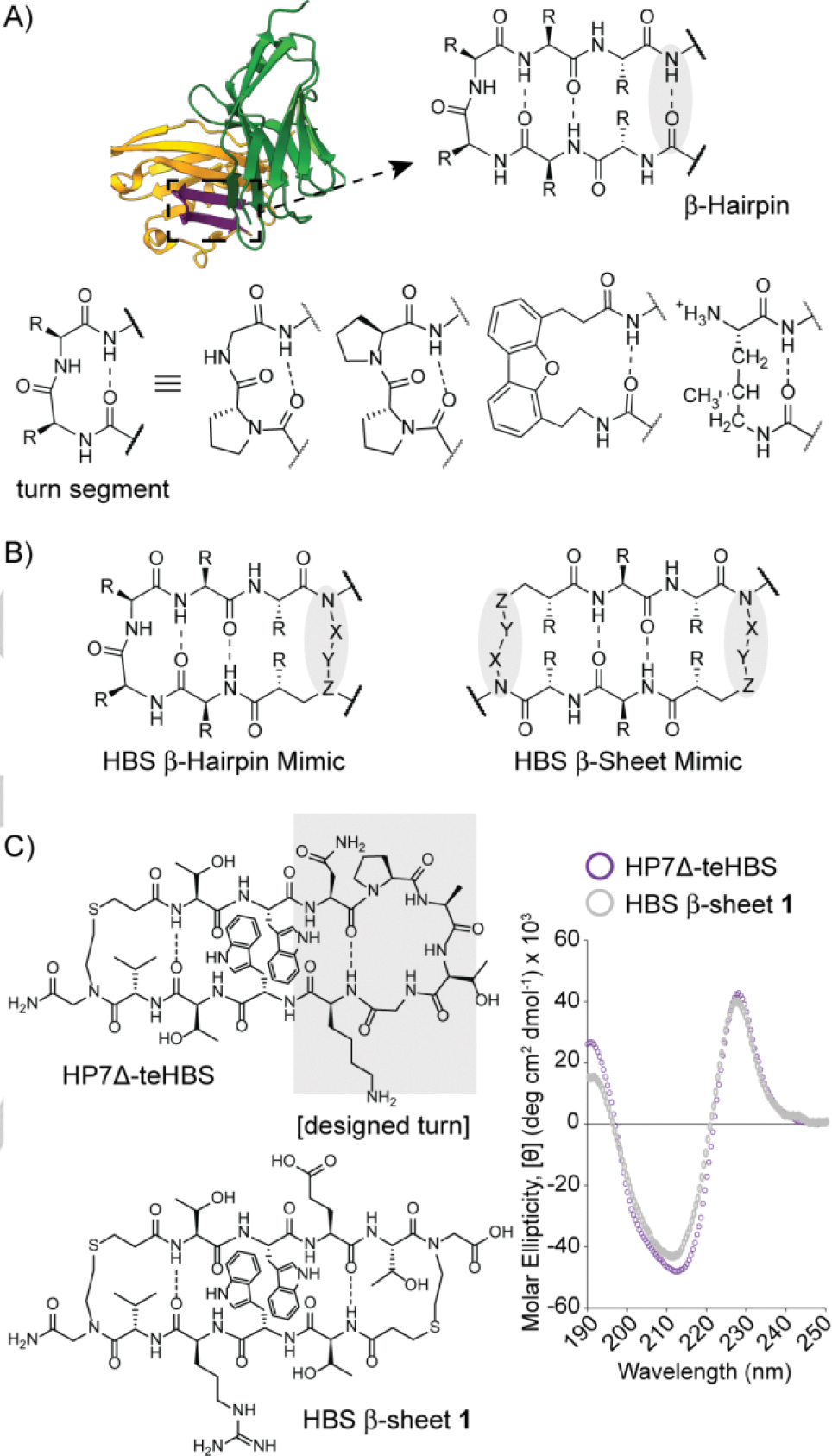
Stabilization of β-hairpins. (A) The turn segment of hairpins has been mimicked by β-turn analogs. Examples of D-Pro-L-Pro, D-Pro-Gly, dibenzofuran, and methylornithine are shown. (B) Replacement of an interstrand hydrogen bond with a covalent bond affords synthetic mimics of protein β-sheets. In this work, we outline synthesis and conformational stability of hydrogen bond surrogate (HBS) stabilized β-sheet derivatives. (C) The conformational stability of a constrained β-sheet (HBS β-sheet 1) is compared with a macrocyclic β-Hairpin (HP7Δ-teHBS), which features a six-residue turn-inducing segment. Circular dichroism spectra were obtained at a concentration of 30 μM peptide in 10 mM potassium fluoride (pH 7.3).
Our goal to create an HBS scaffold that promotes stable β-sheet conformations in short peptide sequences began with the evaluation of the strategy to fold sequences into stable β-hairpin conformations (Figure 1C).[7a, 7b] In these studies, we began with a well-studied β-hairpin model system that features cross-strand aromatic interactions and an optimized hexamer loop (NPATGK).[10] We showed that replacement of one intrastrand hydrogen bond with the HBS surrogate yielded a constrained β-hairpin (HP7Δ-teHBS) with higher conformational and proteolytic stability than the parent β-hairpin. Encouraged by this success, we reasoned that it might be possible to develop macrocyclic β-sheets in which the stabilizing loop is removed in favor of another hydrogen bond surrogate (Figure 1B). The present study describes design, synthesis, and conformational analysis of these minimal antiparallel β-sheet motifs.
We began our exploration by creating a mimic of HP7Δ-teHBS without the hexapeptide loop. HBS β-sheet 1 design contains two HBS constraints to join tetrapeptide strands. These strands consist of a cross-strand tryptophan pair, β-branched residues to enhance strand stability, and charged residues to enhance water solubility. We compared the conformation adopted by double HBS linked strands, HBS β-sheet 1, to the parent β-hairpin HP7Δ-teHBS by circular dichroism (CD) spectroscopy. The minimal macrocyclic β-sheet displays a CD signal that is nearly identical to that of the β-hairpin with the optimized turn segment, suggesting a high degree of conformational ordering. Characteristic features of the β-hairpin and β-sheet mimics include a maximum at 228 nm resulting from exciton coupling between interacting cross-strand Trp residues arranged in an edge-to-face orientation,[11] a minimum at 210–215 nm consistent with β-sheet formation, and a local maximum at 190 nm. The conformational stability of HBS β-sheet 1 is superior to its uncyclized counterpart 1-Linear (Supporting Information), which supports the hypothesis that the double HBS constraint makes an important contribution to rigidify the scaffold.[7a] The correlation between HP7Δ-teHBS and 1 is significant because it is rare for short β-sheet sequences to provide a robust CD signal – importantly the results suggest that the six-residue turn may be jettisoned for a minimal linker.
Encouraged by these preliminary findings with the macrocyclic β-sheet, we sought to develop a method for synthesis of macrocyclic HBS β-sheets. HBS β-sheet 1 contains two thioether bridges. In previous efforts, we have shown that the thioether and olefin-based hydrogen bond surrogates offer roughly equivalent conformational stability to artificial α-helices and β-hairpins.[7a, 8b, 12] We have previously attempted to synthesize macrocyclic HBS β-sheets using the olefin surrogate but these efforts were not successful – a challenge with incorporation of two olefin hydrogen bond surrogates is that the metal catalyst reacted indiscriminately with internal and terminal alkenes. We were able to address this synthetic challenge with thioether hydrogen bond surrogates. We focused on developing a solid-phase methodology for macrocyclic β-sheets that could be readily transferrable to various screening platforms (Figure 2). We tested two routes consisting of thiol alkylation and photo-induced conjugate addition for solid phase synthesis of the thioether linkage (Supporting Information, Figure S1). Both routes provided efficient access to the thioether bridge, but we proceeded with the alkylation method due to its relative ease of setup. The optimized methodology uses an efficient ring closing alkylation reaction and standard solid phase peptide synthesis procedures (Figure 2).
Figure 2.
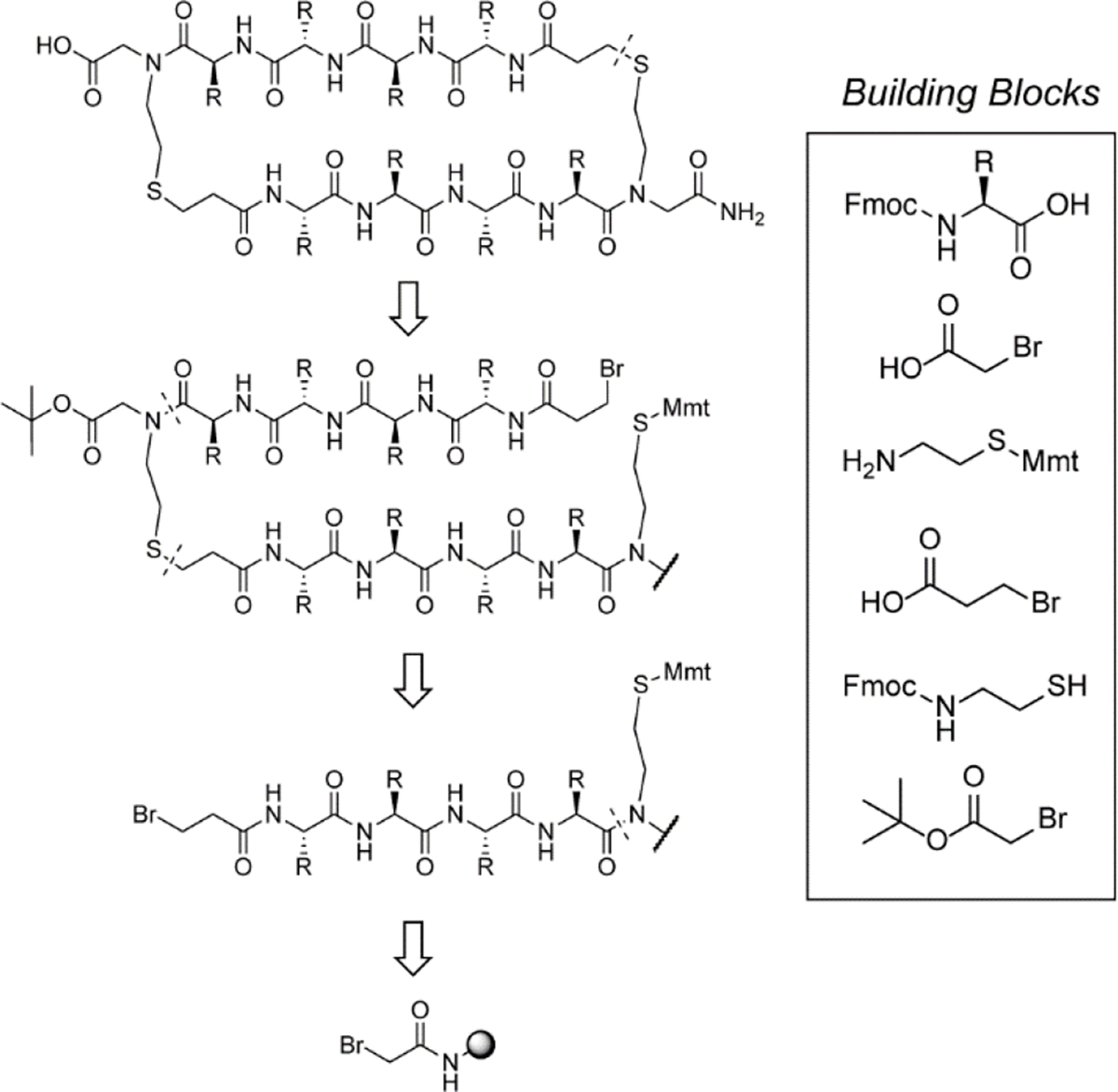
Retrosynthetic analysis of macrocyclic β-sheets. The HBS β-sheets can be readily assembled from simple building blocks on solid phase.
Access to a robust synthetic methodology for macrocyclic β-sheets enabled exploration of the HBS β-sheet scaffold to determine the residue requirement for a stable conformation. The strand sequence in 1 is designed to incorporate a cross-strand tryptophan pair. We conjectured that in the absence of strong turn-inducing units, the macrocycle may require inter-strand interactions for β-sheet stability. This question led us to determine the prevalence of cross-strand interactions in antiparallel β-sheets found within proteins. Analysis of side chain interactions in β-sheets has been previously described; however, these analyses were performed on a older versions of the Protein Data Bank with limited entries.[13] We interrogated the current PDB to understand the prevalence of natural amino acid pairs at non-hydrogen bonded sites.[13c]
Our bioinformatic analysis of antiparallel β-sheets[14] in the PDB shows that cross-strand aromatic, salt-bridge and cation-π interactions are prevalent, in keeping with the earlier studies on β-sheets and proteins overall.[15] Extraction of cross-strand interacting pairs at non-hydrogen bonded sites and normalization for natural occurrence of each residue is plotted as a heat map in Figure 3 (raw and normalized data are included in Supporting Information, Figure S2).[16] We found that tryptophan pairs are over-represented as much as ionic interactions on a normalized basis. Tryptophan pairing has a rich history in β-hairpin/β-sheet design[11a] and aromatic cross-strand interactions have been extensively studied in β-sheets and β-hairpins.[3a] The bioinformatics analysis provides a foundation for understanding important cross-strand contacts in natural β-sheets; we next sought to understand the dependence of the conformational stability of macrocyclic HBS β-sheets on the cross-strand pair.
Figure 3.
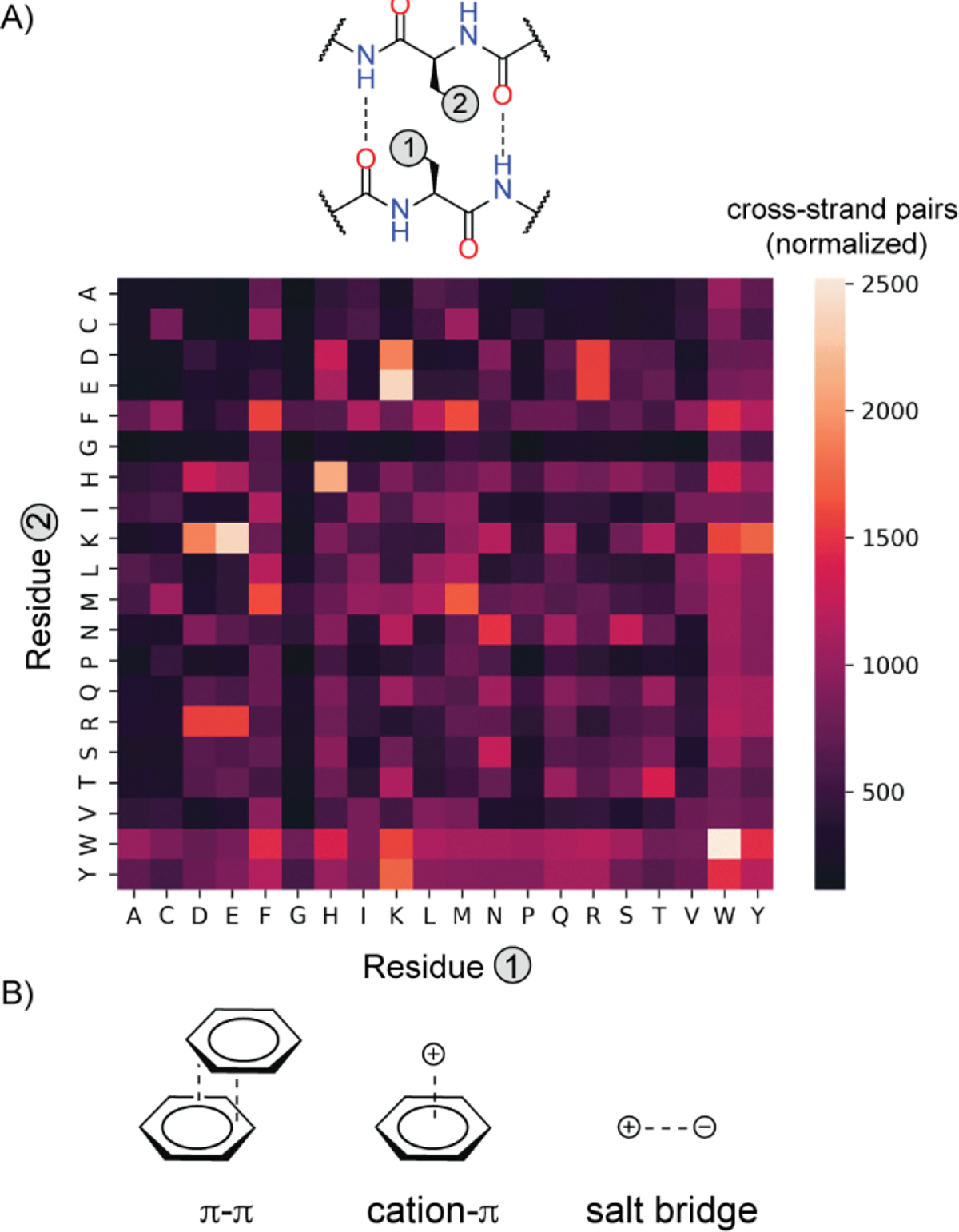
Identification of cross-strand pairs in non-hydrogen-bonded sites within antiparallel β-sheets. (A) Illustration of side-chain interaction between a residue pair at non-hydrogen bonded sites. The plot shows a heatmap indicating prevalence of each pair (normalized for natural occurrence of each amino acid residue). (B) Aromatic interactions, particularly Trp/Trp, cation-π, and ionic interactions are overrepresented in antiparallel β-sheets. Codes and datasets are available on GitHub (see Supporting Information).
We investigated the requirement for the optimal cross-strand pair for the HBS β-sheet by synthesizing and analyzing different aromatic-aromatic, aromatic-aliphatic, and overrepresented combinations (lysine-tyrosine and lysine-glutamic acid) in the PDB (Figure 4A). We compared the impact of the cross-strand pairs on the stability of the macrocyclic β-sheet using circular dichroism (CD) and NMR spectroscopies on five analogs of HBS β-sheet 1. In the first series, we substituted one of the cross-strand tryptophan (Trp) residues with phenylalanine (Phe), cyclohexylalanine (Chx), and tyrosine (Tyr) to obtain macrocycles 2–5, respectively. In the second series, both tryptophan residues were substituted with Tyr/Chx (6) and Tyr/Phe (7) pairings to analyze the impact of tryptophan residues in minimal macrocyclic β-sheets. In the third series, we probed the impact of lysine (Lys) -tyrosine (Tyr) cation-π (8) and lysine-glutamic acid (Glu) salt-bridge (9) interactions that predominate in the heatmap of cross-strand interactions (Figure 3). The stabilities of all nine β-sheets were initially analyzed with circular dichroism spectroscopy. As noted above, CD spectroscopy is not routinely used to evaluate minimal β-sheet models, as it is for α-helices, because either the signal is too weak for analysis or the CD signature does not fit the idealized depiction because of twisting of the strands.[17] Given the robust signal observed for β-sheet 1, we conjectured that the non-tryptophan artificial β-sheets would also display measurable circular dichroism spectra. Figure 4B depicts the 210 nm signal from all spectra for qualitative comparison of the conformational stability of the macrocycles; the full spectra are included in the Supporting Information, Figure S3. We were pleased to observe that the impact of tryptophan substitution could be qualitatively analyzed by CD spectroscopy. HBS β-sheets 1 and 4 with Trp/Trp and Trp/Tyr pairs show the highest degree of stabilization as measured by the minimum at 210 nm. Trp/Phe (2) and Trp/Chx (3) provide substantial signals at 210 nm albeit weaker than 1 and 4. The non-tryptophan β-sheet derivatives 6–7 show smaller CD molar ellipticity values than the aromatic series. The bioinformatics analysis suggests that cation-π and ionic interactions are over-represented in antiparallel β-sheets. In isolated β-sheet scaffolds, the impact of these interactions is not predicted to be as stabilizing as the aromatic interactions due to competition with water. In accordance with this hypothesis, we find that sheets 8 and 9 are minimally structured. Overall, the CD analysis suggests that a cross-strand aromatic pairing between tryptophan and tyrosine provides robust stability for minimal β-sheets; however, aromatic contacts are not required, as cross-strand pairing of Trp/Chx also offers a significant molar ellipticity value. Sheet 5 emerged as the most conformationally defined candidate from these studies. A CD thermal denaturation analysis of this construct shows a temperature-dependent signal change at 228 nm but suggests that the scaffold maintains >50% of its room temperature conformational stability at 95 °C (Figure 4C and S4).
Figure 4.
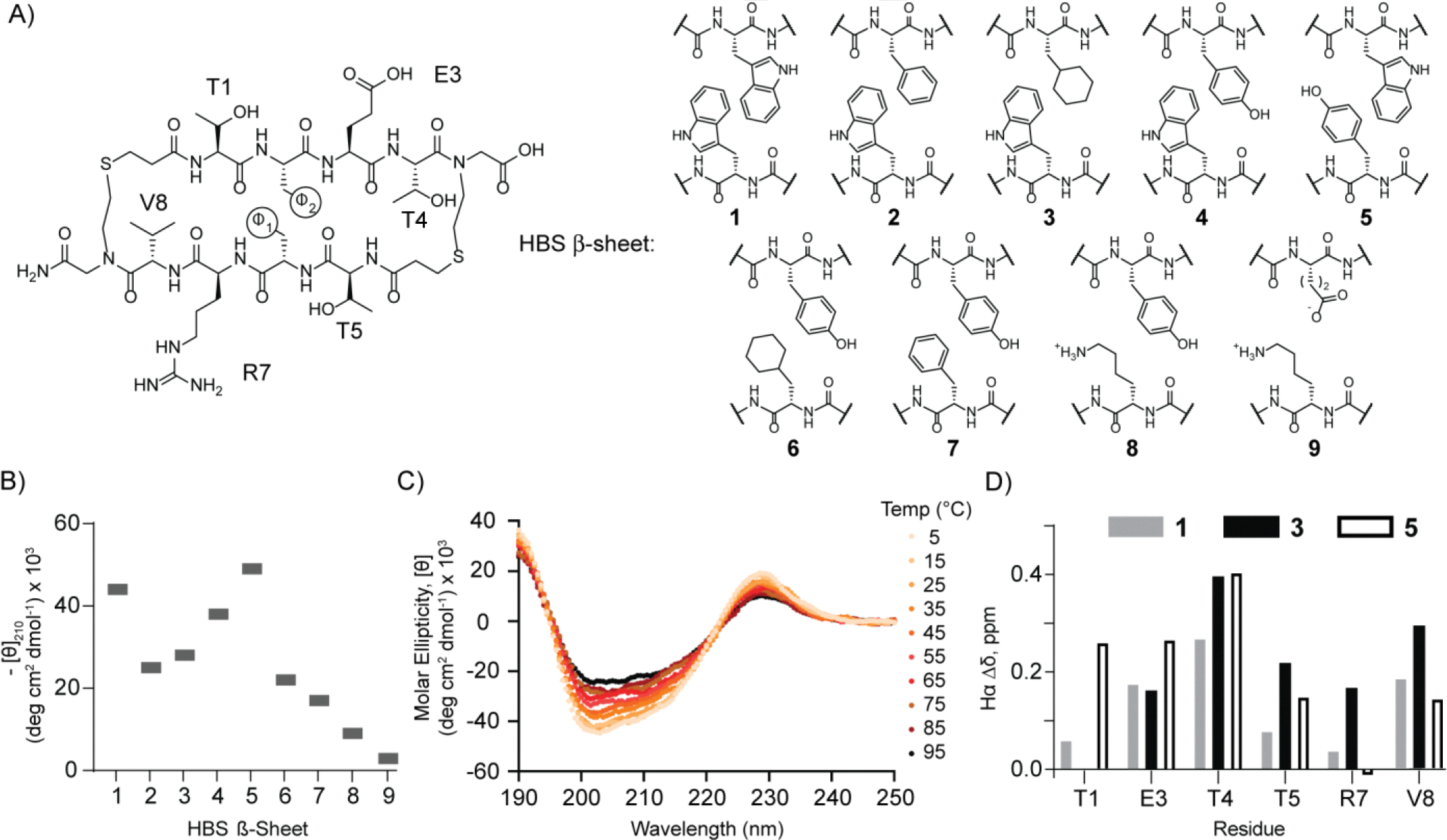
(A) Analysis of the dependence of β-sheet stability on cross-strand pairs in non-hydrogen-bonded sites. Nine pairs representing aromatic-aromatic, aromatic-aliphatic, cation-π, and ionic interactions were investigated as part of the HBS β-sheet scaffold. (B) Summary of circular dichroism results; spectra were obtained at a peptide concentration of 30 μM in 10 mM potassium fluoride (pH 7.3). (C) Thermal denaturation analysis of HBS β-sheet 5 by circular dichroism. (D) Chemical shift differences between the α-protons of each residue in HBS 1 (gray), 3 (black), and 5 (white) and random coil values.[18]
To further evaluate the stability of the HBS β-sheet scaffold, we analyzed the chemical shift of the α-proton (Hα) of each residue. Comparison of the chemical shift of Hα in folded conformations versus unfolded domains provides a metric for identifying different secondary structures.[18] Prior studies have shown that β-sheets exhibit more downfield Hα shifts compared to random coil values, in which a downfield shift of 0.1 ppm or more is indicative of strand formation. The differences in Hα chemical shifts of HBS 1, 3, and 5 as compared to random coil values suggested by Wüthrich are plotted in Figure 4D.[19] This analysis suggests that all three scaffolds adopt geometries expected for β-sheets.
We further analyzed solution and solid-state conformations of HBS 5 through two-dimensional NMR spectroscopy and X-ray crystallography. Combination of 1D NMR, Total Correlation spectroscopy (TOCSY), and Rotating Frame Overhauser effect spectroscopy (ROESY) analyses confirms a robust β-sheet conformation for HBS 5. The sheet signature of 5 is supported by several interstrand NOEs (Figure 5A). We observed strong NOEs across the Tyr/Trp pair and an upfield shift for the H𝛅 proton of Tyr, suggesting an edge-to-face arrangement of the Tyr/Trp cross-strand pair. We also observed cross-strand NH-NH NOEs for both sets of amide protons that point inwards (T1/R7 and E3/T5 residue pairs). Backbone dihedral angles (Φ) define protein secondary structures. We calculated the Φ values from NMR coupling constants, 3JNHCHα – these values fall in the range expected for canonical strands. A structural model of 5 was obtained using 63 NOESY cross-peaks and 8 Φdihedral angles. An overlay of 20 lowest conformations is depicted in Figure 5B. The geometry of the structure was optimized with Schrodinger’s MacroModel software using experimental NOE and coupling constant restraints.[20]
Figure 5.
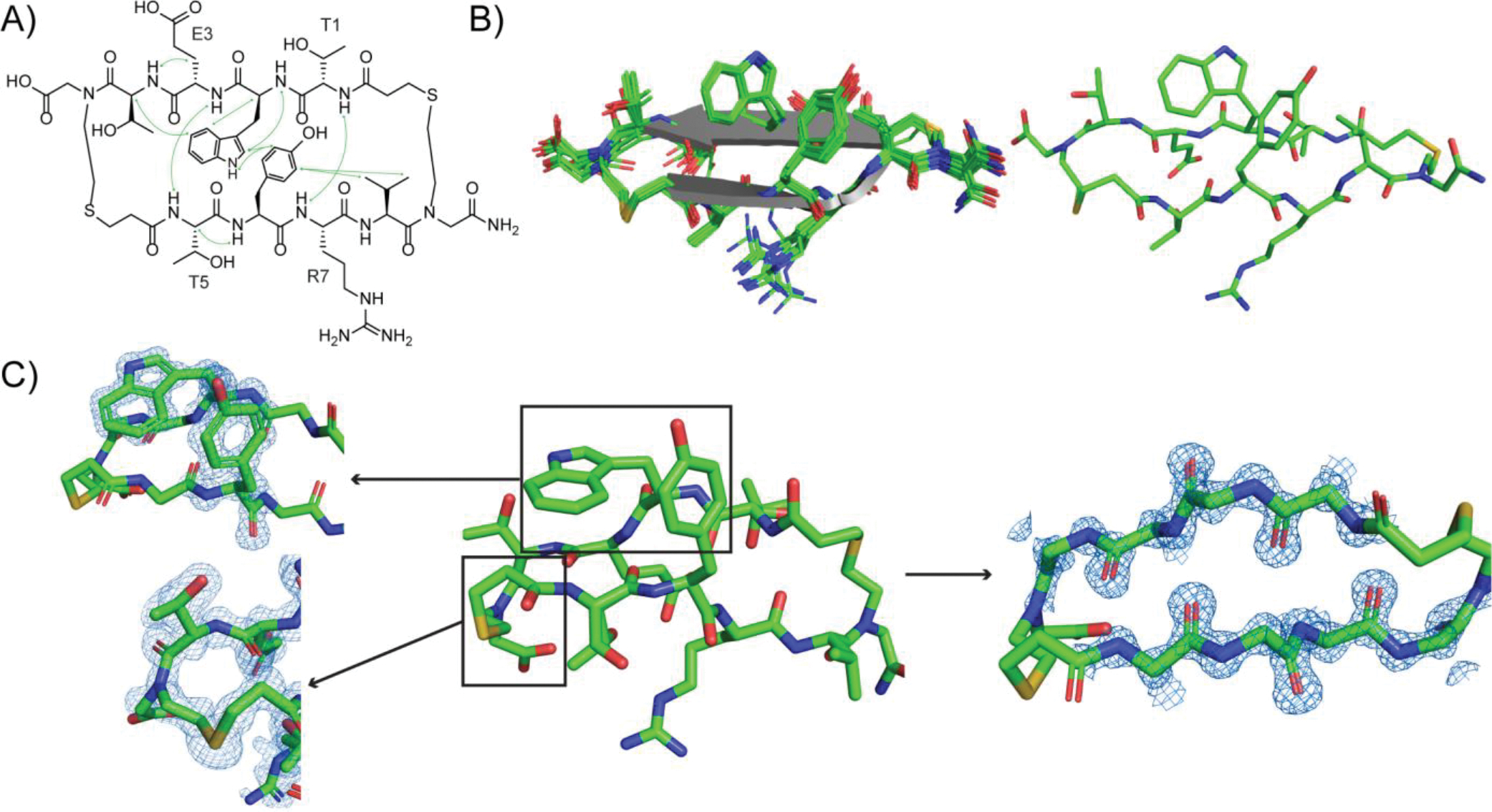
Structural analysis of HBS β-sheet 5. (A) Key NOEs observed by NMR spectroscopy. (B) NMR-derived structures of 5. View of 20 lowest energy structures (left) and lowest energy structure (right). The solution structure was determined from NOESY cross-peaks and 3JNHCHα coupling constants using simulated annealing and energy minimization protocols in Maestro. (C) X-ray crystallographic structure of 5 with electron density map showing the edge-to-face aromatic interactions and the thioether bridge (left) and the hydrogen-bonded strand functionality (right). All carbon, nitrogen, sulfur and oxygen atoms are shown in green, blue, gold and red, respectively. Coordinates have been deposited in the Protein Data Bank, PDB ID: 8DPY.
We were also successful in obtaining a high resolution (0.99 Å) X-ray crystal structure of 5 (Figure 5C). The crystal structure confirms that the individual strands orient into a macrocyclic sheet. The structure shows proper registry of hydrogen bonds and the Tyr/Trp pair interaction predicted from the solution NMR structure. The asymmetric unit cell yields two different edge-to-face orientations of Tyr/Trp (PDB ID: 8DPY). Supporting Information Figure S5 illustrates both T-shaped aromatic interaction observed in the solid-state structure.
In conclusion, we show that a minimal β-sheet can be stabilized by replacement of terminal inter-strand hydrogen bonds with hydrogen bond surrogates. We developed a facile synthesis of thioether-derived hydrogen bond surrogate macrocycles from simple starting materials. The rapid ease of synthesis would enable screening of β-sheet libraries for chosen protein targets. In ongoing efforts, we are investigating the potential of these scaffolds to inhibit protein-protein interactions guided by β-sheet interfaces.[7b, 14] In the current studies, we focused on factors that lead to conformationally-defined HBS β-sheet scaffolds. We interrogated a dataset of antiparallel β-sheets in the PDB to assess cross-strand residues that predominate in protein β-sheets; as expected, aromatic and ionic interactions are over-represented. We next determined if these interactions can also aid the stability of HBS β-sheets. Analysis of a series of cross-strand pairs revealed the minimal requirements for a conformationally-defined β-sheet motif. Solution NMR and X-ray crystallography[21] structures validated our hypothesis that robust β-sheet conformation can be populated in the designed artificial β-sheets.
Supplementary Material
Acknowledgements
We thank the National Institutes of Health (R35GM130333) for financial support of this work. This research used resources 17-ID-1 of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The Center for BioMolecular Structure (CBMS) is primarily supported by the National Institutes of Health through a Center Core P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1607011). We thank Dr. Jean Jakoncic for acquisition of the X-ray diffraction data at the BNL.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Cheng F, Zhao J, Wang Y, Lu W, Liu Z, Zhou Y, Martin WR, Wang R, Huang J, Hao T, Yue H, Ma J, Hou Y, Castrillon JA, Fang J, Lathia JD, Keri RA, Lightstone FC, Antman EM, Rabadan R, Hill DE, Eng C, Vidal M, Loscalzo J, Nature Genetics 2021, 53, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Merritt HI, Sawyer N, Arora PS, Peptide Science 2020, 112, e24145; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pelay-Gimeno M, Glas A, Koch O, Grossmann TN, Angew. Chem. Int. Ed. 2015, 54, 8896–8927; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Horne WS, Grossmann TN, Nat. Chem. 2020, 12, 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Hughes RM, Waters ML, Curr. Opin. Struct. Biol. 2006, 16, 514–524; [DOI] [PubMed] [Google Scholar]; b) Freire F, Gellman SH, J. Am. Chem. Soc. 2009, 131, 7970–7972; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Stanger HE, Gellman SH, J. Am. Chem. Soc. 1998, 120, 4236–4237; [Google Scholar]; d) Haque TS, Gellman SH, J. Am. Chem. Soc. 1997, 119, 2303–2304; [Google Scholar]; e) Robinson JA, Accounts of Chemical Research 2008, 41, 1278–1288; [DOI] [PubMed] [Google Scholar]; f) Li X, Sabol AL, Wierzbicki M, Salveson PJ, Nowick JS, Angew. Chem. Int. Ed. 2021, 60, 22776–22782; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Khakshoor O, Nowick JS, Curr. Opin. Chem. Biol. 2008, 12, 722–729; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Park JH, Waters ML, Org. Biomol. Chem. 2013, 11, 69–77; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Ganesh Kumar M, Mali SM, Raja KMP, Gopi HN, Org. Lett. 2015, 17, 230–233; [DOI] [PubMed] [Google Scholar]; j) Santiveri CM, León E, Rico M, Jiménez MA, Chemistry – A European Journal 2008, 14, 488–499; [DOI] [PubMed] [Google Scholar]; k) Almeida AM, Li R, Gellman SH, J. Am. Chem. Soc. 2012, 134, 75–78; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Kier BL, Anderson JM, Andersen NH, Journal of the American Chemical Society 2015, 137, 5363–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Karle IL, Awasthi SK, Balaram P, Proc. Natl. Acad. Sci. USA 1996, 93, 8189–8193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Diaz H, Tsang KY, Choo D, Espina JR, Kelly JW, J. Am. Chem. Soc. 1993, 115, 3790–3791. [Google Scholar]
- [6].Chen P-Y, Lin C-K, Lee C-T, Jan H, Chan SI, Protein Sci. 2001, 10, 1794–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Sawyer N, Arora PS, ACS Chem. Biol. 2018, 13, 2027–2032; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Blosser SL, Sawyer N, Maksimovic I, Ghosh B, Arora PS, ACS Chem. Biol. 2021, 16, 1518–1525; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sawyer N, Watkins AM, Arora PS, Acc. Chem. Res. 2017, 50, 1313–1322; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Patgiri A, Jochim AL, Arora PS, Acc. Chem. Res. 2008, 41, 1289–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Miller SE, Watkins AM, Kallenbach NR, Arora PS, Proc. Natl. Acad. Sci. USA 2014, 111, 6636–6641; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mahon AB, Arora PS, Chem. Commun. 2012, 48, 1416–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chapman R, Kulp IIIJL, Patgiri A, Kallenbach NR, Bracken C, Arora PS, Biochemistry 2008, 47, 4189–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Andersen NH, Olsen KA, Fesinmeyer RM, Tan X, Hudson FM, Eidenschink LA, Farazi SR, J. Am. Chem. Soc. 2006, 128, 6101–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Cochran AG, Skelton NJ, Starovasnik MA, Proc. Natl. Acad. Sci. USA 2001, 98, 5578; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wu L, McElheny D, Takekiyo T, Keiderling TA, Biochemistry 2010, 49, 4705–4714. [DOI] [PubMed] [Google Scholar]
- [12].Miller SE, Kallenbach NR, Arora PS, Tetrahedron 2012, 68, 4434–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Fooks HM, Martin ACR, Woolfson DN, Sessions RB, Hutchinson EG, J. Mol. Biol. 2006, 356, 32–44; [DOI] [PubMed] [Google Scholar]; b) Lifson S, Sander C, J. Mol. Biol. 1980, 139, 627–639; [DOI] [PubMed] [Google Scholar]; c) Wouters MA, Curmi PMG, Proteins 1995, 22, 119–131; [DOI] [PubMed] [Google Scholar]; d) Hutchinson EG, Sessions RB, Thornton JM, Woolfson DN, Protein Sci. 1998, 7, 2287–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Watkins AM, Arora PS, ACS Chem. Biol. 2014, 9, 1747–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Newberry RW, Raines RT, ACS Chem. Biol. 2019, 14, 1677–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tsutsumi M, Otaki JM, J. Chem. Inf. Model 2011, 51, 1457–1464. [DOI] [PubMed] [Google Scholar]
- [17].Micsonai A, Wien F, Kernya L, Lee Y-H, Goto Y, Réfrégiers M, Kardos J, 2015, 112, E3095–E3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Wishart DS, Case DA, in Methods Enzymol, Vol. 338 (Eds.: James TL, Dötsch V, Schmitz U), Academic Press, 2002, pp. 3–34; [Google Scholar]; b) Wishart DS, Bigam CG, Holm A, Hodges RS, B. D. Sykes, J. Biomol. NMR 1995, 5, 67–81. [DOI] [PubMed] [Google Scholar]
- [19].Pardi A, Billeter M, Wuthrich K, J. Mol. Biol. 1984, 180, 741–751. [DOI] [PubMed] [Google Scholar]
- [20].Macromodel, Version 2021–3 ed., Schrodinger, Inc, New York, 2023. [Google Scholar]
- [21].Crystal coordinates for HBS β-Sheet 5 have been deposited in the Protein Data Bank as PDB ID 8DPY.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.