

Abstract
Prph2 is a photoreceptor-specific tetraspanin with an essential role in the structure and function of photoreceptor outer segments. PRPH2 mutations cause a multitude of retinal diseases characterized by the degeneration of photoreceptors as well as defects in neighboring tissues such as the RPE. While extensive research has analyzed photoreceptors, less attention has been paid to these secondary defects. Here, we use different Prph2 disease models to evaluate the damage of the RPE arising from photoreceptor defects. In Prph2 disease models, the RPE exhibits structural abnormalities and cell loss. Furthermore, RPE functional defects are observed, including impaired clearance of phagocytosed outer segment material and increased microglia activation. The severity of RPE damage is different between models, suggesting that the different abnormal outer segment structures caused by Prph2 disease mutations lead to varying degrees of RPE stress and thus influence the clinical phenotype observed in patients.
Keywords: AB_571111, AB_628459, AB_785511, AB_839504, AB_2285290, AB_2533073, AB_2535792, AB_2535794, AB_2536180, AB_2833006, AB_10003146, macular degeneration, phagocytosis, Prph2, RPE
1 |. INTRODUCTION
Peripherin 2 (Prph2, formerly known as retinal degeneration slow, RDS) is a photoreceptor-specific tetraspanin, which is essential for the development and maintenance of photoreceptor outer segment (OS) structure and function.1–5Prph2 is localized exclusively at the closed rim of discs and lamellae in rod and cones, respectively,1,6 where it oligomerizes with its partner rod outer segment membrane protein 1 (Rom1).2,7 Prph2 and Rom1 assemble into multiple different types of complexes that are essential to the ability of Prph2 to maintain the structure of the OS disc rim.1–6,8–11 Mutations in PRPH2 are the cause of a multitude of retinal diseases with phenotypes ranging from autosomal dominant retinitis pigmentosa (RP) to macular dystrophy (MD) and various forms of pattern dystrophy.12 While PRPH2 expression is restricted to photoreceptors, and thus photoreceptors are the primary cells affected, patients carrying PRPH2 mutations frequently exhibit defects in the retinal pigment epithelium (RPE).12–14
In order to elucidate mechanisms for PRPH2-associated disease, we have previously developed and characterized several mouse models carrying Prph2 disease mutations. These include (1) a heterozygous knockout mouse which models loss-of-function mutations that lead to RP in patients, (2–3) knockin models carrying the Y141C and K153del mutations, both of which exhibit phenotypic heterogeneity and cause RP and/or pattern dystrophy in patients, (4) a knockin model carrying the mutation C213Y, which causes pattern dystrophy in patients, and (5) a transgenic model expressing the mutation R172W, which causes macular disease and cone-dominant defects in patients.12,13,15–26 Our mouse models largely recapitulate photoreceptor-associated phenotypes seen in patients and have demonstrated that different types of defects in Prph2/Rom1 complex assembly lead to different forms of OS structural abnormalities and can differentially affect rod and cone degeneration.15–20 We have not systematically examined the impact of these Prph2 disease mutations on the RPE, in spite of the known presence of RPE-associated defects in patients and the critical role that RPE health plays in maintaining photoreceptors.27,28
The RPE has many essential functions including the maintenance of the outer blood retina barrier, regulation of the transport of ions into the subretinal space, absorption of stray light, and recycling retinoids as part of the visual cycle.27–32 In addition, a key role for the RPE is phagocytosis and the digestion of distal OS material. Photoreceptor OS undergo a daily renewal process in which new discs are synthesized at the base and old discs are shed from the tips. These shed discs are taken up and degraded by the RPE, a process, which is essential for the survival of the photoreceptor cells.33–35 Due to its role in a multitude of processes vital for the photoreceptor function and survival, damage to the RPE is a key part of many retinopathies, including RP, Leber congenital amaurosis, age related macular degeneration, Sorsby’s fundus dystrophy, and Stargardt disease.27,28,36–44
Here, we evaluate the RPE structure and function in mouse models carrying Prph2 disease mutations. We find that the RPE phenotypes mimic those seen in some patients with Prph2 diseases, and that effects on the RPE are not uniform across mutations. Critically, our studies suggest that these defects are associated with impaired digestion and clearance of OS material leading to cellular stress, hypertrophy and multinucleation, and accumulation of microglia.
2 |. MATERIALS AND METHODS
2.1 |. Animal care and use and tissue harvesting
The University of Houston Institutional Animal Care and Use Committee (IACUC) approved all animal experiments and use. Experiments also adhered to the guidelines of the Association for Research in Vision and Ophthalmology and NIH Guide for the Care and Use of Laboratory Animals. Both male and female mice were used and were fed standard rodent diet ad libitum. The R172W transgenic animals have been previously characterized15–17 and carry a Prph2 transgene with the R172W mutation driven by the interphotoreceptor retinoid binding protein (IRBP) promoter, which expresses in both rods and cones. Individual Prph2 knockin lines carrying the Y141C, C213Y, and K153del mutations were made by inGenious Targeting Laboratory, Inc. (Ronkonkoma, New York, USA) and have also been previously characterized.18–20,45 All animals, including wild-type non-transgenic controls are on our in-house “wild-type” background. This strain was created by breeding FVB mice to C57BL/6J, eliminating the rd1 and rd8 mutations and then inbreeding for 10 generations. All the mice used in this study lack the rd8 mutation and carry the RPE65Leu variant. Animals were kept under cyclic light conditions (12 h light, 12 h dark, ~30 lux). Except where otherwise noted, tissues were collected on animals euthanized in late morning, ~12:00 p.m., (5 h after light onset).
2.2 |. Antibodies
Antibodies used are summarized in Table 1.
TABLE 1.
List of antibodies
| Antigen | Species | Clone | Application/Concentration | Source | RRID# |
|---|---|---|---|---|---|
| Prph2 | Rabbit | RDS-CT | 1:1000 (IF) | In house15 | AB_2833006 |
| Prph2 | Mouse | 2B7 | 1:500 (IF) | In house17 | NA |
| Acetylated α-Tubulin | Mouse | Lys40 | 1:2000 (IF) | Invitrogen, Carlsbad, CA (32–2700) | AB_2533073 |
| RPE65 | Mouse | 1:200 (IF) | Fisher Scientific, Waltham, MA (mab5428) | AB_571111 | |
| RPE65 | Rabbit | 1:200 (IF) | Proteintech, Rosemont, IL (17939–1-AP) | AB_2285290 | |
| LC3b | Rabbit | 1:300 (IF) | Novus Biologicals, Littleton, CO (NB100–2220) | AB_10003146 | |
| Lamp-1 | Rabbit | 1:300 (IF) | Abcam, Cambridge, MA (ab208943) | NA | |
| Iba-1 | Rabbit | 1:500 (IF) | Fujifilm, Minato City, Tokyo, Japan (019–19741) | AB_839504 | |
| ZO-1 | Rat | 1:1000(IF) | Santa Cruz, Dallas, TX (sc-33725) | AB_628459 | |
| Rhodopsin | Mouse | 1D4 | 1:2000 (IF) | R. Molday46 | AB_785511 |
| Rabbit IgG 488 | Donkey | 1:1000 (IF) | Invitrogen (A-21206) | AB_2535792 | |
| Mouse IgG 555 | Donkey | 1:1000 (IF) | Invitrogen (A-31570) | AB_2536180 | |
| Rat IgG 488 | Donkey | 1:1000 (IF) | Invitrogen (A-21208) | AB_2535794 |
2.3 |. Preparation of the RPE whole mount and immunofluorescence
To prepare RPE whole mounts, the whole eye was collected from euthanized animals, the cornea was punctured, and the eye was fixed in 4% paraformaldehyde (PFA) for 3–4 h. The cornea was removed, and the neural retina was carefully teased out from the eyecup leaving the RPE, choroid, and sclera behind. The remaining eyecup was then cut equally in four pieces and stored in 1xPBS (137 mM NaCl, 2.7 mM KCl 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) at 4°C until used. For staining, the eyecup piece was allowed to air dry on a glass slide for 10 min. The tissue was washed twice with 1xPBS for 10 min and blocked with blocking solution (5% BSA, 2.5% donkey serum, and 1% Triton X-100 in 1xPBS) for 1 h. Primary antibodies were diluted in blocking solution, added to the whole mount and incubated overnight at 4°C. The tissue was then washed three times in 1xPBS for 10 min, each. Secondary antibodies were diluted in blocking buffer, added to the whole mount and incubated for 1 h at room temperature. The tissue was then washed once in 1xPBS, incubated in DAPI (diluted in blocking buffer) for 20 min, and washed two more times in 1xPBS. Slides were mounted using Prolong Gold mounting media (Thermo Fisher Scientific, Waltham, MA, USA). The whole mounts were imaged using the 40× objective (Num. aperture 0.9) of a Zeiss LSM 800 (Carl Zeiss Microscopy GmbH, Jena, Germany). The cell counter plug-in was used to determine the RPE cells per field in ImageJ. The thresholding function was used to determine the area of individual RPE cells.
2.4 |. Immunofluorescence
After euthanasia, eyes were enucleated, incubated in Davidson fixative (4 ml 20% PFA, 3 ml 100% EtOH, 1 ml acetic acid, 1 ml H2O), and embedded in paraffin using a STP 120 Spin Tissue Processor (Thermo Fisher Scientific). Sections (10 μm) were prepared with a HM 355S Automatic Microtome (Thermo Fisher Scientific). For immunolabeling, the sections were subjected to deparaffinization in xylene followed by rehydration in an ethanol gradient (100%, 90%, 80%, 70%, and 50%) and two washes in water. Antigen retrieval was performed by boiling the section for 30 min in Tris/EDTA buffer (10 mM Tris Base, 1 mM EDTA Solution, 0.05% Tween 20, pH 9.0) followed by 10 min cooling at room temperature. After two additional washes in water the sections were incubated for 3 min in 1% NaBH4 and further washed in water and 1x PBS. For blocking, the slides were incubated for 30 min in blocking solution (as above) at room temperature. Primary antibodies were diluted in blocking solution, added to the sections, and incubated overnight at 4°C. After the incubation with the primary antibodies, the sections were washed three times in 1xPBS and incubated for 1.5 h with the secondary antibodies diluted in blocking solution. After three more washes in 1xPBS, the sections were incubated with DAPI (1:1000, diluted in blocking solution) for 30 min, washed twice in 1xPBS and mounted using Prolong Gold antifade mounting reagent (Thermo Fisher Scientific). The sections were imaged using a Zeiss LSM 800 (Carl Zeiss Microscopy GmbH). The quantification of OS material in the RPE immunofluorescence was performed as described above, with the exception that the antigen retrieval was skipped. The obtained images were quantified using ImageJ. The red channel (RPE65 staining) was converted into a gray scale 8 bit image. This was followed by applying the FFT band pass filter in order to reduce the background signal. The RPE65 stained area was then determined as the region of interest (ROI) by applying a threshold on the image. The green channel (Prph2 staining) was subjected to a color threshold. After the threshold was determined, the “analyze particles tool” was applied in the ROI determined by the RPE65 staining. Particles smaller than 3 pixels as well as particles on the edge of the ROI were excluded. The area of the ROI, as well as the total number of vesicles in the ROI, were measured and subjected to statistical analysis using GraphPad Prism 7.05 (GraphPad Software, San Diego, CA, USA).
2.5 |. Electroretinography
Full-field ERGs were performed as previously described.17 In brief, mice were dark-adapted overnight and were anesthetized with an injection of ketamine and xylazine. Eyes were dilated with 1% cyclopentolate for 5 min after which Gonak (Akorn, Lake Forest, IL, USA) was applied to each eye. Scotopic ERGs were recorded using a strobe flash stimulus of 157 cd-s/m2 followed by a 5 min light adaption at 29.03 cd/m2. Photopic ERGs were recorded using an average from 25 flashes at 157 cd-s/m2, 12.5 cd-s/m2, and 0.79 cds/m2 for white light, green light, and UV light, respectively. C-wave ERGs were performed with mice that were dark-adapted overnight prior to the experiment. C-wave ERGs were recorded from 5 average flashes at 157 cd-s/m2 with an extended time of 4080 ms. All ERGs were done using an UTAS system (LKC, Gaithersburg, MD, USA). The values obtained from both eyes of the animal were averaged. Implicit time and maximum amplitudes were determined using EMWIN software (ver. 9.7.0.12, LKC).
2.6 |. Electron microscopy
All methods used for whole eye collection, fixing, processing, embedding in plastic, and transmission electron microscopy were performed as described previously.47–49 The eyes were collected and fixed between 11:00 a.m. and 12:00 p.m. Measurements of the mitochondrial length were performed using ImageJ.
2.7 |. Statistical analysis
All statistical analyzes were performed using GraphPad Prism 7.05. Numerical data for all experiments are displayed as mean ± SD. Significance was determined using one-way ANOVA with Tukey’s post hoc comparison or two-way ANOVA with Sidak’s post hoc comparison.
3 |. RESULTS
3.1 |. Mouse models of Prph2-related disease exhibit structural anomalies in the RPE
Our goal was to understand how the RPE structure and function are altered in five different Prph2 disease models in which we have previously characterized the ongoing rod and cone degeneration. We chose models that capture a wide range of Prph2 disease phenotypes including RP (Prph2+/−), macular dystrophy (Prph2 R172W), and pattern dystrophy (Prph2C213Y/+). We also included two models carrying mutations associated with broad phenotypic heterogeneity in patients, Prph2Y141C/+ and Prph2K153del/+. The Y141C is a point mutation while the K153 is a three base pair deletion mutation.3,15,18–20 Because human inheritance of these mutations is autosomal dominant, we analyzed all of the mutations in the presence of wild-type (WT) Prph2, either in heterozygous animals (for knockins such as Prph2Y141C/+) or in transgenic animals on the WT background (e.g., for Prph2 R172W animals).
We began by analyzing the ultrastructure of the RPE by performing EM on the outer retina at postnatal day (P) 30 and P180. As expected based on our prior work, at P30 all of the disease models except the Prph2 R172W exhibit abnormal OS characterized by varying degrees of whorl formation (blue arrows, Figure 1A). As the Prph2 R172W model mainly targets cones, the majority of OS (rods) exhibit a largely normal appearance. Focusing on the RPE, at P30, Prph2+/−, Prph2Y141C/+, and Prph2C213Y/+ retinas displayed pronounced accumulation of phagosomes carrying apparent OS material compared to WT mice (red arrows, Figure 1A). We also observed numerous intracellular vacuoles and paracellular openings (green arrowheads, Figure 1A), as well as debris-containing invaginations of the basal RPE membrane (yellow arrows, Figure 1A) leading to an irregular and disrupted interface between adjacent RPE cells and between the RPE and Bruch’s membrane. At P30, the RPE of Prph2 R172W mice is largely normal on EM images. By P180, the accumulation of phagosomes carrying apparent OS material was largely reduced in these disease models (Figure 1B), likely due to progressed retinal degeneration that led to shorter OSs and an overall decrease in the amount of phagocytosed material to be recycled by the RPE. However, pronounced paracellular spaces and debris in basal infoldings were still present (green arrowheads and yellow arrows, Figure 1B) in P180 Prph2+/−, Prph2Y141C/+, and Prph2C213Y/+ retinas. We also evaluated the mitochondrial structure by EM. Mitochondria were largely normal in appearance, with no significant signs of swelling or disorganization. We did observe elongated mitochondria in many of the mutant models at P180 (orange arrows show examples, Figure 1B), which is reflected in our finding that mean mitochondrial length was slightly increased in the Prph2C213Y/+, although the mean mitochondrial length was unchanged in the other models (Figure S1A).
FIGURE 1.
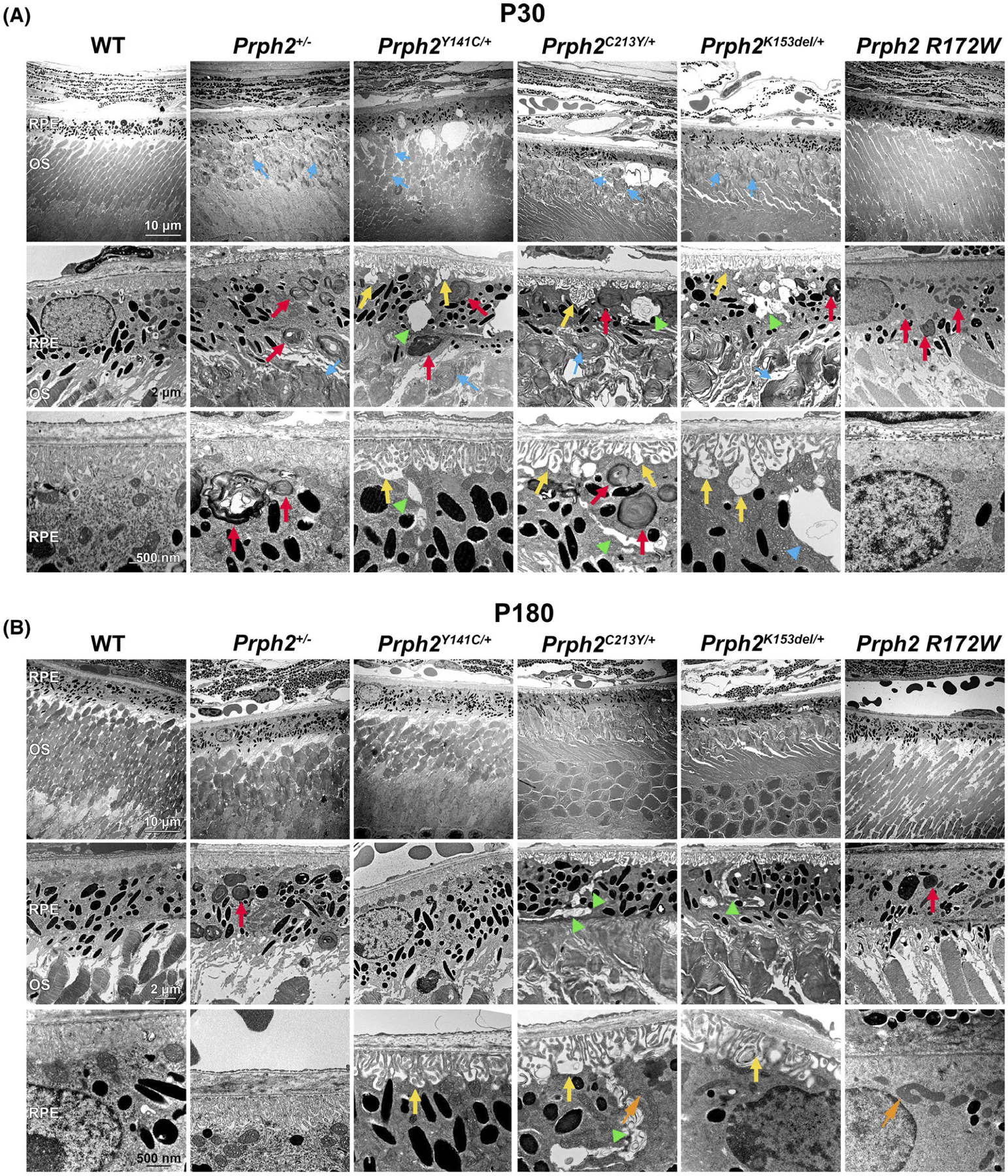
Prph2 disease models exhibit structural anomalies in the RPE. (A, B) EM images were captured from retinal cross sections of the indicated genotypes harvested at P30 (A) or P180 (B). Red arrows highlight OS material in the RPE, yellow arrows highlight invaginations of the basal RPE membrane, green arrowheads highlight intracellular vacuoles and paracellular spaces, orange arrows highlight examples of elongated mitochondria, and blue arrows highlight whorl structures in the OS. Original magnification: 3000× in the upper panels, 10 000× in the middle panels and 25 000× in the lower panels in both (A) and (B). N = 3 eyes per genotype and age. Scale bars: 10 μm (top row) 2 μm (middle) and 500 nm (bottom). OS, outer segment; RPE, retinal pigment epithelium
These EM data suggested that the RPE structure might be impaired in these Prph2 disease models. To evaluate the RPE shape on the light microscopy level, we labeled RPE whole mounts at P30 (Figure 2A) and P180 (Figure 2B) with zonula occludins 1 (ZO-1, a component of tight junctions, green Figure 2).50–52 Healthy RPE cells exhibit fairly even size within the normal hexagonal mosaic, but in aging and retinal degenerative diseases such as age-related macular degeneration, RPE cell loss can lead to hypertrophy and multinucleation of remaining RPE cells.53–57 At P30, the RPE pattern appeared grossly normal (Figure 2A), but by P180, we began to observe increased irregularity in RPE cell size in disease models. At P180, several of our disease models (Prph2K153del/+, Prph2+/−, and Prph2Y141C/+) exhibit enlarged hypertrophic RPE cells (white arrowheads, Figure 2B). To better quantify this phenotype, we asked whether the total number of RPE cells/area was changed, and measured the individual RPE cell area. Consistent with our qualitative observation, we found that the mean number of RPE cells/field trended down in several of the mutant models. This was associated with a significant increase in mean RPE cell area at P180 in Prph2Y141C/+, Prph2C213Y/+, and Prph2K153del/+ (Figure 2C,D). We also observe an increase in multinucleated (more than two) RPE cells in several of our degenerative models at P180. This phenotype is most noticeable in the Prph2K153del/+ but is also present in Prph2+/− and Prph2Y141C/+.
FIGURE 2.
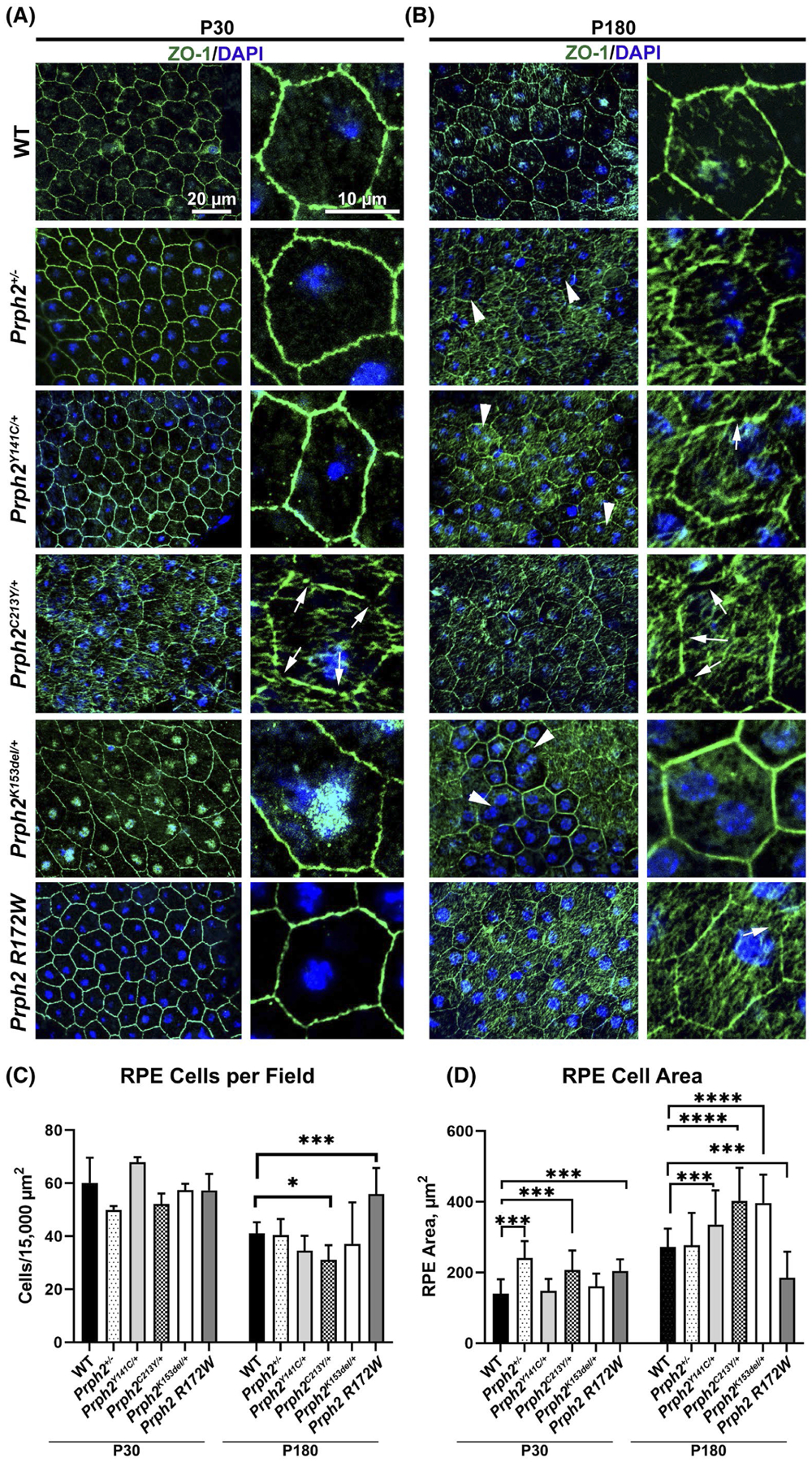
Prph2 disease models exhibit defects in cell size. (A, B) RPE flat mounts prepared from tissues harvested at P30 (A) or P180 (B) were labeled with an antibody for the tight junction protein ZO-1 (green) and counterstained with DAPI (blue). Arrows highlight gaps in ZO-1 labeling. Arrowheads highligh enlarged RPE cells. (C) The number of RPE cells in a 15 000 μm2 field was counted and plotted at P30 and P180. (D) The area of individual RPE cells was measured and plotted. Shown is mean ± SD, *p < .05, ***p < .001 and ****p < .0001 determined by two-way ANOVA, with Sidak’s post hoc test. Original magnification 40×. Scale bars: 20 μm (left panel of each pair), 10 μm (right panel of each pair). N = 3 eyes per genotype and age
While analyzing the RPE shape, we also noticed some changes in the ZO-1 labeling pattern. At P30, all lines except Prph2C213Y/+ and Prph2K153del/+exhibited relatively normal patterns of ZO-1 labeling. In contrast, the examination of flat mounts showed frequent gaps in ZO-1 labeling and increased internalization of ZO-1 in Prph2C213Y/+ (white arrows, Figure 2A) and internalization of ZO-1 in Prph2K153del/+. By P180, the gaps in ZO-1 labeling became more frequent in the Prph2C213Y/+ and were also visible in Prph2Y141C/+ and Prph2 R172W eyes (white arrows, Figure 2B). Combined these data indicate that disease causing mutations in Prph2 lead to structural defects in the RPE, including the accumulation of phagosomes, RPE hypertrophy, and increased multinucleation.
3.2 |. Electroretinography analysis of RPE responses in Prph2 disease models
Having established that Prph2 disease mutations can lead to structural defects in the RPE, we next asked whether we could observe any functional defects in the RPE. We began by performing electroretinography on all our models. The most commonly characterized ERG responses are the a-wave (which originates from photoreceptors) and b-wave (which reflects signals from inner retinal neurons). However, the c-wave can be seen as an indicator of the health and function of the RPE cells.58–60 The c-wave, which follows the scotopic a-and b-wave, represents the sum of two potentials generated in response to the decrease in subretinal [K+] caused by light exposure61 and is seen much longer after the light exposure than the a-and b-waves. We have previously performed ERG on all of these models to record standard a-and b-waves, so here we undertook prolonged recordings in order to capture c-waves as well (Figure 3A).
FIGURE 3.
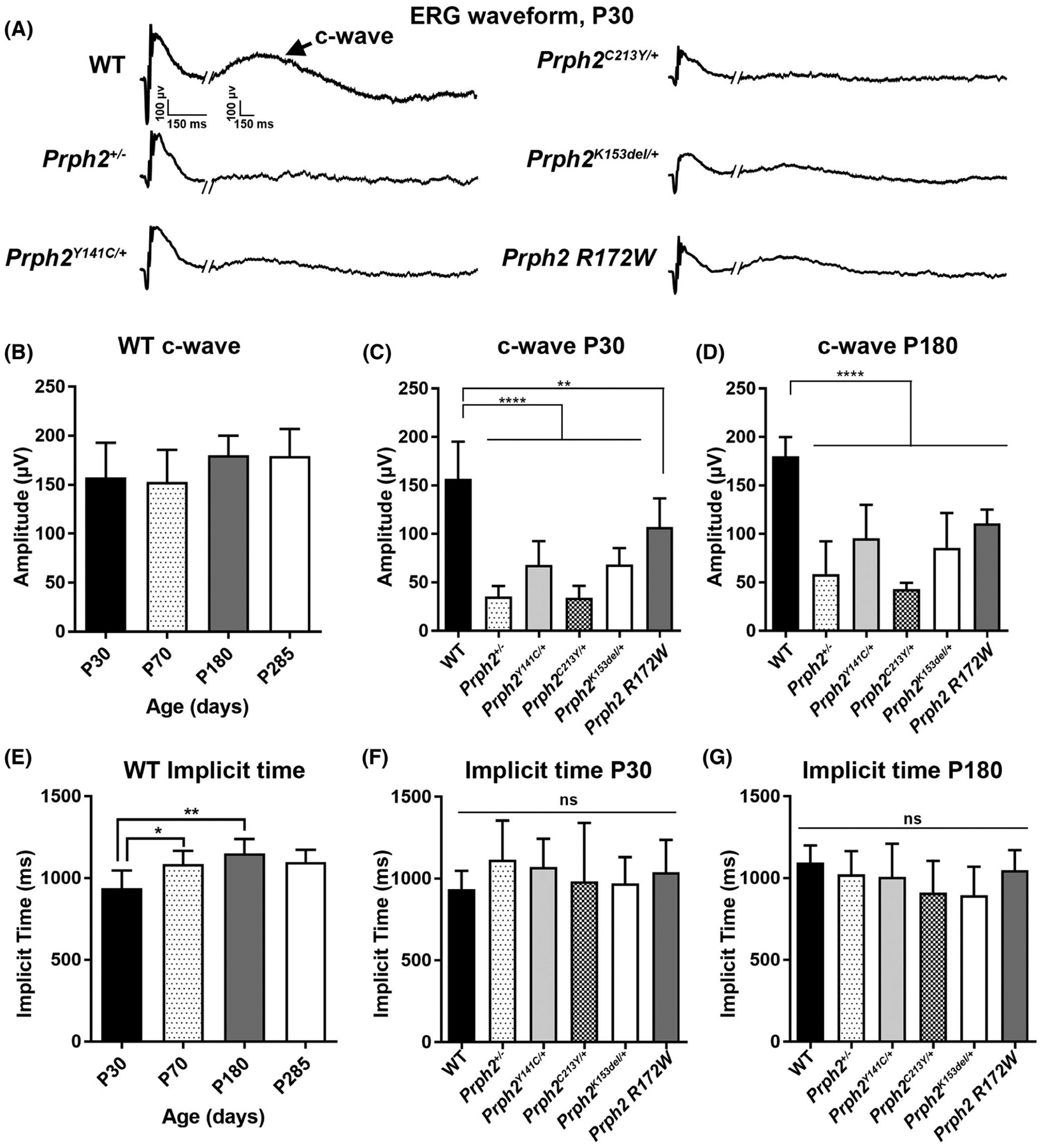
ERG c-wave amplitudes are impaired in Prph2 disease models. (A–G) Prolonged (4080 ms) full field ERGs were performed under scotopic conditions. (A) Representative ERG waveforms from each genotype are shown, arrow highlights c-wave. (B, E) Plotted is maximum c-wave amplitude (B) and implicit time (E) for WT mice over time (B: P30 N = 14; P70 N = 6; P180 N = 5; P285 N = 3. E: P30 N = 13; P70 N = 6; P180 N = 5; P285 N = 3). (C, D, F, G) Plotted is maximum c-wave amplitude (C, D) (C: WT N = 12; Prph2+/− N = 13; Prph2Y141C/+ N = 18; Prph2C213Y/+ N = 9; Prph2k153del/+ N = 10; Prph2 R172W N = 7. D: WT N = 5; Prph2+/− N = 8; Prph2Y141C/+ N = 11; Prph2C213Y/+ N = 5; Prph2k153del/+ N = 7; Prph2 R172W N = 15) and implicit time (F, G) (F: WT N = 13; Prph2+/− N = 14; Prph2Y141C/+ N = 18; Prph2C213Y/+ N = 9; Prph2k153del/+ N = 10; Prph2 R172W N = 6. G: WT N = 6; Prph2+/− N = 8; Prph2Y141C/+ N = 112; Prph2C213Y/+ N = 6; Prph2k153del/+ N = 7; Prph2 R172W N = 15) measured from disease models at P30 (C, F) and P180 (D, G). Shown is mean ± SD, **p < .01 and ****p < .0001 determined by two-way ANOVA, with Sidak’s post hoc test
We first characterized c-wave responses in WT mice over time. There was no significant change in c-wave amplitude up to ~10 months of age, although c-wave implicit times were slightly higher at P70 and P180 compared to P30 (Figure 3B,E). We assessed c-wave amplitudes and implicit times in our disease models at P30 and P180, and while there were no significant differences in implicit time across groups or time points (Figure 3F,G), all of the models displayed significant reductions in c-wave amplitude compared to WT mice at both P30 and P180 (Figure 3C,D). This deficit was most pronounced in the Prph2+/− and Prph2C213Y/+ but was severe in all models. Interestingly, while c-wave amplitudes were significantly reduced in Prph2 mutant models at both P30 and P180 (compared to WT), this phenotype was not progressive: c-wave amplitudes did not worsen further from P30 to P180 (Figure 3C,D), and in some groups (including WT), mean c-wave amplitudes were slightly increased at P180 vs. P30. Because the amplitude of the c-waves is dependent on signals generated by photoreceptors, it can be difficult to separate whether c-wave defects reflect RPE dysfunction or are secondary to photoreceptor dysfunction. To help assess this, we plotted c-waves normalized to a-waves at P30 and P180 (Figure S1B,C). At P30, we observe reduced mean c-waves (after normalizing to a-waves) in several of the mutant models but the difference compared to WT only attained statistical significance in the case of Prph2C213Y/+. At P180, there was no difference in c-waves after normalizing to a-waves, consistent with the idea that the severe progressive photoreceptor dysfunction in these lines masks our ability to separately assess RPE function via c-wave ERG, and suggesting that c-waves may not be an effective way to assess RPE function in cases with pre-existing severe photoreceptor dysfunction.
All of the disease models exhibited a-and b-wave measurements consistent with our previous studies describing these models.15–20 Specifically, all models displayed a significant reduction in scotopic a-and b-wave amplitudes at P30 and P180, indicating a decline in rod function (Figure S2A,B). Furthermore, all models except Prph2Y141C/+ and Prph2+/− exhibited a significant decrease in the amplitude photopic b-wave at both time points, indicating a loss of cone function. (Figure S2A,B).
3.3 |. Mutations in Prph2 result in a delayed processing of shed discs
One of the main functions of the RPE is the phagocytosis and subsequent processing of discs, which are shed during the daily renewal of the photoreceptor OSs.33–35 In our EM studies, we observed the accumulation of OS material in the RPE (Figure 1); therefore, we hypothesized that the digestion of phagocytosed OS material may be delayed or impaired in our Prph2 disease models. OS disc shedding and subsequent RPE phagocytosis and digestion follows a circadian rhythm.62–66 We began by evaluating the presence of OS material in the RPE at various time points after light onset in WT retinas using immunofluorescence labeling for OS markers, Prph2 (green) and rhodopsin (red) (Figure S3). Rhodopsin and Prph2 fluorescent puncta are readily visible in the RPE at two and three hours after light onset but are largely cleared by four hours after light onset (Figure S3).
To evaluate whether the clearance of OS material was delayed or impaired in Prph2 disease models, we collected retinas at P30 and P180 at four hours after light onset. Sections were co-stained for Prph2 (to identify OS, green) and the RPE marker RPE65 (to accurately identify the area of the RPE, red) (Figure 4A,B). Unlike WT retinas, at P30, Prph2 R172W and Prph2C213Y/+ displayed prevalent undigested OS material in the RPE at four hours after light onset (white arrowheads, Figure 4A). At P180, only Prph2 R172W exhibited pronounced vesicles in the RPE at 4 h after light onset (white arrowheads, Figure 4B), suggesting that the phenotype had partially resolved.
FIGURE 4.
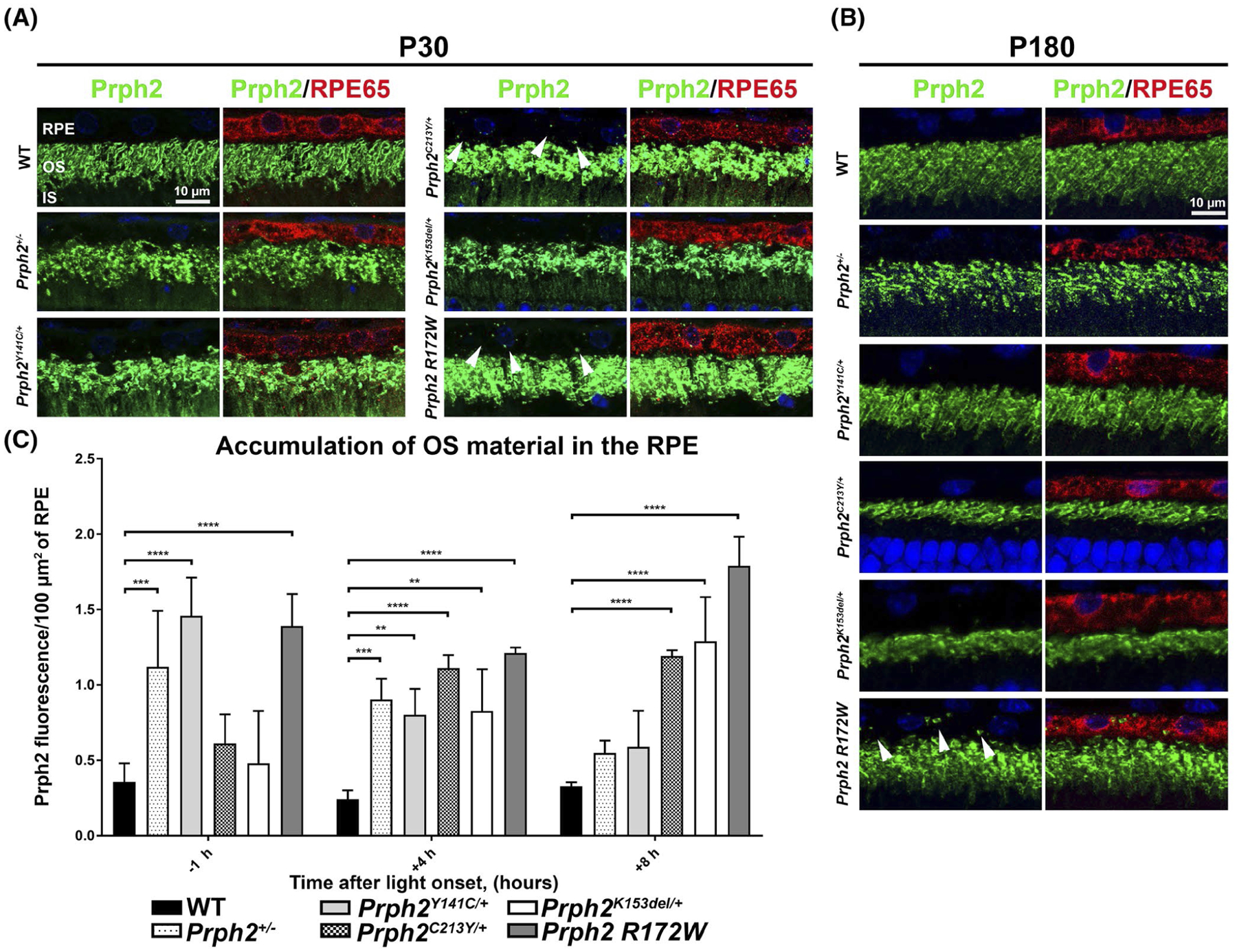
The clearance of OS material from the RPE is delayed in Prph2 disease models. (A, B) Retinal sections harvested at P30 (A) or P180 (B) at 4 h after light onset were co-stained for Prph2 (green, mAB 2B7) and RPE65 (red) and counterstained with DAPI (blue). (C) Retinal sections were collected at P30 at 1 h before light onset and 4, and 8 h after light onset and labeled with Prph2 and RPE65 as in (A, B). The number of Prph2 positive puncta in the RPE was quantified using imageJ and normalized to the area of the RPE. Plotted is mean ± SD. *p < .05, **p < .01, ***p < .001, and ****p < .0001 determined by two-way ANOVA, with Sidak’s post hoc test. IS, inner segments; ONL, outer nuclear layer; OS, outer segments; RPE, retinal pigment epithelium. Magnification at 63×. Scale bar: 10 μm. N = 3 eyes per genotype and for each time point
To more thoroughly characterize this phenotype, tissues were collected at one hour before light onset, and at four and eight hours after light onset. To quantify the amount of OS material in the RPE, we plotted the number of Prph2 fluorescent particles in the RPE normalized to the area of RPE65 fluorescence (as a measure of RPE area, Figure 4C). Consistent with our initial characterization showing that peak OS accumulation in the WT retina occurs approximately two hours after light onset, the WT retinas exhibited very low amounts of OS material in the RPE at all of these time points. Prph2 R172W exhibited the most pronounced elevation in OS material in the RPE of any model. This increase was evident at all time points examined, suggesting that this model has an overall impairment in RPE clearance of OS material (Figure 4C). All of the other models also exhibited significant increases in the presence of OS material in the RPE compared to WT, but not all at the same time points or to the same degree. Prph2K153del/+ and Prph2C213Y/+ were similar to WT at one hour before light onset, but steadily increased after that and were significantly higher than WT at four and eight hours after light onset, suggesting that in these models, the clearance of OS material is delayed but not completely blocked. In contrast, Prph2+/− and Prph2Y141C/+ were the most elevated at one hour before light onset, but steadily dropped over time reaching levels that were not significantly different from WT by eight hours after light onset, suggesting that in these models, the normal rhythm of OS digestion and clearance may be altered. Combined, these data demonstrate that Prph2 mutations can lead to a variety of different abnormalities in the clearance of OS material from the RPE.
3.4 |. The autophagy-lysosomal pathway is disrupted in the Prph2 disease models
To help understand potential mechanisms underlying the accumulation of OS material in the RPE in Prph2 disease models, we analyzed whether the autophagy-lysosomal pathway (ALP) was disrupted. After shed OS discs are phagocytosed by the RPE, the resulting endocytic vesicles enter the autophagy-lysosomal pathway as part of the process of digesting OS material.67–71 The microtubule-associated protein 1 light chain 3 (LC3) plays an essential role in LC3-associated phagocytosis, a process which together with classic phagocytosis mediates the degradation of the shed discs.72–74 The isoform LC3b is the most prevalent in the murine RPE, and its absence causes a delay in LC3-mediated phagocytosis.73 LC3b-i is distributed diffusely in the cell, while the lipidated form, LC3b-ii, is bound to autophagosome membranes and exhibits a more punctate labeling pattern.75 As autophagosomes fuse with lysosomes, the LC3b-ii protein is degraded. We co-stained retinal sections harvested at 12:00 p.m. from our disease models at P30 and P180 for LC3b (green, antibodies recognize LC3b-i and LC3b-ii) and RPE65 (red, Figure 5). At P30, WT retinas exhibit very low levels of LC3b staining, with a few small puncta visible in the RPE. In contrast, RPEs from the disease models exhibit increased LC3b labeling compared to WT as well as aggregation of LC3b into larger clumps (arrows, Figure 5A). Though present in all disease models, this phenotype was most pronounced in Prph2C213Y/+ and Prph2Y141C/+ samples. At P180, there were slightly more LC3b puncta in the WT RPE, but they were still mostly small, and labeling was less pronounced than in the disease models (Figure 5B). The accumulation of LC3b and, particularly, large aggregates suggest that while LC3b-mediated phagocytosis still occurs in the RPE of the disease models, the subsequent processing of the autophagosome-lysosome pathway is impaired. Another observation we made was a trend of LC3b to accumulate in the nuclei of the disease models (Figure 5). The nuclear trapping of LC3b is usually accompanied by a decrease in autophagy efficiency and thus supports a negative impact on autophagy caused by pathological mutation in Prph2.76 To help understand whether Prph2 accumulated in these LC3b positive puncta, we co-labeled retinal sections for Prph2 and LC3b. We observed substantial overlap between Prph2 and LCB3b in the disease models (white arrowheads, Figure 6). This partial co-localization was seen at P30 (Figure 6A) and persisted at P180 (Figure 6B), and suggests that at least a portion of the Prph2 vesicles accumulating in the disease models enter the LC3-associated ALP pathway.
FIGURE 5.
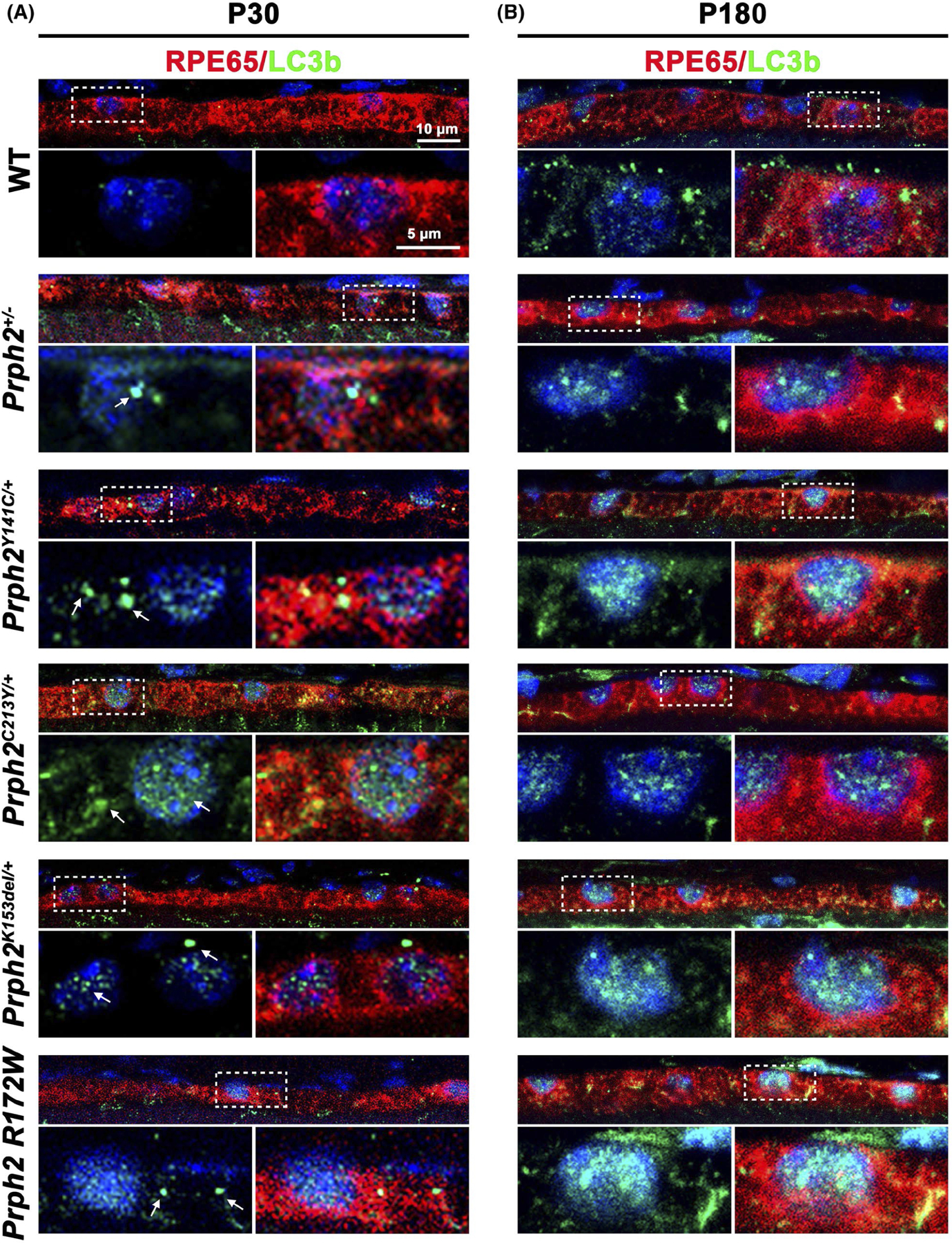
LC3b positive aggregates increase in Prph2 disease models. (A, B) Retinal sections harvested at P30 (A) and P180 (B) were co-labeled with LC3b (green) and RPE65 (red) and counterstained with DAPI (blue). Dotted outlined areas are magnified in panels shown below. Arrows highlight accumulation of LC3b aggregates in (A). Magnification 63×. Scale bars: 10 μm (top images) and 5 μm (insets) N = 3 eyes per genotype and age
FIGURE 6.
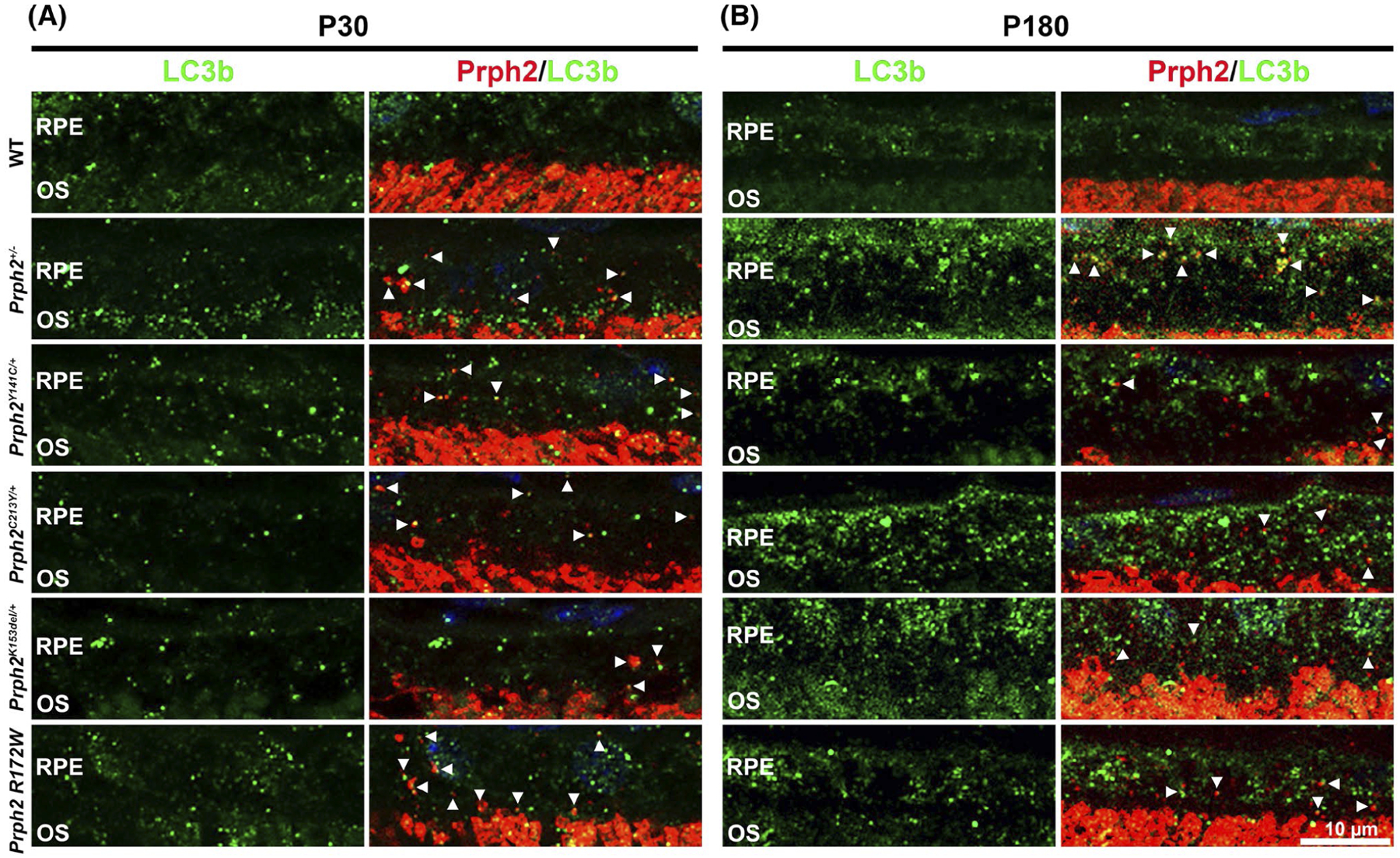
Prph2 positive vesicles co-localize with LC3b. Retina sections harvested at noon from animals aged P30 (A) and P180 (B) were co-labeled with LC3b (green) and Prph2 (red) and counterstained with DAPI (blue). Arrowheads highlight co-localization between Prph2 and LC3b. OS, outer segment; RPE, retinal pigment epithelium. Magnification 63×. Scale bar: 10 μm
Lysosomal associated membrane protein-1 (LAMP-1) is also involved in the ALP. It is found on lysosomes and some other endosomal vesicles and plays a key role in the formation and sorting of phagocytic vesicles77–80 and is thus a good marker to evaluate the accumulation of endocytic pathway components. Immunofluorescence labeling shows that at P30, Prph2C213Y/+ and Prph2Y141C/+ exhibit the accumulation of LAMP-1 signal in the RPE compared to WT (arrows, Figure 7A, brightfield images are shown on the right to help define the RPE). By P180, all of the Prph2 disease models exhibit dramatic increase in LAMP-1 expression in the RPE compared to WT (arrows, Figure 7B). Taken together these results provide evidence for an accumulation of phagocytic vesicles in the RPE of the Prph2 disease models, suggesting that normal processing through the ALP system is impaired.
FIGURE 7.
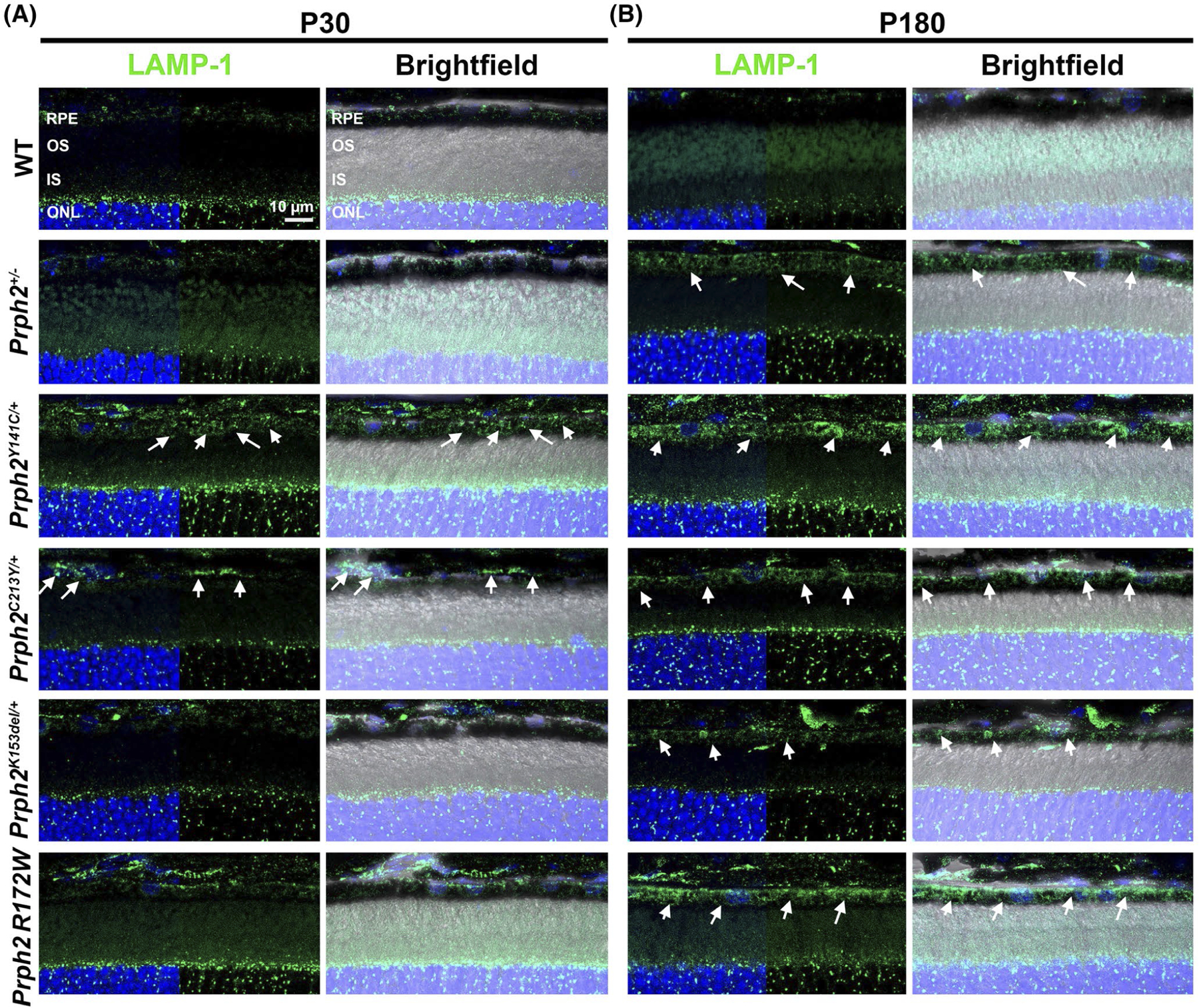
Lysosomes and other LAMP-1 positive vesicles accumulate in the RPE of Prph2 disease models. Retinal sections harvested at P30 (A) and P180 (B) were labeled with LAMP-1 (green) and counterstained with DAPI (blue). Right panels in each pair have brightfield overlay to help localize the RPE layer. Arrows highlight increased expression of LAMP-1. Magnification 63×. Scale bar: 10 μm. N = 3 eyes per genotype and age. IS, inner segments; ONL, outer nuclear layer; OS, outer segments; RPE, retinal pigment epithelium
3.5 |. Microglia are present in the RPE in Prph2 disease models
Delayed clearing of phagocytic vesicles from the RPE can result in an inflammatory reaction,81,82 reflected by an increase in microglia in the outer retina. Given our observed accumulation of phagocytic vesicles and the RPE hypertrophy (typically associated with RPE cell loss83), we asked whether there were signs of increased microglia adjacent to the RPE. We fluorescently labeled RPE flat mounts with the microglia marker Iba1 (red, Figure 8) at P30 and P180. No microglia were observed in the RPE of WT mice; however, all disease models exhibited microglia at the RPE at P30 (Figure 8A). At P180, microglia were even more prevalent in the Prph2Y141C/+ and Prph2K153del/+ models (arrows, Figure 8B) and also exhibited a more activated cell morphology characterized by larger cell size and the presence of lamellipodial extensions (arrowheads, Figure 8B).
FIGURE 8.
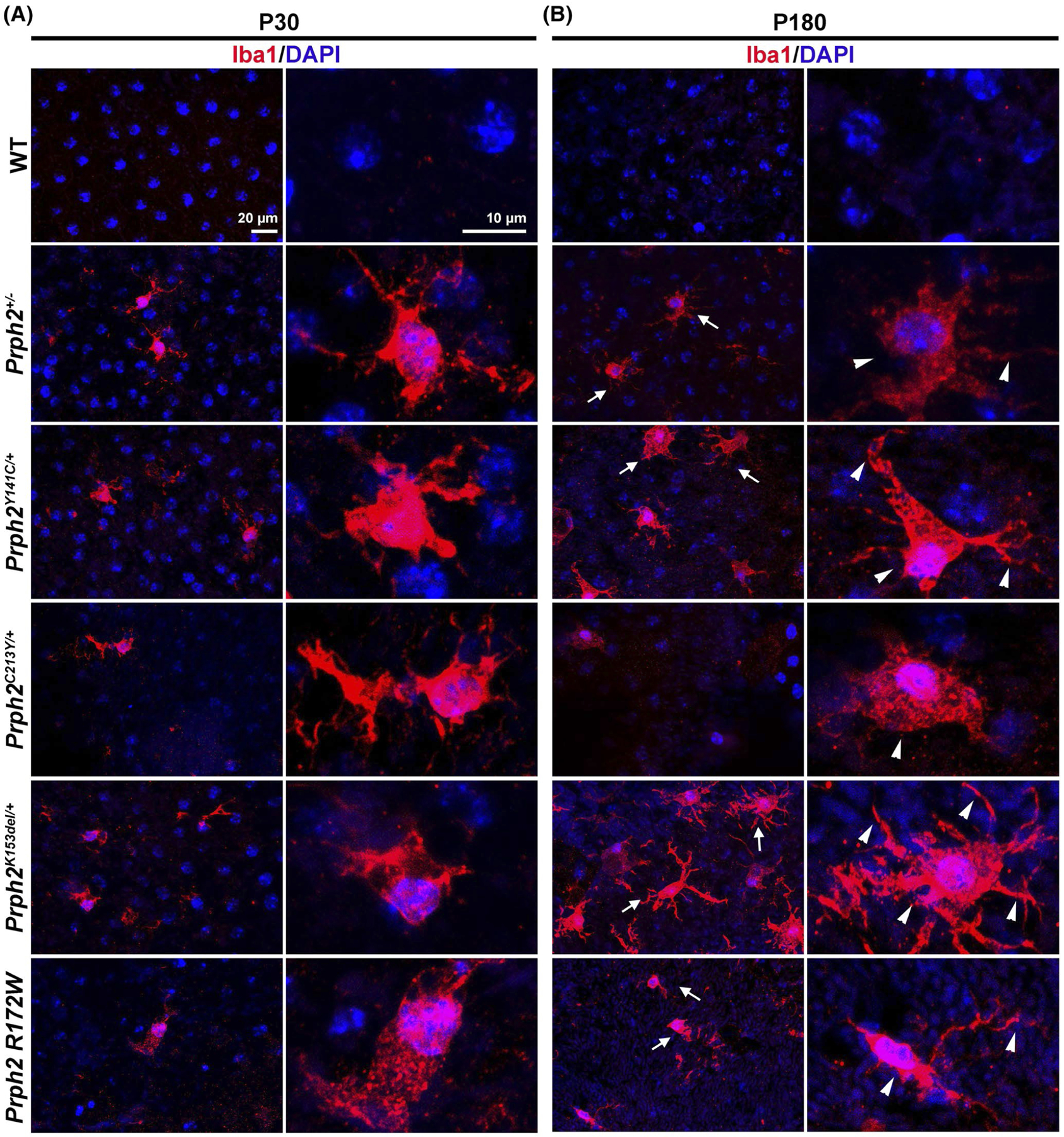
Microglia accumulate in the RPE of the models of Prph2 related dieseases. RPE whole mounts harvested at P30 (A) and P180 (B) were labeled with Iba1 (red) to label microglia and counterstained with DAPI (blue). Arrows in (B) show more prevlance of microglia at P180 in the disease models while arrowheads show larger cell size and the presence of lamellipodial extensions in all P180 models. Right panels in each pair show magnified views from left panels. Magnification, 40×. N = 3 flat mounts per genotype and age
4 |. DISCUSSION
In this study, we demonstrate that, in addition to their well-established effects on photoreceptors, mutations in the photoreceptor-specific gene, Prph2, lead to severe defects in RPE structure and function and promote the recruitment of inflammatory cells to the outer retina. To help understand which of our models exhibit the most severe RPE phenotypes, Table 2 provides a qualitative summary of each of the parameters we examined. While there is variability depending on the experiment, we did observe consistent trends. Prph2C213Y/+ and Prph2Y141C/+ retinas exhibited the most pronounced accumulation of OS material in the RPE by several measures, including the accumulation of LC3b aggregates, LAMP-1 staining, and the presence of OS material in the RPE on EM. Prph2C213Y/+ also exhibited the most pronounced defects in the RPE structure. In contrast, Prph2K153del/+ exhibited more signs of RPE hypertrophy and multinucleation than the other models, a sign of RPE cell loss. Prph2K153del/+ also exhibited the greatest degree of Iba1-positive cells, indicating an accumulation of activated microglia. The models used in this study display different levels of reduction in Prph2 protein levels (compared to WT, summarized in Table 2), but the severity of RPE phenotypes does not directly correlate with Prph2 levels.
TABLE 2.
Summary of phenotypes
| Most severe > Least severe | |
|---|---|
| Prph2 protein levels | |
| Prph2 levelsb | Prph2+/− > C213Y > K153del > R172W = Y141C |
| Structural markers | |
| EM vacuoles/paracellular spaces | C213Y = K153del = Y141C > Prph2+/− = R172Wa |
| ZO-1 gaps | C213Y > Y141C > Prph2+/− = R172W > K153del |
| Hypertrophy and multinucleation | K153del > Prph2+/− = Y141C > R172W > C213Y |
| ERG function | |
| c-wave amplitude | C213Y = Prph2+/− > Y141C = K153del > R172W |
| Impaired phagocytosis | |
| OS material on EM | C213Y = Y141C = Prph2+/− > K153del = R172W |
| Clearance of Prph2-labeled puncta | R172W > Prph2+/− = Y141C > K153del = C213Y |
| LC3b aggregates and levels | C213Y = Y141C > Prph2+/− = R172W > K153del |
| LAMP-1 expression | C213Y = Y141C > Prph2+/− = R172W > K153del |
| Inflammation | |
| Microglia accumulation | K153del > Y141C = R172W > Prph2+/− = C213Y |
The equal sign (=) denotes genotypes that exhibited a similar degree of severity. Genotypes on the left of the “greater than” sign (>) exhibit more severe phenotypes than those to the right.
Prph2 levels were taken from previously published work.18–20 Prph2 is reduced as follows: Prph2+/−, 40% of WT; Prph2C213Y/+, 50% of WT; Prph2K153del/+, 80% of WT; Prph2Y141C/+, no reduction (i.e. 100% of WT).
The transgenic model (Prph2 R172W) does not display a reduction Prph2.
While there are few physiological assays to measure RPE health in vivo, we observed a decrease in the amplitude of the ERG c-wave in all disease models. When the phototransduction cascade is triggered by light, the dark current is inhibited, which decrease the transport of K+ from the OS to the subretinal space, thus decreasing the concentration of extracellular K+ in the subretinal space.58,60,84,85 If K+ concentrations drop too low in the subretinal space, the phototransduction cascade can be disrupted58; therefore, decreased [K+] in the subretinal space triggers the hyperpolarization of the apical membrane of the RPE,58,86,87 and release of K+ from the RPE to the subretinal space compensates for the [K+] loss caused by the end of the dark current.58 This RPE hyperpolarization, together with the activity of the Kir4.1 channel in the Müller glial cells, forms the c-wave observed in the ERG.58,59,88–91 The severe decrease in the amplitude of the c-wave, which we observe in all of our Prph2 disease models, thus suggests that the ability of the RPE cells to respond to changes in the environment may be impaired. This has been found in other models of RPE-associated retinal disease. For example, in a mouse model for Stargardt-like macular dystrophy, as well as an aryl hydrocarbon receptor (AHR) knockout mouse model, the c-wave amplitudes and RPE structures were significantly impaired.92,93 However, because c-waves rely on initial light-evoked signals in photoreceptors, much of the observed reduction in c-waves may be due to overall reductions in phototransduction. As photoreceptor-generated signals drop due to the Prph2 mutations, the overall decrease in extracellular [K+] may be reduced, leading to a smaller compensatory hyperpolarization of the RPE. As a result, c-wave ERG may not be an optimal method for assessment of RPE functional responses in cases where severe defects in photoreceptor function exist.
One of our most striking findings is the observation that OS disc digestion in the RPE is impaired or delayed in Prph2 models. The processing of OS discs requires several steps, including the binding of the shed discs to the apical membrane of the RPE, internalization, and finally digestion of the internalized discs via the endocytic pathway and the ALP.35,94 The accumulation of undigested discs in the RPE in our models suggests that both the binding and internalization steps are unaffected, while the actual digestion of the discs is impaired. Previous in vitro and in vivo experiments have shown that in addition to classical phagocytosis, a portion of the digestion of shed OS is achieved via ALP.72–74 The disease models analyzed in our study all displayed an accumulation of LC3b, a marker for ALP, a partial co-localization of LC3b and Prph2 in the Prph2 models, and an increase in the expression of LAMP-1, a marker of lysosomes and other endocytic vesicles. These results indicate a delay (or impairment) in either the processing/maturation of LC3b positive autophagosomes or their subsequent fusion with lysosomes (after which stage LC3b is degraded along with the other contents of the lysosome). We observed a modest increase in RPE cell size and reduction in RPE cell number, raising the possibility that impaired clearance of OS material contributes to overt RPE toxicity. This situation is complex; while impaired clearance may eventually lead to cell death, a reduction in cell number may then exacerbate defects in OS clearance (as the number of cells to process OS is reduced).
Since the RPE does not express Prph2 itself, delayed RPE clearance of OS material is most likely due to the altered OS structure or protein content in the Prph2 disease models. Recent work has demonstrated that a delay in phagocytic clearance of OS material can be caused by altered content of the phagocytic vesicle.95 A mouse model of Stargardt macular degeneration, in which mutant human elongation of very long chain fatty acid (ELOVL)-4 protein is mislocalized to the OS and displays phagocytic vesicles with an altered content, which causes a delayed processing of resultant vesicles in the RPE.95 This study pinpointed sequestration of proteins involved in vesicular trafficking in the RPE (specifically Rab7a and dynein) in this pathology. We have shown that our various disease models exhibit alterations in the normal quantity and type of Prph2/Rom1 oligomers, and many of the models also exhibit increased signs of extremely large covalently linked Prph2 complexes. These aggregate-like complexes are especially prevalent in the Y141C and K153del models,18,19 both of which exhibit delayed clearance of OS material in the RPE. However, abnormally large Prph2 protein complexes are unlikely to be the sole driver of delayed or impaired clearance of OS material, because other Prph2 models with abnormalities in OS clearance, such as the Prph2+/− and the Prph2C213Y/+ have reduced levels of Prph2 protein complexes,3,20 and the model with the most significantly impaired OS clearance based on our time course studies (the Prph2 R172W) exhibits only very modest alterations in Prph2/Rom1 complex formation and only in cones.15,17
A second possible contributor to delayed clearance of OS material from the RPE is the altered morphology of the OS. The Prph2 mutations we characterize here almost all lead to OSs with significant malformations. The OSs in the Prph2C213Y/+, Prph2Y141C/+, Prph2+/−, and Prph2K153del/+ all exhibit varying degrees of whorl formation and OS shortening. It is possible that some structural aspect of this altered morphology leads to impaired OS clearance. However, again, this is unlikely to be the only mechanism as rod OS (representing the major OS burden in the mouse retina) are largely normal at P30 in the Prph2 R172W retina. A third possibility is that accumulation of OS material in the RPE arises not because clearance is impaired but because uptake of OS material is increased. Normally OS shedding and synthesis are precisely balanced to ensure that OS length is maintained, but it is not clear how this process may be altered in the radically misformed OS of Prph2 mutants. In addition, in most of the mutant models, the malformed OS are much shorter than WT OS, suggesting that the rate of shedding/phagocytosis may be increased compared to the rate of OS synthesis. In the absence of proper Prph2/Rom1 complexes and the accompanying proper disc architecture, the malformed OS may be more fragile than WT OS leading to increased breakage/accumulation of OS debris in the subretinal space. Such excess material would be phagocytosed by the RPE, potentially overwhelming the natural capacity of the RPE to digest it. Yet once again, the presence of prevalent undigested OS material in the RPEs of the Prph2 R172W line, which has virtually normal OS length,15,16 suggests that this third mechanism cannot be the sole contributor. Interestingly, we find that by some measures, the abnormal accumulation of OS material in the RPE is reduced at P180 compared to P30 in most of our models. This may be because ongoing photoreceptor degeneration in these models reduces the total amount of OS material the RPE must process.
The case of the R172W mutation is interesting. This mutation is one of the best characterized, both in mice and in patients,15–17,25,96–99 with data from many different affected families. In contrast to many other PRPH2 mutations, patients carrying the R172W mutation exhibit a relatively low degree of phenotypic variability, usually presenting with macular dystrophy. Similar to other forms of macular degeneration (such as AMD), R172W-associated disease is often linked to pronounced atrophy of the macula99–102 involving photoreceptors, the RPE, and the choroid. Though mice lack a macula, in our animal model, the R172W mutation still preferentially affects cones, and here, we find that it also leads to signs of RPE stress and impaired function. The connection between these last two observations remains unclear however. Though cones are severely affected (both in terms of structure and function in our mouse model), they make up a very small fraction (~3%–5%) of total photoreceptors in the mouse; therefore, even if abnormal cone OS are difficult for the RPE to digest, it is unclear why the levels of undigested OS material in Prph2 R172W RPEs should be so high. Additionally, the decline in cone function in the Prph2 R172W was followed by a decline in rod function, as well as structural abnormalities in the rod OS.15 Thus, the accumulation of Prph2 vesicle could also be contributed to the structural and functional decline of rods following the degeneration of cones.
The signs of RPE stress and/or degeneration we see in Prph2 models are common features of many retinopathies, including AMD. Overall our findings suggest that impaired clearance of accumulated OS material from mutant Prph2 photoreceptors leads to RPE cell stress, signs of inflammation, and ultimately RPE cell loss. The critical supportive role the RPE plays in the retina means that this disruption in RPE cells is likely a key novel mechanism contributing to the complex and debilitating disease pathology associated with PRPH2 mutations.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Robert S. Molday (University of British Columbia) for generously providing the 1D4 antibody against rhodopsin.
Funding information
This work was supported by grants from the National Institutes of Health (EY10609 & EY018656-MIN&MRA, P20 GM125528, R01-AG070915-SMC)
Abbreviations:
- ALP
autophagy-lysosomal pathway
- ELOVL4
elongation of very long chain fatty acids 4
- EM
electron microscopy
- ERG
electroretinography
- IACUC
Institutional Animal Care and Use Committee
- LAMP-1
lysosomal associated membrane protein-1
- LC3
microtubule-associated protein 1 light chain 3
- MD
macular dystrophy
- OS
outer segment
- P
postnatal day
- PBS
phosphate buffered saline
- PFA
paraformaldehyde
- Prph2
Peripherin 2
- Rom1
rod outer segment membrane protein 1
- RP
retinitis pigmentosa
- RPE
retinal pigment epithelium
- WT
wild-type
- ZO-1
zonula occludens 1
Footnotes
DISCLOSURES
The authors declare no conflicts of interest.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the methods and/or supplementary material of this article.
REFERENCES
- 1.Molday RS, Hicks D, Peripherin ML. A rim-specific membrane protein of rod outer segment discs. Invest Ophthalmol Vis Sci. 1987;28(1):50–61. [PubMed] [Google Scholar]
- 2.Arikawa K, Molday LL, Molday RS, Williams DS. Localization of peripherin/RDS in the disk membranes of cone and rod photoreceptors: relationship to disk membrane morphogenesis and retinal degeneration. J Cell Biol. 1992;116(3):659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng T, Peachey NS, Li S, Goto Y, Cao Y, Naash MI. The effect of peripherin/RDS haploinsufficiency on rod and cone photoreceptors. J Neurosci. 1997;17(21):8118–8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boesze-Battaglia K, Lamba OP, Napoli AA Jr, Sinha S, Guo Y. Fusion between retinal rod outer segment membranes and model membranes: a role for photoreceptor peripherin/RDS. Biochemistry. 1998;37(26):9477–9487. [DOI] [PubMed] [Google Scholar]
- 5.Wrigley JD, Ahmed T, Nevett CL, Findlay JB. Peripherin/RDS influences membrane vesicle morphology. Implications for retinopathies. J Biol Chem. 2000;275(18):13191–13194. [DOI] [PubMed] [Google Scholar]
- 6.Conley SM, Cai X, Naash MI. Nonviral ocular gene therapy: assessment and future directions. Curr Opin Mol Ther. 2008;10(5):456–463. [PMC free article] [PubMed] [Google Scholar]
- 7.Han Z, Anderson DW, Papermaster DS. Prominin-1 localizes to the open rims of outer segment lamellae in Xenopus laevis rod and cone photoreceptors. Invest Ophthalmol Vis Sci. 2012;53(1):361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moritz OL, Molday RS. Molecular cloning, membrane topology, and localization of bovine rom-1 in rod and cone photoreceptor cells. Invest Ophthalmol Vis Sci. 1996;37(2):352–362. [PubMed] [Google Scholar]
- 9.Goldberg AF, Fales LM, Hurley JB, Khattree N. Folding and subunit assembly of photoreceptor peripherin/RDS is mediated by determinants within the extracellular/intradiskal EC2 domain: implications for heterogeneous molecular pathologies. J Biol Chem. 2001;276(46):42700–42706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loewen CJ, Molday RS. Disulfide-mediated oligomerization of peripherin/RDS and rom-1 in photoreceptor disk membranes. Implications for photoreceptor outer segment morphogenesis and degeneration. J Biol Chem. 2000;275(8):5370–5378. [DOI] [PubMed] [Google Scholar]
- 11.Chakraborty D, Ding XQ, Fliesler SJ, Naash MI. Outer segment oligomerization of RDS: evidence from mouse models and subcellular fractionation. Biochemistry. 2008;47(4):1144–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boon CJ, den Hollander AI, Hoyng CB, Cremers FP, Klevering BJ, Keunen JE. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog Retin Eye Res. 2008;27(2):213–235. [DOI] [PubMed] [Google Scholar]
- 13.Francis PJ, Schultz DW, Gregory AM, et al. Genetic and phenotypic heterogeneity in pattern dystrophy. Br J Ophthalmol. 2005;89(9):1115–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gocho K, Akeo K, Itoh N, et al. High-resolution adaptive optics retinal image analysis at early stage central areolar choroidal dystrophy with PRPH2 mutation. Ophthalmic Surg Lasers Imaging Retina. 2016;47(12):1115–1126. [DOI] [PubMed] [Google Scholar]
- 15.Ding XQ, Nour M, Ritter LM, Goldberg AF, Fliesler SJ, Naash MI. The R172W mutation in peripherin/RDS causes a cone-rod dystrophy in transgenic mice. Hum Mol Genet. 2004;13(18):2075–2087. [DOI] [PubMed] [Google Scholar]
- 16.Conley S, Nour M, Fliesler SJ, Naash MI. Late-onset cone photoreceptor degeneration induced by R172W mutation in RDS and partial rescue by gene supplementation. Invest Ophthalmol Vis Sci. 2007;48(12):5397–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conley SM, Stuck MW, Burnett JL, et al. Insights into the mechanisms of macular degeneration associated with the R172W mutation in RDS. Hum Mol Genet. 2014;23(12):3102–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stuck MW, Conley SM, Naash MI. The Y141C knockin mutation in RDS leads to complex phenotypes in the mouse. Hum Mol Genet. 2014;23(23):6260–6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakraborty D, Conley SM, Zulliger R, Naash MI. The K153Del PRPH2 mutation differentially impacts photoreceptor structure and function. Hum Mol Genet. 2016;25(16):3500–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakraborty D, Strayve DG, Makia MS, et al. Novel molecular mechanisms for Prph2-associated pattern dystrophy. FASEB J. 2020;34(1):1211–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khani SC, Karoukis AJ, Young JE, et al. Late-onset autosomal dominant macular dystrophy with choroidal neovascularization and nonexudative maculopathy associated with mutation in the RDS gene. Invest Ophthalmol Vis Sci. 2003;44(8):3570–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fossarello M, Bertini C, Galantuomo MS, Cao A, Serra A, Pirastu M. Deletion in the peripherin/RDS gene in two unrelated Sardinian families with autosomal dominant butterfly-shaped macular dystrophy. Arch Ophthalmol. 1996;114(4):448–456. [DOI] [PubMed] [Google Scholar]
- 23.Nichols BE, Sheffield VC, Vandenburgh K, Drack AV, Kimura AE, Stone EM. Butterfly-shaped pigment dystrophy of the fovea caused by a point mutation in codon 167 of the RDS gene. Nat Genet. 1993;3(3):202–207. [DOI] [PubMed] [Google Scholar]
- 24.Weleber RG, Carr RE, Murphey WH, Sheffield VC, Stone EM. Phenotypic variation including retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of the peripherin/RDS gene. Arch Ophthalmol. 1993;111(11):1531–1542. [DOI] [PubMed] [Google Scholar]
- 25.Wroblewski JJ, Wells JA, Eckstein A, et al. Macular dystrophy associated with mutations at codon 172 in the human retinal degeneration slow gene. Ophthalmology. 1994;101(1):12–22. [DOI] [PubMed] [Google Scholar]
- 26.Zhang K, Garibaldi DC, Li Y, Green WR, Zack DJ. Butterfly-shaped pattern dystrophy: a genetic, clinical, and histopathological report. Arch Ophthalmol. 2002;120(4):485–490. [DOI] [PubMed] [Google Scholar]
- 27.Naylor A, Hopkins A, Hudson N, Campbell M. Tight junctions of the outer blood retina barrier. Int J Mol Sci. 2019;21(1):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sparrow JR, Hicks D, Hamel CP. The retinal pigment epithelium in health and disease. Curr Mol Med. 2010;10(9):802–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cunha-Vaz J, Bernardes R, Lobo C. Blood-retinal barrier. Eur J Ophthalmol. 2011;21(Suppl 6):S3–S9. [DOI] [PubMed] [Google Scholar]
- 30.Wald G, Brown PK. Synthesis and bleaching of rhodopsin. Nature. 1956;177(4500):174–176. [DOI] [PubMed] [Google Scholar]
- 31.Wimmers S, Karl MO, Strauss O. Ion channels in the RPE. Prog Retin Eye Res. 2007;26(3):263–301. [DOI] [PubMed] [Google Scholar]
- 32.Lakkaraju A, Umapathy A, Tan LX, et al. The cell biology of the retinal pigment epithelium. Prog Retin Eye Res. 2020;78:100846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young RW. The renewal of photoreceptor cell outer segments. J Cell Biol. 1967;33(1):61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young RW, Bok D. Participation of the retinal pigment epithelium in the rod outer segment renewal process. J Cell Biol. 1969;42(2):392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kevany BM, Palczewski K. Phagocytosis of retinal rod and cone photoreceptors. Physiology (Bethesda). 2010;25(1):8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marneros AG, Fan J, Yokoyama Y, et al. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am J Pathol. 2005;167(5):1451–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bressler SB. Introduction: understanding the role of angiogenesis and antiangiogenic agents in age-related macular degeneration. Ophthalmology. 2009;116(10 Suppl):S1–S7. [DOI] [PubMed] [Google Scholar]
- 38.Farjood F, Vargis E. Physical disruption of cell-cell contact induces VEGF expression in RPE cells. Mol Vis. 2017;23:431–446. [PMC free article] [PubMed] [Google Scholar]
- 39.Gourier HC, Chong NV. Can Novel treatment of age-related macular degeneration be developed by better understanding of Sorsby’s fundus dystrophy. J Clin Med. 2015;4(5):874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu S-M, Thompson DA, Srikumari CRS, et al. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet. 1997;17(2):194–197. [DOI] [PubMed] [Google Scholar]
- 41.Guziewicz KE, Sinha D, Gómez NM, et al. Bestrophinopathy: an RPE-photoreceptor interface disease. Prog Retin Eye Res. 2017;58:70–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones BW, Pfeiffer RL, Ferrell WD, Watt CB, Marmor M, Marc RE. Retinal remodeling in human retinitis pigmentosa. Exp Eye Res. 2016;150:149–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad Sci U S A. 1998;95(6):3088–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taubitz T, Tschulakow AV, Tikhonovich M, et al. Ultrastructural alterations in the retinal pigment epithelium and photoreceptors of a Stargardt patient and three Stargardt mouse models: indication for the central role of RPE melanin in oxidative stress. PeerJ. 2018;6:e5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Conley SM, Stuck MW, Watson JN, Naash MI. Rom1 converts Y141C-Prph2-associated pattern dystrophy to retinitis pigmentosa. Hum Mol Genet. 2017;26(3):509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacKenzie D, Arendt A, Hargrave P, McDowell JH, Molday RS. Localization of binding sites for carboxyl terminal specific anti-rhodopsin monoclonal antibodies using synthetic peptides. Biochemistry. 1984;23(26):6544–6549. [DOI] [PubMed] [Google Scholar]
- 47.Stricker HM, Ding XQ, Quiambao A, Fliesler SJ, Naash MI. The Cys214–>Ser mutation in peripherin/RDS causes a loss-of-function phenotype in transgenic mice. Biochem J. 2005;388(Pt 2):605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farjo R, Skaggs JS, Nagel BA, et al. Retention of function without normal disc morphogenesis occurs in cone but not rod photoreceptors. J Cell Biol. 2006;173(1):59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chakraborty D, Ding XQ, Conley SM, Fliesler SJ, Naash MI. Differential requirements for retinal degeneration slow intermolecular disulfide-linked oligomerization in rods versus cones. Hum Mol Genet. 2009;18(5):797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, Dejana E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem. 2000;275(27):20520–20526. [DOI] [PubMed] [Google Scholar]
- 51.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84(3):869–901. [DOI] [PubMed] [Google Scholar]
- 52.Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol. 2006;22:207–235. [DOI] [PubMed] [Google Scholar]
- 53.Green WR. Histopathology of age-related macular degeneration. Mol Vis. 1999;5:27. [PubMed] [Google Scholar]
- 54.Feher J, Kovacs I, Artico M, Cavallotti C, Papale A, Balacco GC. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol Aging. 2006;27(7):983–993. [DOI] [PubMed] [Google Scholar]
- 55.Song D, Kanu LN, Li Y, et al. AMD-like retinopathy associated with intravenous iron. Exp Eye Res. 2016;151:122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Cross SD, Stanton JB, Marmorstein AD, Le YZ, Marmorstein LY. Early AMD-like defects in the RPE and retinal degeneration in aged mice with RPE-specific deletion of Atg5 or Atg7. Mol Vis. 2017;23:228–241. [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Q, Chrenek MA, Bhatia S, et al. Comparison of histologic findings in age-related macular degeneration with RPE flatmount images. Mol Vis. 2019;25:70–78. [PMC free article] [PubMed] [Google Scholar]
- 58.Strauss O The retinal pigment epithelium in visual function. Physiol Rev. 2005;85(3):845–881. [DOI] [PubMed] [Google Scholar]
- 59.Samuels IS, Sturgill GM, Grossman GH, Rayborn ME, Hollyfield JG, Peachey NS. Light-evoked responses of the retinal pigment epithelium: changes accompanying photoreceptor loss in the mouse. J Neurophysiol. 2010;104(1):391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steinberg RH. Interactions between the retinal pigment epithelium and the neural retina. Doc Ophthalmol. 1985;60(4):327–346. [DOI] [PubMed] [Google Scholar]
- 61.Steinberg RH, Schmidt R, Brown KT. Intracellular responses to light from cat pigment epithelium: origin of the electroretinogram c-wave. Nature. 1970;227(5259):728–730. [DOI] [PubMed] [Google Scholar]
- 62.Young RW. The daily rhythm of shedding and degradation of cone outer segment membranes in the lizard retina. J Ultrastruct Res. 1977;61(2):172–185. [DOI] [PubMed] [Google Scholar]
- 63.Young RW. The daily rhythm of shedding and degradation of rod and cone outer segment membranes in the chick retina. Invest Ophthalmol Vis Sci. 1978;17(2):105–116. [PubMed] [Google Scholar]
- 64.LaVail MM. Rod outer segment disk shedding in rat retina: relationship to cyclic lighting. Science. 1976;194(4269):1071–1074. [DOI] [PubMed] [Google Scholar]
- 65.Terman JS, Reme CE, Terman M. Rod outer segment disk shedding in rats with lesions of the suprachiasmatic nucleus. Brain Res. 1993;605(2):256–264. [DOI] [PubMed] [Google Scholar]
- 66.Grace MS, Wang LM, Pickard GE, Besharse JC, Menaker M. The tau mutation shortens the period of rhythmic photoreceptor outer segment disk shedding in the hamster. Brain Res. 1996;735(1):93–100. [DOI] [PubMed] [Google Scholar]
- 67.Herman KG, Steinberg RH. Phagosome degradation in the tapetal retinal pigment epithelium of the opossum. Invest Ophthalmol Vis Sci. 1982;23(3):291–304. [PubMed] [Google Scholar]
- 68.Herman KG, Steinberg RH. Phagosome movement and the diurnal pattern of phagocytosis in the tapetal retinal pigment epithelium of the opossum. Invest Ophthalmol Vis Sci. 1982;23(3):277–290. [PubMed] [Google Scholar]
- 69.Bosch E, Horwitz J, Bok D. Phagocytosis of outer segments by retinal pigment epithelium: phagosome-lysosome interaction. J Histochem Cytochem. 1993;41(2):253–263. [DOI] [PubMed] [Google Scholar]
- 70.Futter CE. The molecular regulation of organelle transport in mammalian retinal pigment epithelial cells. Pigment Cell Res. 2006;19(2):104–111. [DOI] [PubMed] [Google Scholar]
- 71.Wavre-Shapton ST, Meschede IP, Seabra MC, Futter CE. Phagosome maturation during endosome interaction revealed by partial rhodopsin processing in retinal pigment epithelium. J Cell Sci. 2014;127(Pt 17):3852–3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frost LS, Lopes VS, Bragin A, et al. The contribution of melanoregulin to microtubule-associated protein 1 light chain 3 (LC3) associated phagocytosis in retinal pigment epithelium. Mol Neurobiol. 2015;52(3):1135–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dhingra A, Alexander D, Reyes-Reveles J, Sharp R, Boesze-Battaglia K. Microtubule-associated protein 1 light chain 3 (LC3) isoforms in RPE and retina. Adv Exp Med Biol. 2018;1074:609–616. [DOI] [PubMed] [Google Scholar]
- 74.Kim J-Y, Zhao H, Martinez J, et al. Noncanonical autophagy promotes the visual cycle. Cell. 2013;154(2):365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saadat KASM, Murakami Y, Tan X, et al. Inhibition of autophagy induces retinal pigment epithelial cell damage by the lipofuscin fluorophore A2E. FEBS Open Bio. 2014;4:1007–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baeken MW, Weckmann K, Diefenthäler P, et al. Novel insights into the cellular localization and regulation of the autophagosomal proteins LC3A, LC3B and LC3C. Cells. 2020;9(10):2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med. 2006;27(5–6):495–502. [DOI] [PubMed] [Google Scholar]
- 78.Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007;26(2):313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cook NR, Row PE, Davidson HW. Lysosome associated membrane protein 1 (Lamp1) traffics directly from the TGN to early endosomes. Traffic (Copenhagen, Denmark). 2004;5(9):685–699. [DOI] [PubMed] [Google Scholar]
- 80.Cheng XT, Xie YX, Zhou B, Huang N, Farfel-Becker T, Sheng ZH. Revisiting LAMP1 as a marker for degradative autophagy-lysosomal organelles in the nervous system. Autophagy. 2018;14(8):1472–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Piippo N, Korkmaz A, Hytti M, et al. Decline in cellular clearance systems induces inflammasome signaling in human ARPE-19 cells. Biochem Biophys Acta. 2014;1843(12):3038–3046. [DOI] [PubMed] [Google Scholar]
- 82.Kauppinen A, Paterno JJ, Blasiak J, Salminen A, Kaarniranta K. Inflammation and its role in age-related macular degeneration. Cell Mol Life Sci. 2016;73(9):1765–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Telegina DV, Kozhevnikova OS, Bayborodin SI, Kolosova NG. Contributions of age-related alterations of the retinal pigment epithelium and of glia to the AMD-like pathology in OXYS rats. Sci Rep. 2017;7(1):41533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Steinberg RH, Linsenmeier RA, Griff ER. Three light-evoked responses of the retinal pigment epithelium. Vision Res. 1983;23(11):1315–1323. [DOI] [PubMed] [Google Scholar]
- 85.Dornonville de la Cour M. Ion transport in the retinal pigment epithelium. A study with double barrelled ion-selective microelectrodes. Acta Ophthalmol Suppl. 1993;209:1–32. [PubMed] [Google Scholar]
- 86.Oakley B 2nd. Potassium and the photoreceptor-dependent pigment epithelial hyperpolarization. J Gen Physiol. 1977;70(4):405–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Oakley B 2nd, Miller SS, Steinberg RH. Effect of intracellular potassium upon the electrogenic pump of frog retinal pigment epithelium. J Membr Biol. 1978;44(3–4):281–307. [DOI] [PubMed] [Google Scholar]
- 88.Steinberg RH, Miller S. Aspects of electrolyte transport in frog pigment epithelium. Exp Eye Res. 1973;16(5):365–372. [DOI] [PubMed] [Google Scholar]
- 89.Witkovsky P, Dudek FE, Ripps H. Slow PIII component of the carp electroretinogram. J Gen Physiol. 1975;65(2):119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Oakley B 2nd, Green DG. Correlation of light-induced changes in retinal extracellular potassium concentration with c-wave of the electroretinogram. J Neurophysiol. 1976;39(5):1117–1133. [DOI] [PubMed] [Google Scholar]
- 91.Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20(15):5733–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim S-Y, Yang H-J, Chang Y-S, et al. Deletion of aryl hydrocarbon receptor AHR in mice leads to subretinal accumulation of microglia and RPE atrophy. Invest Ophthalmol Vis Sci. 2014;55(9):6031–6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kuny S, Cho WJ, Dimopoulos IS, Sauve Y. Early onset ultrastructural and functional defects in RPE and photoreceptors of a Stargardt-Like Macular Dystrophy (STGD3) transgenic mouse model. Invest Ophthalmol Vis Sci. 2015;56(12):7109–7121. [DOI] [PubMed] [Google Scholar]
- 94.Nandrot EF. Animal models, in “The quest to decipher RPE phagocytosis”. Adv Exp Med Biol. 2014;801:77–83. [DOI] [PubMed] [Google Scholar]
- 95.Esteve-Rudd J, Hazim RA, Diemer T, et al. Defective phagosome motility and degradation in cell nonautonomous RPE pathogenesis of a dominant macular degeneration. Proc Natl Acad Sci U S A. 2018;115(21):5468–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Poloschek CM, Bach M, Lagreze WA, et al. ABCA4 and ROM1: implications for modification of the PRPH2-associated macular dystrophy phenotype. Invest Ophthalmol Vis Sci. 2010;51(8):4253–4265. [DOI] [PubMed] [Google Scholar]
- 97.Goldberg AF, Molday RS. Expression and characterization of peripherin/RDS-rom-1 complexes and mutants implicated in retinal degenerative diseases. Methods Enzymol. 2000;316:671–687. [DOI] [PubMed] [Google Scholar]
- 98.Wickham L, Chen FK, Lewis GP, et al. Clinicopathological case series of four patients with inherited macular disease. Invest Ophthalmol Vis Sci. 2009;50(8):3553–3561. [DOI] [PubMed] [Google Scholar]
- 99.Michaelides M, Holder GE, Bradshaw K, Hunt DM, Moore AT. Cone-rod dystrophy, intrafamilial variability, and incomplete penetrance associated with the R172W mutation in the peripherin/RDS gene. Ophthalmology. 2005;112(9):1592–1598. [DOI] [PubMed] [Google Scholar]
- 100.Nakazawa M, Wada Y, Tamai M. Macular dystrophy associated with monogenic Arg172Trp mutation of the peripherin/RDS gene in a Japanese family. Retina. 1995;15(6):518–523. [DOI] [PubMed] [Google Scholar]
- 101.Piguet B, Héon E, Munier FL, et al. Full characterization of the maculopathy associated with an Arg-172-Trp mutation in the RDS/peripherin gene. Ophthalmic Genet. 1996;17(4):175–186. [DOI] [PubMed] [Google Scholar]
- 102.Reig C, Serra A, Gean E, et al. A point mutation in the RDS-peripherin gene in a Spanish family with central areolar choroidal dystrophy. Ophthalmic Genet. 1995;16(2):39–44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the methods and/or supplementary material of this article.