

Abstract
Background:
Whereas human leukocyte antigen (HLA) class I mutation-associated neoantigen burden has been linked with response to immune checkpoint blockade (ICB), the role of HLA class II-restricted neoantigens in clinical responses to ICB is less studied. We used computational approaches to assess HLA class II immunogenic mutation (IMM) burden in patients with melanoma and lung cancer treated with ICB.
Patients and methods:
We analyzed whole-exome sequence data from four cohorts of ICB-treated patients with melanoma (n = 110) and non-small-cell lung cancer (NSCLC) (n = 123). MHCnuggets, a neural network-based model, was applied to estimate HLA class II IMM burdens and cellular fractions of IMMs were calculated to assess mutation clonality. We evaluated the combined impact of HLA class II germline genetic variation and class II IMM burden on clinical outcomes. Correlations between HLA class II IMM burden and density of tumor-infiltrating lymphocytes were computed from expression data.
Results:
Responding tumors harbored a significantly higher HLA class II IMM burden for both melanoma and NSCLC (P ≤ 9.6e—3). HLA class II IMM burden was correlated with longer survival, particularly in the NSCLC cohort and in the context of low intratumoral IMM heterogeneity (P < 0.001). HLA class I and II IMM landscapes were largely distinct suggesting a complementary role for class II IMMs in tumor rejection. A higher HLA class II IMM burden was associated with CD4+ T-cell infiltration and programmed death-ligand 1 expression. Transcriptomic analyses revealed an inflamed tumor microenvironment for tumors harboring a high HLA class II IMM burden.
Conclusions:
HLA class II IMM burden identified patients with NSCLC and melanoma that attained longer survival after ICB treatment. Our findings suggest that HLA class II IMMs may impact responses to ICB in a manner that is distinct and complementary to HLA class l-mediated responses.
Keywords: immunogenic mutations, HLA class II neoantigens, immune checkpoint blockade, clinical outcome
INTRODUCTION
Tumor mutational burden (TMB) has emerged as a biomarker for cancer treatments, particularly immune checkpoint blockade (ICB) treatments in solid tumors, but it remains an imperfect biomarker of response.1 Nuanced approaches that focus on the quality rather than the quantity of mutations harbored in coding regions of the tumor genome are therefore needed to enhance the predictive value of TMB.2–7 Historically, antitumor immune responses have been considered to be driven by human leukocyte antigen (HLA) class I-restricted cytotoxic CD8+ T-cell responses, whereas the role of HLA class II-restricted CD4+ T cells remains less understood, especially in the context of ICB.8 To this end, HLA class I immunogenic mutation (IMM) burden has been reported to be strongly associated with increased tumor-infiltrating CD8+ T-cell density and longer overall survival (OS) for patients with a variety of solid tumors.7,9,10 In the immunotherapy setting, previous efforts have focused on investigating the value of HLA class I neoepitope burden in predicting clinical outcomes with ICB.11–13 Preclinical studies have shown that CD4+ T-cell neoepitope vaccination induced complete tumor rejection, tumor microenvironment (TME) re-shaping and antigen spread.14 Furthermore, tumor rejection in the context of immunotherapy has been shown to require the activity of both antigen-specific CD8+ and CD4+ T cells, suggesting the non-overlapping but complementary role of HLA class I and class II-restricted neoantigens.13 Despite the strong biological rationale, the value of HLA class II mutation and neoantigen burden in predicting response to ICB has not been previously systematically assessed across cancer types.
More recently, several HLA class ll-restricted neoantigen computational prediction tools have been developed, leveraging peptides identified by mass spectrometry (MS) and enabling high-throughput systematic evaluation of patients’ HLA class II neoantigen landscapes.16,17 Despite the advantages of MS-based approaches that capture biological processes including protein cleavage, gene expression, and protein transport, MS data do not typically resolve one-to-one peptide-HLA allele relationships. To alleviate this issue, MS data deconvolution can be combined by in vitro HLA allele-specific binding affinity assessments to computationally derive HLA class II-restricted neoantigen predictions.18 We have developed MHCnuggets, an open-source HLA class I and II neoantigen predictor that uses transfer learning to combine MS and HLA binding affinity data.7 Here, we extended the MHCnuggets framework by implementing an HLA affinity ranking system and applied this approach to evaluate the role of HLA class II IMM burden in four independent cohorts of patients with non-small-cell lung cancer (NSCLC) and melanoma treated with immunotherapy. We further integrated HLA class II IMMs with HLA germline variation, tumor clonal architecture and investigated the TME composition of tumors harboring a high class II IMM burden.
METHODS
Cohort data compilation
We analyzed whole exome sequence data of 233 patients with NSCLC and melanoma treated with ICB from four published cohorts: the Anagnostou_NSCLC cohort consisted of 89 patients with NSCLC treated with single agent or combination ICB1; the Rizvi_NSCLC cohort consisted of 34 patients with NSCLC treated with pembrolizumab19; the Anagnostou_Melanoma cohort consisted of 46 patients with melanoma treated with nivolumab or combination nivolumab/ipilimumab as part of the CheckMate 038 clinical trial,20 and the Snyder_Melanoma cohort consisted of 64 patients treated with ipilimumab.21 Demographics were extracted from the original publications. For the NSCLC cohorts, programmed death-ligand 1 (PD-L1) expression values and smoking status (former smoker, current smoker, and never smoker) were retrieved from the original publications (Supplementary Methods, available at https://doi.org/10.1016/j.annonc.2022.03.013). Clinical responses to ICB treatment were classified as durable clinical benefit (DCB) or non durable benefit (NDB). For all four cohorts, DCB was defined as radiographic complete response, partial response, or stable disease for a duration longer than 6 months.
Assessment of IMM
Whole exome sequencing data were analyzed with the Strelka mutation calling pipeline 22 (Supplementary Methods, available at https://doi.Org/10.1016/j.annonc.2022.03.013). All missense mutations identified in each cohort were considered as potential IMMs. Mutant and wild-type peptide sequences surrounding the affected amino acid were extracted, filtering out silent and nonsense mutations with ‘varcode’.23 Windowing around the affected amino acid, all possible 12–20mer24–26 mutant/reference peptide pairs were selected as potential HLA class II neoepitopes. HLA class I candidate neoepitopes were extracted for all possible 8–12mer mutant/reference peptide pairs.26–28 HLA class I were called with OptiType29 and HLA class II genotypes were called with SOAP-HLA30 and x-HLA31 (Supplementary Methods, available at https://doi.org/10.1016/j.annonc.2022.03.013).
To compute IMM burdens, we used MHCnuggets,7 and converted predicted HLA binding affinities for each peptide into a rank-based score {Supplementary Methods, available at https://doi.Org/10.1016/j.annonc.2022.03.013). Predicted neoepitopes were selected using affinity rank thresholds of <0.01, <0.001, and <0.0001. A threshold of 0.001 was used as an estimate of the actual presentation rate in the TME (Supplementary Methods, available at https://doi.org/10.1016/j-annonc.2022.03.013). IMMs were defined as missense mutations with at least one predicted mutation-association neoantigen (MANA) with an HLA affinity ranking at the 0.001 percentile for a given HLA haplotype. We computed IMM burden values for each tumor by counting IMMs, defined as mutations that produce at least one predicted MANA with affinity ranking as described above.
Intratumoral IMM heterogeneity analyses
Intratumoral HLA class I or II IMM heterogeneity was assessed by determining the fraction of subclonal IMMs as follows:
(Supplementary Methods, available at https://doi.org/10.1016/j.annonc.2022.03.013}. Samples with no IMMs were excluded from downstream analyses and IMM heterogeneity thresholds of 0.05 and 0.1 were considered. This approach is similar to that of McGranahan et al.32
Tumor-infiltrating immune cell subsets and T-cell receptor differential abundance analyses
RNA and T-cell receptor (TCR) sequencing data from the Anagnostou_Melanoma cohort were retrieved from the original publication and used to assess the TME composition before—and during—ICB treatment20 (Supplementary Methods, available at https://doi.Org/10.1016/j.annonc.2022.03.013). Normalized expression data were inputted in CIBERSORT vl.06 (https://cibersort.stanford.edu/)33 to deconvolute 22 immune cell types’ absolute composition. Absolute composition values of all 22 immune cell types were analyzed for differential comparison between high and low HLA class II IMM burden tumors. TCR clonotypic productive frequencies for the Anagnostou_Melanoma cohort were retrieved from the original publication.20 The number of total productive TCR clones and TCR repertoire clonality were utilized for differential abundance analyses between high and low HLA class II IMM burden tumors. Gene set enrichment analysis was carried out as described in the Supplementary Methods, available at https://doi.org/10.1016/j.annonc.2022.03.013.
Statistical analysis
The two-sided Mann—Whitney U test (MW) was used to evaluate differences in IMM burden and TMB with respect to DCB and NDB groups across all cohorts. For survival analysis, high IMM burden tumors were ranked in the fourth quartile (top 25%) of their cohort and low IMM burden individuals ranked in the first three quartiles (bottom 75%). NSCLC and melanoma cohorts were grouped by cancer type and analyzed with Bayesian survival models34 to confirm the absence of cohort effects (Supplementary Methods and Supplementary Table S1, available at https://doi.org/10.1016/j.annonc.2022.03.013). Downstream survival analyses were done with cohorts of the same cancer types grouped together to increase statistical power. Kaplan—Meier curves were used to visualize differences in survival for patients with high and low IMM burden tumors. Survival differences between groups were compared with a two-sided log-rank test and hazard ratios (HRs) were calculated by univariate Cox regressions. Multivariate Cox regression was used to evaluate the impact of established factors on patient survival. Hazard ratios and P values from the Wald test were reported. A P value threshold of 0.05 was used as the indication for statistical significance, and Benjamini-Hochberg false discovery rate (FDR) correction was applied. For correlation analyses, we used pairwise Spearman correlations, and P values were reported with Benjamini-Hochberg FDR corrections.
RESULTS
HLA class II IMM burden is linked with clinical benefit from ICB
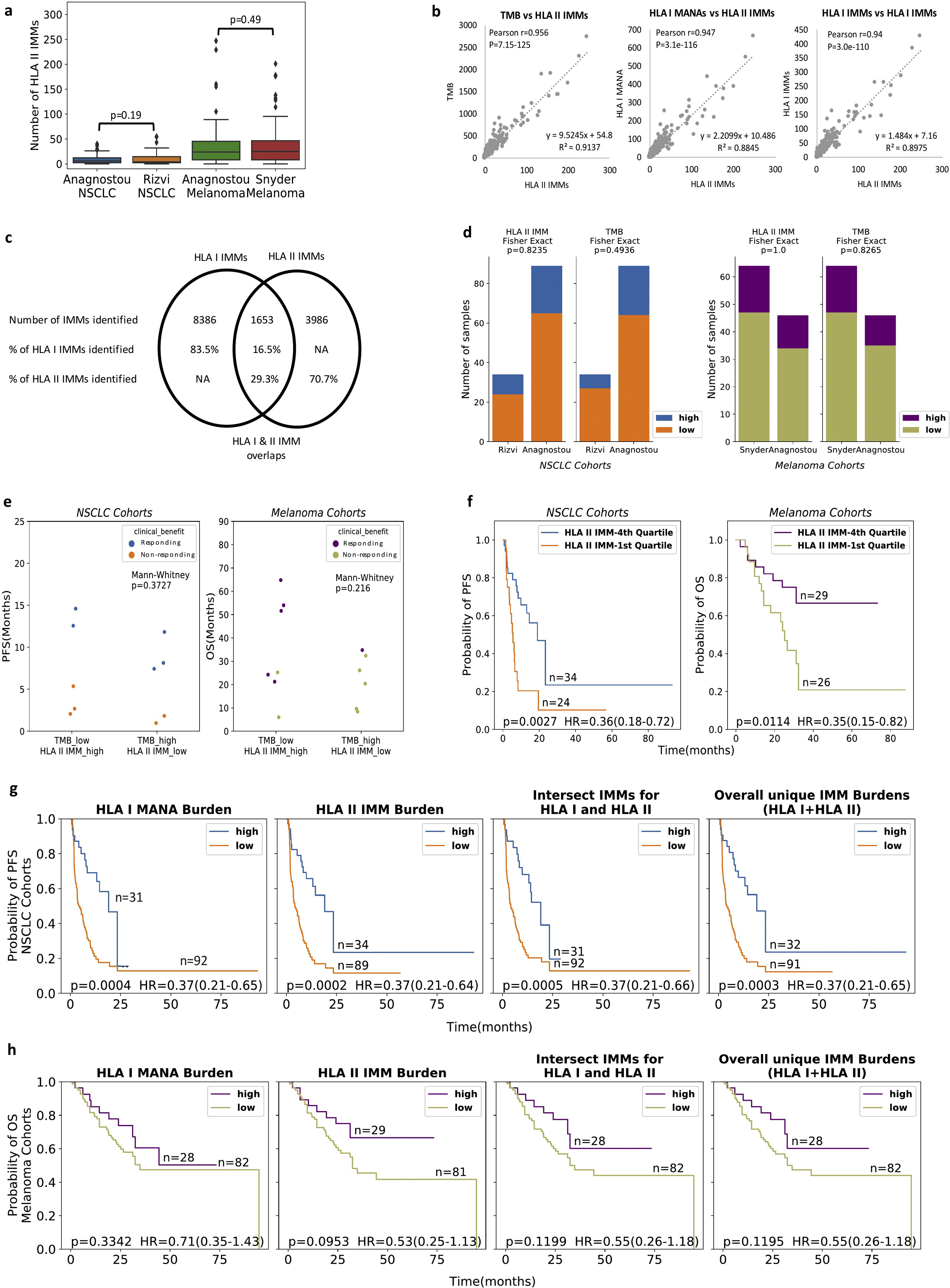
A total of 5639 HLA class II IMMs were identified in tumor samples from 233 individuals across all four NSCLC and melanoma cohorts. In NSCLC, average (± standard error) HLA class II IMM burden was 8.6 ± 0.95 (Anagnostou_NSCLC) and 10.94 ± 2.65 (Rizvi_NSCLC), whereas in melanoma, average HLA class II IMM burden was 41.48 ± 8.25 (Anagnostou_Melanoma) and 40.39 ± 5.96 (Snyder_Melanoma) (Supplementary Tables S2 and S3, available at https://doi.Org/10.1016/j.annonc.2022.03.013). We did not detect a difference in HLA class II IMM burden distributions between cohorts within tumor types (NSCLC cohorts MW P = 0.19; melanoma cohorts MW P = 0.50; Supplementary Figure S1A, available at https://doi.org/10.1016/j.annonc.2022.03.013). HLA class II IMM burdens in the NSCLC (median = 6 mutations) and melanoma cohorts (median = 25 mutations) were an order of magnitude smaller than TMB in NSCLC (median = 97 mutations) and melanoma (median = 283 mutations). Additionally, HLA class II IMM burdens were smaller than HLA class I IMM burdens in NSCLC (median = 13 mutations) and melanoma (median = 43 mutations) (Supplementary Table S2 and Supplementary Figure SIB, available at https://doi.org/10.1016/j.annonc.2022.03.013). In all four cohorts, the HLA class II IMM burden of tumors responding to therapy was consistently higher than that of non-responding tumors (Anagnostou_NSCLC MW P = 0.00032; Rizvi_ NSCLC MW P = 0.0096; Anagnostou_Melanoma MW P = 0.001; Snyder_Melanoma MW P = 0.0011; Figure 1A-D). HLA class II IMM burdens were highly correlated with TMB (Pearson’s r = 0.96, P < 0.0001), as well as with HLA class I IMM burdens (Pearson’s r = 0.95, P < 0.0001) and with HLA class I MANA burdens (Pearson’s r = 0.94, P < 0.0001; Supplementary Figure S1B, available at https://doi.org/10.1016/j.annonc.2022.03.013). Less than one-third of HLA class II IMMs (1653 out of 5639, 29%) overlapped with HLA class I IMMs (Supplementary Figure S1C, available at https://doi.Org/10.1016/j.annonc.2022.03.013).
Figure 1. HLA class II immunogenic mutation burden is a marker for patient response and survival.

(A)-(D) HLA class II IMM burden and TMB both separate responder and non-responder groups in all four cohorts (Anagnostou NSCLC HLA II IMM burden MW P = 0.0003, TMB MW P = 0.0018; Rizvi NSCLC HLA II IMM burden MW P = 0.0096, TMB MW P = 0.00098; Anagnostou melanoma HLA II IMM burden MW P = 0.001, TMB MW P = 0.0019; Snyder melanoma HLA II IMM burden MW P = 0.0011, TMB MW P = 0.0045). HLA class II IMM burden was an order of magnitude smaller than TMB. (E-H) Patients were stratified into high (top 25%} and low (bottom 75%) groups based on their HLA class II IMM burden or TMB for survival analyses. Cohorts of the same tumor type were grouped. (E) High HLA class II IMM burden was associated with longer progression-free survival in NSCLC cohorts (log rank P = 0.0002, HR = 0.37, 95% CI 0.21–0.64). (F) High HLA class II immunogenic mutation burden was trending towards significantly longer OS in melanoma cohorts (log rank P = 0.095, HR = 0.53, CI 0.25–1.13}. (G) TMB-high group was significantly associated with longer PFS in NSCLC cohorts (log rank P = 0.0033, HR = 0.46, CI 0.27–0.78}. (H) No survival benefit was observed in the TMB-high group of the melanoma cohorts (log rank P = 0.67, HR = 0.87, CI 0.44–1.70). P values were calculated based on the log rank test; a P value of 0.05 was used for significance level.
CI, confidence interval; HLA, human leukocyte antigen; HR, hazard ratio; IMM, immunogenic mutation; MW, Mann—Whitney U test; NSCLC, non-small-cell lung cancer; OS, overall survival; PFS, progression-free survival; TMB, tumor mutational burden.
For survival analysis, given the similar HLA class II IMM burden distributions, we grouped the cohorts by cancer type to increase statistical power. First, patients were stratified into high burden (top 25%) and low burden groups (bottom 75%) in their respective tumor types; no cohort bias towards HLA class II IMM-high or -low burden was seen in either tumor type (Fisher’s exact test P = 0.8235 for NSCLC, P = 1.0 for melanoma; Supplementary Figure S1D, available at https://doi.Org/10.1016/j.annonc.2022.03.013). Higher HLA class II IMM burden was associated with longer progression-free survival (PFS) in the NSCLC cohorts [log rank P = 0.0002, HR = 0.37, 95% confidence interval (CI) 0.21–0.64; Figure IE] while a trend towards longer OS was noted in the melanoma cohorts (log rank P = 0.095, HR = 0.53, 95% CI 0.25–1.13; Figure 1F). High TMB was significantly associated with longer PFS in the NSCLC cohorts (log rank P = 0.0033, HR = 0.46, 95% CI 0.27–0.78; Figure 1G), but not in the melanoma cohorts (log rank P = 0.67, HR = 0.87, 95% CI 0.44–1.70; Figure 1H).The improved prognostic value of HLA class II IMMs in the melanoma cohorts can be explained by re-categorization of five responding tumors from the TMB-low group into the HLA class II IMM-high group (Supplementary Figure S1E, available at https://doi.org/10.1016/j.annonc.2022.03.013). Notably, there was an increased survival benefit of patients with HLA class II IMM burdens in the top quartile (top 25%) compared with the lowest quartile (bottom 25%) in the melanoma cohorts (log rank P = 0.01, HR = 0.35, 95% CI 0.15–0.82; Supplementary Figure S1F, available at https://doi.Org/10.1016/j.annonc.2022.03.013).
We next evaluated the impact of clonal HLA class II IMMs on clinical outcomes by computing the intratumor IMM heterogeneity (IMMhet) for each tumor (Methods). Using estimated mutation cancer ceil fractions and excluding samples with low purity (<20%), we assessed HLA class II IMM clonalities for 193 tumor samples (Supplementary Methods, available at https://doi.Org/10.1016/j.annonc.2022.03.013), and found that 84.6% of identified IMMs were clonal {Supplementary Tables S2 and S3, and Supplementary Figure S2A and 8, available at https://doi.org/10.1016/j.annonc.2022.03.013). IMMhet, defined as the fraction of subclonal HLA class II IMMs, was similar in the melanoma (6.3%) and NSCLC (5.8%) cohorts. Whereas NSCLC tumors harboring high numbers of clonal IMMs (top25%) had iower levels of IMMhet (MW P = 0.059, Supplementary Figure S2C, available at https://doi.org/10.1016/j.annonc.2022.03.013), this observation was not apparent for the melanoma tumors analyzed (MW P = 0.32, Supplementary Figure S2D, available at https://doi.org/10.1016/j.annonc.2022.03.013). Patients with NSCLC tumors with high clonal HLA class II IMM burden (top 25%) had a significantly longer PFS (log rank P = 0.0001, HR = 0.3 CI 0.16–0.58; Figure 2A). In considering different IMMhet levels, patients with tumors harboring low IMMhet and high clonal HLA class II IMM burden (top 25%) had longer PFS in the NSCLC cohorts (IMMhet <0.05: log rank P = 0.0041, HR = 0.25, 95% CI 0.09–0.7; IMMhet <0.1: log rank P = 0.0009, HR = 0.32, 95% CI 0.16–0.65; Figure 2B and C). These findings did not reach statistical significance in the melanoma cohorts (clonal HLA class II IMM burden without lMMhet: log rank P = 0.4, HR = 0.72, 95% CI 0.32–1.64; with IMMhet <0.05: log rank P = 0.27, HR = 0.56, 95% CI 0,2–1.57; with IMMhet <0.1: log rank P = 0.49, HR = 0.74, 95% CI 0.31–1.76; Figure 2D-F). lMMhEt alone was not correlated with PFS in the NSCLC cohorts (IMMhet ≥0.05, log rank P = 0.83, HR = 1.05, 95% CI 0.65–1.71; IMMhet ≤0.1, log rank P = 0.22, HR = 0.73, 95% CI 0.44–1.22; Supplementary Figure S2E, available at https://doi.Org/10.1016/j.annonc.2022.03.013) or OS in the melanoma cohorts (IMMhet ≤0.05, log rank P = 0.84, HR = 0.93, 95% CI 0.49–1.79; IMMhet <0.1, log rank P = 0.66, HR = 0.84, 95% CI 0.4–1.78; Supplementary Figure S2F, available at https://doi.org/10.1016/j.annonc.2022.03.013).
Figure 2. Impact of clonality and tumor heterogeneity on patient survival.

Clonality of IMMs was determined using mutation cellular fractions. Samples with low purity (a threshold of 20%) were excluded from this analysis. Patients were stratified into high (top 25%) and low (bottom 75%) groups based on their clonal HLA class II IMM burden. For the NSCLC cohorts, a total of 99 samples passed the purity threshold; for melanoma cohorts, a total of 94 samples passed the purity threshold. Intratumoral IMM heterogeneity (IMMhet) was defined as the fraction of subclonal IMMs. (A) High clonal class II IMM burden without IMMhet thresholds showed significant association with progression-free survival (PFS) in NSCLC cohorts [log rank P = 0.0001, HR = 0.3 confidence interval (CI) 0.16–0.58]. (B), (C) NSCLC tumors harbored high clonaJ HLA class II IMM burden combined with IMMhet thresholds of ≤0.05 or ≤0.1 and had significantly longer PFS: (B) with IMMhet ≤0.05 (log rank P = 0.0041, HR = 0.25, CI 0.09–0.7), (C) with IMMhet ≤ 0.1 (log rank P = 0.0009, HR = 0.32, CI 0.16–0.65). (D)-(F) Clonal HLA class II IMM burden with or without IMMhet thresholds did not show any association with overall survival (OS) in melanoma cohorts: (D) without IMMhet (log rank P = 0.4, HR = 0.72 CI 0.33–1.56), (E) with IMMhet ≤0.05 (log rank P = 0.21, HR = 0.52, CI 0.18–1.46), (F) with IMMhet ≤0.1 (log rank P = 0.33, HR = 0.65, CI 0.27–1.56).
HLA, human leukocyte antigen; HR, hazard ratio; NSCLC, non-small-cell lung cancer.
Impact of germline HLA class II variation in combination with HLA class II IMM burden on clinical outcomes
We first considered the impact of patients’ HLA class II germiine variation combined with HLA class II IMM burden on survival. Whereas we found no correlation between HLA class II IMM burden and the number of HLA class II heterozygous alleles for the HLA-DPA1, DPB1, DQA1, DQB1, or DRB1 genes (NSCLC cohorts: Spearman’s rho = −0.03, P = 0.71; melanoma cohorts: Spearman’s rho = 0.08, P = 0.39), the majority of cases analyzed harbored maximal HLA class II germline heterozygosity, defined as 9 or 10 heterozygous HLA class II alleles (NSCLC cohorts: 78 out of 123, 63%; melanoma cohorts 80 out of 110, 72%). Maximal HLA class II germline heterozygosity, when combined with high HLA class II IMM burden (top 25%), conferred longer PFS in the NSCLC (log rank P = 0.0002, HR = 0.24, 95% CI 0.11–0.54; Figure 3A), with a trend towards longer OS in the melanoma cohorts (log rank P = 0.12, HR = 0.47, 95% CI 0.8–1.26; Figure 3B, Supplementary Table S2 and Supplementary Results, available at https://doi.Org/10.1016/j.annonc.2022.03.013). Next, we controlled for either HLA class II IMM burden or HLA class II allele counts to evaluate their contributions to survival benefit. Within the patient group with maximal HLA class II heterozygosity, those with tumors harboring high HLA class II IMM burden had improved PFS in the NSCLC cohorts (log rank P = 0.0001, HR = 0.24, 95% CI 0.11–0.52; Figure 3C, Supplementary Figure S3A, available at https://doi.Org/10.1016/j.annonc.2022.03.013) and OS in melanoma cohorts (log rank P = 0.1, HR = 0.49, 95% CI 0.2–1.19; Figure 3D, Supplementary Figure S3B and Supplementary Results, available at https://doi.org/10.1016/j.annonc.2022.03.013).
Figure 3. Impact of HLA class II alleles in combination with HLA class II immunogenic mutation burden on patient outcome.

(A), (B) Patients with the maximum heterozygous HLA class II alleles (9 or 10) and high HLA class If immunogenic mutation (IMM) burdens were found to have longer PFS in NSCLC cohorts (log rank P = 0.0002, HR = 0.24, CI 0.11–0.54), but not with OS in melanoma cohorts (log rank P = 0.12, HR = 0.47, CI 0.18–1.26). (C), (D) Within samples with maximum heterozygous HLA class II alleles, samples with high HLA class II IMM burdens had improved PFS in NSCLC (log rank P = 0.0001, HR = 0.24, CI 0.11–0.54) and OS in melanoma cohorts {log rank P = 0.1, HR = 0.47, CI 0.18–1.26). (E), (F) Maximum heterozygosity in at least one of HLA-DPA1 and HLA-DPB1 genes along with high HLA class II IMM was found to improve patient PFS in NSCLC cohorts (log rank P = 0.0005, HR = 0.26, CI 0.12–0.58) and resulted in a weak trend to longer OS in melanoma cohorts (log rank P = 0.099, HR = 0.41, CI 0.14–1.23). (G), (H) Within samples with more than 3 HLA-DP alleles, those with high HLA class II IMM burden had further improvements in PFS of NSCLC cohorts (log rank = 0.0001, HR = 0.27, CI 0.14–0.55) and OS in melanoma cohorts (log rank P = 0.063, HR = 0.45, CI 0.19–1.07). Log rank P values are reported.
CI, confidence interval; HLA, human leukocyte antigen; HR, hazard ratio; NSCLC, non-small-cell lung cancer; OS, overall survival; PFS, progression-free survival.
Of the HLA class II loci studied, heterozygosity at the HLA-OP locus (3 or 4 HLA-DP alleles) combined with high HLA class II IMM burden (top 25%) showed an association with longer PFS in the NSCLC cohorts (log rank P = 0.0005, HR = 0.26, 95% CI 0.12–0.58; Figure 3C, Supplementary Figure S4A and Supplementary Results, available at https://doi.Org/10.1016/j.annonc.2022.03.013) and a weak trend for longer OS in the melanoma cohorts (log rank P = 0.0994, HR = 0,41, 95% CI 014–1.23; Figure 3D, Supplementary Figure S4A and B, and Supplementary Results, available at https://doi.Org/10.1016/j.annonc.2022.03.013). When controlled for HLA-DP heterozygosity, we again noted an improved survival in the HLA class II IMM-high group compared with the low group in the NSCLC (log rank P = 0.0001, HR = 0.27, 95% CI 0.14–0.55; Figure 3E, Supplementary Figure S4C and Supplementary Results, available at https://doi.Org/10.1016/j.annonc.2022.03.013), and melanoma cohorts (log rank P = 0.063, HR = 0.45, 95% CI 0.19–1.07; Figure 3F, Supplementary Figure S4D and Supplementary Results, available at https://doi.Org/10.1016/j.annonc.2022.03.013).
Established factors, such as PD-L1 expression and smoking status, have previously been shown to impact clinical outcomes for patients with NSCLC treated with ICB.35,36 Here, we found that NSCLC tumors with a high HLA class II IMM burden showed positive PD-L1 expression (Fisher exact test P = 0.024; Supplementary Figure S5A, available at https://doi.org/10.1016/j.annonc.2022.03.013), but the levels of PD-L1 expression did not monotonically correlate with levels of HLA class II IMM burdens (Spearman’s rho = 0.07, P = 0.57). A high HLA class II IMM burden was also correlated with current or former smoking status (Fisher exact test P = 5.47e—5; Supplementary Figure S5B, available at https://doi.org/10.1016/j.annonc.2022.03.013), as well as with number of pack-years in a subset of patients (Spearman’s rho = 0.36, P = 0.037). We subsequently analyzed the interactions of HLA class II IMM burden, patient demographics and PD-L1 expression and smoking in a multivariate Cox survival model. For the NSCLC cohorts, continuous HLA class II IMM burden (Wald Test P = 0.01, HR = 0.96, 95% CI 0.93–0.99), age (Wald Test P = 0.03, HR = 0.97, 95% CI 0.94–1.0) and PD-L1 expression (Wald Test P = 0.01, HR = 0.65, 95% CI 0.48–0.89) were independently associated with favorable PFS (Figure 4A). For the melanoma cohorts, we found a trend towards an independent association between HLA class II IMM burden and OS (Wald Test P = 0.06, HR = 0.99, 95% CI 0.98–1.0; Figure 4B). The number of heterozygous HLA class II alleles, as a continuous variable, was not independently associated with survival in either cohort type (Figure 4). Differences in treatment did not have significant impact on patient survival (Supplementary Figures S6 and S7, and Supplementary Results, available at https://doi.org/10.1016/j.annonc.2022.03.013).
Figure 4. Associations between established factors and patient survival.

Established factors of age, smoking status, and PD-L1 expression and HLA class II immunogenic mutation (IMM) burdens were analyzed through multivariate Cox survival models to understand their impact on patient survival. Smoking status was categorized into three groups—never smoker = 0, former smoker = 1, and current smoker = 2. PD-L1 expression levels were assigned into four groups: strong (≥50% membranous staining) = 2, weak (l%−49% membranous staining) = 1, negative (<1% membranous staining) = 0, and unassessed. Patients with unassessed PD-L1 expression were excluded in this analysis. HLA class II IMM burdens, as a continuous variable, indicated the number of IMMs a sample had. Germline HLA class II heterozygosity, as a continuous variable, indicated the numbers of unique HLA class II alleles a sample had. (A) HLA class II IMM burden (Wald test P = 0.01, HR = 0.96, CI 0.93–0.99), age (Wald test P = 0.03, HR = 0.97, CI 0.94–1.0) and PD-L1 tumor expression (Wald test P = 0.01, HR = 0.65, CI 0.48–0.89) were found to reduce survival hazard in NSCLC cohorts. (B) Only HLA class II IMM burden had a trend towards significant effects in reducing survival hazard in melanoma cohorts (Wald test P = 0.06, HR = 0.99, CI 0.98–1.0).
CI, confidence interval; HLA, human leukocyte antigen; HR, hazard ratio; NSCLC, non-small-cell lung cancer; PD-L1, programmed death-ligand 1.
High HLA class II IMM burden is associated with an inflamed TME
In order to better understand the relationship between HLA class II IMMs and composition of the TME before ICB, we leveraged transcriptomic and TCR sequence data from the published Anagnostou_Melanoma cohort.20 RNA sequence data deconvolution revealed that densities of pretreatment activated CD4+ memory T cells and M0 macrophages in the TME were positively correlated with HLA class II IMM burden (Spearman’s rho = 0.39, P = 0.01, FDR P = 0.12 and Spearman’s rho = 0.45, P = 0.003, FDR P = 0.08, respectively; Figure 5A, Supplementary Table S4, available at https://doi.Org/10.1016/j.annonc.2022.03.013). Next, immune cell fractions were compared in the TME of tumors with high (top 25%) versus low HLA class II IMM burdens (bottom 75%). Activated memory CD4+ T cells and T follicular helper cells were significantly more abundant in the TME of high HLA class II IMM burden tumors before ICB (MW P = 0.022 and P = 0.028, respectively; Figure 5B). A trend towards higher abundance of CD8+ T cells and Ml macrophages was also noted in the TME of high HLA class II IMM burden tumors (MW P = 0.081 and 0.1, respectively; Figure 5B).
Figure 5. Correlation between HLA class II immunogenic mutation burden and pretreatment tumor infiltration lymphocytes in melanoma.

(A) Pairwise Spearman correlations between pretreatment immune cell subsets (determined by RNA sequencing data deconvolution or TCR sequencing) and HLA class II IMM load. A positive correlation was found between HLA class II IMM burden and pretreatment MO macrophages (Spearman’s rho = 0.45, P = 0.003, FDR P = 0.08), and CD4+ memory activated T cells (Spearman’s rho = 0.39, P = 0.01, FDR P = 0.12). (B) Tumor infiltration lymphocyte levels in high and low HLA class IMM burden groups were compared. Pretreatment CD4+ memory activated T cells (Mann—Whitney P = 0.022) and T follicular helper cells (Mann—Whitney P = 0.028) were found to have a significantly increased expression level in the high burden group. CD8+ T cells (Mann—Whitney P = 0.081) and Ml macrophages (Mann—Whitney P = 0.1) had higher expression levels in high HLA class II IMM burden group trending towards significance. Thin black lines indicate individual samples in the group of interest. Thick black line indicates the average of the group. Purple shading indicates the distributions of HLA class II IMM burden high groups, and the dark khaki shading indicates the distributions of HLA class El IMM burden low groups.
FDR, false discovery rate; HLA, human leukocyte antigen; IMM, immunogenic mutation; NK, natural killer; TCR, T-cell receptor.
*FDR adjusted P < 0.2.
**FDR adjusted P < 0.1.
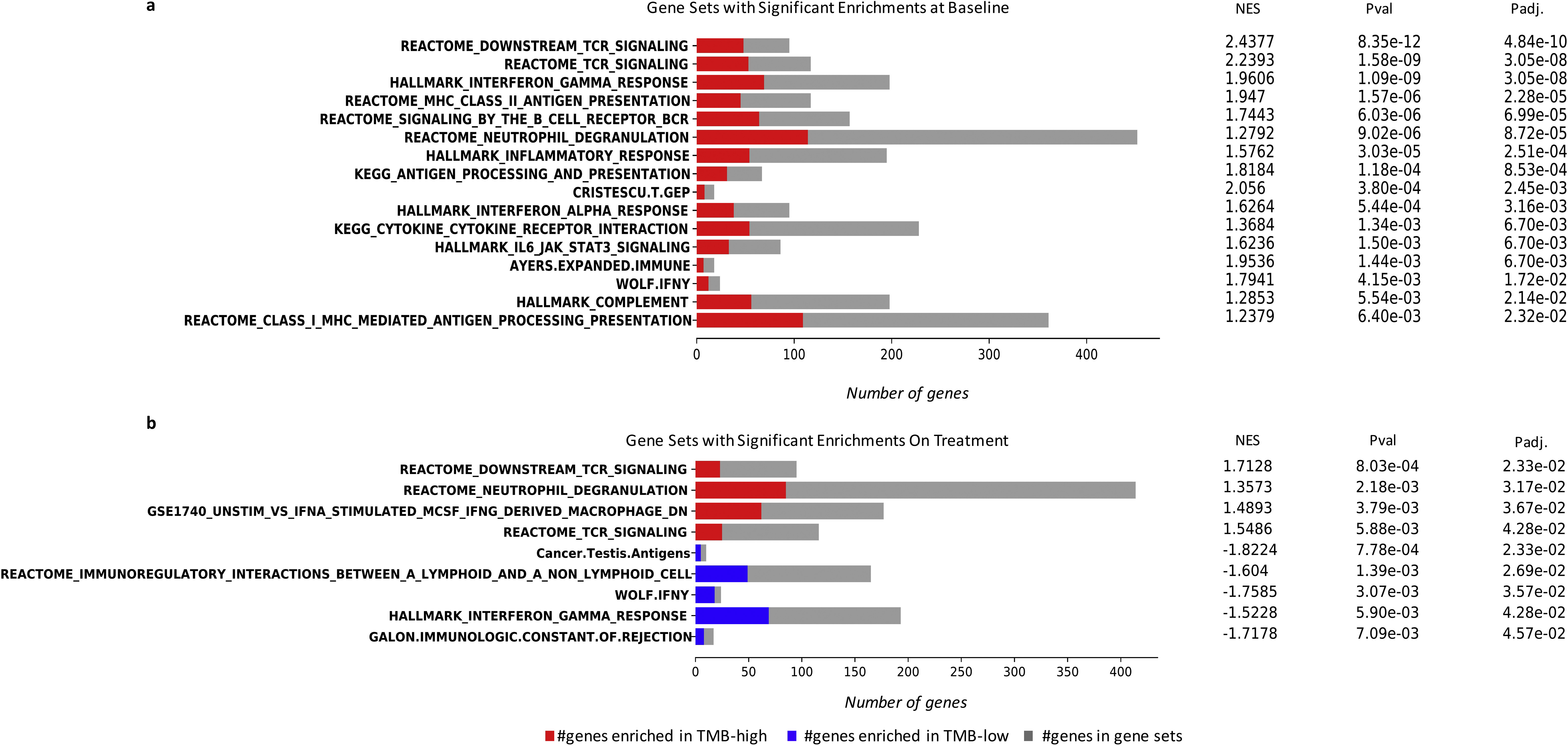
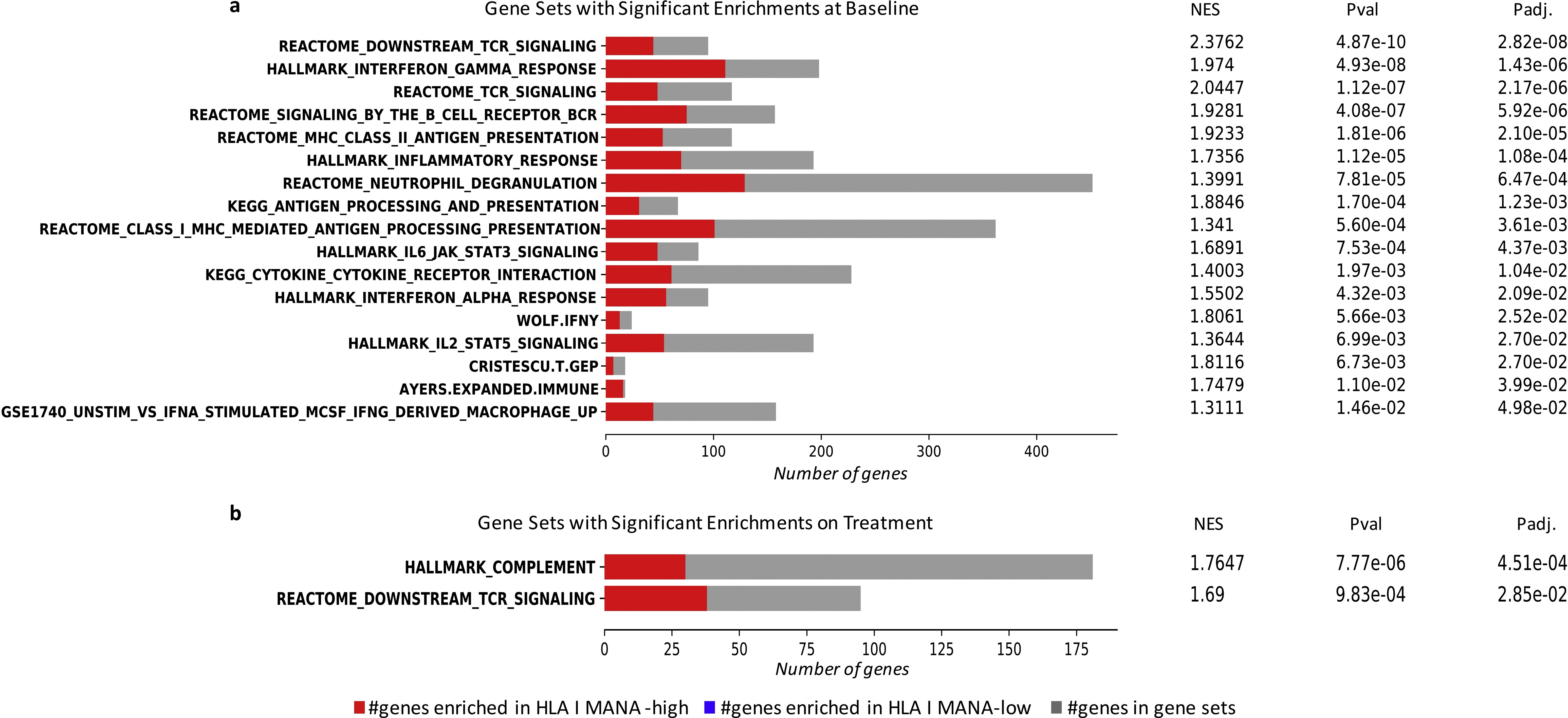
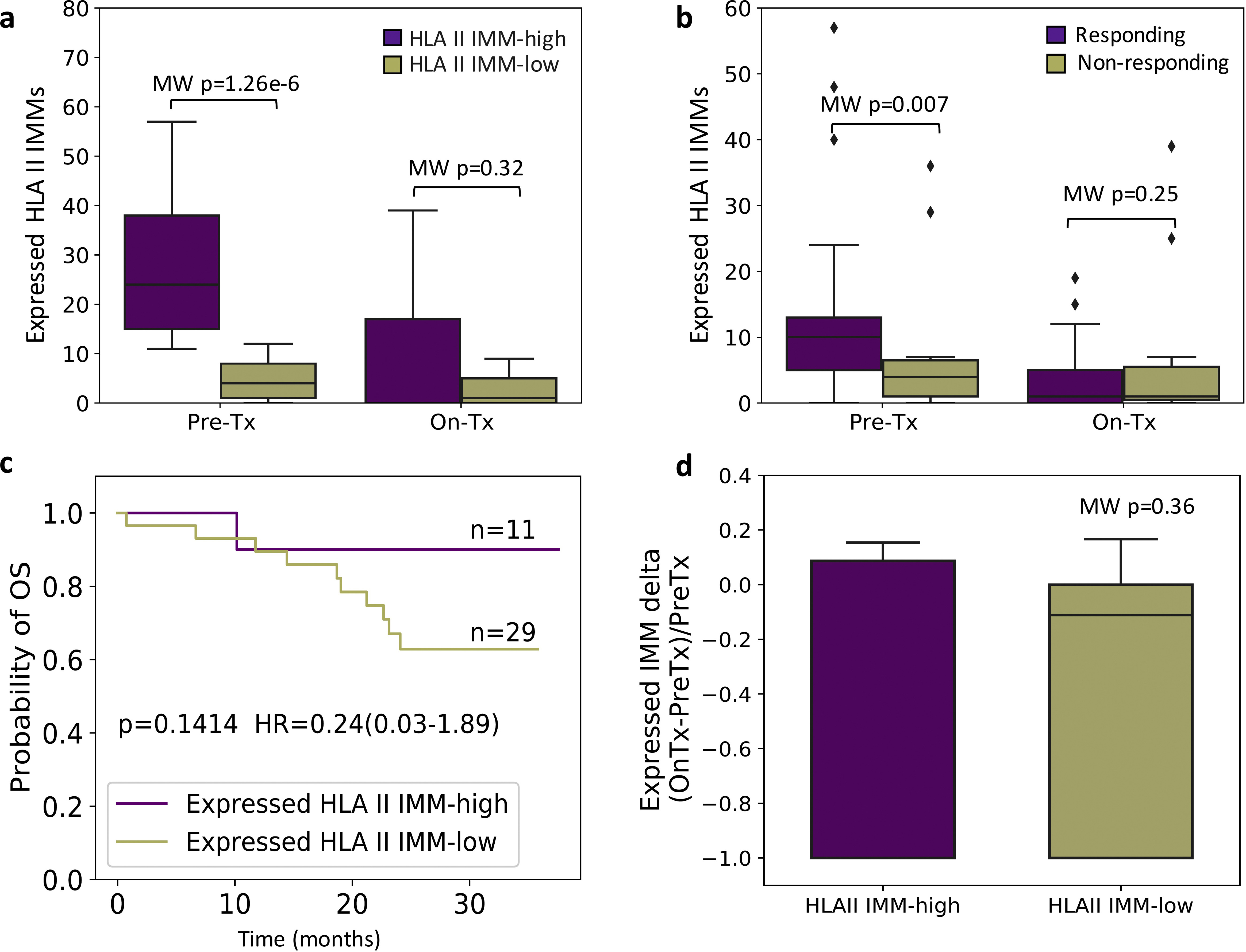
To further interrogate inflammatory responses in the TME, we used gene set enrichment analyses (GSEA) to analyze pathway-level expression differences, using 58 gene sets related to antigen presentation, tumor immune response, and type 2 immunity.37–50 We found an overrepresentation of interferon-γ response gene sets in the HLA class II IMM-high tumors before ICB (FDR P = 5.31e—16). Prominent enrichments were also found in gene sets related to B-cell receptor (BCR)/TCR signaling (FDR P = 2.03e—13 and 1.06e—11, respectively) and antigen presentation (FDR P = 1.12e—7), particularly MHC class II antigen presentation (FDR P = 2.59e—6) in pretreatment tumors with a high HLA class II IMM burden (Figure 6, Supplementary Table S5, available at https://doi.org/10.1016/j.annonc.2022.03.013). Notably, ICB induced an up-regulation of gene sets related to TCR/BCR signaling and IL2_STAT5 TCR activation in the TME of HLA class II IMM-high tumors that persisted during treatment (FDR P = 4.91e—4, 4.91e—4, and P = 0.041, respectively; Figure 6, Supplementary Table S6, available at https://doi.org/10.1016/j.annonc.2022.03.013). These patterns of pre-ICB inflammatory TME phenotype and TME reshaping while observed in TMB and HLA class I MANA burden-high tumors, were less prominent (Supplementary Results, Supplementary Figures S8 and S9, Supplementary Tables S7-10, available at https://doi.Org/10.1016/j.annonc.2022.03.013). Further investigation of HLA class II IMM expression found that on average one-third were expressed at baseline (Supplementary Results, Supplementary Figure S10 and Supplementary Table S11, available at https://doi.org/10.1016/j.annonc.2022.). Collectively, our findings suggest an inflamed TME for tumors harboring a high HLA class II burden with additional induction of adaptive immunity cascades after treatment with ICB.
Figure 6. Gene set enrichment analyses for HLA class II immunogenic mutation burden-high and -low group.

A selection of 58 gene sets related to inflammatory response, antigen presentations, and type 2 immunity were used to assess differences of gene expression in HLA class II IMM burden-high and -low groups. All the gene sets that were significantly enriched in either the high or low groups are shown. The number of genes that overlapped with gene sets overrepresented in the HLA class II IMM burden high group are shown in red, and the number of genes that overlapped with gene sets overrepresented in the HLA class II [MM burden-low group are shown in blue. Overall, 21 gene sets were enriched in the HLA class II IMM burden-high group at baseline. Six gene sets continued to be enriched in this group on treatment. Particularly, strong inflammatory response, antigen presentation, and T-cell receptor/B-cell receptor signaling gene set enrichments can be seen both at baseline and on treatment.
HLA, human leukocyte antigen; IMM, immunogenic mutation; NES, normalized enrichment score; BCR, B-cell receptor; TCR, T-cell receptor; IL, interleukin; JAK, Janus kinase; STAT, signal transducer and activator of transcription 3; MHC, major histocompatibility complex; INFγ, interferon gamma; DC, dendritic cell; TNF, tumor necrosis factor; MCSF, macrophage colony-stimulating factor.
DISCUSSION
In this study, we characterized the HLA class II-restricted IMM and associated neoantigen landscapes for patients with lung cancer and melanoma receiving ICB therapies. While HLA class II IMM burden was, overall, an order of magnitude smaller than TMB, IMM burden was more strongly associated with clinical response and survival. The majority of HLA class II IMMs did not overlap with HLA class I IMMs, suggesting that the former represent a distinct subset of IMMs that may play a critical! role in tumor rejection, complementing HLA class l-restricted neoepitope induction of CDS+ T-cell cytotoxicity. Additionally, the impacts of HLA class II IMM’s clonality and HLA class II heterozygosity were less conclusive (Supplementary Discussion, available at https://doi.Org/10.1016/j.annonc.2022.).
Whereas HLA class I neoantigens have been historically at the epicenter of neoantigen-driven tumor rejection, tumor antigen-specific CD4+ T-cell activity in the TME has been shown to be a prerequisite for spontaneous and immunotherapy-related tumor rejection.15,51 The less stringent sequence and length requirements for neopeptide binding to the HLA class II proteins compared with HLA class I-restricted epitopes, further supports the notion that a sizable fraction of HLA class II epitopes may be presented.52 In murine tumor models, a large fraction of the immunogenic mutanome has been shown to be recognized by CD4+ T cells and subsequent vaccination with such epitopes may confer sustained tumor clearance.14 These observations suggest that HLA class II-restricted IMMs and associated neoantigens may have key and non-over-lapping—with HLA class I neoepitope—functions in anti-tumor immune responses. Collectively, our findings suggest a role for immunogenic HLA class II-restricted mutations and neoantigens in predicting clinical outcomes with ICB.
Importantly, in studying the TME of tumors with a high content of HLA class II IMMs by RNA sequence data deconvolution, we identified an increased density of CD4+ memory activated T cells, T follicular helper cells, and Ml macrophages, which are critical components of successful tumor rejection.53,54 GSEA leveraging transcriptome data before and during ICB revealed a prominent enrichment in adaptive immunity programs, with increased representation of interferon-γ, HLA class II antigen processing and presentation, and TCR/BCR signaling pathway gene sets. Notably, in the NSCLC cohorts, tumors with high HLA class II IMM burden also had higher PD-LI expression and were encountered in individuals with a smoking history. These findings suggest that HLA class II IMM burden is affecting tumor immune surveillance and potentially reshaping the TME towards a more inflamed state, ultimately promoting tumor rejection.
There are several limitations to our study. First, only MANAs derived from missense mutations were considered, therefore this effort did not evaluate putative neoantigens from indels, splice variants, and somatic gene fusions that could also generate immunogenic neoepitopes and drive tumor rejection.55 Furthermore, peptide-HLA binding prediction methods are biased towards the most studied HLA alleles, for which neoantigen predictions are more accurate. As new experimental affinity data become available for rare HLA alleles, we expect that prediction methods will improve for rare HLA-restricted neoantigens. Notably, we used a stringent HLA class II affinity rank of <0.001, to limit false-positive neoantigen selection. The binding affinity for HLA class II-restricted peptides is nevertheless less stringent than the affinity for HLA class I-restricted antigens, allowing for more promiscuous binding.56 Furthermore, driver mutations poorly bound to HLA class II molecules can be positively selected during tumorigenesis.57 The larger effective binding range of MHC class II and effect of HLA class II genotypes in constraining driver mutation probability suggest that a stringent antigen binding and presentation ranking approach may underestimate the HLA class II actual binding residues. Finally, in this study we considered all HLA class II IMMs to be equally contributing to antitumor immune responses; future studies will evaluate the contribution of dual HLA class I and II IMMs and highly expressed HLA class II IMM-derived neoepitopes in driving clinical outcome in the context of immunotherapy.
In conclusion, our work supports the importance of HLA class II IMMs in antitumor immune responses that are reflected in clinical outcomes for patients treated with ICB. Such efforts may inform selection criteria for the increasing number of patients receiving cancer immunotherapy. Furthermore, our findings may inform mutation selection for cancer vaccine approaches, expanding HLA class I-only neoepitope vaccination strategies.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
FUNDING
This work was supported in part by the US National Institutes of Health, United States [grant number CA121113] (to VA, VEV), the ECOG-ACRIN Thoracic Malignancies Integrated Translational Science Center [grant number UG1CA233259] (to VA, VEV), the Bloomberg-Kimmel Institute for Cancer Immunotherapy (no grant number) (to VA, VEV), the V Foundation, United States (no grant number) (to VA, VEV), Swim Across America, United States (no grant number) (to VA, VEV), the Allegheny Health Network — Johns Hopkins Research Fund (no grant number) (to VA), and the LUNGevity Foundation, United States (no grant number) (to VA, VEV).
Footnotes
DISCLOSURE
Under a license agreement between Genentech and the Johns Hopkins University, XMS and RK and the University are entitled to royalty distributions related to technology described in the study discussed in this publication. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies. VA receives research funding to her institution from AstraZeneca and has received research funding to her institution from Bristol Myers Squibb in the past 5 years. VEV is a founder of Delfi Diagnostics and Personal Genome Diagnostics, serves on the Board of Directors and as a consultant for both organizations, and owns Delfi Diagnostics and Personal Genome Diagnostics stock, which are subject to certain restrictions under university policy. Additionally, Johns Hopkins University owns equity in Delfi Diagnostics and Personal Genome Diagnostics. VEV is an advisor to Bristol Myers Squibb, Genentech, Merck, and Takeda Pharmaceuticals. Within the last 5 years, VEV has been an advisor to Daiichi Sankyo, Janssen Diagnostics, and Ignyta (these arrangements have been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies). JRW is founder and owner of Resphera Biosciences LLC, and serves as a consultant to Personal Genome Diagnostics Inc. All other authors have declared no conflicts of interest.
ETHICAL APPROVAL
Regulatory and ethical considerations are described in detail in the original publications from which published data were retrieved to perform the analyses described in this study.
REFERENCES
- 1.Anagnostou V, Niknafs N, Marrone K, et al. Multimodal genomic features predict outcome of immune checkpoint blockade in non-small- cell lung cancer. Nat Cancer. 2020;1:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parkhurst MR, Robbins PF,Tran E, et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov. 2019;9:1022–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tran E, Ahmadzadeh M, Lu YC, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lennerz V, Fatho M, Gentilini C, et al. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci USA. 2005;102:16013–16018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19:133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan TA, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shao XM, Bhattacharya R, Huang J, et al. High-throughput prediction of MHC class I and II neoantigens with MHCnuggets. Cancer Immunol Res. 2020;8:396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tay RE, Richardson EK, Toh HC. Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2021;28:5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown SD, Warren RL, Gibb EA, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24:743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thorsson V, Gibbs DL, Brown SD, et al. The immune landscape of cancer. Immunity. 2018;48:812–830 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghorani E, Rosenthal R, McGranahan N, et al. Differential binding affinity of mutated peptides for MHC class I is a predictor of survival in advanced lung cancer and melanoma. Ann Oncol. 2018;29:271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Allen EM, Miao D, Schilling B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miao D, Margolis CA, Vokes Nl, et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable soiid tumors. Nat Genet. 2018;50:1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kreiter S, Vormehr M, van de Roemer N, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alspach E, Lussier DM, Miceli AP, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574: 696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Racle J, Michaux J, Rockinger GA, et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat Biotechnol. 2019;37:1283–1286. [DOI] [PubMed] [Google Scholar]
- 17.Chen B, Khodadoust MS, Olsson N, et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat Biotechnol. 2019;37:1332–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48:W449–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anagnostou V, Bruhm DC, Niknafs N, et al. Integrative tumor and immune cell multiomic analyses predict response to immune checkpoint blockade in melanoma. Cell Rep Med. 2020;1:100139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371: 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saunders CT, Wong WS, Swamy S, Becq J, Murray LI, Cheetham RK Strelka: accurate somatic small-variant calling from sequenced tumor- normal sample pairs. Bioinformatics. 2012;28:1811–1817. [DOI] [PubMed] [Google Scholar]
- 23.Kodysh J, Rubinsteyn A. OpenVax: An open-source computational pipeline for cancer neoantigen prediction. Methods Mol Biol. 2020;2120:147–160. [DOI] [PubMed] [Google Scholar]
- 24.Abelin JG, Harjanto D, Malloy M, et al. Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction. Immunity. 2019;51:766–779 e717. [DOI] [PubMed] [Google Scholar]
- 25.Chong C, Marino F, Pak H, et al. High-throughput and sensitive immunopeptidomic5 platform reveals profound interferonγ-mediated remodeling of the human leukocyte antigen (HLA) ligandome. Mol Cell Proteomics. 2018;17:533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bassani-Sternberg M, Braun lein E, Klar R, et a I. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun. 2016;7:13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abelin JG, Keskin DB, Sarkizova S, et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity. 2017;46:315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trolle T, McMurtrey CP, Sidney J, et al. The length distribution of class I-restricted T cell epitopes is determined by both peptide supply and MHC allele-specific binding preference. J Immunol. 2016;196:1480–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. 2014;30:3310–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao H, Wu J, Wang Y, et al. An integrated tool to study MHC region: accurate SNV detection and HLA genes typing in human MHC region using targeted high-throughput sequencing. PLoS One. 2013;8:e69 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie C, Yeo ZX, Wong M, et al. Fast and accurate HLA typing from short-read next-generation sequence data with xHLA. Proc Natl Acad Sci U S A. 2017;114:8059–8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity 3nd sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodrich B, Gabry J, AN I, Brilleman S. Rstanarm: bayesian applied regression modeling via Stan. In: Introduction to Bayesian Computation Using the rstanarm Package. 2020. [Google Scholar]
- 35.Davis AA, Patel VG. The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer. 2019;7:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Norum J, Nieder C.Tobacco smoking and cessation and PD-L1 inhibitors in non-small cell lung cancer (NSCLC): a review of the literature. ESMO Open. 2018;3:e000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ayers M, Lunceford J, IMebozhyn M, et al. IFM-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017; 127: 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beltra JC, Manne S, Abdel-Hakeem MS, et al. Developmental relationships of four exhausted CD8(+) T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity. 2020;52:825–841 e828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calabro A, Beissbarth T, Kuner R, et al. Effects of infiltrating lymphocytes and estrogen receptor on gene expression and prognosis in breast cancer. Breast Cancer Res Treat. 2009;116:69–77. [DOI] [PubMed] [Google Scholar]
- 40.Cristescu R, Mogg R, Ayers M, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018: 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39:11–26. [DOI] [PubMed] [Google Scholar]
- 42.Griss J, Viteri G, Sidiropoulos K, Nguyen Vy, Fabregat A, Flermjakob Ft. ReactomeGSA - efficient multi-omics comparative pathway analysis. Mai Cell Prateomics. 2020;19:2115–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanehisa M, Furumichi M, Sato X Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49:D545–D551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khan O, Giles JR, McDonald S, et al. TOX transcriptionally and epigenetically programs CD8(+)T cell exhaustion. Nature. 2019;571:211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liberzon A, Birger C, Thorvaldsdottir FI, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177:7303–7311. [DOI] [PubMed] [Google Scholar]
- 47.Schaefer CF, Anthony K, Krupa S, et al PID: the pathway interaction database. Nucleic Acids Res. 2009;37:D674–D679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szanto A, Balint BL, Nagy ZS, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARy-regulated gene expression in macrophages and dendritic cells. Immunity. 2010;33:699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tassiulas I, Flu X, Ho FI, et al. Amplification of IFN-alpha-induced STAT1 activation and inflammatory function by Syk and ITAM-containing adaptors. Nat Immunol. 2004;5:1181–1189. [DOI] [PubMed] [Google Scholar]
- 50.Wolf DM, Lenburg ME, Yau C, Boudreau A, van ‘t Veer U. Gene coexpression modules as clinically relevant hallmarks of breast cancer diversity. PLoS One. 2014;9:e88309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hunder NN, Wallen H, Cao J, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arnold PY, La Gruta NL, Miller T, et al. The majority of immunogenic epitopes generate CD4+ T cells that are dependent on MHC class II- bound peptide-flanking residues. J Immunol. 2002;169:739–749. [DOI] [PubMed] [Google Scholar]
- 53.Ley K. Ml Means Kill; M2 Means Heal. J Immunol. 2017;199:2191–2193. [DOI] [PubMed] [Google Scholar]
- 54.MacLeod MK, Kappler JW, Marrack P. Memory CD4 T cells: generation, reactivation and re-assignment. Immunology. 2010;130:10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turajlic S, Litchfield K, Xu H, et al. , Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan- cancer analysis. Lancet Oncol. 2017;18:1009–1021. [DOI] [PubMed] [Google Scholar]
- 56.Consogno G, Manici S, Facchinetti V, et al. Identification of immunodominant regions among promiscuous HLA-DR-restricted CD4+ T-cell epitopes on the tumor antigen MAGE-3. Blood. 2003; 101:1038–1044. [DOI] [PubMed] [Google Scholar]
- 57.Marty Pyke R, Thompson WK, Salem RM, Font-Burgada J, Zanetti M, Carter H. Evolutionary pressure against MHC class II binding cancer mutations. Cell. 2018;175:416–428 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.