

Abstract
A large family of polypeptide N-acetylgalactosaminyltransferases (GalNAc-Ts) initiate mucin type O-glycosylation transferring α-GalNAc from a UDP-GalNAc donor to the hydroxyl groups of Ser and Thr residues of peptides and proteins, thereby defining sites of O-glycosylation. Mutations and differential expression of several GalNAc-Ts are associated with many disease states including cancers. The mechanisms by which these isozymes choose their targets and their roles in disease are not fully understood. We previously showed that the GalNAc-Ts possess common and unique specificities for acceptor type, peptide sequence and prior neighboring, and/or remote substrate GalNAc glycosylation. In the present study, the role of flanking charged residues was investigated using a library of charged peptide substrates containing the central -YAVTPGP- acceptor sequence. Eleven human and one bird GalNAc-T were initially characterized revealing a range of preferences for net positive, net negative, or unique combinations of flanking N- and/or C-terminal charge, correlating to each isozyme’s different electrostatic surface potential. It was further found that isoforms with high sequence identity (>70%) within a subfamily can possess vastly different charge specificities. Enzyme kinetics, activities obtained at elevated ionic strength, and molecular dynamics simulations confirm that the GalNAc-Ts differently recognize substrate charge outside the common +/−3 residue binding site. These electrostatic interactions impact how charged peptide substrates bind/orient on the transferase surface, thus modulating their activities. In summary, we show the GalNAc-Ts utilize more extended surfaces than initially thought for binding substrates based on electrostatic, and likely other hydrophobic/hydrophilic interactions, furthering our understanding of how these transferases select their target.
Keywords: electrostatic interactions, GALNT, glycosyltransferase, molecular dynamics, mucin type O-glycosylation
Introduction
Mucin type O-glycosylation is a diverse, complex, and abundant post-translational modification found on ~80% of secreted and membrane bound proteins (Bennett et al. 2012). This type of glycosylation is initiated by the UDP-N-acetyl-D-galactosamine: polypeptide N-acetylgalactosaminyltransferase (GalNAc-T) family of enzymes, for which 20 isozymes are known in humans. GalNAc-Ts initiate mucin type O-glycosylation (henceforth O-glycosylation) by catalyzing the transfer of the α-GalNAc sugar from a UDP-GalNAc donor to the hydroxyl group of serine or threonine residues of their protein substrates. This O-GalNAc residue may be subsequently elongated by additional glycosyltransferases, resulting in a large array of O-glycan structures. O-glycosylation influences protein structure by adding rigidity and protease resistance while adding binding epitopes that may regulate biological signaling cascades (Garner et al. 2001; Hollingsworth and Swanson 2004; Pecori et al. 2020; Tajadura-Ortega et al. 2021; Zhang et al. 2021; Brockhausen et al. 2022). As such, O-glycosylation plays a crucial role in cell–cell interactions contributing to embryonic development, innate immune response, tumorigenesis, and metastasis (Bagdonaite et al. 2021; Beaman et al. 2022; Q. Hu et al. 2022; Tabak 2010; Tian and Ten Hagen 2009; Wu et al. 2020; Xia et al. 2004; Yu et al. 2007). Mutations or changes in the expression of GalNAc-Ts and the core elongating glycosyltransferases are therefore associated with multiple disease states including many cancers, embryonic lethality in the fly (PGANTs) and the mouse (C1GALT1), and the dysregulation of biological pathways (Beaman and Brooks 2014; Y. Hu et al. 2018; Schwientek et al. 2002; Tian and Ten Hagen 2009). For example, O-glycosylation regulates the stability of fibroblast growth factor 23 (FGF23), enhances the binding and uptake of the low-density lipoprotein (LDLR) and the very low density lipoprotein receptors (VLDLR), and modulates death receptor sensitivity (DR4/5) (Wagner et al. 2007; Pedersen et al. 2014; Wang et al. 2018; de las Rivas et al. 2020). Additionally, host O-glycans decorating viral envelope glycoproteins play important roles in host cell recognition and host antibody shielding (Machiels et al. 2011; Bagdonaite and Wandall 2018; Silver et al. 2020). Recently, it was shown that O-glycosylation mediates the furin cleavage of the SARS-CoV-2 spike protein decreasing its infectivity and syncytia formation in cell culture (Zhang et al. 2021; Gonzalez-Rodriguez et al. 2023). Therefore, a clear understanding of how the initiating GalNAc transferases identify and glycosylate specific glycosites is needed to fully understand the biological roles of this important, but poorly understood modification.
The GalNAc-Ts are evolutionarily conserved throughout metazoans. The majority of these isozymes possess shared characteristics, including a C-terminal lectin domain attached to an N-terminal catalytic domain joined together via a flexible linker. Previous studies have shown that both domains, in tandem, control the specificities of these isozymes toward protein and glycoprotein targets (Gerken et al. 2006, 2008, 2011, 2013; Bennett et al. 2012; Revoredo et al. 2016; de las Rivas et al. 2017, 2018, 2019). The catalytic domain generally prefers to glycosylate Thr over Ser residues at varying rates while also recognizing a range of different but overlapping peptide motifs that vary with isozyme (Gerken et al. 2006, 2008, 2011, 2013; Daniel et al. 2020). The most variable motifs are N-terminal of the site of glycosylation, whereas a common C-terminal PXP motif is shared between most but not all GalNAc-Ts. The GalNAc-Ts furthermore exhibit different preferences for sites of both neighboring and remote prior O-GalNAc glycosylation (summarized in Fig. 9; Wandall et al. 2007; Gerken et al. 2013; Revoredo et al. 2016; de las Rivas et al. 2017, 2018, 2019). The lectin domain recognizes long-range prior glycosylation (+/− 6–17 residues from the acceptor) on nearly all isozymes, whereas in a small subset of isozymes, the catalytic domain recognizes prior glycosylation (+/− 1–3 residues) relative to the targeted glycosite.
Fig. 9.
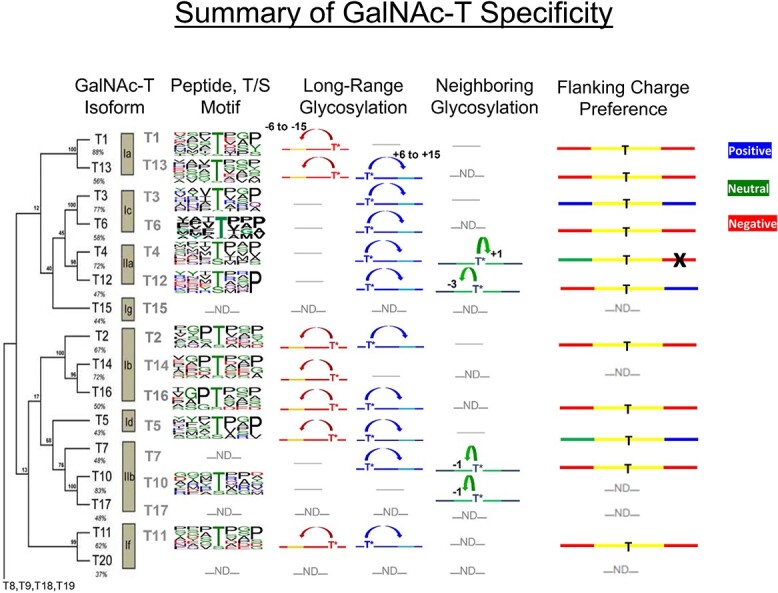
Summary of GalNAc-T specificity. The leftmost column “GalNAc-T Isoform” shows the phylogenetic tree for the human GalNAc-T family of isoforms including subfamilies and percent sequence identities (Bennett et al. 2012). “Peptide T/S Motif” shows the random peptide derived peptide substrate motifs as Sequence Logos (Gerken et al. 2006, 2008, 2011; de las Rivas et al. 2019). “Long Range Glycosylation” and “Neighboring Glycosylation” columns show the prior glycosylation preferences of the lectin and catalytic domains, respectively, where T* represents the position of the initial Ser/Thr-O-GalNAc and the arrows represent the positions of subsequential glycosylation (Gerken et al. 2013; Revoredo et al. 2016). The “Flanking Charge Preference” column summarizes this current work where blue, red, and green represent positive, negative, and neutral flanking residue preferences and the black “X” indicates an inverse preference. Note that “-ND-” indicates not determined, whereas “---” indicates no or weak activity.
Most mammals express ~20 isozymes, whereas the lower animals (Toxoplasma gondii, Caenorhabditis elegans, and Drosophila melanogaster) possess fewer isozymes (5, 9, and 13 isoforms respectfully), suggesting that evolution has assigned specific roles for each of the GalNAc-Ts and/or established the need for transferase redundancy. In the fly, we have shown at least three PGANTs have nearly identical substrate preferences as their mammalian orthologues thus suggesting their function and biological roles may be conserved over evolution (Gerken et al. 2008; Schwientek et al. 2002 and Supplementary Fig. S7). Whether these orthologues play common roles across diverse species remains unknown, but several GalNAc-Ts have been found to have specific biological roles in humans and other mammals. GalNAc-T3 regulates FGF23 furin cleavage and receptor binding stability, GalNAc-T2 controls plasma lipid levels through its glycosylation of apolipoprotein C-III, whereas GalNAc-T11 glycosylation of the LDL and VLDL receptors substantially increases their lipid binding and uptake (de las Rivas et al. 2020; Holleboom et al. 2011; Pedersen et al. 2014; Wang et al. 2018). This, combined with the GalNAc-Ts cell/tissue specific expression levels, suggests that many of the GalNAc-Ts likely possess specific and important biological roles that have not yet been elucidated (Ten Hagen et al. 2003).
Recently, our collaborative studies on the O-glycosylation of FGF23 by GalNAc-T3 revealed that prior remote glycosylation of Thr171 is required for subsequent lectin mediated glycosylation of Thr178 by GalNAc-T3, both in model peptide (~NT171PIPRRHT178RSAEDD~) and cell culture studies (de las Rivas et al. 2020). Biologically, the glycosylation of Thr178 prevents furin cleavage at Arg179, thus stabilizing FGF23 for secretion and active receptor binding. This finding was the first demonstration of Nature using a GalNAc-T’s lectin domain to target and glycosylate a specific site of a protein that modulates its function. In our attempt to further increase the glycosylation of FGF23 at Thr178 by GalNAc-T3, we introduced the optimal GalNAc-T3 isozyme motif (-YAVTPGP-), along with other GalNAc-T motifs, at Thr178 using a series of model FGF23 glycopeptides. Strikingly, the inserted optimal GalNAc-T3 motif did not increase T178 glycosylation, as was initially expected (de las Rivas et al. 2020). Further examination of the sequences revealed that the most active inserted sequence had the least negative charge. It was also noted that the region flanking Thr178 contained a sequence of negatively charged residues (Glu-Asp-Asp) directly C-terminal to the inserted motifs. These findings suggested that flanking charged residues, outside of +/− 3 residues of the glycosylation site could play a significant role in GalNAc-T3 activity.
To systematically access the roles of flanking charged residues on GalNAc-T specificity, we developed a series of charged peptide substrates with different flanking charge distributions around the optimal GalNAc-T3 sequence for study (Table 1). These peptides were recently used to characterize the D. melanogaster PGANT9A and B splice variants (May et al. 2020). Interestingly, the splice variants showed different activities towards the charged substrates that correlated with the differences in their surface electrostatic potentials. Several studies from our lab and others (Biller et al. 2000; de las Rivas et al. 2020; Gerken et al. 2011; May et al. 2020; Nehrke et al. 1996, 1997; O’Connell et al. 1991, 1992) have also suggested that flanking charged residues may impact the overall activity of GalNAc-Ts towards substrates. Here we report a systematic investigation of the effects of flanking charge on 11 of the 20 human GalNAc-Ts and the zebra finch (Taeniopygia guttata) tgGalNAc-T3, filling in another gap in our understanding of GalNAc-T specificity. Our results show each GalNAc-T isozyme has a specific flanking substrate charge preference that correlates to their surface electrostatic charge distributions. These studies further show the GalNAc-T isozymes recognize longer peptide sequences, more remote from the known peptide binding site than we previously understood. These findings add a novel and significantly more complex mechanism for how the GalNAc-Ts recognize and glycosylate their protein substrates, further contributing to our understanding of how GalNAc-Ts linked to disease may choose their in-vivo targets.
Table 1.
Charged peptide substrates.
| GAGAXXXYAVTPGPXXXAGAG | where XXX = 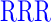 (R), (R), 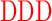 (D), or (D), or 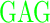 (G) (G) |
|---|---|
| Substrate ID and net charge | Substrate sequence |
| RR (+6) | GAGA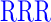 YAVTPGP YAVTPGP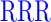 AGAG AGAG |
| RG (+3) | GAGA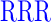 YAVTPGP YAVTPGP AGAG AGAG |
| GR (+3) | GAGA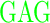 YAVTPGP YAVTPGP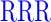 AGAG AGAG |
| RD (0) | GAGA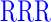 YAVTPGP YAVTPGP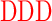 AGAG AGAG |
| GG (0) | GAGA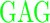 YAVTPGP YAVTPGP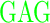 AGAG AGAG |
| DR (0) | GAGA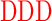 YAVTPGP YAVTPGP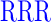 AGAG AGAG |
| GD (−3) | GAGA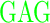 YAVTPGP YAVTPGP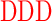 AGAG AGAG |
| DG (−3) | GAGA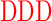 YAVTPGP YAVTPGP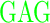 AGAG AGAG |
| DD (−6) | GAGA YAVTPGP YAVTPGP AGAG AGAG |
Results
The role of flanking substrate charge on GalNAc-T specificity
We initially characterized human GalNAc-T1, -T2, -T3, -T4, -T5, -T6, -T7, -T11, -T12, -T13, -T16, and zebra finch tgGalNAc-T3 (representing isozymes from each of the major GalNAc-T subfamilies) against the nine differently charged substrates given in Table 1. The peptides were designed around a previously identified optimal GalNAc-T3 motif (-YAVTPGP-; de las Rivas et al. 2020) likely to be glycosylated by most GalNAc-Ts, although at different rates, and contain all possible N- and C-terminal charge combinations. Positive charged residues were represented by a triad of arginine residues (abbreviated R), negative residues by a triad of aspartic acid residues (abbreviated D), and neutral residues by the Gly-Ala-Gly sequence (abbreviated G) which were placed flanking the GalNAc-T3 motif. The triads of charged residues were chosen to maximize the effects of flanking charge on GalNAc-T activity, mimicking the charged residues around the glycosylated Thr178 of FGF23 (de las Rivas et al. 2020). From the available non-redundant GalNAc-T structures with bound peptide substrates and UDP, we observed that substrate residues +/−3 of the acceptor Ser/Thr largely superimpose, whereas substrate residues outside this range are more likely to be located in unique positions on the enzyme surface (Fig. 1). This observation suggests the central-YAVTPGP-portion of each of our charged substrates will bind to a given GalNAc-T isoform in a similar manner, whereas the extended flanking charged residues could likely bind in different orientations.
Fig. 1.
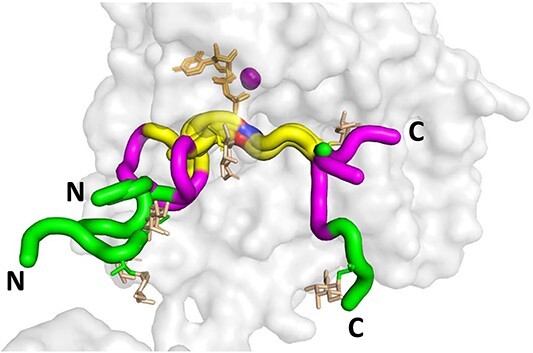
Peptide substrates bound to the GalNAc-Ts superimpose +/− 3 residues of the acceptor Thr or Ser. Shown is the structure of GalNAc-T2 (PDB: 2FFU) with superimposed (glyco)peptide structures as tubes for the seven reported structures of GalNAc-Ts bound to (glyco)peptides in the presence of Mn2+ (purple) and UDP (tan). The acceptor Thr/Ser are colored red/blue, the flanking +/− 3 residues are colored yellow, the flanking −6 to −4 and + 4 to +6 residues are colored purple, > +/− 6 residues colored green. Note the yellow region (+/− 3 residues) largely overlay for all the peptide substrates. The glycopeptide GalNAc residues are colored in orange. Structures are shown such that the bound peptides are oriented from the N- to C-terminal from the left to right. Substrate peptides shown: P3 (tgGalNAc-T3, PDB: 6S24), mEA2 (GalNAc-T2, PDB: 4D0Z), FGF23c (tgGalNAc-T3, PDB: 6S22), DGP6 (GalNAc-T4, PDB: 6H0B), DGP5_17 (GalNAc-T12, PDB: 6PXU), and AC13 (GalNAc-T2, PDB: 5AJP). See Supplementary Table S1 for the (glyco)peptide sequences.
The nine peptides in Table 1 were incubated under identical reaction conditions with each GalNAc-T isoform and the extent of peptide glycosylation determined as shown in Figs 2 and 3. The left panels of Figs 2 and 3 show the relative glycosylation of the nine peptides and a blank control. The middle panels show rearranged data to provide an additional visual for observing the effects of systematically altering N-/C-terminal charge. These two plots reveal that the GalNAc-Ts have different charge preferences. For the ease of discussion, the transferases in Fig. 2(A)–(G) were ordered based on their charge preferences, with isoforms most active against the negative charged substrates at the top of the figure (i.e. GalNAc-T11 and -T16) down to transferases that show activities for the neutral and slightly positive substrates although still preferring net negative charged substrates (i.e. GalNAc-T6 and -T13). The GalNAc-T3 and its homologue the zebra finch tgGalNAc-T3 give the greatest activity towards the most positive charged peptide substrates (Fig. 3A–B), whereas GalNAc-T5, -T12, and -T4 display unique N- and/or C-terminal charge preferences (Fig. 3C–E). These results show that flanking charge is another factor capable of modulating GalNAc-T specificity and that the effects vary greatly among GalNAc-Ts. These studies expand on our previous work on the fly PGANT9A and B splice variants which prefer negative charged substrates (PGANT9A) and positive C-terminal charged substrates (PGANT9B) similar to GalNAc-T2 and -T5, respectively (May et al. 2020).
Fig. 2.
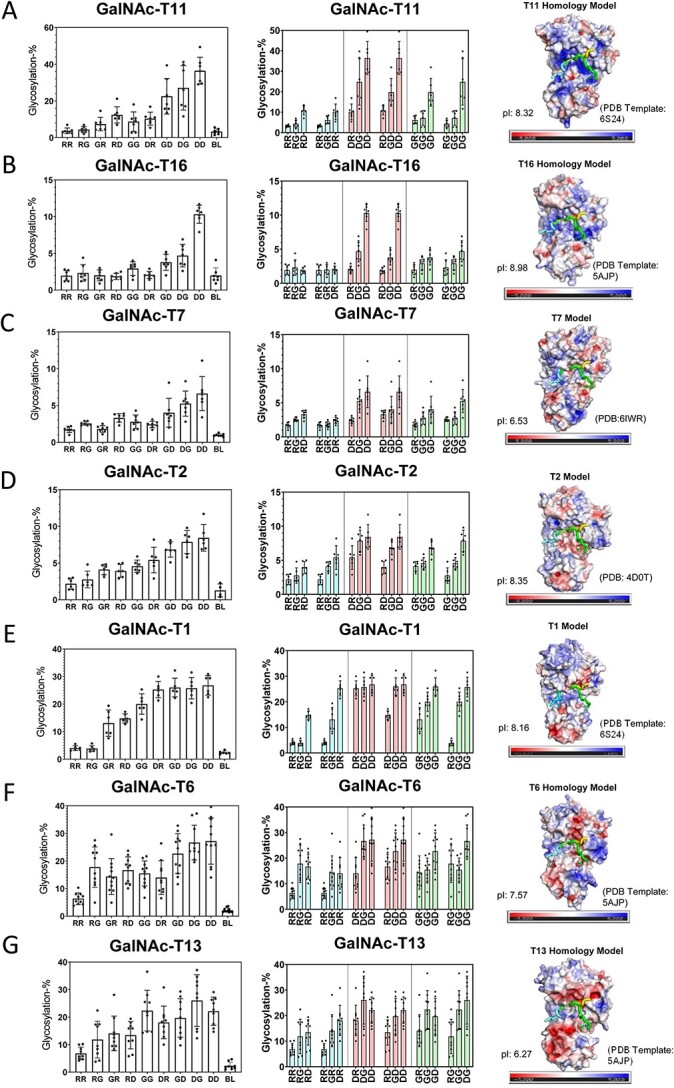
Negative charge preferring GalNAc-Ts and their electrostatic surface potentials. Shown are GalNAc-T activity against the charged peptides in Table 1 (left and center columns) and transferases electrostatic surface potential (right column). Transferases in (A)–(G) are ordered by decreasing negative charge specificity. Transferase activities (left panels) are shown for the library of peptides arranged from the left to right starting from the most positive RR peptide to the most negative DD peptide (see Table 1 for peptide abbreviations, BL represents no peptide control). These data are reordered in the middle panels such that either the N- or C-terminal charge remains constant, whereas the opposite C- or N-terminal charge varies from positive (R), neutral (G), to negative (D). These plots reveal the sensitivity of each GalNAc-T to systematic changes in N- or C- terminal charge. The blue bars correspond to fixed positive N- or C- terminal charge, red bars correspond to fixed negative N- or C- terminal charge, and green bars correspond to fixed neutral N- or C-terminal. GalNAc-T structures and electrostatic surface potentials (right panels). Negative charges are colored red, neutral colored white, and positive colored blue. Bound peptide substrates are oriented left to right N- to C-terminus and are shown as tube structures colored cyan, yellow, and green (taken from tgGalNAc-T3, PDB: 6S24,GalNAc-T2, PDB:2FFU, GalNAc-T2, PDB: 5AJP, respectively); also see Fig. 1. Isoelectric points are shown on the lower left of each structure panel. See Materials and methods for details on the homology modeling and electrostatic surface potential calculations.
Fig. 3.

Positive and unique charge preferring GalNAc-Ts and their electrostatic surface potentials. Shown are GalNAc-T activities against the charged peptides in Table 1 (left and center columns) and transferases electrostatic surface potential (right column). (A) and (B) show positive charge preferring GalNAc-T3 and zebra finch tgGalNAc-T3, and (C)–(E) show unique charge preferring GalNAc-T5, T12, and T4. See the legend of Fig. 2 for a full explanation of the plots.
What is most striking are the different patterns of surface charge shown for each transferase (Figs 2 and 3, right panels and Supplementary Figs S2–S3). Note that the transferases that prefer negatively charged peptides exhibit positively charged surface regions surrounding the likely peptide binding regions (Fig. 2). These patterns roughly follow our initial ordering with the most intensely positive surface charge transferase at the top (GalNAc-T11 in Fig. 2A) to the least positive surface charge transferase towards the bottom (GalNAc-T13 in Fig. 2G). Thus, the GalNAc-T surface charge density may play a factor in selecting charged substrates.
The electrostatic surface potentials of tgGalNAc-T3 and hGalNAc-T3 (Fig. 3A–B) show dense regions of negative charge within the potential peptide binding site consistent with these transferases preferring positively charged substrates. Interestingly, the regions of negative charge are not as dense as the positively charged regions of GalNAc-T11 and -T16, perhaps explaining the more gradual increase in activity of tgGalNAc-T3 and -hT3 as substrate charge becomes positive. GalNAc-T5 has preferences for C-terminal positive flanking charged peptides (Fig. 3C), consistent with its electrostatic surface potential showing a region of negative charge where the C-terminal of the peptides would bind (Fig. 3C). GalNAc-T12 shows its highest activity towards substrates with a negative N-terminal charge and a positive C-terminal charge consistent with its positive and negative electrostatic surface potentials where the flanking N- and C-terminal charged of the peptide would likely interact (Fig. 3D). Finally, GalNAc-T4 shows relatively uniform activity against most peptides, except for reduced activities for peptides with a C-terminal negative charge. This is consistent with GalNAc-T4’s weakly negative electrostatic surface where the C-terminal ends of the peptides would bind (Fig. 3E). Taken together, these results are consistent with the roles of GalNAc-T electrostatic surface potentials dictating their specificities toward charged peptide substrates.
High ionic strength shields electrostatic charge–charge interactions between GalNAc-Ts and their substrates
We next wanted to directly show that the electrostatic surface potentials of the GalNAc-Ts played a role in their specificities towards charged peptide substrates. To test this, we increased the ionic strength of the reactions, from the more physiological ~125 to ~400 mM, to disrupt substrate-transferase charge–charge interactions. If electrostatic interactions between the enzyme surface and the charged regions of our substrates contribute to GalNAc-T specificities, then we would expect to see a leveling of the activities of the different charged peptide substrates at high ionic strength, where the common central binding motif (-YAVTPGP-) would become the major factor dictating specificity. Figure 4 shows the results of parallel low and high ionic strength reactions with GalNAc-T1, tgT3, -T6, -T1, -T11, and -T5. As expected, the activities for each of the transferases characterized at high ionic strength showed decreased charge specificity across all charged substrates (Fig. 4). Also, the activity against the fully neutral peptide substrate (GG) for half of the GalNAc-Ts studied did not significantly change at high ionic strength again suggesting that electrostatic interactions between the substrate and the surface of the enzyme indeed play a role in their specificity. However, for tgGalNAc-T3, GalNAc-T5, and GalNAc-T6, the GG peptide activity significantly decreased at high ionic strength, suggesting that charged residues may be involved in some aspect of their catalysis. Finally, for GalNAc-T1 and possibly for -T11, the glycosylation of the most positive charged substrates (RR, RG, GR) increased at elevated ionic strength, whereas the glycosylation of the neutral GG remained the same. This suggests that charge–charge repulsion may play a role in the lower glycosylation of these positive charged substrates at lower ionic strength (Fig. 4A and E). Overall, these results confirm that the GalNAc-T peptide substrate charge specificity is based on electrostatic charge–charge interactions.
Fig. 4.
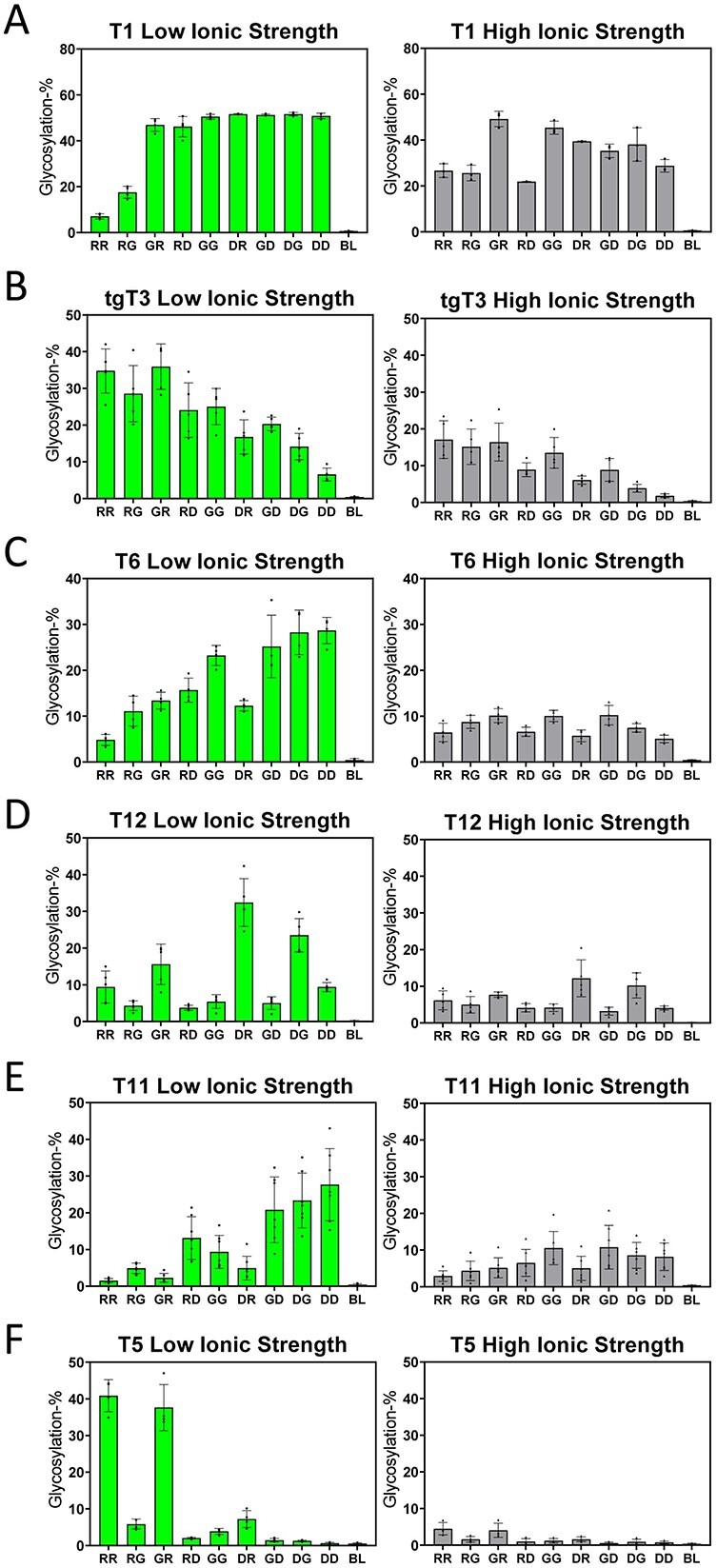
Effects of high ionic strength on GalNAc-T activity against charged substrates. Shown are the low (~125 mM, left panels) and high ionic (~400 mM right panels) strength activities against the charged peptide substrates in Table 1, for GalNAc-T1, tgGalNAc-T3, GalNAc-T6, -T12, -T11, and -T5 (A–F). See the legend of Fig. 2 for a full explanation of the plots.
Peptide kinetics suggests different peptide substrate binding modes
We next wanted to determine if the kinetic parameters (km and kcat) would reveal information on whether the binding modes of our charged peptide substrates differed. For these kinetic studies we chose tgGalNAc-T3 and GalNAc-T2, transferases with opposite charge substrate preferences, and GalNAc-T5 and -T12, transferases with unique N- and/or C- terminal charge preferences. We reasoned that if the binding mode of the central acceptor motif (-YAVTPGP-) is not impacted by the flanking charged residues on each charged peptide (Fig. 1) then the kcat values would likely remain the same, whereas the km values could vary. However, if both kcat and km values are altered, it could be concluded that the binding of the central acceptor motif is differentially impacted by the interactions of the flanking charged residues on the surface of the transferase. As shown below, we found that both kinetic constants changed for the majority of the peptides, and in only a few cases the kcat values remained relatively unchanged (Fig. 5 and Table 2).
Fig. 5.
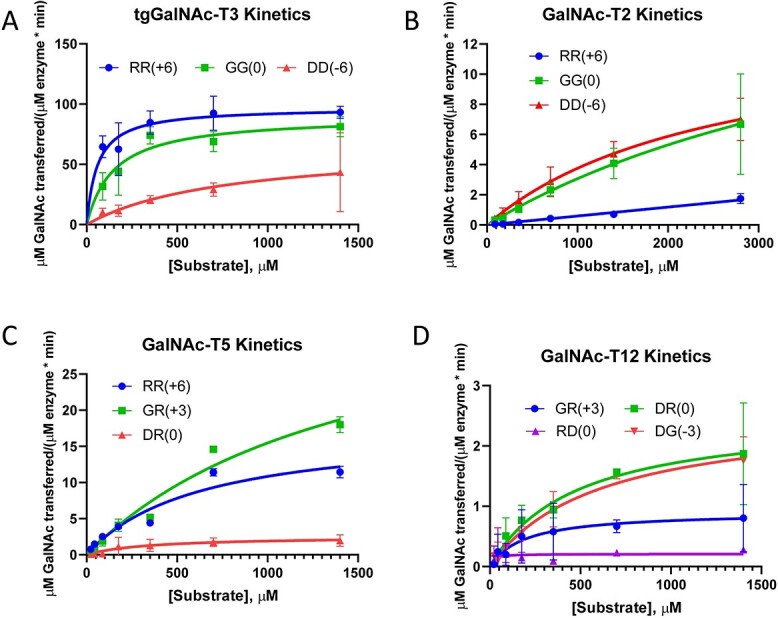
GalNAc-T charged peptide substrate kinetics. Plots for tgGalNAc-T3, GalNAc-T2, -T5, and -T12 (A-D) against selected charged peptide substrates of different relative activities. Data were analyzed and plotted using the Michaelis Menten module in GraphPad. Obtained km and kcat values are given in Table 2. Variations in kcat within a given GalNAc-T suggest different binding modes.
Table 2.
GalNAc-T charged peptide substrate kinetic parameters.
| Kinetic parameters | ||
|---|---|---|
| GalNAc-T charged peptide | km (μM) | kcat (min−1) |
| GalNAc-T2 | ||
| RR (+6) | n.da | n.da |
| GG (0) | 5420 ± 3830 | 78.1 ± 40.2 |
| DD (−6) | 2590 ± 664 | 53.5 ± 8.1 |
| tgGalNAc-T3 | ||
| RR (+6) | 58.5 ± 19.4 | 2860 ± 173 |
| GG (0) | 149 ± 43.9 | 2650 ± 217 |
| DD (−6) | 772 ± 636 | 1930 ± 788 |
| GalNAc-T5 | ||
| RR (+6) | 629 ± 207 | 120 ± 18.5 |
| GR (+3) | 1520 ± 536 | 265 ± 57.7 |
| DR (0) | n.da | n.da |
| GalNAc-T12 | ||
| GR (+3) | 156 ± 124 | 12.5 ± 3.1 |
| DR (0) | 400 ± 162 | 34.1 ± 5.7 |
| RD (0) | n.da | n.da |
| DG (−3) | 576 ± 147 | 36.1 ± 4.2 |
aKinetic parameters unable to be calculated
For tgGalNAc-T3, the RR, GG, and DD peptides were characterized representing the most active to the least active substrates, respectively. From the plots in Fig. 5 and kinetic constants in Table 2, the kcat for the GG peptide is only slightly lower than that of the most active RR peptide, whereas its km value is approximately 2.5-fold higher. An analog of the GG peptide having the GAGA-YAVTPGP-AGAG sequence has been shown to be an optimal human GalNAc-T3 substrate giving one of the fastest turnover rates against both human and tgGalNAc-T3 (de las Rivas et al. 2020). These results, therefore, indicate that the added positive charge of the RR substrate only slightly increases its kcat compared with GG while significantly lowering its km. Thus, tgGalNAc-T3’s negatively charged surface likely increases the apparent binding affinity of the RR peptide in comparison to the GG peptide, whereas not affecting how and where it binds on the transferase (Fig. 3B). On the other hand, the kcat of the DD peptide is approximately one-half of the kcat of the RR and GG peptides, whereas its km is ~5-fold higher compared with the GG peptide and ~ 13-fold higher than the RR peptide. This suggests the flanking negative charge of the DD peptide causes the central -YAPTGPG- sequence to bind differently on to the transferase due to charge–charge repulsion of the flanking residues.
For GalNAc-T2, the DD, GG, and RR peptides were characterized representing GalNAc-T2’s most active, intermediately active, and least active substrates, respectively. The central binding sequence of these peptides (-YAVTPGP-) is predicted by the Isoform Specific Predictor of O-glycosylation (ISOGlyP) to be a poor substrate for GalNAc-T2 (Mohl et al. 2020a) consistent with the high km and low kcat values that we observed (Fig. 5B and Table 2). Due to the high km values (even after obtaining data at ~3 mM), there are significant uncertainties in the actual calculated kinetic constants. However, it is clear that the kcat values of the DD and GG peptides are likely similar (Fig. 5B). On the other hand, the km value of the DD peptide is likely half of that of the GG peptide based on their different activities obtained at low substrate concentration (shown in Fig. 2D), as well as from the estimated km values in Table 2. Thus, the additional negative charge of the DD peptide may not significantly alter kcat, whereas it clearly lowers its km value with respect to GalNAc-T2. Together, this suggests the GG and DD peptide may bind to GalNAc-T2 in an identical manner. This is consistent with the surface electrostatics of GalNAc-T2 that reveal regions of weak positive charge surrounding the likely peptide binding pocket (Fig. 2D). The kinetic parameters for the least active RR peptide could not be determined but the plots clearly suggest a higher km and lower kcat values compared with the GG and DD peptide. These differences suggest that the positive charge of the RR peptide may reduce binding and/or significantly alter how this peptide interacts with the transferase.
For GalNAc-T5, kinetic studies were performed on the GR, RR, and DR peptides representing its only active substrates. These substrates all contain a positive C-terminal charge suggesting that this charge is necessary for GalNAc-T5 activity against our specific substrates. The kcat value of the most active GR peptide is ~2-fold higher than the RR peptide but, surprisingly, its km value is also ~2-fold higher (Fig. 5C and Table 2). This suggests that the RR peptide binds at higher affinity than the GR peptide despite the RR peptide showing a lower kcat (Fig. 3C). The GalNAc-T5 electrostatic surface potential reveals a highly negatively charged cleft where the N-terminus of the substrate would likely bind, suggesting that the binding of the positive charged N-terminus of the RR peptide may be enhanced compared with the neutral N-terminus of the GR peptide, potentially leading to its lower km value. We conclude that these two peptides likely bind in similar but not identical orientations on GalNAc-T5. The kcat of the next least active DR peptide could not be accurately determined but it is significantly lower (~10–15-fold) than that of the GR and RR peptide while its high km was undetermined. Its lower kcat is consistent with GalNAc-T5’s N-terminal negatively charged cleft (as shown in Fig. 3C), which would be unfavorable for binding the DR peptide, thus suggesting the DR peptide binds poorly and/or differently than the GR and RR peptide. Together, these results show that although a positive C-terminal substrate charge is required for activity, the charge of the N-terminus still affects the binding of our substrates to GalNAc-T5.
For GalNAc-T12, kinetic studies were performed on the three most active DR, DG, and GR peptides along with the relatively inactive RD peptide (having the inverse charge of the most active DR peptide). The results show that the two most active peptides (DR and DG) have similar kinetic constants (Table 2 and Fig. 5D) and likely binds identically to the transferase. Interestingly, the next least active GR peptide has a kcat value ~3-fold lower than the fastest DR and DG peptides while seemingly having a km that is also ~3-fold lower (Table 2). However, due to the variability of the experimental data we cannot confirm that its km is in fact significantly lower than the DR or DG peptides. Nevertheless, the low kcat of the GR peptide suggests it binds differently than the more active DR and DG peptides. Together, this suggests the charge–charge interactions between the N-terminal Asp residues and the transferase’s positive charged pocket are important for binding. Finally, the kcat value for the relatively inactive RD peptide could not be obtained but is significantly lower compared with the most active peptides due to substrate-enzyme charge–charge repulsion reducing and/or altering peptide binding. We conclude that both the N-terminal and C-terminal flanking charges are important for GalNAc-T12 activity; however, the nature of the N-terminal charge seems to have a greater impact on substrate activity.
In summary, the relatively large changes of km and kcat observed between the differently charged substrates against a given GalNAc-T reveal that the charged residues binding outside the overlapping common peptide binding site (shown in Fig. 1) can significantly alter the binding mode and subsequent glycosylation of the common -YAVTPGP- acceptor sequence.
Molecular dynamic simulations reveal differences between most active and relatively inactive substrates
To further understand the molecular basis of the charge specificity of the transferases, molecular dynamics (MD) simulations were conducted on GalNAc-T2 and -T12 and tgGalNAc-T3 to evaluate substrate binding stability and interactions. For simplicity we typically performed simulations on the most active and least active substrates. The MDs were performed with the central -YAVTPGP- sequence docked and minimized to the catalytic domain substrate binding site while allowing the flanking charged N- and C- terminal ends to move freely throughout a 250 ns simulation. The results are summarized in Figs 6–7, which show the starting and ending structures along with an overlay structure showing the entire trajectory of the peptide substrate bound to the enzymes. The latter provides a visual of the degree of motion of the bound substrate. To quantitatively assess the trajectories, the root mean squared deviation (RMSD) and radius of gyration (Rg) values of the backbone atoms of the peptide substrate were calculated for each trajectory. In addition, the end-to-end distances (D-end) between the N- and C-terminal α-carbons of the peptide were obtained from the trajectory (Fig. 8).
Fig. 6.

Summary of the MD simulations of tgGalNAc-T3 and GalNAc-T12 against their most active and least active substrates. Shown are the initial structures (left column), overlaid trajectory (middle column), and final structures (right column) for the tgGalNAc-T3 and GalNAc-T12 MD simulations. (A) and (B): for tgGalNAc-T3 for its most active (RR) and least active (DD) charged substrates. (C) and (D): simulations for GalNAc-T12 for its most active (DR) and one of the least active (RD) substrates. Charged peptide residues are color coded blue for the positive Arg residues, red for the negative Asp residues, green for the neutral Gly and Ala residues, and yellow for the central +/− 3 binding motif. Red arrows in (C) and (D) represent movements of the peptide during the simulation. RMSD, Rg, and D-end measurements of the MD simulations are plotted in Fig. 8. See Materials and methods and Supplementary Movies S1–S4.
Fig. 7.

Summary of the MD simulations of GalNAc-T2 against charged substrates of different activities. Shown are the initial structures (left column), overlaid trajectory (middle column), and final structures (right column) for the MD simulations. Simulations were performed for the DD, GG, and RR peptides (A–C, respectively) representing the most active to least active charged substrates, respectively. Charged peptide residues are color coded as described in Fig. 6. Red arrows in (A)–(C) represent movements of the peptide during the simulation. RMSD, Rg, and D-end measurements of the MD simulations are plotted in Fig. 8. See materials and methods and Supplementary Movies S5–S7.
Fig. 8.

RMSD, Rg, and end-to-end distance measurements (D-end) for the MD simulations of tgGalNAc-T3 (A), GalNAc-T12 (B), and -T2 (C) revealing common differences between the most active and least active substrates as discussed in the text. The RMSD (top row), radius of gyration (Rg) (middle row), and end-to-end (D-end) distance measurements (bottom row) were obtained from the simulations in Figs 6, 7 and Supplementary Movies S1–S7. See Materials and methods for details. RMSD values represent the relative of movement of the peptide backbone atoms, Rg values represent the compactness of the peptide backbone atoms, and D-end values are the distances between the N- and C-terminal alpha carbons, also a measure of the compactness of the peptide.
MD simulations for the positive charge substrate preferring tgGalNAc-T3 were performed with the most active (RR) and least active (DD) substrates. The overlaid trajectories of the RR and DD peptides (Fig. 6A and B) show the RR peptide to be considerably less conformationally variable compared with the DD peptide. In the RR peptide simulation, the central -YAVTPGP- sequence remained tightly bound to the enzyme, whereas the positive charged residues of the peptide quickly moved to the negative charge regions of tgGalNAc-T3 and remained in a relatively fixed extended like conformation for the remainder of the simulation (Supplementary Movie S1 and Fig. 6A). Quantitatively, the RMSD values for the RR peptide are relatively low and stable throughout the trajectory (Fig. 8A, top panel), whereas the Rg and D-end values remain high and stable, consistent with the extended orientation of the peptide. The trajectory of the negatively charged least active DD peptide gave more erratic movements where the N-terminal GAGA- residues and the -YAVTPGP- acceptor sequence bound together (~21 ns) for a short time before unbinding and moving away from the catalytic domain (~37 ns) (Supplementary Movie S2). At around 190 ns, the flanking residues bound to patches of positive charge on the surface of the transferase resulting in an extended binding orientation different than the RR peptide (Fig. 6B). Consequently, the RMSD values of the DD peptide are higher, and more variable compared with the RR peptide throughout the simulation until the peptide reaches its final bound position on the surface of the transferase (Fig. 8A, top panel). Likewise, the more flexible and self-interacting DD peptide initially shows lower Rg and D-end values than the extended bound RR peptide (Fig. 8A, middle and bottom panels), which increases only after the DD peptide finds its extended binding site on the enzyme. We conclude that although the DD peptide may eventually bind in an extended conformation on the surface of tgGalNAc-T3, its -YAVTPGP- acceptor sequence remains poorly bound based on our simulations and on our kinetic studies.
MD simulations were performed for the DR peptide preferring GalNAc-T12 with its most active (DR) and least active (RD) peptides substrates (Figs 6C and D, Fig. 8B). In the DR peptide trajectory, the flanking residues remained bound to the transferase with the N- and C-terminal charged residues quickly moving into opposite charged pockets on the enzyme surface as shown by the arrow in Fig. 6(C). These regions of the peptide remained in a fixed orientation throughout the duration of the simulation as shown by its low RMSD, high Rg, and high D-end values (Fig. 8B and Supplementary Movie S3). Interestingly, at ~108 ns, the -YAVTPGP- acceptor sequence is released from the catalytic binding pocket (~4.5 Å), whereas the flanking charged residues remain bound to the enzyme for the rest of the trajectory (Fig. 8B). In contrast, the N- and C-hydrophobic and charged regions for the least active RD peptide, quickly (~8 ns) self-associate and remain bound together for the remainder of the trajectory (as shown by the arrows in the overlaid trajectory) giving higher RMSD, lower Rg, and lower D-end values (Fig. 6D, Fig. 8B, and Supplementary Movie S4). Additionally, the -YAVTPGP- acceptor sequence quickly dissociates (~35 ns) from the catalytic domain remaining unbound throughout the simulation (Fig. 6D). Thus, the differences in the RMSD, Rg, and D-end values observed between the most active and least active substrates are similar for both GalNAc-T12 and tgGalNAc-T3 (Fig. 8A and B).
MD simulations for the negative charge substrate preferring GalNAc-T2 were performed on the most active (DD), neutral (GG), and least active (RR) charged substrates. For all the peptides, the first half of the trajectories were relatively stable following the trends observed for tgGalNAc-T3 and T12 (Fig. 8C). However, during the second half of the trajectories, the trends became quite chaotic as shown in the overlaid structures, RMSD, Rg, and D-end values (Fig. 7 and Fig. 8C). The most active DD peptide showed substantial contact with the surface of the transferase in the initial half of the trajectory, although, for a short period of time (for 13 ns at 112 ns), the -YAVTPGP- sequence dissociated from the catalytic binding site by ~8 Å before rebinding (Fig. 7A and Supplementary Movie S5). This is consistent with the -YAVTPGP- sequence being a relatively poor substrate for GalNAc-T2. Interestingly, at 117 ns, the Tyr residue of the acceptor sequence formed a hydrophobic interaction with the Pro residue directly C-terminal to the acceptor Thr, collapsing the peptide in on itself resulting in increases in RMSD and decreases in Rg and D-end values (Fig. 8C). At ~194 ns, the N-terminal GAGA residues come in contact with the -YAVTPGP- acceptor residues and remain bound until near the end of the trajectory, after which at, ~244 ns, the peptide extends, finding pockets of positive charge on the enzyme’s surface. These movements are shown in the RMSD, Rg, and D-end values of the peptide (Fig. 7A, Fig. 8C, and Supplementary Movie S5). The intermediately active GG peptide initially bound across the surface of the enzyme; however, at ~41 ns, its -YAVTPGP- sequence left the catalytic domain peptide binding site before subsequentially rebinding after ~61 ns. Later in the trajectory, at ~199 ns, its N- and C-terminal GAGA/AGAG residues associated with one another through hydrophobic interactions resulting in a compact peptide configuration that was maintained to the end of the trajectory (Fig. 7B and Supplementary Movie S6). These movements can be observed from the arrows on the overlaid structures of Fig. 7(B) as well as in the RMSD, Rg and D-end values in Fig. 8(C). For the least active RR peptide trajectory, the central -YAVTPGP- remained bound to the catalytic domain although, at ~50 ns, its flanking charged residues and its N- and C-terminal GAGA/AGAG residues moved away from the catalytic domain and associated with one another (Supplementary Movie S7). These contacts were maintained before dissociating at ~162 ns where the N- and C- terminal charged and hydrophobic ends moved freely before finding negatively charged pockets on the lectin domain (~227 ns) resulting in a final “U” shaped, semi-compact, peptide structure (Fig. 7C and Supplementary Movie S7). These erratic movements and variable conformations are also evident in the overlaid trajectory, RMSD, Rg, and D-end values (Fig. 7C and Fig. 8C). Thus, for the most part, the MD simulations of GalNAc-T2 are similar to the results of the tgGalNAc-T3 and GalNAc-T12 simulations where the most active substrates are bound to the enzyme surface, whereas the less active substrates are more loosely bound and tend to self-associate.
In summary, our MD studies reveal that the binding of the -YAVTPGP- acceptor sequence is significantly affected by the interactions of the flanking charge residues at the surface of the transferase which is consistent with our kinetic analysis.
Discussion
More than 20 years ago, the roles of flanking charged residues on GalNAc-T activity were studied by the Lawrence Tabak group revealing that charged residues flanking an acceptor glycosite could significantly influence acceptor glycosylation both in-vitro and in-vivo (Nehrke et al. 1996, 1997; O’Connell et al. 1991, 1992). However, these studies were done prior to our full understanding of the large number of different GalNAc-T isoforms that could be expressed in a cell. Subsequent work using random peptide substrates, detected a weak net substrate charge bias that correlated with GalNAc-T isoform isoelectric point and electrostatic surface charge (Gerken et al. 2011). Our current study was begun based on the unexpected observation of the glycosylation of a series of FGF23 model substrates by GalNAc-T3, where flanking charge 4–6 residues C-terminal of the acceptor seemed to play a profound role in modulating its activity (de las Rivas et al. 2020). In the present study we developed a library of charged substrates designed to systematically characterize the roles of flanking charged residues against a large series of GalNAc-T isoforms. The library consisted of the GalNAc-T3 optimal sequence motif (-YAVTPGP-) flanked by triads of positive, neutral, and negative charged residues (Table 1). Our studies revealed that these remote flanking charged residues have profound effects on glycosylation, with most isoforms preferring negative charged substrates, a small number of isoforms preferring positive charged substrates, and a subset of isozymes possessing unique patterns of N- and C-terminal charge preferences (Figs 2–3). These findings have furthered our understanding of how the GalNAc-Ts select their peptide targets, as summarized in Fig. 9.
To further understand the origins of the charge effects, we analyzed each isoform’s electrostatic surface potential that was found consistent with their substrate charge preferences (Figs 2 and 3). It was also observed that the surface charge density correlated with a transferases’ range of specificity for charged substrates. We also showed that electrostatic interactions were indeed involved, as increasing ionic strength ~3× of physiological levels nearly eliminated the differences in the activities between differently charged substrates (Fig. 4). A kinetic analysis between the most active to least active charged peptide substrates revealed significant differences in the km and kcat values (Fig. 5 and Table 2). These kinetic differences were taken to show how the specificities of the GalNAc-Ts are influenced by charged residues outside of the conserved ±3 binding motif, impacting the overall binding modes and orientations of the peptide substrates. These findings were further supported by our MD analyses of peptides bound to transferases which showed the most active substrates binding in more extended and relatively fixed orientations compared with the least active substrates that bound in more compact and variable orientations as monitored by the RMSD, Rg, and D-end values (Figs 6–8). Thus, the greater stability and extended nature of the most active substrates leads to lower km values and higher turnover rates (kcat) (Fig. 8 and Table 2). On this basis, we have shown that the GalNAc-Ts utilize their surface electrostatics for selecting substrates via charge–charge interactions beyond +/−3 residues of the acceptor thereby affecting substrate binding and overall enzymatic turnover.
One example where remote substrate charge may be playing a specific biological role is the O-glycosylation of the LDL receptor’s LA linkers by GalNAc-T11. These linker regions, which connect the LA modules together, are almost exclusively glycosylated by GalNAc-T11 only in the context of the fully folded LA modules (Pedersen et al. 2014; Wang et al. 2018). Interestingly, the LA modules flanking the linker regions contain several highly conserved aspartic acid residues which are also shown to be required for ligand binding (Wang et al. 2018). Based on our findings of GalNAc-T11’s highly positive surface charge and its strong preference for negatively charged substrates, it is plausible that the highly conserved negative charge of the surface of the LA-modules may facilitate GalNAc-T11’s glycosylation of the LA-linker regions helping to explain the high specificity of GalNAc-T11 for glycosylating these LA linker regions (as shown in Supplementary Fig. S4).
It is further informative to compare the charge preference of the GalNAc-Ts belonging to the same subfamilies. GalNAc-T1 and -T13 in subfamily Ia, and -T2 and -T16 in subfamily Ib (see Fig. 9) prefer negatively charged substrates although with different patterns (Fig. 2). However, GalNAc-T4 and T12 in subfamily IIa demonstrate different N- and C- terminal charge preferences, where GalNAc-T4’s preferences are quite different and not as strict as those observed for GalNAc-T12 (Fig. 3). In addition, GalNAc-T3 and -T6 in subfamily Ic show opposite charge preferences, with GalNAc-T3 preferring positively charge substrates and GalNAc-T6 preferring negatively charge substrates (Figs 2 and 3). The latter observation may help explain the contrasting roles of GalNAc-T3 and -T6 in colorectal cancer (CRC) progression (Duan et al. 2018; Lavrsen et al. 2018; Liu et al. 2019; Nielsen et al. 2022; Ogawa et al. 2022) while having an ~77% sequence identity and exhibiting very similar substrate specificities (see Supplementary Fig. S8 and Supplementary Table S4). Thus, their different charge preferences may play a role in their different physiological effects. These findings may help identify transferase specific CRC substrates, including the potential for uncovering new mechanisms of CRC progression.
The GalNAc-T orthologues of the higher animals are highly conserved (Bennett et al. 2012). For example, the human and the zebra finch GalNAc-T3s, which share ~79% sequence identity, have nearly identical substrate specificities (de las Rivas et al. 2020), and in this work, they have nearly identical flanking charge specificities. We therefore examined whether GalNAc-T charge substrate preferences would be evolutionarily conserved to more distant metazoan species such as for the fly PGANT5, PGANT2, and PGANT35A orthologues of GalNAc-T1, -T2, and -T11, which we have shown to have very similar random peptide specificities (Gerken et al. 2008 and see Supplementary Fig. S7 and Supplementary Table S4). We have recently characterized the charged substrate specificity of the fly PGANT9A, another fly orthologue to the human GalNAc-T1 that shares ~51% sequence identity (May et al. 2020) (Fig. 10A). The electrostatic surfaces of these two orthologues show positive charged surfaces around the peptide binding site; however, PGANT9A shows more intense regions of positive charge perhaps explaining its higher relative glycosylation against the more negatively charge substrates (Fig. 10A and B). We therefore characterized the charge preferences of PGANT35A (dT1), the orthologue to the human GalNAc-T11, which shares 43% sequence identity. Similar to what is observed for GalNAc-T11, the homology model of PGANT35A has a well-defined region of positive charge surrounding the predicted peptide binding site (Fig. 10C). Interestingly, PGANT35A gave slightly different results than GalNAc-T11 with the DG peptide having higher glycosylation than the DD peptide. This may be explained by PGANT35A’s large patch of negative charge located above the region where the C-terminus of our peptides may bind (Fig. 10C and D). The slightly different substrate charge specificity between the human GalNAc-T11 and PGANT35A may also help explain why the human enzyme cannot rescue the PGANT35A knockout in the fly, even though they have nearly identical peptide substrate specificity (Bennett et al. 2010). The remaining PGANTs previously studied by our lab were found to be inactive, nevertheless, we performed electrostatic surface calculations (Supplementary Fig. S5) for the PGANT orthologues of the human GalNAc-Ts that we characterized in Figs 2 and 3. We found very rough similarities in the electrostatic potentials between orthologues PGANT1 and GalNAc-T5 as well as PGANT5 and GalNAc-T1. However, there were no obvious electrostatic surface charge similarities between PGANT2 and GalNAc-T2 despite having nearly identical substrate sequence specificities (Supplementary Fig. S5). This suggests that for only a subset of these orthologues are their charge specificities evolutionarily conserved across metazoan species.
Fig. 10.

Charged peptide preferences and electrostatic distributions for the fly PGANT9A and PGANT35A, orthologues to hGalNAc-T1 and -T11. Transferase activity against the charged peptides in Table 1 (left and center columns) and transferase electrostatic surface potential (right column) for (A) PGANT9A, (B) GalNAc-T1, (C) PGANT35A, and (D) GalNAc-T11. See the legend of Fig. 2 for a full explanation of plots. These data show partial charge preference and electrostatic conservation to their mammalian orthologues in Fig. 2. See Materials and methods for details on the homology modeling and electrostatic charge calculations. Charged peptide glycosylation data for PGANT9A are taken from (May et al. 2020).
To date, the O-glycosylation predictor, ISOGlyP, is the only O-glycosylation prediction tool that is GalNAc-T isoform specific (Mohl et al. 2020a). All other prediction algorithms such as NetOGlyc4.0 (Steentoft et al. 2013), Captor (Zhu et al. 2022), and the O-glycoprotein repository (OGP) (Huang et al. 2021) lack GalNAc-T isoform specificity and rely on known in-vivo O-glycosylation sites reported in glycoprotein databases. Current O-glycosylation databases almost entirely lack knowledge of the GalNAc-T isoform(s) responsible for the glycosylation. In contrast, ISOGlyP utilizes in vitro derived isoform specific random peptide derived positional enhancement values (EV) +/−5 residues from the Ser/Thr acceptor, and as such is considered an independent orthogonal approach compared with the use of the in-vivo O-glycosylation databases. A recent comparison of ISOGlyP and NetOGlyc for predicting in-vivo sites of glycosylation found that both predictors had a similar accuracy of 70–75% (with 15–25% of the predicted sites non-overlapping), suggesting both approaches are lacking features that could improve their prediction (Mohl et al. 2020a, 2020b). Our findings of the roles of flanking charged residues on GalNAc-T specificity suggest that flanking charge could be one such feature that could likely improve the ISOGlyP predictions. It is, furthermore, likely that the diversity of charge effects observed among the GalNAc-Ts could have confounded the predictors that utilize the generalized in-vivo O-glycosylation databases. We, therefore, anticipate that future in-vivo studies may confirm our findings of the role of substrate charge on the specificity of these enzymes.
In summary, we have shown in this work that the GalNAc-T isoforms have different substrate charge specificities determined by each enzyme’s unique electrostatic surface potential. We have also shown that the GalNAc-Ts utilize a much larger portion of their catalytic domain, outside the common peptide binding site to interact with their peptide and protein substrates. Although we have examined flanking substrate charge; it is undoubtable that each transferase will have additional preferences for flanking hydrophobic/hydrophilic residues adding further complexity to this important family of transferases. In conclusion, the GalNAc-Ts are a unique family of enzymes that recognize multiple substrate properties to initiate mucin type O-glycosylation. Fully understanding the multiple specificities of the GalNAc-Ts will be invaluable in understanding how these enzymes choose their in vivo targets and elucidating their roles in disease.
Materials and methods
Reagents and peptide substrates
Charge peptide substrates listed in Table 1 were custom synthesized by RS Synthesis (Louisville, KY). Stock solutions of 4 or 8 mM were prepared for each of the peptides after lyophilization from water multiple times and adjusting the pH to ~7 with 0.1 M NaOH/HCl. Concentrations were confirmed using a DeNovix DS-11 nanodrop spectrophotometer (DeNovix Inc., Wilmington, DE) using the extinction coefficient of 1,490 M−1 cm−1 at 280 nm for a single Tyr residue (Gasteiger et al. 2005). Fully N-acetylated UDP-[3H]GalNAc was obtained from American Radiolabeled Chemicals Inc (St. Louis, MO), whereas nonlabelled UDP-GalNAc was obtained from Millipore-Sigma (St. Louis, MO). Stock solutions of radiolabeled 20 mM UDP-GalNAc were prepared by adding stock UDP-[3H]-GalNAc to 20 mM unlabeled UDP-GalNAc giving ∼6 × 108 DPM/μmole. ScintiVerse BD Cocktail fluid was obtained from Fisher Scientific (Pittsburgh, PA). Liquid scintillation counting was performed on a Beckman LS 6500 Scintillation Counter. BioPureSPN TARGA C18 macro spin columns were obtained from The Nest Group Inc. (Ipswich, MA). Random peptide substrates were obtained from RS synthesis (Louisville, KY) and Sussex Research (Ottawa, CN).
Transferases
As in our previous work (Revoredo et al. 2016; Daniel et al. 2020) GalNAc transferases (using the GalNAc-T naming convention of Bennett et al. 2012) were obtained as purified soluble N-terminal truncated enzymes from multiple sources and expression systems. Human GalNAc-T1 was a gift of Kelley Moremen (CCRC, University of Georgia) and expressed from HEK293F cells (Moremen et al. 2018). Human GalNAc-T2, -T3, -T4, -T6, -T7, -T12, and tgGalNAc-T3 were obtained from Ramon Hertado-Guerrero (University of Zaragosa, SP) and expressed from Pichia pastoris (de las Rivas et al. 2020). Human GalNAc-T5, -T11, -T13, -T16, and dT1 (PGANT35A) were gifts of Henrik Clausen (University of Copenhagen, DK) and expressed from High Five insect cells (Schwientek et al. 2002; Vester-Christensen et al. 2013).
Transferase glycosylation
It should be noted that the GalNAc-Ts are commonly N- and/or O- glycosylated in vivo (see Supplementary Tables S2 and S3), which may potentially alter their specificity against uncharged and charged substrates, particularly for the latter if the glycans are sialylated. The GalNAc-Ts characterized in Figs 2 and 3, except for GalNAc-T1, were expressed from yeast or insect cells thus their glycosylation will differ from the mammalian expressed versions. The transferases expressed from Pichia pastoris (GalNAc-T2, -T3, -T4, -T6, -T7, -T12, and tgGalNAc-T3) are not shown to be O-glycosylated, whereas those transferases containing potential N-glycosylation sites (see Supplementary Table S2) were treated with the MBP-EndoH fusion protein (maltose binding protein- Endo N-acetyl-β-D-glucosaminidase H) to trim potential N-glycans down to Asn-N-GlcNAc (de las Rivas et al. 2020). Transferases expressed from High Five insect cells (GalNAc-T5, -T11, -T13, -T16, and dT1 (PGANT35A)) may contain non-sialylated paucimannose N-glycans (Shi and Jarvis 2007) and non-sialylated short O-linked glycans (Wang et al 2021). As our charged peptide preferences (Figs 2 and 3) correlate well with the GalNAc-T surface electrostatics, we conclude that the different glycosylation states of the expressed transferases minimally affect our findings relative to a non-glycosylated transferase.
Nevertheless, to evaluate whether in vivo glycosylation may alter the charge specificity of the GalNAc-Ts characterized in this work we accessed the GlycoDomainViewer (GlycoDomain Viewer (ku.dk) and Glygen (GlyGen Home | Informatics Resources for Glycoscience | glygen.org) (Steentoft et al. 2013; York et al. 2020) and mapped the known O-glycosylation and known and predicted N-glycosylation sites onto the transferase structures (see Supplementary Fig. S6). For the N-glycosylation sites, GalNAc-T2, -T7, -T12, and -T16 lack predicted sites, GalNAc-T4, -T6, and -T13 have predicted sites that are not presently reported glycosylated, whereas tgGalNAc-T3 and GalNAc-T1, -T3, -T5, and -T11 contain reported N-glycosylation sites (Supplementary Table S2). An analysis of these sites shows that most are located on the opposite side from the peptide binding site of the catalytic domain, in the N-terminal transmembrane stem region, or in the lectin domain (see Supplementary Fig. S6). Generally, the locations of the N-linked glycosites are distant from the peptide binding site and are not present within the patches of surface charge that dictate GalNAc-T substrate charge specificity. This suggests that the GalNAc-Ts expressed in-vivo, which contain N-glycans, will likely possess nearly the same charge specificities as the enzymes used in this work and in particular for GalNac-T1, which was expressed from the HEK293F human cell line. The known GalNAc-T O-glycosylation sites are given in Supplementary Table S3, where the majority of the sites are found on the N-terminal stem domain of the enzymes and not expected to alter specificity. Except for GalNAc-T3, all the transferases also contain O-glycosylation sites in the catalytic domain, flexible linker, and/or the lectin domain. Roughly half of these transferases are O-glycosylated on the catalytic domain in regions that may potentially interfere with peptide substrate binding: T368 of GalNAc-T2; T379 of GalNAc-T4; T648 of GalNAc-T5; T333 of GalNAc-T6; S483 of GalNAc-T7; T136 of GalNAc-T11; and T289 of GalNAc-T12 (see Supplementary Table S3 and Supplementary Fig. S6). Whether these sites are significantly occupied in vivo is currently unknown. The remaining known GalNAc-T in vivo O-glycosylation sites are located in regions remote of the peptide binding sites on both the catalytic and lectin domains and would not be expected to alter substrate specificity (see Supplementary Table S3 and Supplementary Fig. S6).
Transferase reactions
Final reactions consisted of sodium cacodylate buffer, pH 6.8, 1 mM β-mercaptoethanol, 0.1% Triton X-100, UDP-[3H]GalNAc (0.2–2 mM), enzyme (0.002–0.036 μM), peptide substrate (0.35–1.4 mM), and were incubated at 37 °C on a Labnet Vortemp 56 shaking microincubator for 10 min to overnight depending on GalNAc-T activity. Elevated ionic strength reactions included 300 mM NaCl in the final reaction and were performed in parallel with non-elevated ionic strength reactions. After incubation, reactions were quenched with 200 μL of 0.5% TFA in H2O. TARGA-C18 spin columns were prior equilibrated by passing sequentially: acetonitrile (300 μL), 50/50 acetonitrile/H2O in 0.1% TFA (300 μL), and 0.1% TFA in H2O (700 μL). The latter washes were eluted by spinning at 800 rpm in an Eppendorf Minispin Plus tabletop centrifuge. Ten percent (22 μL) of the reaction volume was removed for [3H] scintillation counting (initial DPM), and the remainder was applied to the equilibrated TARGA C18 hydrophobic spin columns and spun for 1 minute. After the sample was eluted, columns were washed with 800 μL of 0.1% TFA in H2O to remove free UDP-[3H]GalNAc and [3H]GalNAc by centrifugation at 800 rpm for 1 min giving the A-eluate. The bound (glyco)peptide products/reactants (B eluate) were eluted using two washes of 200 μL of 50/50 acetonitrile in 0.1% TFA followed by 200 μL of 100% acetonitrile, each spun for 1 min at 800 rpm, and a final 100 μL of 100% acetonitrile spun for 4 min. [3H] scintillation counting was performed on the combined flow through and wash (~998 μL A eluate) and the eluted (glyco)peptide products/reactants (~700 μL B eluate). The extent of peptide glycosylated, in mM, was obtained by dividing the B counts (in DPM) of the eluted (glyco)peptides by the initial total DPM (as well as by the sum of the DPM of the A and B eluates) of the UDP-[3H]GalNAc and by multiplying by the initial mM of UDP-GalNAc. The percent of peptide glycosylation was then obtained from the mM peptide glycosylated and the initial mM concentration of the peptide substrate. Overnight transferase reactions by GalNAc-T1 and -T3, where all the charged peptides were exhaustively glycosylated confirmed that the spin columns could bind the full range of charged peptides by giving 100% glycosylation for all the substrates (data not shown). Reactions were typically repeated three or more times as shown in the plots. GraphPad Prism version 9.2 for Windows (GraphPad Software, San Diego, CA) was used to plot the data.
GalNAc-T enzyme kinetics
Stock concentrations of charged peptide substrates were made to yield final reaction concentrations of 2.8, 1.4, 0.7, 0.35, 0.175, 0.0875, 0.0437, and 0.0218 mM. Reaction conditions and workup were identical to the GalNAc-T activity assays after subtracting a no peptide control. Reaction times varied, depending on substrate concentrations, ranging from 6 to 240 min, depending on enzyme activity to maintain peptide glycosylation to <20%. Percent peptide glycosylation values were then converted to μM of GalNAc transferred/(μM enzyme*min) according to the initial amount of UDP-GalNAc, substrate, and enzyme used. Kcat (μM GalNAc/(μM enzyme*min) or min−1), km (μM), and Vmax (μM GalNAc/min) values and Michaelis Menten plots were obtained using GraphPad Prism software.
Protein structure modeling
Homology structures of GalNAc-Ts lacking known crystal structures were obtained using SWISSMODEL (Waterhouse et al. 2018) utilizing three substrate bound GalNAc-T templates having different lectin domain orientations: the “lectin left compact structure” (tgGalNAc-T3, PDB: 6S24), “lectin extended structure” (GalNAc-T2, PDB:2FFU), and “lectin right compact structure” (GalNAc-T2, PDB: 5AJP), as shown in Supplementary Fig. S1. These templates were chosen due to the uncertainty of the orientation of the lectin domain as demonstrated by these different structures (see Supplementary Fig. S1). These templates allowed for the examination of the three most likely orientations of the lectin domain with bound peptides: P3: GAT*GAGAGAGTTPGPG, EA2: PTTDSTTPAPTTK, and MUC5-AC13:GTTPSPVPTTSTT*SAP, respectively, where T* represents Thr-O-GalNAc. These peptide structures were extracted from the template structures and aligned to the superimposed catalytic domains of the homology models using PyMOL (Supplementary Figs S2–S3). Surface electrostatics were calculated by the Adaptive Poisson–Boltzmann Solver APBS extension in PyMOL. Supplementary Figs S2–S3 show the electrostatic surfaces for the three possible lectin domain orientations for all the GalNAc-Ts studied in this work. Isoelectric point, pI, calculations were performed on the N-terminal truncated sequences using the ExPASy-ProtParam tool (Gasteiger et al. 2005).
MD simulations
tgGalNAc-T3 (PDB: 6S24), GalNAc-T2 (PDB: 5AJP), and GalNAc-T12 (PDB: 6XPU) structures were imported into Schrödinger Maestro from the RCSB Protein Data Bank (PDB), missing loops were added, and the structure energy minimized. Minimized structures were imported into PyMOL where the -YAVTPGP- sequence of the charged peptide substrates were superimposed onto the existing bound peptide. After extending the -YAVTPGP- sequence and minimization, the prepared structures were submitted to CHARMM-GUI Solution Builder (Jo et al. 2008; Brooks et al. 2009; Lee et al. 2016) to obtain a solvated and electrically neutral system in a rectangular periodic boundary water box with dimensions of 108–118 Å. NaCl Ions were added via the Monte Carlo method at a concentration of 150 mM. CHARMM-GUI output files (.pdb solvated structure files, .psf structure files, and .str topology files) were processed by using the Making-It-Rain CHARMM-GUI notebook (Arantes et al. 2021). The Making-It-Rain code (Eastman et al. 2017) was adapted and executed by Google Colaboratory’s computing resources (https://colab.research.google.com). Structures were equilibrated at 310 °K, 1 atm, with a default force constant of 500 kJ/mol for 1,000 steps in 0.1 nanoseconds with an integration time of 2 femtoseconds. The resulting equilibrated structure (.pdb) files, initial atom trajectory (.rst) files, and CHARMM protein structure file (.psf) from CHARMM-GUI were used by the OpenMM engine (Eastman et al. 2017) to perform production MD simulations. Simulations consisted of one-thousand 0.25 ns strides using an integration timestep of 2 femtoseconds for a total simulation time of 250 ns. After completion, strides were concatenated utilizing Pytraj (Roe and Cheatham 2013) to obtain the complete trajectory and to calculate the RMSD, Rg, and D-end measurements.
Transferase specificities from random peptide substrates
The human GalNAc-T6, -T11, and the fly PGANT35A peptide substrate specificities that have not been previously reported by our lab were obtained as previously described (Gerken et al. 2006, 2011; Perrine et al. 2009) using random peptide substrates based on the GAGAXXXXXTXXXXXAGAGK sequence, where X = G,A,P,V,L,Y,E,Q,R,H (PVI), X = G,A,P,I,M,F,D,N,R,K (PVII) and X = G,A,P,VY,E,N,S,R,K (PVIII). Random peptides were partially glycosylated utilizing UDP-[3H]-GalNAc and processed as described previously. Glycopeptide products were isolated on a mixed Tn lectin column and further purified on Sephadex G10 (Gerken et al. 2011). Edman sequencing to determine the compositional changes in the X residues was performed on an Applied Biosystems Procice 494 peptide sequencer (as described in Gerken et al. 2006, 2011) for GalNAc-T11 and PGANT35A and on a Shimadzu PPSQ53A sequencer (Shimadzu Scientific instruments Inc., Columbia, MD) for GalNAc-T6. Positional residue specific enhancement values (EV) were obtained by comparing individual residue mole fractions of the glycopeptide product to the mole fraction in the initially non glycosylated peptide. EV values greater than 1 indicate a preference for glycosylation, values of 1 suggest neutral preference and values less than 1 a reduced preference for glycosylation. Comparisons of the EVs of GalNAc-T3 and -T6 and GalNAc-T11 and PGANT35A are given in Supplementary Figs S7 and S8 and are tabulated in Supplementary Table S4. Note that the EVs for GalNAc-T3 have been reported previously (Gerken et al. 2011). EV values for GalNAc-T3 and -T11 are presently incorporated into the ISOGlyP predictor (https://isoglyp.utep.edu/), whereas those for GalNAc-T6 will be subsequently added.
Abbreviations
GalNAc, N-acetylgalactosamine; GalNAc-T, UDP-GalNAc:polypeptide N-α-acetylgalactosaminyl-transferases; UDP, uridine diphosphate; MD, molecular dynamics; RMSD, root mean square deviation; Rg, radius of gyration; D-end, end-to-end distance measurement; Peptide names (RR, RD, etc.), see Table 1; DPM, disintegrations per minute; TFA, trifluoroacetic acid; O-glycosylation, mucin type O-glycosylation; PGANT, D. melanogaster UDP-GalNAc:polypeptide N-α-acetylgalactosaminyl-transferases; EV, enhancement values; LA repeats, LDLR-type A repeats; CRC, colorectal cancer; pI, isoelectric point
Supplementary Material
Acknowledgements
We would like to acknowledge and thank the Case Western Reserve University Undergraduate Biochemistry students, Dayna Nguyen and Kaitlyn Moore, and Westlake Ohio High School student Mantas Viazmitinas for their participation and help in collecting data for this work. We would also like to thank the following individuals for providing the transferases utilized in this work: Malene Bech Vester-Christensen, Yun Kong and Shengjun Wang (H. Clausen Laboratory); Roy W. Johnson, Shuo Wang and Heather A. Moniz (K. Moremen Laboratory); and Matilde de las Rivas, Erandi, Lira-Navarrete, and Ana García-García (R. Hurtado-Guerrero Laboratory). We also acknowledge Sichun Yang, Center for Proteomics and Bioinformatics at Case Western Reserve Univ. for assisting us in setting up the MD calculations. We thank Francisco Corzana López (Universidad de La Rioja; Departamento de Química) for helpful discussions of our MD simulations.
Contributor Information
Collin J Ballard, Department of Biochemistry, Case Western Reserve University, Cleveland, OH 44106, USA.
Miya R Paserba, Department of Biochemistry, Case Western Reserve University, Cleveland, OH 44106, USA.
Earnest James Paul Daniel, Department of Biochemistry, Case Western Reserve University, Cleveland, OH 44106, USA.
Ramón Hurtado-Guerrero, Department of Biomedical Engineering, The Institute for Biocomputation and Physics of Complex Systems (BIFI), University of Zaragoza, Mariano Esquillor s/n, Campus Rio Ebro, Edificio I+D, Zaragoza 50018, Spain; Copenhagen Center for Glycomics, Department of Cellular and Molecular Medicine, Faculty of Health Sciences, University of Copenhagen, Blegdamsvej 3, DK-2200 Copenhagen, Denmark; Fundación ARAID, Zaragoza 50018, Spain.
Thomas A Gerken, Department of Biochemistry, Case Western Reserve University, Cleveland, OH 44106, USA.
Authors’ contributions
CJB: methodology, investigation, visualization, validation, software, writing-original draft, writing-reviewing and editing, MRP: methodology, investigation, visualization, validation, software, writing-reviewing and editing, EP D: methodology, investigation, visualization, validation, writing-reviewing and editing, RH-G: resources, writing-reviewing and editing, TAG: conceptualization, project administration, funding acquisition, supervision, investigation, methodology, visualization, writing-original draft preparation, writing-review and editing. All authors approved the final manuscript.
CRediT author statement
Collin J Ballard (Investigation-Equal, Methodology-Equal, Software-Equal, Validation-Equal, Visualization-Equal, Writing—original draft-Lead, Writing—review and editing-Lead), Miya R Paserba (Investigation-Equal, Methodology-Equal, Software-Lead, Validation-Equal, Visualization-Equal, Writing—review and editing-Supporting), Earnest James Paul Daniel (Investigation-Equal, Methodology-Equal, Validation-Equal, Visualization-Equal, Writing—review and editing-Supporting), Ramón Hurtado Guerrero (Resources-Lead, Writing—review and editing-Supporting), Thomas Gerken (Conceptualization-Lead, Funding acquisition-Lead, Investigation-Equal, Methodology-Equal, Project administration-Lead, Resources-Lead, Supervision-Lead, Validation-Equal, Visualization-Equal, Writing—original draft-Equal, Writing—review and editing-Equal).
Funding
This work was supported by grants from the National Institutes of Health: (R01-GM113534 to T.A.G.), (P41-GM103390 to K.M., Complex Carbohydrate Research Center, University of Georgia), and (P01-GM107012 to G.J.B., Complex Carbohydrate Research Center, University of Georgia). Funding by the Danish National Research Foundation (DNRF107) is also acknowledged (to H.C., Univ. Copenhagen). We thank ARAID, MEC (PID2019-105451GB-I00 to R.H.-G.), and Gobierno de Aragón (E34_R17 and LMP58_18 to R.H.-G.) with FEDER (2014–2020) funds for “Building Europe from Aragón” for financial support.
Conflict of interest statement. None declared.
Data availability statement
The data underlying this study are available in the paper and in its online supplementary materials. Any additional data not included will be shared on reasonable request to the corresponding author.
References
- Arantes PR, Polêto MD, Pedebos C, Ligabue-Braun R. Making it rain: cloud-based molecular simulations for everyone. J Chem Inf Model. 2021:61(10):4852–4856. 10.1021/acs.jcim.1c00998. [DOI] [PubMed] [Google Scholar]
- Bagdonaite I, Wandall HH. Global aspects of viral glycosylation. Glycobiology. 2018:28(7):443–467. 10.1093/glycob/cwy021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagdonaite I, Pallesen EMH, Nielsen MI, Bennett EP, Wandall HH. Mucin-type O-GalNAc glycosylation in health and disease. In: Lauc G, Trbojević-Akmačić I, Editors, The role of glycosylation in health and disease. Springer International Publishing, Cham, Switzerland; 2021. p. 25–60. 10.1007/978-3-030-70115-4_2 [DOI] [Google Scholar]
- Beaman E-M, Brooks SA. The extended ppGalNAc-T family and their functional involvement in the metastatic cascade. Histol Histopathol. 2014:29(3):293–304. 10.14670/HH-29.293. [DOI] [PubMed] [Google Scholar]
- Beaman E-M, Carter DRF, Brooks SA. GALNTs: master regulators of metastasis-associated epithelial-mesenchymal transition (EMT)? Glycobiology. 2022:32(7):556–579. 10.1093/glycob/cwac014. [DOI] [PubMed] [Google Scholar]
- Bennett EP, Chen Y-W, Schwientek T, Mandel U, Schjoldager K, ter-B. G., Cohen, S. M., & Clausen, H. Rescue of Drosophila melanogaster l(2)35Aa lethality is only mediated by polypeptide GalNAc-transferase pgant35A, but not by the evolutionary conserved human ortholog GalNAc-transferase-T11. Glycoconj J. 2010:27(4):435–444. 10.1007/s10719-010-9290-5. [DOI] [PubMed] [Google Scholar]
- Bennett EP, Mandel U, Clausen H, Gerken TA, Fritz TA, Tabak LA. Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology. 2012:22(6):736–756. 10.1093/glycob/cwr182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biller M, Mardberg K, Hassan H, Clausen H, Bolmstedt A, Bergstrom T, Olofsson S. Early steps in O-linked glycosylation and clustered O-linked glycans of herpes simplex virus type 1 glycoprotein C: effects on glycoprotein properties. Glycobiology. 2000:10(12):1259–1269. 10.1093/glycob/10.12.1259. [DOI] [PubMed] [Google Scholar]
- Brockhausen, I., Wandall, H. H., Hagen, K. G. T., & Stanley, P. (2022). O-GalNAc glycans. In: Essentials of glycobiology [Internet] . 4th edition. Cold Spring Harbor Laboratory Press, New York, NY. 10.1101/glycobiology.4e.10 [DOI] [Google Scholar]
- Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009:30(10):1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel EJP, de las Rivas M, Lira-Navarrete E, García-García A, Hurtado-Guerrero R, Clausen H, Gerken TA. Ser and Thr acceptor preferences of the GalNAc-Ts vary among isoenzymes to modulate mucin-type O-glycosylation. Glycobiology. 2020:30(11):910–922. 10.1093/glycob/cwaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J, Chen L, Gao H, Zhen T, Li H, Liang J, Zhang F, Shi H, Han A. GALNT6 suppresses progression of colorectal cancer. Am J Cancer Res. 2018:8(12):2419–2435. [PMC free article] [PubMed] [Google Scholar]
- Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, Wang L-P, Simmonett AC, Harrigan MP, Stern CD, et al. OpenMM 7: rapid development of high performance algorithms for molecular dynamics. PLoS Comput Biol. 2017:13(7):e1005659. 10.1371/journal.pcbi.1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner B, Merry AH, Royle L, Harvey DJ, Rudd PM, Thillet J. Structural elucidation of the N- and O-glycans of human apolipoprotein(a): role of O-glycans in conferring protease resistance. J Biol Chem. 2001:276(25):22200–22208. 10.1074/jbc.M102150200. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker JM, Editor, The proteomics protocols handbook. Humana Press, Totowa, NJ; 2005. p. 571–607. 10.1385/1-59259-890-0:571 [DOI] [Google Scholar]
- Gerken TA, Raman J, Fritz TA, Jamison O. Identification of common and unique peptide substrate preferences for the UDP-GalNAc:polypeptide α-N-acetylgalactosaminyltransferases T1 and T2 derived from oriented random peptide substrates. J Biol Chem. 2006:281(43):32403–32416. 10.1074/jbc.M605149200. [DOI] [PubMed] [Google Scholar]
- Gerken TA, Ten Hagen KG, Jamison O. Conservation of peptide acceptor preferences between drosophila and mammalian polypeptide-GalNAc transferase ortholog pairs. Glycobiology. 2008:18(11):861–870. 10.1093/glycob/cwn073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerken TA, Jamison O, Perrine CL, Collette JC, Moinova H, Ravi L, Markowitz SD, Shen W, Patel H, Tabak LA. Emerging paradigms for the initiation of mucin-type protein O-glycosylation by the polypeptide GalNAc transferase family of glycosyltransferases. J Biol Chem. 2011:286(16):14493–14507. 10.1074/jbc.M111.218701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerken TA, Revoredo L, Thome JJC, Tabak LA, Vester-Christensen MB, Clausen H, Gahlay GK, Jarvis DL, Johnson RW, Moniz HA, et al. The lectin domain of the polypeptide GalNAc transferase family of glycosyltransferases (ppGalNAc Ts) acts as a switch directing glycopeptide substrate glycosylation in an N- or C-terminal direction, further controlling mucin type O-glycosylation. J Biol Chem. 2013:288(27):19900–19914. 10.1074/jbc.M113.477877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rodriguez E, Zol-Hanlon M, Bineva-Todd G, Marchesi A, Skehel M, Mahoney KE, Roustan C, Borg A, Di Vagno L, Kjær S, et al. O-linked sialoglycans modulate the proteolysis of SARS-CoV-2 spike and likely contribute to the mutational trajectory in variants of concern. ACS Central Science. 2023:9(3):393–404. 10.1021/acscentsci.2c01349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holleboom AG, Karlsson H, Lin R-S, Beres TM, Sierts JA, Herman DS, Stroes ESG, Aerts JM, Kastelein JJP, Motazacker MM, et al. Heterozygosity for a loss-of-function mutation in GALNT2 improves plasma triglyceride clearance in man. Cell Metab. 2011:14(6):811–818. 10.1016/j.cmet.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004:4(1):1. 10.1038/nrc1251. [DOI] [PubMed] [Google Scholar]
- Hu Y, Feng J, Wu F. The multiplicity of polypeptide GalNAc-transferase: assays, inhibitors, and structures. Chembiochem. 2018:19(24):2503–2521. 10.1002/cbic.201800303. [DOI] [PubMed] [Google Scholar]
- Hu Q, Tian T, Leng Y, Tang Y, Chen S, Lv Y, Liang J, Liu Y, Liu T, Shen L, et al. The O-glycosylating enzyme GALNT2 acts as an oncogenic driver in non-small cell lung cancer. Cell Mol Biol Lett. 2022:27(1):71. 10.1186/s11658-022-00378-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Wu M, Zhang Y, Kong S, Liu M, Jiang B, Yang P, Cao W. OGP: a repository of experimentally characterized O-glycoproteins to facilitate studies on O-glycosylation. Genom Proteom Bioinform. 2021:19(4):611–618. 10.1016/j.gpb.2020.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008:29(11):1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- de las Rivas M, Lira-Navarrete E, Daniel EJP, Compañón I, Coelho H, Diniz A, Jiménez-Barbero J, Peregrina JM, Clausen H, Corzana F, et al. The interdomain flexible linker of the polypeptide GalNAc transferases dictates their long-range glycosylation preferences. Nat Commun. 2017:8:1959. 10.1038/s41467-017-02006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de las Rivas M, Lira-Navarrete E, Gerken TA, Hurtado-Guerrero R. Polypeptide GalNAc-Ts: from redundancy to specificity. Curr Opin Struct Biol. 2019:56:87–96. 10.1016/j.sbi.2018.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de las Rivas M, Paul Daniel EJ, Coelho H, Lira-Navarrete E, Raich L, Compañón I, Diniz A, Lagartera L, Jiménez-Barbero J, Clausen H, et al. Structural and mechanistic insights into the catalytic-domain-mediated short-range glycosylation preferences of GalNAc-T4. ACS Cent Sci. 2018:4(9):1274–1290. 10.1021/acscentsci.8b00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de las Rivas M, Paul Daniel EJ, Narimatsu Y, Compañón I, Kato K, Hermosilla P, Thureau A, Ceballos-Laita L, Coelho H, Bernadó P, et al. Molecular basis for fibroblast growth factor 23 O-glycosylation by GalNAc-T3. Nat Chem Biol. 2020:16(3):351–360. 10.1038/s41589-019-0444-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrsen K, Dabelsteen S, Vakhrushev SY, Levann AMR, Haue AD, Dylander A, Mandel U, Hansen L, Frödin M, Bennett EP, et al. De novo expression of human polypeptide N-acetylgalactosaminyltransferase 6 (GalNAc-T6) in colon adenocarcinoma inhibits the differentiation of colonic epithelium. J Biol Chem. 2018:293(4):1298–1314. 10.1074/jbc.M117.812826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput. 2016:12(1):405–413. 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Pan S, Xiao Y, Liu Q, Xu J, Jia L. Correction to: LINC01296/miR-26a/GALNT3 axis contributes to colorectal cancer progression by regulating O-glycosylated MUC1 via PI3K/AKT pathway. J Exp Clin Cancer Res. 2019:38:142. 10.1186/s13046-019-1140-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiels B, Lété C, Guillaume A, Mast J, Stevenson PG, Vanderplasschen A, Gillet L. Antibody evasion by a gammaherpesvirus O-glycan shield. PLoS Pathog. 2011:7(11):e1002387. 10.1371/journal.ppat.1002387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May C, Ji S, Syed ZA, Revoredo L, Daniel EJP, Gerken TA, Tabak LA, Samara NL, Hagen KGT. Differential splicing of the lectin domain of an O-glycosyltransferase modulates both peptide and glycopeptide preferences. J Biol Chem. 2020:295(35):12525–12536. 10.1074/jbc.RA120.014700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohl JE, Gerken TA, Leung M-Y. ISOGlyP: De novo prediction of isoform-specific mucin-type O-glycosylation. Glycobiology. 2020a:31(3):168–172. 10.1093/glycob/cwaa067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohl JE, Gerken T, Leung M-Y. Predicting mucin-type O-glycosylation using enhancement value products from derived protein features. J Theor Comput Chem. 2020b:19(3):2040003. 10.1142/s0219633620400039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moremen KW, Ramiah A, Stuart M, Steel J, Meng L, Forouhar F, Moniz HA, Gahlay G, Gao Z, Chapla D, et al. Expression system for structural and functional studies of human glycosylation enzymes. Nat Chem Biol. 2018:14(2):156–162. 10.1038/nchembio.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehrke K, Hagen FK, Tabak LA. Charge distribution of flanking amino acids influences O-glycan acquisition in vivo(*). J Biol Chem. 1996:271(12):7061–7065. 10.1074/jbc.271.12.7061. [DOI] [PubMed] [Google Scholar]
- Nehrke K, Ten Hagen KG, Hagen FK, Tabak LA. Charge distribution of flanking amino acids inhibits O-glycosylation of several single-site acceptors in vivo. Glycobiology. 1997:7(8):1053–1060. 10.1093/glycob/7.8.1053-c. [DOI] [PubMed] [Google Scholar]
- Nielsen MI, de Haan N, Kightlinger W, Ye Z, Dabelsteen S, Li M, Jewett MC, Bagdonaite I, Vakhrushev SY, Wandall HH. Global mapping of GalNAc-T isoform-specificities and O-glycosylation site-occupancy in a tissue-forming human cell line. Nat Commun. 2022:13(1):1. 10.1038/s41467-022-33806-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell B, Tabak LA, Ramasubbu N. The influence of flanking sequences on O-glycosylation. Biochem Biophys Res Commun. 1991:180(2):1024–1030. 10.1016/S0006-291X(05)81168-4. [DOI] [PubMed] [Google Scholar]
- O’Connell BC, Hagen FK, Tabak LA. The influence of flanking sequence on the O-glycosylation of threonine in vitro. J Biol Chem. 1992:267(35):25010–25018. 10.1016/S0021-9258(19)73998-2. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Tanaka A, Namba K, Shia J, Wang JY, Roehrl MH. Early-stage loss of GALNT6 predicts poor clinical outcome in colorectal cancer. Front Oncol. 2022:12:802548. 10.3389/fonc.2022.802548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecori F, Akimoto Y, Hanamatsu H, Furukawa J, Shinohara Y, Ikehara Y, Nishihara S. Mucin-type O-glycosylation controls pluripotency in mouse embryonic stem cells via Wnt receptor endocytosis. J Cell Sci. 2020:133(20):jcs245845. 10.1242/jcs.245845. [DOI] [PubMed] [Google Scholar]
- Pedersen NB, Wang S, Narimatsu Y, Yang Z, Halim A, Schjoldager KT-BG, Madsen TD, Seidah NG, Bennett EP, Levery SB, et al. Low density lipoprotein receptor class a repeats are O-glycosylated in linker regions. J Biol Chem. 2014:289(25):17312–17324. 10.1074/jbc.M113.545053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrine CL, Ganguli A, Wu P, Bertozzi CR, Fritz TA, Raman J, Tabak LA, Gerken TA. Glycopeptide-preferring polypeptide GalNAc transferase 10 (ppGalNAc T10), involved in mucin-type O-glycosylation, has a unique GalNAc-O-Ser/Thr-binding site in its catalytic domain not found in ppGalNAc T1 or T2. J Biol Chem. 2009:284(30):20387–20397. 10.1074/jbc.M109.017236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revoredo L, Wang S, Bennett EP, Clausen H, Moremen KW, Jarvis DL, Ten Hagen KG, Tabak LA, Gerken TA. Mucin-type O-glycosylation is controlled by short- and long-range glycopeptide substrate recognition that varies among members of the polypeptide GalNAc transferase family. Glycobiology. 2016:26(4):360–376. 10.1093/glycob/cwv108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013:9(7):3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Schwientek T, Bennett EP, Flores C, Thacker J, Hollmann M, Reis CA, Behrens J, Mandel U, Keck B, Schäfer MA, et al. Functional conservation of subfamilies of putative UDP-N-acetylgalactosamine:polypeptide N-acetylgalactosaminyltransferases in Drosophila, Caenorhabditis elegans, and mammals: ONE SUBFAMILY COMPOSED OF l(2)35Aa IS ESSENTIAL INDROSOPHILA. J Biol Chem. 2002:277(25):22623–22638. 10.1074/jbc.M202684200. [DOI] [PubMed] [Google Scholar]
- Shi X, Jarvis DL. Protein N-glycosylation in the baculovirus-insect cell system. Curr Drug Targets. 2007:8(10):1116–1125. 10.2174/138945007782151360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver ZA, Antonopoulos A, Haslam SM, Dell A, Dickinson GM, Seaman MS, Desrosiers RC. Discovery of O-linked carbohydrate on HIV-1 envelope and its role in shielding against one category of broadly neutralizing antibodies. Cell Rep. 2020:30(6):1862–1869.e4. 10.1016/j.celrep.2020.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steentoft C, Vakhrushev SY, Joshi HJ, Kong Y, Vester-Christensen MB, Schjoldager KT-BG, Lavrsen K, Dabelsteen S, Pedersen NB, Marcos-Silva L, et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013:32(10):1478–1488. 10.1038/emboj.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabak LA. The role of mucin-type O-glycans in eukaryotic development. Semin Cell Dev Biol. 2010:21(6):616–621. 10.1016/j.semcdb.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajadura-Ortega V, Gambardella G, Skinner A, Halim A, Van Coillie J, Schjoldager KT-BG, Beatson R, Graham R, Achkova D, Taylor-Papadimitriou J, et al. O-linked mucin-type glycosylation regulates the transcriptional programme downstream of EGFR. Glycobiology. 2021:31(3):200–210. 10.1093/glycob/cwaa075. [DOI] [PubMed] [Google Scholar]
- Ten Hagen KG, Fritz TA, Tabak LA. All in the family: the UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycobiology. 2003:13(1):1R–16R. 10.1093/glycob/cwg007. [DOI] [PubMed] [Google Scholar]
- Tian E, Ten Hagen KG. Recent insights into the biological roles of mucin-type O-glycosylation. Glycoconj J. 2009:26(3):325–334. 10.1007/s10719-008-9162-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vester-Christensen MB, Bennett EP, Clausen H, Mandel U. Generation of monoclonal antibodies to native active human glycosyltransferases. Methods Mol Biol (Clifton, NJ). 2013:1022:403–420. 10.1007/978-1-62703-465-4_30. [DOI] [PubMed] [Google Scholar]
- Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K, Lee D, von Goetz M, Yee SF, Totpal K, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med. 2007:13(9):9. 10.1038/nm1627. [DOI] [PubMed] [Google Scholar]
- Wandall HH, Irazoqui F, Tarp MA, Bennett EP, Mandel U, Takeuchi H, Kato K, Irimura T, Suryanarayanan G, Hollingsworth MA, et al. The lectin domains of polypeptide GalNAc-transferases exhibit carbohydrate-binding specificity for GalNAc: lectin binding to GalNAc-glycopeptide substrates is required for high density GalNAc-O-glycosylation. Glycobiology. 2007:17(4):374–387. 10.1093/glycob/cwl082. [DOI] [PubMed] [Google Scholar]
- Wang S, Mao Y, Narimatsu Y, Ye Z, Tian W, Goth CK, Lira-Navarrete E, Pedersen NB, Benito-Vicente A, Martin C, et al. Site-specific O-glycosylation of members of the low-density lipoprotein receptor superfamily enhances ligand interactions. J Biol Chem. 2018:293(19):7408–7422. 10.1074/jbc.M117.817981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu Z, Hu W, Hao P, Yang S. Impact of expressing cells on glycosylation and glycan of the SARS-CoV-2 spike glycoprotein. ACS Omega. 2021:6(24):15988–15999. 10.1021/acsomega.1c01785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018:46(W1):W296–W303. 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Zhang C, Zhang K, Chen Q, Wu S, Huang H, Huang T, Zhang N, Wang X, Li W, et al. PpGalNAc-T4-catalyzed O-glycosylation of TGF-β type II receptor regulates breast cancer cells metastasis potential. J Biol Chem. 2020:296:100119. 10.1074/jbc.RA120.016345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L, Ju T, Westmuckett A, An G, Ivanciu L, McDaniel JM, Lupu F, Cummings RD, McEver RP. Defective angiogenesis and fatal embryonic hemorrhage in mice lacking core 1–derived O-glycans. J Cell Biol. 2004:164(3):451–459. 10.1083/jcb.200311112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York WS, Mazumder R, Ranzinger R, Edwards N, Kahsay R, Aoki-Kinoshita KF, Campbell MP, Cummings RD, Feizi T, Martin M, et al. GlyGen: computational and informatics resources for glycoscience. Glycobiology. 2020:30(2):72–73. 10.1093/glycob/cwz080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L-G, Andrews N, Zhao Q, McKean D, Williams JF, Connor LJ, Gerasimenko OV, Hilkens J, Hirabayashi J, Kasai K, et al. Galectin-3 interaction with Thomsen-Friedenreich disaccharide on cancer-associated MUC1 causes increased cancer cell endothelial adhesion. J Biol Chem. 2007:282(1):773–781. 10.1074/jbc.M606862200. [DOI] [PubMed] [Google Scholar]
- Zhang J, ten Dijke P, Wuhrer M, Zhang T. Role of glycosylation in TGF-β signaling and epithelial-to-mesenchymal transition in cancer. Protein Cell. 2021:12(2):2. 10.1007/s13238-020-00741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Yin S, Zheng J, Shi Y, Jia C. O-glycosylation site prediction for Homo sapiens by combining properties and sequence features with support vector machine. J Bioinforma Comput Biol. 2022:20(01):2150029. 10.1142/S0219720021500293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the paper and in its online supplementary materials. Any additional data not included will be shared on reasonable request to the corresponding author.