

Summary:
Inflammation can trigger lasting phenotypes in immune and non-immune cells. Whether systemic inflammation, such as that caused by severe coronavirus disease 2019 (COVID-19), triggers innate immune memory in hematopoietic cells is unknown. We found that circulating hematopoietic stem and progenitor cells (HSPC), enriched from peripheral blood, captured the diversity of bone marrow (BM) HSPC, enabling investigation of their epigenomic reprogramming following COVID-19. Alterations in innate immune phenotypes and epigenetic programs of HSPC persisted for months to one year following severe COVID-19 and were associated with distinct transcription factor (TF) activities, altered regulation of inflammatory programs, and durable increases in myelopoiesis. HSPC epigenomic alterations were conveyed, through differentiation, to progeny innate immune cells. Early activity of IL-6 contributed to these persistent phenotypes in human COVID-19 and a mouse coronavirus infection model. Epigenetic reprogramming of HSPC may underly altered immune function following infection and be broadly relevant, especially for millions of COVID-19 survivors.
Keywords: COVID-19, epigenetic memory, trained immunity, hematopoietic stem and progenitor cells, monocytes, single-cell, IL-6, epigenome, transcriptome, peripheral blood mononuclear cell progenitor input enrichment, post-acute sequelae SARS-CoV-2 infection (PASC)
In Brief
Severe COVID-19 can reprogram hematopoiesis and establish epigenetic memory in hematopoietic stem and progenitor cells (HSPC) and progeny myeloid cells for up to one year. These durable alterations, which could affect post-infection immune responses and equilibrium, are controlled in part by the activity of IL-6 during acute disease.
Graphical Abstract
Introduction
COVID-19, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, is characterized by a range of symptoms and severity, and frequently includes long-term complications. Innate immune cell activation together with delayed adaptive immune and interferon (IFN) responses feature prominently in acute severe COVID-191–13. Importantly, the long-term effects of COVID-19 on the immune system and the nature of persistent molecular and cellular changes are poorly understood. These durable changes in the immune system could influence subsequent immune responses to pathogens, vaccines, and contribute to long-term clinical symptom, e.g., post-acute sequelae of SARS-CoV-2 infection (PASC)14–22 and COVID-19-associated multisystem inflammatory syndrome in adults (MIS-A) and children (MIS-C)23–27. While several biomarkers are associated with PASC, with examples including elevated antibody responses to Epstein-Barr virus, persistent SARS-CoV-2 virus, or low cortisol28,29, the molecular and cellular changes following COVID-19 that alter immunity and contribute to ongoing pathology are poorly understood.
Recent studies have established that innate immune cells and their progenitors can maintain durable epigenetic memory of prior infection or inflammation, altering innate immune equilibrium and responses to subsequent challenges30,31. Innate immune memory is primarily attributed to persistent chromatin alterations that modify cell responses, including in long-lived innate immune cells30–35, epithelial stem cells36–38, hematopoietic progenitors, and their mature progeny cells39–48. Altered chromatin accessibility, has been described in mature immune cells one to three months following COVID-1949. Establishing the cellular source of such durable epigenetic memory has been a challenge, especially considering the short lifespan50 of many mature innate immune cells. Studies in mice indicate HSPC can be epigenetically reprogrammed upon exposure to inflammation, leading to long-lasting phenotypic changes in progeny cells42,45–48,51. Underscoring the functional relevance of this epigenetic reprogramming, enhanced granulopoiesis in response to β-glucan drives improved anti-tumor responses47, and IL-1 signaling-induced myelopoiesis contributes to an HSC-transplantable model of inflammatory disease46.
While innate immune memory is well-studied in mouse models, the breadth, relevance, and molecular features of such phenotypes in humans have been more elusive. Recent studies have revealed innate immune memory in humans after receiving Bacillus Calmette-Guérin (BCG)43,44,53, a tuberculosis vaccine. Importantly, certain BM HSPC phenotypes, such as myeloid cell fate priming, persist for at least three months following BCG vaccination44. While controlled vaccine studies have established innate immune memory or trained immunity phenotypes in humans, the limited access to experimental studies on human HSPC has impeded our understanding of dynamic HSPC phenotypes in disease, especially in the context of natural infection.
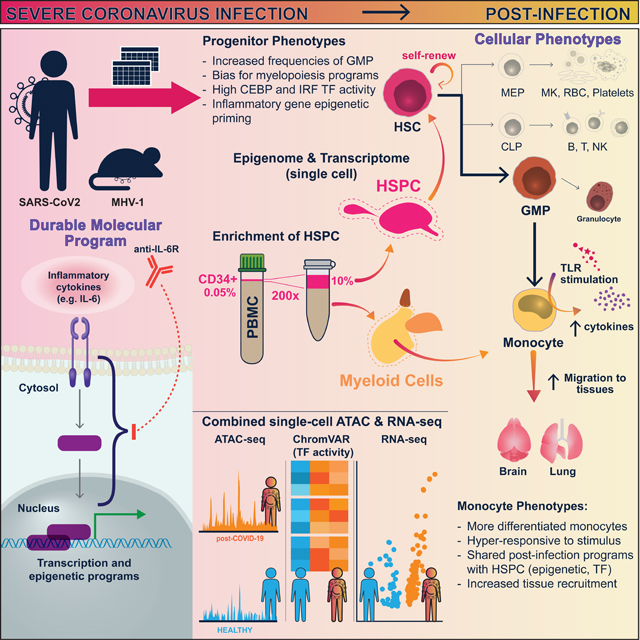
In this study, we identified epigenetic innate immune memory that results from SARS-CoV-2 infection by characterizing the cellular and molecular features during the post-infection period of COVID-19. We focused on analyzing chromatin and transcription at the single-cell level in monocytes and their progenitors, HSPC. High-resolution transcriptomic and chromatin accessibility maps were generated for diverse HSPC subtypes and PBMC following severe COVID-19. We discovered lasting epigenetic and transcription changes in HSPC and monocytes, indicating an altered innate immune response. These changes were associated with increased myelopoiesis in HSPC and led to inflammatory, migratory, and differentiating monocyte phenotypes. While the blockade of IL-6 signaling modestly improved outcomes in critically ill patients54,55, the impact of early activity of this pleiotropic cytokine on recovery remains unclear. We found that disruption of IL-6R signaling during acute infection resulted in a reduction in post-infection HSPC and monocyte inflammatory programs in human patients and mice. Our findings underscore the potential of inflammatory cytokine signaling, like IL-6, during acute viral infections to induce enduring epigenetic changes in HSPC and progeny innate immune cells, potentially influencing various post-infection phenomena in humans, including tissue repair, protection against other infections, chronic inflammation, and long-term sequalae.
Results
Altered chromatin accessibility and durable epigenetic memory in monocytes following COVID-19
To study the molecular features of HSPC and mature immune cells in convalescent severe COVID-19 study participants, we collected blood samples from early convalescent (2–4 months after onset of disease, “Early”) and late convalescent participants (4–12 months after onset, “Late”), and participants recovering from non-COVID-19 critical illness (“nonCoV”, requiring ICU admission) for plasma and epigenomic analysis, including Assay for Transposase-Accessible Chromatin (ATAC)-seq, and single-cell analysis (Figure S1A-B, Table S1). Study participants were enrolled during the first wave of infections in New York City prior to administration of COVID-19 vaccines and were infected with 614D and 614G variants of the virus56. To comprehensively study cellular and molecular features of immune cells in this cohort, we established a multimodal assay and analysis workflow incorporating combined single nuclei (sn) RNA and ATAC-seq (snRNA/ATAC-seq) for PBMC and HSPC, sorted monocyte and HSPC subset “bulk” ATAC-seq, multiplexed immunoassay-based quantitation of plasma proteins, and immunophenotyping by flow cytometry (Figure S1).
To determine if mature circulating CD14+ monocytes from COVID-19 convalescent individuals have distinct and durable epigenetic signatures, we profiled chromatin accessibility by ATAC-seq (either snRNA/ATAC-seq or conventional ATAC-seq) across the four clinical groups: Healthy, nonCoV, Early and Late (n = 57). Bulk and single-nuclei “pseudo-bulk” ATAC-seq were analyzed together when appropriate (Figure 1A, STAR Methods).
Figure 1. Durable epigenetic alterations in monocytes following severe COVID-19.

(A) Overview of the cohort and ATAC-seq workflow for epigenetic profiling of CD14+ monocytes.
(B) PCA representation of CD14+ monocyte ATAC-seq dataset based on a combined set of DAR (n=2029) (left). Boxplots (right) show PC1 and PC2 values for the individuals in each clinical group.
(C) Unsupervised hierarchical clustering of DAR in CD14+ monocytes by clinical group. Normalized peak accessibility for individuals and DAR clustered by chromatin accessibility trends across clinical groups. (FDR < 0.1 for DAR)
(D) GO analysis of genes associated to cluster-specific DAR in CD14+ monocytes. (p<0.05)
(E) Top: Normalized read density for cluster-specific DAR. Each DAR (group average) is represented by a linked line across groups. Bottom: Cluster average of normalized DAR density score per individual across clinical groups.
(F) Representative ATAC-seq genome tracks of DAR from C2 and C4 in CD14+ monocytes. Boxplots display normalized DAR densities for each study participant.
Principal component (PC) analysis of differentially accessible regions (DAR/peaks) revealed that individual datapoints clustered by group, with PC1 capturing chromatin accessibility features associated with early recovery from critical illness (nonCoV, Early; e.g., monocyte differentiation) and PC2 capturing common features of post-COVID states (Early, Late; e.g. response to IL-6) (Figure 1B, Figure S2A).
Across all groups, DAR in monocytes clustered into four major groups based on their accessibility profiles (Figure 1C-E, Table S3). Epigenetic changes between Early and Healthy were most notable, where cluster 2 (C2), cluster 3 (C3), and cluster 4 (C4) DAR had increased accessibility and cluster 1 (C1) had decreased accessibility in Early (Figure 1C, 1E, Table S3). C4 peak accessibility, while increased in Early, returned toward the Healthy baseline in Late and were therefore referred to as “transient” DAR (Figure 1C-E, Table S3). These transient C4 DAR were enriched for gene ontology (GO) categories related to leukocyte activation, cytokine production, and differentiation (Figure 1D) and were annotated to pro-inflammatory and activation molecules (e.g., IL18; CSF1R, the receptor for macrophage differentiation and maintenance factor CSF1; IL17RA, receptor for IL17, which regulates monocyte differentiation and migration) (Figure 1E-F). In contrast, C2 DAR were more accessible in Late than Healthy, though with reduced average magnitude compared to Early and nonCoV. We refer to C2 as “persistent” DAR since these disease-related epigenetic changes lasted 4–12 months after disease onset. Persistent monocyte epigenetic features were annotated to genes including IL21R, MAPKAPK2, TNIP2, CREB1, and MMP1 and GO categories related to monocyte activation and cytokine production (Figure 1D-F). (Figure 1E, boxplots).
Thus, bulk ATAC-seq profiling indicates that circulating monocytes retain altered epigenetic landscapes following recovery from severe COVID-19 with distinct signatures in early and late convalescence. Notably, increased chromatin accessibility at genes encoding cytokines (e.g., IL10, IFNG), adhesion molecules (e.g., ADAM9, ITGAL), and differentiation factors (e.g., KLF13, FOXP1) persisted for one year following acute COVID-19, highlighting either an active maintenance of these programs through ongoing signaling or durable epigenetic memory in monocyte progenitors (Figure 1E).
Altered monocyte phenotypes and transcriptional programs post-COVID-19
Considering that epigenetic signatures of COVID-19 may affect gene expression, we performed snRNA/ATAC-seq analysis of PBMC samples from our cohort (n=32) to study transcriptional and epigenetic changes and cell type distribution (Figure S4). Notably, CD14+ monocytes exhibited the highest number of gene expression changes between post-COVID-19 and Healthy (Figure S4B, Table S4), indicating innate immune memory in these short-lived circulating cells. Given the dysregulation of the myeloid population in acute disease57,58, we focused on myeloid populations and their progenitors for exploration of innate immune memory phenotypes following COVID-19.
We re-clustered and annotated myeloid cells as CD14+ monocytes, CD16+ monocytes, and dendritic cells (DC) (Figure 2A-B). Analysis of CD14+ monocytes uncovered differentially expressed genes (DEG) and differential activity in domains of regulatory chromatin (DORC)59 between post-illness and Healthy groups (Figure 2C, Figure S2B-C, Table S4-5). DORC represent functionally linked non-coding gene regulatory elements, and associated genes are enriched for developmental programs and depleted for housekeeping genes59.
Figure 2. Altered CD14+ monocyte programs and function following severe COVID-19.

(A) snRNA-seq UMAP visualization of myeloid clusters.
(B) Frequency of myeloid subcluster within individual’s total myeloid population by clinical group.
(C) Average normalized expression of top group-defining genes ranked by adjusted p-value in CD14+ monocytes.
(D) Myeloid cluster UMAP showing CD14+ monocyte subclusters.
(E) Expression of myeloid subcluster-defining genes. Red-dashed box highlights similarities between M.SC3 and DC.
(F) Scheme for ex-vivo stimulation of CD14+ monocytes with R848 and IFNα (top). Boxplots show concentration of secreted cytokines 24hr post-stimulation.
(G) GO analysis of upregulated genes in post-COVID CD14+ monocytes at 6hr post-stimulation compared to Healthy.
(H) Correlation of foldchanges in DORC activity (unstimulated) and in gene expression (stimulated with R848+IFNα for 6 hours) between healthy and post-COVID groups (unstimulated, Early or Late or both). Labeled genes are DORC-associated genes with significant upregulation in both DORC and RNA expression (adj.p < 0.05 for both differential DORC and gene expression). Homogeneity between quadrant distribution of statistically significant and non-significant genes shown by chi-square test of independence.
(I) Genome tracks displaying DORC region for IL7R in CD14+ monocytes by group. DORC-associated peaks are connected to the IL7R gene body with loops.
(J) Average DORC score of each study participant across cohorts.
Early monocytes showed enrichment of GO terms related to myeloid cell activation and cytokine production in both differential DORC and DEG (Figure S2D, Table S4-5). Inflammatory programs in Late overlapped with Early and were enriched compared to nonCoV (e.g., “antigen processing and presentation”, “response to virus”; Figure S2D). Late group exhibited persistent DORC signatures and gene expression programs (Figure S2D), congruent with durable bulk-ATAC-seq signatures (Figure 1).
In line with incomplete resolution of inflammation in Early and nonCoV, we found transcriptional upregulation of a broadly defined inflammatory gene module in CD14+ monocytes from both groups (Figure S2F), including genes associated with acute inflammation and anti-viral responses (Figure 2C, S2F, Table S4). Interestingly, while expression of many of these inflammatory genes returned to baseline in Late, other inflammatory molecules (e.g., S100A8, S100A9) and genes related to antigen presentation (e.g., CD74, B2M) remained upregulated in Late compared to Healthy and Early (Figure 2C, S2B, S2D-E, Table S4), suggesting a more differentiated phenotype in Late CD14+ monocytes.
Human monocytes are transcriptionally heterogeneous60. To investigate this heterogeneity in post-COVID-19 monocytes, we annotated three additional subclusters in CD14+ monocytes (M.SC1, M.SC2, and M.SC3), based on marker gene expression (Figure 2D-E, Table S2). While M.SC1 was equally distributed across groups, M.SC2 was enriched in Early and nonCoV, returned to baseline in Late (Figure 2B), and displayed epigenomic signatures associated with inflammatory programs (Figure S2G). Notably, M.SC3 was uniquely enriched in Late and characterized by increased expression of inflammatory monocyte and DC signature genes, including those relating to antigen presentation (e.g., CD74, HLA genes), migration (e.g., ITGB2), and inflammation (e.g., S100A6, LYZ) (Figure 2B-E, Table S2). M.SC3 cells resembled intermediate monocytes61,62, and highly expressed intermediate monocyte transcriptional program62 (Figure S2H). Thus, Late monocytes exhibit distinct characteristics including epigenomic signatures, M.SC3 enrichment, and differential enrichment of epigenetic and transcriptional signatures linked to antigen presentation, activation, differentiation, and anti-viral responses.
To examine whether epigenetic and transcriptional signatures in post-COVID-19 monocytes are associated with functional differences, we stimulated monocytes from PBMC with TLR7/8 agonist R848 and IFNα for 6 and 24 hours to model an anti-viral response (Figure 2F, top). We found significantly higher secretion of IL-6, GM-CSF, and TNFα at 24 hours in post-COVID-19 monocytes compared to Healthy (Figure 2F, bottom; S3A), consistent with a more differentiated and hyper-responsive molecular phenotype (Figure 2D-E, Table S7). We performed RNA-seq to globally assess stimulation-induced transcription (6hr) and found that genes more highly expressed in the post-COVID-19 group were enriched for GO terms related to response to virus and cytokines, and differentiation (Figure 2G, S3B-C, Table S7). To investigate the contribution of epigenetic poising to the hyper-responsiveness of post-COVID-19 monocytes, we compared stimulation-induced gene expression changes with altered chromatin accessibility in unstimulated post-COVID-19 monocytes. Several hyper-responsive genes (e.g., IL7R, CCL5, CXCR4, IFI30, OASL, GBP5) showed increased chromatin accessibility in post-COVID-19 naive monocytes, indicating epigenetic poising (Figure 2H-J, S3D-F). Collectively, our findings suggest persistent epigenetic alterations in post-COVID-19 monocytes, including epigenetic poising associated with hyper-responsiveness to stimulation.
Circulating HSPC reflect BM HSPC composition and phenotypes
Since circulating monocytes have short lifespan and are continuously replenished from HSPC, we hypothesized that the distinguishing characteristics of post-COVID-19 monocytes may stem from altered hematopoiesis or epigenetic phenotypes in HSPC that are conveyed through development.
To overcome the impracticalities of obtaining HSPC from BM to study hematopoiesis and epigenetic memory in human disease, we developed a platform to enrich and analyze circulating CD34+ HSPC (~0.05% of PBMC). We aimed to determine if these circulating HSPC accurately represent the heterogeneity and epigenetic/transcriptional phenotypes of their BM counterparts. To achieve this, we purified CD34+ HSPC from BM mononuclear cells (BMMC) and PBMC of the same donors and reintroduced them into the respective tissues’ total mononuclear cells at enriched frequencies. Using snRNA/ATAC-seq, we comprehensively profiled these cells (Figure 3A, Table S1). The co-clustering of CD34+ HSPC from both BMMC and PBMC was evident in RNA and ATAC-seq UMAP plots, indicating shared transcriptional and epigenomic programs (Figure 3B). Based on known marker genes, we annotated the HSPC as hematopoietic stem cells and multipotent progenitors (HSC/MPP), lymphoidprimed MPP (LMPP), megakaryocyte-erythroid progenitors (MEP), erythroid progenitors (Ery), granulocyte-monocyte progenitors (GMP), and basophil-eosinophil-mast cell progenitors (BEM)63 (Figure 3B-C). The expression of lineage-defining marker genes in both BM and peripheral HSPC subtypes confirmed that circulating HSPC encompass the molecular diversity and characteristics of BM HSPC (Figure 3B-C).
Figure 3. Epigenomic and transcriptomic analysis of rare circulating HSPC establishes their similarity to BM HSPC.

(A) Schema depicting donor-paired PBMC/BMMC analysis with Progenitor Input Enrichment (PBMC- and BMMC-PIE), followed by snRNA/ATAC-seq. Approximate percentage of HSPCs from pre-enrichment and enriched samples indicated.
(B) UMAP for snRNA/ATAC-seq data of paired BMMC- and PBMC-PIE (n=2). Plots annotated for major cell types, and tissue origins. HSPC-only UMAP plots were annotated for HSPC subtypes (bottom).
(C) Expression of HSPC subtype-specific genes in PBMC- (top) and BMMC-PIE (bottom) dataset.
(D) Summary of study participants profiled using PBMC-PIE workflow.
(E) UMAP of PBMC-PIE snRNA-seq data.
(F-G) UMAP of HSPC for RNA- and ATAC-seq data. HSPC subtypes are annotated using two methods: annotation based on cell type-specific gene expression (F), and annotation from ATAC-seq dataset for human BM HSPC subtype64 (G).
Given observed similarity between circulating and BM HSPC, we pursued study of circulating HSPC paired with the mature immune cell populations, using an experimental workflow we termed PBMC analysis with Progenitor Input Enrichment (PBMC-PIE). This approach is particularly suitable for single-cell profiling of peripheral HPSC in infectious and inflammatory diseases, as it utilizes readily available PBMC samples. We applied the PBMC-PIE workflow to samples from our post-COVID-19 cohort to study phenotypes and epigenetic memory in HSPC (Figure 3D, S4C).
Durable chromatin accessibility signatures in HSPC following severe COVID-19
Using snRNA/ATAC-seq, we analyzed PBMC-PIE samples from a subset of our cohort to investigate gene expression and chromatin accessibility. We examined 28,069 peripheral CD34+ HSPC after pre-processing and cell-type annotation. Projecting these RNA-based annotations onto the snATAC-seq HSPC UMAP also labeled distinct subclusters (Figure 3F, right), indicating co-segregating expression and epigenetic profiles among major HSPC subtypes.
Additionally, we annotated peripheral HSPC by incorporating bulk-ATAC-seq data from FACS-sorted BM HSPC subsets in our previous studies64; and scRNA-seq data from BMMC65. Both projections demonstrated substantial consensus, supporting the representative diversity of peripheral HSPC and their similarity to BMMC HSPC (Figure 3F-G, S4K-L). Notably, the HSC/MPP cluster was prominently represented among peripheral HSPC, with high expression of CD164 (an early hematopoietic stem cell marker) in the HSC/MPP subcluster identified through bulk-ATAC-seq-guided annotation (Figure S4L-M).
Regarding HSPC epigenetic programs, PCA analysis of bulk ATAC-seq DAR revealed group-specific clustering of individual samples (Figure 4A-B, Table S3). PC score distributions between groups were significant, with similarities observed between Early and nonCoV (PC1) and distinguishing features in Late (PC2) (Figure 4B, box plots). DAR analysis revealed three clusters including persistent (C1) and transient (C2) epigenetic programs in HSPC (Figure 4C, Table S3). DAR in C1 were annotated to genes enriched for GO terms related to differentiation, migration, activation, and cytokine-mediated signaling, suggesting establishment of long-lasting epigenetic memory in HSPC. DAR in C2 were associated with genes related to myeloid differentiation, activation, and cytokine production (Figure 4C-E, Table S3). Notably, while monocytes from both nonCoV and Early shared some epigenomic features (Figure 1), their HSPC exhibited more distinct profiles (Figure 4B-C, Table S3). The prominence of the transient program (C2) in Early, compared to nonCoV, may reflect the direct effects of increased systemic inflammation on HSPC in severe COVID-19, which could be relevant to the durable epigenetic effects observed in Late monocytes and HSPC, as well as functional differences in stimulated monocytes (Figure 2I). Persistent (C1) and transient (C2) HSPC DAR shared programs for cell migration, activation, and differentiation. while cytokine-mediated signaling was specific to C1, and the coagulation program was specific to C2 (Figure 4D, Table S3).
Figure 4. Sustained epigenetic alterations in CD34+ HSPC following severe COVID-19.

(A) Overview depicting clinical groups and ATAC-seq workflow used for epigenomic profiling of CD34+ HSPC.
(B) PCA plot of HSPC ATAC-seq samples using the combined set of DAR (left). Boxplots (right) show PC1 and PC2 values for each donor across groups.
(C) Top: Normalized read density of all DAR in each clusters by groups. Each DAR (group average) is represented by a linked line across groups (top). Bottom: Cluster average of normalized DAR density score for individuals within groups.
(D) GO analysis of genes associated to cluster-specific DAR in HSPC. (p<0.05)
(E) ATAC-seq genome tracks for representative cluster-specific DAR in HSPC. Boxplots display normalized DAR densities for each donor across groups.
Durably altered hematopoiesis following severe COVID-19
To determine whether epigenomic signatures in post-COVID-19 HSPC correspond to changes in transcriptional programs and altered progenitor subsets, we analyzed differences in RNA and chromatin accessibility (DORC) between Healthy and diseased groups in HSPC. DEG and active DORCs in all convalescent groups compared to Healthy were enriched for erythrocyte differentiation and antigen presentation programs, likely reflecting common responses to inflammation and stress (Figure 5A, S5B-C). In contrast, programs of myeloid differentiation and platelet activation were selectively enriched in the post-COVID-19 state. Differential DORC analysis revealed GO terms related to cellular activation and cytokine production, suggesting epigenetic poising of these programs across disease conditions.
Figure 5. Durably altered phenotypes and programs in HSPC following severe COVID-19.

(A) GO analysis of genes with upregulated expression, and chromatin accessibility (DORC) in HSPC of each clinical group compared to Healthy.
(B) Frequency of HSPC subcluster among total HSPC for individual across groups.
(C) UMAP of HSPC with subtype annotations.
(D) Gene set enrichment analysis (GSEA) between Early and Late HSPC.
(E-F) UMAP displaying GMP module score (E) and DORC scores for the neutrophil module (F) per cell (left). Distribution of the module scores per cell in each clinical group (right).
(G) Heatmap of chromVAR score (Z-score-normalized median) for subtype-defining TFs.
(H-I) chromVAR scores for FOS::JUN and CEBPA in HSPC. TF scores projected on HSPC UMAP (left). Average TF score per individual (middle) and per-cell score (right) across groups.
Dysregulation of hematopoiesis was evident in post-COVID-19 HSPCs, with DEG linked to myelopoiesis, including KLF2 and CEBPD (Figure 5A, S5B-D, Table S4). Notably, GMP frequencies among HSPC were significantly increased in the disease groups, particularly in the Late group, indicating that skewed myelopoiesis is a durable progenitor phenotype, independent of emergency hematopoiesis (Figure 5B, S5E). While gene set signatures in Early and Late were similar when compared to Healthy (enrichment of differentiation and immune response programs; Figure 5A), comparative analysis of gene expression between Early and Late HSPCs distinguished enrichment of differentiation, antigen presentation, and adhesion programs in Late HSPCs, while Early HSPCs exhibited enrichment of viral-response and stimulus-response programs (Figure 5D).
To study transcriptional regulation of granulopoiesis and myelopoiesis we defined a GMP module comprising GMP cluster-defining marker genes and assessed the distribution of this module score among individual cells from each group (Figure 5E, S5F). We also defined a module consisting of DORC accessibility at GMP and neutrophil differentiation associated genes (Figure 5F). Both expression and chromatin modules of GMP and neutrophil differentiation were significantly increased in nonCoV, Early, and Late, indicating a durable epigenetic program driving granulopoiesis and myelopoiesis following severe COVID-19 (Figure 5E-F).
We next explored relationships between durable epigenetic programs in post-COVID-19 HSPC and GMP programming by TFs. The activity of characteristic GMP TFs (AP-1 and CEBPA) was enriched in GMP and low but present in HSC/MPP (Figure 5G-I, Table S6). Notably, AP-1 (FOS/JUN) and CEBPA activity in HSPC was significantly higher in both Early and Late at the level of individuals and cells (Figure 5H-I, Table S6). Together, these findings revealed prominent alterations in HSPC epigenomes and transcriptomes following COVID-19 infection, including programs favoring myelopoiesis and antigen presentation.
Post-COVID-19 epigenetic signatures and TF programs are shared between HSPC and monocytes
In addition to skewing differentiation, durable post-COVID-19 epigenetic programs in HSPC may be inherited by mature progeny cells thereby conferring altered phenotypes. Chromatin accessibility signatures in HSPC (Figure 4) and monocytes (Figure 1) revealed shared epigenetic and transcriptional programs (e.g., Figure S2D, Figure 5A, Table S4-5). Differential TF motif accessibility analysis in Early and Late (compared to Healthy) revealed the prominent activity of inflammation-responsive TFs, including the enrichment of AP-1 (FOS/JUN) and IRF family motifs, in Early HSPC and monocytes (Figure 6A). Late HSPC maintained increased CEBP family and JUN activity, consistent with increased GMP frequencies post-COVID-19 (Figure 5B). While most AP-1 activity diminished in Late, IRF activity remained enriched in both HSPC and monocytes (Figure 6A, S6A-B, Table S6). For example, IRF2 showed activity in HSC/MPP and GMP subclusters among HSPC and in the Late-enriched M.SC3 monocyte subcluster (Figure 6B-C, UMAP plots). IRF2 activity was also increased in HSPC and monocytes from Early and Late (Figure 6B-C, violin plots). Late HSPC and monocytes exhibited increased activity of other TFs associated with monocyte differentiation and activation, including HOX factors, NRF1, and CTCF (Figure 6A, S6B-C, Table S6).
Figure 6. TF programs are durably altered following severe COVID-19 and are shared between HSPC and CD14 + monocytes.

(A) Differentially active TFs in HSPC (left) and CD14+ monocytes (right) between Healthy and Early/Late groups.
(B-C) chromVAR scores for IRF2 in HSPC (B) and CD14+ monocytes (C). Scores projected on myeloid UMAP (left). Per-cell scores across groups (right).
(D) Average expression of representative marker genes of M.SC3 in each individual.
(E-F) M.SC3 module score in myeloid cells (E) and HSPC (F). M.SC3 module score is projected onto UMAP (left). Per-cell M.SC3 module score across groups (right).
Moreover, several monocyte activation-related genes such as CD74 and S100As, showed coordinated upregulation in both HSPC and monocytes, most prominently and consistently in Late (Figure 6D). To investigate a shared epigenetic program from HSPC to monocytes, we generated module scores for the Late-enriched M.SC3 using marker genes and projected the scores onto the HSPC UMAP. This analysis revealed the expression of the M.SC3 module in HSPC, with enrichment in HSC/MPP and GMP subclusters (Figure 6F, left). Further, the distribution of M.SC3 module activity increased in post-COVID-19 HSPC, especially in Late (Figure 6F, right), corresponding to its sustained activity in monocytes up to one year after COVID-19.
IL-6R blockade attenuates post-COVID-19 epigenetic phenotypes
While all aforementioned post-COVID-19 molecular and cellular features showed between-group differences (e.g., Healthy vs. Late), we studied within-group variance in GMP frequencies within the Late group to determine association with any clinical characteristics. We focused on the increased GMP frequency in Late, considering it a foundational program for associated progeny monocyte phenotypes. Strikingly, IL-6R blockade treatment (Tocilizumab) during acute infection was significantly associated with most of the variance in Late GMP frequencies after multiple hypothesis test correction. Importantly, as Tocilizumab administration was near-random during the Spring/Summer 2020 COVID-19 peak in New York City, there were no discernable clinical or demographic features influencing the analyses (Table S1). The untreated Late group (Late: no aIL-6R) had significantly higher GMP frequencies compared to both Healthy and Late that received IL-6R blockade treatment (Late: aIL-6R; Figure 7A, S7A). Clinical features such as comorbidities and variable recovery may link to GMP phenotypes, however, the size and clinical complexity of our post-COVID-19 cohort limited our power to detect such correlations (Figure S7A). In contrast, IL-6R blockade resulted in a clear and significant cellular readout persisting for months to one year. We next evaluated TF activities (inferred by motif accessibility) characteristic of IL-6R signaling and myelopoiesis between the Healthy and Late groups with and without IL-6R blockade. Comparing the untreated group to the Healthy and treated groups, we found increased chromatin accessibility at motifs for STAT3, an IL-6R signal-activated TF (Figure 7B). Anti-IL-6R treatment also reduced accessibility of motifs for IRF and CEBP TF families, which were persistently elevated in the untreated group (Figure 7B). CEBPA and CEBPB program steady state or emergency myelopoiesis, respectively, while CEBPE regulates granule formation in developing granulocytes66,67. Increased chromatin accessibility at STAT3 and other motifs may either represent sustained TF chromatin binding or maintained chromatin accessibility independent of TF binding. Interestingly, the crucial erythropoiesis program TF, GATA1, showed an opposite effect, implying that long-term consequences of IL-6R signaling direct progenitor cell fate towards myelopoiesis over erythropoiesis (Figure 7B). Altered epigenomic and TF programs were complemented by transcriptional changes in Late, with IL-6R blockade decreasing expression of post-COVID-19 programs associated with antigen presentation and inflammation, while increasing expression of genes associated with an HSC/hematopoiesis maintenance program (e.g., MEIS1, CD34; Figure 7C, S7B). Together, these results suggest that IL-6 signaling during acute severe COVID-19 durably programs increased GMP frequencies and myelopoiesis via epigenetic mechanisms involving IL-6R signal-activated TFs. IL-6R signaling blockade during acute COVID-19 appears to mitigate these long-term programs.
Figure 7. IL-6R signaling programs post-infection phenotypes.

(A) GMP frequency in HSPC for each individual across groups. (Wilcoxon’s test, * p < 0.05; Healthy-reference)
(B) Average chromVAR score (row-normalized) for selected TFs across groups.
(C) DEG in Late HSC/MPP between aIL-6R treated and non-treated groups.
(Wilcoxon’s test, * adj.p < 0.05, “Late with aIL-6R”-reference)
(D) Experiment schema of A/J mice MHV-1 infection model.
(E) UMAP of mouse BM lineage-depleted progenitor populations, with celltype annotation and simplified trajectories.
(F) Frequency of progenitor subtypes among lineage-depleted cells across groups.
(G) Average chromVAR score (row-normalized) for selected TFs across groups.
(H) Per-cell GMP module score in the HSC/MPP cluster across groups. (Wilcoxon’s test, * p < 0.05; Recovered-reference)
(I) UMAP of BALF macrophage and color-scaled density of each group.
(J) Cell count ratio between two macrophage subclusters (Mac2/Mac1) in each group.
(K) Absolute cell density (cells/mm2) of selected cell types in the region of interest (ROI) across sample groups in imaging mass cytometry data70 of post-mortem lung tissue. Density per ROI is represented by small transparent dots, while larger dots indicate the average density per study participant. (ROI value, Wilcoxon’s test, * p < 0.05; Recovered-reference)
(L) Monocyte counts in brain tissue across different groups. (ANOVA, * p < 0.05; naïve, n = 5; recovered, n = 9; aIL-6R, n=6)
(M) Representative images from immunofluorescence staining of mouse brain in different groups showing varying degrees of demyelination and a box plot displaying the average intensity of myelin basic protein (MBP) staining per ROI (transparent dot) and per mouse across groups (larger dot). (Wilcoxon’s test, * p < 0.05, Healthy-reference)
IL-6R signaling programs post-infection myelopoiesis in mice
We employed IL-6R blockade during acute coronavirus infection and recovery in mice to investigate the long-term effects of IL-6 on post-infection myelopoiesis. We employed an established mouse model of SARS-CoV-2 convalescence by infecting susceptible A/J mice with murine hepatitis virus 1 (MHV-1). This model was selected because it mimics many aspects of severe COVID-19 and convalescence observed in human68.
In the MHV-1 infection model, mice received MHV-1 with or without anti-IL-6R antibody injection on day 0 (Figure 7D). Convalescent mice cleared virus within two weeks and regained weight by 30 days (Figure S7C). To study post-infection epigenomic and transcriptomic phenotypes, and assess the effects of IL-6R blockade, we performed snRNA/ATAC-seq profiling on lineage-depleted BMMC, comparing naïve mice to recovered mice (with and without anti-IL-6R) at 30 days post-infection.
Analyzing HSPC subclusters and hematopoietic trajectories (Figure 7E, S7D), the frequency of HSC/MPP remained consistent across all conditions, suggesting resolution of acute inflammation and emergency hematopoiesis. In contrast, the frequencies of GMP and monocyte lineage clusters were increased in the recovered groups. Notably, these frequencies were reduced in the anti-IL-6R treated group compared to the recovered mice without treatment (Figure 7F). Interestingly, the frequency of the erythroid lineage cluster exhibited an opposite pattern, decreasing upon infection and normalizing with anti-IL-6R blockade (Figure 7F).
Examining TF scores related to myelopoiesis and erythropoiesis, we observed increased accessibility for STAT3, IRF1, and IRF8 (activated in response to IL-6R signaling) in post-infection HSC/MPP, and these activities were reduced in the IL-6R blockade group (Figure 7G). Post-infection, there was also increased activity of CEBPB and CEBPE, though to a lesser extent in the anti-IL-6R treated group (Figure 7G). Consistent with the establishment of differentiation bias in early progenitors and our human data, accessibility of the erythroid TF GATA1 decreased in post-infection HSC/MPP and was normalized by anti-IL-6R treatment (Figure 7B, 7G). These findings reveal that IL-6R blockade in mice reflect many changes in TF activity and associated modulation of hematopoiesis detected in convalescent patients treated with IL-6R blockade.
To investigate the transcription program of early differentiation bias, we examined the GMP module expression score in HSC/MPP across conditions. The GMP module score was significantly higher in recovered mice compared to both naïve and anti-IL-6R treated groups (Figure 7H), consistent with the GMP frequency distributions observed in our human cohort (Figure 7A). This suggests that MHV-1 infection causes post-infection alterations in hematopoiesis by epigenetically and transcriptionally reprogramming the HSC/MPP population, partially via IL-6 signaling. Overall, our mouse model of coronavirus convalescence recapitulated several key results from our human studies, including increased frequencies of GMP and monocyte progenitors post-infection, enhanced STAT3, IRF, and CEBP activities, and the mitigation of these phenotypes by IL-6R blockade (Figure 7A-C).
Correlation between hematopoietic and tissue phenotypes in coronavirus convalescence
Increased myelopoiesis and epigenetic poising of inflammatory genes in HSPC in the post-infection period may influence tissue recovery and inflammation. To characterize post-infection tissue phenotypes in recovered MHV-1 infected mice, we analyzed bronchoalveolar lavage fluid (BALF) cells using snRNA/ATAC-seq, assessed lung histopathology, and conducted flow cytometric and imaging analysis of the brain. While we focused on BALF macrophage phenotypes and heterogeneity given the established role of monocyte-derived macrophages in respiratory viral infections and their recent derivation from HSPC, we also observed significant increases in T and B cells in total BALF cells post-infection. Using both transcriptome and TF motif accessibility, we identified macrophage subclusters, Mac1 and Mac2 (Figure 7I). Mac1 featured TF activity, including BHLHE41 and PPARγ69, along with gene expression associated with tissue resident phenotypes (Figure S7G-H). Conversely, Mac2 expressed genes related with macrophage activation, differentiation, and migration, and exhibited TF activity from the IRF and AP-1 families (Figure S7G-H). Mac1 (resident macrophages) predominated in the naïve group, while Mac2 (differentiated and monocyte-derived macrophages) predominated in the post-infection groups (Figure 7I-J).
We evaluated the effect of IL-6R blockade on lung macrophages. The proportion of activated/recruited cells increased strikingly in both post-infection groups, though to a lesser extent in the IL-6R blockade condition, suggesting reduced monocyte recruitment and differentiation (Figure 7J). Similar to mouse HSC/MPP data, lung macrophages exhibited increased STAT3 motif accessibility post-infection, which was normalized by IL-6R blockade (Figure S7I). However, unlike HSC/MPP, IRF TF accessibility in lung macrophages increased post-infection and remained unperturbed by IL-6R blockade (Figure S7I).
We defined DEG in macrophages between post-infection groups. Macrophages treated with anti-IL-6R blockade had decreased expression of genes associated with response to stimulus and cell migration (Figure S7J-K). Histopathology analysis of lung tissue aligned with the inflammatory phenotypes observed in the single-cell analysis, revealing alveolar wall widening, extensive inflammatory cell infiltrates, and lymphoid aggregates with macrophages. Notably, mice that received IL-6R blockade showed milder pathology (Figure S7L).
Our findings in the MHV-1 mouse model of post-coronavirus lung pathology are consistent with a recent study conducted on post-mortem lung tissue from post-acute COVID-19 patients. The study revealed persistent alveolar type 2 dysfunction and structural derangement70. Imaging mass spectrometry analysis from this study unveiled increased accumulation of monocytes, neutrophils, and macrophages in the lungs of post-acute COVID-19 patients, compared to healthy controls (Figure 7K, S7M). Although the non-hematopoietic cell composition remained largely unchanged, hematopoietic cells, particularly myeloid cells such as monocytes, neutrophils, and macrophages demonstrated significant alterations (Figure S7M). These findings align with our observations of elevated monocyte-derived macrophages and lung inflammation in mice following recovery from MHV-1 infection (Figure 7I-J).
In contrast to the lung, the brain features delayed (detectable only on day 4) and low-level infection (4–5 logs lower viral titers than lung), that quickly resolves (cleared by day 7)71. We observed notable myeloid infiltration and activation in the brain, consistent with linked effects of hematopoietic and circulating monocyte phenotypes. Flow cytometric analysis revealed increased monocyte numbers in the brain 30 days post-infection, a response reduced significantly by IL-6R blockade (Figure 7L).
Together with myeloid infiltration in MHV-1 recovered mice, we observed a significant reduction in myelin basic protein (MBP) staining, indicating extensive demyelination, and this was reduced by IL-6R blockade (Figure 7M). Additionally, microglia and astrocytes (identified by IBA-1 and GFAP, respectively) increased in recovered mice, and were partially normalized by IL-6R blockade (Figure S7N). These observations collectively suggest a persistent monocyte recruitment to the lungs and brain post-infection, likely driven by enhanced myelopoiesis and tissue migration programmed, at least in part, by IL-6R signaling during acute infection.
Discussion
Persistence of HSPC alterations and molecular phenotypes post-COVID-19
HSPC are long-lived self-renewing precursors to diverse immune cells72 and have unique potential to store inflammation-induced epigenetic memory, impacting hematopoiesis and progeny innate immune cell phenotypes. These programs have been observed in infections and vaccine responses in mice42,73, and in controlled human vaccine studies44. We considered that acute viral infections, particularly SARS-CoV-2 causing severe COVID-19 and systemic inflammation, could trigger such a response. Previous reports noted alterations in HSPC during and immediately following acute COVID-1974,75, consistent with emergency hematopoiesis during infection76–79.
We uncovered linked and persistent epigenetic and transcriptional reprogramming of HSPC and monocytes in convalescent COVID-19 patients up to one-year post-infection. Severe COVID-19 durably increased GMP frequency and expression of GMP signatures in HSC/MPP. Epigenetic poising of inflammatory genes, shared between HSPC and short-lived monocyte progeny correlated with monocyte hyper-responsiveness to stimulation. IL-6R signaling during acute infection contributed to durable epigenetic phenotypes in HSPC and monocytes. These results align with earlier studies of altered myelopoiesis and innate immune memory that derive from HSPC in non-COVID-19-related inflammatory contexts. Our study demonstrates lasting HSPC alterations and epigenetic phenotypes following COVID-19. Enrichment and in-depth single-cell analysis of circulating HSPC from peripheral blood enabled insights into post-infection hematopoiesis and HSPC phenotypes providing a valuable approach for future studies of HSPC in diverse human diseases and conditions.
Implications for post-infection pathology and recovery
Our study indicates persisting post-infection transcriptional and epigenetic programs in monocytes (and their progenitors) associated with activation, differentiation, migration, and antigen presentation. These programs may contribute to ongoing pathology in tissues with inflammation and activated vasculature. Our in vitro activation experiments suggest that these epigenetic changes can augment inflammatory responses. In MHV-1 infected mice, the prolonged monocyte infiltration in the brain and lung is consistent with recent descriptions of PASC, including neurological symptoms and increased inflammatory microglia68. Incomplete recovery after infections or critical illness, including post-ICU syndrome (PICS), lacks clear etiology. These data suggest that hematopoietic reprogramming, including skewed myelopoiesis and epigenetic poising of inflammatory genes, may contribute to persistent myeloid-driven tissue pathology post-infection.
Prior immune stimulation with BCG and b-glucan boosts myelopoiesis and responsiveness of innate immune cells, providing innate immune-driven protection against heterologous infection42,45. Additionally, monocytes develop a long-term “anti-viral resilience” program following adjuvanted influenza vaccine (H5N1+AS03), enhancing responses to Zika and Dengue infection80. Further work is needed to understand the mechanisms of long-term changes in hematopoiesis and immune tone following acute infection, and their association with phenotypes ranging from PASC to protection against heterologous infection.
IL-6 regulates post-infection phenotypes
Our data uncovered activation of the IL-6R signaling pathway as a potential mediator of durably altered hematopoiesis and innate immune memory. IL-6, a pleiotropic cytokine, activates immune cells, promotes cytokine secretion and cell recruitment, and the maintenance and differentiation of HSPC81. Additionally, IL-6 plays beneficial functions in epithelial repair, as a myokine and metabolic regulator, and affects appetite and visceral adipose regulation81–84. Furthermore, maternal IL-6 induces epigenetic memory in neonatal intestinal epithelial stem cells, altering the response of neonates to gastrointestinal infection. This intestinal stem cell IL-6 memory signature shared features with the HSPC IL-6 memory program we describe, including genes associated with antigen presentation (CD74 and MHCII)85. Therefore, IL-6 may program diverse adult stem cell populations for augmented response to future inflammatory or infectious challenge.
In severe cases of COVID-19, elevated IL-6 levels correlate with tissue damage, immune cell recruitment/activation, and acute respiratory distress syndrome (ARDS)86. IL-6R blocking antibodies in critically ill patients with COVID-19 modestly improve outcomes87–89. The lasting impact of high-level IL-6R signaling during acute inflammation on cellular phenotypes is unknown. Surprisingly, IL-6R blockade during acute infection strongly influenced GMP levels up to one-year post-infection; untreated individuals featured higher GMP frequencies, while IL-6R blockade-treated individuals featured lower GMP levels. We validated these findings by modeling IL-6R blockade in a severe mouse coronavirus infection, recapitulating similar results from human. One durable molecular signature of IL-6R signaling during acute infection was sustained chromatin accessibility at STAT3 motifs in both HSPC and monocytes/macrophages.
Notably, a previous study reported that STAT3, in conjunction with AP-1 TFs, can establish and maintain epigenetic memory of inflammation in epidermal stem cells37, highlighting the possibility of an analogous mechanism in post-COVID-19 HSPC. We found that STAT3 motif accessibility corresponds with increased accessibility of motifs for CEBPβ (also named nuclear factor IL-690), a prominent myelopoiesis TF. In addition, both STAT3 and CEBPβ motif accessibility in convalescence was reduced by IL-6R blockade during acute infection.
We also identified sustained chromatin accessibility associated with other TF programs in HSPC and monocytes, including IRFs. Notably, sustained IRF motif accessibility was also described in monocytes following adjuvanted influenza vaccination (H5N1+AS03)80.
Circulating HSPC Analysis: An approach to studying HSPC in a post-COVID-19 cohort
We performed deep characterization of HSPC in a human cohort by enriching rare circulating CD34+ HSPC from PBMC and pairing this with snRNA/ATAC-seq analysis. Paired blood and BMMC samples from the same donors confirmed that rare circulating CD34+ HSPC accurately capture the diversity and major transcriptomic and epigenomic signatures of BM HSPC subsets. Our approach provides a valuable resource for characterizing the effects of diverse challenges and diseases on human HSPC without invasive BM biopsy collection. It enables the evaluation of HSPC and their mature immune cell progeny from PBMC samples facilitating the identification of developmentally linked epigenomic and transcriptomic programs. This has implications for understanding dynamic blood cell development and innate immune memory in inflammatory disease, vaccine responses, vaccine design, and non-genetic variance in immune responses to infection.
While our study focused on blood cells, other cell types also harbor inflammatory epigenetic memory. It is likely that the HSPC and blood cell phenotypes described here interact extensively with tissue resident cells, which may also change in frequency, differentiation programs, and activity within tissue communication circuits38. These interactions may have enduring influence on tissue defense or pathology.
Limitations of the study
We aimed to identify conserved cellular and molecular features post-COVID-19, but lacked sufficient power to directly associate these features with clinical outcomes, including PASC. Future studies should investigate the contribution of altered hematopoiesis, myeloid cell differentiation, and epigenetic priming of inflammatory genes in tissue recovery and pathology in the context of SARS-CoV-2 and PASC. Understanding if mild COVID-19 induces similar programs is an important topic for future work. The role of durable IL-6R-dependent hematopoietic phenotypes in persistent tissue infiltration and their interaction with non-hematopoietic IL-6 targets within tissues remains unknown. The influence of mature granulocytes on post-infection changes and tissue infiltration and pathology, which were minimally studied due to granulocyte depletion in sample preparations, requires dedicated investigation. While we directly assayed BM HSPC in mouse, our human progenitor studies relied on the enrichment of rare circulating HSPC. Further, our datasets indicate the potential for similar progenitor-progeny programs in diverse lineages (e.g., lymphoid lineages) opening avenues for further exploration of these concepts. Understanding the complex contribution of IL-6R signaling to tissue repair and how therapeutic targeting of IL-6 may alter post-infection pathology warrant thorough investigation, especially considering the pleiotropic and potential salutary effects of IL-691.
STAR★Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for the resources should be directed to and will be fulfilled by the lead contact, Steven Josefowicz (szj2001@med.cornell.edu)
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All raw and processed data are deposited on GEO database under accession number GSE196990. All other data types used for analysis in this research are present in the manuscript or the Supplementary Materials. We created a web-based interface to query all multiome data of HSPC, myeloid, and whole PBMC for DORC, expression, and transcription factor activities (https://buenrostrolab.shinyapps.io/covid_myeloid/). (https://buenrostrolab.shinyapps.io/covid_pbmc/, https://buenrostrolab.shinyapps.io/covid_hspc/, https://buenrostrolab.shinyapps.io/covid_myeloid/) The code used for the downstream analysis is available in github repository (https://github.com/sharmasiddhartha231/Final_Covid19_Scripts, 10.5281/zenodo.8097411). The codes for visualizations are available upon request to the lead contact. De-identified clinical cohort information used in this study is available in supplementary table. Any publicly available data that we reanalyzed are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. Additional Supplemental Items are available from Mendeley Data at http://dx.doi.org/10.17632/pfwyrmffdp.1.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| 7-AAD | Biolegend | Cat#420404 | |
| CD34-microbeads | Miltenyi | Cat#130-046-702 | |
| CD34-FITC | Miltenyi | Cat#130-113-178 | |
| CD49f-Pacific Blue | Biolegend | Cat#313620 | |
| CD90-PE | Biolegend | Cat#328110 | |
| CD38-PE/cy7 | Biolegend | Cat#303516 | |
| CD45RA-APC/cy7 | Biolegend | Cat#304128 | |
| CD20-Biotin | Biolegend | Cat#302350 | |
| CD56-Biotin | Biolegend | Cat#362536 | |
| CD14-Biotin | Biolegend | Cat#301826 | |
| Streptavidin-BV605 | BD | Cat#563260 | |
| CD14-APC | BD | Cat#340436 | |
| CD8-FITC | Biolegend | Cat#300906 | |
| CD14-microbeads | Miltenyi | Cat#130-050-201 | |
| Mouse lineage depletion kit | Miltenyi | Cat#130-090-858 | |
| FcX | Biolegend | Cat#156604 | |
| Ly6g-BUV395 | BD | Cat#563978 | |
| MHCII-BV421 | ThermoFisher | Cat#404-5321-82 | |
| CD11c-A488 | ThermoFisher | Cat#53-0114-82 | |
| CD86-PE | Biolegend | Cat#105105 | |
| CD206-BV605 | Biolegend | Cat#141721 | |
| CD4 PE-CF594 | BD | Cat#562285 | |
| CD19-APC | Thermofisher | Cat#17-0193-82 | |
| B220-APC | ThermoFisher | Cat#17-0452-82 | |
| CD45-A700 | Biolegend | Cat#103127 | |
| Ly6c-PerCP-Cy5.5 | Thermofisher | Cat#45-5932-82 | |
| CX3CR1-BV786 | Biolegend | Cat#149029 | |
| CD8-PE-Cy7 | Thermofisher | Cat#25-0081-82 | |
| CD11b-APC-e780 | ThermoFisher | Cat#47-0112-82 | |
| Aqua Live/Dead Viability dye | ThermoFisher | Cat#L34966 | |
| normal donkey serum | Jackson Immunoresearch | RRID: AB_2337258 | |
| Iba1 | Fujifilm | Cat#019-19741 | |
| GFAP | Cell Signaling Technologies | Cat#3657 | |
| MBP | Abcam | Cat#ab40390 | |
| anti-IgG secondary antibodies conjugated to A488 | Jackson Immunoresearch | RRID: AB_2338449 | |
| anti-IgG secondary antibodies conjugated to A594 | Jackson Immunoresearch | RRID: AB_2338873 | |
| anti-IgG secondary antibodies conjugated to A647 | Jackson Immunoresearch | RRID: AB_2338904 | |
| DAPI | Prolong Diamond Antifade | Cat#P36962 | |
| Bacterial and virus strains | |||
| MHV-1 | ATCC | VR-261 | |
| Biological samples | |||
| Paired PBMC and BMMC | AllCells | N/A | |
| Whole blood from study participants | Weill-Cornell Medicine/New York-Presbyterian Hospital | N/A | |
| Chemicals, peptides, and recombinant proteins | |||
| R848 | InvivoGen | Cat#tlrl-r848 | |
| mouse anti-IL-6R blocker | InVivoMab | Cat#BE0047 | |
| IFNα | PBL assay science | Cat#12100 | |
| RPMI medium | Corning | Cat#10-040-CM | |
| Ficoll-Paque PLUS | GE | Cat#17144002 | |
| Collagenase D | Roche | Cat#11088858001 | |
| Critical commercial assays | |||
| Chromium Controller & Next GEM Accessory Kit | 10x Genomics | Cat#1000202 | |
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle | 10x Genomics | Cat#1000285 | |
| NEBNext low input RNA library prep kit for Illumina | NEB | Cat#E6420S | |
| LEGENDplex Human anti-virus response panel (13-plex) | Biolegend | Cat#740390 | |
| 15-plex human pro-inflammatory cytokine assay | Eve Technologies | N/A | |
| TOP-Plus (Pylon 3D analyzer) for antibody level measurement | ET Healthcare | N/A | |
| Lung histology | HistoWiz | N/A | |
| Deposited data | |||
| Imaging mass cytometry | Rendeiro et al.70 | https://doi.org/10.5281/zenodo.4110560 | |
| GRCh38 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.26/ | |
| GRCm38 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/ | |
| Single-cell RNA-seq of human bone marrow | Granja et al.95 | GSE139369 | |
| ATAC-seq data of human HSPC subpopulation | Buenrostro et al.60 | GSE96772 | |
| Experimental models: Organisms/strains | |||
| A/J mouse | The Jackson Laboratory | Cat#000646 | |
| Software and algorithms | |||
| Cell Ranger ARC 1.0.0 | 10X Genomics | https://support.10xgenomics.com/single-cell-multiome-atac-gex/software/downloads/latest | |
| Signac | Stuart et al.92 | https://stuartlab.org/signac/ | |
| Amulet | Thibodeau et al.93 | https://github.com/UcarLab/AMULET | |
| Seurat | Hao et al.94 | https://github.com/satijalab/seurat | |
| Scrublet | Wolock et al.95 | https://github.com/swolock/scrublet | |
| Harmony | Korsunsky et al.96 | https://github.com/immunogenomics/harmony | |
| MACS2 2.1.2 | Zhang et al.97 | https://github.com/macs3-project/MACS/wiki/Install-macs2 | |
| ArchR | Granja et al.98 | https://github.com/GreenleafLab/ArchR | |
| FigR | Kartha et al.99 | https://github.com/buenrostrolab/FigR | |
| Nebulosa | Alquicira-Hernandez et al.100 | https://github.com/powellgenomicslab/Nebulosa | |
| trimmomatic | Bolger et al.101 | https://github.com/timflutre/trimmomatic | |
| BWA | Li et al.102 | https://github.com/lh3/bwa | |
| Samtools | Li et al.103 | https://github.com/samtools | |
| Subread package | Liao et al.104 | https://subread.sourceforge.net/ | |
| IGV | James et al.105 | https://software.broadinstitute.org/software/igv/download | |
| snATACClusteringTools | Ucar Lab | https://github.com/UcarLab/snATACClusteringTools | |
| DiffBind | Stark et al.106 | https://hbctraining.github.io/Intro-to-ChIPseq/lessons/08_diffbind_differential_peaks.html | |
| ChipSeeker | Yu et al.107 | https://github.com/YuLab-SMU/ChIPseeker | |
| EdgeR | Robinson et al.108,109 | https://github.com/StoreyLab/edge | |
| cinaR | Karakaslar et al.110 | https://github.com/eonurk/cinaR | |
| HOMER 4.11 | Heinz et al.111 | http://homer.ucsd.edu/homer/ | |
| HINT | Gusmao et al.112 | https://github.com/CostaLab/reg-gen | |
| SparK | Kurtenbach et al.113 | https://github.com/harbourlab/SparK | |
| ClusterProfiler | Yu et al.114 | https://github.com/YuLab-SMU/clusterProfiler | |
| STAR 2.7.10b | Dobin et al.115 | https://github.com/alexdobin/STAR | |
| DESeq2 | Love et al.116 | https://git.bioconductor.org/packages/DESeq2 | |
| Snakemake | Mölder et al.117 | https://snakemake.readthedocs.io/en/stable/ | |
| Other | |||
| EDTA tubes | BD | Cat#366643 | |
| Heparin tubes | BD | Cat#368480 | |
| NovaSeq6000 | Illumina | N/A | |
| Zeiss LSM 900 | Carl Zeiss Microscopy | N/A | |
| Objective Plan-Apochromat 20x/0.8 M27 | Carl Zeiss Microscopy | N/A | |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Human Study Cohort
A total of 168 study participants were enrolled at Weill-Cornell Medicine/New York-Presbyterian Hospital between March 2020 and March 2021. Participants were recruited from the inpatient division of New York-Presbyterian Hospital and the Weill-Cornell Medicine pulmonary and post-ICU clinics. No statistical methods were used to predetermine sample size. COVID-19 severity scoring was based on the COVID-19 World Health Organization (WHO) Severity Classification118 Study participants were binned into the following groups: i) healthy volunteer donors, ii) recovered severe COVID-19 patients (WHO score 6–7), iii) recovered non-COVID-19 critically ill patients. The recovered severe COVID-19 group was partitioned into an early convalescent group (2–4 months following admission, “Early”) and a late convalescent group (4–12 months following admission, “Late”); this Early and Late delineation of the severe convalescent group was determined based on clinical management and features, namely that patients were monitored in ICU “step down” units until discharged from 2–4 months and then seen as out-patients in post-ICU clinic from 4–12 months. Inclusion criteria for each group were as follows; i) healthy volunteer donors: absence of clinical COVID-19 symptoms at any time prior to blood collection (prior negative SARS-CoV2 PCR and/or seronegative status also considered when available), ii) severe COVID-19 patients: PCR-proven SARS-CoV2 infection with the presence of clinical COVID-19 symptoms requiring admission to ICU-level care and the use of mechanical ventilation, iii) recovered non-COVID-19 critically ill patients: absence of SARS-CoV2 infection as measured by PCR and negative serology on admission and/or throughout hospital admission and non-COVID-19 related critical illness requiring admission to the medical, neurological, or cardiology intensive care unit. Prior infection status in healthy volunteer donors was established by SARS-CoV2 serological testing after donation. There were no specific exclusion criteria other than an inability to provide informed consent or SARS-CoV2 positive serology in asymptomatic healthy volunteers (asymptomatic infection) and non-COVID-19 critically ill participants. Blood was collected in EDTA or sodium heparin-coated vacutainers and kept on gentle agitation until processing, and all blood was processed on the day of collection. Age, sex, and comorbidity data were obtained through EPIC EHR records or when unavailable through a standardized form at the time of donation, and are indicated in Table S1.
The Institutional Review Board at Weill Cornell Medicine granted approval for gathering samples utilized in this research (Protocol #: 21–05023511). During the enrollment phase of the initial studies, all participants gave informed consent, allowing their samples to be used for subsequent research, including the present study.
Human Paired BMMC and PBMC Acquisition
BMMC and PBMC were freshly isolated from the same two adult donors recruited by AllCells (Alameda, CA). The donors gave written consent in accordance with protocols approved by their governing IRB. The isolated BMMC and PBMC were cryopreserved as PBMC from our cohort.
PBMC and Plasma isolation
Whole blood from EDTA or Heparin tubes (BD 366643 and BD 368480, respectively) was spun at 500g for 10 minutes at room temperature with no brake. The undiluted plasma was aliquoted to 1.5 ml microcentrifuge tubes and stored at –80° C for subsequent analysis.
After removal of plasma, the blood was mixed at a 1:1 ratio with room temperature RPMI medium (Corning 10–040-CM), layered over Ficoll-Paque PLUS (GE 17144002), and spun at 700g for 30 minutes at room temperature with minimum acceleration and no brake. The PBMC layer was isolated and washed with RPMI. Cells were then treated with ACK lysis buffer for 3 minutes and counted on a Countess 2 automated cell counter (Thermo Fisher AMQAX1000). Cells were centrifuged again and resuspended in freezing medium (90% FBS + 10% DMSO) and stored in cryogenic vials in a freezing container (Thermo Fisher 5100–0001) at –80° C.
Mice
Female A/J mice were purchased from Jackson Laboratories and were 6 weeks old. Mice were kept under SPF conditions throughout the entire experiment. All experiments were approved by the Sloan Kettering Institute (SKI) Institutional Animal Care and Use Committee. The mouse strain were maintained in the SKI animal facility in accordance with institutional guidelines.
Virus
Parent stock of murine hepatitis virus 1 (MHV-1) was originally obtained from the American Type Culture Collection (ATCC, Manassas, Virginia). It was passaged and maintained as described in our previous study120. Briefly, to propagate virus, murine SR-CDF1-DBT (DBT) cells were infected with the parent stock using a multiplicity of infection of 0.1, virus was allowed to adsorb onto the cells (30 minutes) before media supplementation and culture at 37°C for 24 hours. The cell suspension was harvested, centrifuged, and the resulting supernatant containing the virus particles aliquoted for further use, with virus titers determined by a standard plaque assay. MHV-1 and MHV-1-infected mice were handled under BSL-2 conditions throughout the entire experiment.
METHOD DETAILS
CD34+ and CD14+ Cell Isolation
Frozen PBMC and BMMC were thawed in a 37° C water bath, washed with RPMI, and centrifuged. An aliquot of PBMCs was stained with 7-AAD (Biolegend 420404, 1:20) alone. The rest of the cells were incubated with CD34 microbeads (Miltenyi 130–046-702) and isolated by placing them on a magnetic column (Miltenyi 130–042-201) as per the manufacturer’s specifications. The positive fraction obtained from the magnetic column was stained with the following antibodies – CD34-FITC (Miltenyi 130–113-178, 1:100), CD49f-Pacific Blue (Biolegend 313620, 1:200), CD90-PE (Biolegend 328110, 1:100), CD38-PE/cy7 (Biolegend 303516, 1:100), CD45RA-APC/cy7 (Biolegend 304128, 1:400), Lineage markers (CD20-Biotin {Biolegend 302350, 1:100), CD16-Biotin {Biolegend 302004, 1:100}, CD3-Biotin {Biolegend 344820, 1:100}, CD56-Biotin {Biolegend 362536, 1:100}, and CD14-Biotin {Biolegend 301826, 1:100}), and 7-AAD (Biolegend 420404, 1:20). After incubating in the dark for 30 minutes, cells were washed with PBS and incubated with Streptavidin-BV605 (BD 563260, 1:500) for an additional 30 minutes. CD34+ cells from the positive fraction and viable PBMC from the PBMC aliquot were then sorted on a BD FACSAria cell sorter and mixed at 1:5–1:20 ratios.
The negative fraction from the magnetic column was stained with the following antibodies – CD14-APC (BD 340436, 1:1000), CD8-FITC (Biolegend 300906, 1:400), and 7-AAD (Biolegend 420404, 1:20). After incubating in the dark for 30 minutes, cells were washed with PBS, and CD14+ cells were sorted on a BD FACSAria cell sorter.
Bulk ATAC-seq CD34+ HSPC and CD14+ Monocytes
To perform ATAC-seq, we followed the Omni-ATAC-seq protocol119. We used 50,000 cells for CD14+ monocytes, and 3000~5000 for CD34+ HSPC. HSPC were sorted directly into the PCR tubes prior to following Omni-ATAC-seq protocol.
A/J Mouse MHV-1 Infection
A/J mice were anesthetized with isoflurane and intranasally infected with 5,000 PFU of MHV-1 in a volume of 20ul. Together with the virus, 100 ul of mouse anti-IL-6R blocking antibody (2 ug/ul, InVivoMab) was injected into the anti-IL-6R group. Weight was measured continuously throughout 30 days post-infection and blood was collected on day 4 and day 30 post infection to determine viral titers to see whether the mice were recovered or not. At day 30, we collected bone marrow from tibia and femurs, BALF from lung, and brain tissue. We depleted lineage-positive cells from the bone marrow using microbead-based lineage depletion kit (Miltenyi 130–090-858). For single-nuclei Multiome, we used freshly isolated BALF cells and lineage-depleted bone marrow cells. H&E staining of the lung specimens for histology was performed by HistoWiz.
Single-cell Library Preparation
Nuclei were isolated from a mix of CD34+ cells and PBMC (or BMMC, or mouse progenitor cells or mouse BALF cells) according to ‘Low Cell Input Nuclei Isolation’ protocol (10x Genomics CG000365-Rev B) and were processed using Chromium Controller & Next GEM Accessory Kit (10x Genomics 1000202) and Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle (10x Genomics 1000285) following the manufacturer’s User Guide (10x Genomics CG000338-Rev D). Targeted nuclei recovery ranged from 5,000 to 10,000. The single-cell RNA and ATAC sequencing libraries were prepared using Dual Index Kit TT Set A (10x Genomics 1000215) and Single Index Kit N Set A (10x Genomics 1000212) respectively and sequenced on Illumina NovaSeq6000 or NextSeq platform.
Monocyte Stimulation Assays
CD14 monocytes were isolated from frozen PBMCs using MACS bead separation (Miltenyi Biotec, 130–050-201), and 50,000 cells were resuspended in complete RPMI media before being seeded onto 96-well plates in 100 μl of media. The cells were then stimulated with IFNα (50ng/ml, PBL assay science) and R848 (1 μM, InvivoGen) for 6 and 24 hours. At 6 hours, RNA was extracted to generate RNA-seq libraries using NEBNext low input RNA library prep kit for Illumina (NEB E6420S). After 24 hours of stimulation, supernatants were collected, and Legendplex flow-based cytokine assays were performed without further dilution.
Plasma Cytokine Analysis
Plasma was shipped to Eve Technologies for their 15-plex human pro-inflammatory cytokine assay (Eve Technologies, Calgary, AB, Canada). All samples were analyzed in duplicate.
Antibody Assay
The SARS-CoV-2 total RBD antibody (TAb), surrogate neutralizing antibody (SNAb), and avidity were used to measure plasma antibody levels on the TOP-Plus (Pylon 3D analyzer; ET Healthcare) as previously described121.
Flow Cytometry Analysis of Brain
Mice were euthanized with CO2 and perfused through the pulmonary artery followed by the left ventricle with 20 mL of PBS each. Brain and lung tissues were removed and placed in 2.5 mL of digestion buffer (PBS, 5% FCS, 1 mM Hepes) before finely chopping. 400 U of Collagenase D (Roche) were then added to the mixture which was incubated at 37°C for 30 minutes before the addition of 50 uL of 0.5 M EDTA and an additional 5 minute incubation. Digested tissue was then mashed through a 70 um cell strainer, collected by centrifugation at 700 x g in a swinging bucket centrifuge, and then resuspended in 10 mL of 38% isotonic Percoll. This was then centrifuged at 2000 RPM for 20 minutes with no brake. The resulting debris layer was removed by aspiration and the cell pellet washed once in 10 mL digestion buffer. Cells were then blocked with FcX (BioLegend 156604) at a diliution of 1:100 in FACS buffer (DPBS with 1% BSA and 5mM EDTA): before a 15 minute incubation with the following antibodies at a 1:200 dilution in FACS buffer:
Brain: Ly6g-BUV395 (BD 563978), MHCII-BV421 (ThermoFisher 404–5321-82), CD11c-A488 (ThermoFisher 53–0114-82), CD86-PE (Biolegend 105105), CD206-BV605 (Biolegend 141721), CD4 PE-CF594 (BD 562285), CD19-APC (Thermofisher 17–0193-82), B220-APC (ThermoFisher 17–0452-82), CD45-A700 (Biolegend 103127), Ly6cPerCP-Cy5.5 (Thermofisher 45–5932-82), CX3CR1-BV786 (Biolegend 149029), CD8PE-Cy7 (Thermofisher 25–0081-82), CD11b-APC-e780 (ThermoFisher 47–0112-82) all at 1:1200 in FACS Buffer, and Aqua Live/Dead Viability dye (ThermoFisher L34966) at a concentration of 1:1000.
Brain Immunofluorescent Microscopy
Mice were euthanized with CO2 and perfused through the left ventricle with 20 mL of PBS. Brain tissue was removed, and tissue was post-fixed by immersion in 4% PFA overnight. The following day, tissue was washed 3 times in PBS, and then dehydrarated in 30% sucrose in PBS overnight before freezing in OCT media followed by sectioning at 10 um using a cryotome and collection on charged slides which were stored at –80C before further processing.
Slides were removed from storage, washed once in PBS, and then blocked with PBS containing 0.3% Triton-X-100 and 5% normal donkey serum (Jackson Immunoresearch) for 30 minutes at room temperature. Sections were then incubated with the following primary antibodies at the indicated dilution in blocking buffer overnight at 4 °C: Iba1 (Fujifilm 019–19741), GFAP (Cell Signalling Technologies 3657), or MBP (Abcam ab40390). Sections were then washed in PBS 3 times before incubating in species specific anti-IgG secondary antibodies conjugated to A488, A594, or A647 (Jackson Immunoresearch) diluted 1:500 in blocking buffer for 1 hour at room temperature.
Sections were then washed once in PBS, once in PBS containing DAPI, and a final time in PBS before mounting (Prolong Diamond Antifade, Thermofisher). Coverslipped sections were allowed to dry overnight, sealed with clear nail polish, and imaged on a Zeiss LSM 900 with a Zeiss Plan-Apochromat 20X/0.8 air objective. Images were quantified using ImageJ software by creating max projection z-stacks followed by measurement of the mean grey value of the entire frame. A total of 4 seperate sections of cortical tissue overlying the hipocampus at ~ 1.5 – 2.5 mm posterior to Bregma were captured per mouse.
Imaging Mass Cytometry
We employed imaging mass cytometry data derived from post-mortem lung tissue samples as presented in a previously published study70. For our analysis, we exclusively utilized data from normal lung tissue samples and post-acute COVID-19 samples with negative swab test results.
Single-nuclei ATAC-seq Data Processing
Two single-nuclei ATAC-seq samples were preprocessed using the Cell Ranger ATAC 1.2.0 pipeline and aligned to the GRCh38 (hg38) genome. The cellranger output was processed using Signac92. Individual samples were filtered out (Table S1). Amulet93 was used for filtering out doublets. Post QC and doublet removal, the remaining steps of the Signac pipeline (TF-IDF normalization, SVD, UMAP embedding, and clustering) were completed. UMAP embedding and clustering were done using 30 PCs. The cells were annotated by using a reference PBMC scRNA-seq dataset with Seurat’s anchor transfer functions.
Multiome Data Processing
The Multiome data (ATAC + RNA) (n=30 for the study cohort, and n=2 for paired BMMC and PBMC data, n=5 for mouse Lin- and BALF cell data) were preprocessed using the Cell Ranger ARC 1.0.0 pipeline and aligned to the hg38 genome or mm10. The cellranger output was then processed using the Seurat Weighted Nearest Neighbor Pipeline94. Low QC cells were filtered out (data S2 for details). Doublets were removed using Amulet93 for snATAC-seq and Scrublet95 for snRNA-seq. Initial annotations of cells were carried out using the reference PBMC CITE-seq data in the Seurat package.
The Multiome data from different individuals were pooled using Seurat and Signac for snRNA-seq and snATAC-seq, respectively. Three samples were excluded from the pooling process due to poor quality based on the initial clustering. Low QC cells from individual samples were filtered out from the pooled data. For the snRNA-seq object, sctransform normalization was applied, followed by PCA, and 30 PCs were used for UMAP embedding and clustering. The merged dataset was also batch corrected using Harmony96 with all samples being used as a batch, and the UMAP embedding and clustering were repeated using 20 PCs.
We then pooled snATAC-seq profiles from all human samples and did the first round of cell-type annotation based on snRNA-seq annotations. Then we called peaks on each cell type using MACS2 (version 2.1.2) with the following parameters: `callpeak --nomodel --nolambda --keep-dup all --call-summits. Peak summits from all cell types were combined, extended on both sides by 150 bp. Redundant peaks were removed based on the q-value from MACS2. Using the generated peak region list, the number of reads overlapping a given peak window was determined for each unique cell barcode tag. This generated a peak by cell counts matrix corresponding to ATAC reads in peaks for each cell profiled. High-quality cells are retained with a fraction of reads in peaks (FRiP)>0.4 and sequencing depth > 1000. The cells filtered out in this step were also removed from the snRNA-seq object to ensure the same cells were retained across both modalities.
After QC, the human snRNA-seq object was reprocessed using sctranform, PCA, clustering, UMAP, Harmony, and PBMC CITE-seq reference annotation. In the end, 23 clusters were obtained from the snRNA-seq data using default parameters of Seurat (30 PCs for PCA).
Overall mouse snRNA/ATAC-seq data was processed as above, while skipping Harmony as the data belongs to the same batch.
PBMC Cell Type Annotation
Annotations of these clusters were finalized based on the expression of marker genes for distinct immune cell types. 2 of these clusters were labeled as potential doublets and removed from downstream analyses since they expressed marker genes of more than one immune cell type. In the end, 21 clusters were retained that included 197,260 cells out of 260K cells in total in the snRNA-seq object. We combined and curated to yield 10 annotated cell-type clusters capturing all major PBMC populations based on the expression of cell type-specific marker genes. We observed little to no variation in the frequency of each cell type in PBMC among clinical groups, with the exception of CD4+ T cells and pDCs, which were significantly decreased in the Early group (Figure S4D). The same cells were then retained in the snATAC-seq component of the Multiome data, and the annotations were transferred. Once the cell type annotations for major cell types are finalized, we repeated the peak calling using MACS2 and the aforementioned parameter settings, which are used in the downstream DORC analyses using the ArchR 98 pipeline. The merged snATAC-seq object was also processed using the Signac pipeline with TF-IDF normalization, SVD, UMAP embedding, and clustering. We used Harmony (as part of the Signac pipeline) for batch correction in snATAC-seq data using individual samples as a batch. For both non-corrected and Harmony-batch-corrected UMAP embeddings and clustering, 30 PCs were used.
Human HSPC Annotations
We subseted PBMC-PIE dataset for HSPC-annotated cells. We reprocessed the data as above. We used an unbiased workflow to cluster peripheral CD34+ HSPC and identify differentially expressed genes among the 10 resulting subclusters (Figure S4J). Manual curation of subclusters using marker genes from the literature63–65 led us to merge select subclusters resulting in six HSPC subsets defined by both well-characterized and novel marker genes (Figure 3C, S4I).
While we annotated subclusters of snRNA-seq HSPC data based on the expression of manually curated marker genes in each cluster, the snATAC-seq data was separately annotated for the HSPC subtypes using a previously reported bulk-guided approach64. Briefly, using the bulk ATAC-seq peak set, we generated a peak by cell counts matrix. We identified 27 principal components (PCs) of variation in reference bulk ATAC-seq samples, then scored every single cell by the contribution of each PC. Cells were subsequently clustered using the Euclidean distance between these normalized single-cell PCs scores and PCs of bulk samples. The annotations based on the bulk ATAC-seq dataset were then transferred to the snRNA-seq object based on identical cellular identities. To transfer HSPC subtype annotations from public data to our HSPC data, we used public scRNA-seq data of BMMC from healthy participants and the Seurat package94. The data available from GEO with access code GSE139369. Seurat object was created using BMMC data, normalized, then anchors for transferring annotation was defined by FindTransferAnchors() function of Seurat package using BMMC data as a reference. BMMC annotation information was transferred via anchors to our HSPC data using TransferData().
We observed one isolated cluster with mixed cell type annotation and dominant with certain few samples. We generated HSPC UMAP plot after removing this cluster resulting in 28,069 cells. For downstream analysis of HSPC or its subtype, we used original HSPC subset data.
Mouse Multiome Data Cell Type Annotation
For multiome dataset for lineage-depleted cells, we further removed mature cell populations to increase the resolution of the progenitor population. After removing them, we reprocessed the data as described above. RNA-seq data had low library complexity issues, so we used ATAC-seq data for UMAP and clustering analysis. Based on the ATAC-seq-based clustering we defined the cluster-specific genes. Using these genes we annotated each cluster to subtype. For BALF multiome dataset, we used SingleR for reference-based cell type annotation.
snATAC-seq Motif Analysis
Motif enrichment in the pooled snATAC-seq dataset was conducted using two methods. Before conducting the enrichment analysis, motif information was added to the pooled object using the AddMotifs function in Signac92. Motif information for hg38 was added to the object from the JASPAR2020 database.
For the first approach, we calculated overrepresented motifs in a set of differentially accessible peaks. Differential accessibility (DA) analyses were carried out using the pooled single-nuclei ATAC object (n=32) using Seurat’s FindMarkers function94 by conducting pairwise comparisons among clinical groups for each cell type. The normalized ATAC counts matrix was used for DA analyses using a logistic regression framework with a minimum fold change value of 0.25 and minimum percentage of cells for feature detection at 10%. Differential peaks with an adjusted p-value < 0.05 were kept. We then used these top differential peaks to find overrepresented motifs using the FindMotifs function, which uses a hypergeometric test to find overrepresented motifs in a set of genomic features.
To visualize the chromVAR score as a heatmap, we first took the mean of each TF chromVAR score for each cell. Then median was taken for each group, followed by Z-score normalization. We used select TF families to visualize the heatmap.
We also performed footprinting for a smaller select set of transcription factors using the Footprint function in Signac. This calculated the footprinting information of the motifs for every instance in the genome using the whole genome (Hg38) as a background.
Peak-gene Cis-association and DORC Identification
To calculate peak-gene associations, we used our previously published approach. 59 We considered all the peaks that are located in the +/−50 kb window around annotated TSSs. We used peak counts and imputed gene expression to calculate the observed Spearman correlation (obs) of each peak-gene pair. To estimate the background, we generated 100 background peaks for each peak by matching accessibility and GC content (chromVAR) 122 and calculated the Spearman correlation coefficient between those background peaks and the gene, resulting in a null peak-gene Spearman correlation distribution. We then calculated the expected population mean (pop.mean) and expected population standard deviation (pop.sd) from expected Spearman correlations. The Z score is calculated by z=(obs-pop.mean)/pop.sd. For peaks associated with multiple genes, we only kept peak-gene associations with the smallest p-value.
To define DORCs, we selected genes with at least 8 peaks per gene. The DORC score was calculated at each DORC gene for each cell. We defined the DORC score by summing up all the significantly correlated peak counts per gene per cell. We then normalize the DORC score by dividing the DORC score by the total unique fragments in peaks and obtain a cell DORC score matrix.
Differential Analysis on Single-cell Data
We performed differential analysis for single-nuclei gene expression (RNA) and chromatin accessibility (peaks, DORC, and chromVAR) data using FindMarkers function in Seurat package. We used Wilcoxon’s test and set the log2 fold change threshold to 0. Genes, peaks, or Motifs with adjusted p-value < 0.05 were assigned significant. The result was further filtered to exclude genes that have RPS, RNP, and MT- in their symbols.
Visualizing Clinical Group Density on UMAP Plots
With the processed Seurat RNA-seq object, we extracted shared nearest neighbor graph information94. Using this data, we determined the 50 nearest cells of each cell, then calculated the frequency of each group. We then included group frequency information of each cell in the metadata to visualize it on a UMAP plot using Nebulosa100.
Bulk ATAC-seq Data Processing
ATAC-seq paired-end sequences (n=54 for sorted CD14+, n=47 for sorted CD34+) were trimmed using trimmomatic101 and the trimmed reads were aligned to the GRCh38 (hg38) genome using bwa123. These numbers include convalescent COVID-19 groups with mild symptoms. However, samples from these groups were excluded from differential analysis. Further processing steps were carried out on the aligned data using Picard (http://broadinstitute.github.io/picard/) and Samtools103. MACS297 was used for peak calling on the processed data using the BAMPE option with default parameters. For quality control, FRiP scores were calculated for all samples using the featurecounts program available in the Subread package104. Samples with low FRiP scores (< 0.15) were removed from downstream analysis. To ensure that the samples being removed were indeed poor quality, we conducted visual inspections using IGV105. After FRiP score filtering, we were left with 39 CD14+ samples and 18 CD34+ samples, including Mild groups.
Pseudo-bulk Profiles
The cells that were annotated as CD14+ monocytes (n=32) and HSPC (n=32) from the Multiome ATAC-seq data and single-nuclei ATAC-seq data were used to create ‘pseudo-bulk’ profiles for these cell types using snATACClusteringTools. (https://github.com/UcarLab/snATACClusteringTools). For each sample, we generated bam files for these two cell types. 2 samples were filtered out based on low cell numbers (HDu and jcov124) (only for CD14+), three samples were from the same individual and pooled together (lgtd17, lgtd18, and lgtd19), two samples were filtered out due to clinical reasons (jcov114 from CD14+ only, and jcov49_2 from CD14+ and CD34+), resulting in 28 samples for monocytes and 31 for HSPC. Peak calling was done using MACS2 97 with the BAMPE option.
Differential Accessibility Analysis on Bulk ATAC-seq Data
For differential accessibility (DA) analyses, good quality bulk and pseudobulk ATAC-seq samples for CD14+ monocytes (n=70) and HSPC (n=49) were used to generate consensus peak sets using DiffBind106 by retaining peaks that are detected at least in 2 samples, resulting in 123,477 consensus peaks for Monocytes and 126,672 peaks for HSPC. Consensus peaks that are not called in the single-cell object were discarded from downstream analyses leaving us with 108,370 peaks for Monocytes and 117,871 peaks for HSPC. Consensus peaks were annotated using ChipSeeker107 and the peaks were filtered out based on distance to TSS threshold (<50 KB). Post filtering, we had 96,241 consensus peaks for monocytes and 102,784 peaks for HSPC. These peaks were then used to identify differentially accessible regions among clinical groups using the cinaR R package. For differential accessibility analyses, we used GLM models as implemented in EdgeR108,109 by conducting pairwise comparisons among the clinical groups. Due to the variability of ages across clinical groups, we used age as a covariate. Finally, to account for known and unknown batches, we used Surrogate Variable Analysis using all significant SVs. Differential peaks at FDR 10% were kept for downstream analyses. We identified 2,029 differentially accessible regions (DAR) in monocytes and 1,319 DAR in HSPC between clinical groups (Early, Late, Healthy, and nonCoV).
Functional Enrichment Analyses
The peaks were annotated, including the closest genes using cinaR110 and ChIPSeeker107. The TSS regions were defined as −3KB to 3KB. In cases of overlap, the following order of genomic annotations is used: Promoter > 5’ UTR > 3’ UTR > Exon > Intron > Downstream (defined as downstream of gene end) > Intergenic. Functional enrichment for the differential peak sets was conducted using hypergeometric gene set enrichment tests followed by Benjamini-Hochberg FDR adjustment for P-values (cutoff adjusted p=0.1). For functional enrichment, we used different curated gene sets from the CinaRgenesets package110. For HSPC, Gene Set Enrichment Analysis (GSEA) was carried out in addition to the hypergeometric testing since DA peak counts were smaller.
HOMER111 was used for further functional enrichments for KEGG, GO, REACTOME, etc. (p-value cutoff=0.1). HOMER was also used to identify TF motifs enriched in differential peak sets using default settings both for known and de novo motifs. For motif enrichment analyses, we used the default (whole genome) as a background.
Time Series/Trend Analysis
Differential Peaks from bulk and pseudo-bulk ATAC-seq data were pooled together for the following 4 comparisons: Early vs. Healthy, Late vs. Healthy, and nonCoV vs Healthy for both CD14+ Monocytes and HSPC. These peaks were then used to detect trends across different clinical groups using tcseq. The read counts and log fold change values associated with the differential peaks for each clinical group were used as input, and the clinical groups served as time points for the purpose of our analysis. A various number of clusters were tested before we chose number of clusters as the ideal number to visualize the different patterns in the clinical groups. The differential peaks were then split into clusters, and the Functional Enrichment was conducted for each of the individual clusters.
HINT
Footprints are more reliable with deeply sequenced data. To increase the read depth of our data, we pooled Pseudo-bulk ATAC-seq profiles from each clinical group into a single profile for both CD14+ monocytes and HSPC. Peaks were then called for these pooled profiles using macs2. The HINT (Hmm-based IdeNtification of Transcription factor footprints) framework112 was then used to identify active transcription factor binding sites for each clinical group. We also used HINT to find motifs overlapping with the footprints using the JASPAR database for TF motifs analyzed in Figure 6. We then used HINT to generate average ATAC-seq profiles around binding sites of transcription factors of interest. HINT was also used to calculate the differential changes in TF activity between different clinical groups.
The motif regions overlapping with footprints were annotated using ChipSeeker107, and based on those annotations, hypergeometric geneset enrichment was carried out for the footprinting regions using various genetic datasets. The enrichment results were adjusted using the Benjamini-Hochberg FDR adjustment method (FDR = 10%).
Genome Browsing Tracks
We also visualized Pseudo-bulk profiles using a package called SparK113. Firstly, bedgraph files were generated using deepTools124 with the following command ‘bamCoverage -b bamfile.bam -o outputfilename.bdg -bs 1 -of bedgraph’. Spark was then used to generate genome browser track Figures using the bedgraph files for selected loci. Tracks for DORC-associated genes are generated using sn-ATAC-seq dataset, and CoveragePlot function of Signac was used.
Gene Ontology and GSEA Analysis
We used ClusterProfiler package114 for all Gene Ontology enrichment analyses or GSEA analyses of differentially expressed genes and genes associated to differentially accessible regions.
Bulk RNA-seq Analysis
Reads were quantified using STAR115 with GRCh38 reference genome. Differential gene expression analysis was performed using DESeq2116. Differentially expressed genes were identified by absolute log2FoldChance greater than 1.1 and adjusted p-value less than 0.05. Parallel execution and resources management was carried out by Snakemake117.
Module Analysis
For any module analysis used in this paper, we used function AddModuleScore function in Seurat. Calculated per-cell module score was used for downstream analysis. For GMP and M.SC3 module, we used top highly expressed cluster-specific genes. For neutrophil module, we used genes in neutrophil-related GO terms (“GO:0030223”, “GO:0042119”, “GO:0002283”, “GO:1990266”, “GO:0043312”). For inflammatory module, we used inflammation modules defined in our previous study125.
QUANTIFICATION AND STATISTICAL ANALYSIS
In the relevant Results section and, if applicable, in the figure legends, we provided the details of the statistical tests performed and the number of observations, including the sample sizes compared. In cases where this information was not explicitly mentioned, we utilized the Wilcoxon test to compare the clinical groups with the Healthy group.
Supplementary Material
(A) Summary table of samples used for different analyses.
(B) Swimmer plot showing hospitalization periods, and sample collection time point for each patient in nonCOV, Early, and Late. Early–ICU group is colored with dark red, indicating period of ICU admission.
(C) Viral S protein-specific antibody assays showing antibody level, neutralizing activity, and binding quality. (ANOVA, p*<0.05)
(D-E) A heatmap (D) and boxplots (E) showing plasma cytokine levels measured by Luminex platform. Cytokine level per study participant grouped by group is shown as boxplots (*p<0.05, Wilcoxon’s test, Healthy-reference). For heatmap, median cytokine level per group was normalized by Z-score. (t-test, p *<0.05)
(F) Flow cytometry results showing frequencies of CD14+ monocytes, CD34+ HSPC, and monocyte subsets across groups. (ANOVA, * p<0.05)
(A) GSEA of PC1- and PC2-associated DAR from PCA analysis in CD14+ monocytes. setSize indicate number of DAR-associated genes in each GO term. NES stands for normalized enrichment score, and positive NES shows enrichment of DAR-associated genes with positive PC values (same applies to negative NES and PC value).
(B) Volcano plots showing DEG in CD14+ monocytes between Healthy and Early/Late/nonCov groups.
(C) Volcano plots displaying DORC activity in CD14+ monocyte between the Healthy and Early/Late groups.
(D) GO analysis of significantly upregulated genes (DEG, blue dots), and chromatin accessibility (DORC, grey dots) in CD14+ monocytes of each clinical group compared to Healthy.
(E) GSEA comparing between Early and Late CD14+ monocytes.
(F) Violin plot showing the inflammation score for each clinical group, as obtained from the expression profiles of inflammatory gene sets.
(G) GO analysis of significantly upregulated genes (DEG, blue dots), and chromatin accessibility (DORC, grey dots) in each subcluster of CD14+ monocytes.
(H) Dot plot showing module score of monocyte subtype marker genes fetched from previous study62 in each myeloid subclusters.
(A) Boxplot showing the concentration of cytokines in CD14 + monocytes at 24hrs post-stimulation with R848 and IFNα. Concentrations were measured using a Legendplex 13-plex panel for human viral response.
(B) Volcano plots showing DEGs in CD14 + monocytes after 6 hours of stimulation with R848 and IFNα between the Healthy and post-COVID groups.
(C) Heatmap showing the expression fold change of selected DEGs in CD14 + monocytes between stimulated (R848 and IFNα) and unstimulated groups at the individual level. The Early and Late subgroups in the post-COVID group are annotated on the right.
(D) Genome browser tracks displaying the DORC-associated region linked to the CCL5 gene in CD14 monocytes from healthy and post-COVID groups. Peaks associated with DORC are highlighted and connected to the CCL5 gene body with loops (top).
(E) A box plot showing the comparison of the average CCL5 DORC score of each study participant across cohorts.
(F) Boxplot showing the fold change of module score of DAR C2- and C4-associated genes (from Figure 1) between unstimulated and 6 hours of stimulation in CD14 + monocytes. We observed that gene expression modules for genes annotated to both persistently and transiently accessible DAR clusters exhibited greater induction in post-COVID monocytes compared to healthy monocytes.
(A) UMAP plots displaying unbiased clustering (top) and group distribution (bottom) using snRNA-seq (left) and snATAC-seq (right).
(B) Count matrix showing number of DEGs in various comparisons across cell types.
(C) Stacked bar plot showing cell type composition in each donor.
(D) Boxplots showing percent of major cell type within total PBMC per study participant. (Wilcoxon’s test, * p < 0.05; Healthy-reference)
(E) Schematic describing workflow for analyzing distal regulatory elements and gene expression using snRNA/ATAC-seq data including DORC. ATAC-seq peaks were linked to putative target genes in cis, based on the co-variation of chromatin accessibility and gene expression levels across individual cells developmentally related HSPC and monocytes were then co-clustered based on DORC scores.
(F) Genome track showing chromatin accessibility of CEBPA-associated DORC in HSC/MPP and CD14+ monocytes. Curved lines (loops) above indicate peaks with significant correlation (p-value < 0.05). The grey bar (below gene track) represents peaks.
(G) The number of significant peak-gene connections for all genes.
(H) The number of significantly correlated peaks (p-value < 0.05) for each gene.
(I) Heatmap showing expression of marker genes for each HSPC subcluster.
(J) Unbiased clustering of HSPC UMAP of snRNA-seq data.
(K) UMAP plot of HSPC with annotation transferred from BM single-cell RNA-seq data65.
(L) UMAP plot with subcluster annotation guided by ATAC-seq data of FACS-sorted BM HSPC subpopulations 64.
(M) UMAP plot showing density of CD164-expressing cells in HSPC.
(A) GSEA result of PC1- and PC2-associated DAR from PCA analysis in HSPC. setSize indicate number of DAR-associated genes in each GO term. NES stands for normalized enrichment score, and positive NES shows enrichment of DAR-associated genes with positive PC values (same applies to negative NES and PC value).
(B) Volcano plots showing DEGs in HSPC between Healthy and Early/Late groups. (FDR < 0.05)
(C) Heatmap showing DEGs associated with hematopoietic regulation in different groups. Genes that are discussed in the main text are bolded. (FDR < 0.05)
(D) Box plots showing average expression of select genes per study participant. Genes related to myeloid differentiation are selected from DEGs in Late. (Wilcoxon’s test, * p < 0.05, Healthy-reference)
(E) Projections of group density on the snRNA-seq HSPC UMAP plot.
(F) Dot plot showing average GMP module score in each group.
(A) Volcano plots showing differentially active TFs in HSPC and CD14+ monocytes between Healthy and nonCov. (Wilcoxon’s test, * adjusted p < 0.05, Healthy-reference)
(B) Boxplots for mean chromVAR score of select TFs in HSPC and CD14+ monocytes for study participant.(Wilcoxon’s test, * p < 0.05, Healthy-reference)
(C) TF footprints and violin plots for TF motif-associated chromatin accessibility (chromVAR) activity in HSPC and CD14+ monocytes across groups. (Wilcoxon’s test, * p < 0.05, Healthy-reference).
(A) Dumbbell plot displaying the difference in the average frequency of GMP within HSPC of the Late between two groups, categorized by various clinical parameters. The p-values, obtained using the t-test, are shown along with their adjusted p-values.
(B) GSEA result showing the biological processes that are enriched in HSC/MPP of Late with αIL-6R and Late without αIL-6R.
(C) Weight recovery rate of A/J mice with MHV-1 infection over time (n=7, naïve; n=5, recovered; n=8, aIL-6; top). Viral titers (plaque-forming unit per ul) at day 4 and 30 post-infection (n=10, naïve; n=12, recovered; n=7, αIL-6R; bottom).
(D-E) Dot plots showing the expression of marker genes for different mouse HSPC subtypes (C) and cell types in BALF.
(F) UMAP visualization of the BALF snRNA/ATAC-seq dataset with cell type annotations.
(G) Dot plot showing the expression of marker genes BALF macrophage subclusters.
(H) Heatmap representing the row-normalized average chromVAR scores across different groups of macrophage clusters.
(I) Heatmap representing the row-normalized average chromVAR score for marker TFs in BALF macrophage.
(J) Volcano plots showing the DEGs in macrophages between the anti-IL-6R blockade and recovered groups. (Wilcoxon’s test, * p < 0.05, Recovered-reference).
(K) GSEA showing the biological processes that are enriched in the recovered group compared to the anti-IL-6R blockade group.
(L) Representative images from H&E staining of mouse lung in different groups.
(M) This bar plot shows the fold change in average density of selected hematopoietic cell types from the ROI in lung tissue between post-COVID and healthy samples. Asterisks indicate hematopoietic cell types with significantly different cell densities per ROI between the two groups. (Wilcoxon’s test, * p < 0.05, Healthy-reference)
(N) The box plot displays the average intensity of IBA-1 and GFAP staining per ROI and mouse across groups. Intensity per ROI is represented by small transparent dots, while larger dots indicate the average intensity per mouse. (Wilcoxon’s test, * p < 0.05; Recovered-reference)
Table S1. Demographic, Clinical and QC Data, Related to STAR Methods
Table S2. Cell type-defining marker genes, Related to STAR Methods
Table S7. Legendplex data and normalized gene count matrix of select DEG from monocyte stimulation experiment, Related to Figure 2
Highlights.
Severe COVID-19 programs durable epigenetic changes and hyper-activation in monocytes
Circulating HSPC capture post-COVID changes in hematopoiesis and stem cell programs
Post-COVID HSPC convey epigenetic and transcriptional memory to mature progeny cells
IL-6 contributes to epigenetic reprogramming of mouse and human HSPC and myeloid cells
Acknowledgments
We thank volunteers who donated clinical samples; L. Ivashkiv and T. Holh for discussions; J. Cha and JAX creative department for figure assistance; and S. Sampson for editing. Financial support was provided by:
COVID-19 WCM WCG-COVID-19, NIH-R01AI148416, NIH-U19AI144301, NIH-R01AI148416-S1, NIH-R01AI148416-S2, NIH-RM1GM139738 (WCM, SZJ)
Asan Foundation, ROK (JGC)
ASH-HONORS (AR)
NIH-NCI R25 CA233208 (MSKCC CBSP Training Grant, LP)
HFSP LT000203/2021-L (AL)
NIH/NIAID-R01AI161444 (CMR)
Footnotes
Declaration of Interest
J.D.B. holds patents related to ATAC-seq and scATAC-seq and serves on the Scientific Advisory Board of CAMP4 Therapeutics, seqWell, and CelSee.
S.Z.J. and F.J.B. declare a related patent application: 10203–02-PC; EFS ID: 44924864 Enrichment and Characterization of Rare Circulating Cells, including Progenitor Cells from Peripheral Blood and Uses Thereof.
F.J.B. is a co-founder and scientific advisor of IpiNovyx Bio.
E.J.S. reports personal fees from NIAID through Axle Informatics for the subject matter expert program for the COVID-19 vaccine clinical trials.
R.E.S. is on the scientific advisory board of Miromatrix Inc and Lime Therapeutics and is a paid consultant and speaker for Alnylam Inc.
Inclusion and Diversity
We advocate for inclusive, diverse, and equitable research practices. Our paper includes authors who self-identify as underrepresented ethnic minorities, gender minorities, and members of the LGBTQIA+ community.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, Guo D, Hu W, Yang J, Tang Z, et al. (2020). Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 9, 761–770. 10.1080/22221751.2020.1747363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang D, Chu H, Hou Y, Chai Y, Shuai H, Lee AC-Y, Zhang X, Wang Y, Hu B, Huang X, et al. (2020). Attenuated Interferon and Proinflammatory Response in SARS-CoV-2-Infected Human Dendritic Cells Is Associated With Viral Antagonism of STAT1 Phosphorylation. J. Infect. Dis. 222, 734–745. 10.1093/infdis/jiaa356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang J, Jiang M, Chen X, and Montaner LJ (2020). Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Biol. 108, 17–41. 10.1002/JLB.3COVR0520-272R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, Ellingson MK, Mao T, Oh JE, Israelow B, et al. (2020). Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584, 463–469. 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, Ogishi M, Sabli IKD, Hodeib S, Korol C, et al. (2020). Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370. 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou R, To KK-W, Wong Y-C, Liu L, Zhou B, Li X, Huang H, Mo Y, Luk T-Y, Lau TT-K, et al. (2020). Acute SARS-CoV-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity 53, 864–877.e5. 10.1016/j.immuni.2020.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arunachalam PS, Wimmers F, Mok CKP, Perera RAPM, Scott M, Hagan T, Sigal N, Feng Y, Bristow L, Tak-Yin Tsang O, et al. (2020). Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 369, 1210–1220. 10.1126/science.abc6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Israelow B, Song E, Mao T, Lu P, Meir A, Liu F, Alfajaro MM, Wei J, Dong H, Homer RJ, et al. (2020). Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. J. Exp. Med. 217. 10.1084/jem.20201241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, Péré H, Charbit B, Bondet V, Chenevier-Gobeaux C, et al. (2020). Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724. 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acharya D, Liu G, and Gack MU (2020). Dysregulation of type I interferon responses in COVID-19. Nat. Rev. Immunol. 20, 397–398. 10.1038/s41577-0200346-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, Brewer JR, Han A, Steach HR, Israelow B, et al. (2022). Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature 606, 585–593. 10.1038/s41586-022-04802-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laurent P, Yang C, Rendeiro AF, Nilsson-Payant BE, Carrau L, Chandar V, Bram Y, tenOever BR, Elemento O, Ivashkiv LB, et al. (2022). Sensing of SARS-CoV-2 by pDCs and their subsequent production of IFN-I contribute to macrophage-induced cytokine storm during COVID-19. Sci. Immunol. 7, eadd4906. 10.1126/sciimmunol.add4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Domizio JD, Gulen MF, Saidoune F, Thacker VV, Yatim A, Sharma K, Nass T, Guenova E, Schaller M, Conrad C, et al. (2022). The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature 603, 145–151. 10.1038/s41586-022-04421-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Aly Z, Xie Y, and Bowe B. (2021). High-dimensional characterization of post-acute sequelae of COVID-19. Nature 594, 259–264. 10.1038/s41586-021-03553-9. [DOI] [PubMed] [Google Scholar]
- 15.Del Rio C, Collins LF, and Malani P. (2020). Long-term Health Consequences of COVID-19. JAMA 324, 1723–1724. 10.1001/jama.2020.19719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehandru S, and Merad M. (2022). Pathological sequelae of long-haul COVID. Nat. Immunol. 23, 194–202. 10.1038/s41590-021-01104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sudre CH, Murray B, Varsavsky T, Graham MS, Penfold RS, Bowyer RC, Pujol JC, Klaser K, Antonelli M, Canas LS, et al. (2021). Attributes and predictors of long COVID. Nat. Med. 27, 626–631. 10.1038/s41591-021-01292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bell ML, Catalfamo CJ, Farland LV, Ernst KC, Jacobs ET, Klimentidis YC, Jehn M, and Pogreba-Brown K. (2021). Post-acute sequelae of COVID-19 in a non-hospitalized cohort: Results from the Arizona CoVHORT. PLoS ONE 16, e0254347. 10.1371/journal.pone.0254347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Proal AD, and VanElzakker MB (2021). Long COVID or Post-acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Front. Microbiol. 12, 698169. 10.3389/fmicb.2021.698169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schultheiß C, Willscher E, Paschold L, Gottschick C, Klee B, Henkes S-S, Bosurgi L, Dutzmann J, Sedding D, Frese T, et al. (2022). The IL-1β, IL-6, and TNF cytokine triad is associated with post-acute sequelae of COVID-19. Cell Rep. Med. 3, 100663. 10.1016/j.xcrm.2022.100663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phetsouphanh C, Darley DR, Wilson DB, Howe A, Munier CML, Patel SK, Juno JA, Burrell LM, Kent SJ, Dore GJ, et al. (2022). Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 23, 210–216. 10.1038/s41590-021-01113-x. [DOI] [PubMed] [Google Scholar]
- 22.Choutka J, Jansari V, Hornig M, and Iwasaki A. (2022). Unexplained post-acute infection syndromes. Nat. Med. 28, 911–923. 10.1038/s41591-022-01810-6. [DOI] [PubMed] [Google Scholar]
- 23.Carter MJ, Fish M, Jennings A, Doores KJ, Wellman P, Seow J, Acors S, Graham C, Timms E, Kenny J, et al. (2020). Peripheral immunophenotypes in children with multisystem inflammatory syndrome associated with SARS-CoV-2 infection. Nat. Med. 26, 1701–1707. 10.1038/s41591-020-1054-6. [DOI] [PubMed] [Google Scholar]
- 24.Consiglio CR, Cotugno N, Sardh F, Pou C, Amodio D, Rodriguez L, Tan Z, Zicari S, Ruggiero A, Pascucci GR, et al. (2020). The Immunology of Multisystem Inflammatory Syndrome in Children with COVID-19. Cell 183, 968–981.e7. 10.1016/j.cell.2020.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gruber CN, Patel RS, Trachtman R, Lepow L, Amanat F, Krammer F, Wilson KM, Onel K, Geanon D, Tuballes K, et al. (2020). Mapping Systemic Inflammation and Antibody Responses in Multisystem Inflammatory Syndrome in Children (MIS-C). Cell 183, 982–995.e14. 10.1016/j.cell.2020.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tenforde MW, and Morris SB (2021). Multisystem inflammatory syndrome in adults: coming into focus. Chest 159, 471–472. 10.1016/j.chest.2020.09.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris SB, Schwartz NG, Patel P, Abbo L, Beauchamps L, Balan S, Lee EH, Paneth-Pollak R, Geevarughese A, Lash MK, et al. (2020). Case Series of Multisystem Inflammatory Syndrome in Adults Associated with SARSCoV-2 Infection - United Kingdom and United States, March-August 2020. MMWR Morb Mortal Wkly Rep 69, 1450–1456. 10.15585/mmwr.mm6940e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernández-Castañeda A, Lu P, Geraghty AC, Song E, Lee M-H, Wood J, Yalçın B, Taylor KR, Dutton S, Acosta-Alvarez L, et al. (2022). Mild respiratory SARS-CoV-2 infection can cause multi-lineage cellular dysregulation and myelin loss in the brain. BioRxiv. 10.1101/2022.01.07.475453. [DOI] [Google Scholar]
- 29.Klein J, Wood J, Jaycox J, Lu P, Dhodapkar RM, Gehlhausen JR, Tabachnikova A, Tabacof L, Malik AA, Kamath K, et al. (2022). Distinguishing features of Long COVID identified through immune profiling. medRxiv. 10.1101/2022.08.09.22278592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, Joosten LAB, van der Meer JWM, Mhlanga MM, Mulder WJM, et al. (2020). Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 20, 375–388. 10.1038/s41577-020-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bekkering S, Domínguez-Andrés J, Joosten LAB, Riksen NP, and Netea MG (2021). Trained immunity: reprogramming innate immunity in health and disease. Annu. Rev. Immunol. 39, 667–693. 10.1146/annurev-immunol-102119-073855. [DOI] [PubMed] [Google Scholar]
- 32.Gu H, Zeng X, Peng L, Xiang C, Zhou Y, Zhang X, Zhang J, Wang N, Guo G, Li Y, et al. (2021). Vaccination induces rapid protection against bacterial pneumonia via training alveolar macrophage in mice. eLife 10. 10.7554/eLife.69951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zahalka S, Starkl P, Watzenboeck ML, Farhat A, Radhouani M, Deckert F, Hladik A, Lakovits K, Oberndorfer F, Lassnig C, et al. (2022). Trained immunity of alveolar macrophages requires metabolic rewiring and type 1 interferon signaling. Mucosal Immunol. 15, 896–907. 10.1038/s41385-022-00528-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao Y, Jeyanathan M, Haddadi S, Barra NG, Vaseghi-Shanjani M, Damjanovic D, Lai R, Afkhami S, Chen Y, Dvorkin-Gheva A, et al. (2018). Induction of autonomous memory alveolar macrophages requires T cell help and is critical to trained immunity. Cell 175, 1634–1650.e17. 10.1016/j.cell.2018.09.042. [DOI] [PubMed] [Google Scholar]
- 35.Machiels B, Dourcy M, Xiao X, Javaux J, Mesnil C, Sabatel C, Desmecht D, Lallemand F, Martinive P, Hammad H, et al. (2017). A gammaherpesvirus provides protection against allergic asthma by inducing the replacement of resident alveolar macrophages with regulatory monocytes. Nat. Immunol. 18, 1310–1320. 10.1038/ni.3857. [DOI] [PubMed] [Google Scholar]
- 36.Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, Yuan S, Polak L, Kulukian A, Chai S, and Fuchs E. (2017). Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 550, 475–480. 10.1038/nature24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larsen SB, Cowley CJ, Sajjath SM, Barrows D, Yang Y, Carroll TS, and Fuchs E. (2021). Establishment, maintenance, and recall of inflammatory memory. Cell Stem Cell 28, 1758–1774.e8. 10.1016/j.stem.2021.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niec RE, Rudensky AY, and Fuchs E. (2021). Inflammatory adaptation in barrier tissues. Cell 184, 3361–3375. 10.1016/j.cell.2021.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleinnijenhuis J, Quintin J, Preijers F, Joosten LAB, Ifrim DC, Saeed S, Jacobs C, van Loenhout J, de Jong D, Stunnenberg HG, et al. (2012). Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA 109, 17537–17542. 10.1073/pnas.1202870109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, Jacobs L, Jansen T, Kullberg B-J, Wijmenga C, et al. (2012). Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12, 223–232. 10.1016/j.chom.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kleinnijenhuis J, Quintin J, Preijers F, Joosten LAB, Jacobs C, Xavier RJ, van der Meer JWM, van Crevel R, and Netea MG (2014). BCG-induced trained immunity in NK cells: Role for non-specific protection to infection. Clin. Immunol. 155, 213–219. 10.1016/j.clim.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, Tzelepis F, Pernet E, Dumaine A, Grenier J-C, et al. (2018). BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 172, 176–190.e19. 10.1016/j.cell.2017.12.031. [DOI] [PubMed] [Google Scholar]
- 43.Dos Santos JC, Barroso de Figueiredo AM, Teodoro Silva MV, Cirovic B, de Bree LCJ, Damen MSMA, Moorlag SJCFM, Gomes RS, Helsen MM, Oosting M, et al. (2019). β-Glucan-Induced Trained Immunity Protects against Leishmania braziliensis Infection: a Crucial Role for IL-32. Cell Rep. 28, 2659–2672.e6. 10.1016/j.celrep.2019.08.004. [DOI] [PubMed] [Google Scholar]
- 44.Cirovic B, de Bree LCJ, Groh L, Blok BA, Chan J, van der Velden WJFM, Bremmers MEJ, van Crevel R, Händler K, Picelli S, et al. (2020). BCG vaccination in humans elicits trained immunity via the hematopoietic progenitor compartment. Cell Host Microbe 28, 322–334.e5. 10.1016/j.chom.2020.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitroulis I, Ruppova K, Wang B, Chen L-S, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, et al. (2018). Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 172, 147–161.e12. 10.1016/j.cell.2017.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li X, Wang H, Yu X, Saha G, Kalafati L, Ioannidis C, Mitroulis I, Netea MG, Chavakis T, and Hajishengallis G. (2022). Maladaptive innate immune training of myelopoiesis links inflammatory comorbidities. Cell 185, 1709–1727.e18. 10.1016/j.cell.2022.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalafati L, Kourtzelis I, Schulte-Schrepping J, Li X, Hatzioannou A, Grinenko T, Hagag E, Sinha A, Has C, Dietz S, et al. (2020). Innate Immune Training of Granulopoiesis Promotes Anti-tumor Activity. Cell 183, 771–785.e12. 10.1016/j.cell.2020.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, Scholz CJ, Oosting M, Haendler K, Baßler K, et al. (2018). Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 172, 162–175.e14. 10.1016/j.cell.2017.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.You M, Chen L, Zhang D, Zhao P, Chen Z, Qin E-Q, Gao Y, Davis MM, and Yang P. (2021). Single-cell epigenomic landscape of peripheral immune cells reveals establishment of trained immunity in individuals convalescing from COVID-19. Nat. Cell Biol. 23, 620–630. 10.1038/s41556-021-00690-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, Bigley V, Flavell RA, Gilroy DW, Asquith B, et al. (2017). The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 214, 1913–1923. 10.1084/jem.20170355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jentho E, Ruiz-Moreno C, Novakovic B, Kourtzelis I, Megchelenbrink WL, Martins R, Chavakis T, Soares MP, Kalafati L, Guerra J, et al. (2021). Trained innate immunity, long-lasting epigenetic modulation, and skewed myelopoiesis by heme. Proc Natl Acad Sci USA 118. 10.1073/pnas.2102698118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Netea MG, Quintin J, and van der Meer JWM (2011). Trained immunity: a memory for innate host defense. Cell Host Microbe 9, 355–361. 10.1016/j.chom.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 53.Kong L, Moorlag SJCFM, Lefkovith A, Li B, Matzaraki V, van Emst L, Kang HA, Latorre I, Jaeger M, Joosten LAB, et al. (2021). Single-cell transcriptomic profiles reveal changes associated with BCG-induced trained immunity and protective effects in circulating monocytes. Cell Rep. 37, 110028. 10.1016/j.celrep.2021.110028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hunter CA, and Jones SA (2015). IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 16, 448–457. 10.1038/ni.3153. [DOI] [PubMed] [Google Scholar]
- 55.REMAP-CAP Investigators, Gordon AC, Mouncey PR, Al-Beidh F, Rowan KM, Nichol AD, Arabi YM, Annane D, Beane A, van Bentum-Puijk W, et al. (2021). Interleukin-6 Receptor Antagonists in Critically Ill Patients with Covid-19. N. Engl. J. Med. 384, 1491–1502. 10.1056/NEJMoa2100433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koelle K, Martin MA, Antia R, Lopman B, and Dean NE (2022). The changing epidemiology of SARS-CoV-2. Science 375, 1116–1121. 10.1126/science.abm4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Silvin A, Chapuis N, Dunsmore G, Goubet A-G, Dubuisson A, Derosa L, Almire C, Hénon C, Kosmider O, Droin N, et al. (2020). Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 182, 1401–1418.e18. 10.1016/j.cell.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schulte-Schrepping J, Reusch N, Paclik D, Baßler K, Schlickeiser S, Zhang B, Krämer B, Krammer T, Brumhard S, Bonaguro L, et al. (2020). Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 182, 1419–1440.e23. 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma S, Zhang B, LaFave LM, Earl AS, Chiang Z, Hu Y, Ding J, Brack A, Kartha VK, Tay T, et al. (2020). Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 183, 1103–1116.e20. 10.1016/j.cell.2020.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dutertre C-A, Becht E, Irac SE, Khalilnezhad A, Narang V, Khalilnezhad S, Ng PY, van den Hoogen LL, Leong JY, Lee B, et al. (2019). Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 51, 573–589.e8. 10.1016/j.immuni.2019.08.008. [DOI] [PubMed] [Google Scholar]
- 61.Lee J, Tam H, Adler L, Ilstad-Minnihan A, Macaubas C, and Mellins ED (2017). The MHC class II antigen presentation pathway in human monocytes differs by subset and is regulated by cytokines. PLoS ONE 12, e0183594. 10.1371/journal.pone.0183594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wong KL, Tai JJ-Y, Wong W-C, Han H, Sem X, Yeap W-H, Kourilsky P, and Wong S-C (2011). Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 118, e16–31. 10.1182/blood-2010-12-326355. [DOI] [PubMed] [Google Scholar]
- 63.Pellin D, Loperfido M, Baricordi C, Wolock SL, Montepeloso A, Weinberg OK, Biffi A, Klein AM, and Biasco L. (2019). A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nat. Commun. 10, 2395. 10.1038/s41467-019-10291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buenrostro JD, Corces MR, Lareau CA, Wu B, Schep AN, Aryee MJ, Majeti R, Chang HY, and Greenleaf WJ (2018). Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell 173, 1535–1548.e16. 10.1016/j.cell.2018.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Granja JM, Klemm S, McGinnis LM, Kathiria AS, Mezger A, Corces MR, Parks B, Gars E, Liedtke M, Zheng GXY, et al. (2019). Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol. 37, 1458–1465. 10.1038/s41587-019-0332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, Akashi K, and Tenen DG (2006). C/EBPbeta is required for “emergency” granulopoiesis. Nat. Immunol. 7, 732–739. 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 67.Shyamsunder P, Shanmugasundaram M, Mayakonda A, Dakle P, Teoh WW, Han L, Kanojia D, Lim MC, Fullwood M, An O, et al. (2019). Identification of a novel enhancer of CEBPE essential for granulocytic differentiation. Blood 133, 2507–2517. 10.1182/blood.2018886077. [DOI] [PubMed] [Google Scholar]
- 68.Paidas MJ, Cosio DS, Ali S, Kenyon NS, and Jayakumar AR (2022). Long-Term Sequelae of COVID-19 in Experimental Mice. Mol. Neurobiol. 59, 5970–5986. 10.1007/s12035-022-02932-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.T’Jonck W, Guilliams M, and Bonnardel J. (2018). Niche signals and transcription factors involved in tissue-resident macrophage development. Cell. Immunol. 330, 43–53. 10.1016/j.cellimm.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rendeiro AF, Ravichandran H, Kim J, Borczuk AC, Elemento O, and Schwartz RE (2022). Persistent alveolar type 2 dysfunction and lung structural derangement in post-acute COVID-19. medRxiv. 10.1101/2022.11.28.22282811. [DOI] [Google Scholar]
- 71.De Albuquerque N, Baig E, Ma X, Zhang J, He W, Rowe A, Habal M, Liu M, Shalev I, Downey GP, et al. (2006). Murine hepatitis virus strain 1 produces a clinically relevant model of severe acute respiratory syndrome in A/J mice. J. Virol. 80, 10382–10394. 10.1128/JVI.00747-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seita J, and Weissman IL (2010). Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2, 640–653. 10.1002/wsbm.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brandi P, Conejero L, Cueto FJ, Martínez-Cano S, Dunphy G, Gómez MJ, Relaño C, Saz-Leal P, Enamorado M, Quintas A, et al. (2022). Trained immunity induction by the inactivated mucosal vaccine MV130 protects against experimental viral respiratory infections. Cell Rep. 38, 110184. 10.1016/j.celrep.2021.110184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, McKechnie JL, Ivison GT, Ranganath T, Vergara R, Hollis T, et al. (2020). A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 26, 1070–1076. 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang X, Wen Y, Xie X, Liu Y, Tan X, Cai Q, Zhang Y, Cheng L, Xu G, Zhang S, et al. (2021). Dysregulated hematopoiesis in bone marrow marks severe COVID-19. Cell Discov. 7, 60. 10.1038/s41421-021-00296-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chiba Y, Mizoguchi I, Hasegawa H, Ohashi M, Orii N, Nagai T, Sugahara M, Miyamoto Y, Xu M, Owaki T, et al. (2018). Regulation of myelopoiesis by proinflammatory cytokines in infectious diseases. Cell. Mol. Life Sci. 75, 1363–1376. 10.1007/s00018-017-2724-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boettcher S, and Manz MG (2016). Sensing and translation of pathogen signals into demand-adapted myelopoiesis. Curr. Opin. Hematol. 23, 5–10. 10.1097/MOH.0000000000000201. [DOI] [PubMed] [Google Scholar]
- 78.Paudel S, Ghimire L, Jin L, Jeansonne D, and Jeyaseelan S. (2022). Regulation of emergency granulopoiesis during infection. Front. Immunol. 13, 961601. 10.3389/fimmu.2022.961601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Manz MG, and Boettcher S. (2014). Emergency granulopoiesis. Nat. Rev. Immunol. 14, 302–314. 10.1038/nri3660. [DOI] [PubMed] [Google Scholar]
- 80.Wimmers F, Donato M, Kuo A, Ashuach T, Gupta S, Li C, Dvorak M, Foecke MH, Chang SE, Hagan T, et al. (2021). The single-cell epigenomic and transcriptional landscape of immunity to influenza vaccination. Cell 184, 3915–3935.e21. 10.1016/j.cell.2021.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schaper F, and Rose-John S. (2015). Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 26, 475–487. 10.1016/j.cytogfr.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 82.Ellingsgaard H, Hojman P, and Pedersen BK (2019). Exercise and health — emerging roles of IL-6. Curr. Opin. Physiol. 10, 49–54. 10.1016/j.cophys.2019.03.009. [DOI] [Google Scholar]
- 83.Grivennikov S, Karin E, Terzic J, Mucida D, Yu G-Y, Vallabhapurapu S,Scheller J, Rose-John S, Cheroutre H, Eckmann L, et al. (2009). IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15, 103–113. 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Muñoz-Cánoves P, Scheele C, Pedersen BK, and Serrano AL (2013). Interleukin-6 myokine signaling in skeletal muscle: a double-edged sword? FEBS J. 280, 4131–4148. 10.1111/febs.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lim AI, McFadden T, Link VM, Han S-J, Karlsson R-M, Stacy A, Farley TK, Lima-Junior DS, Harrison OJ, Desai JV, et al. (2021). Prenatal maternal infection promotes tissue-specific immunity and inflammation in offspring. Science 373. 10.1126/science.abf3002. [DOI] [PubMed] [Google Scholar]
- 86.McGonagle D, Sharif K, O’Regan A, and Bridgewood C. (2020). The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 19, 102537. 10.1016/j.autrev.2020.102537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rosas IO, Bräu N, Waters M, Go RC, Hunter BD, Bhagani S, Skiest D, Aziz MS, Cooper N, Douglas IS, et al. (2021). Tocilizumab in Hospitalized Patients with Severe Covid-19 Pneumonia. N. Engl. J. Med. 384, 1503–1516. 10.1056/NEJMoa2028700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rosas IO, Bräu N, Waters M, Go RC, Malhotra A, Hunter BD, Bhagani S, Skiest D, Savic S, Douglas IS, et al. (2022). Tocilizumab in patients hospitalised with COVID-19 pneumonia: Efficacy, safety, viral clearance, and antibody response from a randomised controlled trial (COVACTA). EClinicalMedicine 47, 101409. 10.1016/j.eclinm.2022.101409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zeraatkar D, Cusano E, Martínez JPD, Qasim A, Mangala S, Kum E, Bartoszko JJ, Devji T, Agoritsas T, Guyatt G, et al. (2022). Use of tocilizumab and sarilumab alone or in combination with corticosteroids for covid-19: systematic review and network meta-analysis. bmjmed 1, e000036. 10.1136/bmjmed-2021-000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Akira S, Isshiki H, Sugita T, Tanabe O, Kinoshita S, Nishio Y, Nakajima T, Hirano T, and Kishimoto T. (1990). A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 9, 1897–1906. 10.1002/j.1460-2075.1990.tb08316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rose-John S. (2018). Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol. 10. 10.1101/cshperspect.a028415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stuart T, Srivastava A, Madad S, Lareau CA, and Satija R. (2021). Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341. 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thibodeau A, Eroglu A, McGinnis CS, Lawlor N, Nehar-Belaid D, Kursawe R, Marches R, Conrad DN, Kuchel GA, Gartner ZJ, et al. (2021). AMULET: a novel read count-based method for effective multiplet detection from single nucleus ATAC-seq data. Genome Biol. 22, 252. 10.1186/s13059-021-02469-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wolock SL, Lopez R, and Klein AM (2019). Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 8, 281–291.e9. 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P-R, and Raychaudhuri S. (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, and Greenleaf WJ (2021). ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411. 10.1038/s41588-021-00790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kartha VK, Duarte FM, Hu Y, Ma S, Chew JG, Lareau CA, Earl A, Burkett ZD, Kohlway AS, Lebofsky R, et al. (2022). Functional inference of gene regulation using single-cell multi-omics. Cell Genomics 2. 10.1016/j.xgen.2022.100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Alquicira-Hernandez J, and Powell JE (2021). Nebulosa recovers single-cell gene expression signals by kernel density estimation. Bioinformatics 37, 2485–2487. 10.1093/bioinformatics/btab003. [DOI] [PubMed] [Google Scholar]
- 101.Bolger AM, Lohse M, and Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. [Google Scholar]
- 103.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liao Y, Smyth GK, and Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 105.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yu G, Wang L-G, and He Q-Y (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 108.Ucar D, and Karakaslar EO (2021). cinaR: A comprehensive R package for the dif-ferential analyses and functional interpretation of ATAC-seq data. BioRxiv. 10.1101/2021.03.05.434143. [DOI] [Google Scholar]
- 109.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ready-to-Use Curated Gene Sets for “cinaR” [R package cinaRgenesets version 0.1.1] (2021). https://cran.r-project.org/web/packages/cinaRgenesets/index.html.
- 111.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li Z, Schulz MH, Look T, Begemann M, Zenke M, and Costa IG (2019). Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 20, 45. 10.1186/s13059-019-1642-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kurtenbach S, and Harbour JW (2019). SparK: A Publication-quality NGS Visualization Tool. BioRxiv. 10.1101/845529. [DOI] [Google Scholar]
- 114.Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mölder F, Jablonski KP, Letcher B, Hall MB, Tomkins-Tinch CH, Sochat V, Forster J, Lee S, Twardziok SO, Kanitz A, et al. (2021). Sustainable data analysis with Snakemake. F1000Res. 10, 33. 10.12688/f1000research.29032.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.How to Cite these Guidelines | COVID-19 Treatment Guidelines. https://www.covid19treatmentguidelines.nih.gov/how-to-cite/.
- 119.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Khanolkar A, Hartwig SM, Haag BA, Meyerholz DK, Epping LL, Haring JS, Varga SM, and Harty JT (2009). Protective and pathologic roles of the immune response to mouse hepatitis virus type 1: implications for severe acute respiratory syndrome. J. Virol. 83, 9258–9272. 10.1128/JVI.00355-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Racine-Brzostek SE, Yee JK, Sukhu A, Qiu Y, Rand S, Barone PD, Hao Y, Yang HS, Meng QH, Apple FS, et al. (2021). Rapid, robust, and sustainable antibody responses to mRNA COVID-19 vaccine in convalescent COVID-19 individuals. JCI Insight. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978. 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li H, and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T. (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–91. 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lawlor N, Nehar-Belaid D, Grassmann JDS, Stoeckius M, Smibert P, Stitzel ML, Pascual V, Banchereau J, Williams A, and Ucar D. (2021). Single Cell Analysis of Blood Mononuclear Cells Stimulated Through Either LPS or Anti-CD3 and Anti-CD28. Front. Immunol. 12, 636720. 10.3389/fimmu.2021.636720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Summary table of samples used for different analyses.
(B) Swimmer plot showing hospitalization periods, and sample collection time point for each patient in nonCOV, Early, and Late. Early–ICU group is colored with dark red, indicating period of ICU admission.
(C) Viral S protein-specific antibody assays showing antibody level, neutralizing activity, and binding quality. (ANOVA, p*<0.05)
(D-E) A heatmap (D) and boxplots (E) showing plasma cytokine levels measured by Luminex platform. Cytokine level per study participant grouped by group is shown as boxplots (*p<0.05, Wilcoxon’s test, Healthy-reference). For heatmap, median cytokine level per group was normalized by Z-score. (t-test, p *<0.05)
(F) Flow cytometry results showing frequencies of CD14+ monocytes, CD34+ HSPC, and monocyte subsets across groups. (ANOVA, * p<0.05)
(A) GSEA of PC1- and PC2-associated DAR from PCA analysis in CD14+ monocytes. setSize indicate number of DAR-associated genes in each GO term. NES stands for normalized enrichment score, and positive NES shows enrichment of DAR-associated genes with positive PC values (same applies to negative NES and PC value).
(B) Volcano plots showing DEG in CD14+ monocytes between Healthy and Early/Late/nonCov groups.
(C) Volcano plots displaying DORC activity in CD14+ monocyte between the Healthy and Early/Late groups.
(D) GO analysis of significantly upregulated genes (DEG, blue dots), and chromatin accessibility (DORC, grey dots) in CD14+ monocytes of each clinical group compared to Healthy.
(E) GSEA comparing between Early and Late CD14+ monocytes.
(F) Violin plot showing the inflammation score for each clinical group, as obtained from the expression profiles of inflammatory gene sets.
(G) GO analysis of significantly upregulated genes (DEG, blue dots), and chromatin accessibility (DORC, grey dots) in each subcluster of CD14+ monocytes.
(H) Dot plot showing module score of monocyte subtype marker genes fetched from previous study62 in each myeloid subclusters.
(A) Boxplot showing the concentration of cytokines in CD14 + monocytes at 24hrs post-stimulation with R848 and IFNα. Concentrations were measured using a Legendplex 13-plex panel for human viral response.
(B) Volcano plots showing DEGs in CD14 + monocytes after 6 hours of stimulation with R848 and IFNα between the Healthy and post-COVID groups.
(C) Heatmap showing the expression fold change of selected DEGs in CD14 + monocytes between stimulated (R848 and IFNα) and unstimulated groups at the individual level. The Early and Late subgroups in the post-COVID group are annotated on the right.
(D) Genome browser tracks displaying the DORC-associated region linked to the CCL5 gene in CD14 monocytes from healthy and post-COVID groups. Peaks associated with DORC are highlighted and connected to the CCL5 gene body with loops (top).
(E) A box plot showing the comparison of the average CCL5 DORC score of each study participant across cohorts.
(F) Boxplot showing the fold change of module score of DAR C2- and C4-associated genes (from Figure 1) between unstimulated and 6 hours of stimulation in CD14 + monocytes. We observed that gene expression modules for genes annotated to both persistently and transiently accessible DAR clusters exhibited greater induction in post-COVID monocytes compared to healthy monocytes.
(A) UMAP plots displaying unbiased clustering (top) and group distribution (bottom) using snRNA-seq (left) and snATAC-seq (right).
(B) Count matrix showing number of DEGs in various comparisons across cell types.
(C) Stacked bar plot showing cell type composition in each donor.
(D) Boxplots showing percent of major cell type within total PBMC per study participant. (Wilcoxon’s test, * p < 0.05; Healthy-reference)
(E) Schematic describing workflow for analyzing distal regulatory elements and gene expression using snRNA/ATAC-seq data including DORC. ATAC-seq peaks were linked to putative target genes in cis, based on the co-variation of chromatin accessibility and gene expression levels across individual cells developmentally related HSPC and monocytes were then co-clustered based on DORC scores.
(F) Genome track showing chromatin accessibility of CEBPA-associated DORC in HSC/MPP and CD14+ monocytes. Curved lines (loops) above indicate peaks with significant correlation (p-value < 0.05). The grey bar (below gene track) represents peaks.
(G) The number of significant peak-gene connections for all genes.
(H) The number of significantly correlated peaks (p-value < 0.05) for each gene.
(I) Heatmap showing expression of marker genes for each HSPC subcluster.
(J) Unbiased clustering of HSPC UMAP of snRNA-seq data.
(K) UMAP plot of HSPC with annotation transferred from BM single-cell RNA-seq data65.
(L) UMAP plot with subcluster annotation guided by ATAC-seq data of FACS-sorted BM HSPC subpopulations 64.
(M) UMAP plot showing density of CD164-expressing cells in HSPC.
(A) GSEA result of PC1- and PC2-associated DAR from PCA analysis in HSPC. setSize indicate number of DAR-associated genes in each GO term. NES stands for normalized enrichment score, and positive NES shows enrichment of DAR-associated genes with positive PC values (same applies to negative NES and PC value).
(B) Volcano plots showing DEGs in HSPC between Healthy and Early/Late groups. (FDR < 0.05)
(C) Heatmap showing DEGs associated with hematopoietic regulation in different groups. Genes that are discussed in the main text are bolded. (FDR < 0.05)
(D) Box plots showing average expression of select genes per study participant. Genes related to myeloid differentiation are selected from DEGs in Late. (Wilcoxon’s test, * p < 0.05, Healthy-reference)
(E) Projections of group density on the snRNA-seq HSPC UMAP plot.
(F) Dot plot showing average GMP module score in each group.
(A) Volcano plots showing differentially active TFs in HSPC and CD14+ monocytes between Healthy and nonCov. (Wilcoxon’s test, * adjusted p < 0.05, Healthy-reference)
(B) Boxplots for mean chromVAR score of select TFs in HSPC and CD14+ monocytes for study participant.(Wilcoxon’s test, * p < 0.05, Healthy-reference)
(C) TF footprints and violin plots for TF motif-associated chromatin accessibility (chromVAR) activity in HSPC and CD14+ monocytes across groups. (Wilcoxon’s test, * p < 0.05, Healthy-reference).
(A) Dumbbell plot displaying the difference in the average frequency of GMP within HSPC of the Late between two groups, categorized by various clinical parameters. The p-values, obtained using the t-test, are shown along with their adjusted p-values.
(B) GSEA result showing the biological processes that are enriched in HSC/MPP of Late with αIL-6R and Late without αIL-6R.
(C) Weight recovery rate of A/J mice with MHV-1 infection over time (n=7, naïve; n=5, recovered; n=8, aIL-6; top). Viral titers (plaque-forming unit per ul) at day 4 and 30 post-infection (n=10, naïve; n=12, recovered; n=7, αIL-6R; bottom).
(D-E) Dot plots showing the expression of marker genes for different mouse HSPC subtypes (C) and cell types in BALF.
(F) UMAP visualization of the BALF snRNA/ATAC-seq dataset with cell type annotations.
(G) Dot plot showing the expression of marker genes BALF macrophage subclusters.
(H) Heatmap representing the row-normalized average chromVAR scores across different groups of macrophage clusters.
(I) Heatmap representing the row-normalized average chromVAR score for marker TFs in BALF macrophage.
(J) Volcano plots showing the DEGs in macrophages between the anti-IL-6R blockade and recovered groups. (Wilcoxon’s test, * p < 0.05, Recovered-reference).
(K) GSEA showing the biological processes that are enriched in the recovered group compared to the anti-IL-6R blockade group.
(L) Representative images from H&E staining of mouse lung in different groups.
(M) This bar plot shows the fold change in average density of selected hematopoietic cell types from the ROI in lung tissue between post-COVID and healthy samples. Asterisks indicate hematopoietic cell types with significantly different cell densities per ROI between the two groups. (Wilcoxon’s test, * p < 0.05, Healthy-reference)
(N) The box plot displays the average intensity of IBA-1 and GFAP staining per ROI and mouse across groups. Intensity per ROI is represented by small transparent dots, while larger dots indicate the average intensity per mouse. (Wilcoxon’s test, * p < 0.05; Recovered-reference)
Table S1. Demographic, Clinical and QC Data, Related to STAR Methods
Table S2. Cell type-defining marker genes, Related to STAR Methods
Table S7. Legendplex data and normalized gene count matrix of select DEG from monocyte stimulation experiment, Related to Figure 2
Data Availability Statement
All raw and processed data are deposited on GEO database under accession number GSE196990. All other data types used for analysis in this research are present in the manuscript or the Supplementary Materials. We created a web-based interface to query all multiome data of HSPC, myeloid, and whole PBMC for DORC, expression, and transcription factor activities (https://buenrostrolab.shinyapps.io/covid_myeloid/). (https://buenrostrolab.shinyapps.io/covid_pbmc/, https://buenrostrolab.shinyapps.io/covid_hspc/, https://buenrostrolab.shinyapps.io/covid_myeloid/) The code used for the downstream analysis is available in github repository (https://github.com/sharmasiddhartha231/Final_Covid19_Scripts, 10.5281/zenodo.8097411). The codes for visualizations are available upon request to the lead contact. De-identified clinical cohort information used in this study is available in supplementary table. Any publicly available data that we reanalyzed are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. Additional Supplemental Items are available from Mendeley Data at http://dx.doi.org/10.17632/pfwyrmffdp.1.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| 7-AAD | Biolegend | Cat#420404 | |
| CD34-microbeads | Miltenyi | Cat#130-046-702 | |
| CD34-FITC | Miltenyi | Cat#130-113-178 | |
| CD49f-Pacific Blue | Biolegend | Cat#313620 | |
| CD90-PE | Biolegend | Cat#328110 | |
| CD38-PE/cy7 | Biolegend | Cat#303516 | |
| CD45RA-APC/cy7 | Biolegend | Cat#304128 | |
| CD20-Biotin | Biolegend | Cat#302350 | |
| CD56-Biotin | Biolegend | Cat#362536 | |
| CD14-Biotin | Biolegend | Cat#301826 | |
| Streptavidin-BV605 | BD | Cat#563260 | |
| CD14-APC | BD | Cat#340436 | |
| CD8-FITC | Biolegend | Cat#300906 | |
| CD14-microbeads | Miltenyi | Cat#130-050-201 | |
| Mouse lineage depletion kit | Miltenyi | Cat#130-090-858 | |
| FcX | Biolegend | Cat#156604 | |
| Ly6g-BUV395 | BD | Cat#563978 | |
| MHCII-BV421 | ThermoFisher | Cat#404-5321-82 | |
| CD11c-A488 | ThermoFisher | Cat#53-0114-82 | |
| CD86-PE | Biolegend | Cat#105105 | |
| CD206-BV605 | Biolegend | Cat#141721 | |
| CD4 PE-CF594 | BD | Cat#562285 | |
| CD19-APC | Thermofisher | Cat#17-0193-82 | |
| B220-APC | ThermoFisher | Cat#17-0452-82 | |
| CD45-A700 | Biolegend | Cat#103127 | |
| Ly6c-PerCP-Cy5.5 | Thermofisher | Cat#45-5932-82 | |
| CX3CR1-BV786 | Biolegend | Cat#149029 | |
| CD8-PE-Cy7 | Thermofisher | Cat#25-0081-82 | |
| CD11b-APC-e780 | ThermoFisher | Cat#47-0112-82 | |
| Aqua Live/Dead Viability dye | ThermoFisher | Cat#L34966 | |
| normal donkey serum | Jackson Immunoresearch | RRID: AB_2337258 | |
| Iba1 | Fujifilm | Cat#019-19741 | |
| GFAP | Cell Signaling Technologies | Cat#3657 | |
| MBP | Abcam | Cat#ab40390 | |
| anti-IgG secondary antibodies conjugated to A488 | Jackson Immunoresearch | RRID: AB_2338449 | |
| anti-IgG secondary antibodies conjugated to A594 | Jackson Immunoresearch | RRID: AB_2338873 | |
| anti-IgG secondary antibodies conjugated to A647 | Jackson Immunoresearch | RRID: AB_2338904 | |
| DAPI | Prolong Diamond Antifade | Cat#P36962 | |
| Bacterial and virus strains | |||
| MHV-1 | ATCC | VR-261 | |
| Biological samples | |||
| Paired PBMC and BMMC | AllCells | N/A | |
| Whole blood from study participants | Weill-Cornell Medicine/New York-Presbyterian Hospital | N/A | |
| Chemicals, peptides, and recombinant proteins | |||
| R848 | InvivoGen | Cat#tlrl-r848 | |
| mouse anti-IL-6R blocker | InVivoMab | Cat#BE0047 | |
| IFNα | PBL assay science | Cat#12100 | |
| RPMI medium | Corning | Cat#10-040-CM | |
| Ficoll-Paque PLUS | GE | Cat#17144002 | |
| Collagenase D | Roche | Cat#11088858001 | |
| Critical commercial assays | |||
| Chromium Controller & Next GEM Accessory Kit | 10x Genomics | Cat#1000202 | |
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle | 10x Genomics | Cat#1000285 | |
| NEBNext low input RNA library prep kit for Illumina | NEB | Cat#E6420S | |
| LEGENDplex Human anti-virus response panel (13-plex) | Biolegend | Cat#740390 | |
| 15-plex human pro-inflammatory cytokine assay | Eve Technologies | N/A | |
| TOP-Plus (Pylon 3D analyzer) for antibody level measurement | ET Healthcare | N/A | |
| Lung histology | HistoWiz | N/A | |
| Deposited data | |||
| Imaging mass cytometry | Rendeiro et al.70 | https://doi.org/10.5281/zenodo.4110560 | |
| GRCh38 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.26/ | |
| GRCm38 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/ | |
| Single-cell RNA-seq of human bone marrow | Granja et al.95 | GSE139369 | |
| ATAC-seq data of human HSPC subpopulation | Buenrostro et al.60 | GSE96772 | |
| Experimental models: Organisms/strains | |||
| A/J mouse | The Jackson Laboratory | Cat#000646 | |
| Software and algorithms | |||
| Cell Ranger ARC 1.0.0 | 10X Genomics | https://support.10xgenomics.com/single-cell-multiome-atac-gex/software/downloads/latest | |
| Signac | Stuart et al.92 | https://stuartlab.org/signac/ | |
| Amulet | Thibodeau et al.93 | https://github.com/UcarLab/AMULET | |
| Seurat | Hao et al.94 | https://github.com/satijalab/seurat | |
| Scrublet | Wolock et al.95 | https://github.com/swolock/scrublet | |
| Harmony | Korsunsky et al.96 | https://github.com/immunogenomics/harmony | |
| MACS2 2.1.2 | Zhang et al.97 | https://github.com/macs3-project/MACS/wiki/Install-macs2 | |
| ArchR | Granja et al.98 | https://github.com/GreenleafLab/ArchR | |
| FigR | Kartha et al.99 | https://github.com/buenrostrolab/FigR | |
| Nebulosa | Alquicira-Hernandez et al.100 | https://github.com/powellgenomicslab/Nebulosa | |
| trimmomatic | Bolger et al.101 | https://github.com/timflutre/trimmomatic | |
| BWA | Li et al.102 | https://github.com/lh3/bwa | |
| Samtools | Li et al.103 | https://github.com/samtools | |
| Subread package | Liao et al.104 | https://subread.sourceforge.net/ | |
| IGV | James et al.105 | https://software.broadinstitute.org/software/igv/download | |
| snATACClusteringTools | Ucar Lab | https://github.com/UcarLab/snATACClusteringTools | |
| DiffBind | Stark et al.106 | https://hbctraining.github.io/Intro-to-ChIPseq/lessons/08_diffbind_differential_peaks.html | |
| ChipSeeker | Yu et al.107 | https://github.com/YuLab-SMU/ChIPseeker | |
| EdgeR | Robinson et al.108,109 | https://github.com/StoreyLab/edge | |
| cinaR | Karakaslar et al.110 | https://github.com/eonurk/cinaR | |
| HOMER 4.11 | Heinz et al.111 | http://homer.ucsd.edu/homer/ | |
| HINT | Gusmao et al.112 | https://github.com/CostaLab/reg-gen | |
| SparK | Kurtenbach et al.113 | https://github.com/harbourlab/SparK | |
| ClusterProfiler | Yu et al.114 | https://github.com/YuLab-SMU/clusterProfiler | |
| STAR 2.7.10b | Dobin et al.115 | https://github.com/alexdobin/STAR | |
| DESeq2 | Love et al.116 | https://git.bioconductor.org/packages/DESeq2 | |
| Snakemake | Mölder et al.117 | https://snakemake.readthedocs.io/en/stable/ | |
| Other | |||
| EDTA tubes | BD | Cat#366643 | |
| Heparin tubes | BD | Cat#368480 | |
| NovaSeq6000 | Illumina | N/A | |
| Zeiss LSM 900 | Carl Zeiss Microscopy | N/A | |
| Objective Plan-Apochromat 20x/0.8 M27 | Carl Zeiss Microscopy | N/A | |