

Abstract
All microbial biofilms are initiated through direct physical contact between a bacterium and a solid surface, a step that is controlled by inter- and intramolecular forces. Atomic force microscopy and confocal laser scanning microscopy were used simultaneously to observe the formation of a bond between a fluorescent chimeric protein on the surface of a living Escherichia coli bacterium and a solid substrate in situ. The chimera was composed of a portion of outer membrane protein A (OmpA) fused to the cyan-fluorescent protein AmCyan. Sucrose gradient centrifugation and fluorescent confocal slices through bacteria demonstrated that the chimeric protein was targeted and anchored to the external cell surface. The wormlike chain theory predicted that this protein should exhibit a nonlinear force-extension “signature” consistent with the sequential unraveling of the AmCyan and OmpA domains. Experimentally measured force-extension curves revealed a unique pair of “sawtooth” features that were present when a bond formed between a silicon nitride surface (atomic force microscopy tip) and E. coli cells expressing the OmpA-AmCyan protein. The observed sawtooth pair closely matched the wormlike chain model prediction for the mechanical unfolding of the AmCyan and OmpA substructures in series. These sawteeth disappeared from the measured force-extension curves when cells were treated with proteinase K. Furthermore, these unique sawteeth were absent for a mutant stain of E. coli incapable of expressing the AmCyan protein on its outer surface. Together, these data show that specific proteins exhibit unique force signatures characteristic of the bond that is formed between a living bacterium and another surface.
A nearly universal trait of bacteria is their ability to attach to solid surfaces. Attached cells often form a biofilm, which is defined as a structured community of bacteria enclosed in a self-produced polymeric matrix adherent to an inert or living surface (6). Abundant evidence suggests that proteins on the outer wall of a bacterium play a critical role in initiating contact with another surface. Surface-induced expression of genes coding for various adhesins (e.g., flagella, pili, and fimbriae) have been characterized in the literature (28, 39, 45, 48, 51, 54). Many of these studies rely on the mutation of genes of interest followed by a comparative analysis of the number of cells (wild type versus mutant) that are attached to a solid substrate. While these types of studies provide detailed characterizations of the proteins involved in mediating contact with another surface, the inherent surface-sensing mechanism remains hidden. The actual mechanism that allows a bacterium to sense or perceive another surface is the physical force(s) that exists in the nanometer-scale interface between a cell and that other surface. This type of elementary force, which includes van der Waals, electrostatic, solvation, and steric interactions (21, 22, 26), controls the way in which a protein on a bacterium's cell wall physically touches another surface.
Recently, we used atomic force microscopy to quantitatively probe this form of sensory perception between living bacterial cells and inert surfaces (24, 30, 31). Initial results suggest that bacteria possess the natural ability to modulate intermolecular forces experienced by the cell through the expression of different cell envelope proteins which mediate contact with inorganic surfaces (29). The present work was undertaken to better understand this phenomena by collecting force measurements with a strain of Escherichia coli engineered to express a fluorescent protein on its outer membrane with the well-characterized Lpp-OmpA display system (15). This display system is composed of a fusion protein consisting of the signal sequence and first nine N-terminal amino acids of the mature major E. coli lipoprotein (Lpp) and amino acids 46 to 159 of E. coli outer membrane protein A (OmpA), which contains five membrane-spanning β-sheets with the C terminus exposed to the external side of the outer membrane (15, 16).
Here we have fused the cyan-fluorescent protein AmCyan (32) to the C terminus of Lpp-OmpA and demonstrated that this chimeric protein (Lpp-OmpA-AmCyan) is targeted and anchored to the external cell surface of E. coli. The wormlike chain theory was used to model the theoretical force-extension profile of this chimeric protein when it formed a bond with a solid surface. Comparing the theoretical results with simultaneous force and fluorescence measurements revealed that a specific force signature could be attributed to the formation of a bond between the chimeric protein on a living E. coli cell and a solid substrate in situ. This work demonstrates that specific proteins at the cell-material interface may exhibit unique “force signatures” characteristic of their identity. Furthermore, the simultaneous force and fluorescence measurements provide a novel, in situ perspective of bacterial adhesion that is highly complementary to other methods, such as the use of mutants and tagged proteins, which may also be employed to probe the initial stages of biofilm formation.
MATERIALS AND METHODS
E. coli strains and plasmids.
E. coli TOP10 (Invitrogen, Carlsbad, Calif.) cells were used as a recombinant host for cloning and plasmid propagation, while E. coli strain BL21 Star(DE3) (Invitrogen) was used as a host for expression and surface display of Lpp-OmpA-AmCyan. C. F. Earhart from the University of Texas at Austin graciously provided pTX215 (49), which served as the source of the lpp-ompA gene fusion. The pTX215 plasmid encodes an Lpp-OmpA hybrid protein consisting of the signal sequence and first nine N-terminal amino acids of Lpp, amino acids 46 to 159 of OmpA, and a 13-amino-acid linker segment with the sequence GINSSSVPGDPPW (49). The source of the fluorescent protein gene was pAmCyan (BD Bioscience, San Jose, Calif.), which encodes a variant of the wild-type Anemonia majano cyan-fluorescent protein AmCyan that has been engineered with enhanced emission characteristics (32). The pDNRDual donor vector (BD Bioscience) was used in the cloning procedures, and the pET101DTOPO expression vector (Invitrogen) was used to express the recombinant proteins in E. coli.
E. coli strains bearing plasmids were grown in Luria-Bertani (LB) medium containing 100 μg of ampicillin, except for E. coli bearing pTX215, which was grown in LB medium containing 150 μg of kanamycin per liter. Isopropyl-β-d-thiogalactopyranoside (IPTG; 1 mM) was used as an inducer of gene expression in recombinant bacteria. Cells were grown at 25 to 37°C in an environmental shaker at 200 to 250 rpm.
Construction of Lpp-OmpA-AmCyan fusion protein.
Figure 1 provides a flow diagram summarizing the cloning procedures used in this study. PCR was used to amplify lpp-ompA with pTX215 as the template. The sequences of the forward and reverse primers were 5′-GAAGTTATCAGTCGACATGAAAGCTACTAAACTG-3′ and 5′-ATGGTCTAGAAAGCTTCCATGGGGGATCCCC-3′, respectively. The forward primer was designed with a unique SalI site, while the reverse primer was designed with a unique HindIII site. PCR was carried out for 30 15-s cycles at 92°C, 30 s at 55°C, and 1 min at 72°C with Pfu Turbo DNA polymerase (Stratagene, La Jolla, Calif.). The resulting ≈0.5-kbp PCR product was ligated into the unique SalI and HindIII sites of linearized pDNR-Dual donor vector (BD Biosciences) with the BD In-Fusion Dry-Down PCR cloning kit (BD Biosciences) to make pDNR-Dual/lpp-ompA. Concurrently, PCR was used to amplify amCyan with pAmCyan (BD Bioscience) as the template and the forward and reverse primers 5′-CGGTACCCGGGGATCCCATGGCTCTTTCAAACAAG-3′ and 5′-ATGGTCTAGAAAGCTTTCAGAAAGGGACAACAGA-3′, respectively. The forward primer was designed with a BamHI site, while the reverse primer was designed with a HindIII site. PCR was carried out as described above, resulting in a ≈0.7-kbp product.
FIG. 1.

(A) Flow diagram of the construction of pET101DTOPO/lpp-ompA/Amcyan (used in E. coli Lpp-OmpA-AmCyan). See Materials and Methods for a description. Highlighted is a genetic map of pET101DTOPO/lpp-ompA/Amcyan. Shown across the top are the nucleotide (lowercase) and DNA-derived amino acid (one-letter abbreviation) sequences of Lpp-OmpA-AmCyan. The nucleotide sequence of the expression vector pET101DTOPO is shown in italics; the amino acid sequences of Lpp, OmpA, and AmCyan are in boxes; and the nucleotide and amino acid sequence of the linkers are designated by a line above them. Amp, ampicillin resistance gene; ori, origin of replication; lacI, lactose repressor; RBS, ribosome-binding site; T7, promoter recognized by T7 RNA polymerase. (B) Flow diagram of the construction of pET101DTOPO/Amcyan (used in the control strain of E. coli).
The amCyan PCR product was inserted in frame with and downstream of lpp-ompA by digesting pDNR-Dual/lpp-ompA with BamHI and HindIII and then ligating the amCyan PCR product into the unique BamHI and HindIII sites of the linearized plasmid pDNR-Dual/lpp-ompA. The ligation step was completed with the BD In-Fusion Dry-Down PCR cloning kit (BD Biosciences) to give pDNR-Dual/lpp-ompA/AmCyan. This plasmid was used to transform E. coli strain TOP10 (Invitrogen). The transformed cells were cultured overnight on LB-ampicillin medium, and the plasmid was isolated therefrom. DNA sequence analysis of the cloned DNA was performed to verify the fidelity of the PCR amplification and confirm that each part of the fusion gene was in frame.
Since pDNR-Dual/lpp-ompA/AmCyan is not designed to be used as an expression vector, the lpp-ompA-amCyan gene fusion had to be cloned into the expression vector pET101DTOPO (Invitrogen), which allows expression of recombinant protein with a native N terminus. Briefly, lpp-ompA-amcyan was amplified by PCR as described above with plasmid pDNR-Dual/lpp-ompA/AmCyan as the template and the forward and reverse primers 5′-CACCATGAAAGCTACTAAACTGGTACTG-3′ and 5′-TCAGAAAGGGACAACAGAG-3′, respectively. The resulting ≈1.2-kbp PCR product was ligated into vector pET101DTOPO (Invitrogen) according to the manufacturer's directions. The resulting plasmid, designated pET101DTOPO/lpp-ompA/AmCyan (see highlighted diagram in Fig. 1A), was sequenced to verify the fidelity of the PCR amplification.
Plasmid pET101DTOPO/AmCyan was also constructed as described above with the exception that lpp-ompA was not cloned into it (see Fig. 1B). This plasmid was used as a control since it encodes AmCyan but not the signal sequence and outer membrane domain Lpp-OmpA, and therefore the fluorescent protein should be expressed as a soluble recombinant protein within the cell and not on the surface of E. coli.
Recombinant expression of Lpp-OmpA-AmCyan and growth of cells for force and fluorescence microscopy.
Recombinant expression of Lpp-OmpA-AmCyan as well as AmCyan alone was carried out with E. coli strain BL21 Star(DE3) (Invitrogen). E. coli was transformed with the expression plasmids described above, and expression of recombinant protein was performed with a TOPO TA cloning kit (Invitrogen) as per the manufacturer's instructions. Throughout the text, E. coli containing pET101DTOPO/lpp-ompA/AmCyan will be referred to as Lpp-OmpA-AmCyan, whereas E. coli containing pET101DTOPO/AmCyan will be called the control strain.
The E. coli Lpp-OmpA-AmCyan and control strains were cultured in 100 to 200 ml of LB-ampicillin broth. IPTG (1 mM) was added to the cell-broth mixture when the absorbance reached 0.5 to 1.0 (A600). At an absorbance of 1.0 to 1.5 (A600), the cells were harvested by spinning at 5 000×g for 3 min. Harvested cells were used immediately in force and fluorescence microscopy measurements (discussed below) or stored at −20°C until protein content was analyzed by sucrose gradient centrifugation (discussed below).
Separation of membranes by sucrose gradient centrifugation.
Sucrose gradient centrifugation was performed as described by Francisco et al. (15) to verify that the Lpp-OmpA-AmCyan protein was localized to the outer membrane of E. coli. Cell pellets from 200-ml cultures of E. coli Lpp-OmpA-AmCyan (or the control strain) were thawed and resuspended in 10 ml of 50 mM Tris base, pH 7.5, containing 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, protease inhibitor cocktail (Sigma), and 100 μg of lysozyme per ml, and incubated on ice for 10 min. The cells were then lysed by two passages through a French pressure cell at 12,000 lb/in2. The lysate was centrifuged at 2,500 × g for 10 min at 4°C to remove debris and any remaining whole cells. The membrane fraction was separated from the soluble fraction by centrifugation at 168,000 × g for 1 h at 4°C in a Beckman type 50.2Ti rotor. The membrane fraction was washed, resuspended in 0.8 ml of 25 mM Tris base, pH 7.5, containing 25% (wt/wt) sucrose and loaded onto a step gradient of 4 ml each of 30, 35, 40, 45, 50, and 55% (wt/wt) sucrose in the same buffer. Membranes were centrifuged at 170,000 × g for 24 h at 4°C in a Beckman type 50.2Ti rotor, and 0.5-ml fractions were collected from the bottom of the tube. Buoyant densities (ρ) were determined from refractive index measurements. Because of the visible fluorescence of AmCyan, fractions containing recombinant protein were readily identified with a hand-held lamp that emitted short, visible wavelengths of light.
Preparation of E. coli cells for simultaneous force and fluorescence measurements.
Cells harvested from 200-ml cultures of the Lpp-OmpA-AmCyan strain and control strains of E. coli were washed three times in 50 ml of sterile 0.1 M NaCl. The final washed cell pellet was resuspended in less than 1 ml of sterile 0.1 M NaCl and then blotted onto a hydrophobic glass coverslip with a sterile swab. After several minutes, unattached cells were rinsed from the coverslip with a sterile stream of 0.1 M NaCl. These preparation steps were carried out in a class II biological safety cabinet (Labconco, Kansas City, Mo.).
Hydrophobic coverslips, mentioned above, were created with a self-assembling silane compound called octadecyltrichlorosilane (Sigma-Aldrich). Glass coverslips (#1.5 type; 24 mm by 50 mm) were soaked overnight in piranha solution (1:1 volume ratio of 30% hydrogen peroxide and 70% sulfuric acid [27]) to remove any proprietary coating. These slides were then rinsed in MilliQ water (18.2 MΩ cm), soaked in a fresh piranha solution for a second night, rinsed 10 times in MilliQ water, and finally dried with a stream of purified nitrogen gas. The hydrophobic coating was formed by soaking the coverslips overnight in a 1% solution (by volume) of octadecyltrichlorosilane (in 250 ml of toluene with 0.5 ml of butylamine). The coverslips were rinsed in toluene three times and dried with nitrogen gas.
Force microscopy and confocal laser scanning microscopy.
An integrated atomic force microscope (Veeco/Digital Instruments; Bioscope atomic force microscope with NanoScope IV controller)-confocal laser scanning microscope (Zeiss; Axiovert 200M and LSM 510 Meta) was used to collect simultaneous fluorescence and force measurements on E. coli cells. E. coli cells were located on a coverslip with transmitted light and a 100X/1.45 N.A. objective (α-Plan-Fluar) on the confocal laser scanning microscope. Fluorescence images were collected by exciting cells with a 458/488-nm argon laser and collecting the emission on a photodiode detector after passing the emitted light through a 505-nm long-pass filter.
A detailed description of the use of atomic force microscopy to measure forces between a bacterium and another surface can be found in other publications (24, 30, 31). The following is a brief discussion of how we used atomic force microscopy to measure forces. A silicon nitride cantilever (with an integrated tip) was positioned directly over a monolayer patch of E. coli cells. Force measurements began within 30 to 45 min of the initial harvesting of cells. The tip of a cantilever was brought into contact with a cell (approach force curves) and then pulled from the cell surface (retraction force curves). The velocity of the cantilever was 0.8 to 1.6 μm s−1 with < 0.5 s of contact time between the tip and a cell. The raw data were collected as the output of the photodiode detector (which is directly proportional to the deflection of the cantilever) as a function of the position of the tip, which is translated by a piezoelectric scanner. These raw data were plotted as so-called voltage displacement curves and then converted into force-distance curves according to a well-established protocol (8, 9). The force-distance curves were analyzed with SPIP (Image Metrology) and Igor Pro (WaveMetrics) software. Only retraction curves are shown.
Approximately 10% of the retraction force-distance (or force-extension) curves between E. coli and the silicon nitride tip displayed unique “sawtooth”-like features attributed to the extension of cell surface biopolymers that formed an attractive bond or “bridge” with the tip. These retraction curves were compared to two different theories (see equations 1 and 2 below), which describe the force necessary to extend a linear polymer (e.g., a protein) a certain distance.
To ensure a valid comparison of the observed and predicted force-distance relationships, we measured the spring constant of the cantilevers and also calibrated the movement of the z-piezoelectric scanner used in these experiments. The cantilever spring constant was determined to be 0.02 N m−1 with the hydrodynamic drag method of Craig and Neto (7). Craig and Neto have shown that their method yields spring constant values consistent with the Cleveland method (5), which is one of the most widely used methods for determining the spring constant of an atomic force microscopy cantilever.
The z-piezoelectric scanner was calibrated for accuracy in the 25- to 100-nm range with two National Institute of Standards and Technology (NIST) certified calibration gratings (MikroMasch, Portland, Oreg.): TGZ01C (26.4 ± 0.6 nm, traceable to NIST 821/261141-99) and TGZ02C (102.3 nm ± 1.4 nm, traceable to NIST 821/261141-99). These standards are composed of one-dimensional arrays of rectangular SiO2 steps on an Si wafer. Standards TGZ01C and TGZ02C were measured to be 31.3 ± 0.3 nm and 120.2 ± 0.8 nm, respectively. Averaging the correction factors obtained from these two measurements and propagation of errors yields a divisional correction factor of 1.18 ± 0.02. All raw piezo displacement values were corrected, yielding results with 2% uncertainty (the uncertainty in piezo linearity). It should be noted that the uncertainty of atomic force microscopy force data (force and distance values) is also impacted by the background “noise” in the force curves due to electrical and mechanical noise and thermal fluctuations.
RESULTS AND DISCUSSION
Theoretical force-extension profile of a protein that forms a bond with a solid surface.
The mechanical topology of a linear polymer, such as a protein, can be described by the wormlike chain model, where force (F) is predicted as a function of the distance (x) the polymer is stretched or extended (12). At zero force, polymer chains exist in a coiled state, as this maximizes their conformational freedom or entropy (10). Extending a relaxed polymer generates an opposing force predicted from the reduction of entropy. The wormlike chain model describes the force-extension relationship according to the following equation (1, 34, 35, 41):
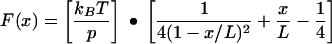 |
(1) |
where p is persistence length (in meters), L is contour length (in meters), kB is Boltzmann's constant (1.381 × 10−23 J K−1), and T is temperature (in degrees Kelvin). The persistence length (p) is a measure of the distance over which a polymer retains the memory of a given direction. As such, the persistence length embodies the stiffness or elasticity of a polymer. Using force microscopy and optical tweezers, others have determined the persistence length of single, purified protein molecules to range between 0.3 and 1.0 nm (4, 13, 23, 34, 35, 37, 38, 42, 50). It is significant that this length is approximately equivalent to the 0.38 nm between α carbon atoms in a polypeptide chain (34). The contour length (L) of a protein can be calculated as the product of the size of an amino acid (0.38 nm) and the total number of amino acids in the protein's primary structure (4, 29, 38).
There is also another theory, called the freely joined chain, model that is commonly used to describe the force-extension profile of linear polymers such as proteins (or nucleic acids). The freely joined chain equation (10, 17, 43, 47) is
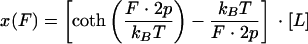 |
(2) |
where the parameters are as defined above for the wormlike chain model. For the freely joined chain model, the elementary segment length, called the Kuhn length, is equal to 2p (18, 20).
Equations 1 and 2 can therefore be used to predict the force topology or “force signature” of a protein for which only the primary structure (number of amino acids) is known. Figure 2A (see dotted black curves) shows the predicted wormlike chain force-extension profiles for three polypeptides, each composed of a different number of amino acids (different contour length L). The persistence length (p) is assumed to be 0.38 nm (the basic length scale of an amino acid) for these theoretical curves. Figures 2B and 2C present a comparison of the wormlike chain and freely joined chain models for two polypeptides of different sizes (L).
FIG. 2.

Theoretical and measured force-extension curves. All forces are attractive and take a negative sign by convention. The dotted and dashed black lines correspond to the theoretical force-extension relationship for a polypeptide of a given size as predicted by the wormlike chain (WLC) or freely joined chain (FJC) model (see equations 1 and 2). The persistence length (p) was taken as 0.38 nm, and the contour length (L) was determined by multiplying the length of a single amino acid residue (0.38 nm) by the total number of amino acids in the polypeptide (228, 252, or 353). (A) Colored traces correspond to the measured force-extension curves for E. coli Lpp-OmpA-AmCyan cells with a fluorescent AmCyan protein targeted to the external surface of the outer membrane. (B) One retraction curve selected from the colored traces presented in A. (C) Measured retraction curves (“flat” black lines that fluctuate around zero force) for E. coli Lpp-OmpA-AmCyan cells treated with proteinase K. The green curve highlights one of the three sawteeth that were observed out of a total of 1,113 retraction curves. (D) Colored traces correspond to measured force-extension curves for the control strain of E. coli that did not have an AmCyan protein in the outer membrane.
The wormlike chain and freely joined chain profiles for any of these proteins are remarkably similar in that each curve can be described in terms of two regions exhibiting a different relationship between force and extension distance. At low force, each profile displays a more or less linear relationship between force and the extension of the polypeptide. The profile dramatically changes to a nonlinear relationship between force and extension as the polypeptide approaches its fully extended length. The main difference between the wormlike chain and freely joined chain models is that the wormlike chain theory predicts that a polypeptide will exhibit more resistance to extension at low force (see Fig. 2B and 2C).
Others have used atomic force microscopy or optical tweezers to obtain force-extension profiles (sometimes referred to as force spectroscopy) for purified proteins adsorbed to a solid substrate such as glass, mica, or gold-coated coverslips. These include a range of proteins from eukaryotes, prokaryotes, and bacteriophages such as carbonic anhydrase B (53), fibronectin (33, 37), fibronectin binding protein (R. Yongsunthon and S. Lower, unpublished data), hexagonally packed intermediate layer (35), myelin basic protein (34), lysozyme (55), outer membrane cytochrome B (S. Lower and R. Yongsunthon, unpublished data), outer surface protein A (19), P-selectin (17), spectrin (44), and titan (3, 4, 23, 38, 42). In every case, the purified protein (adsorbed to a solid substrate) exhibited a characteristic sawtoothlike force-extension profile, which could be described by the wormlike chain or, to a lesser extent, the freely joined chain theory.
However, it has yet to be demonstrated conclusively whether a protein within a native biological membrane of a living cell also yields a characteristic force signature and whether that signature conforms to the wormlike chain or freely joined chain model. If proteins within a membrane (e.g., phospholipid bilayer or lipopolysaccharide layer) display a unique force-extension signature and/or conform to one or both of these models, this might enhance our ability to identify cell wall proteins in bacteria, for example, that mediate contact with other surfaces. Furthermore, this type of information would provide a fundamental appreciation of the elementary forces and mechanical processes that allow a protein on a bacterium's surface to form a physical bond with another surface.
Engineered E. coli strain that expresses a well-characterized fluorescent protein on the external surface of the outer membrane.
To test this hypothesis, we constructed a well-defined chimeric protein in which a fluorescent domain of the chimera was exposed on the external surface of a model gram-negative bacterium. This protein is a tripartite fusion consisting of the signal sequence and first nine amino acids of the major E. coli lipoprotein (Lpp), residues 46 to 159 of outer membrane protein A (OmpA), and a variant of the entire mature cyan-fluorescent protein (AmCyan) from the reef anemone Anemonia majano. We selected the Lpp-OmpA display system because it has been used successfully in previous studies to display large (>250 amino acids), functionally active proteins on the external surface of gram-negative bacteria (14, 15).
A schematic of the expected conformation of the Lpp-OmpA-AmCyan fusion protein in the outer membrane of E. coli is shown in Fig. 3. The positions of the membrane-spanning domains and loops of OmpA are based on the work of Francisco et al. (15) and Koebnik (25). The secondary structure of the AmCyan protein is based on the crystallographic measurements of Wall et al. (52) and Yarbrough et al. (56). The AmCyan protein is an 11-stranded β-barrel (or β-can) with a central α-helix that houses the chromophore (52, 56). The chromophore is composed of three amino acids that fluoresce due to autocatalytic cyclization of residues 66 through 68 (52, 56). The amino and carboxy termini of AmCyan are fully exposed on the outer ends of the β-barrel (see Fig. 3).
FIG. 3.

Schematic of the expected structure of the Lpp-OmpA-AmCyan fusion in the outer membrane of E. coli. This chimeric fusion is composed of two polypeptide domains in series (a portion of the outer membrane protein A, labeled OmpA, and the entire sequence of the cyan-fluorescent protein AmCyan), which are joined to one another by an 11-amino-acid linker and anchored into the outer membrane by a lipoprotein (Lpp). The total length of the fusion is 384 amino acids. The numbers across the top (353, 252, and 228) correspond to the lengths of the polypeptides described by the wormlike chain and freely joined chain models in Fig. 2. Some amino acid residues are labeled with subscript numbers as reference points (e.g., AmCyan228 for the C terminus). The transmembrane strands (purple) of OmpA are numbered sequentially in parentheses. The N and C termini of AmCyan are highlighted in yellow. Membrane-spanning units and loops (blue) are based on the work of Francisco et al. (15) and Koebnik (25). The structure of the AmCyan protein was created with Cn3D software (available from the National Institutes of Health) based on crystallographic measurements from Wall et al. (52) and Yarbrough et al. (56).
Use of sucrose gradient centrifugation and confocal laser scanning microscopy to verify localization of the fluorescent protein to the outer membrane of E. coli.
The location of the AmCyan protein shown in Fig. 3 was confirmed with sucrose gradient centrifugation. Membrane and soluble fractions from E. coli bearing either pET101DTOPO/lpp-ompA/AmCyan (strain Lpp-OmpA-AmCyan) or pET101DTOPO/AmCyan (control strain expressing cytoplasmic AmCyan alone) were prepared as described in Materials and Methods. Because AmCyan has an emission maximum within the visible light range, its purification was readily followed with a hand-held lamp that emitted light in the shorter visible wavelengths.
For protein samples obtained from the control strain of E. coli, a cyan (bluish-green) fluorescence was observed only in the soluble fraction (consisting of cytoplasmic and periplasmic proteins). No fluorescence was observed in membrane fractions. For protein samples obtained from E. coli Lpp-OmpA-AmCyan, the majority of fluorescence was observed in the membrane fraction, while a modest amount of fluorescence was also observed in the soluble fraction. When sucrose gradient centrifugation was performed on the membrane fraction from E. coli Lpp-OmpA-AmCyan, two discrete bands, L (ρ = 1.16 g/ml) and H (ρ = 1.23 g/ml), were observed. Cyan fluoresce was observed only for band H, which had a buoyant density consistent with previously purified outer membrane fractions of gram-negative bacteria (11, 40).
Confocal laser scanning microscopy was also used to verify the location of AmCyan within the two strains of E. coli used in the atomic force microscopy. Figure 4 shows the fluorescence emission of both the control strain (E. coli expressing AmCyan in the cytoplasm) and the Lpp-OmpA-AmCyan strain. The control strain fluoresced throughout the volume of the cell, consistent with the presence of the AmCyan protein in the cytoplasm (see Fig. 4B). Contrary to this, the fluorescence of some E. coli Lpp-OmpA-AmCyan cells was confined to an outer ring along the cell wall (see Fig. 4A). This is consistent with localization of the AmCyan protein to the outer membrane. Not all E. coli Lpp-OmpA-AmCyan cells exhibited a ring of fluorescence. Many of these cells, like the control strain, exhibited fluorescence throughout the cell. This is to be expected, as some of the translated AmCyan protein likely folds into a native structure within the cytoplasm before it has a chance to be exported to the outer membrane. Indeed, supernatant from lysed E. coli Lpp-OmpA-AmCyan cells exhibited modest fluorescence (see above).
FIG. 4.

Confocal laser scanning micrographs of E. coli strains engineered with a fluorescent AmCyan protein targeted to the outer membrane (A) or localized to the cytoplasm (B). Line profiles below each image correspond to the relative intensity along the length of a bacterium highlighted between a pair of arrows. Note the fluorescent ring around the cell envelope of the bacterium highlighted in panel A (E. coli strain Lpp-OmpA-AmCyan). Atomic force microscopy measurements on single cells (e.g., Fig. 2A and 2D) could be conducted at the same time that fluorescence images like these were collected with the confocal laser scanning microscope. Bars, 5 μm.
It should be noted that the outer cell wall emitted a higher degree of fluorescence for E. coli Lpp-OmpA-AmCyan cells placed overnight at 4°C. No such relationship was noted for the control strain of E. coli that localized the AmCyan protein only to the cytoplasm. Presumably, the lower temperature retarded the premature folding of the fusion protein within the cytoplasm, and thus allowed the AmCyan domain of the chimeric protein to be exported more readily to the outer membrane.
While collecting optical and fluorescence images like those shown in Fig. 4, the tip of a force-sensing cantilever was positioned directly over an E. coli bacterium. Force measurements were then initiated by bringing the tip into contact with the surface of the bacterium and then pulling the tip away from the cell while monitoring the deflection of the cantilever. With this procedure we were able to collect fluorescent images of the cell surface while simultaneously measuring forces of interaction between the same cell and a solid substrate (the atomic force microscope [AFM] tip).
Force extension measurements of a fluorescent protein that forms a bond between a living E. coli cell and a solid substrate.
Silicon nitride tips were selected for the atomic force microscopy experiments for several reasons. First, they are commercially available and probably the most widely used tips in atomic force microscopy experiments. Second, these tips are relatively sharp (20 to 60 nm in radius according to the manufacturer, Veeco Instruments), increasing the chance of binding to a single protein. Finally, while silicon nitride may not be a common material in the environment, it promptly undergoes a sequence of hydrolysis reactions in humid air or aqueous solution resulting in a surface layer of silanol groups (SiOH) (46). Silanol groups are the primary surface functional groups on quartz, silica, and glass, which are the most abundant inorganic phases (or minerals) on Earth and arguably the most common substrates used for bacterial adhesion studies.
A silicon nitride substrate (an AFM tip whose surface is composed of silanol as well as some silylamine groups) was brought into contact with an E. coli bacterium expressing AmCyan on its outer surface (strain Lpp-OmpA-AmCyan). The tip was then pulled from the cell while monitoring the force required to retract the tip a certain distance (retraction data). This was repeated on 10 to 15 different cells from two different cultures of E. coli Lpp-OmpA-AmCyan. For these measurements, a bacterium experienced a maximum loading force (the amount of force with which the AFM tip is pressed against a cell) of 0.2 to 6 nN. This force was not enough to dislodge the cells from the coverslip, as verified by continued optical microscopy observations during all phases (before, during, and after) of the atomic force microscopy measurements. Furthermore, this force did not pierce the cells, as confirmed by assessing the viability of the cells subsequent to force measurements. The viability test was carried out by placing an E. coli-coated coverslip onto an LB-ampicillin agar plate and observing the growth of colonies along regions of the coverslip that were probed with the AFM tip.
Figure 2A shows some typical force-extension (or retraction) curves observed for E. coli Lpp-OmpA-AmCyan. All curves exhibited an initial “jump from contact” event corresponding to the amount of adhesion force (observed force at the origin of the x axis) required to initiate separation of the tip from the cell wall (see Fig. 2A). The magnitude of the adhesion force was dependent upon the loading force and was, in general, less than 1 nN. When the tip snapped back towards its resting position, it created a “blind window” in the force curve within which no structure could be observed. Hence, details within the first 20 to 40 nm are hidden by the jump from contact event (see Fig. 2A). This is an unavoidable consequence of using force microscopy because the AFM tip will jump from contact with a surface when the spring constant of the cantilever (0.02 nN/nm for the cantilevers used) exceeds the actual force gradient that exists between the tip and the sample.
Most of the force measurements for E. coli Lpp-OmpA-AmCyan contained only a jump from contact event. However, a significant proportion (10%; n = 1,095) of the retraction curves exhibited unique sawtoothlike profiles or regions where force increased nonlinearly and then suddenly recoiled back towards the line of zero force. Figure 2A (colored traces) shows a number of the retraction curves that contained sawtooth features. These curves were selected to span the range of observations. For clarity, a single curve is also presented in Fig. 2B. Two sawtooth force signatures were observed for interactions between the silicon nitride tip and E. coli Lpp-OmpA-AmCyan (see Fig. 2A and 2B).
Protease accessibility experiments were performed to determine whether proteins exposed on a bacterium's surface were responsible for generating the sawtooth features observed in the retraction curves. Proteinase K was selected because it is a highly reactive protease that has previously been shown to remove proteins exposed on the outer surface of gram-negative bacteria without otherwise altering or damaging the cell envelope (15, 36).
A 54-mg (wet weight) pellet of E. coli Lpp-OmpA-AmCyan (induced with isopropylthiogalactopyranoside) was suspended in a buffer containing proteinase K (27.6 U/ml) for 15 to 20 min. These cells were then washed twice in proteinase-free buffer and three times in 0.1 M NaCl to remove proteinase K, blotted onto a hydrophobic coverslip, and used in atomic force microscopy. The sawtooth features disappeared almost entirely from the retraction curves when E. coli Lpp-OmpA-AmCyan cells were incubated with proteinase K. Of the 1,113 retraction curves collected on 12 different E. coli Lpp-OmpA-AmCyan cells subjected to proteinase K, only 3 curves (0.2%) exhibited sawteeth (see green curve in Fig. 2C). All other retraction curves were featureless (see black curves that fluctuate around zero force in Fig. 2C). This confirmed that the sawtooth features observed in the retraction data originated because of the presence of proteins exposed on the outer surface of E. coli.
The next step was to determine whether specific sawteeth were due to the chimeric protein that was engineered to be expressed on the surface of the E. coli cells. The measured force-extension data were compared to force topology profiles predicted by the wormlike chain model or freely joined chain model for different stretching scenarios involving the fusion protein shown in Fig. 3. The theoretical profiles shown in Fig. 2 describe the extension of a protein composed of 228 amino acids, which is equivalent to the primary length of the fluorescent AmCyan protein; a protein composed of 252 amino acids, which is equivalent to the primary length of AmCyan plus the 11-amino-acid “linker” segment plus the 13 amino acids exposed on the terminal surface segment of OmpA; and a protein composed of 353 amino acids, which is equivalent to the sum of the lengths of the AmCyan protein plus the “linker” segment plus the entire OmpA protein (114 amino acids). See Fig. 3 for reference.
As shown in Fig. 2A, there is excellent agreement between the wormlike chain theory and the observed relationship between force and extension distance for E. coli Lpp-OmpA-AmCyan. It is important to note that the wormlike chain curves were not “fit” to our data. Rather, we selected values for the persistence length and contour length that were equivalent to the physical and structural dimensions of a protein (see the discussion that accompanies equation 1 above). The value of calibrating the z-piezoelectric scanner with NIST-certified standards (see Materials and Methods) cannot be understated. This calibration ensured accurate force and extension data to within 2% uncertainty. In general, the freely joined chain equation also followed the observed measurements. However, the freely joined chain model underestimated the resistance of a polypeptide to extension, particularly at low force (see Fig. 2B).
As shown in Fig. 2A, a range of force signatures were observed for E. coli Lpp-OmpA-AmCyan interacting with the silicon nitride surface. Some of the retraction curves contain a sawtooth profile consistent with the extension of only that portion of the chimeric protein that is exposed to the extracellular solution (AmCyan alone or AmCyan plus the linker plus the outermost amino acids of OmpA; Fig. 2A). Other retraction curves contain a sawtooth profile that corresponds to the extension of the entire fusion protein (Fig. 2A). Finally, some measured curves reveal two distinct sawteeth in agreement with successive unraveling of the extracellular portion of the chimeric protein followed by unraveling of the transmembrane portion of the protein (Fig. 2B).
These three scenarios are consistent with the following explanation of events. As the tip was brought into contact with a cell, the C-terminal portion of the AmCyan protein adsorbed to the tip. It is difficult to know precisely what type of bond formed between the chimeric protein and the surface of the AFM tip. Senden and Drummond (46) conducted a detailed analysis of the surface chemistry of silicon nitride tips manufactured by the same company that supplied the AFM tips for this investigation. It was determined that the silicon nitride surface is close to being electrically neutral from pH 6 to 8.5 because of an equal density of negatively charged silanol groups and positively charged silylamine groups (46). One explanation is that the C terminus of the AmCyan domain formed an electrostatic attraction with a cationic silylamine group on the AFM tip, resulting in a bond that anchored the protein to the silicon nitride tip.
The tip was then retracted from the cell, initiating the mechanical unfolding and extension of the protein. Extension continued until a holding force of 0.1 to 0.3 nN was reached, at which point unfolding of AmCyan became highly probable. Unfolding of the AmCyan domain abruptly reduced the holding force because of an increase in the length of the protein, causing the tip to return to its resting position. This resulted in the first sawtooth profile that correlates with the extension of a polypeptide that is 228 to 252 amino acids in length (Fig. 2A and 2B). At this point, the AmCyan protein could have broken free of the tip, resulting in only one sawtooth. When the protein remained bridged to the tip, continued retraction of the tip again stretched the protein until a force was reached at which the OmpA domain began to unfold. Unfolding of the OmpA domain caused the tip to once again return to its resting position, resulting in the second sawtooth. The result is a series of two sawteeth whose extension matches the successive unraveling of AmCyan and OmpA, which are in mechanical series (Fig. 2B).
A noteworthy gauge of these measurements is the relative distance between two successive sawtooth features detected in the same retraction curve. The wormlike chain model predicts that the domains within the protein fusion Lpp-OmpA-AmCyan should yield force profiles that are separated by 35 to 43 nm at an extending force of −0.3 nN (see relative distance between dotted black curves at −0.3 nN in Fig. 2A). Measured force-extension profiles with E. coli Lpp-OmpA-AmCyan show two successive sawteeth separated by 34 ± 4 nm (Fig. 2B) at this extension force. This is precisely what is expected from the wormlike chain model for these two particular protein domains (OmpA and AmCyan) in series.
Some force-extension measurements show only the second of the two sawtooth features, analogous to unraveling of the entire fusion protein without first an unfolding of the AmCyan domain. This is presumably due to the improper folding or lack of folding of the AmCyan protein. Extension of the AmCyan protein could damage the protein structure so that it did not refold properly before a successive extension of the same protein by the AFM tip. An alternative explanation is that the AmCyan protein may not always fold properly when it is exported to the outer membrane. Indeed, fluorescence observations revealed that the cell envelope of E. coli Lpp-OmpA-AmCyan fluoresced more intensely at lower temperature (see above). This supports the notion that the AmCyan protein was not always properly folded in the outer membrane, as AmCyan should autocatalytically fluoresce upon adopting its native folded structure (52, 56).
Many of the sawtooth signatures shown in Fig. 2A were noted in two or more successive force curves, suggesting that these proteins were not ripped from the outer membrane. Rather, the proteins appear to be stretched to some point at which they break free of the tip and recoil back to the outer membrane of the cell. For the maximum recorded force for these particular sawteeth (0.1 to 0.4 nN from Fig. 2A), the wormlike chain model (equation 1) predicts an extension of roughly 75 to 90% of either domain (AmCyan or OmpA) within the fusion protein. For example, according to equation 1, a force of 0.07 nN would be sufficient to cause the mechanical extension of ≈80% of a polypeptide made of 353 amino acids (where the contour length is 353 amino acids × 0.38 nm per amino acid). This percent extension is equivalent to ≈280 amino acids, or the sum of the entire AmCyan protein (228 amino acids), the linker (11 amino acids), and the two C-terminal-most segments of OmpA (≈40 amino acids) (see Fig. 3 for reference). We were somewhat surprised that a portion of the transmembrane segments of OmpA appeared to be pulled from the outer membrane (see Fig. 2A or 2B). However, others have used an AFM tip to repeatedly pull individual monomers from a purified protein S layer (35).
It is difficult to determine how many individual proteins formed a bond between an E. coli cell and the AFM tip. As stated above, we used a probe with a relatively sharp tip (20 to 60 nm), which would enhance single-molecule interactions. The Derjaguin-Muller-Toporov theory can be used to provide an indirect measure of the number of proteins that formed a bond with the surface of the AFM tip. This theory correlates an adhesion force (F) to the interfacial energy (γ) of an interaction according to the equation F = −4πRγ (21), where R is the radius of the AFM tip in our case. The interfacial energy between E. coli Lpp-OmpA-AmCyan and the silicon nitride surface is calculated to be 0.1 to 1.6 mJ m−2 for R = 20 to 60 nm and F = −0.1 to −0.4 nN (from the maximum force values for the sawteeth shown in Fig. 2A). Interfacial energy values of less than ≈1 mJ m−2 are consistent with specific interactions (e.g., ligand-receptor) between a pair of reactive groups, as opposed to nonspecific interactions involving many functional groups on two surfaces (J. Israelachvili, personal communication). This calculation suggests that one or a few proteins formed a bond between E. coli and the surface of the AFM tip.
Of course, the above discussion would be moot if E. coli naturally expresses proteins composed of 228, 252, or 353 amino acids on its outer membrane. Indeed, wild-type E. coli possess an OmpA protein very similar to the engineered E. coli Lpp-OmpA-AmCyan. Furthermore, the genome of E. coli codes for numerous proteins located on the cell wall (2). Therefore, we collected force-extension measurements on a control strain of E. coli that localizes the AmCyan protein to the cytoplasm. This strain also displayed sawtoothlike profiles in ≈10% of the retraction curves (n = 1,094). However, these sawtooth profiles did not correlate to the theoretical force-extension profiles for any domain within the chimeric Lpp-OmpA-AmCyan protein fusion (see Fig. 2D). Most of the sawteeth noted for the control strain were long range (>100 to 125 nm). Similar long-range sawtooth features were also noted for some force-extension measurements with E. coli Lpp-OmpA-AmCyan (red curve in Fig. 2A). Only two retraction curves for the control strain of E. coli (0.2% of the total number of measurements) vaguely matched any of the three wormlike chain curves plotted in Fig. 2D. It would be speculation to suggest that these two measured curves for the control strain are anything more than outliers.
Relevance to bacterial adhesion and biofilm formation.
These experiments demonstrate that force and fluorescence measurements can be used simultaneously to collect complementary information about macromolecules that form a physical bond between a cell and another surface. In so doing, the actual forces responsible for allowing a bacterium to sense another surface can be probed in situ. Furthermore, the paired force and fluorescence data could provide important clues as to the identification and/or function of biopolymers that are involved in adhesion reactions between a bacterium and an inanimate surface or even another cell. Such experiments would benefit from a “flyfishing” type of technique in which a force microscopy tip is “baited” with the appropriate ligand or lure for a particular protein. Indeed, we have previously shown that inorganic compounds can be used to probe the binding of specific outer membrane proteins on a dissimilatory metal-reducing bacterium (29). Other scenarios could include the use of a tip functionalized with a ligand or antibody.
Care must be taken to select organisms that are well characterized in the literature, as atomic force microscopy measurements alone can be challenging to interpret. For example, our cells were designed to maximize the chance of bonding the AmCyan domain to the AFM tip by the protein's C terminus (see Fig. 3). Some proteins may be exposed on the outer surface of bacteria, such that the termini are buried within the protein's structure, or a significant portion of a protein may be contained within the outer membrane (e.g., integral membrane protein). Resulting force-extension observations may therefore reveal a sawtooth that is shorter than predicted by the wormlike chain theory. However, this shortcoming can be overcome by combining the force measurements with fluorescently tagged proteins and/or using mutants that cannot produce particular proteins of interest (Fig. 2D). Furthermore, the resulting force-extension measurement, even if the protein is not extended by its ends, would provide novel information about the number of amino acids in the primary structure that are actually exposed on the outer surface of the cell in situ.
Another potential challenge deals with the possibility that a single protein might have multiple substructures that unfold at different points along a force-extension profile. For the measurements presented herein, we designed a cell that expresses a protein molecule composed of two domains, a peripheral segment (AmCyan) and an integral membrane segment (OmpA). Each domain within this protein displayed a stable substructure, resulting in up to two sawteeth in the retraction curves (see Fig. 2B). Another group discovered a similar phenomenon when they used an AFM tip to unfold a protein that was adsorbed onto a silicon substrate (19). Hertadi et al. (19) observed that a monomer of outer surface protein A has two stable substructures that mechanically unfold to yield two sawteeth in retraction curves. As we have shown here, a single-sawtooth or multiple-sawtooth signature can be assigned to a unique protein through a comparative analysis of force measurements with a wild-type strain versus a mutant that cannot express (or overexpresses) a protein of interest.
Acknowledgments
This work was supported by a grant from the Department of Energy (DE-FG02-04ER15590) and an instrument grant from the National Science Foundation (EAR-0132433). Partial support was provided by the American Chemical Society for F.P.V. (PRF38107-G2). B.H.L.'s portion of the research was supported by the Natural and Accelerated Bioremediation Research Program (Office of Biological and Environmental Research, Department of Energy). The Pacific Northwest National Laboratory is operated by Battelle Memorial Institute for the U.S. Department of Energy under contract DE-AC06-76RLO 1830.
Louis Bivona and Douglas English assisted in the creation of hydrophobic coverslips. P. Kennelly kindly provided the use of the high-speed ultracentrifuge. Four anonymous reviewers and the editor (D. Ohlendorf) provided constructive criticism that greatly improved the original manuscript. We also thank J. Israelachvili for helpful suggestions. B.H.L. and S.K.L. acknowledge the support of B.C.H.M. and J. Tak, respectively.
REFERENCES
- 1.Baumann, C. G., S. B. Smith, V. A. Bloomfield, and C. Bustamante. 1997. Ionic effects on the elasticity of single DNA molecules. Proc. Natl. Acad. Sci. USA 94:6185-6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blattner, F. R., G. Plunkett, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. ColladoVides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1463. [DOI] [PubMed] [Google Scholar]
- 3.Carrion-Vazquez, M., P. E. Marszalek, A. F. Oberhauser, and J. M. Fernandez. 1999. Atomic force microscopy captures length phenotypes in single proteins. Proc. Natl. Acad. Sci. USA 96:11288-11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carrion-Vazquez, M., A. F. Oberhauser, S. B. Fowler, P. E. Marszalek, S. E. Broedel, J. Clarke, and J. M. Fernandez. 1999. Mechanical and chemical unfolding of a single protein: A comparison. Proc. Natl. Acad. Sci. USA 9:3694-3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cleveland, J. P., S. Manne, D. Bocek, and P. K. Hansma. 1993. A nondestructive method for determining the spring constant of cantilevers for scanning force microscopy. Review of Scientific Instruments. 64:403-405.
- 6.Costerton, J. W., P. S. Stewart, and E. P. Greenberg. 1999. Bacterial biofilms: A common cause of persistent infections. Science 284:1318-1322. [DOI] [PubMed] [Google Scholar]
- 7.Craig, V. S. J., and C. Neto. 2001. In situ calibration of colloid probe cantilevers in force microscopy: Hydrodynamic drag on a sphere approaching a wall. Langmuir 17:6018-6022. [Google Scholar]
- 8.Ducker, W. A., T. J. Senden, and R. M. Pashley. 1991. Direct measurement of colloidal forces using an atomic force microscope. Nature 353:239-241. [Google Scholar]
- 9.Ducker, W. A., T. J. Senden, and R. M. Pashley. 1992. Measurements of forces in liquids using a force microscope. Langmuir 8:1831-1836. [Google Scholar]
- 10.Fisher, T. E., P. E. Marszalek, A. F. Oberhauser, M. Carrion-Vazquez, and J. M. Fernandez. 1999. The micro-mechanics of single molecules studied with atomic force microscopy. J. Physiol. London 520:5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flammann, H. T., and J. Weckesser. 1984. Characterization of the cell-wall and outer-membrane of Rhodopseudomonas capsulata. J. Bacteriology 159:191-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flory, P. J. 1989. Statistical mechanics of chain molecules. Hanser Publishers, Munich, Germany.
- 13.Forbes, J. G., A. J. Jin, and K. Wang. 2001. Atomic force microscope study of the effect of the immobilization substrate on the structure and force-extension curves of a multimeric protein. Langmuir 17:3067-3075. [Google Scholar]
- 14.Francisco, J. A., R. Campbell, B. L. Iverson, and G. Georgiou. 1993. Production and fluorescence-activated cell sorting of Escherichia coli expressing a functional antibody fragment on the external surface. Proc. Natl. Acad. Sci. USA 90:10444-10448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Francisco, J. A., C. F. Earhart, and G. Georgiou. 1992. Transport and anchoring of beta-lactamase to the external surface of Escherichia coli. Proc. Natl. Acad. Sci. USA 89:2713-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Francisco, J. A., C. Stathopoulos, R. A. J. Warren, D. G. Kilburn, and G. Georgiou. 1993. Specific adhesion and hydrolysis of cellulose by intact Escherichia coli expressing surface anchored cellulase or cellulose binding domains. BioTechnology 11:491-495. [DOI] [PubMed] [Google Scholar]
- 17.Fritz, J., A. G. Katopodis, F. Kolbinger, and D. Anselmetti. 1998. Force-mediated kinetics of single P-selectin ligand complexes observed by atomic force microscopy. Proc. Natl. Acad. Sci. USA 95:12283-12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goel, A., M. D. Frank-Kamenetskii, T. Ellenberger, and D. Herschbach. 2001. Tuning DNA “strings”: Modulating the rate of DNA replication with mechanical tension. Proc. Natl. Acad. Sci. USA 98:8485-8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hertadi, R., F. Gruswitz, L. Silver, A. Koide, S. Koide, H. Arakawa, and A. Ikai. 2003. Unfolding mechanics of multiple OspA substructures investigated with single molecule force spectroscopy. J. Mol. Biol. 333:993-1002. [DOI] [PubMed] [Google Scholar]
- 20.Howard, J. 2001. Mechanics of motor proteins and the cytoskeleton. Sinauer Associates, Inc., Sunderland, Mass.
- 21.Israelachvili, J. 1992. Intermolecular and surface forces. Academic Press, London, England.
- 22.Israelachvili, J. N., and P. M. McGuiggan. 1988. Forces between surfaces in liquids. Science 241:795-800. [DOI] [PubMed] [Google Scholar]
- 23.Kellermayer, M. S. Z., S. B. Smith, H. L. Granzier, and C. Bustamante. 1997. Folding-unfolding transitions in single titin molecules characterized with laser tweezers. Science 276:1112-1116. [DOI] [PubMed] [Google Scholar]
- 24.Kendall, T. A., and S. K. Lower. 2004. Forces between minerals and biological surfaces in aqueous solution. Adv. Agronomy 82:1-54. [Google Scholar]
- 25.Koebnik, R. 1999. Structural and functional roles of the surface-exposed loops of the beta-barrel membrane protein OmpA from Escherichia coli. J. Bacteriol. 181:3688-3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leckband, D., and J. Israelachvili. 2001. Intermolecular forces in biology. Q. Rev. Biophys. 34:105-267. [DOI] [PubMed] [Google Scholar]
- 27.Lo, Y.-S., N. D. Huefner, W. S. Chan, P. Dryden, B. Hagenhoff, and T. P. Beebe, Jr. 1999. Organic and inorganic contamination on commercial AFM cantilevers. Langmuir 15:6522-6526. [Google Scholar]
- 28.Low, D., B. Braaten, and M. van der Woude. 1996. Fimbriae, p. 146-157. In F. C. Neidhardt et al. (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed., vol. 1. ASM Press, Washington, D.C. [Google Scholar]
- 29.Lower, S. K., M. F. Hochella, and T. J. Beveridge. 2001. Bacterial recognition of mineral surfaces: nanoscale interactions between Shewanella and α-FeOOH. Science 292:1360-1363. [DOI] [PubMed] [Google Scholar]
- 30.Lower, S. K., C. J. Tadanier, and M. F. Hochella. 2001. Dynamics of the mineral-microbe interface: use of biological force microscopy in biogeochemistry and geomicrobiology. Geomicrobiology J. 18:63-76. [Google Scholar]
- 31.Lower, S. K., C. J. Tadanier, and M. F. Hochella. 2000. Measuring interfacial and adhesion forces between bacteria and mineral surfaces with biological force microscopy. Geochim. Cosmochim. Acta 64:3133-3139. [Google Scholar]
- 32.Matz, M. V., A. F. Fradkov, Y. A. Labas, A. P. Savitsky, A. G. Zaraisky, M. L. Markelov, and S. A. Lukyanov. 1999. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat. Biotechnol. 17:969-973. [DOI] [PubMed] [Google Scholar]
- 33.Meadows, P. Y., J. E. Bemis, and G. C. Walker. 2003. Single-molecule force spectroscopy of isolated and aggregated fibronectin proteins on negatively charged surfaces in aqueous liquids. Langmuir 19:9566-9572. [Google Scholar]
- 34.Mueller, H., H.-J. Butt, and E. Bamberg. 1999. Force measurements on myelin basic protein adsorbed to mica and lipid bilayer surfaces done with the atomic force microscope. Biophys. J. 76:1072-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muller, D. J., W. Baumeister, and A. Engel. 1999. Controlled unzipping of a bacterial surface layer with atomic force microscopy. Proc. Natl. Acad. Sci. USA 96:13170-13174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Myers, C. R., and J. M. Myers. 2004. Cell surface exposure of the outer membrane cytochromes of Shewanella oneidensis MR-1. Lett. Appl. Microbiol. 37:254-258. [DOI] [PubMed] [Google Scholar]
- 37.Oberdorfer, Y., H. Fuchs, and A. Janshoff. 2000. Conformational analysis of native fibronectin by means of force spectroscopy. Langmuir 16:9955-9958. [Google Scholar]
- 38.Oberhauser, A. F., P. E. Marszalek, M. Carrion-Vazquez, and J. M. Fernandez. 1999. Single protein misfolding events captured by atomic force microscopy. Nat. Struct. Biol. 6:1025-1028. [DOI] [PubMed] [Google Scholar]
- 39.Ohmura, N., K. Tsugita, J.-I. Koizumi, and H. Saiki. 1996. Sulfur-binding protein of flagella of Thiobacillus ferrooxidans. J. Bacteriol. 178:5776-5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Osborn, M. J., and R. Munson. 1974. Separation of the inner (cytoplasmic) and outer membranes of Gram-negative bacteria. Methods Enzymol. 31:642-653. [DOI] [PubMed] [Google Scholar]
- 41.Rief, M., J. M. Fernandez, and H. E. Gaub. 1998. Elastically coupled two-level systems as a model for biopolymer extensibility. Phys. Rev. Lett. 81:4764-4767. [Google Scholar]
- 42.Rief, M., M. Gautel, F. Oesterhelt, J. M. Fernandez, and H. E. Gaub. 1997. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science 276:1109-1112. [DOI] [PubMed] [Google Scholar]
- 43.Rief, M., F. Oesterhelt, B. Heymann, and H. E. Gaub. 1997. Single molecule force spectroscopy on polysaccharides by atomic force microscopy. Science 275:1295-1297. [DOI] [PubMed] [Google Scholar]
- 44.Rief, M., J. Pascual, M. Saraste, and H. E. Gaub. 1999. Single molecule force spectroscopy of spectrin repeats: Low unfolding forces in helix bundles. J. Mol. Biol. 286:553-561. [DOI] [PubMed] [Google Scholar]
- 45.Schwarz-Linek, U., M. Hook, and J. R. Potts. 2004. The molecular basis of fibronectin-mediated bacterial adherence to host cells. Mol. Microbiol. 52:631-641. [DOI] [PubMed] [Google Scholar]
- 46.Senden, T. J., and C. J. Drummond. 1995. Surface chemistry and tip-sample interactions in atomic force microscopy. Colloids Surfaces A Physicochem. Eng. Aspects 94:29-51. [Google Scholar]
- 47.Smith, S. B., Y. J. Cui, and C. Bustamante. 1996. Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science 271:795-799. [DOI] [PubMed] [Google Scholar]
- 48.Soutourina, O. A., and P. N. Bertin. 2003. Regulation cascade of flagellar expression in Gram-negative bacteria. FEMS Microbiol. Rev. 27(4):505-523. [DOI] [PubMed] [Google Scholar]
- 49.Stathopoulos, C., G. Georgiou, and C. F. Earhart. 1996. Characterization of Escherichia coli expressing an Lpp-OmpA(46-159)-PhoA fusion protein localized in the outer membrane. Appl. Microbiol. Biotechnol. 45:112-119. [DOI] [PubMed] [Google Scholar]
- 50.Tskhovrebova, L., J. Trinick, J. A. Sleep, and R. M. Simmons. 1997. Elasticity and unfolding of single molecules of the giant muscle protein titin. Nature 387(6630):308-312. [DOI] [PubMed] [Google Scholar]
- 51.Vidal, O., R. Longin, C. Prigent-Combaret, C. Dorel, M. Hooreman, and P. Lejeune. 1998. Isolation of an Escherichia coli K-12 mutant strain able to form biofilms on inert surfaces: involvement of a new ompR allele that increases curli expression. J. Bacteriol. 180:2442-2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wall, M. A., M. Socolich, and R. Ranganathan. 2000. The structural basis for red fluorescence in the tetrameric GFP homolog DsRed. Nat. Struct. Biol. 7:1133-1138. [DOI] [PubMed] [Google Scholar]
- 53.Wang, T., H. Arakawa, and A. Ikai. 2001. Force measurement and inhibitor binding assay of monomer and engineered dimer of bovine carbonic anhydrase B. Biochem. Biophys. Res. Commun. 285:9-14. [DOI] [PubMed] [Google Scholar]
- 54.Whittaker, C. J., C. M. Klier, and P. E. Kolenbrander. 1996. Mechanisms of adhesion by oral bacteria. Annu. Rev. Microbiol. 50:513-552. [DOI] [PubMed] [Google Scholar]
- 55.Yang, G. L., C. Cecconi, W. A. Baase, I. R. Vetter, W. A. Breyer, J. A. Haack, B. W. Matthews, F. W. Dahlquist, and C. Bustamante. 2000. Solid-state synthesis and mechanical unfolding of polymers of T4 lysozyme. Proc. Natl. Acad. Sci. USA 97:139-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yarbrough, D., R. M. Wachter, K. Kallio, M. V. Matz, and S. J. Remington. 2001. Refined crystal structure of DsRed, a red fluorescent protein from coral, at 2.0-angstrom resolution. Proc. Natl. Acad. Sci. USA 98:462-467. [DOI] [PMC free article] [PubMed] [Google Scholar]