

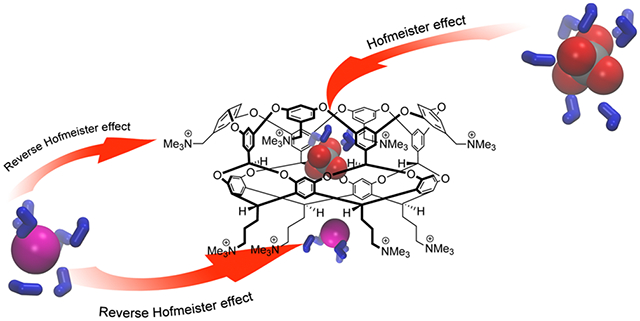
Abstract
A combination of 1H NMR spectroscopy, DLS, and turbidity measurements reveal that polarizable anions engender both the Hofmeister and reverse Hofmeister effects in positand 2. Host 2 possesses two principal and distinctly different binding sites: a “soft” nonpolar pocket and a “hard” crown of ammonium cations. NMR spectroscopy reveals that anion affinity to both sites is comparable, with each site showing characteristic selectivities. NMR spectroscopy also reveals that anions competitively bind to the pocket and induce the Hofmeister effect in host–guest binding at very low concentrations (~2 mM). Furthermore, the suite of techniques utilized demonstrates that anion binding to both sites leads to charge attenuation, aggregation, and finally precipitation (the reverse Hofmeister effect). Anion-induced precipitation generally correlated with affinity, and comparisons between the free host and its adamantane carboxylate (Ada-CO2−) complex reveals that the reverse Hofmeister effect is attenuated by blocking anion binding/charge attenuation at the nonpolar pocket.
Graphical Abstract
INTRODUCTION
In 1888, Franz Hofmeister observed that certain salts induced proteins to precipitate while others caused proteins to become more soluble.1 In the intervening years since, the Hofmeister effect and the attendant Hofmeister series of anions (Figure 1) have been demonstrated across a wide range of molecules, using over 40 different physicochemical measurements.2 Historically, this phenomenon was attributed to salts changing water structure and indirectly affecting a cosolute,2,3 but this is not now believed to be so.4 Rather, the major noncovalent interactions contributing to the Hofmeister effect appear to be direct anion–cosolute interactions involving hydrogen bond donors and nonpolar surfaces of the cosolute.5
Figure 1.

Hofmeister series of anions and an expanded series to include powerful, salting-in anions.
Contrary to the above, in some instances the reverse Hofmeister effect is observed. This phenomenon pertains to the situation whereby instead of increasing the solubility of proteins, nominally salting-in salts actually reduce solubility and induce precipitation. For proteins, this effect is normally observed at a pH lower than the isoelectric point (pI). However, observations can be complex and puzzling. For example, at pH 9.4 it has been shown that lysozyme (pI = 11.35) exhibits a direct Hofmeister effect at high ionic strength (≳300 mM) but the reverse Hofmeister effect at low salt concentrations.6 Regarding the reverse Hofmeister effect, current evidence points to the importance of Coulombic interactions between certain anions and the cationic centers in a protein, leading ultimately to charge neutralization and precipitation.6,7 Tied to this, vibrational sum frequency spectroscopic studies of solid surfaces, and monolayers of protein, surfactant, and polymer, reveal that at low pH, interfacial water structure follows the reverse Hofmeister series.7c,8 Furthermore, a Poisson–Boltzmann model modified with ion-specific potentials suggests it is an entropic contribution by anions which engenders the effect at low salt concentrations, and an entropic contribution from the counter cations that leads to the Hofmeister effect at high salt concentrations.9 These points noted, there are still countless open questions regarding the precise balance of supramolecular interactions responsible for the reverse Hofmeister effect.10
To our knowledge, the reverse Hofmeister effect has not been expressly linked to the solubility of small molecules in water. However, from our perspective these two phenomena are one and the same. Thus, it is common knowledge that to make an ammonium salt more water-soluble, the chloride (Cl−) salt is formed, whereas if poor water solubility is desired (i.e., organic solvent solubility), the perchlorate (ClO4−) salt might be targeted. Small molecule chemists have been content with this knowledge and have not investigated this phenomenon. However, it seems that it is intimately related to the reverse Hofmeister effect and that the study of small molecule solubility could lead to a new level of understanding of this phenomenon that is difficult to access using proteins. With this in mind, the work presented here represents, to our best knowledge, the first attempt to systematically study the reverse Hofmeister effect in a supramolecular system.
Previously, we have demonstrated that in buffered water the Hofmeister effect is manifest11 in the binding of amphiphilic guests to deep-cavity cavitand 1 (Figure 2).12 Octa-acid 1 possesses a nonpolar concavity that readily binds amphiphiles to form the corresponding 1:1 host–guest complex;13 a fact that makes it ideal for projects such as the Statistical Assessment of the Modeling of Proteins and Ligands (SAMPL), a community-wide prediction challenge to evaluate computational methods for affinity determinations in computer-aided drug design.14 We have observed that amphiphile binding to host 1 is attenuated by salting-in anions, i.e., that there is the expected apparent weakening of the hydrophobic effect upon the addition of these salts. Importantly, we determined that this is caused by competitive binding of relatively large, polarizable anions to the nonpolar pocket.11b,d Furthermore, we recently demonstrated using a combination of isothermal titration calorimetry (ITC), quantum calculations, and molecular dynamics (MD) simulations that anion binding to the pocket of 1 is enthalpically favorable and entropically penalized and involves only partial desolvation of the anionic guest.11a
Figure 2.

Anionic and cationic deep-cavity cavitands: the octacarboxylate 1 (counterion = Na+) and the octatrimethylammonium host 2 (positand, counterion = Cl−). In 2, the two principal anion binding sites are shown (cavity and crown).
Curiously, anion binding to the nonpolar pocket of 1 is itself subject to the Hofmeister effect.11c Thus, the affinity of ClO4− for the pocket of 1 is enhanced in the presence of salting-out anions, is attenuated by salting-in anions, and shows a nonmonotonic relationship with salt concentration in the presence of weakly salting-out anions such as chlorate (ClO3−). We determined that the enhanced binding induced by salting-out salts arises from an “active” (binding) cation and an “inactive” (nonbinding) anion. Thus, in the case of NaF, pseudo-specific sodium binding to the carboxylates of the host reduces its net negative charge and increased ClO4− affinity, while the counter fluoride (F−) acts purely as a spectator ion. In contrast, the attenuation of ClO4− affinity with salts such as NaSCN arises because pseudospecific binding of Na+ is weak relative to the SCN− affinity for the pocket that competes with ClO4− binding. However, in cases where the “activity” of cation and anion are comparable, e.g., Na+ versus ClO3−, the attenuation and diminution effects compete and a nonmonotonic relationship between Ka of ClO4− binding and salt concentration is observed.
In related work we recently reported15 the synthesis of water-soluble, octacationic host 2 (positand, Figure 2) bearing eight nonionizable trimethylammonium groups on its outer surface. This significant difference aside, hosts 1 and 2 possess identical structures, and we hypothesized that positand 2 would not only possess stronger binding of anions to its nonpolar cavity but also could bind anions to its “crown” of four ammonium cations in its pendent groups (Figure 2). Furthermore, considering the high positive charge density, we surmised that with additional pseudospecific association of anions to the trimethylammonium groups, positand 2 might also demonstrate the reverse Hofmeister effect. Our studies described here confirm that this is the case; in buffered water host 2 demonstrates anion–nonpolar surface interactions fundamental to the Hofmeister effect, as well as a salting-out phenomenon that follows the reverse Hofmeister effect.
RESULTS AND DISCUSSION
To determine which anions bound to positand 2 in aqueous solution, we used 1H NMR spectroscopy to screen 31 monovalent sodium salts in unbuffered water. This screening led us to select a total of 14 anions for binding constant (Ka) determinations (Table 1). To determine the affinity for the crown of four pendent trimethylammonium groups (), we used the shift of Hj (Figure 2) to the complex of positand 2 with adamantane carboxylate (Ada-CO2−, Ka ≈ 7.21 × 106 M−1 by ITC) blocking the pocket. Only in the case of trichloroacetate (CCl3CO2−) was this strategy unsuccessful. This anion was found to bind so strongly to the cavity (7.03 × 104 M−1 by ITC) that it competed effectively with the adamantyl guest. The data from the determinations were then used to fit a 1:2 model for anion binding to the free host. By this approach, monitoring Hb (Figure 2) allowed the determination of the anion affinity for the nonpolar cavity (, Figures S33-S35 in Supporting Information). The NMR data from hexafluorophosphate (PF6−) did not fit this model well, indicating that pseudospecific binding to the ammonium groups was significant. Additionally, only limited data for the binding of triflate (CF3SO3−) could be collected because of peak coincidence of the Hb and benzyl signals of the host. In both cases a minimum Ka value is quoted.
Table 1.
Anion Affinities for Hosts 1 and 2
| anion | host 1,a KNMR (M−1) |
host 1,a KITC (M−1) |
host 2,b
(M−1) |
host 2,b (M−1) |
|---|---|---|---|---|
| PF6− | 1560 | 2303 | >5000c | 3600 |
| CF3SO3− | 67 | 314 | >5000d | 710 |
| ReO4− | 322 | 371 | 7200 | 1800 |
| ClO4− | 95 | 160 | 4300 | 2400 |
| BH3CN− | 67 | 152 | 3600 | 1300 |
| BF4− | e | e | 35 | 1200 |
| SCN− | 33 | 44 | 2000 | 830 |
| I− | 11 | 17 | 930 | 3200f |
| CCl3CO2− | 5383 | 6337 | 70300g | h |
| CHCl2CO2− | 50 | 52 | 1500 | e |
| Br− | e | e | 17 | 740 |
| NO3− | e | e | 95 | 180 |
| Cl− | e | e | e | 120 |
| F− | e | e | e | e |
Affinities for 1 determined using 1H NMR spectroscopy and ITC are taken from an earlier report.11a
Affinities to 2 were determined using 1H NMR spectroscopy (10 mM phosphate buffered D2O, pD = 7.3). Determinations involving 2 were carried out in at least triplicate, with errors (CV) in anion affinity generally <10% for most anions (see Supporting Information for details). Unless otherwise noted, determinations of utilized the Hb (Figure 2) signal of free 2, whereas determinations utilized the Hj (Figure 2) signal of the 2·Ada-CO2− complex.
Hb signal shifts indicated strong binding, but pseudospecific binding to the trimethylammonium groups prevented accurate determination.
Hb signal shifts indicated strong binding, but due to overlap with the host benzylic signal, insufficient data could be collected.
Binding too weak to determine accurately.
Ka with 1:1 model of the Hl (Figure 2) signal fitting to the titration using the 2·Ada-CO2− complex.
Determined using ITC.
Unable to estimate 1:1 binding to crown due to competitive displacement of guest of 2·Ada-CO2− complex.
The nonpolar cavity of host 2 is primarily composed of eight aromatic rings, has four weak hydrogen bond donors near its base (Hb), and possesses a “soft” positive electrostatic potential (remote charge groups). The switch from negative (1) to positive electrostatic potential (2) means that all anions observed to bind to 1 bind more strongly to host 2. Neither F− nor Cl− showed evidence of complexation; however the mid-Hofmeister series anions bromide (Br−), and nitrate (NO3−) did. For example, NO3− bound with an affinity of ΔG = 2.7 kcal mol−1. Neither Br− nor NO3− has an affinity for 1.
More “hydrophobic” anions bound more strongly still. For example, ClO4− affinity for 2 is ΔG = 5.0 kcal mol−1 (cf. 3.0 kcal mol−1 for host 1), while the affinity of perrhenate (ReO4−) for the pocket was measured to be ΔG = 5.3 kcal mol−1. Strong pseudospecific binding of PF6− led to a poor fit of data for pocket binding. However, a plot of the Ka values for the binding of small inorganic anions to host 1, against the corresponding affinities for 2, is approximately linear (R2 = 0.90) and projects an affinity for PF6− to the cavity of 2 of 16 000 M−1 ΔG = 5.7 kcal mol−1). Thus, switching from a net negative (1) to a net positive electrostatic potential (2) increases affinity by ~1.5 to 2.3 kcal mol−1, with smaller anions showing the larger increase. These results demonstrate that a range of anions bind to the pocket of 2, with affinities ranging from 1.7 to 5.7 kcal mol−1. Furthermore, the diffuse nature of the electrostatic potential and the nonpolar surfaces of the pocket results in a selectivity for large polarizable anions, with species such as ReO4−, PF6−, and CCl3CO2− possessing the strongest affinities.
The downfield shift of the Hb signal induced by anion binding indicates that the ions are bound to the base of the cavity of 2. In silico studies with 111a reveal that when bound this way, anions remain solvated on their “portal side”. This picture is consistent with related data. For example Br− has a solvation shell of ~6.5 waters and a hydration free energy of ΔGhyd ≈ 77 kcal mol−1.16 This is too high to be compensated for by noncovalent interactions with the host. Therefore, only a fraction of the first hydration shell is lost upon binding. Additionally, the fact that polarizable anions accumulate at the surface of water clusters17 and the fact that the stabilization of bound water inside free 1 relies heavily on hydrogen bonding to the bulk18 both support the idea that the “upper” half of the anion and pocket are fully solvated. Furthermore, as the host pocket can contain up to 7 waters,18 approximately half of the solvation shell of Br− must be removed upon binding.
Anion affinity for the nonpolar cavity induces the salting-in Hofmeister effect observed with host 1 at relatively low salt concentrations (10–100 mM).11 The stronger anion affinities for positand 2 mean that the apparent weakening of the hydrophobic effect can be observed at much lower salt concentrations. For example, titrating a 0.5 mM solution of 1:1 complex of 2 and 4-Me-benzoate (ΔG = −6.2 kcal mol−1 by ITC) with NaReO4 results in 50% displacement of the guest at a salt concentration of only 2 mM. Of course, for guests that bind more strongly to 2, e.g., Ada-CO2− ΔG = −9.3 kcal mol−1), the anion affinity must be relatively extreme to observe competition at relatively low salt concentrations. Hence, only in the case of CCl3CO2− is there an apparent weakening of the hydrophobic effect and guest displacement (vide supra). That the Hofmeister effect is evident at such low salt concentrations relative to those typically used in protein work (1–10 M) is due to three factors: (1) the relatively simple structures of these hosts eliminates competing/countering effects, (2) the preorganized nature of the hosts enhances anion affinity, and (3) the use of noncoordinating anions19 (to use the language of organometallic chemists) as strongly coordinating anions in water ensures strong binding (Figure 1).
The values for the anions are also listed in Table 1. The crown differs qualitatively from the cavity by allowing direct ion─ion interactions; the ammonium groups engender a more intense electrostatic potential field in the binding site. F− did not bind to the crown, but the other halides did ΔG = 2.8, 3.9, and 4.8 kcal mol−1 for Cl−, Br−, and iodide (I−), respectively). I− affinity was only slightly weaker than that of the strongest binding guest PF6−. These results lead to a surprising and important conclusion: overall binding to the crown and cavity is quantitatively comparable. For anion recognition in water,20 the assumption has been that strong Coulombic attractions and hydrogen bonding are needed to counter ion solvation. If very tight binding is required, this most likely holds true. However, host 2 demonstrates that anion binding to a “soft”, charge-diffuse pocket can be as strong as binding to a “hard” tetracationic binding site.
Figure 3 compares anion affinity for the crown and cavity ordered by decreasing affinity for the crown. Cl− binds exclusively to the crown, while CCl3CO2− and CHCl2CO2− bind exclusively in the cavity. Between these extremes, specific ratios lie between 44:1 (Br−) and 1:4 (ReO4−). It is interesting to consider “non-coordinating” ClO4−, with its next generation isostere tetrafluoroborate (BF4−). Although frequently treated as interchangeable, they have very different affinity preferences in water: for BF4− is 34:1, and for ClO4− it is 1:1.8. Because BF4− and ClO4− have similar volumes (V = 50 and 51 nm3, respectively) and free energies of hydration (ΔGhyd = 48 and 51 kcal mol−1, respectively), these selectivities demonstrate that it is the nature of their partially hydrated states, rather than the naked anion, that dictates their affinities for the two different sites. Finally, for its relatively strong affinity to the crown, I− has a surprisingly low affinity for the cavity (). Interestingly, ClO4− and I− are often adjacent in the Hofmeister series, but Figure 3 shows that they have essentially the opposite preferences when given the choice of binding to the “soft” and “hard” sites of positand 2. Again, we believe that it is the nature of the hydrated anion that engenders these differences in binding. More generally, it is clear there are multiple competing factors specific to each anion that need to be taken into account.
Figure 3.

Differences in the affinity of anion guests for the crown of ammonium groups and nonpolar cavity of host 2. Anions are shown in the order of decreasing affinity for the crown. The black arrows for the PF6− and CF3SO3− data indicate that binding to the crown is stronger than the shown value (5000 M−1).
Overall, Cl−, NO3−, Br−, BF4−, and I− have a preference for the crown, whereas CF3SO3−, thiocyanate (SCN−), cyanoborohydride (BH3CN−), ReO4−, ClO4−, and PF6− have a preference for the nonpolar cavity. However, there is no clear relationship between these preferences and the (limited) available physical properties of the anions. Thus, although the larger anions tend to prefer the pocket, smaller SCN− preferentially binds to the pocket while larger BF4− and I− prefer the crown. That noted, the strongest correlation we observed (R2 = 0.83) was between ΔG of crown binding and one measure of ion hydration number (Figure S73).16b These results highlight that in order to understand the Hofmeister series, it is necessary to determine the supramolecular profile of each anion: How many waters can it readily lose from its hydration shell? What are the preferred symmetry groups of these hydrated clusters? Given a range of different binding sites, what are the affinity preferences of each anion?
In contrast to host 1, many of these binding anions induce aggregation of 2 at higher concentrations. To investigate this further, we utilized dynamic light scattering (DLS) to examine solutions of the host at different salt concentrations. Figure 4 shows the effects of the halides. Host 2 is monodispersed across all concentrations of NaF and NaCl. However, a degree of aggregation is observed with NaBr, with the observed hydrodynamic diameter increasing from 2.2 nm (monomer) to 3.0 nm (dimer) at 200 mM NaBr. In contrast, the difference between Br− and I− is relatively extreme. Precipitation is observed at 140 mM NaI, but substantial aggregation is observed well below this level; at 12 mM NaI (6 equiv) dimers dominate, while at 78 mM the measured particle diameter corresponds to a ~23-mer aggregate. In combination with the NMR spectroscopy studies, these results show how the more poorly hydrated I− (ΔGhyd values for F−, Cl−, Br−, and I− are respectively 113, 83, 77, and 59 kcal mol−1)16b binds with the host, attenuates the charge groups at the “top” (cavity) and “bottom” (crown) faces of the host, and therefore induces aggregation.
Figure 4.

DLS data for 2.0 mM positand 2 (40 mM phosphate buffered 18 MΩ·cm H2O, pH = 7.3) in the presence of (a) F−, (b) Cl−, (c) Br−, and (d) I−. The x, y, and z axes are respectively particle diameter, salt concentration, and intensity (volume weighted distribution). The time scale for collecting each set of data was kept constant to ensure continuity across different salts.
Overall, for those anions resulting in host aggregation and eventual precipitation, the concentration at which very large sizes (ø > 1000 nm) were observed by DLS showed a reverse Hofmeister ordering: CF3SO3− < PF6− < ReO4− < ClO4− < BH3CN− < BF4− < SCN− < CCl3CO2− < I− (Table 2). To examine this further, we also compared the particle size and aggregation of the host to the complex with Ada-CO2−. In the complex the adamantane moiety prevents anion binding to the pocket, while the strongly solvated carboxylate of the guest helps ensure this face of the host is relatively hydrophilic. We therefore envisioned that aggregation would be attenuated. This was hard to quantify accurately with DLS because in each case aggregation of the complex was attenuated until a sudden, stepwise precipitation occurred (Figure S77). Evidently, however, the kinetics of aggregation of the free host and the complex are markedly different.
Table 2.
CPC Values for Positand 2 and Its Complex with Ada-CO2− Determined by Turbidity and DLS Assaysa
| anion | CPCTurb (mM),b host 2 | CPCDLS (mM),c host 2 | CPCTurb (mM),b 2·Ada-CO2− | CPCDLS (mM),c 2·Ada-CO2− |
|---|---|---|---|---|
| PF6− | 4.0 | 12 | 3.0 | 6.6 |
| CF3SO3− | 9.2 | 5.6 | 23 | 30 |
| ReO4− | 9.4 | 18 | 9.6 | 16 |
| ClO4− | 12 | 30 | 12 | 18 |
| BH3CN− | 19 | 30 | 19 | 27 |
| BF4− | 35 | 54 | 34 | 48 |
| SCN− | 63 | 72 | 65 | 96 |
| I− | 100 | 140 | 120 | 160 |
| CCl3CO2− | 110 | 100 | d | 200 |
| Cl2CHCO2− | d | d | d | d |
| Br− | d | d | d | d |
| NO3− | d | d | d | d |
| Cl− | d | d | d | d |
| F− | d | d | d | d |
Turbidity determinations were carried out in at least triplicate (errors (CV) < 10%). DLS determinations utilized four measurements on single samples.
0.5 mM 2 or its complex in 10 mM phosphate buffer (pH = 7.3).
2.0 mM 2 or its complex in 40 mM phosphate buffer (pH = 7.3).
No observed precipitation.
To better quantify the reverse Hofmeister effect in the host and host–guest complex, we turned to solution phase turbidity measurements21 using a 96-well format assay (Figures S41-S44). Table 2 lists the obtained critical precipitation concentration (CPC) values, along with those derived from DLS data. The results are in close agreement, with the differences attributed to the different concentrations/ionic strengths required for each experiment.
The CPC values from the turbidity assay ranged from 110 mM in the case of CCl3CO2− to 4 mM in the case of PF6−, with the order of precipitating strength again tracking the reverse Hofmeister series: PF6− > CF3SO3− ~ ReO4− > ClO4− > BH3CN− > BF4− > SCN− > I− > CCl3CO2−. With one noticeable exception, the ion ordering for the corresponding Ada-CO2− complex was the same to that of the free host. Thus, CF3SO3− was a much poorer precipitator of the complex than the free host (5th versus second best). Additionally, CCl3CO2− failed to precipitate the complex. We attribute these changes in CPC to the importance of cavity binding for these guests. Between these two anions, CF3SO3− is the better precipitator of the free host because it has affinity for two faces (cavity and crown), whereas CCl3CO2− has no measurable affinity for the crown and so cannot easily induce aggregation along this direction. With the complex, however, these anions have little or no residency time at the cavity site. This means that CF3SO3− can only weakly (Table 1) induce aggregation at the crown face, while CCl3CO2− loses its only principle charge neutralization site and so does not induce precipitation.
A graph of anion affinity to the cavity and crown () against the reciprocal of the CPC value of host 2 (Figure S69) shows that there is a stronger correlation with (R2 = 0.84, versus 0.39 for ). One other noteworthy correlation was that between the partial molar entropy for the aqueous ions and the CPC of 2 and its complex (both R2 = 0.96, Figure S72). Care is warranted here however. First, entropy data are only available for five anions.16b More fundamentally, partial molar entropies are generally obtained by standard electrode potentials reliant on classical models (extended Debye–Hückel theory and Poisson–Boltzmann distribution) to describe ionic profiles near the electrode surface. Such measurements cannot by their nature account for the types of ion specificity observed here.22
General comparisons are again restricted because of limited physical data, but three similarly sized tetrahedral anions, ReO4−, ClO4−, and BF4− (V = 55, 50, and 51 nm3, respectively), are revealing. These anions have free energies of hydration: ΔGhyd = 80.8, 51.1, and 48 M−1, respectively, and it might be expected that these free energy values correlate with their corresponding affinities. However, this is not the case: , 4300, and 35 M−1 and , 2400, and 1200 M−1, respectively. Again, this poor correlation is most certainly rooted in the fact that each anion is solvated differently and each hydrated species responds differently to the pocket or crown. Furthermore, these same three anions demonstrated CPC values of 9.4, 12, and 35 mM, respectively. So while these weakly correlate with the sum of and , they are not directly linked to ΔGhyd.
We also examined the induction-delayed precipitation of the same salts over a one-week period, with salt concentrations 50–95% of the CPC. As anticipated, only those salts with defined CPC values induced precipitation. In all cases precipitation by each salt was found to be dependent on both the nature and concentration of the anion. Figure 5 shows representative examples covering the gamut of response of host 2 to three anions: CF3SO3−, CCl3CO2−, and PF6−. In the broadest of terms the salts can be classified as those inducing precipitation at concentrations well below the CPC (Figure 5a and Figure 5b) and those salts that induced precipitation over a narrow concentration range close to the CPC (Figure 5c). The former include CF3SO3−, CCl3CO2−, and SCN−, while the latter are PF6−, ReO4−, ClO4−, BH3CN−, BF4−, and I−. Overall, anions induced precipitation over an increasingly narrow concentration range in the order CF3SO3− > CCl3CO2−, > SCN− > I− ~ BF4− ~ BH3CN− > ReO4− ~ ClO4− ~ PF6−.
Figure 5.

Representative surface plots of the absorbance intensity as a function of salt concentration (% CPC) and time (hours) for the free host 2 (10 mM sodium phosphate buffer, pH = 7.3) in the presence of (a) CF3SO3−, (b) CCl3CO2−, (c) PF6−.
There was no obvious correlation between these data and the CPC values for each salt. Thus, the anions precipitating over a wide range of concentration cover the range of CPC values, 9.2, 63, and 110 mM, as did those that induced slow precipitation over a narrow concentration range. Similarly, there was no evident correlation between the induction-delayed precipitation data and and . Considering the multiple mechanisms of precipitation that are possible, this is perhaps not surprising, but it is a reminder that this most obvious manifestation of the reverse Hofmeister effect may not be the ideal probe for detailed investigation of the phenomenon.
In the case of 2 complexed with Ada-CO2−, only CF3SO3− (5th best precipitator) and PF6− (1st precipitator) induced precipitation below the CPC value; the other salts did not. As with the DLS and CPC data for 2 and its complex, we attribute this to the latter possessing a strongly solvated anionic center (R–CO2−) at the portal of the cavity turning off aggregation pathways involving this face of the host. Only PF6−, CF3SO3−, and CCl3CO2− bind strongly enough to the pocket to compete with the guest and potentially open up this face to aggregation, but as CCl3CO2− (worst precipitator) has no affinity for the crown, displacement of the guest still only leaves one major aggregation route. Hence it too fails to induce precipitation under the conditions employed.
CONCLUSIONS
A combination of approaches reveal that even relatively simple host 2 possesses complex Hofmeister and reverse Hofmeister properties. Positand 2 possesses two principal, distinctly different binding sites: a “soft” polarizable pocket and a “hard” crown of trimethylammonium groups (Figure 2). Importantly, anion affinity for the two sites is quantitatively comparable (Table 1). However, each site is qualitatively quite distinct, with anion selectivities that are not always self-evident.
Anion binding to the cavity of 2 results in competitive exchange with amphiphiles and hence the apparent reduction in the hydrophobic effect typical of the normal Hofmeister effect. Concomitantly, binding of polarizable anions to either crown or cavity induces the reverse Hofmeister effect by charge attenuation and a decrease in the hydrophilicity of the face of the host that the binding site occupies. In general terms, increased anion affinity correlates with increased ability to induce aggregation. However, because of a multitude of reasons, there is no simple relationship between anion affinity and the resulting CPC value of free host 2 or its complex with adamantane carboxylate (Ada-CO2−). That stated, the differences in the series of CPC values for host and host–guest complex are revealing; for the complex, the reverse Hofmeister effect induced by anions that selectively bind to the cavity of 2 is attenuated or disappears entirely. Thus, the binding of a high-affinity guest with a strongly solvated carboxylate group maintains strong solvation of what otherwise would be a major aggregation face of the host. Likewise, a carboxylate-induced switching-off or reduction in the residency time of a polarizable anion in the cavity also affects the slow precipitation of host 2.
These results demonstrate several key points. First, because anion affinity for soft and hard binding sites is comparable and because anion affinity for both types of sites can induce the normal and reverse Hofmeister effects, these effects are frequently competing. Second, it is almost certainly the case that the reverse Hofmeister effect in proteins is based not only on direct anion–cation binding but also on relatively “soft” sites quite remote from any charge group. Third, given sufficient understanding of the properties of a solute, the reverse Hofmeister effect can be attenuated or turned off by blocking select anion-binding sites. Finally, except in the simplest of systems, using CPC values or analogous macroscale phenomena is unlikely to lead to an improved understanding of the reverse Hofmeister effect. Rather, the focus must be on qualifying and quantify ion recognition sites in a molecule or biomacromolecule. Hence, one of the key, long-term endeavors for supramolecular chemistry is to understand the supramolecular profiles of inorganic anions. Further studies into the reverse Hofmeister effect are ongoing.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge the support of the National Institutes of Health (Grant GM 098141). B.C.G. and T.P. also acknowledge the assistance of the National Science Foundation for a Research Experiences for Undergraduates award (Grant DMR-1460637).
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Hofmeister F Naunyn-Schmiedeberg's Arch. Pharmacol 1888, 24, 247–260. [Google Scholar]; (b) Kunz W; Henle J; Ninham BW Curr. Opin. Colloid Interface Sci 2004, 9 (1–2), 19–37. [Google Scholar]
- (2).Collins KD; Washabaugh MW Q. Rev. Biophys 1985, 18, 323–422. [DOI] [PubMed] [Google Scholar]
- (3).(a) Hamaguchi K; Geiduschek EP J. Am. Chem. Soc 1962, 84, 1329–1338. [Google Scholar]; (b) Marcus Y Chem. Rev 2009, 109 (3), 1346–70. [DOI] [PubMed] [Google Scholar]; (c) Poiseuille JML Ann. Chim. Phys 1847, 21, 76. [Google Scholar]; (d) Cox WM; Wolfenden JH Proc. R. Soc. London, Ser. A 1934, 145 (855), 475–488. [Google Scholar]; (e) Gurney RW Ionic Processes in Solution; McGraw-Hill: New York, 1953. [Google Scholar]; (f) Jenkins HDB; Marcus Y Chem. Rev 1995, 95, 2695–2724. [Google Scholar]
- (4).(a) Omta AW; Kropman MF; Woutersen S; Bakker HJ Science 2003, 301, 347–349. [DOI] [PubMed] [Google Scholar]; (b) Tielrooij KJ; Garcia-Araez N; Bonn M; Bakker HJ Science 2010, 328, 1006–1009. [DOI] [PubMed] [Google Scholar]; (c) Bakker HJ Nature 2012, 491, 533–534. [DOI] [PubMed] [Google Scholar]
- (5).(a) Jungwirth P; Cremer PS Nat. Chem 2014, 6 (4), 261–3. [DOI] [PubMed] [Google Scholar]; (b) Lo Nostro P; Ninham BW Chem. Rev 2012, 112 (4), 2286–322. [DOI] [PubMed] [Google Scholar]; (c) Zhang YJ; Cremer PS Annu. Rev. Phys. Chem 2010, 61, 63–83. [DOI] [PubMed] [Google Scholar]; (d) Xie WJ; Gao YQ J. Phys. Chem. Lett 2013, 4, 4247–4252. [DOI] [PubMed] [Google Scholar]; (e) Thormann E. RSC Adv. 2012, 2 (22), 8297. [Google Scholar]; (f) Zangi RJ Phys. Chem. B 2010, 114, 643–650. [DOI] [PubMed] [Google Scholar]; (g) Pegram LM; Wendorff T; Erdmann R; Shkel I; Bellissimo D; Felitsky DJ; Record MTJ Proc. Natl. Acad. Sci. U. S. A 2010, 107, 7716–7721. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Tobias DJ; Hemminger JC Science 2008, 319 (5867), 1197–8. [DOI] [PubMed] [Google Scholar]; (i) Li Y; Wang Y; Huang G; Ma X; Zhou K; Gao J Angew. Chem., Int. Ed 2014, 53 (31), 8074–8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Deyerle BA; Zhang Y Langmuir 2011, 27 (15), 9203–9210. [DOI] [PubMed] [Google Scholar]
- (6).Zhang Y; Cremer PS Proc. Natl. Acad. Sci. U. S. A 2009, 106, 15249–15253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Schwierz N; Horinek D; Netz RR Langmuir 2013, 29 (8), 2602–2614. [DOI] [PubMed] [Google Scholar]; (b) Schwierz N; Horinek D; Netz RR Langmuir 2010, 26 (10), 7370–7379. [DOI] [PubMed] [Google Scholar]; (c) Flores SC; Kherb J; Cremer PS J. Phys. Chem. C 2012, 116 (27), 14408–14413. [DOI] [PubMed] [Google Scholar]; (d) Paterová J; Rembert KB; Heyda J; Kurra Y; Okur HI; Liu WR; Hilty C; Cremer PS; Jungwirth P J. Phys. Chem. B 2013, 117 (27), 8150–8. [DOI] [PubMed] [Google Scholar]; (e) Lund M; Jungwirth P J. Phys.: Condens. Matter 2008, 20, 494218. [Google Scholar]
- (8).Chen X; Flores SC; Lim SM; Zhang Y; Yang T; Kherb J; Cremer PS Langmuir 2010, 26 (21), 16447–54. [DOI] [PubMed] [Google Scholar]
- (9).Boström M; Parsons DF; Salis A; Ninham BW; Monduzzi M Langmuir 2011, 27 (15), 9504–9511. [DOI] [PubMed] [Google Scholar]
- (10).Schwierz N; Horinek D; Sivan U; Netz RR Curr. Opin. Colloid Interface Sci 2016, 23, 10–18. [Google Scholar]
- (11).(a) Sokkalingam P; Shraberg J; Rick SW; Gibb BC J. Am. Chem. Soc 2016, 138 (1), 48–51. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gibb CL; Oertling EE; Velaga S; Gibb BC J. Phys. Chem. B 2015, 119 (17), 5624–38. [DOI] [PubMed] [Google Scholar]; (c) Carnegie R; Gibb CLD; Gibb BC Angew. Chem., Int. Ed 2014, 53 (43), 11498–11500. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gibb CL; Gibb BC J. Am. Chem. Soc 2011, 133, 7344–7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Gibb CLD; Gibb BC J. Am. Chem. Soc 2004, 126, 11408–11409. [DOI] [PubMed] [Google Scholar]; (b) Liu S; Whisenhunt-Ioup SE; Gibb CLD; Gibb BC Supramol. Chem 2011, 23 (6), 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Sullivan MR; Sokkalingam P; Nguyen T; Donahue JP; Gibb BC J. Comput.-Aided Mol. Des 2017, 31 (1, SAMPL5 Special Issue), 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gibb CLD; Gibb BC J. Comput.-Aided Mol. Des 2014, 28 (4), 319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yin J; Henriksen NM; Slochower DR; Shirts MR; Chiu MW; Mobley DL; Gilson MK J. Comput.-Aided Mol. Des 2017, 31 (1), 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hillyer MB; Gibb CLD; Sokkalingam P; Jordan JH; Ioup SE; Gibb BC Org. Lett 2016, 18 (16), 4048–51. [DOI] [PubMed] [Google Scholar]
- (16).(a) Lamoureux G; Roux B J. Phys. Chem. B 2006, 110 (7), 3308–3322. [DOI] [PubMed] [Google Scholar]; (b) Marcus Y. Ion Properties, 1st ed.; Marcel Dekker: New York, 1997. [Google Scholar]
- (17).Jungwirth P; Tobias DJ J. Phys. Chem. B 2002, 106, 6361–6373. [Google Scholar]
- (18).Ewell J; Gibb BC; Rick SW J. Phys. Chem. B 2008, 112, 10272–10279. [DOI] [PubMed] [Google Scholar]
- (19).Krossing I; Raabe I Angew. Chem., Int. Ed 2004, 43 (16), 2066–90. [DOI] [PubMed] [Google Scholar]
- (20).(a) Langton MJ; Serpell CJ; Beer PD Angew. Chem., Int. Ed 2016, 55 (6), 1974–87. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kubik S Chem. Soc. Rev 2010, 39 (10), 3648–63. [DOI] [PubMed] [Google Scholar]; (c) Sessler JL; Gale PA; Cho W-S Anion Receptor Chemistry; Royal Society of Chemistry: Cambridge, U.K., 2006. [Google Scholar]; (d) Gale PA; Busschaert N; Haynes CJ; Karagiannidis LE; Kirby IL Chem. Soc. Rev 2014, 43, 205–241. [DOI] [PubMed] [Google Scholar]; (e) O’Neil EJ; Smith BD Coord. Chem. Rev 2006, 250 (23–24), 3068–3080. [Google Scholar]; (f) Hirsch AKH; Fischer FR; Diederich F Angew. Chem., Int. Ed 2007, 46, 338–352. [DOI] [PubMed] [Google Scholar]; (g) Berryman O; Johnson DW Chem. Commun 2009, 3143–3153. [DOI] [PubMed] [Google Scholar]; (h) Schmidtchen FP Chem. Soc. Rev 2010, 39 (10), 3916–35. [DOI] [PubMed] [Google Scholar]; (i) Caballero A; Zapata F; Beer PD Coord. Chem. Rev 2013, 257 (17–18), 2434–2455. [Google Scholar]; (j) Dutta R; Ghosh P Chem. Commun 2014, 50 (73), 10538–54. [DOI] [PubMed] [Google Scholar]; (k) García-España E; Díaz P; Llinares JM; Bianchi A Coord. Chem. Rev 2006, 250 (23–24), 2952–2986. [Google Scholar]; (l) Schmuck C; Wich P The development of artificial receptors for small pepticles using combinatorial approaches. In Creative Chemical Sensor Systems; Schrader T, Ed.; Springer-Verlag Berlin: Berlin, 2007; Vol. 277, pp 3–30. [Google Scholar]
- (21).Wang T; Liu G; Zhang G; Craig VSJ Langmuir 2012, 28 (3), 1893–1899. [DOI] [PubMed] [Google Scholar]
- (22).Buck RP; Rondinini S; Covington AK; Baucke FGK; Brett CMA; Camoes MF; Milton MJT; Mussini T; Naumann R; Pratt KW; Spitzer P; Wilson GS Pure Appl. Chem 2002, 74, 2169–2200. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.