


Abstract
The Msh2–Msh3 mismatch repair (MMR) complex in Saccharomyces cerevisiae recognizes and directs repair of insertion/deletion loops (IDLs) up to ∼17 nucleotides. Msh2–Msh3 also recognizes and binds distinct looped and branched DNA structures with varying affinities, thereby contributing to genome stability outside post-replicative MMR through homologous recombination, double-strand break repair (DSBR) and the DNA damage response. In contrast, Msh2–Msh3 promotes genome instability through trinucleotide repeat (TNR) expansions, presumably by binding structures that form from single-stranded (ss) TNR sequences. We previously demonstrated that Msh2–Msh3 binding to 5′ ssDNA flap structures interfered with Rad27 (Fen1 in humans)-mediated Okazaki fragment maturation (OFM) in vitro. Here we demonstrate that elevated Msh2–Msh3 levels interfere with DNA replication and base excision repair in vivo. Elevated Msh2–Msh3 also induced a cell cycle arrest that was dependent on RAD9 and ELG1 and led to PCNA modification. These phenotypes also required Msh2–Msh3 ATPase activity and downstream MMR proteins, indicating an active mechanism that is not simply a result of Msh2–Msh3 DNA-binding activity. This study provides new mechanistic details regarding how excess Msh2–Msh3 can disrupt DNA replication and repair and highlights the role of Msh2–Msh3 protein abundance in Msh2–Msh3-mediated genomic instability.
Graphical Abstract
Graphical Abstract.

Introduction
Mismatch repair (MMR) is a specialized DNA repair pathway known for its role in identifying and directing the correction of errors that evade the intrinsic fidelity mechanisms of the replication machinery, thereby increasing the fidelity of replication ∼100–1000-fold (1–4). In Saccharomyces cerevisiae, two heterodimeric MutS homolog (Msh) complexes initiate MMR with distinct but partially overlapping binding affinities (5,6). Msh2–Msh6 predominantly binds and directs the repair of single base mispairs (with the exception of C–C mismatches) and 1–2 nucleotide insertion-deletion loops (IDLs) (6–10). Msh2–Msh3 binds and directs repair of some mispairs, including A–A, C–C and T–G (10–12), as well as both short and longer IDLs of up to 17 nucleotides in length (13–16). Following mismatch recognition, MutL homologs (Mlh), Mlh1–Mlh3 and/or Mlh1–Pms1 (Pms2 in humans) are recruited by MSH–DNA complexes in an ATP-dependent manner. The endonuclease activity of Mlh homologs is directed to cleave the nascent strand distal to the mismatch, an activity that requires a Msh complex. Mlh1–Pms1 is also activated by proliferating cell nuclear antigen (PCNA) (17–19), while Mlh1–Mlh3 is not (20,21). Mlh1–Mlh2 lacks endonuclease activity and acts as an accessory factor (22,23). Subsequently, Exo1 and replicative DNA polymerase delta (Pol δ), or epsilon (Pol ϵ), are recruited to remove the error and resynthesize the DNA to restore the structure of the double helix (24–26).
Msh2–Msh3 is a structure-specific DNA-binding protein that binds a variety of different DNA intermediates, with a preference for substrates with double-strand (ds)/single-strand (ss) DNA junctions (13,14,27,28). This allows Msh2–Msh3 to initiate several pathways in DNA metabolism in addition to MMR. During 3′ non-homologous tail removal (3′NHTR), a step that occurs in a sub-class of DSBR (29–32), Msh2–Msh3 binds to ds/ssDNA junctions with 3′ ssDNA non-homologous tails to stabilize them and recruits the structure-specific endonuclease Rad1-Rad10/Saw1, thereby promoting cleavage of the unannealed tails, allowing repair to proceed via DNA synthesis (29,30,33). Msh2–Msh3 also promotes heteroduplex rejection, preventing recombination between homoleogous sequences (34,35). In this context, Msh2–Msh3 binds IDL structures, similar to MMR, but recruits Sgs1 to unwind the D-loop. (36,37). Given these known functions, it is not surprising that loss of Msh2–Msh3 is associated with an increase in genomic instabilities that contribute to hereditary and sporadic cancers in humans (38–50). At the same time, Msh2–Msh3 also promotes genome instability in structure-specific contexts. One notable example is trinucleotide repeat (TNR) sequences; Msh2–Msh3 binding promotes the expansion of (CNG) tracts and likely other repeat sequences that form secondary structures (51–57), including in S. cerevisiae (28,58,59). Similarly, Msh2–Msh3 binding to B-DNA/Z-DNA junctions promotes mutation (60). Thus, Msh2–Msh3 binding to non-canonical DNA structures can compromise genome stability. Overexpression of MSH3 also results in a base-base mismatch repair deficiency that has been attributed to an imbalance of the relative protein ratios between Msh2–Msh3 and Msh2–Msh6 (61,62).
Msh2–Msh3 ATPase activity is required to promote genome stability through MMR, 3′ NHTR and heteroduplex rejection (63,64) and to promote genome instability through TNR expansions (65), although it is dispensable for DNA structure binding (14,64,66). Like MutS and Msh2–Msh6, Msh2–Msh3 contains two composite ATP-binding/hydrolysis sites with highly conserved Walker A and Walker B adenosine nucleotide-binding sites that are essential for ATP binding and hydrolysis, respectively (67–70). In S. cerevisiae, amino acid substitutions of the Walker A (G796 in Msh3), predicted to prevent ATP binding, abolished Msh2–Msh3-mediated MMR (64). Notably, Msh2–Msh3 binding to different DNA structures alters the kinetics of Msh2–Msh3 ATP binding, hydrolysis and nucleotide turnover, which impact downstream steps such as Msh2–Msh3 turnover and recruitment of partner proteins, promoting genome stability or instability (14,27,28,58,66,68,71,72).
We previously demonstrated that Msh2–Msh3 binds 5′ ssDNA flap structures, albeit with lower affinity than 3′ ssDNA flaps (14), to form a specific complex that interacts with the ss/dsDNA junction (28). 5′ ssDNA flaps are generated in at least two DNA metabolic pathways: Okazaki fragment maturation (OFM) during DNA replication and long-patch base excision repair (LP-BER). Polymerase (Pol) δ extends the initiator primer in OFM and primer upstream of the abasic site in LP-BER, eventually encountering the preceding DNA fragment. Pol δ proceeds with synthesis, displacing the 5′-end of the downstream segment, forming a single-stranded 5′ flap structure (73–75). This intermediate is cleaved by endonuclease Rad27 (Fen1 in mammals), leaving a nick that is sealed by DNA ligase Cdc9LigI (DNA Ligase I (LigI) in humans). We demonstrated that Msh2–Msh3 competes with both Rad27FEN1 and Cdc9LigI for binding to DNA substrates. This resulted in the inhibition of Rad27 FEN1 endonuclease activity, ligation and a significant reduction of Okazaki fragment processing in vitro (28). Given that 5′ flap processing is essential for DNA metabolism in vivo, uncontrolled binding of Msh2–Msh3 to 5′ flap intermediates poses a potential risk for normal DNA metabolism.
Here we present evidence that elevated levels of Msh2–Msh3 interfere with DNA metabolism in vivo through multiple pathways, likely as a result of binding to non-canonical DNA structures. The cell responds to Msh2–Msh3’s interference with a checkpoint-like response. Msh3 is present at low levels in yeast, ∼4–10 times lower than Msh2 or Msh6 (76,77). Nonetheless, even low levels of Msh2–Msh3 overexpression increased sensitivity to the alkylating drug methyl methanesulfonate (MMS), which generates lesions typically repaired by base excision repair (BER). Msh2–Msh3 overexpression also induced defects in cell cycle progression that are likely a result of Okazaki fragment stress. Our results support a model in which elevated levels of Msh2–Msh3 interfere with normal DNA metabolism, not simply by binding DNA substrates but by engaging in aberrant signaling in an ATP binding-dependent manner. This work provides novel, mechanistic information demonstrating how Msh2–Msh3, known primarily to promote genome stability, can disrupt DNA replication and repair. Our data provide a more robust understanding of how the DNA metabolic pathways that generate a variety of DNA intermediates with double-strand/single-strand junctions are affected by elevated Msh2–Msh3 levels and provides a model for how the cell responds. Furthermore, they suggest that tight regulation of the Msh2–Msh3 expression levels is important in vivo to prevent interference with both DNA synthesis and the processing of a variety of DNA structures.
Materials and methods
Plasmids and yeast strains
To generate low-copy and high-copy plasmids expressing MSH3, a SacII–PstI fragment from pEAI215 (71), which includes DNA sequence from ∼1 kb upstream of MSH3 to ∼ 500 bp downstream of MSH3 from its endogenous chromosomal location, was ligated into pRS423 (78) digested with SacII and PstI to generate pSP1. This is a high copy 2μ plasmid with a LEU2 marker. From this plasmid, a SacII–SalI fragment was excised, containing the entire MSH3 sequence from pEAI215 and ligated into pRS424 (78) and pRS414 (79) digested with SacII and SalI to generate pSP15 and pSP18, respectively. Both carry a TRP1 marker; pSP15 is a 2μ plasmid, while pSP18 is a single copy ARS CEN plasmid. Plasmids were transformed into a msh3Δ yeast strain background using the lithium acetate method (80).
Overexpression plasmids of MSH2 (pMMR8) and msh2G693D (pEAE270) (Supplementary Table S1) have been described previously (13,14,81). Galactose-inducible overexpression plasmids of MSH3 (pMMR20), msh3Y925A (pCK94 or pMME2), msh3G796A (pCK42), MSH6 (pEAE218) and empty vector (pJAS104) were described previously (Supplementary Table S1) (13,14,33,64,81). We generated msh3D870A in pEAI218 (71) by site-directed mutagenesis. A Bsu36I–MluI fragment from this plasmid, containing the msh3D870A, was sub-cloned into pMMR20 (13), to generate a galactose-inducible msh3D870A overexpression plasmid (pMME3). MSH3, msh3, or empty vector plasmids (all carrying the leu2D nutritional marker) were co-transformed with the MSH2 or msh2 overexpression plasmid (TRP1 marker) into various yeast strains using the lithium acetate method (80). For the His-PCNA (POL30) and His-pcna (pol30) mutant experiments, yb2062 (His-POL30), yb2063 (His-pol30K164R), yb2064 (His-pol30K242R), yb2066 (His-pol30K164R/K242R)(82) were made trp1- and leu2- by sequential marker swap by hisG-URA3-hisG pop-out with pNKY85 (targeting LEU2) and pNKY1009 (targeting TRP1) (83,84) to generate JSY4937-4945 (His-POL30), JSY5007-5012 (His-pol30K164R). JSY5013-14 (His-pol30K242R) and JSY5015-27 (His-pol30K164R/K242R). These strains were co-transformed with pMMR8 and pMMR20 or pJAS104.
All strains used in this study are described in Supplementary Table S2.
Galactose inducible overexpression
Cultures of a msh3Δ (JSY1505 or JSY905) or His-POL30/pol30 backgrounds carrying both pMMR8 (MSH2) or pMMR8-derived (msh2G693D) and pMMR20 (MSH3) or pMMR20-derived (msh3 alleles) (13) were grown to mid-log phase in synthetic complete (SC) medium in the presence of 2% lactate and 2% glycerol as carbon sources. Protein expression was induced by the addition of 2% galactose for 17 h. Uninduced and induced cells were collected for flow cytometry, quantitative real-time PCR (qRT-PCR), and/or western blotting to analyze PCNA modification. Cells harvested for flow cytometry were washed with sterile deionized water and fixed in 70% ethanol at 4°C for a minimum of 1 h (up to 1 week) before flow cytometry analysis. Cells harvested for RNA extraction were washed with UltraPure™ DNase/RNase-Free Distilled Water (Invitrogen), harvested by centrifugation and resuspended in β-mercaptoethanol/Buffer RLT solution as described by RNeasy Mini Kit (QIAGEN) guidelines. Aliquots from each time point were snap-frozen in liquid nitrogen and stored at -80°C until ready for processing.
RNA isolation and quantitative real-time PCR
Total RNA was isolated from cultured yeast cells using the QIAGEN RNeasy Mini Kit. As recommended by the manufacturer, residual DNA was removed by on-column DNase I digestion was carried out for 15 min. RNA concentration was determined via nanodrop. One μg of total RNA was reverse transcribed using a mix of oligo(dT) and random hexamer primers following the manufacturer's instructions of the iScriptTM cDNA synthesis kit (BioRad). Primers to detect endogenous transcript levels of MSH2, MSH3, MSH6 and PDA1 are described in Supplementary Table S3. The RT-PCR was performed at 95°C for 3 min, 40 cycles of amplification consisting of denaturation at 95°C for 15 s, annealing at 55°C for 30 s, extension at 72°C for 30 s, followed by melting curve analysis, using a CFX-95 Touch Multiplex instrument. Each RNA extraction was performed a minimum of three times and each qPCR experiment was performed in triplicate. Each PCR included a standard curve with genomic DNA, and the levels of target transcripts were normalized to that of the reference gene that encodes Pyruvate Dehydrogenase Alpha 1 (PDA1). The generation of specific PCR products was confirmed by gel electrophoresis. Experiments were done in the absence of reverse transcriptase and visualized by electrophoresis to determine lack of DNA contamination. The relative starting quantities (SQ) of mRNAs for MSH genes and PDA1 were calculated from corresponding standard curves. Standard curves had an average R2> 0.98.
Canavanine resistance assays
Mutation rates were measured at the CAN1 locus as previously described (85,86). Briefly, strains were grown on SC–Trp–Leu plates until colonies reached 2 mm in size. The carbon source in the plates was 2% glucose or 2% galactose for MSH3 uninduced or induced, respectively. Colonies were then suspended in 100 μl of 1× TE (10 mM Tris–HCl, pH 7.4; 1 mM EDTA) and diluted 1:10 000. Twenty μl of the undiluted colony suspension was plated on SC–Trp–Leu–Arg + Canavanine, and 100 μl of the 10−4 dilution was plated on SC–Trp–Leu–Arg. Both permissive and selective plates contained 2% glucose as a carbon source. The plates were incubated at 30°C until colonies reached ∼1–2 mm in size. Colonies were counted, and mutation rates and 95% confidence intervals were calculated through FluCalc fluctuation analysis software (87). Assays were performed on multiple independent isolates for each genotype on separate days.
Methyl methanesulfonate survival assays
Cultures were grown to the mid-log phase in liquid SC–Trp media for assays performed with high and low-copy plasmids. Cells were diluted and plated on appropriate SC–Trp plates or SC–Trp plates containing 0.005, 0.01, 0.015, 0.0175 or 0.020% MMS. To avoid degradation of MMS and ensure consistent results, cells were plated within hours of pouring the plates. After incubation at 30°C for 4 days, percent survival was calculated as the ratio of the number of colonies that grew in the presence of MMS relative to the no MMS control. Assays were repeated a minimum of three times with at least two independent isolates.
For assays performed with overexpression strains, cultures were grown to the mid-log phase in SC media in the presence of lactate and glycerol as carbon sources. As described above, MSH3 or msh3 expression was induced by adding 2% galactose. Cells were diluted and plated into appropriate SC–Trp–Leu plates or SC–Trp–Leu plates containing 0.005% MMS.
Cell cycle analysis by flow cytometry
After fixation in 70% ethanol, yeast cells were washed with sodium citrate/EDTA solution (50 mM sodium citrate and 1 mM EDTA [pH 8.0]) and treated with 0.06 mg RNase A (Invitrogen) at 50°C for 2 h. This was followed by adding 0.25 mg of Proteinase K (Sigma-Aldrich) and incubation at 50°C for 1–2 h. Cells were mixed with a sodium citrate/EDTA solution containing 1 μM SYTOX® Green. Stained DNA was then analyzed for chromosomal content using a BD Fortessa Flow Cytometer at an excitation wavelength of 488 nm. Data shown were analyzed using BD FACSDiva™ and FlowJo™ software.
Detection of PCNA modification by western blot
Total protein extracts from yeast strains overexpressing Msh2–Msh3 were TCA precipitated and analyzed by Western blot with an anti-PCNA antibody as described previously (88,89). Blots were imaged with Bio-Rad Chemi-Doc Touch Imaging System. Linear changes in exposure were applied to entire blots. PCNA signal intensity was quantified using ImageJ software.
DNA substrates
Oligonucleotides were synthesized by Integrated DNA Technologies (IDT, Coralville, IA) or Midland Certified Reagents Company (TX). For ATPase assays, the homoduplex (LS1/LS2) and +8 loop MMR (LS2/LS8) substrates sequences, assembly and purification were as described previously (14,66,71). Radiolabeled isotope was purchased from Perkin Elmer Life Sciences. Synthetic oligonucleotides were labeled with [γ 32P]-ATP and T4 polynucleotide kinase (New England Biolabs) at their 5′ end, as previously described (90). Following gel purification, substrate annealing was performed in a 1:2:4 ratio (labeled oligonucleotide: template: second oligonucleotide), as previously described (90). The oligonucleotides used in this study are listed in Supplementary Table S4.
Protein purification
S. cerevisiae Msh2–Msh3 (66) and Pol δ (91) were expressed and purified as previously described.
Pol δ strand extension assay
Two DNA substrates were used in the strand extension assays: the synthesis substrate and the strand displacement substrate. The synthesis substrate was created by annealing 5′ 32P-labeled 44mer to 110nt template sequence in a 1:2 ratio, respectively. The strand displacement substrate was formed by annealing a 5′ 32P-labeled 44 nucleotides upstream primer to a 110nt template containing a 60nt downstream primer in a 1:2:4 ratio. Five nM of the synthesis substrate was incubated with Pol δ (75 nM with the synthesis substrate and 150 nM for the strand displacement substrate) and increasing concentrations of Msh2–Msh3 (50, 100, 250 nM) for 30 min at 30°C. Reactions were performed in 20 μl volume in reaction buffer containing 50 mM Tris–HCl pH 8.0; 2 mM DTT, 0.25 mg/ml bovine serum albumin (BSA), 8 mM MgCl2, 1 mM ATP, 0.1 mM dNTPs, 25 mM NaCl. Reactions were terminated using 2× termination dye containing 90% formamide (v/v), 10 mM EDTA, with 0.01% bromophenol blue and xylene cyanole. Samples were then resolved on a 12% polyacrylamide gel containing 7 M urea. Products were analyzed by a PhosphorImager (Typhoon 9500) and quantified using ImageQuant version 5.2 (Molecular Dynamics). All experiments were done at least in triplicate. Representative gels are shown.
ATPase assays
Hydrolysis of ATP was monitored using a coupled spectrophotometric assay as described previously (66,92). In this assay, the conversion of ATP to ADP and Pi is linked to the oxidation of NADH to NAD+ and is monitored as a decrease in absorbance at 340 nm. Assays were performed at 30°C and monitored using a Varian, Cary-50 Bio UV–vis spectrophotometer. The reactions contained 20 mM Tris-acetate (pH 7.5), 0.3 mM NADH, 5 mM PEP, 20 U/ml pyruvate kinase, 20 U/ml lactate dehydrogenase, 2 mM magnesium acetate, DNA (250 nM) and Msh2–Msh3 (50 nM) and up to 2.5 mM ATP. Msh2–Msh3 was pre-bound to DNA, followed by adding ATP in small increments. Approximately 80 data points were fit to a linear curve. The rate of ATP hydrolysis at each ATP concentration was calculated by multiplying the slope of the line by 159 (the change in absorbance of NADH per unit time) (92).
Results
Characterization of MSH3 expression levels
Msh2–Msh3 binding interferes with the processing of 5′ ssDNA flap intermediates by Rad27FEN1 and Cdc9LigIin vitro, competing with Rad27FEN1 for binding the DNA intermediate (28). Because 5′ ssDNA flaps are generated in multiple DNA metabolic pathways, such as lagging strand synthesis, and long-patch BER, we were intrigued by the possibility that Msh2–Msh3 might interfere in these processes in vivo, leading to genome instability and replication stress. Notably, elevated levels of Msh2–Msh3 are a key driver of genome instability in eukaryotes via TNR expansions (93,94). Based on these results, we set out to test the hypothesis that elevated Msh2–Msh3 levels can drive genomic instability via 5′ flap DNA intermediates.
To address this hypothesis, we established a series of plasmids to express MSH3 at different levels. First, we generated a single copy ARS CEN plasmid carrying MSH3 under the control of its endogenous promoter (pSP18, MSH3-LC). Second, we constructed a high copy 2μ plasmid carrying MSH3 under the control of its endogenous promoter (pSP15, MSH3-HC). Finally, MSH2 and MSH3 were co-overexpressed as described previously; MSH2 (pMMR8) expression was under the control of the ADC1 promoter, expressed constitutively and at elevated levels, and MSH3 (pMMR20) was expressed under the control of a galactose-inducible GAL-PGK promoter (13,14).
The endogenous Msh3 protein levels are not detectable by western blot (64). Instead, we measured expression levels by qRT-PCR, although we note that there is not necessarily a linear relationship between gene expression and protein levels. RNA was extracted from wild-type and msh3Δ cells. Consistent with low endogenous Msh3 protein levels, we observed endogenous MSH3 mRNA levels to be six times lower than endogenous MSH2 or MSH6 expression levels (Figure 1A, B). The MSH3 mRNA levels were four and sixty-four times higher than MSH3 endogenous levels when expressed from MSH3-LC or MSH3-HC plasmids, respectively (Figure 1A, B). No significant differences in MSH2 or MSH6 expression levels were observed in the presence of MSH3-LC or MSH3-HC. (Figure 1B).
Figure 1.

Expression levels of MSH2, MSH3 and MSH6 in different cellular contexts. Endogenous mRNA levels of MSH2 (red), MSH3 (blue) and MSH6 (green) were measured using RT-qPCR. (A) RNA was isolated from MSH3 or msh3Δ yeast cells at mid-log phase. (B) RNA was isolated at mid-log phase from msh3Δ strains carrying either an empty vector or either a low copy number (ARS CEN; LC) or a high copy number (2 micron; HC) plasmid, bearing MSH3 under the control of the endogenous MSH3 promoter. (C) RNA was isolated from strains co-overexpressing MSH2, under a constitutive promoter, and MSH3, under a galactose-inducible promoter. Culture aliquots were collected at indicated hours (h) after induction with galactose. All RNA levels were normalized to the reference gene PDA1. Data represents the mean of at least three independent experiments. Error bars represent SEM.
The msh3Δ strain was co-transformed with MSH2 (pMMR8), and a second plasmid bearing either a galactose-inducible copy of MSH3 (pMMR20) or an empty vector control derived from pMMR20 (pJAS104 [EV]). Prior to galactose induction, we observed an increase in MSH3 mRNA of approximately ∼2300-fold over MSH3 endogenous levels in these strains (Figure 1A, C), indicating read-through transcription in the absence of galactose. No MSH3 mRNA was observed in the control msh3Δ strain co-expressing MSH2 and the empty vector (pJAS104) (Supplementary Figure S1). After induction by galactose for 17 h, MSH3 expression levels increased to ∼14-fold compared to pre-induction (0h) levels, ∼32 000 times higher than the endogenous levels of MSH3 mRNA (Figure 1A, C). Using these constructs, we previously demonstrated detectable Msh3 or msh3 protein levels following induction, including msh3 alleles tested here, although protein levels are still low (64).
Studies in human cell lines demonstrated a strong Msh2–Msh6-specific mutator phenotype when MSH3 is overexpressed, presumably because excess Msh3 competes with Msh6 for interactions with Msh2 (61,62). To functionally test MSH3 overexpression in yeast, we performed canavanine resistance assays, a Msh2–Msh6-specific mutator assay following galactose-induced overexpression of MSH3 (Table 1). MSH3 overexpression (O/E) in galactose increased the mutation rate ∼9-fold over the rate observed in MSH3 O/E grown in glucose, similar to the elevated mutation rate in msh6Δ, consistent with the presence of elevated Msh3 protein. Growth of WT strains in galactose in the absence of MSH3 O/E did not have this effect (Table 1). MSH3-HC also increased the mutation rate, albeit to a lesser extent (∼2-fold), while MSH3-LC had no effect (Table 1).
Table 1.
Canavanine resistance mutation rates
| Genotype | Carbon source | Rate of canavanine resistance [95% confidence intervals] | Change (relative to glucose) |
|---|---|---|---|
| WT # (n = 22) | Glucose | 3.9 × 10−7 [2.5 × 10−7–5.5 × 10−7] | 1 |
| WT# (n = 22) | Galactose | 2.6 × 10–7 [1.7 × 10−7–3.7 × 10−7] | 0.7 |
| WT + MSH3 O/E* (n = 66) | Glucose | 7.9 × 10−7 [6.5 × 10−7–9.3 × 10−7] | 1 |
| WT + MSH3 O/E* (n = 121) | Galactose | 7.2 × 10−6 [6.6 × 10−6–7.9 × 10−6] | 9.1 |
| Genotype | Carbon source | Rate of canavanine resistance [95% confidence intervals] | Change (relative to empty vector) |
| WT+ HC& (n = 77) | glucose | 4.2 × 10−7 [3.3 × 10−7–5.1 × 10−7] | 1 |
| WT+ MSH3-HC% (n = 77) | glucose | 8.0 × 10−7 [6.6 × 10−7–9.5 × 10−7] | 1.9 |
| WT+ LC$) (n = 77) | glucose | 4.3 × 10−7 [3.4 × 10−7–5.2 × 10−7] | 1 |
| WT+ MSH3-LC+ (n = 77) | glucose | 4.3 × 10−7 [3.5 × 10−7–5.1 × 10−7] | 1 |
#FY23; * JSY321-323; & JSY263; % JSY264; $ JSY265; + JSY266.
Overexpression of MSH2 MSH3 leads to MMS sensitivity
Given that Msh2–Msh3 interferes with 5′ flap processing in vitro, we tested whether elevated MSH3 expression sensitized the cells to MMS, a monofunctional alkylation agent that methylates DNA at N7-deoxyguanine and N3-deoxyadenine (95), a lesion typically repaired by the BER pathway. msh3Δ yeast cells carrying MSH3-LC or MSH3-HC plasmids were grown to mid-log phase, serially diluted and grown on selective (SC-trp) plates in the absence or presence of MMS. At 0.015%, 0.0175% and 0.02% concentrations of MMS, cells carrying either MSH3-LC or MSH3-HC exhibited mild but significant sensitivity to MMS compared to empty vector controls (Figure 2A). When MSH2 and MSH3 were co-overexpressed (MSH2 MSH3 O/E), yeast cells exhibited significant sensitivity to 0.005% MMS compared to overexpression of MSH2 alone (Figure 2B). With higher MSH3 overexpression, cells were sensitive to much lower MMS concentrations, consistent with a correlation between Msh3 levels and MMS sensitivity. These results indicate that excess Msh2–Msh3 compromises LP-BER, suggesting interference with 5′ flap processing in vivo. MSH2 MSH6 overexpression has also been shown to promote MMS sensitivity (96).
Figure 2.

Elevated MSH3 expression renders cells sensitive to methyl methanesulfonate (MMS). (A) MMS sensitivity of msh3Δ strains carrying either a low-copy (MSH3 LC, light shade) or high-copy number (MSH3 HC, dark shade) plasmid containing either empty vector (EV, gray lines) or MSH3 (blue lines) were grown in increasing concentrations of MMS. Plotted graphs represent at least two independent experiments done in triplicate and with at least two independent transformants. Error bars were calculated by SEM; P values (two-way ANOVA) were calculated in Prism (MSH3 LC versus EV LC P < 0.0001; MSH3 HC versus EV HC, P = 0.0317). (B) MMS sensitivity of msh3Δ strains co-overexpressing MSH2 and MSH3. Cultures were grown to mid-log phase and MSH3 expression was induced by the addition of galactose. Time point 17 h after induction was collected, serial diluted and plated onto SC agar plates in the absence or presence of 0.005% MMS. Plotted data represents the results of at least three independent experiments. Error bars represent the SEM; P values (two-way ANOVA) were calculated in Prism (*P = 0.0297).
Overexpression of MSH2 MSH3 interferes with cell cycle progression
Okazaki fragment maturation (OFM) also requires the processing of a displaced 5′ ssDNA flap; interference with OFM causes delays in cell cycle progression (82,89,97–100). Therefore, we assessed cell cycle progression by flow cytometry in cells overexpressing MSH2 and MSH3. After MSH2 and MSH3 co-overexpression, MSH2 MSH3 O/E shown in Figure 1C, we observed substantial defects in cell cycle progression, which were not observed in the presence of the empty vector or MSH2 and MSH6 co-overexpression (Figure 3). Under these conditions, Msh3 protein is not visible in cleared lysates, whereas Msh6 is (data not shown;(7)), indicating high levels of Msh2–Msh6 upon overexpression compared with Msh2–Msh3. The cell cycle profile shifted dramatically, with an apparent accumulation of cells in the early S phase and a complete loss of defined G1 (1C) and G2/M (2C) populations. We quantified cell populations in G1, S or G2/M phases of the Cell Cycle function in FlowJo software (see Supplementary Figure S2). Upon MSH2 MSH3 co-overexpression, we observed a substantial increase in the number of cells in the S phase with a concomitant decrease in cells in G1 and, to a lesser extent, in G2/M (Figure 3, Supplementary Figure S3). This accumulation of cells in the S phase is typically associated with slowed fork progression and/or DNA damage (101–103). These data suggest that overexpression of Msh2–Msh3 interferes with normal DNA metabolism, slowing down or inhibiting S phase. Notably, when the cells were released back into glucose, reversing the MSH3 induction, they eventually resumed normal cell cycle progression (Figure 4). Notably, the cell cycle profile of cells co-overexpressing MSH2 MSH3 resembled that of rad27Δ cells at elevated temperature (100), consistent with a model in which excess Msh2–Msh3 outcompetes Rad27FEN1in vivo and MSH2 MSH3 overexpression makes the cells functionally RAD27 null.
Figure 3.

MSH2 MSH3 overexpression induces a delay in cell cycle progression. MSH2 and MSH3 were overexpressed following galactose induction in a msh3Δ background. Aliquots were collected at 0 and 17 h after induction. (A) Histograms are shown of chromosomal content of asynchronous populations of MSH2 + empty vector (EV), MSH2 + MSH3 and MSH2 + MSH6 at several timepoints following addition of galactose. 1C indicates 1× DNA content; 2C indicates 2x DNA content. (B) Quantification of relative proportion of cells in different phases of the cell cycle for MSH2 + EV, MSH2 + MSH3 or MSH2 + MSH6. The percentage of cells in G1 (1C), G2/M (2C) or S (between 1C and 2C) phases was determined using FlowJo software (see Supplementary Figure S2 for details). Plotted values correspond to data collected from at least three independent experiments from at least two independent isolates. Error bars represent SEM. Pairwise t-tests indicated significant differences in the number of cells in G1 and S phase between MSH2 EV and MSH2 MSH3 O/E (G1, P< 0.0001; S, P = 0.0026) and between MSH2 MSH3 O/E and MSH2 MSH6 O/E (G1, P = 0.0023; S, P = 0.0011).
Figure 4.

Recovery from MSH2 MSH3 overexpression. MSH2 and MSH3 were overexpressed and induced as previously described. Cells were released from induction by transfer into glucose-containing media. Samples were collected and fixed for flow cytometry analysis (A) before induction (–17 h), after induction (0 min), after release into glucose every 30–60 min for 8 h, and (B) 24 h after release. Histograms show the distribution of chromosomal content at each time point. (C) Cell cycle profiles were quantified using FlowJo software (see Supplementary Figure S2).
MSH3 overexpression enhances PCNA post-translational modification
Interference with Okazaki fragment processing leads to cell cycle delays and DNA damage responses, signaling cascades that are partly mediated by post-translational modifications in PCNA (88,89,100). We examined whether MSH2 MSH3 overexpression triggered PCNA modifications, which would support the hypothesis that a DNA damage response is activated. Following MSH2 MSH3 overexpression, whole-protein TCA precipitation was performed, and the resulting cell extracts were analyzed by western blot with α-PCNA antibody (88,89). We observed two PCNA-specific bands, one consistent with the size of unmodified PCNA (∼29 kDa) (Figure 5A) and a PCNA band with lower mobility (∼49 kDa), consistent with a post-translationally modified form of the protein. This modified PCNA band was significantly increased following MSH2 MSH3 overexpression compared to the empty vector (Figure 5B) and MSH2 MSH6 overexpression (Supplementary Figure S4).
Figure 5.

MSH2 MSH3 overexpression induces post-translational modification of PCNA. MSH2 and MSH3 or empty vector (EV) were overexpressed in a msh3Δ background, as previously described. Following induction, TCA protein extracts were prepared, and the proteins were separated by 12% SDS-PAGE, transferred onto a membrane and then probed with anti-PCNA (P4D1), a gift from the Stillman lab. (A) Western blot for PCNA. Modified PCNA is marked as M-PCNA. The asterisk indicates non-specific bands. These data are from a single blot, but not adjacent lanes, as indicated by the vertical line. (B) PCNA western blot quantification, shown as relative band densities of modified versus unmodified PCNA in terms of the percentage of total PCNA signal (measured as the sum of unmodified and modified bands). A pairwise t-test was performed to test significance of the difference in the proportion of M-PCNA; P< 0.0001.
PCNA is differentially modified in response to distinct signaling pathways (88,104,105). To determine the residue at which PCNA is modified following MSH2 MSH3 overexpression, we compared the modification of His-PCNA, His-pcnaK164R, His-pcnaK242R and His-pcnaK164R/K242R. Mutation of these highly conserved lysines to arginine renders them unmodifiable with either SUMO or ubiquitin. When MSH2 MSH3 was overexpressed in the presence of His-pol30-K164R or His-pol30-K164/RK242R, modified PCNA was no longer detectable. The His-pol30-K242R exhibited modification levels similar to His-PCNA (Figure 6C). These results indicated that the MSH2 MSH3 overexpression-dependent post-translational modification of PCNA occurs at K164 PCNA and not at K242.
Figure 6.

PCNA post-translational modification in the context of MSH2 MSH3 overexpression occurs at the K164 residue. MSH2 and MSH3 or MSH2 and empty vector were overexpressed following galactose induction in a His-POL30, His-pol30K164R, His-pol30K242R, or His-pol30K164R + K242R, background. Aliquots were collected and fixed for flow cytometry analysis before (0h) and after (17h) induction. After induction, cells were harvested for TCA extraction and anti-PCNA western blot. (A) Histograms are shown of chromosomal content of asynchronous populations before and after induction with galactose. Flow cytometry experiments were repeated at least three times, with at least two independent transformants. 1C indicates 1× DNA content; 2C indicates 2× DNA content. (B) Quantification of relative proportion of cells in different phases of the cell cycle. The percentage of cells in G1 (1C), G2/M (2C) or S (between 1C and 2C) phases was determined using BD FlowJo software (see Supplementary Figure S2 for details). Plotted values correspond to data collected from at least three independent experiments. Error bars represent SEM. Pairwise t-tests comparing the number of cells in G1 and S phase following MSH2 MSH3 O/E in His-POL30 versus His-pol30 alleles were performed. The counts were significantly different for His-POL30 versus His-pol30K164R (P< 0.0001 for G1 and S phase cells) or His-pol30K164R + K242R (GI: P = 0.0098, S: P< 0.0001). In contrast, the cells counts were not significantly different between His-POL30 and His-pol30K242R (G1: P = 0.8497; S: P = 0.8314). (C) PCNA western Blot. Note that the PCNA and M-PCNA of his-POL30 strains migrates at a higher weight due to the His-tag. These data are from a single blot, but not adjacent lanes, as indicated by the vertical lines. (D) PCNA western Blot Quantification as described above. Pairwise t-tests indicated significant difference in the proportion of M-PCNA in His-POL30 compared to His-pol30K164R or His-pol30K164R + K242R (p<0.0001) but not compared to His-pol30-K242R (P = 0.4152).
We analyzed TCA-precipitated His-PCNA by western blot, using an α-ubiquitin (α-Ub) antibody (Supplementary Figure S5A) and were unable to detect the MSH2 MHS3 overexpression-dependent modified PCNA band. In contrast, the MSH2 MSH3 overexpression-dependent PCNA modification exhibited the same mobility as the PCNA modification induced by high MMS levels (Supplementary Figure S5B), which are known to promote PCNA sumoylation (105,106). Finally, the PCNA modification was abrogated in the absence of SIZ1 (Figure 7A, B), which encodes the SUMO E3 ligase responsible for K164 sumoylation (107). Notably, the MSH2 MSH3 overexpression-dependent cell cycle phenotype was also dependent on SIZ1. There was no S phase accumulation following MSH2 MSH3 overexpression in siz1Δ (Figure 7C, D, Supplementary Figure S6).
Figure 7.

MSH2 MSH3 overexpression-induced cell cycle defects are dependent on SIZ1. MSH2 and MSH3 were overexpressed in a siz1Δ background, as previously described. Samples were collected for flow cytometry analysis before and after induction, and cells were harvested for western blot after induction. (A) Western blot for PCNA. Modified PCNA is marked as M-PCNA. These data are from a single blot, but not adjacent lanes, as indicated by the vertical lines. (B) PCNA western blot quantification as previously described. The proportion of M-PCNA is significantly different in the SIZ1 versus siz1Δ backgrounds (P< 0.0001) determined by a pairwise t-test. (C) Flow cytometry analysis before and after induction. (D) Quantification of cell cycle analysis based on flow cytometry data. Pairwise t-tests indicated significant difference in cell counts in G1 (P< 0.0001) and S (P = 0.0026).
The cell cycle analysis of these strains indicated that K164 is also required for the MSH2 MSH3 overexpression-dependent accumulation of cells in S phase, while K242 was not essential. We note that the His-PCNA strain had a distinct cell cycle profile, with fewer cells in G2/M. Nonetheless, the accumulation of cells in the S phase remained clearly observable following MSH2 MSH3 overexpression in this HIS-POL30 background (Figures 6A, B).
The MSH2 MSH3 overexpression phenoype is RAD9-dependent
MSH2 MSH3 overexpression leads to cell cycle delays, from which the cells recover, and post-translational modification of PCNA, all suggestive of activation of a cell cycle checkpoint response. Rad9, a cell cycle checkpoint protein involved in DNA damage signaling, is activated in response to impaired Okazaki fragment processing (89,108). We, therefore, overexpressed MSH2 MSH3 in a rad9Δ. Deletion of RAD9 was sufficient to suppress the cell cycle disruption caused by MSH2 MSH3 overexpression (Figure 8A, B). The proportion of PCNA modified following MSH2 MSH3 overexpression was also reduced in a rad9Δ background (Figure 8C, D), although there was some residual modification, ∼25% of what was observed in RAD9 cells. These results indicated that a Rad9-mediated cell cycle checkpoint response becomes activated in the presence of excess Msh2–Msh3, leading to cell cycle arrest and PCNA modification, potentially as a result of Msh2–Msh3 interference with OFM.
Figure 8.

MSH2 MSH3 overexpression-induced cell cycle defects are dependent on RAD9. MSH2 and MSH3 were overexpressed in a rad9Δ background, as previously described. Samples were collected for flow cytometry analysis before and after induction. (A) Flow cytometry analysis before and after induction. (B) Quantification of cell cycle analysis based on flow cytometry data. Pairwise t-tests indicated significant difference in cell counts in G1 and S phases (P< 0.0001) in RAD9 versus rad9Δ. (C) Western blot for PCNA. Modified PCNA is marked as M-PCNA. (D) PCNA western blot quantification as previously described. The proportion of M-PCNA is significantly different in the RAD9 versus rad9Δ backgrounds (P< 0.0001) determined by a pairwise t-test.
ELG1 is required for PCNA modification and cell cycle defects caused by MSH2 MSH3 overexpression
During lagging strand synthesis, PCNA must be loaded onto DNA at each Okazaki fragment by Replication Factor C (RFC), while PCNA unloading is carried out by a related complex in which Elg1 replaces the primary subunit in RFC, known as the Elg1-Replication Factor C-like Complex (Elg1-RLC) (109). Elg1 unloads both unmodified and SUMOylated PCNA but preferentially binds to SUMOylated PCNA (110). Elg1 unloading of PCNA at each Okazaki fragment is dependent on successful processing and ligation of the Okazaki fragment (111). Therefore, we decided to test the effects of MSH2 MSH3 overexpression in an elg1Δ background. We found that the MSH2 MSH3 overexpression-dependent cell cycle defect was abrogated in elg1Δ (Figure 9, Supplementary Figure S6). We also observed reduced modification of PCNA (Figure 9). The requirement of ELG1 for the cell cycle defect is consistent with the hypothesis that MSH2 MSH3 overexpression interferes with Okazaki fragment processing in vivo.
Figure 9.

MSH2 MSH3 overexpression-induced cell cycle defects are dependent on ELG1. MSH2 and MSH3 were overexpressed in an elg1Δ background, as previously described. Samples were collected for flow cytometry analysis before and after induction. (A) Flow cytometry analysis before and after induction. (B) Quantification of cell cycle analysis based on flow cytometry data. Pairwise t-tests indicated significant difference in cell counts in G1 (P = 0.0005) and S (P = 0.0022) in ELG1 versus elg1Δ. (C) Western blot for PCNA. Modified PCNA is marked as M-PCNA. These data are from a single blot, but not adjacent lanes, as indicated by the vertical line. (D) PCNA western blot quantification as previously described. The proportion of M-PCNA is significantly different in the ELG1 versus elg1Δ backgrounds (P = 0.0003) determined by a pairwise t-test.
msh3 alleles that disrupt ATP-binding and hydrolysis activities suppress disruption of cell cycle progression and PCNA post-translational modification
One possibility was that in vivo, excess Msh2–Msh3 binding to 5′ ssDNA flaps sterically interfered with Okazaki fragment maturation. Alternatively, Msh2–Msh3 bound to 5′ ssDNA flaps could promote aberrant Msh2–Msh3 activity that contributed to a signaling cascade leading to genome instability, similar to what is thought to occur in the presence of TNR structures (65,93). To distinguish between these possibilities, we tested the ability of ATPase-deficient msh2 and msh3 mutants to affect cell cycle progression. ATP is essential for the function and regulation of Msh2–Msh3 but is not required for DNA binding (13,14,63,64,66). Therefore, if Msh2–Msh3 simply binds to DNA structures and blocks Rad27FEN1, ATP binding, and/or hydrolysis should be dispensable for inducing the cell cycle defect. In fact, defects in these activities might even exacerbate the phenotype, as ATP hydrolysis contributes to Msh2–Msh3 turnover on the DNA (14,66). Alternatively, functional ATPase activity may be required to observe this effect, e.g. to recruit downstream proteins, as is the case for promoting TNR expansions (65,93).
To test these possibilities, we co-overexpressed MSH2 and either msh3G796A or msh3D870A (64,93). These mutations disrupt the highly conserved Walker A motif (msh3G796), which mediates ATP binding, or the Walker B motif (msh3D870), which mediates ATP hydrolysis (64,68). Notably, the cell cycle defect was significantly less pronounced when either msh3 allele was overexpressed compared to the overexpression of wild-type MSH3 (Figure 10A, C). This suggests that full induction of the cell cycle defect requires Msh3 ATP binding and hydrolysis. Disruption of the Msh2 Walker A motif (msh2G693D MSH3) also suppressed the cell cycle defects when overexpressed (Figure 10E), indicating that ATP binding to both Msh2 and Msh3 is required for Msh2–Msh3 to disrupt cell cycle progression.
Figure 10.

MSH2 MSH3 overexpression-mediated cell cycle defect depends on Msh3 ATP binding and hydrolysis. MSH2 (A–D) or msh2G693D (E) was co-overexpressed with MSH3 (B, D) or msh3 alleles (A, C) in a msh3Δ (A, C) or mlh1Δ (B, D) background. MSH2 was constitutively overexpressed; for all others, expression was induced with galactose. Aliquots were collected at 0 and 17 h after induction. Harvested cells were fixed, stained and processed by flow cytometry. Histograms of the asynchronous population are shown. Pairwise t-tests were performed to determine whether observed differences in G1 and S cell counts were significantly different. We observed significant differences between MSH2 MSH3 and MSH2 msh3G796A (G1: P = 0.0105; S: P = 0.0351) or MSH2 msh2D870A (G1: P< 0.0001; S P = 0.0007), but not MSH2 msh3Y925A (G1; P = 0.9298; S: P = 0.5414). We also observed significant differences between MLH1 and mlh1Δ backgrounds (G1 and S, P< 0.0001).
We overexpressed MSH2 msh3Y925A, a separation-of-function allele that is defective in MMR but functional in 3′NHTR (64). Based on the human Msh2–Msh3 crystal structure, Y925 is predicted to regulate nucleotide occupancy of the nucleotide-binding pocket by pushing a conserved phenylalanine (F940) into the nucleotide-binding pocket (68). In vitro, Msh2-msh3Y925A retains ATP hydrolysis activity, but the kinetics of hydrolysis are significantly altered, indicating a defect in the regulation of ATP binding, ATP hydrolysis, and/or nucleotide turnover (Supplementary Figure S7). When overexpressed, the msh3Y925A allele (Supplementary Figure S4) conferred cell cycle defects indistinguishable from the wild-type MSH3 overexpression profile (Figure 10A, C). This is consistent with the hypothesis that ATP binding/hydrolysis by Msh2–Msh3 is required to impose cell cycle defects. However, regulation of this activity is less important, as observed for 3′ NHTR (64).
We also analyzed the effects of these msh3 alleles on PCNA post-translational modification. When either the Walker A (msh3G796A) or Walker B (msh3D870A) motif was disrupted, post-translational modification of PCNA in response to MSH2 msh3 co-overexpression was decreased to a similar extent (Figure 11A,B). Co-overexpression of MSH2 msh3Y925A promoted PCNA modification (Supplementary Figure S4) at a level similar to wild-type, indicating that misregulated ATPase activity is sufficient for this effect. These results correlated with the intermediate effects of the Walker A and Walker B mutations and the wild-type effect of MSH2 msh3Y925A overexpression on the cell cycle progression phenotype (Figure 10C). MSH2 MSH6 overexpression did not induce enhanced PCNA modification (Supplementary Figure S4).
Figure 11.

MSH2 MSH3 overexpression induces PCNA post-translational modification in an ATP binding-and hydrolysis-dependent manner. MSH2 and MSH3 or msh3 alleles were overexpressed in a msh3Δ (A) or mlh1Δ (C) background, as previously described. Western blot for PCNA. Modified PCNA is marked as M-PCNA. The data in (A) are from a single blot, but not adjacent lanes, as indicated by the vertical line. (B, D) PCNA western blot quantification as previously described. Pairwise t-tests were performed to determine whether the differences in the proportion of M-PCNA were significant. We observed significant differences between MSH2 MSH3 and MSH2 msh3G796A or MSH2 msh2D870A (P< 0.0001), but not MSH2 msh3Y925A (P = 0.6901). We also observed significant differences between MLH1 and mlh1Δ backgrounds (P = 0.0332).
These data indicate that an active Msh2–Msh3-mediated pathway alters cell-cycle progression and induces post-translational modification of PCNA that might indicate replication stress and/or activation of a DNA damage response with Msh2–Msh3 levels are elevated.
Downstream steps in MMR are required for cell cycle progression defects when MSH3 is overexpressed
The observation that Msh2–Msh3 ATP binding and hydrolysis activities are required to observe defects in cell cycle progression indicated that downstream steps in a Msh2–Msh3-mediated pathway might also be required to observe this phenotype. In MMR, Msh2–Msh3 DNA-binding leads to the recruitment of Mlh complexes and activation of their latent endonuclease activity (20,21,112–116). We tested whether MLH1 is required for the cell cycle defect when MSH2 and MSH3 are co-overexpressed. We created a mlh1Δ strain, effectively inhibiting any downstream MMR activity by eliminating all three Mlh complexes: Mlh1–Pms1, Mlh1–Mlh2 and Mlh1–Mlh3. The MSH2 MSH3 overexpression-dependent cell cycle defect was eliminated in the mlh1Δ background (Figure 10B,D; Supplementary Figure S6). These results indicate that one or more Mlh complexes contribute to Msh2–Msh3-mediated replication stress that disrupts the cell cycle. Notably, we still observed enhanced PCNA modification under these conditions (Figure 11C, D), indicating the elevated Msh2–Msh3 is sufficient for this effect.
Msh2-Msh3 modulates DNA polymerase δ synthesis activity in vitro
We previously demonstrated that Msh2–Msh3 interferes with Rad27FEN1 and Cdc9LigI activity when allowed to bind the relevant DNA substrates (28). Given that Msh2–Msh3 binds to ss/dsDNA junctions, we considered the possibility that Msh2–Msh3 interacting with different DNA structures might also impact DNA polymerase activity. As Pol δ is likely recruited in both Okazaki fragment processing and LP-BER, we tested Msh2–Msh3’s effect on Pol δ activity in vitro in the presence of a simple primer-template (synthesis) DNA substrate or a strand displacement substrate (Figure 12). We compared Msh2–Msh3 substrate binding efficiency on both DNA structures and found Msh2–Msh3 binds efficiently to both, albeit with lower affinity on the strand displacement substrate compared to the synthesis substrate (compare lanes 3 and 6, Figure 12A). Next, we assessed the ability of Msh2–Msh3 to modulate Pol δ synthesis on the synthesis substrate. Titration of Msh2–Msh3 into a primer extension reaction inhibited synthesis by Pol δ (Figure 12A). We observed similar inhibition, albeit to a lesser degree, on the strand displacement substrate [observe substrate retention (44nt) in lanes 8–10, containing Msh2–Msh3] (Figure 12B). However, in contrast to synthesis inhibition, we also observed a stimulation in the strand displacement synthesis products (lanes 8–10). We hypothesize this stimulation to occur on account of Msh2–Msh3 binding to the 5′ primer terminus of the ssDNA/dsDNA junction and transiently opening up the downstream primer allowing for increased Pol δ strand displacement synthesis on a small subset of substrates. These results supported the hypothesis that Msh2–Msh3 can modify DNA metabolism pathways in a DNA structure-dependent manner, with variable impacts on genome stability.
Figure 12.

Msh2–Msh3 modulate DNA polymerase δ activity in a DNA substrate-dependent manner. (A) Msh2–Msh3 binds to the synthesis substrate (left) and the strand displacement substrate (right) with different affinities. Quantification is shown in right panel. (B) Msh2–Msh3 inhibits DNA polymerase δ in the presence of synthesis substrate (left) and stimulates its strand displacement synthesis activity (right).
Discussion
Msh2–Msh3 binds to a wide range of DNA structures (13,14,27,28,68,117,118). In this study, we demonstrated that Msh2–Msh3 could potentially interfere with in vivo DNA metabolism pathways that involve distinct DNA structures, including 5′ ssDNA flap intermediates. Even low levels of MSH3 overexpression increased in vivo MMS sensitivity, and higher levels compromised progression through the S phase. This effect was not simply due to Msh2–Msh3 binding to DNA but rather required downstream steps, including ATPase activity and Mlh complexes. MSH3 overexpression appeared to trigger a RAD9-dependent DNA damage checkpoint response and modification of PCNA at K164.
Maintaining the ‘right’ amount of Msh2-Msh3 is important for genome stability
Altered Msh2–Msh3 expression, up or down, can be deleterious. Downregulation of Msh2–Msh3 is linked to tumorigenesis and cancer (38,47,96), while upregulation of Msh2–Msh3 promotes TNR expansions (93,94). Elevated Msh2 Msh3 has not been associated with human cancers (96). At the same time, Msh complexes are differentially expressed among organisms and tissues (61,94,96,119,120). For instance, analysis of the abundance of different Msh complexes in actively proliferating murine tissues (including testis, spleen and thymus) showed high expression of Msh6 but no Msh3; on the other hand, low proliferative tissues such as muscle, heart and brain showed high MSH3 expression levels (119). Msh2–Msh3 protein levels were ∼10× lower than Msh2–Msh6 in human fibroblasts cell lines (121). These differences suggest that proliferating cells produce enough Msh2–Msh3 to participate in processes that maintain genome stability (e.g. MMR and 3′NHTR) but keep Msh3 at lower levels relative to Msh2 and Msh6 to limit aberrant DNA metabolic processes.
We previously demonstrated approximately equal levels of Msh2 and Msh6 levels in logarithmically growing cells, by quantitative immunoblotting (122). While Msh2 and Msh6 expression seem to depend on one another for stability, Msh3 is not stabilized by the presence of either Msh2 or Msh6 (123). Other studies measuring relative levels of Msh2, Msh3 and Msh6 by mass spectrometry (77) or quantitative immuno-purification (76) have observed an excess of Msh2 and Msh6 relative to Msh3. In this study, we assessed the transcript levels of untagged, endogenous MSH2, MSH6 and MSH3 by qRT-PCR. Our results indicate that a wild-type yeast strain has a relative endogenous mRNA expression pattern of: Msh6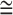 Msh2
Msh2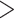 Msh3. Overexpression of Msh3, which generates an imbalance of this distribution, was previously linked with strong mutator phenotypes in human cells (61,62). Consistent with this, when we overexpressed MSH3 alone, we observed an increase in canavanine resistance, which primarily measures Msh2–Msh6 activity, indicating a decrease in Msh2–Msh6 function (Table 1). Decreased Msh2–Msh6 activity when Msh3 is overexpressed is presumably caused by a reduction in the formation of Msh2–Msh6 protein complexes, disrupting the ‘balance’ between Msh2–Msh6 and Msh2–Msh3 complex formation. When we co-overexpressed MSH2 and MSH3 in budding yeast, we observed distinct phenotypes that included sensitivity to alkylating DNA damage (Figure 2), cell cycle delays (Figures 3 and 4) and an enhancement of post-translationally modified PCNA (Figure 5). Our results suggest that controlling the abundance of Msh3 is a mechanism by which cells can limit the interactions of Msh2–Msh3 with DNA structures, including 5′ ssDNA flaps, that modify Msh2–Msh3 function to promote genome instability. A similar correlation between Msh3 levels and TNR expansions was previously observed in a mouse model (94) and human cell lines (93). We predict that both the absolute levels of Msh2–Msh3 and the relative levels of Msh2–Msh3 versus Msh2–Msh6 are important to maintain the ‘right’ balance of MMR activities in different cellular and genomic contexts. Overexpression of MSH2 MSH6 in yeast disrupted Msh2–Msh6-mediated MMR and heteroduplex rejection, perhaps via sequestration of partner proteins (96). Similarly, overexpression of yeast MLH1 (124) or PMS1 (125) disrupted MMR and was predicted to promote formation of non-functional MMR complexes (126).
Msh3. Overexpression of Msh3, which generates an imbalance of this distribution, was previously linked with strong mutator phenotypes in human cells (61,62). Consistent with this, when we overexpressed MSH3 alone, we observed an increase in canavanine resistance, which primarily measures Msh2–Msh6 activity, indicating a decrease in Msh2–Msh6 function (Table 1). Decreased Msh2–Msh6 activity when Msh3 is overexpressed is presumably caused by a reduction in the formation of Msh2–Msh6 protein complexes, disrupting the ‘balance’ between Msh2–Msh6 and Msh2–Msh3 complex formation. When we co-overexpressed MSH2 and MSH3 in budding yeast, we observed distinct phenotypes that included sensitivity to alkylating DNA damage (Figure 2), cell cycle delays (Figures 3 and 4) and an enhancement of post-translationally modified PCNA (Figure 5). Our results suggest that controlling the abundance of Msh3 is a mechanism by which cells can limit the interactions of Msh2–Msh3 with DNA structures, including 5′ ssDNA flaps, that modify Msh2–Msh3 function to promote genome instability. A similar correlation between Msh3 levels and TNR expansions was previously observed in a mouse model (94) and human cell lines (93). We predict that both the absolute levels of Msh2–Msh3 and the relative levels of Msh2–Msh3 versus Msh2–Msh6 are important to maintain the ‘right’ balance of MMR activities in different cellular and genomic contexts. Overexpression of MSH2 MSH6 in yeast disrupted Msh2–Msh6-mediated MMR and heteroduplex rejection, perhaps via sequestration of partner proteins (96). Similarly, overexpression of yeast MLH1 (124) or PMS1 (125) disrupted MMR and was predicted to promote formation of non-functional MMR complexes (126).
An MMR-like response is required for Msh2-Msh3-mediated cell cycle genomic instabilities
Initially, the in vivo effects of elevated MSH3 or MSH2 MSH3 expression suggested a simple model in which Msh2–Msh3 recognized and bound 5′ ssDNA flap structures as previously demonstrated in vitro (14,28), thereby blocking Rad27FEN1 activity in vivo. This would explain the MMS sensitivity, the defect in cell cycle progression through the S phase and the modification of PCNA. However, two key observations, specifically the requirement for (i) Msh2–Msh3 ATPase activity and (ii) the requirement for MLH1, indicated an active, Msh2–Msh3-mediated aberrant MMR-like response reminiscent of current models for Msh2–Msh3’s role in promoting TNR expansions (54,55,65,93,127).
Msh2–Msh3-mediated S phase accumulation and PCNA modification depended on Msh2–Msh3 ATP binding (Walker A) and hydrolysis (Walker B) activity. We and others have previously demonstrated that Msh2–Msh3 DNA binding does not require ATP (13,14,27,72). In fact, the presence of ATP promotes dissociation from DNA in vitro (14,27,72,118). Notably, the msh2 and msh3 Walker A mutations have dominant-negative effects in vivo (63,64); Msh2-msh3G693D inhibited ATP-dependent dissociation from DNA substrates in vitro (14), an observation interpreted to be a result of reduced Msh2–Msh3 turnover on the DNA. Therefore, DNA binding alone is not sufficient to produce the adverse effects of MSH2 MSH3 overexpression. In contrast, msh3Y925A, predicted to alter the regulation of Msh2–Msh3 ATPase activity, did not reduce Msh2–Msh3-mediated interference with cell cycle progression. msh3Y925A also exhibited a dominant negative effect on MMR in vivo (64), indicating that this allele interfered with MMR but likely through a distinct mechanism. These observations are consistent with a model in which Msh2–Msh3 is not simply binding to 5′ ssDNA flaps and sterically hindering Rad27FEN1-mediated processing, although we predict that this capacity contributes to the cellular phenotypes.
Msh2–Msh3 ATPase activity is required for both Msh2–Msh3-mediated MMR and 3′ NHTR (64,66,128), although there are differential molecular requirements for the regulation of ATPase activity in these two pathways; msh3Y925A was defective in MMR but functional in 3′ NHTR (64). Further, Msh2–Msh3 nucleotide binding, hydrolysis and turnover are differentially modulated by MMR versus 3′ NHTR DNA substrates. Msh2–Msh3 ATPase activity is similarly required to promote TNR expansions; mutations in the Msh2 Walker A or Msh3 Walker B motif disrupted TNR expansions in mammalian systems (65,93). Msh2–Msh3 binding to TNR DNA substrates altered nucleotide (ADP and ATP) binding and hydrolysis (27,72,93). We hypothesize that Msh2–Msh3 binding to non-canonical 5′ ssDNA flaps similarly alters Msh2–Msh3’s nucleotide binding/hydrolysis/turnover cycle and initiates an MMR-like response that disrupts OFM. Notably, Msh2–Msh3 may also be directly affecting DNA synthesis by Pol δ in OFM and/or LP-BER. We observed that Msh2–Msh3 either stimulated (synthesis substrate) or inhibited (strand displacement substrate) DNA Pol δ activity in vitro in a DNA structure-dependent manner. These observations, coupled with a differential affinity of Msh2–Msh3 for binding these substrates, predict a model in which Msh2–Msh3 binds and alters that conformation of the DNA structures to enhance or inhibit Pol δ activity. In vivo, Msh2–Msh3 could, when in sufficient quantities, similarly modulate Pol δ activity, altering and disrupting the kinetics of DNA synthesis.
Loss of Msh2–Msh3 ATPase activity also compromises its recruitment of Mlh complexes. Therefore, the requirement for ATP binding and, to a lesser extent, hydrolysis, may be related to the ability to recruit Mlh complexes. This is consistent with the requirement for MLH1 to observe the MSH2 MSH3 overexpression cell cycle phenotype. Loss of MLH1 eliminates all three Mlh complexes. This suggests a model in which Msh2–Msh3 recruits one or more MLH complexes when bound to 5′ ssDNA flaps and that this interferes with Rad27FEN1-mediated pathways. Notably, all three Mlh complexes play a role in Msh2–Msh3-mediated TNR expansion (66,127,129–132). Based on our data, we suggest a model in which Msh2–Msh3 bound to a 5′ ssDNA flap intermediate recruits Mlh complexes, but Msh2–Msh3’s altered ATP binding/hydrolysis activity misregulates Mlh activation, similar to what has been proposed in TNR expansion studies, and interferes with Rad27-mediated OFM.
A possible role of modified PCNA in Msh2–Msh3-mediated genome instability
The MSH2 MSH3 overexpression-dependent modification of PCNA at K164 and the abrogation of this phenotype, as well as the cell cycle phenotype, in elg1Δ highlights a role for PCNA and its loading/unloading dynamics when the cells responds to elevated Msh2–Msh3 levels. PCNA plays a central role in DNA damage tolerance and repair signaling via post-translational modification. Monoubiquitination of PCNA at K164, catalyzed by the Rad6-Rad18 complex, recruits low-fidelity translesion synthesis polymerases (Pol η, Rev1 and Pol ζ) for potentially mutagenic lesion bypass (105,133–135). PCNA can be further poly-ubiquitinated at K164 by Ubc13-Mms2 and Rad5 to promote high-fidelity recombination (105,136,137). Alternatively, PCNA can be SUMOylated on residues K127 and/or K164, by the Ubc9-Siz1 complex, to prevent recombination through the recruitment of the anti-recombinase Srs2 (105–107,138–140). In this study, we showed that when excess Msh2–Msh3 is present in vivo, PCNA post-translational modification at K164 is enhanced (Figure 5, 6). We were unable to detect PCNA ubiquitination by western blot (Supplementary Figure S5) or by mass spectrometry (data not shown). In contrast, the MSH2 MSH3 overexpression-dependent modification exhibited the same mobility as PCNA following MMS treatment that induces PCNA sumoylation (Supplementary Figure S5B). Further, deletion of SIZ1, which is required for PCNA sumoylation, resulted in loss of both the cell cycle phenotype and the PCNA modification (Figure 7). The siz1Δ phenotypes and the requirement of ELG1 for the cell cycle defect to be observed in the context of Msh2–Msh3 expression indicate that the modification is SUMO (Figures 7, 9; Supplementary Figure S5). SUMOylated PCNA is associated with the recruitment of Elg1(110).
The requirement for ELG1, which unloads unmodified and sumoylated PCNA to recycle PCNA during lagging strand synthesis (109–111,141), indicated that an alteration in PCNA cycling may be contributing to the MSH2 MSH3 overexpression-dependent phenotype. Okazaki fragment ligation is required for PCNA recycling by Elg1 (111). We reason that if Msh2–Msh3 is interfering with cell cycle progression by blocking 5′ flap processing, it might also be interrupting the normal cycling of PCNA. Such an interruption could result in the accumulation of SUMOylated PCNA at the replication fork, which would explain the enhancement of modified PCNA observed in Figure 5 upon MSH2 MSH3 overexpression.
We also note that PCNA interacts with Msh complexes via their PIP-box motifs (142). The PCNA-Msh6 interaction recruits Msh2–Msh6 to the replication fork and plays a critical role in Exo1-independent MMR (19,122). Over-retention of PCNA on the DNA in elg1Δ recruits increased Msh2–Msh6, trapping it, accumulating MMR intermediates and resulting in elevated mutation rates and genomic instability (143,144). Excess PCNA might similarly retain and trap excess Msh2–Msh3 in elg1Δ, preventing it from interfering with OFM and/or DNA polymerase activity. Excess PCNA may also block Msh2–Msh3 interactions with 5′ flap intermediates, preventing Msh2–Msh3 binding. This could block Msh2–Msh3’s ability to recruit Mlh complexes; work in human Msh2–Msh3 demonstrated overlapping PCNA and MLH interaction motifs such that PCNA and Mlh complexes compete for binding to Msh2–Msh3 (145). As noted above, MLH1 is required for the MSH2 MSH3 overexpression phenotypes. (146).
Model for MSH2 MSH3 overexpression-mediated genome instability
In this study, we demonstrated that overexpression of the Msh2–Msh3 complex in budding yeast could induce alkylation sensitivity (Figure 2), cell cycle progression delays (Figures 3 and 4) and a putative PCNA-mediated DNA damage response (Figure 8). These phenotypes required functional Msh2–Msh3 ATPase activity (Figures 10 and 11) and were abrogated in rad9Δ, elg1Δ and mlh1Δ backgrounds (Figures 8–11). These data indicate that Msh2–Msh3 has the potential to disrupt many pathways in DNA metabolism, likely through its broad DNA binding capacity (Figure 12). Our findings further support the idea that Msh2–Msh3 binding alone is insufficient to determine between genome stability or instability outcomes. These features are similar to the requirements of Msh2–Msh3 in promoting TNR expansions (27,28,72,147,148), possibly suggesting a common mechanism for promoting genomic instability.
We propose the following model for the effects of excess Msh2–Msh3 (Figure 13): (i) Increased Msh2–Msh3 abundance increases its ability to bind 5′ ssDNA flap intermediates during OFM or LP-BER, in addition to MMR loop structures, blocking efficient OFM and potentially activating a RAD9-dependent checkpoint response. (ii) Excess Msh2–Msh3 would also bind DNA substrates for Pol δ, either inhibiting (simple primer-template substrate) or enhancing (gapped substrate) its activity and altering the kinetics of DNA synthesis. A direct interaction between Msh2–Msh3 and DNA Pol δ is possible, analogous to a Msh2–Msh3/DNA Pol β interaction in the human system (149). (iii) Binding of a non-canonical DNA structure alters the ATP cycle within the Msh2–Msh3, potentially impacting (promoting?) its interaction with Mlh complexes, as well as turnover of the Msh2–Msh3 complex on the DNA. (iv) DNA-bound Msh2–Msh3 recruits Mlh complexes, which in the presence of non-canonical Msh2–Msh3 DNA structures leads to aberrant activation of the Mlh via repeated rounds of nicking in an attempt to repair the DNA, leading to a checkpoint response. Alternatively, Msh2–Msh3 bound to 5′ ssDNA flap structures may misdirect Mlh endonuclease activity to the wrong strand, as demonstrated in vitro in the presence of TNR structures (150). The cells responds by slowing progression through the cell cycle, with cells accumulating in S phase, until levels of Msh2–Msh3 are reduced. It is clear that careful regulation of Msh2–Msh3 is critical for preventing aberrant or pathogenic outcomes in DNA metabolism, while retaining its advantageous genome stability functions.
Figure 13.

Model of Msh2–Msh3 activity at the replication fork at endogenous and elevated MSH2 MSH3 expression levels. (A) Normal DNA Replication and Repair [i]: Normal structures observed during DNA replication (a mismatch intermediate or flap generated during Okazaki fragment maturation (OFM) or repair (5′ flaps created during long-patch base excision repair (LP-BER); [ii]: Recognition of the mismatch by Msh2–Msh3 and normal Okazaki fragment maturation and 5′ flap synthesis repair during LP-BER–at endogenous protein levels, Msh2–Msh3 is NOT predicted to interact with 5′ flap structures, [iii]: Mismatch binding by Msh2–Msh3 leads to ATP binding and subsequent interaction with Mlh complex. Flaps generated during OFM or LP-BER are cleaved by Rad27FEN1, [iv]: Msh2–Msh3 hydrolyzes ATP and the MSH-MLH complex dissociates from the mismatch allowing action from the recruited downstream MMR factors. The nick that is generated by flap cleavage is ligated by Cdc9LigI [v]: Successful completion of MMR, OFM and LP-BER. (B) Aberrant DNA Replication and Repair on account of elevated Msh2-Msh3 levels [i]: Normal structures observed during DNA replication (a mismatch intermediate or nick generated during OFM or LP-BER, [ii]: increased local concentration of Msh2–Msh3 allows Msh2–Msh3 binding to primer termini and slow down polymerase activity, elevated Msh2–Msh3 may lead to altered MMR loop repair via Msh2–Msh3 oligomerization [iii]: Increased levels Msh2–Msh3 interferes with polymerase synthesis and strand displacement, flap cleavage and nick ligation. Non-canonical DNA structure binding may alter ATP binding and hydrolysis kinetics and possibly recruit MLH complexes. In each scenario, OFM and LP-BER are hindered, MMR may be affected–excess Msh2–Msh3 interferes with Msh2–Msh6 function [iv]: In response to incomplete MMR, OFM and LP-BER, PCNA modification is triggered, [v]: which leads activation of cell cycle checkpoints and delay in cell cycle progression.
Supplementary Material
Acknowledgements
We are grateful for the support of the UB Confocal Microscope and Flow Cytometry Facility (CMFCF). We thank Katherine Casazza for technical assistance and for helpful discussions. We are grateful to Sarah Piacente and Jaime O’Connor for performing canavanine resistance assays and to Michaela (Cornaire) Radel for early MMS sensitivity assays. We thank Dr Bruce Stillman for the gift of α-PCNA antibody used in this study.
Notes
Present address: Melisa Medina-Rivera, Division of Nutritional Sciences, Cornell University, Ithaca, NY 14850, USA.
Present address: Natalie A. Lamb, Molecular Biology and Strain Development, National Renewable Energy Laboratory, Golden, CO 80401, USA.
Present address: Charanya Kumar, Department of Molecular Biology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
Present address: Anja Bielinsky, Department of Biochemistry and Molecular Genetics, University of Virginia, Charlottesville, VA 22908, USA.
Contributor Information
Melisa Medina-Rivera, Department of Biochemistry, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo NY, 14203, USA.
Samantha Phelps, Department of Biochemistry, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo NY, 14203, USA.
Madhumita Sridharan, Department of Biology, Indiana University Purdue University Indianapolis, Indianapolis, IN, 46202, USA.
Jordan Becker, Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN, 55455, USA.
Natalie A Lamb, Department of Biochemistry, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo NY, 14203, USA.
Charanya Kumar, Department of Biochemistry, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo NY, 14203, USA.
Mark D Sutton, Department of Biochemistry, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo NY, 14203, USA.
Anja Bielinsky, Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN, 55455, USA.
Lata Balakrishnan, Department of Biology, Indiana University Purdue University Indianapolis, Indianapolis, IN, 46202, USA.
Jennifer A Surtees, Department of Biochemistry, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo NY, 14203, USA.
Data availability
The data underlying this article are available in the article and in its online supplementary material. Further data underlying this article will be shared on reasonable request to the corresponding author.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
NIH IMSD training grant [R25 GM095459 to M.M.R]; NIH diversity supplement [GM066094 to M.M.R.]; National Science Foundation [1929346 to L.B.]; NIH [R35GM141805 and R01GM134681 to A.K.B.]; NIH [GM087459]; American Cancer Society Research Scholar Grant [RSG-14-2350-01 to J.A.S.]; J.A.S. is also grateful for support from the University at Buffalo's Genome, Environment and Microbiome Community of Excellence. Funding for open access charge: University at Buffalo's Genome, Environment and Microbiome Community of Excellence.
Conflict of interest statement. None declared.
References
- 1. Modrich P., Lahue R.. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 1996; 65:101–133. [DOI] [PubMed] [Google Scholar]
- 2. Iyer R.R., Pluciennik A., Burdett V., Modrich P.L.. DNA mismatch repair: functions and mechanisms. Chem. Rev. 2006; 106:302–323. [DOI] [PubMed] [Google Scholar]
- 3. McCulloch S.D., Kunkel T.A.. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008; 18:148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ganai R.A., Johansson E.. DNA replication—a matter of fidelity. Mol. Cell. 2016; 62:745–755. [DOI] [PubMed] [Google Scholar]
- 5. Acharya S., Wilson T., Gradia S., Kane M.F., Guerrette S., Marsischky G.T., Kolodner R., Fishel R.. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:13629–13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marsischky G.T., Filosi N., Kane M.F., Kolodner R.. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996; 10:407–420. [DOI] [PubMed] [Google Scholar]
- 7. Alani E. The Saccharomyces cerevisiae Msh2 and Msh6 proteins form a complex that specifically binds to duplex oligonucleotides containing mismatched DNA base pairs. Mol. Cell. Biol. 1996; 16:5604–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iaccarino I., Palombo F., Drummond J., Totty N.F., Hsuan J.J., Modrich P., Jiricny J.. MSH6, a Saccharomyces cerevisiae protein that binds to mismatches as a heterodimer with MSH2. Curr. Biol. 1996; 6:484–486. [DOI] [PubMed] [Google Scholar]
- 9. Marsischky G.T., Kolodner R.D.. Biochemical characterization of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 complex and mispaired bases in DNA. J. Biol. Chem. 1999; 274:26668–26682. [DOI] [PubMed] [Google Scholar]
- 10. Lamb N.A., Bard J.E., Loll-Krippleber R., Brown G.W., Surtees J.A.. Complex mutation profiles in mismatch repair and ribonucleotide reductase mutants reveal novel repair substrate specificity of MutS homolog (MSH) complexes. Genetics. 2022; 221:iyac092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harrington J.M., Kolodner R.D.. Saccharomyces cerevisiae Msh2–Msh3 acts in repair of base-base mispairs. Mol. Cell. Biol. 2007; 27:6546–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Srivatsan A., Bowen N., Kolodner R.D.. Mispair-specific recruitment of the Mlh1–Pms1 complex identifies repair substrates of the Saccharomyces cerevisiae Msh2–Msh3 complex. J. Biol. Chem. 2014; 289:9352–9364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Habraken Y., Sung P., Prakash L., Prakash S.. Binding of insertion/deletion DNA mismatches by the heterodimer of yeast mismatch repair proteins MSH2 and MSH3. Curr. Biol. 1996; 6:1185–1187. [DOI] [PubMed] [Google Scholar]
- 14. Surtees J.A., Alani E.. Mismatch repair factor MSH2-MSH3 binds and alters the conformation of branched DNA structures predicted to form during genetic recombination. J. Mol. Biol. 2006; 360:523–536. [DOI] [PubMed] [Google Scholar]
- 15. Sia E.A., Kokoska R.J., Dominska M., Greenwell P., Petes T.D.. Microsatellite instability in yeast: dependence on repeat unit size and DNA mismatch repair genes. Mol. Cell. Biol. 1997; 17:2851–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jensen L.E., Jauert P.A., Kirkpatrick D.T.. The large loop repair and mismatch repair pathways of Saccharomyces cerevisiae act on distinct substrates during meiosis. Genetics. 2005; 170:1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Plotz G., Welsch C., Giron-Monzon L., Friedhoff P., Albrecht M., Piiper A., Biondi R.M., Lengauer T., Zeuzem S., Raedle J.. Mutations in the MutSalpha interaction interface of MLH1 can abolish DNA mismatch repair. Nucleic Acids Res. 2006; 34:6574–6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pluciennik A., Dzantiev L., Iyer R.R., Constantin N., Kadyrov F.A., Modrich P.. PCNA function in the activation and strand direction of MutLα endonuclease in mismatch repair. Proc. Natl. Acad. Sci. 2010; 107:16066–16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goellner E.M., Smith C.E., Campbell C.S., Hombauer H., Desai A., Putnam C.D., Kolodner R.D.. PCNA and Msh2–Msh6 activate an Mlh1–Pms1 endonuclease pathway required for Exo1-independent mismatch repair. Mol. Cell. 2014; 55:291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rogacheva M.V., Manhart C.M., Chen C., Guarne A., Surtees J., Alani E.. Mlh1–Mlh3, a meiotic crossover and DNA mismatch repair factor, is a Msh2–Msh3-stimulated endonuclease. J. Biol. Chem. 2014; 289:5664–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manhart C.M., Ni X., White M.A., Ortega J., Surtees J.A., Alani E.. The mismatch repair and meiotic recombination endonuclease Mlh1–Mlh3 is activated by polymer formation and can cleave DNA substrates in trans. PLoS Biol. 2017; 15:e2001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harfe B.D., Minesinger B.K., Jinks-Robertson S.. Discrete in vivo roles for the MutL homologs Mlh2p and Mlh3p in the removal of frameshift intermediates in budding yeast. Curr. Biol. 2000; 10:145–148. [DOI] [PubMed] [Google Scholar]
- 23. Campbell C.S., Hombauer H., Srivatsan A., Bowen N., Gries K., Desai A., Putnam C.D., Kolodner R.D.. Mlh2 is an accessory factor for DNA mismatch repair in Saccharomyces cerevisiae. PLoS Genet. 2014; 10:e1004327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kunkel T.A., Erie D.A.. Eukaryotic mismatch repair in relation to DNA replication. Annu. Rev. Genet. 2015; 49:291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kunkel T.A., Erie D.A.. DNA mismatch repair. Annu. Rev. Biochem. 2005; 74:681–710. [DOI] [PubMed] [Google Scholar]
- 26. Bowen N., Kolodner R.D.. Reconstitution of Saccharomyces cerevisiae DNA polymerase ϵ-dependent mismatch repair with purified proteins. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:3607–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Owen B.A., Yang Z., Lai M., Gajec M., Badger J.D., Hayes J.J., Edelmann W., Kucherlapati R., Wilson T.M., McMurray C.T.. (CAG)(n)-hairpin DNA binds to Msh2–Msh3 and changes properties of mismatch recognition. Nat. Struct. Mol. Biol. 2005; 12:663–670. [DOI] [PubMed] [Google Scholar]
- 28. Kantartzis A., Williams G.M., Balakrishnan L., Roberts R.L., Surtees J.A., Bambara R.A.. Msh2–Msh3 interferes with Okazaki fragment processing to promote trinucleotide repeat expansions. Cell Rep. 2012; 2:216–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li F., Dong J., Eichmiller R., Holland C., Minca E., Prakash R., Sung P., Yong Shim E., Surtees J.A., Eun Lee S. Role of Saw1 in Rad1/Rad10 complex assembly at recombination intermediates in budding yeast. EMBO J. 2013; 32:461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sugawara N., Pâques F., Colaiácovo M., Haber J.E.. Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:9214–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Evans E., Sugawara N., Haber J.E., Alani E.. The Saccharomyces cerevisiae Msh2 mismatch repair protein localizes to recombination intermediates in vivo. Mol. Cell. 2000; 5:789–799. [DOI] [PubMed] [Google Scholar]
- 32. Saparbaev M., Prakash L., Prakash S.. Requirement of mismatch repair genes MSH2 and MSH3 in the RAD1-RAD10 pathway of mitotic recombination in Saccharomyces cerevisiae. Genetics. 1996; 142:727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eichmiller R., Medina-Rivera M., DeSanto R., Minca E., Kim C., Holland C., Seol J.H., Schmit M., Oramus D., Smith J.et al.. Coordination of Rad1-Rad10 interactions with Msh2–Msh3, Saw1 and RPA is essential for functional 3' non-homologous tail removal. Nucleic Acids Res. 2018; 46:5075–5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jiang J., Bai L., Surtees J.A., Gemici Z., Wang M.D., Alani E.. Detection of high-affinity and sliding clamp modes for MSH2-MSH6 by single-molecule unzipping force analysis. Mol. Cell. 2005; 20:771–781. [DOI] [PubMed] [Google Scholar]
- 35. Refsland E.W., Livingston D.M.. Interactions among DNA ligase I, the flap endonuclease and proliferating cell nuclear antigen in the expansion and contraction of CAG repeat tracts in yeast. Genetics. 2005; 171:923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sugawara N., Goldfarb T., Studamire B., Alani E., Haber J.E.. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:9315–9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chakraborty U., Alani E.. Understanding how mismatch repair proteins participate in the repair/anti-recombination decision. FEMS Yeast Res. 2016; 16:fow071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Edelmann W., Umar A., Yang K., Heyer J., Kucherlapati M., Lia M., Kneitz B., Avdievich E., Fan K., Wong E.et al.. The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Res. 2000; 60:803–807. [PubMed] [Google Scholar]
- 39. van Oers J.M., Edwards Y., Chahwan R., Zhang W., Smith C., Pechuan X., Schaetzlein S., Jin B., Wang Y., Bergman A.et al.. The MutSβ complex is a modulator of p53-driven tumorigenesis through its functions in both DNA double-strand break repair and mismatch repair. Oncogene. 2014; 33:3939–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Santos L.S., Silva S.N., Gil O.M., Ferreira T.C., Limbert E., Rueff J.. Mismatch repair single nucleotide polymorphisms and thyroid cancer susceptibility. Oncol. Lett. 2018; 15:6715–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ashktorab H., Azimi H., Varma S., Tavakoli P., Nickerson M.L., Brim H.. Distinctive DNA mismatch repair and APC rare variants in African Americans with colorectal neoplasia. Oncotarget. 2017; 8:99966–99977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raskin L., Guo Y., Du L., Clendenning M., Rosty C., Lindor N.M., Gruber S.B., Buchanan D.D.. Targeted sequencing of established and candidate colorectal cancer genes in the Colon Cancer Family Registry Cohort. Oncotarget. 2017; 8:93450–93463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morak M., Käsbauer S., Kerscher M., Laner A., Nissen A.M., Benet-Pagès A., Schackert H.K., Keller G., Massdorf T., Holinski-Feder E.. Loss of MSH2 and MSH6 due to heterozygous germline defects in MSH3 and MSH6. Fam. Cancer. 2017; 16:491–500. [DOI] [PubMed] [Google Scholar]
- 44. Takahashi M., Koi M., Balaguer F., Boland C.R., Goel A.. MSH3 mediates sensitization of colorectal cancer cells to cisplatin, oxaliplatin, and a poly(ADP-ribose) polymerase inhibitor. J. Biol. Chem. 2011; 286:12157–12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Park J.M., Huang S., Tougeron D., Sinicrope F.A.. MSH3 mismatch repair protein regulates sensitivity to cytotoxic drugs and a histone deacetylase inhibitor in human colon carcinoma cells. PLoS One. 2013; 8:e65369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nogueira G.A.S., Costa E.F.D., Lopes-Aguiar L., Lima T.R.P., Visacri M.B., Pincinato E.C., Lourenço G.J., Calonga L., Mariano F.V., Altemani A.et al.. Polymorphisms in DNA mismatch repair pathway genes predict toxicity and response to cisplatin chemoradiation in head and neck squamous cell carcinoma patients. Oncotarget. 2018; 9:29538–29547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miao H.K., Chen L.P., Cai D.P., Kong W.J., Xiao L., Lin J.. MSH3 rs26279 polymorphism increases cancer risk: a meta-analysis. Int. J. Clin. Exp. Pathol. 2015; 8:11060–11067. [PMC free article] [PubMed] [Google Scholar]
- 48. Dietlein F., Thelen L., Jokic M., Jachimowicz R.D., Ivan L., Knittel G., Leeser U., van Oers J., Edelmann W., Heukamp L.C.et al.. A functional cancer genomics screen identifies a druggable synthetic lethal interaction between MSH3 and PRKDC. Cancer Discov. 2014; 4:592–605. [DOI] [PubMed] [Google Scholar]
- 49. Carethers J.M. Microsatellite instability pathway and EMAST in colorectal cancer. Curr. Colorectal. Cancer Rep. 2017; 13:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Adam R., Spier I., Zhao B., Kloth M., Marquez J., Hinrichsen I., Kirfel J., Tafazzoli A., Horpaopan S., Uhlhaas S.et al.. Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. Am. J. Hum. Genet. 2016; 99:337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Iyer R.R., Pluciennik A., Napierala M., Wells R.D.. DNA triplet repeat expansion and mismatch repair. Annu. Rev. Biochem. 2015; 84:199–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Massey T.H., Jones L.. The central role of DNA damage and repair in CAG repeat diseases. Dis. Model Mech. 2018; 11:dmm031930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jones L., Houlden H., Tabrizi S.J.. DNA repair in the trinucleotide repeat disorders. Lancet Neurol. 2017; 16:88–96. [DOI] [PubMed] [Google Scholar]
- 54. Schmidt M.H.M., Pearson C.E.. Disease-associated repeat instability and mismatch repair. DNA Repair (Amst.). 2016; 38:117–126. [DOI] [PubMed] [Google Scholar]
- 55. Usdin K., House N.C., Freudenreich C.H.. Repeat instability during DNA repair: insights from model systems. Crit. Rev. Biochem. Mol. Biol. 2015; 50:142–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Du J., Campau E., Soragni E., Ku S., Puckett J.W., Dervan P.B., Gottesfeld J.M.. Role of mismatch repair enzymes in GAA·TTC triplet-repeat expansion in Friedreich ataxia induced pluripotent stem cells. J. Biol. Chem. 2012; 287:29861–29872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McGinty R.J., Mirkin S.M.. Cis- and trans-modifiers of repeat expansions: blending model systems with human genetics. Trends Genet. 2018; 34:448–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Williams G.M., Surtees J.A.. MSH3 promotes dynamic behavior of trinucleotide repeat tracts in vivo. Genetics. 2015; 200:737–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Williams G.M., Petrides A.K., Balakrishnan L., Surtees J.A.. Tracking expansions of stable and threshold length trinucleotide repeat tracts in vivo and in vitro using Saccharomyces cerevisiae. Methods Mol. Biol. 2020; 2056:25–68. [DOI] [PubMed] [Google Scholar]
- 60. McKinney J.A., Wang G., Mukherjee A., Christensen L., Subramanian S.H.S., Zhao J., Vasquez K.M.. Distinct DNA repair pathways cause genomic instability at alternative DNA structures. Nat. Commun. 2020; 11:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Marra G., Iaccarino I., Lettieri T., Roscilli G., Delmastro P., Jiricny J.. Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:8568–8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Drummond J.T., Genschel J., Wolf E., Modrich P.. DHFR/MSH3 amplification in methotrexate-resistant cells alters the hMutSalpha/hMutSbeta ratio and reduces the efficiency of base-base mismatch repair. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:10144–10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Studamire B., Price G., Sugawara N., Haber J.E., Alani E.. Separation-of-function mutations in Saccharomyces cerevisiae MSH2 that confer mismatch repair defects but do not affect nonhomologous-tail removal during recombination. Mol. Cell. Biol. 1999; 19:7558–7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kumar C., Williams G.M., Havens B., Dinicola M.K., Surtees J.A.. Distinct requirements within the Msh3 nucleotide binding pocket for mismatch and double-strand break repair. J. Mol. Biol. 2013; 425:1881–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tomé S., Holt I., Edelmann W., Morris G.E., Munnich A., Pearson C.E., Gourdon G.. MSH2 ATPase domain mutation affects CTG*CAG repeat instability in transgenic mice. PLoS Genet. 2009; 5:e1000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kumar C., Eichmiller R., Wang B., Williams G.M., Bianco P.R., Surtees J.A.. ATP binding and hydrolysis by Saccharomyces cerevisiae Msh2–Msh3 are differentially modulated by mismatch and double-strand break repair DNA substrates. DNA Repair (Amst.). 2014; 18:18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Obmolova G., Ban C., Hsieh P., Yang W.. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000; 407:703–710. [DOI] [PubMed] [Google Scholar]
- 68. Gupta S., Gellert M., Yang W.. Mechanism of mismatch recognition revealed by human MutSβ bound to unpaired DNA loops. Nat. Struct. Mol. Biol. 2011; 19:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lamers M.H., Perrakis A., Enzlin J.H., Winterwerp H.H., de Wind N., Sixma T.K.. The crystal structure of DNA mismatch repair protein MutS binding to a G x T mismatch. Nature. 2000; 407:711–717. [DOI] [PubMed] [Google Scholar]
- 70. Warren J.J., Pohlhaus T.J., Changela A., Iyer R.R., Modrich P.L., Beese L.S.. Structure of the human MutSalpha DNA lesion recognition complex. Mol. Cell. 2007; 26:579–592. [DOI] [PubMed] [Google Scholar]
- 71. Lee S.D., Surtees J.A., Alani E.. Saccharomyces cerevisiae MSH2-MSH3 and MSH2-MSH6 complexes display distinct requirements for DNA binding domain I in mismatch recognition. J. Mol. Biol. 2007; 366:53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lang W.H., Coats J.E., Majka J., Hura G.L., Lin Y., Rasnik I., McMurray C.T.. Conformational trapping of mismatch recognition complex MSH2/MSH3 on repair-resistant DNA loops. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:E837–E844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bambara R.A., Murante R.S., Henricksen L.A.. Enzymes and reactions at the eukaryotic DNA replication fork. J. Biol. Chem. 1997; 272:4647–4650. [DOI] [PubMed] [Google Scholar]
- 74. Liu Y., Zhang H., Veeraraghavan J., Bambara R.A., Freudenreich C.H.. Saccharomyces cerevisiae flap endonuclease 1 uses flap equilibration to maintain triplet repeat stability. Mol. Cell. Biol. 2004; 24:4049–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Burgers P.M. Polymerase dynamics at the eukaryotic DNA replication fork. J. Biol. Chem. 2009; 284:4041–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ghaemmaghami S., Huh W.K., Bower K., Howson R.W., Belle A., Dephe N., O'Shea E.K., Weissman J.S.. Global analysis of protein expression in yeast. Nature. 2003; 425:737–741. [DOI] [PubMed] [Google Scholar]
- 77. Kulak N.A., Pichler G., Paron I., Nagaraj N., Mann M.. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods. 2014; 11:319–324. [DOI] [PubMed] [Google Scholar]
- 78. Christianson T.W., Sikorski R.S., Dante M., Shero J.H., Hieter P.. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992; 110:119–122. [DOI] [PubMed] [Google Scholar]
- 79. Sikorski R.S., Hieter P.. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989; 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gietz D., St Jean A., Woods R.A., Schiestl R.H.. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992; 20:1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kijas A.W., Studamire B., Alani E.. Msh2 separation of function mutations confer defects in the initiation steps of mismatch repair. J. Mol. Biol. 2003; 331:123–138. [DOI] [PubMed] [Google Scholar]
- 82. Becker J.R., Gallo D., Leung W., Croissant T., Thu Y.M., Nguyen H.D., Starr T.K., Brown G.W., Bielinsky A.K.. Flap endonuclease overexpression drives genome instability and DNA damage hypersensitivity in a PCNA-dependent manner. Nucleic Acids Res. 2018; 46:5634–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Alani E., Cao L., Kleckner N.. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics. 1987; 116:541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Voth W.P., Jiang Y.W., Stillman D.J.. New ‘marker swap’ plasmids for converting selectable markers on budding yeast gene disruptions and plasmids. Yeast. 2003; 20:985–993. [DOI] [PubMed] [Google Scholar]
- 85. Xu X., Page J.L., Surtees J.A., Liu H., Lagedrost S., Lu Y., Bronson R., Alani E., Nikitin A.Y., Weiss R.S.. Broad overexpression of ribonucleotide reductase genes in mice specifically induces lung neoplasms. Cancer Res. 2008; 68:2652–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lamb N.A., Bard J.E., Buck M.J., Surtees J.A.. A selection-based next generation sequencing approach to develop robust, genotype-specific mutation profiles in Saccharomyces cerevisiae. G3 (Bethesda). 2021; 11:jkab099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Radchenko E.A., McGinty R.J., Aksenova A.Y., Neil A.J., Mirkin S.M.. Quantitative analysis of the rates for repeat-mediated genome instability in a yeast Experimental System. Methods Mol. Biol. 2018; 1672:421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Das-Bradoo S., Nguyen H.D., Wood J.L., Ricke R.M., Haworth J.C., Bielinsky A.K.. Defects in DNA ligase I trigger PCNA ubiquitylation at Lys 107. Nat. Cell Biol. 2010; 12:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nguyen H.D., Becker J., Thu Y.M., Costanzo M., Koch E.N., Smith S., Myung K., Myers C.L., Boone C., Bielinsky A.K.. Unligated Okazaki fragments induce PCNA ubiquitination and a requirement for Rad59-dependent replication fork progression. PLoS One. 2013; 8:e66379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Stewart J.A., Campbell J.L., Bambara R.A.. Flap endonuclease disengages Dna2 helicase/nuclease from Okazaki fragment flaps. J. Biol. Chem. 2006; 281:38565–38572. [DOI] [PubMed] [Google Scholar]
- 91. Langston L.D., O’Donnell M. DNA polymerase delta is highly processive with proliferating cell nuclear antigen and undergoes collision release upon completing DNA. J. Biol. Chem. 2008; 283:29522–29531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bianco P.R., Xu C., Chi M.. Type I restriction endonucleases are true catalytic enzymes. Nucleic Acids Res. 2009; 37:3377–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Keogh N., Chan K.Y., Li G.M., Lahue R.S.. MutSβ abundance and Msh3 ATP hydrolysis activity are important drivers of CTG•CAG repeat expansions. Nucleic Acids Res. 2017; 45:10068–10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tomé S., Manley K., Simard J.P., Clark G.W., Slean M.M., Swami M., Shelbourne P.F., Tillier E.R., Monckton D.G., Messer A.et al.. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington's disease mice. PLoS Genet. 2013; 9:e1003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Pegg A.E. Methylation of the O6 position of guanine in DNA is the most likely initiating event in carcinogenesis by methylating agents. Cancer Invest. 1984; 2:223–231. [DOI] [PubMed] [Google Scholar]
- 96. Chakraborty U., Dinh T.A., Alani E.. Genomic instability promoted by overexpression of mismatch repair factors in yeast: a model for understanding cancer progression. Genetics. 2018; 209:439–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Vallen E.A., Cross F.R.. Mutations in RAD27 define a potential link between G1 cyclins and DNA replication. Mol. Cell. Biol. 1995; 15:4291–4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Reagan M.S., Pittenger C., Siede W., Friedberg E.C.. Characterization of a mutant strain of Saccharomyces cerevisiae with a deletion of the RAD27 gene, a structural homolog of the RAD2 nucleotide excision repair gene. J. Bacteriol. 1995; 177:364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tishkoff D.X., Boerger A.L., Bertrand P., Filosi N., Gaida G.M., Kane M.F., Kolodner R.D.. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:7487–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Becker J.R., Pons C., Nguyen H.D., Costanzo M., Boone C., Myers C.L., Bielinsky A.-K.. Genetic interactions implicating postreplicative repair in Okazaki fragment processing. PLos Genet. 2015; 11:e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Darzynkiewicz Z., Zhao H., Halicka H.D., Rybak P., Dobrucki J., Wlodkowic D. DNA damage signaling assessed in individual cells in relation to the cell cycle phase and induction of apoptosis. Crit. Rev. Clin. Lab. Sci. 2012; 49:199–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tanaka S., Umemori T., Hirai K., Muramatsu S., Kamimura Y., Araki H.. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature. 2007; 445:328–332. [DOI] [PubMed] [Google Scholar]
- 103. Huang X., Halicka H.D., Traganos F., Tanaka T., Kurose A., Darzynkiewicz Z.. Cytometric assessment of DNA damage in relation to cell cycle phase and apoptosis. Cell Prolif. 2005; 38:223–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Stelter P., Ulrich H.D.. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003; 425:188–191. [DOI] [PubMed] [Google Scholar]
- 105. Hoege C., Pfander B., Moldovan G.L., Pyrowolakis G., Jentsch S.. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002; 419:135–141. [DOI] [PubMed] [Google Scholar]
- 106. Pfander B., Moldovan G.L., Sacher M., Hoege C., Jentsch S.. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature. 2005; 436:428–433. [DOI] [PubMed] [Google Scholar]
- 107. Parker J.L., Bucceri A., Davies A.A., Heidrich K., Windecker H., Ulrich H.D.. SUMO modification of PCNA is controlled by DNA. EMBO J. 2008; 27:2422–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Barbour L., Ball L.G., Zhang K., Xiao W.. DNA damage checkpoints are involved in postreplication repair. Genetics. 2006; 174:1789–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kubota T., Nishimura K., Kanemaki M.T., Donaldson A.D.. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol. Cell. 2013; 50:273–280. [DOI] [PubMed] [Google Scholar]
- 110. Parnas O., Zipin-Roitman A., Pfander B., Liefshitz B., Mazor Y., Ben-Aroya S., Jentsch S., Kupiec M.. Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. EMBO J. 2010; 29:2611–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kubota T., Katou Y., Nakato R., Shirahige K., Donaldson A.D.. Replication-coupled PCNA unloading by the Elg1 complex occurs genome-wide and requires Okazaki fragment ligation. Cell Rep. 2015; 12:774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kadyrov F.A., Dzantiev L., Constantin N., Modrich P.. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006; 126:297–308. [DOI] [PubMed] [Google Scholar]
- 113. Kadyrov F.A., Holmes S.F., Arana M.E., Lukianova O.A., O’Donnell M., Kunkel T.A., Modrich P.. Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J. Biol. Chem. 2007; 282:37181–37190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kadyrova L.Y., Kadyrov F.A.. Endonuclease activities of MutLα and its homologs in DNA mismatch repair. DNA Repair (Amst.). 2016; 38:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Smith C.E., Mendillo M.L., Bowen N., Hombauer H., Campbell C.S., Desai A., Putnam C.D., Kolodner R.D.. Dominant mutations in S. cerevisiae PMS1 identify the Mlh1–Pms1 endonuclease active site and an exonuclease 1-independent mismatch repair pathway. PLos Genet. 2013; 9:e1003869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Al-Sweel N., Raghavan V., Dutta A., Ajith V.P., Di Vietro L., Khondakar N., Manhart C.M., Surtees J.A., Nishant K.T., Alani E.. mlh3 mutations in baker's yeast alter meiotic recombination outcomes by increasing noncrossover events genome-wide. PLoS Genet. 2017; 13:e1006974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Young S.J., Sebald M., Shah Punatar R., Larin M., Masino L., Rodrigo-Brenni M.C., Liang C.C., West S.C.. MutSβ stimulates Holliday junction resolution by the SMX complex. Cell Rep. 2020; 33:108289. [DOI] [PubMed] [Google Scholar]
- 118. Wilson T., Guerrette S., Fishel R.. Dissociation of mismatch recognition and ATPase activity by hMSH2-hMSH3. J. Biol. Chem. 1999; 274:21659–21664. [DOI] [PubMed] [Google Scholar]
- 119. Tomé S., Simard J.P., Slean M.M., Holt I., Morris G.E., Wojciechowicz K., te Riele H., Pearson C.E.. Tissue-specific mismatch repair protein expression: MSH3 is higher than MSH6 in multiple mouse tissues. DNA Repair (Amst.). 2013; 12:46–52. [DOI] [PubMed] [Google Scholar]
- 120. Inokuchi K., Ikejima M., Watanabe A., Nakajima E., Orimo H., Nomura T., Shimada T.. Loss of expression of the human MSH3 gene in hematological malignancies. Biochem. Biophys. Res. Commun. 1995; 214:171–179. [DOI] [PubMed] [Google Scholar]
- 121. Tian L., Gu L., Li G.M.. Distinct nucleotide binding/hydrolysis properties and molar ratio of MutSalpha and MutSbeta determine their differential mismatch binding activities. J. Biol. Chem. 2009; 284:11557–11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kumar C., Piacente S.C., Sibert J., Bukata A.R., O’Connor J., Alani E., Surtees J.A.. Multiple factors insulate Msh2–Msh6 mismatch repair activity from defects in Msh2 domain I. J. Mol. Biol. 2011; 411:765–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Arlow T., Kim J., Haye-Bertolozzi J.E., Martínez C.B., Fay C., Zorensky E., Rose M.D., Gammie A.E.. MutSα mismatch repair protein stability is governed by subunit interaction, acetylation, and ubiquitination. G3 (Bethesda). 2021; 11:jkaa065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Shcherbakova P.V., Kunkel T.A.. Mutator phenotypes conferred by MLH1 overexpression and by heterozygosity for mlh1 mutations. Mol. Cell. Biol. 1999; 19:3177–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kramer W., Fartmann B., Ringbeck E.C.. Transcription of mutS and mutL-homologous genes in Saccharomyces cerevisiae during the cell cycle. Mol. Gen. Genet. 1996; 252:275–283. [DOI] [PubMed] [Google Scholar]
- 126. Shcherbakova P.V., Hall M.C., Lewis M.S., Bennett S.E., Martin K.J., Bushel P.R., Afshari C.A., Kunkel T.A.. Inactivation of DNA mismatch repair by increased expression of yeast MLH1. Mol. Cell. Biol. 2001; 21:940–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Pinto R.M., Dragileva E., Kirby A., Lloret A., Lopez E., St Claire J., Panigrahi G.B., Hou C., Holloway K., Gillis T.et al.. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington's disease mice: genome-wide and candidate approaches. PLoS Genet. 2013; 9:e1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Studamire B., Quach T., Alani E.. Saccharomyces cerevisiae Msh2p and Msh6p ATPase activities are both required during mismatch repair. Mol. Cell. Biol. 1998; 18:7590–7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Gomes-Pereira M., Fortune M.T., Ingram L., McAbney J.P., Monckton D.G.. Pms2 is a genetic enhancer of trinucleotide CAG.CTG repeat somatic mosaicism: implications for the mechanism of triplet repeat expansion. Hum. Mol. Genet. 2004; 13:1815–1825. [DOI] [PubMed] [Google Scholar]
- 130. Pluciennik A., Burdett V., Baitinger C., Iyer R.R., Shi K., Modrich P.. Extrahelical (CAG)/(CTG) triplet repeat elements support proliferating cell nuclear antigen loading and MutLα endonuclease activation. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:12277–12282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Roy J.C.L., Vitalo A., Andrew M.A., Mota-Silva E., Kovalenko M., Burch Z., Nhu A.M., Cohen P.E., Grabczyk E., Wheeler V.C.et al.. Somatic CAG expansion in Huntington's disease is dependent on the MLH3 endonuclease domain, which can be excluded via splice redirection. Nucleic Acids Res. 2021; 49:3907–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Miller C.J., Kim G.Y., Zhao X., Usdin K.. All three mammalian MutL complexes are required for repeat expansion in a mouse cell model of the Fragile X-related disorders. PLoS Genet. 2020; 16:e1008902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Waters L.S., Minesinger B.K., Wiltrout M.E., D'Souza S., Woodruff R.V., Walker G.C. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 2009; 73:134–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Watanabe K., Tateishi S., Kawasuji M., Tsurimoto T., Inoue H., Yamaizumi M.. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004; 23:3886–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Broomfield S., Chow B.L., Xiao W.. MMS2, encoding a ubiquitin-conjugating-enzyme-like protein, is a member of the yeast error-free postreplication repair pathway. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:5678–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Hofmann R.M., Pickart C.M.. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell. 1999; 96:645–653. [DOI] [PubMed] [Google Scholar]
- 137. Pâques F., Haber J.E.. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999; 63:349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Papouli E., Chen S., Davies A.A., Huttner D., Krejci L., Sung P., Ulrich H.D.. Crosstalk between SUMO and Ubiquitin on PCNA Is Mediated by Recruitment of the Helicase Srs2p. Mol. Cell. 2005; 19:123–133. [DOI] [PubMed] [Google Scholar]
- 139. Arbel M., Liefshitz B., Kupiec M.. How yeast cells deal with stalled replication forks. Curr. Genet. 2020; 66:911–915. [DOI] [PubMed] [Google Scholar]
- 140. Fan L., Zhang W., Rybchuk J., Luo Y., Xiao W.. Genetic dissection of budding yeast PCNA mutations responsible for the regulated recruitment of Srs2 helicase. mBio. 2023; 14:e0031523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Aroya S.B., Kupiec M.. The Elg1 replication factor C-like complex: a novel guardian of genome stability. DNA Repair (Amst.). 2005; 4:409–417. [DOI] [PubMed] [Google Scholar]
- 142. Clark A.B., Valle F., Drotschmann K., Gary R.K., Kunkel T.A.. Functional interaction of proliferating cell nuclear antigen with MSH2-MSH6 and MSH2-MSH3 complexes. J. Biol. Chem. 2000; 275:36498–36501. [DOI] [PubMed] [Google Scholar]
- 143. Paul Solomon D., Lovely J., Gaubitz C., Lundblad V., Kelch B.A., Kubota T.. Effective mismatch repair depends on timely control of PCNA retention on DNA by the Elg1 complex. Nucleic Acids Res. 2019; 47:6826–6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Johnson R.E., Kovvali G.K., Guzder S.N., Amin N.S., Holm C., Habraken Y., Sung P., Prakash L., Prakash S.. Evidence for involvement of yeast proliferating cell nuclear antigen in DNA mismatch repair. J. Biol. Chem. 1996; 271:27987–27990. [DOI] [PubMed] [Google Scholar]
- 145. Iyer R.R., Pluciennik A., Genschel J., Tsai M.S., Beese L.S., Modrich P.. MutLalpha and proliferating cell nuclear antigen share binding sites on MutSbeta. J. Biol. Chem. 2010; 285:11730–11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zhu Q., Chang Y., Yang J., Wei Q.. Post-translational modifications of proliferating cell nuclear antigen: a key signal integrator for DNA damage response (Review). Oncol. Lett. 2014; 7:1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. McMurray C.T. Hijacking of the mismatch repair system to cause CAG expansion and cell death in neurodegenerative disease. DNA Repair (Amst.). 2008; 7:1121–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. McMurray C.T. Mechanisms of trinucleotide repeat instability during human development. Nat. Rev. Genet. 2010; 11:786–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lai Y., Budworth H., Beaver J.M., Chan N.L., Zhang Z., McMurray C.T., Liu Y.. Crosstalk between MSH2-MSH3 and polβ promotes trinucleotide repeat expansion during base excision repair. Nat. Commun. 2016; 7:12465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Kadyrova L.Y., Gujar V., Burdett V., Modrich P.L., Kadyrov F.A.. Human MutLγ, the MLH1-MLH3 heterodimer, is an endonuclease that promotes DNA expansion. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:3535–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material. Further data underlying this article will be shared on reasonable request to the corresponding author.