

Abstract
Efficient treatment of acute myeloid leukemia (AML) patients remains a challenge despite recent therapeutic advances. Here, using a CRISPRi screen targeting chromatin factors, we identified the nucleosome‐remodeling factor (NURF) subunit BPTF as an essential regulator of AML cell survival. We demonstrate that BPTF forms an alternative NURF chromatin remodeling complex with SMARCA5 and BAP18, which regulates the accessibility of a large set of insulator regions in leukemic cells. This ensures efficient CTCF binding and boundary formation between topologically associated domains that is essential for maintaining the leukemic transcriptional programs. We also demonstrate that the well‐studied PHD2‐BROMO chromatin reader domains of BPTF, while contributing to complex recruitment to chromatin, are dispensable for leukemic cell growth. Taken together, our results uncover how the alternative NURF complex contributes to leukemia and provide a rationale for its targeting in AML.
Keywords: acute myeloid leukemia, BPTF, chromatin remodeling, insulator regions, SMARCA5
Subject Categories: Cancer; Chromatin, Transcription & Genomics; Molecular Biology of Disease
Chromatin remodeling factors BPTF, SMARCA5 and BAP18 form a complex that regulates insulator accessibility and higher‐order genome organization in acute myeloid leukemia.
Introduction
In eukaryotic cells, the DNA inside the nucleus is hierarchically packaged as chromatin. The nucleosome represents the first level in this hierarchy. It consists of a DNA segment wrapped around an octamer of four histone proteins with side chains subject to extensive post‐translational modifications. At higher organizational levels, nucleosomes are assembled into fibers, loops, domains, and compartments, forming a dynamic structure regulating all the DNA‐dependent processes and safeguarding genome integrity (Misteli, 2020). Chromatin dysregulation is known to lead to various disorders ranging from developmental abnormalities to cancer (Mirabella et al, 2015; Valencia & Kadoch, 2019), and targeting epigenetic regulators represents a very attractive therapeutic strategy.
Acute myeloid leukemia (AML) is a malignant blood disorder characterized by the uncontrolled proliferation of myeloid progenitors in the bone marrow and peripheral blood. It is the most common type among acute leukemias (incidence of 1:25,000) and is very aggressive, with only 24% of patients demonstrating 5‐year survival (Shallis et al, 2019). In patients suffering from AML, genes encoding chromatin regulators are among the most mutated (Papaemmanuil et al, 2016). One example of such mutation is the rearrangement of the mixed lineage leukemia histone methyltransferase MLL, which leads to the generation of highly oncogenic MLL‐fusion proteins (Winters & Bernt, 2017). These fusions drive leukemia in part by dysregulation of the genes controlling hematopoiesis and hematopoietic stem cell maintenance, such as the HOXA cluster genes, HOX co‐factor MEIS1 and MYC (Krivtsov & Armstrong, 2007; Faber et al, 2009; Delgado & León, 2010; Bahr et al, 2018). To identify new vulnerabilities in human MLL‐rearranged AML, we performed a CRISPR interference (CRISPRi) screen against all chromatin‐associated factors. This revealed Bromodomain PHD finger Transcription Factor (BPTF), a subunit of the Nucleosome Remodeling Factor (NURF) complex, as an essential AML regulator.
BPTF is a single, 340 kDa polypeptide featuring five defined structural domains: DDT, WHIM, and PHD1 at the N‐terminus, and PHD2 and BROMO at the C‐terminus (Alkhatib & Landry, 2011). The chromatin reader specificities of the C‐terminal tandem PHD2‐BROMO domain have been extensively studied and proposed as a recruitment mode for the NURF complex (Li et al, 2006; Wysocka et al, 2006; Ruthenburg et al, 2011; preprint: Marunde et al, 2022). The ISWI family ATPase SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin, Subfamily A 1 (SMARCA1) is a catalytic component, with Retinoblastoma‐binding proteins 4 and 7 (RBBP4 and RBPP7) and BPTF Associated Protein 18 (BAP18) as auxiliary subunits of the complex (Barak et al, 2003; Vermeulen et al, 2010). The NURF complex catalyzes nucleosome sliding (Hamiche et al, 1999; Schwanbeck et al, 2004), which was shown to be necessary for regulating multiple signaling pathways in Drosophila (Badenhorst et al, 2002, 2005; Kwon et al, 2008). Bptf KO mice die at gastrulation (Landry et al, 2008), but the precise functions of the NURF complex in mammals are not understood. Here, we show that BPTF, via the formation of an alternative complex, regulates chromatin accessibility of insulator regions, ensuring CTCF binding, which is required for maintaining leukemogenic gene expression and AML cell proliferation. Unexpectedly, we show that the PHD2 and BROMO chromatin module of BPTF, while contributing to the efficient chromatin targeting, is not essential for the NURF complex function.
Results
BPTF is required for the proliferation of acute myeloid leukemia cells
To identify novel chromatin‐associated factors essential for leukemic cells, we designed a pooled CRISPR interference (CRISPRi) library targeting 1,046 genes previously associated with chromatin regulation (Fig EV1A and Dataset EV1). The sgRNAs were designed using our previously defined optimized parameters (Radzisheuskaya et al, 2016) (see Materials and Methods for details). For each gene, the two most frequently used transcription start sites were targeted with a total of six sgRNAs per gene. The final library size was 6,234 sgRNAs, of which 334 were negative and 37 were positive controls. Using this library, we performed the screen in human MLL‐AF9 rearranged leukemic cell lines, THP‐1 and MOLM‐13, and defined hits as genes for which at least two sgRNAs had a log2 fold change associated with a q‐value of less than 0.05. This resulted in 66 hits in THP‐1 dCas9 cells and 169 in MOLM‐13 dCas9 cells, with 63 of the identified hits shared between the two cell lines (Figs 1A and B, and EV1B, and Dataset EV2). Among the shared hits, there were multiple common essential genes, and several factors previously reported to sustain the proliferation of leukemic cells (e.g., KDM1A, MEN1, PRMT5, and SWI/SNF complex components) (Yokoyama et al, 2004; Caslini et al, 2007; Shi et al, 2013; Cruickshank et al, 2015; Radzisheuskaya et al, 2019; Ravasio et al, 2020; Rago et al, 2022; Chambers et al, 2023). We focused the further investigation on BPTF (Figs 1C and EV1C) since it has not been previously studied in the context of AML.
Figure EV1. The alternative NURF complex is required for the proliferation of acute myeloid leukemia cells.
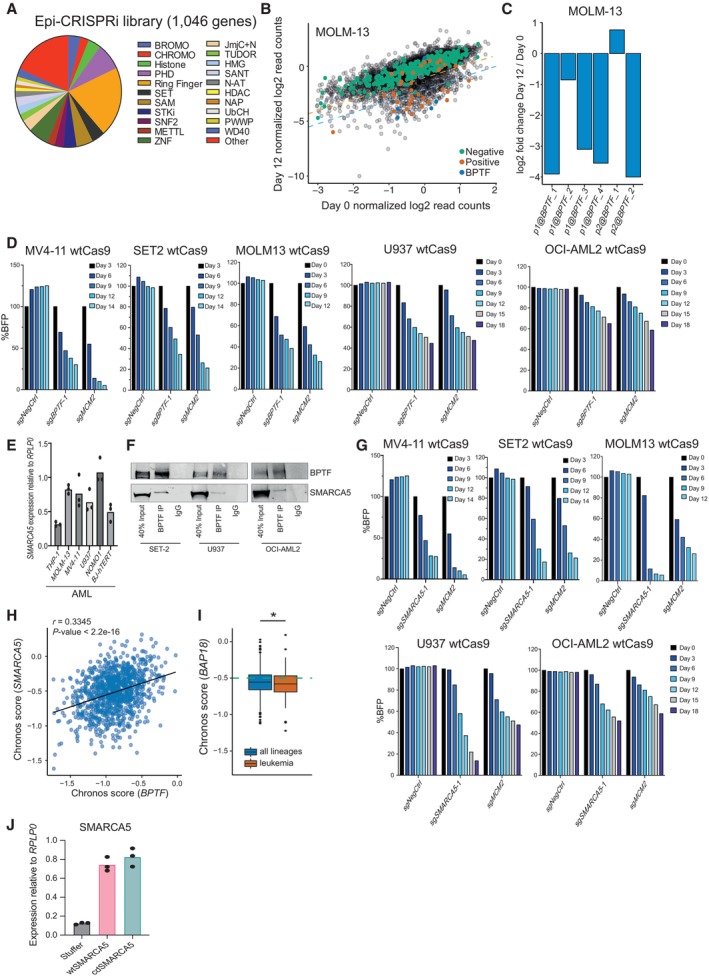
- Overview of the CRISPRi epi‐library composition.
- Scatterplot indicating log2 normalized read counts for each sgRNA in MOLM‐13 cells at day 0 and day 12 of the experiment. The orange and blue dashed lines indicate 2‐ and 4‐fold depletion in the screen, respectively. The screen was performed in two biological replicates, i.e., independent cell transductions (n = 6,184).
- Log2 fold changes for all the sgRNAs against BPTF in the screen of the MOLM‐13 cells. “p1@” and “p2@” in the sgRNA name indicate that it targets either the first (p1) or second (p2) most frequently used promoter for this gene according to the FANTOM5 consortium data. The data are the mean of two biological replicates, i.e., independent cell transductions.
- Competition assay after BPTF KO in MOLM‐13, MV4‐11, SET2, U937, and OCI‐AML2 wtCas9 cells. The cells were transduced with the lentiviral cassettes expressing the corresponding sgRNAs and BFP and mixed with untransduced cells. The percentage of BFP‐positive cells was measured over time. The data were normalized to Day 3 for MV4‐11 and SET2 cells, and to Day 0 for MOLM‐13, U937 and OCI‐AML2 cells. The experiment was performed at least twice with similar results.
- qRT‐PCR analysis for SMARCA5 expression in AML cell lines, values are normalized to RPLP0 and shown as mean (n = 3 technical replicates, i.e., independent qRT‐PCR reactions). The experiment was performed at least twice with similar results in a selection of cell lines.
- Western blot analysis of BPTF and SMARCA5 after the BPTF immunoprecipitation (IP) in U937, SET2, and OCI‐AML2 cells.
- Competition assay after SMARCA5 KO in MOLM‐13, MV4‐11, SET2, U937, and OCI‐AML2 wtCas9 cells. The cells were transduced with the lentiviral cassettes expressing the corresponding sgRNAs and BFP and mixed with untransduced cells. The percentage of BFP‐positive cells was measured over time. The data were normalized to Day 3 for MV4‐11 and SET2 cells and to Day 0 for MOLM‐13, U937, and OCI‐AML2 cells. The experiment was performed at least twice with similar results.
- Correlation between the BPTF and SMARCA5 Chronos dependency scores across all the cancer cell lines in the DepMap portal (version 22Q2) (n = 1,086). The correlation statistics were calculated using the Pearson's product–moment correlation.
- Chronos scores for BAP18 in all the lineages and leukemic cell lines. In the box plot, the middle line shows the median. The lower and upper hinges represent the first and third quartiles. The upper whisker stretches from the hinge to the largest value, but only up to 1.5 times the interquartile range. The lower whisker extends from the hinge to the smallest value, but only up to 1.5 times the interquartile range. Any data beyond the whiskers are plotted separately. The dashed green line indicates the essentiality threshold defined by the DepMap (version 22Q2). Welch two sample t‐test was used to assess the statistical significance. P < 0.0001. n = 970 for “all lineages” and n = 116 for “leukemia”.
- qRT‐PCR analysis for SMARCA5 expression in THP‐1 cells overexpressing wild‐type (wt) or catalytic dead (cd) SMARCA5 (n = 3 technical replicates, i.e., independent qRT‐PCR reactions). Data are normalized to RPLP0 and shown as mean.
Source data are available online for this figure.
Figure 1. The alternative NURF complex is required for the proliferation of acute myeloid leukemia cells.
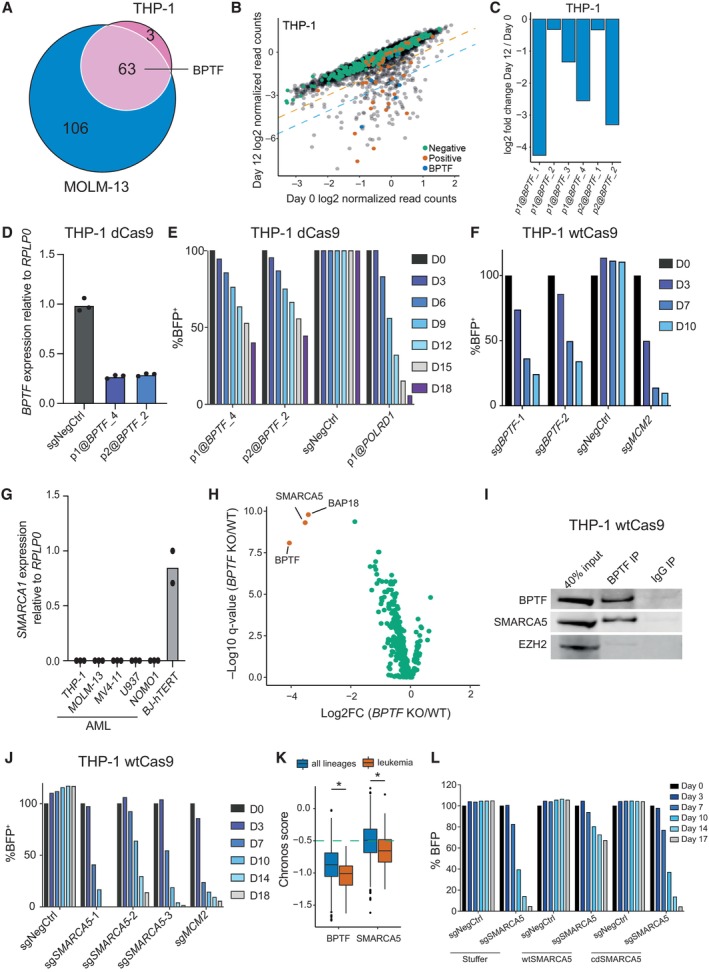
- Venn diagram indicating the overlap between the hits identified in the screened cell lines.
- Scatterplot indicating log2 normalized read counts for each sgRNA in THP‐1 cells at day 0 and day 12 of the experiment (n = 6,184). The orange and blue dashed lines indicate 2‐fold and 4‐fold depletion in the screen, respectively. “Negative”—non‐targeting sgRNAs; “positive”—sgRNAs targeting common essential genes. The screen was performed in two biological replicates, i.e., independent cell transductions.
- Log2 fold changes for all the sgRNAs against BPTF in the screen of the THP‐1 cells. “p1@” and “p2@” in the sgRNA name indicate that sgRNA targets either the first (p1) or the second (p2) most frequently used promoter for this gene according to the FANTOM5 consortium data. The data are mean of two biological replicates, i.e., independent cell transductions.
- qRT‐PCR analysis of BPTF expression in THP‐1 dCas9 cells after the knockdown with two BPTF‐targeting sgRNAs, values are normalized to RPLP0 and shown as mean (n = 3 technical replicates, i.e., three qRT‐PCR reactions). The experiment was independently performed three times with similar results.
- Competition assay after BPTF knockdown in THP‐1 dCas9 cells. The cells were transduced with the lentiviral cassettes expressing the corresponding sgRNAs and BFP and mixed with untransduced cells. The percentage of BFP‐positive cells was measured over time. The data were normalized to Day 0 and NegCtrl sgRNA. The experiment was validated with a growth curve assay, see Source data.
- Competition assay after BPTF KO in THP‐1 wtCas9 cells. The cells were transduced with the lentiviral cassettes expressing the corresponding sgRNAs and BFP and mixed with untransduced cells. The percentage of BFP‐positive cells was measured over time. The data were normalized to Day 0. The experiment was independently performed four times with similar results.
- qRT‐PCR analysis of SMARCA1 expression in AML cell lines and BJ‐hTERT fibroblasts, values are normalized to RPLP0 and shown as mean ± SD (n = 3 technical replicates, i.e., independent qRT‐PCR reactions). The experiment was repeated in a selection of cell lines with similar results.
- Volcano plot illustrating BPTF IP‐MS data in THP‐1 wtCas9 cells. The interactors with log2 fold change over 2 and a q‐value less than 0.01 are highlighted in orange.
- Western blot analysis of BPTF and SMARCA5 after the BPTF immunoprecipitation (IP) in THP‐1 cells. EZH2 was used as a negative control for IP. The experiment was independently performed twice with similar results.
- Competition assay after SMARCA5 KO in THP‐1 wtCas9 cells. The cells were transduced with the lentiviral cassettes expressing the corresponding sgRNAs and BFP and mixed with untransduced cells. The percentage of BFP‐positive cells was measured over time. The data were normalized to Day 0. The experiment was independently performed four times with similar results.
- Depmap dependency scores (Chronos scores) for BPTF and SMARCA5 in all lineages and leukemic cell lines. In the box plot, the middle line shows the median. The lower and upper hinges represent the first and third quartiles. The upper whisker stretches from the hinge to the largest value but only up to 1.5 times the inter‐quartile range. The lower whisker extends from the hinge to the smallest value but only up to 1.5 times the inter‐quartile range. Any data beyond the whiskers are plotted separately. The dashed green line indicates the essentiality threshold defined by the DepMap (version 22Q2). Welch two sample t‐test was used to assess the statistical significance. *P < 0.0001. n = 970 for “all lineages” and n = 116 for “leukemia”.
- Competition assay after SMARCA5 KO in THP‐1 wtCas9 cells overexpressing wild‐type (wt) or catalytic dead (cd) SMARCA5. The cells were transduced with the lentiviral cassettes expressing the sgRNA and BFP and mixed with untransduced cells. The percentage of BFP‐positive cells was measured over time. The data were normalized to Day 0.
Source data are available online for this figure.
To validate the requirement of BPTF for leukemic cells, we performed proliferation‐based competition assays using both CRISPRi and CRISPR knockout (KO) systems. Knockdown of BPTF in THP‐1 dCas9 cells efficiently reduced BPTF expression (Fig 1D) and impacted cell proliferation, as measured in a competition assay with untransduced cells (Fig 1E). Moreover, BPTF KO significantly impacted the growth of multiple human AML cell lines (Figs 1F and EV1D), further confirming BPTF as an essential AML regulator.
SMARCA5 associates with BPTF in AML cells and is required for their proliferation
To understand the mechanism by which BPTF contributes to the proliferation of AML cells, we first investigated if the catalytic nucleosome remodeling subunit of NURF, SMARCA1, was required. Surprisingly, SMARCA1 was not expressed in any of the leukemic cell lines analyzed (Fig 1G). Therefore, to explore the composition of the NURF complex in AML cells, we affinity‐purified BPTF‐associated proteins in THP‐1 wtCas9 cells transduced with either a non‐targeting sgRNA or an sgRNA against BPTF and determined the BPTF‐associated proteins by mass spectrometry. This led to the identification of SMARCA5 as the top BPTF interactor, along with BAP18 (Fig 1H and Dataset EV3). SMARCA5 has been found to interact with BPTF in HeLa and 293FT cells in a previous study (Oppikofer et al, 2017), while BAP18 was also previously proposed as a component of the NURF complex (Vermeulen et al, 2010). We found that SMARCA5 was expressed in leukemic lines lacking SMARCA1 (Fig EV1E). We also independently verified SMARCA5 and BPTF interaction by co‐immunoprecipitation in multiple AML cell lines (Figs 1I and EV1F).
To test if SMARCA5 is required for the proliferation of leukemic cells, we knocked it out using specific sgRNAs. As shown in Figs 1J and EV1G, there was a strong selection against the cells depleted for SMARCA5 in multiple AML cell lines. SMARCA5 was also identified as a hit in our CRISPRi screen in MOLM‐13 cells but did not reach the hit threshold in THP‐1 cells (Dataset EV1), likely due to the low efficiency of the CRISPRi sgRNAs. Moreover, SMARCA5 exhibits the highest co‐dependency with BPTF according to the DepMap portal of cancer vulnerabilities (https://depmap.org/portal/) (Fig EV1H). Despite BPTF, SMARCA5, and BAP18 being classified as common essential genes by the DepMap portal, leukemic cell lines demonstrate higher sensitivity to their loss than all other cell types (Figs 1K and EV1I), suggesting a potential therapeutic window for targeting the NURF complex in AML. To test whether the catalytic activity of SMARCA5 is required for the function of the NURF complex in AML cells, we ectopically expressed either wild‐type or catalytic dead (K112R) versions of SMARCA5 in THP‐1 wtCas9 cells (Fig EV1J) and transduced them with SMARCA5 KO sgRNA. While wild‐type SMARCA5 rescued the SMARCA5 KO phenotype, the catalytic dead version did not (Fig 1L), demonstrating the importance of the SMARCA5 ATPase activity for the complex function.
Together, our results show that only the alternative NURF complex, consisting of BPTF, SMARCA5 and BAP18, is present in AML cells and is required for AML cell proliferation.
BPTF and SMARCA5 are essential for maintaining AML in vivo
Next, we tested if BPTF, SMARCA1, and SMARCA5 were required for mouse MLL‐AF9 (MA9) leukemic cell proliferation. Similar to the results obtained in human cells, Bptf and Smarca5 KO decreased proliferation (Fig 2A and B), while Smarca1 was dispensable for mouse MLL‐AF9 cells (Appendix Fig S1A).
Figure 2. BPTF and SMARCA5 are essential for maintaining AML in vivo .
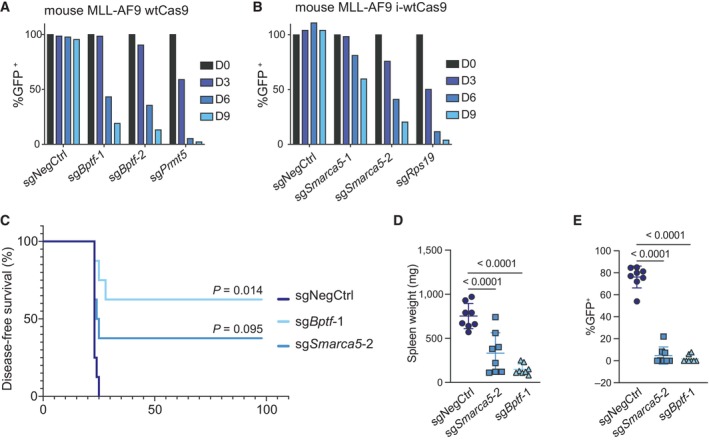
- Competition assay after Bptf KO in mouse MLL‐AF9 wtCas9 cells. The cells were transduced with the lentiviral cassettes expressing the corresponding sgRNAs and GFP and mixed with untransduced cells. The percentage of GFP‐positive cells was measured over time. The data were normalized to Day 0. The experiment was independently performed twice with similar results.
- Competition assay after Smarca5 KO in mouse MLL‐AF9 cells with dox‐inducible wtCas9 expression. The cells were transduced with the lentiviral cassettes expressing the corresponding sgRNAs and GFP and mixed with untransduced cells. The percentage of GFP‐positive cells was measured over time. The data were normalized to Day 0.
- Kaplan Meier survival curve comparing Bptf KO, Smarca5 KO and the negative control. n = 8 animals per group. P‐values comparing either Bptf KO or Smarca5 KO to the negative control after Bonferroni correction for multiple comparisons are 0.014 and 0.095, respectively, according to a Log‐rank Mantel‐Cox test.
- Spleen weight upon mice sacrifice. Data are shown as mean ± SD. The one‐way analysis of variance (ANOVA) was performed to determine statistical significance, n = 8 per group (total of 24).
- Percentage of transduced (GFP+) donor cells in the spleens of the experimental mice upon sacrifice. Data are shown as mean ± SD. The one‐way analysis of variance (ANOVA) was performed to determine statistical significance, n = 8 per group (total of 24).
Source data are available online for this figure.
To test if BPTF and SMARCA5 were also required in vivo, we transduced MLL‐AF9 cells, expressing leukocyte marker isoform CD45.2 and doxycycline‐inducible Cas9 (MLL‐AF9 i‐wtCas9), with the sgRNAs of interest (in a GFP‐expressing vector). The transduced cells were then sorted based on GFP expression and transplanted into sublethally irradiated mice (expressing leukocyte marker isoform CD45.1). Four days after injection, mice were given doxycycline to induce Cas9 expression. Mice injected with the non‐targeting sgRNA‐transduced cells succumbed to disease within 25 days post‐injection. In contrast, three out of eight mice injected with the sgSmarca5‐transduced cells and five out of eight animals injected with the sgBptf‐transduced cells lived for the entire duration of the experiment (Fig 2C). Before termination of the experiment, peripheral blood analysis was performed on the surviving mice to test if any leukemic cells were detectable. We found that the eight surviving mice had between 0.004 and 0.033% CD45.2+ (donor) cells in peripheral blood (Appendix Fig S1B), indicating that BPTF and SMARCA5 loss efficiently eliminated the donor cells. The spleens of all the experimental mice were weighed and analyzed for GFP expression. Mice transplanted with the non‐targeting sgRNA‐transduced cells had significantly larger spleens than those injected with sgBptf‐ or sgSmarca5‐transduced cells (Fig 2D). Notably, the mice injected with sgBptf‐ or sgSmarca5‐transduced cells exhibited very low GFP‐positivity in the spleens (Fig 2E), illustrating that AML arising in the KO groups was caused by escaper cells that had lost expression of the KO sgRNAs or were untransduced in the first place. Taken together, our results show that BPTF and SMARCA5 are required for leukemic progression in vivo and could represent potential targets for AML treatment.
BPTF and SMARCA5 sustain the expression of MYC and MYC‐regulated genes
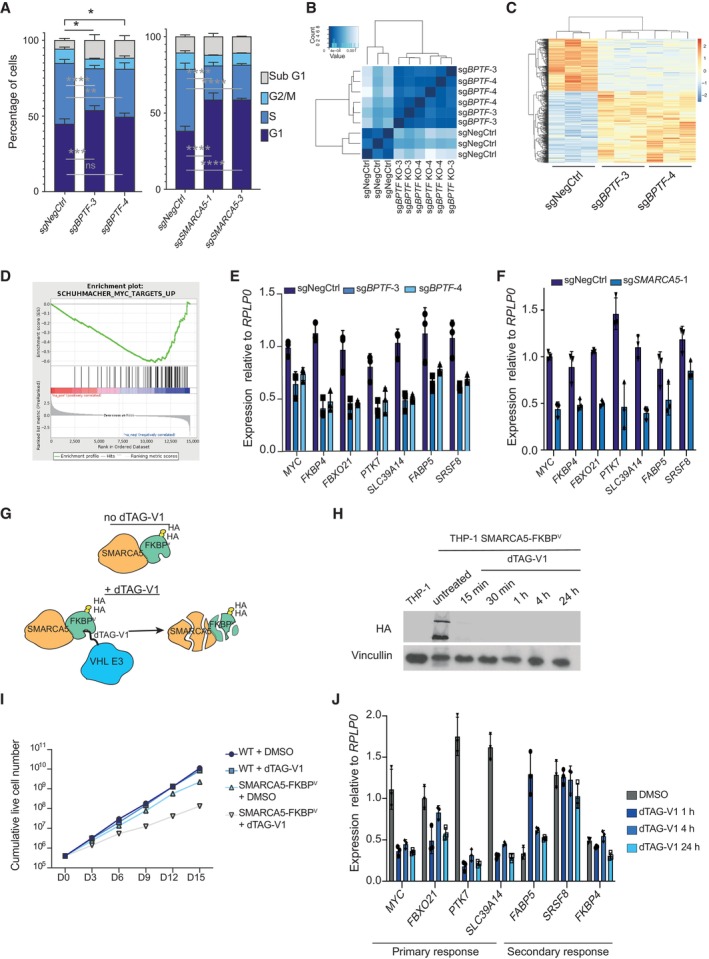
To understand how BPTF and SMARCA5 downregulation impacts leukemic cell proliferation, we first assessed the consequences of either BPTF or SMARCA5 depletion on cell cycle progression. THP‐1 wtCas9 cells were transduced with sgRNAs against BPTF or SMARCA5, and the cells were subjected to EdU staining 5 days after. Loss of BPTF resulted in an increase of the apoptotic sub‐G1 fraction, a moderate G1 arrest and a decrease in the proportion of cells in the S phase (Fig 3A). SMARCA5 KO led to a more prominent cell cycle arrest in the G1 phase and reduced the S phase cell population (Fig 3A). The involvement of SMARCA5 in several other complexes (Li et al, 2021) may explain the phenotypic differences between the depletion of the two genes. We next examined the impact of losing the NURF complex on AML cell differentiation. We measured the myeloid‐monocytic lineage differentiation marker CD11b levels after BPTF or SMARCA5 KO in three AML cell lines: THP‐1, SET2, and U937. These cell lines express CD11b (Fig EV2A) and are known to exhibit significant CD11b upregulation during differentiation (Chanput et al, 2014; Fiskus et al, 2021; Skopek et al, 2023). Interestingly, while BPTF and SMARCA5 KO did not affect CD11b levels in THP‐1 and SET2 cells, they did induce differentiation in U937 cells (Fig EV2B). These findings indicate that while NURF complex KO induces strong proliferation defects in most of the AML cell lines (Figs 1K, and EV1D and F), its effect on cell differentiation can vary depending on the cell type.
Figure 3. BPTF and SMARCA5 sustain the expression of MYC and MYC‐regulated genes.
-
ACell cycle analysis of THP‐1 wtCas9 cells transduced with either a non‐targeting sgRNA, BPTF KO or SMARCA5 KO sgRNAs (n = 3 biological replicates, i.e., independent cell transductions). Two‐way ANOVA followed by Dunnett's multiple comparisons test was performed to assess significance *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). Data are shown as mean ± SD.
-
BHierarchical clustering of the RNA‐sequencing samples in THP‐1 wtCas9 cells upon BPTF KO (n = 3 biological replicates, i.e., independent cell transductions).
-
CHeatmap of the differentially expressed genes upon the BPTF KO (n = 3 biological replicates, i.e., independent cell transductions).
-
DEnrichment plot for the MYC target gene set (GSEA: SCHUMACHER_MYC_TARGETS_UP).
-
E, FqRT‐PCR validation of the downregulation of MYC and its target genes upon BPTF (E) or SMARCA5 (F) KO (n = 3 biological replicates, i.e., independent cell transductions). Data are shown as mean ± SD.
-
GSchematic illustration of the SMARCA5 degradation system. FKBPV stands for FKBPF36V mutation.
-
HHA‐SMARCA5 Western blot in SMARCA5 degron knock‐in cells after dTAG‐V1 (100 nM) treatment for the indicated time points.
-
IGrowth curve for the wild‐type and SMARCA5 degron knock‐in cells (n = 3 biological replicates, i.e., independent cell treatments). Cells were treated with DMSO or 100 nM dTAG‐V1 as indicated. The experiment was performed twice with similar results.
-
JqRT‐PCR validation of the downregulation of MYC and its target genes upon SMARCA5 degradation (n = 3 biological replicates, i.e., independent cell treatments). Data are shown as mean ± SD.
Source data are available online for this figure.
Figure EV2. BPTF and SMARCA5 sustain the expression of MYC and MYC‐regulated genes.
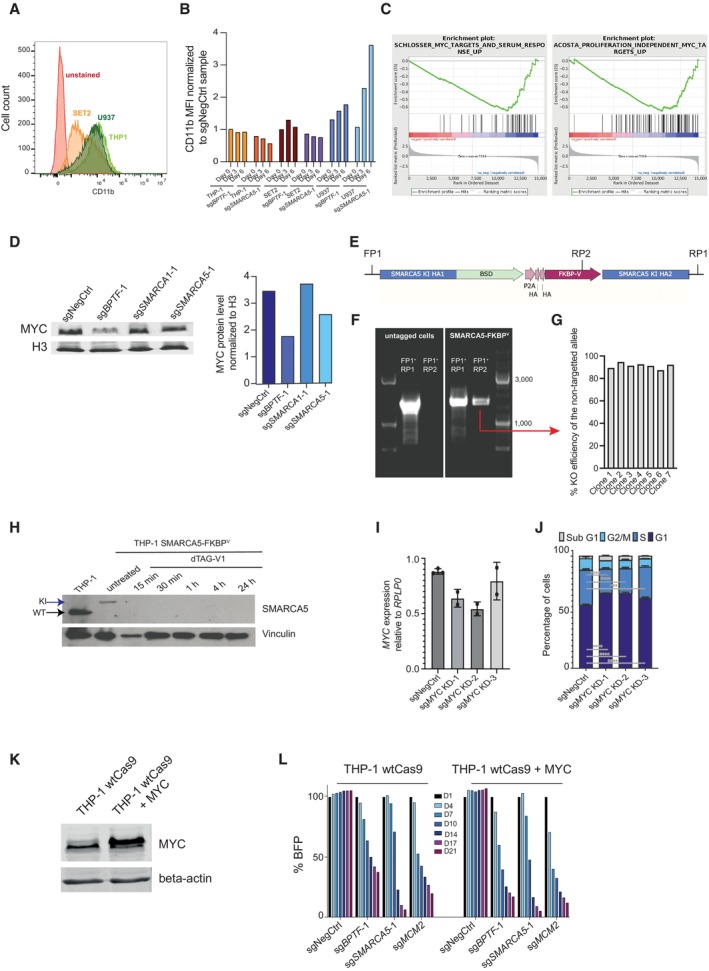
- Flow cytometry histogram depicting CD11b levels in three cell lines: SET2, U937, and THP‐1.
- The median fluorescent intensity of CD11b in THP‐1, SET2, and U937 cells after knocking out either BPTF or SMARCA5. The levels were normalized to the cells that were transduced with the non‐targeting sgRNA.
- Enrichment plots for MYC target genes among the downregulated genes after BPTF KO (GSEA categories: SCHLOSSER_MYC_TARGETS_AND_SERUM_RESPONSE_UP and ACOSTA_PROLIFERATION_INDEPENDENT_MYC_TARGETS_UP).
- Western blotting analysis of MYC protein levels (left) and their quantification (right) after BPTF, SMARCA1 and SMARCA5 KOs.
- Schematic representation of genotyping primers used to verify the successful knock‐in.
- Agarose gel image after the PCR using the genotyping primers represented in (E) on either wild‐type THP‐1 cells or the SMARCA5 degron knock‐in cells.
- KO efficiency graph generated using TIDE for the untargeted SMARCA5 locus in the individual clones generated from the SMARCA5 degron knock‐in cell pool.
- Western blotting analysis of SMARCA5 protein expression in wild‐type and SMARCA5 degron knock‐in cells upon treatment with dTAG‐V1 (100 nM). WT stands for wild‐type and KI—for knock‐in.
- qRT‐PCR analysis for the expression of MYC in THP‐1 cells transduced with a non‐targeting sgRNA or three different mixes of two MYC KD sgRNAs, as downregulation of MYC was insufficient using only one sgRNA, n = 3 biological replicates (independent cell transductions) for negative control and n = 2 biological replicates (independent cell transductions) for the remaining samples. Data are normalized to RPLP0 and shown as mean ± SD (one‐way ANOVA followed by Dunnett's multiple comparisons test was performed to assess significance, *P < 0.05). The experiment was performed twice with similar results.
- Cell cycle analysis of the THP‐1 cells transduced with a nontargeting sgRNA or MYC KD sgRNAs (n = 3 biological replicates, i.e., independent cell transductions). Data are shown as mean ± SD.
- Western blot for MYC and beta‐actin in wild‐type THP‐1 cells and THP‐1 cells with MYC overexpression.
- Competition assay after BPTF or SMARCA5 KO in wild‐type or MYC‐overexpressing THP‐1 cells. The cells were transduced with the lentiviral cassettes expressing the corresponding sgRNAs and BFP and mixed with untransduced cells. The percentage of BFP‐positive cells was measured over time. The data were normalized to Day 1.
Source data are available online for this figure.
To analyze gene expression changes induced in leukemic cells by the BPTF loss, we performed RNA sequencing (RNA‐seq) in THP‐1 wtCas9 cells 4 days after transduction with two independent sgRNAs targeting BPTF. This time point was chosen to allow for efficient BPTF depletion while collecting the cells before the onset of cell death. Analysis of gene expression changes demonstrated excellent agreement between the independent sgRNAs with 346 genes up‐ and 252 genes downregulated after BPTF KO (absolute log2 FC > 1, q value < 0.05) (Fig 3B and C, and Dataset EV4). Significant enrichment for MYC target genes was observed among the downregulated genes, including the downregulation of MYC itself (Figs 3D and EV2C), which we confirmed by reverse transcription‐quantitative PCR (RT‐qPCR) (Fig 3E and F). We also verified the decrease in MYC protein levels by western blotting (49% downregulation for BPTF KO and 25% – for SMARCA5 KO) (Fig EV2D).
To test whether the downregulation of MYC target genes was a direct consequence of the NURF loss, we generated cells with endogenously FKBPF36V‐tagged (Nabet et al, 2018) SMARCA5 (Figs 3G and EV2E). One of the SMARCA5 alleles contained the desired knock‐in in these cells, while the second allele was knocked out (Fig EV2F and G). The established SMARCA5‐FKBPF36V cells showed efficient and quick degradation of SMARCA5 (Figs 3H and EV2H) and displayed a proliferation defect upon adding dTAG‐V1 (Fig 3I). When compared to the untagged cells, the SMARCA5‐FKBPF36V cells showed reduced expression of SMARCA5 and slightly decreased proliferation (Figs 3I and EV2H). This could be because of the knockout of the untagged allele or the self‐degradation of the fusion protein. Nevertheless, these cells were suitable for studying the effects of short‐term downregulation of the SMARCA5 protein. Analysis of MYC and MYC target gene expression following degradation of SMARCA5 indicated that MYC and some of its targets, e.g., FBXO21, PTK7, and SLC39A14, were downregulated after just 1 h of treatment, suggesting this is likely a primary effect of SMARCA5 depletion. In contrast, it took significantly longer to downregulate other MYC target genes like FABP5, SRSF8, and FKBP4, suggesting a potential secondary effect (Fig 3J).
Next, we investigated if the reduction in cell proliferation caused by the depletion of NURF in AML cells can be attributed to the downregulation of MYC. We established CRISPRi conditions to mildly downregulate MYC in THP‐1 cells (Fig EV2I). We found that a 30–40% knockdown of MYC resulted in cell cycle changes similar to those observed with the loss of NURF, such as increased G1 and decreased S phase cell numbers (Fig EV2J). However, overexpression of MYC did not restore the proliferation defects caused by the BPTF or SMARCA5 KOs in THP‐1 cells (Fig EV2K and L).
Taken together, our results show that the alternative NURF complex regulates MYC transcriptional network in leukemic cells, which is consistent with previous studies reporting the downregulation of MYC target genes upon BPTF knockdown in fibroblasts and glioblastoma cells (Richart et al, 2016; Green et al, 2020). However, MYC downregulation does not fully account for the phenotype of the NURF complex loss in AML cells.
BPTF and SMARCA5 together remodel insulator regions in AML cells
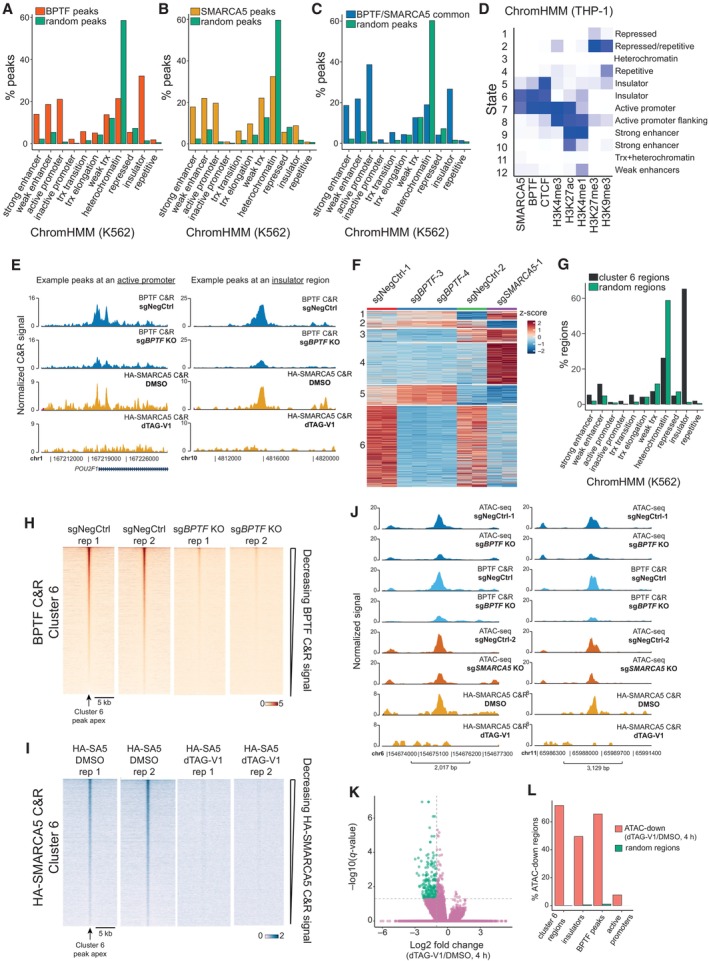
To understand the mechanism by which BPTF and SMARCA5 regulate gene expression, we determined their genomic occupancy in AML cells. For BPTF, we used THP‐1 wtCas9 cells transduced with either a non‐targeting sgRNA or a BPTF KO sgRNA and performed Cut&Run (Skene & Henikoff, 2017) followed by next‐generation sequencing 4 days after transduction. This resulted in the identification of 22,505 BPTF‐specific peaks (Dataset EV5). When overlapping those with the chromatin‐based genome annotation from another leukemia cell line, K562 (ChromHMM) (Ernst & Kellis, 2012), we found that BPTF preferentially binds to enhancers, active promoters, and insulators and is depleted in regions of low gene density (“heterochromatin” category). The enrichment was assessed by comparing the occurrence of the BPTF peaks in different categories to a distribution of a random peak set of the same sequence length and region number (Fig 4A). To profile SMARCA5 occupancy, we used the generated dTAG knock‐in cell line, in which a DNA sequence coding for the HA tag was also introduced into the SMARCA5 locus (Fig 3G). We treated the cells with dTAG‐V1 for 24 h and performed Cut&Run analysis using the anti‐HA antibody. This resulted in 28,399 SMARCA5‐specific peaks called (Dataset EV5). SMARCA5 peaks were enriched at enhancers, active promoters, and insulators and depleted in low‐gene density regions (Fig 4B). Based on the significant peaks called with a log2FC > 2 and an FDR of < 0.00001, 30% of BPTF peaks overlapped or mapped within 1 kb of SMARCA5 peaks (Fig EV3A), and the common peaks were predominantly located in the active promoter (48%) and insulator (30%) regions (Fig 4C). BPTF and SMARCA5 co‐bind a significant portion of active promoters and insulators (Fig EV3B). However, only a small subset of enhancers demonstrates NURF complex binding (Fig EV3B). Since K562 cells are of lymphoblast origin and could have differences in their chromatin state organization in comparison to THP‐1 cells, we used publicly available histone modifications data (H3K4me3, H3K27ac, H3K4me1, H3K27me3, and H3K9me3) and our BPTF, SMARCA5, and CTCF Cut&Run data to build a 12‐state ChromHMM model for THP‐1 cells (Figs 4D and EV3D). Due to the limited histone modification data available for THP‐1 cells, our model did not efficiently resolve transcribed regions and heterochromatin categories. However, it allowed us to further confirm that BPTF and SMARCA5 binding is mostly confined to insulators and active promoters in THP‐1 cells (states 5, 6, and 7) (Fig 4D and E).
Figure 4. BPTF and SMARCA5 together remodel insulator regions in AML cells.
-
A, BBar plots illustrating the percentage of BPTF (A) or HA‐SMARCA5 (B) Cut&Run peaks overlapping different chromatin domain categories defined by the ChromHMM in K562 cells. For both BPTF and SMARCA5, we generated a corresponding set of random peaks of the same average length and size (shown in green). Some peaks may overlap several categories; hence, the total percentage does not equal 100. “trx” stands for transcription.
-
CBar plot illustrating the percentage of common BPTF and SMARCA5 Cut&Run peaks overlapping different chromatin domain categories defined by the ChromHMM in K562 cells. A corresponding set of random peaks of the same average length and size was generated and shown in green. Some peaks may overlap several categories; hence, the total percentage does not equal 100. “trx” stands for transcription.
-
DTHP‐1 ChromHMM heatmap of emission parameters. The darker the blue color, the greater the probability of observing the mark/factor binding in the state.
-
EExamples of common BPTF and HA‐SMARCA5 Cut&Run peaks.
-
Fk‐means clustering of the differential ATAC‐seq peaks after the NURF KO.
-
GBar plot illustrating the percentage of regions in cluster 6 overlapping chromatin domain categories defined by the ChromHMM in K562 cells. A corresponding set of random regions of the same average length and size was generated and shown in green. Some regions may overlap several categories; hence, the total percentage does not equal 100. “trx” stands for transcription.
-
H, IHeatmaps of BPTF (H) and SMARCA5 (I) Cut&Run signal across the differential ATAC‐seq cluster 6.
-
JExamples of differential ATAC‐seq peaks in cluster 6.
-
KVolcano plots comparing ATAC‐seq changes between the DMSO‐ and dTAG‐V1‐treated cells (4 h). The peaks with an absolute log2 fold change over 1 and a q‐value < 0.01 are highlighted in green. n = 215,576.
- L
Data information: All the presented Cut&Run and ATAC‐seq experiments were performed in two biological replicates, i.e., independent cell transductions or treatments.
Source data are available online for this figure.
Figure EV3. BPTF and SMARCA5 bind active promoters and insulators in AML cells.
-
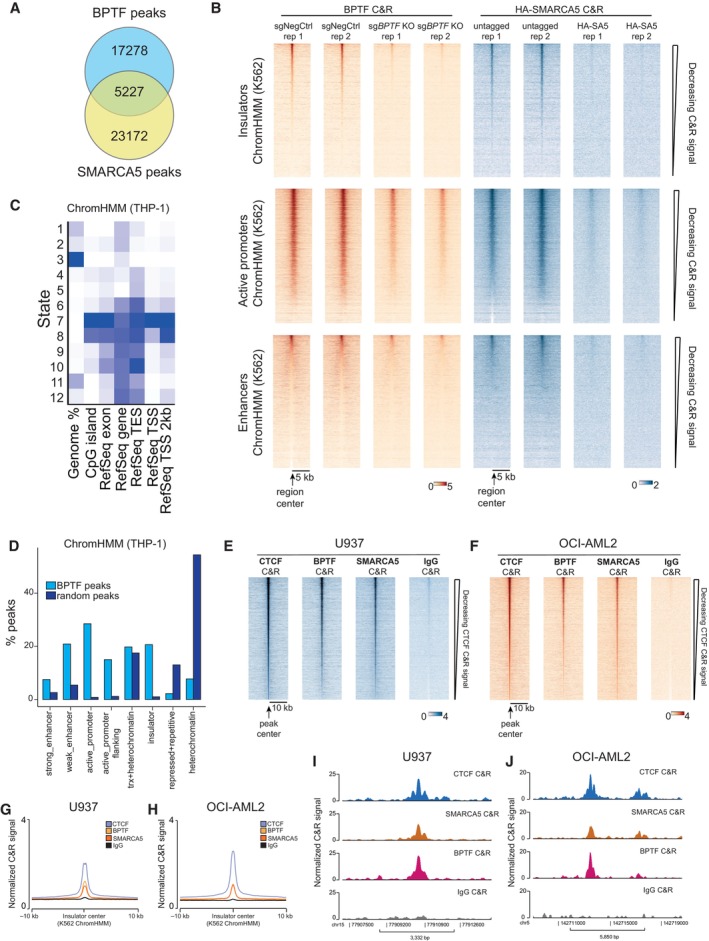
AVenn diagram of the overlap between the BPTF and HA‐SMARCA5 peaks. Each peak was extended by 500 bps on each side to estimate this overlap. 701 common regions are expected by chance.
-
BHeatmap of the BPTF and SMARCA5 Cut&Run signal across all the insulators, active promoters, and enhancers predicted by the ChromHMM in K562 cells.
-
CHeatmap displaying the relative percentage of the genome represented by each state (first column) and relative fold enrichment for: CpG islands, RefSeq exons, genes, transcription end sites (TES), transcription start sites (TSS), and 2,000 base pair regions around the TSS. The darker the blue color, the higher the fold enrichment.
-
DBar plots illustrating the percentage of BPTF Cut&Run peaks overlapping different chromatin domain categories defined by the ChromHMM in THP‐1 cells. Some peaks may overlap several categories; hence, the total percentage does not equal 100. “trx” stands for transcription.
-
E, FHeatmaps of the CTCF, BPTF, and SMARCA5 Cut&Run signal across all the CTCF peaks in U937 (E) and OCI‐AML2 (F) cells.
-
G, HAverage profiles of the CTCF, BPTF, and SMARCA5 Cut&Run signal in U937 (G) and OCI‐AML2 (H) cells at the insulator regions predicted by the ChromHMM in K562 cells.
-
I, JExamples of the common CTCF, SMARCA5, and BPTF insulator peaks in U937 (I) and OCI‐AML2 (J) cells.
Data information: All the Cut&Run experiments in THP‐1 cells were performed in two biological replicates, i.e., independent cell transductions or treatments.
Source data are available online for this figure.
To investigate if the NURF complex displays similar genomic occupancy in other AML cell lines, we conducted Cut&Run experiments for CTCF, BPTF, and SMARCA5 in U937 and OCI‐AML2 cells. This showed that BPTF and SMARCA5 are co‐localized with the majority of CTCF peaks in these cells (Fig EV3E and F). Overlapping these data with the insulator and active promoter regions predicted by the ChromHMM model in K562 cells, we further confirmed the NURF complex localization at these genomic categories in AML cells (Fig EV3G–J and Appendix Fig S2).
Since NURF is a nucleosome remodeling complex, we next determined how its loss affected chromatin accessibility in AML cells. THP‐1 wtCas9 cells were transduced with either a non‐targeting sgRNA or sgRNAs inducing BPTF or SMARCA5 KO, and the cells were subjected to ATAC‐sequencing 4 days after transduction. The experiments were performed independently for BPTF and SMARCA5 KO, and two negative controls are therefore displayed in Fig 4F and Appendix Fig S3A. BPTF and SMARCA5 KO significantly changed chromatin accessibility patterns (Appendix Fig S3B and C). BPTF KO predominantly induced loss of accessibility (16,346 peaks down and 424 peaks up at least twofold). In comparison, SMARCA5 KO led to decreases and increases in the ATAC‐seq signal (5,032 peaks down and 4,593 peaks up at least twofold) (Dataset EV6). Random clustering of the differential ATAC‐seq peaks revealed that the largest cluster (cluster 6) contained 16,726 ATAC peaks showing reduced signal upon the KO of both BPTF and SMARCA5 (Fig 4F and Appendix Fig S3D). The majority (65.3%) of ATAC‐seq peaks in this cluster overlap insulator regions defined by ChromHMM (Fig 4G and Appendix Fig S3E) and, consistently, this region set is significantly enriched for the binding motif of CTCF, a central regulator of higher‐order chromatin organization (57.91% of regions with motif, q‐value < 0.0001) (Appendix Fig S3F). Overall, cluster 6 contained 27% of all the insulators defined by the ChromHMM. Importantly, we also observed significant enrichment for BPTF and SMARCA5 bound regions among the cluster 6 sites (Fig 4H–J, and Appendix Fig S3G and H).
To determine whether the loss of the NURF complex regulates chromatin accessibility directly, we performed ATAC‐sequencing after rapid degradation of SMARCA5 in SMARCA5‐FKBPF36V knock‐in cell line. After only 4 h of dTAG‐V1 treatment, we observed a significant loss of chromatin accessibility at 212 genomic regions, with no gain in accessibility observed (Fig 4K). 62% of the regions with reduced accessibility overlapped with BPTF binding sites and 72% – with the downregulated ATAC regions detected in the KO experiments (cluster 6, Fig 4L). Importantly, 50% of the significantly downregulated ATAC‐seq peaks in the dTAG‐V1‐treated cells represented insulator regions (Fig 4L). This provides strong supporting evidence that the NURF complex remodels insulator regions in AML cells.
Taken together, we have found that the NURF complex binds active promoters, enhancers and insulators AML cells, and its loss reduces chromatin accessibility of a large set of insulator regions.
The NURF complex facilitates CTCF binding and chromatin insulation
To assess whether NURF‐mediated chromatin remodeling of the insulator regions affects CTCF binding, we performed Cut&Run‐sequencing in THP‐1 wtCas9 cells 4 days after transduction with either a non‐targeting sgRNA or sgRNAs against BPTF and SMARCA5. This revealed that SMARCA5 and BPTF KO reduced CTCF binding at the NURF‐remodeled insulator regions (ATAC‐seq cluster 6) (Fig 5A and B), but not at the other differential ATAC‐seq clusters (Fig EV4A and B).
Figure 5. The NURF complex facilitates CTCF binding and chromatin insulation.
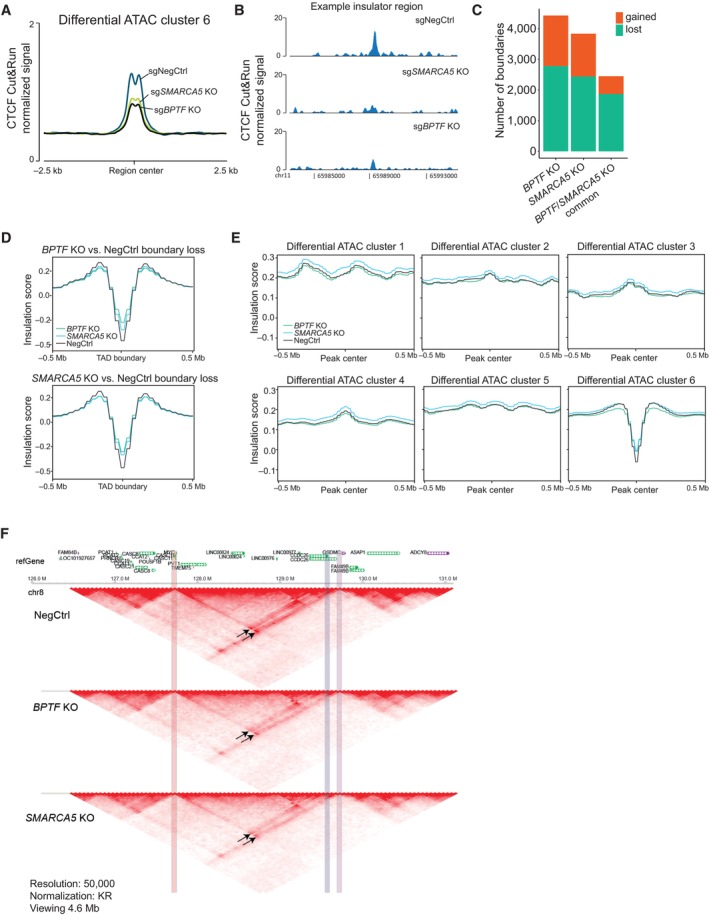
- Average profile of CTCF Cut&Run signal across the ATAC‐seq peaks decreased after the NURF KO (cluster 6) in THP‐1 wtCas9 cells.
- CTCF binding at a representative insulator region in THP‐1 wtCas9 cells transduced with the indicated sgRNAs.
- The number of changed TAD boundaries between the BPTF or SMARCA5 KO and wild‐type, and the number of boundaries commonly changed in the two KOs.
- Average insulation score profiles across all the boundaries lost upon BPTF (top) and SMARCA5 (bottom) KO.
- Average insulation score profiles across the differential ATAC‐seq clusters.
- Hi‐C data snapshots at 25 kb resolution from negative control, BPTF and SMARCA5 KO samples. MYC promoter region is highlighted in red. MYC enhancer sites are highlighted in blue (interaction loop 1) and purple (interaction loop 2). The interaction loops are indicated with arrows.
Data information: The CTCF Cut&Run experiment was performed three times with similar results, each in two biological replicates. The HiC experiment was performed in two biological replicates (independent cell transductions) that were pooled together at the analysis stage to increase sequencing depth.
Source data are available online for this figure.
Figure EV4. The NURF complex facilitates CTCF binding and chromatin insulation.
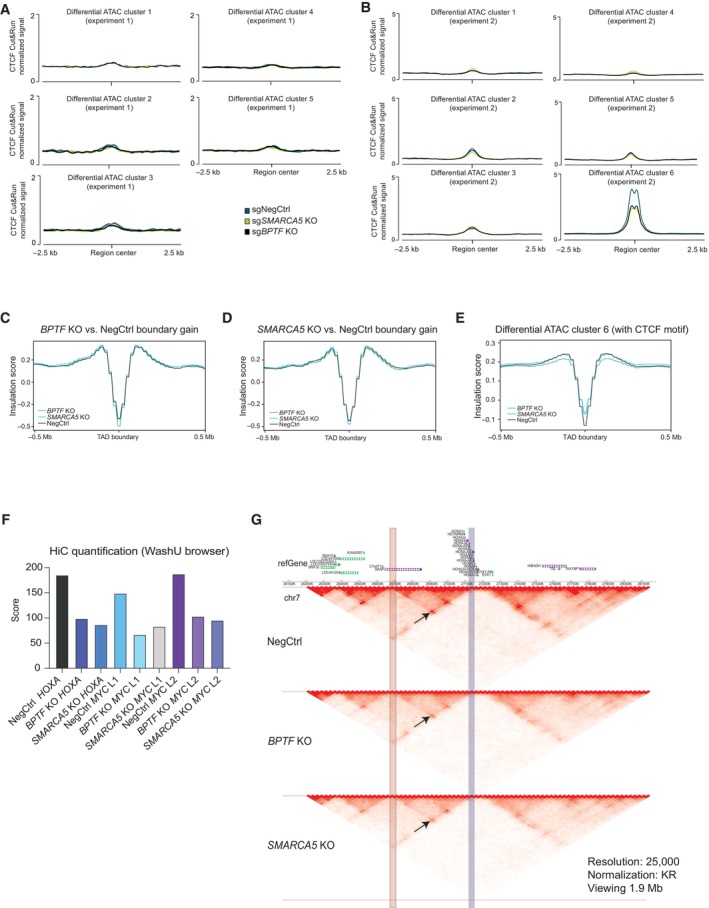
-
A, BAverage profile of CTCF binding at differential ATAC‐seq clusters in two independent experiments.
-
C, DAverage insulation score profiles across all the boundaries gained upon BPTF (C) and SMARCA5 (D) KO.
-
EAverage insulation score profile across the differential ATAC‐seq cluster 6 regions with CTCF motifs.
-
FWashU Browser score at the indicated interaction points.
-
GHi‐C data snapshots at 25 kb resolution from negative control, BPTF and SMARCA5 KO samples. Anchor points for the indicated loop (arrow) at the HOXA cluster are highlighted in red and blue.
Data information: The CTCF Cut&Run experiment was performed three times with similar results, each in two biological replicates. The HiC experiment was performed in two biological replicates that were pooled together at the analysis stage to increase sequencing depth.
Source data are available online for this figure.
CTCF regulates the formation of topologically associated domains (TADs) and chromatin loops by binding to boundary elements, or insulators, and blocking the Cohesin‐mediated loop extrusion (Dekker & Mirny, 2016). Since we observed a loss of accessibility and decreased CTCF binding at the inferred insulator regions, we next asked how the loss of the NURF complex affects TAD organization in AML cells. For that, we performed Hi‐C experiments in THP‐1 wtCas9 cells transduced with either a non‐targeting sgRNA or sgRNAs against BPTF or SMARCA5. Calling TAD boundaries using the HiCExplorer tool (Crane et al, 2015), we found that BPTF and SMARCA5 KOs led to gain and loss of boundaries, with approximately double the number of lost versus gained boundaries in the KO samples (Fig 5C and Dataset EV7). Differentially called boundaries common between the two KOs were predominantly in the “lost” category and comprised around 27% of all the TAD boundaries (Fig 5C). Consistently, in the SMARCA5 KO samples, local insulation score minimums (used to define TAD boundaries) (Crane et al, 2015) were increased in the regions defined as lost boundaries in BPTF KO, and vice versa (Fig 5D). This was not true for the gained boundaries (Fig EV4C and D). Next, we analyzed average insulation scores across the differential ATAC‐seq clusters described in Fig 4F. Consistently with the enrichment of insulator regions in cluster 6, we found that only this cluster demonstrated a characteristic dip in the Hi‐C insulation score (Fig 5E). Importantly, KO of BPTF and SMARCA5 led to reduced insulation of these sites, which was even more pronounced at the regions within this cluster having a CTCF binding motif (Fig EV4E). This further links the remodeling activity of the NURF complex to the regulation of genome topology.
Recently, a specific blood enhancer cluster (BENC), located at approximately 1.8 Mb from the MYC gene, was identified as essential for MLL‐AF9‐driven AML (Bahr et al, 2018). Interestingly, we found that NURF depletion led to a decreased interaction between the MYC promoter and BENC (Figs 5F and EV4F), suggesting a potential mechanism leading to the downregulation of MYC expression in BPTF and SMARCA5 KO cells. We also found reduced interaction within the HOXA cluster TAD (Fig EV4F and G), which could explain the downregulation of several HOXA cluster genes in the KO cells and further contribute to the loss of AML cell viability.
In summary, we have shown that BPTF and SMARCA5 remodel a large subset of TAD boundaries in AML cells, facilitating CTCF binding and insulation at these regions. BPTF and SMARCA5 also promote the long‐range interaction between MYC and one of its enhancers (BENC), allowing for the maintenance of the MYC transcriptional program in leukemic cells.
Tandem PHD2 and BROMO reader domains of BPTF are not required for the proliferation of AML cells
The chromatin reader specificities of the tandem C‐terminal PHD2/BROMO domains of BPTF have been extensively profiled in vitro and proposed to mediate BPTF recruitment to chromatin (Li et al, 2006; Wysocka et al, 2006; Ruthenburg et al, 2011; preprint: Marunde et al, 2022), while the functions of the N‐terminal BPTF domains (DDT, WHIM and PHD1) are unknown.
To determine which regions of BPTF were required for the growth of leukemic cells, we performed a CRISPR KO tiling screen, in which the entire coding sequence of the gene of interest is targeted with a library of sgRNAs. We designed a library comprising 806 different sgRNAs targeting BPTF, 111 negative control sgRNAs, and 243 positive control sgRNAs, which were then used to transduce THP‐1 wtCas9 cells. This screen identified two critical regions in BPTF for which sgRNAs were preferentially depleted: the first 800 amino acids, comprising the N‐terminal domains DDT, WHIM, and PHD1, and a second region between 1,600–1,800 amino acids with no known coding structural domains (Fig 6A and Dataset EV8). In contrast, sgRNAs targeting the well‐characterized PHD2 and BROMO coding regions were only depleted to the same extent as sgRNAs targeting throughout the gene body. To confirm the results of the tiling screen, we ran a proliferation‐based competition assay with sgRNAs targeting the different BPTF regions coding for its structural domains in both human and mouse AML cells. This corroborated that targeting the first three N‐terminal domains has a more significant effect on cell proliferation than targeting the C‐terminal domains (Figs 6B and EV5A).
Figure 6. Tandem PHD2 and BROMO reader domains of BPTF contribute to NURF recruitment but are not required for the proliferation of the AML cells.
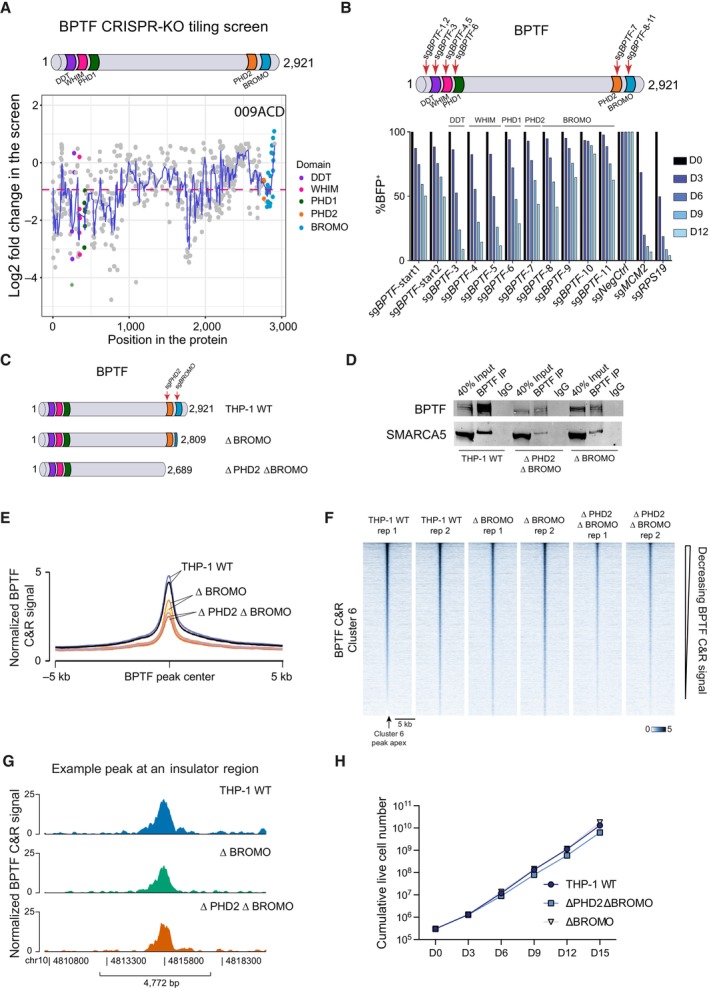
- Overview of the BPTF tiling screen results in THP‐1 cells. Pink dashed line represents average dropout across the whole gene. Blue line represents a simple moving average of five sgRNAs. The tiling screen was performed in two biological replicates, i.e., independent cell transductions.
- Competition assay after targeting BPTF with different sgRNAs in THP‐1 wtCas9 cells. The cells were transduced with the lentiviral cassettes expressing the corresponding sgRNAs and BFP and mixed with untransduced cells. The percentage of BFP‐positive cells was measured over time. The data were normalized to Day 0 and NegCtrl sgRNA.
- Schematic representation of the generated endogenous BPTF truncation cell lines.
- Western blot analysis of BPTF and SMARCA5 after BPTF immunoprecipitation in wild‐type and BPTF truncation THP‐1 wtCas9 cell lines.
- Average profile of BPTF binding in wild‐type and BPTF truncation cell lines across all the BPTF peaks defined in Fig 4A. The Cut&Run experiment was performed in two biological replicates, i.e., independent cell cultures.
- Heatmap of BPTF binding in wild‐type and BPTF truncation cell lines across the ATAC‐seq peaks decreased after the NURF KO (cluster 6) in THP‐1 wtCas9 cells.
- BPTF binding in wild‐type and BPTF truncation THP‐1 wtCas9 cell lines at a representative insulator region that is directly remodeled by the NURF complex in AML cells.
- Growth curve of the wild‐type and BPTF truncation cell lines, n = 3 biological replicates, i.e., independent cell cultures. Data are shown as mean ± SD.
Source data are available online for this figure.
Figure EV5. Tandem PHD2 and BROMO reader domains of BPTF are not required for the proliferation of the AML cells.
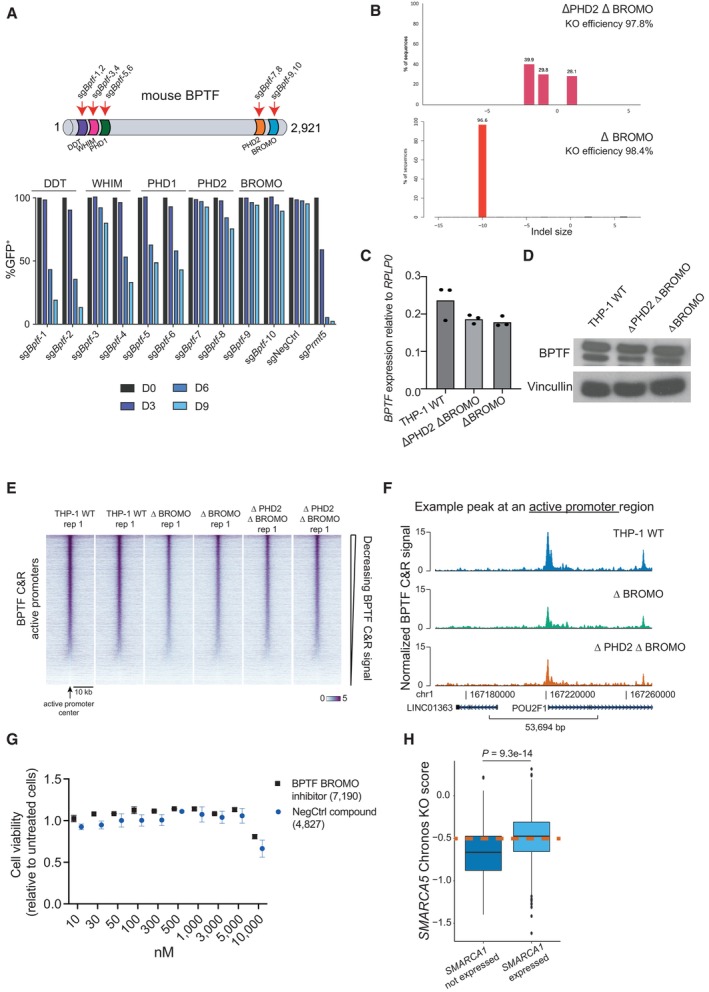
-
ACompetition assay after targeting Bptf with different sgRNAs in mouse MLL‐AF9 wtCas9 cells. The cells were transduced with the lentiviral cassettes expressing the corresponding sgRNAs and GFP and mixed with untransduced cells. The percentage of GFP‐positive cells was measured over time. The data were normalized to Day 0.
-
BIndel analysis of the mutations induced by the sgRNAs in the selected BPTF truncation clones.
-
C, DBPTF expression at the mRNA (n = 3 technical replicates, i.e., independent qRT‐PCR reactions; the data are normalized to RPLP0 and presented as mean) (C) and protein (D) levels in the wild‐type and BPTF truncation clones.
-
EBPTF binding in wild‐type and BPTF truncation clones across all the active promoter regions defined by the ChromHMM in K562 cells. The Cut&Run was performed in two biological replicates.
-
FBPTF binding in wild‐type and BPTF truncation clones at a representative promoter region.
-
GRelative cell viability of the THP‐1 cells after treatment with increasing concentrations of either a BPTF BROMO domain inhibitor or a structurally related negative control compound. Data are shown as mean ± SD, n = 3 biological replicates, i.e., independent cell treatments.
-
HBox plot indicating SMARCA5 DepMap KO score in cancer cell lines expressing and not expressing SMARCA1. In the box plot, the middle line shows the median. The lower and upper hinges represent the first and third quartiles. The upper whisker stretches from the hinge to the largest value, but only up to 1.5 times the interquartile range. The lower whisker extends from the hinge to the smallest value, but only up to 1.5 times the interquartile range. Any data beyond the whiskers are plotted separately. The dashed line indicates the essentiality threshold defined by the DepMap (version 22Q2). P < 0.0001 based on Wilcoxon rank sum test with continuity correction. n = 833 for “expressed” and n = 172 for “not expressed”.
Source data are available online for this figure.
To further validate that the PHD2 and BROMO domains are not essential for supporting the proliferation of AML cells, we generated THP‐1 wtCas9 cell lines with truncated BPTF by expressing an sgRNA targeting either a part of the gene just upstream of the PHD2 coding region or right at the beginning of the BROMO domain (Fig 6C). TIDE analysis (Brinkman et al, 2014) of each of these clones showed KO efficiency of 97.8% and 96.6% for the cells lacking the PHD2‐BROMO or the BROMO domain only, respectively (Fig EV5B). Both ΔPHD2 ΔBROMO and ΔBROMO cell lines showed comparable BPTF expression levels to unedited cells (Fig EV5C and D), and the truncated proteins retained their interaction with SMARCA5 (Fig 6D). To assess whether PHD2‐BROMO domains are required for BPTF genomic occupancy, we mapped the BPTF binding in the truncated clones by Cut&Run. These experiments showed that the mutant BPTF proteins still bind to the previously mapped BPTF binding sites, albeit with a decreased efficiency (Figs 6E–G, and EV5E and F). Importantly, AML proliferation was not significantly affected in the cells expressing only mutant BPTF without the PHD2 and BROMO domains (Fig 6H). Consistently, we could not detect an effect on the proliferation of THP‐1 cells treated with a recently developed BPTF BROMO‐domain inhibitor (Martinelli et al, 2023) (Fig EV5G). In summary, while the BPTF PHD2‐BROMO domains can aid in effectively directing NURF to chromatin, they are not necessary for forming a functional NURF complex or sustaining the growth of AML cells.
Discussion
We have shown that BPTF and SMARCA5 form an alternative NURF complex in AML cells, which is essential for their proliferation in vitro and in vivo. Furthermore, we demonstrated that BPTF and SMARCA5 regulate chromatin accessibility and CTCF binding at a large set of insulator regions, which is essential to maintain proper TAD insulation and gene expression. Specifically, we found that NURF promotes the interaction between the BENC enhancer and the MYC promoter, facilitating MYC expression (Fig 7).
Figure 7. NURF inactivation leads to AML growth arrest and apoptosis.
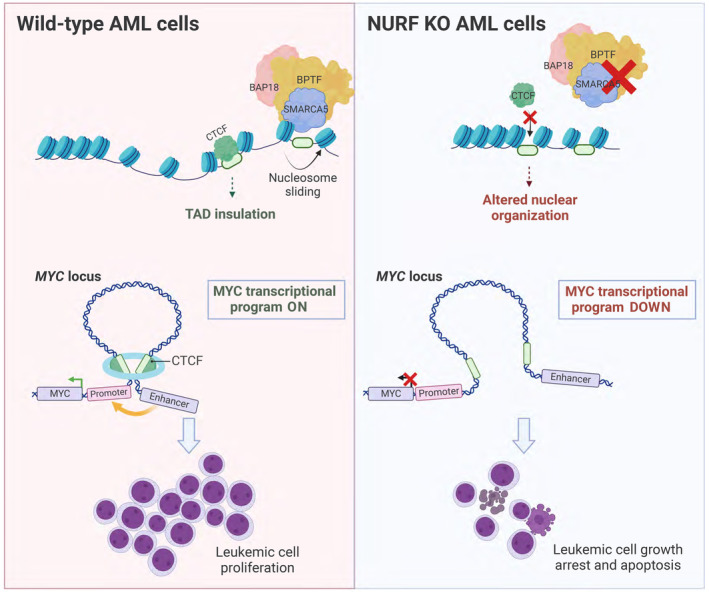
In AML cells, the NURF complex consists of BPTF, SMARCA5, and BAP18, which remodels chromatin at insulator regions by means of nucleosome sliding, thereby facilitating access of CTCF to its binding sites. This ensures TAD insulation and enhancer‐promoter loop formation, essential to maintain leukemic expression patterns, with MYC transcriptional program representing an important example (left). Upon the KO of the NURF components, the accessibility of the insulator regions is decreased, leading to changes in higher‐order genome organization, altered gene expression and loss of AML cell proliferative capacity (right). Created with BioRender.com.
We found that the canonical NURF catalytic subunit SMARCA1 (Barak et al, 2003; Alkhatib & Landry, 2011) is not expressed in AML cell lines, suggesting that only the alternative BPTF/SMARCA5 NURF complex is functional in these cells. Such an alternative complex is unlikely to be specific to leukemic cells since BPTF was shown to interact with SMARCA5 in HeLa and HEK293T cells (Oppikofer et al, 2017). However, the absence of SMARCA1 expression could potentially sensitize AML cells to SMARCA5 loss. Indeed, based on the data from the Depmap project, we found that SMARCA1 expression is a good predictor of SMARCA5 sensitivity of cancer cell lines (Fig EV5H).
Consistent with our results, two previous studies reported a loss of chromatin accessibility at insulator regions and a concomitant decrease in CTCF binding at those sites upon the knockout of Smarca5 (Barisic et al, 2019) or Bptf (Qiu et al, 2015) in mouse ES cells. Barisic et al also found a global decrease in insulation scores in Smarca5 KO ES cells, while we observed this for a large subset but not all the TADs (Fig 5C). This difference could be explained by either species or cell type differences and likely additional remodelers contributing to insulator regulation in human AML. Despite not incurring an effect on all TADs in leukemic cells, NURF activity is required for their proliferation (Fig 1). In contrast, the knockout of Smarca5 and Bptf in ES cells only leads to mild proliferative defects (Qiu et al, 2015; Barisic et al, 2019), further highlighting the specific functions of the NURF complex in the context of hematopoietic cells.
The oncogenic transcription factor MYC is a known driver of AML development and progression (Luo et al, 2005; Delgado & León, 2010; Kim et al, 2010; Fauriat & Olive, 2014; Li et al, 2014). Our Hi‐C data showed decreased long‐range interaction between the MYC promoter and the previously described BENC enhancer (Bahr et al, 2018) upon NURF depletion. Since the BENC enhancer has a critical role in maintaining MLL‐AF9 rearranged leukemia in vivo (Bahr et al, 2018), the loss of this interaction could explain the decreased expression of MYC upon the loss of the NURF complex. Consistent with our results and supporting the functional link between the NURF and CTCF occupancy, it was recently shown that targeted degradation of CTCF leads to diminished interaction between the MYC promoter and BENC enhancer and decreased MYC expression in B‐ALL cells (Hyle et al, 2019). Several previous reports demonstrated the importance of another chromatin remodeling complex, SWI/SNF, for the BENC enhancer function (Chambers et al, 2023) and MYC expression in AML cells (Shi et al, 2013; Rago et al, 2022). While both remodeling complexes (NURF and SWI/SNF) localize to the promoter, enhancer, and insulator regions (Barisic et al, 2019; Centore et al, 2020), their loss affects the accessibility of these regions differently. We found that NURF loss does not impact the accessibility of promoters and enhancers, while it reduces the accessibility of the insulator regions and impacts CTCF binding. Similar findings were observed upon Smarca5 KO in mouse ES cells (Barisic et al, 2019). SWI/SNF loss mainly affects the accessibility of enhancer regions, which leads to decreased binding of lineage‐specific transcription factors and reduced target gene expression (Barisic et al, 2019; Centore et al, 2020; Chambers et al, 2023). In the AML context, the SWI/SNF complex mediates the enhancer function of BENC, enabling the binding of RUNX1, LMO2 and MEIS1 at the locus. SWI/SNF loss leads to a substantial reduction in MYC expression in AML cells, and the decreased proliferation induced by SWI/SNF depletion can be rescued by MYC overexpression (Shi et al, 2013; Rago et al, 2022; Chambers et al, 2023). In contrast, we found that the NURF complex has a global role in ensuring efficient TAD insulation in AML cells, including at the BENC‐MYC locus. The NURF complex loss leads to a moderate (~30–40%) downregulation of MYC expression, consistent with TAD formation modulating transcriptional outputs rather than determining them (Misteli, 2020). We also found that MYC overexpression could not rescue the phenotype of NURF depletion in AML cells, further supporting the role of the NURF complex as a global regulator of higher‐order chromatin structure.
It has been proposed that BPTF interacts with chromatin via its C‐terminal PHD2 and BROMO domains (Li et al, 2006; Wysocka et al, 2006; Ruthenburg et al, 2011; preprint: Marunde et al, 2022). An elegant recent study extensively profiled the specificity of this tandem domain module in a nucleosome context and found that it preferentially recognizes H3K4me3‐H3K18ac or H3K4me3‐H3K14ac nucleosomes (preprint: Marunde et al, 2022). This is consistent with our findings that BPTF localizes to active chromatin regions and is present at most active promoter regions (Figs 4 and EV3). Interestingly, we found that while PHD2‐BROMO domains contribute to the efficient NURF binding, they do not determine it, as BPTF lacking these domains can bind chromatin and sustain the proliferation of leukemic cells. Similar findings were reported for BRD4, where chemical inhibition of its BROMO domain was shown to reduce chromatin localization but was insufficient to confer a phenotype comparable to the degradation of the protein (Winter et al, 2017; Zheng et al, 2023). This demonstrates that a combination of mechanisms determines the recruitment of the multidomain chromatin regulators. In the case of BPTF, chromatin binding by the PHD1 domain could represent an additional mode of chromatin recognition. Importantly, it has been demonstrated that BPTF can directly bind to DNA in vitro (Jordan‐Sciutto et al, 1999). This binding activity is primarily found in the first 400 amino acids of the protein, which include the DDT and WHIM domains (Jordan‐Sciutto et al, 1999). While these domains are known to mediate the interaction with the ISWI ATPases (Eberharter et al, 2004; Aravind & Iyer, 2012; Dong et al, 2013; Sharif et al, 2021), they were also proposed to have DNA binding functions in other proteins (Doerks et al, 2001; Aravind & Iyer, 2012). To deepen our understanding of how the NURF complex is directed to chromatin, it will be important to analyze the functional significance of the different segments of the N‐terminal BPTF region. This will also help identifying the most effective strategy for inhibiting BPTF function.
Our finding that AML cells can proliferate without the BPTF BROMO domain weakens the premise for current efforts to target this domain with small molecules for cancer treatment (Urick et al, 2015; Xu et al, 2019; Lu et al, 2021; Zahid et al, 2021). Indeed, we found that a BPTF BROMO domain inhibitor did not affect the proliferation of AML cells (Fig EV5G). Thus, depleting BPTF or SMARCA5 with proteolysis‐targeting chimaeras (PROTACs) to induce complete protein degradation or designing small molecule inhibitors to the N‐terminal part of BPTF could represent better therapeutic strategies. Although BPTF and SMARCA5 are necessary for normal hematopoiesis (Kokavec et al, 2017; Xu et al, 2018), leukemic cells show a high dependency on NURF components (Figs 1K and EV1I) and there may therefore be a therapeutic window for targeting the NURF complex. Notably, BPTF has also been shown to be required for melanoma (Koludrovic et al, 2015; Laurette et al, 2020), high‐grade glioma (Green et al, 2020), pancreatic cancer (Muñoz Velasco et al, 2022), lymphoma (Richart et al, 2020; Bai et al, 2022) and ovarian cancer (Miao et al, 2021). In addition, a recent study has shown that deleting a single BPTF allele can sustain B‐cell maturation while delaying lymphomagenesis in mice (Richart et al, 2020).
In summary, we have shown that BPTF and SMARCA5 are present in an alternative NURF complex in AML, where both proteins are required for CTCF binding, sustaining genomic architecture and driving transcriptional programs required for leukemic cell survival. Our results highlight NURF as a potential therapeutic target in AML.
Materials and Methods
Cell culture
THP‐1 (male, RRID:CVCL_0006) cells were cultured in RPMI 1640 with GlutaMAX and 1 mM sodium pyruvate (Gibco), supplemented with 10% heat‐inactivated FBS and 1× Penicillin/Streptomycin (Gibco). MV4‐11 (male, RRID:CVCL_0064), MOLM‐13 (male, RRID:CVCL_2119), SET2 (female, RRID:CVCL_2187), U937 (male, RRID:CVC_0007) and OCI‐AML2 (male, RRID:CVCL_1619) cells were grown in RPMI 1640 with GlutaMAX and 1 mM sodium pyruvate (Gibco) supplemented with 20% heat‐inactivated FBS and 1× Penicillin/Streptomycin (Gibco). HEK293‐FT packaging cells (female, RRID:CVCL_6911) cells were grown in Dulbecco's Modified Eagle's Medium – high glucose (Gibco) supplemented with 10% FBS, GlutaMAX, 1 mM sodium pyruvate and 1× Penicillin–Streptomycin (Gibco). Mouse MLL‐AF9 secondary leukemia cells (female) were cultured in RPMI 1640 with GlutaMAX and 1 mM sodium pyruvate (Thermo Fisher Scientific), supplemented with 20% heat‐inactivated FBS, 1× Penicillin/Streptomycin (Gibco) and 20% of culture media supernatant from the IL‐3‐secreting cell line (made in‐house). All cell lines were tested for mycoplasma regularly and maintained at 37°C, with 5% CO2 and 95% humidity. Cell lines expressing wtCas9 and the SMARCA5‐dTAG knock‐in cell line were grown with 5 μg/ml blasticidin. THP‐1 cells with truncated BPTF were grown in media with 2 μg/ml puromycin and 5 μg/ml blasticidin, sgRNA‐expressing cells—in 2 μg/ml puromycin.
Lentiviral production and transduction
2 × 106 HEK293‐FT cells were plated in a 10 cm culture dish 24 h pretransfection. The next day, when cells reached a confluency of about 70%, they were transfected using a standard calcium phosphate method. The transfection mix included 8 μg of packaging vector (psPAX2), 4 μg of envelope vector (pCMV‐VSV), and 10 μg of the plasmid of interest. Twenty‐four hours post‐transfection, the medium was changed. Seventy‐two hours post‐transfection, the virus‐containing supernatant was harvested and filtered using 0.45 μm filters. For transduction, the virus‐containing supernatant was spun down onto RetroNectin‐coated (Takara Biotech, Cat No. T100B) 6‐well plates for 2 h at 2,000 g. The supernatant was then removed, and the cell suspension (1 × 106 cells per well) was added. Twenty‐four hours post‐transduction, the required selection reagent was added.
sgRNA design and cloning
CRISPR KO sgRNAs were designed using the Broad Institute CRISPR design tool (https://portals.broadinstitute.org/gppx/crispick/public). CRISPRi sgRNAs were designed as previously stated in Radzisheuskaya et al (2016). The U6‐sgRNA‐SFFV‐puro‐P2A‐EGFP sgRNA vector was generated by substituting Cas open reading frame with a puromycin resistance cassette in the pL‐CRISPR.SFFV.GFP plasmid (Addgene cat. no 57827). For sgRNA cloning into pU6‐sgRNA‐EF1α‐puro‐T2A‐BFP, oligos were annealed in annealing buffer (200 mM potassium acetate, 60 mM HEPES‐KOH pH 7.4, 4 mM magnesium acetate) and ligated into BstXI + BlpI (NEB) digested pU6‐sgRNA‐EF1α‐puro‐T2A‐BFP. For sgRNA cloning into U6‐sgRNA‐SFFV‐puro‐P2A‐EGFP, the oligos were phosphorylated by T4 PNK (NEB) and annealed in the T4 ligation buffer (NEB). The oligos and plasmid mixture was then subjected to digestion by BsmBI (NEB) and ligation by T4 ligase (NEB) (4 cycles of 42°C—5 min and 16°C—5 min, inactivation 55°C—15 min). For a complete list of sgRNAs used in this study, see Dataset EV9. Bacteria used for cloning: Escherichia coli DH5alpha, Stbl3 and HST08.
After selection, the cells were counted, and a spike‐in of approximately 20% untransduced cells was added to each sample. The percentage of BFP+ or GFP+ (transduced) cells was then recorded on a flow cytometer at the specified time points.
Cell cycle analysis
Cell cycle analysis was conducted 5 days after transduction with the sgRNA of interest. Twenty‐four hours before analysis, cells were seeded in triplicates at a density of 2.5 × 105/ml. EdU labeling was performed using the Click‐iT EdU Alexa Fluor 488/6 47 Flow Cytometry Assay Kit (Thermo Fisher Cat no: C10425 and Cat. no: C10634, respectively) following manufacturer's instructions but using 40% of the recommended volumes for the Click‐iT reaction mix. Cells were labeled with 10 μM EdU for 50 min. Before the analysis, cells were stained with DAPI (1 μg/ml).
Cell viability assay
2000 THP‐1 wtCas9 cells were plated in 100 μl media. The indicated concentrations of BPTF BROMO inhibitor (7190) or negative control compound (4827) were added to the cells (Martinelli et al, 2023). Both molecules were obtained from https://opnme.com/. 1% SDS (final concentration) was used as a positive control. Seventy‐two hours later, cell viability was measured using CellTiter‐Glo® Luminescent Cell Viability Assay (Promega) according to the manufacturer's instructions. For analysis, background luminescence was subtracted. Finally, luminescence values of the treated cells were normalized to values obtained from non‐treated cells.
Cell differentiation assay
The samples were first washed with 3% FBS/PBS before being mixed with 200 μl of the staining solution, which contained APC anti‐mouse/human CD11b antibody (Biolegend, 101212) in 3% FBS/PBS (1:200) per sample and left on ice for 30 min. Afterwards, the samples were washed twice with 3% FBS/PBS and then resuspended in 250 μl 3% FBS/PBS for acquisition on a flow cytometer.
Flow cytometry
Flow cytometry was performed using a BD LSR II flow cytometer, a BD FACSMelody™ Cell Sorter, BD LSRFortessa™ X‐20, and Beckman Coulter CytoFlex. Flow cytometry data were analyzed in FlowJo.
Growth curves
3 × 105 or 5 × 105 cells/ml were seeded and counted every 72 h using Invitrogen™ Countess II Automated Cell Counter (Thermo Fisher Scientific). 3 × 105 or 5 × 105 cells/ml (respectively) were used for replating. For growth curves performed using dTAG‐V1, new dTAG‐V1 (100 nM final concentration) was added to the culture every 48 h.
Generation of SMARCA5 degron knock‐in line
The generation of the SMARCA5 knock‐in line involved methods as in Damhofer et al (2021). Briefly, the targeting construct was assembled from PCR products or synthetic DNA blocks (IDT) using the In‐Fusion cloning kit (Takara). This construct contained: a 500 bp left homology arm, blasticidin resistance gene, P2A, 2× HA tags, FKBPF36V, a GSG linker, and a 500 bp right homology arm. 5 × 105 THP‐1 cells were nucleofected with the targeting construct, together with the synthetic sgRNA targeting the desired region (ordered from IDT) and SpCas9 protein (100 pmol) using the Amaxa 4D nucleofector system (Lonza) with the SG Cell Line 4D‐NucleofectorTM X Kit S (Lonza Cat. no: V4XC‐3032) following manufacturer's instructions. The program of choice for nucleofection was FF‐100. The transfected cells were selected with blasticidin (5 μg/ml). PCRs were run to confirm the knock‐in at the correct locus, and the cells were used as a pool. The target protein was degraded using 100 nM dTAG‐V1 (Tocris Cat. No. 6914) at the indicated time intervals. For a complete list of sequences used to generate this knock‐in line, refer to Dataset EV3.
Generation of the truncated BPTF cell lines
THP‐1 expressing wtCas9 cells were transduced with sgRNAs targeting the gene upstream from the regions coding for the PHD2 or BROMO domains. After selection with puromycin, single‐cell clones were established by sorting and analyzed using TIDE (Brinkman et al, 2014) (https://tide.nki.nl/). The sgRNA and primer sequences used for generating these cell lines can be found in Dataset EV9.
CRISPRi library design
Based on the literature and database search, we generated a list of 1,046 chromatin‐associated factors and designed the sgRNAs against them using the principles defined in Radzisheuskaya et al (2016). In detail, CAGE promoter predictions were taken from this FANTOM5 file: http://fantom.gsc.riken.jp/5/datafiles/latest/extra/CAGE_peaks/hg19.cage_peak_phase1and2combined_ann.txt.gz. New 300 bp regions were defined 50 bp upstream and 250 bp downstream from the center of the 201,802 original CAGE regions. The corresponding DNA sequences were extracted. The sequences were trimmed and filtered as follows: rm‐only seqs were removed, rm subsequences in the beginning or the end of the original 300 bp seq were removed (trimmed), a rm subsequence occurring inside the sequence, caused the sequence to be split in two sequences at that position with the rm subsequence removed, rm trimmed sequences shorter than 20 bp were removed. The promoters of the target 1,046 chromatin‐associated factors were defined as 4.5 kb upstream and 0.5 kb downstream of the TSS and were overlapped in a strand‐specific manner with the 300 bp CAGE regions. All CAGE regions that overlapped a promoter were used for sgRNA prediction. Next, all the sgRNAs starting with G were ranked based on the following criteria: (i) whether the sgRNA falls within the optimal window (−20, + 90) of the TSS defined by the FANTOM5 project; (ii) SSC on‐target score (Xu et al, 2015); (iii) CDF off‐target scores (Doench et al, 2016). For each gene, the 4 best‐scoring sgRNAs against the most commonly used promoter (p1@) and 2 best‐scoring sgRNAs against the second most commonly used promoter (p2@) were selected. 6,234 sgRNAs comprised the library, of which 334 were negative controls and 37 were positive controls (POLR2A, POLR1D, GTF2B, HSPA9, RPA3, SPC24, AURKB) selected from (Gilbert et al, 2014). Dataset EV1 contains the sequences of the sgRNAs in the library.
Cloning of the CRISPRi library
The oligonucleotide pool consisting of 6,234 unique sequences was synthesized by CustomArray. Each oligonucleotide consisted of a unique 20 nucleotide sgRNA sequence flanked on each side by the overhangs containing restriction sites for BstXI and BlpI enzymes. The pool was PCR‐amplified for 20 cycles with Phusion Polymerase (ThermoFisher). Cycling conditions: Initial denaturation 98°C (30 s); denaturation 98°C (10 s); annealing and elongation 72°C (30 s); final extension 72°C (10 min). The PCR product was purified using the PCR Purification Kit (Qiagen), digested with BstXI and BlpI enzymes and ligated into the BstXI/BlpI digested pU6‐sgRNA EF1Alpha‐puro‐T2A‐BFP vector. To preserve the diversity of the library, colonies were scraped from 30 × 15 cm plates after the transformation of the ligation mixture, which allowed to achieve 170× coverage of the library. For a complete list of overhangs and primers used for the screen, refer to Dataset EV9.
CRISPRi screen execution and data analysis
The CRISPRi screen was conducted in two cell lines (THP‐1 dCas9 or MOLM‐13 dCas9) with two biological replicates for each cell line. Briefly, 12 × 106 THP‐1 dCas9 or MOLM‐13 dCas9 cells were transduced with the Epi‐CRISPRi library at approximately 30% transduction efficiency. Twenty‐four hours post‐transduction cells were selected with puromycin (2 μg/ml), and 48 h after selection, 6 × 106 for each replicate were collected for genomic DNA (gDNA) extraction (day 0 timepoint). The remaining cells were replated in media containing puromycin (2 μg/ml). For each replicate, the cells were passaged to maintain at least 500× of the library representation. After 12 days of culture (day 12 timepoint), the pellets for gDNA extraction were collected again. Genomic DNA was extracted using DNeasy and Blood Tissue Kit (QIAGEN) according to the manufacturer's protocol, and 30 ug for each sample was amplified for 18 cycles. Cycling conditions: Initial denaturation 98°C (2 min); denaturation 98°C (30 s); annealing 60°C (15 s); elongation 72°C (20 s); and final extension 72°C (10 min). The resulting PCR product was then used for the second round of amplification for 20 cycles introducing Illumina adaptor sequences and different barcodes for each sample for the subsequent Illumina sequencing. Cycling conditions: Initial denaturation 98°C (2 min); denaturation 98°C (30 s); annealing 60°C (15 s); elongation 72°C (20 s); and final extension 72°C (10 min). The resulting PCR products were run on a 2% agarose gel and extracted from a gel using Illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare) and then purified with the PCR Purification Kit (Qiagen) according to the manufacturer's instructions. The DNA was then quantified by Qubit and KAPA qPCR. Subsequently, the fragments were pooled, denatured, diluted, and sequenced using NextSeq500 according to the manufacturer's protocols. The samples were demultiplexed, and the reads were mapped with bowtie, allowing two mismatches (bowtie ‐m 1 ‐v 2 ‐ ‐best ‐ ‐strata). Mapped reads per sgRNAs were counted for all the conditions. The number of reads for each sgRNA was then normalized to the mean number of reads in a sample, and the sgRNAs with the read numbers in the bottom 2% of the library on day 0 were removed from the analysis. 334 negative control gRNAs were included in the analysis, and we sampled this set to generate a set of negative control genes (four randomly selected negative control sgRNA constituted a negative control gene). Next, all the sgRNAs for a particular gene were ranked by decreasing depletion. Then, for sgRNAs of each rank, we chose a log2 fold change significance cutoff value so that no more than 5% of sgRNAs in this set were from the negative control category. We called a gene a hit if its first and second‐ranked sgRNAs met the log2 fold change significance threshold.
Tiling screen design and cloning
The tiling library was designed to contain 806 different sgRNAs targeting BPTF. Negative control sgRNAs were designed based on non‐targeting sequences (111 sgRNAs). Positive control sgRNAs were designed to target regions coding for the catalytic domains of DOT1L (86 sgRNAs) and KDM1A (157 sgRNAs), known essential regulators of leukemic cell growth. The sgRNAs were designed using the Broad Institute web tool (https://portals.broadinstitute.org/gppx/crispick/public). Oligonucleotides encoding the sgRNAs described were synthesized as a pooled library and cloned using the same protocol as described for the CRISPRi library. The screen was conducted and analyzed as described above for the CRISPRi screen. Additional sgRNA filtering was performed to remove the non‐functional sgRNAs, as described in He et al (2019) (using k = 6). The cell numbers were adjusted to maintain the library representation of 1,000× throughout the experiment.
Protein extraction and Western blotting
Before lysis, 6 × 106 cells were washed once using cold PBS. After pelleting, cells were lysed using 120 μl Lysis buffer (50 mM Tris–HCl, pH 7.4; 150 mM NaCl; 1 mM EDTA, 1% Triton X‐100) supplemented with cOmplete™ Protease Inhibitor Cocktail (11697498001) and Benzonase® Nuclease (Millipore Cat no: 70664). Samples were incubated for 30 min at 4°C on a rotator and then sonicated for 5 min (30 s ON/30 s OFF) using a Bioruptor (Diagenode). After centrifugation at 12,000 g for 10 min at 4°C, the supernatants were transferred to a new tube. Protein concentration was determined by Bradford using protein assay dye concentrate (Bio‐Rad Cat. no: 5000006) according to the manufacturer's instructions.
Between 50 and 60 μg of total protein was then separated on an SDS‐PAGE gel or a NuPAGE™ 3 to 8% Tris‐Acetate (ThermoFisher Cat. No: EA03755BOX) using 1 × TGS buffer (Bio‐Rad Cat. No: 1610732) or NuPAGE™ Tris‐Acetate SDS (Thermo Fisher Cat. No: LA0041), respectively, as running buffer. The gels were run at 140 V for 2 to 3 h. The transfer was done onto nitrocellulose (Amersham™ Protran® Premium Western blotting membranes, nitrocellulose. Merck Cat.no: GE10600003) or PVDF membranes (Amersham Hybond P 0.45 PVDF Cat.no: 10600069) in a Bio‐Rad wet‐tank blotting system using 1 x TGS buffer (Bio‐Rad Cat. no: 1610772) supplemented with 5% EtOH as transfer buffer at a constant amperage of 370 mA for 3 h. For smaller proteins of interest, transfer was done in 1× TG buffer (Bio‐Rad Cat no: 1610771) with 10% EtOH at a constant voltage of 100 V for 1 h. After transfer, membranes were blocked for 1 h in 5% milk in 1× Phosphate‐Buffered Saline, 0.1% Tween® 20 Detergent (PBS‐T) at room temperature, followed by overnight incubation at 4°C with the primary antibody. The membrane was washed three times (5, 10, and 5 min) with PBS‐T before incubation with the secondary antibody for 1 h at room temperature. HRP‐labeled antibodies were detected using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Cat.no: 34580) according to the manufacturer's instructions and developed on an X‐ray developer machine (Valsøe). Antibodies labeled with Infrared Fluorescent Dyes were developed using Odyssey® imager. For a complete list of antibodies used in this study, see Dataset EV9.
Immunoprecipitation followed by western blotting
6 × 106 cells were lysed following the protocol for protein extraction described above. After centrifugation at 12,000 g, the supernatants were transferred to a new tube. For IPs using Protein A Sepharose beads, the samples were precleared for 1 h on a rotator at 4°C using 20 μl of packed Protein A beads previously washed twice in TBS buffer (50 mM Tris HCl, pH 7.4; 150 mM NaCl). After preclearing, the beads were precipitated, and the supernatant was then transferred to another tube. Before immunoprecipitation, 40% input was separated (as indicated). Immunoprecipitation was performed by adding the desired antibody at a concentration of 2 μg per sample. Samples were left rotating with the antibody for 1 h at 4°C before adding washed BSA‐blocked beads (20 μl packed beads per sample) or Protein A Dynabeads™ (ThermoFisher Cat. no: 10002D) (previously washed once in PBS and twice in TBS; 30 μl/sample) and left rotating overnight. The next day, samples were washed twice in TBS buffer. Elution was done by adding 20 μl of 2× LSB buffer with DTT directly onto the washed beads and incubating the samples for 10 min at 50°C; eluents were recovered by spinning for 2 min at 1,000 g. The elution step was repeated once or twice for the Sepharose or the Dynabeads™ IPs, respectively, and eluents were pooled (final volume 40 or 60 μl, respectively). Before loading, samples were boiled at 95°C for 5 min. For a complete list of antibodies used for this study, see Dataset EV9.
Animal studies
The mice were of the following genotype: B6.SJL‐Ptprc a /BoyAiTac (ordered from Taconic with number 4000‐F). This study was approved by the Danish Veterinary and Food Administration (Fødevarestyrelsen) under the license number 2017‐15‐0201‐01176 and the project number P21‐074. All protocols involving mouse experiments were authorized by the Department of Experimental Medicine (Afdeling for Eksperimentel Medicin, AEM) of the University of Copenhagen. Mice were kept in cages by Tecniplast (Italy) in a 12 h/12 h light/dark cycle with a 15 to 30‐min twilight period at a temperature of 22°C (±2°C) and a relative humidity: 55 ± 10%. Animals were fed ad libitum with Altromin 1314 (extruded form) from Altromin GmbH & Co. KG (Germany) and provided tap water acidified with citric acid to pH 3–4. Aspen chip from Tapvei (Denmark) was used as bedding material. Red plastic JAKO shelter designed by AEM and made by Molytex (Denmark) and a gnawing stick made by Tapvej (Denmark) were used for enrichment. Shredded paper from Lillico (UK) was used as nesting material. The mice were kept in a specific pathogen‐free environment. Only female mice were involved in the study, and no blinding was conducted. All mice were included in the analysis and randomly chosen for injections.
Tail vein injection
MLL‐AF9 i‐wtCas9 cells were transduced with the sgRNAs of interest and sorted for GFP to select the cells carrying the desired sgRNA. 5 × 105 live GFP‐positive cells were transplanted into sub‐lethally irradiated (4.5 Gy) B6.SJL mice by tail vein injections. Immediately after injection, mice were given medicated water (ciprofloxacin 0.1 g/l) until doxycycline treatment was started. Induction of Cas9 was started 4 days post‐transplantation by including doxycycline (2 mg/ml) and 2% sucrose in the water. Mice were monitored every other day and euthanized as soon as AML symptoms or signs of uneasiness were observed. The presence of an enlarged spleen confirmed the emergence of AML. The spleens were weighed, measured, and further processed and analyzed by flow cytometry to check for GFP presence in the tissue.
RNA extraction
Before RNA extraction, cells were washed once in cold PBS. RNA extraction was performed using the Rneasy Mini Kit (Qiagen Cat no: 74016) according to the manufacturer's instructions. RNA quality and concentration were determined using Nanodrop 2000 and Bioanalyzer.
RNA‐sequencing and analysis
After RNA extraction, libraries for sequencing were prepared with TruSeq RNA Library Prep Kit (Illumina) according to the manufacturer's instructions but halving the Illumina recommended volumes. After quality assessment, libraries were sequenced on a NextSeq 500.
RNA sequencing reads were 3′ trimmed for base quality 15 and adapter sequences using version 0.4.5 of TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore) and then aligned to human assembly hg38 with STAR v2.4 using default parameters. Data quality and transcript coverage were assessed using the Picard tool CollectRNASeqMetrics (http://broadinstitute.github.io/picard/). Read count tables were generated with HTSeq v0.9.1. Normalization and differential expression were evaluated with DESeq2 using the default parameters, and outliers were assessed by sample grouping in principal component analysis. Gene set enrichment analysis (GSEA, http://software.broadinstitute.org/gsea) was run against MsigDB v6 using the preranked option and log2 fold change for pairwise comparisons.
Reverse transcription and quantitative PCR
500 ng of RNA was used to perform reverse transcription with Transcriptor Universal cDNA Master (Roche Cat. no. 5893151001) according to the manufacturer's instructions. Quantitative PCR with reverse transcription (qRT‐PCR) was performed in technical triplicates using 5 ng of cDNA and 1× LightCycler 480 SYBR Green I Master Mix (Roche Cat. no. 04707516001) per reaction on a LightCycler 480 Instrument II (Roche). Levels of gene expression were measured by relative quantification to the expression of a housekeeping gene (RPLP0). For a complete list of qRT‐PCR primers used in this study, refer to Dataset EV9.
Hi‐C
Hi‐C was performed using the Arima Hi‐C Kit according to the manufacturer's protocol. The samples were sequenced using Novaseq. Hi‐C data were processed using HiC‐Pro v2.11.1 1 (Servant et al, 2015). The sequence reads were mapped onto the reference genome hg38 with a minimum mapping quality of 30 and binned at 10, 25, and 40 kb. A genome coordinate file in Bed format on DpnII and HinfI restriction fragments was generated using the genome digestion tool from HiC‐Pro. Other settings associated with HiC‐Pro were kept as default. Uniquely mapped paired‐end reads were retained and mapped to DpnII and HinfI‐restricted fragments. Replicates were pooled for each sample to obtain 267/223/431 M validate interactions in sgBPTF KO/sgSMARCA5 KO/sgNegCtrl samples. The pooled interactions were subsequently rendered into the .hic format by the tool provided by HiC‐Pro. The final Hi‐C matrices were binned at 25 kb, and the median counts per bin are ~2,700 in the range of ~2,600–2,850. TADs were called using hicFindTADs from HiCExplorer (3.4.3) (Ramírez et al, 2018). Quantifying specific loops and visualizing Hi‐C data were done in the WashU Epigenome Browser (https://epigenomegateway.wustl.edu/) with hg38 as the reference genome. Knight‐Ruiz normalization was used, and visualization was at a fixed score scale (max 150).
ATAC‐seq
ATAC‐seq was carried out following the protocol from Corces et al (2017).
Cut&Run
Cut&Run was performed using the CUTANA™ ChIC/CUT&RUN Assay kit (EpiCypher) according to the manufacturer's instructions. All antibodies were used at a concentration of 0.5 mg/ml, and antibody binding was carried out overnight. For additional details on antibodies used for this assay, refer to Dataset EV9.
ATAC‐ and Cut&Run data analysis
The sequencing reads were 3′ trimmed and filtered for quality and adapter content using TrimGalore (v0.4.5), with a quality setting of 15, and running version 1.15 of cutadapt and version 0.11.5 of FastQC. For some of the sequencing runs, adaptor trimming was performed during the bcl2 to fastq conversion. Reads were aligned to human assembly hg38 with version 2.3.4.1 of bowtie2 (http://bowtie‐bio.sourceforge.net/bowtie2/index.shtml) and were deduplicated using MarkDuplicates in Picard Tools (v2.16.0). The BEDTools suite (http://bedtools.readthedocs.io) was used to create normalized read density profiles, using a read extension of 0 bp for ATAC samples. Subsequent Cut&Run data visualization and analysis were performed using EaSeq software (Lerdrup et al, 2016). For BPTF and HA‐SMARCA5 Cut&Run analysis, to call peaks, the ALT tool within the Easeq software was used with default parameters and using BPTF KO or dTAG‐treated cells as background controls, respectively. To call CTCF peaks in U937 and OCI‐AML2 cells, the ALT tool within the Easeq software was used with default parameters and using the IgG sample as a background control. To analyze peak distribution across different chromatin states, Chromatin State Segmentation by HMM data for K562 cells from ENCODE/Broad (Ernst & Kellis, 2010; Ernst et al, 2011) was downloaded from the UCSC Genome Browser and converted to the GRCh38/hg38 genome assembly using the liftOver tool available through the UCSC Genome Browser. Motif signatures were obtained using Homer v4.5 (http://homer.ucsd.edu).
For ATAC‐seq, a global peak atlas was created by first removing blacklisted regions (http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/hg38‐human/hg38.blacklist.bed.gz), then merging all peaks within 500 bp, and counting reads with version 1.6.1 of featureCounts (http://subread.sourceforge.net). Comparison of intra vs intergroup clustering in principle component analysis was used to determine normalization strategy, using either the median ratio method of DESeq2 or a sequencing depth‐based factor normalized to ten million uniquely mapped fragments. Differential enrichment was scored using DESeq2 for all pairwise group contrasts. Peak‐gene associations were created using linear genomic distance to the transcription start site.
Generation of ChromHMM states for THP‐1 cells
The following datasets were used: CTCF, BPTF, and HA‐SMARCA5 Cut&Run data (generated in this study), H3K27me3, H3K27ac, H3K4me1, H3K4me3, and H3K9me3 ChIP‐seq data obtained from Data ref: Lin et al (2022) (GSE208043 (GSM6333440‐50)). Fastq files were retrieved with SRAtools (2.10.8) using prefetch and fastrq‐dump from SRA run number SRR20106860‐70 and then compressed with gzip. Fastq files were hard trimmed to leave 50 bp on the 5′ side with Trim Galore (0.6.6) in paired mode, then trimmed again with Trim Galore in paired mode with the default settings to remove adaptors. Trimmed fastq files were mapped to GRCh38 (iGenomes NCBI) genome with Bowtie2 (2.4.2) (Langmead & Salzberg, 2012) in paired mode with—end‐to‐end—sensitive setting. The Bowtie2 output was sorted by read name with samtools sort ‐n, then the mate coordinates, insert size, and mate score tags of the pairs were added using samtools fixmate ‐m and then sorted again according to chromatin coordinates with samtools sort, and, finally, the duplicates were removed with samtools markdup ‐r.
To generate the binary for ChromHMM to learn the model, bam files were binarized with ChromHMM (1.24) (Ernst & Kellis, 2012) BinarizeBam in ‐mixed mode. ChIP input sample was used as a control for all the histone modification ChIP samples. For CTCF Cut&Run, no control was used. For HA‐SMARCA5 and BPTF Cut&Run, dTAG‐treated and BPTF KO Cut&Run samples were used as controls, respectively. The Binarized Bam was then used for ChromHMM LearnModel with 12 states and hg38 genome.
BPTF interactomics analysis
For each of the four replicates of THP‐1 cells transduced with either sgNegCtrl of sgBPTF KO sgRNA, 10 million cells were lysed in 50 mM EPPS, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, complete protease inhibitors, and 25 U/ml of benzonase. The lysate was kept on ice for 5 min to allow DNA digestion and cleared by centrifugation at 20,000 g for 5 min and filtering through acropep advance 96‐well 2 ml, 1 μm glass fiber filter plate. Immunoprecipitation was performed for 1 h at 4°C with anti‐BPTF antibody (for details, see Dataset EV9) in Eppendorf™ Deepwell™ Plates 96 with shaking at 1,300 rpm. After immunoprecipitation, the beads were washed five times in wash buffer (50 mM EPPS, pH 7.4, 150 mM NaCl) and subjected to trypsin digestion for 2 h at 37°C with 15 μl of 10 ng/μl LysC and 20 ng/μl Trypsin in 10 mM EPPS pH 8.5 on Orochem OF1100 plates. The digest was labeled with 4 μl 20 g/l TMTPro tags, as the manufacturer recommended. The material was fractionated by Pierce™ High pH Reversed‐Phase Peptide Fractionation Kit, concatenating 2 fractions into a superfraction (e.g., 1 and 5). After fractionation, the samples were evaporated using a vacuum centrifuge, resuspended in 20 μl 0.1% TFA and 4.5 μl was analyzed by Waters nanoAcquity M Class UPLC on 2 μm particle size, 75 μm × 500 mm easyspray column in direct injection mode. The samples were separated using the following gradient of buffer A (0.1% formic acid in water) and buffer B (0.1% formic acid in acetonitrile): 0–7% in 5 min, 7–30% in 90 min, 30–50% in 20 min. Eluting peptides were analyzed on Orbitrap Fusion Lumos instrument using the MS3 SPS method with the settings recommended by the instrument manufacturer with the following modifications: (i) CID NCE for MS2 was set at 32; (ii) HCD NCE for MS3 was set at 45; (iii) C series exclusion was disabled, since the TMTPro reagent was not enabled in C‐series exclusion. Data were analyzed in Proteome Discoverer 2.4 software. A database search was performed with the Sequest HT search engine using the Homo Sapiens UniProt database containing only reviewed entries and canonical isoforms (retrieved on 14/06/2019). Oxidation (M) was set as a variable modification, while TMTPro was set as a fixed modification. A maximum of two missed cleavages were permitted. The precursor and fragment mass tolerances were 10 ppm and 0.6 Da, respectively. PSMs were validated by percolator with a 0.01 posterior error probability (PEP) threshold. Only PSMs with isolation interference < 25% and at least five MS2 fragments selected for MS3 matched to peptide sequence were considered. The quantification results of peptide spectrum matches were combined into protein‐level quantitation using the MSstatsTMT R package (Choi et al, 2014) with at least two peptides per protein.
Author contributions
Kristian Helin: Conceptualization; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing. Aliaksandra Radzisheuskaya: Conceptualization; formal analysis; supervision; funding acquisition; investigation; visualization; methodology; writing – original draft; writing – review and editing. Isabel Peña‐Rømer: Conceptualization; formal analysis; investigation; visualization; writing – original draft; writing – review and editing. Eugenia Lorenzini: Investigation; writing – review and editing. Richard Koche: Formal analysis; investigation; visualization; writing – review and editing. Yingqian Zhan: Formal analysis; investigation; visualization; writing – review and editing. Pavel V Shliaha: Formal analysis; investigation; writing – review and editing. Alexandra J Cooper: Investigation; writing – review and editing. Daria Shlyueva: Investigation; methodology; writing – review and editing. Jens V Johansen: Formal analysis; investigation; methodology; writing – review and editing. Ronald C Hendrickson: Supervision. Zheng Fan: Formal analysis; investigation; writing – review and editing.
Disclosure and competing interests statement
KH is a co‐founder of Dania Therapeutics Aps and a scientific advisor for Hannibal Health Innovation. KH was recently a scientific advisor for Inthera Bioscience AG and MetaboMed Inc. Kristian Helin is a member of the Advisory Editorial Board of The EMBO Journal. This has no bearing on the editorial consideration of this article for publication.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Source Data for Expanded View and Appendix
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Reagent availability
The cell lines and plasmids generated in this study are available upon request.
Acknowledgements
We thank all members of the Helin lab for helpful discussions; Jan‐Erik Messling and Ann Sophie Moroni for their assistance with the in vivo experiments; Sarah Teed, Sudeep Sahadeva, and Hayley Porter for technical assistance. Flow cytometry experiments were performed with the assistance of the flow cytometry core facilities at BRIC and the ICR. AR and DS were partly funded by the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska‐Curie grant agreements 659171 and 749362, respectively. The work in the Helin laboratory was supported by the Kirsten and Freddy Johansen Foundation, the Neye Foundation, the Brain Tumor Charity, a center grant from the Novo Nordisk Foundation to the NNF Center for Stem Cell Biology (no. NNF17CC0027852, KH), the Memorial Sloan Kettering Cancer Center Support Grant (no. NIH P30 CA008748, KH) and startup funding from the ICR. The work in the Radzisheuskaya laboratory was supported by the ICR fellow startup grant from the ICR. Figure 7 and Synopsis figure were created with Biorender.com.
The EMBO Journal (2023) 42: e114221
Contributor Information
Aliaksandra Radzisheuskaya, Email: alex.radzisheuskaya@icr.ac.uk.
Kristian Helin, Email: kristian.helin@icr.ac.uk.
Data availability
All the next‐generation sequencing data is available at GEO under the accession number GSE226688 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE226688). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD044656 (http://www.ebi.ac.uk/pride/archive/projects/PXD044656).
References
- Alkhatib SG, Landry JW (2011) The nucleosome remodeling factor. FEBS Lett 585: 3197–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Iyer LM (2012) The HARE‐HTH and associated domains: novel modules in the coordination of epigenetic DNA and protein modifications. Cell Cycle 11: 119–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badenhorst P, Voas M, Rebay I, Wu C (2002) Biological functions of the ISWI chromatin remodeling complex NURF. Genes Dev 16: 3186–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badenhorst P, Xiao H, Cherbas L, Kwon SY, Voas M, Rebay I, Cherbas P, Wu C (2005) The Drosophila nucleosome remodeling factor NURF is required for Ecdysteroid signaling and metamorphosis. Genes Dev 19: 2540–2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW, Murison A, Langenfeld K, Petretich M, Scognamiglio R et al (2018) A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 553: 515–520 [DOI] [PubMed] [Google Scholar]
- Bai D, Zhou Y, Shen F, Gao D, Suo W, Zhang H, Li H (2022) BPTF activates the MAPK pathway through coexpression with Raf1 to promote proliferation of T‐cell lymphoma. Oncol Lett 24: 223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak O, Lazzaro MA, Lane WS, Speicher DW, Picketts DJ, Shiekhattar R (2003) Isolation of human NURF: a regulator of Engrailed gene expression. EMBO J 22: 6089–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barisic D, Stadler MB, Iurlaro M, Schübeler D (2019) Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature 569: 136–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, Van Steensel B (2014) Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42: e168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caslini C, Yang Z, El‐Osta M, Milne TA, Slany RK, Hess JL (2007) Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res 67: 7275–7283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centore RC, Sandoval GJ, Soares LMM, Kadoch C, Chan HM (2020) Mammalian SWI/SNF chromatin remodeling complexes: emerging mechanisms and therapeutic strategies. Trends Genet 36: 936–950 [DOI] [PubMed] [Google Scholar]
- Chambers C, Cermakova K, Chan YS, Kurtz K, Wohlan K, Lewis AH, Wang C, Pham A, Dejmek M, Sala M et al (2023) SWI/SNF blockade disrupts PU.1‐directed enhancer programs in normal hematopoietic cells and acute myeloid leukemia. Cancer Res 83: 983–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanput W, Mes JJ, Wichers HJ (2014) THP‐1 cell line: an in vitro cell model for immune modulation approach. Int Immunopharmacol 23: 37–45 [DOI] [PubMed] [Google Scholar]
- Choi M, Chang CY, Clough T, Broudy D, Killeen T, MacLean B, Vitek O (2014) MSstats: an R package for statistical analysis of quantitative mass spectrometry‐based proteomic experiments. Bioinformatics 30: 2524–2526 [DOI] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott‐Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B et al (2017) An improved ATAC‐seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14: 959–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane E, Bian Q, Patton Mccord R, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, Meyer BJ (2015) Condensin‐driven remodelling of X chromosome topology during dosage compensation. Nature 523: 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshank AV, Sroczynska P, Sankar A, Miyagi S, Rundsten CF, Johansen JV, Helin K (2015) SWI/SNF subunits SMARCA4, SMARCD2 and DPF2 collaborate in MLL‐rearranged leukaemia maintenance. PLoS ONE 10: e0142806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damhofer H, Radzisheuskaya A, Helin K (2021) Generation of locus‐specific degradable tag knock‐ins in mouse and human cell lines. STAR Protoc 2: 100575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Mirny L (2016) The 3D genome as moderator of chromosomal communication. Cell 164: 1110–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado MD, León J (2010) Myc roles in hematopoiesis and leukemia. Genes Cancer 1: 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R et al (2016) Optimized sgRNA design to maximize activity and minimize off‐target effects of CRISPR‐Cas9. Nat Biotechnol 34: 184–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerks T, Copley R, Bork P (2001) DDT – a novel domain in different transcription and chromosome remodeling factors. Trends Biochem Sci 26: 145–146 [DOI] [PubMed] [Google Scholar]
- Dong J, Gao Z, Liu S, Li G, Yang Z, Huang H, Xu L (2013) SLIDE, the protein interacting domain of imitation switch remodelers, binds DDT‐domain proteins of different subfamilies in chromatin remodeling complexes. J Integr Plant Biol 55: 928–937 [DOI] [PubMed] [Google Scholar]
- Eberharter A, Vetter I, Ferreira R, Becker PB (2004) ACF1 improves the effectiveness of nucleosome mobilization by ISWI through PHD‐histone contacts. EMBO J 23: 4029–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M (2010) Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol 28: 817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M (2012) ChromHMM: automating chromatin state discovery and characterization. Nat Methods 9: 215–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M et al (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber J, Krivtsov AV, Stubbs MC, Wright R, Davis TN, van den Heuvel‐Eibrink M, Zwaan CM, Kung AL, Armstrong SA (2009) HOXA9 is required for survival in human MLL‐rearranged acute leukemias. Blood 113: 2375–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauriat C, Olive D (2014) AML drug resistance: c‐Myc comes into play. Blood 123: 3528–3530 [DOI] [PubMed] [Google Scholar]
- Fiskus W, Mill CP, Nabet B, Perera D, Birdwell C, Manshouri T, Lara B, Kadia TM, DiNardo C, Takahashi K et al (2021) Superior efficacy of co‐targeting GFI1/KDM1A and BRD4 against AML and post‐MPN secondary AML cells. Blood Cancer J 11: 98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC et al (2014) Genome‐scale CRISPR‐mediated control of gene repression and activation. Cell 159: 647–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AL, DeSisto J, Flannery P, Lemma R, Knox A, Lemieux M, Sanford B, O'Rourke R, Ramkissoon S, Jones K et al (2020) BPTF regulates growth of adult and pediatric high‐grade glioma through the MYC pathway. Oncogene 39: 2305–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamiche A, Sandaltzopoulos R, Gdula DA, Wu C (1999) ATP‐dependent histone octamer sliding mediated by the chromatin remodeling complex NURF. Cell 97: 833–842 [DOI] [PubMed] [Google Scholar]
- He W, Zhang L, Villarreal OD, Fu R, Bedford E, Dou J, Patel AY, Bedford MT, Shi X, Chen T et al (2019) De novo identification of essential protein domains from CRISPR‐Cas9 tiling‐sgRNA knockout screens. Nat Commun 10: 4541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyle J, Zhang Y, Wright S, Xu B, Shao Y, Easton J, Tian L, Feng R, Xu P, Li C (2019) Acute depletion of CTCF directly affects MYC regulation through loss of enhancer–promoter looping. Nucleic Acids Res 47: 6699–6713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan‐Sciutto KL, Dragich JM, Rhodes JL, Bowser R (1999) Fetal Alz‐50 clone 1, a novel zinc finger protein, binds a specific DNA sequence and acts as a transcriptional regulator. J Biol Chem 274: 35262–35268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB, Orkin SH (2010) A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 143: 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokavec J, Zikmund T, Savvulidi F, Kulvait V, Edelmann W, Skoultchi AI, Stopka T (2017) The ISWI ATPase Smarca5 (Snf2h) is required for proliferation and differentiation of hematopoietic stem and progenitor cells. Stem Cells 35: 1614–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koludrovic D, Laurette P, Strub T, Keime C, Le Coz M, Coassolo S, Mengus G, Larue L, Davidson I (2015) Chromatin‐remodelling complex NURF is essential for differentiation of adult melanocyte stem cells. PLoS Genet 11: e1005555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Armstrong SA (2007) MLL translocations, histone modifications and leukaemia stem‐cell development. Nat Rev Cancer 7: 823–833 [DOI] [PubMed] [Google Scholar]
- Kwon SY, Xiao H, Glover BP, Tjian R, Wu C, Badenhorst P (2008) The nucleosome remodeling factor (NURF) regulates genes involved in Drosophila innate immunity. Dev Biol 316: 538–547 [DOI] [PubMed] [Google Scholar]
- Landry J, Sharov AA, Piao Y, Sharova LV, Xiao H, Southon E, Matta J, Tessarollo L, Zhang YE, Ko MSH et al (2008) Essential role of chromatin remodeling protein bptf in early mouse embryos and embryonic stem cells. PLoS Genet 4: e1000241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurette P, Coassolo S, Davidson G, Michel I, Gambi G, Yao W, Sohier P, Li M, Mengus G, Larue L et al (2020) Chromatin remodellers Brg1 and Bptf are required for normal gene expression and progression of oncogenic Braf‐driven mouse melanoma. Cell Death Differ 27: 29–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerdrup M, Johansen JV, Agrawal‐Singh S, Hansen K (2016) An interactive environment for agile analysis and visualization of ChIP‐sequencing data. Nat Struct Mol Biol 23: 349–357 [DOI] [PubMed] [Google Scholar]
- Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ (2006) Molecular basis for site‐specific read‐out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 442: 91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Osdal T, Ho Y, Chun S, McDonald T, Agarwal P, Lin A, Chu S, Qi J, Li L et al (2014) SIRT1 activation by a c‐MYC oncogenic network promotes the maintenance and drug resistance of human FLT3‐ITD acute myeloid leukemia stem cells. Cell Stem Cell 15: 431–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gong H, Wang P, Zhu Y, Peng H, Cui Y, Li H, Liu J, Wang Z (2021) The emerging role of ISWI chromatin remodeling complexes in cancer. J Exp Clin Cancer Res 40: 1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Xu W, Hong P, Wu C, Zhang Z, Zhang S, Xing L, Yang B, Zhou W, Xiao Q et al (2022) Decoding the spatial chromatin organization and dynamic epigenetic landscapes of macrophage cells during differentiation and immune activation. Nat Commun 13: 1–19 (GSE208043 (GSM6333440‐50) https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE208043). [DATASET] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Lu H, Duan Z, Wang J, Han J, Xiao S, Chen H, Jiang H, Chen Y, Yang F et al (2021) Discovery of high‐affinity inhibitors of the BPTF bromodomain. J Med Chem 64: 12075–12088 [DOI] [PubMed] [Google Scholar]
- Luo H, Li Q, O'Neal J, Kreisel F, Le Beau MM, Tomasson MH (2005) c‐Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma‐associated antiapoptotic mutations. Blood 106: 2452–2461 [DOI] [PubMed] [Google Scholar]
- Martinelli P, Schaaf O, Mantoulidis A, Martin L, Fuchs JE, Bader G, Gollner A, Wolkerstorfer B, Rogers C, Balıkçı E et al (2023) Discovery of a chemical probe to study implications of BPTF bromodomain inhibition in cellular and in vivo experiments. ChemMedChem 18: e202200686 [DOI] [PubMed] [Google Scholar]
- Marunde MR, Fuchs HA, Burg JM, Popova IK, Vaidya A, Hall NW, Meiners MJ, Watson R, Howard SA, Novitzky K et al (2022) Nucleosome conformation dictates the histone code. BioRxiv 10.1101/2022.02.21.481373 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Miao J, Zhang M, Huang X, Xu L, Tang R, Wang H, Han S (2021) Upregulation of bromodomain phd finger transcription factor in ovarian cancer and its critical role for cancer cell proliferation and survival. Biochem Cell Biol 99: 304–312 [DOI] [PubMed] [Google Scholar]
- Mirabella AC, Foster BM, Bartke T (2015) Chromatin deregulation in disease. Chromosoma 125: 75–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T (2020) The self‐organizing genome: principles of genome architecture and function. Cell 183: 28–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz Velasco R, Jiménez Sánchez P, García García A, Blanco Martinez‐Illescas R, Pastor Senovilla Á, Lozano Yagüe M, Trento A, García‐Martin RM, Navarro D, Sainz B et al (2022) Targeting BPTF sensitizes pancreatic ductal adenocarcinoma to chemotherapy by repressing ABC‐transporters and impairing multidrug resistance (MDR). Cancers (Basel) 14: 1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A et al (2018) The dTAG system for immediate and target‐specific protein degradation. Nat Chem Biol 14: 431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppikofer M, Bai T, Gan Y, Haley B, Liu P, Sandoval W, Ciferri C, Cochran AG (2017) Expansion of the ISWI chromatin remodeler family with new active complexes. EMBO Rep 18: 1697–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N et al (2016) Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 374: 2209–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Song C, Malakouti N, Murray D, Hariz A, Zimmerman M, Gygax D, Alhazmi A, Landry JW (2015) Functional interactions between NURF and Ctcf regulate gene expression. Mol Cell Biol 35: 224–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzisheuskaya A, Shlyueva D, Müller I, Helin K (2016) Optimizing sgRNA position markedly improves the efficiency of CRISPR/dCas9‐mediated transcriptional repression. Nucleic Acids Res 44: e141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzisheuskaya A, Shliaha PV, Grinev V, Lorenzini E, Kovalchuk S, Shlyueva D, Gorshkov V, Hendrickson RC, Jensen ON, Helin K (2019) PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat Struct Mol Biol 26: 999–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rago F, Rodrigues LU, Bonney M, Sprouffske K, Kurth E, Elliott GN, Ambrose J, Aspesi P, Oborski J, Chen JT et al (2022) Exquisite sensitivity to dual BRG1/BRM ATPase inhibitors reveals broad SWI/SNF dependencies in acute myeloid leukemia. Mol Cancer Res 20: 361–372 [DOI] [PubMed] [Google Scholar]
- Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, Habermann B, Akhtar A, Manke T (2018) High‐resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun 9: 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasio R, Ceccacci E, Nicosia L, Hosseini A, Rossi PL, Barozzi I, Fornasari L, Zuffo RD, Valente S, Fioravanti R et al (2020) Targeting the scaffolding role of LSD1 (KDM1A) poises acute myeloid leukemia cells for retinoic acid–induced differentiation. Sci Adv 6: eaax2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richart L, Pau E C‐DS, Río‐Machín A, De Andrés MP, Cigudosa JC, Lobo VJSA, Real FX (2016) BPTF is required for c‐MYC transcriptional activity and in vivo tumorigenesis. Nat Commun 7: 10153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richart L, Felipe I, Delgado P, de Andrés MP, Prieto J, Del Pozo N, García JF, Piris MA, Ramiro A, Real FX (2020) Bptf determines oncogenic addiction in aggressive B‐cell lymphomas. Oncogene 39: 4884–4895 [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Milne TA, Dewell S, McGinty RK, Yuen M, Ueberheide B, Dou Y, Muir TW, Patel DJ et al (2011) Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell 145: 692–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanbeck R, Xiao H, Wu C (2004) Spatial contacts and nucleosome step movements induced by the NURF chromatin remodeling complex. J Biol Chem 279: 39933–39941 [DOI] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, Barillot E (2015) HiC‐Pro: an optimized and flexible pipeline for Hi‐C data processing. Genome Biol 16: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shallis RM, Wang R, Davidoff A, Ma X, Zeidan AM (2019) Epidemiology of acute myeloid leukemia: recent progress and enduring challenges. Blood Rev 36: 70–87 [DOI] [PubMed] [Google Scholar]
- Sharif SB, Zamani N, Chadwick BP (2021) BAZ1B the protean protein. Genes 12: 1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Whyte WA, Zepeda‐Mendoza CJ, Milazzo JP, Shen C, Roe JS, Minder JL, Mercan F, Wang E, Eckersley‐Maslin MA et al (2013) Role of SWI/SNF in acute leukemia maintenance and enhancer‐mediated Myc regulation. Genes Dev 27: 2648–2662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S (2017) An efficient targeted nuclease strategy for high‐resolution mapping of DNA binding sites. Elife 6: e21856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skopek R, Palusińska M, Kaczor‐Keller K, Pingwara R, Papierniak‐Wyglądała A, Schenk T, Lewicki S, Zelent A, Szymański Ł (2023) Choosing the right cell line for acute myeloid leukemia (AML) research. Int J Mol Sci 24: 5377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urick AK, Hawk LML, Cassel MK, Mishra NK, Liu S, Adhikari N, Zhang W, Dos Santos CO, Hall JL, Pomerantz WCK (2015) Dual screening of BPTF and Brd4 using protein‐observed fluorine NMR uncovers new bromodomain probe molecules. ACS Chem Biol 10: 2246–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia AM, Kadoch C (2019) Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat Cell Biol 21: 152–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG et al (2010) Quantitative interaction proteomics and genome‐wide profiling of epigenetic histone marks and their readers. Cell 142: 967–980 [DOI] [PubMed] [Google Scholar]
- Winter GE, Mayer A, Buckley DL, Kelliher MA, Churchman LS, Bradner JE (2017) BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol Cell 67: 5–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters AC, Bernt KM (2017) MLL‐rearranged leukemias—an update on science and clinical approaches. Front Pediatr 5: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P et al (2006) A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442: 86–90 [DOI] [PubMed] [Google Scholar]
- Xu B, Cai L, Butler JM, Chen D, Lu X, Allison DF, Lu R, Rafii S, Parker JS, Zheng D et al (2018) The chromatin remodeler BPTF activates a stemness gene‐expression program essential for the maintenance of adult hematopoietic stem cells. Stem Cell Reports 10: 675–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wang Q, Leung ELH, Li Y, Fan X, Wu Q, Yao X, Liu L (2019) Compound C620‐0696, a new potent inhibitor targeting BPTF, the chromatin‐remodeling factor in non‐small‐cell lung cancer. Front Med 14: 60–67 [DOI] [PubMed] [Google Scholar]
- Xu H, Xiao T, Chen CH, Li W, Meyer CA, Wu Q, Wu D, Cong L, Zhang F, Liu JS et al (2015) Sequence determinants of improved CRISPR sgRNA design. Genome Res. 25: 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML (2004) Leukemia proto‐oncoprotein MLL forms a SET1‐like histone methyltransferase complex with menin to regulate hox gene expression. Mol Cell Biol 24: 5639–5649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahid H, Olson NM, Pomerantz WCK (2021) Opportunity knocks for uncovering the new function of an understudied nucleosome remodeling complex member, the bromodomain PHD finger transcription factor, BPTF. Curr Opin Chem Biol 63: 57–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Gold S, Iwanaszko M, Howard BC, Wang L, Shilatifard A (2023) Distinct layers of BRD4‐PTEFb reveal bromodomain‐independent function in transcriptional regulation. Mol Cell 83: 2896–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]