

Abstract
Background
Diabetic cardiomyopathy (DCM) causes the myocardium to rely on fatty acid β-oxidation for energy. The accumulation of intracellular lipids and fatty acids in the myocardium usually results in lipotoxicity, which impairs myocardial function. Adipsin may play an important protective role in the pathogenesis of DCM. The aim of this study is to investigate the regulatory effect of Adipsin on DCM lipotoxicity and its molecular mechanism.
Methods
A high-fat diet (HFD)-induced type 2 diabetes mellitus model was constructed in mice with adipose tissue-specific overexpression of Adipsin (Adipsin-Tg). Liquid chromatography-tandem mass spectrometry (LC–MS/MS), glutathione-S-transferase (GST) pull-down technique, Co-immunoprecipitation (Co-IP) and immunofluorescence colocalization analyses were used to investigate the molecules which can directly interact with Adipsin. The immunocolloidal gold method was also used to detect the interaction between Adipsin and its downstream modulator.
Results
The expression of Adipsin was significantly downregulated in the HFD-induced DCM model (P < 0.05). Adipose tissue-specific overexpression of Adipsin significantly improved cardiac function and alleviated cardiac remodeling in DCM (P < 0.05). Adipsin overexpression also alleviated mitochondrial oxidative phosphorylation function in diabetic stress (P < 0.05). LC–MS/MS analysis, GST pull-down technique and Co-IP studies revealed that interleukin-1 receptor-associated kinase-like 2 (Irak2) was a downstream regulator of Adipsin. Immunofluorescence analysis also revealed that Adipsin was co-localized with Irak2 in cardiomyocytes. Immunocolloidal gold electron microscopy and Western blotting analysis indicated that Adipsin inhibited the mitochondrial translocation of Irak2 in DCM, thus dampening the interaction between Irak2 and prohibitin (Phb)-optic atrophy protein 1 (Opa1) on mitochondria and improving the structural integrity and function of mitochondria (P < 0.05). Interestingly, in the presence of Irak2 knockdown, Adipsin overexpression did not further alleviate myocardial mitochondrial destruction and cardiac dysfunction, suggesting a downstream role of Irak2 in Adipsin-induced responses (P < 0.05). Consistent with these findings, overexpression of Adipsin after Irak2 knockdown did not further reduce the accumulation of lipids and their metabolites in the cardiac myocardium, nor did it enhance the oxidation capacity of cardiomyocytes expose to palmitate (PA) (P < 0.05). These results indicated that Irak2 may be a downstream regulator of Adipsin.
Conclusions
Adipsin improves fatty acid β-oxidation and alleviates mitochondrial injury in DCM. The mechanism is related to Irak2 interaction and inhibition of Irak2 mitochondrial translocation.
Supplementary Information
The online version contains supplementary material available at 10.1186/s40779-023-00493-5.
Keywords: Diabetic cardiomyopathy, Mitochondrial translocation, Mitochondrial function, Fatty acid β-oxidation
Background
The prevalence of diabetes mellitus is increasing rapidly worldwide and has become one of the serious public health problems [1, 2]. Cardiac dysfunction, a common sequela of diabetes, is considered a major cause of morbidity and mortality in patients with diabetes mellitus [3]. Among them, progressive structural and functional remodeling of the heart unrelated to coronary arteries and hypertension, is termed diabetic cardiomyopathy (DCM) [4]. Similar to the clinical classification of diabetes mellitus, the DCM models used in the study are also divided into type 1 and type 2, with type 2 accounts for more than 90% of human diabetic patients [5]. However, effective therapeutic interventions for type 2 DCM are currently limited.
DCM, as a metabolic disease, initially exposes the heart to a high-fat lipotoxic environment, which leads to its pathophysiology [6]. The diabetic heart maintains cardiac output by increasing the utilization of fatty acids, which is thought to be an adaptive process [7, 8]. In the pathogenesis of DCM, the excessive utilization and uptake of fatty acids and lipids exceed the adaptive regulation, leading to the accumulation of fatty acids and lipids in cardiomyocytes, eventually producing lipotoxicity and affecting myocardial contractile function [9].
Adipocytes are considered active endocrine organs and secrete a variety of bioactive factors called adipokines, which are mainly used to control the metabolism of fat homeostasis [10]. Dysregulation of adipokines is an important mechanism of metabolic diseases [11]. A large body of evidence suggests a strong link between adipokines and increased cardiovascular risks in obese patients [12, 13]. In recent years, adipokine-mediated crosstalk between adipose tissue and the heart has been widely recognized [14]. Normal physiological levels of multiple adipokines are essential to maintenance of normal cardiovascular function [15, 16]. Among the numerous adipokines, Adipsin has attracted attention due to its reduced levels in animal models of obesity and type 2 diabetes mellitus [17]. Our previous work showed that Adipsin has a cardioprotective role in myocardial viability, cardiac dysfunction and adverse remodeling during myocardial infarction (MI) injury [18]. Adipsin is mainly encapsulated in extracellular vesicles and taken up by target cells, thereby exerting its respective effects [19]. The aim of this study is to investigate the effect of Adipsin on DCM induced by high-fat diet (HFD) and its possible mechanisms.
In particular, we constructed an animal model with adipose tissue overexpression of Adipsin to evaluate whether Adipsin has cardioprotective effects. This study also aimed to examine how Adipsin regulates myocardial metabolism and thus affects myocardial function and its specific molecular mechanisms. Our observation helped elucidate a novel mechanism of Adipsin alteration in DCM pathophysiology, revealing the reversal of mitochondrial dysfunction as a therapeutic target for DCM.
Materials and methods
Animal
All animal experiments were carried out in accordance with the guidelines for the use of experimental animals of the National Institutes of Health and were approved by the Animal Experiment Ethics Committee of Air Force Medical University (No. 20201017). The Cre-LoxP system was used to regulate the expression of the target gene Adipsin. A recombinant vector Rosa26-(SA/PCAG)-loxp-stop-loxp-cDNA-PA was constructed and inserted into the first intron of Rosa26. The knock-in mice were crossed with adipose-specific Cre mice (Adipoq-cre) to generate a conditional overexpression mouse model with tissue-specific expression of foreign genes. The obtained Rosa26LSL/+ :Cre was successfully constructed as adipose tissue-specific overexpression mice, and the transgenic mice were named Adipsin-Tg, while the Rosa26LSL/+ mice failed to construct adipose tissue-specific overexpression Adipsin and were named nontransgenic mice (NTg) [20]. Eighty 6-week-old male C57BL/6J mice were obtained from the Animal Center of the Air Force Medical University (Xi’an, China), and divided into 4 groups: Adipsin-Tg mice (n = 32), NTg mice (n = 32), and wild-type (WT) mice (C57BL/6, n = 16). In addition, newborn mice (1–3 days old, n = 20), and adult mice (8–12 weeks old, n = 10) were also used in the present study.
These mice were given a normal chow diet (CHD) or a high-fat diet (HFD), and were divided into CHD + Adipsin-Tg group (n = 16), CHD + NTg group (n = 16), CHD + WT (n = 16), HFD + Adipsin-Tg (n = 16), and HFD + NTg (n = 16) group. Diabetic mice were fed with a HFD (5.24 kcal/g; 60% of calories from fat, 20% of calories from protein, and 20% of calories from carbohydrate; D12492, Research Diets, USA), whereas control mice received a CHD (3.85 kcal/g; 10% of calories from fat, 20% of calories from protein, and 70% of calories from carbohydrate; D12450J, Research Diets, USA). Mice were kept in a light/dark cycle at 23 °C for 12 h and given adequate water. Six-week-old Adipsin-Tg or NTg mice were given a HFD for 6 months to establish a DCM mouse model for subsequent experiments [21]. Target gene expression was downregulated in mouse myocardium by myocardial spot injection. Anesthesia with 2% N-isoflurane was used during the operation process. After exposing the heart, adenovirus (1 × 1010 GC/ml, 40 μl per mouse) (50 μl needle, 705RN, Hamilton, USA) was injected into the free wall of the left ventricle. White adipose tissue (WAT), brown adipose tissue (BAT), heart, lungs, spleen, liver, kidney, and muscle tissues were then rapidly removed for subsequent experiments. At the end of the experiment, mice were euthanized using 30% VD/min of 100% carbon dioxide in accordance with the 2020 AVMA Euthanasia Guidelines.
Primary isolation and culture of neonatal and adult mouse cardiomyocytes, neonatal fibroblasts and endothelial cells
As described earlier [22], the chest wall of newborn mice (1–3 days old, n = 20) was dissected and the hearts were removed. The ventricles were cut into pieces and digested with 0.1% type II collagenase. After centrifugation, the precipitate containing cardiomyocytes and cardiac fibroblasts was preserved, and primary cardiomyocytes and cardiac fibroblasts were obtained by differential adherence method. For primary endothelial cells, we sequentially digested with 0.2% collagenase II and 0.25% trypsin in the ventricle, followed by centrifugation at 1000 g for 5 min to produce endothelial cell precipitates [19]. Adult mice (8–12 weeks old, n = 10) were anesthetized with N-isoflurane (2%) and hearts were rapidly excised. The hearts were perfused using the Langendorff system (PanLab, Barcelona, Spain), and cardiomyocytes were isolated and cultured according to previously described methods [23].
Immunogold staining and transmission electron microscope analysis
For immunocolloidal gold electron microscopy studies, mice were anesthetized with N-isoflurane (2%) and hearts were rapidly excised. Heart tissue samples with a volume of less than 1 mm3 were rapidly fixed in an immunocolloidal gold electron microscopy fixative (G1124-100ML, Servicebio, China). Samples were then dehydrated with gradient ethanol, wrapped in resin, polymerized and sectioned with an ultraminiform microtome (70–80 nm), and coated with nickel mesh foil. The grids were hydrated with phosphate buffer saline (PBS) and blocked with PBS containing 0.1% bovine serum albumin (BSA) to block nonspecific reactions. Anti-interleukin (IL)-1 receptor-associated kinase-like 2 (Irak2) antibodies (bs-1427R, Bioss, China) were diluted to 1:500 with blocking solution and incubated overnight at 4 °C. Samples were then rinsed with trisbuffered saline (TBS) at room temperature followed by incubation with secondary antibody (10 nm colloidal gold goat anti-mouse, G7777, Sigma-Aldrich, Germany). Samples were fixed with 1% glutaraldehyde in PBS for 7 min and incubated with acetate uranyl and lead citrate. Images were acquired under a transmission electron microscope (HT7700, HITACHI, Japan). Black gold particles (10 nm in size) were considered as positive expression [24].
For routine transmission electron microscope examination, fresh heart tissue with a volume less than 1 mm3 was quickly placed into an electron microscope fixation device (G1102, Servicebio, China) for internal fixation, rinsed, re-fixed, dehydrated in a gradient, permeable, embedded, and cut into 60–80 nm ultrathin sections with an ultrathin microtome. Images were obtained with a transmission electron microscope (HT7700, HITACHI, Japan) at 300 kV. Mitochondrial images were analyzed using ImageJ software [25].
Measurement of mitochondrial oxygen consumption rate (OCR)
A Seahorse XF Cell Mitochondrial Stress Test kit (103015-100, Agilent, USA) was used. In brief, primary mouse cardiomyocytes were seeded at a density of 1 × 105/ml in Agilent Seahorse XF24 cell culture microplate (V7-PS, 100777-004, Agilent, USA) on an XF24 extracellular flux analyzer (Agilent Seahorse Bioscience, USA). The Seahorse XF Cell Mitochondrial Stress Test kit was used to provide inhitiors in sequence on plates containing culture medium, including oligomycin (1 μmol/L), fluoro-carbonyl cyanide phenylhydrazone (FCCP; 0.5 μmol/L), rotenone (1 μmol/L), and antimycin A (1 μmol/L). Wave software (Agilent Seahorse Bioscience, USA) was employed for data collection and calculated basic and maximum OCR and other parameters [26].
Small interfering RNA (siRNAs) and recombinant adenovirus vectors construction
Construction of Irak2 siRNAs and recombinant adenovirus vector expressing Irak2 short hairpin RNA (shRNA)
To downregulate Irak2 expression, three Irak2 siRNAs were designed by BLOCK-iT™ RNAi Designer and synthesized by Tsingke Biotechnology Company (Beijing, China) (Additional file 1: Table S1). For in vitro studies, primary cardiomyocytes were transfected with Lipofectamine 2000 (Thermo Fisher Scientific, Michigan, USA), the one with the highest knockdown efficiency was selected to convert the selected siRNA sequence to an shRNA sequence. The empty adenovirus vector (Ad-shControl) and the recombinant adenovirus vector expressing shIrak2 (Ad-shIrak2) were constructed by Tsingke Biotechnology Company (Beijing, China). The sequences of shRNA for Irak2 were 5′-CCGGGCACCTTTGCCGATATCTCTCGAGTTTTTT-3′ and 5′-AATTAAAAAAGCACCTTTGCCGATATCTCTCGAGAGATATCGGCAAAGGTGC-3′. In vivo experiments, the target gene Irak2 was introduced into the hearts of Adipsin-Tg or NTg mice by myocardial spot injection (NTg + Ad-shControl, NTg + Ad-shIrak2, Adipsin-Tg + Ad-shControl, Adipsin-Tg + Ad-shIrak2).
Construction of cell line overexpressing Adipsin
To enhance Adipsin expression, Adipsin was delivered in vitro using adenovirus carrying Adipsin (Ad-Adipsin) and a control vector (Ad-Control). After cells had grown to 50–60% confluence as described previously [27], adenovirus was transduced into cardiomyocytes at 100:1 multiplicity of infection in FBS-free medium, and various subsequent experiments were performed.
Echocardiography
M-mode echocardiography was performed using a VEVO 2100 echocardiography system (VisualSonics Inc., Toronto, Canada). Briefly, mice were anesthetized with 2.5% isoflurane. Anesthesia was maintained by inhalation of 2% isoflurane, and heart rate was recorded (approximately 400 bpm). End-systolic left ventricular internal diameters (LVIDs) and end-diastolic left ventricular internal diameters (LVIDd) were measured. Left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were calculated by software algorithm. Early diastolic (E) and late diastolic (A) mitral cross flow velocity and the ratio of early diastolic (E) to late diastolic (A) mitral flow velocity (E/A) were measured by Doppler echocardiography. Pulsed-wave Doppler images of mitral E-wave and A-wave velocities was obtained from a four-chamber cardiac view. The E/A ratio was measured to evaluate diastolic function. The parameters were derived from five consecutive cardiac cycles in M-mode. Early-diastolic peak velocity (e’) of mitral valve ring was also measured, and E/e’ was calculated to reflect left ventricular diastolic function. All echocardiographic images were analyzed with the use of the Vevo 2100 software (Toronto, Canada). Vevo Strain Software (Toronto, Canada) was used to perform speckle tracking-based strain analyses of the parasternal long-axis B-mode loops [22].
Real-time quantitative PCR
Total RNA was extracted from WAT, BAT, heart, lung, spleen, liver, kidney and muscle tissue, as well as cardiomyocytes, fibroblasts and endothelial cells using TRIzol (Invitrogen, 15596026, USA) and methylene chloride. Quantification was performed with Nano Drop2000 nucleic acid (Thermo Fisher Scientific, Michigan, USA). cDNA was then synthesized with the PrimeScript RT reagent kit and gDNA Eraser (rr047q, TaKaRa, Japan), and real-time quantitative-PCR was carried out by SYBR Premix Ex Taq II (rr820l, TaKaRa, Japan). All procedures were performed in accordance with the manufacturer’s guidelines [27]. Primer sequences are shown in the Additional file 1: Table S2.
Co-immunoprecipitation (Co-IP)
Cardiomyocytes antigen samples were prepared using protein A/G immunoprecipitation magnetic beads (B23201, Bimake, USA), preprocessed with magnetic beads, conjugated with anti-Adipsin and anti-Irak2 antibodies, precipitated and eluted antigens. Experiments were performed according to the manufacturer’s protocol. Finally, 20–50 μl of 1 × sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) loading buffer was added to the prepared antigen samples, thoroughly stirred, heated at 95 °C for 5 min. Magnetic separation was performed again. The supernatant was collected and placed in a new Eppendorf tube. After centrifugation at 13,000 g for 10 min at room temperature, the supernatant was collected for subsequent detection [18].
Western blotting
Proteins or mitochondria were extracted from heart, epididymal WAT, BAT, etc., then protein loading buffer was added, boiled and subjected to electrophoresis by 10% or 12% SDS–PAGE (Cwbiotech, Beijing, China). Samples were transferred to 0.22-μm polyvinylidene difluoride absorbent membranes (Millipore, MA, USA). Membranes were blocked with 10% milk for 1 h at 37 °C and then incubated with specified primary antibodies overnight at 4 °C, followed by incubation with the corresponding secondary antibody at room temperature. To visualize the enhanced chemiluminescence (ECL) color bands using chemiluminescence technology. Densitometry was performed using ImageJ software. Protein density was normalized by β-actin, voltage-dependent anion channel 1 (VDAC1) and transferrin (TF) levels [27]. The primary antibodies used for Western blotting are shown in the Additional file 1: Table S3.
Glutathione-S-transferase (GST) pull down and mass spectrometry analysis
Experiments were performed using the pierce GST Protein Interaction Pull Down kit (21516, Thermo Fisher Scientific, USA) according to the manufacturer’s manual. Firstly, the GST-tagged fusion protein GST-Adipsin (pET-21b + BamHI + Adipsin + GST + HindIII) was constructed as a protein bait (constructed and synthesized by Tsingke Biotechnology Company, Beijing, China). Prey proteins for primary cardiomyocyte lysis were obtained by immobilizing and binding bait proteins to immobilized bait proteins using balanced glutathione agarose. Then, the eluted protein complexes were employed for mass spectrometry analysis and subsequent immunoblotting [28].
Immunohistochemistry
Mouse hearts were fixed overnight in 4% paraformaldehyde (pH 7.4), embedded in paraffin, and serially sectioned (5-mm-thick sections) for histological analysis. Sections were deparaffinized and endogenous peroxidase was blocked with antigen repair. After sealing with 3% BSA for 1 h, sections were incubated overnight with the specified primary antibodies in a wet box at 4 °C, followed by incubation with the corresponding secondary antibodies (HRP-labeled) and staining using diaminobenzidine (DAB) and hematoxylin. After dehydration and sealing, sections were visualized and images were collected. DAB-positive expression was brown yellow. At least 10 fields were randomly selected from each heart for immunohistochemistry analysis [29].
JC-1 staining and flow cytometry
To detect mitochondrial membrane potential, cardiomyocytes were incubated with JC-1 (Beyotime, Shanghai, China) for 20 min at 37 °C. Cardiomyocytes were analyzed within 1 h using flow cytometry (CytoFLEX LX, Beckman, USA). All data were analyzed using EXPO32 ADC analysis software (Beckman, Washington, USA). For flow cytometry analysis, unstained cells were used as controls to identify the forward scatter/side scatter (FSC/SSC) gates used in the same experiments. A total of 10,000 cells was detected in each sample [30].
Determination of malondialdehyde (MDA) and triglyceride (TG) content
The lipid oxidation MDA assay kit was used to detect the level of MDA in cardiac tissues (BC0025, Solarbio, China) [31]. Tissue TG assay kit (BC0625, Solarbio, China) was employed to monitor TG content in cardiac myocardium [32]. MDA and TG contents were calculated according to standard curve.
Fatty acid β-oxidation (FAO) analysis
In adult mouse cardiomyocytes, FAO was measured according to the previously described method [21]. In brief, isolated adult cardiomyocytes were incubated with analysis medium containing fatty acids for 10 min, and at the end of the reaction the supernatant was added to 4 ml scintillation cocktail (Sigma-Aldrich, Germany) and radioactivity was measured using a scintillation counter [33].
XF palmitate (PA) oxidative stress test
FAO levels were determined using the Seahorse XF PA Oxidative Stress Test kit (102720-100, Agilent, USA). Mouse adult cardiomyocytes were incubated at a density of 1 × 105/ml in Agilent Seahorse XF24 Cell Culture Microplates (V7-PS, 100777-004, Agilent, USA). OCR was measured in the presence and absence of Etomoxir (HY-50202, MCE, USA), an inhibitor of carnitine palmitoyltransferase-1 (CPT-1), which is essential for FAO and is located at the inner surface of the outer mitochondrial membrane. Cells were stored overnight at 37 °C according to the manufacturer’s FAO assay protocol (Agilent, CA, USA). The PA oxidative stress test was performed in the presence of Etomoxir and PA-BSA or control-BSA, and each substance was added 0 and 15 min before the start of the assay, respectively. The final concentrations of the drugs used were: etomoxir 4 µmol/L, oligomycin 1.5 µmol/L, FCCP 1 µmol/L, and rotenone/antimycin A 0.5 µmol/L, respectively. FAO was determined by calculating the difference in OCR between Etomoxir treated and untreated cells. OCR values were expressed as pmol/(min·µg) protein [34].
Enzyme-linked immunosorbent assay (ELISA)
Serum Adipsin levels were detected using the mouse Adipsin ELISA kit according to the manufacturer’s procedure (E-ELM0335c, Elabscience, China). The absorbance (OD) value was measured at 450 nm by spectrophotometer.
Mitochondria biological function
To assess the functional status of mitochondria, citrate synthase (CS) and electron transport chain complex activities (complexes I/II/III/IV/V) were measured using commercially available kits (Sigma-Aldrich, Germany).
Statistical analysis
Data are expressed as mean ± standard deviation (SD). SPSS software (version 23; Inc., Chicago, IL, USA) and GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA) software were used. Statistical analyses between groups were done by two-tailed Student’s t test or one-way ANOVA, with Fisher’s post hoc comparison test when appropriate. P < 0.05 was considered to be statistically significant.
Results
Adipsin is significantly downregulated in HFD-induced DCM
To identify the possible role of Adipsin in the pathophysiology of DCM, we used a mouse model of HFD-induced DCM (Additional file 1: Tables S4, S5). The serum Adipsin level began to decrease at the 2nd month of HFD feeding and reached the lowest level at the 6th month (P < 0.05; Fig. 1a, Additional file 1: Fig. S1a). After 6 months of HFD feeding, the expression of Adipsin mRNA was decreased in both WAT and BAT compared with CHD mice (WAT: 6.29 ± 0.45 vs. 10.79 ± 0.39, P < 0.05; BAT: 4.34 ± 0.23 vs. 7.63 ± 0.28, P < 0.05; Additional file 1: Fig. S1b). However, at 6 months, Adipsin mRNA expression was largely undetectable in heart, lung, spleen, liver, kidney and muscle tissues, as well as cardiomyocytes, cardiac fibroblasts and endothelial cells of CHD and HFD-fed mice (Additional file 1: Fig. S1b, c). In addition, Western blotting showed that Adipsin levels in WAT, BAT, and heart of DCM mice were significantly lower than those of normal mice (WAT: 0.18 ± 0.12 vs. 1.00 ± 0.24, P < 0.05; BAT: 0.54 ± 0.14 vs. 1.00 ± 0.14, P < 0.05; heart: 0.32 ± 0.08 vs. 1.00 ± 0.16, P < 0.05; Fig. 1b). Therefore, we speculated that adipose tissues express and secrete the adipokine Adipsin, which enters the diabetic heart and plays an important protective role. In support of these findings, immunohistochemical staining and immunofluorescence staining of myocardial tissues showed that the content of Adipsin was significantly decreased in HFD-fed mice (P < 0.05; Fig. 1c, d). These results suggest that Adipsin play an important role in the pathophysiology of DCM.
Fig. 1.

Adipsin is significantly downregulated in HFD-induced DCM. a Western blotting results and quantitative analysis of serum Adipsin levels in the treatment groups at 0, 2, 4, and 6 months (n = 4). b Western blotting results and quantitative analysis of Adipsin expression in WAT, interscapular BAT and heart tissue lysates from CHD- and HFD-fed mice (n = 6). c, d Immunohistochemical/immunofluorescence stainings and quantitative analysis of Adispin in mouse hearts from CHD- and HFD-fed groups (n = 5). Scale bar = 50 μm. Statistical significance was determined by two-tailed Student’s t test. All data are represented with mean ± SD. *P < 0.05 vs. CHD; CHD chow diet, HFD high-fat diet, WAT white adipose tissue, BAT brown adipose tissue, TF transferrin, IOD integrated option density, DAPI 4’,6-diamidino-2-phenylindole
Overexpression of Adipsin can improve cardiac function, reduce mitochondrial cristae damage, and improve mitochondrial function
To further examine the effects of Adipsin on DCM, heterozygous mice with conditionally overexpressing Adipsin were generated. Primers 1 and 2 were used to amplify the mouse genome. In WT mice, primer 1 showed a positive band at 994 bp and primer 2 showed a negative band at 749 bp. Heterozygous mice (Rosa26LSL/+) had a positive band at both primer 1 (994 bp) and primer 2 (749 bp). Heterozygous mice (Rosa26LSL/+) were crossed with adipose tissue-specific Cre mice (Adipoq-cre) to generate Adipsin-Tg (Rosa26LSL/+ :Cre) and their corresponding control mice (NTg). Adipsin-Tg (Rosa26LSL/+ :Cre) mice showed a positive band at primer Cre (400 bp) (Additional file 1: Fig. S1d). Compared with NTg mice, Adipsin mRNA levels in WAT and Adipsin protein levels in WAT and cardiac myocardium were significantly increased in Adipsin-Tg mice (P < 0.05; Additional file 1: Fig. S1e, f).
Echocardiographic analysis showed that LVEF, LVFS, and E/A ratio of HFD-fed mice were significantly decreased, and LVIDd and LVIDs were increased (P < 0.05; Fig. 2a, b), indicating that both systolic and diastolic functions were severely impaired in DCM mice. Adipsin overexpression reversed these effects. These results indicate that Adipsin improves cardiac function in DCM mice. In addition, swollen and structurally abnormal myocardial mitochondria were observed in DCM mice compared with control mice. Adipsin-Tg diabetic mice showed reduced cardiomyocyte mitochondrial structure damage, evidenced by mitochondrial cristae fragmentation (P < 0.05; Fig. 2c). In this context, Adipsin attenuates myocardial mitochondrial cristae damage and preserves mitochondrial structure. Moreover, Adipsin significantly improved mitochondrial complex I/II/III/IV/V activities in diabetic hearts (P < 0.05; Additional file 1: Fig. S2a).
Fig. 2.

Overexpression of Adipsin can improve cardiac function, reduce mitochondrial cristae damage, and improve mitochondrial function. a Echocardiographic results and quantitative analysis of echocardiographic data including LVEF, LVFS, LVIDd and LVIDs (n = 8). b Pulse-wave Doppler results and quantitative analysis of the E/A ratio (n = 8). c Transmission electron microscopic images, and quantitative analysis of cristae amount per μm2 and the proportion of mitochondria with disorganized cristae of myocardium from NTg and Adipsin-Tg groups with CHD- or HFD-feeding (n = 5). Scale bar = 500 nm. d OCR in cardiomyocytes from various treatment groups, and quantitative statistical analysis of OCR including basal respiration, maximal respiration, ATP production, and spare respiration capacity. Statistical significance was determined using one-way ANOVA analysis. All data are represented with mean ± SD. †P < 0.05 vs. CHD + NTg; ‡P < 0.05 vs. CHD + Adipsin-Tg; ¶P < 0.05 vs. HFD + NTg; *P < 0.05 vs. Control + Ad-Control; §P < 0.05 vs. Control + Ad-Adipsin; ||P < 0.05 vs. PA + Ad-Control. CHD chow diet, HFD high-fat diet, Adipsin-Tg Adipsin tissue-specific transgenic mice, NTg nontransgenic mice, LVEF left ventricular ejection fraction, LVFS left ventricular fractional shortening, LVIDd end-diastolic left ventricular internal diameters, LVIDs end-systolic left ventricular internal diameters, E/A the ratio of mitral peak velocity of early filling (E) to mitral peak velocity of late filling (A), OCR oxygen consumption rate, OM oligomycin, FCCP fluoro-carbonyl cyanide phenylhydrazone, Rot/AA rotenone and antimycin A, PA palmitate
To further verify the effect of Adipsin on mitochondria, we performed in vitro experiments in which cardiomyocytes were treated with Ad-Adipsin and Ad-Control for 8 h and then assessed for 24 h mitochondrial stress in the presence or absence of PA (300 μmol/L). The basal respiration, maximal respiration, ATP production, and spare respiration capacities of PA-treated cardiomyocytes were significantly decreased, while the effects were alleviated by Ad-Adipsin (P < 0.05; Fig. 2d). Meanwhile, the mitochondrial membrane potential of Ad-Adipsin treated cells was significantly increased (P < 0.05; Additional file 1: Fig. S2b). This finding suggests a protective effect of Adipsin on myocardial contractile and mitochondrial function in DCM.
Adipsin and Irak2 bind directly in cardiomyocytes
To investigate the mechanism by which Adipsin induces myocardial protection, we used the GST fusion protein sink technology (GST pull-down) to search for potential proteins that interact with Adipsin (Fig. 3a). We constructed a GST-Adipsin fusion protein to capture the protein(s) interacting with Adipsin in cardiomyocytes. Thus, the protein that interacts with adipsin, Irak2, was identified by liquid chromatography-tandem mass spectrometry (LC–MS/MS) analysis of captured prey proteins (Additional file 1: Table S6). We performed immunoblotting of prey proteins and found that Irak2 bands directly to Adipsin (Fig. 3b). In addition, Co-IP assays revealed a direct interaction between Adipsin and Irak2 in Ad-Adipsin treated cardiomyocytes (Fig. 3c, d, Additional file 1: Fig. S3). The interaction between Adipsin and Irak2 was enhanced in PA stimulated cardiomyocytes (2.26 ± 0.20 vs. 1.00 ± 0.22, P < 0.05; Fig. 3e). Moreover, the co-localization of Adipsin and Irak2 in cardiomyocytes was further verified by immunofluorescence (Fig. 3f). These data support that Adipsin enters cardiomyocytes and binds directly to Irak2.
Fig. 3.

Adipsin and Irak2 bind directly in cardiomyocytes. a Schematic representation of the experimental protocol. The cardiomyocytes lysis prey protein was obtained by immobilizing GST-Adipsin with equilibrated glutathione agarose. Then, the eluted protein complexes were employed for mass spectrometry analysis and subsequent immunoblotting. b Proteins bound and pulled down by purified GST-Adipsin in cardiomyocytes. c, d Co-IP results indicating the interaction between Adipsin and Irak2 following the infection with Ad-Adipsin. e Co-IP results indicating the interaction between Adipsin and Irak2 with or without PA challenge following the infection with Ad-Adipsin (n = 5). f Colocalization images and analysis of Adipsin (green) and Irak2 (red) proteins in cardiomyocytes. Scale bar = 50 μm. Statistical significance was determined using a two-tailed Student’s t test. All data are represented with mean ± SD. *P < 0.05 vs. Control. LC–MS/MS liquid chromatography-tandem mass spectrometry, Co-IP Co-immunoprecipitation, DAPI 4′,6-diamidino-2-phenylindole, Irak2 interleukin-1 receptor-associated kinase-like 2, PA palmitate, GST glutathione-S-transferase, IB immunoblotting
Overexpression of Adipsin inhibits mitochondrial translocation of Irak2
To further evaluate the interaction between Adipsin and Irak2, we also examined changes in Irak2 expression in the myocardium of Adipsin-Tg mice. Our results showed that Adipsin overexpression decreased mitochondrial Irak2 expression and increased cytoplasmic Irak2 expression in DCM mouse myocardium, but the total Irak2 level was not changed (P < 0.05; Fig. 4a), indicating that Adipsin inhibited Irak2 translocation from cytoplasm to mitochondria. Similarly, overexpression of Adipsin reduced the number of Irak2-positive spots on mitochondria in diabetic hearts using immunocolloidal gold electron microscopy (P < 0.05; Fig. 4b). These observations further confirm that Adipsin reduces the mitochondrial translocation of Irak2. In addition, to explore the effect of Adipsin on Irak2, we examined Irak2 downstream molecules localized to myocardial mitochondria. Consistent with previous report [35], this study also demonstrated that Irak2 is exclusively expressed in the cytoplasm in the non-diabetes state, while Irak2 is able to translocate to the inner mitochondrial membrane and interact with prohibitin (Phb) and optic atrophy protein 1 (Opa1) upon PA challenge (Additional file 1: Fig. S4). Furthermore, the expression of Phb and Opa1 were downregulated in the myocardium of DCM mice induced by HFD. Adipsin overexpression significantly increased the expression of Phb and Opa1 in HFD-induced DCM mice (P < 0.05; Fig. 4c). The results further suggest that Phb and Opa1, as downstream of Irak2, are regulated by Adipsin overexpression in the animal model of DCM. In conclusion, Adipsin inhibits the mitochondrial translocation of Irak2 in DCM, thereby reducing the interaction between Irak2 and Phb-Opa1 on mitochondria and improving the structural integrity and function of mitochondria.
Fig. 4.

Overexpression of Adipsin inhibits mitochondrial translocation of Irak2. a Western blotting results and quantitative analysis of Irak2 expression in cytoplasm and mitochondria of cardiomyocytes in the CHD + NTg, CHD + Adipsin-Tg, NTg + HFD, and Adipsin-Tg + HFD groups (n = 6). b Electron microscopy images of Irak2 stained with immunogold and quantitative analysis of Irak2-positive dots (red arrow) in the myocardial mitochondria of NTg or Adipsin-Tg mice fed with CHD or HFD (n = 5). Scale bar = 200 nm. c Western blotting results and quantitative analysis of Phb and Opa1 in myocardial mitochondria of NTg or Adipsin-Tg mice fed with CHD or HFD (n = 6). Statistical significance was determined using one-way ANOVA. All data are represented with mean ± SD. †P < 0.05 vs. CHD + NTg; ‡P < 0.05 vs. CHD + Adipsin-Tg; ¶P < 0.05 vs. HFD + NTg. Mito mitochondria, Cyto cytosol, CHD chow diet, HFD high-fat diet, Adipsin-Tg Adipsin tissue-specific transgenic mice, NTg nontransgenic mice, Irak2 interleukin-1 receptor-associated kinase-like 2, VDAC1 voltage-dependent anion channel 1, Phb prohibitin, Opa1 optic atrophy protein 1
Adipsin improves cardiac function, reduces mitochondrial cristae damage, and improves mitochondrial function through Irak2 signaling
To further test whether the cardioprotective effect of Adipsin was mediated through Irak2, Irak2 was knocked down in myocardial tissues. We found that mice receiving myocardial spot injection of Ad-shIrak2 successfully reduced Irak2 expression in myocardial tissues at the both mRNA and protein levels (P < 0.05; Additional file 1: Fig. S5a-c). Subsequently, mice were immediately placed on a HFD for 6 months and then examined for DCM-related phenotypes. Echocardiography analysis showed that compared with DCM mice, Adipsin-Tg DCM mice had significantly improved cardiac function, as indicated by increased LVEF, LVFS, and E/A ratio, as well as decreased LVIDd and LVIDs (P < 0.05; Fig. 5a, b). Interestingly, Adipsin overexpression did not further alleviate cardiac dysfunction in DCM mice under Irak2 knockdown conditions, suggesting a downstream role of Irak2 in Adipsin-induced responses (P < 0.05; Fig. 5a, b). Our data further revealed the protective role of Adipsin overexpression against DCM-induced mitochondrial destruction. Similarly, overexpression of Adipsin attenuated mitochondrial destruction, enhanced mitochondrial complex I/II/III/IV/V activity in diabetic myocardial tissues. Consistently, mitochondrial damage was not further attenuated by Adipsin overexpression (P < 0.05; Fig. 5c, Additional file 1: Fig. S6a), and improved mitochondrial complex I/II/III/IV/V activities (P < 0.05; Additional file 1: Fig. S6b) after Irak2 knockdown. Next, intracellular Irak2 was knocked down with siRNA prior to Ad-Adipsin and PA treatment of cardiomyocytes. Assessment of mitochondrial stress revealed a significant increase in basal respiration, maximal respiration, ATP production, and spare respiration capacity in cells treated with Ad-Adipsin in response to lipotoxic PA stimulation. Consistently, overexpression of Adipsin did not further improve oxidative phosphorylation (P < 0.05; Fig. 5d). Additionally, Adipsin overexpression did not further induce mitochondrial membrane potential benefit after Irak2 knockdown (P < 0.05; Additional file 1: Fig. S6c). Taken together, these results indicate that Irak2 mitochondrial translocation plays an important role in DCM-induced contractile dysfunction, mitochondrial cristae damage, and mitochondrial injury in Adipsin-mediated myocardial protection.
Fig. 5.

Adipsin improves cardiac function, reduces mitochondrial cristae damage, and improves mitochondrial function through Irak2 signaling pathway. a Echocardiographic results and quantitative analysis of echocardiographic data including LVEF, LVFS, LVIDd and LVIDs (n = 8). b Pulse-wave Doppler results and quantitative analysis of the E/A ratio (n = 8). c Transmission electron microscopic images of myocardium from different groups (n = 8). Scale bar = 500 nm. d OCR in cardiomyocytes from various treatment groups, and quantitative statistical analysis of OCR including basal respiration, ATP production, maximal respiration and spare respiration capacity. Statistical significance was determined using one-way ANOVA. All data are represented with mean ± SD. †P < 0.05 vs. CHD + WT; ‡P < 0.05 vs. HFD + NTg + Ad-shControl; *P < 0.05 vs. Con; §P < 0.05 vs. PA + Ad-Control + siControl. CHD chow diet, HFD high-fat diet, Adipsin-Tg Adipsin tissue-specific transgenic mice, NTg nontransgenic mice, LVEF left ventricular ejection fraction, LVFS left ventricular fractional shortening, LVIDd end-diastolic left ventricular internal diameters, LVIDs end-systolic left ventricular internal diameters, E/A the ratio of mitral peak velocity of early filling (E) to mitral peak velocity of late filling (A), OCR oxygen consumption rate, OM oligomycin, FCCP fluoro-carbonyl cyanide phenylhydrazone, Rot/AA rotenone and antimycin A, PA palmitate, Irak2 interleukin-1 receptor-associated kinase-like 2, siControl control small interfering RNA
Adipsin improves cardiomyocyte FAO through the Irak2 signaling pathway
Electron microscopic analysis showed that Adipsin overexpression significantly reduced the size of lipid droplets in the cardiac myocardium of DCM mice. After Irak2 knockdown, overexpression of Adipsin did not further reduce myocardial lipid deposition (P < 0.05; Fig. 6a). The contents of TG and MDA were measured in myocardial tissues. The results showed that TG and MDA contents in myocardial tissue of DCM mice were significantly higher than those of WT mice, and these effects were reversed by Adipsin overexpression (P < 0.05). Interestingly, in the presence of Irak2 knockdown, Adipsin overexpression did not further reduce TG and MDA contents in diabetic hearts (P < 0.05; Fig. 6b). These findings suggest that Adipsin inhibits the accumulation of lipids and their metabolites in the myocardium via Irak2.
Fig. 6.

Adipsin improves cardiomyocyte FAO through the Irak2 signaling pathway. a Transmission electron microscopic images and quantitative analyses of mouse hearts depicting larger lipid droplets accumulation in various treatment groups (n = 6). Lipid droplets were marked by asterisks. Scale bar = 1 μm. b TG and MDA content in myocardial tissues (n = 8). c FAO evaluating with [3H]-oleic acid uptake (n = 6). d Oxidation pressure test of palmitate in cardiomyocytes from various treatment groups, and evaluation of FAO using quantitative analysis of maximum ΔOCR. Statistical significance was determined using one-way ANOVA. All data are represented with mean ± SD. †P < 0.05 vs. CHD + WT; ‡P < 0.05 vs. HFD + NTg + Ad-shControl. CHD chow diet, HFD high-fat diet, Adipsin-Tg Adipsin tissue-specific transgenic mice, NTg nontransgenic mice, TG triglyceride, MDA malonaldehyde, FAO fatty acid β-oxidation, BSA bovine serum albumin, ETO etomoxir, PA palmitate, OCR oxygen consumption rate, OM oligomycin, FCCP fluoro-carbonyl cyanide phenylhydrazone, Rot/AA rotenone and antimycin A, △OCR oxygen consumption rate[PA-(PA+ETO)]—oxygen consumption rate[BSA-(BSA+ETO)], Irak2 interleukin-1 receptor-associated kinase-like 2
Cardiomyocytes were isolated from diabetic mice overexpressing Adipsin (HFD + Adipsin-Tg) and corresponding controls. Our results indicated that FAO is significantly reduced in cardiomyocytes stressed by diabetes. Adipsin overexpression increased the level of FAO in cardiomyocytes. After Irak2 knockdown, overexpression of Adipsin did not further increase the level of FAO in cardiomyocytes under diabetic stress (P < 0.05; Fig. 6c). To further test our hypothesis, we used the palmitic acid oxidative stress assay to assess FAO in diabetes-damaged cardiomyocytes. The dependence of cardiomyocytes on FAO was evaluated by measuring acute response to an FAO inhibitor (Etomoxir), also known as maximum ΔOCR. Our results indicated the maximum ΔOCR of cardiomyocytes was significantly reduced in HFD-fed mice, indicating that the extent of FAO was reduced in mice suffering from diabetic damage. In diabetic cardiomyocytes overexpressing Adipsin, the maximum ΔOCR of cells was significantly increased, indicating that Adipsin significantly improved FAO in diabetic hearts. As expected, Adipsin overexpression did not further increase maximum ΔOCR in diabetic hearts after Irak2 knockdown (P < 0.05; Fig. 6d). These findings indicated that Adipsin reduced the accumulation of TG and MDA in diabetic myocardium, while improving FAO in diabetic cardiomyocytes. Irak2 may be involved in the development of DCM as a downstream regulator of Adipsin.
Discussion
DCM is a major public health threat with few effective prevention and treatment options [36], so there is a need to elucidate the underlying mechanisms of this devastating comorbidity. Preservation of mitochondrial integrity and function has been shown to protect against the development of DCM [37, 38]. Here, we found that adipose tissue-specific overexpression of Adipsin improves cardiac dysfunction, mitochondrial structure disruption and mitochondrial damage in DCM. Adipsin, the first adipokine identified, is a serine protease homolog that is synthesized and secreted by adipocytes, and its synthesis is closely associated with metabolic status, especially the lipid profile [39, 40]. Previous studies have shown that Adipsin exerts a protective effect on pancreatic β cells, thereby improving hyperglycemia. At the same time, Adipsin levels were significantly reduced in obese mice [17, 41]. In parallel, our results showed that Adipsin levels were significantly reduced in DCM mice. Our further study showed that Adipsin had a cardioprotective effect to significantly improve cardiac function in DCM mice. These findings provide strong evidence for Adipsin as a novel target for DCM treatment.
DCM exposes the heart to metabolic stress, such as a high-fat environment, in our current experimental setting [42]. In order to adapt to the high-fat environment, the heart starts to switch to fatty acids as its main energy source [43]. With the continuous progress of metabolic pathology, excessive utilization and uptake of fatty acids/lipids cannot be compensated, leading to an imbalance of fatty acid supply and uptake, lipid overload (steatosis), accumulation of fatty acids and lipids in cardiomyocytes, causing lipotoxicity, poor FAO and impaired myocardial function [7, 9], which is consistent with our current experimental results in DCM mice. Interestingly, our data revealed that increased Adipsin levels significantly reduced myocardial TG accumulation and intracellular MDA content in cardiomyocytes of DCM mice, suggesting a beneficial role of Adipsin in intracellular lipid accumulation and lipotoxicity. Our study revealed that adipose tissue-specific overexpression of Adipsin promoted intracellular Adipsin content and improved FAO, suggesting the improvement of myocardial FAO may be involved in Adipsin induced responses in DCM. Not surprisingly, many preclinical studies have described the efficacy of targeting cardiac FAO to treat DCM [44, 45], suggesting the promise of FAO and metabolic adaptation in the treatment of DCM [42].
To investigate the specific molecular mechanisms by which Adipsin protects cardiomyocytes, we found that Adipsin binds directly to Irak2 in cardiomyocytes. Irak2 is a member of the IL-1R-related kinase (IRAK) family [46]. Recent studies have shown that Toll-like receptor/IL-1 receptor (TLR/IL-1R) is closely related to cell metabolism, especially lipid accumulation, cellular growth, and ER stress, in which Irak2 plays a crucial regulatory role [47–49]. Importantly, defects in Irak2 or its kinase, in addition to demonstrating that translocation of Irak2 into the mitochondrial membrane space and inner membrane plays a role, also reduced HFD-induced metabolic disorders [24, 50]. In our study, Adipsin improved myocardial fatty acid metabolism and protected myocardial function by inhibiting Irak2 mitochondrial translocation. Irak2 has been shown to translocate into mitochondrial space and to locate to the inner mitochondrial membrane, where it interacts with Phb, resulting in the recruitment of Opa1 by Phb from cristae junctions, thereby disrupting the supercomplex and mitochondrial integrity [24]. Opa1 is an important molecule that regulates mitochondrial fusion and cristae integrity to control kinesin-related proteins, which favors mitochondrial cristae attachment [51]. Our results showed that increased Adipsin inhibits Irak2 mitochondrial translocation in diabetic myocardium, thereby eliminating the interaction between Irak2 and Phb-Opa1, reducing mitochondrial cristae damage, improving mitochondrial function, and myocardial function. Our further experiment showed that Irak2 plays an important role in the myocardial protection provided by Adipsin in DCM. Inhibition of Irak2 mitochondrial translocation by Adipsin can improve the structure and function of myocardial mitochondria, inhibit the accumulation of TG and MDA in cardiomyocytes, and improve FAO and the function of myocardial contraction, thereby blocking the pathogenesis of DCM. Importantly, we found that inhibition of the mitochondrial translocation of myocardial Irak2 through Adipsin action reduced the interaction of Irak2 with Phb-Opa1, resulting in improved mitochondrial integrity and function, and reduced TG and MDA accumulation in cardiomyocytes, leading to the improved FAO and myocardial function (Fig. 7).
Fig. 7.
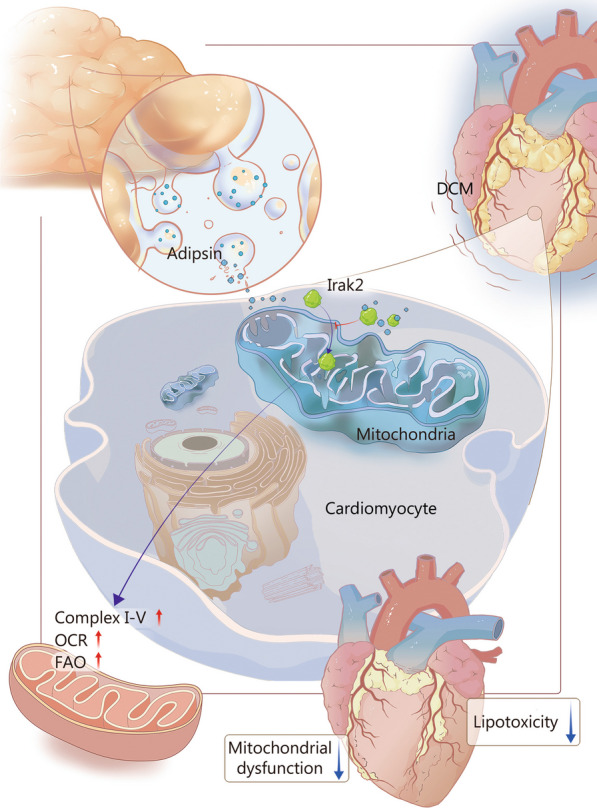
Schematic illustration of Adipsin-induced alleviation of diabetic cardiomyopathy (DCM) by inhibiting Irak2 mitochondrial translocation. Adipsin is secreted by adipose tissues and enters cardiomyocytes. Under HFD conditions, Adipsin enters cardiomyocytes to bind with Irak2 and inhibit its mitochondrial translocation. Reduction in Irak2 entry into mitochondria improves FAO, protects mitochondrial structure and function, reduces myocardial lipid accumulation, further improves cardiac function, and prevents DCM. HFD high-fat diet, OCR oxygen consumption rate, FAO fatty acid β-oxidation, Irak2 interleukin-1 receptor-associated kinase-like 2
Several limitations of this study should be considered. First, the induction of DCM using a HFD alone may not adequately mimic the situation of type 2 diabetes mellitus in humans. We need to elucidate the role of Adipsin in different types and stages of diabetes by inducing type 2 DCM with a combination of HFD and streptozotocin (STZ). Second, the present study showed that Adipsin could alleviate HFD-induced mitochondrial dysfunction and cardiac dysfunction. Since multiple cell types may be involved in the development and progression of DCM, further studies are needed to determine which specific cell types contribute to DCM cardiac remodeling. Third, our study illustrated that Adipsin effectively improved cardiomyocyte FAO. However, DCM is known to constitute complex substrate utilization and immune metabolic processes [42, 52], it is necessary to gain insight into the role of Adipsin on different substrate utilization and immune metabolism is warranted. Fourth, although we confirmed that Irak2 is an indispensable relevant molecule in the protective response induced by Adipsin, Irak2 may not be the only essential molecule for this process. Future studies should aim to elucidate the complex underlying mechanisms. Despite these obvious limitations, the present study provides novel therapeutic targets for DCM and compelling evidence for the mitochondrial protective properties of Adipsin.
Conclusions
Our findings reveal an important cardioprotective role for Adipsin in DCM, where Adipsin provides a powerful beneficial complex response to myocardial and mitochondrial function in DCM. Importantly, Adipsin induced cardiac contractile benefits was closely associated with improved mitochondrial FAO and reduced myocardial lipotoxicity and lipid storage, thereby delaying the progression of DCM. Future work is needed to extend the possible cardioprotective properties of Adipsin in other cardiovascular diseases as well as to develop small molecules targeting Adipsin.
Supplementary Information
Additional file 1: Table S1 siRNA sequences for Irak2knockdown. Table S2 Primer sequences for real-time quantitative-PCR. Table S3 Primary antibodies used for Western blotting, immunoprecipitation, and immunohistochemistry studies. Table S4 Blood glucose and body weight of experimental animals (mean ± SD, n = 9). Table S5 Echocardiographic metrics of experimental animals (mean ± SD, n = 9). Table S6 Mass spectrum analysis of GST-Adipsin-interacting proteins. Fig. S1 Evaluation of Adipsin expression in myocardium and validation of successful construction of Adipsin-Tg mice. Fig. S2 Adipsin overexpression improves mitochondrial function in the hearts of DCM. Fig. S3 Interaction between Adipsin and Irak2 following administration of Ad-Adipsin in cardiomyocytes. Fig. S4 Co-IP detected the interaction between Irak2 and Phb-Opa1 in myocardial mitochondria. Fig. S5 Construction of AAV9-shIrak2 and verification of transfection effectiveness. Fig. S6 Impact of Irak2 knockdown on Adipsin-Tg induced protective effects of mitochondrial morphology and function.
Acknowledgements
The authors thank Jialin Si, Department of Pediatric Dentistry, School of Stomatology, Air Force Medical University, Xi’an, China, for his contribution to the image collection.
Abbreviations
- DCM
Diabetic cardiomyopathy
- HFD
High-fat diet
- CHD
Chow diet
- LC–MS/MS
Liquid chromatography-tandem mass spectrometry
- GST
Glutathione-S-transferase
- Co-IP
Co-immunoprecipitation
- Irak2
Interleukin-1 receptor-associated kinase-like 2
- PA
Palmitate
- BSA
Bovine serum albumin
- TBS
Tris-buffered saline
- LVIDs
End-systolic left ventricular internal diameters
- LVIDd
End-diastolic left ventricular internal diameters
- LVEF
Left ventricular ejection fraction
- LVFS
Left ventricular fractional shortening
- E/A
The ratio of mitral peak velocity of early filling (E) to mitral peak velocity of late filling (A)
- WAT
White adipose tissue
- BAT
Brown adipose tissue
- VDAC1
Voltage-dependent anion channel 1
- TF
Transferrin
- MDA
Malondialdehyde
- TG
Triglyceride
- FAO
Fatty acid β-oxidation analysis
- OCR
Oxygen consumption rate
- TLR/IL-1R
Toll-like receptor/interleukin-1 receptor
- Phb
Prohibitin
- Opa1
Optic atrophy protein 1
- Mito
Mitochondria
- Cyto
Cytosol
- STZ
Streptozotocin
- MI
Myocardial infarction
- ELISA
Enzyme-linked immunosorbent assay
- IOD
Integrated option density
- Adipsin-Tg
Adipsin tissue-specific transgenic mice
- NTg
Nontransgenic mice
- OM
Oligomycin
- FCCP
Fluoro-carbonyl cyanide phenylhydrazone
- Rot/AA
Rotenone and antimycin A
- DAPI
4′,6-Diamidino-2-phenylindole
- IB
Immunoblotting
- ETO
Etomoxir
- 2D Echo
Conventional echocardiography
- E/e’
The ratio between mitral inflow and mitral annular excursion velocity during early diastole
- Strain
Strain echocardiography
- CS
Citrate synthase
Author contributions
DDS and XZ defined the study concept and design and carefully revised the manuscript. MYJ, WRM, and XBZ made substantial contributions to the study concept and design, data analysis and interpretation, drafting, and revision. XHZ made substantial contributions to data acquisition, data analysis, and data interpretation. YD, JL, YZ, YC, DXW, XFS, CYL, and XMG were responsible for the animal models. LX, HW, and XZ were responsible for translating documentation and drawing schematics. DDS is the guarantor of this work and therefore has full access to all data in the study and is responsible for the integrity of the data and the accuracy of the data analysis. The study received final approval from all authors prior to submission. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (82070398, 81922008), the Key Basic Research Projects of Basic Strengthening Plan (2022-JCJQ-ZD-095-00), and the Top Young Talents Special Support Program in Shaanxi Province (2020).
Availability of data and materials
The datasets analysed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
All experimental animal procedures were approved by the Animal Care and Use Committee of the Air Force Medical University and followed the Animal Research Advisory Committee of the National Institutes of Health guidelines (No. 20201017).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Meng-Yuan Jiang, Wan-Rong Man, and Xue-Bin Zhang contributed equally to this work.
Contributor Information
Xuan Zhang, Email: zhangxuan@301hospital.com.cn.
Dong-Dong Sun, Email: wintersun3@fmmu.edu.cn.
References
- 1.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Razavi M, Wei YY, Rao XQ, Zhong JX. DPP-4 inhibitors and GLP-1RAs: cardiovascular safety and benefits. Mil Med Res. 2022;9(1):45. doi: 10.1186/s40779-022-00410-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knapp M, Tu X, Wu R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharmacol Sin. 2019;40(1):1–8. doi: 10.1038/s41401-018-0042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chavali V, Tyagi SC, Mishra PK. Predictors and prevention of diabetic cardiomyopathy. Diabetes Metab Syndr Obes. 2013;6:151–160. doi: 10.2147/DMSO.S30968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nathan DM. Diabetes: advances in diagnosis and treatment. JAMA. 2015;314(10):1052–1062. doi: 10.1001/jama.2015.9536. [DOI] [PubMed] [Google Scholar]
- 6.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90(1):207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 7.Taegtmeyer H, Mcnulty P, Young ME. Adaptation and maladaptation of the heart in diabetes: part I: general concepts. Circulation. 2002;105(14):1727–1733. doi: 10.1161/01.cir.0000012466.50373.e8. [DOI] [PubMed] [Google Scholar]
- 8.Pang L, Jiang X, Lian X, Chen J, Song EF, Jin LG, et al. Caloric restriction-mimetics for the reduction of heart failure risk in aging heart: with consideration of gender-related differences. Mil Med Res. 2022;9(1):33. doi: 10.1186/s40779-022-00389-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young ME, Mcnulty P, Taegtmeyer H. Adaptation and maladaptation of the heart in diabetes: part II. Circulation. 2002;105(15):1861–1870. doi: 10.1161/01.cir.0000012467.61045.87. [DOI] [PubMed] [Google Scholar]
- 10.Waki H, Tontonoz P. Endocrine functions of adipose tissue. Annu Rev Pathol. 2007;2:31–56. doi: 10.1146/annurev.pathol.2.010506.091859. [DOI] [PubMed] [Google Scholar]
- 11.Mcgown C, Birerdinc A, Younossi ZM. Adipose tissue as an endocrine organ. Clin Liver Dis. 2014;18(1):41–58. doi: 10.1016/j.cld.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 12.Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol. 2005;115(5):911–919. doi: 10.1016/j.jaci.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 13.Saxton SN, Clark BJ, Withers SB, Eringa EC, Heagerty AM. Mechanistic links between obesity, diabetes, and blood pressure: role of perivascular adipose tissue. Physiol Rev. 2019;99(4):1701–1763. doi: 10.1152/physrev.00034.2018. [DOI] [PubMed] [Google Scholar]
- 14.Zhao S, Kusminski CM, Scherer PE. Adiponectin, leptin and cardiovascular disorders. Circ Res. 2021;128(1):136–149. doi: 10.1161/CIRCRESAHA.120.314458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith CC, Mocanu MM, Davidson SM, Wynne AM, Simpkin JC, Yellon DM. Leptin, the obesity-associated hormone, exhibits direct cardioprotective effects. Br J Pharmacol. 2006;149(1):5–13. doi: 10.1038/sj.bjp.0706834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito S, Hashimoto H, Yamakawa H, Kusumoto D, Akiba Y, Nakamura T, et al. The complement C3-complement factor D-C3a receptor signalling axis regulates cardiac remodelling in right ventricular failure. Nat Commun. 2022;13(1):5409. doi: 10.1038/s41467-022-33152-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lo JC, Ljubicic S, Leibiger B, Kern M, Leibiger IB, Moede T, et al. Adipsin is an adipokine that improves beta cell function in diabetes. Cell. 2014;158(1):41–53. doi: 10.1016/j.cell.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Man W, Song X, Xiong Z, Gu J, Lin J, Gu X, et al. Exosomes derived from pericardial adipose tissues attenuate cardiac remodeling following myocardial infarction by Adipsin-regulated iron homeostasis. Front Cardiovasc Med. 2022;9:1003282. doi: 10.3389/fcvm.2022.1003282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Duan Y, Zhang X, Jiang M, Man W, Zhang Y, et al. Adipsin alleviates cardiac microvascular injury in diabetic cardiomyopathy through Csk-dependent signaling mechanism. BMC Med. 2023;21(1):197. doi: 10.1186/s12916-023-02887-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duan Y, Zhang X, Zhang X, Lin J, Shu X, Man W, et al. Inhibition of macrophage-derived foam cells by Adipsin attenuates progression of atherosclerosis. Biochim Biophys Acta Mol Basis Dis. 2022;1868(12):166533. doi: 10.1016/j.bbadis.2022.166533. [DOI] [PubMed] [Google Scholar]
- 21.Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. 2019;124(9):1360–1371. doi: 10.1161/CIRCRESAHA.118.314607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S, Zhao Z, Fan Y, Zhang M, Feng X, Lin J, et al. Mst1 inhibits Sirt3 expression and contributes to diabetic cardiomyopathy through inhibiting Parkin-dependent mitophagy. Biochim Biophys Acta Mol Basis Dis. 2019;1865(7):1905–1914. doi: 10.1016/j.bbadis.2018.04.009. [DOI] [PubMed] [Google Scholar]
- 23.Sun Y, Stenson K, Mohan ML, Gupta MK, Wanner N, Asosingh K, et al. Hypoxia sensing of β-adrenergic receptor is regulated by endosomal PI3Kγ. Circ Res. 2023;132(6):690–703. doi: 10.1161/CIRCRESAHA.122.321735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou H, Wang H, Yu M, Schugar RC, Qian W, Tang F, et al. IL-1 induces mitochondrial translocation of IRAK2 to suppress oxidative metabolism in adipocytes. Nat Immunol. 2020;21(10):1219–1231. doi: 10.1038/s41590-020-0750-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang M, Lin J, Wang S, Cheng Z, Hu J, Wang T, et al. Melatonin protects against diabetic cardiomyopathy through Mst1/Sirt3 signaling. J Pineal Res. 2017;63(2):e12418. doi: 10.1111/jpi.12418. [DOI] [PubMed] [Google Scholar]
- 26.Xu H, Yu W, Sun M, Bi Y, Wu NN, Zhou Y, et al. Syntaxin17 contributes to obesity cardiomyopathy through promoting mitochondrial Ca2+ overload in a Parkin-MCUb-dependent manner. Metabolism. 2023;143:155551. doi: 10.1016/j.metabol.2023.155551. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Cao Y, Zheng R, Xiong Z, Zhu Z, Gao F, et al. Fibroblast-specific activation of Rnd3 protects against cardiac remodeling in diabetic cardiomyopathy via suppression of Notch and TGF-β signaling. Theranostics. 2022;12(17):7250–7266. doi: 10.7150/thno.77043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Z, Guo D, Wang Q, Liu P, Xiao Y, Wu P, et al. Lgr5-mediated p53 Repression through PDCD5 leads to doxorubicin resistance in hepatocellular carcinoma. Theranostics. 2019;9(10):2967–2983. doi: 10.7150/thno.30562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taube JM, Akturk G, Angelo M, Engle EL, Gnjatic S, Greenbaum S, et al. The society for immunotherapy of cancer statement on best practices for multiplex immunohistochemistry (IHC) and immunofluorescence (IF) staining and validation. J Immunother Cancer. 2020;8(1):e000155. doi: 10.1136/jitc-2019-000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perelman A, Wachtel C, Cohen M, Haupt S, Shapiro H, Tzur A. JC-1: alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 2012;3(11):e430. doi: 10.1038/cddis.2012.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang H, Wu X, Zhao G, Feng K, Ni K, Sun X. Renal clearable ultrasmall single-crystal Fe nanoparticles for highly selective and effective ferroptosis therapy and immunotherapy. J Am Chem Soc. 2021;143(38):15812–15823. doi: 10.1021/jacs.1c07471. [DOI] [PubMed] [Google Scholar]
- 32.Tao L, Yi Y, Chen Y, Zhang H, Orning P, Lien E, et al. RIP1 kinase activity promotes steatohepatitis through mediating cell death and inflammation in macrophages. Cell Death Differ. 2021;28(4):1418–1433. doi: 10.1038/s41418-020-00668-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116(2):264–278. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 34.Wu D, Jian C, Peng Q, Hou T, Wu K, Shang B, et al. Prohibitin 2 deficiency impairs cardiac fatty acid oxidation and causes heart failure. Cell Death Dis. 2020;11(3):181. doi: 10.1038/s41419-020-2374-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Del Dotto V, Mishra P, Vidoni S, Fogazza M, Maresca A, Caporali L, et al. OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep. 2017;19(12):2557–2571. doi: 10.1016/j.celrep.2017.05.073. [DOI] [PubMed] [Google Scholar]
- 36.Rawshani A, Rawshani A, Franzen S, Eliasson B, Svensson AM, Miftaraj M, et al. Mortality and cardiovascular disease in type 1 and type 2 diabetes. N Engl J Med. 2017;376(15):1407–1418. doi: 10.1056/NEJMoa1608664. [DOI] [PubMed] [Google Scholar]
- 37.Jin L, Geng L, Ying L, Shu L, Ye K, Yang R, et al. FGF21-Sirtuin 3 axis confers the protective effects of exercise against diabetic cardiomyopathy by governing mitochondrial integrity. Circulation. 2022;146(20):1537–1557. doi: 10.1161/CIRCULATIONAHA.122.059631. [DOI] [PubMed] [Google Scholar]
- 38.Li X, Hong Y, He H, Jiang G, You W, Liang X, et al. FGF21 mediates mesenchymal stem cell senescence via regulation of mitochondrial dynamics. Oxid Med Cell Longev. 2019;2019:4915149. doi: 10.1155/2019/4915149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11(2):85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cook KS, Min HY, Johnson D, Chaplinsky RJ, Flier JS, Hunt CR, et al. Adipsin: a circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science. 1987;237(4813):402–405. doi: 10.1126/science.3299705. [DOI] [PubMed] [Google Scholar]
- 41.Ryu KY, Jeon EJ, Leem J, Park JH, Cho H. Regulation of Adipsin expression by endoplasmic reticulum stress in adipocytes. Biomolecules. 2020;10(2):314. doi: 10.3390/biom10020314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roberts NW, González-Vega M, Berhanu TK, Mull A, García J, Heydemann A. Successful metabolic adaptations leading to the prevention of high fat diet-induced murine cardiac remodeling. Cardiovasc Diabetol. 2015;14:127. doi: 10.1186/s12933-015-0286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, et al. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci. 2000;97(4):1784–1789. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fragasso G, Palloshi A, Puccetti P, Silipigni C, Rossodivita A, Pala M, et al. A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J Am Coll Cardiol. 2006;48(5):992–998. doi: 10.1016/j.jacc.2006.03.060. [DOI] [PubMed] [Google Scholar]
- 45.Fragasso G, Perseghin G, De Cobelli F, Esposito A, Palloshi A, Lattuada G, et al. Effects of metabolic modulation by trimetazidine on left ventricular function and phosphocreatine/adenosine triphosphate ratio in patients with heart failure. Eur Heart J. 2006;27(8):942–948. doi: 10.1093/eurheartj/ehi816. [DOI] [PubMed] [Google Scholar]
- 46.Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465(7300):885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15(4):323–332. doi: 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferraro F, Steinle A, Narasimhan H, Bleilevens A, Stolzenberg PM, Braunschweig T, et al. IRAK2 downregulation in triple-negative breast cancer cells decreases cellular growth in vitro and delays tumour progression in murine models. Int J Mol Sci. 2023;24(3):2520. doi: 10.3390/ijms24032520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu J, Chen Y, Huang Q, Liu W, Ji X, Hu F, et al. IRAK2 counterbalances oncogenic Smurf1 in colon cancer cells by dictating ER stress. Cell Signal. 2018;48:69–80. doi: 10.1016/j.cellsig.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 50.Hu YH, Wang Y, Wang F, Dong YM, Jiang WL, Wang YP, et al. SPOP negatively regulates Toll-like receptor-induced inflammation by disrupting MyD88 self-association. Cell Mol Immunol. 2021;18(7):1708–1717. doi: 10.1038/s41423-020-0411-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glytsou C, Calvo E, Cogliati S, Mehrotra A, Anastasia I, Rigoni G, et al. Optic atrophy 1 is epistatic to the core MICOS component MIC60 in mitochondrial cristae shape control. Cell Rep. 2016;17(11):3024–3034. doi: 10.1016/j.celrep.2016.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puri K, Lal N, Shang R, Ghosh S, Flibotte S, Dyer R, et al. Diabetes mellitus severity and a switch from using lipoprotein lipase to adipose-derived fatty acid results in a cardiac metabolic signature that embraces cell death. J Am Heart Assoc. 2019;8(21):e014022. doi: 10.1161/JAHA.119.014022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1 siRNA sequences for Irak2knockdown. Table S2 Primer sequences for real-time quantitative-PCR. Table S3 Primary antibodies used for Western blotting, immunoprecipitation, and immunohistochemistry studies. Table S4 Blood glucose and body weight of experimental animals (mean ± SD, n = 9). Table S5 Echocardiographic metrics of experimental animals (mean ± SD, n = 9). Table S6 Mass spectrum analysis of GST-Adipsin-interacting proteins. Fig. S1 Evaluation of Adipsin expression in myocardium and validation of successful construction of Adipsin-Tg mice. Fig. S2 Adipsin overexpression improves mitochondrial function in the hearts of DCM. Fig. S3 Interaction between Adipsin and Irak2 following administration of Ad-Adipsin in cardiomyocytes. Fig. S4 Co-IP detected the interaction between Irak2 and Phb-Opa1 in myocardial mitochondria. Fig. S5 Construction of AAV9-shIrak2 and verification of transfection effectiveness. Fig. S6 Impact of Irak2 knockdown on Adipsin-Tg induced protective effects of mitochondrial morphology and function.
Data Availability Statement
The datasets analysed during the current study are available from the corresponding author on reasonable request.