

SUMMARY
Fully understanding Autism Spectrum Disorder (ASD) genetics requires whole-genome sequencing (WGS). We present the latest release of the Autism Speaks MSSNG resource, which includes WGS data from 5,100 individuals with ASD and 6,212 non-ASD parents and siblings (total n=11,312). Examining a wide variety of genetic variants in MSSNG and the Simons Simplex Collection (SSC; n=9,205), we identified ASD-associated rare variants in 718/5,100 individuals with ASD from MSSNG (14.1%) and 350/2,419 from SSC (14.5%). Considering genomic architecture, 52% were nuclear sequence-level variants, 46% were nuclear structural variants (including copy number variants, inversions, large insertions, uniparental isodisomies, and tandem repeat expansions), and 2% were mitochondrial variants. Our study provides a guidebook for exploring genotype-phenotype correlations in families who carry ASD-associated rare variants and serves as an entry point to the expanded studies required to dissect the etiology in the ~85% of the ASD population that remain idiopathic.
Keywords: Autism Spectrum Disorder, neurodevelopmental disorders, whole-genome sequencing, copy number variation, structural variation, rare variants, polygenic risk scores, phenotype measures
In Brief
The latest release of the Autism Speaks MSSNG resource provides an expanded sample size and facilitates the comprehensive examination of the roles of many types of genetic variation in Autism Spectrum Disorder.
Graphical Abstract
INTRODUCTION
Autism Spectrum Disorder (ASD) is a neurodevelopmental condition whose core symptoms are social and communication difficulties, repetitive behaviors, and a restricted set of interests1,2. ASD is observed in ~1–2% of individuals and is about four times more common in males than females3,4. ASD is clinically heterogeneous, with some individuals exhibiting mild challenges and others experiencing severe symptoms and a range of co-occurring physical and mental health conditions5. Twin studies have estimated its heritability to be 64–91%6. ASD is also genetically heterogeneous, with a multitude of genes implicated7–10. Rare or de novo high-impact genetic variants are typically identified in 5–20% of individuals with ASD, and more often in those with complex medical presentations11,12. Different perspectives exist in the ASD community regarding the preferred language to refer to individuals with ASD, or autistic people; here we use the former term, although we recognize other preferences13–15. Previous whole-genome sequencing (WGS) studies have examined the contributions of different classes of variants (Table S1A); however, they typically analyzed only one or a few classes at a time and thus incompletely capture the genomic architecture of ASD.
This study had two objectives. The first was to introduce a substantial update to the Autism Speaks MSSNG resource (Figure 1). Compared to the previous release7, MSSNG now contains WGS data from twice as many individuals with ASD (5,100 versus 2,613) and total individuals (including family members; 11,312 versus 5,152), expanded genetic variant data, a redesigned web portal allowing the exploration of genotype and phenotype data, integration with the Terra cloud platform (https://terra.bio), and other enhancements (Table 1). Our second objective was to leverage MSSNG’s WGS data to comprehensively examine the roles of many types of genetic variation in ASD, including common single nucleotide polymorphisms (SNPs), as well as rare and de novo single nucleotide variants (SNVs), short insertions/deletions (indels), mitochondrial DNA (mtDNA) variants, and structural variants (SVs, including copy number variants (CNVs), inversions, larger insertions, uniparental disomies (UPDs), and tandem repeat expansions (TREs)). These analyses encompass both coding and non-coding regions and both dominant and recessive modes of inheritance. We also used WGS data from 9,205 individuals from the Simons Simplex Collection (SSC)16 for replication and 2,504 unrelated population controls from the 1000 Genomes Project (1000G)17, for a total of >23,000 WGS samples analyzed (Table S1B).
Figure 1. Data Processing and Analysis Workflow.
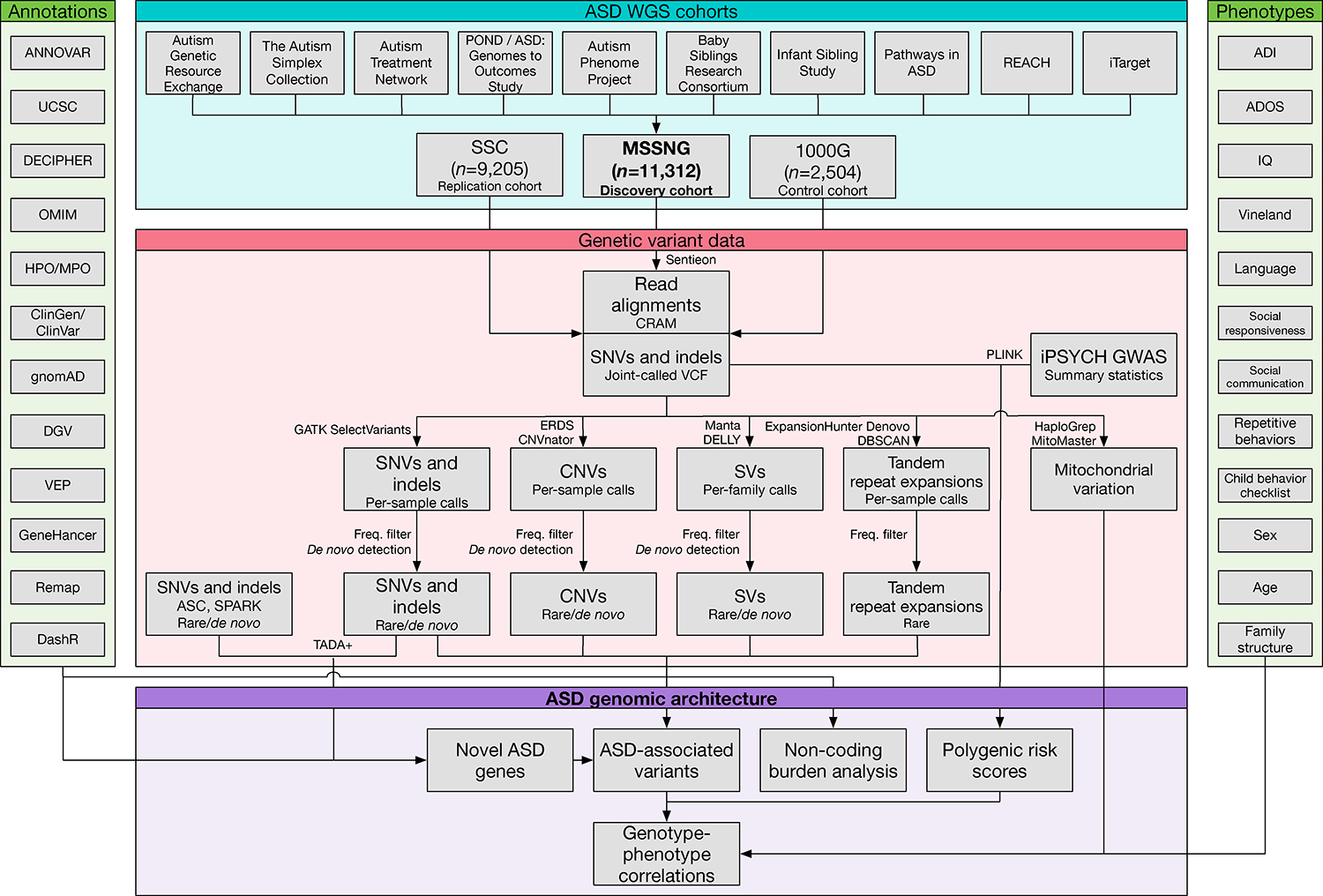
The Genetic Variant Data section applies to all samples except 1,738 sequenced by Complete Genomics, which used proprietary software. Although CNVs and TREs are types of SVs, they are shown separately because different methods were used for their detection. CNVs include deletions and duplications ≥1 kb, while SVs include deletions, duplications, insertions, and inversions ≥50 bp.
Table 1. Comparison Between the Previous Version of MSSNG7 and the Current Release.
Multiplex families are defined as those with two or more children with ASD and do not include families having non-sibling members with ASD.
| Previous version | Current version | |
|---|---|---|
| Participants | ||
| Total individuals | 5,152 | 11,312 |
| Individuals with ASD | 2,613 | 5,100 |
| Families | 2,063 | 4,258 |
| Multiplex families | 489 | 777 |
| Families having non-sibling members with ASD | 1 | 10 |
| Genetic variant data | ||
| Reference genome | GRCh37 | GRCh38 |
| Single-nucleotide variants and indels | Single sample | Joint-called* |
| Copy number variants | Not available | Available |
| Structural variants | Not available | Available |
| Tandem repeat expansions | Not available | Available* |
| Polygenic risk scores | Not available | Available* |
| Phenotype data | ||
| Consensus phenotype measures | Not available | Available |
| Data access | ||
| Phenotype Data Explorer | Not available | Available |
| Terra integration | Not available | Available |
| Researchers approved for access | ||
| Researchers | 75 | 342 |
| Institutions | 17 | 65 |
| Countries | 4 | 20 |
Only for samples sequenced on Illumina platforms.
RESULTS
Overview of MSSNG
All individuals with ASD in MSSNG meet diagnostic criteria according to the Diagnostic and Statistical Manual of Mental Disorders (DSM)1, often supported by the Autism Diagnostic Interview-Revised (ADI-R)18,19 and/or the Autism Diagnostic Observation Schedule (ADOS)20,21. MSSNG aggregates data from several cohorts and studies (Table S1C), including the Province of Ontario Neurodevelopmental Network (POND; https://pond-network.ca). Individuals of non-European ancestry comprise ~25% of MSSNG, including >2% each Admixed American, African, East Asian, and South Asian (Figure S1). For 3,565 of the individuals with ASD, genome sequences for both parents are available. MSSNG contains many multiplex (MPX) families, including 696 having two individuals with ASD and 88 with ≥3. MSSNG also includes 263 non-ASD siblings. Early samples (n=1,738) were sequenced using Complete Genomics technology22, while the rest were sequenced on Illumina platforms. Phenotype data based on 121 different tests are available (Table S1D).
MSSNG access is controlled by a Data Access Committee, and researchers can apply by submitting an ASD-related proposal7. As of July 2022, 342 researchers from 65 institutions in 20 countries have access, with many publishing their results (Table S1E). Data are stored on the Google Cloud Platform (GCP); large flat files (e.g., CRAM) are accessible via Cloud Storage buckets, while variant calls, annotations, sample metadata, and phenotype data are stored as BigQuery tables. The MSSNG portal (https://research.mss.ng), designed for the medical genomics community, allows variants to be queried based on sample, gene, or genomic region.
Enhancements to MSSNG
This release of MSSNG contains numerous improvements (Table 1). Reads are now aligned to GRCh38, and small variants are joint-called for improved accuracy (Illumina samples only). Also available are polygenic risk scores (PRS) and calls for many types of SVs. The MSSNG portal interface has been redesigned to accommodate the additional variant types (e.g., CNVs; Figure S2A), and the Integrative Genomics Viewer (IGV)23-based read viewer displays CNV and SV calls for the entire family being examined. The new Phenotype Data Explorer allows users to analyze data at the level of the entire dataset, subsets (e.g., stratified by sex), or specific individuals (Figure S2B). Finally, MSSNG now supports accessing data via Terra (Figure S2C).
Exploring phenotype data can be challenging for users who lack extensive knowledge of the various tests, compounded by the fact that the tests (or versions thereof) may vary depending on when and where they were administered. To address these challenges, we developed several consensus phenotype measures, each encapsulating multiple data points into one easy-to-understand measure. For example, the Global Ability Consensus Estimate is calculated based on several measures related to IQ, verbal and nonverbal ability, and motor skills. Other consensus measures include the Adaptive Behaviour Standard Score, Socialization Standard Score, Full Scale IQ, and measures for common co-occurring conditions.
Discovery of ASD-associated genes
Previously, the Autism Sequencing Consortium (ASC) developed TADA+, an enhanced version of the transmission and de novo association (TADA) test24, and applied it to whole-exome sequencing (WES) data from 6,430 trios, 5,556 cases, and 8,809 controls, identifying 102 ASD-associated genes with false discovery rate (FDR) <0.18. To increase power, here we incorporated de novo variants from 12,375 additional trios from MSSNG and the Simons Foundation Powering Autism Research (SPARK)25,26 WES cohort (Table S2A–C). The number of exonic de novo variants per child were similar for ASC, MSSNG, and SPARK (Figure 2A). After incorporating the additional trios, we detected 134 ASD-associated genes with FDR <0.1 (Table S2D). Of these, 67 were identified by the previous TADA+ analysis, 67 were new, and 35 from the previous analysis no longer met the FDR threshold (Figure 2B, Table S2E). Many of the new genes (27/67) are not currently in the Simons Foundation Autism Research Initiative (SFARI) Gene database27, providing novel molecules for study (Figure 2C–D). For most of the new genes, the evidence constituted a mix of de novo protein-truncating variants (PTVs), de novo damaging missense (DMis) variants, and excess PTVs in cases compared with controls (Figure 2E). However, for some genes the evidence consisted exclusively of PTVs (e.g., MED13, TANC2, DMWD) or de novo DMis variants (e.g., ATP2B2, DMPK, PAPOLG), providing insight into potential molecular mechanisms (e.g., haploinsufficiency for PTV-biased genes and gain-of-function or dominant-negative mechanisms for DMis-biased genes). Most new genes had high pLI scores (Figure 2F), suggesting haploinsufficiency as a common mechanism. To determine the contribution of MSSNG, we repeated this analysis using only ASC and SPARK data, which gave 120 ASD-associated genes (23 lost and nine gained relative to the full list) (Table S2D).
Figure 2. Identification of ASD-Associated Genes Using TADA+.
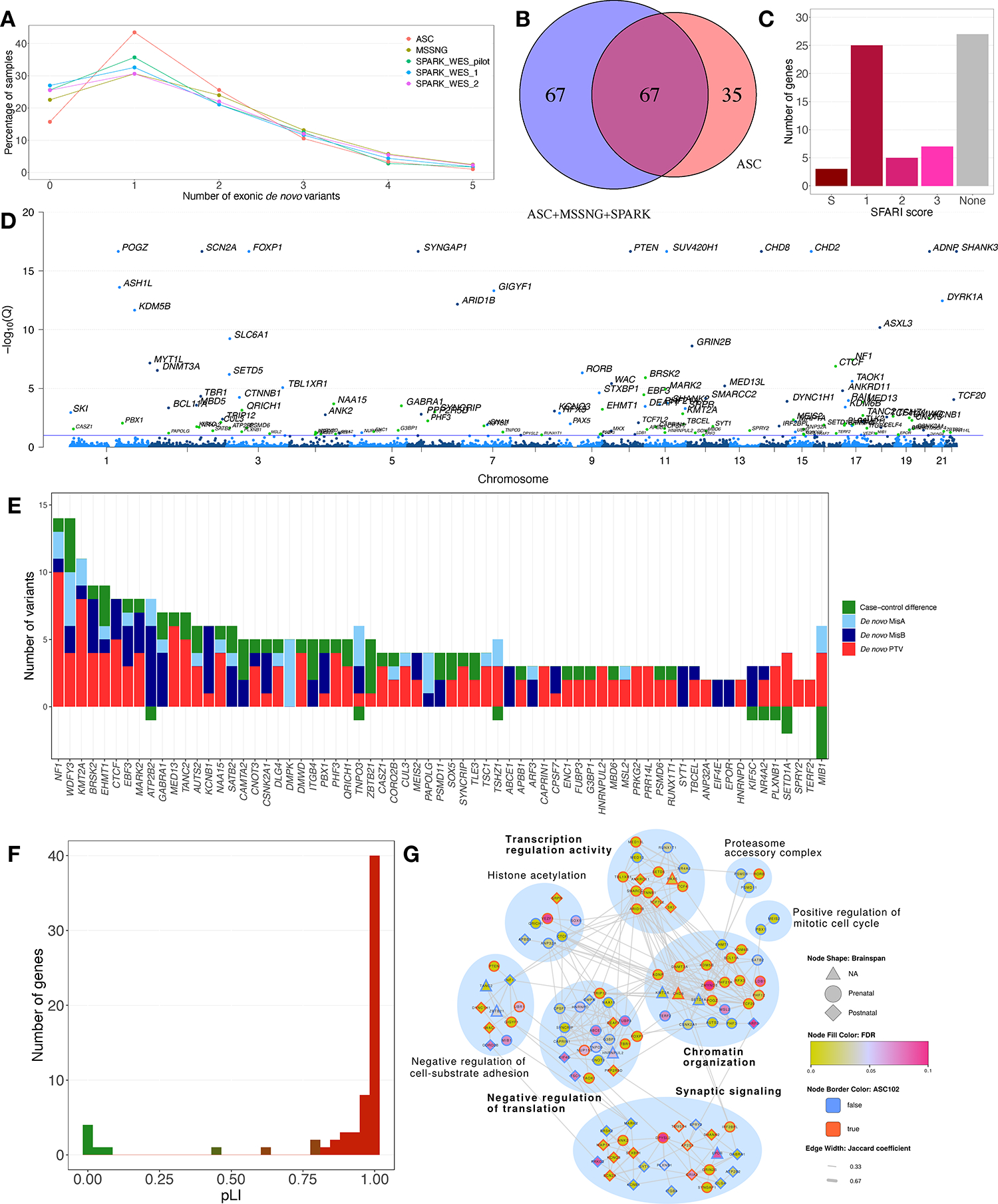
(A) Exonic de novo variants per individual in ASC, MSSNG, and SPARK. (B) ASD-associated genes discovered only in the previous TADA+ analysis8 (“ASC”), only in the current analysis (“ASC+MSSNG+SPARK”), or both. (C) Distribution of SFARI Gene scores for the newly discovered genes. (D) FDRs for the 134 ASD-associated genes. Blue dots: genes also identified in the previous TADA+ analysis; green dots: new genes. Genes with FDR calculated as zero were assigned a value of 10−17. The blue line represents FDR=0.1. (E) Evidence supporting the new ASD-associated genes. “Case-control difference” represents PTVs in cases minus controls. The y-axis is truncated at −3; the value for MIB1 is −9. (F) pLI values for the new genes. (G) Network diagram of TADA+ genes. Only genes connected to gene networks are shown. Nodes represent genes, and edges represent protein-protein and pathway interactions between gene pairs. Modules are indicated by blue circles, with module labels derived from GO term enrichment tests (bold: significantly enriched).
One new gene of interest was DMPK. RNA toxicity stemming from TREs in the 3′-UTR of DMPK cause myotonic dystrophy type I (DM1)28, and individuals with DM1 have a higher incidence of ASD29,30. In addition, we recently identified DMPK as one of the top candidate genes for association of TREs with ASD31. Here, five de novo DMis variants were detected in DMPK (Table S2D), suggesting that they may represent another mechanism by which this gene contributes to ASD susceptibility. Another interesting ASD-candidate gene was GABRA1, which encodes the most abundant α subunit of the GABAA receptor, which mediates fast inhibitory neurotransmission32. Synaptic protein dysregulation and other alterations in the GABAergic system, including in the GABAA receptors, have been implicated in ASD33,34. Further, mice treated with valproic acid, one of the few suspected environmental susceptibility factors for ASD35, exhibit ASD-like phenotypes and significantly decreased GABRA1 expression36.
Several new ASD-associated genes were located within CNV regions previously associated with neurodevelopmental disorders (NDDs). For example, PRKG2, HNRNPD, and HNRDPL are three of the five genes in the critical region for 4q21 microdeletion syndrome, which is associated with intellectual disability (ID) and impaired speech37,38. TSHZ1, although primarily associated with a non-NDD phenotype, is in the 18q deletion region, which is characterized by ID and ASD39. Other TADA+ genes within ASD-associated CNV regions included CASZ1 (1p36), TBCEL (11q23.3), PSMD11 (17q11.2), RUNX1T1 (8q21.3), ABCE1 (4q31.21-q31.22), and PAPOLG (2p16.1-p15), potentially pinpointing which genes in these regions contribute to their associated NDDs.
One of the highest-confidence genes from the original TADA+ analysis to “drop out” was NRXN1. In ASC, NRXN1 had three de novo PTVs, one de novo DMis variant, and one PTV in a case (versus none in controls). However, zero de novo PTVs or DMis variants were found in NRXN1 in MSSNG or SPARK. This reflects the limitation that TADA+ considers only sequence-level variants. Previous evidence for the involvement of NRXN1 in ASD and other NDDs has largely been from deletions40,41. NRXN1 was also identified in our recent study of TREs in ASD31, underscoring the idea that all types of genetic variation must be considered, as is the case when using the EAGLE protocol for scoring ASD-relevant genes42.
Of the 134 genes, 106 were involved in gene networks. Based on gene connectivity, we classified the genes into eight functional modules (Figure 2G). The four largest modules were significantly enriched in the Gene Ontology (GO) terms synaptic signaling, chromatin organization, transcription co-regulator activity, and negative regulation of translation (Table S2F); 14/21 genes in the latter module were newly identified. Permutation tests showed that the connectivity of all pairs of genes in our network were greater than expected at random (Table S2G). To assess the specific contribution of MSSNG, we generated a network using the TADA+ genes derived from only ASC and SPARK. The resulting network was similar, except that the negative regulation of transcription module was no longer significant (Table S2F).
Detecting new ASD-associated genes may facilitate the identification of links with other disorders. While the overlap between ASD and attention deficit/hyperactivity disorder (ADHD), obsessive-compulsive disorder (OCD), and ID are well-established43,44, links between ASD and other disorders are less clear. Interestingly, several new ASD-associated genes have tentative links with neurodegenerative disorders, such as Alzheimer’s disease, including NR4A2, ATP2B2, EIF4E, EBF3, MARK2, EPOR, G3BP1, PLXNB1, APBB1, and ANP32A. ASD and neurodegenerative diseases have common clinical features, including language, executive function, and motor impairments45, and share some molecular pathomechanisms (e.g., synaptic deregulation).
Recessive events
To identify potential recessive events, we searched for homozygous and compound heterozygous (CH) PTVs and DMis variants. Only biallelic events with PTVs on both alleles (PTV-PTV events) were enriched in ASD (see subsequent section “Genomic architecture of rare coding variants in ASD”), so we focused on PTV-PTV events. A total of 198 genes harbored ≥ 1 PTV-PTV event and 33 had them in ≥ 2 unrelated individuals with ASD. In six multiplex families from MSSNG, two siblings with ASD shared the same PTV-PTV event (2 homozygous and 4 CH) in 6 distinct genes (CASP5, DNAH14, DCHS2, C1orf229, DZANK1, and SMTNL1). Interestingly, PTV-PTV events in DNAH14 were found in another six individuals with ASD. DNAH14 encodes axonemal dyneins, which are microtubule-associated motor protein complexes, and a recent study reported three unrelated individuals with NDDs having CH events in DNAH1446. Ten genes with PTV-PTV events overlapped genes from the curated Genomics England neurology and neurodevelopmental disorders panel classified as having biallelic modes of inheritance (ABCC6, BCAS3, CCDC40, DEAF1, GPR179, MMACHC, OBSCN, PRSS12, SORD, and TELO2). DEAF1 was also identified in the TADA+ analysis, suggesting that it may confer ASD susceptibility in a dose-dependent manner.
Structural variation
We used two pipelines to detect SVs: a read depth-based workflow47, which detects CNVs ≥1 kb, and another employing split read and paired-end mapping-based algorithms, which detects deletions, duplications, insertions, and inversions ≥50 bp. We identified rare SVs falling into five categories: chromosomal abnormalities, genomic disorders, large or gene-rich CNVs not overlapping known genomic disorder regions, UPDs, and smaller SVs disrupting ASD/NDD genes (including the 134 TADA+ genes). Pathogenic SVs were detected in 6% of individuals with ASD. Most (96%) were deletions or duplications ≥1 kb; we found comparatively few pathogenic deletions or duplications <1 kb, large insertions, and inversions.
The WGS data also allowed deeper investigation of inversions and large insertions, and here we highlight two examples of such pathogenic variants. The first is a 71 bp de novo frameshift insertion in SYNGAP1 comprised of 65 bp of mtDNA from MT-CO3 and a 6 bp microduplication (Figure 3A–C). The insertion affects all known splice variants of SYNGAP1 and to our knowledge is the first pathogenic mtDNA insertion reported in ASD. The second is a 13,472 bp inversion of unknown inheritance (parental samples unavailable) affecting SCN2A (Figure 3D–E).
Figure 3. Examples of Pathogenic SVs.
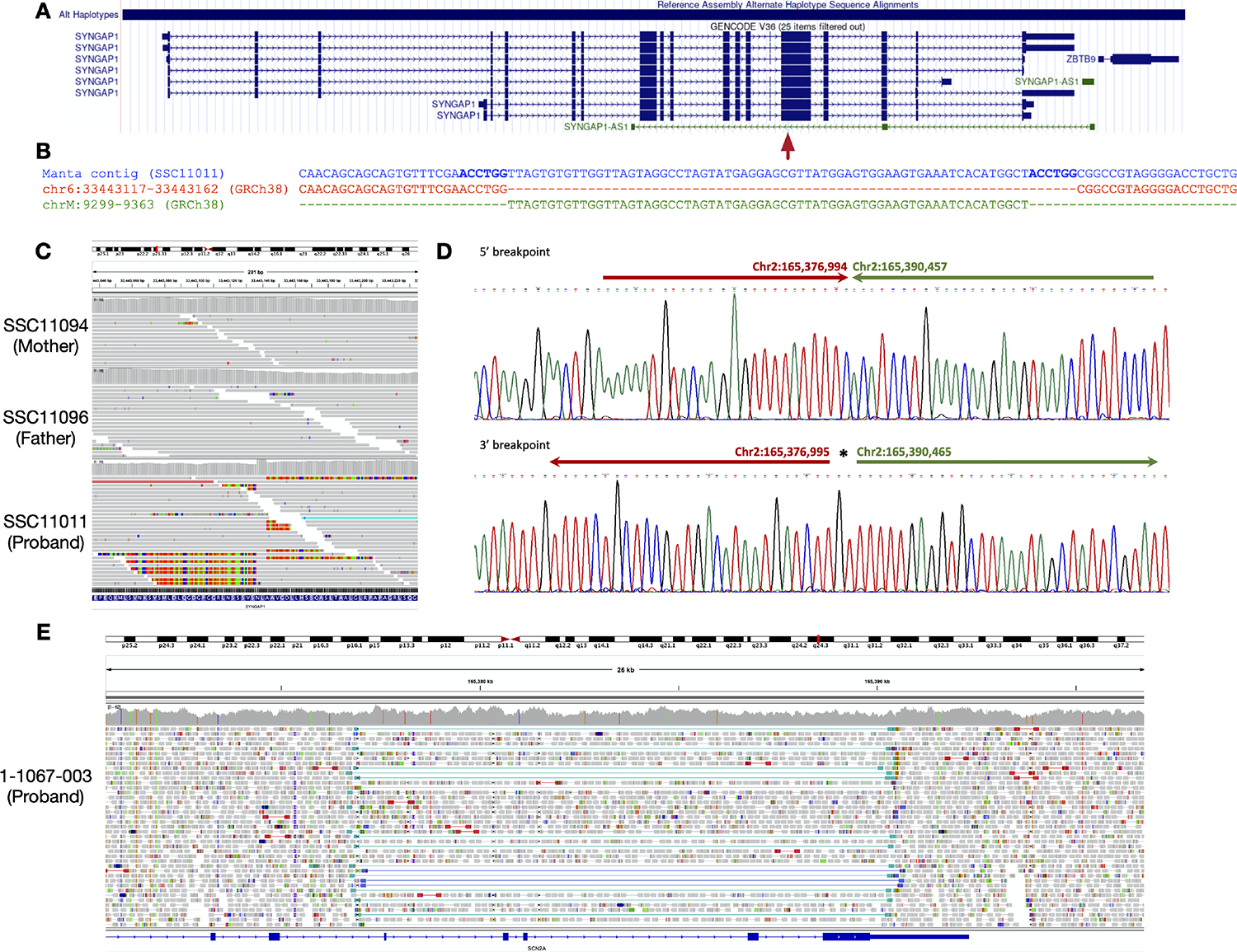
(A-C) 71 bp de novo frameshift insertion in SYNGAP1 comprising 65 bp of mtDNA and a 6 bp microduplication. (A) Schematic of SYNGAP1, with the insertion indicated by a red arrow. (B) Alignment of the insertion-containing contig sequence assembled by Manta, the reference sequence, and the mtDNA sequence inserted into chromosome 6. The microduplication is indicated in bold. (C) IGV visualization. The colored portions of reads represent mismatched bases, allowing precise breakpoint identification. The read depth increase reflects the 6 bp microduplication. (D-E) 13.4 kb inversion overlapping SCN2A. (D) Sequence trace showing the 5’ and 3’ breakpoints. (E) IGV visualization. The dark and light blue lines indicate anomalously mapped read pairs exhibiting the signature of an inversion.
Isodisomic UPD (isoUPD) results in homozygosity spanning some (partial) or all (complete) of a chromosome, potentially unmasking recessive variants. We identified two partial and three complete isoUPDs in individuals with ASD, with none in non-ASD siblings. None were within known imprinted domains. We lack platform-matched data to determine if this UPD rate is higher than in the general population; however, an increase was observed compared to a population study of 23andMe data48 (ASD: 5/15,038 child-parent duos [0.033%]; population: 129/916,712 duos [0.014%]; Fisher’s Exact Test: odds ratio (OR)=2.4, p=0.07). No heterodisomies were detected.
To demonstrate how WGS can aid interpretation of copy number gains, we resolved breakpoint junctions of duplications impacting ASD candidate genes in a subset of individuals with ASD in MSSNG. This process is largely manual, so we selected a subset of duplications and showed how their impact on gene structure and function can be evaluated by visualizing reads in IGV (STAR Methods). By fine-mapping the breakpoints of 375 duplications identified by read depth-based algorithms in 332 individuals, we identified 248 unique duplications ranging from 4.3 kb to 6.5 Mb (Table S3A). Most events remained at or near their locus of origin, including tandem duplications (192/248), likely non-allelic homologous recombination (NAHR)-mediated events (6/248), and complex duplications involving sequence transposition <100 kb from their locus of origin or inverted triplications without sequence transposition (14/248). Some complex duplications involved large-scale (>100 kb) transpositions (14/248) or interchromosomal transpositions (2/248), and 20/258 remained unresolved. Several duplications of uncertain clinical significance increased dosage of NDD genes, including de novo tandem events at USP7 (OMIM: 602519) and MEF2C (OMIM: 600662).
Mitochondrial variation
We evaluated pathogenic variants, haplogroups, and heteroplasmy (variants present in only some copies of an individual’s mtDNA genome) to study their association with ASD (STAR Methods). We identified 23 instances of known pathogenic variants with >5% heteroplasmy in individuals with ASD (Table S4A). Of these, 17 were de novo, defined as when maternal heteroplasmy was undetectable or <5%. The highest de novo heteroplasmy value was a 47% load of the m.13513G>A variant associated with Leigh Disease. The frequency of newly observed pathogenic heteroplasmies >5% in individuals with ASD (17/6,044) was significantly greater than the frequency of pathogenic heteroplasmies in mothers only (0/5,320) or fathers only (4/5,295) (χ2 test: p=6 × 10−5). Two pathogenic heteroplasmies were identified in individuals without maternal sequencing data, so inheritance status could not be determined. We also identified four families for which a pathogenic variant was present at >5% heteroplasmy in both a mother and her child with ASD. In one, the child had a 49% load of the m.3243A>G variant associated with mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS) (maternal load: 12%). Both mother and child had clinical symptoms consistent with MELAS. The average intergenerational change in heteroplasmy for pathogenic variants was an 11% shift toward the pathogenic allele in children with ASD. We also evaluated mtDNA variants causing homoplasmic disorders generally affecting vision and hearing only but found no association with ASD (Table S4B).
Certain macrohaplogroups have previously been associated with ASD susceptibility49. However, we observed no significant difference in haplogroup distribution when comparing individuals with ASD with their fathers (n=4,821 duos) (Table S4C).
Finally, we evaluated the inheritance of heteroplasmy at all mtDNA positions. A prior study suggested increased transmission of heteroplasmy to children with ASD50. However, we found no significant difference between the shifts from mother to children with ASD and the shifts from mother to non-ASD siblings in either MSSNG or SSC (Figure S3).
Genomic architecture of rare coding variants in ASD
We integrated the data above to give both high-level and detailed views of the genomic architecture of rare coding variants in ASD. We begin at a high level by comparing the burden of different variant types in individuals with ASD from either MSSNG or SSC with non-ASD siblings from SSC. De novo PTVs in constrained genes (gnomAD loss-of-function observed/expected upper bound fraction (LOEUF) <0.35) were significantly enriched in individuals with ASD, as were de novo DMis variants (Figure 4A). We observed no significant enrichment in inherited PTVs in constrained genes, but they were enriched in the 134 TADA+ genes for individuals with ASD in MSSNG, suggesting that PTVs in some TADA+ genes may have incomplete penetrance. No enrichment of inherited DMis variants was observed. Consistent with previous findings51, PTV-PTV biallelic events were significantly enriched in individuals with ASD, although we did not observe enrichment for events involving DMis variants (Figure 4A). Significant enrichment in ASD was observed for nearly all categories of SVs (Table S5A), including TREs31.
Figure 4. Genomic Architecture of Rare Variants in ASD.
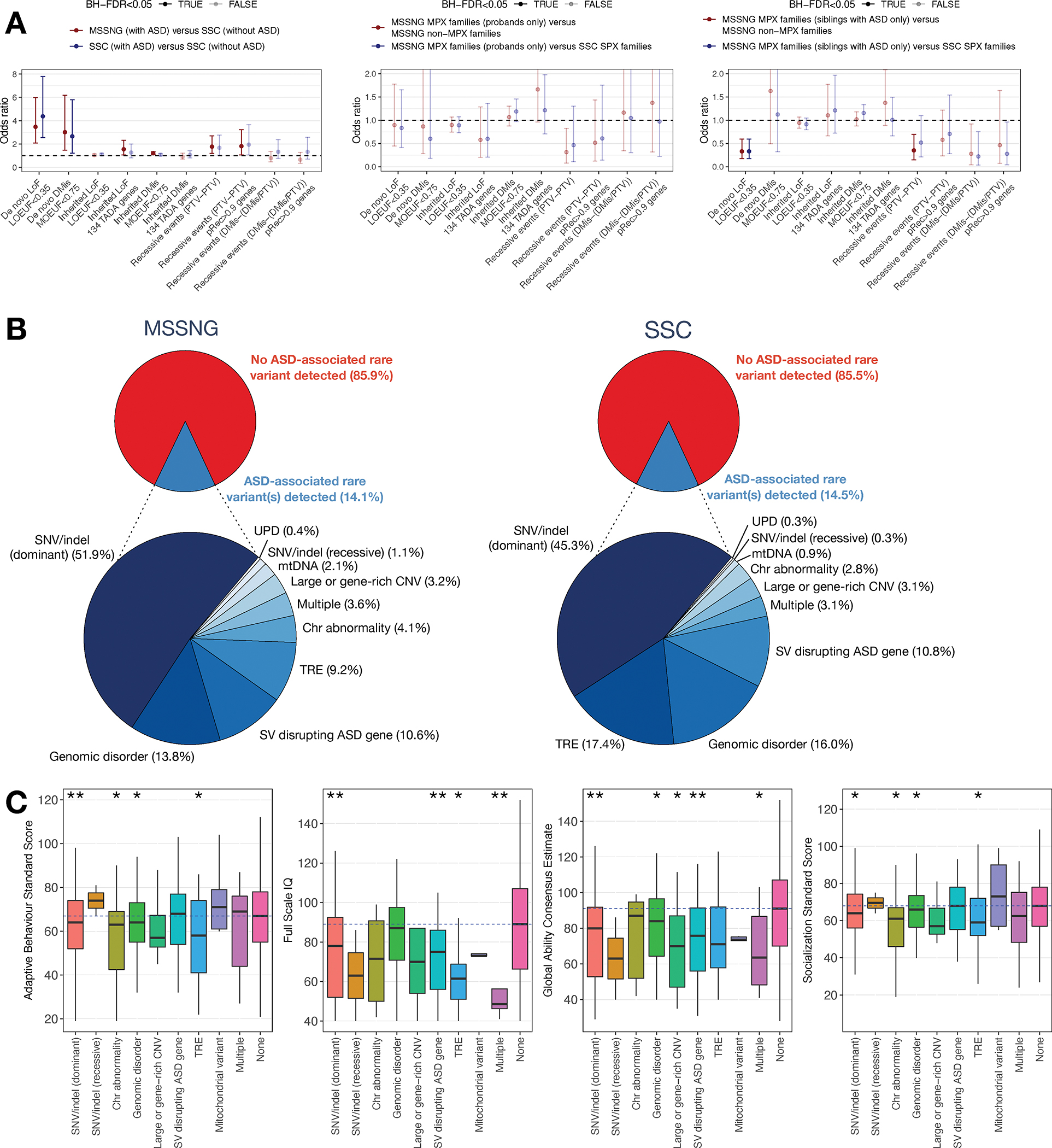
(A) Burden analysis of sequence-level rare coding variants. Left, individuals with ASD versus non-ASD siblings. Middle, probands from MSSNG MPX families versus those from either SSC SPX families (having exactly one individual with ASD and at least one sibling without) or MSSNG non-MPX families (having exactly one individual with ASD and no siblings). Right, same as middle except siblings with ASD instead of probands. Burdens for other rare ASD-associated variants are given in Table S5A. Compared with sequence-level variants, many SV types had very high ORs, including chromosomal abnormalities (OR=4.9), genomic disorders (OR=8.3), and SVs impacting ASD genes (OR=24) (comparisons between MSSNG individuals with ASD and SSC non-ASD siblings). (B) Yield of ASD-associated rare variants (top) and stratified by variant type (bottom). “Multiple” indicates individuals with ASD-associated variants in more than one category. (C) Distributions of consensus phenotype measures for individuals with ASD having each type of ASD-associated rare variant, or no variant. *Nominally significant (p<0.05); **: significant after multiple testing correction.
Next, we performed burden comparisons in MPX families (where inherited variation may have a larger role) versus simplex (SPX) (de novo variation). As expected, de novo PTVs in constrained genes were significantly depleted in MPX families (Table S5A). However, much of this signal came from siblings with ASD in MPX families rather than probands (the first child in the family to be diagnosed with ASD) (Figure 4B–C). This may reflect an ascertainment bias in which families having one child with ASD are more likely to have subsequent children evaluated and diagnosed52. We also observed a non-significant depletion of recessive events in MPX families. In some MPX families, individuals with ASD shared an ASD-associated rare variant, and in others, they had different variants (Figure S4), consistent with our previous findings53.
Overall, ASD-associated rare variants were detected in ~14% of individuals with ASD, with the yield similar in MSSNG and SSC (Figure 4B). The largest contributor was dominant (where only one copy of the gene must be affected) sequence-level variants (51%). We observed no significant difference between ancestry groups in the overall prevalence of ASD-associated rare variants (Table S5B). Testing whether any class of rare variant differed in prevalence among groups, we detected an enrichment of TREs in individuals of African descent (OR=5.0, FDR=0.0004) (Table S5C).
To examine genotype-phenotype associations, we compared the distributions of four consensus phenotype measures—Adaptive Behavior Standard Score, Full Scale IQ, Global Ability Consensus Estimate, and Socialization Standard Score—in individuals with ASD from MSSNG having each type of ASD-associated rare variant versus those with no such variant. Nearly all categories of ASD-associated rare variants were associated with lower scores for all four measures (Figure 4C), although an important caveat is that the four measures are correlated (Table S5D). Using logistic regression, dominant SNVs/indels were significantly associated with lower scores across all four measures, and TREs for all measures except the Global Ability Consensus Estimate. Some variant types, such as large or gene-rich CNVs, were consistently associated with lower scores but were not significant, possibly due to lack of power.
To give a detailed view of genomic architecture, we enumerated the genes and regions impacted (Figure 5 and Table S5E–M). Of the genes in the TADA+ list or that were ranked definitive by EAGLE curation42, those most frequently affected by dominant PTVs (de novo and inherited) and DMis variants (de novo only) in individuals with ASD included PTEN, KDM5B, MIB1, and CHD8. In addition to trisomy 21 and sex chromosome aneuploidies, several other chromosomal abnormalities were observed, including multiple translocations. The most frequently detected genomic disorders included CNVs at 16p11.2, 1q21.1, 15q11-q13, and 22q11.21. A variety of large or gene-rich CNVs not overlapping canonical genomic disorder regions were identified, including two each at 8p23.3-p23.1, 5p15.31-p15.2, and 2q23.3-q24.1. ASD-associated genes most affected by SVs included NRXN1, PTCHD1-AS, and AUTS2. Top genes for TREs were described previously31 and are recapitulated in Figure 5. Finally, the mtDNA genes most frequently affected by pathogenic variants were MT-TL1 (MELAS) and MT-ND6 (Leigh Disease).
Figure 5. Detailed View of the Genomic Architecture of Rare Variants in ASD.
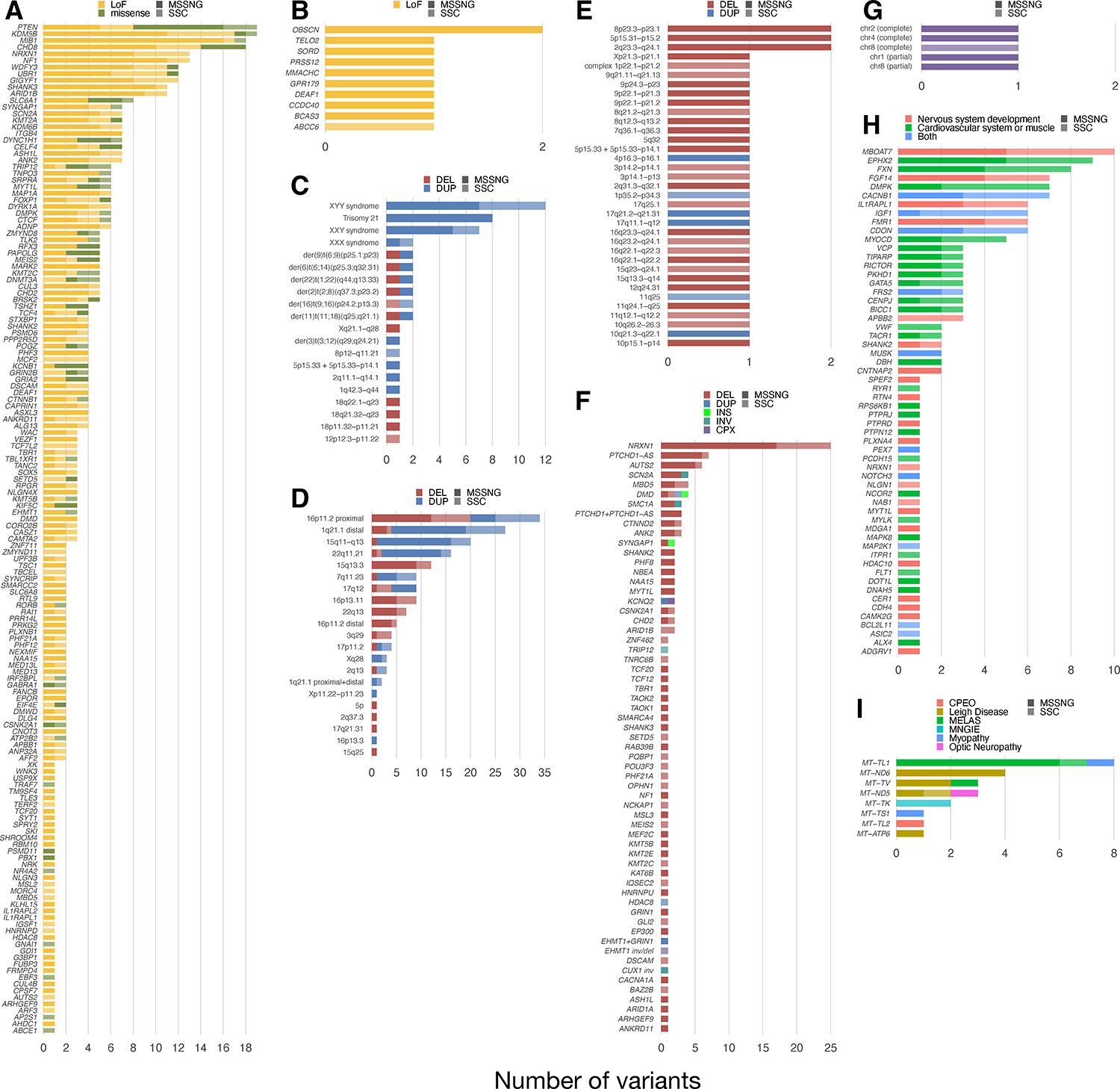
(A) De novo or rare inherited (gnomAD allele frequency <10−4) PTVs and de novo DMis variants in autosomal genes identified by the TADA+ analysis or evaluated as definitive by EAGLE42. For X-linked genes, we identified hemizygous PTVs in males with frequency <10−4 in constrained genes (pLI > 0.95) found in the Genomics England neurology and neurodevelopmental disorders panel. (B) Recessive events (PTV-PTV events in genes from the Genomics England panel). (C) Chromosomal abnormalities. (D) Genomic disorders. (E) Large or gene-rich CNVs other than genomic disorders. (F) SVs disrupting ASD-associated genes. (G) UPDs. (H) TREs identified previously31. (I) Pathogenic mtDNA variants. CPEO, chronic progressive external ophthalmoplegia; MNGIE, mitochondrial neurogastrointestinal encephalomyopathy.
Non-coding variants
We annotated rare sequence-level variants according to their impact on enhancers, promoters, topologically associating domains (TADs), and other regulatory elements (STAR Methods). We first performed transmission bias tests to compare the number of transmitted versus non-transmitted singleton variants (private to a particular family) impacting each non-coding element. The underlying hypothesis is that variants affecting elements related to ASD susceptibility will be over-transmitted from parents to individuals with ASD. After correcting for multiple testing, little enrichment was observed; however, in MSSNG, variants predicted to damage promoters were over-transmitted to individuals with ASD (Figure S5). In SSC, no over-transmission was observed in individuals with ASD or in non-ASD siblings. Similar results were obtained with variants having frequency <0.1% (Table S6A).
We also performed burden tests, in which the number of variants in a non-coding element were compared between individuals with ASD and non-ASD siblings. After correcting for multiple tests, no annotation classes were significantly enriched in ASD for rare, singleton, or de novo variants (Table S6B).
Polygenic risk scores
Much of our knowledge of ASD genetics is from studies of rare-inherited and de novo gene-disrupting variants. However, common variation also contributes to ASD heritability54,55. Here, we used summary statistics from a recent ASD genome-wide association study (GWAS)56 to calculate PRS in MSSNG, SSC, and 1000G. To assess technical reproducibility, we compared PRS in 10 monozygotic twin pairs from MSSNG. Relative to the overall PRS range, all between-twin differences were small (Figure 6A). PRS distributions were similar in MSSNG, SSC, and 1000G (Figure 6B). Using individuals with ASD from MSSNG and SSC as cases and 1000G as controls, higher PRS was weakly associated with ASD susceptibility (OR=1.03, p=2.5 × 10−3; Nagelkerke’s R2=0.0039). Adjusting for sex, individuals with ASD in both MSSNG and SSC had higher mean PRS than non-ASD siblings in SSC (Figure 6C). No significant difference in PRS was observed between individuals with ASD in MSSNG versus SSC. Next, we performed a polygenic transmission disequilibrium test (pTDT), finding that PRS was significantly over-transmitted in individuals with ASD in both SSC (consistent with previous results57) and MSSNG, but not in non-ASD siblings (Figure 6D). Stratifying by sex, PRS was significantly over-transmitted in males with ASD in both MSSNG and SSC as well as females in SSC (Figure S6B).
Figure 6. PRS Analysis.
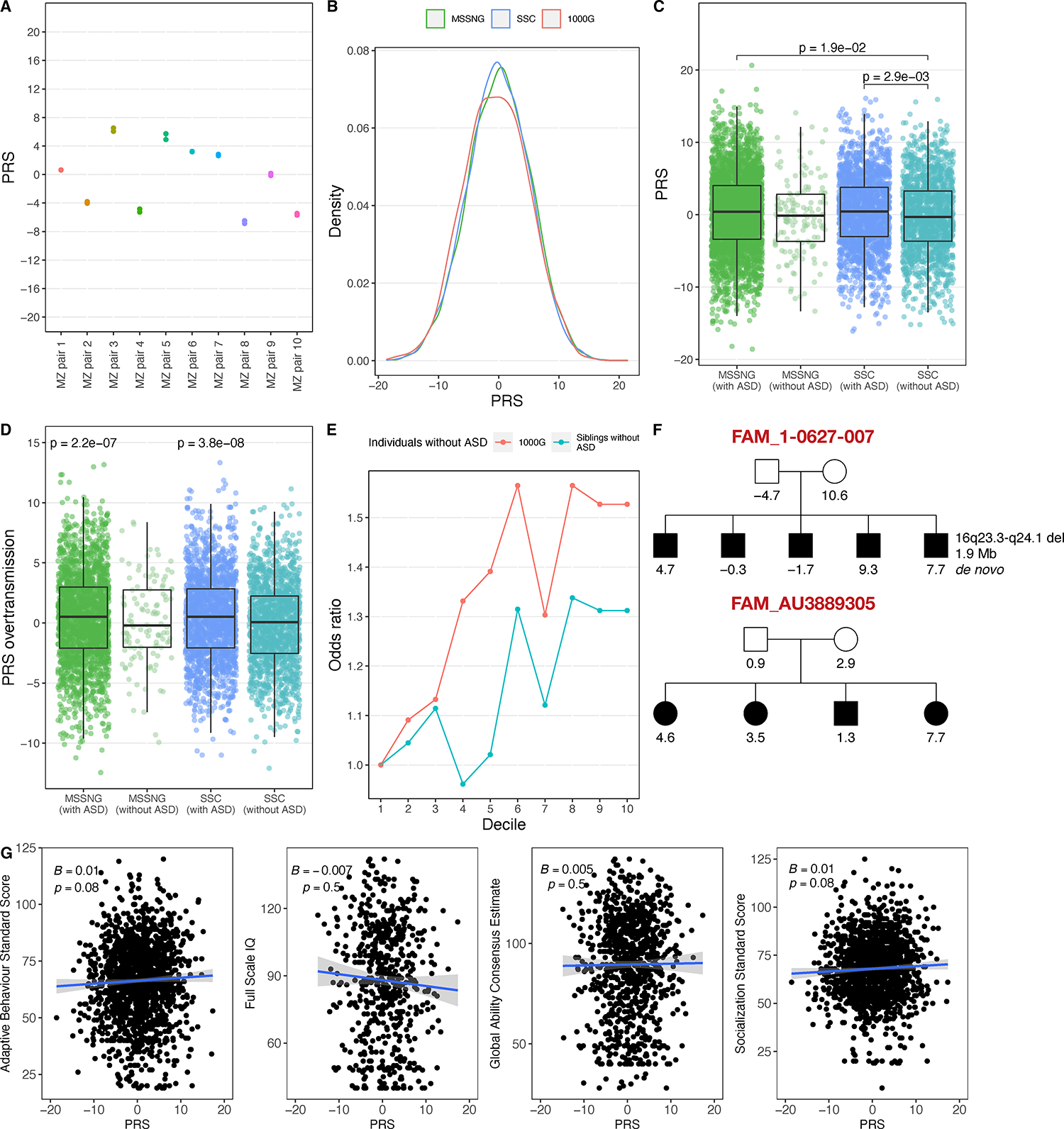
(A) Reproducibility in ten pairs of monozygotic (MZ) twins from MSSNG. (B) PRS distributions in MSSNG, SSC, and 1000G. (C) Comparison between individuals with ASD and non-ASD siblings. (D) Over-transmission of polygenic risk from parents to children. (E) Odds ratio of individuals with ASD to those without in each PRS decile, relative to decile 1, in MSSNG and SSC combined. (F) Pedigrees showing the PRS for each individual in two large MPX families. (G) Association between PRS and the four consensus phenotype measures.
As similar trends were observed in MSSNG and SSC, we combined them to explore the extremes of the PRS distribution. We partitioned the children with ASD and individuals without ASD into PRS deciles and then computed ORs relative to the lowest decile. This was done using two control sets: non-ASD siblings from MSSNG and SSC (n=666 total individuals per decile) and individuals from 1000G (n=551 total individuals per decile). The ORs for the highest PRS decile were 1.32 and 1.53, respectively, relative to the lowest decile (Figure 6E).
MSSNG contains several large MPX families, affording the unique opportunity to explore PRS at the family level. We highlight two families, one with five children with ASD (FAM_1-0627-007) and the other with four (FAM_AU3889305) (Figure 6F). Other than one child in FAM_1-0627-007 with a 1.9 Mb de novo deletion at 16q23.3-q24.1, no ASD-associated rare variants were detected in the children. In both families, the children have a wide range of PRS (FAM_1-0627-007: −1.7 to 9.3; FAM_AU3889305: 1.3 to 7.7). Scores of 7.7 and 9.3 are in the 93rd and 96th percentiles, respectively, so if these children had been the only ones in their respective families, it may have been tempting to attribute their ASD to high polygenic risk. Given the additional context afforded by the lower PRS in the other children with ASD from these families, along with the non-ASD mother having the highest PRS in FAM_1-0627-007, there does not appear to be a basis for associating the ASD in these families solely with polygenic risk.
We leveraged the family structures in MSSNG and SSC to explore additional trends related to polygenic risk. In SSC sibling pairs (one with ASD and one without) whose PRS scores differed by more than one standard deviation, the sibling with ASD had the higher score 58% of the time (binomial test: p=6.8 × 10−4), and similarly for two standard deviations (65%; p=0.01). We detected no significant difference in mean PRS in individuals with ASD from MPX families compared with SPX families (Figure S6C), suggesting that ASD in MPX families may be more attributable to rare, high-impact variants. No significant difference was observed between PRS in mothers of children with ASD versus fathers (Figure S6D). We hypothesized that individuals in 1000G may have a lower mean PRS than parents in MSSNG and SSC, but no significant difference was observed (Figure S6E). No significant association was detected between PRS and our consensus phenotype measures (Figure 6G).
ASD open science using Terra
Online data repositories have many advantages, including data persistence, format standardization, searchability, security, and version control. However, they are poorly suited to large files (e.g., CRAM) due to the time and expense required to download and store local copies of the data. Online code deposition also presents problems58, as the code often fails to reproduce the published results or cannot be executed at all59. With data storage, compute capabilities, and code in one place, cloud computing eliminates the need for each research group to store its own copy of the data and removes the disconnect between data and code60. However, using and sharing cloud-based data can be technically challenging.
Terra (https://terra.bio) is a platform for researchers to collaboratively use cloud-based data. Its fundamental unit of organization is the “workspace”, each of which can contain multiple notebooks (code interleaved with its output) and workflows (chains of programs linked together, typically for computationally intensive operations). As MSSNG data are already stored in GCP, Terra is a natural fit for exploring cloud-based ASD research. To illustrate its use, we created two Terra workspaces. The first includes eight notebooks written in Python or R that help researchers get started accessing MSSNG data via Terra. It also contains two workflows that illustrate how to run ExpansionHunter61 and ExpansionHunter Denovo62. The second contains two notebooks that generate figures associated with our non-coding and PRS analyses. Researchers can inspect the underlying data, run code on those data themselves, and modify or extend the code by cloning the workspace. All notebooks and workflows are described in more detail in Table S1F.
DISCUSSION
For the latest release of MSSNG, we generated a rich resource of genetic data, including sequence-level, structural, and mitochondrial variants, tandem repeats, and polygenic risk scores. To meet the needs of researchers with different hypotheses or expertise, this information is accessible via several interfaces. The MSSNG portal is suitable for users without programming experience or who are interested in variants from a few genes or regions. The Terra integration is aimed at users with more complex research questions. Finally, advanced users can access MSSNG via BigQuery tables or flat files in GCP.
Importantly, MSSNG’s governance places its participants at the forefront of all decision-making. Clinicians who enrol families are heavily involved in the research study, and input is regularly sought from a Participant Advisory Committee. Annual meetings are held with hundreds of participating families discussing new developments, consents are regularly updated with participant input, and the most appropriate methods for interpreting and communicating genomic findings are contemplated by multidisciplinary teams42,63,64. Research findings that meet standard clinical reporting criteria are returned to families accompanied by genetic counselling.
Our analysis of MSSNG yielded numerous findings that strengthen (or challenge) previous results or provide new insight into ASD genomic architecture. We identified 134 ASD-associated genes, as well as numerous ASD-associated variants that would have been difficult to detect without WGS. We found evidence challenging previous claims that certain mitochondrial haplogroups are associated with ASD and that heteroplasmy transmission is increased in children with ASD. No significant difference in the yield of ASD-associated rare variants among ancestry groups was observed. ASD-associated rare variation was related to lower scores in our consensus phenotype measures. We found that rare, dominant variation may play a greater role in MPX ASD, given the trend toward the depletion of rare, damaging recessive events and lack of enrichment for polygenic risk in these families. Finally, we report a comprehensive description of the contribution of different variant types to the genomic architecture of ASD, laying the groundwork for future studies into the ~85% of families who remain genetically unresolved (recognizing genetics may not be the only contributor).
One advantage of WGS compared with WES is the ability to explore non-coding variation. However, our analysis revealed limited enrichment of rare variants impacting non-coding elements in individuals with ASD (Figure S5), which is consistent with previous findings65. In contrast to previous results66, we observed no enrichment in de novo variants impacting promoter regions in SSC. Such observations highlight the challenges inherent in detecting robust and reproducible non-coding signals in a disorder as genetically heterogeneous as ASD, as different trends can be observed depending on the methodology used. They also highlight the need for even more ASD WGS data.
It has been suggested that PRS has, or will have, clinical or predictive utility for many different conditions67,68. However, our PRS analysis (in particular, the low OR and R2, the small difference in mean PRS between individuals with and without ASD, and the modest enrichment of individuals with ASD in the highest PRS deciles) suggests that ASD PRS should be interpreted with caution and currently may not be informative at the level of individuals or families. Despite its current limitations, ASD PRS may eventually add to a more complete understanding of the mix of rare and common variant contributions to ASD69,70. For instance, larger ASD GWASs, along with comparisons involving larger control cohorts such as the UK BioBank, may increase the proportion of variance explained. Further, PRS will likely become an important part of frameworks that model the multifactorial nature of ASD, such as liability threshold models55.
This paper provides a significant new resource and analysis roadmap for ASD studies as genome sequencing begins to take its place in precision medicine applications71. With increasing emphasis being placed on genotype-phenotype correlations, individual-level understanding will be key to making earlier diagnoses, which may improve outcomes by allowing earlier treatment72. The molecular targets arising from this study may also eventually inform pharmacological interventions73,74.
Limitations of the Study
By examining many types of genetic variants, we detected rare ASD-associated variants in ~14% of individuals with ASD. However, our study did not examine all possible genetic factors. We did not investigate somatic variants or epigenetic contributors, although these have been investigated in smaller studies75–78. We did not investigate recessive events other than those involving two sequence-level variants, such as a large deletion on one allele and a damaging sequence-level variant on the other. Finally, we were unable to identify genetic variants in complex regions of the genome that may only be accessible with long-read sequencing.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Stephen Scherer (stephen.scherer@sickkids.ca).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Access to MSSNG and SFARI data can be obtained by completing data access agreements at https://research.mss.ng and https://www.sfari.org/resource/sfari-base, respectively. The 1000 Genomes Project WGS data are publicly available via Amazon Web Services (https://docs.opendata.aws/1000genomes/readme.html).
All original code has been deposited at Zenodo and is publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cohorts comprising MSSNG
MSSNG contains data from several cohorts and studies (Table S1C). These include the ASD: Genomes to Outcomes Study, Autism Genetic Resource Exchange (AGRE), Autism Phenome Project, The Autism Simplex Collection (TASC), Autism Treatment Network, Baby Siblings Research Consortium, Infant Sibling Study, iTARGET, Pathways in ASD, the Province of Ontario Neurodevelopmental Network (POND), and Relating genes to Adolescent and Child mental Health (REACH). A breakdown of the samples corresponding to each study is given in Table S1C. The sex of each individual in MSSNG is given in Table S1B. The sample size of MSSNG (total number of individuals, including individuals with and without ASD) is 11,312, and the number of individuals with ASD is 5,100. Individuals meeting DSM diagnostic criteria for ASD were assigned to the ASD group, while individuals not meeting diagnostic criteria were assigned to the non-ASD group.
Ethics approval
Informed consent was obtained from all MSSNG participants. Approval for use of the AGRE data was obtained from WCG IRB (https://www.wcgirb.com). Approval for other cohorts was obtained from the Research Ethics or Institutional Review Board of each recruiting site, including Montreal Children’s Hospital-McGill University Health Centre, McMaster University-Hamilton Integrated, Memorial University-Eastern Health, Holland Bloorview Kids Rehabilitation Hospital, Queen’s University, University of Alberta, University of British Columbia, IWK Health Centre, University of California Davis, University of California San Diego, University of Miami, and The Hospital for Sick Children.
METHOD DETAILS
Whole-genome sequencing of new MSSNG samples
Samples added to MSSNG since the previous release7 were sequenced on Illumina platforms. DNA was extracted from whole blood, lymphoblastoid cell lines, or (for a small number of samples) saliva. DNA quality was assessed using gel electrophoresis. Sample purity was assessed using the Nanodrop OD260/280 ratio, and DNA was quantitated using the Qubit High Sensitivity Assay. DNA libraries were prepared using the TruSeq Nano DNA Library Preparation Kit (Illumina), the same kit except omitting the PCR step, or the NxSeq® AmpFREE Low DNA Fragment Library Kit (Lucigen) following the manufacturers’ instructions. Sequencing was performed on either the HiSeq 2000, HiSeq 2500, or HiSeq X platforms.
Detection of sequence-level variants in MSSNG
Alignment of reads against the GRCh38 reference sequence was performed using a pipeline conforming to the Centers for Common Disease Genomics (CCDG) functional equivalence standard79. Sentieon v201808.0180, which includes an optimized implementation of BWA81, was used to perform alignment, base quality score recalibration, and marking of duplicate reads, producing alignment files in CRAM format. The Sentieon implementation of the Genome Analysis Toolkit (GATK)82 was used to generate genomic VCF (gVCF) files for each sample. The gVCF files were combined to produce joint-genotyped VCF files (one per chromosome) containing variant calls from all samples. The gVCF files were divided into shards of ~50 Mb each to facilitate parallelization of the joint genotyping step. Variant metrics were generated using the CollectVariantCallingMetrics function of Picard. Workflows for read alignment and small variant detection are available as workflow description language (WDL) files on Dockstore (https://dockstore.org/search?organization=DNAstack).
Replication and population control datasets
The Simons Simplex Collection (SSC)16, which includes WGS data from individuals with ASD and their family members, was used as a replication dataset. Alignment files and small variant calls for 9,205 SSC samples were downloaded from SFARI Base (https://base.sfari.org). Samples from the 1000 Genomes Project (1000G)17 were used as population control data. Alignment files for high-coverage sequencing data from 2,504 unrelated 1000G samples83 were downloaded from Amazon Web Services, and small variant calls were downloaded from the European Bioinformatics Institute (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20190425_NYGC_GATK). Variant detection and analysis of the SSC and 1000G data were performed using identical or near-identical pipelines as for MSSNG, allowing for true comparability across datasets. Specifically, the read alignment and small variant detection pipelines used to generate the downloaded data above were nearly identical to those used for MSSNG, as they used BWA and GATK (version 3.5-0-g36282e) according to the CCDG functional equivalence standard. All other analyses of SSC and 1000G data (CNV detection, SV detection, mtDNA variant detection, PRS calculations, and variant annotations) were performed by our group using the same pipelines as used for MSSNG.
Experimental design
MSSNG was used as the discovery dataset and SSC as the replication dataset. Sample randomization and stratification, blinding, and sample-size estimation were not applicable in this study. All families having at least one child with ASD can be included in MSSNG; there are no exclusion criteria.
Small variant annotation, filtering, and de novo detection
Small variants were annotated using a custom ANNOVAR-based pipeline84 using the parameters --neargene 1 --splicing_threshold 2 --dbtype refseq --buildver hg38. The database versions or download dates of the various annotation resources used are given in Table S7A.
To be labeled as high quality, calls were required to have FILTER=“PASS” and depth (DP) >=10. Other criteria based on the genotype quality (GQ) and allelic fraction (AF) also had to be satisfied depending on the variant type. For heterozygous SNVs, also required were GQ ≥ 99 and 0.3 ≥ AF > 0.8. For heterozygous indels, also required were GQ ≥90 and 0.3 ≥ AF > 0.8. For homozygous SNPs and indels, also required were GQ ≥25 and AF ≥0.8.
De novo variants were detected using DeNovoGear v1.1.1-313-geac367485. The Sentieon/GATK VCF file for each family was used as input to DeNovoGear. Autosomes and chromosome X in male and female children were called using separate models. Variants that failed Sentieon/GATK FILTER, had a non-reference parental genotype, or had filtering allele frequency (faf95) >1% in GnomAD (exomes v2.1.1 and genomes v3.0) or frequency >1% in parents were removed. Poorly sequenced sited (<95% of parent samples genotyped) were also excluded. Putative de novo SNVs were defined as those with pp_DNM >0.95 and GQ 99. Putative de novo indels were defined as those with GQ >90 for autosomes and X chromosome in females. To reduce the number of false positive de novo indels, we removed those with <30% of reads supporting the indel call in the child or >10% of reads supporting the alternative allele in a parent. For chromosome X in males, only the read support for the alternative allele in the mother was considered for filtering indels.
Comparison of variant counts
To assess the comparability of the sequence-level variants detected in the WGS samples used in this study, we stratified the samples into categories based on dataset (MSSNG, SSC, or 1000G), sequencing platform (Complete Genomics or Illumina HiSeq 2000/2500/X), and DNA library preparation method (PCR-based or PCR-free) and then examined the distributions of SNV and indel counts for various classes of variants (all, rare, rare exonic, rare DMis, rare PTVs, or de novo). Only high-quality variants (as defined in Methods) were included. All categories had similar distributions, except that Complete Genomics samples differed from the others in terms of indel counts, in particular with fewer indels overall but more de novo indels (Figure S7).
Ancestry determination
We extracted the genotypes for 57,984 positions from a previously published list (https://www.tcag.ca/tools/1000genomes.html) and retained ~43,000 positions for ADMIXTURE analysis86 after removing variants with missing calls (genotyping rate <99%) in MSSNG, SSC, and 1000G. We refined population clusters by applying K-means clustering with K=7. Ancestry labels of 1000G samples were reassigned based on the population majority in the refined clusters. We then trained a random forest classifier using the 1000G samples with the reassigned labels. Ancestry labels in MSSNG and SSC were assigned using separate random forest classifiers. The ancestry assignment for the MSSNG samples was a consensus of the ADMIXTURE analysis and Google’s genomic ancestry inference using deep learning (https://cloud.google.com/blog/products/gcp/genomic-ancestry-inference-with-deep-learning). Self-reported ancestry was used in cases where ADMIXTURE and the deep learning method were discrepant; if no self-reported ancestry was available, the individual was tagged as OTH (other).
Except for those involving PRS, all analyses in this paper used individuals from all ancestry groups, with population principal components being used to adjust for potential ancestry-related confounding effects where applicable. PRS analyses were performed only in individuals of European ancestry, because that is the population in which the original GWAS was performed (see the “Calculation of polygenic risk scores” section for more details).
Identification of damaging biallelic events
We identified damaging homozygous variants and compound heterozygous events in autosomes and classified each into one of two categories: PTV-PTV (PTVs on both alleles) or DMis- (DMis/PTV) (a DMis variant on one allele and either a PTV or a DMis variant on the other allele). Homozygous variants were identified in all individuals, while compound heterozygous events were identified only in individuals having both parents sequenced so that phase could be determined. Only variants with frequency <1% and that were high quality as defined previously were used. DMis variants were defined as those with MPC scores >= 2, equivalent to MisB in the TADA+ analysis.
When evaluating the burden of damaging biallelic events, only individuals with both parents sequenced were considered. Potential ASD-associated damaging biallelic events were defined as those that (1) met the criteria described above; (2) were in a category (PTV-PTV or DMis- (DMis/PTV)) found to be enriched in individuals with ASD in our burden analysis; (3) were in genes in the Genomics England neurology and neurodevelopmental disorders panel labeled as having biallelic models of inheritance (n=3,149). In testing genes enriched in recessive events, we did not specifically test the subset of genes with low LOEUF scores, as this does not apply to recessive inheritance87.
Detection of copy number variants
For Illumina data, CNVs >=1 kb were detected using a pipeline47 involving the algorithms ERDS88 and CNVnator89. CNV frequencies were calculated separately for three groups of samples—MSSNG parents sequenced by HiSeq X, MSSNG parents sequenced by HiSeq 2000/2500, and 1000 Genomes Project individuals. To avoid reliance on an external ASD cohort, we did not compute SSC parent frequencies. Samples with anomalous CNV counts (more than three standard deviations higher than the mean) were not included in frequency calculations. A threshold of 50% reciprocal overlap was used when calculating CNV frequencies. That is, if sample A contained a CNV having at least 50% reciprocal overlap with some CNV B in the sample being annotated, then sample A was considered to have CNV B for frequency purposes.
Rare CNVs were defined as those with less than 1% frequency according to both ERDS and CNVnator in each of the three groups, as well as in MSSNG parents sequenced by Complete Genomics. High-quality CNVs were defined as those that were detected by both ERDS and CNVnator with at least 50% reciprocal overlap and for which less than 70% of the CNV overlapped assembly gaps, centromeres, and segmental duplications. “High-quality rare” (HQR) CNVs were defined as those that were both high-quality and rare. HQR CNVs were used for subsequent analysis. De novo CNVs were defined as HQR CNVs that were not detected by either ERDS or CNVnator in either parent. Further sample-level quality control was performed after identifying HQR and de novo CNVs. Samples with HQR counts more than three standard deviations higher than the mean were tagged as outliers. Due to the smaller number of individuals of non-European ancestry, this was done in European individuals only. The de novo CNV counts appeared to have a Poisson distribution, so we applied the Anscombe transformation to normalize the distribution. Samples with transformed counts more than three standard deviations higher than the mean (in any ancestry group) were tagged as outliers.
For Complete Genomics data, CNVs were detected using a proprietary pipeline provided by the company. Frequencies were calculated and rare CNVs were identified in the same way as for Illumina samples. All Complete Genomics CNVs were considered high-quality for the purposes of identifying HQR CNVs. Subsequent sample-level quality control was performed using the same method as for Illumina samples.
Detection of structural variants
Unlike for CNVs, we did not have an existing workflow for the detection of SVs from Illumina sequencing data. Thus, we developed a workflow specifically for this study by identifying promising candidate algorithms, evaluating the concordance and accuracy of those algorithms, selecting the most accurate combination of algorithms, and then determining effective filtering criteria to attain high sensitivity and specificity.
Dozens of algorithms for detecting SVs from short-read WGS data have been developed90. We chose an initial set of algorithms for further evaluation based on the following criteria: all algorithms were required to predict most types of SVs (deletions, duplications, insertions, and inversions) and to have high accuracy based on a previous evaluation90; at least some had to be under active development; and collectively use a variety of detection strategies (e.g., split reads, anomalous paired-end mapping, local assembly). The algorithms selected for further evaluation were DELLY91, GRIDSS92, LUMPY93, Manta94, SoftSV95, SvABA96, and Wham97 (Table S7B).
We evaluated the accuracy of each algorithm using two methods. The first involved running the algorithms on WGS data from the HuRef98, NA1287817, and HG00299 genomes, and then comparing the SVs detected by each caller to SV benchmarks detected by orthogonal technologies (i.e., other than Illumina short-read sequencing). The HuRef benchmark was the same as used previously47, but adding PBSV (https://github.com/PacificBiosciences/pbsv) and Sniffles100 calls from in-house 100x Pacific Biosciences long-read data. The NA12878 and HG002 benchmarks were derived from previous studies90,99. The second method involved performing CRAM confirmation47 on randomly selected calls from each algorithm in order to more fully evaluate specificity. Based on our evaluation, we found that Manta had the best combination of sensitivity and specificity. Although it made more false positives than Manta, we found that DELLY calls that overlapped with Manta calls were useful for giving added confidence to the Manta calls and were also useful for detecting inversions. Thus, we selected Manta and DELLY as the basis of our SV-detection workflow.
After algorithm selection, we determined whether the number of anomalously mapped paired-end reads and split reads supporting a given variant call could be used as filters to distinguish true calls from false ones. We found that neither variable was useful for this purpose. Next, for each variant type, we evaluated the sensitivity and specificity of various caller and stringency combinations (Manta PASS, DELLY PASS, Manta any, or DELLY any), and developed criteria that optimized sensitivity while maintaining good specificity (Table S7C).
Prior to calculating SV frequencies, we computed the number of SVs detected for each sample in 15 categories based on a combination of SV type (five categories: deletion, duplication, insertion, inversion, or overlapping deletion/duplication) and algorithm (three categories: detected by DELLY only, by Manta only, or by both DELLY and Manta). To reduce the impact of false positives, samples for which the call count was more than three standard deviations higher than the mean in two or more categories were excluded from the frequency calculations. As with CNVs, frequencies were calculated separately for MSSNG HiSeq X parents, MSSNG HiSeq 2000/2500 parents, and 1000 Genomes Project individuals. Frequencies were calculated separately for DELLY and Manta. A threshold of 90% reciprocal overlap was used when calculating SV frequencies. Rare CNVs were defined as those with less than 1% frequency in each of the three groups according to both Manta and DELLY, as well as in Complete Genomics MSSNG parents. SVs were defined as high-quality when they satisfied the filtering criteria derived as described above. HQR SVs were defined as for CNVs. For further quality control (QC) tagging, HQR and de novo counts were subjected to the Anscombe transformation, and samples were tagged as failing QC if they were an outlier for at least one of the five variant types (SVs detected by DELLY alone, Manta alone, or both Manta and DELLY were aggregated for each SV type prior to detecting outliers). QC based on HQR was performed only in samples of European ancestry, as other ancestry groups had fewer samples for comparison and generally had more rare SVs due to the European bias of the reference genome.
SVs in Complete Genomics samples were detected using the company’s proprietary pipeline. Samples having an SV count that was an outlier in at least one SV category (deletion, distal-duplication, distal-duplication-inversion, interchromosomal, inversion, probable-inversion, and tandem-duplication) were excluded from the frequency calculation. Complete Genomics outliers were determined separately for different versions of the variant-detection software.
Inheritance assignment for copy number and structural variants
CNVs and SVs in children with both parents sequenced were tagged according to their putative inheritance. For CNVs, the child calls were compared to the father and mother calls independently. For each parent, for a given child call, all overlapping calls in the parent were identified. If the size of the child CNV was between half and twice the sum of the lengths of the overlapping parent calls, and the CNV type (deletion or duplication) matched, then the CNV was tagged as inherited from that parent. CNV calls in the child with no overlapping CNVs in the parent were tagged as having the potential to be de novo. If some overlapping calls were found in the parent, but the above size condition was not satisfied, then the child call was tagged for manual inspection of its de novo status. The inheritance assignments from both parents were then consolidated, with CNVs tagged as inherited for both parents retagged as ambiguous. Only CNVs tagged as potentially de novo for both parents were tagged as de novo after consolidation.
To minimize potential false positive de novo tags, child samples sequenced on Illumina platforms were compared with both the ERDS and CNVnator calls from the parent samples. For samples sequenced by Complete Genomics, a second CNV caller was not available, so additional filters (based on copy number estimates of the CNVs with flanking regions in the child and both parents) were used to minimize false positive de novo tagging.
SV inheritance was determined similarly, with the following differences designed to account for the more precise sizes and breakpoints afforded by the SV-detection methodology. As the members of a given family were joint-called for samples sequenced on Illumina platforms, a stringent 90% overlap threshold was used as the cutoff. Because samples sequenced by Complete Genomics were processed on a per-sample basis (i.e., not joint-called), a less stringent 50% overlap threshold was used.
Annotation and interpretation of copy number and structural variants
CNVs and SVs were annotated using a custom R script (v3.6.1) employing the GenomicRanges and data.table libraries. Gene annotations, genomic features, phenotype ontologies, and disease information were downloaded from various sources (Table S7D). SVs were interpreted according to ACMG and ClinGen guidelines101,102. SVs classified as likely pathogenic or pathogenic (LP/P) are described as pathogenic. All pathogenic SVs were verified by CRAM confirmation47, overlap with microarray findings, and/or Sanger sequencing. For de novo SVs, we verified the variant’s absence in parents.
Resolution of duplication structures
To investigate the potential to use WGS data to resolve the structures of duplications and understand their impact on genes, we identified rare duplications in a subset of individuals with ASD in MSSNG that overlapped exons from a broad list of ASD candidate genes (Table S3B). Known recurrent genomic disorders (e.g., 15q11-q13 duplications) were excluded. Duplication structures were analyzed by visualizing CRAM files using IGV23 and were classified as tandem or complex through manual inspection of paired-end reads and split reads at the duplication breakpoint junctions. Duplications having breakpoints mapping within homologous segmental duplications or LINE elements were considered likely NAHR events. Breakpoint junctions that could not be classified as tandem, complex, or likely NAHR events were considered unresolved. The sequence-level breakpoint coordinates of tandem and complex duplications were determined through analysis of split read sequence in IGV and using BLAT103. The last nucleotide of a split read before a deviation from the reference sequence was considered the sequence-level breakpoint of the duplication. Breakpoints that could not be resolved to the nucleotide level using split reads were estimated from the location of paired-end reads and read depth changes. The ERDS coordinates were used as an approximation of the true duplication breakpoints for likely NAHR and unresolved duplications. Duplications with identical breakpoints that were found in multiple related or unrelated individuals were deemed to represent a single unique event. The impact of each duplication on the ASD candidate gene(s) was annotated manually using the “NCBI RefSeq genes, curated subset” track of the UCSC Genome Browser104. A duplication was considered to increase gene dosage when all RefSeq isoforms were fully contained within the duplication. The reading frame of intragenic duplications and fusion genes created at the breakpoints of a duplication were assessed using the UCSC Genome Browser.
Detection of uniparental disomies
Whole-chromosome and large segmental uniparental disomies (UPDs) were identified using SNPs and CNVs. For a subset of common SNPs, Mendelian errors were calculated using PLINK105, and samples with excessive errors on a given chromosome were identified. Candidate regions were compared with CNV calls to exclude regions overlapping deletions. Putative isodisomies were further verified by checking that the SNP genotypes were mostly homozygous. Log R ratio (LRR) and B-allele frequency (BAF) plots were generated for the entire family to visualize UPDs.
Non-coding annotations
We used Ensembl Variant Effect Predictor (VEP) v102106 and the October 2019 release of ANNOVAR84 to perform non-coding variant annotations based on information from several databases. The variant data from each ASD WGS cohort were converted into ANNOVAR- and VEP-compatible format, with multi-allelic variants split and indels normalized to ensure correct matching of variants. Selected files from the non-coding variant databases (see below) were also converted to ANNOVAR-compatible format.
Regulatory features from Ensembl Regulatory Build (http://useast.ensembl.org/info/genome/funcgen/regulatory_build.html)107 were added using VEP. Annotations related to promoters, enhancers, and their target genes were obtained from the GeneHancer database (geneHancerInteractionsDoubleElite, UCSC update January 2019)108. Transcription factor binding sites and other regulatory elements were derived from the ReMap2020 non-redundant peak file (http://remap.univ-amu.fr)109. Long non-coding RNA annotations were obtained from LNCipedia v5.2 (https://lncipedia.org)110. Small non-coding RNA annotations were acquired from DASHR v2.0 (https://dashr2.lisanwanglab.org)111. Retrotransposon insertion polymorphism annotations were obtained from dbRIP (https://dbrip.brocku.ca)112.
Topologically associating domain (TAD) boundaries were derived from a previous publication113, and five annotations were generated using the TAD boundaries. The first three annotations had possible values of 0, 1, or 2, depending on whether neither TAD flanking the boundary contained genes from a given gene list (0), one of the two TADs did (1), or both did (2). The three gene lists used were the ASD-associated genes from our TADA+ analysis (134 genes), genes associated more generally with NDDs (1,250 genes), or genes intolerant to loss of function (pLI >0.9) (2,867 genes)114. The 1,250 NDD-associated genes were derived from the literature (excluding those having autosomal recessive inheritance)8,115,116, SFARI genes (score 1–3 plus syndromic), genes with ClinGen haploinsufficiency or triplosensitivity scores of 2 or 3117, and the Geisinger Developmental Brain Disorder Gene Database (https://dbd.geisingeradmi.org). The remaining two annotations were based on the “brain expression specificity” of the left and right TAD—that is, the fraction of genes in each TAD that are expressed in the brain based on Illumina Body Map 2.0 RNA-seq data. Specifically, let BL and BR be the proportion of genes that are brain-expressed in the left and right TAD, respectively. Then the first annotation represented the difference in brain expression specificity (|BL − BR|) and the second the sum of brain expression specificity (BL + BR).
All non-coding annotations and their sources are listed in detail in Table S6C.
Mitochondrial analysis
For samples sequenced on Illumina platforms, reads aligning to the mitochondrial genome were extracted and realigned to the revised Cambridge Reference Sequence (NC_012920) using BWA v0.7.8. Pileups were generated with SAMtools mpileup v1.1118, requiring the program to include duplicate reads and retaining all positions in the output. Custom scripts were developed to parse the mpileup output to determine the most frequently occurring non-reference base at each position. The heteroplasmic fractions were calculated and VCF files were generated. For samples sequenced by Complete Genomics, mitochondrial variants detected by the proprietary software were extracted. For both platforms, FASTA files replacing mitochondrial reference bases with alternative bases at sites where the heteroplasmic fraction was greater than or equal to 0.5 were also generated and haplogroups were predicted using HaploGrep v2.1.1119. The VCF files were annotated using ANNOVAR-based custom scripts with annotations from MitoMaster120 (April 2019) and Ensembl v96.
Consensus phenotype measures
Consensus scores across several domains were calculated to facilitate the use of varied behavioral data across cohorts. These measures are not meant to replace the detailed and strategic work that a clinical psychologist or a computational scientist may choose to do with these data, but rather to provide reasonable ways of summarizing cognitive/behavioral data across domains of interest, especially for researchers who have less familiarity with the underlying tests. These measures represent a work in progress, and we welcome feedback on their design and utility as well as ideas for additional consensus measures. Below, we describe the consensus measures currently available in MSSNG.
Adaptive Behaviour Standard Score:
Two measures capture this domain within MSSNG: Adaptive Behavior Assessment System (ABAS) and Vineland Adaptive Behavior Scales Survey Interview Form (Vineland). The computed score is the global adaptive composite (GAC) score from the ABAS or the adaptive behavior standard score from the Vineland. If more than one instrument is available, the most recent score is used. There is active work being done to create a reliable correction strategy that will allow the use of scores from both instruments to be used as a continuous measure121.
Socialization Standard Score:
This score is also calculated from the ABAS or Vineland, but the social composite score of the ABAS or the socialization standard score of the Vineland are used. If more than one instrument is available, the most recent score is used.
Full Scale IQ:
While there are several IQ instruments in MSSNG, we include only Wechsler scales and Stanford-Binet under this category. Full Scale IQs (FSIQs) are prioritized over abbreviated IQs and Wechsler scales are prioritized over Stanford-Binet. Among FSIQs, the algorithm selects the most recent one if they are more than two years apart.
Global Ability Consensus Estimate:
This score is intended to estimate global abilities when gold standard full IQ measures are not available. If a Full Scale IQ is available as per above, then this category is populated by that value. Otherwise, the following measures are used to populate this variable, in descending order of preference. The first choices are the Mullen Scales of Early Learning composite scores, followed by Merrill-Palmer (only when all three subdomain scores are available). Subsequently, nonverbal IQ can be used: Leiter International Performance Scale, followed by Raven’s Progressive Matrices. Lastly, verbal standard scores from language assessments can be used in the following order: Clinical Evaluation of Language Fundamentals (CELF), Oral and Written Language Scales (OWLS), Preschool Language Scale (PLS), Expressive Vocabulary Test (EVT), and the Peabody Picture Vocabulary Test (PPVT). This is the category with the most noise and involving a clinical psychologist is highly recommended, depending on the research aim. Within each category, the most recent test is selected.
Co-occurring conditions (ADHD, anxiety, seizures, and gastrointestinal conditions):
The available data across cohorts is extremely variable and may include everything from single items (e.g., parental report of a condition) to standardized instruments (e.g., Child Behavior Checklist (CBCL), Symptoms and Normal Behaviors (SWAN) score, Conners Comprehensive Behavior Rating Scales, Revised Children’s Anxiety and Depression Scale (RCADS), and Spence Children’s Anxiety Scale). In all categories, a non-stringent approach was taken so that if any available scores across any measures reflect clinical concern, then the co-occurring condition variable is labeled as true (e.g., ADHD=true). If none of the available measures suggest clinical concern, then the co-occurring condition variable is labeled as false (e.g., ADHD=false). These measures should not be considered evidence of diagnoses, but rather simply of clinical concern.
ADOS calibrated severity scores:
To score past versions of item-level ADOS data, past protocol items corresponding to the most recent ADOS-2 version for each module were matched up and used. One item, “Hand and Finger and Other Complex Mannerisms”, was originally two separate items in previous ADOS versions. For this item, the higher score of the two was used. For “Unusual Eye Contact”, anything above 1 was recoded as 2, and zeros remained the same. After applying these transformations, the ADOS-2 algorithm122,123 was used to create domain-level sums and standardized severity scores. Missing items were treated as zeros; however, no more than two missing items were allowed when computing the algorithm total. Furthermore, no more than two missing items were allowed when computing the Social Communication Domain total and no more than one missing item was allowed when computing the Restricted and Repetitive Behavior domain total. The ADOS calibrated severity scores are a work in progress and will be released once fully implemented (please contact the authors for more information).
QUANTIFICATION AND STATISTICAL ANALYSIS
ASD gene list generation
We generated a new ASD gene list by adding more trio data to a previous study by the ASC8, which used an enhanced version of the original TADA approach24 to discover ASD-associated genes based on case-control data and de novo variants in trios. Specifically, we added de novo variants detected in individuals with ASD in trios from the MSSNG and SPARK25,26 cohorts. Variants from MSSNG and SPARK were annotated with Missense badness, PolyPhen-2, and constraint (MPC) scores124 using dbNSFP125. Genes with FDR <0.1 were considered to be ASD-associated.
Because the ASC variants were annotated using Variant Effect Predictor (VEP), whereas we annotated the MSSNG and SPARK variants using ANNOVAR, we compared the two annotation methods to ensure their classifications of PTVs and DMis variants were concordant. Upon re-annotating the ASC de novo variants with ANNOVAR, we observed only minor differences between the VEP and ANNOVAR annotations (Table S7E). We also observed high concordance between which missense variants had MPC scores in the ASC data and which were annotated with MPC scores using dbNSFP125 (Table S7E). Finally, for missense variants having MPC scores from both methods (the vast majority), only 0.14% differed by more than 0.1. Collectively, the variant annotations for ASC and for MSSNG/SPARK were highly concordant.
Because variant quality score recalibration was not applied to the SPARK variant calls, we applied the hard filters recommended by the GATK development team (https://gatk.broadinstitute.org/hc/en-us/articles/360035890471-Hard-filtering-germline-short-variants). We considered recurrent de novo variants more likely to be false, so we manually inspected all recurrent de novo variants in IGV to verify their correctness and removed those deemed to be false. We also verified a randomly selected subset of non-recurrent de novo variants in IGV to verify their correctness and de novo status.
ASC variant coordinates were converted from GRCh37 to GRCh38 using the NCBI liftover tool. Sample overlap between MSSNG and SPARK was determined using PLINK105. Because we only had access to de novo variants for ASC, sample overlap between ASC and MSSNG/SPARK could not be determined using PLINK. Thus, we conservatively removed any sample in MSSNG or SPARK that shared a de novo variant with an ASC sample. However, when the MSSNG/SPARK sample and the ASC sample were different sexes, both variants were retained. To ensure that there was no overlap between the MSSNG/SPARK de novo PTVs and PTVs in cases from the ASC case-control data, we downloaded the full set of variant-level data (https://atgu-exome-browser-data.s3.amazonaws.com/ASC/ASC_variant_results.tsv.bgz) from the ASC website (https://asc.broadinstitute.org), and then identified PTVs in ASC cases that overlapped with de novo PTVs from MSSNG/SPARK. Because the TADA+ model gives more weight to de novo variants than case-control variants, we retained the MSSNG/SPARK de novo variants and discarded the overlapping ASC case-control variants. Due to the lack of sample-level information for the ASC case-control data, it was not possible to compare sample sex in order to potentially retain overlapping variants.
Gene network generation
A network diagram representing the ASD-associated genes identified in the TADA+ analysis was generated. For each gene, we used GeneMANIA126 (dataset version gmdata-2021-04-29) to identify the 200 gene neighbors with the closest association based on protein-protein interactions and biological pathways using the “gene ontology biological process” weighting option. To select genes strongly connected to others, we retained only nodes connected by edges with a weighted Jaccard coefficient ≥ 0.33. The network was visualized using Cytoscape127.
Based on the gene connectivity, we partitioned the network into several modules. Each module was tested for pathway enrichment using Fisher’s Exact Tests to compare the sets of genes in each module with sets of genes described by pathway-related GO terms. To avoid GO terms with too few or too many genes, we used only terms with between 5 and 800 genes.
To determine whether the connectivity of the genes in our network was greater than expected by chance, we performed the following procedure for each edge E. For genes A and B connected by E, we calculated their Jaccard index (the fraction of genes connected to either A or B that are connected to both). Let NA represent the number of genes connected to A, and similarly for NB. We performed 1999 permutations, where each permutation involved randomly selecting a set of NA genes and a set of NB genes and calculating the Jaccard index of those sets. The random selections were made from the set of all genes connected to our TADA+ genes in GeneMania (n=7,112). The p-value for the permutation test was the proportion of the 2000 Jaccard indices (1999 random permutations plus the actual Jaccard index for A and B) that were greater than or equal to the actual Jaccard index.
Calculation of polygenic risk scores
We calculated PRS in individuals of European ancestry from the MSSNG, SSC, and 1000G cohorts using PLINK v1.9105. For MSSNG, PRS was calculated only for individuals sequenced on Illumina platforms. We used summary statistics from a previously-published ASD meta-GWAS, which included the iPSYCH and Psychiatric Genomics Consortium (PGC) cohorts56. Due to sample overlap between PGC and both MSSNG and SSC, we used summary statistics derived only from iPSYCH (13,076 cases and 22,664 controls).
We included only SNPs with minor allele frequency >0.05 (iPSYCH controls) and imputation quality score (INFO) >0.9. To avoid potential strand conflicts, complementary SNPs were excluded. We also excluded SNPs with FILTER != PASS and those within the broad MHC region (chr6:25,000,000–35,000,000). Data from MSSNG, SSC and 1000G were merged prior to clumping, and only SNPs common to all three cohorts were retained. Clumping was performed with an r2 threshold and radius of 0.1 and 500 kb, respectively. PRS values were generated by including SNPs with p-value ≤ 0.1, weighting by the additive scale effect (log10 OR) of each variant, and then summing over the variants. Scores were centred to a mean of zero. In analyzing the PRS data, families in which a non-sibling member had an ASD diagnosis were excluded. PRS values for all P-value thresholds are given in Table S7F.
Non-coding transmission bias tests
We defined the odds ratio for over-transmission of non-coding variants with annotation A as OR = (AT/ANT) / (OT/ONT), where AT is the number of transmitted non-coding variants with annotation A, ANT is the corresponding number of non-transmitted variants, and OT and ONT are the total number of transmitted and non-transmitted non-coding variants, respectively, for all annotations other than A. To reduce noise, only variants with PhastCons score >0 were considered. To avoid the case where the expected transmission rate is not equal to 50% (for example, when both parents were heterozygous for a given variant, or one or both parents were homozygous), we included only variants for which one parent was heterozygous and the other did not have the variant. Complete Genomics samples were not included in the non-coding transmission tests because their genome-wide transmission rate differed from the expected 50%.
Burden tests
We used logistic regression to compare individuals with ASD with non-ASD siblings. The covariates used were sex, presence or absence of an ASD-associated rare variant, and population structure variables from ADMIXTURE86. To correct for potential technical differences in variant detection between sequencing platforms (for example, HiSeq 2000 versus HiSeq X), we also used as a covariate the total number of variants of the type being tested. For instance, when testing the burden of de novo variants in promoters, the number of de novo variants genome-wide was used as a covariate. FDRs were calculated using the Benjamini-Hochberg (BH) method.
Supplementary Material
Figure S1. Genotype-Computed Ancestry of Individuals with WGS Data Used in This Study, Related to Table 1. Distributions are shown for (A) MSSNG, (B) SSC, and (C) 1000 Genomes Project.
Figure S2. Screenshots of Interfaces for Accessing MSSNG Data, Related to Table 1. Screenshots are shown for (A) CNV querying, (B) the Phenotype Data Explorer, and (C) a Terra notebook.
Figure S3. Distributions of Heteroplasmy Deltas, Related to STAR Methods. Deltas are defined as child heteroplasmy minus mother heteroplasmy for a given position in the mtDNA sequence. MSSNG ASD: n=35,279 variants; MSSNG non-ASD: n=2,171; SSC ASD: n=29,005; SSC non-ASD: n=18,328. No significant differences were observed between the shifts from mother to children with ASD and from mother to non-ASD siblings in either MSSNG (ASD: 0.0026 ± 0.046; non-ASD: 0.0043 ± 0.058; p=0.18) or SSC (ASD: 0.0012 ± 0.042; non-ASD: 0.0014 ± 0.048; p=0.65).
Figure S4: Examples of Multiplex Families from MSSNG with ASD-Associated Rare Variants, Related to Figure 4.
Figure S5. Non-coding Transmission Tests, Related to STAR Methods. Odds ratios are shown for the number of singleton variants that were transmitted versus non-transmitted in (A) individuals with ASD in MSSNG, (B) individuals with ASD in SSC, and (C) non-ASD siblings in SSC.
Figure S6. PRS Analysis, Related to Figure 6. (A) PRS distribution when samples are ranked from highest to lowest score. (B) Sex-stratified polygenic transmission disequilibrium tests. (C) Comparison between individuals with ASD from MPX families in MSSNG and individuals with ASD in SSC (which are all SPX families). (D) Comparison between fathers and mothers without ASD, stratified by dataset (MSSNG or SSC). (E) Comparison between parents without ASD and 1000 Genomes Project population controls.
Figure S7. Distributions of Different Categories of Variants, Stratified by Sequencing Platform and DNA Library Preparation Method, Related to STAR Methods. Distributions are shown for (A) SNVs and (B) indels.
Table S2: ASD Gene Discovery, Related to Figure 2.
Table S3: Duplication Structure Resolution, Related to STAR Methods.
Table S4: Mitochondrial Variation, Related to STAR Methods.
Table S6: Non-Coding Analysis, Related to STAR Methods.
Table S7: Methods Details, Related to STAR Methods.
New MSSNG release contains WGS from 11,312 individuals from families with ASD
Extensive variant data available, including SNVs/indels, SVs, tandem repeats & PRS
Annotation reveals 134 ASD-associated genes, plus SVs not detectable without WGS
Rare, dominant variation has a prominent role in multiplex ASD
ACKNOWLEDGMENTS
We thank the families participating in MSSNG, as well as the generosity of the donors who support this program. We acknowledge the resources of Autism Speaks and The Centre for Applied Genomics. We thank Anders Børglum and Jakob Grove for providing the iPSYCH-only summary statistics and Kyle Satterstrom for providing TADA+ scripts. We are grateful to all of the families at the participating SSC and SPARK sites, as well as the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R. Goin-Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, E. Wijsman). We appreciate obtaining access to genetic and phenotypic data on SFARI Base. Approved researchers can obtain the SSC and SPARK datasets by applying at https://base.sfari.org. Funding was provided by the University of Toronto McLaughlin Centre, Genome Canada/Ontario Genomics, Genome BC, the Government of Ontario, the Canadian Institutes of Health Research (CIHR), the Canada Foundation for Innovation, Autism Speaks, Autism Speaks Canada, Brain Canada, Kids Brain Health Network, Qatar National Research Fund (NPRP10-0202-170320), Ontario Brain Institute, SickKids Foundation, and SFARI (570794, N.S. and S.S.). We thank Roche for the use of the HuRef PacBio sequencing data. B. Trost was supported by the CIHR Banting Postdoctoral Fellowship and the Canadian Open Neuroscience Platform Research Scholar Award. L.O.L. held the Lap-Chee Tsui Fellowship for Research Excellence. M.H.Z. acknowledges the support of the Fonds de recherche du Québec–Santé Junior 1 Research Scholar programme. J.S. and the REACH project were supported by grants from the National Institutes of Health (MH113715, MH119746, 1MH109501) and SFARI (606768). L.Z. holds the Stollery Children’s Hospital Foundation Chair in Autism. M.E.S.L. holds a BC Children’s Hospital Research Institute Investigator Grant Award. S.W.S. holds the Northbridge Chair in Pediatric Research at The Hospital for Sick Children and the University of Toronto.
INCLUSION AND DIVERSITY STATEMENT
We worked to ensure sex balance in the recruitment of human subjects (the male:female ratio for individuals with ASD in MSSNG closely mirrors the well-established 4:1 sex bias in ASD). We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure that the study questionnaires were prepared in an inclusive way. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Footnotes
DECLARATION OF INTERESTS
E. Anagnostou has received consultation fees from Roche, Quadrant, and Oron; grant funding from Roche; in-kind supports from AMO Pharma and CRR; editorial honoraria from Wiley; and book royalties from APPI and Springer. She co-holds a patent for the device Anxiety Meter (patent # US20160000365A1). S.W.S. is on the Scientific Advisory Committee of Population Bio, serves as a Highly Cited Academic Advisor for King Abdulaziz University, and intellectual property from aspects of his research held at The Hospital for Sick Children are licensed to Athena Diagnostics and Population Bio.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.American Psychiatric Association (2013). Diagnostic and Statistical Manual of Mental Disorders 5th ed. (American Psychiatric Publishing; ). [Google Scholar]
- 2.Lord C, Brugha TS, Charman T, Cusack J, Dumas G, Frazier T, Jones EJH, Jones RM, Pickles A, State MW, et al. (2020). Autism spectrum disorder. Nat Rev Dis Primers 6, 5. 10.1038/s41572-019-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Public Health Agency of Canada (2021). Autism Spectrum Disorder: Highlights from the 2019 Canadian Health Survey on Children and Youth (https://www.canada.ca/content/dam/phac-aspc/documents/services/publications/diseases-conditions/autism-spectrum-disorder-canadian-health-survey-children-youth-2019/autism-spectrum-disorder-canadian-health-survey-children-youth-2019.pdf).
- 4.Centers for Disease Control and Prevention (2022). Autism and Developmental Disabilities Monitoring (ADDM) Network (https://www.cdc.gov/ncbddd/autism/addm.html). Centers for Disease Control and Prevention. https://www.cdc.gov/ncbddd/autism/addm.html. [Google Scholar]
- 5.Doshi-Velez F, Ge Y, and Kohane I (2014). Comorbidity clusters in autism spectrum disorders: An electronic health record time-series analysis. Pediatrics 133. 10.1542/peds.2013-0819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tick B, Bolton P, Happé F, Rutter M, and Rijsdijk F (2016). Heritability of autism spectrum disorders: a meta-analysis of twin studies. J Child Psychol Psychiatry 57, 585–595. 10.1111/jcpp.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuen RKC, Merico D, Bookman M, Howe JL, Thiruvahindrapuram B, Patel RV, Whitney J, Deflaux N, Bingham J, Wang Z, et al. (2017). Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci 20, 602–611. 10.1038/nn.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, Peng M, Collins R, Grove J, Klei L, et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23. 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu JM, Satterstrom FK, Peng M, Brand H, Collins RL, Dong S, Wamsley B, Klei L, Wang L, Hao SP, et al. (2022). Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat Genet 54, 1320–1331. 10.1038/s41588-022-01104-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou X, Feliciano P, Shu C, Wang T, Astrovskaya I, Hall JB, Obiajulu JU, Wright JR, Murali SC, Xu SX, et al. (2022). Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat Genet 54, 1305–1319. 10.1038/s41588-022-01148-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tammimies K, Marshall CR, Walker S, Kaur G, Thiruvahindrapuram B, Lionel AC, Yuen RKC, Uddin M, Roberts W, Weksberg R, et al. (2015). Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA 314, 895–903. 10.1001/jama.2015.10078. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez BA, and Scherer SW (2017). Syndromic autism spectrum disorders: moving from a clinically defined to a molecularly defined approach. Dialogues Clin Neurosci 19, 353–371. 10.31887/DCNS.2017.19.4/sscherer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vivanti G (2020). Ask the editor: what is the most appropriate way to talk about individuals with a diagnosis of autism? J Autism Dev Disord 50, 691–693. 10.1007/s10803-019-04280-x. [DOI] [PubMed] [Google Scholar]
- 14.Bury SM, Jellett R, Spoor JR, and Hedley D (2020). “It defines who I am” or “It’s something I have”: what language do [autistic] Australian adults [on the autism spectrum] prefer? J Autism Dev Disord. 10.1007/s10803-020-04425-3. [DOI] [PubMed] [Google Scholar]
- 15.Botha M, Hanlon J, and Williams GL (2021). Does language matter? identity-first versus person-first language use in autism research: a response to Vivanti. J Autism Dev Disord. 10.1007/s10803-020-04858-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischbach GD, and Lord C (2010). The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron 68, 192–195. 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 17.1000 Genomes Project Consortium (2015). A global reference for human genetic variation. Nature 526, 68–74. 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lord C, Rutter M, and Le Couteur A (1994). Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord 24, 659–685. 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- 19.Kim SH, and Lord C (2012). New autism diagnostic interview-revised algorithms for toddlers and young preschoolers from 12 to 47 months of age. J Autism Dev Disord 42, 82–93. 10.1007/s10803-011-1213-1. [DOI] [PubMed] [Google Scholar]
- 20.Lord C, Rutter M, Goode S, Heemsbergen J, Jordan H, Mawhood L, and Schopler E (1989). Autism diagnostic observation schedule: a standardized observation of communicative and social behavior. J Autism Dev Disord 19, 185–212. 10.1007/BF02211841. [DOI] [PubMed] [Google Scholar]
- 21.Lord C, Risi S, Lambrecht L, Cook EH, Leventhal BL, DiLavore PC, Pickles A, and Rutter M (2000). The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord 30, 205–223. [PubMed] [Google Scholar]
- 22.Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. (2010). Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327, 78–81. 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 23.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He X, Sanders SJ, Liu L, De Rubeis S, Lim ET, Sutcliffe JS, Schellenberg GD, Gibbs RA, Daly MJ, Buxbaum JD, et al. (2013). Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet 9, e1003671. 10.1371/journal.pgen.1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.SPARK Consortium (2018). SPARK: A US cohort of 50,000 families to accelerate autism research. Neuron 97, 488–493. 10.1016/j.neuron.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feliciano P, Zhou X, Astrovskaya I, Turner TN, Wang T, Brueggeman L, Barnard R, Hsieh A, Snyder LG, Muzny DM, et al. (2019). Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom Med 4, 19. 10.1038/s41525-019-0093-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banerjee-Basu S, and Packer A (2010). SFARI Gene: an evolving database for the autism research community. Dis Model Mech 3, 133–135. 10.1242/dmm.005439. [DOI] [PubMed] [Google Scholar]
- 28.Sznajder ŁJ, and Swanson MS (2019). Short tandem repeat expansions and RNA-mediated pathogenesis in myotonic dystrophy. Int J Mol Sci 20, E3365. 10.3390/ijms20133365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ekström A-B, Hakenäs-Plate L, Samuelsson L, Tulinius M, and Wentz E (2008). Autism spectrum conditions in myotonic dystrophy type 1: a study on 57 individuals with congenital and childhood forms. Am J Med Genet B Neuropsychiatr Genet 147B, 918–926. 10.1002/ajmg.b.30698. [DOI] [PubMed] [Google Scholar]
- 30.Lagrue E, Dogan C, De Antonio M, Audic F, Bach N, Barnerias C, Bellance R, Cances C, Chabrol B, Cuisset J-M, et al. (2019). A large multicenter study of pediatric myotonic dystrophy type 1 for evidence-based management. Neurology 92, e852–e865. 10.1212/WNL.0000000000006948. [DOI] [PubMed] [Google Scholar]
- 31.Trost B, Engchuan W, Nguyen CM, Thiruvahindrapuram B, Dolzhenko E, Backstrom I, Mirceta M, Mojarad BA, Yin Y, Dov A, et al. (2020). Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature 586, 80–86. 10.1038/s41586-020-2579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu S, Noviello CM, Teng J, Walsh RM, Kim JJ, and Hibbs RE (2018). Structure of a human synaptic GABAA receptor. Nature 559, 67–72. 10.1038/s41586-018-0255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fatemi SH, Reutiman TJ, Folsom TD, and Thuras PD (2009). GABA(A) receptor downregulation in brains of subjects with autism. J Autism Dev Disord 39, 223–230. 10.1007/s10803-008-0646-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di J, Li J, O’Hara B, Alberts I, Xiong L, Li J, and Li X (2020). The role of GABAergic neural circuits in the pathogenesis of autism spectrum disorder. Int J Dev Neurosci 80, 73–85. 10.1002/jdn.10005. [DOI] [PubMed] [Google Scholar]
- 35.Christensen J, Grønborg TK, Sørensen MJ, Schendel D, Parner ET, Pedersen LH, and Vestergaard M (2013). Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 309, 1696–1703. 10.1001/jama.2013.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chau DK-F, Choi AY-T, Yang W, Leung WN, and Chan CW (2017). Downregulation of glutamatergic and GABAergic proteins in valproic acid associated social impairment during adolescence in mice. Behav Brain Res 316, 255–260. 10.1016/j.bbr.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Bonnet C, Andrieux J, Béri-Dexheimer M, Leheup B, Boute O, Manouvrier S, Delobel B, Copin H, Receveur A, Mathieu M, et al. (2010). Microdeletion at chromosome 4q21 defines a new emerging syndrome with marked growth restriction, mental retardation and absent or severely delayed speech. J Med Genet 47, 377–384. 10.1136/jmg.2009.071902. [DOI] [PubMed] [Google Scholar]
- 38.Hu X, Chen X, Wu B, Soler IM, Chen S, and Shen Y (2017). Further defining the critical genes for the 4q21 microdeletion disorder. Am J Med Genet A 173, 120–125. 10.1002/ajmg.a.37965. [DOI] [PubMed] [Google Scholar]
- 39.O’Donnell L, Soileau B, Heard P, Carter E, Sebold C, Gelfond J, Hale DE, and Cody JD (2010). Genetic determinants of autism in individuals with deletions of 18q. Hum Genet 128, 155–164. 10.1007/s00439-010-0839-y. [DOI] [PubMed] [Google Scholar]
- 40.Autism Genome Project Consortium (2007). Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet 39, 319–328. 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lowther C, Speevak M, Armour CM, Goh ES, Graham GE, Li C, Zeesman S, Nowaczyk MJM, Schultz L-A, Morra A, et al. (2017). Molecular characterization of NRXN1 deletions from 19,263 clinical microarray cases identifies exons important for neurodevelopmental disease expression. Genet Med 19, 53–61. 10.1038/gim.2016.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaaf CP, Betancur C, Yuen RKC, Parr JR, Skuse DH, Gallagher L, Bernier RA, Buchanan JA, Buxbaum JD, Chen C-A, et al. (2020). A framework for an evidence-based gene list relevant to autism spectrum disorder. Nat Rev Genet 21, 367–376. 10.1038/s41576-020-0231-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kushki A, Anagnostou E, Hammill C, Duez P, Brian J, Iaboni A, Schachar R, Crosbie J, Arnold P, and Lerch JP (2019). Examining overlap and homogeneity in ASD, ADHD, and OCD: a data-driven, diagnosis-agnostic approach. Transl Psychiatry 9, 318. 10.1038/s41398-019-0631-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thurm A, Farmer C, Salzman E, Lord C, and Bishop S (2019). State of the field: differentiating intellectual disability from autism spectrum disorder. Front Psychiatry 10, 526. 10.3389/fpsyt.2019.00526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khan SA, Khan SA, Narendra AR, Mushtaq G, Zahran SA, Khan S, and Kamal MA (2016). Alzheimer’s disease and autistic spectrum disorder: is there any association? CNS Neurol Disord Drug Targets 15, 390–402. 10.2174/1871527315666160321104303. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Yuan Y, Liu C, Xu Y, Xiao N, Long H, Luo Z, Meng S, Wang H, Xiao B, et al. (2022). DNAH14 variants are associated with neurodevelopmental disorders. Hum Mutat 43, 940–949. 10.1002/humu.24386. [DOI] [PubMed] [Google Scholar]
- 47.Trost B, Walker S, Wang Z, Thiruvahindrapuram B, MacDonald JR, Sung WWL, Pereira SL, Whitney J, Chan AJS, Pellecchia G, et al. (2018). A comprehensive workflow for read depth-based identification of copy-number variation from whole-genome sequence data. Am J Hum Genet 102, 142–155. 10.1016/j.ajhg.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakka P, Pattillo Smith S, O’Donnell-Luria AH, McManus KF, 23andMe Research Team, Mountain JL, Ramachandran S, and Sathirapongsasuti JF (2019). Characterization of prevalence and health consequences of uniparental disomy in four million individuals from the general population. Am J Hum Genet 105, 921–932. 10.1016/j.ajhg.2019.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chalkia D, Singh LN, Leipzig J, Lvova M, Derbeneva O, Lakatos A, Hadley D, Hakonarson H, and Wallace DC (2017). Association between mitochondrial DNA haplogroup variation and autism spectrum disorders. JAMA Psychiatry 74, 1161–1168. 10.1001/jamapsychiatry.2017.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, Picard M, and Gu Z (2016). Genetic evidence for elevated pathogenicity of mitochondrial DNA heteroplasmy in autism spectrum disorder. PLoS Genet 12, e1006391. 10.1371/journal.pgen.1006391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doan RN, Lim ET, De Rubeis S, Betancur C, Cutler DJ, Chiocchetti AG, Overman LM, Soucy A, Goetze S, Autism Sequencing Consortium, et al. (2019). Recessive gene disruptions in autism spectrum disorder. Nat Genet 51, 1092–1098. 10.1038/s41588-019-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ozonoff S, Young GS, Carter A, Messinger D, Yirmiya N, Zwaigenbaum L, Bryson S, Carver LJ, Constantino JN, Dobkins K, et al. (2011). Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics 128, e488–95. 10.1542/peds.2010-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuen RKC, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, Chrysler C, Nalpathamkalam T, Pellecchia G, Liu Y, et al. (2015). Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med 21, 185–191. 10.1038/nm.3792. [DOI] [PubMed] [Google Scholar]
- 54.Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, Mahajan M, Manaa D, Pawitan Y, Reichert J, et al. (2014). Most genetic risk for autism resides with common variation. Nat Genet 46, 881–885. 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klei L, McClain LL, Mahjani B, Panayidou K, De Rubeis S, Grahnat A-CS, Karlsson G, Lu Y, Melhem N, Xu X, et al. (2021). How rare and common risk variation jointly affect liability for autism spectrum disorder. Mol Autism 12, 66. 10.1186/s13229-021-00466-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R, et al. (2019). Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51, 431–444. 10.1038/s41588-019-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weiner DJ, Wigdor EM, Ripke S, Walters RK, Kosmicki JA, Grove J, Samocha KE, Goldstein JI, Okbay A, Bybjerg-Grauholm J, et al. (2017). Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet 49, 978–985. 10.1038/ng.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perkel JM (2021). Reactive, reproducible, collaborative: computational notebooks evolve. Nature 593, 156–157. 10.1038/d41586-021-01174-w. [DOI] [PubMed] [Google Scholar]
- 59.Pimentel JF, Murta L, Braganholo V, and Freire J (2019). A large-scale study about quality and reproducibility of Jupyter notebooks. In 2019 IEEE/ACM 16th International Conference on Mining Software Repositories (MSR), pp. 507–517. 10.1109/MSR.2019.00077. [DOI] [Google Scholar]
- 60.Stein LD, Knoppers BM, Campbell P, Getz G, and Korbel JO (2015). Data analysis: Create a cloud commons. Nature 523, 149–151. 10.1038/523149a. [DOI] [PubMed] [Google Scholar]
- 61.Dolzhenko E, Deshpande V, Schlesinger F, Krusche P, Petrovski R, Chen S, Emig-Agius D, Gross A, Narzisi G, Bowman B, et al. (2019). ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35, 4754–4756. 10.1093/bioinformatics/btz431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dolzhenko E, Bennett MF, Richmond PA, Trost B, Chen S, van Vugt JJFA, Nguyen C, Narzisi G, Gainullin VG, Gross AM, et al. (2020). ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol 21, 102. 10.1186/s13059-020-02017-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hoang N, Cytrynbaum C, and Scherer SW (2018). Communicating complex genomic information: A counselling approach derived from research experience with Autism Spectrum Disorder. Patient Educ Couns 101, 352–361. 10.1016/j.pec.2017.07.029. [DOI] [PubMed] [Google Scholar]
- 64.Vorstman J, and Scherer SW (2021). What a finding of gene copy number variation can add to the diagnosis of developmental neuropsychiatric disorders. Curr Opin Genet Dev 68, 18–25. 10.1016/j.gde.2020.12.017. [DOI] [PubMed] [Google Scholar]
- 65.Werling DM, Brand H, An JY, Stone MR, Zhu L, Glessner JT, Collins RL, Dong S, Layer RM, Markenscoff-Papadimitriou E, et al. (2018). An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nature Genetics 50, 727–736. 10.1038/s41588-018-0107-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.An J-Y, Lin K, Zhu L, Werling DM, Dong S, Brand H, Wang HZ, Zhao X, Schwartz GB, Collins RL, et al. (2018). Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362, eaat6576. 10.1126/science.aat6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lambert SA, Abraham G, and Inouye M (2019). Towards clinical utility of polygenic risk scores. Hum Mol Genet 28, R133–R142. 10.1093/hmg/ddz187. [DOI] [PubMed] [Google Scholar]
- 68.Lewis CM, and Vassos E (2020). Polygenic risk scores: from research tools to clinical instruments. Genome Med 12, 44. 10.1186/s13073-020-00742-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.D’Abate L, Walker S, Yuen RKC, Tammimies K, Buchanan JA, Davies RW, Thiruvahindrapuram B, Wei J, Brian J, Bryson SE, et al. (2019). Predictive impact of rare genomic copy number variations in siblings of individuals with autism spectrum disorders. Nat Commun 10, 5519. 10.1038/s41467-019-13380-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Antaki D, Guevara J, Maihofer AX, Klein M, Gujral M, Grove J, Carey CE, Hong O, Arranz MJ, Hervas A, et al. (2022). A phenotypic spectrum of autism is attributable to the combined effects of rare variants, polygenic risk and sex. Nat Genet. 10.1038/s41588-022-01064-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Costain G, Cohn RD, Scherer SW, and Marshall CR (2021). Genome sequencing as a diagnostic test. CMAJ 193, E1626–E1629. 10.1503/cmaj.210549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bradshaw J, Steiner AM, Gengoux G, and Koegel LK (2015). Feasibility and effectiveness of very early intervention for infants at-risk for autism spectrum disorder: a systematic review. J Autism Dev Disord 45, 778–794. 10.1007/s10803-014-2235-2. [DOI] [PubMed] [Google Scholar]
- 73.Baribeau D, and Anagnostou E (2022). Novel treatments for autism spectrum disorder based on genomics and systems biology. Pharmacol Ther 230, 107939. 10.1016/j.pharmthera.2021.107939. [DOI] [PubMed] [Google Scholar]
- 74.Paulsen B, Velasco S, Kedaigle AJ, Pigoni M, Quadrato G, Deo AJ, Adiconis X, Uzquiano A, Sartore R, Yang SM, et al. (2022). Autism genes converge on asynchronous development of shared neuron classes. Nature 602, 268–273. 10.1038/s41586-021-04358-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Krupp DR, Barnard RA, Duffourd Y, Evans SA, Mulqueen RM, Bernier R, Rivière J-B, Fombonne E, and O’Roak BJ (2017). Exonic mosaic mutations contribute risk for autism spectrum disorder. Am J Hum Genet 101, 369–390. 10.1016/j.ajhg.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lim ET, Uddin M, De Rubeis S, Chan Y, Kamumbu AS, Zhang X, D’Gama AM, Kim SN, Hill RS, Goldberg AP, et al. (2017). Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat Neurosci 20, 1217–1224. 10.1038/nn.4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Siu MT, Butcher DT, Turinsky AL, Cytrynbaum C, Stavropoulos DJ, Walker S, Caluseriu O, Carter M, Lou Y, Nicolson R, et al. (2019). Functional DNA methylation signatures for autism spectrum disorder genomic risk loci: 16p11.2 deletions and CHD8 variants. Clin Epigenetics 11, 103. 10.1186/s13148-019-0684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Siu MT, Goodman SJ, Yellan I, Butcher DT, Jangjoo M, Grafodatskaya D, Rajendram R, Lou Y, Zhang R, Zhao C, et al. (2021). DNA methylation of the oxytocin receptor across neurodevelopmental disorders. J Autism Dev Disord 51, 3610–3623. 10.1007/s10803-020-04792-x. [DOI] [PubMed] [Google Scholar]
- 79.Regier AA, Farjoun Y, Larson DE, Krasheninina O, Kang HM, Howrigan DP, Chen B-J, Kher M, Banks E, Ames DC, et al. (2018). Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat Commun 9, 4038. 10.1038/s41467-018-06159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kendig KI, Baheti S, Bockol MA, Drucker TM, Hart SN, Heldenbrand JR, Hernaez M, Hudson ME, Kalmbach MT, Klee EW, et al. (2019). Sentieon DNASeq variant calling workflow demonstrates strong computational performance and accuracy. Front Genet 10, 736. 10.3389/fgene.2019.00736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, Corvelo A, Clarke WE, Musunuri R, Nagulapalli K, et al. (2022). High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185, 3426–3440.e19. 10.1016/j.cell.2022.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164. 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ramu A, Noordam MJ, Schwartz RS, Wuster A, Hurles ME, Cartwright RA, and Conrad DF (2013). DeNovoGear: de novo indel and point mutation discovery and phasing. Nat Methods 10, 985–987. 10.1038/nmeth.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Alexander DH, Novembre J, and Lange K (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19, 1655–1664. 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Harrison SM, Biesecker LG, and Rehm HL (2019). Overview of specifications to the ACMG/AMP variant interpretation guidelines. Curr Protoc Hum Genet 103, e93. 10.1002/cphg.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhu M, Need AC, Han Y, Ge D, Maia JM, Zhu Q, Heinzen EL, Cirulli ET, Pelak K, He M, et al. (2012). Using ERDS to infer copy-number variants in high-coverage genomes. Am J Hum Genet 91, 408–421. 10.1016/j.ajhg.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abyzov A, Urban AE, Snyder M, and Gerstein M (2011). CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21, 974–984. 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kosugi S, Momozawa Y, Liu X, Terao C, Kubo M, and Kamatani Y (2019). Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol 20, 117. 10.1186/s13059-019-1720-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, and Korbel JO (2012). DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, 333–339. 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cameron DL, Schröder J, Penington JS, Do H, Molania R, Dobrovic A, Speed TP, and Papenfuss AT (2017). GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res 27, 2050–2060. 10.1101/gr.222109.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Layer RM, Chiang C, Quinlan AR, and Hall IM (2014). LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15, R84. 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, and Saunders CT (2016). Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 95.Bartenhagen C, and Dugas M (2016). Robust and exact structural variation detection with paired-end and soft-clipped alignments: SoftSV compared with eight algorithms. Brief Bioinform 17, 51–62. 10.1093/bib/bbv028. [DOI] [PubMed] [Google Scholar]
- 96.Wala JA, Bandopadhayay P, Greenwald NF, O’Rourke R, Sharpe T, Stewart C, Schumacher S, Li Y, Weischenfeldt J, Yao X, et al. (2018). SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res 28, 581–591. 10.1101/gr.221028.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kronenberg ZN, Osborne EJ, Cone KR, Kennedy BJ, Domyan ET, Shapiro MD, Elde NC, and Yandell M (2015). Wham: identifying structural variants of biological consequence. PLoS Comput Biol 11, e1004572. 10.1371/journal.pcbi.1004572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. (2007). The diploid genome sequence of an individual human. PLoS Biol 5, e254. 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zook JM, Hansen NF, Olson ND, Chapman L, Mullikin JC, Xiao C, Sherry S, Koren S, Phillippy AM, Boutros PC, et al. (2020). A robust benchmark for detection of germline large deletions and insertions. Nat Biotechnol 38, 1347–1355. 10.1038/s41587-020-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, and Schatz MC (2018). Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 15, 461–468. 10.1038/s41592-018-0001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med 22, 245–257. 10.1038/s41436-019-0686-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kent WJ (2002). BLAT-the BLAST-like alignment tool. Genome Res 12, 656–664. 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, Lee BT, Learned K, Karolchik D, Hinrichs AS, et al. (2016). The UCSC Genome Browser database: 2016 update. Nucleic Acids Res 44, D717–725. 10.1093/nar/gkv1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575. 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, and Cunningham F (2016). The Ensembl Variant Effect Predictor. Genome Biol 17, 122. 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zerbino DR, Wilder SP, Johnson N, Juettemann T, and Flicek PR (2015). The Ensembl Regulatory Build. Genome Biol 16, 56. 10.1186/s13059-015-0621-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, Rosen N, Kohn A, Twik M, Safran M, et al. (2017). GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017. 10.1093/database/bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chèneby J, Ménétrier Z, Mestdagh M, Rosnet T, Douida A, Rhalloussi W, Bergon A, Lopez F, and Ballester B (2020). ReMap 2020: a database of regulatory regions from an integrative analysis of human and arabidopsis DNA-binding sequencing experiments. Nucleic Acids Res 48, D180–D188. 10.1093/nar/gkz945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Volders P-J, Helsens K, Wang X, Menten B, Martens L, Gevaert K, Vandesompele J, and Mestdagh P (2013). LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res 41, D246–251. 10.1093/nar/gks915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kuksa PP, Amlie-Wolf A, Katanić Ž, Valladares O, Wang L-S, and Leung YY (2019). DASHR 2.0: integrated database of human small non-coding RNA genes and mature products. Bioinformatics 35, 1033–1039. 10.1093/bioinformatics/bty709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang J, Song L, Grover D, Azrak S, Batzer MA, and Liang P (2006). dbRIP: a highly integrated database of retrotransposon insertion polymorphisms in humans. Hum Mutat 27, 323–329. 10.1002/humu.20307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z, et al. (2014). Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet 94, 677–694. 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233. 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Riggs ER, Church DM, Hanson K, Horner VL, Kaminsky EB, Kuhn RM, Wain KE, Williams ES, Aradhya S, Kearney HM, et al. (2012). Towards an evidence-based process for the clinical interpretation of copy number variation. Clin Genet 81, 403–412. 10.1111/j.1399-0004.2011.01818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li H (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Weissensteiner H, Pacher D, Kloss-Brandstätter A, Forer L, Specht G, Bandelt H-J, Kronenberg F, Salas A, and Schönherr S (2016). HaploGrep 2: mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res 44, W58–63. 10.1093/nar/gkw233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lott MT, Leipzig JN, Derbeneva O, Xie HM, Chalkia D, Sarmady M, Procaccio V, and Wallace DC (2013). mtDNA variation and analysis using Mitomap and Mitomaster. Curr Protoc Bioinformatics 44, 1.23.1–26. 10.1002/0471250953.bi0123s44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dupuis A, Moon MJ, Brian J, Georgiades S, Levy T, Anagnostou E, Nicolson R, Schachar R, and Crosbie J (2021). Concurrent validity of the ABAS-II questionnaire with the Vineland II interview for adaptive behavior in a pediatric ASD sample: high correspondence despite systematically lower scores. J Autism Dev Disord 51, 1417–1427. 10.1007/s10803-020-04597-y. [DOI] [PubMed] [Google Scholar]
- 122.Lord C, Rutter M, DiLavore P, Risi S, Gotham K, and Bishop S (2012). Autism Diagnostic Observation Schedule–2nd Edition (ADOS-2). (Western Psychological Services; ). [Google Scholar]
- 123.Hus V, and Lord C (2014). The autism diagnostic observation schedule, module 4: revised algorithm and standardized severity scores. J Autism Dev Disord 44, 1996–2012. 10.1007/s10803-014-2080-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Samocha KE, Kosmicki JA, Karczewski KJ, O’Donnell-Luria AH, Pierce-Hoffman E, MacArthur DG, Neale BM, and Daly MJ (2017). Regional missense constraint improves variant deleteriousness prediction. bioRxiv, 148353. 10.1101/148353. [DOI] [Google Scholar]
- 125.Liu X, Wu C, Li C, and Boerwinkle E (2016). dbNSFP v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat 37, 235–241. 10.1002/humu.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. (2010). The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 38, W214–220. 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Genotype-Computed Ancestry of Individuals with WGS Data Used in This Study, Related to Table 1. Distributions are shown for (A) MSSNG, (B) SSC, and (C) 1000 Genomes Project.
Figure S2. Screenshots of Interfaces for Accessing MSSNG Data, Related to Table 1. Screenshots are shown for (A) CNV querying, (B) the Phenotype Data Explorer, and (C) a Terra notebook.
Figure S3. Distributions of Heteroplasmy Deltas, Related to STAR Methods. Deltas are defined as child heteroplasmy minus mother heteroplasmy for a given position in the mtDNA sequence. MSSNG ASD: n=35,279 variants; MSSNG non-ASD: n=2,171; SSC ASD: n=29,005; SSC non-ASD: n=18,328. No significant differences were observed between the shifts from mother to children with ASD and from mother to non-ASD siblings in either MSSNG (ASD: 0.0026 ± 0.046; non-ASD: 0.0043 ± 0.058; p=0.18) or SSC (ASD: 0.0012 ± 0.042; non-ASD: 0.0014 ± 0.048; p=0.65).
Figure S4: Examples of Multiplex Families from MSSNG with ASD-Associated Rare Variants, Related to Figure 4.
Figure S5. Non-coding Transmission Tests, Related to STAR Methods. Odds ratios are shown for the number of singleton variants that were transmitted versus non-transmitted in (A) individuals with ASD in MSSNG, (B) individuals with ASD in SSC, and (C) non-ASD siblings in SSC.
Figure S6. PRS Analysis, Related to Figure 6. (A) PRS distribution when samples are ranked from highest to lowest score. (B) Sex-stratified polygenic transmission disequilibrium tests. (C) Comparison between individuals with ASD from MPX families in MSSNG and individuals with ASD in SSC (which are all SPX families). (D) Comparison between fathers and mothers without ASD, stratified by dataset (MSSNG or SSC). (E) Comparison between parents without ASD and 1000 Genomes Project population controls.
Figure S7. Distributions of Different Categories of Variants, Stratified by Sequencing Platform and DNA Library Preparation Method, Related to STAR Methods. Distributions are shown for (A) SNVs and (B) indels.
Table S2: ASD Gene Discovery, Related to Figure 2.
Table S3: Duplication Structure Resolution, Related to STAR Methods.
Table S4: Mitochondrial Variation, Related to STAR Methods.
Table S6: Non-Coding Analysis, Related to STAR Methods.
Table S7: Methods Details, Related to STAR Methods.
Data Availability Statement
Access to MSSNG and SFARI data can be obtained by completing data access agreements at https://research.mss.ng and https://www.sfari.org/resource/sfari-base, respectively. The 1000 Genomes Project WGS data are publicly available via Amazon Web Services (https://docs.opendata.aws/1000genomes/readme.html).
All original code has been deposited at Zenodo and is publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.