

Abstract
Expansion of a triplet repeat tract in exon 1 of the HTT gene causes Huntington’s disease (HD). The mutant HTT protein (mHTT) has numerous aberrant interactions with diverse, pleiomorphic effects. Lowering mHTT is a promising approach to treat HD, but it is unclear when lowering should be initiated, how much is necessary, and what duration should occur to achieve benefits. Furthermore, the effects of mHTT lowering on brain lipids have not been assessed. Using a mHtt-inducible mouse model, we analyzed mHtt lowering initiated at different ages and sustained for different time-periods. mHTT protein in cytoplasmic and synaptic compartments of the striatum was reduced 38–52%; however, there was minimal lowering of mHTT in nuclear and perinuclear regions where aggregates formed at 12 months of age. Total striatal lipids were reduced in 9-month-old LacQ140 mice and preserved by mHtt lowering. Subclasses important for white matter structure and function including ceramide (Cer), sphingomyelin (SM), and monogalactosyldiacylglycerol (MGDG), contributed to the reduction in total lipids. Phosphatidylinositol (PI), phosphatidylserine (PS), and bismethyl phosphatidic acid (BisMePA) were also changed in LacQ140 mice. Levels of all subclasses except ceramide were preserved by mHtt lowering. mRNA expression profiling indicated that a transcriptional mechanism contributes to changes in myelin lipids, and some but not all changes can be prevented by mHtt lowering. Our findings suggest that early and sustained reduction in mHtt can prevent changes in levels of select striatal proteins and most lipids, but a misfolded, degradation-resistant form of mHTT hampers some benefits in the long term.
Keywords: Huntington’s disease, Striatum, Lipidomics, Myelin, Sphingolipid
1. Introduction
Huntington’s disease (HD) is a heritable neurodegenerative disease caused by a CAG expansion in the huntingtin (HTT) gene. The protein product, huntingtin (HTT), is ubiquitously expressed but enriched in neurons (Difiglia et al., 1995; Sharp et al., 1995) and has been implicated in numerous molecular functions including vesicle trafficking, autophagy, transcription and DNA repair (Reviewed by (Caviston and Holzbaur, 2009; Gao et al., 2019; Saudou and Humbert, 2016)). HTT is expressed in various cell types throughout the body (Li et al., 1993; Strong et al., 1993) and has also been shown to have essential functions during development (Barron et al., 2021; Duyao et al., 1995; McKinstry et al., 2014; Nasir et al., 1995; O’Kusky et al., 1999; Zeitlin et al., 1995). The CAG-repeat expansion encodes a polyglutamine expansion in HTT, which causes protein accumulation and aggregation and has pleiomorphic effects that contribute to HD pathology ranging from mitochondrial dysfunction, transcriptional defects, cholesterol mishandling, altered palmitoylation, and metabolic changes from altered signaling in the hypothalamus (Nopoulos, 2016; Petersen and Bjorkqvist, 2006).
HTT transcription lowering strategies have become central to HD translational studies (reviewed by (Tabrizi et al., 2019)). Interventional trials have targeted total HTT (that is, normal and mutant HTT) using antisense oligonucleotides (ASOs) (reviewed by (Caron et al., 2018)) or HTT pre-mRNA using an oral drug, branaplam (LMI070) (Keller et al., 2022). In animal models (mice, sheep, and nonhuman primates), additional strategies of gene therapy using transcriptional repressors to target expression from the mutant allele (Zeitler et al., 2019) and modified interfering RNAs (RNAi) (Alterman et al., 2019) or AAV expressing microRNAs (miRNAs) or short hairpin RNAs (shRNAs) (Keeler et al., 2016; McBride et al., 2011) to target RNA levels are actively being pursued (Boudreau et al., 2009; Datson et al., 2017; DiFiglia et al., 2007; Drouet et al., 2009; Kordasiewicz et al., 2012; Rodriguez-Lebron et al., 2005; Stanek et al., 2014; Stanek et al., 2013; Zeitler et al., 2019). Although lowering total HTT in humans was hoped to be generally safe (Leavitt et al., 2020), insufficient evidence currently exists to make this conclusion. There is support for selective lowering of mHTT based on data in mice that show loss of wild type (WT) HTT may affect neuronal function including synaptic connectivity (Barron et al., 2021; Dragatsis et al., 2000; McKinstry et al., 2014; Mehler et al., 2019).
Proteins known to change with HD progression may serve as useful readouts for investigating the efficacy of HTT lowering. For instance, DARPP32 is enriched in striatal projection neurons and is progressively reduced in HD patient postmortem brain and in HD mouse models (Bibb et al., 2000). The cAMP phosphodiesterase PDE10A is reduced early and sustainably in HD mouse striatum measured both by western blot and mass spectrometry (Sapp et al., 2020; Skotte et al., 2018; Zeitler et al., 2019) or ligand binding (Bertoglio et al., 2022; Zeitler et al., 2019). Microarray analysis in neurons derived from human stem cells (Mehta et al., 2018), mass spectrometry and western blot analyses in striatal synaptosomes (Sapp et al., 2020) and immunofluorescence (IF) studies in mice (Oyama et al., 2006) showed SCN4B is lowered in HD models. ATP5A protein levels are altered in numerous mass spectrometry studies (Agrawal and Fox, 2019; Langfelder et al., 2016; McQuade et al., 2014; Sapp et al., 2020).
The intracellular location of HTT positions it to affect lipids in membranes. HTT normally associates with membranes where it interacts directly with lipid bilayers (Kegel et al., 2009b). Lipids comprise ~50% of the total dry weight in brain and have diverse cellular roles as membrane structural components, signaling molecules, and sources of energy (Hyuk Yoon et al., 2022). Disruptions to lipid composition/metabolism have been linked to disease (Harayama and Riezman, 2018); thus, their study may provide insight into disease mechanism or lead to discovery of new biomarkers. We and others have described alterations to lipid levels in HD mouse models (Carroll et al., 2015; Di Pardo et al., 2017; Farzana et al., 2023; Iuliano et al., 2021; Vodicka et al., 2015) and post-mortem HD brain tissue (Hunter et al., 2018; Hunter et al., 2021; Phillips et al., 2022). Specific glycerophospholipids showed differential associations with mHTT (Kegel et al., 2009a) and mHTT can alter the biophysical properties of lipid bilayers (Chaibva et al., 2018; Kegel et al., 2009b) underscoring the potential for direct consequences of the presence of mHTT on cellular lipids.
The effects of lowering total Htt or mHtt alone on striatal proteins and behavioral and psychiatric measures have been investigated in HD mouse models after delivery of reagents to the striatum or lateral ventricle (Southwell et al., 2018; Zeitler et al., 2019). Biomarkers (mHTT levels and aggregation) that are responsive to total or mHtt lowering have been investigated in HD mouse models (Bertoglio et al., 2022; Caron et al., 2022; Southwell et al., 2015). However, the impact of lowering mHtt on lipids has not been examined. Here we used the inducible HD knock-in mouse model, LacQ140, in which the expression of mHtt alone is regulated by adding or withdrawing the lactose analog isopropyl b-D-1-thiogalactopyranoside (IPTG) in their drinking water (Coffey et al., 2020; Cronin et al., 2001; Marchionini et al., 2022; Scrable, 2002), to study the effects of mHtt lowering for different time periods on proteins known to be affected in HD models and using a survey of lipids. We also tracked changes in levels of soluble and aggregated mHTT protein in different subcellular compartments. The results show that early and sustained reduction in mHtt in this HD mouse model can delay select protein changes and prevent numerous lipid derangements. Using a systems biology approach, we integrated our lipidomic findings with a previously generated transcriptomic dataset, identifying dysregulation of pathways relating to myelin and white matter including genes directly involved in biosynthetic pathways for lipids we found changed in LacQ140 mice. An SDS-soluble misfolded form of mHTT persists, however, and benefits eventually dwindled.
2. Materials and methods
2.1. Animals
The LacO/LacIR-regulatable HD mouse model (LacQ140) was generated by crossing the HttLacQ140/+ mouse to the TgACTB-lacI*Scrb mouse as previously described (Cronin et al., 2001; Marchionini et al., 2022). The default state of the LacQ140 mouse is global repression of mHtt due to Lac Repressor binding to the Lac operators. The continuous administration of IPTG starting from E5 interrupts the binding between the Lac repressor and operators, resulting in a de-repressed state, and maximal expression of mHtt in LacQ140. All WT mice were HttLacO+/+; b-actin-LacIR tg. Mice were provided with enrichment (envirodry, play tunnels, Bed-o’cobs and plastic bones) and housed uniform for genotype, gender, and treatment. Mice were fed ad libitum. The lactose analog IPTG was provided in drinking water (at 10mM) which de-represses the LacQ140 allele and keeps normal mHtt expression. During embryonic development, mHtt expression levels were maintained at normal levels by administering IPTG to pregnant dams starting at embryonic day 5 (E5). IPTG was administered never (mHtt always repressed), continuously (mHtt continuously expressed) or withdrawn at 2 or 8 months (mHtt expressed normally then repressed at 2 or 8 months). The CAG repeat length range in HttLacO-Q140/+ mice was 143–157 with average of 148 and median of 148 CAG. Our biochemical data from 12-month-old mice come from the same animals used for longitudinal behavioral studies reported previously (Marchionini et al., 2022), whereas data from 6- and 9-month-old mice come from animals born at the same time, housed in the same room, and aged together as a parallel cohort.
2.2. Sample preparation
The striatum from one hemisphere for each mouse was homogenized in 750 μl 10mM HEPES pH7.2, 250mM sucrose, 1uM EDTA + protease inhibitor tablet (Roche Diagnostics GmbH, Mannheim, Germany) + 1mM NaF + 1mM Na3VO4. A 150 μl aliquot of this crude homogenate was removed and protein concentration was determined using the Bradford method (BioRad, Hercules, CA). Subcellular fractionation by density gradient ultracentrifugation using Optiprep was performed on remaining 600 μl sample for the 6- and 12-month-old mice as previously described (Iuliano et al., 2021).
2.3. Capillary immunoassay
Equal amounts of protein from the crude homogenates were analyzed using the automated simple western system, Wes (ProteinSimple, Bio-Techne, San Jose, CA), which utilizes a capillary-based immunoassay. The protocol described in the manual was followed to detect HTT, GFAP and DARPP32 using 0.6 μg of sample. Quantitative analysis of protein levels is done automatically using the Compass for Simple Western Software (ProteinSimple) on electropherogram data. The peak area (using automatic “dropped line” option in software) of each protein of interest was normalized to the peak area of the vinculin loading control. Figures show protein bands similar to traditional western blots using “lane view” option in the Compass software to create a blot-like image from the electropherogram data.
2.4. Western blot analysis
Equal amounts of protein from the crude homogenates were analyzed by western blot for levels of HTT and other proteins of interest as previously described (Sapp et al., 2020). Briefly, 10 μg of protein were separated by SDS-PAGE, transferred to nitrocellulose, and probed with primary antibody overnight. Nitrocellulose membranes were cut into horizonal strips to maximize the number of antibodies that could be probed on one blot. Peroxidase labeled secondary antibodies were used with the SuperSignal West Pico Chemiluminescent substrate (Thermo-Scientific, Rockford, IL, #34580) and digital images were captured with a CCD camera (AlphaInnotech, Bayern, Germany). For western blot analysis of subcellular fractions, equal volumes of each sample (15 μl) were separated by SDS-PAGE. Pixel intensity quantification of the western blot signals on the digital images was determined using ImageJ software (NIH) by manually circling each band and multiplying the area by the average signal intensity. The total signal intensity was normalized to vinculin or GAPDH loading controls.
2.5. Antibodies
The following antibodies and dilutions were used in this study: Anti-HTT Ab1 (aa1–17,(Difiglia et al., 1995)) 1:50 for capillary immunoassay and 1:2000 for western blot; anti-HTT EPR5526 (Abcam, Waltham, MA, ab109115, 1:2000 for western blot); anti-polyQ MW1 (MilliporeSigma, Burlington, MA, MABN2427, 1:50 for capillary immunoassay); anti-polyQ PHP3 (generous gift from Dr. Ali Khoshnan, 1:2000 for western blot); Anti-PDE10A (Abcam, Waltham, MA, #ab177933, 1:2000 for western blot); Anti-DARPP32 (Abcam, #ab40801, 1:2000 for capillary immunoassay); Anti-GFAP (MilliporeSigma, Burlington, MA, AB5804, 1:3000 for capillary immunoassay); Anti-GAPDH (MilliporeSigma, Burlington, MA, #MAB374, 1:10000 for western blot); Anti-Sodium channel subunit beta-4 (Abcam, Waltham, MA, #ab80539, 1:500 for western blot); Anti-vinculin (Sigma, St. Louis, MO, #V9131, 1:5000 for capillary immunoassay, 1:2000 for western blot); Anti-ATP5A (Abcam, Waltham, MA, #ab14748, 1:2000 for western blot); Anti-HTT MW8 (University of Iowa Developmental Studies Hybridoma Bank, 1:1000 for filter trap); Anti-HTT S830 (generous gift from Dr. Gillian Bates, (Landles et al., 2010) 1:8000), HDAC1 (Abcam, Waltham, MA, ab32369–7, 1:4000).
2.6. Filter trap assay
Based on protocol described in (Scherzinger et al., 1997; Weiss et al., 2008), equal protein amounts for each sample (40 μg) were brought up to 50 μl volume with PBS and 50 μl 4% SDS in PBS was added to each sample to make final concentration 2% SDS. A cellulose acetate membrane was wet in 2% SDS/PBS and placed in dot blot apparatus. The 100 μl samples were added to each well and pulled through the membrane with a vacuum then washed 3 times with 200 μl 2% SDS/PBS. The membrane was removed from the apparatus, washed in Tris buffered saline + 0.1% Tween-20 (TBST) then processed for western blot using MW8 or S830 antibodies. The total signal intensity of each dot was measured in ImageJ by circling the entire dot and multiplying the area by the average signal intensity minus the background signal from an empty dot.
2.7. Statistical analysis of protein data
One-way ANOVA with Tukey’s multiple comparison test was performed to determine significance between groups. Asterisks on graphs show p values and are described in the figure legends.
2.8. Lipid extraction
Lipids were extracted using methyl tert-butyl ether (MTBE) as previously described and analyzed using ion switching and molecular assignment as previously described (Breitkopf et al., 2017; Iuliano et al., 2021; Matyash et al., 2008). Each age group was processed together. Crude homogenates (100 μl) of dissected mouse striatum (see Sample preparation, 2.2) were transferred into 20 ml glass scintillation vials. 750 μl of HPLC grade methanol was added to each sample, then vials were vortexed. 2.5 ml of MTBE was then added to each sample and incubated on a platform shaker for 1 hour. After incubation, 625 μl of water was added to induce polar and non-polar phase separation. The non-polar lipid containing (upper) phase was collected into a new vial, and the polar (lower) phase was subsequently re-extracted with 1 ml of MTBE/methanol/water (10/3/2.5, v/v/v). Following re-extraction, the lipid containing phases were combined and allowed to dry on a platform shaker, then further dried with nitrogen gas. Extracted lipids were hand delivered to the Beth Israel Deaconess Medical Center Mass Spectrometry Core Facility.
2.9. Lipid annotation
Data for each timepoint was classified by LIPID MAPS category: glycerophospholipids, glycerolipids, sphingolipids, sterol lipids, fatty acyls, and prenol lipids were detected (Liebisch et al., 2020). Each category contains distinct subclasses as annotated below. Glycerophospholipids: Phosphatidylcholine (PC), Phosphatidylethanolamine (PE), Phosphatidylserine (PS), Phosphatidylinositol (PI), Methylphosphocholine (MePC), Phosphatidic acid (PA), Bis-methyl phosphatidic acid (BisMePA), Dimethyl phosphatidylethanolamine (dMePE), Phosphatidylgylcerol (PG), Bis-methylphosphatidylserine (BisMePS), Bis-methyl phosphatidyl ethanolamine (BisMePE), Cardiolipin (CL), Phosphatidylethanol (PEt), Biotinyl-phosphoethanolamine (BiotinylPE), Phosphatidylmethanol (PMe), Phosphatidylinositol-bisphosphate (PIP2), Phosphatidylinositol-monophosphate (PIP), Lysophosphatidylcholine (LPC), Lysophosphatidylethanolamine (LPE), Lysophosphatidylserine (LPS), Lysophosphatidylinositol (LPI), Lysophosphosphatidylgylcerol (LPG), Lysodimethyl phosphatidyl ethanolamine (LdMePE). Glycerolipids: Triglyceride (TG), Monogalactosyldiacylglycerol (MGDG), Monogalactosylmonoacylglycerol (MGMG), Diglyceride (DG), Sulfoquinovosylmonoacylglycerol (SQMG), Sulfoquinovosyldiacylglycerol (SQDG). Sphingolipids: Hexosylceramides (Hex1Cer), Simple Glc series (CerG1), Sphingomyelin (SM), Ceramide (Cer), Ceramide phosphate (CerP), Sulfatide (ST), Sphingoid base (So), Sphingomyelin phytosphingosine (phSM), Simple Glc series (CerG2GNAc1), Ceramide phosphorylethanolamine (CerPE), Sphingosine (SPH), Dihexosylceramides (Hex2Cer). Sterol lipids: Cholesterol ester (ChE), Zymosterol (ZyE). Fatty acyls: Fatty acid (FA), Acyl Carnitine (AcCa). Prenol lipids: Coenzyme (Co).
Individual lipid species were annotated according to sum composition of carbons and double bonds in the format lipid subclass (total number of carbons: total number of double bonds). If distinct fatty acid chains could be identified, they were annotated separated by an underscore (ex. PC 32:1, or PC (16:0_16:1). Using this approach, we cannot determine the sn-1 or sn-2 positions of the fatty acid chains. Lipid species within the sphingolipid category contain prefixes ‘d’ or ‘t’ to denote dihydroxy or tri-hydroxy bases. For example, SM(d18:1_23:0) contains 2 hydroxyl groups. The Hex1Cer subclass is comprised of both glucosylceramide (GlcCer) and galactosylceramide (GalCer); the orientation of one of the hydroxyl groups in Glc differs from in Gal, and thus cannot be resolved by these methods (Reza et al., 2021). Plasmanyl lipid species (ether linked) are annotated by ‘e’ and plasmenyl/plasmalogen (vinyl ether linked) lipid species are annotated by ‘p’ (ex. PC (36:5e) or PE (16:0p_20:4) (Koelmel et al., 2017).
2.10. Lipidomics statistics and data visualization
Heatmap and hierarchical clustering in Fig. 4 were generated using Morpheus from the Broad Institute (Cambridge, MA, https://software.broadinstitute.org/morpheus). Hierarchical clustering was performed across all rows (lipid subclasses) using the one minus Pearson correlation distance metric. Rows determined to be the most similar are merged first to produce clusters, working iteratively to merge rows into clusters. The dendrogram displays the order of clustering with the most similar rows shown in closest proximity. Lipid expression values are assigned to colors based on the minimum (blue, low relative expression) and maximum (red, high relative expression) values for each independent row. Heatmap in Fig. 5 was generated in R using the ComplexHeatmap package v2.16.0 (Gu, 2022). Each column represents data from one animal. Statistical significance was determined by one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test between treatment groups for lipid subclasses (6mo: N = 36, 9mo: N = 24, 12mo: N = 29) or lipid species (6mo: N = 800, 9mo: N = 632, 12mo: N = 735). The two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli was applied to ANOVA p-values to control the false discovery rate (FDR) with significance accepted at q<0.05 (Q=5%).
2.11. RNA-sequencing analysis
The raw counts matrix derived from striatal samples in the LacQ140 mouse model was accessed from Gene Expression Omnibus (GEO GSE156236). The 6-month cohort included n = 10 mice per group (WT, LacQ140, LacQ140_2M, LacQ140_A) and the 12-month cohort included n = 10 WT, n = 9 LacQ140, n = 9 LacQ140_8M, n = 10 LacQ140_2M, and n = 10 LacQ140_A. Analysis was performed using R v4.3.0 in the RStudio IDE (2023.03.0+386). Transcripts with fewer than 10 total counts were removed from the analysis. Differential expression analysis was performed using DESeq2 v1.40.1 (Love et al., 2014). P-values were computed with the Wald test and were corrected for multiple testing using the Benjamini and Hochberg procedure (Benjamini and Hochberg, 1995). Genes with a fold change greater than 20% in either direction and with an adjusted p-value < 0.05 were considered differentially expressed. Functional enrichment analysis was conducted using the ClusterProfiler package v4.8.1. Over-representation analysis was performed for upregulated or downregulated differentially expressed genes (DEGs) with the enrichGO function (org.Mm.eg.db v3.17.0). Gene set enrichment analysis was performed with the gseGO function using a ranked list of genes (Wald statistic) for each comparison (LacQ140 vs WT, LacQ140_8M vs WT, LacQ140_2M vs WT, and LacQ140_A vs WT) at 6 and 12 months. Heatmaps were plotted using ComplexHeatmap package v2.16.0 with rows representing individual animals and rows representing genes.
3. Results
3.1. Time course of mHTT protein lowering with regulated transcriptional repression using LacQ140 mouse striatum
We used the inducible HD knock-in mouse model, LacQ140, in which the expression of mHtt throughout the body was regulated by adding or withdrawing the lactose analog isopropyl β-D-1-thiogalactopyranoside (IPTG) in drinking water using an established Lac operator/repressor system (Fig. 1a) (Marchionini et al., 2022; Scrable, 2002). We used the model to lower mHtt from conception, starting at 2 or 8 months of age. To control for the effects of IPTG, WT mice also received IPTG treatment over their lifetime. The striatum was examined at 6, 9, and 12 months of age (Fig. 1b). Antibodies against varied epitopes within HTT detect different pools of normal and mutant HTT by immunofluorescence and immunohistochemistry (Difiglia et al., 1995; Kegel et al., 2002; Sapp et al., 1997; Tousley et al., 2019; Trettel, 2000). Numerous conformations (over 100 distinct classes) of purified HTT and mHTT have been identified by electron microscopy in vitro (Seong et al., 2010). Polyglutamine (polyQ) expansion induces confirmational changes particular to mHTT detectible even after SDS-PAGE and/or western blot with several antibodies to regions in the poly-Q region (MW1 (Ko et al., 2001; Legleiter et al., 2009) and 3B5H10 (Miller et al., 2011)) and in the adjacent polyproline region (PHP1–4 (Ko et al., 2018) and S830 (Sathasivam et al., 2010)).
Fig. 1.
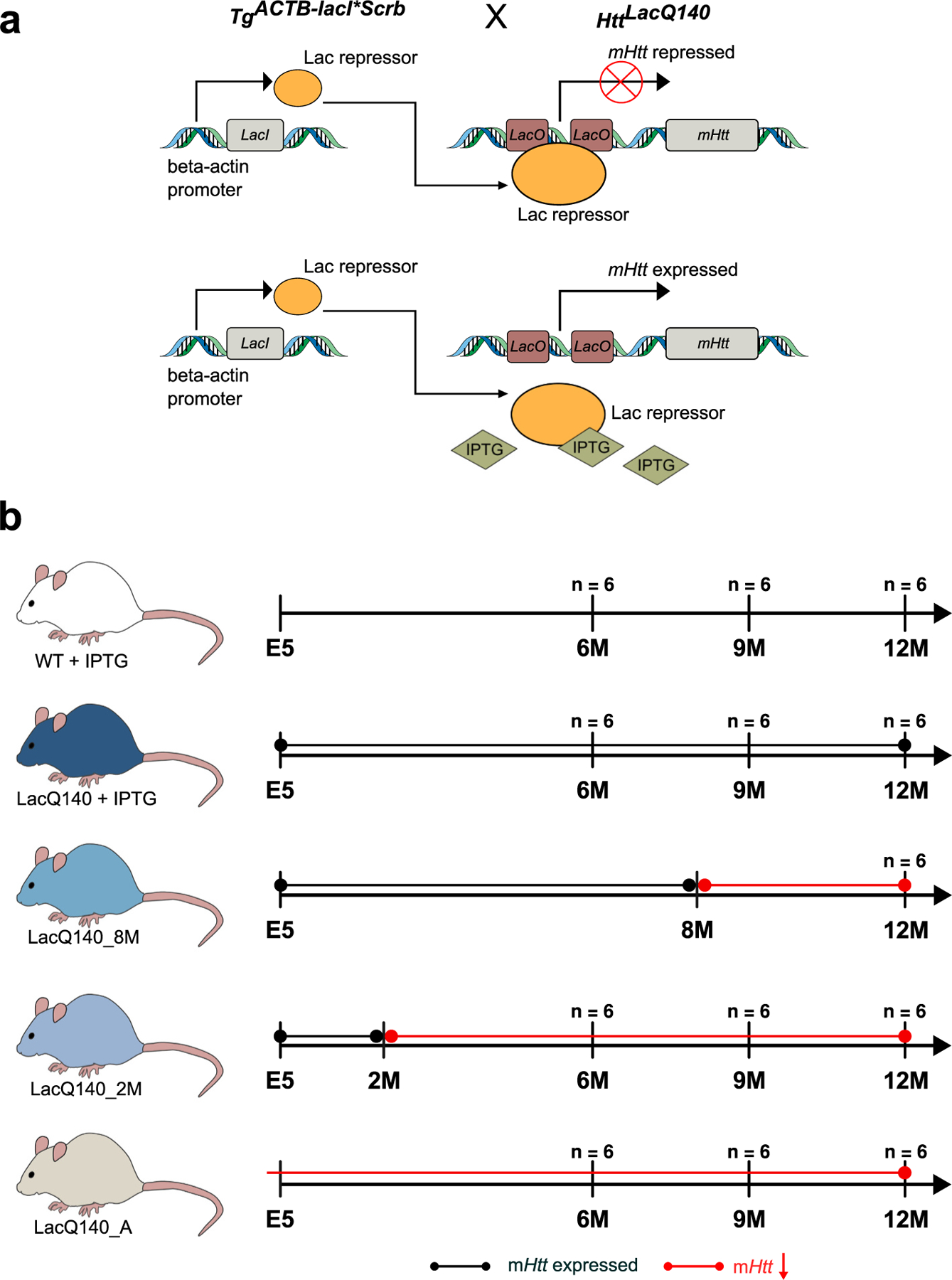
Generation of LacQ140 mice and treatment paradigm.
(a) The LacO/LacIR-regulatable HD mouse model (LacQ140) was generated by crossing the HttLacQ140/+ mouse to the TgACTB-lacI*Scrb mouse (Cronin et al., 2001) as previously described (Marchionini et al., 2022). The default state of the LacQ140 mouse is global repression of mHtt due to Lac Repressor binding to the Lac operators. Administration of IPTG starting from embryonic day 5 (E5) interrupts the binding between the Lac repressor and operators, resulting in a de-repressed state, and maximal expression of mHtt in LacQ140. All WT mice were HttLacO+/+; b-actin-LacIR tg. (b) Mice were fed ad libitum; the lactose analog IPTG was provided in drinking water (at 10mM) which de-represses the LacQ140 allele and keeps normal mHtt expression. During embryonic development, mHtt expression levels were maintained at normal levels by administering IPTG to pregnant dams starting at embryonic day 5 (E5). IPTG was continuously administered to WT mice. IPTG was administered continuously (mHtt continuously expressed, LacQ140), withdrawn at 8 months (mHtt repressed beginning at 8 months, LacQ140_8M), withdrawn 2 at months (mHtt repressed beginning at 2 months, LacQ140_2M), or never administered (mHtt always repressed, LacQ140_A). Tissue for each group (except LacQ140_8M) was collected at 6, 9, and 12 months of age.
To assess lowering, full length HTT and mHTT proteins were detected by capillary immunoassay using anti-HTT antibody Ab1 targeting aa 1–17 and anti-polyQ antibody MW1 (Fig. 2), and by western blot using anti-HTT antibody EPR5526, and anti-mHTT antibody PHP3, which recognizes an altered conformer of polyproline in mHTT (Ko et al., 2018) (Fig. S1). Overall, results showed lowering of 35–52% of mHTT detected with Ab1, MW1 and PHP3 in 6- and 9-month mice with transcriptional repression (Fig. 2a, Fig. S1a) and (Fig. 2b, Fig. S1b).
Fig. 2.
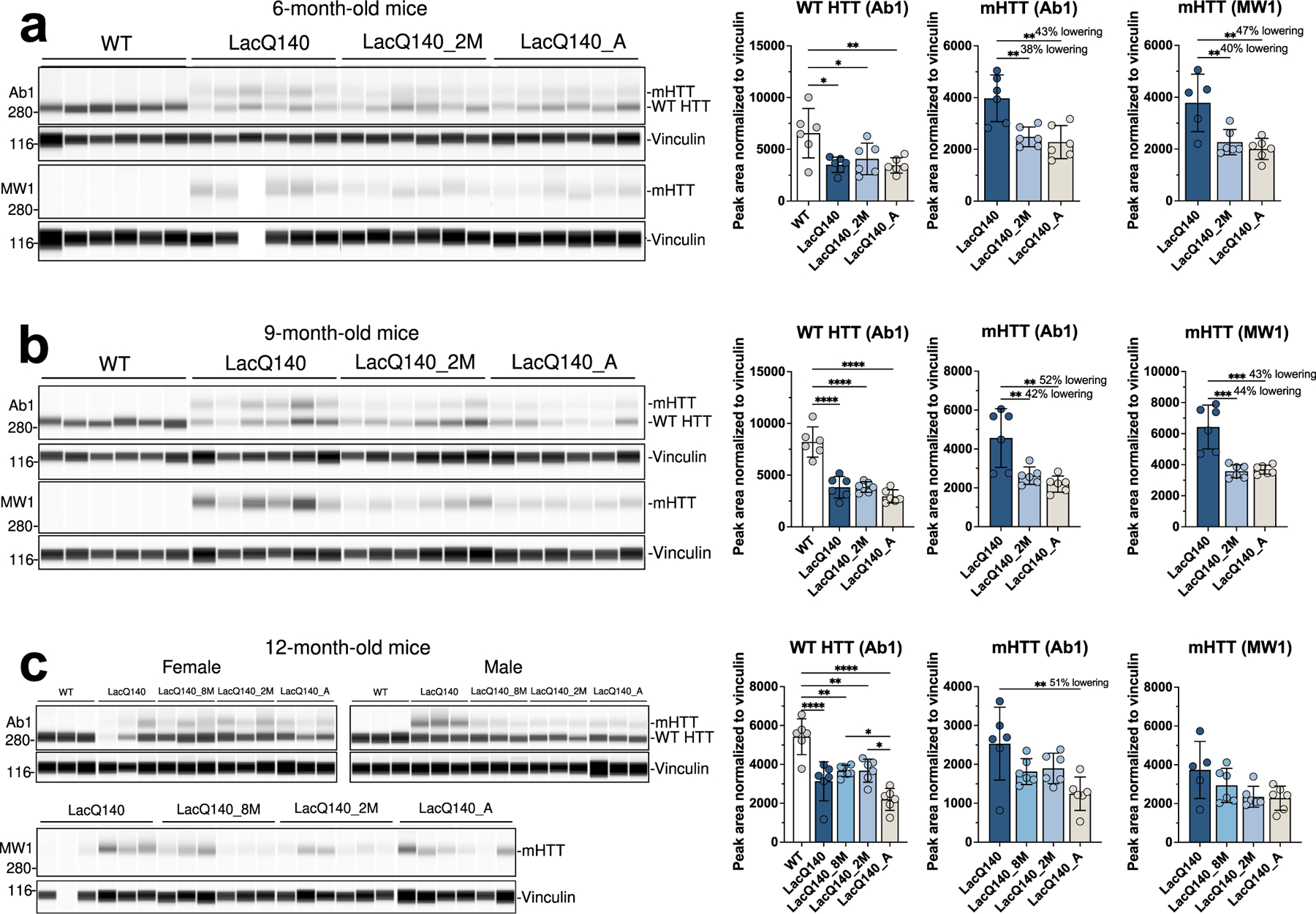
Analysis of mHTT protein levels in crude homogenates of 6-, 9- and 12-month old mice.
HTT levels were analyzed by capillary immunoassay on equal amounts of protein (0.6 μg) using anti-HTT antibody Ab1 and anti-polyQ antibody MW1. (a) Peak area analysis performed using Compass software in 6-month-old mice shows a significant decrease in WT HTT as detected with Ab1 in all LacQ140 mice compared to WT mice (F(3, 20) = 5.674, **P=0.0056, One-way ANOVA with Tukey’s multiple comparison test, n=6). mHTT levels are significantly lower in LacQ140_2M and LacQ140_A as detected with both Ab1 and MW1 compared to LacQ140 (a, Ab1: F(2, 15) = 11.25, **P=0.0010, −38% and −43% respectively; MW1: F(2, 14) = 9.879, **P=0.0021, −40% and −47% respectively). (b) Peak area analysis in 9-month-old mice shows a significant decrease in WT HTT as detected with Ab1 in all LacQ140 mice compared to WT mice (F(3, 20) = 34.67, ****P<0.0001, One-way ANOVA with Tukey’s multiple comparison test, n=6). mHTT levels are significantly lower in LacQ140_2M and LacQ140_A, as detected with both Ab1 and MW1, compared to LacQ140 (b, Ab1: F(2, 15) = 10.82, **P=0.0012, −42% and −52% respectively; MW1: F(2, 15) = 20.82, ****P<0.0001, −44% and −43% respectively). (c) Peak area analysis in 12-month-old mice shows a significant decrease in WT HTT as detected with Ab1 in all LacQ140 mice compared to WT mice (F(4, 25) = 15.81, ****P<0.0001, One-way ANOVA with Tukey’s multiple comparison test, n=6). WT HTT was significantly lower in LacQ140_A compared to LacQ140_8M and LacQ140_2M mice. mHTT levels are significantly lower in LacQ140_A mice, as detected with Ab1, compared to LacQ140 (c, Ab1: F(3, 20) = 5.017, **P=0.0094, −51%, One-way ANOVA with Tukey’s multiple comparison test, n=6). Asterisks on graphs represent Tukey’s multiple comparison test, n=6 mice per group (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Although EPR5526 recognizes both WT and mHTT, no significant lowering of mHTT protein was measured with mHtt gene repression using this antibody in 6-month mice. In the 12-month mice, mHTT was significantly reduced in LacQ140_A (51%) mice compared to LacQ140 mice (Fig. 2c). However, this was observed only in the capillary immunoassay using antibody Ab1 but not with antibodies MW1, EPR5526, or PHP3 (Fig. S1c). mHtt repression in the LacQ140_2M and LacQ140_8M mice yielded no significant reduction of mHTT protein levels compared to LacQ140 mice using any antibody. These results show that systemic regulated repression of mHtt transcription in LacQ140 mice results in partial lowering of mHTT protein levels in the striatum (38–52%), but a soluble form of full-length mHTT remains as mice age.
3.2. Effects of mHtt lowering on its distribution in different subcellular cytoplasmic compartments
We next looked at the effects of mHtt repression on its protein levels in different subcellular compartments. Density gradient fractionation and ultracentrifugation for subcellular fractionation of cytoplasmic components was performed as shown in Fig. S2a. The schematic in Fig. S2b indicates representative proteins that are enriched in different cytoplasmic compartments. In 6-month-old mice, the mHTT/WT HTT ratio was significantly lower in LacQ140_2M mice in fractions 13 and 14 compared to LacQ140 mice (Fig. S2c and d). Similar results were observed in 12-month-old mice, where the mHTT/WT HTT ratio in fractions 1, 3, 13, and 14 in LacQ140 mice was significantly lower with mHtt repression for different periods of time compared to LacQ140 mice (Fig. S2e and f). There was no change in the distribution of HTT and mHTT in the fractions between groups in 6-month-old mice (Fig. S3a and b) or in 12-month-old mice (Fig. S3c and d). Altogether, these results show that mHTT is lowered in cytoplasmic fractions through 12 months.
3.3. Effects of mHtt lowering on its distribution in crude nuclear fractions
In 12-month-old LacQ140 mice, repressing mHtt transcription only partially reduced levels of mHTT protein in crude homogenates (Fig. 2c) even though mHTT was efficiently lowered in the sub cytoplasmic compartments contained in the S1 fraction (Fig. S2f). We speculated that mHTT may reside in other compartments where it is more resistant to removal by transcript repression. To address this idea, we examined P1 fractions which contain nuclei, ER, large perinuclear structures such as the recycling compartment, some mitochondria and autophagosomes (Kegel et al., 2000). HTT was detected with antibody Ab1 in WT mice (2 alleles worth) and LacQ140 mice (1 allele worth) and mHTT (1 allele worth) in LacQ140 mice at both 6 and 12 months (Fig. 3a and b). Significant lowering of mHTT protein was observed in 6-month-old LacQ140_A but not LacQ140_2M mice using antibody Ab1 (Fig. 3a). In 12-month mice, repression of the mHtt allele at any age or duration failed to lower mHTT protein levels in the P1 fraction (Fig. 3b).We queried whether forms of mHTT with altered migration with SDS-PAGE could be detected in the P1 fractions using antibody S830, which has been reported by us and others to detect a smear using SDS-PAGE and western blot (Landles et al., 2010; Vodicka et al., 2014). At 12-months, LacQ140 mice showed an S830-positive smear above the HTT/mHTT bands which was not lowered in the LacQ140_8M, LacQ140_2M, or LacQ140_A mice (Fig. 3c). Filter trap assay showed lowering of an SDS-insoluble aggregated species (recognized by S830) in both crude homogenate and P1 fractions; slightly less lowering of aggregated mHTT occurred in the P1 fraction when transcript repression was initiated at 2 or 8 months (Fig. S4a and b). Altogether, these results show that a soluble species of mHTT in the P1 fraction is more resistant to lowering after transcriptional repression.
Fig. 3.
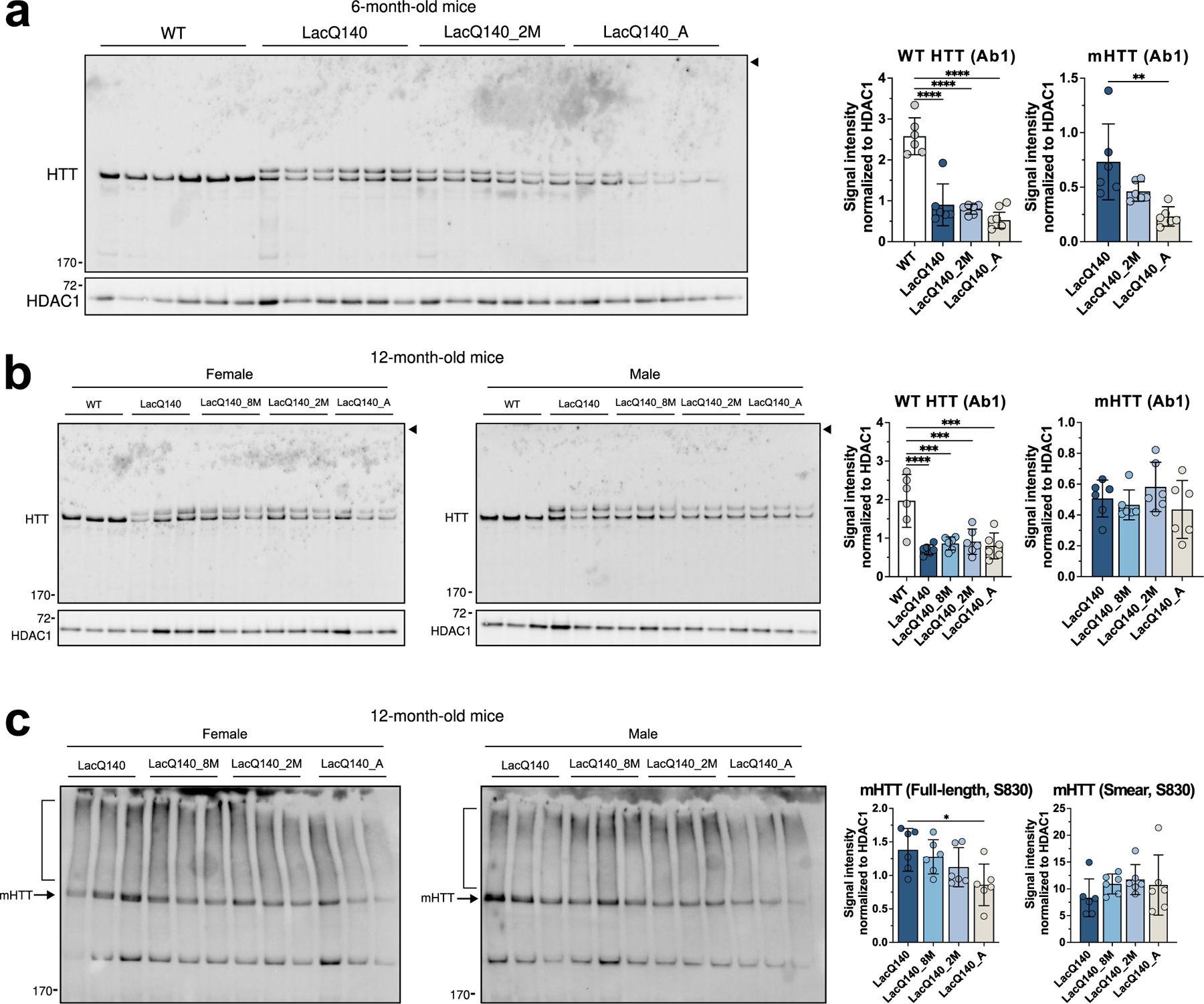
HTT protein levels in P1 fractions.
Equal protein (10 μg) from P1 fractions from 6-month-old (a) and 12-month-old (b) LacQ140 and WT mice were analyzed by western blot for HTT levels with anti-HTT Ab1. No aggregated protein was observed at the top of the gel (arrowhead). Total pixel intensity quantification for each band was measured using ImageJ software and normalized to HDAC1 signal. There was a significant decrease in WT HTT signal in all the treatment conditions for LacQ140 mice compared to WT mice in both (a) 6-month-old mice (F(3, 20) = 40.34, ****P<0.0001, One-way ANOVA with Tukey’s multiple comparison test, n=6) and (b) 12-month-old mice (F(4, 25) = 11.01, ****P<0.0001, One-way ANOVA with Tukey’s multiple comparison test, n=6). There were significantly lower levels of mHTT in the 6-month-old LacQ140_A mice compared to LacQ140 (a, F(2, 15) = 8.233, **P=0.0039, One-way ANOVA with Tukey’s multiple comparison test, n=6) but no changes in mHTT levels were detected in the 12-month-old LacQ140 mice (F(3, 20) = 1.137, P=0.3583, n.s., One-way ANOVA), (b). Equal protein (10 μg) from P1 fractions from 12-month-old LacQ140 and WT mice were analyzed by western blot for HTT levels with anti-HTT S830 (c). The S830 antibody detected a smear of HTT signal (bracket) as well as full-length mHTT (arrow). There were significantly lower levels of full length mHTT in the 12-month-old LacQ140_A mice compared to LacQ140 (F(3, 20) = 3.548, *P=0.0330, One-way ANOVA with Tukey’s multiple comparison test, n=6) and no changes detected in the HTT smear in all LacQ140 mice (F(3, 20) = 0.9281, P=0.4453, n.s., One-way ANOVA). Asterisks on graphs represent Tukey’s multiple comparison test, n=6 mice per group (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
3.4. Effects of mHtt lowering on levels of GFAP, DARPP32, SCN4B, PDE10A, and ATP5A
Prior studies in different mouse models of HD have shown that levels of some neuronal proteins are altered in the mouse striatum, namely DARPP32, PDE10A, SCN4B, and ATP5A (Agrawal and Fox, 2019; Bibb et al., 2000; Gu et al., 1996; Langfelder et al., 2016; McQuade et al., 2014; Oyama et al., 2006; Sapp et al., 2020; Skotte et al., 2018). To assess levels of these proteins and that of the astrocyte protein, glial acidic fibrillary protein (GFAP), in LacQ140 mice without and with mHtt gene repression, crude homogenates from 6, 9, and 12-month-old mice were analyzed by capillary immunoassay or western blot.
In agreement with previous studies in HD mouse models (Sapp et al., 2020; Skotte et al., 2018; Zeitler et al., 2019) striatum from the LacQ140 exhibited a significant reduction in PDE10A at all ages examined. Early mHtt lowering starting at 2 months of age statistically preserved PDE10A levels at 6 and 9 months (Fig. S5a and b), but the effect was lost by 12 months of age when transcript repression was initiated at 2 or 8 months (Fig. S5c). Transcript repression initiated embryonically (LacQ140_A) and examined at 6 and 12 months preserved PDE10A levels, although no protection was observed at 9 months. SCN4B expression was reduced in the striatum of LacQ140 at 6 and 9 months of age and levels were preserved with early mHtt lowering (Fig. S5a and b). DARPP32 levels were significantly lower in LacQ140 compared to WT mice at 9 months but not at 6 or 12 months, and there were no differences in levels of GFAP and ATP5A between WT and LacQ140 at any age examined (Fig. S6a–c).
3.5. Effects of mHtt lowering on lipids detected by mass spectrometry
We surveyed for lipid changes in LacQ140 striatum compared to WT and the effects of lowering mHtt using liquid chromatography and mass spectrometry (LC-MS) as previously described (Iuliano et al., 2021). For each age, lipids were extracted from crude homogenates of striatum from each treatment group/genotype and analyzed as a set. The total lipids per group were compared. Our MS intensity measurements were relative measurements so only samples processed together can be compared (i.e., by age group). The sum of lipids for each genotype and/or treatment group were reported as a proportion of WT within each age group (Fig. 4a–c). No changes in total lipid were observed at 6 months or 12 months (Fig. 4a and c). However, at 9 months LacQ140 mice had significantly lower levels of total lipids compared to WT or to LacQ140_A mice (Fig. 4b). A heat map and hierarchical clustering of lipid changes by subclass at 9 months revealed two major groups wherein subclasses moved in the same direction even if all weren’t statistically significant (Fig. 4d). The top cluster delimited in blue shows subclasses that decreased in LacQ140 mice compared to WT and were corrected by lowering. In contrast, the cluster marked in red shows subclasses that were increased in LacQ140 mice compared to WT and were improved by mHtt lowering. Other subclasses marked in black did not change. Summaries of lipid subclasses and number of species detected for each age group are in Table 1, and an overview of changes in individual species across ages is shown in Table 2. Data and graphs for 6-month-old mice subclasses and species can be found in Fig. S7 & S8 and Supplemental File 2; 9-month-old mice in Fig. S9 & S10 and Supplemental File 2; and 12-month-old mice in Fig. S11 & S12 and Supplemental File 2.
Fig. 4.
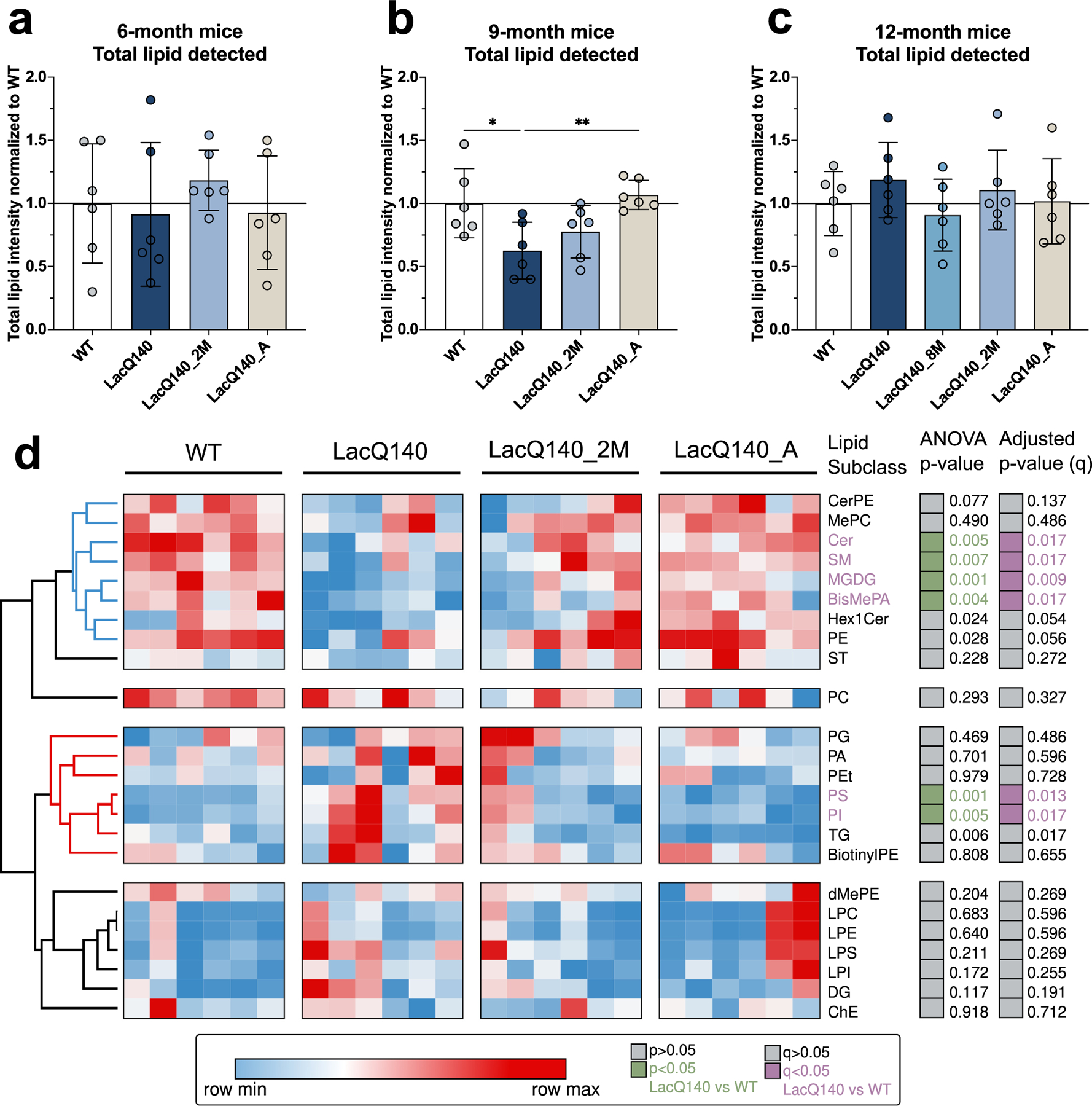
Analyses of lipids in crude homogenates LacQ140 caudate putamen by mass spectrometry.
(a) Total lipid intensity detected at 6 months normalized to WT; no significant difference between groups (one-way ANOVA: F(3, 20) = 0.4604, P=0.7130, n.s., n=6). (b) Total lipid intensity detected at 9 months normalized to WT; LacQ140 mice have decreased total lipid intensity which is reversed in LacQ140_A mice (one-way ANOVA & Tukey’s multiple comparison test: F(3, 20) = 5.474, **P=0.0065, n=6). (c) Total lipid intensity detected at 12 months normalized to WT; no significant difference between groups (one-way ANOVA: F (4, 25) = 0.7504, P=0.5671, n.s., n=6). (d) Heatmap depicts the lipid subclass composition for WT, LacQ140, and treatment groups at 9 months. Hierarchical clustering was performed across lipid subclasses (rows) and columns (animals) using the one minus Pearson correlation distance metric. ANOVA p-value column indicates lipid subclasses significantly changed between LacQ140 and WT mice in green (p<0.05, One-way ANOVA with Tukey’s multiple comparison test, n=6). Lipid subclasses with adjusted p-values (q) < 0.05 LacQ140 vs WT are indicated in purple (q<0.05, two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli, N=24 lipid subclasses, n=6 mice). Source data and full statistical details can be found in Supplemental Files 2, 5, & 6.
Table 1.
Overview of lipid subclass changes at 6, 9, and 12 months
| |
|
6-month-old mice |
9-month-old mice |
12-month-old mice |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Category | Subclass | Species detected | ANOVA p-val | Adj p-val (q), N =36 | Species detected | ANOVA p-val | Adj p-val (q), N =24 | Species detected | ANOVA p-val | Adj p-val (q), N =29 |
| Glycerophospholipids | PC | 134 | 0.7415 | 1 | 107 | 0.2928 | 0.3267 | 160 | 0.4243 | 0.4627 |
| PE | 152 | 0.1975 | 1 | 154 | 0.0284* | 0.0563 | 119 | 0.5653 | 0.5372 | |
| PS | 62 | 0.1171 | 1 | 58 | 0.0014 | 0.0125 | 25 | 0.46 | 0.4627 | |
| PI | 19 | 0.0189 | 0.7144 | 17 | 0.0054 | 0.0168 | 34 | 0.0086* | 0.0338* | |
| MePC | 17 | 0.9859 | 1 | 7 | 0.4901 | 0.486 | 15 | 0.7552 | 0.6329 | |
| PA | 13 | 0.9596 | 1 | 6 | 0.7008 | 0.5957 | 9 | 0.0025* | 0.0184* | |
| BisMePA | 4 | 0.1589 | 1 | 1 | 0.0041 | 0.0168 | 6 | 0.2812 | 0.3647 | |
| dMePE | 9 | 0.195 | 1 | 2 | 0.2041 | 0.269 | 0 | N/A | N/A | |
| PG | 19 | 0.6255 | 1 | 4 | 0.4685 | 0.486 | 4 | 0.859 | 0.6765 | |
| BisMePE | 1 | 0.8072 | 1 | 0 | N/A | N/A | 0 | N/A | N/A | |
| BisMePS | 0 | N/A | N/A | 0 | N/A | N/A | 2 | 0.0001* | 0.0022* | |
| CL | 22 | 0.5812 | 1 | 0 | N/A | N/A | 0 | N/A | N/A | |
| PEt | 7 | 0.3261 | 1 | 2 | 0.9792 | 0.7283 | 0 | N/A | N/A | |
| PMe | 2 | 0.8695 | 1 | 0 | N/A | N/A | 0 | N/A | N/A | |
| PIP2 | 2 | 0.1393 | 1 | 0 | N/A | N/A | 3 | 0.0161* | 0.0394* | |
| PIP | 1 | 0.7034 | 1 | 0 | N/A | N/A | 0 | N/A | N/A | |
| LPC | 30 | 0.7943 | 1 | 10 | 0.6835 | 0.5957 | 7 | 0.2805 | 0.3647 | |
| LPE | 23 | 0.9947 | 1 | 16 | 0.6396 | 0.5957 | 4 | 0.0127* | 0.035* | |
| LPS | 7 | 0.9061 | 1 | 5 | 0.211 | 0.269 | 2 | 0.0817 | 0.1287 | |
| LPI | 7 | 0.7746 | 1 | 1 | 0.1715 | 0.2551 | 0 | N/A | N/A | |
| LPG | 4 | 0.8888 | 1 | 0 | N/A | N/A | 0 | N/A | N/A | |
| LdMePE | 1 | 0.4552 | 1 | 0 | N/A | N/A | 0 | N/A | N/A | |
| BiotinylPE | 0 | N/A | N/A | 1 | 0.8078 | 0.6554 | 1 | 0.0225* | 0.0451* | |
| Glycerolipids | TG | 107 | 0.5998 | 1 | 44 | 0.0057* | 0.0168* | 72 | 0.0002* | 0.0022* |
| MGDG | 3 | 0.8693 | 1 | 2 | 0.0005 | 0.0089 | 5 | 0.0214* | 0.0451* | |
| MGMG | 6 | 0.7537 | 1 | 0 | N/A | N/A | 6 | 0.0497* | 0.0887 | |
| DG | 13 | 0.1381 | 1 | 70 | 0.1174 | 0.1905 | 59 | 0.6614 | 0.5834 | |
| SQMG | 2 | 0.5986 | 1 | 0 | N/A | N/A | 0 | N/A | N/A | |
| SQDG | 1 | 0.9316 | 1 | 0 | N/A | N/A | 0 | N/A | N/A | |
| Sphingolipids | Hex1Cer | 0 | N/A | N/A | 85 | 0.0243* | 0.0542 | 104 | 0.4617 | 0.4627 |
| CerG1 | 50 | 0.5466 | 1 | 0 | N/A | N/A | 0 | N/A | N/A | |
| SM | 47 | 0.6398 | 1 | 23 | 0.0066 | 0.0168 | 36 | 0.3243 | 0.3973 | |
| Cer | 14 | 0.9804 | 1 | 6 | 0.0048 | 0.0168 | 21 | 0.4374 | 0.4627 | |
| CerP | 0 | N/A | N/A | 0 | N/A | N/A | 17 | 0.0035* | 0.0193* | |
| ST | 10 | 0.3977 | 1 | 9 | 0.2283 | 0.2717 | 14 | 0.2176 | 0.3199 | |
| So | 3 | 0.4465 | 1 | 0 | N/A | N/A | 0 | N/A | N/A | |
| CerPE | 0 | N/A | N/A | 1 | 0.0766 | 0.1367 | 0 | N/A | N/A | |
| phSM | 4 | 0.3619 | 1 | 0 | N/A | N/A | 0 | N/A | N/A | |
| CerG2GNAc1 | 1 | 0.5784 | 1 | 0 | N/A | N/A | 0 | N/A | N/A | |
| SPH | 0 | N/A | N/A | 0 | N/A | N/A | 1 | 0.775 | 0.6329 | |
| Hex2Cer | 0 | N/A | N/A | 0 | N/A | N/A | 1 | 0.0112 | 0.035 | |
| Sterol Lipids | ChE | 1 | 0.5341 | 1 | 1 | 0.918 | 0.7124 | 2 | 0.9151 | 0.6958 |
| ZyE | 0 | N/A | N/A | 0 | N/A | N/A | 3 | 0.0092* | 0.0338* | |
| Fatty Acyls | FA | 2 | 0.5893 | 1 | 0 | N/A | N/A | 0 | N/A | N/A |
| AcCa | 0 | N/A | N/A | 0 | N/A | N/A | 2 | 0.5847 | 0.5372 | |
| Prenol Lipids | Co | 0 | N/A | N/A | 0 | N/A | N/A | 1 | 0.0523 | 0.0887 |
| Total | 800 | 632 | 735 | |||||||
Bold text indicates lipid subclasses significantly different between WT and LacQ140 mice (p<0.05 or q<0.05).
Asterisk indicates lipid subclasses significantly different among other groups (p<0.05 or q<0.05).
One way ANOVA was conducted followed by Tukey’s multiple comparisons test, two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli, q<0.05.
Table 2.
Overall comparison of lipid species results across ages.
| Age (months) | # Species detected | # Species changed (p<0.05)* | # Species changed WT vs. LacQ140 (p<0.05)* | # Species changed (q<0.05)** | # Species changed WT vs. LacQ140 (q<0.05)** | # Species recovered with lowering (q<0.05)** |
|---|---|---|---|---|---|---|
| 6 | 800 | 9 | 4 | 0 | 0 | 0 |
| 9 | 632 | 192 | 72 | 17 | 14 | 14 |
| 12 | 735 | 162 | 36 | 72 | 26 | 1 |
p value for ANOVA;
Adjusted p value determined using Benjamini, Krieger, and Yekutieli procedure.
We assessed if there were more subtle alterations at the subclass or individual species level that did not influence the overall lipid levels. In striatum of LacQ140 mice at 6-months-of-age, we detected a modest increase in the subclass phosphatidylinositol (PI) compared to WT (Fig. S7). Individual species of PI (18:0_20:4, 16:0_22:6), and phosphatidylserine PS (18:0_20:4, 22:6_22:6) were increased in LacQ140 mice compared to WT (Fig. S8). However, no subclass or species changes survived correction using the Benjamini, Krieger, and Yekutieli procedure with a 5% false discovery rate (FDR) of q <0.05, N=36 subclasses (Table 1) and N=800 species (Supplemental File 2). Consistent with our observations at 6 months, PS and PI were increased in 9-month-old LacQ140 mice compared to WT (Fig. 5a and b). A limited number of species are modestly increased at 6 months, which progresses to more robust increases in these glycerophospholipids at the subclass level at 9 months. A significant reduction in bismethyl phosphatidic acid (BisMePA) also occurred (Fig. 5c). Functionally, PI is the precursor for PIPs which are important for protein kinase c (PKC) signaling at the synapse (Berridge, 2016) and can also act as important docking and activating molecules for membrane associated proteins, including HTT (Kegel et al., 2009a). PS is an abundant anionic glycerophospholipid necessary for activation of several ion channels (Hirt and Leist, 2003), fusion of neurotransmitter vesicles, regulation of AMPA signaling, and coordination of PKC, Raf-1, and AKT signaling (Kim et al., 2014).
Fig. 5.
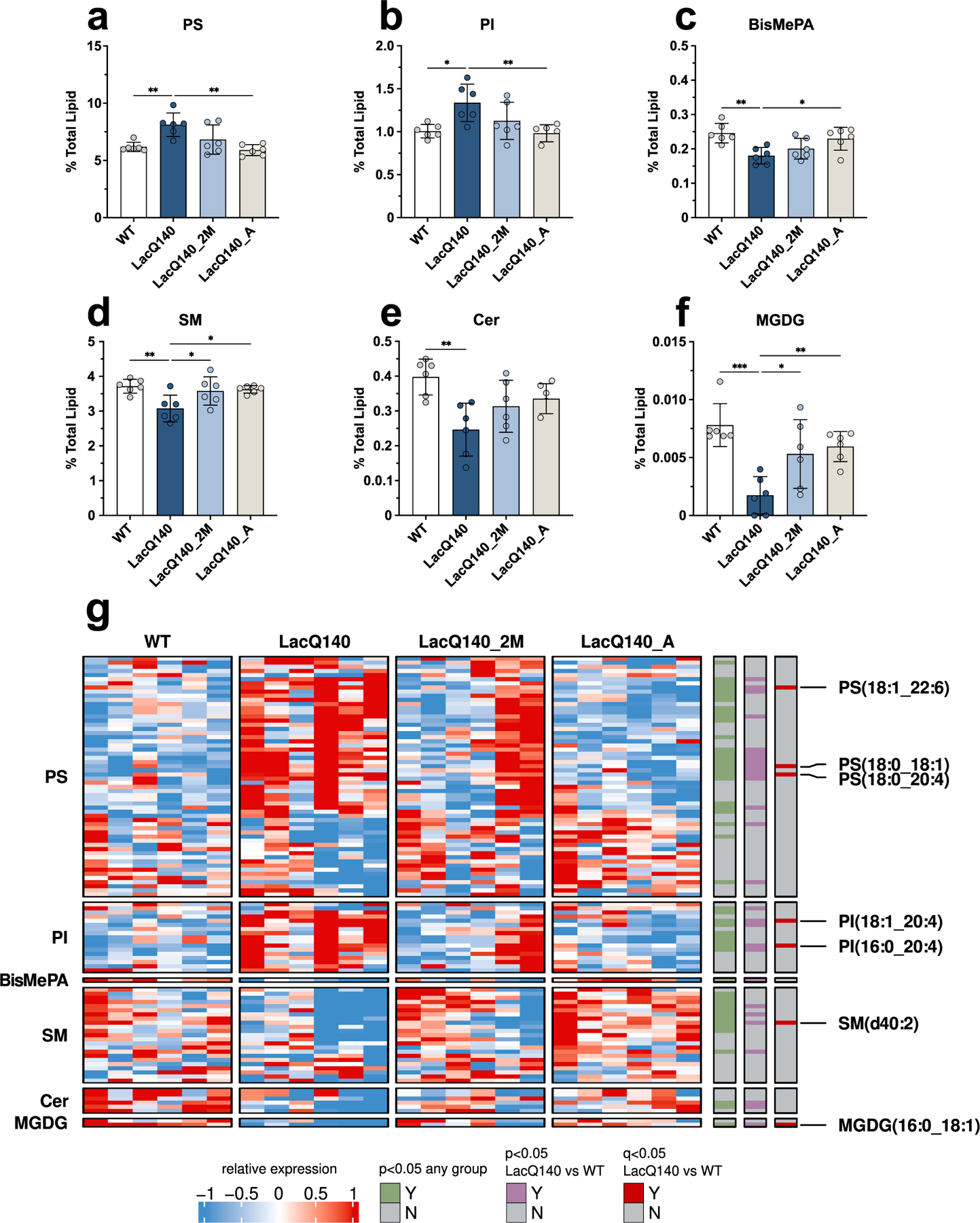
Restoration of dysregulated lipid subclasses with lowering of mHtt.
Graphs show relative intensities for lipid subclasses at 9 months of age expressed as a percent of total lipid intensity detected (bars = mean, error bars = ± SD). (a) PS increased in LacQ140 mice and is reversed in LacQ140_A mice; F(3, 20) = 7.601, **P=0.0014, q=0.0125, (b) PI increased in LacQ140 mice and reversed in LacQ140_A mice; F(3, 20) = 5.707, **P=0.0054, q=0.0168, (c) BisMePA decreased in LacQ140 mice and is reversed in LacQ140 mice; F(3, 20) = 6.086, **P=0.0041, q=0.0168, (d) SM decreased in LacQ140 mice and is reversed in LacQ140_2M and LacQ140_A mice; F(3, 20) = 5.465, **P=0.0066, q=0.0168, (e) Cer decreased in LacQ140 mice; F(3, 20) = 5.883, **P=0.0048, q=0.0168, (f) MGDG decreased in LacQ140 mice and is reversed in LacQ140_2M and LacQ140_A mice; F(3, 20) = 9.350, ***P=0.0005, q=0.0089. Statistics are one-way ANOVA and asterisks on graphs represent Tukey’s multiple comparison test, n=6 mice per group. (g) Heatmap shows individual lipid species that comprise each subclass at 9 months of age. Hierarchical clustering was performed across individual lipid species (rows) and animals (columns) using the one minus Pearson correlation distance metric. Individual lipid species significantly changed between any group are indicated in green (p<0.05, one-way ANOVA), lipid species significantly changed between LacQ140 and WT are indicated in purple (p<0.05, one-way ANOVA, n=6) and red (q<0.05, one-way ANOVA & two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli, N=632 lipid species, n=6 mice). Source data and full statistical details can be found in Supplemental Files 2, 5, & 6.
In contrast to the increases observed in glycerophospholipids, 9-month-old LacQ140 mice exhibit significant reductions in the subclasses sphingomyelin (SM) and ceramide (Cer) (Fig. 5d and e), lipids central in sphingolipid metabolism and important for myelination. Cer is the backbone for SM as well as more complex glycosphingolipids, which are highly enriched in myelin (Chrast et al., 2011). Moreover, the low abundance glycerolipid monogalactosyldiacylglycerol (MGDG) was strikingly reduced at 9 months (Fig. 5f). MGDG regulates oligodendrocyte differentiation and is considered a marker of myelination (Inoue et al., 1971; Schmidt-Schultz and Althaus, 1994).
All subclasses (PI, PS, BisMePA, SM, Cer, MGDG) with changes between WT and LacQ140 had q values<0.05 (N=24 subclasses) (Table 1; Fig. 4d). Early lowering of mHtt (LacQ140_A) restored levels of each of these subclasses except for Cer and lowering mHtt starting at 2 months (LacQ140_2M) was sufficient to correct levels of SM and MGDG (Fig. 5a–f). To assess the effects of mHtt lowering on the distinct lipid species that comprise each subclass, lipid species were plotted in a heatmap (Fig. 5g). The broad changes at the subclass level can be attributed to large numbers of individual lipid species changing within each subclass even if each species is not significantly different (Fig. 5g & Fig. S9). Alternatively, subclasses can remain unchanged but still contain individual lipid species with differences. For example, the subclass Hex1Cer was not significantly changed (Fig. S9), but many individual species were significantly altered in LacQ140 mice (Fig. S10). These species included 3 that were significantly decreased in LacQ140 mice and normalized to WT levels with lowering of mHtt: Hex1Cer (d18:1_18:0), Hex1Cer (t18:0_18:0), Hex1Cer (d18:1_25:1) (Fig. S10). Combined, these 3 species represent approximately 1% of total Hex1Cer detected. Hex1Cer is comprised of galactosylceramide and glucosylceramide and cannot be distinguished by LC-MS/MS since they are isomers with the same mass and charge (Reza et al., 2021); however, previous studies have determined that the majority of Hex1Cer in the brain is galactosylceramide, which is highly enriched in myelin (Vanier and Svennerholm, 1975). Galactosylceramides are major constituents of the myelin sheath and contribute to its stabilization and maintenance (Schmitt et al., 2015).
In total, we identified 14 lipid species significantly altered between LacQ140 and WT mice using a stringent FDR cutoff (FDR 5%/q<0.05, Benjamini, Krieger, and Yekutieli procedure, N=632 lipids) and all 14 showed improvement with mHtt lowering at 9 months (Fig. S10). This included increases in PI (16:0_20:4) & PI (18:1_20:4) and PS (18:0_20:4), PS (18:0_18:1) and PS (18:1_22:6). Lipid species decreased in LacQ140 mice and restored with mHtt lowering included Hex1Cer species as detailed above, SM (d40:2), PE (16:0p_20:4) and MGDG (16:0_18:1) (Fig. 5g, Fig. S10). Taken together, these results indicate a profound disruption of myelin lipids in LacQ140 mice, which is attenuated with lowering of mHtt.
In the striatum of LacQ140 mice at 12 months, no significant differences at the subclass level were found compared to WT (Table 1; Fig. S11) although 29 subclasses were measured. At the individual lipid species level, we identified 26 lipid species significantly altered between LacQ140 and WT mice (FDR 5%/q<0.05, Benjamini, Krieger, and Yekutieli procedure, N = 735 lipids). (Fig. S12). Contrary to the reversal of lipid dysregulation observed at 9 months, the only lipid species that was responsive to lowering mHtt at 12 months was CerP (t40:3), which was increased in LacQ140 and decreased with early mHtt lowering (LacQ140_A) (Fig. S12). Myelin enriched lipids, including 6 species of Hex1Cer (d18:1_18:0, d:40:2, d18:2_22:0, d41:2, t18:0_22:1 & d18:1_22:1) and ST (d18:1_22:1), decreased and were not improved with mHtt lowering (Fig. S12). Similarly, nine triacylglycerol (TG) species, eight of which contain oleic acid (C18:1) were decreased in LacQ140 mice and unchanged with mHtt lowering (Fig. S12). Overall, these results demonstrate that most individual lipid changes were no longer reversible by 12 months.
3.6. Bioinformatic analysis of transcriptional alterations to lipid metabolism and myelination
To determine if any of the lipid changes could be explained by altered transcription of genes regulating lipid-related metabolic pathways, we re-analyzed the previously generated RNAseq dataset in the LacQ140 striatum (GEO GSE156236). Differential expression analysis (Love et al., 2014) detected 1360 upregulated and 1431 downregulated genes at 6 months and 1375 upregulated and 1654 downregulated genes at 12 months (FC>20%, adjusted p-value <0.05) (Fig. S13a). Overrepresentation analysis against GO Biological Processes (GO BP) revealed that downregulated genes were enriched in terms related to lipid metabolism and signaling as well as myelination. In 6-month LacQ140 downregulated genes, GO BP terms ensheathment of neurons (padj<0.01), axon ensheathment (padj<0.01), myelination (padj<0.05), cellular lipid metabolic process (padj<0.05), lipid metabolic process (padj<0.05), phospholipase C-activating G protein-coupled receptor signaling pathway (padj<0.05), response to lipid (padj<0.05) were significantly over-represented (Fig. 6a). In LacQ140 upregulated genes, there was no over-representation of terms that could directly explain changes to lipid levels (Supplemental Files 3 & 4, complete list of DEGs and pathways).
Fig. 6.
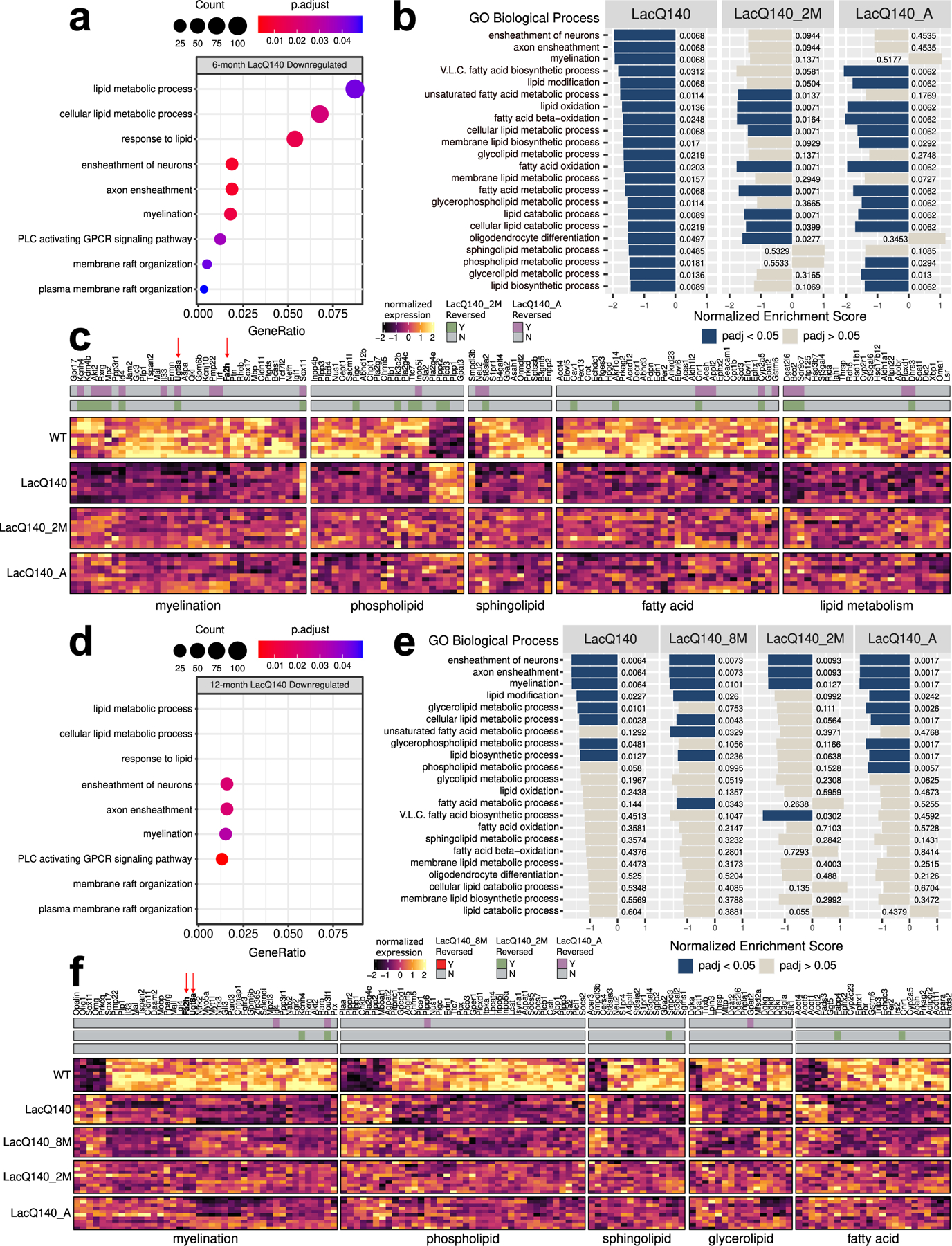
Lipid metabolism and myelin associated transcriptional changes and reversal with mHtt lowering.
(a) Dotplot (clusterProfiler) of lipid or myelin related GO BP terms overrepresented (one sided hypergeometric test, padj<0.05) in 6-month LacQ140 downregulated genes (padj<0.05, FC>20%). GeneRatio represents the number of genes associated with each GO term/number of downregulated genes. Dots are sized by count of genes associated with respective terms. (b) Gene set enrichment analysis (clusterProfiler) of 6-month LacQ140, LacQ140_2M and LacQ140_A groups compared to WT. All lipid and myelin associated GO BP terms significantly enriched in LacQ140 compared to WT (padj<0.05) are displayed. X axis for each respective group represents the normalized enrichment score (NES) and bars are colored by significance (padj<0.05 = blue, padj>0.05 = tan). Adjusted p-values are displayed adjacent to bars. (c) Heatmap shows differentially expressed genes in 6-month-old LacQ140 mice compared to WT (padj<0.05, FC > ±20%). Gene expression is shown as median ratio normalized counts (DESeq2), scaled by respective gene (columns). Bars above the heatmap indicate DEGs reversed with mHtt lowering. LacQ140_2M = green, LacQ140_A = purple; padj<0.05, FC > 20% opposite of LacQ140. (d) Dotplot of lipid or myelin related GO BP terms overrepresented in 12-month downregulated genes. GO BP terms significantly overrepresented at 6-months (a) are displayed for comparison. 4/9 terms (ensheathment of neurons, axon ensheathment, myelination, and phospholipase C activating G-protein coupled receptor signaling pathway) are significantly overrepresented in 12-month LacQ140 downregulated genes (one sided hypergeometric test, padj < 0.05). (e) Gene set enrichment analysis of LacQ140, LacQ140_8M, LacQ140_2M and LacQ140_A groups compared to WT. GO BP terms enriched at 6-months (b) are displayed for comparison. 8/22 GO BP terms are significantly negatively enriched (padj < 0.05) at 12-months, shown in blue. (f) Heatmap shows differentially expressed genes in 12-month-old LacQ140 mice compared to WT (padj<0.05, FC > ±20%). Gene expression is shown as median ratio normalized counts (DESeq2), scaled by respective gene (columns). Bars above the heatmap DEGs reversed with mHtt lowering (LacQ140_8M = bottom bar, no reversal, LacQ140_2M = green, LacQ140_A = purple; padj<0.05, FC >20% opposite of LacQ140). GO BP terms associated with genes, fold changes, and exact FDR values can be found in Supplemental Files 3 & 4.
Next, Gene Set Enrichment Analysis (GSEA) was conducted (Subramanian et al., 2005; Wu et al., 2021) to examine the impact of mHtt repression on the enrichment profiles of terms related to lipid metabolism and myelination. At 6 months, 3 of the top 20 enriched GO BP terms were related to myelination and all were negatively enriched: ensheathment of neurons, myelination, and axon ensheathment (Supplemental File 3). Further, every significantly enriched myelin or lipid related GO BP term was negatively enriched in LacQ140 compared to WT (padj <0.05) (Fig. 6b, Supplemental File 3). Repression of mHtt in LacQ140_2M and LacQ140_A groups was sufficient to reverse the negative enrichment of many GO BP terms including myelination/axon ensheathment/ensheathment of neurons, and sphingolipid/phospholipid metabolic processes suggesting some beneficial impact of mHtt lowering (Fig. 6b).
Specifically, 6-month LacQ140 mice had reduced expression of genes encoding proteins involved in oligodendrocyte development (Akt2, Bcas1, Ptgds, Cldn11, Trf, Tcf7l2, Qki), myelin structure and compaction (Mbp, Plp1, Tspan2, Mal), and myelin lipid biosynthesis (Ugt8a, Fa2h, Aspa) (Fig. 6c). LacQ140 mice also exhibited dysregulation of genes encoding enzymes including phospholipases (Pla2g7, Pla2g4c, Pla2g4e, Plb1, Plcd3, Plcd4), phosphatases & kinases (Plpp1, Plpp3, Plppr2, Inpp4b, Pik3c2b), and regulators of phospholipid biosynthesis (Gpat2, Gpat3, Chpt1) (Fig. 6c). Enzymes responsible for biosynthesis and metabolism of sphingolipids were altered in LacQ140 mice: Sptssb, Ormdl2, Gba2, Asah, were downregulated and Smpdl3b was upregulated. Genes encoding enzymes responsible for fatty acid elongation (Elovl1, Elovl5, Elovl6) were all downregulated (Fig. 6c). Of the DEGs related to lipid metabolism and myelination 23 were reversed in LacQ140_2M mice, 23 were reversed in LacQ140_A mice and 15 were reversed in both (padj < 0.05, FC > 20% opposite direction of LacQ140) (Fig. 6c & Fig. S13b). Three of the nine GO BP terms significantly over-represented in 6-month LacQ140 downregulated genes were also over-represented in 12-month downregulated genes: ensheathment of neurons (padj <0.01), axon ensheathment (padj <0.01), myelination (padj <0.05), and phospholipase C-activating G protein-coupled receptor signaling pathway (padj<0.01) (Fig. 6d). Similarly, GSEA showed that only a subset (8/22) of the GO BP terms related to lipid metabolism or myelination were enriched at both timepoints (Fig. 6e). Terms including glycerophospholipid metabolic process and glycerolipid metabolic processes showed improvement with lowering of mHtt, whereas terms related to myelination remained negatively enriched with mHtt repression (Fig. 6e).
DEGs associated with myelin and lipid biosynthesis were both downregulated and upregulated in 12-month LacQ140 mice. LacQ140 mice had reduced (Cntnap1, Mal, Tspan2, Plp1, Mobp) and increased (Omg, Opalin) expression of genes encoding proteins involved in myelin structure and compaction (Fig. 6f). Similarly, oligodendrocyte development related genes were both decreased (Akt2, Cldn11, Myt1l) and increased (Prkcq, Olig1, Sox11). Genes related to myelin lipid biosynthesis (down: Ugt8a, Fa2h, Selenoi), phospholipid synthesis/metabolism (up: Chkb, Agpat2, Mboat2, down: Agpat1, Lpcat4), phosphatases (up: Plpp1, Plpp2, Plppr2, down: Plpp3, Plpp6, Inpp5a), phospholipases (up: Pla2g4e, down: Plcb1, Plcb3, Plce1, Plcl1) and sphingolipid synthesis/metabolism (up: Cers4, Smpdl3b, Acer3, down: Gba2, St3gal2, Smpd3, Sgpp2, Sgms1, A4galt, Fads3) were differentially expressed in both directions (Fig. 6f). In 12-month LacQ140 mice, glycerolipid synthesis/metabolism genes (up: Dgat1, down: Dgat2, Dgat2l6, Lpin3, Dagla) and DAG kinase genes (up: Dgka, Dgkg, down: Dgkb, Dgki) were differentially expressed (Fig. 6f). Consistent with the enrichment results at this timepoint, fewer genes were reversed with repression of mHtt (5 genes LacQ140_2M, 4 genes LacQ140_A) (Fig. 6f & Fig. S13c).
DEGs were also manually curated for genes that could directly impact levels of PS, PI, sphingolipids, and glycerolipids. Phosphatidylserine synthase enzymes (Ptdss1/Ptdss2) (Kim et al., 2014) and rate limiting enzymes for PI biosynthesis (CDP-Diacylglycerol Synthases: Cds1/Cds2) (Blunsom and Cockcroft, 2020) were unchanged in 6- and 12-month LacQ140 mice (Supplemental Files 3 & 4). For glycerolipids, the only known enzymes that catalyze TG biosynthesis (Harris et al., 2011) were increased (Dgat1) and decreased (Dgat2) at 12 months; mRNA levels remained unchanged with mHtt repression (Fig. 6f).
Lipid changes might be explained by altered cellular composition of tissue. To address this possibility, we surveyed for changes in transcript levels for cellular markers. No change in mRNA expression levels for the microglial markers Iba1 and Cd68 or for the reactive astrocyte marker Gfap were observed that might indicate upregulation of these cell types could account for the lipid changes. However, our transcriptional profiling did reveal an expression signature consistent with altered oligodendrocyte development and alterations to myelin structure or compaction in both 6- and 12-month-old mice (Fig. 6c and f). UDP-galactose-ceramide galactosyltransferase (Ugt8a) is expressed in oligodendrocytes where it catalyzes the formation of GalCer (Hex1Cer) from Cer; Ugt8a mRNA was decreased at both 6 and 12 months in the LacQ140 mice (Fig. 6c & Fig. 7a). Embryonic repression of mHtt and repression beginning at 2-months of age preserved Ugt8a mRNA measured at 6-months; however, the effect was lost by 12-months of age (Fig. 6c and f). These mRNA results mirror our findings of Hex1Cer lipid levels measured using LC-MS in LacQ140 striatal lysates. GalCer and GlcCer are both members of the Hex1Cer subclass; the gene catalyzing the transfer of glucose to ceramide (Ugcg) was not changed at the mRNA level, suggesting lower levels of Hex1Cer in LacQ140 mice are due to lower levels of GalCer. Ugt8a can also transfer galactose to diacylglycerol to form the lipid MGDG (van der Bijl et al., 1996) which we found was decreased with mHtt expression and improved with mHtt lowering (Fig. 5f and g). In contrast, Fa2h mRNA was reduced at 6 and 12-months but not improved with mHtt lowering (Fig. 6c and f). Fatty acid 2-hydroxylase, which is encoded by Fa2h, modifies fatty acids substrates with a hydroxyl (-OH) group, producing 2-hydroxylated fatty acids that are incorporated into myelin sphingolipids and provide stability (Fig. 7b and c) (Alderson et al., 2006). Overall, these data reveal transcriptomic changes in key lipid metabolic and biosynthetic genes in LacQ140 striatum, which directly impact the lipidomic profile. Critically, many of these lipidomic and transcriptomic alterations can be attenuated with lowering of mHtt.
Fig. 7.
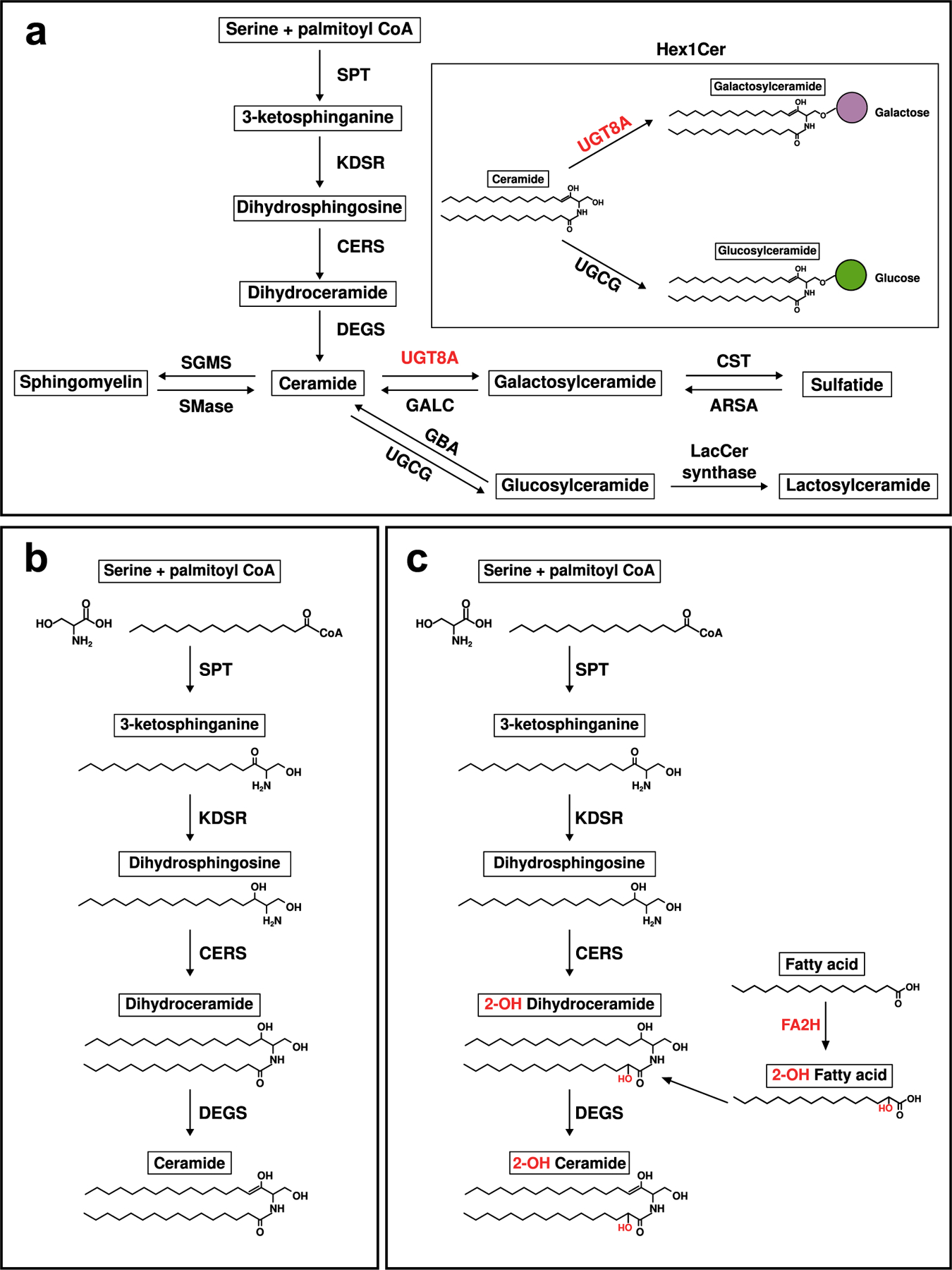
Simplified de novo sphingolipid biosynthesis pathway.
(a) Serine and palmitoyl CoA are condensed by serine palmitoyltransferase (SPT) to generate 3-ketosphinganine. 3-ketophinganine is reduced to dihydrosphingosine by 3-ketodihydrosphingosine reductase (KDSR). Dihydrosphingosine is acetylated by ceramide synthases (CERS) and further desaturated by ceramide desaturase (DEGS) to generate ceramide. Ceramide is the substrate for generation of other sphingolipids (sphingomyelin, galactosylceramide, glucosylceramide, sulfatide, and lactosylceramide). Abbreviations: SGMS = sphingomyelin synthase, SMase = sphingomyelinase, UGT8A = UDP galactosyltransferase 8A, GALC = galactosylceramidase. CST = galactosylceramide sulfotransferase, ARSA = arylsulfatase, GBA = glucosylceramidase, UGCG = UDP-glucose ceramide glucosyltransferase, LacCer synthase = lactosylceramide synthase. (b) Biosynthesis of non-hydroxylated sphingolipids. (c) Biosynthesis of 2-hydroxy sphingolipids. Fatty acid 2-hydroxylase (FA2H) catalyzes hydroxylation of fatty acids in the C2 position, which can be incorporated into sphingolipid precursors (i.e., dihydroceramide) in the acylation step of de novo synthesis.
4. Discussion
Here we used the LacQ140 inducible HD mouse model to initiate whole body mHtt reduction at different ages and evaluate effects on proteins and lipids. Lowering mHTT protein by 38–52% in the LacQ140 caudate-putamen, starting from conception up to 12 months of age, was sufficient to prevent mHtt induced changes in the levels of some proteins and lipids. However, a degradation-resistant, soluble species of the protein detected in older mice limited long term benefit of mHtt lowering. Our lipid data show clear evidence of changes impacting myelin which mechanistically is due in part to aberrant transcription as evidenced by differentially expressed genes regulating some of the same lipids that were found altered. The correction of lipid changes we identified here correlates with the behavioral changes measured previously in LacQ140 mice using a comprehensive set of unbiased, high-content assays (Marchionini et al., 2022) in that behavior was closer to normal at 6 and 9 months with early mHTT lowering, but the protective effect of lowering degraded by 12 months.
In humans and mice, HTT and mHTT are expressed throughout the body with highest expression in brain and testis but both are detected in almost all cell types (Li et al., 1993; Strong et al., 1993). In this mouse model of HD, mHtt repression was measured in various tissues including brain (cortex, striatum, cerebellum, hippocampus, thalamus), liver, pancreas, white adipose tissue, brown adipose tissue, and gonads (Marchionini et al., 2022). We focused on the striatum for this study because it manifests a high degree of pathology in HD mouse models and in human patients. Whole body lowering of mHTT may have provided essential benefits superior to regional lowering. While intrastriatal delivery of AAV1/2 encoding a transcriptional repressor (ZFP-HTT) engineered to target the mutant allele expressed under the human synapsin promoter (Zeitler et al., 2019), or a miRNA against the huntingtin gene (Stanek et al., 2014) could powerfully lower mHTT levels, only modest recovery of phenotypes was observed. ASOs introduced to the CNS through intracerebroventricular or intraparenchymal injection, which theoretically limits effects to CNS but targets many cell types, improved some behaviors and brain volume (Kordasiewicz et al., 2012; Southwell et al., 2018). In contrast, our findings show that modest whole body lowering of mHTT was able to prevent lipid changes in striatum up to 9 months. These benefits may be due to mHTT lowering in numerous cell types within striatum not reached by AAV1/2, or due to effects on other brain regions such as cortex which provides cortical input to striatum. There may also be beneficial effects due to lowering in the periphery, which might for instance impact the microbiome and transit of nutrients across the blood brain barrier, or communication of endocrine signals. However, lowering in the periphery with ASOs had no effect on neurodegeneration in Q111 HD mice (Coffey et al., 2017). While the AAV-HTT-ZFP study described above treated as early as 2 months, our best preservation of markers occurred when mHTT was lowered embryonically, indicating early mHTT lowering provided the most favorable outcome.
We identified forms of mHTT protein that were resistant to mHtt lowering detected by various HTT antibodies. These resistant forms of mHTT identified by immunoblot may correspond to mHTT aggregates or foci found to be resistant to lowering in LacQ140 mice using MSD and immunofluorescence methods (Marchionini et al., 2022). Others have described aggregated and soluble forms of mHTT that resist degradation in distinct cellular compartments, including full length mHTT in the nucleus (Diaz-Hernandez et al., 2005; Fox et al., 2020; Wheeler, 2000; Zeitler et al., 2019). In the LacQ140 striatum, the SDS-soluble degradation-resistant form of full-length mHTT, detected by us using anti-bodies S830, MW1 and EPR5526, resides in a perinuclear or nuclear compartment which might correspond to the “juxtanuclear quality control compartment (JUNQ)” described by Frydman and colleagues (Kaganovich et al., 2008). Accrual of misfolded mHTT in the nucleus likely contributes to transcriptional interference and the eventual failure of prolonged benefits of modest mHTT lowering. Targeting these resistant fractions of misfolded mHTT by a chaperone activity to aid in its degradation may be beneficial in combination with gene therapy HTT lowering strategies.
We found that early, continuous partial lowering of mHTT protein for up to 12 months fully or partially preserved PDE10A and SCN4B protein levels with initiation of lowering embryonically providing the highest benefits. These data agree with previous findings showing preservation of Pde10a mRNA in the LacQ140 model after early mHtt lowering (Marchionini et al., 2022) and preservation of the Pde10a PET signal in the Q175 model after striatal injection of AAV-HTT-ZFP (Zeitler et al., 2019). However, our data here indicate that the time points chosen for post-treatment analysis are important and that changes do not follow a linear neurodegenerative trajectory in mice. The greatest number of protein and lipid changes in LacQ140 mice occurred at 9 months, with few changes detected at 12 months. Similarly, Langfelder et al. found a greater number of differentially expressed genes at 6 months compared to 10 months in zQ175 mice (Langfelder et al., 2016). This suggests that in the mouse brain, adverse responses to mHTT oscillate or go through waves of degeneration and regeneration. Therefore, to appreciate any benefits afforded by mHtt lowering, frequent or continuous monitoring should be conducted.
In this study, mass spectrometry of lipids identified numerous alterations in LacQ140 striatum, many of which were prevented with modest mHtt lowering. To our surprise, lipidomic analysis showed that LacQ140 mice had increased levels in species of glycerophospholipids PI and PS starting at 6 months and progressing to a significant change in the PI and PS subclass level at 9 months. Proteomics in Q140 synaptosomes revealed changes in proteins that regulate PI levels including PKC signaling and PIP2 hydrolysis, changes in two isoforms of DGKs, and alterations in one of the rate-limiting enzymes in PI synthesis (CDS2) (Sapp et al., 2020) all of which impact PI levels (Blunsom and Cockcroft, 2020). Transcriptomic profiling of LacQ140 mice also showed a plethora of altered mRNA levels for enzymes that impact PI and PIPs (Fig. 6c and f; Supplemental Files 2 and 3). Normally found on the inner leaflet of the plasma membrane, PS can be externalized by apoptotic cells to signal for their demolition (Williamson and Schlegel, 2002) and in neuronal synapses, externalized PS signals to microglia for synaptic pruning (Scott-Hewitt et al., 2020).Microglial activation occurs in HD post-mortem brain (Sapp et al., 2001) and increased pruning by microglia is hypothesized to contribute to synaptic loss in R6/2 HD striatum (Savage et al., 2020) and in zQ175 and BACHD mice, as well as in HD brain (Wilton et al., 2021). An overall increase in PS could inadvertently mark synapses or myelin (Djannatian et al., 2023) for engulfment by microglia. The ratio of PS to PE impacts autophagy (Polyansky et al., 2022) which may in turn impair mHTT removal (Qin et al., 2003; Yamamoto et al., 2006). Both PI and PS are abundant in astrocytes as well as neurons (Fitzner et al., 2020) so it is unclear which cell type(s) is producing the changes in these lipids. We cannot rule out the possibility that lipid changes are due to altered cellular composition of the brain.
Changes in white matter detected through imaging are one of the first signs of disease in people with HD (PwHD) (Bourbon-Teles et al., 2019; Di Paola et al., 2012; Faria et al., 2016; Hobbs et al., 2010; Matsui et al., 2015; Odish OF et al., 2015; Oh et al., 2021; Paulsen, 2009; Paulsen et al., 2008; Phillips et al., 2013; Poudel et al., 2015; Rosas et al., 2006; Singh et al., 2013; Sweidan et al., 2020; Tereshchenko et al., 2019). Morphometric studies of postmortem HD brains showed reduced cross-sectional area of white matter as well as gray matter atrophy (de la Monte et al., 1988; Vonsattel and DiFiglia, 1998). In HD post-mortem brain tissue, a dramatic shift in the profile of various sphingolipids including Cer, SM, hexosylceramides, and sulfatides occurred (Phillips et al., 2022). Here, we demonstrate in the striatum of 9-month-old LacQ140 mice significant reductions (compared to WT mice) of relative levels of total lipids and the lipid subclasses SM and Cer, and three individual species of Hex1Cer, all important for myelin (O’Brien and Sampson, 1965). Our data are in alignment with data from R6/1 mice showing changes in cerebroside and sulfatide levels (Desplats et al., 2007), from the R6/2 mouse model showing reductions in components of the sphingolipid biosynthesis pathway (Di Pardo et al., 2017), and findings in a transgenic sheep model OVT73, similarly showing decreased levels of numerous species of SM (Skene et al., 2017). A salient finding was a profound reduction in LacQ140 striatum of the low abundance signaling lipid MGDG, which regulates oligodendrocyte differentiation (Inoue et al., 1971). MGDG is considered a marker of myelination and stimulates PKC-alpha activity in oligodendrocytes to support process formation (Schmidt-Schultz and Althaus, 1994). We previously reported reduced levels of MGDG in subcellular fractions of Q175/Q7 HD striatum at 2 and 6 months (Iuliano et al., 2021). Crucially, in LacQ140 mice, lowering mHtt improved the loss of SM and MGDG suggesting protection against white matter pathology.
Altered oligodendrocyte differentiation or survival due to direct effects of mHTT protein on transcription in the nucleus may account for many of the lipid changes we observed. RNA transcripts of genes important for oligodendrocyte differentiation and myelin sphingolipid biosynthesis were altered in LacQ140 mice. The transcription factor Tcf7l2, which was lower at 6 months in LacQ140 mice, was recently implicated in altered myelin formation in R6/2 and Q175 mice (Benraiss et al., 2022). The basic helix-loop-helix transcription factor Olig1, which was increased at 12 months in LacQ140 mice compared to WT, is important for commitment of cells to CNS oligodendrocyte identity (Dai et al., 2015). Critically, Lim et al. presented evidence showing abnormal oligodendrocyte maturation in multiple HD postmortem brain regions, as well as R6/2 brain, with single-nucleus RNAseq showing changes in OLIG1 and OLIG2 (Lim et al., 2022). Altered levels of myelin transcripts were found in human embryonic stem cells differentiated along an oligodendrocyte pathway (Osipovitch et al., 2019) and an epigenic etiology for changes in myelin gene expression in human oligodendrocyte precursors that was blocked by inactivation of mHTT allele was described (Bardile et al., 2019).
Changes in particular enzyme levels that regulate lipid biosynthesis that were changed at the transcriptional level in LacQ140 mice can have dire consequences and result directly in myelin defects. Both Ugt8a and Fa2h mRNA were lower in the striatum of LacQ140 mice; Ugt8a mRNA levels were protected by mHtt lowering at 6 months, but Fa2h mRNA was not. Work by others showed that mice deficient in the Ugt8a gene exhibited abnormal myelin maturation and structure (Bosio et al., 1996; Coetzee et al., 1996). In humans, mutations in FA2H are associated with leukodystrophy and hereditary spastic paraplegia type SPG35 (Edvardson et al., 2008; Rattay et al., 2019) highlighting the importance of hydroxylated sphingolipids in myelin integrity. Adult Fa2h-deficient mice have normal oligodendrocyte differentiation with normal appearing myelin that later degenerates, showing “splitting of lammelae” by 18 months (Zöller et al., 2008). This is similar to the Ki140CAG mouse model (Pérot et al., 2022) where myelin appears to be quite normal into early adulthood, but then may start to degenerate with disease progression.
Although transcriptional deregulation clearly impacts lipid modifying enzymes, levels of myelin-related lipids could be caused by an interaction of mHTT with oligodendrocyte membranes or a be a consequence of Wallerian degeneration of cortical-striatal axons of the neurons. Interestingly, the presence of SM increased permeabilization of monolayers by mHTT in vitro (Chaibva et al., 2018), suggesting mHTT could have particular effects on myelin lipids. Crucially, mHTT was localized within myelin sheaths using immunogold EM in 9-month-old Q175 striatum (Ko et al., 2018). Moreover, mHTT can be secreted by neurons in culture and in the brain as a soluble free form (Caron et al., 2021). We speculate that mHTT could insert directly into myelin bilayers to disrupt myelin architecture.
Observing white matter changes in animal models has been challenging. White matter loss was reported in R6/1 mice (Rattray et al., 2013), and changes in myelination have been described in Yac128 (Teo et al., 2019; Teo et al., 2016) and HdhQ250 mice (Jin et al., 2015), but experiments designed to look for white matter changes in the Q150 HD mouse model showed brain atrophy but no white matter abnormalities (Steventon et al., 2016). A recent imaging study of OVT73 sheep brain reported changes in diffusivity in the internal capsule at 9–10 years, suggesting changes in white matter microstructure (Taghian et al., 2022). Our biochemical experiments here show that mHTT effects on striatal lipid homeostasis in HD mouse models are complex. We and others have reported lipidomic and metabolomic studies on knock-in Q140/Q140 HD mice at single time points (Vodicka et al., 2015) and Q111 HD mice (Carroll et al., 2015) but did not observe loss of lipids important for white matter. Curiously, the lipid differences in LacQ140 mice measured at 9 months disappeared at 12 months suggesting that, even in the absence of mHtt lowering, the mouse brain insulted with mHTT attempts to heal itself and succeeds at some level. Consistent with our lipidomic findings, a longitudinal imaging study over 18 months showed transient changes in diffusivity/fractional anisotropy of corpus collosum in Q140 mouse brain (Pérot et al., 2022). These results echo imaging data from presymptomatic PwHD suggesting attempted remyelination (Phillips et al., 2014). Hence, if HD mouse models undergo a series of degeneration and regeneration cycles, observations at one or two time points may be misleading. By 12 months, although changes at the lipid subclass level were annulled in the LacQ140 mice, a detailed analysis of the individual lipid species comprising these subclasses shows a shift in species within each subclass. The altered composition of subclasses may modify function, weaken HD brains, or predispose them to further stress. In fact, behavioral analysis of these mice showed loss of benefit by mHTT lowering at 12 months (Marchionini et al., 2022) indicating that although levels of lipids by subclasses are restored at 12 months (with or without mHTT lowering), the changes in individual lipid species which compose each subclasses correlate with behavioral degradation. Because the behavioral analyses were conducted as longitudinal studies (n=23–32 mice per group examined at 6, 9, and 12-months of age), it precluded biochemical experiments which require animal sacrifice; instead, cross sectional biochemical studies were performed in parallel for 6 and 9-month timepoints.
Not all lipid changes were reversed with mHtt lowering. Consistent with metabolic defects in HD, here we report reduced levels of species of TG, glycerolipid molecules used for energy storage which can be metabolized by mitochondria, in LacQ140 compared to WT. Of note, reductions in species of TG were not reversed by mHtt lowering. Interestingly, the LacQ140 mice exhibited reciprocal changes in the two biosynthetic enzymes Dgat1 and Dgat2 at the transcriptional level, suggesting the ability to store energy may arise in part from this variation.
5. Conclusions
Collectively, our studies advocate early lowering of mHTT for greatest benefit, and in this context, modest lowering is sufficient to delay some protein and lipid changes. Furthermore, our work shows readily detectable but transient changes in lipids highly enriched in myelin, consistent with possible white matter damage and regeneration occurring in the LacQ140 mouse model.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2023.106313.
Supplementary Material
Acknowledgements
Illustrations of DNA in Fig. 1a and mouse in Fig. 1b were adapted from Vivek Kumar and Heath Robinson respectively. In graphical abstract: mouse brain adapted from Federico Claudi, Eppendorf tubes adapted from Diogo Losch De Oliveira, and repeating pipettor adapted from Shuhan He. Graphics listed above were sourced from scidraw.io.
Funding
This work was funded by CHDI Foundation, Inc., a nonprofit biomedical research organization exclusively dedicated to developing therapeutics that will substantially improve the lives of HD-affected individuals, the Dake Family Fund to MD and KBK-G, and NIH 1S10RR023594S10 to MD.
Abbreviations:
- HD
Huntington’s disease
- HTT
Huntingtin
- ASOs
Antisense oligonucleotide
- miRNA
MicroRNA
- shRNA
Short hairpin RNA
- DARPP32
Dopamine And CAMP-Regulated Neuronal Phosphoprotein 32
- PDE10A
Phosphodiesterase 10A
- SCN4B
Sodium Voltage-Gated Channel Beta Subunit 4
- ATP5A
ATP Synthase F1 Subunit Alpha
- PIP
Phosphatidylinositol phosphate
- IPTG
Isopropyl b-D-1-thiogalactopyranoside
- Poly-Q
Polyglutamine
- GFAP
Glial fibrillary acidic protein
- LC-MS
Liquid chromatography – mass spectrometry
- PS
Phosphatidylserine
- PI
Phosphatidylinositol
- BisMePA
Bis-methyl phosphatidic acid
- PKC
Protein kinase C
- SM
Sphingomyelin
- Cer
Ceramide
- MGDG
Monogalactosyldiacylglycerol
- Hex1Cer
Monohexosylceramide
- GalCer
Galactosylceramide
- GlcCer
Glucosylceramide
- TG
Triacylglycerol
- GO BP
Gene Ontology Biological Processes
- GSEA
Gene Set Enrichment Analysis
- Ugt8a
UDP-Galactose Ceramide Galactosyltransferase
- Fa2h
Fatty acid 2-hydroxylase
Footnotes
Ethics approval
Mice were housed at Psychogenics (Paramus, NJ) and all treatments and procedures were conducted with oversight by Psychogenics Institutional Animal Care and Use Committee.
Declaration of Competing Interest
KBK-G spouse owns less than 0.1% stock in the following companies: Advanced Microdevices, Aveo Pharmaceuticals, Inc, Boston Scientific Corporation, Bristol-Myers Squibb Company, Cisco Systems, Inc., Fate Therapeutics, GE Healthcare Life Sciences, Generex Biotechnology Corporation, Idera Pharmaceuticals, Inc., Nante Health, Neurometrics, Inc., NuGenerex, Repligen Corporation, Sesen Bio, Inc., T2 Biosystems, and Vericel Corporation. Other authors have no declarations of interest.
Data availability
All datasets generated are included in this article and can be found in Supplemental Files 2–6. RNA-sequencing data analyzed is available at Gene Expression Omnibus (GEO) accession number GSE156236.
References
- Agrawal S, Fox JH, 2019. Novel proteomic changes in brain mitochondria provide insights into mitochondrial dysfunction in mouse models of Huntington’s disease. Mitochondrion 47, 318–329. 10.1016/j.mito.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderson NL, Maldonado EN, Kern MJ, Bhat NR, Hama H, 2006. FA2H-dependent fatty acid 2-hydroxylation in postnatal mouse brain. J. Lipid Res 47, 2772–2780. 10.1194/jlr.M600362-JLR200. [DOI] [PubMed] [Google Scholar]
- Alterman JF, Godinho BMDC, Hassler MR, Ferguson CM, Echeverria D, Sapp E, Haraszti RA, Coles AH, Conroy F, Miller R, Roux L, Yan P, Knox EG, Turanov AA, King RM, Gernoux G, Mueller C, Gray-Edwards HL, Moser RP, Bishop NC, Jaber SM, Gounis MJ, Sena-Esteves M, Pai AA, DiFiglia M, Aronin N, Khvorova A, 2019. A divalent siRNA chemical scaffold for potent and sustained modulation of gene expression throughout the central nervous system. Nat. Biotechnol 37, 884–894. 10.1038/s41587-019-0205-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardile CF, Garcia-Miralles M, Caron NS, Rayan NA, Langley SR, Harmston N, Rondelli AM, Yi Teo RT, Waltl S, Anderson LM, Bae HG, Jung S, Williams A, Prabhakar S, Petretto E, Hayden MR, Pouladi MA, 2019. Intrinsic mutant HTT-mediated defects in oligodendroglia cause myelination deficits and behavioral abnormalities in Huntington disease. Proc. Natl. Acad. Sci. U. S. A 116, 9622–9627. 10.1073/pnas.1818042116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron JC, Hurley EP, Parsons MP, 2021. Huntingtin and the Synapse. Front. Cell. Neurosci 15, 689332. 10.3389/fncel.2021.689332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y, 1995. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- Benraiss A, Mariani JN, Tate A, Madsen PM, Clark KM, Welle KA, Solly R, Capellano L, Bentley K, Chandler-Militello D, Goldman SA, 2022. A TCF7L2-responsive suppression of both homeostatic and compensatory remyelination in Huntington disease mice. Cell Rep 40, 111291. 10.1016/j.celrep.2022.111291. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, 2016. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev 96, 1261–1296. 10.1152/physrev.00006.2016. [DOI] [PubMed] [Google Scholar]
- Bertoglio D, Bard J, Hessmann M, Liu L, Gärtner A, De Lombaerde S, Huscher B, Zajicek F, Miranda A, Peters F, Herrmann F, Schaertl S, Vasilkovska T, Brown CJ, Johnson PD, Prime ME, Mills MR, Van der Linden A, Mrzljak L, Khetarpal V, Wang Y, Marchionini DM, Skinbjerg M, Verhaeghe J, Dominguez C, Staelens S, Munoz-Sanjuan I, 2022. Development of a ligand for in vivo imaging of mutant huntingtin in Huntington’s disease. Sci. Transl. Med 14, 3682. [DOI] [PubMed] [Google Scholar]
- Bibb JA, Yan Z, Svenningsson P, Snyder GL, Pieribone VA, Horiuchi A, Nairn AC, Messer A, Greengard P, 2000. Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc. Natl. Acad. Sci. U. S. A 97, 6809–6814. 10.1073/pnas.120166397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blunsom NJ, Cockcroft S, 2020. CDP-Diacylglycerol Synthases (CDS): Gateway to Phosphatidylinositol and Cardiolipin Synthesis. Front. Cell Development. Bio 8 10.3389/fcell.2020.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosio A, Binczek E, Stoffel W, 1996. Functional breakdown of the lipid bilayer of the myelin membrane in central and peripheral nervous system by disrupted galactocerebroside synthesis. Proc. Natl. Acad. Sci 93, 13280–13285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RL, McBride JL, Martins I, Shen S, Xing Y, Carter BJ, Davidson BL, 2009. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol. Ther 17, 1053–1063. 10.1038/mt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourbon-Teles J, Bells S, Jones DK, Coulthard E, Rosser A, Metzler-Baddeley C, 2019. Myelin Breakdown in Human Huntington’s Disease: Multi-Modal Evidence from Diffusion MRI and Quantitative Magnetization Transfer. Neuroscience 403, 79–92. 10.1016/j.neuroscience.2017.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitkopf SB, Ricoult SJH, Yuan M, Xu Y, Peake DA, Manning BD, Asara JM, 2017. A relative quantitative positive/negative ion switching method for untargeted lipidomics via high resolution LC-MS/MS from any biological source. Metabolomics 13. 10.1007/s11306-016-1157-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron NS, Dorsey ER, Hayden MR, 2018. Therapeutic approaches to huntington disease: From the bench to the clinic. Nat. Rev. Drug Discov 17, 729–750. 10.1038/nrd.2018.133. [DOI] [PubMed] [Google Scholar]
- Caron NS, Banos R, Yanick C, Aly AE, Byrne LM, Smith ED, Xie Y, Smith SEP, Potluri N, Findlay Black H, Casal L, Ko S, Cheung D, Kim H, Seong IS, Wild EJ, Song JJ, Hayden MR, Southwell AL, 2021. Mutant Huntingtin Is Cleared from the Brain via Active Mechanisms in Huntington Disease. J. Neurosci 41, 780–796. 10.1523/JNEUROSCI.1865-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron NS, Banos R, Aly AE, Xie Y, Ko S, Potluri N, Anderson C, Black HF, Anderson LM, Gordon B, Southwell AL, Hayden MR, 2022. Cerebrospinal fluid mutant huntingtin is a biomarker for huntingtin lowering in the striatum of Huntington disease mice. Neurobiol. Dis 166 10.1016/j.nbd.2022.105652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JB, Deik A, Fossale E, Weston RM, Guide JR, Arjomand J, Kwak S, Clish CB, MacDonald ME, 2015. HdhQ111 mice exhibit tissue specific metabolite profiles that include striatal lipid accumulation. PLoS One 10. 10.1371/journal.pone.0134465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviston JP, Holzbaur ELF, 2009. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol 19, 147–155. 10.1016/j.tcb.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaibva M, Gao X, Jain P, Campbell WA, Frey SL, Legleiter J, 2018. Sphingomyelin and GM1 Influence Huntingtin Binding to, Disruptionof, and Aggregation on Lipid Membranes. ACS Omega 3, 273–285. 10.1021/acsomega.7b01472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrast R, Saher G, Nave KA, Verheijen MHG, 2011. Lipid metabolism in myelinating glial cells: Lessons from human inherited disorders and mouse models. J. Lipid Res 52, 419–434. 10.1194/jlr.R009761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Suzuki K, Popko B, 1996. Myelination in the Absence of Galactocerebroside and Sulfatide: Normal Structure with Abnormal Function and Regional Instability. Cell 86, 209–219. [DOI] [PubMed] [Google Scholar]
- Coffey SR, Bragg RM, Minnig S, Ament SA, Cantle JP, Glickenhaus A, Shelnut D, Carrillo JM, Shuttleworth DD, Rodier JA, Noguchi K, Bennett CF, Price ND, Kordasiewicz HB, Carroll JB, 2017. Peripheral huntingtin silencing does not ameliorate central signs of disease in the B6.HttQ111/+ mouse model of Huntington’s disease. PLoS One 12, e0175968. 10.1371/journal.pone.0175968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey SR, Andrew M, Ging H, Hamilton J, Flower M, Kovalenko M, Bragg RM, Cantle JP, McHugh CA, Carrillo JM, Rodier J-A, Marchionini DM, Wilkinson HA, Kwak S, Howland DS, Frank Bennett C, Mouro Pinto R, Auburger G, Zeitlin SO, Kordasiewicz HB, Tabrizi SJ, Wheeler VC, Carroll JB, 2020. Huntingtin lowering reduces somatic instability at CAG-expanded loci. bioRxiv 10.1101/2020.07.23.218347. [DOI]
- Cronin CA, Gluba W, Scrable H, 2001. The lac operator-repressor system is functional in the mouse. Genes Dev 15, 1506–1517. 10.1101/gad.892001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Bercury KK, Ahrendsen JT, Macklin WB, 2015. Olig1 function is required for oligodendrocyte differentiation in the mouse brain. J. Neurosci 35, 4386–4402. 10.1523/JNEUROSCI.4962-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datson NA, Gonzalez-Barriga A, Kourkouta E, Weij R, van de Giessen J, Mulders S, Kontkanen O, Heikkinen T, Lehtimaki K, van Deutekom JC, 2017. The expanded CAG repeat in the huntingtin gene as target for therapeutic RNA modulation throughout the HD mouse brain. PLoS One 12, e0171127. 10.1371/journal.pone.0171127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, Vonsattel JP, Richardson EP Jr., 1988. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington’s disease. J. Neuropathol. Exp. Neurol 47, 516–525. 10.1097/00005072-198809000-00003. [DOI] [PubMed] [Google Scholar]
- Desplats PA, Denny CA, Kass KE, Gilmartin T, Head SR, Sutcliffe JG, Seyfried TN, Thomas EA, 2007. Glycolipid and ganglioside metabolism imbalances in Huntington’s disease. Neurobiol. Dis 27, 265–277. 10.1016/j.nbd.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paola M, Luders E, Cherubini A, Sanchez-Castaneda C, Thompson PM, Toga AW, Caltagirone C, Orobello S, Elifani F, Squitieri F, Sabatini U, 2012. Multimodal MRI analysis of the corpus callosum reveals white matter differences in presymptomatic and early Huntington’s disease. Cereb. Cortex 22, 2858–2866. 10.1093/cercor/bhr360. [DOI] [PubMed] [Google Scholar]
- Di Pardo A, Basit A, Armirotti A, Amico E, Castaldo S, Pepe G, Marracino F, Buttari F, Digilio AF, Maglione V, 2017. De novo synthesis of sphingolipids is defective in experimental models of Huntington’s disease. Front. Neurosci 11 10.3389/fnins.2017.00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Hernandez M, Torres-Peraza J, Salvatori-Abarca A, Moran MA, Gomez-Ramos P, Alberch J, Lucas JJ, 2005. Full motor recovery despite striatal neuron loss and formation of irreversible amyloid-like inclusions in a conditional mouse model of Huntington’s disease. J. Neurosci 25, 9773–9781. 10.1523/JNEUROSCI.3183-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel J-P, Carraway R, Reeves SA, Boyce FM, Aronint N, 1995. Huntingtin Is a Cytoplasmic Protein Associated with Vesicles in Human and Rat Brain Neurons. Neuron 14, 1075–1081. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sena-Esteves M, Chase K, Sapp E, Pfister E, Sass M, Yoder J, Reeves P, Pandey RK, Rajeev KG, Manoharan M, Sah DW, Zamore PD, Aronin N, 2007. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc. Natl. Acad. Sci. U. S. A 104, 17204–17209. 10.1073/pnas.0708285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djannatian M, Radha S, Weikert U, Safaiyan S, Wrede C, Deichsel C, Kislinger G, Rhomberg A, Ruhwedel T, Campbell DS, van Ham T, Schmid B, Hegermann J, Möbius W, Schifferer M, Simons M, 2023. Myelination generates aberrant ultrastructure that is resolved by microglia. J. Cell Biol 222. 10.1083/jcb.202204010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S, 2000. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet 26, 300–306. [DOI] [PubMed] [Google Scholar]
- Drouet V, Perrin V, Hassig R, Dufour N, Auregan G, Alves S, Bonvento G, Brouillet E, Luthi-Carter R, Hantraye P, Deglon N, 2009. Sustained effects of nonallele-specific Huntingtin silencing. Ann. Neurol 65, 276–285. 10.1002/ana.21569. [DOI] [PubMed] [Google Scholar]
- Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL, et al. , 1995. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 269, 407–410. 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- Edvardson S, Hama H, Shaag A, Gomori JM, Berger I, Soffer D, Korman SH, Taustein I, Saada A, Elpeleg O, 2008. Mutations in the Fatty Acid 2-Hydroxylase Gene Are Associated with Leukodystrophy with Spastic Paraparesis and Dystonia. Am. J. Hum. Genet 83, 643–648. 10.1016/j.ajhg.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria AV, Ratnanather JT, Tward DJ, Lee DS, van den Noort F, Wu D, Brown T, Johnson H, Paulsen JS, Ross CA, Younes L, Miller MI, Investigators P-H, Coordinators of the Huntington Study G, 2016. Linking white matter and deep gray matter alterations in premanifest Huntington disease. Neuroimage Clin 11, 450–460. 10.1016/j.nicl.2016.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzana F, McConville MJ, Renoir T, Li S, Nie S, Tran H, Hannan AJ, Hatters DM, Boughton BA, 2023. Longitudinal spatial mapping of lipid metabolites reveals pre-symptomatic changes in the hippocampi of Huntington’s disease transgenic mice. Neurobiol. Dis 176, 105933. 10.1016/j.nbd.2022.105933. [DOI] [PubMed] [Google Scholar]
- Fitzner D, Bader JM, Penkert H, Bergner CG, Su M, Weil MT, Surma MA, Mann M, Klose C, Simons M, 2020. Cell-type- and brain-region-resolved mouse brain lipidome. Cell Rep 32. 10.1016/j.celrep.2020.108132. [DOI] [PubMed] [Google Scholar]
- Fox LM, Kim K, Johnson CW, Chen S, Croce KR, Victor MB, Eenjes E, Bosco JR, Randolph LK, Dragatsis I, Dragich JM, Yoo AS, Yamamoto A, 2020. Huntington’s Disease Pathogenesis Is Modified In Vivo by Alfy/Wdfy3 and Selective Macroautophagy. Neuron 105, 813–821.e6. 10.1016/j.neuron.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Chakraborty A, Geater C, Pradhan S, Gordon KL, Snowden J, Yuan S, Dickey AS, Choudhary S, Ashizawa T, Ellerby LM, La Spada AR, Thompson LM, Hazra TK, Sarkar PS, 2019. Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. eLife 10.7554/eLife.42988.001. [DOI] [PMC free article] [PubMed]
- Gu Z, 2022. Complex heatmap visualization. iMeta 10.1002/imt2.43. [DOI] [PMC free article] [PubMed]
- Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH, 1996. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol 39, 385–389. 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- Harayama T, Riezman H, 2018. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol 19, 281–296. 10.1038/nrm.2017.138. [DOI] [PubMed] [Google Scholar]
- Harris CA, Haas JT, Streeper RS, Stone SJ, Kumari M, Yang K, Han X, Brownell N, Gross RW, Zechner R, Farese JRV, 2011. DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes [S]. J. Lipid Res 52, 657–667. 10.1194/jlr.M013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt UA, Leist M, 2003. Rapid, noninflammatory and PS-dependent phagocytic clearance of necrotic cells. Cell Death Differ 10, 1156–1164. 10.1038/sj.cdd.4401286. [DOI] [PubMed] [Google Scholar]
- Hobbs NZ, Henley SM, Ridgway GR, Wild EJ, Barker RA, Scahill RI, Barnes J, Fox NC, Tabrizi SJ, 2010. The progression of regional atrophy in premanifest and early Huntington’s disease: a longitudinal voxel-based morphometry study. J. Neurol. Neurosurg. Psychiatry 81, 756–763. 10.1136/jnnp.2009.190702. [DOI] [PubMed] [Google Scholar]
- Hunter M, Demarais NJ, Faull RLM, Grey AC, Curtis MA, 2018. Subventricular zone lipidomic architecture loss in Huntington’s disease. J. Neurochem 146, 613–630. 10.1111/jnc.14468. [DOI] [PubMed] [Google Scholar]
- Hunter M, Demarais NJ, Faull RLM, Grey AC, Curtis MA, 2021. An imaging mass spectrometry atlas of lipids in the human neurologically normal and Huntington’s disease caudate nucleus. J. Neurochem 157, 2158–2172. 10.1111/jnc.15325. [DOI] [PubMed] [Google Scholar]
- Hyuk Yoon J, Seo Y, Suk Jo Y, Lee S, Cho E, Cazenave-Gassiot A, Shin Y-S, Hee Moon M, Joo An H, Wenk MR, Suh P-G, 2022. Brain lipidomics: From functional landscape to clinical significance. Sci. Adv 8, 9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Deshmukh DS, Pieringer RA, 1971. The Association of the Galactosyl Diglycerides of Brain with Myelination. J. Biol. Chem 246, 5688–5694. 10.1016/S0021-9258(18)61860-5. [DOI] [PubMed] [Google Scholar]
- Iuliano M, Seeley C, Sapp E, Jones EL, Martin C, Li X, DiFiglia M, Kegel-Gleason KB, 2021. Disposition of proteins and lipids in synaptic membrane compartments is altered in Q175/Q7 Huntington’s disease mouse striatum. Front. Synapt. Neurosci 13 10.3389/fnsyn.2021.618391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Peng Q, Hou Z, Jiang M, Wang X, Langseth AJ, Tao M, Barker PB, Mori S, Bergles DE, Ross CA, Detloff PJ, Zhang J, Duan W, 2015. Early white matter abnormalities, progressive brain pathology and motor deficits in a novel knock-in mouse model of Huntington’s disease. Hum. Mol. Genet 24, 2508–2527. 10.1093/hmg/ddv016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaganovich D, Kopito R, Frydman J, 2008. Misfolded proteins partition between two distinct quality control compartments. Nature 454, 1088–1095. 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeler AM, Sapp E, Chase K, Sottosanti E, Danielson E, Pfister E, Stoica L, DiFiglia M, Aronin N, Sena-Esteves M, 2016. Cellular Analysis of Silencing the Huntington’s Disease Gene Using AAV9 Mediated Delivery of Artificial Micro RNA into the Striatum of Q140/Q140 Mice. J. Huntingtons Dis 5, 239–248. 10.3233/JHD-160215. [DOI] [PubMed] [Google Scholar]
- Kegel KB, Kim M, Sapp E, Mcintyre C, Castañ JG, Aronin N, Difiglia M, 2000. Huntingtin Expression Stimulates Endosomal-Lysosomal Activity, Endosome Tubulation, and Autophagy. J. Neurosci 20, 7268–7278. 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, Sapp E, Wang Y, Qin ZH, Chen JD, Nevins JR, Aronin N, DiFiglia M, 2002. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J. Biol. Chem 277, 7466–7476. 10.1074/jbc.M103946200. [DOI] [PubMed] [Google Scholar]
- Kegel KB, Sapp E, Alexander J, Valencia A, Reeves P, Li X, Masso N, Sobin L, Aronin N, DiFiglia M, 2009a. Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J. Neurochem 110, 1585–1597. 10.1111/j.1471-4159.2009.06255.x. [DOI] [PubMed] [Google Scholar]
- Kegel KB, Schewkunow V, Sapp E, Masso N, Wanker EE, DiFiglia M, Goldmann WH, 2009b. Polyglutamine expansion in huntingtin increases its insertion into lipid bilayers. Biochem. Biophys. Res. Commun 387, 472–475. 10.1016/j.bbrc.2009.07.039. [DOI] [PubMed] [Google Scholar]
- Keller CG, Shin Y, Monteys AM, Renaud N, Beibel M, Teider N, Peters T, Faller T, St-Cyr S, Knehr J, Roma G, Reyes A, Hild M, Lukashev D, Theil D, Dales N, Cha JH, Borowsky B, Dolmetsch R, Davidson BL, Sivasankaran R, 2022. An orally available, brain penetrant, small molecule lowers huntingtin levels by enhancing pseudoexon inclusion. Nat. Commun 13 10.1038/s41467-022-28653-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-Y, Huang BX, Spector AA, 2014. Phosphatidylserine in the brain: Metabolism and function. Prog. Lipid Res 56, 1–18. 10.1016/j.plipres.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Ou S, Patterson PH, 2001. New anti-huntingtin monoclonal antibodies: implications for huntingtin conformation and its binding proteins. Brain Res. Bull 56, 319–329. 10.1016/s0361-9230(01)00599-8. [DOI] [PubMed] [Google Scholar]
- Ko J, Isas JM, Sabbaugh A, Yoo JH, Pandey NK, Chongtham A, Ladinsky M, Wu WL, Rohweder H, Weiss A, Macdonald D, Munoz-Sanjuan I, Langen R, Patterson PH, Khoshnan A, 2018. Identification of distinct conformations associated with monomers and fibril assemblies of mutant huntingtin. Hum. Mol. Genet 27, 2330–2343. 10.1093/hmg/ddy141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelmel JP, Ulmer CZ, Jones CM, Yost RA, Bowden JA, 2017. Common cases of improper lipid annotation using high-resolution tandem mass spectrometry data and corresponding limitations in biological interpretation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 766–770. 10.1016/j.bbalip.2017.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, Hung G, Bennett CF, Cleveland DW, 2012. Sustained Therapeutic Reversal of Huntington’s Disease by Transient Repression of Huntingtin Synthesis. Neuron 74, 1031–1044. 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landles C, Sathasivam K, Weiss A, Woodman B, Moffitt H, Finkbeiner S, Sun B, Gafni J, Ellerby LM, Trottier Y, Richards WG, Osmand A, Paganetti P, Bates GP, 2010. Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J. Biol. Chem 285, 8808–8823. 10.1074/jbc.M109.075028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, Deverasetty S, Tebbe A, Schaab C, Lavery DJ, Howland D, Kwak S, Botas J, Aaronson JS, Rosinski J, Coppola G, Horvath S, Yang XW, 2016. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci 19, 623–633. 10.1038/nn.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt BR, Kordasiewicz HB, Schobel SA, 2020. Huntingtin-Lowering Therapies for Huntington Disease: A Review of the Evidence of Potential Benefits and Risks. JAMA Neurol 77, 764–772. 10.1001/jamaneurol.2020.0299. [DOI] [PubMed] [Google Scholar]
- Legleiter J, Lotz GP, Miller J, Ko J, Ng C, Williams GL, Finkbeiner S, Patterson PH, Muchowski PJ, 2009. Monoclonal Antibodies Recognize Distinct Conformational Epitopes Formed by Polyglutamine in a Mutant Huntingtin Fragment *. J. Biol. Chem 284, 21647–21658. 10.1074/jbc.M109.016923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S-H, Schilling G, Young WS, Li X, Margolis RL, Stine OC, Wagster MV, Abbott MH, Franz ML, Ranen NG, Folstein SE, Hedreen JC, Ross CA, 1993. Huntington’s disease gene (IT15) is widely expressed in human and rat tissues. Neuron 11, 985–993. 10.1016/0896-6273(93)90127-D. [DOI] [PubMed] [Google Scholar]
- Liebisch G, Fahy E, Aoki J, Dennis EA, Durand T, Ejsing CS, Fedorova M, Feussner I, Griffiths WJ, Köfeler H, Merrill AH, Murphy RC, O’Donnell VB, Oskolkova O, Subramaniam S, Wakelam MJO, Spener F, 2020. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J. Lipid Res 61, 1539–1555. 10.1194/jlr.S120001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim RG, Al-Dalahmah O, Wu J, Gold MP, Reidling JC, Tang G, Adam M, Dansu DK, Park H-J, Casaccia P, Miramontes R, Reyes-Ortiz AM, Lau A, Hickman RA, Khan F, Paryani F, Tang A, Ofori K, Miyoshi E, Michael N, McClure N, Flowers XE, Vonsattel JP, Davidson S, Menon V, Swarup V, Fraenkel E, Goldman JE, Thompson LM, 2022. Huntington disease oligodendrocyte maturation deficits revealed by single-nucleus RNAseq are rescued by thiamine-biotin supplementation. Nat. Commun 13, 7791. 10.1038/s41467-022-35388-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S, 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchionini DM, Liu J-P, Ambesi-Impiombato A, Kerker K, Cirillo K, Bansal M, Mushlin R, Brunner D, Ramboz S, Kwan M, Kuhlbrodt K, Tillack K, Peters F, Rauhala L, Obenauer J, Greene JR, Hartl C, Khetarpal V, Lager B, Rosinski J, Aaronson J, Alam M, Signer E, Muñoz-Sanjuán I, Howland D, Zeitlin SO, 2022. Benefits of global mutant huntingtin lowering diminish over time in a Huntington’s disease mouse model. JCI Insight 7. 10.1172/jci.insight.161769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui JT, Vaidya JG, Wassermann D, Kim RE, Magnotta VA, Johnson HJ, Paulsen JS, Investigators P-H, Coordinators of the Huntington Study G, 2015. Prefrontal cortex white matter tracts in prodromal Huntington disease. Hum. Brain Mapp 36, 3717–3732. 10.1002/hbm.22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, Schwudke D, 2008. Lipid extraction by methyl-terf-butyl ether for high-throughput lipidomics. J. Lipid Res 49, 1137–1146. 10.1194/jlr.D700041-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, Ojeda SR, Davidson BL, 2011. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol. Ther 19, 2152–2162. 10.1038/mt.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinstry SU, Karadeniz YB, Worthington AK, Hayrapetyan VY, Ozlu MI, Serafin-Molina K, Risher WC, Ustunkaya T, Dragatsis I, Zeitlin S, Yin HH, Eroglu C, 2014. Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J. Neurosci 34, 9455–9472. 10.1523/JNEUROSCI.4699-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade LR, Balachandran A, Scott HA, Khaira S, Baker MS, Schmidt U, 2014. Proteomics of Huntington’s disease-affected human embryonic stem cells reveals an evolving pathology involving mitochondrial dysfunction and metabolic disturbances. J. Proteome Res 13, 5648–5659. 10.1021/pr500649m. [DOI] [PubMed] [Google Scholar]
- Mehler MF, Petronglo JR, Arteaga-Bracho EE, Gulinello ME, Winchester ML, Pichamoorthy N, Young SK, DeJesus CD, Ishtiaq H, Gokhan S, Molero AE, 2019. Loss-of-Huntingtin in Medial and Lateral Ganglionic Lineages Differentially Disrupts Regional Interneuron and Projection Neuron Subtypes and Promotes Huntington’s Disease-Associated Behavioral, Cellular, and Pathological Hallmarks. J. Neurosci 39, 1892–1909. 10.1523/JNEUROSCI.2443-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SR, Tom CM, Wang Y, Bresee C, Rushton D, Mathkar PP, Tang J, Mattis VB, 2018. Human Huntington’s Disease iPSC-Derived Cortical Neurons Display Altered Transcriptomics, Morphology, and Maturation. Cell Rep 25 (1081–1096), e6 10.1016/j.celrep.2018.09.076. [DOI] [PubMed] [Google Scholar]
- Miller J, Arrasate M, Brooks E, Libeu CP, Legleiter J, Hatters D, Curtis J, Cheung K, Krishnan P, Mitra S, Widjaja K, Shaby BA, Lotz GP, Newhouse Y, Mitchell EJ, Osmand A, Gray M, Thulasiramin V, Saudou F, Segal M, Yang XW, Masliah E, Thompson LM, Muchowski PJ, Weisgraber KH, Finkbeiner S, 2011. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat. Chem. Biol 7, 925–934. 10.1038/nchembio.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR, 1995. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 81, 811–823. 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- Nopoulos PC, 2016. Huntington disease: a single-gene degenerative disorder of the striatum. Dialogues Clin. Neurosci 18, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JS, Sampson EL, 1965. Lipid composition of the normal human brain: gray matter, white matter, and myelin. J. Lipid Res 6, 537–544. 10.1016/S0022-2275(20)39619-X. [DOI] [PubMed] [Google Scholar]
- O’Kusky JR, Nasir J, Cicchetti F, Parent A, Hayden MR, 1999. Neuronal degeneration in the basal ganglia and loss of pallido-subthalamic synapses in mice with targeted disruption of the Huntington’s disease gene. Brain Res 818, 468–479. 10.1016/s0006-8993(98)01312-2. [DOI] [PubMed] [Google Scholar]
- Odish OF, Leemans A, Reijntjes RH, van den Bogaard SJ, Dumas EM, Wolterbeek R, Tax CM, Kuijf HJ, Vincken KL, van der Grond J, Roos RA, 2015. Microstructural brain abnormalities in Huntington’s disease: A two-year follow-up. Hum. Brain Mapp 36, 2061–2074. 10.1002/hbm.22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SL, Chen CM, Wu YR, Valdes Hernandez M, Tsai CC, Cheng JS, Chen YL, Wu YM, Lin YC, Wang JJ, 2021. Fixel-Based Analysis Effectively Identifies White Matter Tract Degeneration in Huntington’s Disease. Front. Neurosci 15, 711651. 10.3389/fnins.2021.711651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipovitch M, Asenjo Martinez A, Mariani JN, Cornwell A, Dhaliwal S, Zou L, Chandler-Militello D, Wang S, Li X, Benraiss SJ, Agate R, Lampp A, Benraiss A, Windrem MS, Goldman SA, 2019. Human ESC-Derived Chimeric Mouse Models of Huntington’s disease reveal cell-intrinsic defects in glial progenitor cell differentiation. Cell Stem Cell 24, 107–122.e7. 10.1016/j.stem.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama F, Miyazaki H, Sakamoto N, Becquet C, Machida Y, Kaneko K, Uchikawa C, Suzuki T, Kurosawa M, Ikeda T, Tamaoka A, Sakurai T, Nukina N, 2006. Sodium channel beta4 subunit: down-regulation and possible involvement in neuritic degeneration in Huntington’s disease transgenic mice. J. Neurochem 98, 518–529. 10.1111/j.1471-4159.2006.03893.x. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, 2009. Functional imaging in Huntington’s disease. Exp. Neurol 216, 272–277. 10.1016/j.expneurol.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, Guttman M, Johnson S, MacDonald M, Beglinger LJ, Duff K, Kayson E, Biglan K, Shoulson I, Oakes D, Hayden M, Predict HDI, Coordinators of the Huntington Study G, 2008. Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J. Neurol. Neurosurg. Psychiatry 79, 874–880. 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérot JB, Célestine M, Palombo M, Dhenain M, Humbert S, Brouillet E, Flament J, 2022. Longitudinal multimodal MRI characterization of a knock-in mouse model of Huntington’s disease reveals early gray and white matter alterations. Hum. Mol. Genet 31, 3581–3596. 10.1093/hmg/ddac036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen A, Bjorkqvist M, 2006. Hypothalamic-endocrine aspects in Huntington’s disease. Eur. J. Neurosci 24, 961–967. 10.1111/j.1460-9568.2006.04985.x. [DOI] [PubMed] [Google Scholar]
- Phillips O, Sanchez-Castaneda C, Elifani F, Maglione V, Di Pardo A, Caltagirone C, Squitieri F, Sabatini U, Di Paola M, 2013. Tractography of the corpus callosum in Huntington’s disease. PLoS One 8, e73280. 10.1371/journal.pone.0073280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips O, Squitieri F, Sanchez-Castaneda C, Elifani F, Caltagirone C, Sabatini U, Di Paola M, 2014. Deep white matter in Huntington’s disease. PLoS One 9. 10.1371/journal.pone.0109676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips G, Saville JT, Hancock SE, Brown SHJ, Jenner AM, McLean C, Fuller M, Newell KA, Mitchell TW, 2022. The long and the short of Huntington’s disease: how the sphingolipid profile is shifted in the caudate of advanced clinical cases. Brain Communicat 4. 10.1093/braincomms/fcab303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyansky A, Shatz O, Fraiberg M, Shimoni E, Dadosh T, Mari M, Reggiori FM, Qin C, Han X, Elazar Z, 2022. Phospholipid imbalance impairs autophagosome completion. EMBO J 10.15252/embj.2022110771. [DOI] [PMC free article] [PubMed]
- Poudel GR, Stout JC, Dominguez DJ, Churchyard A, Chua P, Egan GF, Georgiou-Karistianis N, 2015. Longitudinal change in white matter microstructure in Huntington’s disease: The IMAGE-HD study. Neurobiol. Dis 74, 406–412. 10.1016/j.nbd.2014.12.009. [DOI] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Kegel KB, Kazantsev A, Apostol BL, Thompson LM, Yoder J, Aronin N, DiFiglia M, 2003. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet 12, 3231–3244. 10.1093/hmg/ddg346. [DOI] [PubMed] [Google Scholar]
- Rattay TW, Lindig T, Baets J, Smets K, Deconinck T, Söhn AS, Hörtnagel K, Eckstein KN, Wiethoff S, Reichbauer J, Döbler-Neumann M, Krägeloh-Mann I, Auer-Grumbach M, Plecko B, Münchau A, Wilken B, Janauschek M, Giese A-K, De Bleecker JL, Ortibus E, Debyser M, Lopez de Munain A, Pujol A, Bassi MT, D’Angelo MG, De Jonghe P, Züchner S, Bauer P, Schöls L, Schüle R, 2019. FAHN/SPG35: a narrow phenotypic spectrum across disease classifications. Brain 142, 1561–1572. 10.1093/brain/awz102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattray I, Smith EJ, Crum WR, Walker TA, Gale R, Bates GP, Modo M, 2013. Correlations of behavioral deficits with brain pathology assessed through longitudinal MRI and histopathology in the R6/1 mouse model of Huntington’s disease. PLoS One 8, e84726. 10.1371/journal.pone.0084726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reza S, Ugorski M, Suchański J, 2021. Glucosylceramide and galactosylceramide, small glycosphingolipids with significant impact on health and disease. Glycobiology 31, 1416–1434. 10.1093/glycob/cwab046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Lebron E, Denovan-Wright EM, Nash K, Lewin AS, Mandel RJ, 2005. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington’s disease transgenic mice. Mol. Ther 12, 618–633. 10.1016/j.ymthe.2005.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas HD, Tuch DS, Hevelone ND, Zaleta AK, Vangel M, Hersch SM, Salat DH, 2006. Diffusion tensor imaging in presymptomatic and early Huntington’s disease: Selective white matter pathology and its relationship to clinical measures. Mov. Disord 21, 1317–1325. 10.1002/mds.20979. [DOI] [PubMed] [Google Scholar]
- Sapp E, Schwarz C, Chase K, Bhide PG, Young AB, Penney J, Vonsattel JP, Aronin N, Difiglia M, 1997. Huntingtin Localization in Brains of Normal and Huntington’s Disease Patients. Ann. Neurol 42, 604–612. [DOI] [PubMed] [Google Scholar]
- Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP, DiFiglia M, 2001. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol 60, 161–172. 10.1093/jnen/60.2.161. [DOI] [PubMed] [Google Scholar]
- Sapp E, Seeley C, Iuliano M, Weisman E, Vodicka P, DiFiglia M, Kegel-Gleason KB, 2020. Protein changes in synaptosomes of Huntington’s disease knock-in mice are dependent on age and brain region. Neurobiol. Dis 141 10.1016/j.nbd.2020.104950. [DOI] [PubMed] [Google Scholar]
- Sathasivam K, Lane A, Legleiter J, Warley A, Woodman B, Finkbeiner S, Paganetti P, Muchowski PJ, Wilson S, Bates GP, 2010. Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington’s disease. Hum. Mol. Genet 19, 65–78. 10.1093/hmg/ddp467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F, Humbert S, 2016. The Biology of Huntingtin. Neuron 89, 910–926. 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Savage JC, St-Pierre MK, Carrier M, El Hajj H, Novak SW, Sanchez MG, Cicchetti F, Tremblay ME, 2020. Microglial physiological properties and interactions with synapses are altered at presymptomatic stages in a mouse model of Huntington’s disease pathology. J. Neuroinflammation 17, 98. 10.1186/s12974-020-01782-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE, 1997. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558. 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- Schmidt-Schultz T, Althaus HH, 1994. Monogalactosyl Diglyceride, a Marker for Myelination, Activates Oligodendroglial Protein Kinase C. J. Neurochem 62, 1578–1585. [DOI] [PubMed] [Google Scholar]
- Schmitt S, Cantuti Castelvetri L, Simons M, 2015. Metabolism and functions of lipids in myelin. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1851, 999–1005. 10.1016/j.bbalip.2014.12.016. [DOI] [PubMed] [Google Scholar]
- Scott-Hewitt N, Perrucci F, Morini R, Erreni M, Mahoney M, Witkowska A, Carey A, Faggiani E, Schuetz LT, Mason S, Tamborini M, Bizzotto M, Passoni L, Filipello F, Jahn R, Stevens B, Matteoli M, 2020. Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J 39 10.15252/embj.2020105380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrable H, 2002. Say when: reversible control of gene expression in the mouse by lac. Semin. Cell Dev. Biol 13, 109–119. 10.1016/s1084-9521(02)00017-4. [DOI] [PubMed] [Google Scholar]
- Seong IS, Woda JM, Song J-J, Lloret A, Abeyrathne PD, Woo CJ, Gregory G, Lee J-M, Wheeler VC, Walz T, Kingston RE, Gusella JF, Conlon RA, MacDonald ME, 2010. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet 19, 573–583. 10.1093/hmg/ddp524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AH, Loev SJ, Schilling G, Li SH, Li XJ, Bao J, Wagster MV, Kotzuk JA, Steiner JP, Lo A, et al. , 1995. Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron 14, 1065–1074. 10.1016/0896-6273(95)90345-3. [DOI] [PubMed] [Google Scholar]
- Singh S, Mehta H, Fekete R, 2013. Altered Fractional Anisotropy in Early Huntington’s Disease. Case Rep. Neurol 5, 26–30. 10.1159/000348399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene DJ, Middleton B, Fraser CK, Pennings JLA, Kuchel TR, Rudiger SR, Bawden CS, Morton AJ, 2017. Metabolic profiling of presymptomatic Huntington’s disease sheep reveals novel biomarkers. Sci. Rep 7 10.1038/srep43030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skotte NH, Andersen JV, Santos A, Aldana BI, Willert CW, Nørremølle A, Waagepetersen HS, Nielsen ML, 2018. Integrative Characterization of the R6/2 Mouse Model of Huntington’s Disease Reveals Dysfunctional Astrocyte Metabolism. Cell Rep 23, 2211–2224. 10.1016/j.celrep.2018.04.052. [DOI] [PubMed] [Google Scholar]
- Southwell AL, Smith SE, Davis TR, Caron NS, Villanueva EB, Xie Y, Collins JA, Ye ML, Sturrock A, Leavitt BR, Schrum AG, Hayden MR, 2015. Ultrasensitive measurement of huntingtin protein in cerebrospinal fluid demonstrates increase with Huntington disease stage and decrease following brain huntingtin suppression. Sci. Rep 5, 12166. 10.1038/srep12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwell AL, Kordasiewicz HB, Langbehn D, Skotte NH, Parsons MP, Villanueva EB, Caron NS, Østergaard ME, Anderson LM, Xie Y, Dal Cengio L, Findlay-Black H, Doty CN, Fitsimmons B, Swayze EE, Seth PP, Raymond LA, Frank Bennett C, Hayden MR, 2018. Huntingtin suppression restores cognitive function in a mouse model of Huntington’s disease. Sci. Transl. Med 10, 3959. [DOI] [PubMed] [Google Scholar]
- Stanek LM, Yang W, Angus S, Sardi PS, Hayden MR, Hung GH, Bennett CF, Cheng SH, Shihabuddin LS, 2013. Antisense oligonucleotide-mediated correction of transcriptional dysregulation is correlated with behavioral benefits in the YAC128 mouse model of Huntington’s disease. J. Huntingtons Dis 2, 217–228. 10.3233/JHD-130057. [DOI] [PubMed] [Google Scholar]
- Stanek LM, Sardi SP, Mastis B, Richards AR, Treleaven CM, Taksir T, Misra K, Cheng SH, Shihabuddin LS, 2014. Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum. Gene Ther 25, 461–474. 10.1089/hum.2013.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steventon JJ, Trueman RC, Ma D, Yhnell E, Bayram-Weston Z, Modat M, Cardoso J, Ourselin S, Lythgoe M, Stewart A, Rosser AE, Jones DK, 2016. Longitudinal in vivo MRI in a Huntington’s disease mouse model: Global atrophy in the absence of white matter microstructural damage. Sci. Rep 6 10.1038/srep32423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong TV, Tagle DA, Valdes JM, Elmer LW, Boehm K, Swaroop M, Kaatz KW, Collins FS, Albin RL, 1993. Widespread expression of the human and rat Huntington’s disease gene in brain and nonneural tissues. Nat. Genet 5, 259–265. 10.1038/ng1193-259. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, 2005. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweidan W, Bao F, Bozorgzad NS, George E, 2020. White and Gray Matter Abnormalities in Manifest Huntington’s Disease: Cross-Sectional and Longitudinal Analysis. J. Neuroimaging 30, 351–358. 10.1111/jon.12699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Ghosh R, Leavitt BR, 2019. Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron 101, 801–819. 10.1016/j.neuron.2019.01.039. [DOI] [PubMed] [Google Scholar]
- Taghian T, Gallagher J, Batcho E, Pullan C, Kuchel T, Denney T, Perumal R, Moore S, Muirhead R, Herde P, Johns D, Christou C, Taylor A, Passler T, Pulaparthi S, Hall E, Chandra S, O’Neill CA, Gray-Edwards H, 2022. Brain Alterations in Aged OVT73 Sheep Model of Huntington’s Disease: An MRI Based Approach. J. Huntington’s Disease 1–16. 10.3233/jhd-220526. [DOI] [PMC free article] [PubMed]
- Teo RTY, Hong X, Yu-Taeger L, Huang Y, Tan LJ, Xie Y, To XV, Guo L, Rajendran R, Novati A, Calaminus C, Riess O, Hayden MR, Nguyen HP, Chuang KH, Pouladi MA, 2016. Structural and molecular myelination deficits occur prior to neuronal loss in the YAC128 and BACHD models of Huntington disease. Hum. Mol. Genet 25, 2621–2632. 10.1093/hmg/ddw122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo RTY, Ferrari Bardile C, Tay YL, Yusof NABM, Kreidy CA, Tan LJ, Pouladi MA, 2019. Impaired Remyelination in a Mouse Model of Huntington Disease. Mol. Neurobiol 56, 6873–6882. 10.1007/s12035-019-1579-1. [DOI] [PubMed] [Google Scholar]
- Tereshchenko A, Magnotta V, Epping E, Mathews K, Espe-Pfeifer P, Martin E, Dawson J, Duan W, Nopoulos P, 2019. Brain structure in juvenile-onset Huntington disease. Neurology 92, e1939–e1947. 10.1212/WNL.0000000000007355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tousley A, Iuliano M, Weisman E, Sapp E, Richardson H, Vodicka P, Alexander J, Aronin N, DiFiglia M, Kegel-Gleason KB, 2019. Huntingtin associates with the actin cytoskeleton and alpha-actinin isoforms to influence stimulus dependent morphology changes. PLoS One 14, e0212337. 10.1371/journal.pone.0212337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, 2000. Dominant phenotypes produced by the HD mutation in STHdhQ111 striatal cells. Hum. Mol. Genet 9, 2799–2809. 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- van der Bijl P, Strous GJ, Lopes-Cardozo M, Thomas-Oates J, van Meer G, 1996. Synthesis of non-hydroxy-galactosylceramides and galactosyldiglycerides by hydroxy-ceramide galactosyltransferase. Biochem. J 317, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanier MT, Svennerholm L, 1975. Chemical pathology of Krabbe’s disease. III. Ceramide-hexosides and gangliosides of brain. Acta Paediatr. Scand 64, 641–648. 10.1111/j.1651-2227.1975.tb03896.x. [DOI] [PubMed] [Google Scholar]
- Vodicka P, Lim J, Williams DT, Kegel KB, Chase K, Park H, Marchionini D, Wilkinson S, Mead T, Birch H, Yates D, Lyons K, Dominguez C, Beconi M, Yue Z, Aronin N, DiFiglia M, 2014. Assessment of chloroquine treatment for modulating autophagy flux in brain of WT and HD mice. J. Huntingtons Dis 3, 159–174. 10.3233/JHD-130081. [DOI] [PubMed] [Google Scholar]
- Vodicka P, Mo S, Tousley A, Green KM, Sapp E, Iuliano M, Sadri-Vakili G, Shaffer SA, Aronin N, DiFiglia M, Kegel-Gleason KB, 2015. Mass Spectrometry Analysis of Wild-Type and Knock-in Q140/Q140 Huntington’s Disease Mouse Brains Reveals Changes in Glycerophospholipids Including Alterations in Phosphatidic Acid and Lyso-Phosphatidic Acid. J. Huntington’s Disease 4, 187–201. 10.3233/JHD-150149. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M, 1998. Huntington disease. J. Neuropathol. Exp. Neurol 57, 369–384. 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Weiss A, Klein C, Woodman B, Sathasivam K, Bibel M, Regulier E, Bates GP, Paganetti P, 2008. Sensitive biochemical aggregate detection reveals aggregation onset before symptom development in cellular and murine models of Huntington’s disease. J. Neurochem 104, 846–858. 10.1111/j.1471-4159.2007.05032.x. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, 2000. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum. Mol. Genet 9, 503–513. 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- Williamson P, Schlegel RA, 2002. Transbilayer phospholipid movement and the clearance of apoptotic cells. Biochim. Biophys. Acta 1585, 53–63. 10.1016/s1388-1981(02)00324-4. [DOI] [PubMed] [Google Scholar]
- Wilton D, Mastro K, Gergits F, Willing CR, Frouin A, Daggett A, Gu X, Kim A, Yang X, Stevens B, Stevens B, 2021. Microglia mediate early corticostriatal synapse loss and cognitive dysfunction in Huntington’s Disease through complement-dependent mechanisms. bioRxiv 10.1101/2021.12.03.471180. [DOI]
- Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, Fu X, Liu S, Bo X, Yu G, 2021. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The Innovat 2 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Cremona ML, Rothman JE, 2006. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J. Cell Biol 172, 719–731. 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitler B, Froelich S, Marlen K, Shivak DA, Yu Q, Li D, Pearl JR, Miller JC, Zhang L, Paschon DE, Hinkley SJ, Ankoudinova I, Lam S, Guschin D, Kopan L, Cherone JM, Nguyen HOB, Qiao G, Ataei Y, Mendel MC, Amora R, Surosky R, Laganiere J, Vu BJ, Narayanan A, Sedaghat Y, Tillack K, Thiede C, Gärtner A, Kwak S, Bard J, Mrzljak L, Park L, Heikkinen T, Lehtimäki KK, Svedberg MM, Häggkvist J, Tari L, Tóth M, Varrone A, Halldin C, Kudwa AE, Ramboz S, Day M, Kondapalli J, Surmeier DJ, Urnov FD, Gregory PD, Rebar EJ, Muñoz-Sanjuán I, Zhang HS, 2019. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med 25, 1131–1142. 10.1038/s41591-019-0478-3. [DOI] [PubMed] [Google Scholar]
- Zeitlin S, Liu J-P, Chapman DL, Papaioannou VE, Efstratiadis A, 1995. Increased apoptosis and early embryonic lethality in mice nullizygou s for the Huntington ’s disease gene homologu e. Nat. Genet 11, 155–163. [DOI] [PubMed] [Google Scholar]
- Zöller I, Meixner M, Hartmann D, Büssow H, Meyer R, Gieselmann V, Eckhardt M, 2008. Absence of 2-hydroxylated sphingolipids is compatible with normal neural development but causes late-onset axon and myelin sheath degeneration. J. Neurosci 28, 9741–9754. 10.1523/JNEUROSCI.0458-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets generated are included in this article and can be found in Supplemental Files 2–6. RNA-sequencing data analyzed is available at Gene Expression Omnibus (GEO) accession number GSE156236.