

Abstract
Background and Aims:
Genome-wide association studies (GWAS) of opioid use disorder (OUD) and cannabis use disorder (CUD) have lagged behind those of alcohol use disorder (AUD) and smoking, where many more loci have been identified. We sought to identify novel loci for substance use traits (SUTs) in both African- (AFR) and European- (EUR) ancestry individuals to enhance our understanding of the traits’ genetic architecture.
Design:
We used multi-trait analysis of GWAS (MTAG) to analyze four SUTs in EUR subjects (OUD, CUD, AUD and smoking initiation [SMKinitiation]), and three SUTs in AFR subjects (OUD, AUD and smoking trajectory [SMKtrajectory]). We conducted gene-set and protein–protein interaction analyses and calculated polygenic risk scores (PRS) in two independent samples.
Setting:
This study was conducted in the United States.
Participants:
A total of 5692 EUR and 4918 AFR individuals in the Yale-Penn sample and 29 054 EUR and 10 265 AFR individuals in the Penn Medicine BioBank sample.
Findings:
MTAG identified genome-wide significant (GWS) single nucleotide polymorphisms (SNPs) for all four traits in EUR: 41 SNPs in 36 loci for OUD; 74 SNPs in 60 loci for CUD; 63 SNPs in 52 loci for AUD; and 183 SNPs in 144 loci for SMKinitiation. MTAG also identified GWS SNPs in AFR: 2 SNPs in 2 loci for OUD; 3 SNPs in 3 loci for AUD; and 1 SNP in 1 locus for SMKtrajectory. In the Yale-Penn sample, the MTAG-derived PRS consistently yielded more significant associations with both the corresponding substance use disorder diagnosis and multiple related phenotypes than the GWAS-derived PRS.
Conclusions:
Multi-trait analysis of genome-wide association studies boosted the number of loci found for substance use traits, identifying genes not previously linked to any substance, and increased the power of polygenic risk scores. Multi-trait analysis of genome-wide association studies can be used to identify novel associations for substance use, especially those for which the samples are smaller than those for historically legal substances.
Keywords: alcohol use disorder, cannabis use disorder, GWAS, opioid use disorder, polygenic risk scores, smoking initiation
INTRODUCTION
The etiology of substance use traits (SUTs) involves both environmental and genetic factors, with estimates of heritability ~50%. Genome-wide association studies (GWAS) of SUTs have successfully identified hundreds of genome-wide significant (GWS) risk variants [1–7], providing insight into the underlying biology that influences substance use and misuse. However, because common genetic variants have small effects on trait susceptibility, large samples are needed to identify GWS associations, with recent GWAS of SUTs requiring case sample sizes of over 20 000 to identify variants. Difficulty in recruiting large study samples has, therefore, limited gene discovery efforts [8], with variant discovery for some traits (e.g. opioid use disorder [OUD] and cannabis use disorder [CUD]) lagging behind that of more common SUTs such as alcohol and smoking-related phenotypes. Furthermore, underrepresentation of non-European-ancestry individuals has limited genetic discovery and exacerbated health inequalities [9]. In addition to identifying relevant biology, GWAS results are also necessary to calculate polygenic risk scores (PRS), which may have clinical use [10]. Increasing the power of GWAS results for SUTs in multiple ancestries will improve our understanding of their underlying biology and inform diagnostic and treatment efforts.
Multi-trait analysis of GWAS (MTAG) was developed to boost the statistical power of GWAS by incorporating information from effect estimates across traits [11]. It enables the joint analysis of multiple, genetically correlated traits, generating trait-specific estimates of the effects of each single nucleotide polymorphism (SNP). MTAG is also valuable because it can be applied to summary statistics of GWAS rather than requiring individual genotypes, addressing the sample overlap often present across GWAS discovery samples for different traits using linkage disequilibrium (LD) score regression [11].
Because SUTs have shown high degrees of genetic correlation in GWAS and twin and family studies [12], they are ideally suited to MTAG. MTAG has been used to boost genetic findings for SUTs, both for traits involving the same substance and for cross-substance traits. In a meta-analysis of problematic alcohol use (PAU) [13], MTAG analysis of PAU and a measure of weekly alcohol consumption [4] increased the number of independent loci for PAU from 29 to 76. MTAG analysis of OUD with AUD and CUD [14] increased the number of GWS loci for OUD to 18 from three in the initial GWAS meta-analysis. Because MTAG for multiple genetically correlated SUTs yields greater statistical and interpretive power than individual-trait GWAS, the availability of large GWAS of alcohol- and smoking-related traits could enhance findings from GWAS of traits with smaller accumulated samples (e.g. CUD and OUD).
Here, we conduct an MTAG analysis of the largest available GWAS for three SUTs in African-ancestry (AFR) individuals: OUD, alcohol use disorder (AUD) and smoking trajectory (SMKtrajectory); and four SUTs in European-ancestry (EUR) individuals: OUD, CUD, AUD and smoking initiation (SMKinitiation). We integrated information from the GWAS summary statistics to identify novel loci for each SUT. We also conducted gene prioritization, gene-set and protein–protein interaction (PPI) analyses to characterize the underlying biology of the novel genes in the context of SUTs. Finally, we generated polygenic risk scores (PRS) to examine the power increment for each set of MTAG-GWAS summary statistics in two independent samples.
METHODS
GWAS summary statistics
We identified available GWAS summary statistics for opioid (use disorder) [1], cannabis (use disorder and lifetime use) [3, 5], alcohol (use disorder and drinks per week) [2, 4] and tobacco (smoking initiation, Fagerström Test for Nicotine Dependence [FTND], smoking trajectory) [4, 6, 7] in both AFR and EUR individuals. Other ancestries were not included because of lack of data.
Genetic correlation
Genetic correlation (rg) measures the genetic similarity between two different traits [15]. We calculated pairwise genetic correlations for the SUTs within each ancestry using linkage disequilibrium score regression (LDSC) [16] and HapMap 3 SNPs. Pre-computed EUR linkage disequilibrium (LD) scores and weights were downloaded from the LDSC GitHub website (https://github.com/bulik/ldsc). We computed AFR LD scores and weights in Million Veteran Program (MVP) genotype data using cov-LDSC [17].
Phenotype selection
To maximize the power of the joint analysis, for each substance we selected the trait with the strongest genetic correlations with other SUTs (Table S1). We selected three SUTs for MTAG analysis in AFR samples: OUD [1], AUD [2] and SMKtrajectory [7] and four SUTs for MTAG analysis in EUR samples: OUD [1], CUD [3], AUD [2] and SMKinitiation [4]. Because our downstream PRS analysis used the Yale-Penn dataset and the CUD GWAS included genotype data from Yale-Penn subjects, we avoided sample overlap by generating summary statistics for CUD using a ‘leave-one-out’ meta-analysis that excluded Yale-Penn subjects. All other GWAS were independent of the Yale-Penn dataset.
Multi-trait analysis of GWAS summary statistics (MTAG)
We used trait-specific effective sample sizes and transformed Z-scores. SNPs present in all of the SUT summary statistics were included in the MTAG calculation (AFR n = 2 648 506; EUR n = 2 888 727), which used default MTAG parameters (i.e. each SNP’s sample size was larger than two thirds of the 90th percentile of all SNPs’ sample sizes). We calculated maximum False Discovery Rate (maxFDR) [11] as the upper bound of False Discovery Rate (FDR) for each MTAG result. We used PLINK1.9 to perform clumping procedures on the four MTAG results across a range of 3000 kb and r2 > 0.1. GWS variants located within 1 Mb were merged into a single locus. Loci were annotated with the nearest protein-coding gene (within 1 Mb) using SNPsnap [18].
Identification of novel variants for each SUT
Loci that were not previously GWS for any SUT were labeled as ‘novel.’ To ascertain this, we systematically evaluated whether variants identified in the MTAG analysis were previously associated with either the primary SUT or other SUTs. First, for each lead variant, we determined whether any nearby variant (within 1 Mb) was GWS in the initial GWAS or in the other three contributing GWAS. We, then, determined whether the locus had prior SUT associations using the GWAS catalog [19] implemented in FUMA [20], annotating all lead variants with prior associations with any SUT (not limited to the GWAS included in this study). For completeness, we also labeled the variant with the closest protein-coding gene (within 1 Mb) and searched the GWAS catalog [19] for prior associations of that gene with SUTs.
Gene-set analysis
To determine whether the identified genes are involved in important biological processes, GWS SNPs from the EUR sample were mapped to genes by ANNOVAR [21] implemented in FUMA [20]. Next, we curated gene-set enrichment and Gene Ontology (GO) terms using the GO annotation database [22, 23], with gene-set enrichment P-values adjusted using a Bonferroni correction for each test (adjustment P-value <0.05).
PPI
To further investigate the relationship between the prioritized protein-coding genes from each MTAG result in EUR, we used STRING database v11.5 s [24] to conduct PPI analyses. For each SUT MTAG result, we used annotated GWS genes as input to query the PPIs in the database. PPI enrichment P-value and pairwise interaction scores were reported by the STRING database. We used a cut-off interaction score >0.4 to identify PPIs with high confidence.
Yale-Penn dataset
The Yale-Penn sample was recruited for genetic studies of substance use disorders (SUDs) [25]. It was deeply phenotyped using the Semi-Structured Assessment for Drug Dependence and Alcoholism (SSADDA), a comprehensive psychiatric interview schedule [26, 27]. The Yale-Penn phenome-wide association studies (PheWAS) dataset includes 4917 unrelated AFR and 5692 unrelated EUR genotyped individuals and over 650 summarized phenotypes categorized into 20 substance, medical, demographic and psychiatric sections [25].
Penn Medicine BioBank Dataset
The Penn Medicine BioBank (PMBB) is an electronic health recordlinked biobank at the University of Pennsylvania [28]. Patients at Penn Medicine are recruited under a single umbrella institutional review board (IRB) protocol that provides consent for blood collection, genotyping and access to medical records for research purposes. The PMBB dataset includes genotype data for 10 383 AFR and 29 355 EUR individuals. International Classification of Diseases (ICD)-9 and -10 codes were extracted from the electronic health record. Case/control status for each substance use disorder (OUD, CUD, AUD and TUD) was determined based on the presence or absence of relevant ICD-9 and -10 codes (Table S2).
PRS and phenotype association test
We used PRS-continuous shrinkage (PRS-CS) [29] to generate PRS in both the Yale-Penn [25] and PMBB datasets, pre-computed LD reference for HapMap3 SNPs in AFR and EUR samples to account for local LD, and an optimal global shrinkage parameter learned from the data. We generated a GWAS-based PRS and a MTAG-based PRS for each of the SUTs in both AFR and EUR individuals. To ensure comparability between GWAS-based and MTAG-based PRS, only the SNPs used in the MTAG calculation were included in PRS calculation. Effective sample sizes were used to generate GWAS- and MTAG-based PRS.
To compare the power of GWAS-based PRS with MTAG-based PRS, we tested the associations of each PRS in the Yale-Penn dataset with phenotypes in the corresponding substance section (e.g. AUD PRS were tested with alcohol-related phenotypes). We used the PheWAS package in R [30] with linear or logistic regression models as appropriate. We used age, sex and 10 genetic principal components (PCs) as covariates [31]. Each substance section includes Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV and DSM-5 diagnoses for the corresponding SUD. For diagnoses, we calculated the incremental pseudo R2 value after adding the polygenic score to the logistic regression models. To account for multiple testing, we used a significance P-value <0.05 divided by the number of phenotypes in each substance.
In the PMBB dataset, we tested the associations of each PRS with case/control status for the corresponding substance use disorder using logistic regression in R, including sex, age and the top 10 PCs as covariates.
The analysis described here was not preregistered, and as such the results should be considered exploratory.
RESULTS
Genetic correlation between SUTs
Pairwise rg calculated using LDSC were significant and moderate or high between OUD, CUD, cannabis lifetime use, AUD, drinks per week, SMKinitiation and SMKtrajectory in EUR; and OUD, AUD and SMKtrajectory in AFR (Figure 1, Table S1). In EUR, CUD had stronger genetic correlations with other SUTs than cannabis lifetime use, and AUD had stronger genetic correlations than drinks per week. FTND was not significantly genetically correlated with CUD, cannabis lifetime use or drinks per week in EUR. There was not enough power for CUD to compute genetic correlations with other SUTs in AFR. We therefore selected OUD [1] (effective n = 74 635), CUD [3] (effective n = 48 900), AUD [2] (effective n = 171 601) and SMKinitiation [4] (effective n = 632 802) for MTAG in EUR; and OUD [1] (effective n = 32 240), AUD [2] (effective n = 67 686) and smoking trajectory (SMKtrajectory; effective n = 40 736) [7] for MTAG in AFR.
FIGURE 1.
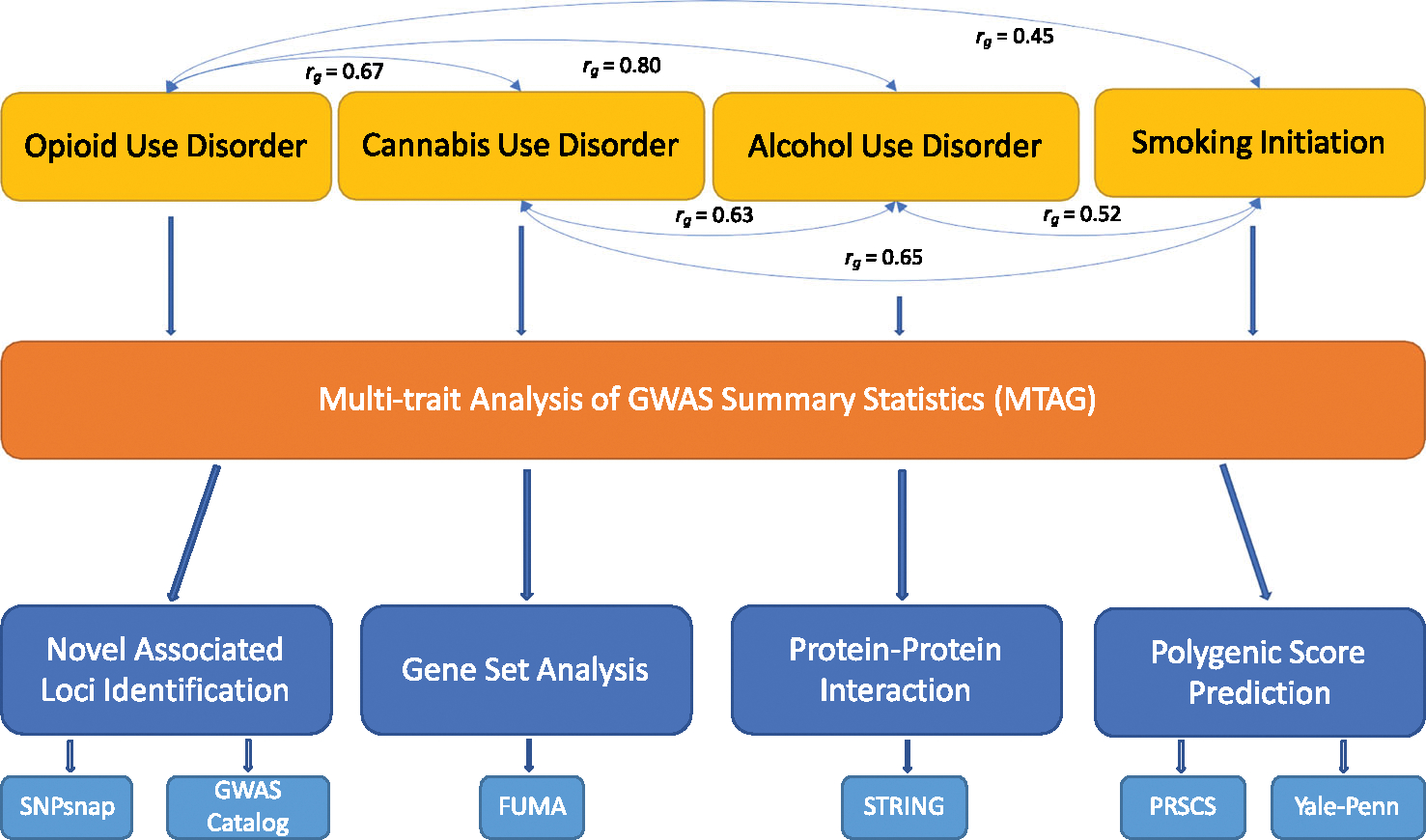
Overview of the analysis. The four substance use traits (SUTs) that were used in multi-trait analysis of genome-wide association studies (MTAG) analysis have pairwise genetic correlations between 0.45 and 0.80. For each SUT MTAG-genome-wide association studies (GWAS) result, we identified novel loci, performed gene-set analysis, protein–protein interaction analysis and examined the increased predictive power of the corresponding polygenic risk score.
MTAG SUTs and locus discovery
The pairwise genetic correlations for the SUTs ranged from 0.45 to 0.80 in EUR and 0.61 to 0.66 in AFR, supporting the use of MTAG to conduct a joint analysis of these traits. MTAG increased the effective sample size and the number of independent loci identified for each SUT, with low maxFDR values supporting the robustness of the results (Table 1). In EUR, the number of independent loci increased from 4 to 36 for OUD, 2 to 60 for CUD, 12 to 52 for AUD and 74 to 144 for SMKinitiation. In AFR, the number of independent loci increased from 1 to 2 for OUD and 0 to 1 for SMKtrajectory, whereas the number of independent loci for AUD remained the same. Among the four sets of MTAG results in EUR (Figure 2, Tables S3–S5, S7), 20 loci were significantly associated with all four SUTs, the most significant of which was the locus containing NCAM1, supported by two intronic variants in complete LD (rs1940701 for OUD and AUD and rs4479020 for CUD and SMK). Other loci associated with all four SUTs in EUR included intronic variants in DPP4 and CADM2 and an intergenic variant near ZNF184. In AFR no loci were associated with all three SUTs.
TABLE 1.
Summary of MTAG results.
| EUR |
AFR |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OUD |
CUD |
AUD |
SMK |
OUD |
AUD |
SMK |
||||||||
| GWAS | MTAG | GWAS | MTAG | GWAS | MTAG | GWAS | MTAG | GWAS | MTAG | GWAS | MTAG | GWAS | MTAG | |
|
| ||||||||||||||
| SNPs used for MTAG | 2 888 846 | 2 888 846 | 2 888 846 | 2 888 846 | 2 648 505 | 2 648 505 | 2 648 505 | |||||||
| SigSNPs | 18 | 1465 | 7 | 2547 | 363 | 1854 | 3049 | 5380 | 3 | 7 | 59 | 45 | 0 | 3 |
| SigSNPs (after clumping) | 4 | 41 | 2 | 74 | 20 | 63 | 105 | 183 | 1 | 2 | 4 | 3 | 0 | 1 |
| Independent locus | 4 | 36 | 2 | 60 | 12 | 52 | 74 | 144 | 1 | 2 | 2 | 2 | 0 | 1 |
| χ2 | 1.159 | 1.376 | 1.099 | 1.455 | 1.281 | 1.463 | 1.771 | 1.865 | 1.021 | 1.045 | 1.053 | 1.055 | 1.044 | 1.049 |
| Effective sample size | 74 635 | 176 876 | 48 900 | 223 956 | 171 601 | 282 208 | 632 802 | 709 603 | 32 240 | 68 047 | 67 686 | 71 092 | 40 736 | 45 743 |
Abbreviations: AFR, African ancestry; AUD, alcohol use disorder; CUD, cannabis use disorder; EUR, European ancestry; GWAS, genome-wide association studies; OUD, opioid use disorder; MTAG, multi-trait analysis of GWAS; SMK, smoking; SNPs, single nucleotide polymorphisms, SigSNPs, SNPs that reach a genome wide significant p-value threshold < 5 × 10−8.
FIGURE 2.
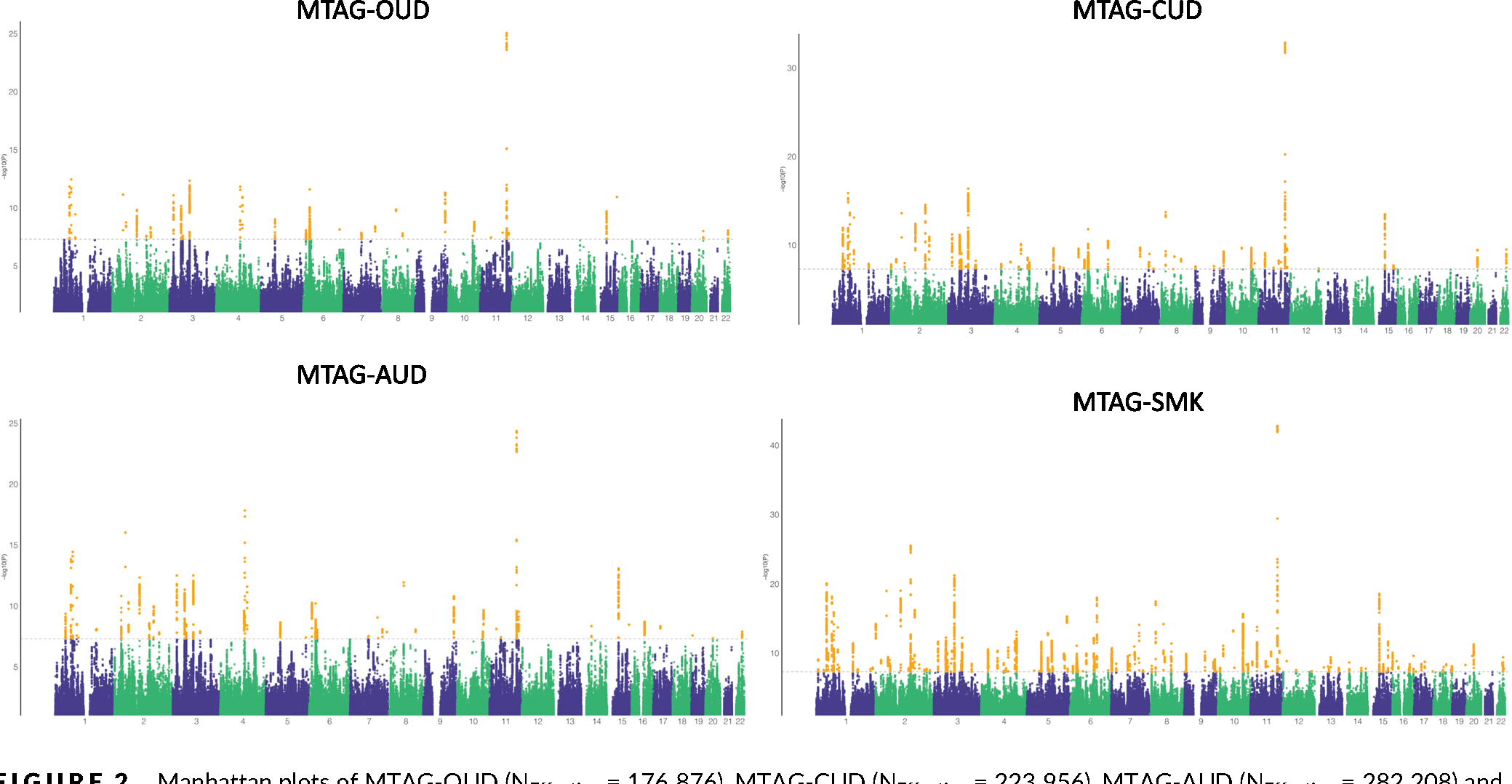
Manhattan plots of MTAG-OUD (NEffective = 176 876), MTAG-CUD (NEffective = 223 956), MTAG-AUD (NEffective = 282 208) and MTAG-SMK (NEffective = 709 603). Dashed lines indicate genome-wide significance (P < 5 × 10−8) and yellow dots indicate genome-wide significance single nucleotide polymorphisms. AUD, alcohol use disorder; CUD, cannabis use disorder; OUD, opioid use disorder; MTAG, multi-trait analysis of genome-wide association studies; SMK, smoking.
Opioid use disorder (MTAG-OUD)
In EUR, of the 41 independent GWS SNPs in 36 loci (Table S3), three were GWS in the OUD GWAS, 17 were GWS in at least one of the other of the three SUT GWAS used for MTAG, and 30 were GWS in other GWAS of SUTs, not limited to the GWAS included in this study. Variants including exonic SNPs in OPRM1 and FURIN were GWS in previous OUD GWAS [1,14]. Five GWS loci are novel (i.e. no prior associations with OUD or any SUT). These include a variant in the 3′ untranslated regions of POR, intronic variants in CNOT4 and MTMR2 and intergenic variants near TMEM170B and SNAI1. In AFR, one of the two independent GWS SNPs in two loci (Table S3) is novel—an intronic variant in GIHCG.
Cannabis use disorder (MTAG-CUD)
MTAG yielded 74 independent GWS SNPs in 60 genomic risk loci in EUR (Table S4). Of the 60 loci, three were GWS in the CUD GWAS, 43 were GWS in at least one of the other three SUT GWAS used for MTAG, and 51 were GWS in other GWAS of SUTs. We replicated previously associated loci for CUD [3], including an intergenic variant near CLU in the same locus identified in the input CUD GWAS, an intronic variant in GBF1, and an exonic variant in FURIN. We identified four novel GWS loci, including three intronic variants—one each in CNOT4, TMEM245 and MTMR2—and an intergenic variant near TMEM170B.
Alcohol use disorder (MTAG-AUD)
We identified 63 independent GWS SNPs in 52 genomic risk loci for AUD in EUR (Table S5). Of these, 9 loci were GWS in the AUD GWAS, 23 were GWS in at least one of the other three SUT GWAS used for MTAG, and 40 were GWS in other GWAS of SUTs. These included multiple previously reported AUD-associated variants—exonic variants in GCKR and SLC39A8, and intronic variants in ANKK1 and FTO [2]. We also identified 10 novel GWS loci, including exonic variants in POR and SYNGAP1; intronic variants in DNM3, CSMD3, CNOT4, LRFN5, ZNF804A and TCF20; and two intergenic variants near TMEM170B and SORCS3. Of note, SNP rs1229984, located in the alcohol dehydrogenase gene ADH1B, did not pass the default MTAG quality control parameter in EUR as it was not present in at least two thirds of the sample. Because of the well-known strong association of rs1229984 with alcohol phenotypes [2, 13], we conducted a separate analysis with a less stringent filter to include that SNP (Table S6). In AFR, we identified three independent SNPs in two loci, all which had prior associations with SUTs (Table S5).
Smoking initiation and trajectory (MTAG-SMK)
We identified 183 independent GWS SNPs in 144 genomic risk loci (Table S7). Of these, 86 were GWS in the SMKinitiation GWAS, seven were GWS in at least one of the other three SUT GWAS used for MTAG, and 130 were GWS in other GWAS of SUTs. Eight GWS loci were novel, including a variant within TNRC6B and seven intergenic variants: one each near WDR12, PCDH7, ITGA1, TMEM170B, SP4, CTDP1 and SORCS3. In AFR, one novel locus for SMKtrajectory was identified in GIHCG (Table S7).
Gene-set analysis
Enriched gene sets were identified for all studied traits in EUR: 28 for MTAG-OUD, 73 for MTAG-CUD, 51 for MTAG-AUD and 70 for MTAG-SMK (Tables S8–S11). The gene-set ‘regulation of cell differentiation’, which contains the novel gene POR and the opioid-specific gene OPRM1, showed significant enrichment for OUD (PBon = 0.001). ‘Protein dimerization activity’, one of the significantly enriched gene-sets for CUD (PBon = 3.7 × 10−9), harbors the novel genes MTMR2 and FOXP2. For AUD, the enriched gene set ‘regulation of synapse structure or activity’ (PBon = 0.012) contains the novel genes DNM3, LRFN5 and SYNGAP1 along with DRD2. For SMK, 70 gene sets showed significant enrichment, with the novel gene TNRC6B mapping to 12 of them. Of note, the gene set ‘neuron differentiation’, which includes NCAM1, is significantly enriched (PBon = 0.031).
Protein–protein interaction
We observed significant PPI enrichment (P < 0.05) for protein-coding genes that were GWS for MTAG-AUD and MTAG-SMKinitiation in EUR, whereas PPI enrichment was nonsignificant for GWS genes in MTAG-OUD (P = 0.175) and MTAG-CUD (P = 0.175) (Tables S12–S15). For GWS genes in MTAG-AUD, we identified high PPI for NF1 and SYNGAP1 (interaction score = 0.697), NF1 and CSMD3 (interaction score = 0.42) and NCAM1 and SEMA6D (interaction score = 0.467). For GWS genes in MTAG-SMK, TNRC6B and FOXO3, two smoking-associated genes, showed high PPI (interaction score = 0.9).
PRS associations with primary phenotypes
For EUR, all four MTAG-based PRS (PRSMTAG) showed stronger associations with the primary diagnosis than the individual-trait GWAS-based PRS (PRSGWAS) in the Yale-Penn dataset (Figure 3, Tables S16–S19) and the PMBB dataset (Table S20). This difference was most evident for CUD, where in the Yale-Penn dataset the Bonferroni-corrected association with DSM-IV cannabis dependence was nonsignificant for PRSGWAS-CUD (OR = 1.16, P = 1.94 × 10−3), but it was significant with PRSMTAG-CUD (OR = 1.30, P = 2.18 × 10−9). This was replicated in the PMBB dataset, where the association with CUD was nonsignificant for PRSGWAS-CUD (OR = 1.14, P = 0.08), but was significant with PRSMTAG-CUD (OR = 1.29, P = 7.86 × 10−4).
FIGURE 3.

Comparison of GWAS-based PRS and MTAG-based PRS. Red and blue dots in each plot represent the GWAS-based PRS and MTAG-based PRS, respectively. Vertical dashed lines indicate significance threshold after Bonferroni correction. GWAS, genome-wide association studies; MTAG, multi-trait analysis of GWAS; PRS, polygenic risk scores.
The Yale-Penn dataset contains additional related phenotypes for each substance. For each of the SUTs, there were more Bonferroni-corrected significant associations with PRSMTAG than PRSGWAS (Tables S16–S19). Notably, the number of significantly associated phenotypes for PRSGWAS-CUD was 6, whereas for PRSMTAG-CUD it was 25. Phenotypes that were significantly associated when using PRSMTAG-CUD included age of first use of marijuana, the DSM-IV cannabis dependence diagnosis and the DSM-5 CUD criterion count.
In addition, the incremental R2 values both for diagnoses and related phenotypes were higher for PRSMTAG than PRSGWAS (Table 2). The greatest improvement was between PRSMTAG-CUD and PRSGWAS-CUD, where the incremental R2 for ‘ever used cannabis’ was 1.62% and 0.38%, respectively. Moderate improvement was also observed for PRSMTAG-SMK compared with the well-powered PRSGWAS-SMK.
TABLE 2.
PRS increment R2.
| GWAS-PRS | MTAG-PRS | ||
|---|---|---|---|
|
| |||
| Opioid | Ever used opioid | 0.97% | 2.31% |
| DSM-IV diagnosis | 1.03% | 2.26% | |
| DSM-5 diagnosis | 1.01% | 2.16% | |
| Cannabis | Ever used cannabis | 0.38% | 1.62% |
| DSM-IV diagnosis | 0.19% | 0.91% | |
| DSM-5 diagnosis | 0.23% | 1.13% | |
| Alcohol | Ever used alcohol | 0.26% | 1.11% |
| DSM-IV diagnosis | 0.91% | 1.68% | |
| DSM-5 diagnosis | 0.84% | 1.72% | |
| Smoking | Ever used smoking | 1.81% | 2.04% |
| DSM-IV diagnosis | 3.43% | 4.16% | |
Abbreviations: DSM, Diagnostic and Statistical Manual of Mental Disorders; GWAS, genome-wide association studies; MTAG, multi-trait analysis of GWAS; PRS, polygenic risk scores.
Findings were less consistent in the AFR datasets. For OUD, neither the PRSGWAS-OUD nor the PRSMTAG-OUD showed significant associations in the Yale-Penn dataset or the PMBB dataset. For SMKtrajectory, the PRSGWAS-SMK performed slightly better than the PRSMTAG-SMK in the Yale-Penn dataset, whereas the opposite was true in the PMBB dataset. For AUD, PRSMTAG-AUD showed stronger associations than PRSGWAS-AUD in both datasets, and there were more Bonferroni-corrected significant associations with PRSMTAG-AUD (n = 13) than PRSGWAS-AUD (n = 6).
DISCUSSION
We performed a joint analysis of SUTs using MTAG, which yielded an effective sample size up to fourfold that of the individual GWAS. This yielded novel associated variants in loci not previously linked to any SUT, including a novel locus in AFR associated with both OUD and SMKtrajectory. Interestingly, one of our novel associations, CNOT4, was recently identified in a GWAS of maximum alcohol use [32] and a GWAS of smoking initiation [33], which supports our discovery here. A prior MTAG analysis of PAU [13], which leveraged information from a GWAS of drinks per week, identified 119 GWS variants. Of the 45 SNPs in EUR in common between that analysis and ours, 14 were associated with MTAG-AUD in our sample. In our EUR MTAG-OUD analysis, we also replicated 6 of 9 variants in common with a recent OUD MTAG analysis of OUD, CUD and AUD [14]. Although OPRM1 was not significantly associated in the prior MTAG, our EUR MTAG-OUD analyses identified the OPRM1 SNP rs1799971 as a lead variant, potentially because of our inclusion of the smoking initiation GWAS or the larger input OUD GWAS sample in our analysis.
Among the most significant risk loci for SUTs are those that encode proteins with clear connections to the substance involved. This includes the μ-opioid receptor for OUD [34], various nicotinic cholinergic receptor subunits for smoking initiation [4] and alcohol metabolizing enzymes for alcohol consumption and AUD [35]. However, beyond the substance-specific proteins that directly interact with the drug a wide range of biological mechanisms are common to all addictive behaviors—these include reward pathways, learning and memory, withdrawal and other functions [36]. We, therefore, expected that the greater statistical power of MTAG would reveal novel genes with associations to multiple substances based on common mechanisms of risk (i.e. an addiction factor) [37, 38]. In fact, 5 of 19 novel loci identified in our EUR analysis were significantly associated with two or more substances. The use of multiple substances is common and has been associated with poorer treatment outcomes in individuals with SUDs [39]. These shared genes may represent targets for therapies aimed at treating co-occurring SUDs.
Patients with SUDs also have higher rates of comorbid psychiatric disorders than the general population [40] PheWAS using electronic health record (EHR) data have shown associations between PRS for SUTs and non-substance use psychiatric diagnostic codes [13], suggesting genetic overlap between them. Consistent with this hypothesis, 9 of 19 novel genes identified in our EUR MTAG analysis were significant in GWAS of depression or schizophrenia [41–45]. Furthermore, several of the novel genes (SYNGAP1, ZNF804A, CSMD3, DNM3, LRFN5, TCF20 and TNRC6B) have been previously associated with neurodevelopmental disorders [46–52]. Mutations in SYNGAP1 reduce protein activity, leading to impaired synaptic plasticity, a key factor in brain development [46]. Autism patients have lower expression of ZFN804A in the anterior cingulate gyrus [47], and CSMD4 has been identified as a risk gene for autism spectrum disorder [48]. Our results suggest a connection between SUTs and neurodevelopmental disorders.
We functionally annotated the EUR GWS loci in all four SUTs to explore plausible underlying biological processes. We found that associations for AUD were enriched in genes (including CNOT4) involved in regulating synaptic structure and activity, an enrichment not previously observed in the AUD GWAS [2]. In addition, we find evidence for neuronal differentiation in smoking initiation, in line with findings in the original GWAS [4]. In the PPI analysis, we observed significantly enriched protein interaction networks for MTAG-AUD and MTAG-SMK. In addition, we observed multiple interactions between novel genes identified by MTAG and previously identified SUT-associated genes, which provide biological support for the MTAG results.
In PRS analyses, the PRSMTAG outperformed the PRSGWAS for all SUTs in EUR, but only for AUD in AFR. This was evident for the CUD PRS, where in EUR the PRSMTAG was significantly associated with the diagnosis, whereas the PRSGWAS was not. As PRS for many traits are being considered as potential biomarkers for disorders [53], the use of MTAG may yield more powerful PRS without having to recruit larger samples for GWAS.
Our study has some limitations. Careful evaluation is required to assess the broader impact of MTAG-based PRS on phenotypic associations beyond the primary phenotype. A loss of specificity may result from the inclusion of multiple genetically correlated traits, potentially confounding PheWAS of MTAG-PRS. Because of smaller sample sizes for GWAS of tobacco use disorder, we selected GWAS for smoking traits that reflect tobacco use more generally, in contrast to the selection of substance use disorder for our other traits. Finally, although we included data for AFR individuals, the smaller sample size meant that our results were less conclusive, and we were unable to include a GWAS of cannabis use in this population. Future GWAS should prioritize recruitment of non-European datasets.
In summary, in an MTAG analysis of SUTs we identified 19 novel loci in EUR and 1 in AFR, and, in two independent datasets, found that the associations of PRSMTAG with relevant traits were more significant than with PRSGWAS. As the size of GWAS samples continues to increase, MTAG analyses could provide a complementary method to leverage more powerful GWAS to boost the findings of risk variants for genetically correlated traits, particularly where case ascertainment is more challenging, thereby enhancing our understanding of the biology underlying these phenotypes and its clinical applicability.
Supplementary Material
ACKNOWLEDGEMENTS
Supported by the Veterans Integrated Service Network 4 Mental Illness Research, Education and Clinical Center, the Million Veteran Program (I01 BX004820 and I01 CX001734), National Institute on Drug Abuse P30 DA046345 (H.R.K.) and K01DA051759 (E.C.J.), and National Institute on Alcohol Abuse and Alcoholism R01 AA026364 (J.G.), K01 AA028292 (R.L.K.) and T32 AA028259 (J.D. D.). We acknowledge the Penn Medicine BioBank (PMBB) for providing data and thank the patient-participants of Penn Medicine who consented to participate in this research program. We also thank the Penn Medicine BioBank team and Regeneron Genetics Center for providing genetic variant data for analysis. The PMBB is approved under IRB protocol 813913 and supported by Perelman School of Medicine at University of Pennsylvania, a gift from the Smilow family and the National Center for Advancing Translational Sciences of the National Institutes of Health under CTSA award number UL1TR001878.
Footnotes
DECLARATION OF INTERESTS
H.R.K. is a member of advisory boards for Dicerna Pharmaceuticals, Sophrosyne Pharmaceuticals and Enthion Pharmaceuticals; a consultant to Sobrera Pharmaceuticals; the recipient of research funding and medication supplies for an investigator-initiated study from Alkermes; and a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which was supported in the last 3 years by Alkermes, Dicerna, Ethypharm, Lundbeck, Mitsubishi and Otsuka. J.G. and H.R.K. hold United States Patent 10 900 082: Genotype-guided Dosing of Opioid Receptor Agonists, 26 January 2021. The other authors have no disclosures to make.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The genome-wide summary statistics used for this analysis are available through dbGaP (accession number phs001672), PGC (https://pgc.unc.edu) and GSCAN (https://genome.psych.umn.edu/index.php/GSCAN). MTAG results are available from the corresponding authors upon request.
REFERENCES
- 1.Kember RL, Vickers-Smith R, Xu H, Toikumo S, Niarchou M, Zhou H, et al. Cross-ancestry meta-analysis of opioid use disorder uncovers novel loci with predominant effects in brain regions associated with addiction. Nat Neurosci. 2022;25(10):1279–1287. 10.1038/s41593-022-01160-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kember RL, Vickers-Smith R, Zhou H, Xu H, Dao C, Justice AC, et al. Genetic underpinnings of the transition from alcohol consumption to alcohol use disorder: shared and unique genetic architectures in a cross-ancestry sample [Internet] 2021; 2021.09.08.21263302[cited 2022 Jul 19]. Available from: 10.1101/2021.09.08.21263302v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson EC, Demontis D, Thorgeirsson TE, Walters RK, Polimanti R, Hatoum AS, et al. A large-scale genome-wide association study meta-analysis of cannabis use disorder. Lancet Psychiatry. 2020; 7(12):1032–1045. 10.1016/S2215-0366(20)30339-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.23andMe Research Team, HUNT All-In Psychiatry, Liu M, Jiang Y, Wedow R, Li Y, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51(2):237–244. 10.1038/s41588-018-0307-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasman JA, Verweij KJ, Gerring Z, Stringer S, Sanchez-Roige S, Treur JL, et al. GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal effect of schizophrenia liability. Nat Neurosci. 2018;21(9):1161–1170. 10.1038/s41593-018-0206-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quach BC, Bray MJ, Gaddis NC, Liu M, Palviainen T, Minica CC, et al. Expanding the genetic architecture of nicotine dependence and its shared genetics with multiple traits. Nat Commun. 2020;11(1):5562. 10.1038/s41467-020-19265-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu K, Li B, McGinnis KA, Vickers-Smith R, Dao C, Sun N, et al. Genome-wide association study of smoking trajectory and meta-analysis of smoking status in 842,000 individuals. Nat Commun. 2020;11(1):5302. 10.1038/s41467-020-18489-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palotie A, Widén E, Ripatti S. From genetic discovery to future personalized health research. N Biotechnol. 2013;30(3):291–295. 10.1016/j.nbt.2012.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sirugo G, Williams SM, Tishkoff SA. The missing diversity in human genetic studies. Cell. 2019;177(1):26–31. 10.1016/j.cell.2019.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wray NR, Lin T, Austin J, McGrath JJ, Hickie IB, Murray GK, et al. From basic science to clinical application of polygenic risk scores: a primer. JAMA Psychiatry. 2021;78(1):101–109. 10.1001/jamapsychiatry.2020.3049 [DOI] [PubMed] [Google Scholar]
- 11.Turley P, Walters RK, Maghzian O, Okbay A, Lee JJ, Fontana MA, et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat Genet. 2018;50(2):229–237. 10.1038/s41588-017-0009-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deak JD, Johnson EC. Genetics of substance use disorders: a review. Psychol Med. 2021;51(13):2189–2200. 10.1017/S0033291721000969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou H, Sealock JM, Sanchez-Roige S, Clarke TK, Levey DF, Cheng Z, et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat Neurosci. 2020;23(7):809–818. 10.1038/s41593-020-0643-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deak JD, Zhou H, Galimberti M, Levey DF, Wendt FR, Sanchez-Roige S, et al. Genome-wide association study in individuals of European and African ancestry and multi-trait analysis of opioid use disorder identifies 19 independent genome-wide significant risk loci. Mol Psychiatry. 2022;27(10):3970–3979. 10.1038/s41380-022-01709-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Rheenen W, Peyrot WJ, Schork AJ, Lee SH, Wray NR. Genetic correlations of polygenic disease traits: from theory to practice. Nat Rev Genet. 2019;20(10):567–581. 10.1038/s41576-019-0137-z [DOI] [PubMed] [Google Scholar]
- 16.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–1241. 10.1038/ng.3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo Y, Li X, Wang X, Gazal S, Mercader JM, 23 and Me Research Team, et al. Estimating heritability and its enrichment in tissue-specific gene sets in admixed populations. Hum Mol Genet. 2021; 30(16):1521–1534. 10.1093/hmg/ddab130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pers TH, Timshel P, Hirschhorn JN. SNPsnap: a web-based tool for identification and annotation of matched SNPs. Bioinforma Oxf Engl. 2015;31(3):418–420. 10.1093/bioinformatics/btu655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005–D1012. 10.1093/nar/gky1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. 10.1038/s41467-017-01261-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gene Ontology Consortium. The gene ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49(D1):D325–D334. 10.1093/nar/gkaa1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–D612. 10.1093/nar/gkaa1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kember RL, Hartwell EE, Xu H, Rotenberg J, Almasy L, Zhou H, et al. Phenome-wide association analysis of substance use disorders in a deeply phenotyped sample. Biol Psychiatry. 2023;93(6):536–545. [cited 2023 Feb 1]. Available from: https://www.sciencedirect.com/science/article/pii/S0006322322015153. https://doi.org/10.1016/j.biopsych.2022.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierucci-Lagha A, Gelernter J, Feinn R, Cubells JF, Pearson D, Pollastri A, et al. Diagnostic reliability of the semi-structured assessment for drug dependence and alcoholism (SSADDA). Drug Alcohol Depend. 2005;80(3):303–312. 10.1016/j.drugalcdep.2005.04.005 [DOI] [PubMed] [Google Scholar]
- 27.Pierucci-Lagha A, Gelernter J, Chan G, Arias A, Cubells JF, Farrer L, et al. Reliability of DSM-IV diagnostic criteria using the semi-structured assessment for drug dependence and alcoholism (SSADDA). Drug Alcohol Depend. 2007;91(1):85–90. 10.1016/j.drugalcdep.2007.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verma A, Damrauer SM, Naseer N, Weaver JE, Kripke CM, Guare L, et al. The Penn medicine BioBank: towards a genomics-enabled learning healthcare system to accelerate precision medicine in a diverse population. J Pers Med. 2022;12(12):1974. 10.3390/jpm12121974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ge T, Chen C-Y, Ni Y, Feng YCA, Smoller JW. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun. 2019;10(1):1776. 10.1038/s41467-019-09718-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013;31(12):1102–1111. 10.1038/nbt.2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheutlin AB, Dennis J, Karlsson Linnér R, Moscati A, Restrepo N, Straub P, et al. Penetrance and pleiotropy of polygenic risk scores for schizophrenia in 106,160 patients across four health care systems. Am J Psychiatry. 2019;176(10):846–855. 10.1176/appi.ajp.2019.18091085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deak JD, Levey DF, Wendt FR, Zhou H, Galimberti M, Kranzler HR, et al. Genome-wide investigation of maximum habitual alcohol intake (MaxAlc) in 247,755 European and African Ancestry U.S. Veterans informs the relationship between habitual alcohol consumption and alcohol use disorder 2022; 2022.05.02.22274580 [cited 2022 Jun 10]. Available from: 10.1101/2022.05.02.22274580v2 [DOI] [Google Scholar]
- 33.Saunders GRB, Wang X, Chen F, Jang SK, Liu M, Wang C, et al. Genetic diversity fuels gene discovery for tobacco and alcohol use. Nature. 2022;612(7941):720–724. 10.1038/s41586-022-05477-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou H, Rentsch CT, Cheng Z, Kember RL, Nunez YZ, Sherva RM, et al. Association of OPRM1 functional coding variant with opioid use disorder: a genome-wide association study. JAMA Psychiatry. 2020;77(10):1072–1080. 10.1001/jamapsychiatry.2020.1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kranzler HR, Zhou H, Kember RL, Vickers Smith R, Justice AC, Damrauer S, et al. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat Commun. 2019;10(1):1499. 10.1038/s41467-019-09480-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3(8):760–773. 10.1016/S2215-0366(16)00104-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hatoum AS, Colbert SMC, Johnson EC, Huggett SB, Deak JD, Pathak GA, et al. Multivariate genome-wide association meta-analysis of over 1 million subjects identifies loci underlying multiple substance use disorders. Nat Mental Health. 2022;1(3):210–223. 10.1101/2022.01.06.22268753v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hatoum AS, Johnson EC, Colbert SMC, Polimanti R, Zhou H, Walters RK, et al. The addiction risk factor: a unitary genetic vulnerability characterizes substance use disorders and their associations with common correlates. Neuropsychopharmacology. 2022;47(10):1739–1745. 10.1038/s41386-021-01209-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L, Min JE, Krebs E, Evans E, Huang D, Liu L, et al. Polydrug use and its association with drug treatment outcomes among primary heroin, methamphetamine, and cocaine users. Int J Drug Policy. 2017;49:32–40. 10.1016/j.drugpo.2017.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Langås A-M, Malt UF, Opjordsmoen S. Comorbid mental disorders in substance users from a single catchment area - a clinical study. BMC Psychiatry. 2011;11(1):25. 10.1186/1471-244X-11-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baselmans BML, Jansen R, Ip HF, van Dongen J, Abdellaoui A, van de Weijer MP, et al. Multivariate genome-wide analyses of the well-being spectrum. Nat Genet. 2019;51(3):445–451. 10.1038/s41588-018-0320-8 [DOI] [PubMed] [Google Scholar]
- 42.Levey DF, Stein MB, Wendt FR, Pathak GA, Zhou H, Aslan M, et al. Bi-ancestral depression GWAS in the million veteran program and meta-analysis in >1.2 million individuals highlight new therapeutic directions. Nat Neurosci. 2021;24(7):954–963. 10.1038/s41593-021-00860-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu C, Aragam N, Li X, Villla EC, Wang L, Briones D, et al. BCL9 and C9orf5 are associated with negative symptoms in schizophrenia: meta-analysis of two genome-wide association studies. PLoS ONE. 2013;8(1):e51674. 10.1371/journal.pone.0051674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ikeda M, Takahashi A, Kamatani Y, Momozawa Y, Saito T, Kondo K, et al. Genome-wide association study detected novel susceptibility genes for schizophrenia and shared trans-populations/diseases genetic effect. Schizophr Bull. 2019;45(4):824–834. 10.1093/schbul/sby140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yao X, Glessner JT, Li J, Qi X, Hou X, Zhu C, et al. Integrative analysis of genome-wide association studies identifies novel loci associated with neuropsychiatric disorders. Transl Psychiatry. 2021;11(1):69. 10.1038/s41398-020-01195-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kilinc M, Creson T, Rojas C, Aceti M, Ellegood J, Vaissiere T, et al. Species-conserved SYNGAP1 phenotypes associated with neurodevelopmental disorders. Mol Cell Neurosci. 2018;91:140–150. 10.1016/j.mcn.2018.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anitha A, Thanseem I, Nakamura K, Vasu MM, Yamada K, Ueki T, et al. Zinc finger protein 804A (ZNF804A) and verbal deficits in individuals with autism. J Psychiatry Neurosci. 2014;39(5):294–303. 10.1503/jpn.130126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song W, Li Q, Wang T, Li Y, Fan T, Zhang J, et al. Putative complement control protein CSMD3 dysfunction impairs synaptogenesis and induces neurodevelopmental disorders. Brain Behav Immun. 2022;102:237–250. 10.1016/j.bbi.2022.02.027 [DOI] [PubMed] [Google Scholar]
- 49.Shi G-D, Zhang X-L, Cheng X, Wang X, Fan BY, Liu S, et al. Abnormal DNA methylation in thoracic spinal cord tissue following transection injury. Med Sci Monit Int Med J Exp Clin Res. 2018;24:8878–8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cappuccio G, Attanasio S, Alagia M, Mutarelli M, Borzone R, Karali M, et al. Microdeletion of pseudogene chr14.232.A affects LRFN5 expression in cells of a patient with autism spectrum disorder. Eur J Hum Genet. 2019;27(9):1475–1480. 10.1038/s41431-019-0430-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Torti E, Keren B, Palmer EE, Zhu Z, Afenjar A, Anderson IJ, et al. Variants in TCF20 in neurodevelopmental disability: description of 27 new patients and review of literature. Genet Med Off J Am Coll Med Genet. 2019;21(9):2036–2042. 10.1038/s41436-019-0454-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Granadillo JL, Stegmann A, Guo H, Xia K, Angle B, Bontempo K, et al. Pathogenic variants in TNRC6B cause a genetic disorder characterised by developmental delay/intellectual disability and a spectrum of neurobehavioural phenotypes including autism and ADHD. J Med Genet. 2020;57(10):717–724. 10.1136/jmedgenet-2019-106470 [DOI] [PubMed] [Google Scholar]
- 53.Fullerton JM, Nurnberger JI. Polygenic risk scores in psychiatry: will they be useful for clinicians? F1000Research. 2019;8:1293. 10.12688/f1000research.18491.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genome-wide summary statistics used for this analysis are available through dbGaP (accession number phs001672), PGC (https://pgc.unc.edu) and GSCAN (https://genome.psych.umn.edu/index.php/GSCAN). MTAG results are available from the corresponding authors upon request.