

Abstract
Anterior pituitary cell function requires a high level of protein synthesis and secretion which depend heavily on mitochondrial adenosine triphosphate production and functional endoplasmic reticula. Obesity adds stress to tissues, requiring them to adapt to inflammation and oxidative stress, and adding to their allostatic load. We hypothesized that pituitary function is vulnerable to the stress of obesity. Here, we utilized a 10- to 15-week high-fat diet (HFD, 60%) in a thermoneutral environment to promote obesity, testing both male and female FVB.129P mice. We quantified serum hormones and cytokines, characterized the metabolic phenotype, and defined changes in the pituitary transcriptome using single-cell RNA-sequencing analysis. Weight gain was significant by 3 weeks in HFD mice, and by 10 weeks all HFD groups had gained 20 g. HFD females (15 weeks) had increased energy expenditure and decreased activity. All HFD groups showed increases in serum leptin and decreases in adiponectin. HFD caused increased inflammatory markers: interleukin-6, resistin, monocyte chemoattractant protein-1, and tumor necrosis factorα. HFD males and females also had increased insulin and increased TSH, and HFD females had decreased serum prolactin and growth hormone pulse amplitude. Pituitary single-cell transcriptomics revealed modest or no changes in pituitary cell gene expression from HFD males after 10 or 15 weeks or from HFD females after 10 weeks. However, HFD females (15 weeks) showed significant numbers of differentially expressed genes in lactotropes and pituitary stem cells. Collectively, these studies reveal that pituitary cells from males appear to be more resilient to the oxidative stress of obesity than females and identify the most vulnerable pituitary cell populations in females.
Keywords: pituitary, sc-RNA-seq, high fat diet, obesity, oxidative stress
Obesity is defined by the World Health Organization as excessive fat accumulation, which leads to impaired health (1). This condition is a major risk factor for diabetes, metabolic syndrome, cardiovascular disease, fatty liver disease, and cancer (1‐4).
The anterior pituitary serves a critical regulatory role in the body In addition to a small resident adult stem cell population, the anterior pituitary produces key hormones from distinct specialized secretory cell populations: adrenocorticotropin (ACTH) from corticotropes; follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from gonadotropes; prolactin (PRL) from lactotropes; thyroid-stimulating hormone (TSH) from thyrotropes; and growth hormone (GH) from somatotropes. Of these secretory cell types, the thyrotropes and somatotropes contribute directly to the defenses against metabolic stress. TSH stimulates the secretion of thyroid hormone to increase metabolic rate and burn fat (5‐7), and GH optimizes body composition by building bone and muscle and breaking down fat (8‐10).
However, obesity from a high-fat diet (HFD) (11) or high-sugar diet (12) causes both oxidative and endoplasmic reticulum (ER) stress overall in the pituitary of rodents and pituitary somatotrope functions have been shown to be suppressed in obese humans and rodents (8‐11, 13‐18). In rodents, diet induced obesity increases GH clearance (19, 20) and reduces levels of Gh mRNA (21), and receptors for growth hormone–releasing hormone and ghrelin (22) resulting in lowered GH pulse amplitude (18, 22).
Our overall interest in the metabolic regulation of somatotrope or gonadotrope functions (23‐28) led us to initiate a study of the individual pituitary cell transcriptomic responses to a HFD challenge. We note that previous studies of the impact of HFD on rodent pituitaries have exposed the mice to a HFD at room temperature (22, 29‐32), which is lower than the thermoneutral zone for mice and causes thermal stress and compensatory thermogenic responses (33‐43). To avoid confounding variables from a potential allostatic overload, we exposed both males and females to a 60% HFD in a room which was held at thermoneutrality for mice (28.4 °C) (43). The control diet–fed and HFD-exposed mice were characterized with regard to metabolic health and serum levels of adipokines, cytokines, and pituitary hormones. The transcriptomic responses of the individual pituitary cell populations were determined through single-cell RNA sequencing analyses (RNA-seq).
Here, we report sex-dependent and time-dependent pituitary transcriptomic responses to a HFD feeding at thermoneutrality. Pituitary cells from HFD males may be more resilient to oxidative stress than pituitary cells from females, showing modest changes mainly in stem cell populations. In contrast, differentially expressed genes in females were more numerous and focused in clusters with either lactotrope or stem cell signatures. Gene Ontology (GO) analyses of these female clusters demonstrate downregulation of genes encoding pathways involved with protein translation, oxidative phosphorylation, and upregulation of pathways that signal mitochondrial dysfunction. Collectively our studies show that a relatively short-term HFD causes a reduction in genes encoding proteins that support mitochondrial and ER function in female lactotropes and stem cells.
Materials and Methods
Animals
Strain and diet
All experimental procedures were approved by the Institutional Animal Care and Use committee. We used the FVB hybrid strain of mice, FVB.129P2-Pde6b+Tyr c-ch/AntJ, also called “sighted FVB” strain, which was originally derived to avoid the retinal degeneration in the FVB strain. Each group was first acclimated to the thermoneutral environment for 1 week (beginning at 12 weeks of age) and then exposed to the HFD or control diet for either 10 weeks or 15 weeks. For all groups, the HFD (fat, 60 kcal%; carbohydrate, 36 kcal%; protein, 19 kcal%) was supplied as palatable pellets. The sources of fat were lard and soybean oil (ENVIGO, TD.06414). Mice receiving the control diet received ENVIGO reduced sucrose control diet (10% fat, TD.08806).
Thermoneutral housing
All animals were housed in a dedicated thermoneutral room that had been retrofitted to maintain a temperature of 28.4 °C. This was chosen as an environment that would remove the cold stress of a room temperature environment, which stimulates compensatory responses and potential confounds to interpretation of our data. Our decision to use a thermoneutral environment was informed by many studies and reviews in the past 5 years that discussed the use of a thermoneutral environment and its translational applicability to human disease (33‐43). As the thermoneutrality temperature has not been determined for our specific strain, we chose a temperature in the range recommended for most mouse strains and determined that the temperature inside the mouse cages did not rise above recommended levels. Continuous remote monitoring of the room temperature ensured consistency (Room Alert, AVTECH Software, Inc.). Room temperature, humidity, and animal health status were monitored daily. Along with cage changes, food intake and weight measurements were recorded weekly. Cage changes and weight/food measurements took place in the thermoneutral room.
Metabolic health profiling
One week before the mice were euthanized, their metabolic health was tested in metabolic cages (Comprehensive Lab Animal Monitoring Systems [CLAMS]; Oxymax, Columbus Instruments), as described in previous studies (23, 24). Food and water were available ad libitum, and the metabolic cage system was in the same room where the mice were housed at 28.4 °C on an 11 hours light (07:00 start) and 13 hours dark (18:00 start) cycle. The diet was a powdered diet during their time in the CLAMS unit and pelleted in the cages. Mice were housed in a CLAMS unit 4 days before euthanasia, as described in previous studies (23, 24). Data for the 15-week group were recorded for 48 hours, and data from the 10-week group were collected over a 72-hour recording period (both groups having acclimated for 24 hours before recording). Because we ran this experiment under thermoneutral conditions, we were particularly interested to determine whether the mice showed evidence of metabolic changes.
Glucose tolerance tests
One week before the mice were placed in the metabolic cages, glucose tolerance tests (GTTs) were performed as previously described (23, 24). Basal glycemia levels were determined, followed by an intraperitoneal injection of 1 g/kg of 20% glucose. Glycemia was then monitored at 15, 30, 60, 90, and 120 minutes after the injection. Mice were returned to their cages to acclimate for 1 week before entering the metabolic cages.
Sample collection
On the same morning the mice were removed from the CLAMS unit, they were euthanized for sample collection. The mice were deeply anesthetized with isoflurane and decapitated by guillotine. Pituitaries were removed for dispersion and single-cell RNA-seq, and serum was collected for hormone and cytokine analyses.
Multiplex Assays
Sera were analyzed for pituitary hormones, adipokines, and inflammatory markers as described previously (23, 24) with the use of multiplex mouse adipokine or mouse pituitary hormone enzyme immunoassays (Millipore Sigma). The pituitary hormone enzyme immunoassay kit (MPTMAG-49K-05, RRID:AB_2811194) was used to quantify ACTH, LH, FSH, TSH, and PRL. GH was assayed using RPTMAG-86K-01 (RRID:AB_2716840), which we have found to give the most consistent GH measurements. The mouse adipokine kit (MADKMAG-71K-07, RRID:AB_2801416) was used to detect serum levels of adiponectin, interleukin-6 (IL-6), insulin, leptin, monocyte chemoattractant protein-1 (MCP-1), plasminogen activator inhibitor-1 (PAI-1), tumor necrosis factor-alpha (TNFα), and resistin. Serum leptin was quantified by enzyme-linked immunosorbent assay (Quantikine Elisa Systems, MOB00B, R&D systems, RRID:AB_2943468). All serum assays have been published and are described in previous reports (24, 25, 44‐47).
Single-Cell RNA Sequencing and Analysis
For single-cell RNA-sequencing (scRNA-seq) analyses, we collected 4 to 6 pituitaries per sex/diet/treatment length. All pituitaries were dispersed as previously described (25). Following the final mechanical dispersion/centrifugation, cells were washed gently, centrifuged, and resuspended in Dulbecco’s modified Eagle’s medium with no gentamicin. At this point, individual pituitary cell suspensions were pooled within treatment groups (2-3 pituitaries/pool) and counted. The concentration of the cells within each pool was adjusted to optimally target 10 000 cells according to 10× Genomics guidelines. Samples were then submitted to the UAMS Cancer Institute Genomics Core facility. If visual examination under a microscope (EVOS M7000, Thermo Fisher Scientific) detected cell debris or aggregates, cell suspensions were passed through a Flowmi (SP BEL-ART, catalog # 136800040) cell strainer (40 µm). Cells were then centrifuged at 250g for 10 minutes and resuspended in 1× phosphate-buffered saline (Ca2+/Mg2+ free) containing 0.04% (w/v) bovine serum albumin. To determine cell concentration and viability, cells were stained with ReadyProbes_Cell Viability Imaging Kit, Blue/Green (Thermo Fisher Scientific, catalog # R37609), and manually counted.
Single-cell 3′ library generation was performed using a 10× Genomics Chromium Controller and Chromium next GEM Single Cell 3′ Reagent Kits v 3.1 (Dual index) according to the manufacturer's protocol. Cell suspensions were loaded onto the middle 4 channels of a chromium single-cell G chip according to the manufacturer's instructions, aiming for 8000 cells per channel. Following generation of single-cell gel bead in emulsions, reverse transcription, fragmentation, and complementary DNA (cDNA) amplification, library preparations and bar coding were performed. The concentration and size distribution of the libraries were assessed with a fragment analyzer and a Qubit fluorometer. Libraries were sequenced on the Illumina NovaSeq 6000 platform with paired-end mode (read 1: 28 cycles, read 2 : 90 cycles, i7: 10 cycles, i5: 10 cycles) to generate a minimum of 20 000 read pairs per cell. 10× Genomics Cell Ranger 7.1 mkfastq wrapper and was used to perform sample demultiplexing and generate fastq files.
Quality control, bioinformatics clustering and differential gene expression were performed as before (44). Demultiplexed fastq files generated by the UAMS Genomics Core were analyzed with the 10× Genomics Cell Ranger 3.1.0 count function for sequence alignment and gene counting, a self-contained single-cell RNAseq pipeline developed by 10× Genomics. The reads were aligned to the University of California, Santa Cruz (UCSC) mm10 reference transcriptome using STAR and transcript counts were generated (48, 49). The R package Seurat was used to further analyze the filtered, aggregated, and depth normalized counts generated by cellranger count and cellranger aggr (http://software.10xgenomics.com/single-cell/overview/welcome) (50). Cells with unique feature counts over more the 75th percentile plus 1.5 times the interquartile range (IQR) or less 200 unique features, total gene counts more than more the 75th percentile plus 1.5 times the IQR or less than 1000 counts, and/or mitochondrial feature percentage more the 75th percentile plus 1.5 times the IQR or less than the 25th percentile minus 1.5 times the IQR were filtered out of the data. Next, the 2000 highest variable features were selected. The data were then scaled by a linear transformation and variation associated with cell death and cell cycling where regressed out of the scaled data using mitochondrial feature percentage and cell cycle scoring values. Counts were normalized using the SCTransform and PCA was performed on the scaled data. A Uniform Manifold Approximation and Projection (UMAP) was used to visualize and explore the clustering results. Seurat's FindNeighbors and FindClusters functions were optimized to label clusters based on the visual clustering in the projections. Seurat FindAllMarkers function was used to identify gene markers that define cluster cell types. These gene markers were compared with known markers of expected pituitary cell types (51, 52) in order to assign appropriate cell type labels (53). Differential expression analysis was performed using the FindMarkers to compare similar cell types across diet and gender. The MAST test, a GLM framework that treats cellular detection rate as a covariate, was used to determine statistically different transcription(54). Genes with an false discovery rate adjusted P < .055 and a Log2FC ≥ 0.58 (fold increase 1.5) were considered to be statistically significant.
Statistics and power analyses
We used the latest version of Graphpad PRISM (10) with P < .05 and tests appropriate for the design (eg, 2-way analysis of variance [ANOVA] with Bonferroni post hoc tests (45‐47, 55)) Post hoc power analyses established the number of replicates with the help of the G* Power application: https://www.psychologie.hhu.de/arbeitsgruppen/allgemeine-psychologie-und-arbeitspsychologie/gpower.html. F-test for population variance was performed in R code. Specifics regarding statistical analyses used (except scRNA-seq analysis, which is described above) can be found in individual figure legends. The energy expenditure (EE) analysis of covariance done for this work was provided by the NIDDK Mouse Metabolic Phenotyping Centers (www.mmpc.org) using their EE analysis page (http://www.mmpc.org/shared/regression.aspx) and supported by grants DK076169 and DK115255.
Results
Impact of HFD on Weight Gain
Both males and females on a HFD gained weight compared with control diet–fed mice, as summarized in Table 1. The weight gain was significant by week 6 in females (adjusted P = .01) (Fig. 1A and 1B) and by week 3 in males on the HFD for 15 weeks (adjusted P < .0001) (Fig. 1D) control diet mice. Table 1 shows information complementary to Fig. 1. Notably, it shows that, because the additional weight gain reached a plateau, the average amount of weight gained after 10 weeks on a HFD was not significantly different from that in the group exposed to a HFD for 15 weeks (Table 1).
Table 1.
Comparison of weight changes in all experimental groups (g) (number of animals/group)
| Sex and time on diet | Experimental group | ||
|---|---|---|---|
| Control Diet | HFD | Significance | |
| Females 10 weeks | |||
| Starting weight | 23.3± 0.74 (5) | 23.7± 1 (5) | NS |
| Final weight | 32.2 ± 1.2 | 43.5 ± 2.5 | Adj P = .01 |
| Weight change | 9 ± 0.5 | 19.7 ± 2.9 | Adj P = .01 |
| Females 15 weeks | |||
| Starting weight | 22 ± 1 (4) | 21.9 ± 0.4 (4) | NS |
| Final weight | 28.5 ± 0.89 | 42.5 ± 2.4 | Adj P < .0001 |
| Weight change | 6.25 ± 1.5 | 20.6 ± 2 | Adj P < .0001 |
| Males 10 weeks | |||
| Starting weight | 27 ± 0.5 (6) | 26.8 ± 0.6 (7) | NS |
| Final weight | 40 ± 1.4 | 48 ± 0.66 | Adj P < .0001 |
| Weight change | 13 ± 1.7 | 20 ± 1 | Adj P < .0001 |
| Males 15 weeks | |||
| Starting weight | 26.4 ± 0.48 (5) | 30 ± 1.3 (5) | NS |
| Final weight | 36.3 ± 1.5 | 51 ± 2 | Adj P < .0001 |
| Weight change | 10 ± 1 | 21 ± 2 | Adj P < .0001 |
Figure 1.

Weight gain in FVB.P129 females (A, B) or males (C, D) after 10 weeks (A, C) or 15 weeks (B, D) on a high-fat diet under thermoneutral conditions. Asterisks indicate significantly increased weight in HFD mice when compared with control diet mice. All data were analyzed by 2-way ANOVA and Bonferroni's multiple comparisons test, adjusted P <.05. In the 10-week group, a defective water bottle created a wet cage, which may have stressed the mice causing an initial drop in weight in the control group. They recovered, however.
Impact of HFD on Metabolic Health
At the end of the diet period, the metabolic impact of a HFD was assessed in metabolic cages, in the same thermoneutral room in which the animals were housed. Males and females on a HFD for 15 weeks consumed less food volume (Fig. 2A and 2C) and fewer calories (Fig. 2B and 2D) than control diet–fed mice. Because the groups exposed for 10 weeks showed similar responses, Fig. 2 shows only the data from the 15-week group.
Figure 2.

After 15 weeks on a high-fat diet, female (A, B) and male (C, D) mice were placed in metabolic cages for 48 hours that weighed the total amount of food eaten on a scale in the cage. Total grams consumed in light or dark phases is shown by numbers in the bars. Females (A) and males (C) on a HFD ate less food by weight. However, females (B) consumed more kcal than males (D) on a HFD. Increased food consumption in the dark reflects the fact that the mice are nocturnal. Analysis by Student's t-test, 2-way ANOVA and Bonferroni's multiple comparison test (adjusted P < .05). Both females (E) and males (G) on a HFD showed lower respiratory quotient (RQ) values, which is calculated as the ratio of the volume of carbon dioxide divided by the volume of oxygen during the 3-day period in the metabolic cages. This represents a tendency to metabolize fats as the primary source of fuel. In both sexes, energy expenditure was higher in mice fed a high-fat diet (F, H), especially in females (F). RQ and EE were plotted over time on a 11 hour light/13 hour dark cycle. (I, J). A comparison of the RQ values during the day and night shows striking differences between controls and HFD mice. Analysis of movement in the metabolic cages was done by detecting light beam breaks in the X-axis, indicating ambulation or grooming or Z axis, indicating jumping or rearing. The number of beam breaks is seen in the bars. After 15 weeks on the HFD, females showed significantly lower ambulation and grooming (K), but not rearing or jumping (L). Males showed lower ambulation only in the dark phase (M) with no changes in rearing or jumping (N). Analysis by 2-way ANOVA and Bonferroni's multiple comparison test (adjusted P < .05).
The CLAMS unit measured the volume of CO2 (VCO2) and O2 (VO2), calculating the respiratory quotient (RQ) as the ratio of VCO2/VO2. All groups on a HFD had a significantly lower RQ, which indicates increased burning of fat instead of carbohydrates as the primary fuel source. This agrees with RQ calculations reported by other workers (34, 37, 42), and is represented by the data from the 15 week HFD group in Fig. 2E and 2G. In addition, females on a HFD for 15 weeks have higher levels of EE (kcal burned/hour) especially during the dark phase (Fig. 2F). Males also show a slight increase in EE after 15 weeks on a HFD (Fig. 2H). Analysis of covariance of EE showed that the energy expenditure differences cannot be simply explained by the increase in body mass (Fig. S1 (56)). RQ and EE were plotted over time for females and males (Fig. 2I and 2J). HFD affected the normal rise in RQ leading to the dark phase in both sexes. In both sexes, EE was increased leading to the dark phase and the slight increase by the HFD can be visualized. No changes in EE were seen in the groups on a 10-week HFD (data not shown).
Tests of activity in metabolic cages identified differences between the 10-week and 15-week groups. Activity was detected by the number of light beam breaks in the horizontal plane (walking or grooming, X axis) or vertical plane (rearing or jumping, Z axis). All groups showed increased activity during the dark phase, reflecting nocturnal feeding and activity. In males or females exposed to a HFD for 10 weeks (Fig. S2 (56)), there was no significant impact of a HFD on activity. In contrast, females exposed to a HFD for 15 weeks showed reduced ambulation and grooming (Fig. 2K), but not rearing or jumping (Fig. 2L). Males on a HFD for 15 weeks showed reduced ambulation during the dark phase (Fig. 2M), but no changes in rearing and jumping (Fig. 2N).
Impact of HFD on cytokines and adipokines
Analysis of serum levels of adipokines and cytokines showed little difference between the 10 weeks and 15 weeks HFD groups and the serum values for 10- and 15-week groups were combined. Serum leptin levels were significantly higher (Fig. 3A) and serum adiponectin levels were significantly lower (Fig. 3B) in all groups exposed to a HFD and both females and males on a HFD also showed higher serum levels of insulin (Fig. 3C), with the difference in males being much greater than that in females. Serum GTTs showed a delayed drop in serum glucose in the males after 15 weeks on a HFD (Fig. S3 (56)). Both males and females on a HFD showed higher levels of the inflammatory markers IL-6 (Fig. 3D), MCP-1 (Fig. 3E), TNFα (Fig. 3F), and resistin (Fig. 3G). Females show higher levels of PAI-1 than males, but in neither sex were the levels of PAI-1 affected by a HFD (Fig. 3H).
Figure 3.
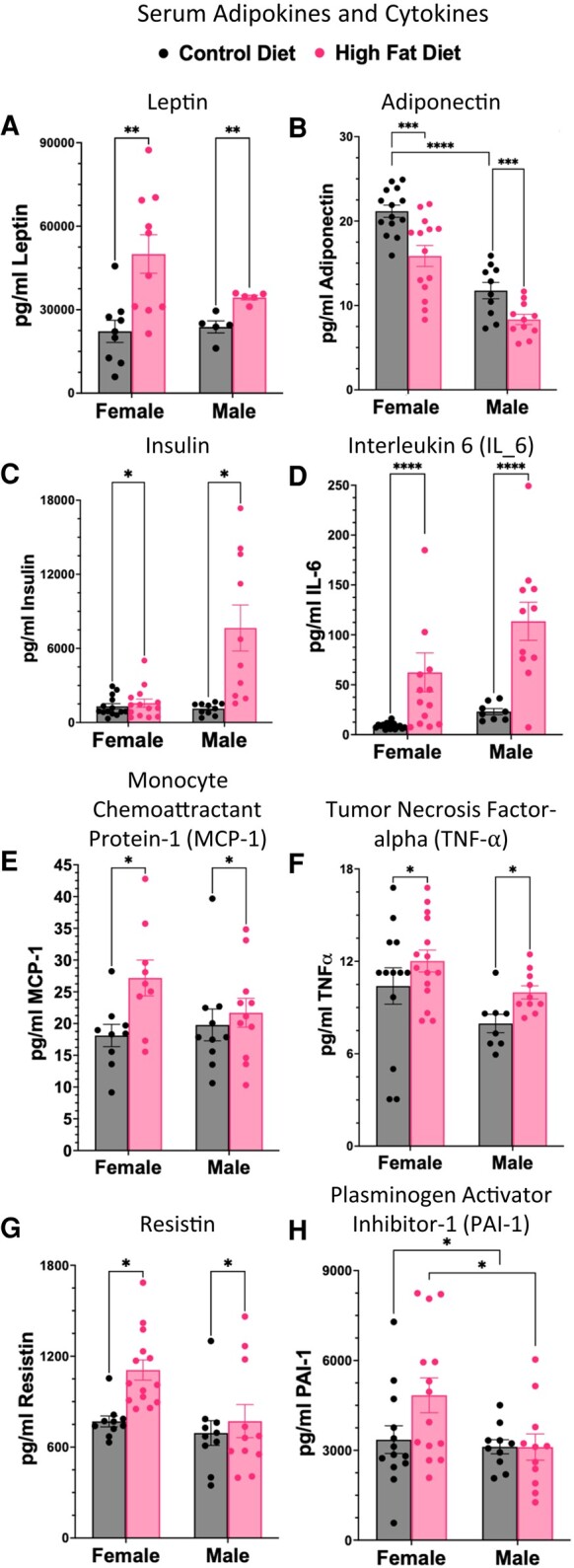
Cytokines and adipokines were assayed in both male and female mice on a HFD for 10 to 15 weeks. Lower levels of adiponectin after a HFD (B) reflect the obese state and the inflammatory response (B). Both males and females showed hyperleptinemia (A), and higher insulin (C); however only males showed hyperinsulinemia (C). The lower levels of adiponectin correlated with higher levels of IL-6 (D) leptin (A), MCP-1 (E), TNFα (F). Other inflammatory markers that are elevated are resistin (G). PAI1 is higher in females than males, but not elevated by HFD (H). Leptin, adiponectin, resistin, and IL-6 analysis by 2-way ANOVA and Bonferroni's multiple comparison test (adjusted P < .05). TNFα, MCP-1, and PAI-1 analysis by 2-way ANOVA and Fisher's least significant difference test (P < .05).
Impact of HFD on serum pituitary hormones
The effect of HFD on the overall serum levels of most pituitary hormones in mice held at thermoneutrality was similar at 10 and 15 weeks, so, except in the case of PRL, we were able to combine the values from the 10- and 15-week groups. The rise in levels of TSH in males and females (Fig. 4A) was a significant HFD-driven change, with greater responses seen in males than females. Levels of TSH were also higher in control males than in control females. Levels of PRL were 100× higher in females than in males, which necessitated plotting on separate graphs (Figs. 4B and 4C). Our statistical analysis showed that the sex differences were significant. Notably, serum PRL was reduced only in females exposed to a HFD for 15 weeks (Fig. 4B). No diet-related PRL changes were seen in males after 15 weeks on a HFD (Fig. 4C). No changes were seen in either male or female serum levels of ACTH with a HFD (Fig. 4D). Although we observed sex differences in serum levels of LH and FSH, these were not significantly altered by HFD (Fig. 4E and 4F) .
Figure 4.
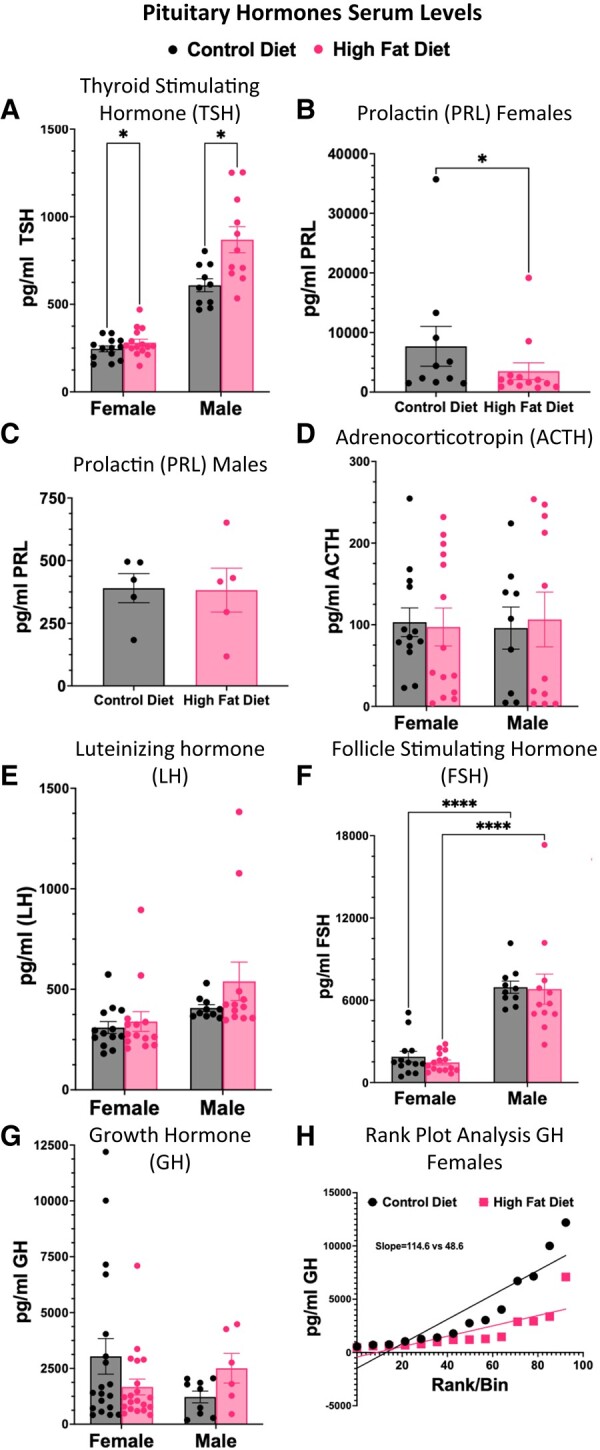
Assays of serum levels of pituitary hormones following a HFD for 10 to 15 weeks. Values were combined as there were no significant changes with time on the diet. The data show elevation only in TSH in males (A) following a HFD. PRL is reduced only in females after a HFD for 15 weeks (B). The overall PRL levels in males (C) are significantly lower than those in females and they are plotted in a separate graph. The sex differences are significant (Tukey's ad hoc tests; adjusted P < .0001). There are no changes in ACTH (D), LH (E), or FSH (F) in either sex with a HFD. Average serum levels of GH did not show a significant change, although variance in GH levels was increased in males (G, test of variance in GH levels of HFD males vs control diet males, F = 35.278, P = 4.975 × 10–05). Furthermore, in females, a rank/plot analysis of individual values showed lower pulses of GH in females on a HFD (H). Except for PRL, analysis of the 4 groups was 2-way ANOVA followed by Tukey's ad hoc tests of significant differences at adjusted P < .05. Analysis of serum PRL showed significance with Student's t and Mann–Whitney tests (P < .02).
In both male and female mice, the averaged GH values showed no significant differences between the control and HFD groups (Fig. 4G). HFD Males showed a trend towards more increased GH pulses and showed a higher variance in GH values than ND males (Fig. 4G, F-value = 35.278, P = 4.975 × 10–05). With respect to females, to account for the known pulsatility of GH secretion, a rank-plot analysis was performed on the collected data. Values from female groups on a HFD showed fewer high amplitude pulses in a linear array, which was detected by rank-plot analysis (Fig. 4H). The slope of the line was significantly different from that of control females on a control diet (P < .001)
Impact of a Thermoneutral Environment on Serum Hormone Levels
Our initial analyses of serum levels of pituitary hormones compared ranges and averages with those collected from groups of mice housed at room temperature. This would determine whether the thermoneutral environment had an impact on secretion from the pituitary cells and also would indirectly test the impact of the environment on the hypothalamic hormones. We compared serum TSH and ACTH in 2 groups of age-matched females housed for 15 weeks in either a room temperature or thermoneutral environment. The range of TSH levels in a group of 5 females held at room temperature was 158 to 336 pg/mL (average = 260 ± 38 pg/mL) and the range of TSH levels for a group of 8 females held at thermoneutral conditions was 157.5 to 292 pg/mL (average 236 ± 15 pg/mL). These averages are not different from one another (P = .76). Similarly, serum ACTH levels for 5 females at room temperature ranged from 66 to 167 pg/mL (average 96.99 ± 18 pg/mL) and the levels for 8 females at thermoneutrality ranged from 22.5 to 153.6 pg/mL for 7 females adding 1 outlier at 254 pg/mL (average 106.8 ± 27 for 8 females). These averaged serum ACTH levels for both groups were not significantly different (P = .76). Our previous published studies of other groups housed at room temperature also show similar ranges for these hormones (24, 25). Altogether, the evidence does not indicate that that thermoneutrality alone has affected the pituitary directly or indirectly through effects upon hypothalamic hormones.
Impact of HFD on Pituitary Transcriptome at the Single Cell Level
We performed scRNA-seq transcriptomics analysis of each experimental group (male or female, control diet or HFD, 10 or 15 weeks) in 2 concurrently processed, duplicate pools. After determining that the duplicate pools produced identical maps, the data were combined, and the cells were clustered computationally based on the shared nearest neighbor–based algorithm with the use of Seurat. Established pituitary endocrine and stem cell markers were used to assign broad population identities (44, 51, 52) (Table S1 (56)) .
The UMAP profiles from the males or females exposed to a HFD for 10 and 15 weeks is provided (Fig. 5). All pituitary cell types were identified in separate UMAP clusters. An analysis of numbers of cells in each cluster showed a 42% to 54% decline in corticotropes in the female and a 12% to 25% decline in corticotropes in the males. Pituitary stem cells from females also showed a 14% to 20% decline and in males, a 10% to 14% decline. However, somatotropes from females after 15 weeks HFD showed a 35% increase in numbers of cells with that gene signature.
Figure 5.

Single cell RNA-Seq UMAP plots showing that we were able to detect near-neighbor clusters representing all major pituitary cell types. Cells were clustered based on the detection of a common set of transcripts which represented the consensus for each cell type (Table S1 shows the top 10 genes expressed for each cluster). A comparison of these plots shows a similar distribution comparing males and females and time on the diet for each sex.
Determination of differentially expressed genes (DEGs) indicated that males exposed to a HFD for 10 weeks showed no DEGs in the pituitary hormone cell types based on the significance threshold utilized (log2FC+0.58; adjusted P ≤ .055, Table S2 (56)). In this group, pituitary stem cells showed a reduction in 7 genes that encode proteins mostly involved with defense and immune functions. Males exposed to a HFD for 15 weeks also showed DEGs mainly in pituitary stem cells. In lactotropes, Gh mRNA was reduced 1.5-fold with an adjusted P < .055 (Table S3 (56)). Stem cell DEGs also included genes encoding proteins involved in defense and immune response. The expression of Prl mRNA in stem cells may indicate that the some of these cells are differentiating towards a more mature hormone cell type.
Females exposed to HFD for 10 weeks showed modest or no transcriptomic changes in most pituitary clusters (Table S4 (56)). At Log2FC of 0.58 (fold increase of 1.5) and adjusted P < .055 thresholds, only the lactotrope cluster and gonadotropes in females showed DEGs. Notably, lactotropes showed a 1.8-fold decrease in Gh mRNA (Table S4 (56)), which correlates with the decrease in serum GH pulses detected by rank/plot analysis. Gonadotropes showed an increase in expression of the gene encoding secretogranin 2 (Scg2), a protein which colocalizes with LH in secretory granules and may play a role in LH trafficking (57).
The most DEG changes were evident in lactotropes and stem cells from females exposed to HFD for 15 weeks (Table S5 (56)). The somatotrope cluster also had 3 downregulated ribosomal protein genes and an upregulation of the actin beta subunit gene and the gene encoding the erythroblast transformation–specific related transcription factor. There were no DEGs in thyrotropes and corticotropes from females exposed to HFD for 15 weeks. The lactotrope cluster in these females showed 20 DEGs; 4 were upregulated and 16 were downregulated. Figure 6A illustrates a bar graph showing the results of the GO analysis of downregulated DEGs in lactotropes (Table S5 (56)), which highlight cytoplasmic translation and ribosomal biogenesis processes. The downregulated Prl mRNA (>2 fold), which correlates with the lower serum PRL seen in females on a HFD for 15 weeks (Fig. 6B) is shown elsewhere (Table S5 (56)). Similarly, the downregulated Gh mRNA in the lactotrope population from females after 10 weeks on the HFD (Table S4 (56)) correlates with the rank plot analysis showing reduced GH pulse amplitude (Fig. 4H). It also points to a multihormonal cell population clustered with the lactotropes. Figure 6B and 6C correlate the downregulation of ribosomal proteins with the network of pathways impacted by the HFD, showing the genes that contribute to each phase of ribosome biogenesis.
Figure 6.

Bar graph showing a Gene Ontology analysis of downregulated DEGs in the lactotrope cluster from females after a HFD (15 weeks). The most abundant DEGs are involved with cytoplasmic translation and ribosomal biogenesis.
The stem cell cluster from females exposed to a HFD for 15 weeks showed the most DEGs (from Table S6 (56)) with the use of the threshold of Log2FC = 0.58 (fold change = 1.5; adjusted P = .055). These DEGs are primarily downregulated. Among these DEGs are representatives of genes encoding the S-100 protein family, which are markers for folliculostellate cells. There are also genes encoding many ribosomal proteins (shared with the DEGs in female lactotropes) and protein subunits belonging to the respiratory chain. A GO analysis of the downregulated genes which met the threshold of Log2FC = .58 (fold change 1.5); adjusted P < .055 is shown in Fig. 7A. As with the lactotrope cluster, the changes are seen in processes related to cytoplasmic translation and ribosome biogenesis. Figure 7B and 7C illustrates the network of genes encoding proteins that affect ribosome biogenesis.
Figure 7.

Bar graph showing a GO analysis of downregulated DEGs in stem cell cluster from females after a HFD (15 weeks). The most abundant DEGs are involved with cytoplasmic translation and ribosomal biogenesis.
DEGs encoding proteins that are related to mitochondrial structure and function are shown elsewhere (Tables S5 and 6 (56)). To determine whether this differential expression indicates a trend towards an overall reduction in mitochondrial function, we ran a GO analysis of all DEGs in lactotropes and stem cells that met the significance threshold of adjusted P < .055 without a minimum fold cutoff. The expression levels of all 443 genes in the lactotrope cluster that were differentially expressed at this significance threshold are listed elsewhere (Table S7 (56)). The major GO terms for these DEGS are listed elsewhere (Tables S8 and 9 (56)). The GO terms for the 167 downregulated genes include processes related to protein synthesis, mitochondrial function, and programmed cell death (Table S8 (56)). The GO terms for the 259 upregulated genes relate to cell differentiation, transcription, cell motility and programmed cell death (Table S9 (56)).
Similarly, in the stem cell cluster, all of the differentially expressed genes meeting the significance threshold of adjusted P < .055 without a minimum fold change cutoff are listed elsewhere (Table S10 (56)). The GO analyses for the 244 downregulated genes is shown elsewhere (Table S11 (56)). These include signaling, translation, cell differentiation, and transcription processes. The 25 upregulated genes in the stem cell cluster represent cell differentiation, programmed cell death, and transcription processes (Table S12 (56)).
Ingenuity pathway analysis was then run on DEGs from both stem cells and lactotropes to detect the canonical pathways represented by all DEGs that met the significance threshold of adjusted P < .055, in females after 15 weeks on the HFD (listed in Tables S7 or S10 (56)). Figure 8 is a heat map summarizing the major canonical pathways, comparing gene expression in lactotropes and stem cells from females after 15 weeks on the HFD with those on a control diet (the z-score is seen in the Y axis and is color coded to highlight downregulated [blue] vs upregulated pathways [orange]). Some of the same pathways in Fig. 8 are enriched in both stem cell and lactotrope cell clusters with the genes in the top 5 pathways listed in Tables 2 and 3. The most prominently downregulated canonical pathways included protein synthesis (EIF2 signaling) and oxidative phosphorylation. The most prominent upregulated pathway signifies mitochondrial dysfunction, which is consistent with the downregulated genes encoding proteins in the respiratory chain (Tables S5, S6, S7, and S10 (56)). As indicated above, all DEGs that met the significance threshold are shown elsewhere (Tables S7 and S10 (56)).
Figure 8.

Heat map developed from ingenuity pathway analysis showing Z scores of canonical pathways represented by upregulated or downregulated genes in female pituitary lactotrope or stem cell clusters after 15 weeks on a HFD. Tables 2 and 3 list the genes in the first 5 pathways in Fig. 8.
Table 2.
Ingenuity pathway analysis of canonical pathways based on DEGs in lactotrope cluster
| Canonical pathway | Genes in canonical pathway |
|---|---|
| EIF2 Signaling | ACTB, EIF4A1, FAU, Gm15483, GSK3B, HNRNPA1, IGF1R, PAIP1, PPP1CB, RPL10, RPL10A, RPL11, RPL12, RPL13, RPL14, RPL15, RPL17, RPL18, RPL18A, RPL19, RPL21, RPL22, Rpl22l1, RPL23, RPL24, RPL26, RPL27A, RPL28, RPL3, RPL30, RPL31, Rpl34, RPL35, RPL35A, RPL36, Rpl36a, RPL36AL, RPL37A, RPL39, RPL4, RPL41 RPL7, RPL7A, RPL9, RPLP0, RPLP1, RPS10, RPS11, RPS14, RPS15, RPS15A, RPS16, RPS18, RPS19, RPS2, RPS20, RPS21, RPS23, RPS24, RPS25, RPS26, RPS27, RPS27A, RPS28,RPS29, RPS3, RPS4Y1, RPS5, RPS6, RPS7, RPS8, RPS9, RPSA |
| Oxidative Phosphorylation | Atp5e, ATP5MC1, ATP5MC2, ATP5MF, ATP5MG, ATP5PD, ATP5PF, COX4I1, COX6A1, COX6B1, Cox6c, Cox7a2/Cox7a2l2, COX7A2L, COX8A, NDUFA1, NDUFA11, NDUFA13, NDUFA4, NDUFAB1, NDUFB11, UQCR11 |
| Mitochondrial Dysfunction | Atp5e, ATP5MC1, ATP5MC2, ATP5MF, ATP5MG, ATP5PD, ATP5PF, Calm1, COX4I1, COX6A1, COX6B1, Cox6c, Cox7a2/Cox7a2l2, COX7A2L, COX8A, NDUFA1, NDUFA11, NDUFA13, NDUFA4, NDUFAB1, NDUFB11, PARK7, PPP3CA, PRKACA, SOD1, SOD2, TOMM34, TOMM7, UQCR11 |
| Sirtuin signaling pathway | ADAM10, ATP5MC1, ATP5PF, FOXO3, GABARAP, GSK3B, H3-3A/H3-3B, NDUFA1, NDUFA11, NDUFA13, NDUFA4, NDUFAB1, NDUFB11, SOD1, SOD2, TOMM34, TOMM7 |
| NRF2-mediated oxidative stress response | ACTB, DNAJA1, FOS, FTH1, FTL, GSK3B, HSP90AB1, SOD1, SOD2, TXN |
Females on HFD, 15 weeks, thermoneutrality; adjusted P < .055.
Table 3.
Ingenuity pathway analysis of canonical pathways based on DEGs in stem cell cluster
| Canonical pathway | Genes in canonical pathway |
|---|---|
| EIF2 signaling | ACTB, FAU, Gm15483, GSK3B, PPP1CB, RPL10, RPL10A, RPL11, RPL12, RPL13, RPL14, RPL17, RPL18, RPL18A, RPL19, RPL21, RPL22, Rpl22l1, RPL23, RPL24, RPL26, RPL27A, RPL28, RPL3, RPL30, Rpl34, RPL35A, RPL36, Rpl36a, RPL36AL, RPL37A, RPL39, RPL41, RPL7, RPL9, RPLP0, RPLP1, RPS10, RPS11, RPS12, RPS14, RPS15, RPS15A, RPS16, RPS18, RPS19, RPS2, RPS20, RPS21, RPS23, RPS24, RPS26, RPS27, RPS27A, RPS28, RPS29, RPS3, RPS4Y1, RPS5, RPS6, RPS7, RPS8, RPS9, RPSA |
| Oxidative phosphorylation | Atp5e, ATP5MC1, ATP5MF, ATP5MG, ATP5PD, ATP5PF, ATP5PO, COX4I1, COX6B1, Cox6c, COX8A, CYB5A, NDUFA13, NDUFA2, NDUFA4, NDUFA7, NDUFB11, NDUFB3, NDUFB7, NDUFB8, NDUFB9, NDUFS8, UQCR10, UQCR11 |
| Mitochondrial dysfunction | Atp5e, ATP5MC1, ATP5MF, ATP5MG, ATP5PD, ATP5PF, ATP5PO, Calm1, COX4I1, COX6B1, Cox6c, COX8A, CYB5A, MGST1, NDUFA13, NDUFA2, NDUFA4, NDUFA7, NDUFB11, NDUFB3, NDUFB7, NDUFB8, NDUFB9, NDUFS8, PARK7, PRDX6, SOD1, TOMM7, UQCR10, UQCR11 |
| Sirtuin signaling pathway | ATP5MC1, ATP5PF, GABARAP, GSK3B, H3-3A/H3-3B, LDHB, MAP1LC3B, NDUFA13, NDUFA2, NDUFA4, NDUFA7, NDUFB11, NDUFB3, NDUFB7, NDUFB8, NDUFB9, NDUFS8, POLR2F, SLC25A4, SOD1, TOMM7, TSPO |
| NRF2-mediated oxidative stress response | ACTB, ACTG 1, AKR1A1, CYP2F1, FTH1, FTL, GSK3B, GSTM5, HSP90AA1, HSP90AB1, MGST1, PPIB, PRDX1, SOD1, TXN |
Females on HFD, 15 weeks, thermoneutrality; adjusted P < .055.
Ingenuity pathway analysis identified additional pathways related to responses to oxidative stress (Fig. 8). Both stem cell and lactotrope cell clusters show increases in genes relevant to Sirtuin signaling, which relates to cell survival under stressful conditions such as obesity. Sirtuins play important roles in inflammation, mitochondrial dysfunction, and insulin resistance (32, 58). In addition, the transcription factor, Nrf2-mediated oxidative stress response pathway is reduced in both clusters, pointing to a vulnerability to reactive oxygen species in these populations.
Figure 8 also shows pathways that differ when the stem cell and lactotrope cell clusters are compared. Notably, stem cells show a reduction in expression of the S100 family of signaling proteins. There is also reduced G-protein coupled receptor and phospholipase C signaling. Lactotropes show no changes in genes encoding proteins that control in these pathways. In contrast, lactotropes show an increase in genes encoding estrogen receptor, NF-κB, and GnRH signaling. Genes encoding proteins in pathways supporting autophagy and microRNA biogenesis are increased in lactotropes, they are decreased in stem cells.
Discussion
Development of the FVB mouse obesity model
This study developed an obesity model under thermoneutral conditions, testing 2 different time points on a HFD, 10 weeks and 15 weeks, in both male and female mice. Several discoveries were made. First, it appears that whereas the additional time on the diet (15 weeks vs 10 weeks) did not significantly impact the amount of weight gained or levels of inflammatory markers, the extra 5 weeks’ exposure to the HFD did result in critical pituitary transcriptomic changes in lactotropes and stem cells in females. In the metabolic cages, females also showed higher EE and were less active if on the HFD for 15 weeks. Second, we report that, in general, male pituitary cells appear to be relatively resilient to the oxidative stress of a HFD compared with females, in spite of the similar rise in inflammatory markers and weight in males and females. Indeed, males also showed higher levels of metabolic stress by an higher increases in levels of insulin and IL-6, and decreased insulin sensitivity (delayed glucose tolerance test).
The third discovery relates to important strain differences with respect to weight gain in female mice. Here we report significant weight gain in a relatively short term period in FVB.129P female mice. A parallel pilot study shows that females in this strain will gain similar amounts of weight at room temperature as well (data not shown). This is significant because reports show that C57Bl/6 females may require as much as 27 weeks on a HFD (at room temperature) for weight gain (59). Male C57Bl6 mice appear to respond well to a HFD (22, 30). We postulate that the female sensitivity to a HFD in our FVB/129P females may reflect a strain difference. The FVP/129P hybrid females now provide a model for studies of the impact of a relatively short term HFD on female mice.
Changes in adipokines and cytokines
The HFD obesity model showed the expected increases in leptin in both sexes, which reflected the additional fat stores (60‐62). Males showed a greater increase in insulin, which correlated with the delayed drop in glucose in a GTT assay (Fig. S1 (56)). The downregulation of adiponectin in all groups exposed to a HFD is consistent with the inverse relationship between plasma adiponectin levels and regulators, which were elevated, including leptin, IL-6, and TNFα (63‐65). Adiponectin plays an anti-inflammatory role in the body, protecting against insulin resistance and cardiovascular disease (63‐69). Proinflammatory cytokines, such as leptin, TNFα, and IL-6, which were elevated in both HFD male and female mice, inhibit adiponectin (63‐69).
Resistin is an adipokine that is also linked to obesity and diabetes or insulin resistance (70‐72) and is elevated in HFD mice groups. Whereas 2 studies have reported that higher levels of resistin stimulate pituitary somatotrope secretion in rats or primates (73, 74) and corticotrope secretion in primates (74), the higher resistin in our HFD mice did not appear to affect secretion from these cell types. Both male and female HFD mice show a slight increase in MCP-1, which is produced by white adipose tissue and is upregulated in obese mice (75). MCP-1 has been reported to induce adipose dedifferentiation and contribute to hyperinsulinemia and type II diabetes (75).
Impact of a HFD on pituitary hormone serum levels
We compared serum levels of pituitary hormones (ranges and averages) with data from past cohorts of FVB/129P mice housed at room temperature (24, 25) as well as a cohort housed during the same time period. We concluded that the thermoneutral environment did not impact basal secretion of any of the pituitary hormones, which would be an indirect measurement of the impact of this environment on the function of their hypothalamic releasing hormones.
Furthermore, except for PRL in females, there was no impact of time on the diet on the changes in serum pituitary hormone levels. Both HFD males and females showed an increase in serum TSH. The higher TSH levels in males could result from hyperinsulinemia, which reduces de-iodinase and thus affects negative feedback by lowering available tri-iodothyronine (76) In addition, because leptin stimulates pro-TRH (Thyrotropin releasing hormone) biosynthesis directly in TRH neurons (77) the hyperleptinemia of obesity could also have stimulated TSH (78, 79).
HFD females showed lower levels of PRL after 15 weeks on a HFD. They also showed fewer high-amplitude serum GH pulses when rank/plot analysis was used to analyze the data. The finding for GH agrees with reports from previous studies by Luque and Kineman in male C57Bl/6 mice, which were HFD-fed at room temperature (22). We did not detect reduced GH pulses in the males in this study although, the high variance in serum GH in the HFD males suggests a dysregulation of these processes. The difference could reflect the strain, or the thermoneutral environment.
Impact of a HFD on Pituitary Transcriptome
We used consensus profiles that identified gene signatures for individual pituitary cell types first developed by others (51, 52) and identified clusters representing each type (Table S1 (56)). This allows comparisons with populations described previously (44, 51, 52). However, as can be seen in Fig. 5, the lactotrope signature identifies a more numerous group of cells than expected, suggesting a broader expression of PRL transcripts including subsets of progenitor cells. We did not detect a separate group of multihormonal cells in these populations; however, we did detect expression of Gh mRNA in lactotropes as well as Prl mRNA in cells belonging to the somatotrope cluster. This agrees with our previous studies of purified somatotropes (by FACS) (46), which reported significant Prl mRNA and protein expression in the purified population (which were identified by immunolabeling as 99% GH-bearing somatotropes).
Analysis of transcriptomics in the female pituitary (15 weeks HFD) shows evidence of mitochondrial dysfunction and oxidative stress in lactotrope and stem cell clusters. This correlates with studies of other organs exposed to the oxidative stress of obesity (2‐4, 80, 81). The pathology includes inflammation (69, 82‐84), mitochondrial dysfunction (2, 85‐88), and apoptosis (89, 90), all of which can compromise the function of cells, tissues, and organs. The inflammatory response associated with a HFD begins in adipose tissue as adipocytes undergo hypertrophy and hyperplasia and increase storage of fatty acids (82). The overproduction of proinflammatory mediators (82, 83) lead to a low-grade chronic inflammation that contributes to the onset and progression of metabolic disease (91). With respect to mitochondrial dysfunction, oxidative stress also results from the inflammation (92) and production of reactive oxygen species (3, 80, 83, 93‐95). The buildup of free fatty acids both reduces mitochondrial adenosine triphosphate production (2, 85‐87, 96‐99) and compromises the capacity of the ER to synthesize, mature, fold, and transport proteins (100, 101) resulting in ER stress (100‐104). Transcriptomic evidence in female lactotrope and stem cell clusters indicates that cells with these gene signatures are most vulnerable to oxidative stress.
Resilience of male pituitary cells
Our analysis of the impact of the HFD on the transcriptome of pituitary hormone–bearing clusters in males showed an unexpected resilience to the toxic impact of a HFD, despite a weight gain, a rise in inflammatory markers similar to that of HFD females, lower adiponectin, and clear evidence of insulin insensitivity (high insulin and delayed glucose tolerance). In addition, the Il-6 and insulin rise in males was much higher than that of females. It is possible that the higher levels of TSH or the lack of reduction in GH could have selectively protected the anterior pituitary cells in the male by promoting production of antioxidant defense pathways. In addition, TSH is known to regulate chaperone and unfolding enzyme production in thyroid follicular cells, thereby preventing the unfolded protein stress response (105).
The transcriptomics data from both 10 and 15-week HFD male groups indicate that the pituitary stem cell cluster is the most reactive cell population in the male mouse pituitary and exhibited DEGs related to the biological processes of immune response and defense. Notably, the interferon gamma–induced GTPase (Igtp) message was reduced in the stem cell cluster from male pituitaries after 10 weeks on the HFD. This gene encodes a protein that is expected to downregulate the activity of PKR-Like Endoplasmic Reticulum Kinase (PERK), which is a key regulator in the unfolded protein response (UPR) to ER stress (102‐104). Although we did not detect changes in the gene that encodes PERK (eukaryotic translation initiation factor 2 alpha kinase 3 [Eifak3]), the reduction in Igtp observed in HFD males might serve as an adaptive response that ultimately prevented PERK downregulation.
As stated above, the relative resilience of the male pituitary cells to a HFD was unexpected. In addition to the higher TSH levels, it is possible that this resilience may reflect the FVB.129P hybrid strain itself. We also suggest that the thermoneutral environment may have reduced the allostatic load allowing the pituitary cells in the male to defend against the metabolic stress and inflammation associated with obesity and maintain homeostasis. However, this response was not seen in the female pituitary cells.
Female pituitary cell populations vulnerable to oxidative stress
Our scRNA-seq analyses are the first to report that a relatively short-term HFD impacts the pituitary transcriptome in the female in specific clusters. The study of the female pituitary transcriptome showed that the group exposed to a HFD for 10 weeks had very modest changes, mostly in lactotropes or stem cells. However, the results for the 15-week group showed that the extra 5 weeks of metabolic stress greatly affected the transcriptome, specifically within both lactotrope and stem cell population clusters. Whereas the changes that we identified in females on a HFD for 15 weeks primarily relate to fundamental processes that support protein translation and mitochondrial function, signature pituitary-specific transcripts, such as Prl in lactotropes and S100 in folliculostellate (stem) cells were also affected by 15 week HFD.
A GO analysis of DEGs that met the most stringent threshold showed that processes associated with protein synthesis were most affected by the HFD (Figs. 6 and 7). In many tissues, cellular ER is perturbed by the obese state (103) resulting in an imbalance between the load of unfolded proteins and the capacity of the cellular machinery to handle this load (100‐104), resulting in ER stress. An ER stress adaptive response is to reduce protein synthesis, which we observe evidence for in the lactotrope and stem cell populations in our study. A secondary adaptive response, the UPR involves the transcriptional activation of target genes, involved in the synthesis of molecular chaperones that support protein folding and re-establish homeostasis (102), and, ultimately, if the stress is not relieved, cell death can be initiated (102). Our data showing reduced expression of genes encoding ribosomal proteins in the lactotrope and stem cell clusters from the HFD-fed females suggests that the 2 clusters in our obese mouse model may have begun to adapt to ER stress by reducing protein synthesis. However, we did not find evidence that the ER has activated target genes in the UPR pathway, nor was this pathway detected in any of our GO or ingenuity pathway analysis analyses. Studies by other groups show that diet induced obesity (at room temperature) will eventually trigger the UPR pathway in male mice (100, 101) or after 27 weeks on the HFD in male rats (11). At the 10- and 15-week HFD timepoints measured in our study, we did not detect UPR upregulation, suggesting that the difference in sex and/or strain and/or length of time on HFD allowed our female pituitaries to remain resilient to this toxic effect of the HFD. As with the males, it is also possible that the thermoneutral environment reduced the allostatic load and allowed the cells to defend against oxidative stress. A longer period on the diet may show activation of UPR pathways.
The initial analysis of DEGs that changed by more than 1.5-fold pointed to reductions in genes encoding subunits of the respiratory chain and we expanded the analysis in to include all DEGs that met the significance threshold without a fold change cutoff (Tables S7-S12 (56)). Using this relaxed threshold, both lactotrope and stem cell clusters showed downregulation of the canonical oxidative phosphorylation pathways, with a corresponding increase in pathways that signaled mitochondrial dysfunction. This result is consistent with studies showing the negative impact of oxidative stress on mitochondrial function (2).
To summarize, our findings indicate that, in females, the populations which appear most vulnerable to oxidative stress are those that bear a lactotrope or stem cell transcriptome signature. We show that these 2 populations are responding to increased time under the metabolic stress of obesity as we do not see these changes after just 10 weeks on the HFD, although the mice are obese at 10 weeks.
The remaining pituitary cell types in the female and most cell types in the male appear resilient even at the 15 week time point and do not show overt responses to the oxidative stress at the transcript level. Other studies suggesting broader changes in the pituitary transcriptome after 15 weeks on HFD at room temperature (10, 22, 30) may reflect strain differences, and/or the increased allostatic load of thermal stress. Future studies will determine if a prolonged exposure to the metabolic stress of obesity under thermoneutral conditions will exacerbate the impact of obesity on the pituitary.
Acknowledgments
The authors appreciate the work of Donald J. Johann, MD, MSc, FACP, and Ping Li, PhD, in the Genomics Core for running the scRNA-seq applications. We also thank Dr. Jordan Bird for Bioinformatics Analyses.
Abbreviations
- ACTH
adrenocorticotropin
- ANOVA
analysis of variance
- DEG
differentially expressed gene
- EE
respiratory quotient
- ER
endoplasmic reticulum
- FSH
follicle-stimulating hormone
- GH
growth hormone
- GO
Gene Ontology
- HFD
high-fat diet
- IL
interleukin
- IQR
interquartile range
- LH
luteinizing hormone
- MCP
monocyte chemoattractant protein
- PAI
plasminogen activator inhibitor
- PRL
prolactin
- RQ
respiratory quotient
- TNF
tumor necrosis factor
- TSH
thyroid-stimulating hormone
- UMAP
Uniform Manifold Approximation and Projection
- UPR
unfolded protein response
Contributor Information
Tiffany K Miles, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Angela K Odle, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Stephanie D Byrum, Department of Biochemistry and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA; Department of Biomedical informatics, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA; Arkansas Children's Research Institute, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Alex Lagasse, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Anessa Haney, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Victoria G Ortega, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Cole R Bolen, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Jewel Banik, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Milla M Reddick, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Ashley Herdman, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Melanie C MacNicol, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Angus M MacNicol, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Gwen V Childs, Department of Neurobiology and Developmental Sciences, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Funding
Sturgis Foundation; UAMS Development Enhancement Award; National Institutes of Health (NIH) 5 R01 DK127723; NIH R01 DK 113776; NIH R01 HD 093461; Center for Translational Pediatric Research funded under the NIH/National Institute of General Medical Sciences (NIH/NIGMS) P20GM121293, National Science Foundation Award No. OIA-1946391, and the UAMS Winthrop P. Rockefeller Cancer Institute.
Disclosures
The authors have nothing to disclose with respect to conflict of interest or relevant business interests except that Dr. Childs is a member of the Endocrinology Editorial board.
Data Availability
The scRNAseq data described in this study have been deposited in the Gene Expression Omnibus under accession number GSE244595. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE244595;!!LFqOYw!tMtqSxyj2rqniozi97aZ7hRL0nF_fl3AH0FnOIkPMORfEaWCeAQlROvFLWwmm9P8xatuQBmSJwW3vmnHPplQ$
References
- 1. World Health Organization Fact Sheet. 06/09/2021. https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&cad=rja&uact=8&ved=2ahUKEwjo--3r67WDAxVVkyYFHTxVCtgQFnoECBMQAQ&url=https%3A%2F%2Fwww.who.int%2Fnews-room%2Ffact-sheets%2Fdetail%2Fobesity-and-overweight&usg=AOvVaw1bNnwAfyzAvwzQsAga1V6x&opi=89978449
- 2. de Mello AH, Costa AB, Engel JDG, Rezin GT. Mitochondrial dysfunction in obesity. Life Sci. 2018;192:26‐32. [DOI] [PubMed] [Google Scholar]
- 3. Marseglia L, Manti S, D'Angelo G, et al. Oxidative stress in obesity: a critical component in human diseases. Int J Mol Sci. 2014;16(1):378‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knight JA. Diseases and disorders associated with excess body weight. Ann Clin Lab Sci. 2011;41(2):107‐121. [PubMed] [Google Scholar]
- 5. Sinha RA, Singh BK, Yen PM. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat Rev Endocrinol. 2018;14(5):259‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takahashi Y. Essential roles of growth hormone (GH) and insulin-like growth factor-I (IGF-I) in the liver. Endocr J. 2012;59(11):955‐962. [DOI] [PubMed] [Google Scholar]
- 7. Poddar M, Chetty Y, Chetty VT. How does obesity affect the endocrine system? A narrative review. Clin Obes. 2017;7(3):136‐144. [DOI] [PubMed] [Google Scholar]
- 8. Pijl H, Langendonk JG, Burggraaf J, et al. Altered neuroregulation of GH secretion in viscerally obese premenopausal women. J Clin Endocrinol Metab. 2001;86(11):5509‐5515. [DOI] [PubMed] [Google Scholar]
- 9. Qu XD, Gaw Gonzalo IT, Al Sayed MY, et al. Influence of body mass index and gender on growth hormone (GH) responses to GH-releasing hormone plus arginine and insulin tolerance tests. J Clin Endocrinol Metab. 2005;90(3):1563‐1569. [DOI] [PubMed] [Google Scholar]
- 10. Ruiz S, Vázquez F, Pellitero S, Puig-Domingo M. ENDOCRINE OBESITY: pituitary dysfunction in obesity. Eur J Endocrinol. 2022;186(6):R79‐R92. [DOI] [PubMed] [Google Scholar]
- 11. Gong Y, Yang J, Wei S, et al. Lipotoxicity suppresses the synthesis of growth hormone in pituitary somatotrophs via endoplasmic reticulum stress. J Cell Mol Med. 2021;25(11):5250‐5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mercau ME, Repetto EM, Perez MN, et al. Moderate exercise prevents functional remodeling of the anterior pituitary gland in diet-induced insulin resistance in rats: role of oxidative stress and autophagy. Endocrinology. 2016;157(3):1135‐1145. [DOI] [PubMed] [Google Scholar]
- 13. Bonert VS, Elashoff JD, Barnett P, Melmed S. Body mass index determines evoked growth hormone (GH) responsiveness in normal healthy male subjects: diagnostic caveat for adult GH deficiency. J Clin Endocrinol Metab. 2004;89(7):3397‐3401. [DOI] [PubMed] [Google Scholar]
- 14. De Marinis L, Bianchi A, Mancini A, et al. Growth hormone secretion and leptin in morbid obesity before and after biliopancreatic diversion: relationships with insulin and body composition. J Clin Endocrinol Metab. 2004;89(1):174‐180. [DOI] [PubMed] [Google Scholar]
- 15. Rasmussen MH, Hvidberg A, Juul A, et al. Massive weight loss restores 24-hour growth hormone release profiles and serum insulin-like growth factor-I levels in obese subjects. J Clin Endocrinol Metab. 1995;80(4):1407‐1415. [DOI] [PubMed] [Google Scholar]
- 16. Rasmussen MH, Juul A, Kjems LL, Skakkebaek NE, Hilsted J. Lack of stimulation of 24-hour growth hormone release by hypocaloric diet in obesity. J Clin Endocrinol Metab. 1995;80(3):796‐801. [DOI] [PubMed] [Google Scholar]
- 17. Vahl N, Jorgensen JO, Skjaerbaek C, Veldhuis JD, Orskov H, Christiansen JS. Abdominal adiposity rather than age and sex predicts mass and regularity of GH secretion in healthy adults. Am J Physiol. 1997;272(6 Pt 1):E1108‐E1116. [DOI] [PubMed] [Google Scholar]
- 18. Williams T, Berelowitz M, Joffe SN, et al. Impaired growth hormone responses to growth hormone-releasing factor in obesity. A pituitary defect reversed with weight reduction. N Engl J Med. 1984;311(22):1403‐1407. [DOI] [PubMed] [Google Scholar]
- 19. Ahmad I, Steggles AW, Carrillo AJ, Finkelstein JA. Obesity- and sex-related alterations in growth hormone messenger RNA levels. Mol Cell Endocrinol. 1989;65(1-2):103‐109. [DOI] [PubMed] [Google Scholar]
- 20. Dubey AK, Hanukoglu A, Hansen BC, Kowarski AA. Metabolic clearance rates of synthetic human growth hormone in lean and obese male rhesus monkeys. J Clin Endocrinol Metab. 1988;67(5):1064‐1067. [DOI] [PubMed] [Google Scholar]
- 21. Zhou X, De Schepper J, De Craemer D, et al. Pituitary growth hormone release and gene expression in cafeteria-diet-induced obese rats. J Endocrinol. 1998;159(1):165‐172. [DOI] [PubMed] [Google Scholar]
- 22. Luque RM, Kineman RD. Impact of obesity on the growth hormone axis: evidence for a direct inhibitory effect of hyperinsulinemia on pituitary function. Endocrinology. 2006;147(6):2754‐2763. [DOI] [PubMed] [Google Scholar]
- 23. Akhter N, Odle AK, Allensworth-James ML, et al. Ablation of leptin signaling to somatotropes: changes in metabolic factors that cause obesity. Endocrinology. 2012;153(10):4705‐4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Allensworth-James M, Odle A, Haney A, Childs GV. Sex differences in somatotrope dependency on leptin receptors in young mice: ablation of LEPR causes severe growth hormone deficiency and abdominal obesity in males. Endocrinology. 2015;156(9):3253‐3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Childs GV, Akhter N, Haney A, et al. The somatotrope as a metabolic sensor: deletion of leptin receptors causes obesity. Endocrinology. 2011;152(1):69‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miles TK, Silva Moreira AR, Allensworth-James ML, et al. Sex differences in somatotrope response to fasting: biphasic responses in male mice. J Endocrinol. 2020;247(3):213‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Crane C, Akhter N, Johnson BW, et al. Fasting and glucose effects on pituitary leptin expression: is leptin a local signal for nutrient status? J Histochem Cytochem. 2007;55(10):1059‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Syed M, Cozart M, Haney AC, et al. Ghrelin restoration of function in vitro in somatotropes from male mice lacking the Janus kinase (JAK)-binding site of the leptin receptor. Endocrinology. 2013;154(4):1565‐1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marques C, Meireles M, Norberto S, et al. High-fat diet-induced obesity rat model: a comparison between wistar and sprague-dawley rat. Adipocyte. 2016;5(1):11‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ruggiero-Ruff RE, Le B, Lainez N, Coss D. Single-Cell Transcriptomics identifies pituitary gland cell type population changes in diet induced obesity. J Endocr Soc. 2023;7(Supplement_1). 10.1210/jendso/bvad114.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tortoriello DV, McMinn J, Chua SC. Dietary-induced obesity and hypothalamic infertility in female DBA/2J mice. Endocrinology. 2004;145(3):1238‐1247. [DOI] [PubMed] [Google Scholar]
- 32. Zhou S, Tang X, Chen HZ. Sirtuins and insulin resistance. Front Endocrinol (Lausanne). 2018;9:748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Benegiamo G, von Alvensleben GVG, Rodríguez-Lopez S, et al. The genetic background shapes the susceptibility to mitochondrial dysfunction and NASH progression. J Exp Med. 2023;220(4):e20221738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dieckmann S, Strohmeyer A, Willershäuser M, et al. Susceptibility to diet-induced obesity at thermoneutral conditions is independent of UCP1. Am J Physiol Endocrinol Metab. 2022;322(2):E85‐E100. [DOI] [PubMed] [Google Scholar]
- 35. Ganeshan K, Chawla A. Warming the mouse to model human diseases. Nat Rev Endocrinol. 2017;13(8):458‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ginting RP, Lee JM, Lee MW. The influence of ambient temperature on adipose tissue homeostasis, metabolic diseases and cancers. Cells. 2023;12(6):881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hankenson FC, Marx JO, Gordon CJ, David JM. Effects of rodent thermoregulation on animal models in the research environment. Comp Med. 2018;68(6):425‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. James CM, Olejniczak SH, Repasky EA. How murine models of human disease and immunity are influenced by housing temperature and mild thermal stress. Temperature (Austin). 2023;10(2):166‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. John LM, Petersen N, Gerstenberg MK, et al. Housing-temperature reveals energy intake counter-balances energy expenditure in normal-weight, but not diet-induced obese, male mice. Commun Biol. 2022;5(1):946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oates JR, Sawada K, Giles DA, et al. Thermoneutral housing shapes hepatic inflammation and damage in mouse models of non-alcoholic fatty liver disease. Front Immunol. 2023;14:1095132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reitman ML. Of mice and men—environmental temperature, body temperature, and treatment of obesity. FEBS Lett. 2018;592(12):2098‐2107. [DOI] [PubMed] [Google Scholar]
- 42. Škop V, Guo J, Liu N, et al. Mouse thermoregulation: introducing the concept of the thermoneutral point. Cell Rep. 2020;31(2):107501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Speakman JR, Keijer J. Not so hot: optimal housing temperatures for mice to mimic the thermal environment of humans. Mol Metab. 2012;2(1):5‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Allensworth-James M, Banik J, Odle A, et al. Control of the anterior pituitary cell lineage regulator POU1F1 by the stem cell determinant musashi. Endocrinology. 2021;162(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Allensworth-James ML, Odle AK, Lim J, et al. Metabolic signalling to somatotrophs: transcriptional and post-transcriptional mediators. J Neuroendocrinol. 2020;32(11):e12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Odle AK, Allensworth-James ML, Akhter N, et al. A sex-dependent tropic role for leptin in the somatotrope as A regulator of POU1F1 and POU1F1-dependent hormones. Endocrinology. 2016;157(10):3958‐3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Odle AK, Beneš H, Melgar Castillo A, et al. Association of gnrhr mRNA with the stem cell determinant musashi: a mechanism for leptin-mediated modulation of GnRHR expression. Endocrinology. 2018;159(2):883‐894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-Seq aligner. Bioinformatics. 2013;29(1):15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zheng GXY, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8(1):14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cheung LYM, George AS, McGee SR, et al. Single-Cell RNA sequencing reveals novel markers of male pituitary stem cells and hormone-producing cell types. Endocrinology. 2018;159(12):3910‐3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ho Y, Hu P, Peel MT, et al. Single-cell transcriptomic analysis of adult mouse pituitary reveals sexual dimorphism and physiologic demand-induced cellular plasticity. Protein Cell. 2020. 11:565‐583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Franzen O, Gan LM, Björkegren JLM. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford). 2019;2019:baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Finak G, McDavid A, Yajima M, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16(1):278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Odle AK, Akhter N, Syed MM, et al. Leptin regulation of gonadotrope gonadotropin-releasing hormone receptors as a metabolic checkpoint and gateway to reproductive competence. Front Endocrinol (Lausanne). 2017;8:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Miles TK. Data from: Anterior Pituitary Transcriptomics Following a High Fat Diet: Impact of oxidative stress on cell metabolism. Supplemental data. doi: 10.5061/dryad.98sf7m0q8, https://datadryad.org/stash/share/b7TECr4uB2pQv6VjJ7VlsOscogRSg0sm1VX6k0w9bsA. [DOI] [PMC free article] [PubMed]
- 57. Nicol L, McNeilly JR, Stridsberg M, Crawford JL, McNeilly AS. Influence of steroids and GnRH on biosynthesis and secretion of secretogranin II and chromogranin A in relation to LH release in LbetaT2 gonadotroph cells. J Endocrinol. 2002;174(3):473‐483. [DOI] [PubMed] [Google Scholar]
- 58. Li Y, Zhou Y, Wang F, et al. SIRT4 is the last puzzle of mitochondrial sirtuins. Bioorg Med Chem. 2018;26(14):3861‐3865. [DOI] [PubMed] [Google Scholar]
- 59. Yang Y, Smith DL Jr, Keating KD, Allison DB, Nagy TR. Variations in body weight, food intake and body composition after long-term high-fat diet feeding in C57BL/6J mice. Obesity (Silver Spring). 2014; 22(10):2147‐2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Frederich RC, Hamann A, Anderson S, Löllmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1(12):1311‐1314. [DOI] [PubMed] [Google Scholar]
- 61. MacDougald OA, Hwang CS, Fan H, Lane MD. Regulated expression of the obese gene product (leptin) in white adipose tissue and 3T3-L1 adipocytes. Proc Natl Acad Sci U S A. 1995;92(20):9034‐9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Maffei M, Halaas J, Ravussin E, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1(11):1155‐1161. [DOI] [PubMed] [Google Scholar]
- 63. Ouchi N, Walsh K. Adiponectin as an anti-inflammatory factor. Clin Chim Acta. 2007;380(1-2):24‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shibata R, Izumiya Y, Sato K, et al. Adiponectin protects against the development of systolic dysfunction following myocardial infarction. J Mol Cell Cardiol. 2007;42(6):1065‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Takemura Y, Ouchi N, Shibata R, et al. Adiponectin modulates inflammatory reactions via calreticulin receptor-dependent clearance of early apoptotic bodies. J Clin Invest. 2007;117(2):375‐386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Arita Y, Kihara S, Ouchi N, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257(1):79‐83. [DOI] [PubMed] [Google Scholar]
- 67. Bruun JM, Lihn AS, Verdich C, et al. Regulation of adiponectin by adipose tissue-derived cytokines: in vivo and in vitro investigations in humans. Am J Physiol Endocrinol Metab. 2003;285(3):E527‐E533. [DOI] [PubMed] [Google Scholar]
- 68. Matsuzawa Y. Adiponectin: a key player in obesity related disorders. Curr Pharm Des. 2010;16(17):1896‐1901. [DOI] [PubMed] [Google Scholar]
- 69. Maury E, Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol. 2010;314(1):1‐16. [DOI] [PubMed] [Google Scholar]
- 70. Steppan CM, Bailey ST, Bhat S, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409(6818):307‐312. [DOI] [PubMed] [Google Scholar]
- 71. Steppan CM, Lazar MA. Resistin and obesity-associated insulin resistance. Trends Endocrinol Metab. 2002;13(1):18‐23. [DOI] [PubMed] [Google Scholar]
- 72. Azuma K, Katsukawa F, Oguchi S, et al. Correlation between serum resistin level and adiposity in obese individuals. Obes Res. 2003;11(8):997‐1001. [DOI] [PubMed] [Google Scholar]
- 73. Rodríguez-Pacheco F, Vázquez-Martínez R, Martínez-Fuentes AJ, et al. Resistin regulates pituitary somatotrope cell function through the activation of multiple signaling pathways. Endocrinology. 2009;150(10):4643‐4652. [DOI] [PubMed] [Google Scholar]
- 74. Sarmento-Cabral A, Peinado JR, Halliday LC, et al. Adipokines (leptin, adiponectin, resistin) differentially regulate all hormonal cell types in primary anterior pituitary cell cultures from two primate Species. Sci Rep. 2017;7(1):43537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci U S A. 2003;100(12):7265‐7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Michalaki MA, Vagenakis AG, Leonardou AS, et al. Thyroid function in humans with morbid obesity. Thyroid. 2006;16(1):73‐78. [DOI] [PubMed] [Google Scholar]
- 77. Nillni EA, Vaslet C, Harris M, Hollenberg A, Bjørbak C, Flier JS. Leptin regulates prothyrotropin-releasing hormone biosynthesis. Evidence for direct and indirect pathways. J Biol Chem. 2000;275(46):36124‐36133. [DOI] [PubMed] [Google Scholar]
- 78. Flier JS, Harris M, Hollenberg AN. Leptin, nutrition, and the thyroid: the why, the wherefore, and the wiring. J Clin Invest. 2000;105(7):859‐861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pujanek M, Bronisz A, Malecki P, Junik R. Pathomechanisms of the development of obesity in some endocrinopathies—an overview. Endokrynol Pol. 2013;64(2):150‐155. [PubMed] [Google Scholar]
- 80. Matsuda M, Shimomura I. Increased oxidative stress in obesity: implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes Res Clin Pract. 2013;7(5):e330‐e341. [DOI] [PubMed] [Google Scholar]
- 81. Volkan G, Abdulsamed K, Emin Ş, Hacı Ahmet D. Leptin and its role in oxidative stress and apoptosis: an overview. In: Venketeshwer R, Leticia R, eds. Role of Obesity in Human Health and Disease. IntechOpen; 2021:Ch. 9. [Google Scholar]
- 82. Galic S, Oakhill JS, Steinberg GR. Adipose tissue as an endocrine organ. Mol Cell Endocrinol. 2010;316(2):129‐139. [DOI] [PubMed] [Google Scholar]
- 83. Johnson AR, Milner JJ, Makowski L. The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev. 2012;249(1):218‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mraz M, Haluzik M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J Endocrinol. 2014;222(3):R113‐R127. [DOI] [PubMed] [Google Scholar]
- 85. Bournat JC, Brown CW. Mitochondrial dysfunction in obesity. Curr Opin Endocrinol Diabetes Obes. 2010;17(5):446‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Heinonen S, Buzkova J, Muniandy M, et al. Impaired mitochondrial biogenesis in adipose tissue in acquired obesity. Diabetes. 2015;64(9):3135‐3145. [DOI] [PubMed] [Google Scholar]
- 87. Putti R, Sica R, Migliaccio V, Lionetti L. Diet impact on mitochondrial bioenergetics and dynamics. Front Physiol. 2015;6:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schneeberger M, Dietrich MO, Sebastian D, et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. 2013;155(1):172‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pintus F, Floris G, Rufini A. Nutrient availability links mitochondria, apoptosis, and obesity. Aging (Albany NY). 2012;4(11):734‐741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tinahones FJ, Coin Aragüez L, Murri M, et al. Caspase induction and BCL2 inhibition in human adipose tissue: a potential relationship with insulin signaling alteration. Diabetes Care. 2013;36(3):513‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11(2):85‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5(1):9‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bondia-Pons I, Ryan L, Martinez JA. Oxidative stress and inflammation interactions in human obesity. J Physiol Biochem. 2012;68(4):701‐711. [DOI] [PubMed] [Google Scholar]
- 94. Fernandez-Sanchez A, Madrigal-Santillan E, Bautista M, et al. Inflammation, oxidative stress, and obesity. Int J Mol Sci. 2011;12(5):3117‐3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Roberts CK, Sindhu KK. Oxidative stress and metabolic syndrome. Life Sci. 2009;84(21-22):705‐712. [DOI] [PubMed] [Google Scholar]
- 96. Yin X, Lanza IR, Swain JM, Sarr MG, Nair KS, Jensen MD. Adipocyte mitochondrial function is reduced in human obesity independent of fat cell size. J Clin Endocrinol Metab. 2014;99(2):E209‐E216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zorzano A, Liesa M, Palacin M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol. 2009;41(10):1846‐1854. [DOI] [PubMed] [Google Scholar]
- 98. Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435(2):297‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kusminski CM, Scherer PE. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol Metab. 2012;23(9):435‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ozcan L, Ergin AS, Lu A. et al. et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009; 9(1):35‐51. [DOI] [PubMed] [Google Scholar]
- 101. Won JC, Jang PG, Namkoong C, et al. Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity (Silver Spring). 2009;17(10):1861‐1865. [DOI] [PubMed] [Google Scholar]
- 102. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519‐529. [DOI] [PubMed] [Google Scholar]
- 103. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135(1):61‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Christis C, Fullaondo A, Schildknegt D, Mkrtchian S, Heck AJR, Braakman I. Regulated increase in folding capacity prevents unfolded protein stress in the ER. J Cell Sci. 2010;123(Pt 5):787‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The scRNAseq data described in this study have been deposited in the Gene Expression Omnibus under accession number GSE244595. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE244595;!!LFqOYw!tMtqSxyj2rqniozi97aZ7hRL0nF_fl3AH0FnOIkPMORfEaWCeAQlROvFLWwmm9P8xatuQBmSJwW3vmnHPplQ$