

Abstract
The Nrf2 transcription factor is a master regulator of the cellular response to oxidative stress, and Keap1 is its primary negative regulator. Activating Nrf2 by inhibiting the Nrf2–Keap1 protein–protein interaction has shown promise for treating cancer and inflammatory diseases. A loop derived from Nrf2 has been shown to inhibit Keap1 selectively, especially when cyclized, but there are no reliable design methods for predicting an optimal macrocyclization strategy. In this work, we employed all-atom, explicit-solvent molecular dynamics simulations with enhanced sampling methods to predict the relative degree of preorganization for a series of peptides cyclized with a set of bis-thioether “staples”. We then correlated these predictions to experimentally measured binding affinities for Keap1 and crystal structures of the cyclic peptides bound to Keap1. This work showcases a computational method for designing cyclic peptides by simulating and comparing their entire solution-phase ensembles, providing key insights into designing cyclic peptides as selective inhibitors of protein–protein interactions.
Graphical Abstract
INTRODUCTION
Cyclic peptides (CPs) are a privileged chemical space in drug design. Their ability to position functional groups into a three-dimensional (3D) binding epitope allows them to modulate protein–protein interactions (PPIs) with high affinity and specificity.1–3 CP natural products and artificial CP libraries are common sources for new PPI inhibitors, yielding exciting new clinical candidates.4 While these approaches can be successful, computational design approaches would greatly accelerate the development of CP drugs. Baker and colleagues have reported methods for designing well-structured head-to-tail CPs and recently extended their methodology to designing CPs with greater passive membrane permeability.5,6 However, these efforts have been limited to well-structured head-to-tail CPs with natural amino acids and their enantiomers, and they have not yet succeeded in the routine design of PPI inhibitors. As an alternative to head-to-tail cyclization, cysteine alkylation reactions allow for improved rational design and library development for CP discovery,7–11 and they can induce a variety of secondary structures in peptides to improve affinity and specificity.12 For example, cross-linking using cysteine alkylation increased α-helicity, bioactivity, and cell permeability for a series of BH3 peptide-derived MCL-1 ligands.13,14 The Kritzer group has also used cysteine alkylation with ortho-, meta-, and para-dibromomethylbenzene linkers, originally described by Timmerman et al.,15 for “diversity-oriented stapling” in the development of helical and nonhelical PPI inhibitors.10,12,15 However, to date, such strategies have rarely been performed without computational methods to guide them. It remains impossible to predict computationally which non-natural linker will best stabilize a desired CP conformation, particularly for nonhelical binding epitopes.
Indeed, accurate prediction of structural ensembles is one of the major challenges in designing CPs as PPI inhibitors. NMR data indicate that even relatively structured CPs interconvert among several conformations in solution.16,17 Thus, beyond just predicting a single low-energy structure, understanding the full structural ensembles is essential for the accurate design of structured CPs. The primary difficulty in simulating CP structural ensembles is in ensuring sufficient sampling of the conformational space because CPs have a uniquely rugged energy landscape.18,19 In recent years, the Lin group has applied molecular dynamics (MD) methods with enhanced sampling to simulate conformational ensembles of head-to-tail CPs.20–22 The work presented here expands this method to CPs with several linkers produced by alkylating cysteines. Also, for the first time, we directly applied these methods to the design of a PPI inhibitor.
Here, we apply these expanded methods to the PPI between nuclear factor-erythroid 2 p45-related factor 2 (Nrf2) and Kelch-like ECH-associated protein-1 (Keap1).23–26 Nrf2 is a transcription factor that binds to the antioxidant response element enhancer sequence and activates more than 200 genes that encode for cytoprotective proteins, which attenuate cellular injury from oxidative stress.23 When no chemical stress is present, Nrf2 is bound to Keap1 in the cytosol. However, oxidative stress induces intramolecular and intermolecular disulfide formation for cysteine-rich Keap1, releasing Nrf2 from Keap1 binding. Nrf2 then translocates to the nucleus and activates the antioxidant response.25,27,28 The Nrf2 activator dimethyl fumarate is used clinically to treat psoriasis and relapsing remitting multiple sclerosis,29 and sulforaphane and bardoxolone methyl are Nrf2 activators currently going through stages of clinical trials for kidney failure and for rheumatoid arthritis.30–32 Nrf2 activators have also been tested as potential therapies for other pathologies including cancer, cardiovascular, respiratory, renal, digestive, metabolic, auto-immune, and neurodegenerative diseases.33,34 Despite their wide use, these Nrf2 activators have limited target specificity because they are electrophilic compounds that react with the redox-sensitive cysteines in Keap1. Therefore, these compounds can also react with cysteines in other proteins, producing off-target effects.26 Inhibitors that can selectively target the Nrf2–Keap1 PPI, rather than targeting redox-sensitive cysteines, could lead to improved treatments with fewer side effects.
Previous studies have used Nrf2–Keap1 as a testing ground for designing CPs and have developed CP inhibitors with nanomolar affinity for Keap1. Begnini et al. were able to improve the potency of a 4 μM macrocyclic, natural product-derived inhibitor to 30–70 nM by first using molecular docking to select smaller sets of compounds for synthesis and then performing iterative structure–activity relationship studies.35 A key loop of Nrf2 at its Keap1 binding interface, sequence 76LDEETGEFL84 with 79ETGE82 as the minimum binding sequence,24,36,37 was used by several groups to develop CP inhibitors of Keap1. Steel et al. used rational design to cyclize the native Nrf2 binding loop with a disulfide bridge or a perfluoroaryl linker.38 They also substituted Glu78 with proline. These changes resulted in a Ki improvement from 1400 to 6.1 nM for the Nrf2–Keap1 interaction. Recently, Whitty, Allen, and colleagues reported the design of CP inhibitors using data from X-ray crystal structures and machine learning, which exposed a trade-off between preorganization and strain.39 These computational methods were able to rationalize structure–activity relationships but not to predict high-affinity binders.
In this study, we sought to develop computational methods that could guide the design of CPs that mimic the bound conformation of the Keap1-binding loop of Nrf2, 77DEETGE82 (Figure 1A).24 Computational alanine scans show that Glu79, Thr80, and Glu82 make relevant contributions to binding with 4.4, 0.5, and 3.8 kcal/mol to the total binding ΔΔG, respectively.40–42 Alanine scanning mutagenesis measurements have estimated the interaction energy for Glu79, Thr80, and Glu82 to be 4.3, 2.0, and 3.0 kcal/mol.43 The amide proton in Gly81 forms an important intramolecular interaction to the backbone carbonyl of Asp77 and stabilizes the bound conformation (Figure 1A).36 Figure 1B shows the backbone torsional angles of DEETGE in its Keap1-bound conformation. These torsional angles represent the desired ETGE backbone conformations for our CP designs. A key assumption for our approach is that CPs with structural ensembles that preorganize the ETGE region toward these specific torsional angles will have better binding affinities for Keap1. Similar assumptions are common in medicinal chemistry and often lead to high-affinity ligands.3
Figure 1.
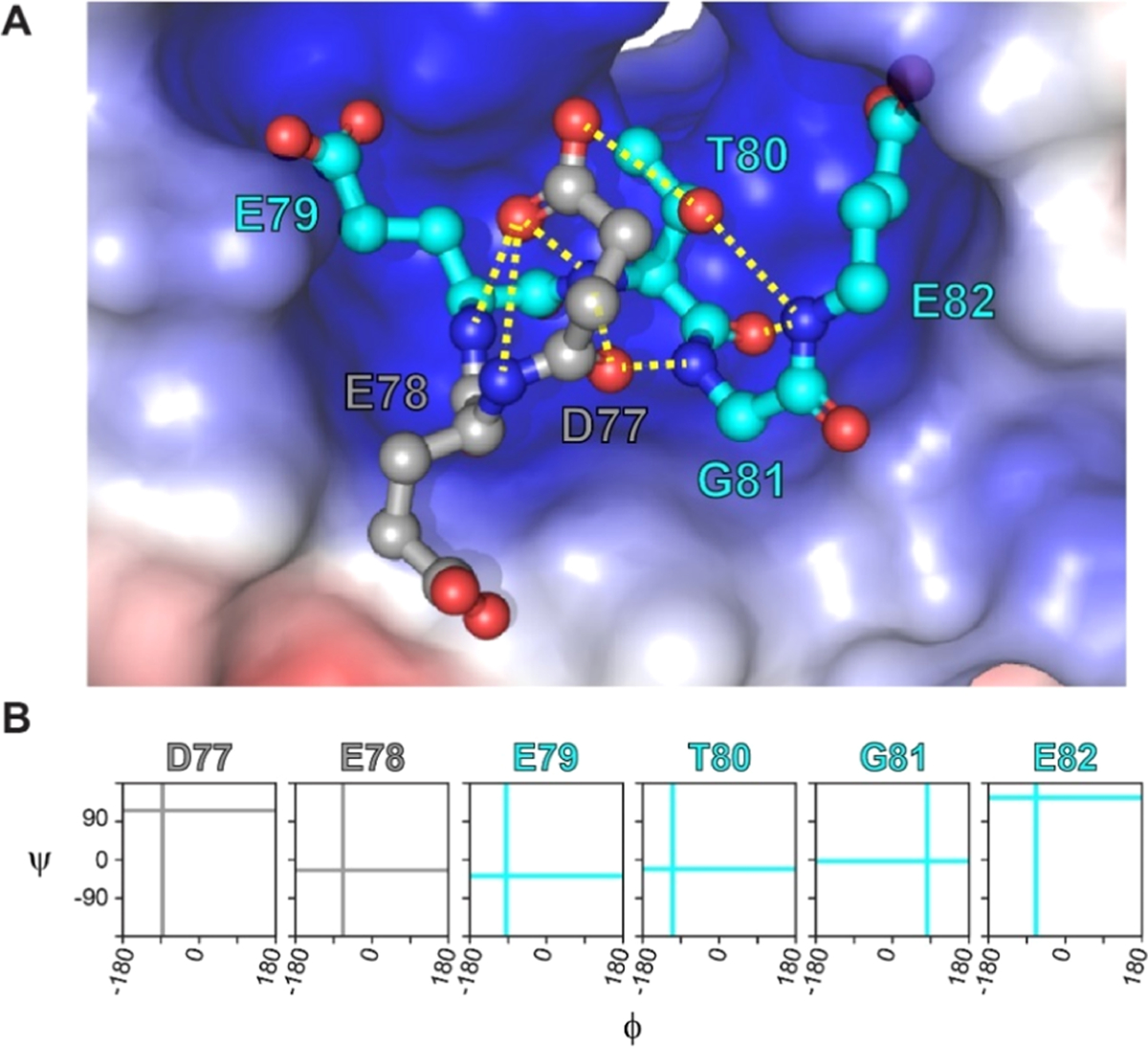
DEETGE loop of Nrf2 binds to Keap1. (A) Three-dimensional visualization of the DEETGE motif of Nrf2 (gray and cyan balls and sticks) occupying the binding pocket of Keap1 (shown in the electrostatic potential surface; PDB 2FLU).24 Residues D77 and E78 are shown in gray, residues E79 to E82 are shown in cyan, and key intramolecular hydrogen bonds are shown as yellow dotted lines. (B) Backbone torsional angles of the DEETGE epitope bound to Nrf2. Cyan lines indicate the (Φ, Ψ) values in the desired conformation for the residues in the core binding sequence ETGE. Gray lines indicate the (Φ, Ψ) values in the desired conformations for D77 and E78.
In this work, we applied all-atom, explicit-solvent MD simulations and enhanced sampling methods to nine designed peptide sequences containing the DEETGE epitope (or derivatives) with various linkers (Figure S1) to predict their relative degree of structural preorganization. We then performed experimental binding assays and X-ray crystallography to verify whether these predictions indeed correlated with the experimentally measured binding affinities and Keap1-bound structures. By employing this newly developed method, we were able to accurately predict high-affinity CP binders to Keap1.
METHOD
Bias-Exchange Metadynamics Molecular Dynamics Simulations.
To overcome the difficulty in conformational sampling of CPs, the Lin group developed a method that incorporates bias-exchange metadynamics (BE-META) simulations44–46,49 tailored to CPs, principal component analysis (PCA),47 and a modified density peak-based cluster analysis to provide a converged set of clusters that describes the entire conformational ensemble.48 Sampling was enhanced by identifying essential transitional motions that involved coupled two-dihedral changes that represent the slowest degrees of freedom for the peptide’s motions.20 As developed in our previous work, we performed BE-META simulations of the CP systems here with two-dimensional (2D) biases of (φi, ψi) and (ψi, φi+1) to enhance the coordinated torsional motions and help the simulations sample thermodynamically relevant conformations in our BE-META simulations. In preliminary simulations, we noticed that CPs were getting stuck in very few and consistent conformations, even when we biased the coordinated backbone torsional motions. This was not observed in prior work, which used head-to-tail cyclic peptides, so we hypothesized that adding replicas with biased torsional motions in the linkers was needed to promote more comprehensive sampling of the conformational landscape. To obtain the equilibrium structural ensembles for an unbiased data analysis, we added five neutral replicas, which were allowed to exchange conformations with other replicas but did not have any biases themselves.
For every simulated CP, two initial random conformations were built and two independent sets of BE-META simulations starting from these conformations were performed. We then performed dihedral principal component analysis47 (see below) and projected the structures in the 2D principal component space. Convergence was considered satisfactory once a normalized integrated product between the density distributions in the principal component space between the two independent trajectories was 0.9 or above.50,51 The simulation duration ranged from 100 to 500 ns, depending on when the duplicate simulations reached convergence.
The GROMACS 2018 package was used to perform all simulations.52 The RSFF2 force field and the TIP3P water model were used to model each CP.53,54 The RSFF2 force field was parametrized using a coil library of the 20 natural amino acids, intending to capture their intrinsic structural preferences.53 A generalized AMBER force field (GAFF) was used for the perfluoroaryl (pfl), ortho-dimethylbenzene (ortho), meta-dimethylbenzene (meta), and para-dimethylbenzene (para) linkers (Figure S1A–D).55 The Antechamber program in AmberTools was used to generate partial atomic charges for each linker (Figure S2) as described in the charge generating protocol described in the Supporting Information.56
Initial structures were solvated in a cubic box of pre-equilibrated water with a minimum distance between the peptide and the walls of the box set to 1.0 nm. The steepest descent algorithm was used to minimize the solvated structure. Upon minimization, the system was equilibrated in two stages: First, all peptide heavy atoms were position-restrained, and 50 ps of NVT simulation at 300 K and 50 ps of NPT simulation at 300 K and 1 bar were performed. In the second stage, the position restraints were removed, and the same sequence of NVT and NPT simulations was performed for a second time, this time for 100 ps each. BE-META production runs were then performed in the NPT ensemble, at 300 K and 1 bar, with a 2 fs time step, and data were recorded every 500 steps (every 1 ps). The last 100 ns of these BE-META production runs was used for data analysis. All simulations were run using the leapfrog algorithm, with water geometry maintained using SETTLE and hydrogen-containing bonds constrained to equilibrium lengths using LINCS. The nonbonded interaction cutoff was set to 1.0 nm, with coulombic interactions beyond the cutoff computed using particle mesh Ewald summation with a Fourier spacing of 0.12 nm and cubic interpolation. Dispersion corrections for both the energy and pressure were applied to the long-range van der Waals interactions. Temperature was controlled by velocity rescaling with a coupling time constant of 0.1 ps. A Parrinello–Rahman barostat was used for pressure control, with a coupling time constant of 2.0 ps and isothermal compressibility of 4.5 × 10−5 bar−1. Files describing how to run BE-META simulations of cyclic peptides can be found on GitHub (https://github.com/ysl-lab/CP_tutorial).
Analysis of Predicted Structural Ensembles.
Dihedral Principal Component Analysis and Cluster Analysis.
Dihedral principal component analysis, followed by cluster analysis, was performed on the neutral replicas in the BE-META simulations to characterize the structural ensemble of the CPs. Principal component analysis (PCA) is a well-established approach for dimensionality reduction without significant information loss. When attempting to account for essential dynamics of the system on a low-dimensional free energy landscape, internal coordinates such as backbone dihedral angles are favored over Cartesian coordinates since the latter tend to reflect the dominant overall motion rather than the much smaller internal motion of the protein.57 Therefore, to employ a sensitive metric to separate conformations for a CP owing to its circular nature, we used the dihedral angle principal component analysis (dPCA)57,58 with the Φ and Ψ angles involved in each CP. First, we calculated the backbone φ and ψ dihedral angles for the entire trajectories. We then performed a dihedral angle PCA (dPCA)57,58 utilizing the cosines and sines of the φ and ψ dihedral angles of all amino acids followed by a grid-based density peak-based clustering to quantify the population of each cluster.59 During the clustering, the 2D principal subspace along principal components 1 and 2 was first divided into 200 × 200 grids. Cluster analysis was performed only on the grid subsections with data populations larger than 0.1%. To describe the convergence behavior of each enhanced sampling simulation, we monitored the overlap of probability densities between the two independent sets of BE-META simulations as a function of the simulation time. The normalized integrated product of the population densities in the 2D principal subspace was obtained to test for convergence. Convergence was considered satisfactory once a normalized integrated product50 between the last 100 ns of the two independent trajectories was found to be 0.9 or above.,51 Scripts to perform dihedral principal component analysis can be found on GitHub (https://github.com/ysl-lab/CP_tutorial).
Mimicry Analysis.
To evaluate how well each CP preorganized the ETGE residues, for each frame sampled in a CP simulation, we first aligned the backbone atoms of the ETGE region of the CP to the backbone atoms of the ETGE region of Nrf2 bound to Keap1 (PDB ID: 2FLU)24 and computed a backbone root-mean-square deviation (RMSD) value for that frame. We then plotted the backbone RMSD distribution for the entire CP ensemble to evaluate how well each CP preorganized the ETGE residues into the desired structure. To further quantify the degree of mimicry for a CP, we calculated the % of frames that had backbone RMSD < 0.5 Å, which we call the “preorganized population” of that ensemble. We also calculated the average backbone RMSD for the preorganized population for each CP simulated.
Clash Analysis.
To analyze whether a CP conformation would result in steric clashes with Keap1, we first positioned the CP conformation within the Keap1 binding pocket by aligning the backbone atoms of the ETGE region of the CP to the backbone atoms of the ETGE region of Nrf2 bound to Keap1 (PDB ID: 2FLU).24 We then used the Chimera molecular modeling package60 to detect steric clashes, defined as the number of heavy atoms in the superimposed CP (excluding the side chain of the hotspot residues), which have overlapping van der Waals radii with atoms in the binding pocket of Keap1. No docking, relaxation, or energy minimization was performed on the positioned structures, leading to a relatively static, conservative clash analysis. The CP conformations that passed the mimicry analysis and the clash analysis thus represent the most ideal situation, i.e., the hotspot residues are preorganized, and no rearrangements of the linker are required for the preorganized hotspot resides to engage Keap1.
Wet-Lab Experiments.
Details on peptide synthesis, protein expression and purification, biolayer interferometry, protein expression and purification for crystallography, and X-ray crystallography can be found in the Supporting Information.
RESULTS
Cyclic Peptide Design and MD Simulations.
To benchmark our computational methodology, we started with control CPs for which there were published binding affinity data.38 The first control was pfl-CDEETGEC (denoted CP1), which contains the Nrf2-derived DEETGE flanked by cysteines, which are further cross-linked using a perfluoroarene (pfl) linker. CP1 was previously developed by Steel et al. and its Ki for the Nrf2–Keap1 protein–protein interaction was measured at 1400 nM using a competition fluorescence polarization assay. Steel et al. also designed an analogue, pfl-CDPETGEC (denoted CP2), replacing the glutamate corresponding to Nrf2 Glu78, which we will renumber as Glu3, with proline. The authors reasoned that Glu3 is the second residue of a β-turn, and thus, proline might stabilize the turn in that position.38 They measured the Ki at 6.1 nM, making CP2 one of the highest-affinity Keap1 inhibitors reported to date. We simulated the conformational ensembles of CP1 and CP2 using the enhanced sampling BE-META methodology described in the Methods. We found that neither of the CPs was predicted to be well-structured in solution. However, some portion of the structural ensembles indeed adopt conformations that preorganize the ETGE residues for Keap1 binding (Figure 2). To better quantify the overall degree of preorganization, specifically how well the entire conformational ensemble mimics the desired structure, we calculated the probability density distribution of the RMSD values, where RMSD is calculated between the CP conformation and the desired conformation using the backbone atoms of the ETGE residues (Figure 3). These data show that a sizable proportion of the conformational ensemble of CP1 has its ETGE sequence preorganized into a conformation similar to the Keap1-bound conformation of the corresponding residues on Nrf2. We define the preorganized population of the ensemble to be the percentage of frames with an RMSD less than 0.5 Å compared to the desired structure. For CP1, the preorganized population represented 15% of the conformational ensemble and the average RMSD for the preorganized population was 0.335 Å (Figure 4A,4B and Table 1). For CP2, the preorganized population was 29% of the conformational ensemble and the average RMSD for the preorganized population was 0.257 Å. Therefore, our simulations predicted that the conformational ensemble of CP2 is more preorganized toward the desired conformation in terms of both the time it spends in similar conformations and how closely those conformations match the desired conformation. Overall, these data helped validate the simulation methods and explained the improvement in binding affinity due to the substitution from glutamate to proline at position 3.
Figure 2.
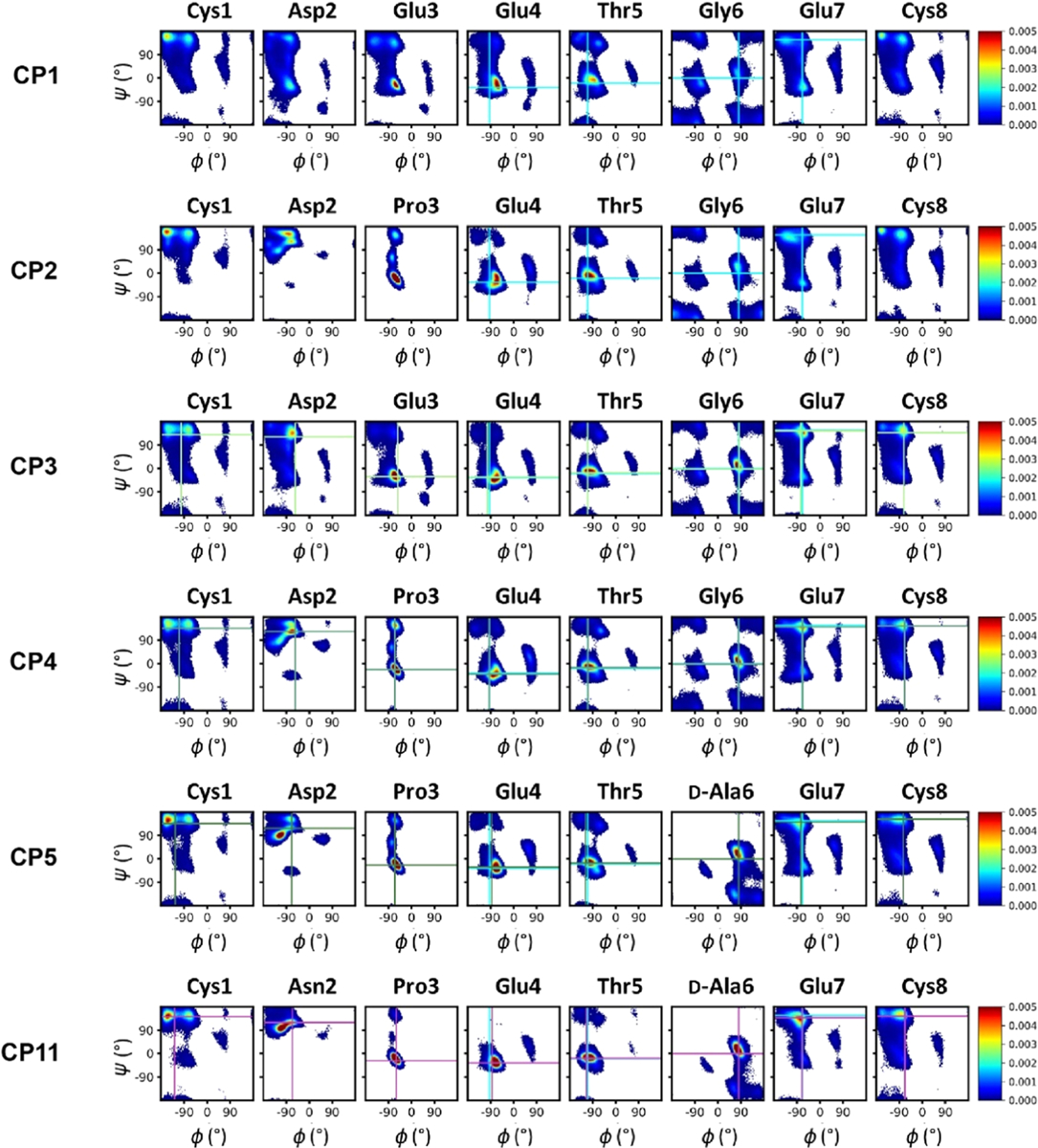
Ramachandran plots showing the entire simulated ensembles for selected CPs. Cyan lines indicate the (φ, ψ) values in the desired conformation for the residues in the core binding sequence ETGE. Light-green, green, dark-green, and magenta lines indicate the (φ, ψ) values of Keap1-bound crystal structures of CP3, CP4, CP5, and CP11, respectively. Similar plots for CP6–10 are shown in Figure S4.
Figure 3.
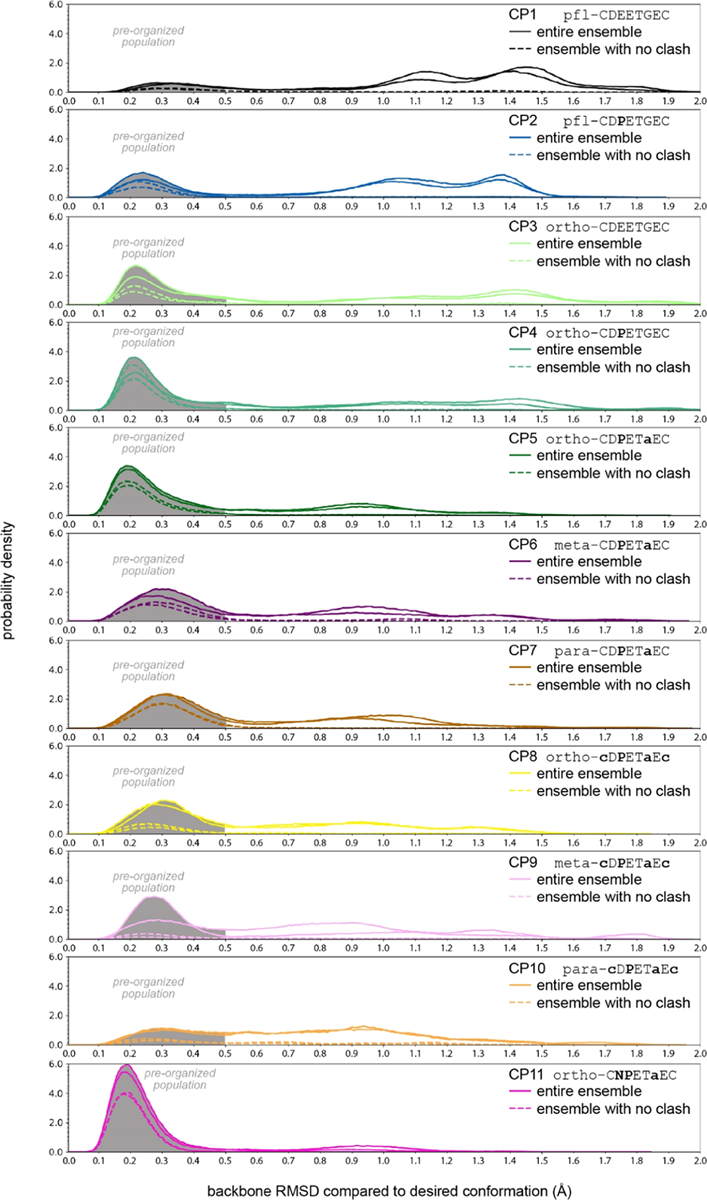
Snapshot of the simulated ensembles of 11 CPs. Plots show probability density versus backbone RMSD to the desired conformation for each of the CPs simulated; two independent simulations were performed for each CPs and results from both are shown. Solid lines show the entire ensemble, and dashed lines show the ensemble with no clash (the entire ensemble excluding any frames that would clash with Keap1 when overlaid with the desired conformation within the binding site). Each plot shows a shaded area for backbone RMSD below 0.5 Å, which denotes the preorganized population, which can be compared across all CPs.
Figure 4.
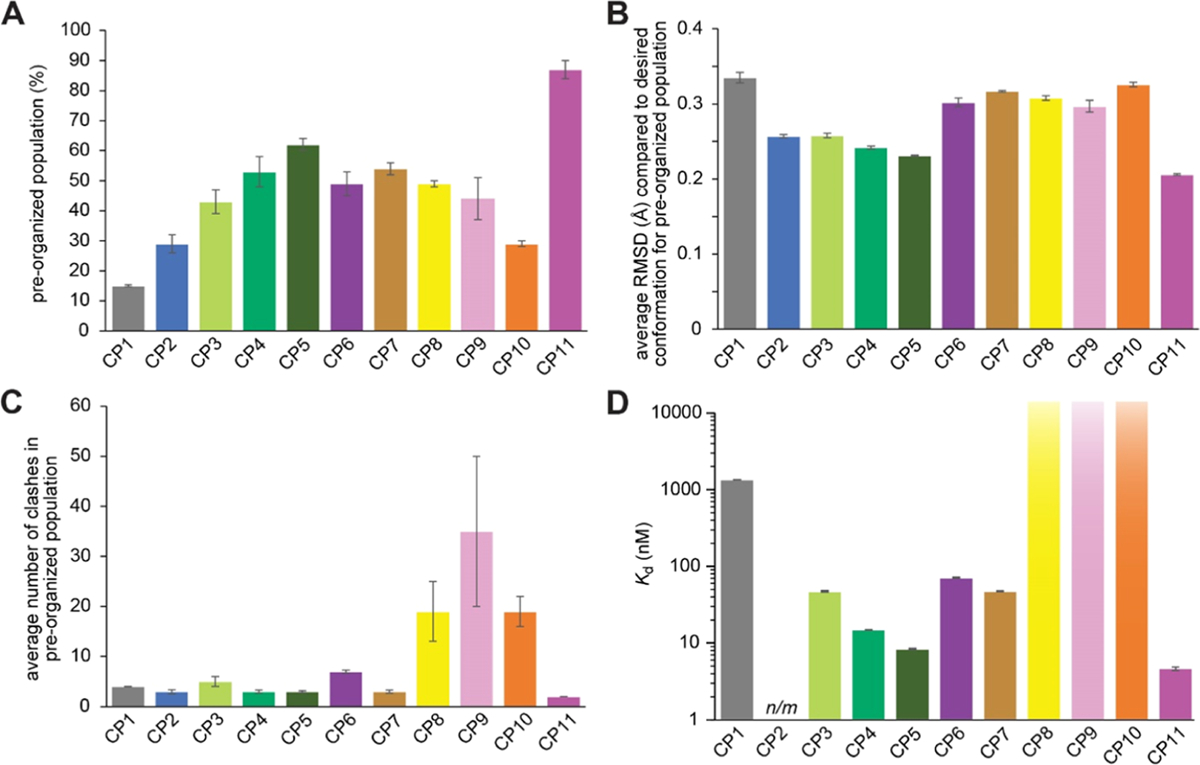
Summary of results from simulations (A–C) and experimental affinity measurements (D). (A) Percentage of each ensemble which has a backbone RMSD of less than 0.5 Å compared to the desired ETGE conformation (preorganized population). (B) Average backbone RMSD compared to the desired ETGE conformation for the preorganized population. (C) Average number of clashes in the preorganized population, defined as the average number of atoms with van der Waals radii that overlap with atoms of Keap1 when each frame is overlaid with the desired ETGE conformation within the Keap1 binding pockets. Error bars for computationally derived values represent the standard error of the mean from two independent simulations using different starting conformations. (D) Binding affinity of each CP, experimentally measured by biolayer interferometry. Error bars for the Kd values represent the standard error of the mean from three independent trials. CP8, CP9, and CP10 showed no detectable binding at concentrations as high as 15 μM. n/m refers to measurements that were not made due to poor synthesis yields.
Table 1.
Summary of the Theoretical and Experimental Resultsa
| CP name | sequence | preorganized population (%) | average backbone RMSD for the preorganized population (Å) | average number of clashes in preorganized population | Kd (nM) |
|---|---|---|---|---|---|
| CP1 | PFL_CDEETGEC | 15 ± 0.4 | 0.335 ± 0.007 | 4 ± 0.1 | 1400 ± 80 |
| CP2 | PFL_CDPETGEC | 29 ± 3 | 0.257 ± 0.002 | 3 ± 0.6 | not meas. |
| CP3 | ortho_CDEETGEC | 43 ± 4 | 0.258 ± 0.003 | 5 ± 0.7 | 48 ± 10 |
| CP4 | ortho_CDPETGEC | 53 ± 5 | 0.242 ± 0.002 | 3 ± 0.3 | 15 ± 1 |
| CP5 | ortho_CDPETaEC | 62 ± 2 | 0.231 ± 0.001 | 3 ± 0.3 | 8.4 ± 0.6 |
| CP6 | meta_CDPETaEC | 49 ± 4 | 0.302 ± 0.006 | 7 ± 0.5 | 72 ± 9 |
| CP7 | para_CDPETaEC | 54 ± 2 | 0.317 ± 0.001 | 3 ± 0.4 | 48 ± 3 |
| CP8 | ortho_cDPETaEc | 49 ± 1 | 0.308 ± 0.003 | 19 ± 6 | not obs. |
| CP9 | meta_cDPETaEc | 44 ± 7 | 0.297 ± 0.008 | 35 ± 15 | not obs. |
| CP10 | para_cDPETaEc | 29 ± 1 | 0.326 ± 0.003 | 19 ± 3 | not obs. |
| CP11 | ortho_CNPETaEC | 87 ± 3 | 0.206 ± 0.001 | 2 ± 0.1 | 4.7 ± 1.7 |
Preorganized population represents the percentage of the ensemble the CP occupies the desired ETGE conformation with a backbone RMSD below 0.5 Å. The average number of clashes in the preorganized population indicates the average number of atoms in Keap1 that overlap in space with the CP when its preorganized population is aligned to the ETGE conformation derived from Nrf2 bound to Keap1. Reported Kd values represent experimentally measured binding affinities, as measured by biolayer interferometry using peptides with biotin and a Trp-Arg spacer. CP2 could not be prepared in enough quantity to measure binding affinities (not measured), and CP8, CP9, and CP10 showed no measurable binding affinity when tested up to 15 μM. Errors for computationally derived values represent the standard error of the mean from two independent simulations using different starting conformations, and errors for Kd values represent the standard error of the mean from three independent trials.
Next, we moved forward with simulations of novel CPs. We found the chemical synthesis of CPs with perfluoroarene linker chemistry to be relatively low-yielding (<10% for CP1 and prohibitively low for CP2; see below).61 We also noted that other chemistries would allow installation of different linkers, which could modulate CP structural preferences and therefore influence the preorganization of the ETGE residues. Therefore, we simulated CPs that would use more robust macrocyclization chemistry, cysteine alkylation with dibromomethylbenzenes.10,15 BE-META MD simulations were performed for ortho-CDEETGEC and ortho-CDPETGEC (CP3 and CP4, respectively). The substitution of the perfluoroaryl linker with the ortho-dimethylbenzene linker led to an increase in predicted preorganized population, from 15% for CP1 to 43% for CP3 (Figure 4A and Table 1). When Glu3 was substituted with proline within the ortho-dimethylbenzene-linked macrocycle (compound CP4), the preorganized population increased to 53%. For CP3 and CP4, the mean backbone RMSD values for the preorganized population were 0.258 and 0.242 Å, respectively (Figure 4B and Table 1). Thus, for both the native sequence and the Glu3 → Pro-substituted sequence, the ortho-dimethylbenzene linker better preorganized the ensemble toward the desired structure, and the preorganized conformations also better matched the desired conformation.
While examining the (φ, ψ) dihedrals for the desired ETGE epitope, we noted that Gly6 had a positive φ torsional angle (Gly81 in Figure 1B). Therefore, we hypothesized that replacing Gly6 with a d-alanine would further shift the conformational ensemble toward the desired structure. Simulations of ortho-CDPETaEC (compound CP5) showed an increase in the preorganized population relative to CP4, which had a Gly in position 6 (62% preorganized compared to 53%, Figure 4A, Table 1). The preorganized population had an average backbone RMSD of 0.231 Å, compared to 0.242 Å for CP4 (Figure 4B, Table 1). Thus, these simulations predicted that CP5 would be more preorganized for Keap1 binding compared to CP1–CP4.
Experimental Binding Data with Recombinant Keap1.
Following the computational studies, we sought to compare our predictions with experimental data. Based on the previous work on CPs that mimic Nrf2′s Keap1-binding loop,38,62 we used binding affinity for recombinant Keap1 as an experimental measure that should correlate with the degree of preorganization of the binding epitope. Synthesis of CPs required us to adapt our design strategies in several ways. First, we had poor yields for macrocyclization using the perfluoroaryl linker.38,61,63 We were able to synthesize enough CP1 to test it experimentally but not CP2, and this further prompted our switch to the more robust dimethylbenzene linker chemistry.10,15 Second, to allow for analysis using biolayer interferometry (BLI), we introduced a biotin on the peptides’ N-termini. BLI is a technique that measures the interference pattern of white light reflected from a layer of biomolecules immobilized on the surface of a streptavidin sensor tip.64 BLI provides a direct Kd rather than an indirect Ki value and also measures binding kinetics in the form of on- and off-rates. Finally, we introduced a Trp-Arg sequence as a spacer for the biotin, to allow for accurate concentration determination by UV–vis spectrophotometry and to allow for robust characterization by mass spectrometry. To confirm that these additions did not substantially alter the simulation results, we performed identical simulations on analogues of CP3 and CP4 with the added N-terminal Trp-Arg. For CP3, the added residues decreased the preorganized population and made it closer to that of CP1, but the effects on the predicted ensembles of CP4 and a later design, CP11, were minimal (see Table S2).
We measured binding affinities for Keap1 for CP1, CP3, and CP4 using BLI (Figures 4D, 5, and Table 1). CP1 had a Kd of 1400 ± 80 nM, which agreed well with the experimental Ki value found by Steel et al. in a competition fluorescence polarization assay (Ki of 1400 nM).38 CP3 had a Kd value of 48 ± 10 nM, which represents a 29-fold improvement over CP1. This trend matched the MD results, which predicted a higher degree of preorganization for CP3 with the ortho-dimethylbenzene linker compared to CP1 with the perfluoroaryl linker. CP4, which included the ortho-dimethylbenzene linker and the Glu3 → Pro substitution, had a Kd of 15 ± 1 nM, a threefold improvement over CP3. This result also matched the simulations, which predicted a further degree of preorganization for CP4 relative to CP3. CP5, which introduced the Gly6 → d-Ala substitution, had a Kd for Keap1 of 8.4 ± 0.6 nM, representing a further twofold improvement in binding affinity compared to CP4 (Figure 4D and Table 1). Overall, the binding affinity data confirmed predictions made by the simulated conformational ensembles. In this initial series of designs, we improved binding by 160-fold compared to CP1 using a simple torsional analysis strategy to guide a series of substitutions and predict the effects of those substitutions using our MD methodology.
Figure 5.
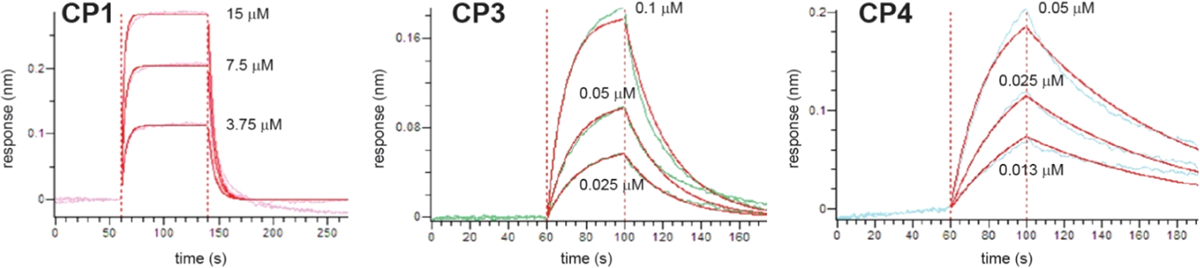
Biolayer interferometry data (various colors) and curve fits (red) for representative CPs binding to Keap1. Each trial tested at least three different protein concentrations (labeled on the plots). These data represent one of three independent trials for each CP. Additional data are listed in Figure S6.
Design, MD Simulations, Binding Data, and Clash Analysis for Analogues with Altered Staple Geometries.
Alkylation of cysteines using the dibromomethylbenzenes allows for diversity-oriented stapling, in which the shape of the linker is varied to sample different macrocycle geometries.10,15 One of the most valuable applications for our methodology would thus be to predict which linker, for example, among ortho-, meta-, or para-dimethylbenzene, would best preorganize the conformational ensemble toward the desired conformation. To address this need, we next simulated isomers of CP5 (ortho-CDPETaEC), which incorporated meta-dimethylbenzene and para-dimethylbenzene linkers. Meta-CDPETaEC and para-CDPETaEC are denoted as CP6 and CP7, respectively. Both had a less preorganized conformational ensemble compared to CP5 and the preorganized population had a greater average backbone RMSD compared to that of CP5 (Figures 3, 4A,B, and Table 1). Thus, we predicted that the ortho linker would outperform the meta- and para-linkers. In addition to different linker isomers, varying the stereo-chemistry of the cysteines is also a common method for diversifying macrocycle geometry.10,12,15 Consequently, we also simulated macrocycles that had ortho-, meta-, and para-dimethylbenzene linkers linked to two d-cysteines, denoted as CP8, CP9, and CP10, respectively. These three CPs all had somewhat less preorganized populations than their l-cysteine-containing counterparts CP5, CP6, and CP7 (Figures 3, 4A,B, and Table 1).
To this point, we had only considered the degree of preorganization within the conformational ensemble. However, given the diversity of the macrocycle linkers we were simulating, we anticipated that these CPs could have different degrees of steric clash in the Keap1-binding pocket. To obtain a simple measure of predicted steric clash for each conformational ensemble, we positioned each frame in the ensemble within the Keap1-binding pocket by overlaying the ETGE epitope with the corresponding residues of the Nrf2-bound structure.24 We then computed the degree of steric clash between that frame’s conformation and Keap1 in terms of the number of atoms in the CP whose van der Waals radii overlapped with those of atoms of Keap1. While this measure of predicted steric clash did not consider the flexibility of the macrocycle and the receptor, it provided a simple measure of the relative degree of predicted clash that could be compared across entire conformational ensembles. Some of the effects of the predicted clash could be visualized across an ensemble by removing all frames with any clash and then replotting the “clash-free” ensemble as a population density distribution with respect to backbone RMSD to the ETGE sequence (Figure 3, dashed lines). Comparing these distributions to the full ensembles, it is clear that the preorganized populations are predominantly clash-free. To make more quantitative comparisons among all the CP ensembles, we calculated the average clash for the preorganized population for each CP (Figure 4C and Table 1). This analysis revealed that the preorganized conformations of CP1–CP5 all had a small degree of clash. CP6 and CP7, isomers of CP5 with meta- and para-dibromomethylbenzene linkers, also had a relatively low degree of clash for their preorganized conformations. However, CP8–CP10, isomers of CP5–CP7 with d-cysteines, had a much higher overall degree of clash (Figure 4C and Table 1). This led us to predict that despite their preorganization of the ETGE conformation, these CPs would be much less compatible with Keap1 binding compared to their l-cysteine-containing isomers.
To test our predictions, we synthesized CP6–CP10 and measured their binding to recombinant Keap1 using BLI. CP6 and CP7 had Kd values of 72 ± 9 nM and 48 ± 3 nM, respectively. These are 10- and 5-fold poorer in affinity compared to CP5, confirming the prediction that the ortho linker would preorganize the macrocycle to a greater degree than the meta- or para-linkers. It is unclear whether the meta-dimethylbenzene-linked CP6 had the poorest affinity among these isomers because it had the least degree of preorganization (Figures 3 and 4A,B) or a somewhat higher degree of clash (Figure 4C) or whether both factors were important. Finally, CP8, CP9, and CP10 all showed no detectable binding to Keap1 by BLI at concentrations as high as 15 μM. These experimental findings are consistent with the relatively high degree of clash precluding binding altogether for CP8 and CP10 (Figure 3C and Table 1).
Design of Analogues with a Neutral Analogue Substituted for Asp2.
As a final demonstration of the new methodology, we examined the role of Asp2 that occupies the first position of the β-turn in the ETGE conformation (Figure 1A). We noticed that in the crystal structure of Nrf2 bound to Keap1, Asp2 (corresponds to Asp77 of Nrf2) is responsible for crucial intramolecular hydrogen bonds between its side chain and other backbone amides (Figure 1A). We hypothesized that aspartate could be substituted for asparagine in this position to provide for the same set of hydrogen bonds without the additional negative charge. We designed a CP identical to CP5 (ortho-CDPETaEC) but with Asp2 substituted with Asn, CP11 (ortho-CNPETaEC). In the simulations, CP11 had a preorganized population that represented 87% of the conformational ensemble and the average backbone RMSD for the preorganized population was 0.206 Å (Figures 3, 4A,B, and Table 1). CP11 had the most preorganized conformational ensemble compared to all other CPs. CP11 also had the smallest degree of clash (Figure 4C and Table 1). Binding assays to recombinant Keap1 revealed a Kd of 4.7 ± 1.7 nM, a twofold improvement from CP5. Overall, the results from CP11 demonstrated that the H-bond interactions and not the charge-mediated interactions were important at position 2 of the binding epitope. Furthermore, the results provide additional support for the predictive nature of the MD simulations.
Crystal Structures of Cyclic Peptides Bound to Keap1.
To further evaluate our design strategy, we crystallized the Keap1 Kelch domain in complex with each of CP3, CP4, CP5, and CP11, using a crystal form reported by the Allen lab.43 The crystals diffracted to resolutions between 1.5 and 1.8 Å, allowing unambiguous placement of the cyclic peptides in the Nrf2-binding pocket, except for the benzene rings of the ortho-dimethylbenzene linkers (Figure S7). All four CPs bound Keap1 with the ETGE binding epitopes in conformations highly similar to the desired conformation of Nrf2 (Figure 6A). Importantly, the conformations of the ETGE epitopes and of the larger cysteine-to-cysteine peptide backbones all matched the preorganized populations from the MD simulations (Figures 2 and 6B) with backbone RMSD values between 0.20 and 0.24 Å for the ETGE epitopes and between 0.37 and 0.67 Å for the entire peptide backbones (Table S5). Because the ortho-dimethylbenzene linkers were not well-defined in the four structures, we infer that there may be multiple binding modes or some flexibility within the binding pocket (Figure S6). To investigate the linker structure further, we clustered the preorganized populations of each CP taking into account backbone torsional angles as well as the eight additional torsional angles in the linker. This analysis revealed subpopulations of these preorganized populations with distinct linker geometries (Figure S8). These results are highly valuable for continued improvements in the accuracy of modeling these commonly used dimethylbenzene linkers.
Figure 6.
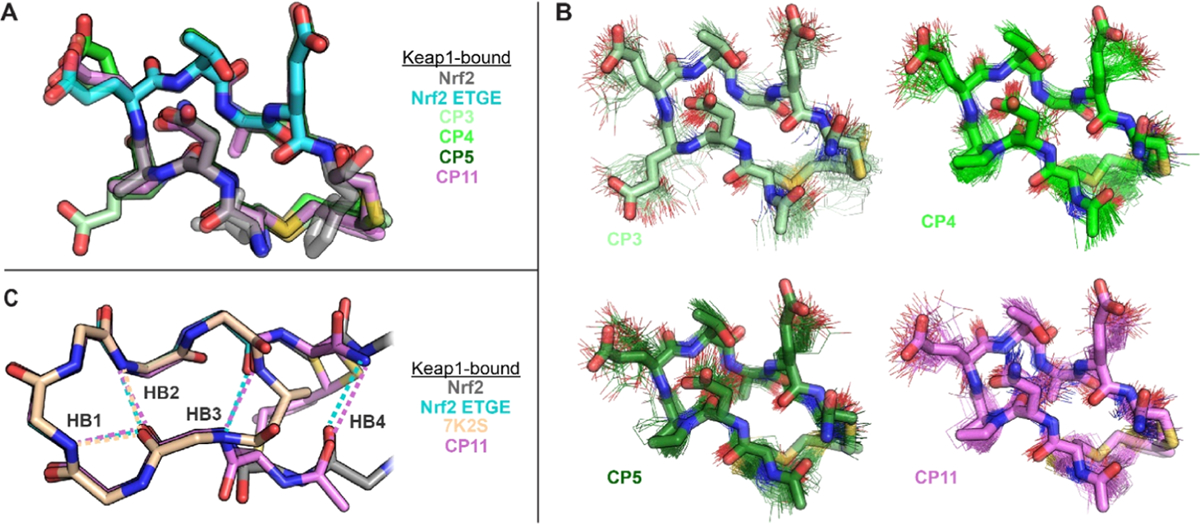
Crystal structures of CP3, CP4, CP5, and CP11 bound to Keap1. (a) Overlay showing excellent match between the ETGE binding epitope of Keap1-bound Nrf2 (cyan) and the Keap1-bound conformations of all four CPs. (b) Overlays showing excellent match between the preorganized populations of each CP with its Keap1-bound structure. 100 randomly selected frames of each CP’s preorganized population are shown in wireframe, and each CP’s Keap1-bound structure is shown in sticks. Portions of the N-terminal arginine and tryptophan residues are omitted for clarity. (c) Comparison of the Keap1-bound structures of a head-to-tail CP reported by Allen, Whitty, and co-workers (PDB 7K2S),39 a Nrf2-derived peptide,24 and CP11. Key intrabackbone hydrogen bonds are shown as dotted lines in the corresponding colors and are labeled HB1–HB4. CPs cyclized with ortho-dimethylbenzene linkers preserve two additional backbone hydrogen bonds present in the native ligand (HB3 and HB4). The image shows only backbone atoms for clarity. Structures have been deposited in the RCSB Protein Data Bank under the accession codes 8PKU (CP3), 8PKV (CP4), 8PKW (CP5), and 8PKX (CP11).
The crystal structures directly revealed the effects of our designed substitutions on the CP geometry and interactions with Keap1. Replacing Glu at position 3 with Pro (producing CP4 from CP3) preorganized the β-turn at positions 2–5 while losing a polar contact to Tyr525 of Keap1. The binding affinity data suggest that the loss of the polar contact was not as important as preorganization of the β-turn. Replacing Gly at position 6 with d-Ala (producing CP5 from CP4) led to a slight rotation of nearby Tyr572 on Keap1, but these residues are not close enough to interact via van der Waals forces, so there was a plenty of room for the d-Ala in the bound structure. Replacing Asp at position 2 with Asn (producing CP11 from CP5) maintained intramolecular hydrogen bonds between the side chain carbonyl of Asp/Asn and backbone amides of residues 4 and 5, further stabilizing the β-turn, and also maintained a water network that connects to polar residues of Keap1 including Arg415 and Arg483. In the bound structure of CP5, the side chain carboxylate of Glu4 is only 5 Å from the side chain carboxylate of Asp2. Thus, it is possible that changing the Asp2 to an Asn in CP11 may reduce repulsive interactions in the bound conformation, leading to greater preorganization and higher binding affinity.
DISCUSSION
Most prior work on all-atom CP simulations has been limited to head-to-tail CPs using only natural amino acids and enantiomers of natural amino acids.21,22,65 In this work, we sought to use a set of commonly used, high-yielding cyclization linkers derived from reacting two cysteines with ortho-, meta-, or para-dibromomethylbenzene. This chemistry, originally termed “CLIPS” by Timmerman and co-workers,15 has been used for over 15 years for rapid and high-yielding peptide macrocyclization reactions.7–12 To allow MD simulations of these commonly used cyclization linkers, we parametrized the charges following a protocol that is compatible with the general Amber force fields. Similar parametrization was also performed for the perfluoroaryl (pfl) linker, originally described by Pentelute and co-workers,61 to allow simulation of a control CP that was previously reported as a Keap1 inhibitor. The development of parameters for each linker was a key roadblock to this work, and the parameters developed here may greatly facilitate continued design efforts using these convenient linkers.
In this work, we applied an enhanced sampling BE-META methodology to test a series of predictions based on individual amino acid substitutions in a Keap1-binding peptide sequence derived from Nrf2. With adjustments for the degree of predicted clash, the methodology successfully predicted the effects of a variety of substitutions including substitutions primarily affecting backbone torsions (Glu3 → Pro and Gly5 → d-Ala), substitutions of different linker geometries (perfluoroarene and ortho-, meta-, and para-dimethylbenzene linkers, as well as substitution of the cross-linked l-Cys with d-Cys), substitutions affecting backbone chirality (Gly5 → d-Ala and l-Cys → d-Cys), and substitutions affecting subtle side chain interactions (Asp2 → Asn). The binding affinity results confirmed predictions made by the simulated conformational ensembles and permitted us to use a simple torsional analysis strategy to design substitutions that better promoted the ETGE conformation.
The ability of our computational methodology to predict the relative binding affinities of peptides cyclized with ortho-, meta-, and para-dimethylbenzene linkers provides evidence that our parametrization of these artificial groups is valid for further studies. The robust yields, low cost, and simple implementation of this cyclization chemistry have ensured its broad usage in early stages of drug design.10,12 Despite this extensive use, the most common method for applying this chemistry is still largely trial and error. Thus, the newly developed ability to perform high-level MD simulations on CPs incorporating these linkers and the opportunity to understand how the linker structure and dynamics affect the macrocycle structure represent significant steps forward for design efforts in this promising chemical space.
This computational method is versatile and highly complementary to alternative methods used to predict the structures of CPs. Notable efforts in this field include recent work by Baker and co-workers, who have used Rosetta66 successfully to design well-structured CPs of lengths 7–14 as well as membrane-permeable CPs of lengths 6–12.5,6 However, these sequences all comprised a very limited number of naturally occurring residues, with an emphasis on hydro-phobic amino acids and high proline content.6 The extent to which these limitations will also limit useful chemical space when this method is applied to the design of PPI inhibitors remains unknown. Furthermore, while tools such as Rosetta have demonstrated great capability in identifying well-structured sequences, this method described herein breaks new ground when it comes to predicting the entire structural landscape of flexible CPs and using that more nuanced view of the CP structure to guide design.
Several prior efforts have sought to use conformational constraints to promote high-affinity binding of Nrf2-derived sequences to Keap1.34,38,39,42,62,67–70 In a particularly notable example, Allen, Whitty, and co-workers recently reported crystal structures of head-to-tail CPs bound to Keap1; the CPs were designed using solution NMR and machine learning to rationalize structure–activity relationships.39 Their highest-affinity peptide was similar to CP4, but with a D-β-homoalanine (Dha) residue linking the termini instead of two cysteines cross-linked with ortho-dimethylbenzene. The Dha-cyclized peptide was unable to preserve a hydrogen bond between Glu7 and Asp2 present in the native ligand (HB3 in Figure 6C), and the authors ascribed this to strain imposed by the Dha cyclization linker.39 By contrast, all four CPs with the ortho-dimethylbenzene linker are able to bind Keap1 while maintaining this hydrogen bond. The native ligand from Nrf2 also has an additional hydrogen bond between the Nrf2 residues Gln75 and Leu84 (HB4 in Figure 6C), which matches closely in the bound structures of all four CPs by a hydrogen bond between the carboxamide at the C-terminus and the backbone carbonyl of the N-terminal arginine residue (Figure 6C). These additional hydrogen bonds likely increase the preorganization of these CPs. Interestingly, our observations suggest that cross-linking two cysteines with an ortho-dimethylbenzene linker is particularly stabilizing within antiparallel β-sheet structures. Despite many applications of this cross-linking chemistry, to the best of our knowledge, this is the first time this cross-link has been shown to stabilize an antiparallel β-sheet or β-hairpin structure.
Finally, the end result of our sequence optimization scheme, CP11, was found to display binding affinity improved by 280-fold over that of the initial compound. In fact, CP11 rivals the most favorable binding affinity for Keap1 reported to date (4.7 nM compared to 2.8 nM for a disulfide-mediated cyclic peptide of similar size).42 Outstanding challenges for developing Keap1 inhibitors and other CP inhibitors of PPIs include designing for improved bioavailability.25,71,72 The Asp2 → Asn mutation described herein presents a good first step toward improving cell penetration, as a negative charge is typically observed to decrease cell penetration.73,74 Future improvements may include the incorporation of N-methylated or noncanonical amino acids to additionally improve the membrane permeability. Despite these challenges, the computational methodology described here represents an exciting new avenue for developing macrocyclic inhibitors of protein–protein interactions.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by GM125856 and GM148407, J.A.K., and GM124160, Y.-S.L. The authors are grateful to Shruthi Shet and Reinhard Albrecht for technical assistance and to the staff of the beamline X06DA at the SLS (Villigen, Switzerland) for excellent technical support.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.3c01337.
Materials and methods, charge generation protocol, peptide synthesis, protein expression and purification, biolayer interferometry, and X-ray crystallography (PDF)
Crystallographic data (ZIP)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jcim.3c01337
The authors declare no competing financial interest.
Contributor Information
Francini Fonseca Lopez, Department of Chemistry, Tufts University, Medford, Massachusetts 02155, United States.
Jiayuan Miao, Department of Chemistry, Tufts University, Medford, Massachusetts 02155, United States;.
Jovan Damjanovic, Department of Chemistry, Tufts University, Medford, Massachusetts 02155, United States;.
Luca Bischof, Department of Protein Evolution, Max Planck Institute for Biology, 72076 Tübingen, Germany;; Interfaculty Institute of Biochemistry, Tübingen University, 72076 Tübingen, Germany
Michael B. Braun, Department of Protein Evolution, Max Planck Institute for Biology, 72076 Tübingen, Germany; Interfaculty Institute of Biochemistry, Tübingen University, 72076 Tübingen, Germany
Yingjie Ling, Department of Chemistry, Tufts University, Medford, Massachusetts 02155, United States.
Marcus D. Hartmann, Department of Protein Evolution, Max Planck Institute for Biology, 72076 Tübingen, Germany; Interfaculty Institute of Biochemistry, Tübingen University, 72076 Tübingen, Germany;
Yu-Shan Lin, Department of Chemistry, Tufts University, Medford, Massachusetts 02155, United States;.
Joshua A. Kritzer, Department of Chemistry, Tufts University, Medford, Massachusetts 02155, United States;
Data Availability Statement
Software packages used and parameters applied are described in the Methods and in the Supporting Information. Details of MD simulation protocols, dihedral principal component analysis, mimicry analysis, and clash analysis are provided in the Supporting Information. Parameter files, input files, output files, and scripts for the MD simulations are provided in the Supporting Information. Files describing how to run BE-META simulations of CPs and scripts to perform dihedral principal component analysis can be found on GitHub (https://github.com/ysl-lab/CP_tutorial). Structures have been deposited in the RCSB Protein Data Bank under the accession codes 8PKU (CP3), 8PKV (CP4), 8PKW (CP5), and 8PKX (CP11).
REFERENCES
- (1).Zorzi A; Deyle K; Heinis C Cyclic Peptide Therapeutics: Past, Present and Future. Curr. Opin. Chem. Biol 2017, 38, 24–29. [DOI] [PubMed] [Google Scholar]
- (2).Wishart DS; Feunang YD; Guo AC; Lo EJ; Marcu A; Grant JR; Sajed T; Johnson D; Li C; Sayeeda Z; Assempour N; Iynkkaran I; Liu Y; Maciejewski A; Gale N; Wilson A; Chin L; Cummings R; Le D; Pon A; Knox C; Wilson M DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res 2018, 46 (D1), D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Jing X; Jin K A Gold Mine for Drug Discovery: Strategies to Develop Cyclic Peptides into Therapies. Med. Res. Rev 2020, 40 (2), 753–810. [DOI] [PubMed] [Google Scholar]
- (4).Bashiruddin NK; Suga H Construction and Screening of Vast Libraries of Natural Product-like Macrocyclic Peptides Using in Vitro Display Technologies. Curr. Opin. Chem. Biol 2015, 24, 131–138. [DOI] [PubMed] [Google Scholar]
- (5).Bhardwaj G; O’Connor J; Rettie S; Huang Y-H; Ramelot TA; Mulligan VK; Alpkilic GG; Palmer J; Bera AK; Bick MJ; Di Piazza M; Li X; Hosseinzadeh P; Craven TW; Tejero R; Lauko A; Choi R; Glynn C; Dong L; Griffin R; van Voorhis WC; Rodriguez J; Stewart L; Montelione GT; Craik D; Baker D Accurate de Novo Design of Membrane-Traversing Macrocycles. Cell 2022, 185 (19), 3520–3532.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hosseinzadeh P; Bhardwaj G; Mulligan VK; Shortridge MD; Craven TW; Pardo-Avila F; Rettie SA; Kim DE; Silva D-A; Ibrahim YM; Webb IK; Cort JR; Adkins JN; Varani G; Baker D Comprehensive Computational Design of Ordered Peptide Macrocycles. Science 2017, 358 (6369), 1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cerulli RA; Shehaj L; Brown H; Pace J; Mei Y; Kritzer JA Stapled Peptide Inhibitors of Autophagy Adapter LC3B. ChemBioChem 2020, 21 (19), 2777–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Heinis C; Rutherford T; Freund S; Winter G Phage-Encoded Combinatorial Chemical Libraries Based on Bicyclic Peptides. Nat. Chem. Biol 2009, 5 (7), 502–507. [DOI] [PubMed] [Google Scholar]
- (9).Jo H; Meinhardt N; Wu Y; Kulkarni S; Hu X; Low KE; Davies PL; DeGrado WF; Greenbaum DC Development of α-Helical Calpain Probes by Mimicking a Natural Protein–Protein Interaction. J. Am. Chem. Soc 2012, 134 (42), 17704–17713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Peraro L; Siegert TR; Kritzer JA Conformational Restriction of Peptides Using Dithiol Bis-Alkylation. In Methods in Enzymology; Elsevier, 2016; Vol. 580, pp 303–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Peraro L; Zou Z; Makwana KM; Cummings AE; Ball HL; Yu H; Lin Y-S; Levine B; Kritzer JA Diversity-Oriented Stapling Yields Intrinsically Cell-Penetrant Inducers of Autophagy. J. Am. Chem. Soc 2017, 139 (23), 7792–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Brown H; Chung M;Üffing A; Batistatou N; Tsang T; Doskocil S; Mao W; Willbold D; Bast RC; Lu Z; Weiergräber OH; Kritzer JA Structure-Based Design of Stapled Peptides That Bind GABARAP and Inhibit Autophagy. J. Am. Chem. Soc 2022, 144 (32), 14687–14697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Muppidi A; Wang Z; Li X; Chen J; Lin Q Achieving Cell Penetration with Distance-Matching Cysteine Cross-Linkers: A Facile Route to Cell-Permeable Peptide Dual Inhibitors of Mdm2/Mdmx. Chem. Commun 2011, 47 (33), 9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Muppidi A; Zhang H; Curreli F; Li N; Debnath AK; Lin Q Design of Antiviral Stapled Peptides Containing a Biphenyl Cross-Linker. Bioorg. Med. Chem. Lett 2014, 24 (7), 1748–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Timmerman P; Beld J; Puijk WC; Meloen RH Rapid and Quantitative Cyclization of Multiple Peptide Loops onto Synthetic Scaffolds for Structural Mimicry of Protein Surfaces. ChemBioChem 2005, 6 (5), 821–824. [DOI] [PubMed] [Google Scholar]
- (16).Yang C-H; Brown JN; Kopple KD Crystal Structure and Solution Studies of the Molecular Conformation of the Cyclic Hexapeptide Cyclo-(Gly-L-His-Gly-L-Ala-L-Tyr-Gly). J. Am. Chem. Soc 1981, 103 (7), 1715–1719. [Google Scholar]
- (17).Kopple KD; Wang YS; Cheng AG; Bhandary KK Conformations of Cyclic Octapeptides. 5. Crystal Structure of Cyclo(Cys-Gly-Pro-Phe)2 and Rotating Frame Relaxation (T1.Rho.) NMR Studies of Internal Mobility in Cyclic Octapeptides. J. Am. Chem. Soc 1988, 110 (13), 4168–4176. [Google Scholar]
- (18).Morrison C Constrained Peptides’ Time to Shine? Nat. Rev. Drug Discovery 2018, 17 (8), 531–533. [DOI] [PubMed] [Google Scholar]
- (19).Damjanovic J; Miao J; Huang H; Lin Y-S Elucidating Solution Structures of Cyclic Peptides Using Molecular Dynamics Simulations. Chem. Rev 2021, 121 (4), 2292–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).McHugh SM; Rogers JR; Yu H; Lin Y-S Insights into How Cyclic Peptides Switch Conformations. J. Chem. Theory Comput 2016, 12 (5), 2480–2488. [DOI] [PubMed] [Google Scholar]
- (21).McHugh SM; Yu H; Slough DP; Lin Y-S Mapping the Sequence–Structure Relationships of Simple Cyclic Hexapeptides. Phys. Chem. Chem. Phys 2017, 19 (4), 3315–3324. [DOI] [PubMed] [Google Scholar]
- (22).Slough DP; McHugh SM; Lin Y-S Understanding and Designing Head-to-Tail Cyclic Peptides. Biopolymers 2018, 109 (10), No. e23113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Itoh K; Chiba T; Takahashi S; Ishii T; Igarashi K; Katoh Y; Oyake T; Hayashi N; Satoh K; Hatayama I; Yamamoto M; Nabeshima Y An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun 1997, 236 (2), 313–322. [DOI] [PubMed] [Google Scholar]
- (24).Lo S-C; Li X; Henzl MT; Beamer LJ; Hannink M Structure of the Keap1:Nrf2 Interface Provides Mechanistic Insight into Nrf2 Signaling. EMBO J 2006, 25 (15), 3605–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wakabayashi N; Itoh K; Wakabayashi J; Motohashi H; Noda S; Takahashi S; Imakado S; Kotsuji T; Otsuka F; Roop DR; Harada T; Engel JD; Yamamoto M Keap1-Null Mutation Leads to Postnatal Lethality Due to Constitutive Nrf2 Activation. Nat. Genet 2003, 35 (3), 238–245. [DOI] [PubMed] [Google Scholar]
- (26).Kobayashi M; Li L; Iwamoto N; Nakajima-Takagi Y; Kaneko H; Nakayama Y; Eguchi M; Wada Y; Kumagai Y; Yamamoto M The Antioxidant Defense System Keap1-Nrf2 Comprises a Multiple Sensing Mechanism for Responding to a Wide Range of Chemical Compounds. Mol. Cell. Biol 2009, 29 (2), 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Fourquet S; Guerois R; Biard D; Toledano MB Activation of NRF2 by Nitrosative Agents and H2O2 Involves KEAP1 Disulfide Formation. J. Biol. Chem 2010, 285 (11), 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Clarke JL; Murray JB; Park BK; Copple IM Roles of Nrf2 in Drug and Chemical Toxicity. Curr. Opin. Toxicol 2016, 1, 104–110. [Google Scholar]
- (29).Mrowietz U; Christophers E; Altmeyer P Treatment of Psoriasis with Fumaric Acid Esters: Results of a Prospective Multicentre Study. Br. J. Dermatol 1998, 138 (3), 456–460. [DOI] [PubMed] [Google Scholar]
- (30).Hao H; Xie F; Xu F; Wang Q; Wu Y; Zhang D LipoxinA4 Analog BML-111 Protects Podocytes Cultured in High-Glucose Medium against Oxidative Injury via Activating Nrf2 Pathway. Int. Immunopharmacol 2022, 111, No. 109170. [DOI] [PubMed] [Google Scholar]
- (31).Hao W; Li M; Cai Q; Wu S; Li X; He Q; Hu Y Roles of NRF2 in Fibrotic Diseases: From Mechanisms to Therapeutic Approaches. Front. Physiol 2022, 13, No. 889792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Manda G; Milanesi E; Genc S; Niculite CM; Neagoe IV; Tastan B; Dragnea EM; Cuadrado A Pros and Cons of NRF2 Activation as Adjunctive Therapy in Rheumatoid Arthritis. Free Radicals Biol. Med 2022, 190, 179–201. [DOI] [PubMed] [Google Scholar]
- (33).Cuadrado A; Manda G; Hassan A; Alcaraz MJ; Barbas C; Daiber A; Ghezzi P; León R; López MG; Oliva B; Pajares M; Rojo AI; Robledinos-Antón N; Valverde AM; Guney E; Schmidt HHHW Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev 2018, 70 (2), 348–383. [DOI] [PubMed] [Google Scholar]
- (34).Crisman E; Duarte P; Dauden E; Cuadrado A; Rodríguez-Franco MI; López MG; León R KEAP1-NRF2 Protein–Protein Interaction Inhibitors: Design, Pharmacological Properties and Therapeutic Potential. Med. Res. Rev 2022, No. med.21925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Begnini F; Geschwindner S; Johansson P; Wissler L; Lewis RJ; Danelius E; Luttens A; Matricon P; Carlsson J; Lenders S; König B; Friedel A; Sjö P; Schiesser S; Kihlberg J Importance of Binding Site Hydration and Flexibility Revealed When Optimizing a Macrocyclic Inhibitor of the Keap1–Nrf2 Protein–Protein Interaction. J. Med. Chem 2022, 65 (4), 3473–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chen Y; Inoyama D; Kong A-NT; Beamer LJ; Hu L Kinetic Analyses of Keap1-Nrf2 Interaction and Determination of the Minimal Nrf2 Peptide Sequence Required for Keap1 Binding Using Surface Plasmon Resonance: Kinetic Analyses of Keap1-Nrf2 Peptide Interactions. Chem. Biol. Drug Des 2011, 78 (6), 1014–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Gavenonis J; Sheneman BA; Siegert TR; Eshelman MR; Kritzer JA Comprehensive Analysis of Loops at Protein-Protein Interfaces for Macrocycle Design. Nat. Chem. Biol 2014, 10, 716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Steel RJ; O’Connell MA; Searcey M Perfluoroarene-Based Peptide Macrocycles That Inhibit the Nrf2/Keap1 Interaction. Bioorg. Med. Chem. Lett 2018, 28 (16), 2728–2731. [DOI] [PubMed] [Google Scholar]
- (39).Ortet PC; Muellers SN; Viarengo-Baker LA; Streu K; Szymczyna BR; Beeler AB; Allen KN; Whitty A Recapitulating the Binding Affinity of Nrf2 for KEAP1 in a Cyclic Heptapeptide, Guided by NMR, X-Ray Crystallography, and Machine Learning. J. Am. Chem. Soc 2021, 143 (10), 3779–3793. [DOI] [PubMed] [Google Scholar]
- (40).Siegert TR; Bird MJ; Makwana KM; Kritzer JA Analysis of Loops That Mediate Protein–Protein Interactions and Translation into Submicromolar Inhibitors. J. Am. Chem. Soc 2016, 138 (39), 12876–12884. [DOI] [PubMed] [Google Scholar]
- (41).Siegert TR; Bird M; Kritzer JA Identifying Loop-Mediated Protein–Protein Interactions Using LoopFinder. In Modeling Peptide–Protein Interactions; Schueler-Furman O; London, N., Eds.; Methods in Molecular Biology; Springer: New York, NY, 2017; Vol. 1561, pp 255–277. [DOI] [PubMed] [Google Scholar]
- (42).Lu M-C; Chen Z-Y; Wang Y-L; Jiang Y-L; Yuan Z-W; You Q-D; Jiang Z-Y Binding Thermodynamics and Kinetics Guided Optimization of Potent Keap1–Nrf2 Peptide Inhibitors. RSC Adv 2015, 5 (105), 85983–85987. [Google Scholar]
- (43).Zhong M; Lynch A; Muellers SN; Jehle S; Luo L; Hall DR; Iwase R; Carolan JP; Egbert M; Wakefield A; Streu K; Harvey CM; Ortet PC; Kozakov D; Vajda S; Allen KN; Whitty A Interaction Energetics and Druggability of the Protein–Protein Interaction between Kelch-like ECH-Associated Protein 1 (KEAP1) and Nuclear Factor Erythroid 2 Like 2 (Nrf2). Biochemistry 2020, 59 (4), 563–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Piana S; Laio A A Bias-Exchange Approach to Protein Folding. J. Phys. Chem. B 2007, 111 (17), 4553–4559. [DOI] [PubMed] [Google Scholar]
- (45).Todorova N; Yarovsky I Molecular Modelling of Peptide Folding, Misfolding and Aggregation Phenomena. Procedia Comput. Sci 2010, 1 (1), 1185–1193. [Google Scholar]
- (46).Baftizadeh F; Cossio P; Pietrucci F; Laio A Protein Folding and Ligand-Enzyme Binding from Bias-Exchange Metadynamics Simulations. Curr. Phys. Chem 2012, 2 (1), 79–91. [Google Scholar]
- (47).Ichiye T; Karplus M Collective Motions in Proteins: A Covariance Analysis of Atomic Fluctuations in Molecular Dynamics and Normal Mode Simulations. Proteins 1991, 11 (3), 205–217. [DOI] [PubMed] [Google Scholar]
- (48).Yu H; Lin Y-S Toward Structure Prediction of Cyclic Peptides. Phys. Chem. Chem. Phys 2015, 17 (6), 4210–4219. [DOI] [PubMed] [Google Scholar]
- (49).Cossio P; Marinelli F; Laio A; Pietrucci F Optimizing the Performance of Bias-Exchange Metadynamics: Folding a 48-Residue LysM Domain Using a Coarse-Grained Model. J. Phys. Chem. B 2010, 114 (9), 3259–3265. [DOI] [PubMed] [Google Scholar]
- (50).Damas JM; Filipe LCS; Campos SRR; Lousa D; Victor BL; Baptista AM; Soares CM Predicting the Thermodynamics and Kinetics of Helix Formation in a Cyclic Peptide Model. J. Chem. Theory Comput 2013, 9 (11), 5148–5157. [DOI] [PubMed] [Google Scholar]
- (51).Miao J; Descoteaux ML; Lin Y-S Structure Prediction of Cyclic Peptides by Molecular Dynamics + Machine Learning. Chem. Sci 2021, 12 (44), 14927–14936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hess B; Kutzner C; van der Spoel D; Lindahl E GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput 2008, 4 (3), 435–447. [DOI] [PubMed] [Google Scholar]
- (53).Zhou C-Y; Jiang F; Wu Y-D Residue-Specific Force Field Based on Protein Coil Library. RSFF2: Modification of AMBER Ff99SB. J. Phys. Chem. B 2015, 119 (3), 1035–1047. [DOI] [PubMed] [Google Scholar]
- (54).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79 (2), 926–935. [Google Scholar]
- (55).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and Testing of a General Amber Force Field. J. Comput. Chem 2004, 25 (9), 1157–1174. [DOI] [PubMed] [Google Scholar]
- (56).Case DA; Aktulga HM; Belfon K; Ben-Shalom IY; Berryman JT; Brozell SR; Cerutti DS; Cheatham TE III; Cisneros GA; Cruzeiro VWD; Darden TA; Duke RE; Giambasu G; Gilson MK; Gohlke H; Goetz AW; Harris R; Izadi S; Izmailov SA; Kasavajhala K; Kaymak MC; King E; Kovalenko A; Kurtzman T; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Machado M; Man V; Manathunga M; Merz KM; Miao Y; Mikhailovskii O; Monard G; Nguyen H; O’Hearn KA; Onufriev A; Pan F; Pantano S; Qi R; Rahnamoun A; Roe DR; Roitberg A; Sagui C; Schott-Verdugo S; Shajan A; Shen J; Simmerling CL; Skrynnikov NR; Smith J; Swails J; Walker RC; Wang J; Wang J; Wei H; Wolf RM; Wu X; Xiong Y; Xue Y; York DM; Zhao S; Kollman PA Amber2022, reference manual, 2022.
- (57).Sittel F; Jain A; Stock G Principal Component Analysis of Molecular Dynamics: On the Use of Cartesian vs. Internal Coordinates. J. Chem. Phys 2014, 141 (1), No. 014111. [DOI] [PubMed] [Google Scholar]
- (58).Mu Y; Nguyen PH; Stock G Energy Landscape of a Small Peptide Revealed by Dihedral Angle Principal Component Analysis. Proteins 2005, 58 (1), 45–52. [DOI] [PubMed] [Google Scholar]
- (59).Rodriguez A; Laio A Clustering by Fast Search and Find of Density Peaks. Science 2014, 344 (6191), 1492–1496. [DOI] [PubMed] [Google Scholar]
- (60).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera?A Visualization System for Exploratory Research and Analysis. J. Comput. Chem 2004, 25 (13), 1605–1612. [DOI] [PubMed] [Google Scholar]
- (61).Spokoyny AM; Zou Y; Ling JJ; Yu H; Lin Y-S; Pentelute BL A Perfluoroaryl-Cysteine S N Ar Chemistry Approach to Unprotected Peptide Stapling. J. Am. Chem. Soc 2013, 135 (16), 5946–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Lu M-C; Jiao Q; Liu T; Tan S-J; Zhou H-S; You Q-D; Jiang Z-Y Discovery of a Head-to-Tail Cyclic Peptide as the Keap1-Nrf2 Protein-Protein Interaction Inhibitor with High Cell Potency. Eur. J. Med. Chem 2018, 143, 1578–1589. [DOI] [PubMed] [Google Scholar]
- (63).Fadzen CM; Wolfe JM; Cho C-F; Chiocca EA; Lawler SE; Pentelute BL Perfluoroarene-Based Peptide Macrocycles to Enhance Penetration Across the Blood–Brain Barrier. J. Am. Chem. Soc 2017, 139 (44), 15628–15631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Kumaraswamy S; Tobias R Label-Free Kinetic Analysis of an Antibody–Antigen Interaction Using Biolayer Interferometry. In Protein–Protein Interactions; Meyerkord CL; Fu H, Eds.; Methods in Molecular Biology; Springer: New York, NY, 2015; Vol. 1278, pp 165–182. [DOI] [PubMed] [Google Scholar]
- (65).Cummings AE; Miao J; Slough DP; McHugh SM; Kritzer JA; Lin Y-S β-Branched Amino Acids Stabilize Specific Conformations of Cyclic Hexapeptides. Biophys. J 2019, 116 (3), 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Leaver-Fay A; Tyka M; Lewis SM; Lange OF; Thompson J; Jacak R; Kaufman KW; Renfrew PD; Smith CA; Sheffler W; Davis IW; Cooper S; Treuille A; Mandell DJ; Richter F; Ban Y-EA; Fleishman SJ; Corn JE; Kim DE; Lyskov S; Berrondo M; Mentzer S; Popović Z; Havranek JJ; Karanicolas J; Das R; Meiler J; Kortemme T; Gray JJ; Kuhlman B; Baker D; Bradley P Rosetta3. In Methods in Enzymology; Elsevier, 2011; Vol. 487, pp 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Georgakopoulos ND; Talapatra SK; Gatliff J; Kozielski F; Wells G Modified Peptide Inhibitors of the Keap1–Nrf2 Protein–Protein Interaction Incorporating Unnatural Amino Acids. ChemBioChem 2018, 19 (17), 1810–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Hancock R; Schaap M; Pfister H; Wells G Peptide Inhibitors of the Keap1–Nrf2 Protein–Protein Interaction with Improved Binding and Cellular Activity. Org. Biomol. Chem 2013, 11 (21), 3553. [DOI] [PubMed] [Google Scholar]
- (69).Jiang Z-Y; Lu M-C; Xu L; Yang T-T; Xi M-Y; Xu X-L; Guo X-K; Zhang X-J; You Q-D; Sun H-P Discovery of Potent Keap1–Nrf2 Protein–Protein Interaction Inhibitor Based on Molecular Binding Determinants Analysis. J. Med. Chem 2014, 57 (6), 2736–2745. [DOI] [PubMed] [Google Scholar]
- (70).Mou Y; Wen S; Li Y-X; Gao X-X; Zhang X; Jiang Z-Y Recent Progress in Keap1-Nrf2 Protein-Protein Interaction Inhibitors. Eur. J. Med. Chem 2020, 202, No. 112532. [DOI] [PubMed] [Google Scholar]
- (71).Deprey K; Becker L; Kritzer J; Plückthun A Trapped! A Critical Evaluation of Methods for Measuring Total Cellular Uptake versus Cytosolic Localization. Bioconjugate Chem 2019, 30 (4), 1006–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Peraro L; Deprey KL; Moser MK; Zou Z; Ball HL; Levine B; Kritzer JA Cell Penetration Profiling Using the Chloroalkane Penetration Assay. J. Am. Chem. Soc 2018, 140 (36), 11360–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Bernal F; Tyler AF; Korsmeyer SJ; Walensky LD; Verdine GL Reactivation of the P53 Tumor Suppressor Pathway by a Stapled P53 Peptide. J. Am. Chem. Soc 2007, 129 (9), 2456–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Yang NJ; Hinner MJ Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. In Site-Specific Protein Labeling; Gautier A; Hinner MJ, Eds.; Methods in Molecular Biology; Springer: New York, NY, 2015; Vol. 1266, pp 29–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Software packages used and parameters applied are described in the Methods and in the Supporting Information. Details of MD simulation protocols, dihedral principal component analysis, mimicry analysis, and clash analysis are provided in the Supporting Information. Parameter files, input files, output files, and scripts for the MD simulations are provided in the Supporting Information. Files describing how to run BE-META simulations of CPs and scripts to perform dihedral principal component analysis can be found on GitHub (https://github.com/ysl-lab/CP_tutorial). Structures have been deposited in the RCSB Protein Data Bank under the accession codes 8PKU (CP3), 8PKV (CP4), 8PKW (CP5), and 8PKX (CP11).