

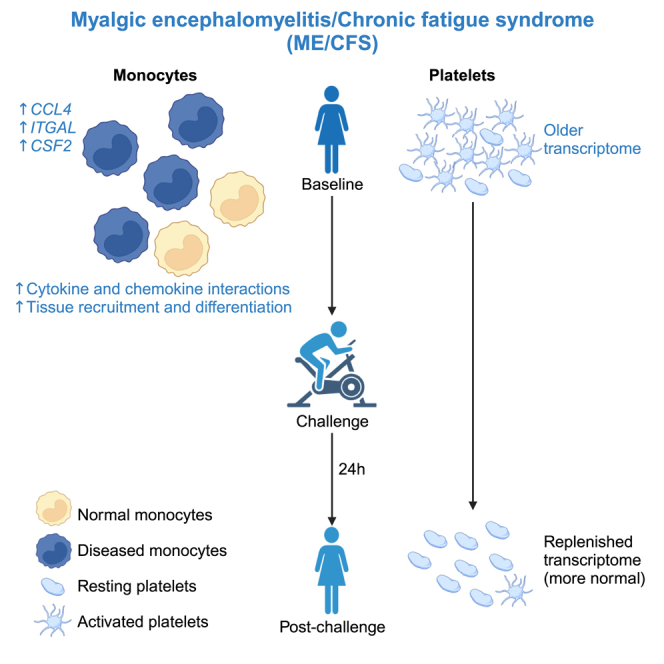
Summary
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is a serious and poorly understood disease. To understand immune dysregulation in ME/CFS, we use single-cell RNA sequencing (scRNA-seq) to examine immune cells in patient and control cohorts. Postexertional malaise (PEM), an exacerbation of symptoms following strenuous exercise, is a characteristic symptom of ME/CFS. To detect changes coincident with PEM, we applied scRNA-seq on the same cohorts following exercise. At baseline, ME/CFS patients display classical monocyte dysregulation suggestive of inappropriate differentiation and migration to tissue. We identify both diseased and more normal monocytes within patients, and the fraction of diseased cells correlates with disease severity. Comparing the transcriptome at baseline and postexercise challenge, we discover patterns indicative of improper platelet activation in patients, with minimal changes elsewhere in the immune system. Taken together, these data identify immunological defects present at baseline in patients and an additional layer of dysregulation in platelets.
Keywords: myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), scRNA-seq, monocytes, platelets, long COVID, exercise challenge, CPET
Graphical abstract
Highlights
-
•
The ME/CFS immune system is profiled by scRNA-seq at baseline and after provocation
-
•
Monocyte dysregulation is prominent and dysregulation correlates with disease severity
-
•
Platelets are also dysregulated, but this dysregulation resolves after provocation
Vu et al. examine changes in gene expression in immune cells of ME/CFS patients. The authors highlight monocyte dysregulation as a prominent feature of the immune system in ME/CFS, and this dysregulation correlates with disease severity.
Introduction
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is a serious human disease that lacks effective treatment options and affects ∼65 million individuals worldwide.1,2 Our minimal understanding of ME/CFS hinders diagnosis, rational approaches to treatment, and development of a cure. Multiple lines of evidence implicate a role for the immune system in ME/CFS.3 For example, transforming growth factor β, a cytokine that confers both pro- and anti-inflammatory signals depending on the microenvironment,4 has been reported to be upregulated in the plasma of ME/CFS patients.5,6 Elevated levels of multiple proinflammatory cytokines have been correlated with disease severity, and activation patterns of pro- and anti-inflammatory cytokines differentiate early cases versus those of longer duration.7 Together, these results suggest that ME/CFS involves the dysregulation of immune cells.
Immune cells of both the innate and adaptive systems are thought to be dysregulated in ME/CFS. Monocytes and natural killer (NK) cells have been reported to exhibit alterations in proportions and surface markers in ME/CFS.8,9 Neutrophils exhibit elevated apoptosis in ME/CFS patients.10 T lymphocytes also display abnormal metabolism and cytokine production,11 and other studies have implicated B cell alterations in ME/CFS.12,13 However, previous studies have examined immune cells in isolation and often across small cohorts. Moreover, heterogeneous immune cell populations have typically been characterized as bulk samples, compromising the ability to distinguish changes that affect only specific cell subsets. These limitations, together with the heterogeneous nature of the disease,14 have likely contributed to the contradictory results between studies.15 Thus, at present, it is far from clear which components of the immune system are the most relevant to ME/CFS.
A defining symptom of ME/CFS is postexertional malaise (PEM), an exacerbation of symptoms resulting from exertion. Understanding changes that occur in immune cell populations during PEM could provide useful insights into the disease, and the development of potential treatment and preventive approaches. An established method of inducing PEM in ME/CFS is with cardiopulmonary exercise tests (CPETs), which monitor multiple parameters during a controlled exercise period with increasing intensity until exhaustion or limiting symptoms appear.16 Incorporating CPET into ME/CFS studies provides objective physiological parameters, increasing confidence in diagnosis.
ME/CFS has been proposed to result from infection by an unknown virus (or other pathogen), leading to the disease in susceptible individuals. This theory derives from the many occurrences of clustered cases of the disease,17 together with the observation that many patients reported experiencing a flu-like or other infection before disease onset. It is unknown whether only specific viruses can trigger ME/CFS or whether many viruses can induce the disease, although accumulating evidence implicates enteroviruses.18,19 Notably, ME/CFS and long COVID (coronavirus disease 2019) share many, although not all, symptoms20; thus, there may be commonalities between the diseases, including a viral origin. However, the degree to which molecular signatures are shared by ME/CFS and long COVID is unknown. Therefore, the knowledge gained in understanding ME/CFS may be of significance regarding long COVID and vice versa.
Here, we used single-cell RNA sequencing (scRNA-seq) to examine immune cells within peripheral blood mononuclear cells (PBMCs) from ME/CFS patients and matched controls. Our goals were to identify immune cell types with dysregulated transcriptomes in ME/CFS, and also recognize cell types that show no substantial differences between the patient and control cohorts to rule out their involvement in the disease state. Finally, because PEM is a defining symptom of ME/CFS, we performed scRNA-seq both before and after exercise challenge (CPET), with the goal of characterizing changes that occur during PEM. We observed extensive alterations in the transcriptome of a subset of monocytes in ME/CFS patients, with largely identical alterations present at baseline (BL) and 24 h after the exercise challenge. Indeed, most gene dysregulation across the ME/CFS immune system was consistent between the BL and postexercise conditions. In contrast to this general property of the ME/CFS immune system, we observed marked differences in platelet transcriptomes from patients at BL compared to post-CPET (PC), suggesting that aberrant platelet activity is associated with PEM. Taken together, this study identifies cell types with distinctive patterns of transcriptome dysregulation; these data suggest new hypotheses for understanding ME/CFS and the role of PEM.
Results
Single-cell transcriptomics of the ME/CFS immune system
Despite evidence that immune dysregulation is a major feature of ME/CFS, the components of the immune system most involved in the disease are unknown. To address this question, we used the 10X platform to perform scRNA-seq to profile ∼5,000 PMBCs per sample from a cohort of 30 patients and 28 controls, matched for sex and BMI and differing in parameters characteristic of ME/CFS (Figures 1A and 1B; Data S1). Samples were obtained before the COVID-19 pandemic, eliminating the possibility that any patients had long COVID rather than ME/CFS. Because PEM is a defining symptom of ME/CFS, we profiled samples from all of the individuals at BL and 24 h after a strenuous exercise challenge21,22 (PC; Figure 1A). This design has the potential to define gene dysregulation in immune cells at BL in patients as well as dysregulation associated with PEM. We used a two-step strategy to sequence each library to equivalent coverage,. First, we sequenced each scRNA-seq library at low coverage (averaging 57 million reads) and used these data to determine the sequencing depth required to obtain a depth of over 4,000 unique molecular identifiers per cell, for a total average sequencing depth of 136 million reads per sample. Datasets were integrated with Seurat version 423 to generate a landscape for cell-type annotation and comparisons between samples. Standard scRNA-seq metrics indicated robust profiling across all of the samples (Figures 1C and S1A–S1C; Data S2). To associate scRNA-seq clusters with cell identities, we used Seurat to identify marker genes, and also examined the expression of established marker genes, which allowed us to identify the majority of the 28 clusters (Figures 1D and 1E).
Figure 1.

Single-cell transcriptomics of the ME/CFS immune system
(A) Study design. PBMCs were collected at BL and 24 h PC for sedentary controls and ME/CFS subjects and used for single-cell gene expression profiling (scRNA-seq).
(B) Demographic and clinical parameters for both cohorts. Oxygen consumption was measured during CPET. The change in maximal oxygen consumptions (VO2 at peak) between the VO2 peaks at BL and PC is indicated. Using the SF-36 Version 2 Health Survey, the general health score was self-evaluated, with 100 as perfect health and 0 as worst health. Graphs represent mean ± SEM. ∗∗p < 0.01, ∗∗∗ p < 0.001.
(C) Quality control metrics, showing genes and transcripts per cell (left and center, respectively) and percentage of mitochondrial reads per cell (right), compared between indicated cohorts.
(D) Integrated uniform manifold approximation and projection (UMAP). Clusters are labeled in order of decreasing number of cells.
(E) Relative expression of marker genes for immune cells (x axis) across clusters (y axis); dots indicate average expression and percentage of cells with detected expression (color and size, respectively).
(F) Cell types with significant differences in relative cell numbers between cohorts. ∗p < 0.05. Panels represent data from 28 healthy controls and 30 ME/CFS cases.
Previous studies have suggested that immune cell composition is altered in ME/CFS.9,24,25 No clusters were specific to the patient cohort, nor were any largely underrepresented in this cohort (Figures S1C and S1D). We compared the proportions of each cell type (cluster) between patients and controls, analyzing BL and PC samples separately. As expected,26 we observed interindividual heterogeneity in the proportions of immune cells, with individual profiles highly consistent at both time points (Data S3). However, there were only limited differences in proportions when we compared patient and control cohorts (Figures 1F and S1D). Proportions of regulatory NK cells (cluster 15) were elevated in patients (1.1- and 1.3-fold at BL and PC, respectively; p = 0.23 and 0.02, after multiple comparison correction), as were γδ T cells (1.3-fold at BL and PC; p = 0.05 and 0.04, respectively). No other proportions of immune cells were significantly altered, except for a marginal decrease in effector/memory CD8+ T cells in patients (0.9-fold, at both time points; p = 0.04 and 0.09). We used flow cytometry to quantify the proportions of major subsets of monocytes and T cells in patients and controls, which demonstrated no substantial changes between cohorts (Figure S2). We conclude that cell types present in PBMCs show very little difference in their relative frequencies in ME/CFS, and that any differences that do exist are minor compared to normal interindividual variations.
Dysregulation within the ME/CFS immune system
To examine transcriptome dysregulation in the ME/CFS immune system, we used Seurat version 423 to determine the number of dysregulated genes per cell type, comparing all of the cells from patients to those from controls, and performing the comparisons separately at BL and PC. Certain cell types exhibit strong signals of transcriptome dysregulation in patients, with CD4+ T cells (naive and effector/memory subsets; clusters 0 and 1, respectively), monocytes (clusters 2, 9, and 10) and cytotoxic NK cells (cluster 3) being the most prominent (Figure 2A). There are also multiple cell types, including those present in high proportions, that show very little evidence of dysregulation associated with ME/CFS. This approach has more power to detect changes in gene expression for cell types present in higher proportions, and may have high false discovery rates.27 As a gauge with which to contrast dysregulation associated with ME/CFS, we also compared control samples between the two time points, anticipating that any signal detected for such a comparison would represent noise, at least for most cell types. This comparison revealed negligible signal, in terms of numbers of differentially expressed genes (Figure 2A), increasing confidence that the signal detected in patient versus control comparisons is meaningful.
Figure 2.

Dysregulation of immune cells in ME/CFS
(A) Counts of differentially expressed (DE) genes (y axis) per immune cell cluster, comparing case and control cells at BL and PC, and compared between control cells at BL and PC.
(B) Counts of the strongest significantly enriched gene sets (excluding those related to translation and with an absolute normalized enrichment score [NES] <2) across the largest cell clusters.
(C) Representative GSEA gene set (GOBP cytoplasmic translation) showing lower detection of ribosomal proteins in major clusters (NES <0) at both BL and PC in ME/CFS (red and purple, respectively). Dots are sized to denote significance (q values); x axis indicates NES.
(D) GSEA results for classical monocytes (cluster 2) comparing patient and control cohorts at BL and PC (red and purple, respectively), focusing on gene sets related to chemokine/cytokine signaling.
(E) Single sample scores generated using GSEA of leading-edge genes from (D). ∗∗p < 0.01.
(F) Differential expression of genes associated with monocyte migration and differentiation at BL and PC in classical monocytes. ∗p < 0.05; ∗∗p < 0.01. Panels represent data from 28 healthy controls and 30 ME/CFS cases.
Multiple frameworks exist to detect differential gene expression in scRNA-seq. Recent approaches have indicated that “pseudobulk” methods (aggregating expression across cells per cluster) can outperform Seurat.27 However, we found that this approach had limited power to identify individual genes with significant differences between case and control samples in different cell types, likely because interindividual variation in gene expression unrelated to disease state is a large confounding factor, and the observation that the largest variation in the dataset can be attributed to sex (Figure S3A). Previous studies have identified sex-specific changes in RNA or microRNA profiles from PBMCs in ME/CFS.28,29 We also performed correlation analyses between gene expression profiles of classical monocytes from all of the female samples and identified higher correlations within the same subject (self) over interindividual comparisons. Notably, interindividual variations among cases were higher than controls, consistent with ME/CFS heterogeneity (Figure S3B).
As an alternative approach to search for coordinated shifts in gene expression reflecting alterations in pathway activation or cell state in ME/CFS, we performed gene set enrichment analysis (GSEA) on each cluster, focusing on C1: HALLMARK, C2: REACTOME, C2: KEGG, and C5: GOBP (biological process) catalogs from the Molecular Signatures collection.30,31 This analysis (Figure 2B; Data S4) indicated two general features: first, expression of ribosomal protein genes and core translational machinery is downregulated in ME/CFS in multiple cell types (Figure 2C), and second, that monocytes exhibited the strongest signals of dysregulation, especially in cluster 2 (classical monocytes) but also in clusters 9 and 10, nonclassical and intermediate monocytes, respectively (Figures 2B and S3C; Data S5). In particular, gene sets associated with chemokine signaling, migration, and activation are expressed at elevated levels in patient classical monocytes (Figure 2D). Furthermore, we observed the suppression of genes associated with interferon gamma (IFN-γ) signaling in patient classical monocytes. These results suggest that classical monocytes from ME/CFS individuals are biased toward a profile that promotes migration of monocytes to tissue and increased progression toward a macrophage fate. However, we also observed the activation of genes associated with interleukin-10 (IL-10), an anti-inflammatory cytokine. Thus, monocytes in ME/CFS patients may undergo a combination of conflicting inputs.
To explore signals that may be differentially affecting monocytes in patients, we examined the identities and expression levels of genes driving the enrichments observed. Using the genes most responsible for enriched chemokine and cytokine signaling gene sets (Data S6), we calculated a composite score for classical monocytes from each sample (Figure 2E; monocyte clusters in Figure S3D). At the level of individual samples, classical monocytes from ME/CFS have higher scores for these genes compared to controls, and these scores are consistent between BL and PC time points for each individual, indicating no impact from the exercise challenge (Figure S3E). We also looked at the differential expression of genes involved in monocyte response, recruitment, and differentiation at BL and PC. Specifically, we observed the upregulation of CCL4, CX3CR1, SELPLG, and ITGAL in patients, whereas CXCR4 expression was suppressed at BL (Figure 2F). CCL4 is a chemoattractant for monocyte recruitment to inflamed and adipose tissue32; CCL4 was upregulated regardless of exercise challenge. CX3CR1, the CX3CL1 receptor, is a marker for the differentiation of classical monocytes to nonclassical counterparts and tissue repair and regeneration.33,34 SELPLG and ITGAL play roles in the tissue recruitment of leukocytes.35 CXCR4, a receptor for CXCL12, regulates monocyte-macrophage differentiation.36 We also detected the upregulation of both inflammatory (TNFRSF1A, TLR4, TLR5) and anti-inflammatory (IL10RB) receptor genes PC. Taken together, these results suggest that monocytes in ME/CFS are aberrantly promigratory and inflammatory at BL and that ME/CFS is characterized by a persistent state of monocyte activation.
Profiling of classical monocytes from ME/CFS patients and controls
To generate a comprehensive profile of classical monocyte dysregulation in ME/CFS and validate our scRNA-seq results, we turned to RNA-seq of purified monocytes. Starting with PBMCs from four female patients and four female controls isolated PC, with individuals distinct from those profiled by scRNA-seq, we isolated classical monocytes (CD14+CD16−) and confirmed that the isolation strategy was effective (Figure S4A). Following RNA isolation, we generated RNA-seq libraries; principal-component analysis (PCA) demonstrated separation between patients and controls, although the patient samples were more dispersed than the controls (Figure 3A). We compared expression profiles of classical monocytes from patients and controls (Figure S4B) and identified enrichment of similar GSEA terms and leading-edge genes between the RNA-seq (Figure 3B; Data S7) and scRNA-seq data (Figure 2D; Data S5), such as regulation of cell migration by cytokines and chemokines and IL-10 signaling. These observations confirmed a pattern of monocyte activation and migration in ME/CFS in an independent cohort of samples. We also examined expression levels for a set of genes associated with monocyte migration and differentiation (Figures 3C and S4C). Perhaps due to heterogeneity in ME/CFS cases (Figure 3A), we found consistent changes only in CCL4 upregulation and CXCR4 downregulation, when examining both the RNA-seq for classical monocytes and the corresponding scRNA-seq cluster (Figures 2F and 3C). However, using the RNA-seq data alone, we identified additional changes, including most prominently upregulation of CSF2 (granulocyte-monocyte colony-stimulating factor, an important monocyte differentiation cytokine). Notably, CSF2 is upregulated in plasma and extracellular vesicles of ME/CFS patients and this upregulation correlated with disease burdens.6,37 Transcripts encoding multiple additional chemokines important for monocyte recruitment (CCL20, CCL5, and CCL3)38 were also upregulated in ME/CFS classical monocytes. These analyses support the hypothesis that monocytes in ME/CFS patients are aberrantly primed to migrate to tissues.
Figure 3.

Complete transcriptomics of classical monocytes
(A) PCA of 4 patient and 4 control transcriptomes from classical monocytes.
(B) Significantly enriched gene sets between cohorts by GSEA. Dots are sized to denote significance (adjusted p values); x axis indicates NES.
(C) Differentially expressed genes between cohorts. Dots are sized to denote significance (p values).
(D) PCA of 3 patient and 4 control proteomes from classical monocytes.
(E) Significantly enriched gene sets between cohorts by GSEA comparing proteome profiles of cases and controls; otherwise as in (B).
(F) Volcano plot of differentially expressed proteins between cohorts. Cohorts of 4 healthy controls and 4 ME/CFS cases (all females) at BL and PC were chosen for proteome (D–F) and transcriptome (A–C) profiling, respectively.
To further validate dysregulation of classical monocytes in ME/CFS, we examined the proteome of classical monocytes from three female patients and four female controls at BL. PCA of the proteomic data showed a clear separation between cases and controls coincident with PC2 (Figure 3D). Interestingly, PC1 separated a single case from all of the other samples, and this case had the highest metrics of disease severity. Proteins driving PC1 include EPX, PRG2, RNASE2, and RNASE3, factors and components of secretory vesicles (Figure S4D). This trend was observed in the RNA-seq dataset in which the genes driving the differences within ME/CFS cases were mostly made up of membrane-associated and secretory protein transcripts (CCL4, CCL20) (Figure S3E).
GSEA comparing the proteomes of classical monocytes from controls and cases confirmed the elevated activation state of classical monocytes in ME/CFS, with the upregulation of pathways such as defense response to bacteria, secretory vesicle formation, and degranulation (Figure 3E; Data S8). We also observed downregulation of translational machinery in the proteome data (Figure 3E). Looking at individual proteins, we found the upregulation of proteins involved in iron metabolism (FTL1), monocyte-to-macrophage differentiation (CNN2, SERPINA1, DCTN1, NUDT21, PLD3), immune cell recruitment (ALDH3B1) and activation (HVCN1, NCF1, PPIA), and platelet activation (STXBP5) (Figure 3F) in cases. Iron metabolism, a tightly regulated process, is associated with immune response; FTL1 is central to this process, functioning to control Fe2+ levels during inflammation, thereby preventing the formation of reactive oxygen species.39 CNN2, SERPINA1, DCTN1, NUDT21, and PLD3 regulate monocyte differentiation into macrophages in different tissues and diseases.40,41,42,43,44 ALDH3B1 induces ITGB1 expression and promotes tissue recruitment.45 HVCN1 upregulation is associated with CNS injury,46 whereas NCF1 is a factor in autoimmune encephalomyelitis.47 Notably, we observed the upregulation of STXBP5, which correlates with platelet activation.48 However, we found proteins involved in translation (RTRAF, RPS2), mitochondrial functions (HADHB), and cell-to-cell interaction (JUP, TPD52L2) to be downregulated in ME/CFS classical monocytes (Figure 3F). To confirm that the results are not driven by a single outlier sample, we repeated these analyses, omitting the patient sample coincident with PC1. As expected, PCA showed a clear separation of controls and cases (Figure S4F), and the GSEA results were largely identical (Figure S4G; Data S9). Similarly, when we examined the changes in the expression of individual proteins, we found that almost all of the upregulated proteins were congruent with and without the outlying sample (Figures 3F and S4H, respectively; Data S10), except for HVCN1. However, when we examined the proteins downregulated in ME/CFS classical monocytes, only RTRAF was found to be significantly downregulated in the more stringent dataset (Data S10). In summary, the upregulation of activation status in monocytes in ME/CFS is recapitulated at the protein level, validating inferences made from the transcriptome analysis.
Monocyte dysregulation is heterogeneous within and between ME/CFS patients
Our data identify classical monocyte dysregulation as a prominent feature of ME/CFS. However, it is unknown whether this feature derives from consistent alterations across monocytes, or alternatively, is restricted to a subset of cells per individual. Similarly, transcriptome profiling of monocytes from patients suggests more extensive variation between patients than between controls (Figure 3B), consistent with patients possessing a heterogeneous monocyte population, in contrast to a more homogeneous population in controls. To explore these possibilities using the classical monocytes single-cell data, we used a machine learning approach, positive unlabeled learning,49 which accommodates mixed populations (Figure 4A). Starting with BL case and control female samples, the algorithm was optimized to label cells as diseased or normal (Figures 4B and S5A). The results indicated that ME/CFS patients possess a heterogeneous classical monocyte population, only some of which are diseased, with the remainder comparable to those in controls (Figures 4A and S5A). The percentage of cells classified as diseased within patients was variable (Figure 4C), but consistent per individual when compared between BL and PC (Figure 4D). We calculated the Calinski-Harabasz (CH) index applied within PCA space, as a metric of performance for the split of predicted diseased and normal groups in comparison with other sample metadata. The CH index analysis demonstrated that cells predicted as diseased are more highly related to one another than those partitioned by disease status, sex, or the identity of an individual (Figure 4E), suggesting that the prediction recovered latent signal within the data.
Figure 4.

Heterogeneity in classical monocyte cells from ME/CFS patients
Data represent BL female samples, unless described otherwise.
(A) Schema describing positive unlabeled learning strategy to stratify single-cell patient transcriptomes.
(B) UMAP for classical monocyte cells, tiled and colored by pD state.
(C) Percentage of pD cells per individual.
(D) Correlation (Spearman) between pD cells per individual, compared between BL and PC.
(E) CH index comparing clustering performance across different stratifications of the single-cell dataset (y axis).
(F) Correlation (Spearman) between MFI-20 score and percentage of pD cells.
(G) Gene sets most differentially enriched between pD and pN cells from cases. Dots are color-coded to indicate enrichment in pD (blue) or pN (yellow) cells; sizes indicate corrected p values.
(H) Genes (y axis) most differentially expressed (x axis) between pD and pN cells in an intrasample paired analysis.
(I) Expression of CCL4 (y axis) from individual samples, aggregating expression over pD and pN cells from cases and over all cells from controls (x axis and color-coded). ∗∗ p < 0.01, ∗∗∗ p < 0.001.
(J) Top gene sets differentially enriched between pD and pN cells, based on GSEA between each subset of cells paired by sample.
(K) Pseudobulk PCA from aggregated cells from control samples (yellow); pN and pD cells from case samples (green and blue, respectively), partitioned by sex.
(L) Genes contributing to negative values for PC2 (top) and PC3 (bottom) in (K).
(E, I, K, and L) Data from 28 healthy controls and 30 ME/CFS cases at BL, with partition based on sex in (I) and (K). (B–D) and (F–H) Data from the female cohort with 20 healthy controls and 20 ME/CFS cases at BL.
We examined correlations between the proportion of aberrant monocytes per individual and patient health parameters. We observed a correlation between the fraction of classical monocytes identified as diseased and patients’ Multidimensional Fatigue Inventory (MFI)-20 score50 (Figure 4F), an evaluation of fatigue. Similar correlations were observed with other metrics, including general health, Short Form (SF)-36 Physical Component Summary scores,51 and PEM severity (Figures S5D–S5F). Similar trends were observed for male samples (Figures S5B, S5C, S5G, and S5H). These observations indicate a relationship between the symptoms that ME/CFS patients experience and the fraction of diseased monocytes.
To examine the dysregulation of predicted diseased (pD) cells, we used GSEA to examine patterns of differential expression between patient cells predicted as normal (pN) versus pD. The transcriptome profiles from pD cells compared to pN cells exhibited changes in the expression of pathways involving cytosolic DNA sensing, cytokine–cytokine receptor interaction, and chemokine signaling (Figure 4G), which were similarly observed when comparing profiles of all patient cells to healthy controls (Figure S5I). In addition, ribosomal gene sets were downregulated in pD cells (Figures 4G and S5J), consistent with observations across multiple cell types (Figure 2C).
To minimize the impact of interindividual variation in gene expression unrelated to disease state and confidently identify genes dysregulated due to ME/CFS, we performed a paired analysis by individual of origin. We calculated the pseudobulk gene expression changes between pD and pN monocytes from the same ME/CFS individual, and then averaged these ratios across individuals. In this analysis, CCL4 (C-C motif chemokine ligands 4) exhibited the strongest change, with elevated expression in pD cells within the same individuals (Figure 4H). We also observed differences in CCL4 expression levels when comparing profiles of pD and pN from patients to controls (Figure 4I). Elevated expression of CCL4 in pD cells was more prominent in female patients than in male patients (Figure 4I). Furthermore, TMEM176B (Transmembrane Protein 176B) was downregulated in pD cells compared to pN cells in ME/CFS individuals as well as control cells (Figures 4H and S5K). TMEM176B is involved in maintaining the immature state of dendritic cells (together with TMEM176A), which is anti-inflammatory.52 Other genes upregulated in pD cells included GIMAP7 and TRIM7, which may contribute to cell survival by suppressing apoptosis53 and promoting inflammation.54 The identities of genes downregulated in pD cells, such as OLR1, G0S2, ATF3, RGCC, and TMEM176B contain both proinflammatory and anti-inflammatory factors,55,56,57,58,59 suggesting that the environment eliciting inflammatory responses is complex, with multiple ME/CFS-specific changes in signaling. For example, ATF3, downregulated in pD cells, is a regulator of IFN responses and able to suppress CCL4 in animal models.57,60 We also performed GSEA using the paired analysis data (Figure 4J). The IFN-γ pathway was upregulated in pN cells, suggesting reduced inflammatory responses of pD cells through this pathway (as in Figure 3C).
Finally, we used PCA to visualize sex-specific pseudobulk transcriptomes generated from control monocytes and from patient cells partitioned into pD and pN. Transcriptomes derived from pD cells clustered away from control cells and from pN cells (Figure 4K). In particular, PC1 reflected the sex of the individuals (Figure S5L), with PC2 and PC3 coincident with pD state for males and females, respectively (Figure 4K). These observations suggest that the case cells were separated into two groups: pD cells distinct from control cells and pN cells that are less different from but still show some deviation from control cells. Analysis of principal-component loadings (Figure 4L) show that CCL4 has a strong negative contribution to both PC2 and PC3, indicating that dysregulation of CCL4 contributes strongly to discrimination between pD and pN cells in males and females. Thus, within patients, a subset of classical monocytes exhibited a high expression of cytokine receptor and chemokine signaling genes, in particular, CCL4. Overall, these analyses demonstrated that classical monocyte populations are heterogeneous within and variable across ME/CFS patients. The machine learning algorithm partitioned monocytes in ME/CFS patients into two groups, which enabled us to identify pD cells in patients and establish that these cells upregulate the expression of specific cytokine receptor and chemokine signaling genes indicative of aberrant monocyte recruitment to tissue. A key observation is that the percentage of pD cells per individual correlated with metrics of disease severity (Figures 4F and S5F).
Signaling pathways affecting monocytes in ME/CFS
Because monocytes in ME/CFS patients show dysregulation in proinflammatory chemokine and cytokine signaling pathways, they may be intrinsically biased toward this premigratory transcriptome; alternatively, they may be responding to changes in intercellular signaling. To assess whether intercellular signaling is altered in ME/CFS, we used CellChat61 to analyze scRNA-seq data to model intercellular communication as a function of ligand and receptor expression levels62 (Figure 5A). To remove confounding factors such as sex, exercise, and cluster size, we focused on the larger female-only cohort at BL and downsampled large clusters to 500 cells before calculating communication probabilities, a recommended approach.63 Comparing the interactomes of patient and control cells, we found evidence suggesting alterations in inferred communication to and from several cell types. In particular, monocyte signaling to certain T and NK cells is predicted to be elevated in ME/CFS, while also increasing signaling interactions among themselves (Figures 5B and S6A). These results suggest that monocyte dysregulation in ME/CFS in part derives from alterations to established intercellular communication pathways.
Figure 5.

Intercellular signaling in the ME/CFS immune system
(A) Schema depicting CellChat strategy.
(B) Circle plot showing differential number of interactions (case minus control), aggregating clusters of similar cell types. Blue indicates case cells exhibit more interactions than control cells; orange indicates control cells exhibit more interactions.
(C) Scatterplot of differential incoming versus outgoing interaction strength in classical monocytes (cluster 2). Positive values indicate increased signaling strength in patients and vice versa.
(D) Heatmap of overall signaling for pathways dysregulated (y axis) for classical monocytes receiving signaling from different cells (y axis; cluster identifiers from Figure 1). Top bar plot indicates aggregate interaction strength of incoming signals; right bar plot indicates aggregate interaction strength of outgoing signals.
(E) Communication probabilities between specific ligand-receptor pairs in the CCL pathway, for case (blue) and control (orange) cells. Panels represent data from the female cohort, with 20 healthy controls and 20 ME/CFS cases at BL.
We next investigated the pathway identities that may contribute to alterations in the interactome of ME/CFS classical monocytes. Among the patients, we found evidence of increased signaling in pathways that regulate monocyte survival and localization to inflamed tissue (Galectin, Resistin, CSF3, CCL),7,64,65 with the strongest dysregulation observed in genes associated with the C-C motif chemokine (CCL) pathway (Figures 5C, 5D, and S6B). The upregulation of CSF2, CFS3, RETN, CCL3, CCL4, and CCL5 was also observed in the independent cohort analyzed by RNA-seq (Figure S6C). To identify the ligand-receptor pairs contributing to changes in signaling in ME/CFS and with cell-type resolution, we compared communication probabilities between the patient and healthy cohorts, finding 782 upregulated and 1,046 downregulated ligand-receptor pairs in patients, aggregated across all pairwise combinations of cell types (Data S11). In particular, for ligand-receptor pairs associated with the CCL pathway, we identified increased signaling from classical monocytes to platelets in ME/CFS patients; this signal derived from elevated CCL3/CCL5 expression in monocytes (Figure 5E). Monocyte–platelet interactions have been demonstrated to be a potent marker of inflammation.66,67 Thus, classical monocytes in ME/CFS patients may exist in an inflammatory state in part due to altered cross-talk between platelets and monocytes.
An abnormal platelet state is coincident with PEM
PEM is a defining symptom of ME/CFS. To investigate whether PEM is associated with changes in immune cells, we compared expression changes across cells between BL and 24 h after CPET. Conventional analysis of differential gene expression comparing the average PC expression to BL did not identify significant genes within cases or controls. Because our study includes samples collected before and after exercise from the same individual, we could leverage a paired analysis to reduce interindividual variation, which can compromise signal detection. For each cell type (cluster), we calculated the expression ratio for each gene per individual in response to exercise, retaining genes that met detection criteria. This approach generates a ΔRNA metric per individual, which we compared between patient and control cohorts (Figure 6A). Tests for differential expression of individual genes again failed to reach significance. However, GSEA, which can detect coordinated shifts in gene expression across genes, showed a marked number of enriched gene sets in platelets, with minimal signal or no signal in other cell types (Figure 6B).
Figure 6.

Aberrant platelet transcriptomes coincident with PEM in ME/CFS
(A) Schema depicting paired analysis (intraindividual expression) of gene expression altered by strenuous exercise in ME/CFS patients compared to controls.
(B) Number of significantly enriched gene sets across clusters (x axis) in a paired analysis comparing case and control ΔRNA measurements with GSEA.
(C) GSEA results for significantly enriched gene sets related to platelet function. GSEA analyses included the comparison of paired ΔRNA measurements between cases and controls (dark purple for scRNA-seq and pink for RNA-seq), as well as comparing group averages at BL (red for scRNA-seq and blue for RNA-seq) and PC (none detected).
(D) Enrichment plot depicting representative significantly enriched gene sets related to platelet function and translation using the intraindividual paired analysis for platelets (cluster 19) from scRNA-seq.
(E) Heatmap of leading-edge genes taken from the gene set “GOBP regulation of platelet activation” between 3 healthy controls (HC) and 3 ME/CFS cases (ME) at BL and PC; expression values row-normalized.
(F) Enrichment plot depicting the same gene sets shown in (D) using the intraindividual paired analysis of bulk RNA-seq of platelet-derived plasma particles.
(B–D) Data from a cohort of 28 healthy controls and 30 ME/CFS cases. (E and F) Data from 3 healthy controls and 3 ME/CFS cases (all females) at both time points.
Gene sets related to platelet function were significantly enriched in the paired analysis of platelets, with positive enrichment in case ratios (PC/BL) compared to controls (Figures 6C and 6D). To examine the behavior of platelets at each time point, we repeated the GSEA analysis directly comparing ME/CFS patients and control cohorts at BL and PC. This analysis revealed reduced expression of platelet-function gene sets at BL in ME/CFS (Figure 6C). Interestingly, these enrichments were not statistically significant in the PC samples. These analyses revealed that the platelet cluster manifests a strong signature of dysregulation only before exercise in ME/CFS, an observation which suggests that strenuous exercise alters platelets in ME/CFS individuals.
Other notable gene sets in the paired GSEA analysis for platelets shared a high number of ribosomal protein genes (Figure 6D; Data S12). Again, this enrichment derived from differences at BL, with no significant enrichment in the PC cohorts. Therefore, the negative enrichment scores for the paired analysis reflects an increase in the detection of ribosomal proteins and translation machinery at BL in ME/CFS. Notably, no other cell types exhibited a significant change due to exercise in the paired GSEA analysis (Figure 6B).
To validate the scRNA-seq findings, we isolated large particles from frozen plasma samples from 3 healthy controls and 3 ME/CFS cases at BL and PC (Figure S7A) for further analysis. Samples were centrifuged at low speed to enrich platelets and platelet-derived aggregates and eliminate any contamination from other, smaller circulating vesicles. We confirmed the identity of the isolated particles by flow cytometry, which revealed that the particles were positive for the platelet marker CD41 and devoid of leukocyte (CD45) and erythrocyte (CD235a) markers (Figure S7B). We conducted RNA-seq on these isolated plasma particles. Consistent with the scRNA-seq GSEA results, we found the transcriptomes of the platelet-derived plasma particles from ME/CFS cases are different from healthy controls at BL (Data S13). However, no significant differential expression was observed between PC case and control samples. GSEA analyses of the platelet-derived plasma particle RNA-seq data recapitulated results observed in the scRNA-seq analysis. In particular, platelet function–related terms were downregulated at BL and upregulated in a paired analysis controlling for individual of origin (Figures 6C and 6F; Data S14). These trends were consistent across patients and demonstrated that the transcriptomes at the PC time point from patients shifted toward gene expression profiles more similar to controls for genes relating to platelet function (Figure 6E). We also detected a consistent trend of increased translation activity in the plasma particles of ME/CFS patients at BL relative to PC, appearing as negative enrichment in the paired GSEA analysis (Figures 6F and S7C). Thus, the ME/CFS platelet transcriptome at BL is abnormal, but following strenuous exercise, platelets in the circulation manifest markedly less dysregulated transcriptomes.
Discussion
This study provides a resource to investigate immune dysregulation in ME/CFS. Here, because classical monocytes manifested the strongest signal of dysregulation in PBMCs in ME/CFS patients, we focused on exploring changes in their gene expression program, as a novel aspect of the disease. However, alterations to additional immune cells are also evident, with the strongest signal observed in certain T cell subsets. Patterns of dysregulation limited to adaptive immune cells specific to a particular antigen are difficult to detect in scRNA-seq data. Nevertheless, it is worth noting that we observe changes in gene expression in clonally diverse antigen-experienced CD4+ T cells and in other adaptive cell populations, an observation consistent with the dysregulation of adaptive immune cells occurring via cytokine-mediated bystander regulation68 rather than via antigen-specific interactions. Nevertheless, the largest alterations in ME/CFS PBMCs are found in monocytes.
Multiple lines of evidence implicate the dysregulation of classical monocytes in ME/CFS. Our analysis discovered the upregulation of chemokine/cytokine pathway genes in patient-derived monocytes as well as a correlation between the proportion of pD monocytes and disease severity. Future studies investigating macrophages from tissue biopsies from ME/CFS patients will be important to investigate the impact of altered monocytes. CellChat analyses implicated alterations in intercellular communication within the ME/CFS immune system. In contrast to controls, in which a diverse network of intercellular interactions is predicted, monocytes are predicted to dominate the overall information exchange in ME/CFS. This observation may be a consequence of prolonged exposure to an inflamed environment in ME/CFS, which can alter cellular metabolism and functions.69 We also detected conflicting pathways related to immune response as upregulated in classical monocytes—specifically, both proinflammatory and anti-inflammatory responses. These observations call for more extensive characterization of classical monocytes in ME/CFS to identify the overall response of such cells in an abnormal disease state. Future studies that integrate plasma cytokine analysis with monocyte/macrophage function will be particularly valuable.
How might aberrant monocyte activation contribute to symptoms experienced by ME/CFS patients? Monocytes express multiple chemokine receptors, which in response to different chemokines direct monocytes to a variety of tissues. We observed an increased expression of CCL3 and CCL4, which direct monocytes to joints and adipose tissues in osteoarthritis and adipose tissue in obesity.32,38 Thus, our observations suggest that ME/CFS patients experience continual improper recruitment of monocytes to one or more tissues. If this hypothesis is correct, then it will be important to examine the balance of pro- and anti-inflammatory macrophages in patient tissues, and whether the macrophages themselves are altered.
A previous study identified aberrant platelet activity in ME/CFS,70 although another study did not observe platelet alterations.10 There is evidence of aberrant platelet activation in long COVID.71 Our analysis suggests a refinement to the hypothesis linking platelet activation to ME/CFS: although aberrant activation may be present at BL, it appears that the platelet population in circulation undergoes a substantial change in response to strenuous exercise, and therefore a clear association with PEM. At BL, the patient platelet transcriptome is biased toward a lower expression program of genes important in platelet activation and an increase in transcripts encoding translational machinery. Following exercise, the platelet transcriptome in patients shifts toward the normal profile, indicating either a loss of platelets harboring defective transcriptomes or an infusion of new platelets. We envision two models to explain these observations. In the first model, perhaps a subset of platelets in patients are susceptible to activation, and strenuous exercise induces microclot formation, removing them from circulation; thus, PC, only normal platelets remain. The second model envisions that exercise induces an influx of normal platelets, perhaps in combination with clearance (or clot formation) of older platelets.
Platelets, lacking a nucleus, have unconventional transcriptomes. Upon their release into circulation, platelets inherit the transcriptome of their megakaryocytes of origin. Therefore, the platelet transcriptome degrades without replenishment as platelets circulate. The rate of degradation in vitro shows a fast degradation, with half of the RNA lost after 6 h and almost all at 24 h.72 Selective degradation of the platelet transcriptome has been reported, in which transcripts encoding the translational machinery degrade slower than others.73 Our analysis shows that platelets at BL in ME/CFS subjects possess a transcriptome indicative of older platelets, with a reduction in transcript levels for genes essential to platelet activation and function and an increase in ribosomal protein genes and other genes relevant to translation. However, the exercise challenge erases such signals. In healthy individuals, acute exercise has been reported to activate platelets74 and upregulate both proinflammatory and anti-inflammatory cytokines,75 but prior studies have not explored the effect of exercise on platelet function in ME/CFS.
We note that aberrant platelet activation and fibrin amyloid microclots have been reported in patients with long COVID76 as well as in ME/CFS patients, although the microclot load was found to be smaller in the ME/CFS cohort.70 These studies correspond most closely to the BL state assessed here, and our results suggest that dysregulation, as judged by transcriptome analysis, occurs at BL. Nevertheless, it is clear that the CPET induces a marked change in the transcriptomes of platelets in ME/CFS individuals.
Distinct from monocyte dysregulation, one of the most prominent features of transcriptome dysregulation in ME/CFS is repression of translational machinery and ribosomal protein genes, which we observed across multiple cell types. Regulation of such genes is complex, involving multiple pathways, including p53 and target of rapamycin,77 although in general, the repression we observe suggests that multiple immune cells exist in a more quiescent or less proliferative state than normal. For example, normal CD8+ T cell activation requires increased translation.78 It is worth noting that both CD4+ and CD8+ T cells in ME/CFS exhibit reduced glycolysis,11 and patient NK cells also have impaired cytotoxic activity.9 Future studies could be designed to systematically test for correlations between these molecular changes, and if such correlations exist, seek a mechanistic understanding of them.
ME/CFS and long COVID, together with other postviral diseases, have been suggested to share common molecular alterations.79 At present, this suggestion is based on an overlapping (although not identical) set of symptoms, rather than molecular data. Our data, describing the circulating immune system in ME/CFS, will be an ideal comparison set for future studies of long COVID, with the potential to identify both congruent and divergent aspects of immune function in ME/CFS and long COVID. In this regard, future studies will determine whether classical monocyte dysregulation is also the most prominent signal in the circulating immune system of long COVID patients, and more important, whether the genes and gene sets affected in ME/CFS are also observed in monocytes from long COVID patients.
Limitations of the study
In this study, we used scRNA-seq to profile the circulating immune system in ME/CFS at BL, and also after symptom provocation. However, PEM can last over an extended amount of time, and our data are limited to analysis only 24 h PC. Moreover, by focusing on immune cells found within PBMCs, we were unable to determine whether ME/CFS results in alterations to tissue resident immune cells. Our profiling of classical monocytes reveals gene expression signatures indicative of aberrant tissue recruitment of monocytes in ME/CFS. Thus, it will be important to perform functional assays on circulating monocytes and on macrophages from tissues to rigorously test this hypothesis. Moreover, although monocytes exhibit the most pronounced dysregulation in ME/CFS, other immune cells also show significant alterations in ME/CFS. In particular, we see evidence of ME/CFS-specific changes in gene expression in multiple T cell subsets, highlighting the need for further analysis of these cells.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC anti-human CD3 Antibody | Biolegend | Cat#300440; RRID: AB_314060 |
| PerCP anti-human CD45 Antibody | Biolegend | Cat#304026; RRID: AB_893337 |
| PE anti-human CD16 Antibody | Biolegend | Cat#302008; RRID: AB_314207 |
| APC/Cyanine7 anti-human CD14 Antibody | Biolegend | Cat#325620; RRID: AB_830693 |
| Human TruStain FcX™ (Fc Receptor Blocking Solution) | Biolegend | Cat#422302; RRID: AB_2818986 |
| FITC anti-human CD4 Antibody | Biolegend | Cat#357406; RRID: AB_2562357 |
| BV785 anti-human CD8a Antibody | Biolegend | Cat#301046; RRID: AB_11219195 |
| BV650 anti-human CD45RA Antibody | Biolegend | Cat#304136; RRID: AB_2563653 |
| APC/Cyanine7 anti-human CCR7 Antibody | Biolegend | Cat#353212; RRID: AB_10916390 |
| PE/Cyanine7 anti-human CD56 Antibody | Biolegend | Cat#362510; RRID: AB_2563927 |
| PE anti-human CD41 Antibody | Biolegend | Cat#303706; RRID: AB_314376 |
| APC/Cyanine7 anti-human CD235a Antibody | Biolegend | Cat#349116; RRID: AB_2650978 |
| Biological samples | ||
| Human Normal Peripheral Blood Mononuclear cells (PBMCs), 20 mil/vial, Frozen | Human Cells Biosciences | Cat#PBMC-C20M |
| Human Normal Peripheral Blood Mononuclear cells and plasma | Center for Enervating NeuroImmune Disease | Frozen PBMC and plasma |
| Critical commercial assays | ||
| Chromium Single Cell 3ʹ v3 | 10x Genomics | Cat#CG000183 Rev A and Rev B |
| Chromium Single Cell 3ʹ GEM, Library & Gel Bead Kit v3 | 10x Genomics | Cat#1000075 |
| Chromium Single Cell B Chip Kit, 16 rxns | 10x Genomics | Cat#1000074 |
| Classical Monocyte Isolation Kit, human | Miltenyi Biotech | Cat#130-117-337 |
| CD14 MicroBeads, human | Miltenyi Biotech | Cat#130-050-201 |
| Ultra II Directional RNAseq | NEB | Cat#E7760 |
| CD3 Microbeads, human | Miltenyi Biotech | Cat#130-097-043 |
| Deposited data | ||
| Analyzed single cell and bulk RNA seq data | [GEO SuperSeries Accession]: [GSE214284] | |
| Software and algorithms | ||
| Cell Ranger mkfastq v6 | 10x Genomics | RRID:SCR_017344 |
| R package | https://www.R-project.org | RRID:SCR_001905 |
| Seurat v4.1.023 | https://doi.org/10.1016/j.cell.2021.04.048 | RRID:SCR_016341 |
| prcomp | https://doi.org/10.1038/s41592-019-0619-0 | RRID:SCR_014676 |
| DoubletFinder v2.0.380 | https://doi.org/10.1016/j.cels.2019.03.003 | RRID:SCR_018771 |
| DEseq2 (v2.03)81 | https://genomebiology.biomedcentral.com/articles/10.1186/s13059-014-0550-8 | RRID:SCR_015687 |
| DESeq2 (v1.34.0)81 | https://genomebiology.biomedcentral.com/articles/10.1186/s13059-014-0550-8 | RRID:SCR_015687 |
| clusterProfiler (v3.18.1)82 | https://doi.org/10.1089/omi.2011.0118 | RRID:SCR_016884 |
| fgsea (v.1.20.0)83 | https://doi.org/10.1101/060012 | RRID:SCR_020938 |
| Singscore (v1.20.0)84 | https://bioconductor.org/packages/release/bioc/html/singscore.html | N/A |
| ggpubr (v0.4.0) | https://rpkgs.datanovia.com/ggpubr/ | N/A |
| CellChat v1.1.361 | https://doi.org/10.1016/j.celrep.2022.111155 | RRID:SCR_021946 |
| CellChatDB62 | https://doi.org/10.1038/s41467-021-21246-9 | RRID:SCR_021946 |
| MSigDB database (v7.5.1)30 | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp) | RRID:SCR_016863 |
| XGBoost (Python, v0.90) | https://doi.org/10.1145/2939672.2939785 | RRID:SCR_021361 |
| FlowJo | BD Bioscience | RRID:SCR_008520 |
| Cutadapt85 | https://doi.org/10.14806/ej.17.1.200 | RRID:SCR_011841 |
| STAR (v2.7) | dx.doi.org/10.17504/protocols.io.3byl4bjr2vo5/v1 | RRID:SCR_004463 |
| ANOVA | RRID:SCR_002427 | |
| Proteome Discoverer (PD) 2.5 | https://doi.org/10.3390/proteomes9010015 | RRID:SCR_014477 |
| Other | ||
| RPMI Medium 1640 | ThermoFisher | Cat#11875093 |
| Fetal Bovine Serum | ThermoFisher | Cat#10437028 |
| Phosphate Buffered Saline 1x | ThermoFisher | Cat#10010023 |
| DMSO | Sigma Aldrich | Cat#D8418 |
| Histopaque 1077 | Sigma Aldrich | Cat#10771 |
| SepMate Tubes | Stemcell Technologies | Cat#85450 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Andrew Grimson (agrimson@cornell.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
(1)
De-identified human single-cell and bulk RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. The superseries accession number is listed in the key resources table.
-
(2)
Analyzed proteomics data provided by the Cornell BRC Proteomics Facility is included as a supplemental table.
-
(3)
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
(4)
This paper does not report original code.
Experimental model and subject details
Human subjects
The human subjects research described in this publication was approved by the Weill Cornell Medical College and Ithaca College Institutional Review Boards and participants provided written informed consent prior to participation as previously described.86 Participants at three sites participated in cardiopulmonary exercise tests (CPETs), donated blood, and provided demographic information as part of the Cornell ME/CFS Collaborative Research Center. The specific cohort in this study was selected using age, BMI, sex, and peak VO2 from CPETs. Age and BMI ranges were matched to minimize confounding factors between cases and controls (Figure 1B). Sex was considered to ensure that the cohort matched disease prevalence where considerably more females than males report having ME/CFS.87,88 Cases were selected that demonstrated a considerable decrease in peak VO2 over the course of 2 CPETs conducted as part of the larger study86 (Figure 1B), as an objective basis for physiological dysfunction within the ME/CFS participants.21
The Multidimensional Fatigue Inventory (MFI-20)50 was used to assess the level of fatigue in cases. The SF-36v2 Health Survey89 was used to compare general health and quality of life between cases and controls. A modified version of the Chronic Fatigue Syndrome severity score90 was used to measure post-exertional malaise (PEM). The severity score measured PEM on a 0–10 scale over the past month. The MFI-20, SF-36v2, and PEM severity score are all self-reported. The MFI-20, SF-36v2, and past month-PEM severity were completed before visiting the test site to perform the CPETs. Current PEM severity was then serially measured at the test site prior to CPET and then every two days post CPET for at least ten days to measure recovery.
In order to preserve anonymity in the public data repository, the subjects’ demographic information at the time of sample collection is coded into the following bins: age bins (1: ≥18 to ≤35 years, 2: >35 to ≤45 years, 3: >45 to ≤55 years, 4: >55 to ≤70 years); BMI bins (1: ≥0 to ≤18.5, 2: >18.5 to ≤25, 3: >25 to ≤27, 4: >27 to ≤30, 5: >30 to ≤100); duration bins (1: ≥0 to ≤5 years, 2: >5 to ≤100 years); general health bins, MFI20 total bins, and physical component score bins (1: ≥0 to ≤20, 2: >20 to ≤40, 3: >40 to ≤60, 4: >60 to ≤80, 5: >80 to ≤100); post-exertional malaise maximum delta bins (1: ≥0 to ≤2, 2: >2 to ≤10). For speed of ME/CFS onset (mecfs_sudden_gradual), 1 indicates "sudden" and 2 indicates "gradual".
Method details
PBMC isolation
ME/CFS cases and healthy sedentary controls participated in a larger study conducted by the Cornell ME/CFS Collaborative Research Center as previously described.86 This study utilized PBMCs processed from whole blood collected from each participant over the course of two days, separated by a cardiopulmonary exercise test (CPET (Figure 1A).
Whole blood was collected in EDTA tubes and centrifuged on the day of collection for 5 min at 500 rcf prior to removing the plasma fraction. The remaining sample was diluted with equal parts PBS and transferred to a SeptMate Tube (Stemcell Technologies) containing Histopaque-1077 (Sigma-Aldrich). SeptMate Tubes were then centrifuged for 10 min at 1,200 rcf and the buffy coat layer was transferred to a new tube. After two washes with PBS (first wash was centrifuged for 10 min at 120 rcf for without brake; the second was centrifuged for 8 min at 300 rcf), the resulting pellet was resuspended in PBMC storage media (60% RPMI 1640, 30% heat inactivated FBS, and 10% DMSO), counted, and divided into aliquots of ∼1–10 million cells per mL. PBMC aliquots were promptly transferred to −80°C for slow freeze down in a Mr. Frosty Freezing Container (Thermo Scientific). After 24 h, the PBMCs were transferred to liquid nitrogen for long-term storage.
Single-cell gene expression profiling
Samples were co-processed for single-cell RNAseq (scRNAseq) in batches of 4–8 PBMC aliquots, such that each batch contained paired baseline and post-CPET samples from the same individual and a mix of cases and controls. PBMCs were prepared for scRNAseq using the 10x Genomics Demonstrated Protocol: Fresh Frozen Human Peripheral Blood Mononuclear Cells for Single Cell RNA Sequencing (CG00039) as a guide. Briefly, vials were rapidly thawed in a 37°C water bath and 500 μL was transferred to a 50 mL centrifuge tube using a wide-bore tip. Cells were serially diluted 1:1 with RPMI 1640 (Gibco # 11875093) plus 10% heat inactivated FBS (Gibco #10438026) in 1-min increments until the total volume reached 32 mL. Cells were centrifuged in a swinging bucket rotor at 300 rcf for 5 min at room temperature. After discarding the majority of the supernatant, cells were gently resuspended in the residual volume with a regular-bore tip to achieve single cell suspension, transferred to a 15 mL centrifuge tube and the volume brought up to 10 mL with RPMI 160 plus 10% heat inactivated FBS. Cells were centrifuged again at 300 rcf for 5 min at room temperature, and the pellet was gently resuspended in 1 mL of 1x PBS (BioWhittaker #17-516F) plus 0.04% BSA (Invitrogen #AM2616) with a wide-bore tip. The cells were transferred to a 2 mL microfuge tube, centrifuged at 300 rcf for 5 min in a swinging bucket centrifuge at room temperature, and resuspended in 1x PBS plus 0.04% BSA to a specified cell concentration to load ∼8,000 cells onto the 10x Chromium chip for a target capture of 5,000 cells. Cells were counted on a TC-20 cell counter (Bio-Rad) multiple times to track total and live cell counts. In total, 120 PBMC samples were processed in 18 batches and only two paired samples from a control individual failed the minimum viability tests and were excluded from the study.
Viable cells were submitted to the Cornell BRC Genomics Facility for processing with the Chromium Single Cell 3ʹ v3 kit (CG000183 Rev A and Rev B, 10x Genomics). The Facility prepared a total of 120 single-cell RNAseq libraries; all underwent quality checks for size distribution on a Fragment Analyzer 5200 (Agilent) and molarity on a QX100 Digital Droplet PCR Machine (Bio-Rad). Libraries were sequenced in an initial run to generate preliminary data quality metrics, and additional sequencing depth was generated as required to meet the target coverage for each sample. The libraries generated in batches 1–13 were primarily sequenced on a NextSeq500 (R1:28bp, R2:55bp) and the libraries from batches 14–18 were sequenced on a HiSeq2000 followed by a NovaSeq6000 (both PE 2x150bp). A final sequencing run on a NextSeq2000 brought all samples to the minimum target depth (see below).
Monocyte isolation and profiling
Magnetic enrichment
Samples from individuals in the larger Cornell Center study that were not included in the scRNA-seq cohort were selected for monocyte profiling. For bulk RNAseq, we used PBMCs from eight post-CPET females (4 cases with low SF36v2 PCS scores and 4 controls, matched by age and BMI). For proteomics, we used PBMCs from eight baseline females (4 cases and 4 controls with similar criteria as described above). Vials containing cryopreserved PBMCs were rapidly thawed in a 37°C water bath and serially diluted 1:1 with RPMI 1640 plus 10% heat inactivated FBS in 1-min increments until the total volume reached 32 mL. Cells were centrifuged in a swinging bucket rotor at 300 rcf for 5 min at room temperature and the pellet was gently resuspended in 10 mL of MACS buffer (Miltenyi Biotech Cat# 130-091-376). After two washes in MACS buffer, centrifuging at 300 rcf for 5 min at room temperature, the pellet was resuspended in 1 mL of MACS buffer and cells were counted with a TC-20 cell counter (Bio-Rad). Monocytes were initially purified with the Classical Monocyte Isolation kit, human (Miltenyi Biotech Cat#130-117-337), then additionally enriched for CD14+ cells using the CD14 MicroBeads (Miltenyi Biotec Cat#130-050-201). After counting, 20,000–140,000 cells were removed to 250 μL of MACs buffer and 750 μL of Trizol LS (ThermoFisher) added to lyse cells for bulk RNAseq profiling. Trizol lysates were frozen at −80°C prior to RNA extraction and submitted to the Cornell Transcriptional Regulation and Expression (TREx) Facility for RNA extraction and RNAseq. Remaining cells were immediately analyzed with flow cytometry.
Flow cytometry
Human TruStain FcX Fc Receptor Blocking Solution (BioLegend Cat#422302) was added according to the number of cells, mixed well and incubated for 10 min at room temperature. Cells were divided into separate microfuge tubes for staining with fluorescently labeled antibodies (CD3-FITC, CD45-PerCP, CD16-PE, and CD14-APC-Cy7 from BioLegend) for flow cytometry and incubated on ice for 30 min. 1 mL of FACS buffer (heat inactivated FBS, 0.5M EDTA pH8.0 1x PBS) was added to the cells, mixed well and centrifuged at 300 rcf for 5 min at 4°C. The supernatant was removed and the cells were washed once more. The final cell pellet was resuspended in 100 μL of FACS buffer and kept on ice in the dark. Sytox blue (0.2 μL/100 μL or less) was added directly before flow cytometry analysis and analyzed on a Thermo Fisher Attune NxT at the Cornell BRC Flow Cytometry Facility.
Bulk RNAseq
At the TREx Facility, RNA was extracted from Trizol following the manufacturer’s protocol with the following exceptions: the aqueous fraction was re-extracted with an equal volume of chloroform in Phase Lock Gel Heavy tubes (QuantaBio) and 2 μL of GlycoBlue (Thermo) was added prior to precipitation to improve RNA recovery. Total RNA samples were quantified with the Qubit HS RNA assay (Thermo) and integrity assessed on a Fragment Analyzer (Agilent) to confirm RQN values ≥7. Bulk polyA + RNAseq libraries were generated from 25 ng total RNA with the NEBNext Ultra II Directional RNA kit (New England Biolabs). Libraries were quantified with a Qubit HS DNA assay (Thermo) and sequenced on a NovaSeq6000 (Illumina) at Novogene to generate a minimum of 20M PE 2x150bp reads per sample.
Proteomics
Classical monocytes isolated by MACS were washed 3 times with PBS to reduce BSA contamination by centrifugation. After the last wash, cells were resuspended in RIPA buffer (Thermo) with protease inhibitor cocktail (Thermo). Cell lysates were submitted to Cornell BRC Proteomics and Metabolomics Facility for quality check and label free quantification. 1.4 μg of proteins from each sample was digested and analyzed by nanoLC-Orbitrap mass spectrometer. Yeast enolase digests (100 fmol) were spiked into each sample for normalization.
Flow cytometry of PBMCs
PBMCs from 4 cases and 4 controls at baseline were thawed as described above. PBMCs were processed for monocyte subset analysis was described above. For T lymphocytes, CD3 cells were enriched using the CD3 MicroBeads (Miltenyi Biotec Cat#130-097-043). After enrichment, cells were incubated with Aqua dead cell stain kit (Thermo) with Human TruStain FcX Fc Receptor Blocking Solution (Biolegend) for 10 min at room temperature. Fluorescently labeled antibodies (CD4-FITC, CD8a-BV785, CD45RA-BV650, CCR7-APC-Cy7, and CD56-PE-Cy7 from BioLegend) were subsequently added to the cells and incubated on ice for 30 min. 1 mL of FACS buffer (heat inactivated FBS, 0.5M EDTA pH8.0 1x PBS) was added to the cells, mixed well and centrifuged at 300 rcf for 5 min at 4°C. The supernatant was removed and the cells were washed once more. The final cell pellet was resuspended in 100μL of FACS buffer and kept on ice in the dark. Flow cytometry analysis was performed on a FACSARia Fusion at the Cornell BRC Flow Cytometry Facility.
Plasma particle isolation and profiling
Enrichment
Frozen plasma samples from 3 cases and 3 controls were thawed at room temperature. 1 mL of plasma were mixed with 2 mL of DMEM and centrifuged at 800 rcf for 20 min. After centrifugation, media was discarded and the pellet were washed in 1 mL of Tyrode’s buffer and centrifuged. The washed pellet was resuspended in 400 μL of PBS. 250 μL of the plasma particles was mixed with 750 μL of Trizol-LS (Thermo) for RNA extraction. The remaining samples were processed for flow cytometry analysis.
Flow cytometry
Plasma particles were incubated with 20 μL of Human TruStain FcX Fc Receptor Blocking Solution (Biolegend) for 10 min at room temperature. Fluorescently labeled antibodies (CD41-PE, CD45-PerCP, and CD235a-APC-Cy7 from BioLegend) were subsequently added to the samples and incubated at room temperature for 30 min in the dark. Samples were washed twice with Tyrode’s buffer by centrifugation at 800 rcf for 5 min at room temperature. Flow cytometry analysis was conducted on a Thermo Fisher Attune NxT at the Cornell BRC Flow Cytometry Facility.
Bulk RNAseq
RNA extraction, polyA + RNAseq library preparation, and sequencing was conducted by the TREx Facility as described above for monocytes.
Quantification and statistical analysis
Single-cell RNAseq
Data processing
Fastq files were generated with cellranger mkfastq (10x Genomics) by the sequencing facility (BRC Genomics Facility or Novogene). Raw count tables were generated with cellranger count v6 (10x Genomics) [cellranger count --id = ID --transcriptome = /path/to/refdata-gex-GRCh38-2020-A/--fastqs = /path/to/directories --sample = list --expect-cells = 5000 --r1-length = 28 --r2-length = 55 –nosecondary]. Because sequencing read lengths from different instruments varied, and this was observed to contribute to bias in the count tables, reads were trimmed to match the minimum length across the dataset (R1 = 28nt, R2 = 55nt).
Single-cell integration and clustering
Count tables from all samples were imported into R to analyze with Seurat (v4.1.0)23. Initial filtering removed cells that did not meet minimum quality criteria (nFeature_RNA >500 & nFeature_RNA <5000 & percent.mt < 30 & log10GenesPerUMI >0.80). A pair of samples from the same control individual were discarded due to an excess of counts for mitochondrial genes indicating a sample quality problem, leaving a total of 116 samples in the final dataset. Normalization of UMI counts for each library was performed using the SCTransform function [SCTransform(sobj, method = "glmGamPoi", vars.to.regress = "percent.mt", return.only.var.genes = FALSE)]. Samples were integrated with the Seurat function RunHarmony (sobj, reduction = "harmony", dims = 1:50), a wrapper for Harmony.91 Clustering with Seurat [FindNeighbors(sobj, reduction = "harmony", dims = 1:50) %>% FindClusters(resolution = 0.6)] generated a total of 29 clusters. Cell doublets within each sample were determined with DoubletFinder (v2.0.3)80 and removed from the dataset. The FindMarkers function in Seurat was used to determine marker genes between clusters and genes that distinguish case and control cells within each cluster.
Pseudobulk analysis
Raw pseudobulk counts were extracted from the Seurat object as the sum of counts for each gene per sample, per cluster. Normalization of pseudobulk matrices with DEseq2 (v1.30.0)81 generated normalized counts for downstream analyses, and was rerun as the cell assignments or cohorts were altered for different analyses. DEseq2 was used to detect differentially expressed genes between groups, using ‘minReplicatesForReplace = Inf’ to reduce the contribution from spurious outliers.
The log2-fold change calculation from DEseq2 was used for gene set enrichment analysis (using R packages clusterProfiler (default parameters: v3.18.1)82 or fgsea83 (minimal gene set size 5, maximum gene set size 2000, eps 0: v1.20.0) to run the GSEA30,92 algorithm, after filtering out genes with low coverage. For example, the GSEA analysis for clusters with more than 10,000 cells was filtered to retain only the top quartile of genes based on the median normalized counts across all samples. The Hallmark, C2:CP and C5 catalogs from the MSigDB database30 were used for enrichment tests.
For the paired analysis controlling for the individual of origin, the post-CPET/BL ratio was calculated for each gene and each individual when normalized counts were available for both timepoints and only for individuals with at least 4 cells contributing to pseudobulk counts at both timepoints. Normalized counts were floored to 1 to reduce the contribution from poorly detected genes. The geometric means of the ratios of all cases and all controls were used to calculate the final log2-fold change metric for the paired analysis; only genes with at least 6 ratios for cases and for controls were retained in the final rank list for GSEA. The Wilcoxon rank-sum test, using the wilcox.test function in base R was used to assess genes that change significantly in response to the CPET challenge.
Scores were calculated per sample (or per ratio) with the R package singscore (v1.20.0),84 using a unique list of genes derived from the GSEA leading edge genes from gene sets related to chemokine/cytokine signaling (listed in Figure 2D), and the Wilcoxon rank-sum test was used to compare groups.
Cell demographics analysis
Cell counts per cluster per individual were normalized to the total cell counts per sample. Wilcoxon rank-sum tests were used to compare normalized cell counts in the case versus control cohorts at baseline and post-exercise respectively, as well as for normalized cell counts at baseline versus post-exercise separately for cases and controls.
Positive unlabeled learning algorithm
Positive unlabeled learning was performed based on publicly available methods and codes with the scRNA-seq dataset as input.49,93 Briefly, the algorithm took the normalized single cell gene expression matrix of cluster 2 (classical monocytes), using data from either females or males at baseline or post-CPET (data slot of the RNA assay in the Seurat object) as input. Cells from control individuals are labeled as positive, and cells from case individuals are unlabeled. Twenty percent of labeled cells (i.e., cells from control individuals) were held out, and the remaining 80% labeled cells together with all unlabeled cells (i.e., cells from case individuals) were used to train an XGB classifier (using Python package XGBoost,94 with default parameters) separating labeled and unlabeled cells. Next, the reserved labeled cells were projected onto the labeled/unlabeled classifier to estimate the probability of cells being labeled if they are positive (control-like). In the following step, all labeled and unlabeled cells were projected onto the labeled/unlabeled classifier. Finally, based on the theorem of conditional probability, the probability of unlabeled cells being positive can be estimated by the probability of the cells being labeled divided by the probability of cells being positive when labeled. Predicted probabilities were averaged across 24 iterations.
To select the probability threshold that determines if a cell is predicted as healthy or diseased, the Calinski-Harabasz Index of predicted diseased and predicted control cells was calculated based on the top 50 principal components of the single cell expression matrix across thresholds 0.1–0.9 (Figure S4A). The threshold with the highest Calinski-Harabasz Index was selected (0.4 for classical monocytes in females at baseline). Cells with probabilities higher than the threshold are considered as predicted control, and cells with probabilities lower than the threshold are considered as predicted diseased.
To correlate the predictions with other metrics, the percentage of cells predicted as diseased were calculated for each individual. Spearman correlation was calculated by R package ggpubr v0.4.0 between percentage of cells predicted diseased at baseline compared to post-CPET in cases and controls, or between percentage predicted-diseased cells and demographic metrics (MFI-20 total score, general health score, SF-36 physical component score, and PEM maximum change) across individuals. PEM maximum change was calculated as the largest delta for the PEM symptom severity scores (from baseline to each of the survey timepoints post-CPET). Significance of difference between case and control for PEM maximum change was determined using Fisher’s z-transformation for correlation coefficients.95
To evaluate the performance of the predictions, Calinski-Harabasz Indexes of cells partitioned by predictions and other sample metadata (sex, case/control, individual of origin) were calculated based on top 50 principal components of the partitioned single cell expression matrix in PCA space, generated with RunPCA from Seurat.
Differential expression between cells predicted diseased and control was performed by FindMarkers function in Seurat. All genes are included. GSEA on log2-fold changes of genes between predicted diseased and predicted control groups of cells was performed using R package fgsea v1.20.0,83 with minimal gene set size = 5, maximum gene set size = 2000 and eps = 0. The Hallmark gene sets and KEGG subset from the C2 catalog in MSigDB database v7.5.130 were used for enrichment tests.
Pseudobulk counts were calculated by aggregating gene expression counts of cells partitioned by the predicted labels. Vst function in DESeq2 v1.34.081 was used to normalize the pseudobulk counts. PCA was performed on normalized pseudobulk counts using the R function prcomp and the loadings were then extracted from rotation of the PCA result. Paired analysis controlling the individual of origin was performed by calculating the log2-fold changes between normalized psuedobulk counts of predicted-diseased and predicted-normal cells for each ME/CFS patient. Ratios for each gene were then averaged across each predicted cohort.
Inference of cell-cell communication
CellChat (v1.1.3)61 was used to infer cell-cell communication probabilities and identify signaling changes across healthy and diseased cell populations. We down-sampled each cluster to 500 cells from females, at baseline only, for a balanced comparison. We discarded clusters with fewer than 500 cells. Briefly, we first identified, for each cluster, differentially over-expressed ligands, receptors and cofactors in the human CellChatDB database,62 then their average expression values were used to calculate communication probabilities between all cell groups. Interaction strength along specific intercellular signaling pathways was calculated by summarizing the communication probabilities of associated ligand-receptor pairs across all clusters. Comparison of healthy and diseased signaling networks was performed as described previously,61 with statistical significance of changes in communication probabilities determined by Wilcoxon rank-sum test and using only cells collected from females at baseline.
Flow cytometry analysis
Raw compensated flow cytometry files (.fcs) were analyzed using Flowjo (v10.8.1). Cells were first gated by size and granularity. Single cells were selected by looking at forward scatter area and height and side scatter area and height. Live cells were selected by gating on Sytox blue or Aqua negative events. Bulk monocytes were selected as CD45+CD3−cells. Monocyte subsets were analyzed by investigating surface expression of CD14 and CD16. Classical monocytes (CD14+CD16−) were gated based on fluorescent-minus-one (FMO) controls. Bulk T lymphocytes were selected as CD4+CD56−or CD8+CD56−cells. T lymphocyte subsets were analyzed by investigating surface expression of CD45RA and CCR7. Naive (CD45RA + CCR7+) and effector/memory (CD45RA-CCR7+/−) T lymphocytes were gated based on FMO controls. Plasma particles were analyzed based on surface expression of CD41, CD45, and CD235a. Platelet-derived particles (CD41+CD45−CD235a-) were gated based on FMO controls. For a complete list of antibodies, see key resources table.
Bulk RNAseq
Fastq files were trimmed to remove 3′ low quality and adaptor sequences with TrimGalore (v0.6),96 a wrapper for cutadapt85 and fastQC,97 retaining reads ≥50bp. Trimmed reads were mapped to the reference genome (GRCh38 with Ensembl gene annotations) with STAR (v2.7) [--outSAMstrandField intronMotif, --outFilterIntronMotifs RemoveNoncanonical, --outSAMtype BAM SortedByCoordinate, --quantMode GeneCounts], which outputs a count table of reads per gene for each sample. DESeq281 was used to normalize raw counts, generate PCA (normalized by rlog) and MA plots, and analyze differential expression, using genes with more than 10 read counts in more than or equal to 3 libraries. The log2-fold change values from DEseq2 were used for GSEA analysis with R package fgsea v1.20.083 as described above, with minimal gene set size = 5, maximum gene set size = 2000, eps = 0 and gseaParam = 0. Paired analysis controlling the individual of origin was conducted as described above for the single-cell pseudobulk profiles for platelets; only genes with at least 2 ratios for cases and for controls were retained in the final rank list for GSEA, Hallmark, C2:KEGG, C2:Reactome, and C5:GO_BP gene sets from the MSigDB database30 were used for enrichment tests.
Proteomics
We omitted one case from the cohort due to low input and MS signal intensity. The raw files were searched against Human UNiProt database using Proteome Discoverer (PD) 2.5 software at the BRC Proteomics Facility. Proteins with at least 2 peptides identified were used for further analysis. Protein abundances were normalized to the abundance of the spike-in, yeast enolase. Function aov() in R was used to perform ANOVA test to identify differentially expressed proteins between monocytes of controls and cases. Benjamini & Hochberg method was used to calculate adjusted p values. Log2-fold change values between patients and controls were used for GSEA analysis with R package fgsea v1.20.083 (minSize = 0, maxSize = 10000, eps = 0, gseaParam = 1, nPermSimple = 100000). C2:Reactome and C5:GO gene sets from the MSigDB database v7.5.130 were used for enrichment tests.
Acknowledgments
This study was supported by U54NS105541 and U54AI178855 to M.R.H., A.G., and D.C.S.; an NIH grant cofunded by the National Institute of Neurological Disorders and Stroke, National Institute of Allergy and Infectious Diseases, National Institute on Drug Abuse, National Heart, Lung, and Blood Institute, National Human Genome Research Institute, and the Office of the Director; and by UL1 TR 002384 from the National Center for Advancing Translational Sciences. We thank the Cornell Biotechnology Resource Center (BRC) Genomics, Flow Cytometry, Transcriptional Regulation and Gene Expression, and Proteomics (RRID: SCR_021727; SCR_021740; SCR_022532; SCR_021743) facilities for support. Thanks to Dr. John Chia for support in recruiting and screening ME/CFS individuals and controls. We send our heartfelt thanks to the ME/CFS subjects who participated in this study. Illustrations were created with Biorender.com.
Author contributions
Conceptualization, A.G. Methodology and software, F.A., H.Z., and J.K.G. Validation, L.T.V. and E.A.F. Formal analysis, F.A., H.Z., D.S.H.I., Y.K., W.C., P.R.M., and J.K.G. Investigation, F.A., L.T.V., H.Z., E.A.F., D.S.H.I., Y.K., W.C., A.E.T., P.R.M., and J.K.G. Resources, C.J.F., G.E.M., S.M.L., J.K.G., B.A.K., J.S., M.R.H., X.M., and D.C.S. Data curation, F.A. and J.K.G. Writing – original draft, A.G. Writing – review & editing, L.T.V., F.A., H.Z., D.S.H.I., E.A.F., M.R.H., J.K.G., and A.G. Visualization, F.A., L.T.V., H.Z., D.S.H.I., and J.K.G. Supervision and project administration, A.G. and J.K.G. Funding acquisition, A.G., M.R.H., and D.C.S.
Declaration of interests
The authors declare no competing interests.
Published: January 16, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101373.
Contributor Information
Jennifer K. Grenier, Email: jgrenier@cornell.edu.
Andrew Grimson, Email: agrimson@cornell.edu.
Supplemental information
References
- 1.Hanson M.R., Germain A. Letter to the Editor of Metabolites. Metabolites. 2020;10:216. doi: 10.3390/metabo10050216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim E.-J., Ahn Y.-C., Jang E.-S., Lee S.-W., Lee S.-H., Son C.-G. Systematic review and meta-analysis of the prevalence of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME) J. Transl. Med. 2020;18:100. doi: 10.1186/s12967-020-02269-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Komaroff A.L., Buchwald D.S., MD Chronic Fatigue Syndrome: An Update. Annu. Rev. Med. 1998;49:1–13. doi: 10.1146/annurev.med.49.1.1. [DOI] [PubMed] [Google Scholar]
- 4.Sanjabi S., Zenewicz L.A., Kamanaka M., Flavell R.A. Anti- and Pro-inflammatory Roles of TGF-β, IL-10, and IL-22 In Immunity and Autoimmunity. Curr. Opin. Pharmacol. 2009;9:447–453. doi: 10.1016/j.coph.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blundell S., Ray K.K., Buckland M., White P.D. Chronic fatigue syndrome and circulating cytokines: A systematic review. Brain Behav. Immun. 2015;50:186–195. doi: 10.1016/j.bbi.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Montoya J.G., Holmes T.H., Anderson J.N., Maecker H.T., Rosenberg-Hasson Y., Valencia I.J., Chu L., Younger J.W., Tato C.M., Davis M.M. Cytokine signature associated with disease severity in chronic fatigue syndrome patients. Proc. Natl. Acad. Sci. USA. 2017;114:E7150–E7158. doi: 10.1073/pnas.1710519114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hornig M., Montoya J.G., Klimas N.G., Levine S., Felsenstein D., Bateman L., Peterson D.L., Gottschalk C.G., Schultz A.F., Che X., et al. Distinct plasma immune signatures in ME/CFS are present early in the course of illness. Sci. Adv. 2015;1:e1400121. doi: 10.1126/sciadv.1400121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maher K.J., Klimas N.G., Fletcher M.A. Chronic fatigue syndrome is associated with diminished intracellular perforin. Clin. Exp. Immunol. 2005;142:505–511. doi: 10.1111/j.1365-2249.2005.02935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eaton-Fitch N., du Preez S., Cabanas H., Staines D., Marshall-Gradisnik S. A systematic review of natural killer cells profile and cytotoxic function in myalgic encephalomyelitis/chronic fatigue syndrome. Syst. Rev. 2019;8:279. doi: 10.1186/s13643-019-1202-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kennedy G., Spence V., Underwood C., Belch J.J.F. Increased neutrophil apoptosis in chronic fatigue syndrome. J. Clin. Pathol. 2004;57:891–893. doi: 10.1136/jcp.2003.015511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mandarano A.H., Maya J., Giloteaux L., Peterson D.L., Maynard M., Gottschalk C.G., Hanson M.R. Myalgic encephalomyelitis/chronic fatigue syndrome patients exhibit altered T cell metabolism and cytokine associations. J. Clin. Invest. 2020;130:1491–1505. doi: 10.1172/JCI132185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato W., Ono H., Matsutani T., Nakamura M., Shin I., Amano K., Suzuki R., Yamamura T. Skewing of the B cell receptor repertoire in myalgic encephalomyelitis/chronic fatigue syndrome. Brain Behav. Immun. 2021;95:245–255. doi: 10.1016/j.bbi.2021.03.023. [DOI] [PubMed] [Google Scholar]
- 13.Milivojevic M., Che X., Bateman L., Cheng A., Garcia B.A., Hornig M., Huber M., Klimas N.G., Lee B., Lee H., et al. Plasma proteomic profiling suggests an association between antigen driven clonal B cell expansion and ME/CFS. PLoS One. 2020;15:e0236148. doi: 10.1371/journal.pone.0236148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Committee on the Diagnostic Criteria for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome, Board on the Health of Select Populations, and Institute of Medicine . National Academies Press (US); 2015. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness. [PubMed] [Google Scholar]
- 15.Noor N., Urits I., Degueure A., Rando L., Kata V., Cornett E.M., Kaye A.D., Imani F., Narimani-Zamanabadi M., Varrassi G., Viswanath O. A Comprehensive Update of the Current Understanding of Chronic Fatigue Syndrome. Anesth. Pain Med. 2021;11:e113629. doi: 10.5812/aapm.113629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stevens S., Snell C., Stevens J., Keller B., VanNess J.M. Cardiopulmonary Exercise Test Methodology for Assessing Exertion Intolerance in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Pediatr. 2018;6:242. doi: 10.3389/fped.2018.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasa S., Nora-Krukle Z., Henning N., Eliassen E., Shikova E., Harrer T., Scheibenbogen C., Murovska M., Prusty B.K., European Network on ME/CFS EUROMENE Chronic viral infections in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) J. Transl. Med. 2018;16:268. doi: 10.1186/s12967-018-1644-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Neal A.J., Hanson M.R. The Enterovirus Theory of Disease Etiology in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Critical Review. Front. Med. 2021;8:688486. doi: 10.3389/fmed.2021.688486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanson M.R. The viral origin of myalgic encephalomyelitis/chronic fatigue syndrome. PLoS Pathog. 2023;19:e1011523. doi: 10.1371/journal.ppat.1011523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong T.L., Weitzer D.J. Long COVID and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)—A Systemic Review and Comparison of Clinical Presentation and Symptomatology. Medicina (Mex.) 2021;57:418. doi: 10.3390/medicina57050418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keller B.A., Pryor J.L., Giloteaux L. Inability of myalgic encephalomyelitis/chronic fatigue syndrome patients to reproduce VO2peak indicates functional impairment. J. Transl. Med. 2014;12:104. doi: 10.1186/1479-5876-12-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davenport T.E., Stevens S.R., Stevens J., Snell C.R., Van Ness J.M. Properties of measurements obtained during cardiopulmonary exercise testing in individuals with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Work Read Mass. 2020;66:247–256. doi: 10.3233/WOR-203170. [DOI] [PubMed] [Google Scholar]
- 23.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitami T., Fukuda S., Kato T., Yamaguti K., Nakatomi Y., Yamano E., Kataoka Y., Mizuno K., Tsuboi Y., Kogo Y., et al. Deep phenotyping of myalgic encephalomyelitis/chronic fatigue syndrome in Japanese population. Sci. Rep. 2020;10:19933. doi: 10.1038/s41598-020-77105-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brenu E.W., Huth T.K., Hardcastle S.L., Fuller K., Kaur M., Johnston S., Ramos S.B., Staines D.R., Marshall-Gradisnik S.M. Role of adaptive and innate immune cells in chronic fatigue syndrome/myalgic encephalomyelitis. Int. Immunol. 2014;26:233–242. doi: 10.1093/intimm/dxt068. [DOI] [PubMed] [Google Scholar]
- 26.Patel A.A., Yona S. Inherited and Environmental Factors Influence Human Monocyte Heterogeneity. Front. Immunol. 2019;10:2581. doi: 10.3389/fimmu.2019.02581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thurman A.L., Ratcliff J.A., Chimenti M.S., Pezzulo A.A. Differential gene expression analysis for multi-subject single-cell RNA-sequencing studies with aggregateBioVar. Bioinformatics. 2021;37:3243–3251. doi: 10.1093/bioinformatics/btab337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheema A.K., Sarria L., Bekheit M., Collado F., Almenar-Pérez E., Martín-Martínez E., Alegre J., Castro-Marrero J., Fletcher M.A., Klimas N.G., et al. Unravelling myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): Gender-specific changes in the microRNA expression profiling in ME/CFS. J. Cell Mol. Med. 2020;24:5865–5877. doi: 10.1111/jcmm.15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gamer J., Van Booven D.J., Zarnowski O., Arango S., Elias M., Kurian A., Joseph A., Perez M., Collado F., Klimas N., et al. Sex-Dependent Transcriptional Changes in Response to Stress in Patients with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Pilot Project. Int. J. Mol. Sci. 2023;24:10255. doi: 10.3390/ijms241210255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinforma. Oxf. Engl. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sindhu S., Kochumon S., Shenouda S., Wilson A., Al-Mulla F., Ahmad R. The Cooperative Induction of CCL4 in Human Monocytic Cells by TNF-α and Palmitate Requires MyD88 and Involves MAPK/NF-κB Signaling Pathways. Int. J. Mol. Sci. 2019;20:E4658. doi: 10.3390/ijms20184658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Getzin T., Krishnasamy K., Gamrekelashvili J., Kapanadze T., Limbourg A., Häger C., Napp L.C., Bauersachs J., Haller H., Limbourg F.P. The chemokine receptor CX3CR1 coordinates monocyte recruitment and endothelial regeneration after arterial injury. EMBO Mol. Med. 2018;10:151. doi: 10.15252/emmm.201707502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feng X., Szulzewsky F., Yerevanian A., Chen Z., Heinzmann D., Rasmussen R.D., Alvarez-Garcia V., Kim Y., Wang B., Tamagno I., et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget. 2015;6:15077–15094. doi: 10.18632/oncotarget.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muller W.A. Getting leukocytes to the site of inflammation. Vet. Pathol. 2013;50:7–22. doi: 10.1177/0300985812469883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sánchez-Martín L., Estecha A., Samaniego R., Sánchez-Ramón S., Vega M.Á., Sánchez-Mateos P. The chemokine CXCL12 regulates monocyte-macrophage differentiation and RUNX3 expression. Blood. 2011;117:88–97. doi: 10.1182/blood-2009-12-258186. [DOI] [PubMed] [Google Scholar]
- 37.Giloteaux L., Li J., Hornig M., Lipkin W.I., Ruppert D., Hanson M.R. Proteomics and cytokine analyses distinguish myalgic encephalomyelitis/chronic fatigue syndrome cases from controls. J. Transl. Med. 2023;21:322. doi: 10.1186/s12967-023-04179-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao X., Gu M., Xu X., Wen X., Yang G., Li L., Sheng P., Meng F. CCL3/CCR1 mediates CD14+CD16- circulating monocyte recruitment in knee osteoarthritis progression. Osteoarthritis Cartilage. 2020;28:613–625. doi: 10.1016/j.joca.2020.01.009. [DOI] [PubMed] [Google Scholar]
- 39.Cronin S.J.F., Woolf C.J., Weiss G., Penninger J.M. The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front. Mol. Biosci. 2019;6:116. doi: 10.3389/fmolb.2019.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang Q.-Q., Hossain M.M., Wu K., Parai K., Pope R.M., Jin J.-P. Role of H2-calponin in Regulating Macrophage Motility and Phagocytosis. J. Biol. Chem. 2008;283:25887–25899. doi: 10.1074/jbc.M801163200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hume D.A. The Many Alternative Faces of Macrophage Activation. Front. Immunol. 2015;6:370. doi: 10.3389/fimmu.2015.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee Y.D., Kim B., Jung S., Kim H., Kim M.K., Kwon J.-O., Song M.-K., Lee Z.H., Kim H.-H. The dynactin subunit DCTN1 controls osteoclastogenesis via the Cdc42/PAK2 pathway. Exp. Mol. Med. 2020;52:514–528. doi: 10.1038/s12276-020-0406-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brumbaugh J., Di Stefano B., Wang X., Borkent M., Forouzmand E., Clowers K.J., Ji F., Schwarz B.A., Kalocsay M., Elledge S.J., et al. Nudt21 Controls Cell Fate by Connecting Alternative Polyadenylation to Chromatin Signaling. Cell. 2018;172:106–120.e21. doi: 10.1016/j.cell.2017.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rizzo G., Gropper J., Piollet M., Vafadarnejad E., Rizakou A., Bandi S.R., Arampatzi P., Krammer T., DiFabion N., Dietrich O., et al. Dynamics of monocyte-derived macrophage diversity in experimental myocardial infarction. Cardiovasc. Res. 2023;119:772–785. doi: 10.1093/cvr/cvac113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y., Li K., Zhao W., Liu Z., Liu J., Shi A., Chen T., Mu W., Xu Y., Pan C., Zhang Z. Aldehyde dehydrogenase 3B2 promotes the proliferation and invasion of cholangiocarcinoma by increasing Integrin Beta 1 expression. Cell Death Dis. 2021;12:1158. doi: 10.1038/s41419-021-04451-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang F., Ma X.-R., Wu Y., Xu Y.-C., Gu H.-M., Wang D.-X., Dong Z.-J., Li H.-L., Wang L.-B., Zhao J.-W. Neutralization of Hv1/HVCN1 With Antibody Enhances Microglia/Macrophages Myelin Clearance by Promoting Their Migration in the Brain. Front. Cell. Neurosci. 2021;15:768059. doi: 10.3389/fncel.2021.768059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong J., Yau A.C.Y., Holmdahl R. Independent and inter-dependent immunoregulatory effects of NCF1 and NOS2 in experimental autoimmune encephalomyelitis. J. Neuroinflammation. 2020;17:113. doi: 10.1186/s12974-020-01789-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ye S., Huang Y., Joshi S., Zhang J., Yang F., Zhang G., Smyth S.S., Li Z., Takai Y., Whiteheart S.W. Platelet secretion and hemostasis require syntaxin-binding protein STXBP5. J. Clin. Invest. 2014;124:4517–4528. doi: 10.1172/JCI75572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elkan C., Noto K. Proceedings of the 14th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining KDD ’08. Association for Computing Machinery; 2008. Learning Classifiers from Only Positive and Unlabeled Data; pp. 213–220. [Google Scholar]
- 50.Smets E.M., Garssen B., Bonke B., De Haes J.C. The Multidimensional Fatigue Inventory (MFI) psychometric qualities of an instrument to assess fatigue. J. Psychosom. Res. 1995;39:315–325. doi: 10.1016/0022-3999(94)00125-o. [DOI] [PubMed] [Google Scholar]
- 51.Nacul L.C., Lacerda E.M., Campion P., Pheby D., Drachler M.d.L., Leite J.C., Poland F., Howe A., Fayyaz S., Molokhia M. The functional status and well being of people with myalgic encephalomyelitis/chronic fatigue syndrome and their carers. BMC Publ. Health. 2011;11:402. doi: 10.1186/1471-2458-11-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Condamine T., Le Texier L., Howie D., Lavault A., Hill M., Halary F., Cobbold S., Waldmann H., Cuturi M.-C., Chiffoleau E. Tmem176B and Tmem176A are associated with the immature state of dendritic cells. J. Leukoc. Biol. 2010;88:507–515. doi: 10.1189/jlb.1109738. [DOI] [PubMed] [Google Scholar]
- 53.Limoges M.-A., Cloutier M., Nandi M., Ilangumaran S., Ramanathan S. The GIMAP Family Proteins: An Incomplete Puzzle. Front. Immunol. 2021;12:679739. doi: 10.3389/fimmu.2021.679739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu M., Zhu X., Yang Z., Zhang W., Sun Z., Ji Q., Chen X., Zhu J., Wang C., Nie S. E3 ubiquitin ligase tripartite motif 7 positively regulates the TLR4-mediated immune response via its E3 ligase domain in macrophages. Mol. Immunol. 2019;109:126–133. doi: 10.1016/j.molimm.2019.01.015. [DOI] [PubMed] [Google Scholar]
- 55.Arslan C., Bayoglu B., Tel C., Cengiz M., Dirican A., Besirli K. Upregulation of OLR1 and IL17A genes and their association with blood glucose and lipid levels in femoropopliteal artery disease. Exp. Ther. Med. 2017;13:1160–1168. doi: 10.3892/etm.2017.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Okabe M., Takarada S., Miyao N., Nakaoka H., Ibuki K., Ozawa S., Watanabe K., Tsuji H., Hashimoto I., Hatasaki K., et al. G0S2 regulates innate immunity in Kawasaki disease via lncRNA HSD11B1-AS1. Pediatr. Res. 2022;92:378–387. doi: 10.1038/s41390-022-01999-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Labzin L.I., Schmidt S.V., Masters S.L., Beyer M., Krebs W., Klee K., Stahl R., Lütjohann D., Schultze J.L., Latz E., De Nardo D. ATF3 Is a Key Regulator of Macrophage IFN Responses. J. Immunol. 2015;195:4446–4455. doi: 10.4049/jimmunol.1500204. [DOI] [PubMed] [Google Scholar]
- 58.Kwok I., Becht E., Xia Y., Ng M., Teh Y.C., Tan L., Evrard M., Li J.L.Y., Tran H.T.N., Tan Y., et al. Combinatorial Single-Cell Analyses of Granulocyte-Monocyte Progenitor Heterogeneity Reveals an Early Uni-potent Neutrophil Progenitor. Immunity. 2020;53:303–318.e5. doi: 10.1016/j.immuni.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 59.Picotto G., Morse L.R., Nguyen N., Saltzman J., Battaglino R. TMEM176A and TMEM176B Are Candidate Regulators of Inhibition of Dendritic Cell Maturation and Function after Chronic Spinal Cord Injury. J. Neurotrauma. 2020;37:528–533. doi: 10.1089/neu.2019.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khuu C.H., Barrozo R.M., Hai T., Weinstein S.L. Activating transcription factor 3 (ATF3) represses the expression of CCL4 in murine macrophages. Mol. Immunol. 2007;44:1598–1605. doi: 10.1016/j.molimm.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 61.Vu R., Jin S., Sun P., Haensel D., Nguyen Q.H., Dragan M., Kessenbrock K., Nie Q., Dai X. Wound healing in aged skin exhibits systems-level alterations in cellular composition and cell-cell communication. Cell Rep. 2022;40:111155. doi: 10.1016/j.celrep.2022.111155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.-H., Myung P., Plikus M.V., Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021;12:1088. doi: 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andrijevic D., Vrselja Z., Lysyy T., Zhang S., Skarica M., Spajic A., Dellal D., Thorn S.L., Duckrow R.B., Ma S., et al. Cellular recovery after prolonged warm ischaemia of the whole body. Nature. 2022;608:405–412. doi: 10.1038/s41586-022-05016-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yıldırım C., Vogel D.Y.S., Hollander M.R., Baggen J.M., Fontijn R.D., Nieuwenhuis S., Haverkamp A., de Vries M.R., Quax P.H.A., Garcia-Vallejo J.J., et al. Galectin-2 induces a proinflammatory, anti-arteriogenic phenotype in monocytes and macrophages. PLoS One. 2015;10:e0124347. doi: 10.1371/journal.pone.0124347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hollmén M., Karaman S., Schwager S., Lisibach A., Christiansen A.J., Maksimow M., Varga Z., Jalkanen S., Detmar M. G-CSF regulates macrophage phenotype and associates with poor overall survival in human triple-negative breast cancer. OncoImmunology. 2016;5:e1115177. doi: 10.1080/2162402X.2015.1115177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Passacquale G., Vamadevan P., Pereira L., Hamid C., Corrigall V., Ferro A. Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. PLoS One. 2011;6:e25595. doi: 10.1371/journal.pone.0025595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stephen J., Emerson B., Fox K.A.A., Dransfield I. The uncoupling of monocyte-platelet interactions from the induction of proinflammatory signaling in monocytes. J. Immunol. 2013;191:5677–5683. doi: 10.4049/jimmunol.1301250. [DOI] [PubMed] [Google Scholar]
- 68.Shim C.-H., Cho S., Shin Y.-M., Choi J.-M. Emerging role of bystander T cell activation in autoimmune diseases. BMB Rep. 2022;55:57–64. doi: 10.5483/BMBRep.2022.55.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lacourt T.E., Vichaya E.G., Chiu G.S., Dantzer R., Heijnen C.J. The High Costs of Low-Grade Inflammation: Persistent Fatigue as a Consequence of Reduced Cellular-Energy Availability and Non-adaptive Energy Expenditure. Front. Behav. Neurosci. 2018;12:78. doi: 10.3389/fnbeh.2018.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nunes J.M., Kruger A., Proal A., Kell D.B., Pretorius E. The Occurrence of Hyperactivated Platelets and Fibrinaloid Microclots in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) Pharm. Basel Switz. 2022;15:931. doi: 10.3390/ph15080931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pretorius E., Vlok M., Venter C., Bezuidenhout J.A., Laubscher G.J., Steenkamp J., Kell D.B. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc. Diabetol. 2021;20:172. doi: 10.1186/s12933-021-01359-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Angénieux C., Maître B., Eckly A., Lanza F., Gachet C., de la Salle H. Time-Dependent Decay of mRNA and Ribosomal RNA during Platelet Aging and Its Correlation with Translation Activity. PLoS One. 2016;11:e0148064. doi: 10.1371/journal.pone.0148064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mills E.W., Green R., Ingolia N.T. Slowed decay of mRNAs enhances platelet specific translation. Blood. 2017;129:e38–e48. doi: 10.1182/blood-2016-08-736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heber S., Volf I. Effects of Physical (In)activity on Platelet Function. BioMed Res. Int. 2015;2015:165078. doi: 10.1155/2015/165078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Docherty S., Harley R., McAuley J.J., Crowe L.A.N., Pedret C., Kirwan P.D., Siebert S., Millar N.L. The effect of exercise on cytokines: implications for musculoskeletal health: a narrative review. BMC Sports Sci. Med. Rehabil. 2022;14:5. doi: 10.1186/s13102-022-00397-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pretorius E., Venter C., Laubscher G.J., Kotze M.J., Oladejo S.O., Watson L.R., Rajaratnam K., Watson B.W., Kell D.B. Prevalence of symptoms, comorbidities, fibrin amyloid microclots and platelet pathology in individuals with Long COVID/Post-Acute Sequelae of COVID-19 (PASC) Cardiovasc. Diabetol. 2022;21:148. doi: 10.1186/s12933-022-01579-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kang J., Brajanovski N., Chan K.T., Xuan J., Pearson R.B., Sanij E. Ribosomal proteins and human diseases: molecular mechanisms and targeted therapy. Signal Transduct. Target. Ther. 2021;6:323. doi: 10.1038/s41392-021-00728-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Araki K., Morita M., Bederman A.G., Konieczny B.T., Kissick H.T., Sonenberg N., Ahmed R. Translation is actively regulated during the differentiation of CD8+ effector T cells. Nat. Immunol. 2017;18:1046–1057. doi: 10.1038/ni.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tate W., Walker M., Sweetman E., Helliwell A., Peppercorn K., Edgar C., Blair A., Chatterjee A. Molecular Mechanisms of Neuroinflammation in ME/CFS and Long COVID to Sustain Disease and Promote Relapses. Front. Neurol. 2022;13:877772. doi: 10.3389/fneur.2022.877772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2021:60012. Preprint at. [Google Scholar]
- 84.Foroutan M., Bhuva D.D., Lyu R., Horan K., Cursons J., Davis M.J. Single sample scoring of molecular phenotypes. BMC Bioinf. 2018;19:404. doi: 10.1186/s12859-018-2435-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. j. 2011;17:10–12. [Google Scholar]
- 86.Germain A., Giloteaux L., Moore G.E., Levine S.M., Chia J.K., Keller B.A., Stevens J., Franconi C.J., Mao X., Shungu D.C., et al. Plasma metabolomics reveals disrupted response and recovery following maximal exercise in myalgic encephalomyelitis/chronic fatigue syndrome. JCI Insight. 2022;7:e157621. doi: 10.1172/jci.insight.157621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jason L.A., Richman J.A., Rademaker A.W., Jordan K.M., Plioplys A.V., Taylor R.R., McCready W., Huang C.F., Plioplys S. A community-based study of chronic fatigue syndrome. Arch. Intern. Med. 1999;159:2129–2137. doi: 10.1001/archinte.159.18.2129. [DOI] [PubMed] [Google Scholar]
- 88.Bakken I.J., Tveito K., Gunnes N., Ghaderi S., Stoltenberg C., Trogstad L., Håberg S.E., Magnus P. Two age peaks in the incidence of chronic fatigue syndrome/myalgic encephalomyelitis: a population-based registry study from Norway 2008-2012. BMC Med. 2014;12:167. doi: 10.1186/s12916-014-0167-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ware J.E., Jr. Quality Metric; 2007. User’s Manual for the SF-36v2 Health Survey. [and others] [Google Scholar]
- 90.Baraniuk J.N., Adewuyi O., Merck S.J., Ali M., Ravindran M.K., Timbol C.R., Rayhan R., Zheng Y., Le U., Esteitie R., Petrie K.N. A Chronic Fatigue Syndrome (CFS) severity score based on case designation criteria. Am. J. Transl. Res. 2013;5:53–68. [PMC free article] [PubMed] [Google Scholar]
- 91.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.-R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mootha V.K., Lindgren C.M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 93.Agmon A. 2022. A-Agmon/Pu-Learn. [Google Scholar]
- 94.Chen T., Guestrin C. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining KDD ’16. Association for Computing Machinery; 2016. XGBoost: A Scalable Tree Boosting System; pp. 785–794. [Google Scholar]
- 95.Soper H.E. On the Probable Error of the Correlation Coefficient to a Second Approximation. Biometrika. 1913;9:91–115. [Google Scholar]
- 96.Babraham Bioinformatics - Trim Galore! https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
- 97.Babraham Bioinformatics - FastQC A Quality Control Tool for High Throughput Sequence Data https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
(1)
De-identified human single-cell and bulk RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. The superseries accession number is listed in the key resources table.
-
(2)
Analyzed proteomics data provided by the Cornell BRC Proteomics Facility is included as a supplemental table.
-
(3)
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
(4)
This paper does not report original code.