

Abstract
Objective:
Since the role of white matter (WM) degenerating microglia in myelination failure is unclear, we sought to define the core features of this novel population of aging human microglia.
Methods:
We analyzed post-mortem human brain tissue to define a population of degenerative microglia (DM) in aging white matter lesions. We employed immunofluorescence staining and gene expression analysis to investigate molecular mechanisms related to the degeneration of DM.
Results:
We found that DM accumulated myelin debris, were selectively enriched in the iron-binding protein light chain ferritin, and accumulated PLIN2-labeled lipid droplets. DM displayed lipid peroxidation injury and enhanced expression for TOM20, a mitochondrial translocase, and a sensor of oxidative stress. DM also displayed enhanced expression of the DNA fragmentation marker phospho-histone H2A.X. We identified a unique set of ferroptosis-related genes involving iron-mediated lipid dysmetabolism and oxidative stress that were preferentially expressed in white matter injury relative to gray matter neurodegeneration.
Interpretation:
Ferroptosis appears to be a major mechanism of white matter injury in Alzheimer’s disease and vascular dementia. White matter DM are a novel therapeutic target to potentially reduce the impact of white matter injury and myelin loss on the progression of cognitive impairment.
INTRODUCTION
During brain aging, recurrent cerebral white matter injury (WMI) disrupts white matter integrity and myelination.1,2 WMI is typically microvascular in origin and associated with an enhanced risk for stroke,3–6 but is also commonly comorbid with Alzheimer’s disease (AD).1,7–10 WMI is a core feature of both AD8 and VCID (vascular contributions to cognitive impairment and dementia), the second leading cause of dementia.1,2,11,12 Microvascular WMI is present in ~85% of cognitively normal adults 75 years or older and greater than 95% of adults with cognitive impairment or dementia.13,14 The susceptibility to repeated episodes of microvascular ischemia thus creates a unique microenvironment where aging human WMI is rendered vulnerable to recurrent oxidative stress. Hence, the risk factors for remyelination failure differ considerably from other demyelinating disorders in younger adults.15 Aging-associated WMI commonly contributes to progressive decline in cognitive function,2,11,16,17 which presents as disturbances in motor performance, executive function, processing speed, and memory.18–23
Microglia play critical roles in the progression of dementia. AD risk genes are highly expressed by gray matter-associated activated microglia, which phagocytose degenerating neurons and contribute to early synaptic loss in AD.24–29 However, the putative role of microglia in WMI progression and cognitive decline is unclear.30,31 Vascular injury with disruption of blood-brain barrier integrity in VCID and neuroaxonal degeneration in AD are two complementary factors that initiate pro-inflammatory microglial activation in WMI.32,33 A potentially regenerative role of phagocytic microglia in WMI has been proposed in some demyelinating disorders.29,31 Microglial phagocytosis is essential for the clearance of myelin debris,34–36 which may reduce inhibitory signals that disrupt oligodendrocyte progenitor cell (OPC) proliferation, recruitment, and differentiation for myelin regeneration.35 However, in aging human WMI, we found that inhibitory signals continue to promote arrested maturation of OPCs to myelinating oligodendrocytes.15,37 To address this paradox, we analyzed the fate of microglia in aging human WMI to determine if disturbances in the microglial clearance of myelin debris might contribute to myelination failure. We unexpectedly found numerous WM microglia where phagocytosis of myelin debris was associated with degenerative features.
Since the role of white matter degenerating microglia (WM DM) in myelination failure is unclear, we sought to define the core features of this novel population of aging human microglia. We employed recently developed approaches to permit fluorescent immunohistochemical multi-labeling in the aging brain,38 which allowed a rigorous analysis of the markers and fate of WM DM and their contribution to remyelination failure. Here we demonstrate that WM DM comprise the major population of microglia in aging human WMI in AD and VCID. WM DM were enriched in myelin debris and PLIN2-associated lipid droplets, which appeared to provide a toxic source of labile iron since DM specifically labeled with the iron-binding protein, light chain ferritin (FTL). Surprisingly, lipid droplet accumulation was accompanied by significant lipid peroxidation injury suggesting that DM sustained iron-mediated oxidative stress. The striking susceptibility of aging microglia to oxidative stress appeared to be related to enhanced mitochondrial oxidative stress and aging-associated microglial senescence. Susceptibility to oxidative stress coincided with enhanced expression of the mitochondrial translocase and oxidative stress sensor Tom20, labeling of DM with the lipid peroxidation marker 4-hydroxynonenol (4HNE), and evidence of senescence-related nuclear DNA fragmentation visualized with phosphohistone H2A.X.
Our findings suggest that myelination failure from recurrent microvascular ischemia in aging human WMI involves a vulnerable population of iron-enriched DM, which display enhanced susceptibility to lipid peroxidation injury and ferroptotic degeneration due to pathological accumulation of myelin debris, a rich source of iron and lipid.39 Consistent with this notion, human WM lesions displayed enhanced expression of ferroptosis-related genes that were distinct from those expressed in gray matter. Aging human WMI in AD and VCID thus involves pronounced susceptibility to ferroptosis-mediated degeneration of iron-rich senescent microglia where lipid peroxidation injury appears to be promoted by dysfunctional microglial clearance of a high burden of myelin debris.
MATERIALS AND METHODS
Study design
Written informed consent was obtained from all individual study participants. All procedures involving human subjects were approved by the University of Washington (UW) Institutional Review Board following the Helsinki Declaration of the World Medical Association. Of the 40 human specimens, 34 were donated by participants in the Adult Changes in Thought (ACT) study, an ongoing population-based study of aging and incident dementia among men and women in Seattle, Washington.14 An additional six cases were from the Seattle Longitudinal Study (SLS), a longitudinal population-based study of psychometric changes from young adulthood to old age.40 There were no differences in the demographic or neuropathologic data for these 40 cases. A systematic brain tissue slicing technique was employed where the brain was embedded in agarose and sliced at 4-5 mm on a modified deli-slicer. Slices were laid out and photographed, and samples were routinely collected from each case. All tissues were either fixed in 10% formalin or 4% paraformaldehyde for three days before storage in sodium oxide solution at 4°C until sectioning. Samples prepared for autopsy were formalin-fixed and paraffin-embedded.
All cases were reviewed in the UW neuropathology core and assigned to a CMI (n=18) or a no-CMI group (nCMI; n=22) based on the extent of microvascular WMI defined by chronic cerebral microinfarcts (CMIs). Standard sections were screened in every brain using H&E or H&E/LFB for cortical and white matter CMIs. Cases with more than one CMI on autopsy review of six standardized brain regions were assigned to the CMI group, as previously described.14 The presence of degenerating microglia was confirmed in all 40 cases. A subset of 18 cases were selected (CMI=9; nCMI=9 unless otherwise noted), for which both fixed tissue for stereology studies and frozen tissues optimal for qPCR studies were available. As summarized in Table 1, neuropathological features of AD and cerebral amyloid angiopathy (CAA) were common to both cohorts and did not differ significantly. The postmortem interval was less than 12 hours for all cases (Table 1). Microscopic examination also was performed for diseases that commonly contribute to cognitive impairment or dementia, as previously described.15 These included combined Hematoxylin and Eosin with Luxol Fast Blue (H&E/LFB) staining to assess for microinfarcts and myelin integrity. Consensus evaluation of ADNC used Bielschowsky silver stain and Aβ (6E10), pTau (Tau2, AT8), and α-synuclein (LB509) immunohistochemical stains to determine Thal phase for Aβ deposits, Braak’s stage for neurofibrillary tangles (NFTs), Consortium to Establish a Registry for AD (CERAD) score for neuritic plaques, and Lewy body disease as described in the NIA-AA (National Institute on Aging Alzheimer’s Association) guidelines for the assessment of ADNC.41 Some cases predated NIA-AA criteria, so the Thal phase was not included in our analyses. Prefrontal white matter pallor was determined using H&E/LFB stains and an overall rating scale of none, mild, moderate, or severe (assessed by an observer blinded to clinical, radiographic, and neuropathologic diagnosis).
Table 1:
Patient Demographics for Cases with or Without Vascular Brain Injury
| Clinicopathological Details | nCMI | CMI | p* |
|---|---|---|---|
| No. (N) of cases | 22 | 18 | |
| Age (yr) | 91.00±0.40 | 87.75±0.48 | 0.3569 |
| Female: Male | 6:4 | 4:5 | |
| Postmortem interval (h) | 6.80±0.15 | 9.82±0.23 | 0.1088 |
| Brain weight (g) | 1112±45.84 | 1170±35.65 | 0.3365 |
| Cystic infarcts, N, (%) | 3 (13.6) | 3 (16.7) | |
| Acute or subacute infarcts, N, (%) | 8 (36.4) | 6 (33.3) | |
| CAA None, N, (%) | 6 (27.3) | 4 (22.2) | |
| CAA Mild, N, (%) | 5 (22.7) | 5 (27.8) | |
| BRAAK Score | 3.75±0.13 | 3.50±0.10 | 0.8996 |
p-value is based on Fisher’s exact test for categorical variables and two-sample t-test for continuous variables. Data are expressed as mean ± standard error of mean.
Tissue processing
The samples were processed as coronal sections cut free-floating (50 μm) in ice-cold PBS using a Leica VTS-1000 vibrating microtome (Leica Microsystems Inc., Bannockburn, IL). Tissue sections first underwent a photobleaching (photoQ) protocol to markedly reduce tissue autofluorescence from lipofuscin, as we recently described.38 After 72 hr photoQ, antigen retrieval was done at 90°C for 10 minutes using Tris-EDTA Buffer with 0.05% Tween 20, pH 9. Three tissue sections per case were incubated for 48 hours at 4°C in a combination of the following primary antibodies as described in the text: goat polyclonal antibody against ionized calcium-binding adaptor molecule 1 (Iba-1; 1:200; Novus; NB100-1028), mouse monoclonal antibody against GFAP (Millipore, #MAB360, 1:500), rabbit polyclonal antibody against light chain ferritin (FTL; 1:200; Abcam, #AB201975), mouse monoclonal antibody against myelin basic protein (MBP(SMI99); Calbiochem #NE1019; 1:500), mouse monoclonal antibody against translocase of outer membrane 20 (TOM20; 1:50; Santa Cruz, #SC-17764), rabbit polyclonal antibody 4-Hydroxynonenal (4-HNE; 1:1000, Abcam, #AB46545), rabbit anti-human perilipin 2 (PLIN2; 1:200, Sigma, HPA016607) or mouse monoclonal antibody against phosphorylated H2A.X (pH2A.X; 1:250, BioLegend, #613402). Hyperphosphorylated Tau was visualized using two antibodies: phospho-Tau monoclonal antibody (AT8; 1:1000; ThermoFisher Scientific, Rockford, IL) following antigen retrieval for 10 minutes at pH 6.0 and monoclonal anti-tau clone Tau-2 (1:1000; Sigma-Aldrich, St. Louis, MO) following antigen retrieval for 10 minutes at pH 9.0.42 Tissue sections were incubated in the appropriate Alexa fluor secondary antibody (Thermo Fisher Scientific) raised against mouse, rabbit, or goat.43 Apoptotic cell death was visualized by immunohistochemical detection of activated caspase-344 or TUNEL staining.42
Visualization of Myelin with FluoroMyelin™ (FM) Green Fluorescent Myelin Stain
FluoroMyelin™ (FM) (F34651, Invitrogen) staining was performed to visualize intact myelin and degenerating myelin debris detected in or around microglia. The FM was prepared by diluting the stock solution 300-fold into PBS. Tissue sections were stained in solution for 20 minutes at room temperature. A subset of four cases with extensive myelin debris was analyzed using double staining for FM and light chain ferritin (see above).
Visualization of brain tissue iron by Perl’s Prussian blue stain.
We visualized brain tissue iron by histochemical detection in a subset of 18 cases (see above) using Perl’s Prussian blue staining kit (StatLab; #KTIRO; McKinney, Texas) with liver tissue processed as a positive control. All cases were also processed for hematoxylin and eosin (H&E)/Luxol Fast Blue to visualize myelin. In four of these 18 cases, we also visualized brain tissue iron using an optimized Perl’s Prussian blue staining kit per the manufacturer’s instructions (Hitobiotec; HTKMS1002; Kingsport, Tennessee).
Stereological quantification of numerical cell densities (Nv)
The Nv for immunolabeled somata micrographs were acquired (image stack of 15-25 μm thick and 1 μm dissector height) using a Nikon Eclipse Ti laser confocal microscope with pinhole (1.2), camera sensitivity (HV), laser power, offset, and camera settings kept constant for all image acquisitions within a given experimental analysis. The blue, green, and red channels were captured at 405 nm, 488 nm, and 561 nm excitation wavelengths. The 10X objective was used for drawing ROI contours. A minimum of 15 ROIs per section was captured from at least three sections per case. For MBP inclusion counts we used a 60x oil immersion objective for cell counts while other immunolabels were counted using a 40X objective. Images were captured with a distance interval of 1.0 μm between z-sections (~30 μm). The xy view of the images is presented as maximal projections of z stacks, and xz or yz slice views of the regions of interest were reconstructed to illustrate the colocalization of markers. Estimates of Nv for microglia (FTL+, Iba1+) and astrocytes (GFAP+) were obtained using detailed protocols for (1) delineation and systematic random sampling of ROIs; (2) section thickness measurements; (3) counting brick construction; (4) Acquisition of the micrographs; and (5) counting procedures, as described.43
Classification of microglial morphology
Three morphological subtypes of microglia (ramified, reactive, or degenerative) were identified based on previously described criteria45,46 by two independent observers blinded to clinical, radiographic, or neuropathologic characteristics related to each tissue sample/case (Table 2). Cells that displayed a ramified phenotype typically had a small round cell body with thin, long, and highly ramified processes, whereas reactive microglia cells displayed a more hypertrophic cell body with shorter thicker processes. Degenerative microglia displayed a spectrum of degenerative features ranging from the condensation of the cell body to fragmentation of processes.
Table 2:
Morphological features of reactive, ramified, and degenerative microglia.
| Morphological Feature | Reactive | Ramified | Degenerative |
|---|---|---|---|
| Soma | ↑ | ↓ | ↑↓ |
| Atrophy | − | − | ± |
| Hypertrophy | ++ | − | ± |
| Ramification | − | + | − |
| Deramification | ± | − | +++ |
| Beading | − | − | ++ |
| Spheroids | ± | − | ++ |
| Fragmentation | − | − | ++ |
| Inclusion | − | − | ++ |
The distinguishing features of each microglial subtype were defined based on a review of our cohort of 40 cases (see Materials and Methods, Study Design). Symbols: −, absent; ±, less pronounced; +, present; ++, often present; +++, always present ↑, big; ↓, small; ↑↓, irregular. Data are expressed as mean ± standard error of mean.
Real-time PCR analysis of ferroptosis markers 47
Human white matter tissue samples were micro-dissected and resuspended in TRIzol (Life Technologies) to extract total RNA. 250 ng RNA was used to make cDNA using the TaqMan Reverse Transcription Kit (Applied Biosystems). cDNA was diluted 1:1 before real-time PCR (qPCR) experiments using TaqMan Gene Expressions Master Mix (Applied Biosystems). All qPCR probes were FAM-MGB (Applied Biosciences): TFRC (Hs00951083-m1), IREB2 (Hs0121790-m1), GCLC (Hs00155249-m1), SLC1A5 (Hs01056542-m1), MT1G (Hs04401199-s1), AKR1C1 (Hs04230636-sH), HMOX1 (Hs01110250-m1). 18S RNA (Rn03928990-g1) was the endogenous control. Gene expression is presented as relative quantity or fold change versus control and calculated using the 2–ΔΔCt method. Statistical analysis was performed on the ΔCt values for each gene.
Analysis of bulk RNAseq data from the Allen Institute for Brain Science
We analyzed a publicly available bulk RNAseq data set from the Allen Institute for Brain Science (https://aging.brain-map.org/rnaseq/search). We analyzed frontal cerebral white matter samples relative to both parietal and temporal gray matter, which provided expression data for 25,434 genes from each of 110 cases diagnosed with a spectrum of ADNC and VBI.48 The cases analyzed were demographically similar to our primary cohort of 40 human cases (see Subjects, above) because they were from the same ACT cohort from the Seattle Metropolitan area collected by the Department of Pathology, University of Washington under the direction of C.D.K. These cases are over-represented by a history of traumatic brain injury as a pathogenetic risk factor for dementia but are otherwise similar to our cohort.
Statistical analysis
Data are expressed as mean ± standard error of mean. Statistical analysis of demographic and stereological data used Prism 9.5.1 statistical software (GraphPad Software Inc., La Jolla, CA). RNA-seq data extracted from the Allen Institute for Brain Science website (above) was analyzed using R (version 4.1.1). Group analyses were tested for independent effects of age, sex, postmortem interval, Braak, and CERAD, and several cystic and acute/subacute infarcts. Group means were compared by one-way or two-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. A 2-tailed Student’s t-test was employed for two group comparisons. P < 0.05 was considered statistically significant. r and r2 > 0.5 – 0.7 and > 0.7 - 1.0 were considered high and very high correlation/association respectively. Data distribution was tested for normality using the Kolmogorov-Smirnov test.
RESULTS
Degenerative microglia are enriched in myelin debris
Since the injury to myelinated axons is a central neuropathological feature of human aging associated WMI, we first sought to quantify by 3-D laser scanning confocal microscopy the density and percentage of Iba1-labeled microglia that accumulated myelin debris (MBPd), as visualized by staining for myelin basic protein (MBP; Fig. 1A, B)44 and confirmed with the myelin stain FluoroMyelin™ (FM; Fig. 1C).49 We compared cases with cerebral microinfarcts (CMI) and microvascular brain injury with those that lacked CMIs (nCMI) but had confirmed AD neuropathological change (ADNC). Unexpectedly, WM lesions not only contained reactive-appearing microglia but also contained an extensive population of microglia that had morphological features consistent with degenerative microglia (DM) (Fig. 1A) including a swollen phagosome (Fig. 1B; Supplemental Fig. 1). Since under conditions of significant myelin damage, astrocytes may also phagocytose myelin debris50 we also co-labeled astrocytes for GFAP and MBP. In contrast to microglia, even in heavily demyelinated lesions, low levels of myelin debris are uncommonly localized to astrocytes (Fig. 1D).
Fig. 1.
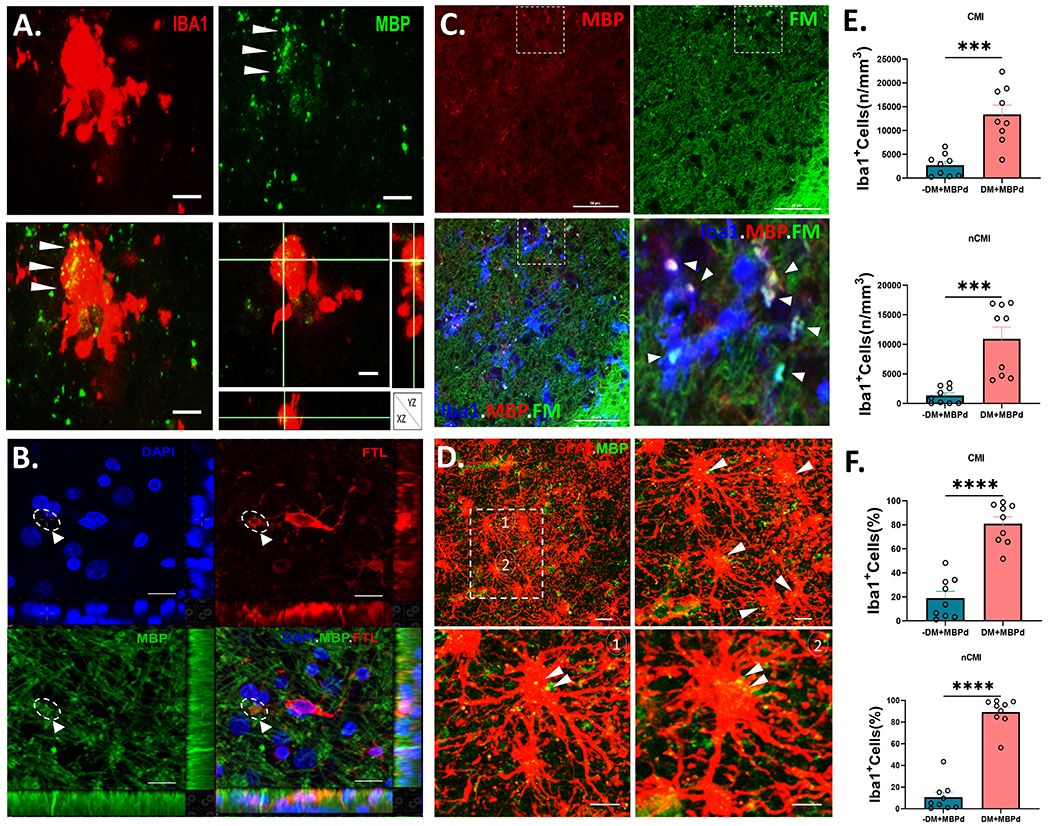
Degenerative microglial (DM) phagocytose myelin debris. (A) A typical Iba1+ DM (red) with a paucity of shrunken processes. Myelin debris (MBPd; green), visualized by staining for myelin basic protein (MBP), was extensively colocalized in the DM soma (merged; arrowheads), as confirmed in the orthogonal views. Scale bars, 20 μm. (B) Example of a DM visualized with ferritin light chain (FTL; red) that extends a prominent phagosome containing myelin debris visualized with MBP (green; arrowhead). DAPI counterstain (blue). See also supplemental figure 2 for a video reconstruction of this cell visualized by 3-D confocal microscopy. Scale bars, 50 μm. (C) Typical example of extensive co-localization of myelin debris in Iba1+ microglia (blue) double-labeled with FluoroMyelin™ (FM; green) and immunostaining for MBP (red). The boxed region in the triple-labeled panel at lower left is shown in higher detail in the lower right panel and shows the extensive distribution of myelin debris in and around the microglia (arrowheads). Scale bars, 10 μm. (D) Typical appearing reactive astrocytes visualized with GFAP (red) in a demyelinated white matter lesion with a striking lack of myelinated fibers labeled with MBP (green). Inset shows two hypertrophic astrocytes (1 and 2) shown at higher magnifications, that contain scattered puncta of MBPd (yellow; arrowheads). Scale bars, 30 μm. (E and F) Stereological counts of the density and percentage of total Iba1+ microglia that co-localized MBPd among nondegenerative microglia (-DM; teal) vs. DM (salmon) for cases diagnosed with cerebral microinfarcts (CMI) in E and cases lacking CMI’s (nCMI) in F. Student’s t-test, ***p<0.001; ****p<0.0001.
Since DM have previously been described in human gray matter lesions with ADNC,46 we first employed a set of morphological criteria to distinguish non-degenerative microglia from DM in our cohort of cases (Table 2). The density and percentage of microglia with MBPd was similar from a quantitative analysis of 18 cases diagnosed either with CMI (n=9) or nCMI (n=9) (Supplemental Fig. 2). Surprisingly, for both CMI (Fig. 1E) and nCMI (Fig. 1F) cases, we found a significantly higher density of DM with MBP+ myelin debris compared to non-degenerative microglia (p<0.0001). The percentage of DM with myelin debris was also significantly higher (~80-85% of total DM; p<0.0001). We next asked whether the accumulation of myelin debris was significantly associated with a degenerative phenotype. The association between the density of Iba1+ microglia with myelin debris and total DM was highly significant for CMI cases with microvascular brain injury (r2=0.8519; p=0.0004) as well as for AD cases (nCMI) (r2= 0.7903; p=0.0013) (Supplemental Fig. 3). Since MBP is a highly charged cationic protein, we also visualized myelin and myelin debris by a non-immunohistochemical method to avoid non-specific myelin interactions, as may potentially occur with some negatively charged antibodies. We visualized myelin and myelin debris with FluoroMyelin™ (FM). We found that FM colocalized with essentially all myelin debris labeled with MBP, including debris engulfed by microglia (Fig. 1C).
The iron-binding protein, light chain ferritin (FTL), was previously found to identify dystrophic microglia in gray matter lesions with ADNC.51 We first determined whether DM with myelin debris could be visualized with FTL and found that phagosomes of FTL-labeled microglia contained MBP+ myelin debris (Fig. 1B; Supplemental Fig. 1). Hence, the accumulation of myelin debris in microglia was highly associated with a degenerative phenotype and suggested that microglial clearance of myelin debris in aging human WMI was significantly associated with microglial degeneration related to vascular brain injury.
White matter DM are distinct from ramified and reactive microglia and enriched in the iron-binding protein light chain ferritin (FTL)
We next determined how the density and percentage of DM differed from that of ramified and reactive microglia. Numerous Iba1+FTL+DM were diffusely distributed in white matter lesions and displayed distinct morphological features that distinguished them from ramified or reactive MG that were only labeled with Iba1 (Fig. 2A; Table 2). The density and percentage of Iba1+ ramified or reactive microglia was significantly less than that of DM, which were the major population of microglia in white matter lesions (Fig. 2B).
Fig. 2.
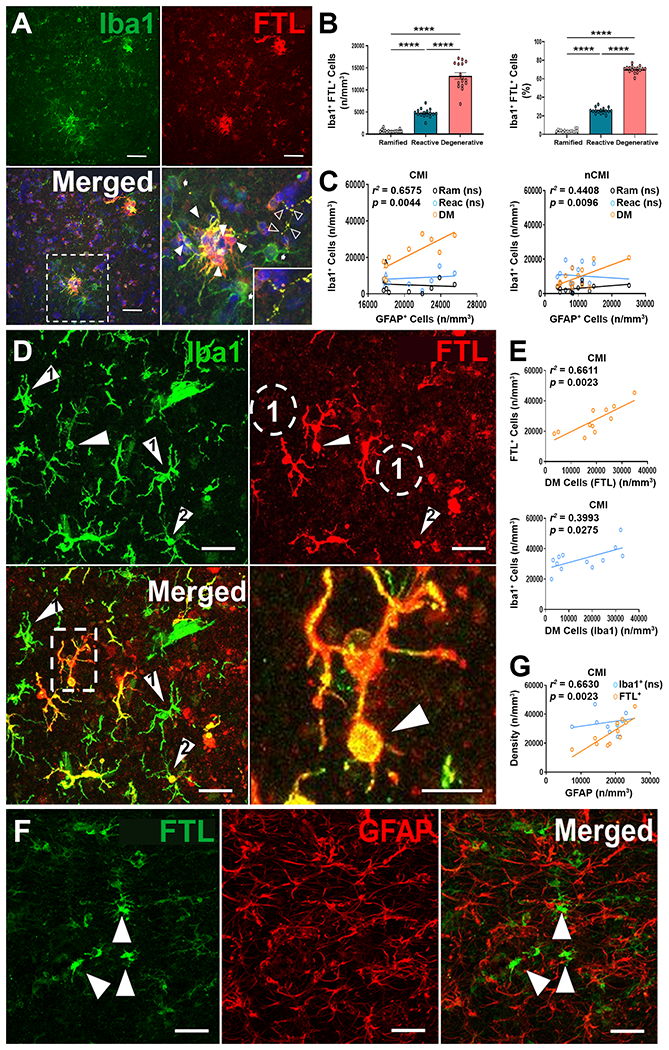
Degenerative microglia (DM) are iron-enriched and selectively express light chain ferritin (FTL). (A) A subset of Iba1+ microglia (green) label for the iron-binding protein light chain ferritin (FTL; red). A higher magnification detail of the boxed region in the merge image is shown on right. Note clusters of DM (closed arrowheads), which label for FTL, whereas non-degenerative microglia (arrows) label for Iba1 but not for FTL. An intensely labeled fragmented DM process is seen at the upper right (open arrowheads) and shown in the inset at lower left. Scale bars, 50 μm. (B) Density and percentage of ramified (Ram), reactive (Reac), and Degenerative (DM) microglia that were Iba1+FTL+. Note that FTL+DM comprised the major population of microglia in WM lesions. ****p<0.0001; ANOVA with Tukey’s post hoc test. (C) DM but not ramified or reactive microglia were significantly associated with the magnitude of WMI quantified as the density of GFAP+ astrocytes (r2=0.6575; p=0.0044). (D) Microglia labeled for Iba1 (green) and FTL (red) display considerable heterogeneity in terms of iron labeling. Note examples from the subset of non-degenerative microglial (green only; arrowhead #1) that do not label with FTL, with their location noted in the ferritin image as dashed circles (#1). In contrast, in the merge image, numerous DM are visualized (red/yellow) which varied from FTL labeling restricted to the soma (arrowhead #2) to extensive labeling of the processes. Note FTL+ DM with swollen phagosome (dashed box), which is shown at right at higher magnification (arrowhead indicates the phagosome). Scale bar, 50 μm. (E) FTL is a superior marker for DM when compared to DM identified by morphology using Iba1 alone. A plot of total FTL+ microglia vs. FTL+ DM yielded a more significant association (r2=0.6611; p=0.0023) when compared with analysis using Iba1 alone (r2=0.3993; p=0.0275). (F) FTL (green) does not label GFAP+ astrocytes (red) in WM lesions. Note in the merged image that FTL+ DM (green; arrowheads) were distinguished from astrocyte labeling (red). Scale bars, 50 μm. (G) Total FTL+ DM are significantly associated with the magnitude of WMI, defined as the density of GFAP+ astrocytes (r2=0.0.6630; p=0.0023), whereas no significant association is seen for total Iba1+ microglia.
We next determined how the distribution of microglial subtypes was influenced by the severity of WMI. We previously showed that the density of GFAP-labeled astrocytes was a sensitive marker of the burden of WMI across a wide range of injury severity.43 The density of Iba1+ DM was significantly associated with the magnitude of WMI defined by GFAP+ astrocyte density for both CMI cases (r2= 0.6575; p<0.0044) and nonCMI cases (r2= 0.4408 p<0.0096) (Fig. 2C). However, neither ramified nor reactive microglia density was significantly associated with the magnitude of astrogliosis.
We next asked if FTL was a marker specifically expressed by DM in white matter lesions. Although DM have been previously described in gray matter, their presence in aging human white matter is poorly defined.52 We analyzed microglia, which were double-labeled for Iba1 and FTL (Fig. 2D). A large subpopulation of FTL+ microglia strongly labeled for ferritin. FTL+ microglia displayed degenerative features that varied from mild to severe. Some cells appeared to be in the early stages of degeneration with FTL labeling restricted to the soma. Cells with more advanced degeneration displayed diffuse labeling of the soma and processes. A prominent feature of some of the cells was a dilated apparent phagosome, perhaps related to the accumulation of myelin debris (Fig. 1D).
In CMI cases (Fig. 2E), we found a significant association between the density of total FTL+ microglia and FTL+ cells that displayed a degenerative microglial phenotype (r2=0.6611; p=0.0023), which supported that DM are enriched in FTL-bound iron. Moreover, we found that cells labeled with Iba1 were less predictive of a degenerative microglial phenotype (r2=0.3993; p=0.0275). Hence, it appears that progression to microglial dystrophy in WMI coincides with FTL-bound iron accumulation.
We next determined how the distribution of DM was influenced by the severity of WMI defined by astrogliosis. We first confirmed that FTL+ cells were not labeled for GFAP (Fig. 2F). The density of FTL+ cells in CMI cases was significantly associated with the magnitude of WMI defined by GFAP+ astrocyte density (Fig. 2G; r2= 0.663; p<0.0023), whereas FTL+ DMs were not significantly associated with WMI in nCMI cases (r2= 0.25; p=0.20). Hence, DM were the major population of microglia in white matter lesions and were more significantly associated with white matter lesions related to the microvascular injury. White matter DM were enriched in iron bound to FTL, which distinguished DM from ramified or reactive microglia, which did not label with FTL. We also analyzed whether the white matter accumulation of iron identified by FTL staining also could be visualized by histochemical detection using an optimized Perl’s Prussian blue stain (see Materials and Methods). In contrast to FTL staining, iron-labeled cells were rarely visualized histochemically in white matter (Supplemental Fig. 4), consistent with prior studies of aging human cerebral gray matter.53,54
White matter DM accumulate lipid droplets and sustain lipid peroxidation injury
Lipid metabolism is integral to microglial phagocytosis.55 A pro-inflammatory population of lipid droplet-accumulating microglia (LDAM) was recently described in rodents and human aging brains that are defective in phagocytosis and produce elevated levels of reactive oxygen species.56 Since we found that DM accumulate myelin debris, we asked if DM also display lipid dysmetabolism and excess lipid droplet accumulation. We visualized the lipid droplet protein PLIN2, a member of the perilipin family of lipid storage proteins, which is a hypoxia-sensitive constitutively expressed marker of cellular lipid droplet accumulation.57,58 We employed 3-D confocal microscopy to visualize PLIN2 accumulation in FTL+Iba1+ DM. PLIN2+ lipid droplets were largely restricted to the cytoplasm of the soma (Fig. 3A). The density and percentage of PLIN2+ DM was similar between CMI and nCMI cases (Fig. 3B). For both cohorts, the majority of DM labeled for PLIN2 (~75%). When we evaluated the association between total microglial labeled with Iba1 and those containing lipid droplets, FTL+PLIN2+ DM were significantly elevated in CMI cases, (r2=0.63; p<0.011; Fig. 3C), but not in nCMI cases (r2=0.38; p=0.19; Fig. 3D). Hence, lipid droplets were selectively enriched in DM and not significantly associated with non-degenerative microglia.
Fig. 3.
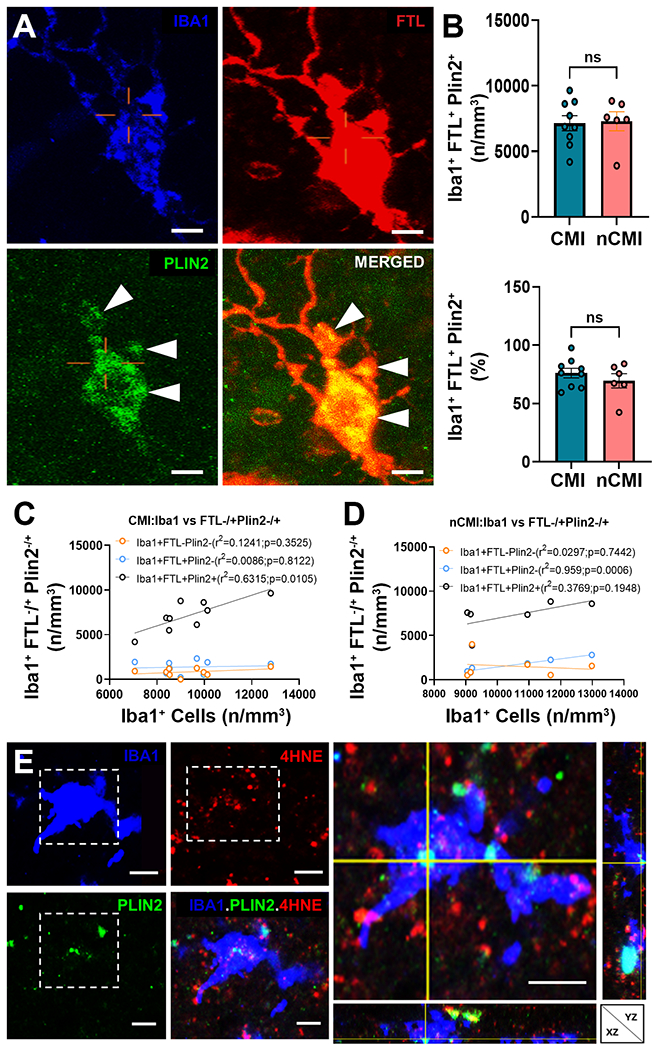
DM accumulate lipid droplets visualized with PLIN2. (A) Detail of a DM triple-labeled for Iba1 (blue), FTL (red) and PLIN2 (green). Note the extensive cytoplasmic labeling (arrowheads) of PLIN2 visualized in the merged image of FTL and PLIN2. Scale bars, 30 μm. (B) The density and percentage of Iba1+FTL+PLIN2+DM were similar in CMI and nCMI cases. (C and D) The density of lipid droplets containing DM (Iba1+FTL+PLIN2+; black) was significantly associated with total microglia in white matter lesions (r2=0.6315; p=0.0105). Nondegenerative microglia that lacked PLIN2 (Iba1+FTL−, salmon) and DM that lacked PLIN2 (Iba1+FTL+; blue) were not significantly associated with the lesion burden of microgliosis. (E) Detail of an Iba1+ (blue), PLIN2+ (green) microglial cell with lipid peroxidation injury visualized by staining for 4-hydroxynonenol (4-HNE; red). Orthogonal view at right. Scale bars, 30 μm.
Since microvascular white matter injury involves ischemia and oxidative stress, we next asked if lipid droplet accumulation in DM is associated with lipid peroxidation injury. We double-labeled for PLIN2 and the lipid peroxidation marker 4-hydroxynonenol (4-HNE).59 PLIN2+ DM labeled strongly for 4-HNE (Fig. 3E). To quantify oxidative stress in nondegenerative and degenerative microglia, we next triple-labeled for Iba1, 4-HNE and Tom20, a mitochondrial outer membrane translocase and sensor of mitochondrial oxidative stress.60,61 The cytoplasm of DM was typically enriched in 4-HNE and Tom20, consistent with robust oxidative stress (Fig. 4A). Since DM displayed no significant differences in either the density or percentage of cells in CMI versus nCMI cases (Fig. 4B), cases were combined for quantitative analysis of microglial localization of 4-HNE, Tom20 or both. Consistent with the percentage of myelin debris containing DM (Fig. 1C), the density of DM was nearly 80% of total Iba1+ microglia (Fig. 4C). In contrast to non-degenerative microglia, DM displayed significant elevations in the density and percentage of cells that labeled for 4-HNE, Tom20 or both (Fig. 4D-F). Within the DM population, ~35% were labeled for lipid peroxidation injury with 4-HNE, and a similar percentage was labeled for Tom20. Hence, DM were significantly enriched in lipid droplets and displayed evidence of pronounced lipid peroxidation injury and mitochondrial oxidative stress.
Fig. 4.
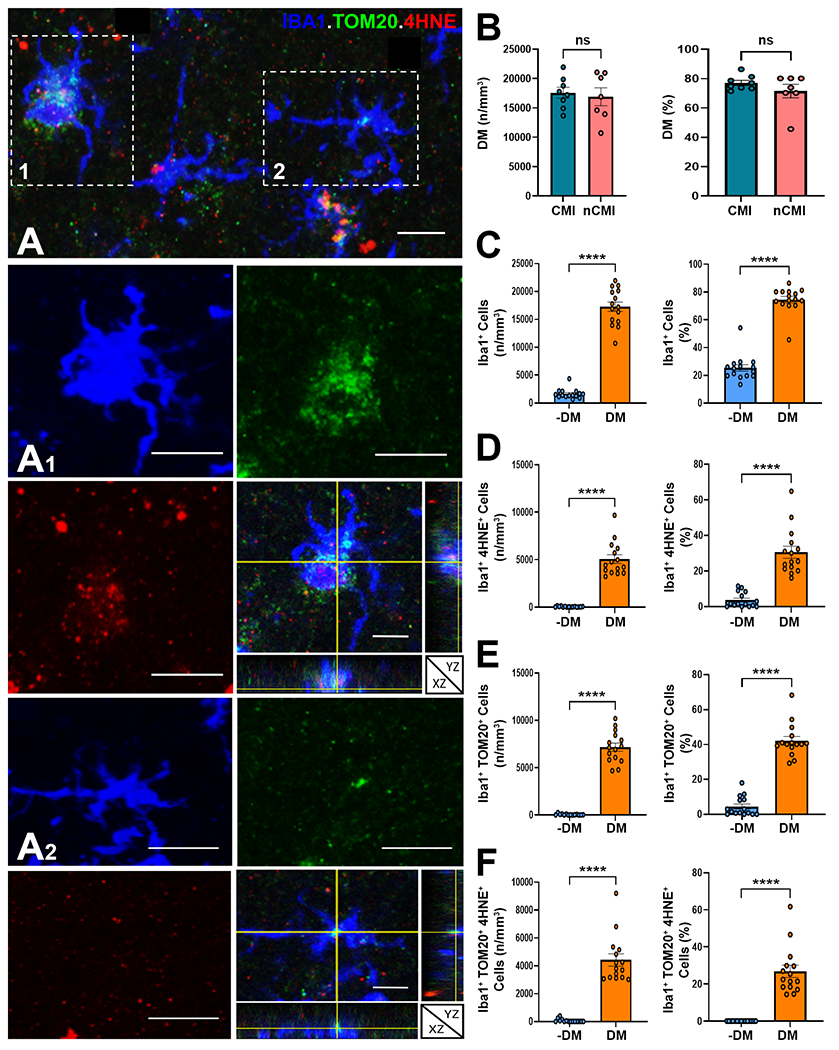
Microglial lipid accumulation is a target for lipid peroxidation injury and mitochondrial oxidative stress in white matter lesions. (A) Example of lesion associated Iba1+ microglia (blue) with varying degrees of lipid peroxidation injury, visualized with 4-hydroxynonenol (4-HNE; red), and mitochondrial oxidative stress, visualized with Tom20 (green). Insets of cells 1 and 2 are shown at higher magnification below together with orthogonal views. Scale bars, 30 μm. (B) The density and percentage of DM were similar in CMI and nCMI cases. (C) The density and percentage of DM in CMI cases was significantly higher than nondegenerative microglia (−DM). (D to F) The density and percentage of DM in CMI cases that labeled for 4-HNE (D), TOM20 (E) and both 4-HNE and TOM20 (F) was significantly higher compared to nondegenerative microglia (−DM). Student’s t-test, ****p<0.0001 in C to F.
Lipid peroxidation injury is accompanied by DNA damage
One of the major consequences of lipid peroxidation injury is the generation of secondary DNA adducts formed by the reaction of reactive aldehydes with DNA.62 We thus asked whether DM also displayed evidence of DNA damage. To detect DNA damage, we stained for phospho-histone H2A.X, which is a sensitive marker of DNA double-strand breaks.63 A broad spectrum of H2A.X staining was observed, which appeared to reflect the spectrum of microglial degenerative change. Cells with minimal signs of degeneration displayed light H2A.X staining (Fig. 5A), consistent with senescence, whereas FTL+ microglia with advanced stages of degeneration displayed more intense highly condensed staining (Fig. 5B). The percentage of microglia that labeled for DNA double-strand breaks with H2A.X was ~20% (Fig. 2C) and was of similar magnitude to DM that labeled for lipid peroxidation injury with 4-HNE (Fig. 4D). In contrast, we found that the majority of astrocytes in WM lesions did not label for H2A.X; but scattered clusters of reactive astrocytes labeled for H2A.X, consistent with a role for astroglial senescence in the aging brain.64 We also stained for the apoptosis markers, activated caspase-3, and TUNEL, and found that neither marker was detected in DM or reactive astrocytes (data not shown).
Fig. 5.
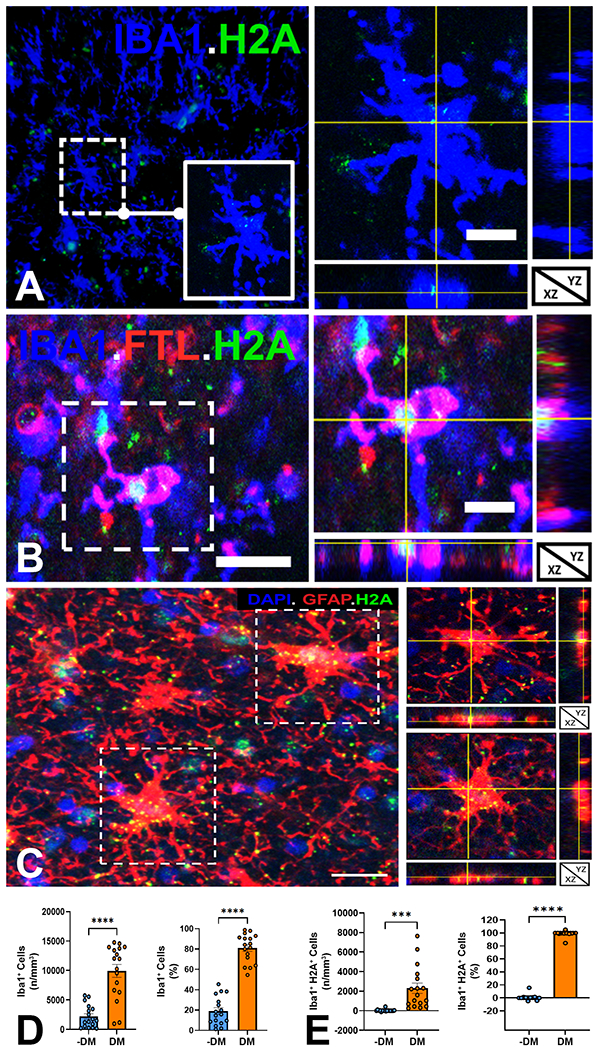
Microglia in white matter lesions sustain DNA damage. (A) Low power view of Iba1+ microglia (blue) stained for DNA double strand breaks with phospho-histone H2A.X (H2A, green). Inset and orthogonal views of a microglia cell with scattered H2A staining consistent with a low level of DNA damage. Scale bars, 30 μm. (B) Detail and orthogonal views of a typical Iba1+ (blue) FTL+ (red) DM with truncated and shrunken processes and intense H2A labeling (arrowheads) consistent with a high level of DNA damage. Scale bars, 30 μm. (C) Low power view of a lesion with several reactive astrocytes (GFAP+, red). Orthogonal views of two cells (dashed boxes) with H2A labeling (green) and DAPI (blue). Scale bars, 30 μm. (D) The density and percentage of DM in white matter lesions was significantly higher than nondegenerative microglia (-DM). (E) The density and percentage of microglia that stained for DNA damage with H2A in white matter lesions was significantly higher for DM compared to nondegenerative microglia (-DM). Because there were no significant differences between CMI and nCMI cases, data was analyzed as the full cohort of 17 cases in D and E. Student’s t-test; ***p< 0.001; ****p<0.0001 in D and E.
White matter DM are enriched in markers of ferroptosis
Since iron-mediated lipid peroxidation injury is a central feature of ferroptosis, we next asked whether the enrichment of DM in our cohort of WMI cases might be accompanied by the enhanced expression for markers of ferroptosis. We employed qRT-PCR to quantify the expression of seven markers that are associated with distinct ferroptosis pathways (Fig. 6A). Five of the seven markers (GCLC, HMOX1, TFRC, IREB2, and SLC1A5) displayed enhanced expression across the cohort, whereas AKRC1c and MT1G were more variably expressed. However, our cohort size was too small to detect significant differences in expression between CMI and nCMI cases.
Fig. 6.
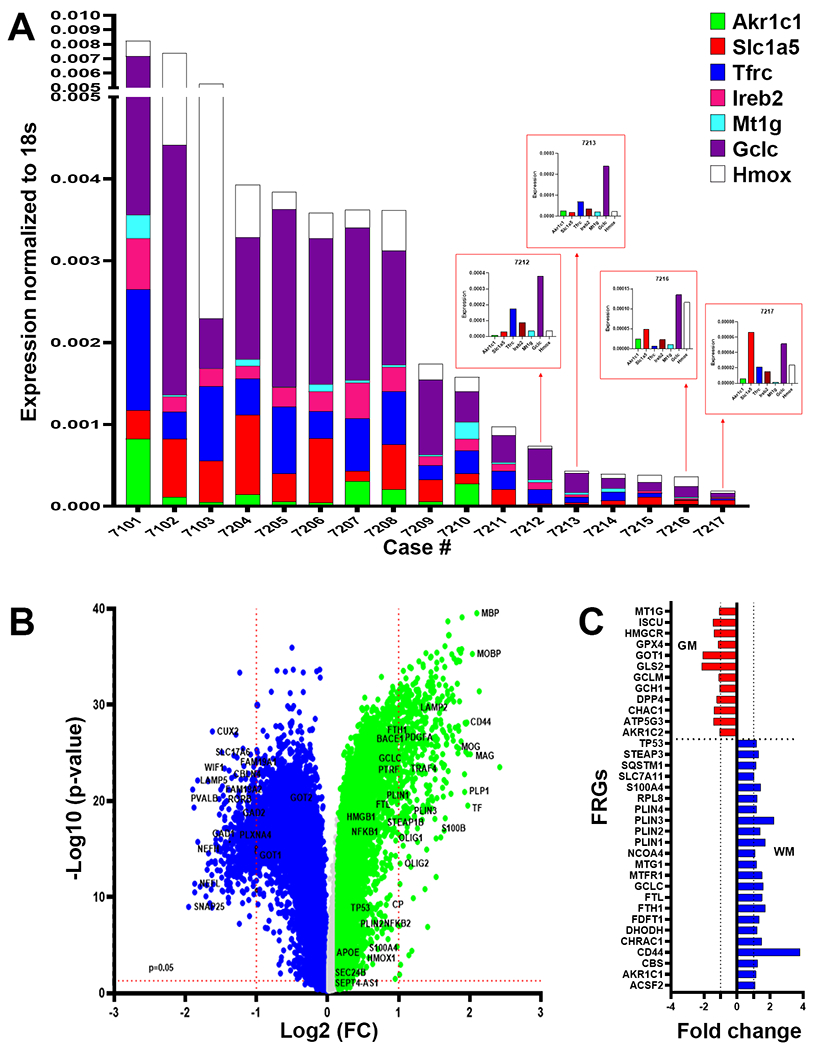
DM are enriched in markers of ferroptosis. (A) Analysis of white matter lesions from 17 cases enrolled in the ACT study diagnosed with dementia-related to microvascular brain injury (mVBI) or ADNC. Gene expression was variably enhanced for seven markers associated with distinct ferroptosis pathways (GCLC, HMOX1, TFRC, IREB2, MR1G, AKRC1c and SLC1A5) quantified by qRT-PCR and normalized to 18S rRNA. (B) Analysis of bulk RNAseq data set from the Allen Institute for Brain Science of gene expression in 110 cases enrolled in the ACT study diagnosed with a spectrum of mVBI or ADNC. Volcano plot of the p-value (−Log 10) and fold change of genes with significantly higher expression in white matter relative to gray matter (green; 21,879 genes) or gray matter relative to white matter (blue; 22,569 genes). A unique set of white matter-associated ferroptosis-related genes were significantly upregulated together with highly expressed genes linked to myelination. (C) Comparison of the fold expression changes for ferroptosis-related genes in white matter (blue) vs. gray matter (red).
We next analyzed a publicly available bulk RNAseq data set from the Allen Institute for Brain Science that analyzed gene expression in 110 cases diagnosed with a spectrum of ADNC and VBI.48 Importantly, these cases were demographically similar to the cases analyzed by qRT-PCR because they derived from the same ACT cohort (See Materials and Methods). A volcano plot of differentially expressed genes demonstrated that a unique set of ferroptosis genes were enriched and significantly upregulated in the white matter in association with highly expressed genes linked to myelination (Fig. 6B). Figure 6C compares the expression of ferroptosis-related genes (FRGs) in white vs. gray matter and demonstrates that a unique FRG set were upregulated in frontal white matter samples compared to parietal gray matter samples, including genes independently identified in our qRT-PCR screen of FRGs (e.g., AKR1C1, FTL, PLIN genes, MTG1 and GCLC). A pathway analysis supported that WMI was associated with a distinctly different group of FRGs when compared to gray matter-associated FRGs (Supplemental Fig. 5). WMI-associated FRGs localized to pathways regulating lipid metabolism (e.g., PUFA, TP53), iron transport/ metabolism (e.g., TFRC, FTL, IREB2) and glutamine and cysteine uptake for glutathione synthesis (e.g., SLC1A5 and SLC7A11). Gray matter injury-associated FRGs included the major checkpoint for ferroptosis (i.e., GPX4/HMGCR).
Discussion
Aging human cerebral WMI appears to be highly susceptible to microglial degeneration from ferroptosis, because of several complementary factors that compromise microglial responses to WMI. Major defining features of ferroptotic aging human white matter microglia were the accumulation of myelin debris in iron-enriched senescent microglia, which displayed mitochondrial metabolic stress, lipid peroxidation injury, and DNA damage.65–67 Recurrent microvascular ischemia to aging WM enhances the burden of iron-rich myelin debris to be cleared by WM microglia.68 Moreover, axonal injury related to Wallerian degeneration in AD may also constitute another significant mechanism that contributes to myelin loss.69 In contrast to nondegenerative microglia, myelin debris was readily visualized in the majority of DM, which suggests that dysfunctional clearance of myelin debris is a key factor that contributes to iron-mediated microglial degeneration. Aging human microglia are iron-rich cells.54 Myelin debris appears to be a potent source of iron enrichment in microglia. Recent in vitro studies support that iron loading in human IPSC-derived microglia not only dampened microglial activation and inflammation, but activated cellular stress response and oxidative stress pathways, enhanced labile free iron, and promoted ferritin iron storage.111 Consistent with these observations, we found that WM DM were enriched in light chain ferritin (FTL), which selectively distinguished DM from activated microglia. Ferritin is the major iron binding protein in the aging brain and is currently the most sensitive means to detect intracellular iron storage in microglia in contrast to histochemical detection of iron, which appears to markedly underestimate iron levels in microglia.53,54 Future studies may be useful to determine if more sensitive means can be developed to detect labile-free iron in aging human microglia using DAB enhancement, for example, as has been described for inflammatory lesions in MS.112
Myelin debris not only enhances microglial iron content but constitutes a potent source of toxic lipid debris that accelerates microglial senescence and impairs remyelination.49,70 Myelin debris further contributes to lipid dysmetabolism within cytoplasmic lipid droplet organelles that dysregulate mitochondrial bioenergetics and augment oxidative lipid peroxidation injury.71 Hence, recurrent microvascular ischemia in aging human WM appears to cause significant oxidative damage to metabolically challenged senescent microglia via iron-mediated lipid peroxidation injury, a key trigger for human microglial ferroptosis.
In addition to microglia, astrocytes may also phagocytose myelin debris under conditions of significant myelin damage.50 In heavily demyelinated WM lesions, astrocytes contained only low levels of myelin debris. Moreover, in contrast to microglia, aging WM astrocytes did not display degenerative changes, were not enriched in light chain ferritin, and displayed low levels of DNA fragmentation consistent with astroglial senescence. These findings suggest that a lack of iron-mediated lipid peroxidation may account for astrocyte resistance to ferroptotic degeneration. Notably, microglia appear to be more susceptible than astrocytes or neurons to ferroptosis in vitro.72,73 Our data may also be an underestimate of microglial degeneration in some WM lesions where DM were only detected as fragmented remnants.
Emerging data support that microglial contact with cerebral vessels contributes to cerebrovascular homeostasis, including the regulation of vasomotor function and remodeling of WM arterioles.74–76 DM were the major population of microglia in our cases of vascular brain injury or AD, which may disrupt microglial-vascular interactions that support white matter integrity. Microvascular ischemia to human white matter is strongly associated with chronic hypertension, diabetes, and heart disease, which contribute to brain-specific vascular dysfunction in both vascular dementia and AD.2 Our analysis of human penetrating white matter arterioles from aging human brain donors found significant cholinergic and bradykinin-mediated vasodilator dysfunction that may contribute to the risk for recurrent WM ischemia.37,77 Notably, we recently found that AD and VCID interact synergistically to impair arteriolar dilation.77 Future studies are needed to determine if the degeneration of microglia contributes to a positive feedback loop that may further exacerbate microvascular ischemia and WMI in vascular dementia and AD.
Diffuse ischemic injury to myelin enhances lipid dysmetabolism. WMI imposes significant metabolic stress on the clearance capacity of lipid-laden WM microglia, given the high burden of myelin debris. Aging microglia display disturbances in mitochondrial bioenergetics, cholesterol clearance, and lipid metabolism,78,79 which likely contributed to impaired phagocytosis of myelin debris and enhanced lipid peroxidation injury in WM DM. Accumulation of myelin debris coincided with a significant increase in DM that labeled PLIN2, a marker of lipid droplet accumulation. PLIN2 is a member of the perilipin family of lipid storage proteins, which is a hypoxia-sensitive constitutively expressed marker of cellular lipid droplet accumulation.57,58 Lipid droplet accumulating microglia (LDAM) display dysfunctional phagocytosis, ROS accumulation and promote a pro-inflammatory state of neurodegeneration.56 We observed robust DM labeling for the lipid peroxidation injury marker, 4-HNE.
Lipid peroxidation injury was accompanied by enhanced expression of Tom20, a mitochondrial outer membrane translocase, which is a sensor of mitochondrial iron-mediated reactive oxygen species generation. 60,61 Mitochondria are centrally involved in regulating responses to ferroptosis.80 Notably, overexpression of Tom20 in vivo was shown to promote neurodegeneration,81 suggestings that enhanced Tom20 expression in DM may contribute to microglial degeneration.
Clearance of myelin debris in DM thus appears to contribute to a state of labile-free iron overload related to ferritin disruption, which catalyzes lipid-targeted superoxide generation via Fenton chemistry.82 Notably, we previously reported that aging human WM sustains significant oxidative stress, as quantified with F2-adrenoprostanes and F4-neuroprostanes—arachidonic acid metabolites, which are sensitive markers of lipid peroxidation injury to myelin and axons, respectively.83 Our findings support that lipid droplet accumulation is central to microglial degeneration by promoting oxidative stress-mediated lipid peroxidation injury, which mediates DNA fragmentation and is a hallmark of ferroptosis.
Microglial senescence may be a key feature related to the susceptibility of human microglia to degeneration. Senescent microglia not only display impaired phagocytosis,56,84,85 but express senescence-associated genes, have shortened telomeres, and display distinct aging-associated transcriptomic changes.86–90 Senescent microglia also display altered secretory profiles with enhanced generation of pro-inflammatory cytokines,91,92 which may disrupt axonal and OPC integrity and repair processes required to promote remyelination. Microglial senescence may account for the pronounced association of DM with aging WM but not with WMI earlier in life. Although WMI is the major form of injury to the preterm brain, DM have not been detected in acute or chronic early human 44,93,94 or experimental WMI.95 Nor have DM been reported in younger adults in association with major demyelinating diseases such as multiple sclerosis. Consistent with microglial senescence, DM displayed fragmented and shortened processes and was labeled for DNA fragmentation with phospho-histone H2A.X.96 Posttranslational histone modification by phosphorylation of H2A promotes double-strand break recognition and repair and is a sensitive marker of DNA double-strand breaks and CNS senescence.63,97 Hence, microglial senescence appears to be another contributing factor related to microglial ferroptosis in aging human WM.
Although we observed numerous DM with features of cell death, WM DM displayed no evidence of apoptotic degeneration. Rather, our findings support that aging-associated WMI involves a mechanism of microglial degeneration consistent with ferroptosis (Summary Fig. 7). Multiple markers of ferroptosis have been recently identified that participate in several key pathways that regulate iron-mediated cell death.98 Although ferroptosis has been recently linked to Parkinson’s disease99 and is implicated in AD,82 a direct role for ferroptosis in aging-associated WMI in AD and VCID has been unclear. Our pathway analysis of AD and VCID cases supports that ferroptosis-related genes are differentially upregulated in gray and white matter lesions (Fig. 6C and Supplemental Fig. 4). Notably, WMI was associated with selective enhanced expression of several ferroptosis-related iron transport proteins, which serve integral roles to sequester intracellular free iron, which readily catalyzes oxygen radicals including superoxide as well as nitric oxide radicals.54,100,101 Transferrin receptor C (TFRC) was recently proposed as a marker of ferroptosis,102 and its expression was elevated in the majority of our cases. Ireb2 (iron response element binding protein 2), which was detected in most of our cases, is also a key regulator of ferrous iron accumulation and an inhibitor of ferroptosis in spinal cord white matter injury.103 Microglia are enriched in ferritin104 and FTL was found to be a selective marker of WM DM.
Fig. 7.
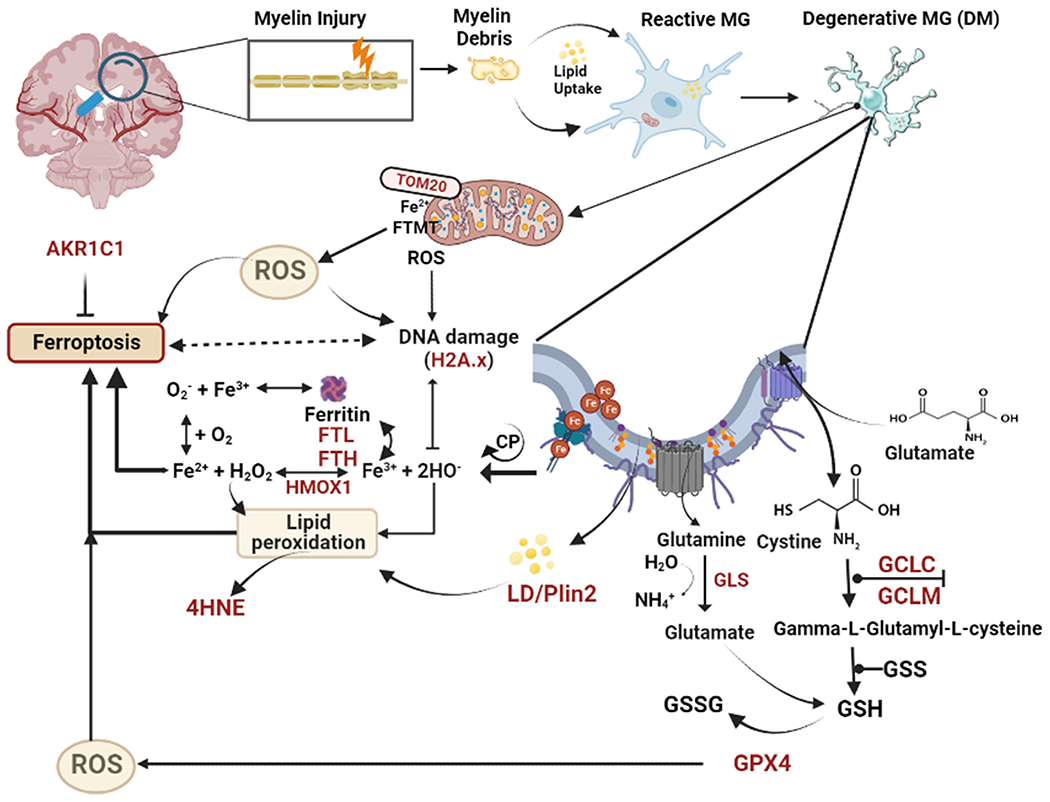
Summary of data supporting a role for ferroptosis in microglial degeneration in human WMI related to vascular dementia or AD. Injury to myelin promotes the accumulation of myelin debris and lipid uptake by senescent reactive microglia (MG). Disturbances in multiple molecules and genes (brown text) appear to contribute to pronounced oxidative stress that leads to a pronounced increase in the number of DM. Central factors appear to involve dysregulation of iron metabolism, impaired generation of glutathione anti-oxidants and mitochondrial metabolic stress, which together contribute to lipid peroxidation injury and DNA damage. Abbreviations: AKR1C1, Aldo-keto reductase family 1 member C1; DM, degenerative microglia; FTL, Ferritin light chain; FTH, Ferritin heavy chain; FTMT, Ferritin mitochondrial; GPX4, Glutathione peroxidase 4; GCLC, Glutamate-cysteine ligase catalytic subunit; GCLM, Glutamate-cysteine ligase modifier subunit; GSH, reduced glutathione; GSSG, oxidized glutathione; 4-HNE, 4-hydxoynonenol; HMOX1, Heme oxygenase 1; H2A.X, phospho-histone H2A.X; LD, lipid droplets; MG, microglia; PLIN2, perilipin 2; ROS, reactive oxygen species; Tom 20, translocase of outer membrane 20.
Enhanced expression of anti-oxidant defenses was also observed in association with WMI. SLC1A5 is a membrane-associated glutamine transporter through which glutaminolysis regulates ferroptosis105 and has been linked to malignant glioma status.106 The gene GCLC encodes gamma-glutamylcysteine synthetase, which is the first rate-limiting enzyme of glutathione synthesis. Glutathione is a central regulator of oxidative stress responses in ferroptosis.107 Expression of GCLC was consistently elevated in our cohort and has been recently identified to protect against ferroptosis.108 Elevation of heme oxygenase 1 (HMOX1) is also consistent with its key antioxidant role in ferroptosis.99 AKRC1c encodes one of several aldoketoreductases that degrade lipid peroxides, promoting resistance to ferroptosis.109 Notably, PLIN2 was significantly enriched in WM DM and aging white matter displayed enhanced expression of PLIN1-4, which regulates lipid droplet metabolism. Taken together, our results provide further support that aging-associated WMI involves significant oxidative stress and lipid peroxidation injury2 and may provide an explanation for the enhanced expression of key anti-oxidant defense mechanisms observed here as a potential compensatory response to ferroptosis.
Although these ex vivo human data are collectively consistent with a mechanism of oxidative stress mediated WMI involving ferroptosis, confirmation in an animal model was not feasible to definitively establish the role of ferroptosis in aging-associated WMI. Animal models of aging-related human microvascular brain injury currently do not exist that replicate the spectrum of human white matter pathology. Currently, available models generate cortical microinfarcts but do not generate the diffuse white matter lesions seen in humans.108 Future studies are needed to determine whether DM are a unique feature of the vascular supply of aging human WMI or also occurs in other species. Such animal models would also be valuable to define the temporal progression of ferroptosis in response to oxidative stress or other processes such as Wallerian degeneration and compare the magnitude of ferroptotic degeneration relative to other cell death mechanisms including apoptosis.
We likely underestimated the magnitude of microglial degeneration, since the analysis of progressive cell death was not feasible. We typically visualized the remnants of degenerating microglia, which were not quantifiable by stereology. Nevertheless, confidence in our findings is supported by the short post-mortem intervals achieved with our rapid autopsy protocol, which provided tissue of high integrity for immunohistochemical and molecular studies.
In conclusion, ferroptosis appears to be a significant mechanism of WMI associated with microvascular brain injury and AD. Microglial degeneration appears to be mediated by a novel form of oxidative stress-associated microglial injury that triggers ferroptosis in cells that sustain iron-mediated lipid peroxidation injury related to the accumulation of myelin debris. Future studies are needed to determine whether WM DM derive from activated microglia110 and the extent to which ferroptotic WMI is preventable as a therapeutic strategy to reduce the impact of microglial dysfunction on vascular dementia and AD.
Supplementary Material
Summary for Social Media If Published, and provide the information below:
1. If you and/or a co-author has a Twitter handle that you would like to be tagged, please enter it here.
@Adephiladex
2. What is the current knowledge on the topic? (one to two sentences)
White matter injury and microglial dysfunction are associated with cognitive impairment in Alzheimer’s disease and vascular dementia. However, the mechanisms of the vulnerability of microglia to injury are not well understood.
3. What question did this study address? (one to two sentences)
We investigated mechanisms related to the clearance of myelin debris in human white matter lesions in AD and vascular dementia cases. We determined whether microglial clearance of myelin debris was related to ferroptosis.
4. What does this study add to our knowledge? (one to two sentences)
This study identified that white matter microglia are highly susceptible to a form of cell death related to the clearance of iron-rich myelin debris. Microglial degeneration involves lipid peroxidation injury and cell death via ferroptosis.
5. How might this potentially impact on the practice of neurology? (one to two sentences)
This study identifies a role for microglial degeneration in white matter injury as a new therapeutic target for AD and vascular dementia.
Acknowledgments:
Supported by grants from the National Institute on Aging (AG054651 to ZB, AG065406 to SAB, AG031892, U01 AG006781, and U19 AG066567 which supports the ACT study, p50 AG005136 and p30AG066509, which support the UW Alzheimer’s disease Research Center), the National Institute of Neurological Disorders and Stroke (NS105984 to SAB) and by the Nancy and Buster Alvord Endowment (to C.D.K). We thank Allison Beller and Aimee Schantz for their superb administrative support, Marta Balogh, Kim Howard, Lisa Keene, Amanda Keen, John Douglas Haswell, and William Dustin-Reed for outstanding technical support. We are grateful to Daniel Perl, M.D. for his invaluable advice and expertise regarding the histopathological analysis of iron accumulation in the aging human brain. We are very grateful to all the ACT participants and families without whose dedication to supporting critical human research this work would be impossible.
Footnotes
Potential Conflicts of Interest: All of the authors declare that they have no potential conflicts of interest.
Data Availability:
All data related to this study are present in the report or the supplemental materials. The bulk RNA sequencing data set are available online through the Allen Institute for Brain Science (see Materials and Methods). Please contact the corresponding author for additional details on materials and methods.
REFERENCES
- 1.Attems J, Jellinger KA. The overlap between vascular disease and Alzheimer’s disease--lessons from pathology. BMC Med 2014;12:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iadecola C. The pathobiology of vascular dementia. Neuron 2013;80:844–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg GA, Wallin A, Wardlaw JM, et al. Consensus statement for diagnosis of subcortical small vessel disease. J Cereb Blood Flow Metab 2016;36:6–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sam K, Crawley AP, Conklin J, et al. Development of white matter hyperintensity is preceded by reduced cerebrovascular reactivity. Ann Neurol 2016;80:277–285. [DOI] [PubMed] [Google Scholar]
- 5.Chen A, Akinyemi RO, Hase Y, et al. Frontal white matter hyperintensities, clasmatodendrosis and gliovascular abnormalities in ageing and post-stroke dementia. Brain 2016;139:242–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Brien JT, Thomas A. Vascular dementia. Lancet 2015;386:1698–1706. [DOI] [PubMed] [Google Scholar]
- 7.Ramirez J, McNeely AA, Berezuk C, Gao F, Black SE. Dynamic progression of white matter hyperintensities in Alzheimer’s disease and normal aging: Results from the Sunnybrook Dementia Study. Front Aging Neurosci 2016;8:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee S, Viqar F, Zimmerman ME, et al. White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited Alzheimer network. Ann Neurol 2016;79:929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alosco ML, Sugarman MA, Besser LM, et al. A Clinicopathologicalinvestigation of white matter hyperintensities and Alzheimer’s disease neuropathology. J Alzheimers Dis 2018;63(4):1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McAleese KE, Firbank M, Dey M, et al. Cortical tau load is associated with white matter hyperintensities. Acta Neuropathol Commun 2015;3:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corriveau RA, Bosetti F, Emr M, et al. The Science of Vascular Contributions to Cognitive Impairment and Dementia (VCID): A framework for advancing research priorities in the cerebrovascular biology of cognitive decline. Cell Mol Neurobiol 2016;36:281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wardlaw JM, Valdes Hernandez MC, Munoz-Maniega S. What are white matter hyperintensities made of? Relevance to vascular cognitive impairment. J Am Heart Assoc 2015;4:001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sonnen J, Santa Cruz K, Hemmy L, et al. Ecology of the aging human brain. Arch Neurol 2011;68:1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sonnen JA, Larson EB, Crane PK, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol 2007;62:406–413. [DOI] [PubMed] [Google Scholar]
- 15.Back SA, Kroenke CD, Sherman LS, et al. White matter lesions defined by diffusion tensor imaging in older adults. Ann Neurol 2011;70:465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalaria RN. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol 2016;131:659–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skrobot OA, Black SE, Chen C, et al. Progress toward standardized diagnosis of vascular cognitive impairment: Guidelines from the vascular impairment of cognition cassification consensus study. Alzheimers Dement 2018;14:280–292. [DOI] [PubMed] [Google Scholar]
- 18.Silbert LC, Nelson C, Howieson DB, Moore MM, Kaye JA. Impact of white matter hyperintensity volume progression on rate of cognitive and motor decline. Neurology 2008;71:108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silbert LC, Dodge HH, Perkins LG, et al. Trajectory of white matter hyperintensity burden preceding mild cognitive impairment. Neurology 2012;79:741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt R, Ropele S, Enzinger C, et al. White matter lesion progression, brain atrophy, and cognitive decline: the Austrian stroke prevention study. Ann Neurol 2005;58:610–6. [DOI] [PubMed] [Google Scholar]
- 21.Sachdev P, Wen W, Chen X, Brodaty H. Progression of white matter hyperintensities in elderly individuals over 3 years. Neurology 2007;68:214–222. [DOI] [PubMed] [Google Scholar]
- 22.Moscufo N, Wakefield DB, Meier DS, et al. Longitudinal microstructural changes of cerebral white matter and their association with mobility performance in older persons. PLoS One 2018;13:e0194051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosario BL, Rosso AL, Aizenstein HJ, et al. Cerebral White Matter and Slow Gait: Contribution of Hyperintensities and Normal-appearing Parenchyma. J Gerontol A Biol Sci Med Sci 2016;71:968–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017;47:566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grubman A, Choo XY, Chew G, et al. Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nat Commun 2021;12:3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wendimu MY, Hooks SB. Microglia phenotypes in aging and neurodegenerative diseases. Cells 2022;11:2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou Y, Song WM, Andhey PS, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 2020;26:131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016;352:712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci 2018;21(10):1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gefen T, Kim G, Bolbolan K, et al. Activated microglia in cortical white matter across cognitive aging trajectories. Front Aging Neurosci 2019;11:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amor S, McNamara NB, Gerrits E, et al. White matter microglia heterogeneity in the CNS. Acta Neuropathol 2022;143:125–141. [DOI] [PubMed] [Google Scholar]
- 32.Spiteri AG, Wishart CL, Pamphlett R, Locatelli G, King NJC. Microglia and monocytes in inflammatory CNS disease: integrating phenotype and function. Acta Neuropathol 2022;143:179–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Y, Zhao X, Zhu Z, Zhang L. Vascular dementia: A microglia’s perspective. Ageing research reviews. Nov 2022;81:101734. doi: 10.1016/j.arr.2022.101734 [DOI] [PubMed] [Google Scholar]
- 34.Cignarella F, Filipello F, Bollman B, et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol 2020;140:513–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 2009;132:288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berghoff SA, Spieth L, Sun T, et al. Microglia facilitate repair of demyelinated lesions via post-squalene sterol synthesis. Nat Neurosci 2021;24:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bagi Z, Brandner DD, Le P, et al. Vasodilator dysfunction and oligodendrocyte dysmaturation in aging white matter. Ann Neurol 2018;83:142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adeniyi PA, Fopiano KA, Banine F, et al. Multispectral LEDs eliminate lipofuscin-associated autofluorescence for immunohistochemistry and CD44 variant detection by in situ hybridization in aging human, non-human primate, and murine brain. ASN Neuro 2022;14:17590914221123138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheli VT, Correale J, Paez PM, Pasquini JM. Iron metabolism in oligodendrocytes and astrocytes, implications for myelination and remyelination. ASN Neuro 2020;12:1759091420962681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaie KW, Willis SL, Caskie GI. The Seattle longitudinal study: relationship between personality and cognition. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn 2004;11(2-3):304–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Back SA, Luo NL, Mallinson RA, et al. Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann Neurol 2005;58:108–120. [DOI] [PubMed] [Google Scholar]
- 43.McNeal DW, Brandner DD, Gong X, et al. Unbiased stereological analysis of reactive astrogliosis to estimate age-associated cerebral white matter injury. J Neuropathol Exp Neurol 2016;75:539–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buser J, Maire J, Riddle A, et al. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann Neurol. 2012;71:93–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol 2009;118:475–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Streit WJ, Xue QS, Tischer J, Bechmann I. Microglial pathology. Acta Neuropathol Commun 2014;2:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Srivastava T, Diba P, Dean JM, et al. A TLR/AKT/FoxO3 immune tolerance-like pathway disrupts the repair capacity of oligodendrocyte progenitors. J Clin Invest 2018;128:2025–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller JA, Guillozet-Bongaarts A, Gibbons LE, et al. Neuropathological and transcriptomic characteristics of the aged brain. Elife 2017;6:e31126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Safaiyan S, Kannaiyan N, Snaidero N, et al. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat Neurosci 2016;19(8):995–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ponath G, Ramanan S, Mubarak M, et al. Myelin phagocytosis by astrocytes after myelin damage promotes lesion pathology. Brain 2017;140:399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lopes KO, Sparks DL, Streit WJ. Microglial dystrophy in the aged and Alzheimer’s disease brain is associated with ferritin immunoreactivity. Glia 2008;56(10):1048–1060. [DOI] [PubMed] [Google Scholar]
- 52.Ahn K, Lee SJ, Mook-Jung I. White matter-associated microglia: New players in brain aging and neurodegenerative diseases. Ageing Res Rev 2022;75:101574. [DOI] [PubMed] [Google Scholar]
- 53.Connor JR, Menzies SL, St. Martin SM, Mufson EJ. Cellular distribution of transferrin, ferritin, and iron in normal and aged human brains. J Neurosci Res 1990;27:595–611. [DOI] [PubMed] [Google Scholar]
- 54.Kenkhuis B, Somarakis A, de Haan L, et al. Iron loading is a prominent feature of activated microglia in Alzheimer’s disease patients. Acta Neuropathol Commun 2021;9:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loving BA, Bruce KD. Lipid and lipoprotein metabolism in microglia. Front Physiol. 2020;11:393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marschallinger J, Iram T, Zardeneta M, et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci 2020;23:194–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McIntosh AL, Senthivinayagam S, Moon KC, et al. Direct interaction of Plin2 with lipids on the surface of lipid droplets: a live cell FRET analysis. Am J Physiol Cell Physiol 2012;303(7):C728–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bildirici I, Schaiff WT, Chen B, et al. PLIN2 Is essential for trophoblastic lipid droplet accumulation and cell survival during hypoxia. Endocrinology 2018;159(12):3937–3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang W, Zhao F, Ma X, Perry G, Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener 2020;15:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Joshi AU, Minhas PS, Liddelow SA, et al. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci 2019;22:1635–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou B, Zhang JY, Liu XS, et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res 2018;28(12):1171–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marnett LJ. Oxy radicals, lipid peroxidation and DNA damage. Toxicology 2002;181-182:219–222. [DOI] [PubMed] [Google Scholar]
- 63.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem 2000;275(13):9390–9395. [DOI] [PubMed] [Google Scholar]
- 64.Cohen J, Torres C. Astrocyte senescence: Evidence and significance. Aging Cell 2019;18(3):e12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao M, Yi J, Zhu J, et al. Role of mitochondria in ferroptosis. Mol Cell 2019;73(2):354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell 2022;82:2215–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 2021;22:266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Connor JR, Menzies SL. Altered distribution of iron in the central nervous system of myelin deficient rats. Neuroscience 1990;34:265–271. [DOI] [PubMed] [Google Scholar]
- 69.McAleese KE, Walker L, Graham S, et al. Parietal white matter lesions in Alzheimer’s disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta Neuropathol 2017;134(3):459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lampron A, Larochelle A, Laflamme N, et al. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med 2015;212:481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol 2019;20(3):137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jiao L, Li X, Luo Y, et al. Iron metabolism mediates microglia susceptibility in ferroptosis. Front Cell Neurosci 2022;16:995084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kapralov AA, Yang Q, Dar HH, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol 2020;16(3):278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bisht K, Okojie KA, Sharma K, et al. Capillary-associated microglia regulate vascular structure and function through PANX1-P2RY12 coupling in mice. Nat Commun 2021;12(1):5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Joost E, Jordão MJC, Mages B, Prinz M, Bechmann I, Krueger M. Microglia contribute to the glia limitans around arteries, capillaries and veins under physiological conditions, in a model of neuroinflammation and in human brain tissue. Brain Struct Funct 2019;224:1301–1314. [DOI] [PubMed] [Google Scholar]
- 76.Császár E, Lénárt N, Cserép C, et al. Microglia modulate blood flow, neurovascular coupling, and hypoperfusion via purinergic actions. J Exp Med 2022;219: e20211071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bagi Z, Kroenke CD, Fopiano KA, et al. Association of cerebral microvascular dysfunction and white matter injury in Alzheimer’s disease. Geroscience 2022;44:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chausse B, Kakimoto PA, Kann O. Microglia and lipids: how metabolism controls brain innate immunity. Semin Cell Dev Biol 2021;112:137–144. [DOI] [PubMed] [Google Scholar]
- 79.Cantuti-Castelvetri L, Fitzner D, Bosch-Queralt M, et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 2018;359:684–688. [DOI] [PubMed] [Google Scholar]
- 80.Gan B Mitochondrial regulation of ferroptosis. J Cell Biol 2021;220:e202105043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Franco-Iborra S, Cuadros T, Parent A, Romero-Gimenez J, Vila M, Perier C. Defective mitochondrial protein import contributes to complex I-induced mitochondrial dysfunction and neurodegeneration in Parkinson’s disease. Cell Death & Disease 2018;9(11):1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maher P, Currais A, Schubert D. Using the oxytosis/ferroptosis pathway to understand and treat age-associated neurodegenerative diseases. Cell Chem Biol 2020;27:1456–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Back S, Kroenke C, Sherman L, et al. White matter lesions defined by diffusion tensor imaging in older adults. Ann Neurol 2011;70:465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Caldeira C, Cunha C, Vaz AR, et al. Key Aging-associated alterations in primary microglia response to beta-amyloid stimulation. Front Aging Neurosci 2017;9:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gabandé-Rodríguez E, Keane L, Capasso M. Microglial phagocytosis in aging and Alzheimer’s disease. J Neurosci Res 2020;98:284–298. [DOI] [PubMed] [Google Scholar]
- 86.Hu Y, Fryatt GL, Ghorbani M, et al. Replicative senescence dictates the emergence of disease-associated microglia and contributes to Aβ pathology. Cell Rep 2021;35(10):109228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ximerakis M, Lipnick SL, Innes BT, et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat Neurosci 2019;22:1696–1708. [DOI] [PubMed] [Google Scholar]
- 88.Spittau B Aging Microglia-phenotypes, functions and implications for age-related neurodegenerative diseases. Front Aging Neurosci 2017;9:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Olah M, Patrick E, Villani AC, et al. A transcriptomic atlas of aged human microglia. Nat Commun 2018;9(1):539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Flanary BE, Streit WJ. Progressive telomere shortening occurs in cultured rat microglia, but not astrocytes. Glia 2004;45:75–88. [DOI] [PubMed] [Google Scholar]
- 91.Koellhoffer EC, McCullough LD, Ritzel RM. Old Maids: Aging and its impact on microglia function. Int J Mol Sci 2017;18:769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Angelova DM, Brown DR. Altered processing of beta-Amyloid in SH-SY5Y cells induced by model senescent microglia. ACS Chem Neurosci 2018;9:3137–3152. [DOI] [PubMed] [Google Scholar]
- 93.Supramaniam V, Vontell R, Srinivasan L, Wyatt-Ashmead J, Hagberg H, Rutherford M. Microglia activation in the extremely preterm human brain. Pediatr Res 2013;73(3):301–9. [DOI] [PubMed] [Google Scholar]
- 94.Haynes RL, Folkerth RD, Trachtenberg FL, Volpe JJ, Kinney HC. Nitrosative stress and inducible nitric oxide synthase expression in periventricular leukomalacia. Acta Neuropathol 2009;118:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Back SA. White matter injury in the preterm infant: pathology and mechanisms. Acta Neuropathol 2017;134:331–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Streit WJ. Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci 2006;29(9):506–10. [DOI] [PubMed] [Google Scholar]
- 97.Barral S, Beltramo R, Salio C, Aimar P, Lossi L, Merighi A. Phosphorylation of histone H2AX in the mouse brain from development to senescence. Int J Mol Sci 2014;15:1554–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ratan RR. The chemical biology of ferroptosis in the central nervous system. Cell Chem Biol 2020;27:479–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ryan SK, Zelic M, Han Y, et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat Neurosci 2023;26:12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ashraf A, Jeandriens J, Parkes HG, So PW. Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer’s disease: Evidence of ferroptosis. Redox Biol 2020;32:101494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yan N, Zhang J. Iron metabolism, ferroptosis, and the links With Alzheimer’s disease. Front Neurosci 2019;13:1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Feng H, Schorpp K, Jin J, et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep 2020;30(10):3411–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ge H, Xue X, Xian J, et al. Ferrostatin-1 alleviates white matter injury via decreasing ferroptosis following spinal cord injury. Mol Neurobiol 2022;59:161–176. [DOI] [PubMed] [Google Scholar]
- 104.Mehlhase J, Gieche J, Widmer R, Grune T. Ferritin levels in microglia depend upon activation: modulation by reactive oxygen species. Biochim Biophys Acta 2006;1763:854–859. [DOI] [PubMed] [Google Scholar]
- 105.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 2015;59:298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Han L, Zhou J, Li L, et al. SLC1A5 enhances malignant phenotypes through modulating ferroptosis status and immune microenvironment in glioma. Cell Death Dis 2022;13:1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jenkins NL, James SA, Salim A, et al. Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging Caenorhabditis elegans. Elife 2020;9:e56580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kang YP, Mockabee-Macias A, Jiang C, et al. Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab 2021;33:174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gagliardi M, Cotella D, Santoro C, et al. Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis 2019;10:902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee J, Hamanaka G, Lo EH, Arai K. Heterogeneity of microglia and their differential roles in white matter pathology. CNS Neurosci Ther 2019;25:1290–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kenkhuis B, van Eekeren M, Parfitt DA, et al. , Stem Cell Rep 2022; 17:1351–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hametner S, Wimmer I, Haider L, et al. Iron and neurodegeneration in the multiple sclerosis brain. Ann Neurol 2013;74:848–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data related to this study are present in the report or the supplemental materials. The bulk RNA sequencing data set are available online through the Allen Institute for Brain Science (see Materials and Methods). Please contact the corresponding author for additional details on materials and methods.