

Abstract
Objective:
Alcohol-associated liver disease is accompanied by microbial dysbiosis, increased intestinal permeability and hepatic exposure to translocated microbial products that contribute to disease progression. A key strategy to generate immune protection against invading pathogens is the secretion of immunoglobulin A (IgA) in the gut. Intestinal IgA levels depend on the polymeric immunoglobulin receptor (pIgR), which transports IgA across the epithelial barrier into the intestinal lumen and hepatic canaliculi. Here we aimed to address the function of pIgR during ethanol-induced liver disease.
Design:
pIgR and IgA were assessed in livers from patients with alcohol-associated hepatitis and controls. Wildtype and pIgR-deficient (pIgR−/−) littermates were subjected to the chronic-binge (NIAAA model) and Lieber-DeCarli feeding model for 8 weeks. Hepatic pIgR re-expression was established in pIgR−/− mice using adeno-associated virus serotype 8 (AAV8)-mediated pIgR expression in hepatocytes.
Results:
Livers of patients with alcohol-associated hepatitis demonstrated an increased colocalization of pIgR and IgA within canaliculi and apical poles of hepatocytes. pIgR-deficient mice developed increased liver injury, steatosis and inflammation after ethanol feeding compared with wildtype littermates. Furthermore, mice lacking pIgR demonstrated increased plasma LPS levels and more hepatic bacteria, indicating elevated bacterial translocation. Treatment with non-absorbable antibiotics prevented ethanol-induced liver disease in pIgR−/− mice. Injection of AAV8 expressing pIgR into pIgR−/− mice prior to ethanol feeding increased intestinal IgA levels and ameliorated ethanol-induced steatohepatitis compared with pIgR−/− mice injected with control-AAV8 by reducing bacterial translocation.
Conclusion:
Our results highlight that dysfunctional hepatic pIgR enhances alcohol-associated liver disease due to impaired antimicrobial defense by IgA in the gut.
Keywords: Alcohol-associated liver disease, mucosal IgA, microbiome, gut-liver axis, pIgR
INTRODUCTION
Alcohol-associated liver disease, which ranges from mild steatosis to cirrhosis and alcohol-associated hepatitis, is the most prevalent chronic liver disease worldwide and represents a leading cause of morbidity and mortality [1]. Within this disease spectrum, alcohol-associated hepatitis is a severe acute-on-chronic liver failure syndrome, which is associated with 90-day mortality rates of 20%–50% [2]. Despite major advances, the treatment for alcohol-associated hepatitis remains suboptimal, with early liver transplantation being the only curative therapy. Accordingly, a better understanding of the molecular mechanisms underlying alcohol-associated liver disease is necessary.
A key feature contributing to the progression of alcohol-associated liver disease is the presence of inflammation in the liver as a result of increased exposure and recognition of microbial products originating from the gut [3, 4]. These bacterial ligands (i.e. lipopolysaccharide) reach the liver via the portal vein due to increased intestinal permeability resulting from alcohol-induced disruption of tight junctions, thereby allowing increased bacterial translocation to activate inflammatory pathways in liver macrophages. This in turn causes the recruitment of other immune cells such as neutrophils, stimulates apoptosis and necrosis, and facilitates crosstalk to hepatic stellate cells, thereby influencing collagen production and fibrosis [5].
In conjunction with antimicrobial peptides, mucus and defense molecules, secretory immunoglobulins are essential for protection of the mucosal surface by binding and neutralizing harmful pathogens [6]. While immunoglobulin type M (IgM) might also coat invading bacteria, IgA is the predominant antibody isotype secreted into the intestinal lumen and plays a critical role in the defence against pathogens and in the maintenance of intestinal homeostasis [7]. Studies in germfree mice demonstrated that microbiota are necessary for developing IgA antibodies while IgA-deficient mice have altered microbiome composition, indicating a bidirectional communication between microbiota and IgA-mediated host immune responses [8, 9]. Intestinal IgA secretion greatly depends on the activity of the polymeric immunoglobulin receptor (pIgR), which transports dimeric IgA and pentameric IgM across the epithelial barrier into the intestinal lumen and hepatic canaliculi [10]. In addition to our findings showing reduced fecal IgA levels in cagemate, but not littermate mice after ethanol feeding [11], livers from mice with ethanol-induced injury contain increased numbers of gut-derived IgA-secreting cells and have IgA deposits in sinusoids [12]. In fact, IgA deposits in the liver is an exclusive feature of patients with alcohol-associated liver disease as compared with chronic liver diseases of a different etiology [13]. Nevertheless, using global IgA-deficient mice, we previously found that complete absence of IgA does not influence ethanol-induced liver disease, most likely due to compensatory mechanisms by increased IgM in the gut [11]. Yet, the specific functional properties of pIgR, particularly its role in maintaining intestinal IgA levels via IgA secretion in the liver in the context of alcohol-associated liver disease are not described.
RESULTS
IgA and pIgR expression increase and compartmentally shift in the liver of patients with alcohol-associated hepatitis.
To investigate pIgR-mediated transcytosis of IgA in the liver of patients with alcohol-associated hepatitis, IgA and pIgR expression was assessed by immunofluorescence in liver samples from seven patients with alcohol-associated hepatitis and five controls, and evaluated by a board-certified pathologist. In control livers, IgA was detected in hepatocytes and in endothelial and subendothelial compartments, where IgA often colocalized with pIgR (Figure 1A). In addition to pIgR expression in canaliculi and apical poles of hepatocytes, pIgR was highly expressed in the cytoplasm of cholangiocytes (Figure 1A). In line, analysing single cell RNA sequencing data from the publicly available Liver Cell Atlas (www.livercellatlas.org; version October 2022), we observed that PIGR is primarily expressed in cholangiocytes and to a lesser extent in hepatocytes in the healthy human liver (Figure S1A–C).
Figure 1. H&E staining and immunofluorescent detection of IgA and pIgR in alcohol-associated liver disease.
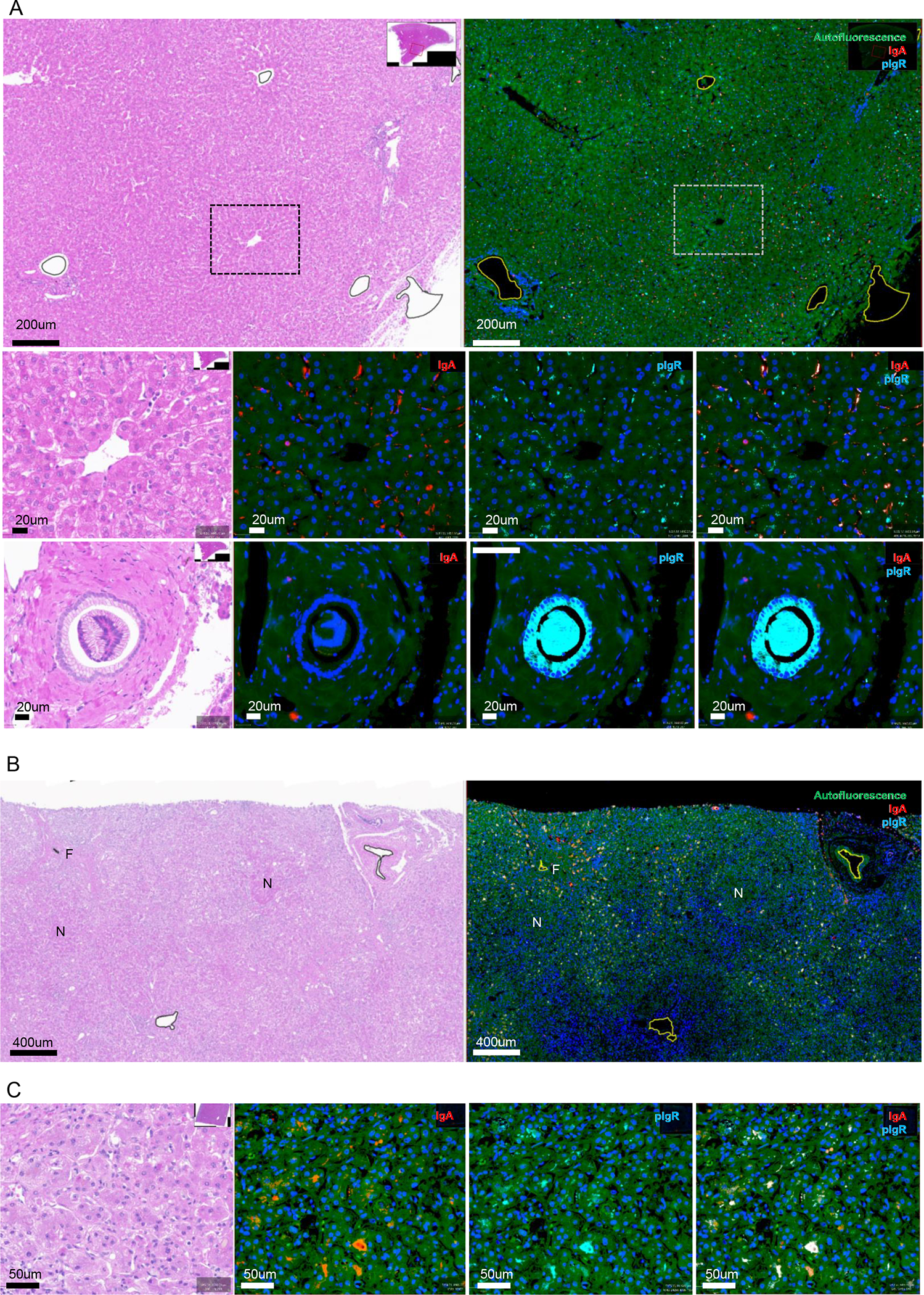
A) Representative images of H&E staining and staining for IgA (red) and pIgR (cyan) of non-alcoholic control liver slides. Low amounts of pIgR in canaliculi and apical poles of hepatocytes, and minimal IgA in hepatocytes. Large amounts of pIgR in the the cytoplasm of cholangiocytes.
B) Representative images of low magnification view of H&E staining and staining for IgA (red) and pIgR (cyan) in patients with alcohol-associated hepatitis. Increased IgA and pIgR staining localizing to zones of active fibroplasia, and away from hepatocytic nodules and end-stage fibrosis.
C) High magnification view of increased IgA and pIgR staining in patients with alcohol-associated hepatitis, with both IgA and pIgR localizing to canaliculi and apical poles of hepatocytes.
N, hepatocytic nodule. F, end-stage fibrosis. Green fluorescence, autofluorescence. Blue fluorescence, nuclei. Red fluorescence, IgA. Cyan fluorescence, polymeric immunoglobulin receptor (pIgR).
Immunofluorescent detection in livers of patients with alcohol-associated hepatitis indicated that both IgA and pIgR levels were increased in the vascular lumen (plasma; not shown) and regions of active fibroplasia compared to controls (Figure 1B, Figure S1D–E). Notably, less IgA and pIgR protein expression was seen within hepatocytic nodules and areas of end-stage fibrosis (Figure 1B). Morphologic localization of immunostaining demonstrated increased pIgR expression within canaliculi and apical poles of hepatocytes. Moreover, IgA deposition shifted to canaliculi and the apical poles of hepatocytes, resulting in increased colocalization of IgA and pIgR compared to control livers (Figure 1C, Figure S1F–I). Taken together, these data indicate that under homeostatic conditions pIgR is mainly functional on cholangiocytes in the liver, while alcohol-associated hepatitis associates with pIgR (and IgA) accumulation within hepatocytes, suggesting dysfunctional pIgR and pIgR-mediated secretion of IgA in the human liver.
Mice lacking pIgR have increased bacterial translocation and ethanol-induced liver disease.
To study the functional consequences of dysfunctional pIgR for alcohol-associated liver disease, female and male pIgR−/− mice were subjected to the chronic-binge ethanol feeding model (NIAAA [14]) and compared with their wildtype (Wt) littermates (Figure 2A). pIgR−/− mice had significantly less fecal IgA levels, while systemic IgA titers were highly elevated compared with Wt mice (Figure 2B–C). Independent of the sex, pIgR−/− mice had increased liver weights, more liver injury as indicated by higher plasma alanine transaminase (ALT) levels, and developed increased hepatic steatosis while food intake was comparable during chronic-binge ethanol feeding (Figure 2D–I, Figure S2A). Moreover, pIgR deficiency in female and male mice resulted in increased hepatic inflammation as indicated by gene expression levels of inflammatory chemokines and markers of immune cells (Cxcl1, Cxcl2, Ccr2, Cd11b; Figure 2J–M, Figure S2F–G), and more accumulation of infiltrating macrophages and neutrophils (CD11b+) in the liver (Figure 2N–O). Plasma levels of ethanol and acetate, the end product of ethanol metabolism, and hepatic expression of Cyp2e1 and Adh1 mRNAs, which regulate hepatic metabolism of ethanol, were unchanged between Wt and pIgR−/− mice (Figure S2B–G), indicating that pIgR deficiency does not affect ethanol metabolism.
Figure 2. pIgR-deficient mice develop more steatohepatitis after chronic-binge ethanol feeding.
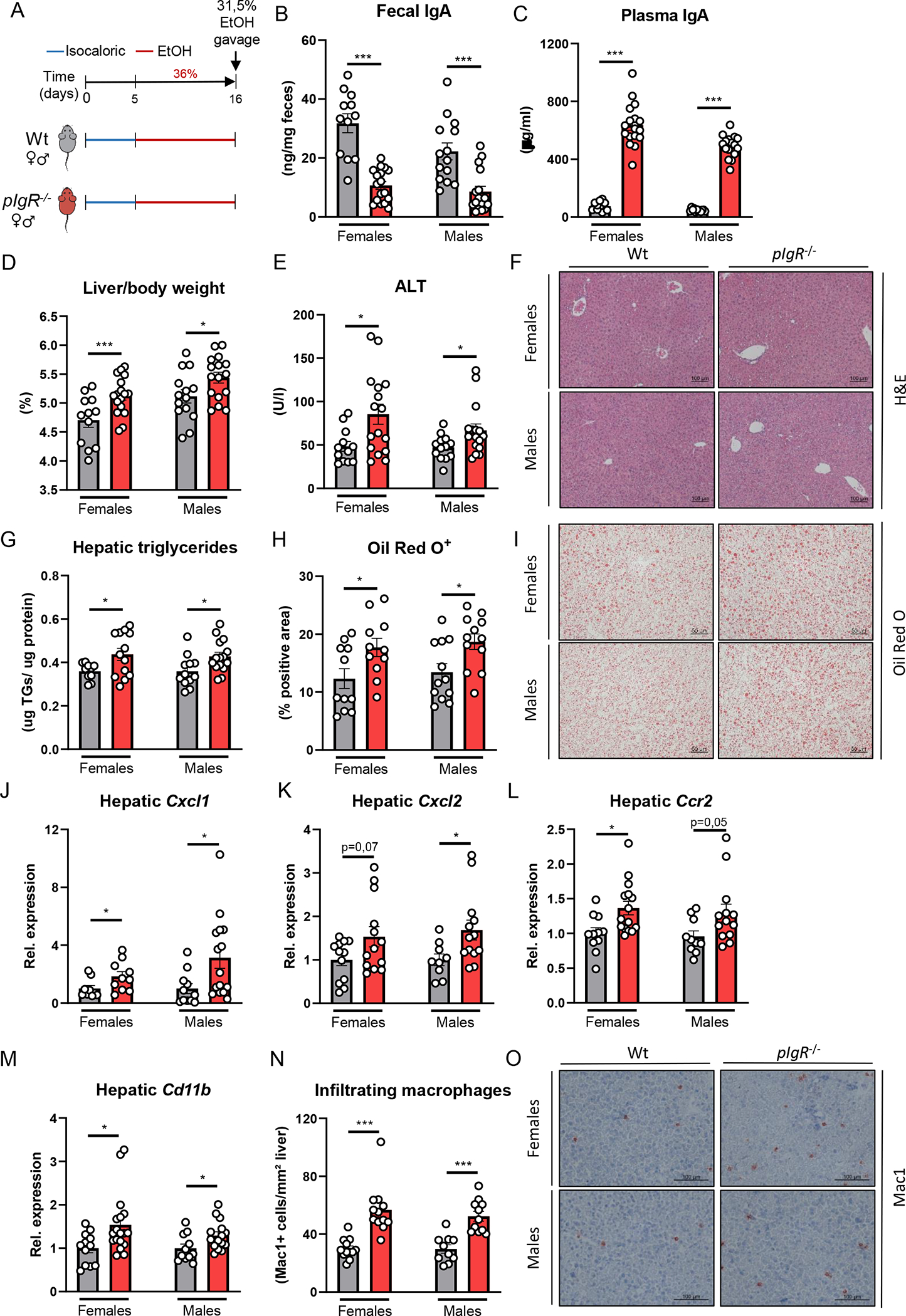
A) Schematic of chronic-binge ethanol feeding study in female and male pIgR−/− mice (red) and wildtype littermates (grey).
B) Fecal IgA levels at the end of the study. </p/> C) Plasma IgA levels at the end of the study. </p/> D) Liver to body weight ratio.
E) Plasma ALT levels.
F) Representative images showing H&E staining of liver sections.
G) Hepatic triglyceride content.
H) Quantification of immunohistochemical staining for lipids using Oil Red O.
I) Representative images showing Oil Red O staining of liver sections.
J-M) mRNA levels of indicated genes (Cxcl1, Cxcl2, Ccr2, Cd11b) in livers of ethanol-fed mice as shown in Figure 2A, assessed by qPCR. Data are shown relative to the respective wildtype mice and normalized to 18S.
N) Quantification of immunohistochemical staining for infiltrating macrophages and neutrophils in the liver using Mac-1.
O) Representative images showing Mac-1 staining of liver sections.
Data shown as mean ± SEM of n=12–18/group from 6 independent experiments. * indicates p≤0.05, ** p≤0.01, *** p≤0.001. Exact p-values are provided in Table S2.
To validate that these findings are not exclusive during chronic-binge ethanol feeding, female pIgR−/− mice and their Wt littermates were fed the Lieber-DeCarli diet or an isocaloric control diet for 8 weeks followed by a single gavage of ethanol (Figure 3A). While chronic Lieber-DeCarli feeding did not affect fecal IgA titers compared with control-fed littermate mice, pIgR-deficiency resulted in diminished fecal IgA and higher systemic IgA titers (Figure 3B–C). In agreement with our observations in the chronic-binge feeding studies, mice lacking pIgR developed more ethanol-induced liver injury and steatosis, and had increased hepatic inflammatory gene expression levels of Cxcl1, Cxcl2, Ccr2 and Cd11b (Figure 3D–M, Figure S3F). Moreover, pIgR−/− mice showed signs of increased fibrosis as indicated by mRNA levels of Col1a1 and Timp1, and fibrotic Sirius Red staining (Figure 3N–Q, Figure S3F). No differences were observed in food intake, plasma ethanol and acetate concentrations, and hepatic mRNA levels of Adh1 and Cyp2e1 between ethanol-fed pIgR−/− and Wt mice (Figure S3A–F). Since mucosal IgA is essential for neutralization of invading pathobionts and pIgR−/− mice have lower levels of intestinal IgA, we assessed bacterial translocation in ethanol-fed mice. Interestingly, ethanol feeding in mice with reduced intestinal IgA levels due to pIgR-deficiency was associated with increased plasma lipopolysaccharide (LPS) levels and bacterial translocation to the liver, as indicated by significantly more hepatic 16S expression in pIgR−/− mice compared with littermate controls (Figure 3R–S).
Figure 3. Increased ethanol-induced liver disease in mice lacking pIgR after chronic ethanol feeding with single gavage of ethanol is associated with elevated bacterial translocation.
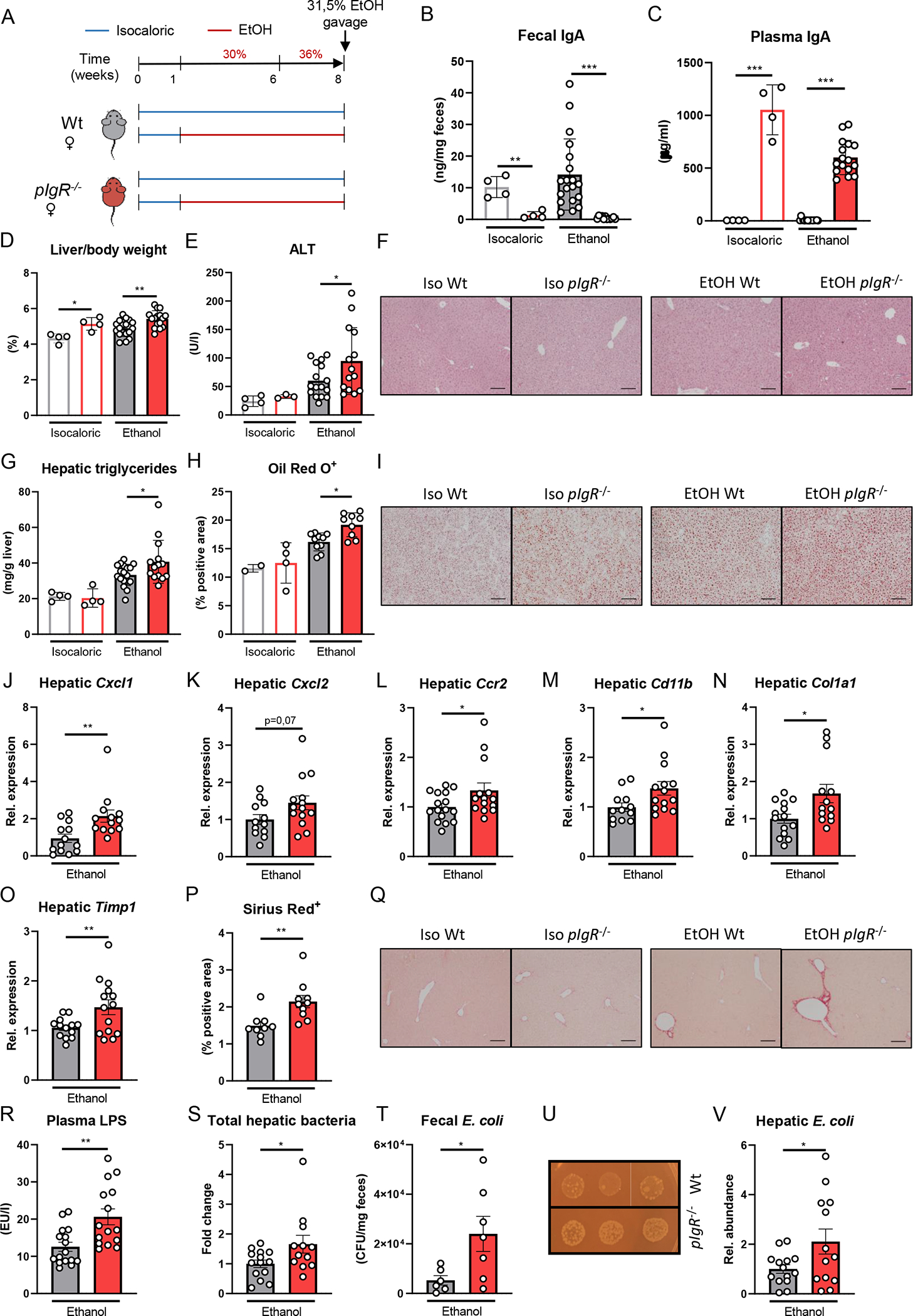
A) Schematic of 8 weeks Lieber-DeCarli ethanol diet or isocaloric control diet feeding study with a single gavage of ethanol in female pIgR−/− mice (red) and wildtype littermates (grey).
B) Fecal IgA levels after dietary intervention.
C) Plasma IgA levels after dietary intervention.
D) Liver to body weight ratio.
E) Plasma ALT levels.
F) Representative images showing H&E staining of liver sections.
G) Hepatic triglyceride content.
H) Quantification of immunohistochemical staining for lipids using Oil Red O.
I) Representative images showing Oil Red O staining of liver sections.
J-O) mRNA levels of indicated genes (Cxcl1, Cxcl2, Ccr2, Cd11b, Col1a1, Timp1) in livers of ethanol-fed mice as shown in Figure 3A, assessed by qPCR. Data are shown relative to the isocaloric control-fed mice and normalized to 18S.
P) Quantification of sirius red staining of liver sections.
Q) Representative images showing sirius red staining of liver sections.
R) Plasma LPS levels after dietary intervention.
S) Total hepatic bacteria assessed by 16S levels in ethanol-fed mice as shown in Figure 3A. Data are shown relative to wildtype mice after normalization to 18S.
T) Colony forming units of viable E. coli in feces of ethanol-fed mice as shown in Figure 3A (n=6–7 mice/group).
U) Representative images showing fecal E. coli cultures in ethanol-fed mice.
V) Hepatic E. coli in ethanol-fed mice as shown in Figure 3A. Data are shown relative to wildtype mice after normalization to 16S.
Data shown as mean ± SEM of n=4 mice/isocaloric group and n=17–16/group for ethanol-fed mice from 4 independent experiments. * indicates p≤0.05, ** p≤0.01, *** p≤0.001, **** p≤0.0001. Exact p-values are provided in Table S3.
To further characterize bacterial translocation to livers of pIgR-deficient mice after 8 weeks of Lieber-DeCarli diet, IgA-coated bacteria were isolated from the feces of Wt mice after ethanol diet (Figure S3G–H). Since E. coli was previously identified as a primary target of IgA antibodies in the gut [15, 16], we performed targeted qPCR for the presence of E. coli in IgA+ versus IgA− sorted bacterial fractions. Compared to IgA− bacteria, the relative abundance of E. coli was significantly increased in IgA-coated bacteria (Figure S3I), indicating E. coli is an important target of IgA in the gut during ethanol-induced liver disease in mice. Hence, we assessed whether low intestinal IgA levels during pIgR-deficiency would affect the presence of E. coli in the gut. In line with our expectations, we observed that mice lacking pIgR have increased amounts of viable E. coli in the gut compared with Wt mice (Figure 3T–U). Moreover, the relative abundance of E. coli in the liver was significantly increased in pIgR−/− mice after ethanol feeding (Figure 3V). Taken together, these data indicate that lack of pIgR results in more severe ethanol-induced liver disease, likely arising from enhanced bacterial translocation to the liver as a consequence of impaired intestinal IgA levels that are required to sustain overgrowth of pathobionts such as E.coli.
Treatment with non-absorbable antibiotics reduces ethanol-induced liver disease in pIgR−/− mice.
To confirm the contribution of the microbiota to increased hepatic inflammation in ethanol-induced liver disease in pIgR-deficient mice, we designed a study in which pIgR−/− mice were treated daily with the non-absorbable antibiotics polymyxin B and neomycin by oral gavage during the last two weeks of Lieber-DeCarli feeding for 8 weeks (Figure 4A). This mixture of antibiotics predominantly targets aerobic Gram-negative bacteria (mostly Enterobacteriaceae) and was shown to reduce alcohol-associated intestinal bacterial overgrowth and dysbiosis in wildtype mice [17].
Figure 4. Treatment with non-absorbable antibiotics reduces ethanol-induced liver disease in pIgR−/− mice.
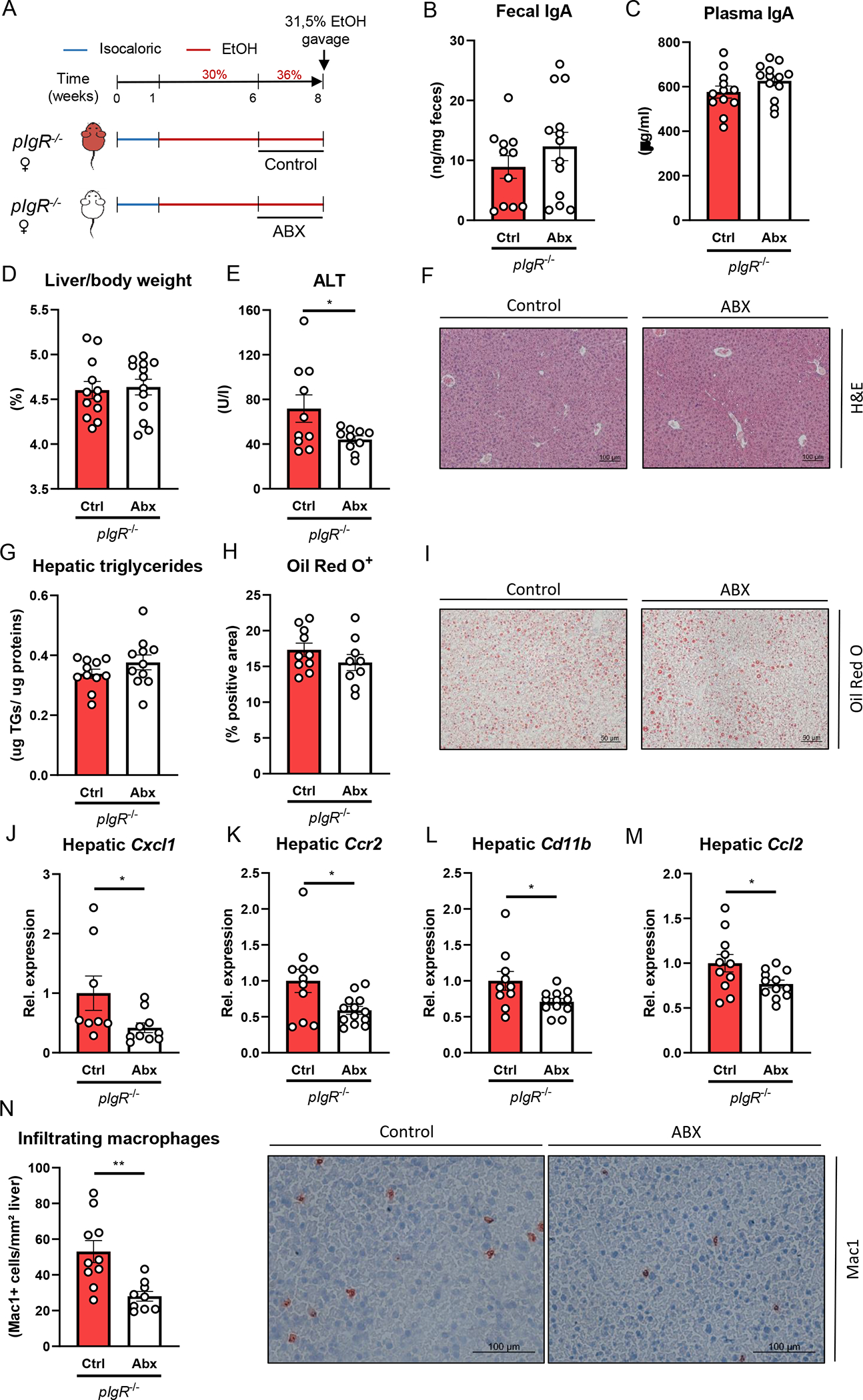
A) Schematic of intervention with antibiotics (white) or vehicle control (red) during the last two weeks of 8 weeks Lieber-DeCarli ethanol diet plus single gavage of ethanol in female pIgR−/− mice.
B) Fecal IgA levels at the end of the study.
C) Plasma IgA levels at the end of the study.
D) Liver to body weight ratio.
E) Plasma ALT levels.
F) Representative images showing H&E staining of liver sections.
G) Hepatic triglyceride content.
H) Quantification of immunohistochemical staining for lipids using Oil Red O.
I) Representative images showing Oil Red O staining of liver sections.
J-M) mRNA levels of indicated genes (Cxcl1, Ccr2, Cd11b, Ccl2) in livers of ethanol-fed mice as shown in Figure 4A, assessed by qPCR. Data are shown relative to the control-treated pIgR−/− mice and normalized to 18S.
N) Quantification and representative images of immunohistochemical staining for infiltrating macrophages and neutrophils in the liver using Mac-1.
Data shown as mean ± SEM of n=11–13 mice/group from 3 independent experiments. * indicates p≤0.05, ** p≤0.01. Exact p-values are provided in Table S4.
While antibiotic treatment during ethanol feeding in pIgR−/− mice did not alter fecal or plasma IgA levels, liver to body weight ratio or hepatic triglyceride levels, plasma levels of ALT were significantly reduced (Figure 4B–I), indicating less severe liver injury in pIgR−/− mice with reduction of intestinal bacteria. Moreover, compared with control-treated pIgR−/− mice, the expression levels of several proinflammatory genes (Cxcl1, Ccr2, Cd11b, Ccl2) in the liver was lower after antibiotic treatment (Figure 4J–M, Figure S4F). In line, we observed less accumulation of infiltrating macrophages and neutrophils (CD11b+) in the liver (Figure 4N). Antibiotic treatment did not cause any changes in food intake or ethanol absorption and metabolism, as plasma ethanol and acetate concentrations, and hepatic mRNA levels of Adh1 and Cyp2e1 were similar to controls (Figure S4A–F). Therefore, our data indicate that bacteria play a role in mediating increased ethanol-induced steatohepatitis during pIgR deficiency.
Hepatic overexpression of pIgR restores gut IgA levels and ameliorates ethanol-induced steatohepatitis in pIgR−/− mice.
Given our findings that patients diagnosed with alcohol-associated hepatitis accumulate pIgR-IgA colocalization within hepatocytes, we sought to investigate the hepatocyte-specific function of pIgR during ethanol-induced liver disease in mice, where pIgR is expressed on cholangiocytes and hepatocytes to a similar extent (Figure S5A–C; www.livercellatlas.org). To address this, we employed an adeno-associated virus serotype 8 expressing pIgR (AAV8-pIgR) to re-express murine pIgR specifically in hepatocytes in the setting of pIgR deficiency [18, 19]. Two weeks prior to feeding Lieber-DeCarli diet for 8 weeks, female pIgR−/− mice were injected with AAV8-pIgR or an AAV8-GFP control vector via tail vein injection. In addition, one group of Wt littermates also received the AAV8-GFP control vector (Figure 5A). Administration of AAV8-pIgR successfully restored hepatic pIgR expression in pIgR-deficient mice (Figure S5D), which resulted in partial restoration of fecal IgA levels compared with control-injected mice, while plasma IgA levels were unaffected (Figure 5B, C). Importantly, while pIgR−/− mice receiving control vectors showed more ethanol-induced liver disease compared with wildtype littermates, recipients of AAV8-pIgR developed significantly less liver injury and steatosis, as indicated by reduced plasma ALT levels and hepatic triglyceride content (Figure 5E–I, Figure S5I). Liver to body weight ratio, plasma ethanol levels, plasma acetate levels and gene expression of Adh1 and Cyp2e1 in the liver was unchanged between the groups (Figure 5D, Figure S5E–I). Hepatic expression levels of inflammatory genes Cxcl1, Cxcl2 and Ccr2 showed less inflammation in the livers of AAV8-pIgR treated mice (Figure 5J–L, Figure S5I). Moreover, treatment with AAV8-pIgR prevented the increased plasma LPS levels in mice lacking pIgR (Figure 5M). Overall, these findings indicate that functional pIgR in hepatocytes is important to secrete IgA in the gut and to maintain gut barrier function, thereby protecting against the development of ethanol-induced liver disease. Moreover, these data further suggest that the high enrichment of pIgR in hepatocytes in human alcohol-associated hepatitis might represent a dysfunctional protein, resulting in diminished transcytosis.
Figure 5. Hepatic pIgR overexpression restores gut IgA levels and ameliorates steatohepatitis in pIgR−/− mice.
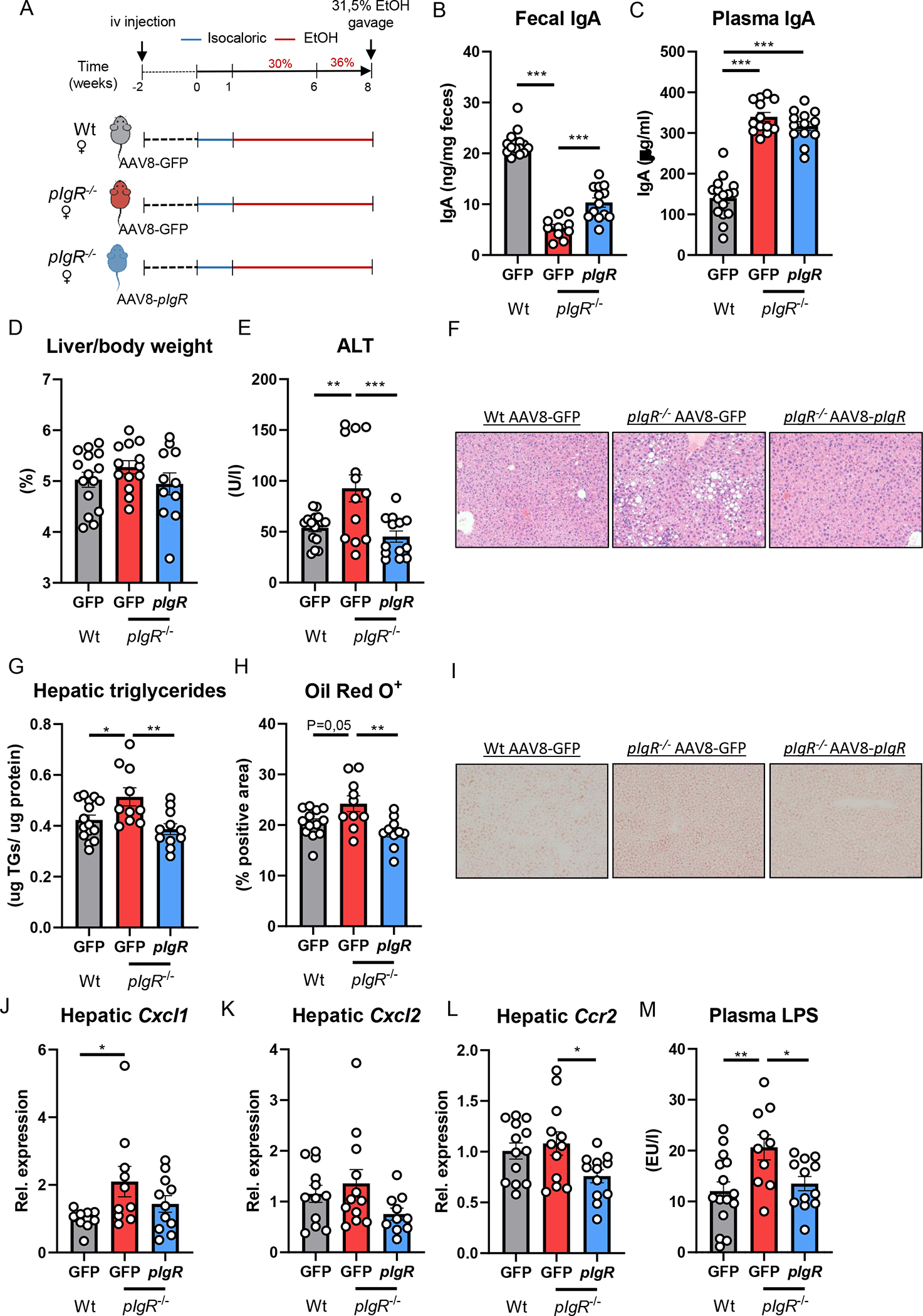
A) Schematic of study layout. Female pIgR−/− mice received a single intravenous injection of GFP-expressing AAV8 control vector (red) or pIgR-expressing AAV8 (blue). Female wildtype littermates received GFP-expressing AAV8 control vector (grey). After 2 weeks, mice were placed on Lieber-DeCarli diet for 8 weeks followed by single gavage of ethanol.
B) Fecal IgA levels at the end of the study.
C) Plasma IgA levels at the end of the study.
D) Liver to body weight ratio.
E) Plasma ALT levels.
F) Representative pictures of H&E staining of liver sections (20x magnification).
G) Hepatic triglyceride levels, normalized to liver protein content.
H) Quantification of immunohistochemical staining for lipids using Oil Red O.
I) Representative images showing Oil Red O staining of liver sections.
J-L) mRNA levels of indicated genes (Cxcl1, Cxcl2, Ccr2) in whole liver tissue of mice as shown in Figure 5A, assessed by qPCR. Data are shown relative to the control-treated wildtype mice after normalization to 18S.
M) Plasma LPS levels at the study endpoint.
Data shown as mean ± SEM of n=13–15 mice per group from 5 independent experiments. * indicates corrected p≤0.05, ** corrected p≤0.01, *** corrected p≤0.001. Exact p-values are provided in Table S5.
DISCUSSION
Harmful drinking of alcohol and alcohol-associated liver disease represent an increasingly prevalent cause of chronic liver disease with mortality rates exceeding those caused by diabetes. Since insights into factors and mechanisms driving the pathogenesis and progression of alcohol-associated steatohepatitis are poorly understood, preventive and therapeutic strategies besides abstinence are currently lacking, making alcohol-associated liver cirrhosis the primary cause of liver transplantation [20]. Here, we investigated the function of pIgR in alcohol-associated liver disease. Using human liver samples and various murine models we demonstrate that functional pIgR in the liver is required for the secretion of IgA into the gut to prevent bacterial translocation and protect against ethanol-induced steatohepatitis.
Our current findings further highlight the importance of antimicrobial defence mechanisms in the gut to protect against ethanol-associated liver injury. Specifically, although multiple layers of protection exist to avoid invasion of harmful pathobionts including the production of mucins by intestinal goblet cells and the secretion of antimicrobial molecules by enterocytes and Paneth cells [21, 22], our novel data show that failure to maintain intestinal IgA (and IgM) levels due to impaired pIgR function is sufficient to exacerbate ethanol-induced liver damage. While increased gut IgM concentrations might also coat invading bacteria and compensate for the loss of IgA [23], functional pIgR is also required for IgM transport. Besides influencing intestinal immunoglobulin levels and mucosal homeostasis, dysfunctional pIgR enhances systemic IgA titers and results in IgA accumulation in the liver due to impaired secretion into the bile. Importantly, patients with alcohol-associated cirrhosis displayed a significant lower rate of IgA secretion into the gut [24], and increased IgA titers in circulation have been described for patients with alcohol-associated liver disease [25]. Moreover, increased IgA deposition in the liver is described as an exclusive feature of alcohol-associated hepatitis among chronic liver diseases [13]. Despite IgA’s primary function to protect against invading unwanted pathogens and pathobionts, excessive IgA can lead to the engagement of Fc receptors, resulting in cellular activation of immune cells and acting pro-inflammatory [26]. Especially upon formation of IgA immune complexes which occurs when IgA aggregates are formed, i.e. when invading bacteria become opsonized, IgA can switch from immune suppression to inflammation leading to production of various inflammatory cytokines [27]. Nevertheless, since hepatic pIgR restoration in our murine model was sufficient to reduce ethanol-induced liver disease without affecting systemic IgA levels, our data suggests that pIgR-mediated maintenance of intestinal IgA levels are primarily important to provide protection against ethanol-induced liver disease, while the potential inflammatory effect of higher systemic IgA might be less important.
In agreement with our current data indicating impaired pIgR-mediated transcytosis of IgA, a recent study identified increased PIGR and IGHA upon integration of liver and plasma proteomics in patients with alcohol-related liver disease, which significantly correlated with the degree of liver fibrosis [28]. Moreover, plasma PIGR levels were recently discovered as promising novel marker for non-alcoholic fatty liver disease [29]. The question remains what renders hepatic pIgR dysfunctional during chronic liver disease, thereby potentially causing dysregulated antibody levels and altered immune responses. Previous studies have shown that pIgR expression and antibody secretion is modulated by multiple mechanisms, including hormonal, microbial and immunological regulation [30]. In fact, increased proinflammatory signaling via interferon γ (IFNγ) and tumor necrosis factor alpha (TNFα), in addition to activation of toll-like receptors (TLRs), processes involved in the inflammatory response observed during alcohol-associated liver disease [31], directly upregulate pIgR gene expression [32]. Thus, the persistent inflammatory environment in the livers of patients with alcohol-associated hepatitis might explain increased PIGR protein levels in their livers. One might hypothesize that inflammation-induced cellular damage (direct or indirectly by ethanol) disrupts intracellular signaling pathways and alter protein trafficking, resulting in profound defects in the pIgR–IgA transcytosis. Whole genome or ATAQ sequencing remains a valuable approach for future studies to assess direct genetic or epigenetic regulation of pIgR by alcohol. Interestingly, a recent study demonstrated that smokers with alcohol abuse have decreased IgA transport across airway epithelium due to improper pIgR function in the lungs as a result of adduct formation by malondialdehyde-acetaldehyde [33]. Since acetaldehyde is the first metabolite formed by ethanol metabolism and malondialdehyde is increasingly detectable in the serum and liver of patients with alcohol-associated liver disease and ethanol-fed rodents [34, 35], malondialdehyde-acetaldehyde-mediated adduct formation might impair proper PIGR function in the liver of patients with alcohol-associated liver disease. Further studies are required to gain more insights into the mechanisms by which alcohol consumption alters pIgR during alcohol-associated liver disease.
In conclusion, we showed an essential role for hepatic pIgR in regulating alcohol-associated hepatic injury, lipid accumulation and inflammation through affecting intestinal IgA levels and the prevention of bacterial translocation. Moreover, hepatocyte-specific overexpression of pIgR was sufficient to ameliorate ethanol-induced steatohepatitis in pIgR-deficient mice.
METHODS
Human cohort
Liver explants from seven patients with severe alcohol-associated hepatitis (subject characteristics in Table 1), who underwent liver transplantation, and five liver tissues from donors (control) were obtained from Clinical Resource for Alcoholic Hepatitis Investigations (NIH R24 AA025017) at Johns Hopkins University (Baltimore, MD, USA). Tissues were excised from explanted livers in patients with severe alcohol-associated hepatitis during liver transplantation, or wedge biopsies from the donor livers. The protocol was approved by the Johns Hopkins Medicine Institutional Review Boards (IRB00107893) and patients were enrolled after written informed consent was obtained.
Table 1.
Data of alcoholic hepatitis patients.
| Patient # | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Age | 34 | 50 | 48 | 39 | 62 | 45 | 32 |
| Sex | F | F | F | F | M | M | M |
| ALT (IU/L) | 49 | 14 | 86 | 30 | 81 | 53 | 108 |
| AST (IU/L) | 144 | 55 | 158 | 62 | 84 | 55 | 162 |
| Total bilirubin (mg/dl) | 27.7 | 39.9 | 28 | 24.9 | 36.2 | 25.3 | 46.9 |
| Creatinine (mg/dl) | 1.5 | 2.4 | 4.3 | 1.8 | 4.5 | 0.6 | 6.9 |
| PT (sec) | 17 | 24 | 15.4 | 22.7 | 24.7 | 25.9 | 25.6 |
| INR | 1.7 | 2.5 | 1.5 | 2.3 | 2.4 | 2.5 | 2.5 |
| MELD | 34 | 38 | 36 | 32 | 44 | 24 | 50 |
Animal experiments
pIgR−/− mice [10, 36] were originally provided by Dr. Charlotte Kaetzel, University of Kentucky, USA. All mice were on a C57BL/6 background. Age- and sex-matched littermate controls aged 8 weeks or older were used for all experiments and housed according to genotype following weaning. Mice were bred under specific pathogen-free conditions at the Department of Biomedical Research or the Department of Laboratory Animal Science and Genetics of the Medical University of Vienna, Austria or at the mouse facilities at University of California San Diego, USA. The studies shown in Figure 2A and Figure 4A were performed with mice from the animal facilities at the Medical University of Vienna, Austria. The studies shown in Figure 3A and Figure 5A were performed with mice from the animal facilities at University of California San Diego, USA. In the antbiotic administration experiment, pIgR−/− littermates received daily a mixture of Polymyxin B (150 mg/kg BW) and Neomycin (200 mg/kg BW) via oral gavage for two weeks as previously described [37] or an equal volume of vehicle (water, control treatment) during the last two weeks of ethanol feeding. To ensure rigor and reproducibility, female pIgR−/− mice from the same litter in multiple breeding cages were assigned to the control or treatment group by random selection and allocation. Additionally, female pIgR−/− mice from different breeding cages were put together in experimental cages receiving the respective treatment. Order of cages and the order of mice within a cage to which a specific treatment was applied, was random. In a similar approach, after allocation of littermate wildtype and pIgR−/− mice in to the different treatment groups by random selection, AAV8-TBG-m-PIGR (AAV-268625, Vector Biolabs; 3.75×10^11 Gc/mouse in 50μl) and control (AAV8-TBG-eGFP (VB1743, Vector Biolabs), 3.75×10^11 Gc/mouse in 50μl) was administered intravenously via the tail vein. Vector expression was driven by the hepatocyte specific thyroxine binding globulin (TBG) promoter. Order of cages and the order of mice within a cage to which a specific treatment was applied, was random. For all animal studies, at least three independent feeding experiments were performed, as indicated in the figure legends. All experimental studies and interventions were approved by the Animal Ethics Committee of the Medical University of Vienna and the Austrian Federal Ministry of Education, Science and Research, and the UCSD (CA, USA), and were performed according to Good Scientific Practice guidelines (License numbers: 335/19 and S09042).
Dietary interventions
For chronic-binge ethanol diet model (NIAAA [14]), mice were fed with Lieber–DeCarli diet for 15 days. At day 16, mice were gavaged with one dose of ethanol (5 g/kg BW) and sacrificed 9 hours later as described. For chronic feeding, mice received Lieber–DeCarli diet for 8 weeks before receiving a single gavage of ethanol (5 g/kg BW) and sacrificing 9 hours later. Liquid diet was freshly prepared 3 times/week with irradiated diet and was administered ad libitum. Mice on isocaloric diet were pair-fed to ethanol-fed mice. Sacrifice of experimental mice occurred randomly with alternating order of treatment/genotype groups to prevent confounding effects of time of harvest. For all further analysis, measurements were done in random order and in a blinded fashion.
Biochemical analyses
Blood was collected in EDTA collection tubes (Greiner Bio-One, Germany) and plasma obtained by centrifugation at 2000xg for 10 minutes. Plasma levels of ALT were determined using Reflotron ALT strips on a Reflotron Plus (Roche). Hepatic triglyceride levels were measured according to manufacturer’s instructions using Liquid Reagents kit (GPO-PAP Triglyceride Liquicolor kit, HUMAN Biochemica and Diagnostica mbH, Wiesbaden, Germany). Protein content was measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Levels of LPS in plasma were determined using the Mouse LPS ELISA kit according to manufacturer’s protocol (CSB-E13066m, Cusabio, Wuhan, China). Plasma ethanol levels were determined using the Ethanol assay kit (MAK076–1KT, Sigma-Aldrich, Vienna, Austria) according to manufacturer’s protocol. Plasma acetate levels were determined using the Acetate colorimetric assay kit (MAK086–1KT, Sigma-Aldrich, Vienna, Austria) according to manufacturer’s protocol. For fecal antibody measurements, snap-frozen fecal samples were smashed in PBS (100mg/ml) using sterile woodsticks followed by short centrifugation to pellet debris. IgA antibody titers in plasma and fecal supernatans are measured by a chemiluminescent-based sandwich ELISA using anti-mouse IgA (Clone C10–3; 556969, Becton Dickinson, Austria) for coating and biotinylated anti-mouse IgA (Clone C10–1; 556978, Becton Dickinson, Austria) for detection as described before [38].
Immunohistochemistry
Human liver tissues were fixed, embedded in paraffin and 4μm sections were cut. Immunofluorescence staining of FFPE sections was performed using a standard protocol (https://dx.doi.org/10.17504/protocols.io.b49yqz7w). IgA was labeled with a rabbit polyclonal antibody (A0262, 1:3000) and detected with AlexaFluor 555Plus (AF555) goat anti-rabbit secondary (A32732, ThermoFisher, 1:500); pIgR was labeled with a rat monoclonal antibody (ab170321, 1:150) and detected with AlexaFluor 647Plus (AF647) goat anti-rat secondary (A48272, ThermoFisher, 1:500). Slides were counterstained with Hoechst 33342 (1:1000). Slides were imaged with a Zeiss AxioscanZ.1 slidescanner using a 20x (0.8) dry objective. Images were evaluated by a board-certified pathologist using QuPath software [39].
Mouse liver sections were embedded in OCT compound and 7μm frozen sections were stained and quantified for CD11b (Mac-1; Clone: M1/70; 550282, Becton Dickinson, Austria) for infiltrating macrophages and neutrophils, and with Oil Red O (Sigma-Aldrich, Vienna, Austria) to determine lipid content as done previously [40]. Formalin-fixed liver samples were embedded in paraffin and 4 μm sections were stained for hematoxylin and eosin for liver morphology and with Sirius Red for liver fibrosis detection. At least four high-power fields of the liver per mouse were randomly selected for quantification of positive-stained area and positive cells per liver. To quantifiy positive-stained area, samples were analyzed using NIH Image J software. The results are presented as percentage area positively stained.
IgA+ bacterial sorting
To isolate and quantify IgA-coated stool bacteria, 1–2 fecal pellets were collected into 1ml of sterile PBS. Pellets were homogenized by vortexing, then centrifuged at 300g for 1 minute to pellet large debris. The supernatant was collected and centrifuged at 12,000xg for 5 minutes. The pellet was resuspended in 1ml of PBS with 1μl SYTO BC (S34855; ThermoFisher) to stain bacterial nucleic acid and incubated for 20 minutes at 4°C. After incubation, the tube was centrifuged (12,000xg, 3 minutes). The supernatant was discarded, and the pellet washed with PBS and centrifuged (12,000xg, 3 minutes). The pellet was resuspended in 1mL of PBS with anti-mouse IgA-PE (1:200 dilution) (Clone: mA-6E1; 12–4204-82; ThermoFisher) and incubated for 20 minutes at 4°C, then centrifuged (12,000xg, 3 minutes), and washed with PBS. Next, the pellet was incubated with anti-PE microbeads (130–048-801; Miltenyi Biotec) for 20 minutes at 4°C, centrifuged (12,000xg, 3 minutes), and washed with PBS. Finally, the pellet was resuspended in 4ml of PBS and applied on a LS magnetic column (Miltenyi Biotec) for IgA+ and IgA− selection, according to manufacturer’s instructions. Collected suspensions were centrifuged (12,000xg, 3 minutes), resuspended in PBS and snap-froozen for gDNA isolation. Purity (IgA+) of sorted bacterial fractions was assessed on a BD Fortessa, and data was analyzed with FlowJo software.
E. coli culture
To determine fecal E. coli levels, mouse fecal pellets were resuspended in 1ml sterile PBS and serial dilutions were made. Ten μl of each dilution from each sample were spotted onto a plate with selective selective LB broth medium (Sigma), and the plates were then incubated at 37°C overnight. Colony numbers of each sample were counted and CFUs were calculated per mg of feces.
gDNA isolation
Genomic DNA was isolated from a sterile liver part and the IgA− and IgA+ sorted bacterial fractions as previously described [37]. gDNA content and quality were assessed using Nanodrop (Peqlab).
RNA isolation
For whole liver RNA isolation, 50mg tissue pieces of left lateral liver lobes were snap frozen in liquid nitrogen. Frozen tissue in QIAzol lysis reagent was homogenized mechanically using stainless steel beads in a Tissue Lyser II (Qiagen, Hilden, Germany). RNA was extracted by QIAzol according to manufacturer’s instructions. RNA content and quality were assessed using Nanodrop (Peqlab).
cDNA generation and qPCR
For quantitative real-time PCR, up to 1μg of RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Thermo Fisher, Waltham, MA, USA) to generate cDNA. Real-time PCR was performed on a CFX96 Real-Time PCR System (Bio-Rad Laboratories, Hercules, CA, USA) using the KAPA SYBR FAST kit (Thermo Fisher, Waltham, MA, USA) and the primers indicated below. Gene expression was normalized to mouse 18S rRNA, Cyclophilin B (Ppib) or Gapdh as indicated. For relative E. coli levels in the sorted bacterial fractions and livers, expression was normalized to bacterial 16S rRNA gene levels. The qPCR value of the 16S rRNA gene was normalized to host 18S rRNA for assessing total bacteria. Expression of target genes in relation to reference gene was determined using the 2−ΔΔCT method. Primer sequences are provided in Table 2.
Table 2.
Primer details.
| Gene | Forward sequence 5’-3’ | Reverse sequence 5’-3’ | Ref. |
|---|---|---|---|
| Adh1 | GGGTTCTCAACTGGCTATGG | ACAGACAGACCGACACCTCC | [41] |
| Ccl2 | AGGTCCCTGTCATGCTTCTG | TCTGGACCCATTCCTTCTTG | [42] |
| Ccr2 | CAGGTGACAGAGACTCTTGGAATG | GAACTTCTCTCCAACAAAGGCATAA | [43] |
| Cd11b | ATGGACGCTGATGGCAATACC | TCCCCATTCACGTCTCCCA | [44] |
| Col1a1 | AACCCTGCCCGCACATG | CAGACGGCTGAGTAGGGAACA | [42] |
| Cxcl1 | GCTGGGATTCACCTCAAGAA | TCTCCGTTACTTGGGGACAC | [41] |
| Cxcl2 | AGTGAACTGCGCTGTCAATG | TTCAGGGTCAAGGCAAACTT | [41] |
| Cyp2e1 | CTTAGGGAAAACCTCCGCAC | GGGACATTCCTGTGTTCCAG | [41] |
| Gapdh | TTGATGGCAACAATCTCCAC | CGTCCCGTAGACAAAATGGT | [45] |
| PIgR | ATGAGGCTCTACTTGTTCACGC | CGCCTTCTATACTACTCACCTCC | [46] |
| Ppib | CAGCAAGTTCCATCGTGTCATCAAGG | GGAAGCGCTCACCATAGATGCTC | [47] |
| Timp1 | TCTTGGTTCCCTGGCGTACTCT | GTGAGTGTCACTCTCCAGTTTGC | [42] |
| 18S | AGTCCCTGCCCTTTGTACACA | CGATCCCAGGGCCTCACTA | [41] |
| Universal bacteria (16S) | GTGSTGCAYGGYTGTCGTCA | ACGTCRTCCMCACCTTCCTC | [48] |
| E. coli | CATGCCGCGTGTATGAAGAA | CGGGTAACGTCAATGAGCAAA | [48] |
Statistical analysis
Data comparing two groups were assessed as appropriate by two-tailed unpaired Student’s t-test following evaluation of Gaussian distribution. A p-value ≤0.05 was considered statistically significant when comparing two groups. Comparison of multiple datasets was done using one-way analysis of variance (ANOVA) with Bonferroni correction and a corrected p≤0.05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism v9.1.2. Results are expressed as mean ± standard error (SEM) unless stated otherwise.
Supplementary Material
Supplementary Figure 1. Human hepatic pIgR expression.
A) UMAP showing distinct clusters among total CD45− live cells in human livers, obtained from the Liver Cell Atlas dataset (www.livercellatlas.org). (ECs: endothelial cells; LSEC: liver sinusoidal endothelial cells, VSMC: vascular smooth muscle cells)
B) Expression of pIgR across the different clusters.
C) Violin plot showing pIgR expression in the different clusters (CD45−) in human livers. (LSEC: liver sinusoidal endothelial cells, VSMC: vascular smooth muscle cells)
D) Quantification of PIGR-positive area in liver sections from controls (n=5) and patients with alcohol-associated hepatitis (n=7).
E) Quantification of IgA-positive area in liver sections from controls (n=5) and patients with alcohol-associated hepatitis (n=7).
F) Quantification of PIGR-IgA double-positive area in liver sections from controls (n=5) and patients with alcohol-associated hepatitis (n=7).
G) Scoring for the amount of PIGR localization in hepatocytes in liver sections from controls (n=5) and patients with alcohol-associated hepatitis (n=7).
H) Scoring for the amount of positive biliary diffuse PIGR localization in liver sections from controls (n=5) and patients with alcohol-associated hepatitis (n=7).
I) Scoring for the amount of positive biliary apical/luminal PIGR localization in liver sections from controls (n=5) and patients with alcohol-associated hepatitis (n=7).
* indicates p≤0.05, ** p≤0.01. Exact p-values are provided in Table S1.
Supplementary Figure 2. Chronic-binge ethanol feeding to pIgR−/− mice and wildtype littermates.
A) Average food intake of female and male pIgR−/− mice and their wildtype littermates after chronic-binge ethanol diet as shown in Figure 2A.
B) Plasma ethanol levels at the study endpoint.
C) Plasma acetate levels at the study endpoint.
D) Gene expression of Adh1 in whole liver tissue, assessed by qPCR and normalized to 18S, shown relative to respective Wt littermates.
E) Gene expression of Cyp2e1 in whole liver tissue, assessed by qPCR and normalized to 18S, shown relative to respective Wt littermates.
F) Gene expression of indicated genes in whole liver tissue of female mice, assessed by qPCR and normalized to Ppib, shown relative to respective Wt littermates.
G) Gene expression of indicated genes in whole liver tissue of male mice, assessed by qPCR and normalized to Ppib, shown relative to respective Wt littermates.
Data shown as mean ± SEM of n=12–18/group from 6 independent experiments. * indicates p≤0.05, ** p≤0.01. Exact p-values are provided in Table S2.
Supplementary Figure 3. Lieber-DeCarli ethanol feeding to pIgR−/− mice and wildtype littermates.
A) Average food intake of female pIgR−/− mice and their wildtype littermates after isocaloric control or Lieber-DeCarli ethanol diet for 8 weeks as shown in Figure 3A.
B) Plasma ethanol levels at the study endpoint in ethanol-fed mice.
C) Plasma acetate levels at the study endpoint in ethanol-fed mice.
D) Gene expression of Adh1 in whole liver tissue of ethanol-fed mice, assessed by qPCR and normalized to 18S, shown relative to Wt littermates.
E) Gene expression of Cyp2e1 in whole liver tissue of ethanol-fed mice, assessed by qPCR and normalized to 18S, shown relative to Wt littermates.
F) Gene expression of indicated genes in whole liver tissue of ethanol-fed mice, assessed by qPCR and normalized to Ppib, shown relative to Wt littermates.
G) Representative FACS plots of IgA+ SYTO BC+ bacteria in pre-sorted and sorted fractions (Negative selection; Positive selection) from feces of wildtype mice after Lieber-DeCarli ethanol diet for 8 weeks.
H) Percentage of IgA+ bacteria in the different sorted bacterial fractions from feces of ethanol-fed wildtype mice (n= 8 mice).
I) E. coli assessed by qPCR in sorted bacterial fractions from feces of ethanol-fed wildtype mice. Data are shown relative to IgA− bacteria after normalization to 16S.
Data shown as mean ± SEM of n=4 mice/isocaloric group and n=17–16/group for ethanol-fed mice from 4 independent experiments. * indicates p≤0.05. Exact p-values are provided in Table S3.
Supplementary Figure 4. Antibiotic treatment of pIgR−/− mice during Lieber-DeCarli ethanol feeding.
A) Average food intake of female pIgR−/− mice treated with antibiotics or vehicle control during the last two weeks of Lieber-DeCarli ethanol diet for 8 weeks as shown in Figure 4A.
B) Plasma ethanol levels at the study endpoint.
C) Plasma acetate levels at the study endpoint.
D) Gene expression of Adh1 in whole liver tissue, assessed by qPCR and normalized to 18S, shown relative to control-treated mice.
E) Gene expression of Cyp2e1 in whole liver tissue, assessed by qPCR and normalized to 18S, shown relative to control-treated mice.
F) Gene expression of indicated genes in whole liver tissue, assessed by qPCR and normalized to Ppib, shown relative to control-treated mice.
Data shown as mean ± SEM of n=11–13 mice/group from 3 independent experiments. * indicates p≤0.05. Exact p-values are provided in Table S4.
Supplementary Figure 5. pIgR-AAV8 administration in pIgR−/− mice.
A) UMAP showing distinct clusters among total CD45− live cells in wildtype mouse livers, obtained from the Liver Cell Atlas dataset (www.livercellatlas.org). (ECs: endothelial cells; HpSCs: hepatic progenitor/stem cells; LECs: lymphatic endothelial cells; LSECs: liver sinusoidal endothelial cells; Nucseq: single-nuclei sequencing)
B) Expression of pIgR across the different clusters.
C) Violin plot showing pIgR expression in the different clusters (CD45−) in wildtype mouse livers. (Nucseq: single-nuclei sequencing; HsPCs: hepatic stem/progenitor cells; LSECs: liver sinusoidal endothelial cells)
D) Induction of pIgR expression in the livers of pIgR−/− mice injected with pIgR-AAV8 as shown in Figure 5A, assessed by qPCR and normalized to 18S.
E) Plasma ethanol levels at the study endpoint.
F) Plasma acetate levels at the study endpoint.
G) Gene expression of Adh1 in whole liver tissue, assessed by qPCR and normalized to 18S, shown relative to wildtype littermates.
H) Gene expression of Cyp2e1 in whole liver tissue, assessed by qPCR and normalized to 18S, shown relative to wildtype littermates.
I) Gene expression of indicated genes in whole liver tissue, assessed by qPCR and normalized to Gapdh, shown relative to wildtype littermates.
Data shown as mean ± SEM of n=13–15 mice per group from 5 independent experiments. * indicates corrected p≤0.05, ** corrected p≤0.01, *** corrected p≤0.001. Exact p-values are provided in Table S5.
What is already known on this topic
Alcohol-associated liver disease is characterized by IgA deposits in the liver and higher circulatory IgA titers.
IgA secretion into bile and the intestinal lumen is regulated via the polymeric immunoglobulin receptor (pIgR).
Mucosal IgA is important to maintain eubiosis and protect against invading pathobionts and pathogens.
What this study adds
Patients with alcohol-associated hepatitis have pIgR and IgA accumulation within hepatocytes, indicating improper IgA transcytosis and secretion.
Low intestinal IgA levels during pIgR-deficiency results in aggravated ethanol-induced steatohepatitis associated with increased bacterial translocation.
Hepatocyte-specific re-expression of pIgR is sufficient to partially restore intestinal IgA levels and ameliorate ethanol-induced liver disease in mice lacking pIgR via the prevention of bacterial translocation.
How this study might affect research, practice or policy
Enhancing hepatic pIgR and intestinal IgA levels might be promising targets to improve therapy for alcohol-induced liver disease.
Acknowledgements
We thank all animal care takers from the Medical University of Vienna and UC San Diego responsible for the animals used in this project. We thank Katarzyna Dobaczewska and Brett Laffey from La Jolla Institute Microscopy and Histology Core Facility for expert immunofluorescence staining.
Sources of Funding
T.H. was supported by a Veni grant (ZonMw, NWO; 91619012), ‘Right-On-Time’ grant (MLDS; W019–28) and Zukunftskollegs grant (FWF; ZK81B). S.L. was supported in part by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) fellowship (LA 4286/1–1) and the “Clinical and Translational Research Fellowship in Liver Disease” by the American Association for the Study of Liver Diseases (AASLD) Foundation. This study was supported in part by NIH grants R01 AA24726, R37 AA020703, U01 AA026939, U01 AA026939–04S1, S10OD021831, by Award Number BX004594 from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development, and a Harrington Discovery Institute Foundation Grant (to B.S.) and services provided by NIH centers P30 DK120515 and P50 AA011999.
Abbreviations
- AAV8
Adeno-associated virus serotype 8
- ADH1
Alcohol Dehydrogenase 1
- ALD
Alcohol-associated liver disease
- ALT
Alanine transaminase
- CCL2
C-C Motif Chemokine Ligand 2
- CCR2
C-C Motif Chemokine Receptor 2
- CD11B
Integrin alpha M
- COL1A1
Collagen type I alpha 1
- CXCL1
C-X-C Motif Chemokine Ligand 1
- CXCL2
C-X-C Motif Chemokine Ligand 2
- CYP2E1
Cytochrome P0 Family 2 Subfamily E Member 1
- H&E
Hematoxylin and Eosin
- IgA
Immunoglobulin type A
- LPS
Lipopolysaccharide
- PIGR
Polymeric immunoglobulin receptor
- TIMP1
TIMP metallopeptidase inhibitor 1
Footnotes
Conflict of interest
B.S. has been consulting for Ambys Medicines, Ferring Research Institute, Gelesis, HOST Therabiomics, Intercept Pharmaceuticals, Mabwell Therapeutics, Patara Pharmaceuticals and Takeda. B.S. is founder of Nterica Bio. UC San Diego has filed several patents with S.L. and B.S. as inventor related to this work. B.S.’s institution UC San Diego has received research support from Artizan Biosciences, Axial Biotherapeutics, BiomX, CymaBay Therapeutics, NGM Biopharmaceuticals, Prodigy Biotech and Synlogic Operating Company.
Study data: Data are available upon reasonable request to the corresponding authors
REFERENCES
- 1.Seitz HK, Bataller R, Cortez-Pinto H, Gao B, Gual A, Lackner C, et al. Alcoholic liver disease. Nat Rev Dis Primers 2018;4:16. [DOI] [PubMed] [Google Scholar]
- 2.Serste T, Cornillie A, Njimi H, Pavesi M, Arroyo V, Putignano A, et al. The prognostic value of acute-on-chronic liver failure during the course of severe alcoholic hepatitis. J Hepatol 2018;69:318–24. [DOI] [PubMed] [Google Scholar]
- 3.Bajaj JS. Alcohol, liver disease and the gut microbiota. Nat Rev Gastroenterol Hepatol 2019;16:235–46. [DOI] [PubMed] [Google Scholar]
- 4.Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol 2015;12:231–42. [DOI] [PubMed] [Google Scholar]
- 5.Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol 2018;15:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bunker JJ, Bendelac A. IgA Responses to Microbiota. Immunity 2018;49:211–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okai S, Usui F, Yokota S, Hori IY, Hasegawa M, Nakamura T, et al. High-affinity monoclonal IgA regulates gut microbiota and prevents colitis in mice. Nat Microbiol 2016;1:16103. [DOI] [PubMed] [Google Scholar]
- 8.Nakajima A, Vogelzang A, Maruya M, Miyajima M, Murata M, Son A, et al. IgA regulates the composition and metabolic function of gut microbiota by promoting symbiosis between bacteria. J Exp Med 2018;215:2019–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peterson DA, McNulty NP, Guruge JL, Gordon JI. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe 2007;2:328–39. [DOI] [PubMed] [Google Scholar]
- 10.Shimada S, Kawaguchi-Miyashita M, Kushiro A, Sato T, Nanno M, Sako T, et al. Generation of polymeric immunoglobulin receptor-deficient mouse with marked reduction of secretory IgA. J Immunol 1999;163:5367–73. [PubMed] [Google Scholar]
- 11.Inamine T, Yang AM, Wang L, Lee KC, Llorente C, Schnabl B. Genetic Loss of Immunoglobulin A Does Not Influence Development of Alcoholic Steatohepatitis in Mice. Alcohol Clin Exp Res 2016;40:2604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moro-Sibilot L, Blanc P, Taillardet M, Bardel E, Couillault C, Boschetti G, et al. Mouse and Human Liver Contain Immunoglobulin A-Secreting Cells Originating From Peyer’s Patches and Directed Against Intestinal Antigens. Gastroenterology 2016;151:311–23. [DOI] [PubMed] [Google Scholar]
- 13.van de Wiel A, Delacroix DL, van Hattum J, Schuurman HJ, Kater L. Characteristics of serum IgA and liver IgA deposits in alcoholic liver disease. Hepatology 1987;7:95–9. [DOI] [PubMed] [Google Scholar]
- 14.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 2013;8:627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, et al. IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci Transl Med 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moll JM, Myers PN, Zhang C, Eriksen C, Wolf J, Appelberg KS, et al. Gut Microbiota Perturbation in IgA Deficiency Is Influenced by IgA-Autoantibody Status. Gastroenterology 2021;160:2423–34 e5. [DOI] [PubMed] [Google Scholar]
- 17.Yan AW, Fouts DE, Brandl J, Starkel P, Torralba M, Schott E, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011;53:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A 2002;99:11854–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Wang H, Bell P, McCarter RJ, He J, Calcedo R, et al. Systematic evaluation of AAV vectors for liver directed gene transfer in murine models. Mol Ther 2010;18:118–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singal AK, Mathurin P. Diagnosis and Treatment of Alcohol-Associated Liver Disease: A Review. JAMA 2021;326:165–76. [DOI] [PubMed] [Google Scholar]
- 21.Bruellman R, Llorente C. A Perspective Of Intestinal Immune-Microbiome Interactions In Alcohol-Associated Liver Disease. Int J Biol Sci 2021;17:307–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macpherson AJ, Slack E, Geuking MB, McCoy KD. The mucosal firewalls against commensal intestinal microbes. Semin Immunopathol 2009;31:145–9. [DOI] [PubMed] [Google Scholar]
- 23.van der Waaij LA, Kroese FG, Visser A, Nelis GF, Westerveld BD, Jansen PL, et al. Immunoglobulin coating of faecal bacteria in inflammatory bowel disease. Eur J Gastroenterol Hepatol 2004;16:669–74. [DOI] [PubMed] [Google Scholar]
- 24.Pelletier G, Briantais MJ, Buffet C, Pillot J, Etienne JP. Serum and intestinal secretory IgA in alcoholic cirrhosis of the liver. Gut 1982;23:475–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van de Wiel A, van Hattum J, Schuurman HJ, Kater L. Immunoglobulin A in the diagnosis of alcoholic liver disease. Gastroenterology 1988;94:457–62. [DOI] [PubMed] [Google Scholar]
- 26.Heineke MH, van Egmond M. Immunoglobulin A: magic bullet or Trojan horse? Eur J Clin Invest 2017;47:184–92. [DOI] [PubMed] [Google Scholar]
- 27.Hansen IS, Baeten DLP, den Dunnen J. The inflammatory function of human IgA. Cell Mol Life Sci 2019;76:1041–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niu L, Thiele M, Geyer PE, Rasmussen DN, Webel HE, Santos A, et al. Noninvasive proteomic biomarkers for alcohol-related liver disease. Nat Med 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niu L, Geyer PE, Wewer Albrechtsen NJ, Gluud LL, Santos A, Doll S, et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol Syst Biol 2019;15:e8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaetzel CS. Cooperativity among secretory IgA, the polymeric immunoglobulin receptor, and the gut microbiota promotes host-microbial mutualism. Immunol Lett 2014;162:10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li S, Tan HY, Wang N, Feng Y, Wang X, Feng Y. Recent Insights Into the Role of Immune Cells in Alcoholic Liver Disease. Front Immunol 2019;10:1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turula H, Wobus CE. The Role of the Polymeric Immunoglobulin Receptor and Secretory Immunoglobulins during Mucosal Infection and Immunity. Viruses 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wyatt TA, Warren KJ, Wetzel TJ, Suwondo T, Rensch GP, DeVasure JM, et al. Malondialdehyde-Acetaldehyde Adduct Formation Decreases Immunoglobulin A Transport across Airway Epithelium in Smokers Who Abuse Alcohol. Am J Pathol 2021;191:1732–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez-Hernandez O, Gonzalez-Reimers E, Quintero-Platt G, Abreu-Gonzalez P, Vega-Prieto MJ, Sanchez-Perez MJ, et al. Malondialdehyde as a Prognostic Factor in Alcoholic Hepatitis. Alcohol Alcohol 2017;52:305–10. [DOI] [PubMed] [Google Scholar]
- 35.Albano E Role of adaptive immunity in alcoholic liver disease. Int J Hepatol 2012;2012:893026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rogier EW, Frantz AL, Bruno ME, Wedlund L, Cohen DA, Stromberg AJ, et al. Secretory antibodies in breast milk promote long-term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proc Natl Acad Sci U S A 2014;111:3074–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hendrikx T, Duan Y, Wang Y, Oh JH, Alexander LM, Huang W, et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut 2019;68:1504–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gruber S, Hendrikx T, Tsiantoulas D, Ozsvar-Kozma M, Goderle L, Mallat Z, et al. Sialic Acid-Binding Immunoglobulin-like Lectin G Promotes Atherosclerosis and Liver Inflammation by Suppressing the Protective Functions of B-1 Cells. Cell Rep 2016;14:2348–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7:16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Busch CJ, Hendrikx T, Weismann D, Jackel S, Walenbergh SM, Rendeiro AF, et al. Malondialdehyde epitopes are sterile mediators of hepatic inflammation in hypercholesterolemic mice. Hepatology 2017;65:1181–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duan Y, Llorente C, Lang S, Brandl K, Chu H, Jiang L, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019;575:505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Demir M, Lang S, Hartmann P, Duan Y, Martin A, Miyamoto Y, et al. The fecal mycobiome in non-alcoholic fatty liver disease. J Hepatol 2022;76:788–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeurissen MLJ, Walenbergh SMA, Houben T, Gijbels MJJ, Li J, Hendrikx T, et al. Prevention of oxLDL uptake leads to decreased atherosclerosis in hematopoietic NPC1-deficient Ldlr(−/−) mice. Atherosclerosis 2016;255:59–65. [DOI] [PubMed] [Google Scholar]
- 44.Hendrikx T, Porsch F, Kiss MG, Rajcic D, Papac-Milicevic N, Hoebinger C, et al. Soluble TREM2 levels reflect the recruitment and expansion of TREM2(+) macrophages that localize to fibrotic areas and limit NASH. J Hepatol 2022. [DOI] [PubMed] [Google Scholar]
- 45.Yang H, Youm YH, Dixit VD. Inhibition of thymic adipogenesis by caloric restriction is coupled with reduction in age-related thymic involution. J Immunol 2009;183:3040–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moon C, VanDussen KL, Miyoshi H, Stappenbeck TS. Development of a primary mouse intestinal epithelial cell monolayer culture system to evaluate factors that modulate IgA transcytosis. Mucosal Immunol 2014;7:818–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kandasamy RK, Vladimer GI, Snijder B, Muller AC, Rebsamen M, Bigenzahn JW, et al. A time-resolved molecular map of the macrophage response to VSV infection. NPJ Syst Biol Appl 2016;2:16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arthur JC, Gharaibeh RZ, Muhlbauer M, Perez-Chanona E, Uronis JM, McCafferty J, et al. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat Commun 2014;5:4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Human hepatic pIgR expression.
A) UMAP showing distinct clusters among total CD45− live cells in human livers, obtained from the Liver Cell Atlas dataset (www.livercellatlas.org). (ECs: endothelial cells; LSEC: liver sinusoidal endothelial cells, VSMC: vascular smooth muscle cells)
B) Expression of pIgR across the different clusters.
C) Violin plot showing pIgR expression in the different clusters (CD45−) in human livers. (LSEC: liver sinusoidal endothelial cells, VSMC: vascular smooth muscle cells)
D) Quantification of PIGR-positive area in liver sections from controls (n=5) and patients with alcohol-associated hepatitis (n=7).
E) Quantification of IgA-positive area in liver sections from controls (n=5) and patients with alcohol-associated hepatitis (n=7).
F) Quantification of PIGR-IgA double-positive area in liver sections from controls (n=5) and patients with alcohol-associated hepatitis (n=7).
G) Scoring for the amount of PIGR localization in hepatocytes in liver sections from controls (n=5) and patients with alcohol-associated hepatitis (n=7).
H) Scoring for the amount of positive biliary diffuse PIGR localization in liver sections from controls (n=5) and patients with alcohol-associated hepatitis (n=7).
I) Scoring for the amount of positive biliary apical/luminal PIGR localization in liver sections from controls (n=5) and patients with alcohol-associated hepatitis (n=7).
* indicates p≤0.05, ** p≤0.01. Exact p-values are provided in Table S1.
Supplementary Figure 2. Chronic-binge ethanol feeding to pIgR−/− mice and wildtype littermates.
A) Average food intake of female and male pIgR−/− mice and their wildtype littermates after chronic-binge ethanol diet as shown in Figure 2A.
B) Plasma ethanol levels at the study endpoint.
C) Plasma acetate levels at the study endpoint.
D) Gene expression of Adh1 in whole liver tissue, assessed by qPCR and normalized to 18S, shown relative to respective Wt littermates.
E) Gene expression of Cyp2e1 in whole liver tissue, assessed by qPCR and normalized to 18S, shown relative to respective Wt littermates.
F) Gene expression of indicated genes in whole liver tissue of female mice, assessed by qPCR and normalized to Ppib, shown relative to respective Wt littermates.
G) Gene expression of indicated genes in whole liver tissue of male mice, assessed by qPCR and normalized to Ppib, shown relative to respective Wt littermates.
Data shown as mean ± SEM of n=12–18/group from 6 independent experiments. * indicates p≤0.05, ** p≤0.01. Exact p-values are provided in Table S2.
Supplementary Figure 3. Lieber-DeCarli ethanol feeding to pIgR−/− mice and wildtype littermates.
A) Average food intake of female pIgR−/− mice and their wildtype littermates after isocaloric control or Lieber-DeCarli ethanol diet for 8 weeks as shown in Figure 3A.
B) Plasma ethanol levels at the study endpoint in ethanol-fed mice.
C) Plasma acetate levels at the study endpoint in ethanol-fed mice.
D) Gene expression of Adh1 in whole liver tissue of ethanol-fed mice, assessed by qPCR and normalized to 18S, shown relative to Wt littermates.
E) Gene expression of Cyp2e1 in whole liver tissue of ethanol-fed mice, assessed by qPCR and normalized to 18S, shown relative to Wt littermates.
F) Gene expression of indicated genes in whole liver tissue of ethanol-fed mice, assessed by qPCR and normalized to Ppib, shown relative to Wt littermates.
G) Representative FACS plots of IgA+ SYTO BC+ bacteria in pre-sorted and sorted fractions (Negative selection; Positive selection) from feces of wildtype mice after Lieber-DeCarli ethanol diet for 8 weeks.
H) Percentage of IgA+ bacteria in the different sorted bacterial fractions from feces of ethanol-fed wildtype mice (n= 8 mice).
I) E. coli assessed by qPCR in sorted bacterial fractions from feces of ethanol-fed wildtype mice. Data are shown relative to IgA− bacteria after normalization to 16S.
Data shown as mean ± SEM of n=4 mice/isocaloric group and n=17–16/group for ethanol-fed mice from 4 independent experiments. * indicates p≤0.05. Exact p-values are provided in Table S3.
Supplementary Figure 4. Antibiotic treatment of pIgR−/− mice during Lieber-DeCarli ethanol feeding.
A) Average food intake of female pIgR−/− mice treated with antibiotics or vehicle control during the last two weeks of Lieber-DeCarli ethanol diet for 8 weeks as shown in Figure 4A.
B) Plasma ethanol levels at the study endpoint.
C) Plasma acetate levels at the study endpoint.
D) Gene expression of Adh1 in whole liver tissue, assessed by qPCR and normalized to 18S, shown relative to control-treated mice.
E) Gene expression of Cyp2e1 in whole liver tissue, assessed by qPCR and normalized to 18S, shown relative to control-treated mice.
F) Gene expression of indicated genes in whole liver tissue, assessed by qPCR and normalized to Ppib, shown relative to control-treated mice.
Data shown as mean ± SEM of n=11–13 mice/group from 3 independent experiments. * indicates p≤0.05. Exact p-values are provided in Table S4.
Supplementary Figure 5. pIgR-AAV8 administration in pIgR−/− mice.
A) UMAP showing distinct clusters among total CD45− live cells in wildtype mouse livers, obtained from the Liver Cell Atlas dataset (www.livercellatlas.org). (ECs: endothelial cells; HpSCs: hepatic progenitor/stem cells; LECs: lymphatic endothelial cells; LSECs: liver sinusoidal endothelial cells; Nucseq: single-nuclei sequencing)
B) Expression of pIgR across the different clusters.
C) Violin plot showing pIgR expression in the different clusters (CD45−) in wildtype mouse livers. (Nucseq: single-nuclei sequencing; HsPCs: hepatic stem/progenitor cells; LSECs: liver sinusoidal endothelial cells)
D) Induction of pIgR expression in the livers of pIgR−/− mice injected with pIgR-AAV8 as shown in Figure 5A, assessed by qPCR and normalized to 18S.
E) Plasma ethanol levels at the study endpoint.
F) Plasma acetate levels at the study endpoint.
G) Gene expression of Adh1 in whole liver tissue, assessed by qPCR and normalized to 18S, shown relative to wildtype littermates.
H) Gene expression of Cyp2e1 in whole liver tissue, assessed by qPCR and normalized to 18S, shown relative to wildtype littermates.
I) Gene expression of indicated genes in whole liver tissue, assessed by qPCR and normalized to Gapdh, shown relative to wildtype littermates.
Data shown as mean ± SEM of n=13–15 mice per group from 5 independent experiments. * indicates corrected p≤0.05, ** corrected p≤0.01, *** corrected p≤0.001. Exact p-values are provided in Table S5.