

Abstract
Purpose:
PTEN loss-of-function/PI3K pathway hyperactivation is associated with poor therapeutic outcomes and immune checkpoint inhibitor resistance across multiple malignancies. Our prior studies in Pb-Cre;PTENfl/flTrp53fl/fl genetically engineered mice (GEM) with aggressive-variant prostate cancer (AVPC) demonstrated tumor growth control in 60% mice following androgen deprivation therapy (ADT)/PI3K inhibitor (PI3Ki)/PD-1 antibody combination, via abrogating lactate cross-talk between cancer cells and tumor-associated macrophages (TAM), and suppression of histone lactylation (H3K18lac)/phagocytic activation within TAM. Here, we targeted immunometabolic mechanism(s) of PI3Ki resistance, with the goal of durable tumor control in AVPC.
Experimental design:
Pb-Cre;PTENfl/flTrp53fl/fl GEM were treated with PI3Ki (copanlisib), MEK inhibitor (trametinib) or Porcupine inhibitor (LGK`974) singly or their combinations. MRI was used to monitor tumor kinetics and immune/proteomic profiling/ex vivo co-culture mechanistic studies were performed on GEM tumors or corresponding tumor-derived cell lines.
Results:
Given our proteomic profiling showing persistent MEK signaling within tumors of PI3Ki-resistant GEM, we tested whether addition of trametinib to copanlisib enhances tumor control in GEM, and observed 80% overall response rate via additive suppression of lactate within TME and H3K18lac within TAM, relative to copanlisib (37.5%) monotherapy. The 20% resistant mice demonstrated feedback Wnt/β-catenin activation, resulting in restoration of lactate secretion by tumor cells and H3K18lac within TAM. Co-targeting Wnt/β-catenin signaling with LGK’974 in combination with PI3Ki/MEKi, demonstrated durable tumor control in 100% mice via H3K18lac suppression and complete TAM activation.
Conclusions:
Abrogation of lactate-mediated cross-talk between cancer cells and TAM results in durable ADT-independent tumor control in PTEN/p53-deficient AVPC, and warrants further investigation in clinical trials.
STATEMENT OF TRANSLATIONAL RELEVANCE
PTEN loss-of-function occurs in ~50% of mCRPC patients, and associated with poor prognosis, and immune checkpoint inhibitor resistance across multiple malignancies. Our prior studies have demonstrated that ADT/PI3Ki/PD-1 triplet combination therapy controls PTEN/p53-deficient PC in 60% of mice via enhancement of TAM phagocytosis. Here, we discovered that resistance to PI3Ki-monotherapy occurred via feedback Wnt/MEK signaling, restoration of tumor cell-lactate secretion, and resultant histone lactylation and inhibition of phagocytosis within TAM. Critically, co-targeting of PI3K/MEK/Wnt signaling pathways using an intermittent dosing schedule of corresponding targeted agents resulted in complete tumor control and significantly prolonged survival without significant long-term toxicity. Collectively, our findings provide “proof-of-concept” that targeting lactate as a macrophage phagocytic checkpoint controls growth of murine PTEN/p53-deficient PC and warrant further investigation in AVPC clinical trials.
INTRODUCTION
Prostate cancer (PC) is diagnosed in 283,000 US men annually, with an estimated 34,700 death every year (1). While androgen deprivation therapy (ADT) has remained the cornerstone of therapy for advanced disease, progression to metastatic castrate-resistant prostate cancer (mCRPC) is inevitable. Several FDA approached therapies, including taxane chemotherapy (docetaxel, cabazitaxel), androgen receptor signaling inhibitors (abiraterone, enzalutamide and apalutamide), dendritic cell vaccine (sipuleucel-T) and radiopharmaceuticals (Lutetium-177 vipivotide tetraxetan), have demonstrated modest improvement in overall survival of mCRPC patients (2, 3). However, the disease remains incurable with high morbidity and mortality. Recent intensified approaches with upfront chemohormonal therapies in the metastatic castration-sensitive PC setting have demonstrated survival benefit (2–5). However, it has led to an increased frequency of aggressive-variant forms of the disease in the mCRPC setting, with loss-of-function (LOF) alterations in transformation-related protein 53 (TP53), retinoblastoma (RB) phosphatase and tensin homolog (PTEN) genes, that can drive lineage plasticity and neuroendocrine/small cell histopathological features (6, 7). The development of definitive medicines to treat these aggressive-variant prostate cancers (AVPC) represents an area of critical unmet need.
PTEN LOF genetic alteration occurs in ~50% of mCRPC patients, which contributes to poor prognosis, therapeutic outcomes and resistance to immune checkpoint inhibitors (ICI) (8–12). One of the molecular consequences of PTEN loss is hyperactivation of the PI3K pathway, which drives increased aerobic glycolysis via the Warburg effect (13) and enhanced proliferation (14), survival (15), and metastasis (16). In addition, PI3K-AKT pathway activation downstream of PTEN loss stimulates immunosuppressive cytokine release, such as CCL2 and IL-10 (10, 11, 17), resulting in infiltration of regulatory T cells (Treg cells), type-2 helper T cells (Th2 cells), M2-polarized tumor-associated macrophages (M2-TAM), myeloid-derived suppressive cells (MDSC) (10, 11, 18), and decreased frequency of memory CD8+ T cells and type-1 helper T cells (Th1 cells) within TME (10, 18). Furthermore, PTEN loss induces expression of programmed death-ligand 1 (PD-L1) and indoleamine 2, 3-dioxygenase 1 (IDO1), which suppress anti-tumor T cell responses within TME (11, 18). Collectively, these PTEN loss-induced immunosuppressive mechanisms contribute to the lack of responsiveness to ICI efficacy across multiple cancers.
Prior preclinical and clinical trial studies have demonstrated limited responses with single-agent PI3K inhibitors (PI3Ki) across multiple solid tumor malignancies, independent of PTEN status (19–22). Furthermore, recent studies of AKT inhibitor ipatasertib in combination with ADT and abiraterone in mCRPC patients also demonstrated limited efficacy (23). Our recent studies have demonstrated that PI3K inhibitor (PI3Ki) suppresses lactate production from PTEN/p53-deficient PC cells, resulting in restoration of TAM phagocytosis and partial suppression of Pb-Cre;PTENfl/flTrp53fl/fl GEM tumor growth, which was enhanced by the addition of ADT/PD-1 blockade (24). In the current study, we discovered that resistance to PI3Ki-monotherapy occurred via feedback Wnt/MEK signaling, restoration of tumor cell-lactate secretion, and resultant histone lactylation and inhibition of phagocytosis within TAM. Critically, co-targeting of PI3K/MEK/Wnt signaling pathways using an intermittent dosing schedule of corresponding targeted agents resulted in complete tumor control and significantly prolonged survival without significant long-term toxicity. Collectively, our findings provide “proof-of-concept” that targeting lactate as a macrophage phagocytic checkpoint controls growth of murine PTEN/p53-deficient PC and warrant further investigation in AVPC clinical trials.
MATERIALS AND METHODS
In vivo murine treatment and prostate tumor growth kinetic studies.
All studies performed in mice were approved by the Institutional Animal Care and Use Committee (IACUC) at University of Chicago and performed in compliance with NIH guidelines. Prostate-specific PTEN/p53-deficient (Pb-Cre;PTENfl/flTrp53fl/fl) mice were screened for autochthonous prostate tumor development at 16 weeks of age by ultrasound. Following the development of solid tumors (5 mm long-axis diameter of solid tumor with ultrasound imaging), the mice were treated with either copanlisib (14mg/kg, iv, every alternate day, Selleckchem; BAY 80-6946), trametinib (3mg/kg, po, every alternate day, Selleckchem; GSK 1120212), LGK`974 (3mg/kg, po, every alternate day, Selleckchem; WNT`974) single agents or their combinations (as indicated in Figure legends). Tumor volumes were monitored using MRI and quantified using Amira software (RRID:SCR_007353), as previously described (24). If solid tumor volume did not grow >20% (stable disease, SD) at indicated time point following treatment, relative to baseline, mice were considered treatment responsive. Percent Partial response (PR) was calculated based on a proportion of total mice that exhibited >30% decrease in solid tumor volume following treatment, relative to baseline. % Overall Response Rate (ORR) was calculated based on number of responder mice (SD + PR), relative to total mice enrolled in a specific treatment.
Cell lines and culture conditions:
AC1 (adenocarcinoma type) and SC1 (sarcomatoid type) cancer cells were derived from PTEN/p53-deficient prostate GEMM tumor. These cells were obtained from and authenticated by Kelly Laboratory at National Cancer Institute (25). AC1 and SC1 cells were cultured in prostate epithelial growth media (PrEGM), which was generated by adding necessary supplements (Lonza; CC-3166) to Prostate Epithelial Basal Media (PrEBM, Lonza; CC-4177), as per manufacturer’s protocol, in the absence and presence of 10% fetal bovine serum (FBS, Gemini; 100500), respectively (25). Experiments were performed on passage 12-18 of AC1/SC1 cells. Prior to experimental use, AC1/SC1 cells were confirmed to be mycoplasma-free using PCR-based testing kit (ATCC; 30-1012K).
Western blot analysis:
Established prostate tumors from Pb-Cre;PTENfl/flTrp53fl/fl mice were harvested following treatment at timespoints indicated in Figure legends, lysed in T-PER buffer (ThermoFisher; 78510) and 10 μg of total protein extracts for each sample underwent sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by Western Blot analysis. AC1 or SC1 cells were treated with indicated drug(s), lysed in RIPA buffer (ThermoFisher; 89900) and 10 μg of total protein extracts underwent SDS-PAGE followed by Western blotting, and probed for pAKT-S473 (Cell Signaling Technology; 4060S), pAKT-T308 (Cell Signaling Technology; 2965S), total AKT (Cell Signaling Technology; 4691S), active-β-catenin (Cell Signaling Technology; 8814S), H3K18lac (PTM biolabs; PTM-1406RM) pERK-T202/Y204 (Cell Signaling Technology; 9101S), total ERK (Cell Signaling Technology; 4691S) or GAPDH (Cell Signaling Technology; 4695S) as indicated in Figure legends.
Proliferation and apoptosis assays:
AC1/SC1 cells were treated with copanlisib (100 nM), trametinib (5 nM), LGK`974 (50 nM) or their combinations in presence of AC1/SC1 for indicated time points. Following treatment, proliferation rate was calculated by dividing the number of cells at a given time point with the seeding density. Hemocytometer method was utilized to count cells at any given time point. Furthermore, cells were stained with anti-Annexin V antibodies and propidium iodide (PI, DNA marker dye) as per protocol supplied with kit (BD biosciences; 556547). Flow cytometry analysis was performed for annexin V+/PI− (apoptotic) and annexin V+/PI+ (necrotic) cells. % cell death was defined as the sum total of both apoptotic and necrotic cells frequencies.
Flow cytometric analysis.
Pb-Cre;PTENfl/flTrp53fl/fl mice were treated with the indicated drug(s). Single cell suspensions were prepared from treated tumors using liberase (Sigma; 5401020001) as previously described (24), and stained with either myeloid or lymphoid antibody cocktails at a density of 1x106 cells/mL in PBS. Briefly, myeloid antibody cocktails were prepared by resuspending 10 μL of anti-CD45 (Biolegend; 103138, RRID:AB_2563061), CD11b (Biolegend; 101257, RRID:AB_2565431), CD11c (Biolegend; 117339, RRID:AB_2562414), Ly6c (Biolegend; 128006), Ly6g (Tonbo Biosciences; 80-5931-U100), MHC-II (Biolegend; 107631), F4/80 (Biolegend; 123147), CD206 (Biolegend; 141708), PD-1 (Biolegend; 135228), PD-L1 antibodies (Invitrogen; 25-5982-82, RRID:AB_2573509) and 1 μL of Ghost-viability dye (Tonbo Biosciences; 13-0870-T100) in 1 mL PBS. Lymphoid antibody cocktails were prepared by resuspending 10 μL of anti-CD45, CD3 (Biolegend; 100216, RRID:AB_493697), CD4 (Biolegend; 100451, RRID:AB_2564591), CD8 (Biolegend; 100748, RRID:AB_2721611), CD19 (Biolegend; 115541, RRID:AB_11204087), PD-1, PD-L1 antibodies and 1 μL of Ghost-viability dye in 1 mL PBS. Following 30 minutes of surface staining, single cell suspensions were washed with PBS and incubated with fix/perm buffer (Biolegend; 421401) for 15 minutes. After fixation, single cell suspensions of samples incubated with myeloid antibody cocktail were washed and resuspended in PBS for flow cytometry run whereas lymphoid antibody cocktail-stained cells were incubated overnight with 1 mL perm buffer (Biolegend; 421402) containing 10 μL of anti-FoxP3 (Biolegend; 126408, RRID:AB_1089115) and Ki67 (Biolegend; 652404) antibodies for intracellular staining. Following completion of staining, cells were washed, resuspended in PBS and flow cytometry was performed for lymphoid markers. Flow cytometry data were gated for myeloid and lymphoid subsets and their activation status using FlowJo v 10.7 software (RRID:SCR_008520).
Conditioned media (CM) collection:
As described above, single cell suspensions were prepared using liberase digestion method using established prostate tumors from untreated Pb-Cre;PTENfl/flTrp53fl/fl mice, and were plated at density of 3x106 cells per P100-culture dish. These suspensions were treated ex vivo with copanlisib (100 nM), trametinib (5 nM), LGK`974 (50 nM) or their combinations under AC1 media conditions, as indicated in Figure legends. Following treatment, supernatants were collected, named as “ex vivo CM” and utilized for TAM functionality experiments. For in vitro CM, supernatants were collected following treatment of AC1/SC1 cells with copanlisib (100 nM), trametinib (5 nM), LGK`974 (50 nM) or their combinations in presence of AC1/SC1 media, as described in Figure legends.
Lactate determination:
Lactate was quantified using colorimetry kit (Biovision; K627-100) on both ex vivo and in vitro CM.
Ex vivo phagocytosis assay:
TAM or their subsets were sorted from untreated PTEN/p53-deficient established prostate tumors using FACS and utilized for two distinct phagocytosis assay approaches. (i) TAM were either pre-treated directly with copanlisib (100 nM), trametinib (5 nM), LGK`974 (50 nM) or their combinations in presence of normal AC1/SC1 media or (ii) indirectly with CM (as described in above subsection) for 24 hours to dissect mechanism of TAM activation/phagocytosis. To investigate role of lactate on macrophage suppression, FACS-sorted TAM or BMDM were pre-treated for 24 hours with ex vivo or in vitro CM, in the presence or absence of lactate (Sigma; L7022), which was added at a final concentration of either 50 or 100 nmol/μL After these pre-treatments, TAM were co-cultured with CTV dye stained AC1/SC1 cells at Tumor cells:TAM ratio of 2:1 for 2 hours. At the end of phagocytosis, cells were then washed twice with PBS and stained with anti-CD45, MHC-II (M1-anti-tumor/activated macrophage marker), PD-1, (1:100 dilution for each), H3K18lac antibodies (1:100 dilution for each) and Ghost-viability dye (1:1000 dilution) in 1 mL PBS. Histone lactylation status and phagocytic activity and of each TAM subset (MHC-IIhi/PD-1lo, MHC-IIhi/PD-1hi, MHC-IIlo/PD-1lo, and MHC-IIlo/PD-1hi TAM) were calculated by normalizing H3K18lac staining and MFI of CTV dye , respectively, relative to untreated groups,. Baseline phagocytic activity of TAM subsets was normalized relative to untreated MHC-IIloPD-1hi TAM.
In vivo macrophage depletion studies:
To elucidate a role for macrophages in driving anti-tumor responses in vivo, Pb-Cre; PTENfl/fl Trp53fl/fl mice with established prostate tumors were treated concurrently with clodronate (200 μg/mouse, ip, once weekly, Encapsula Nano Sciences; SKU-CDL8909), which is known to deplete macrophages in vivo, and combination of copanlisib (14 mg/kg, iv, every alternate day), trametinib (3mg/kg, po, every alternate day) and LGK`974 (3mg/kg, po, every alternate day) for 28 days. Prostate tumor growth kinetics and immune profiling were done, as described in relevant subsections.
Histopathological and survival analysis.
Power calculation was done using published data of Pb-Cre;PTENfl/flTrp53fl/fl mice (n = 32, mortality = 100% and median survival time = 5.5 months) relative to wild-type mice (n=33, mortality = 0% with 10 months follow-up), with hazard ratio = 0.03099 (26). Based on these estimates, the required number of mice using α = 0.05 and β = 0.2 was n=3 in treated group with no mortality to 11 months, relative to n=3 untreated mice. Pb-Cre;PTENfl/flTrp53fl/fl mice with established prostate tumors were treated with copanlisib (14 mg/kg, iv, every alternate day) + trametinib (3 mg/kg, po, every day) + LGK`974 (3 mg/kg, po, every day) until signs of distress post-9 weeks of treatment, such as mild hunched, incision, rough hair coat, squinted eye, sudden body weight loss appeared (27). After mice recovered from visible signs of distress following 3 weeks of drug holiday, treatment schedule was resumed again until mice reached 11 months of age, relative to untreated historic controls (100% mortality by 7 months) (26). Tumors were harvested and approximately 5-μm thick formalin-fixed-paraffin-embedded sections were prepared. Hematoxylin and Eosin (H&E) staining was performed on these sections, and histopathological assessments were carried out by a blinded pathologist for anti-tumor efficacy.
Data analysis.
Data was analyzed using GraphPad Prism (RRID:SCR_002798). All experiments were replicated as indicated in figure legends. Statistical analysis was performed using student t-test, one-way analysis of variance (ANOVA) and Bonferroni post-test with p<0.05 level of significance. Chi-square test was performed to compare % ORR and % partial response.
Data and materials availability.
The authors declare that the data and materials supporting the findings of this study are available within the Article and its Supplementary Information. All raw data are available from corresponding author upon request.
RESULTS
Persistent MEK pathway activation in Pb-Cre;PTENfl/flTrp53fl/fl mice resistant to PI3Ki monotherapy.
Our recent studies have demonstrated anti-tumor responses in subsets of Pb-Cre;PTENfl/flTrp53fl/fl mice following PI3Ki singly or in combination with ADT/aPD-1 therapy, resulting from inhibition of tumor cell intrinsic lactate production and corresponding TAM histone lactylation and phagocytic activation (24). Furthermore, prior studies have demonstrated feedback activation of MEK signaling as one potential mechanism for resistance to PI3Ki monotherapy (28–30). As a first step towards understanding whether MEK signaling drives resistance to PI3Ki monotherapy, we performed western blot analysis on PI3K and MEK signaling pathways in prostate tumors of copanlisib-treated responder/non-responder Pb-Cre;PTENfl/flTrp53fl/fl mice. Interestingly, we observed near-complete inhibition of both p-AKT and p-ERK expression, indicative of suppression of PI3K and MEK pathways, respectively, within tumors of mice responding to copanlisib, relative to untreated control (Supplementary Fig. 1A). In contrast, non-responder mice had persistent MEK activation within tumors, similar to untreated group, despite suppression of PI3K signaling, (Supplementary Fig. 1A). On the other hand, trametinib (MEK inhibitor) completely abolished MEK signaling in PTEN/p53-deficient tumor-derived AC1/SC1 cancer cells (Supplementary Fig. 1B), but demonstrated corresponding feedback activation of PI3K signaling (Supplementary Fig. 1C). Furthermore, copanlisib alone inhibited PI3K signaling in AC1/SC1 cells with partial upregulation of MEK signaling only in AC1 cells (Supplementary Fig. 1C). Combination of copanlisib/trametinib showed a decrease in both PI3K/MEK signaling in AC1/SC1 cells, relative to untreated control (Supplementary Fig. 1C) and similar to copanlisib treated responder mice (Supplementary Fig. 1A). Collectively, these findings suggest that persistent MEK signaling is a potential resistance mechanism to PI3Ki monotherapy in PTEN/p53-deficient PC.
Combination of PI3K and MEK inhibitors control tumor growth in 80% of Pb-Cre; PTENfl/fl Trp53fl/fl mice via suppression of histone lactylation within TAM
As a first step to elucidate the therapeutic impact of dual PI3K/MEK inhibition in PTEN/p53-deficient PC, we treated PTEN/p53-deicient tumor-derived AC1 and SC1 cells with copanlisib, singly and in combination with trametinib in vitro. We observed a modest decrease in proliferation and survival of AC1 cells with trametinib alone, which was not accentuated by combination copanlisib/trametinib. On the other hand, there was no change in proliferation or survival of SC1 cells with copanlisb or tramentinib as single agents or their combination. (Supplementary Fig. S2A–B). In contrast, the combination of trametinib and copanlisib demonstrated an overall response rate (ORR) of 80% in Pb-Cre; PTENfl/fl Trp53fl/fl mice in vivo, which was significantly higher relative to untreated. In contrast, trametinib and copanlisib single-agent treatments led to non-significant increase in ORR of 50% (Fig. 1A and Supplementary Fig. S3A–B) and 37.5% (24) respectively, relatively to untreated, at doses that pharmacodynamically inhibit target activity (Supplementary Fig. 4, (24)). Collectively, these findings suggest that PI3Ki/MEKi combination exerts its anticancer activity via a tumor cell–extrinsic mechanism in PTEN/p53–deficient prostate GEMM.
Figure 1. PI3Ki/MEKi combination therapy induces activated TAM-mediated tumor growth control in 80% of Pb-Cre;PTENfl/flTrp53fl/fl mice.
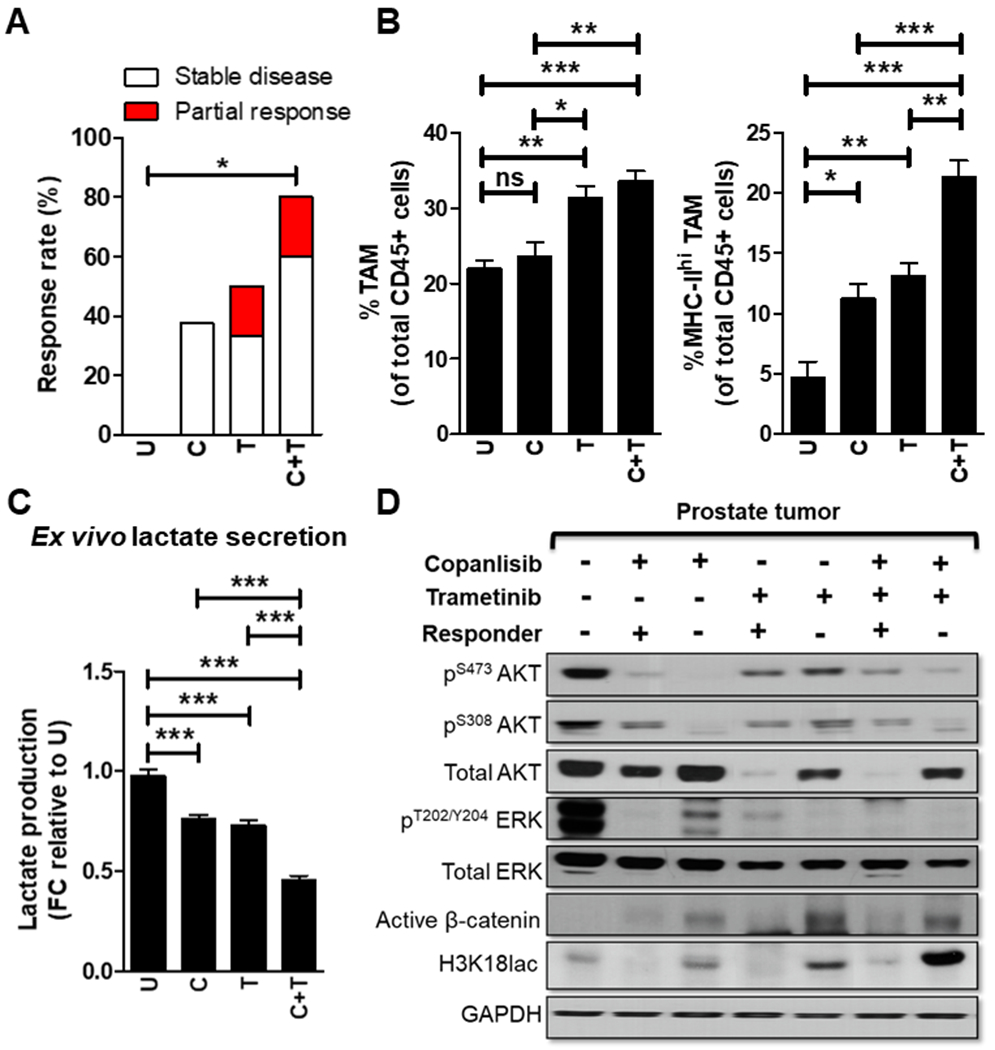
(A) Pb-Cre;PTENfl/flTrp53fl/fl mice with established prostate tumors were treated with trametinib (3 mg/kg, po, every day), singly and in combination with copanlisib (14 mg/kg, iv, every alternate day) for 28 days. Tumor growth was monitored using MRI and response rates (Partial response plus Stable disease) were quantified as described in Methods. (B) At the end of treatment, prostate tumors were profiled using flow cytometry and analyzed for % frequency of total and MHC-II expressing TAM (CD45+CD11b+F4/80+ cells). (C) Single cell suspensions of PTEN/p53-deficient prostate GEM tumors were treated with copanlisib (C, 100 nM), trametinib (T, 5 nM) or their combination for 24 hours as described in Methods. Ex vivo CM were collected from these groups and analyzed for lactate content using colorimetry kits. (D) Prostate tumor extracts harvested from PI3Ki, MEKI and combinatorial treatment groups underwent western blot analysis for the indicated proteins. For in vivo studies, n=5-6 mice per group (experimental male mice with the correct genotype were obtained following screening of 79 littermates derived from 11 breeding pairs and randomized until the indicated number of mice achieved in each group). For ex vivo studies, n=2 independent biological experiments. Significances/p-values were calculated by Chi-square test (panel A), one-way ANOVA (panel B-C) and indicated as follows, *p<0.05, **p<0.01 and ***p<0.001; ns = not statistically significant.
To elucidate potential tumor cell extrinsic mechanism(s) for anti-tumor efficacy of PI3Ki/MEKi combination therapy, we performed immune profiling and observed a 2.8-fold increase in frequency of activated TAM within prostate tumors from trametinib-treated mice, which increased to a 4.6-fold with trametinib/copanlisib, relative to untreated control (Fig. 1B). We have previously demonstrated that ADT/PI3Ki/aPD-1 combination controls tumor growth in Pb-Cre;PTENfl/flTrp53fl/fl mice via suppression of histone lactylation and corresponding enhancement of TAM phagocytosis, particularly in MHC-IIhi/PD-1hi/lo TAM subsets (24). To determine whether PI3Ki/MEKi combination-induced tumor control is mediated via TAM innate immune phagocytic mechanism, we analyzed ex vivo CM collected from single cell suspensions of tumors following treatment with trametinib alone at IC90 concentrations (Supplementary Fig. S1B), and observed a 24.7% decrease in lactate levels that was further decreased to 72% in combination with copanlisib (Fig. 1C). Consistently, in vivo treatment with PI3Ki/MEKi combination demonstrated a decrease in histone lactylation relative to untreated control (Fig. 1D and Supplementary Fig. 4). Importantly, the combination of copanlisib and trametinib demonstrated a 17.2 and 21-fold suppression of histone lactylation in MHC-IIhi/PD-1lo TAM and MHC-IIhi/PD-1hi TAM, respectively, relative to untreated. In contrast, single-agent trametinib or copanlisib led to 3.9- and 4-fold suppression of histone lactylation in MHC-IIhi/PD-1lo and MHC-IIhi/PD-1hi TAM, respectively, relative to untreated (Fig. 2A–B), Critically, the combination of copanlisib and trametinib demonstrated a 7.2- and 7.1-fold increase in phagocytic capacity of MHC-IIhi/PD-1lo and MHC-IIhi/PD-1hi TAM, respectively, relative to untreated controls. In contrast, single agent trametinib or copanlisib led to a 3.8- and 3.7-fold induction in phagocytic capacity of MHC-IIhi/PD-1lo and MHC-IIhi/PD-1hi TAM, respectively, relative to untreated controls (Fig. 2C). In contrast, conditioned media from PI3Ki/MEKi-treated PC cells did not alter MHC-IIlo TAM histone lactylation and phagocytosis (Supplementary Fig. S5A–B). Collectively, these data demonstrate that the PI3Ki/MEKi combination potently suppresses histone lactylation and enhances phagocytosis within activated TAM, relative to untreated or single agent controls. In contrast, direct treatment with copanlisib, trametinib or their combination did not alter TAM phagocytic capacity (Supplementary Fig. S6A–B). Collectively, these data suggest that the majority of the anti-cancer response observed with PI3Ki/MEKi combination in Pb-Cre;PTENfl/flTrp53fl/fl mice s mediated via a tumor cell extrinsic TAM-mediated innate immune mechanism that requires cross-talk with tumor cells within the TME.
Figure 2. PI3Ki/MEKi combination therapy potently suppresses histone lactylation and enhances phagocytosis within activated TAM, relative to single agent controls.
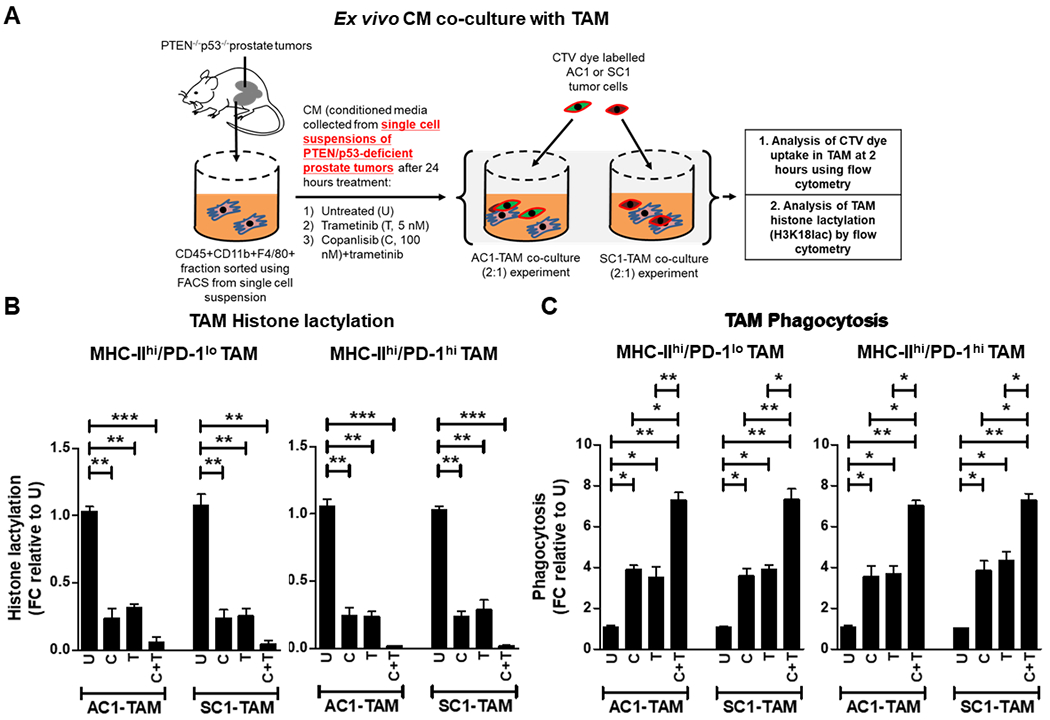
(A) Single cell suspensions of PTEN/p53-deficient prostate GEM tumors were treated with copanlisib (C, 100 nM), trametinib (T, 5 nM) or their combination for 24 hours, and conditioned media (CM) was collected at the end of treatment. FACS-sorted TAM were incubated ex vivo in CM for 24 hours, followed by co-culture with CTV dye stained-AC1/SC1 cells for 2 hours. Bar graphs demonstrate histone lactylation status (B) and phagocytic activity (C) of MHC-IIhi/PD-1hi/lo expressing TAM, relative to untreated group. FC = fold change. n=2 independent biological experiments. Significances/p-values were calculated by one-way ANOVA and indicated as follows, *p<0.05, **p<0.01 and ***p<0.001.
Feedback activation of Wnt/β-catenin signaling observed in non-responder Pb-Cre;PTENfl/flTrp53fl/fl mice following PI3Ki/MEKi combination treatment restores histone lactylation (H3K18lac) and suppresses macrophage phagocytosis.
Our prior studies have demonstrated feedback Wnt/β-catenin signaling as a resistance mechanism to ADT/PI3Ki/aPD1 combination therapy, resulting in restoration of TAM histone lactylation (24). To assess whether a similar mechanism of resistance occurs in 20% of mice treated with PI3Ki/MEKi combination therapy, we performed Western blot analysis on whole tumor extracts, which demonstrated an increase in active β-catenin expression and histone lactylation within tumor from non-responder outlier mouse, relative to responder and untreated groups (Fig. 1D). Consistent with these observations, in vitro western blot analysis of PTEN/p53-deficient tumor-derived AC1/SC1 cells following 72-hours treatment of copanlisib/trametinib combination demonstrated a concomitant increase in active β-catenin expression, relative to 24-hours treatment (Supplementary Fig. S7A–B). This feedback activation of Wnt/β-catenin signaling was accompanied by an increase in lactate production at 72 hours, following an initial decrease with acute copanlisib/trametinib treatment in vitro (Fig. 3A). Collectively, these data suggest that resistance to PI3Ki/MEKi combination therapy is driven by upregulation of Wnt-β-catenin signaling and restoration of tumor cell lactate production/TAM histone lactylation.
Figure 3. Addition of PORCNi overcomes resistance to PI3Ki/MEKi combination therapy and induces activated TAM-mediated 100% response rate in Pb-Cre;PTENfl/flTrp53fl/fl mice.
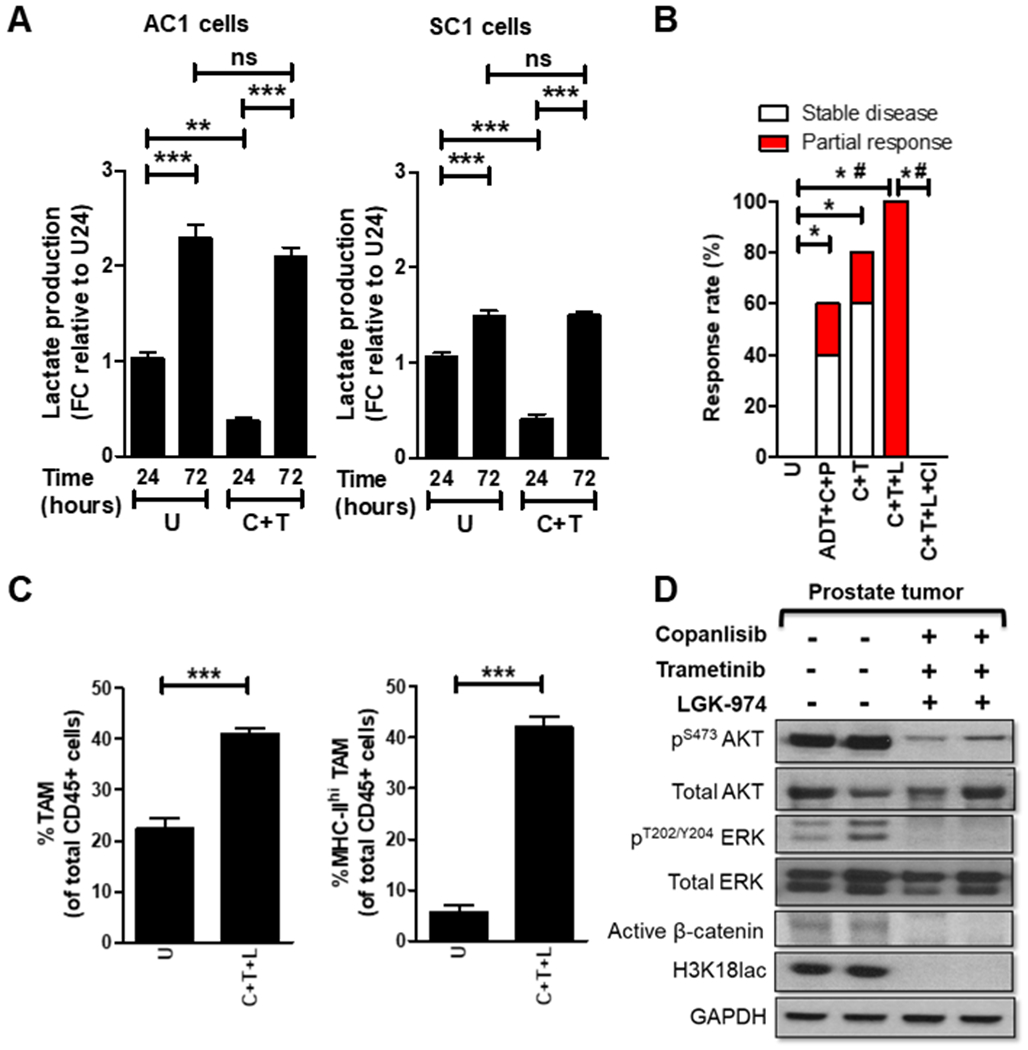
(A) PTEN/p53-deficient GEM tumor-derived AC1/SC1 cells were treated with copanlisib (C, 100 nM), trametinib (T, 5 nM) or their combination for 24 and 72 hours. In vitro CM were collected following treatment and analyzed for lactate content using colorimetry kits. (B) Pb-Cre;PTENfl/flTrp53fl/fl mice with established prostate tumors were treated with indicated combinations of trametinib (T, 3 mg/kg, po, every day), copanlisib (C, 14 mg/kg, iv, every alternate day), LGK`974 (L, 3 mg/kg, po, every day), PD-1 antibody (aPD-1, 100μg/mouse, ip, every alternate day) and ADT (degarelix, 0.625 mg, single dose) for 28 days. Tumors were monitored with MRI and response rates/partial response/stable disease were determined as described in Methods. (C-D) Tumors from untreated or copanlisib+trametinib+LGK`974-treated mice were analyzed by flow cytometry for % frequency of total and MHC-II expressing TAM (CD45+CD11b+F4/80+ cells) (C) or analyzed for the indicated proteins by Western blotting (D). n = 5-6 mice per group for in vivo studies (experimental male mice with the correct genotype were obtained following screening of 92 littermates derived from 15 breeding pairs and were randomized until the indicated number of mice achieved in each group). For in vitro experiments, n = 3 independent biological experiments. Significances/p-values were calculated by one-way ANOVA (panel A), Chi-square test (panel B), student t-test (panel C) and indicated as follows, *p<0.05, **p<0.01, ***p<0.001 and #p<0.05 for partial response (panel B); ns = not statistically significant.
PI3Ki + MEKi + PORCNi combination therapy drives tumor shrinkage in 100% of Pb-Cre;PTENfl/flp53fl/fl mice via complete TAM activation
To overcome Wnt/β-catenin-mediated resistance to PI3Ki/MEKi combination therapy, Pb-Cre;PTENfl/flTrp53fl/fl mice with established tumors were treated with copanlisib + trametinib + LGK`974, which resulted in a 100% response rate/tumor shrinkage, relative to untreated group (Fig. 3B and Supplementary Fig. S8A). This anti-tumor response was rescued by concurrent clodronate treatment (Fig. 3B and Supplementary Fig. S8B) and accompanied by a 2-fold increase in total TAM frequency, relative to untreated. Strikingly, copanlisib + trametinib + LGF’974 resulted in activation of 100% TAM within the TME, relative to approx. 20% activated TAM in untreated tumors (Fig. 3C) and a corresponding suppression of histone lactylation within the TME (Fig. 3D). Analysis of in vitro CM collected following treatment of AC1 and SC1 cells with copanlisib + trametinib + LGK`974 at pre-determined IC90 concentrations (Supplementary Fig. S1B and Supplementary Fig. S9A–B) showed a 78% and 50% decrease in lactate levels, respectively (Supplementary Fig. S10A). Furthermore, in vitro treatment with copanlisib + trametinib + LGK`974 modestly inhibited the proliferation and survival of PTEN/p53-deficient GEM tumor-derived AC1 cells but not SC1 cells (Supplementary Fig. S10B–C), suggesting a predominantly tumor cell extrinsic mechanism by which the combination exerts its anti-tumor effects in vivo. Direct treatment of TAM with copanlisib + trametinib + LGK`974 did not alter TAM phagocytic capacity or histone lactylation status (Supplementary Fig. S11A–C). Critically, co-culture of TAM with CM collected following copanlisib + trametinib + LGK`974 treatment of AC1 and SC1 cells demonstrated a 3.9-fold and 3.1-fold reduction in histone lactylation, with a corresponding 6.8 and 6.3-fold increase in respective MHC-IIhi/PD-1lo and MHC-IIhi/PD-1hi TAM phagocytic capacity, respectively, relative to untreated controls. Lactate “add-back” to the CM collected following copanlisib + trametinib + LGK`974 treatment restored histone lactylation and suppressed phagocytic activity of TAM. Furthermore, the addition of LGK’974 to CM following collection from tumor cells treated with copanlisib + trametinib did not result in suppression of histone lactylation or increase in phagocytic activity, relative to untreated, thus demonstrating that tumor cell intrinsic Wnt/β-catenin signaling and lactate production is responsible for histone lactylation and immunosuppression within TAM (Fig. 4A–B). In contrast, conditioned media from PI3Ki + MEKi + PORCNi treated PTEN/p53-deficient tumor derived cells does not alter histone lactylation and phagocytic activity of MHC-IIlo TAM (Supplementary Fig. S12A–B).
Figure 4. PI3Ki/MEKi/PORCNi combination therapy suppresses lactate production from PTEN/p53-deficient GEM tumor-derived PC cells and secondary histone lactylation within activated TAM, resulting in enhanced TAM phagocytosis.
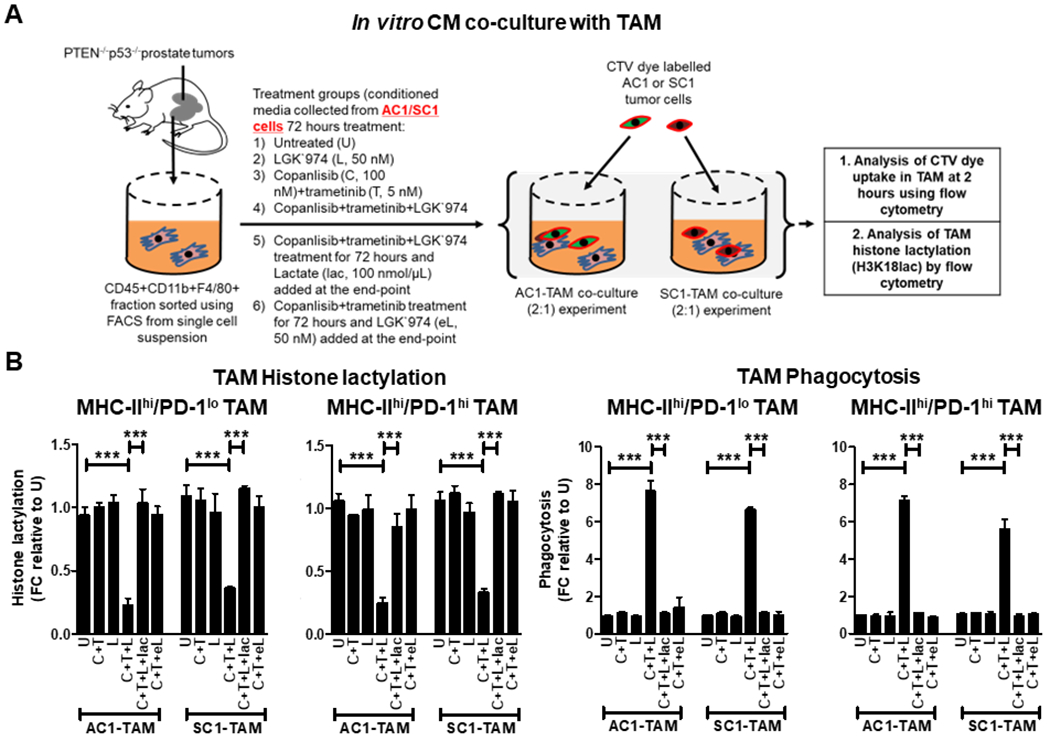
(A) AC1/SC1 cells were treated with copanlisib (C, 100 nM), trametinib (T, 5 nM), LGK`974 (L, 50 nM) or their combination for 72 hours. For mechanistic dissection, lactate (lac, 100 nmol/μL) and LGK`974 (eL, 50nM) were added to the CM collected after C+T+L and C+T treatments of AC1/SC1 cells, respectively. FACS-sorted TAM were incubated with the indicated CM for 24 hours followed by co-culture with CTV dye stained-AC1/SC1 cells for 2 hours. (B) Bar graphs demonstrate fold change (FC) in histone lactylation and phagocytic activity of MHC-IIhi/PD-1hi/lo expressing TAM, relative to untreated group. n = 2 independent biological experiments. Significances/p-values were calculated by one-way ANOVA and indicated as follows, ***p<0.001.
Given the toxicity concerns with combinatorial PI3Ki/MEKi/Porcupine inhibitor targeted therapy, we evaluated efficacy and safety of copanlisib + trametinib + LGK`974 with long-term treatment. We observed dermatitis and conjunctivitis in Pb-Cre;PTENfl/flp53fl/fl mice following 9 weeks of continuous triplet therapy. However, a 3-week drug holiday was sufficient for resolution of dermatitis and conjunctivitis, so we decided to administer the copanlisib + trametinib + LGK`974 regimen on a 9-weeks on/3 weeks off intermittent dosing schedule (Fig. 5A), which significantly improved survival in the Pb-Cre;PTENfl/flp53fl/fl mice with 100% survival at 11 months (statistically pre-determined endpoint), relative to untreated (100% mortality by 7 months, Fig. 5B) (26). Critically, we observed a near-complete pathologic response in responsive mice at the end of both short-term (4 weeks) and long-term (approx. 7 months) treatment (Fig. 5C and Supplementary Table S1). Collectively, these data demonstrate that the decrease in lactate production from PTEN/p53-deficient PC cells in response to PI3Ki + MEKi + Porcupine inhibitor treatment overcomes histone lactylation mediated TAM suppression and stimulates phagocytosis of PC cells by TAM, resulting in long-term tumor control in 100% of the mice, with manageable toxicity in the context of an intermittent dosing schedule (Fig. 5D).
Figure 5. Intermittent long-term dosing of PI3Ki + MEKi + PORCNi results in tumor clearance of established tumors and significant prolongation of survival in Pb-Cre;PTENfl/fl Trp53fl/fl mice.
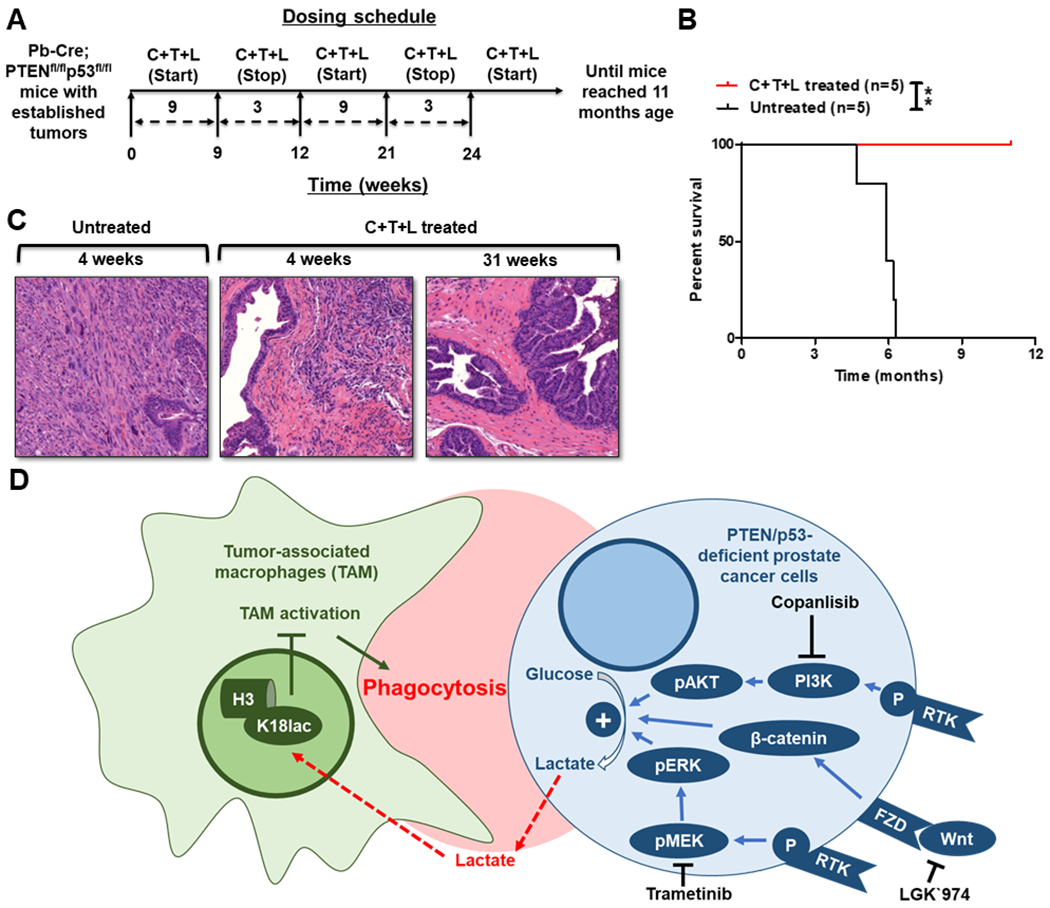
(A) Schema illustrating Pb-Cre;PTENfl/fl Trp53fl/fl mice dosed intermittently with copanlisib (C, 14 mg/kg, iv, every alternate day) + trametinib (T, 3 mg/kg, po, every day) + LGK`974 (L, 3 mg/kg, po, every day). (B) Kaplan-Meier survival curves were plotted for C+T+L treatment, relative to untreated control. (C) Tumors were harvested following short-term and long-term C+T+L treatment, and representative histopathologic H&E staining of formalin-fixed paraffin-embedded tissue is depicted. (D) Model illustrating the decrease in lactate production from PTEN/p53-deficient PC cells in response to PI3Ki+MEKi+PORCNi treatment suppresses histone lactylation and enhances phagocytosis of PC cells within activated TAM. n=5 mice per group (experimental male mice with the correct genotype were obtained following screening of 33 littermates derived from 4 breeding pairs and randomized until the indicated number of mice achieved in each group). Significances/p-values were calculated by Log-rank test and indicated as follows, *p<0.05.
DISCUSSION
Although ICI have revolutionized the landscape of cancer therapeutics, most mCRPC patients are de novo resistant to these medicines, in part due to a paucity of T cells within the TME and/or a tumor cell intrinsic genomic alterations that evade anti-cancer immunity (11, 31). PTEN LOF is present in ~50% of mCRPC patients that positively correlates with poor prognosis, worse therapeutic outcomes, and ICI resistance (8, 9, 11). Prior studies have demonstrated that hyperactivated PI3K signaling in the context of PTEN LOF TME upregulates GLUT1/4 and hexokinase-2, which enhances aerobic glycolysis. We have previously demonstrated that increased lactate production from PTEN/p53-deficient prostate cancer cells epigenetically reprograms TAM via histone lactylation, and polarizes them towards an immunosuppressive phenotype (M2/MHC-IIlo TAM) (24). Furthermore, we identified a negative correlation between TAM M1 activity and tumor cell aerobic glycolysis within the TME of mCRPC patients (24). Metabolically, lactate decreases glycolysis and increases TCA cycle within M1-/M2-TAM, respectively, ultimately reprogramming TAM into a tumor-promoting M2 phenotype (32, 33). Furthermore, lactate augments M2-TAM functionality by upregulating Arg1 expression, resulting in degradation of arginine and decrease in T cell receptor expression and T cell proliferation/effector function (32).
PI3Ki monotherapy has demonstrated limited anti-cancer efficacy in solid tumor malignancies, with or without PTEN/PI3K pathway alterations (19, 20, 22). In PTEN-deficient mCRPC patients, co-targeting PI3K and AR signaling with ipatasertib and abiraterone (androgen receptor signaling inhibitor), respectively, demonstrated a modest improvement in progression-free survival (34). Furthermore, our recent preclinical studies in Pb-Cre;PTENfl/flp53fl/fl mice demonstrated enhanced anti-tumor responses with addition of aPD-1 blockade to the ADT/PI3Ki treatment backbone (24). However, resistance was observed in 40% of mice due to feedback upregulation of Wnt/β-catenin signaling, which restored lactate production and histone lactylation and suppressed phagocytosis within TAM (24). Here, we discovered that PI3K, MEK and Wnt/β-catenin signaling pathways-mediated cross-talk preserves tumor cell-intrinsic lactate production with corresponding TAM histone lactylation/phagocytosis suppression and resultant evasion of anti-cancer innate immunity within PTEN/p53-deficient PC. Concurrent inhibition of these pathways abrogated lactate production from PC cells, resulting in suppression of histone lactylation and phagocytosis-mediated tumor control by activated TAM in 100% of mice. Collectively, we have discovered the importance of lactate as a novel TAM phagocytosis inhibitory checkpoint that evades anti-cancer innate immunity in the context of PTEN/p53-deficient PC. From a translational standpoint, co-targeting oncogenic signaling pathways that preserve lactate production, represents a novel TAM-dependent innate immunomodulatory strategy, that warrants further investigation in combination with ICI in PTEN/p53-deficient AVPC.
Based on prior preclinical studies demonstrating reciprocal feedback relationship between PI3K and MEK signaling pathways in certain cancers (28–30), several PI3Ki/MEKi combinations have been tested in clinical trials, but met with challenges related to low therapeutic index and dose-limiting toxicities, including hyperglycemia, loss of appetite, diarrhea, fatigue, rash, vomiting, and increase in lipase and creatine phosphokinase levels (29, 35, 36). A prior preclinical study has demonstrated that transient/intermittent exposure to PI3Ki/MEKi combination is sufficient to induce tumor cell apoptosis, relative to continuous administration (15). In this study, sustained pharmacodynamic elevation of Bim protein was found to be critical for tumor cell-intrinsic apoptosis induction within TME despite recovery of pAKT and pERK following transient exposure to PI3Ki/MEKi combination. These data demonstrated the feasibility of an intermittent administration approach that may be effective to achieve an anti-tumor response while decreasing drug-induced toxicity (15). In our current preclinical in vivo studies, an intermittent dosing schedule of PI3Ki/MEKi/Wnti combination (~9 weeks on and ~3 weeks off) for at least 7 months led to significant tumor cell extrinsic TAM activation/phagocytosis-mediated tumor control in PTEN/p53-deficient murine PC growth, without significant toxicities or mortality. These findings suggest a potential therapeutic window that is achievable with an intermittent dosing approach of PI3Ki/MEKi/PORCNi, which will need further evaluation in Phase I clinical trials. The availability of hyperpolarized 13C-pyruvate magnetic resonance spectroscopy imaging approaches would enable in vivo pharmacodynamic monitoring of lactate suppression/restoration to optimize intermittent dosing approaches (37). Furthermore, combining imaging with 13C-glucose infusion/isotope tracing would provide a safe and effective approach to understand heterogeneity in tumor metabolism, and unravel the immunometabolic mechanism(s) that underly response vs resistance (38).
Alternatively, direct activation of TAM phagocytosis could be an alternative strategy to treat PTEN-deficient mCRPC/AVPC patients. This can be achieved by either inhibiting TAM phagocytic checkpoints (24) or blocking lactate entry within TAM (39). In prior published work, we discovered an important role for TAM PD-1 expression in evasion of phagocytosis in Pb-Cre;PTENrl/flp53fl/fl mice (24). There are several additional TAM checkpoints under investigation, such as SIRPα/CD47 (40), LILRB1/MHC-I (41) and Siglec-10/CD24 (42). Elucidating the functional role of these “don’t eat me” signals in the lactate-rich TME downstream of PTEN loss is an area of unmet need. Furthermore, monocarboxylate transport (MCT) inhibitors which block MCT-1 (enables lactate import within TAM) and/or MCT-4 (enables lactate export from tumor cells), which are in early drug discovery and development, could represent promising approaches to directly target lactate-mediated immunometabolic cross-talk within the PTEN-deficient TME of AVPC (39, 43), thereby mitigating potential toxicities encountered with kinase inhibitor cocktails (43).
Clinical trial studies with the porcupine inhibitor, LGK`974, have been limited due to the observed toxicity of increased bone resorption (44, 45). This is of particular concern in mCRPC patients who are on life-long ADT that results in a decreased bone mineral density and increased risk of skeletal-related events (46, 47). Given the complexity of the Wnt/β-catenin signaling pathway, which involves multiple Wnt-ligands, frizzled receptors, co-receptors and extracellular regulators (48), a deeper understanding of Wnt signaling in tumor vs bone cells would provide new avenues to effectively treat AVPC patients and mitigate known toxicities. For example, DKK1 blocking agent, which would specifically enhance osteoblast Wnt//β-catenin signaling (49), in combination with PI3Ki/MEKi/PORCNi therapy could be a potential strategy for treating bone resorption in PTEN-deficient mCRPC/AVPC patients. Collectively, our findings provide a mechanistic foundation for a metronomic dose optimization trial to determine immunogenic or TAM activating doses of PI3Ki, MEKi or Wnt/β-catenin targeting agents for treatment of PTEN/p53-deficient AVPC.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs. Andrew Hsieh, Scott Oakes and Sam Grimaldo for providing helpful suggestions for this manuscript. This work was supported by 16CHAL12 (Prostate Cancer Foundation Challenge Award to A. Patnaik), P50CA180995 (A. Patnaik), V Foundation Translational Cancer Research Award (A. Patnaik) and P30CA014599 (E. Markiewicz, M. Zamora, X. Fan)
Author conflict of interest:
Dr. Patnaik has received research funding from Bristol Myers Squibb.
REFERENCES
- 1.Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin 2023;73(1):17–48 doi 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 2.Li Y, Malapati S, Lin YT, Patnaik A. An integrative approach for sequencing therapies in metastatic prostate cancer. The American Journal of Hematology/Oncology 2017;13(12):26–31. [Google Scholar]
- 3.Thomas TS, Pachynski RK. Treatment of Advanced Prostate Cancer. Mo Med 2018;115(2):156–61. [PMC free article] [PubMed] [Google Scholar]
- 4.Sydes MR, Spears MR, Mason MD, Clarke NW, Dearnaley DP, de Bono JS, et al. Adding abiraterone or docetaxel to long-term hormone therapy for prostate cancer: directly randomised data from the STAMPEDE multi-arm, multi-stage platform protocol. Ann Oncol 2018;29(5):1235–48 doi 10.1093/annonc/mdy072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke NW, Ali A, Ingleby FC, Hoyle A, Amos CL, Attard G, et al. Addition of docetaxel to hormonal therapy in low- and high-burden metastatic hormone sensitive prostate cancer: long-term survival results from the STAMPEDE trial. Ann Oncol 2019;30(12):1992–2003 doi 10.1093/annonc/mdz396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conteduca V, Ku SY, Fernandez L, Dago-Rodriquez A, Lee J, Jendrisak A, et al. Circulating tumor cell heterogeneity in neuroendocrine prostate cancer by single cell copy number analysis. NPJ Precis Oncol 2021;5(1):76 doi 10.1038/s41698-021-00211-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montironi R, Cimadamore A, Lopez-Beltran A, Scarpelli M, Aurilio G, Santoni M, et al. Morphologic, Molecular and Clinical Features of Aggressive Variant Prostate Cancer. Cells 2020;9(5) doi 10.3390/cells9051073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jamaspishvili T, Berman DM, Ross AE, Scher HI, De Marzo AM, Squire JA, et al. Clinical implications of PTEN loss in prostate cancer. Nat Rev Urol 2018;15(4):222–34 doi 10.1038/nrurol.2018.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161(5):1215–28 doi 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov 2016;6(2):202–16 doi 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vidotto T, Melo CM, Castelli E, Koti M, Dos Reis RB, Squire JA. Emerging role of PTEN loss in evasion of the immune response to tumours. Br J Cancer 2020;122(12):1732–43 doi 10.1038/s41416-020-0834-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017;46(2):197–204 doi 10.1016/j.immuni.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell 2017;170(4):605–35 doi 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen C, Cai Q, He W, Lam TB, Lin J, Zhao Y, et al. AP4 modulated by the PI3K/AKT pathway promotes prostate cancer proliferation and metastasis of prostate cancer via upregulating L-plastin. Cell Death Dis 2017;8(10):e3060 doi 10.1038/cddis.2017.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoeflich KP, Merchant M, Orr C, Chan J, Den Otter D, Berry L, et al. Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res 2012;72(1):210–9 doi 10.1158/0008-5472.CAN-11-1515. [DOI] [PubMed] [Google Scholar]
- 16.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003;4(3):209–21 doi 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 17.Caforio M, de Billy E, De Angelis B, Iacovelli S, Quintarelli C, Paganelli V, et al. PI3K/Akt Pathway: The Indestructible Role of a Vintage Target as a Support to the Most Recent Immunotherapeutic Approaches. Cancers (Basel) 2021;13(16) doi 10.3390/cancers13164040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin Z, Huang L, Li SL, Gu J, Cui X, Zhou Y. PTEN loss correlates with T cell exclusion across human cancers. BMC Cancer 2021;21(1):429 doi 10.1186/s12885-021-08114-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Awada AH, Morschhauser F, Machiels JP, Salles G, Rottey S, Rule S, et al. PI3K inhibition and modulation of immune and tumor microenvironment markers by copanlisib in patients with non-Hodgkin’s lymphoma or advanced solid tumors. Annals of Oncology 2018;29:viii20 doi 10.1093/annonc/mdy269.065. [DOI] [Google Scholar]
- 20.Liu N, Haike K, Glaeske S, Paul J, Mumberg D, Kreft B, et al. Copanlisib in combination with anti-PD-1 induces regression in animal tumor models insensitive or resistant to the monotherapies of PI3k and checkpoint inhibitors. Hematological Oncology 2017;35(S2):257–8 doi 10.1002/hon.2438_123. [DOI] [Google Scholar]
- 21.Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer 2019;18(1):26 doi 10.1186/s12943-019-0954-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armstrong AJ, Halabi S, Healy P, Alumkal JJ, Winters C, Kephart J, et al. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur J Cancer 2017;81:228–36 doi 10.1016/j.ejca.2017.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sweeney C, Bracarda S, Sternberg CN, Chi KN, Olmos D, Sandhu S, et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. Lancet 2021;398(10295):131–42 doi 10.1016/S0140-6736(21)00580-8. [DOI] [PubMed] [Google Scholar]
- 24.Chaudagar K, Hieromnimon HM, Khurana R, Labadie B, Hirz T, Mei S, et al. Reversal of lactate and PD-1-mediated macrophage immunosuppression controls growth of PTEN/p53-deficient prostate cancer. Clin Cancer Res 2023;29(10):1952–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu YN, Abou-Kheir W, Yin JJ, Fang L, Hynes P, Casey O, et al. Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor beta-initiated prostate cancer epithelial-mesenchymal transition. Mol Cell Biol 2012;32(5):941–53 doi 10.1128/MCB.06306-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005;436(7051):725–30 doi 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burkholder T, Foltz C, Karlsson E, Linton CG, Smith JM. Health Evaluation of Experimental Laboratory Mice. Curr Protoc Mouse Biol 2012;2:145–65 doi 10.1002/9780470942390.mo110217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008;14(12):1351–6 doi 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jokinen E, Koivunen JP. MEK and PI3K inhibition in solid tumors: rationale and evidence to date. Ther Adv Med Oncol 2015;7(3):170–80 doi 10.1177/1758834015571111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qu GP, Shi M, Wang D, Wu JH, Wang P, Gong ML, et al. Dual targeting of MEK and PI3K effectively controls the proliferation of human EGFR-TKI resistant non-small cell lung carcinoma cell lines with different genetic backgrounds. BMC Pulm Med 2021;21(1):208 doi 10.1186/s12890-021-01571-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Antonarakis ES, Piulats JM, Gross-Goupil M, Goh J, Ojamaa K, Hoimes CJ, et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J Clin Oncol 2020;38(5):395–405 doi 10.1200/JCO.19.01638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geeraerts X, Fernandez-Garcia J, Hartmann FJ, de Goede KE, Martens L, Elkrim Y, et al. Macrophages are metabolically heterogeneous within the tumor microenvironment. Cell Rep 2021;37(13):110171 doi 10.1016/j.celrep.2021.110171. [DOI] [PubMed] [Google Scholar]
- 33.Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The Metabolic Signature of Macrophage Responses. Front Immunol 2019;10:1462 doi 10.3389/fimmu.2019.01462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Bono JS, De Giorgi U, Rodrigues DN, Massard C, Bracarda S, Font A, et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin Cancer Res 2019;25(3):928–36 doi 10.1158/1078-0432.CCR-18-0981. [DOI] [PubMed] [Google Scholar]
- 35.Ramanathan RK, Von Hoff DD, Eskens F, Blumenschein G Jr., Richards D, Genvresse I, et al. Phase Ib Trial of the PI3K Inhibitor Copanlisib Combined with the Allosteric MEK Inhibitor Refametinib in Patients with Advanced Cancer. Target Oncol 2020;15(2):163–74 doi 10.1007/s11523-020-00714-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res 2015;21(4):730–8 doi 10.1158/1078-0432.CCR-14-1814. [DOI] [PubMed] [Google Scholar]
- 37.Bok R, Lee J, Sriram R, Keshari K, Sukumar S, Daneshmandi S, et al. The Role of Lactate Metabolism in Prostate Cancer Progression and Metastases Revealed by Dual-Agent Hyperpolarized (13)C MRSI. Cancers (Basel) 2019;11(2) doi 10.3390/cancers11020257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnston K, Pachnis P, Tasdogan A, Faubert B, Zacharias LG, Vu HS, et al. Isotope tracing reveals glycolysis and oxidative metabolism in childhood tumors of multiple histologies. Med 2021;2(4):395–410 doi 10.1016/j.medj.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lemberg KM, Gori SS, Tsukamoto T, Rais R, Slusher BS. Clinical development of metabolic inhibitors for oncology. J Clin Invest 2022;132(1) doi 10.1172/JCI148550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim D, Wang J, Willingham SB, Martin R, Wernig G, Weissman IL. Anti-CD47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia 2012;26(12):2538–45 doi 10.1038/leu.2012.141. [DOI] [PubMed] [Google Scholar]
- 41.Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol 2018;19(1):76–84 doi 10.1038/s41590-017-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019;572(7769):392–6 doi 10.1038/s41586-019-1456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Payen VL, Mina E, Van Hee VF, Porporato PE, Sonveaux P. Monocarboxylate transporters in cancer. Mol Metab 2020;33:48–66 doi 10.1016/j.molmet.2019.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wall JA, Klempner SJ, Arend RC. The anti-DKK1 antibody DKN-01 as an immunomodulatory combination partner for the treatment of cancer. Expert Opin Investig Drugs 2020;29(7):639–44 doi 10.1080/13543784.2020.1769065. [DOI] [PubMed] [Google Scholar]
- 45.Funck-Brentano T, Nilsson KH, Brommage R, Henning P, Lerner UH, Koskela A, et al. Porcupine inhibitors impair trabecular and cortical bone mass and strength in mice. J Endocrinol 2018;238(1):13–23 doi 10.1530/JOE-18-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wallace K, Landsteiner A, Bunner S, Engel-Nitz N, Luckenbaugh A. Epidemiology and mortality of metastatic castration-resistant prostate cancer (mCRPC) in a managed care population in the United States. Journal of Clinical Oncology 2020;38(15_suppl):e13592–e doi 10.1200/JCO.2020.38.15_suppl.e13592. [DOI] [Google Scholar]
- 47.So A, Chin J, Fleshner N, Saad F. Management of skeletal-related events in patients with advanced prostate cancer and bone metastases: Incorporating new agents into clinical practice. Can Urol Assoc J 2012;6(6):465–70 doi 10.5489/cuaj.12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grainger S, Willert K. Mechanisms of Wnt signaling and control. Wiley Interdiscip Rev Syst Biol Med 2018:e1422 doi 10.1002/wsbm.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinzone JJ, Hall BM, Thudi NK, Vonau M, Qiang YW, Rosol TJ, et al. The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood 2009;113(3):517–25 doi 10.1182/blood-2008-03-145169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.