

Abstract
Purpose of Review
Parkinson's disease (PD) is a common neurodegenerative disease, which can cause progressive deterioration of motor function causing muscle stiffness, tremor, and bradykinesia. In this review, we hope to describe approaches that can improve the life of PD patients through modifications of energy metabolism.
Recent Findings
The main pathological features of PD are the progressive loss of nigrostriatal dopaminergic neurons and the production of Lewy bodies. Abnormal aggregation of α‐synuclein (α‐Syn) leading to the formation of Lewy bodies is closely associated with neuronal dysfunction and degeneration. The main causes of PD are said to be mitochondrial damage, oxidative stress, inflammation, and abnormal protein aggregation. Presence of abnormal energy metabolism is another cause of PD. Many studies have found significant differences between neurodegenerative diseases and metabolic decompensation, which has become a biological hallmark of neurodegenerative diseases.
Summary
In this review, we highlight the relationship between abnormal energy metabolism (Glucose metabolism, lipid metabolism, and amino acid metabolism) and PD. Improvement of key molecules in glucose metabolism, fat metabolism, and amino acid metabolism (e.g., glucose‐6‐phosphate dehydrogenase, triglycerides, and levodopa) might be potentially beneficial in PD. Some of these metabolic indicators may serve well during the diagnosis of PD. In addition, modulation of these metabolic pathways may be a potential target for the treatment and prevention of PD.
Keywords: amino acid metabolism, glucose metabolism, lipid metabolism, Parkinson's disease
Improvement of glucose metabolism, lipid metabolism, and amino acid metabolism is potentially useful for the treatment of Parkinson’s disease.
1. INTRODUCTION
Parkinson's disease (PD), also known as paralysis agitans, is a common neurological disorder of the middle‐aged and elderly people, most of whom present with symptoms of PD after the age of 60. 1 It is currently believed that the pathogenesis of PD is mainly due to the loss of dopaminergic neurons from the substantia nigra (SN) of the midbrain, leading to the decreased activity of the dopamine transmitter system in the nigrostriatal area. 2 In addition, Lewy bodies formed by abnormal aggregation of α‐synuclein (α‐Syn) are closely associated with the viability of dopaminergic neurons. At present, the detailed molecular mechanism of PD remains unmapped.
The main causes of PD are aging, environment factors, and genetic factors. These factors can induce apoptosis, 3 autophagy dysfunction, 4 mitochondrial dysfunction, 5 and neuroinflammation, 6 which ultimately causes neuronal death. Irrespective of the etiology of PD, disturbances in intracellular energy metabolism are observed during the development of PD. 7 , 8
It is well known that the main source of energy for any cell includes sugars, fats, and proteins. Previous studies have suggested that obesity can increase the risk of PD. 9 , 10 Most people with PD have been found to weigh less than normal and healthy individuals. 11 Obviously, metabolic abnormalities of these factors (sugars, fats, and proteins) have been known to increase levels of cellular reactive oxygen species (ROS). The accumulation of ROS is said to be one of the main causes for induction of neurological diseases. Therefore, abnormalities in energy metabolism can induce changes in various indicators that can thereby behave as a molecular marker for PD. 8 A previous study had reported that the incidence rate of PD was 1.23 times higher in individuals with metabolic syndrome when compared with individuals with no presentation of metabolic syndrome. 12
The brain consumes around 25% of the body's glucose. 13 Since the brain has no stored energy, glucose regularly crosses the blood–brain barrier to provide energy and aids in the synaptic transmission to neurons through glycolysis or mitochondrial oxidative phosphorylation. Evidence from postmortem tissues of PD patients indicates mitochondrial dysfunctions, compromised electron transport, 14 and damaged tricarboxylic acid cycle (TCA). 15 Aging is a major risk factor for developing PD. It has been shown that proteasomal and autophagic degradation of neurons is common during aging. 16 , 17 , 18 Previous studies have reported that aging causes more vulnerability of the dopaminergic neurons when compared to the other brain cell. 19 With aging, the decreased cellular metabolic activity triggers the accumulation of ROS, which leads to abnormal protein aggregation and finally results in damage to organelles. 20 Eventually, it leads to the dysfunction of ubiquitin‐proteasome pathway and autophagy. Therefore, we suggest that it is the disruption of energy metabolism due to aging that promotes the accumulation of ROS, which inhibits autophagy and promotes apoptosis. Ultimately, it leads to neuronal loss and induces PD. Metabolic abnormalities accelerate brain aging.
High‐fat or/and high‐sugar diets can stimulate brain aging in mice and rats by triggering autophagy impairment, 21 disturbance of Ca2+ homeostasis, 22 and oxidative damage. 23 Athauda and Foltynie had shown that insulin signaling normally regulates a variety of pathway in the brain, but it was found to be disrupted in individuals with PD. 24 Metabolomics profiling of lipids in PD patients revealed that sebum may be identified as a potential biomarker for PD. 25 A recent meta‐analysis indicates that the lipid serum triacylglycerols (TAGs) had a correlation with PD, and the levels of TAG in PD patients are significantly lower when compared to healthy individuals. 26 Except for sugar and fat, amino acid metabolism also has a very important role in PD. Many amino acids, such as glutamate, γ‐aminobutyric acid (GABA), and glycine, are known to act as neurotransmitters used to regulate neuronal activity in a variety of neurological diseases. This is because the abnormal metabolism of these amino acids leads to neurotoxicity and neuronal death. In this review, we focus on the potential role of energy metabolism in the treatment of PD.
2. PARKINSON'S DISEASE AND GLUCOSE METABOLISM
Glucose is the main source of energy for the brain, and it is the main consumer of blood sugar in the resting state. Anandhan et al. have indicated that there is a relationship between alterations in glucose metabolism, oxidative stress, autophagy, and apoptosis caused by PD‐related risk factors. 27 A recent study has reported that patients with diabetes mellitus have a higher risk of developing PD compared with healthy controls. 28 This may be due to the disruption of glucose metabolism during diabetes, which is associated with dopamine dysfunction, especially in PD. For example, the level of dopamine is lower in a rat model of diabetes mellitus when compared to normal rat models. 29 Low dopamine transporter binding in striatal and high cerebrospinal fluid α‐Syn (CSF) levels are found in patients with diabetes mellitus rather than in patients with PD. 28 All these studies have illustrated that disorders in glucose metabolism of the body are significantly related to PD.
2.1. Parkinson's disease and glucose transporters
Glucose crosses the blood–brain barrier to enter the brain tissue; therefore, glucose transporters (GLUTs) play a very important role in brain energy metabolism. There are many different types of GLUTs, out of which, GLUT1, plays a key role in the functioning of the brain. 30 GLUT3 is mainly expressed in neurons, while GLUT1 is mainly expressed in astrocytes. 31 Puchades et al. had reported that the localization and densities of GLUT1 remain unaffected in a mouse model of PD, 32 while Sarkar et al. had reported that GLUT1 is decreased in the striatum of a PD model of mice which was induced with (1‐Methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine) MPTP. 33 In addition, Burks et al. also had demonstrated a significant decrease of GLUT1 in the immunoreactive cells of the striatum after MPTP administration. 34 Upregulating the expression of GLUT3 can inhibit the neurotoxicity induced in dopaminergic N27 cells which was treated with (1‐methyl‐4‐phenylpyridinium) MPP+ during in vitro research. 35 Administration of STF‐31 (GLUT inhibitors) in dopaminergic cells can decrease paraquat toxicity which is used to induce PD models. 36 Currently, there are limited reports on the potential use of GLUTs in the treatment and/or prevention of PD. The precise function of GLUTs in relation to PD is still unmapped. Recent studies have reported that antidiabetic medicines like exenatide may have a potential role in the treatment of PD. 37 , 38 The above‐mentioned findings have indicated that glucose transport and metabolism have a notable relation with PD.
2.2. Parkinson's disease and glycolysis
Disturbances in energy metabolism and reduced adenosine triphosphate (ATP) levels are common in PD. 39 By and large, cortical glucose consumption is low during the early stages of PD. 40 , 41 A previous study has reported that brain aerobic glycolysis is necessary for synapse formation and growth; 42 besides, enhanced glycolysis can alleviate PD. 43 Although aerobic glycolysis is less efficient, it produces ATP faster than oxidative phosphorylation. During acute energy demand, glycolysis will temporarily exceed oxidative phosphorylation in neurons. 44 Previous studies have reported that phosphoglycerate kinase 1 (PGK‐1) is a key metabolic enzyme for glycolysis, and deficiency of PGK‐1 is associated with PD. 45 , 46 Cai et al. had reported that terazosin can increase, ATP levels of brain and alleviate neuronal loss by enhancing the activity of PGK‐1. It can also increase dopamine levels and partially restore motor function in PD models of rats, mice, and flies. 43 It has also been found that individuals who were administered terazosin or any of its related medicines have slower disease progression, fewer PD‐related complications, and less frequent PD diagnoses. 47 Yang et al. proved that upregulation of leptin induced by NaHS can promote glycolysis and inhibit the loss of dopamine neurons in PD model of rats. 48 Glucose can be catalyzed by hexokinase and transformed into glucose‐6‐phosphate (G6P). According to many in vivo and in vitro studies over the past decade, overexpression of hexokinase 2 (HK2) is found to alleviate the symptoms of PD by promoting glycolysis. 49 , 50 Meclizine activates the glycolytic enzyme phosphofructokinase (PFK), thus elevating glycolysis to resist PD. 51 Doxazosin and alfuzosin can increase tyrosine hydroxylase level in a PD model of mice by activating glycolysis. 43 Discontinuous energy metabolism and reduced ATP levels can increase the risk of PD. 43 Promoting glycolysis may slow the progression of PD. Therefore, all these findings illustrate that glycolysis may play a key role in nervous system diseases (Figure 1).
FIGURE 1.
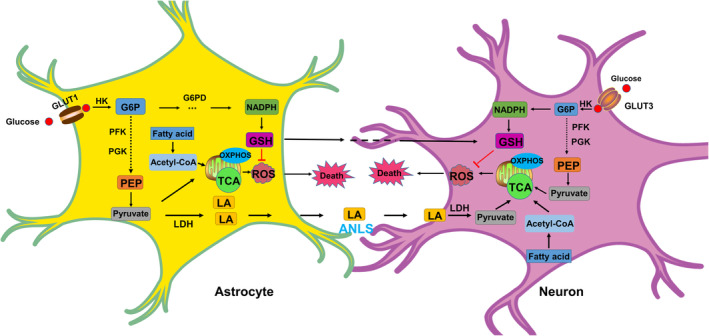
Overview of the glucose metabolism and fatty acids metabolism in neuronal and astrocytic compartments. Neurons and astrocytes can take up glucose respectively through GLUT3 and GLUT1. Glucose is phosphorylated by HK to generate G6P, which is subsequently routed in glycolysis and PPP. The end product of glycolysis is pyruvate. Acetyl‐CoA, derived from pyruvate or fatty acid oxidation, is channeled into the TCA cycle coupled with OXPHOS and ATP synthesis. Impaired mitochondrial energy metabolism leads to ROS accumulation which results in cell death. ATP generation is dependent on OXPHOS, while glucose metabolism is mainly directed toward the PPP to generate NADPH. NADPH is crucial for the GSH, which can reduce the accumulation of ROS. GSH can be shuttled to neurons to maintain redox homeostasis. LA enters the neuron by ANLS, and it can be converted to pyruvate by LDH, which provide energy for neurons. ANLS, astrocyte‐neuron‐lactate shuttle; G6P, glucose‐6‐phosphate; G6PD, glucose‐6‐phosphate dehydrogenase; GLUT1, glucose transporter 1; GLUT3, glucose transporter 3; GSH, glutathione; HK, hexokinase; LA, lactic acid; LDH, lactate dehydrogenase; OXPHOS, oxidative phosphorylation; PEP, phosphoenolpyruvate; PFK, phosphofructokinase; PGK, phosphoglycerate kinase; PPP, pentose phosphate pathway; ROS, reactive oxygen species; TCA, tricarboxylic acid cycle.
2.3. Parkinson's disease and oxidative phosphorylation (OXPHOS)
The main site of oxidative phosphorylation (OXPHOS) is in the mitochondria of eukaryotic cells. ATP production is mainly dependent on OXPHOS pathway (consisting of five complexes I, II, III, IV, and V). Many genes such as Parkin, Pink1, DJ‐1, SNCA, and LRRK2 have been proved to be involved in PD. Mutation or deletion of these genes will lead to decrease in complex I activity and mitochondrial homeostasis, which results in cell death. 52 Previous studies have demonstrated that mitochondrial dysfunction is an important cause of PD. 53 , 54 The susceptibility of DA neurons to mitochondrial dysfunction can be attributed to their high metabolic demand such as neurotransmitter release of high and long‐branched axon. 55 , 56 OXPHOS is the primary approach by which dopaminergic neurons obtain energy. This causes a sustained stimulation of mitochondrial OXPHOS, which increases oxidative damage to mitochondria. 57 Current environmental toxicants, such as 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine and Rotenone, induces PD by damaging the mitochondrial electron transport chain (ETC), resulting in impaired oxidative phosphorylation. 58 , 59 It is found that mtDNA mutations in SN neurons of patients with PD and aged humans are accompanied by a deficiency in OXPHOS. This results in the accumulation of ROS, which ultimately leads to mitochondrial dysfunction causing PD. 60 , 61 It is obvious that the accumulation of ROS induces neuronal apoptosis and autophagy inhibition in PD. 62 The mitochondrial permeability transition (MPT) has a critical role in PD. 63 Previous studies have reported that when MPT occurs, the mitochondrial membrane potential collapses, leading to disrupted oxidative phosphorylation and cell death. 64 , 65 Visch et al. have shown that influx of Ca2+ in mitochondria can promote OXPHOS and ATP production by regulating mitochondrial dehydrogenase enzymes. 66 Imbalance of Ca2+ influx in the mitochondria is one of the causes of PD. 67 Different types of cells respond to stress by enhancing oxidative phosphorylation such as inflammation, starvation, 68 , 69 and ionic imbalance. 70 This generates large amounts of ROS. Therefore, we can suggest that OXPHOS dysfunction‐induced impairment of energy metabolism is one of the major reasons for PD by influencing MPT and Ca2+ signaling, which eventually leads to neuronal apoptosis or autophagy inhibition.
2.4. Parkinson's disease and gluconeogenesis
Gluconeogenesis is the conversion of a variety of non‐sugar substances into glucose or glycogen. The major sites of gluconeogenesis are the liver and kidneys. Mazzio et al. proved that intermediates of gluconeogenesis, such as pyruvate, malate, and phosphoenolpyruvate, have neuroprotective effects against MPP+‐induced neurotoxicity. These intermediates promote anaerobic substrate‐level phosphorylation, thereby promoting glycolysis. 71 In addition, Kim et al. reported that MPTP triggered the expression of glycolysis and gluconeogenesis‐related proteins in mice. 72 At present, there are few reports on the relationship between gluconeogenesis and PD. According to the above data, we suggest that the intermediate products of gluconeogenesis have a regulatory role in PD. This regulatory role is achieved by triggering glycolysis which in turn triggers oxidative phosphorylation.
2.5. Parkinson's disease and pentose phosphate pathway
The pentose phosphate pathway (PPP) is one of the mechanisms for oxidative breakdown of glucose. Previous studies have reported that the level of glucose‐6‐phosphate dehydrogenase (G6PD), a key enzyme of the PPP, is low during the early stages of PD. 73 PPP is the main source of nicotinamide adenine dinucleotide phosphate (NADPH) in cells when compared to normal energy supply. 74 Mejías et al. reported that overexpression of G6PD in DA neurons can alleviate the neurotoxicity induced by MPTP in mice. 75 Therefore, the most important function of PPP in neurons is to regulate redox homeostasis by consuming glucose. 76 This indicates that elevated NADPH levels resist PD by reducing the accumulation of ROS. However, Tu et al. reported that the expression of G6PD is increased in all the four different in vivo PD models. 77 In addition, Lei et al. demonstrated that overexpression of G6PD using paraquat promotes toxicity, while inhibition of G6PD using 6‐aminonicotinamide alleviates mitochondrial functional impairment induced by paraquat in DA neurons. 78 These findings suggest that excessive NADPH aggravates the neurotoxicity effect of paraquat, lipopolysaccharide, and MPTP in the mitochondria. It is the presence of NADPH during the oxidative cycle of these toxicants that promotes mitochondrial death. Therefore, the relationship between G6PD and PD is still controversial. 79 , 80 From the above‐mentioned evidences, four major reasons can be found accountable for this phenomenon. (1) Drug‐induced PD and idiopathic PD may have a difference in pathogenic mechanisms. (2) The role of G6PD may be different during different stages of PD such as the early stage of PD and the late stage of PD. (3) Influenced by different factors such as heredity, environment, and aging, the activity of G6PD during PD may be different. (4) G6PD can influence the neurons or/and astrocytes during PD because astrocytes provide both energy and nutritional support for neurons. Ohno et al. suggested that the PPP activity in astrocytes is five to seven times higher than neurons. 81 In addition, a previous study has reported that the maintenance of neuronal redox homeostasis is largely dependent on the supply of reduced glutathione (GSH) by astrocytes because GSH or its metabolites can be shuttled to neurons. 82 Therefore, all the above evidences indicate that the PPP is closely associated with PD. This association is made possible by the involvement of PPP in neuronal or/and astrocyte metabolism 75 , 77 , 83 (Figure 1).
3. PARKINSON'S DISEASE AND LIPID METABOLISM
Some of the most common lipids that are consumed by humans include triacylglycerols (TAGs), cholesterols, and phospholipids. 84 Lipids in the brain play an important component role in the structural functioning and physiological functioning of neurons. It is essential for the development and maintenance of the central nervous system (CNS). There has been an increasing number of evidences which has reported that the lipid metabolism is highly involved in the pathological progression of neurodegenerative diseases, such as AD 85 and PD. 86 Outeiro et al. proved that α‐Syn aggregation induces abnormal accumulation of lipids. 87 Mitochondrial dysfunction is an important reason for the onset of PD. The accumulation of intracellular lipids induces mitochondrial dysfunction and thus reduces the number of mitochondria, which further promotes lipid accumulation. 88 Lipidomics is an emerging field that can provide new insights and new answers to improve early diagnosis and to track disease progression. 89 It also plays an important role in the study of lipids in energy conversion and biofilm structure. 86 Recently, a growing body of research suggests that disorders of lipid metabolism are strongly associated with PD. 90 , 91 Lipid metabolism is involved in the formation of a variety of biofilms in the cell.
3.1. Parkinson's disease and triacylglycerols
TAG is an ester, derived from three fatty acids and glycerol. Many studies have shown that the level of TAG in the blood of patients with PD is low when compared to healthy individuals. 92 , 93 High levels of TAG in the blood have been proven to have protective effects during PD. 94 , 95 Mice overexpressing α‐SynA53T cause decreased levels of TAG in PD mice model. 96 Recently, more and more reports have highlighted that the level of intracellular TAG is significantly low in both PD rat model and patients with PD. 93 , 97 This association between TAG and PD risk could be linked to the level of DA. Rada et al. demonstrated that repeated intake of sugar increases the extracellular DA levels. 98 High consumption of sugar can increase serum TAG. 99
Faning et al. reported that TAG has a protective role against α‐Syn‐cytotoxicity, which is by inhibiting the accumulation of oleic acid and diglyceride (DG). 100 Accumulation of diglyceride in the endoplasmic reticulum results in α‐Syn trafficking defects. High levels of oleic acid promote α‐Syn membrane binding, which enhances membrane‐associated toxicity. 100 In other words, inhibition of TAG formation makes neurons more vulnerable to α‐Syn toxicity. A previous study had reported that mutation of the human gene ATP13A2 causes Parkinsonism with dementia. 101 Marcos et al. have proven that overexpression of ATP13A2 in SH‐SY5Y cells reduces the levels of TAG. 102 TAG is necessary for the synthesis of new membranes; therefore, it can be observed that probable overexpression of ATP13A2 disrupts the homeostasis of TAG, which causes PD.
Polyunsaturated fatty acids (PUFA) are an important component for the synaptic and mitochondrial membrane formations, and it is often susceptible to damage by reactive oxygen species (ROS). Previous studies have suggested that PUFA can promote the binding of α‐Syn and mitochondrial membrane, as well as promote the pathological aggregation of α‐Syn. 103 , 104 , 105 Recent studies have reported that administration of omega‐3 PUFA is effective in alleviating the effects of PD. 106 Interestingly, the levels of omega‐6 PUFA are higher in patients with PD compared with healthy control. 107 Meng et al. found that administration of omega‐3 PUFA to mice model of PD successfully inhibited the neurotoxicity caused by MPP+, and omega‐6 PUFA is upregulated by the induction of MPP+. 108 However, the specific molecular mechanism remains unclear. Campos et al. have shown that the esterification of arachidonic acid (a kind of omega‐6 PUFA) into TAG may cause a protective effect during Fe‐induced dopamine neuron injury. 109 Therefore, with the above‐mentioned evidences, it can be suggested that excess PUFA forms TAG, which are stored in lipid droplets through esterification and prevents ROS damage to neurons caused by fatty acid β‐oxidation. This may explain the low levels of TAG in patients with PD. Excess PUFA generates ROS, which disrupts the membrane system, especially the cell membrane and mitochondria membrane. Mutation of α‐syn leads to an increase in TAG hydrolysis, resulting in a low level of TAG in cells. 110 , 111 This signifies an association between abnormal TAG metabolism and α‐syn aggregation. (Figure 2).
FIGURE 2.
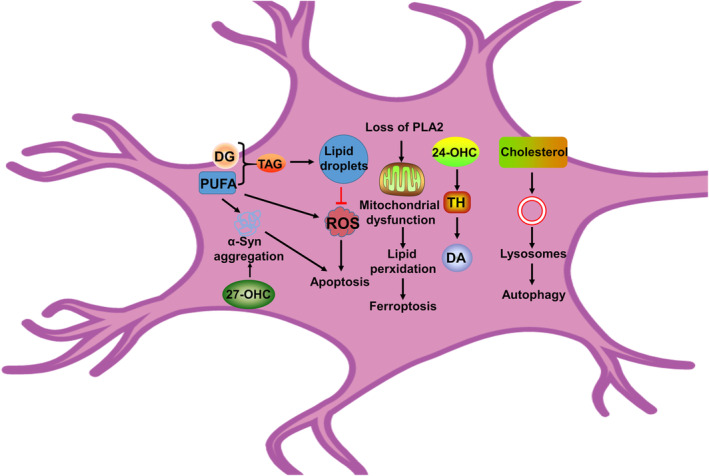
Overview of the lipid metabolism dysregulations in PD brain. Loss of PLA2 gene function results in lipid peroxidation and impaired mitochondrial function, which is associated with ferroptosis. 24‐OHC can activate tyrosine hydroxylase and promote dopamine synthesis. 27‐OHC can increase the level of α‐Syn, which induces apoptosis and eventually leads to neuronal death. The formation of lipid droplets can decrease the levels of ROS, which can inhibit apoptosis. Excess PUFA can increase the levels of α‐syn and ROS, which leads to apoptosis. PUFA forms TAG, which can be stored in lipid droplets. Inhibition of TAG formation makes neurons more vulnerable to α‐Syn toxicity. Disruption of cholesterol metabolism causes lysosomal dysfunction, which leads to autophagy dysfunction. PD, Parkinson's disease; PLA2, phospholipase A2; PUFA, polyunsaturated fatty acids; ROS, reactive oxygen species; TAG, triacylglycerol; α‐Syn: α‐synuclein.
3.2. Parkinson's disease and cholesterols
Cholesterol plays a very important role in maintaining the integrity and fluidity of the cell membrane. It is mainly synthesized in the astrocytes through endoplasmic reticulum. 112 It has a crucial role in regulating variety of physiological functions such as synthesis of various steroid hormones, vitamin D, and maintenance of neuronal development. 113 Cholesterol is mainly distributed in lipid rafts, which are involved in a variety of signal regulation. Although the human brain makes up only about 2% of the body weight, it contains 25% of cholesterol and cholesterol derivatives. 114 , 115 , 116 A previous study has reported that lipid rafts are strongly associated with PD‐related proteins such as α‐syn, LRRK2, Parkin, and DJ‐1. 117 Cholesterol is present in synapses and can interact directly with neurotransmitter receptors. It is necessary for synaptic transmission and formation. Therefore, disturbances in cholesterol metabolism can cause changes in the functioning of brain neurons, leading to various neurological disorders. 118 , 119 An interesting phenomenon is seen when cholesterol synthesis in cells can produce lipoproteins which can be obtained through blood; however, in the brain, cholesterol must be synthesized de novo because lipoproteins cannot cross the blood–brain barrier. 114 , 120 Studies have suggested that changes in the level of cholesterol is closely associated with PD. 121 , 122
Cholesterol can be modified into various derivatives, that is, 24‐OHC (24‐hydroxy cholesterol), 25‐OHC (25‐hydroxy cholesterol), and 27‐OHC (27‐hydroxy cholesterol). 24‐OHC can activate tyrosine hydroxylase and promote dopamine synthesis. 116 27‐OHC can increase the level of α‐syn, which induces apoptosis and eventually leads to neuronal death. 123 Studies have reported that hypercholesterolemia aggravates MPTP‐induced loss of dopaminergic neurons in the SN mice model of PD having motor dysfunction. 124 At present, there are few reports on the role of cholesterol in PD. 124 The specific mechanisms of cholesterol remain controversial. 125 Eriksson et al. have proven that high levels of cholesterol have a dual effect on PD. 126 The increased levels of cholesterol give rise to selective the permeability of lysosomal membranes and thus inhibit cell death while promoting the accumulation of α‐syn. The lysosomal function is strongly associated with PD. 127 Studies have also proven that high levels of cholesterol may be a sign of immature or damaged lysosomes. 121 , 128 García‐Sanz et al. reported that the accumulation of lysosomal cholesterol alters the interaction between α‐syn and lipid rafts, promoting the oligomerization of α‐Syn. Eventually, these α‐Syn, which cannot be degraded by lysosomes, get fibrillated. 112
Mutations in the Glucocerebrosidase (GBA) gene lead to an increased risk of PD. GBA knockout mice increase the levels of cholesterols. 121 In addition, the high levels of cholesterols can promote the accumulation of autophagosomes, which can damage autophagy. 129 Therefore, we suggest that the disruption of cholesterol metabolism causes lysosomal dysfunction, which leads to autophagy dysfunction. Eventually, it leads to neuronal death.
3.3. Parkinson's disease and phospholipids
Phospholipids are the main components of intracellular biofilms. It can be divided into two categories: glycerophospholipids and sphingolipids, which are the precursors for lipid mediators involved in signal transduction. 130 Recent studies have reported that the accumulation of lipid peroxides can lead to membrane damage and thus induce cell death. Pan et al. reported that heterozygous deletion of synaptojanin1 (a phosphoinositide phosphatase) results in PD‐like symptoms in mice. It is caused by upregulating of phosphatidylinositol‐bis‐4,5‐phosphate (PI(4,5)P2). This may be because of the damage (PI(4,5)P2) causes the synaptic vesicle endocytosis, which results in the nerve terminals to selectively eliminate in the midbrain neurons. 131 , 132
Previous studies have identified that calcium‐independent phospholipase A2 (PLA2) is the causative gene for PD. 133 , 134 , 135 Mori et al. mentioned that PLA2 is necessary for the maintenance and survival of dopaminergic neurons and for α‐Syn stability. 136 In a mice model of PD, overexpression of the mutant A53T α‐Syn gene causes reduction in expression of PLA2, which leads to the accumulation of oxidized phospholipids and eventually ferroptosis. 137 Loss of PLA2 gene function leads to lipid peroxidation and impaired mitochondrial function. 138 Sánchez et al. suggested that downregulation of the gene PLA2G6 in the nervous system of zebrafish will result in the loss of dopaminergic neurons and parkinsonism. 139 Therefore, lipid metabolism is one of the most important factors in maintaining normal neuronal function. Phospholipid peroxidation produced by disrupted phospholipid metabolism makes dopaminergic neurons susceptible to ferroptosis. 137 Studies have revealed that ferroptosis is involved in PD. Lipid peroxide accumulation resulting in cell death is an important feature of ferroptosis. Hence, it is worth mentioning that ferroptosis induced by disorders in the phospholipid metabolism may be one of the causes of PD (Figure 2).
4. PARKINSON'S DISEASE AND AMINO ACIDS
The pathophysiology of PD is deeply associated with amino acid metabolism. Glutamate metabolism plays a significant role in the progress of PD. A metabolomics study has shown that alanine and phenylalanine levels in the cerebrospinal fluid are decreased in PD patients compared with normal subjects. 140 Gliomas are known to release large amounts of glutamate through the cystine/glutamate antiporter system Xc‐. 141 Glutamine is converted to glutamate and is taken into presynaptic terminals of glutamatergic neurons by excitatory amino acid transporters (EAATs). Increased extracellular glutamate concentration induces abnormal synaptic signaling leading to excitotoxicity and death of neurons. 142
As a dopamine precursor, levodopa is used to increase dopaminergic neurotransmission in patients with PD. However, long‐term usage of levodopa leads to involuntary movements such as levodopa‐induced dyskinesia (LID) and overactivity of glutamatergic cortico‐striatal projections. 143 Glutamate‐induced oxidative toxicity is closely associated with ferroptosis. 144 , 145 Many studies have reported that ferroptosis is involved in PD. 146 Safinamides target the glutamatergic system selectively and reversibly by inhibiting monoamine oxidase‐B (MAO‐B). This restores the striatal dopaminergic tone and reduces the subthalamic/nigral glutamatergic hyperactivity through use‐dependent sodium channel blockade, which prevents calcium channel opening and results in the inhibition of abnormal glutamate release in a PD model. 147 , 148 Therefore, it can be mentioned that abnormalities in glutamate metabolism produce neurotoxicity and oxidative toxicity, which induce dopaminergic neuronal death.
Cysteine is one of the three amino acids that make up GSH. Studies have reported that the total contents of glutathione (GSH) and GSH/oxidized glutathione (GSSG) ratio is extremely low in the temporal cortex and cerebellum of patients with PD. 149 , 150 , 151 , 152 , 153 This is accompanied by a higher susceptibility to oxidative stress. This finding suggests that the concentration of GSH, GSH/GSSG cycle, and genetic modifications in GSH homeostasis affects the ROS/reactive nitrogen species (RNS) of patients with PD. 151 Low concentration of GSH results in oxidative stress and consequently may induce mitochondrial dysfunction, oxidative damage in DNA, and proteins, and triggers neurodegeneration resulting in PD. 154 , 155 GSH depletion can cause an accumulation of extracellular glutamate in astrocyte cultures. 156 Reduced cysteine uptake leads to a reduction in GSH, which is associated with glutamate neurotoxicity. 157 , 158 In addition, glutathione peroxidase 4 (GPX4) depends on GSH a key enzyme of ferroptosis. 159 All these findings indicate that the activity and concentration of cysteine are involved in PD. This involvement is seen during regulation of the redox homeostasis and bioenergetic metabolism.
It has been reported that the level of γ‐aminobutyric acid (GABA), an inhibitory neurotransmitter, is low in the cerebrospinal fluid of patients with PD. 160 It is also involved in the occurrence and development of PD. 161 GABA can control the activity of DA neurons of the SN. Loss of GABA or its synthesizing enzyme glutamic acid decarboxylase (GAD) has been observed in patients with PD. 162 Kuruvilla et al. reported that combination of GABA, serotonin, and autologous bone marrow cells can inhibit 6‐hydroxydopamine‐induced (6‐OHDA) PD. 163 Glycine is another inhibitory neurotransmitter that acts as a co‐agonist with glutamate at the site of glutamate receptors. 164 Inhibition of glycine transport can promote the function of dopamine axons. 165 Therefore, controlling the metabolism of GABA and glycine may be a potential target for the treatment of PD.
5. DISCUSSION
In this review, we summarize the relationship between different energy metabolisms (glucose metabolism, lipid metabolism, and amino acid metabolism) with PD (Figures 1 and 2). Besides, we also summarize the key finding of metabolic pathway (Table 1). Sugar is the main energy supplier for the human body. Patients with PD prefer to consume more sugar than normal people. 166 This is because an increase in sugar consumption increases the level of dopamine in the brain, which may be a compensatory mechanism in patients with PD. 166 , 167 , 168 Recent studies have proven that diabetes can increase the risk of PD. Many drugs used for targeting glucose metabolism, such as terazosin, meclizine, and alfuzosin, have been identified as possible treatment of options for PD (Table 2). However, the potential role of GLUTs and PPP remains unmapped. Promoting glycolysis gluconeogenesis contributes to alleviating PD. Through recent findings, it has been shown that disruptions in glucose metabolism by regulating the concerned pathways eventually lead to alterations in ATP and ROS levels. Therefore, improving these intracellular pathways associated with apoptosis, autophagy, and ferroptosis by regulating the balance of energy metabolism may be an innovative method to treat PD. Previous studies have proven that astrocyte‐neuron lactate shuttle (ANLS) can consume sugar through anaerobic glycolysis and provide energy metabolic support to neurons. 169 , 170 , 171 Neurons then produce ATP through oxidative phosphorylation. Therefore, there must be a relationship between neurons and astrocytes in the relevant pathways of sugar metabolism or their intermediate products by regulating the accumulation of ROS and thus inducing PD. It is worth to investigating whether astrocytes or/and neurons play any role in dopamine neuron damage. For example, while studying about the relationship between PD and sugar transport, more focus needs to be diverted toward astrocytes. This is because, in the brain, glycogen is mainly stored in the astrocytes. Jia et al. demonstrated that neuronal metabolism of excess lactate leads to more mitochondrial reactive oxygen species (mtROS) production. 172
TABLE 1.
Summary of key findings in each section of metabolic pathway.
| Metabolic pathway | Study | Key finding | Reference |
|---|---|---|---|
| Glucose transporter | MPTP‐induced mouse model of PD | GLUT1 is decreased in the striatum | 31, 34 |
| MPP+‐induced N27 cell model of PD | Upregulating the expression of GLUT3 can inhibit the neurotoxicity induced by MPP+ | 35 | |
| Glycolysis | Human | PGK‐1 mutations contribute to vulnerability to parkinsonism in humans | 45, 46 |
| 6‐OHDA‐induced rat model of PD | Upregulation of leptin induced by NaHS can promote glycolysis and alleviate PD | 48 | |
| Rotenone‐induced SH‐SY5Y cell model of PD; rotenone/MPTP‐induced mouse model of PD | Overexpression of HK2 can alleviate the symptoms of PD by promoting glycolysis | 49, 50 | |
| OXPHOS | Human; Human | mtDNA mutations in SN neurons of patients with PD and aged humans are accompanied by a deficiency in OXPHOS | 60, 61 |
| Gluconeogenesis | MPP+‐induced N2A cell model of PD | Intermediates of gluconeogenesis have a neuroprotective effect against MPP+‐induced neurotoxicity | 71 |
| MPTP‐induced mouse model of PD | Gluconeogenesis‐related proteins are involved in PD | 72 | |
| PPP | MPTP‐induced mouse model of PD; Human; Human | G6PD plays an important role in PD, however, the role of G6PD in PD is still controversial | 75, 79, 80 |
| TAG | Human; Human | The level of TAG in the blood of patients with PD is low, when compared to healthy individuals | 92, 93 |
| Human; Human | High level of TAG in the blood has protective effects in PD | 94, 95 | |
| MPP+‐induced mouse model of PD | Administration of omega‐3 PUFA is effective in alleviating the effects of PD | 108 | |
| Cholesterol | SH‐SY5Y cells | 24‐OHC can activate tyrosine hydroxylase and promote dopamine synthesis | 116 |
| SH‐SY5Y cells | 27‐OHC can increase the level of α‐syn, which induces apoptosis and eventually leads to neuronal death | 123 | |
| MPP+‐induced BE(2)‐M17 cell model of PD | High level of cholesterol has a dual effect on PD | 126 | |
| Phospholipids | iPLA2‐VIA‐deficient Drosophila | PLA2 is necessary for the maintenance and survival of dopaminergic neurons and for α‐Syn stability | 136 |
Abbreviations: 24‐OHC, 24‐hydroxy cholesterol; 27‐OHC, 27‐hydroxy cholesterol; 6‐OHDA, 6‐hydroxydopamine; G6PD, glucose‐6‐phosphate dehydrogenase; GLUT, glucose transporter; HK2, hexokinase 2; MPP+, 1‐methyl‐4‐phenylpyridinium; MPTP, 1‐Methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine; OXPHOS, oxidative phosphorylation; PGK‐1, phosphoglycerate kinase 1; PLA2, calcium‐independent phospholipase A2; PPP, pentose phosphate pathway; PUFA, Polyunsaturated fatty acids; SN, substantia nigra; TAG, triacylglycerols.
TABLE 2.
Drugs targeting glucose metabolism for the potential treatment of Parkinson's disease.
| Drug | Study | Target | Pathway | Reference |
|---|---|---|---|---|
| Terazosin | MPTP‐induced mouse model of PD; 6‐OHDA‐induced rat model of PD; rotenone‐induced fly model of PD | PGK‐1 | Glycolysis | 43 |
| Meclizine | 6‐OHDA‐induced SH‐SY5Y cell model of PD | PFK | Glycolysis | 51 |
| Alfuzosin | MPTP‐induced mouse model of PD; a decreased risk of developing PD in human | PGK‐1 | Glycolysis | 43, 184 |
| Doxazosin | MPTP‐induced mouse model of PD; a decreased risk of developing PD in human | PGK‐1 | Glycolysis | 43, 184 |
| 6‐aminonicotinamide | Paraquat‐induced SK‐N‐SH cell model of PD | G6PD | PPP | 78 |
| Hydrazine sulfate | MPP+‐induced N2A cell model of PD | PEPCK | Gluconeogenesis | 71 |
| STF‐31 | Paraquat‐induced N27 cell model of PD | GLUT | Glucose transport | 36 |
Abbreviations: PEPCK, Phosphoenolpyruvate carboxykinase; MPP+, 1‐methyl‐4‐phenylpyridinium; PGK‐1, phosphoglycerate kinase 1; 6‐OHDA, 6‐hydroxydopamine; PFK, phosphofructokinase; G6PD, glucose‐6‐phosphate dehydrogenase; PPP, pentose phosphate pathway; GLUT, glucose transporter; MPTP, 1‐Methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine; STF‐31, 4‐[[[[4‐(1,1‐dimethylethyl)phenyl]sulfonyl]amino]methyl]‐N‐3‐pyridinyl‐benzamide.
Neurons are highly susceptible to oxidative damage because of their high unsaturated fatty acid content, high oxygen consumption, and relatively weak antioxidant defense mechanisms. The formation of TAG not only protects the neurons from α‐Syn, but also inhibits fatty acid accumulation thereby preventing β‐oxidation. Excessive production of ROS causes disruption of mitochondrial function as well as cell membrane integrity, which induces apoptosis or ferroptosis. Hantikainen et al. demonstrated that high consumption of saturated fats may increase the risk of PD, and a diet low in saturated fat may be beneficial in preventing PD. 173 It is known that cholesterol plays a key role in maintaining the integrity and fluidity of cell membrane. Therefore, we suggest that abnormalities in cholesterol metabolism may affect cellular autophagy and thus neurological function.
It is worthwhile to propose that administration of omega‐3 PUFA helps to alleviate PD. Eating more omega‐3 PUFA than other fats may be beneficial for patients with PD. It is certain that abnormalities in lipid metabolism can increase the chances of PD. 174 , 175 , 176 Food therapy has a good chance of improving PD. A variety of amino acid neurotransmitters, such as glutamate, GABA, and glycine, is said to have its effect on PD. Neurons use a lot of energy to release neurotransmitters; however, excessive excitation of neurons can cause neurotoxicity. Therefore, the balance of amino acid neurotransmitter metabolism is essential in order to maintain a perfect neuronal functioning. Magistretti et al. reported that glutamate can stimulate astrocytes to utilize glucose and release lactic acid (LA) through glycolysis. When energy in the cell is insufficient, the cell produces energy by breaking down amino acids. We can conclude that abnormality of amino acid metabolism affects PD by regulating neuronal excitability.
In summary, abnormalities in energy metabolism are mainly involved in the pathological process of PD by disrupting mitochondrial dysfunction or damaging membrane integrity in neurons. Inhibiting the accumulation of intracellular ROS is an important approach to alleviate PD. Since the functioning of various enzymes can effectively control intracellular energy metabolism through glucose metabolism and lipid metabolism, inhibitors or activators of related enzymes have potential roles in alleviating PD. This study points out that oxidative stress may be the underlying cause of induction for neuronal death, which may be associated with apoptosis, autophagy, and ferroptosis.
6. CONCLUSION
PD patients exhibit disturbances in glycolysis and oxidative phosphorylation, decreased triglyceride levels, disrupted phospholipid metabolism, loss of GABA neurons, and other changes in energy metabolism. Disturbances in metabolism affect the progression of PD by triggering autophagy, apoptosis, and ferroptosis. Patients with multiple metabolic disorders like diabetes are at high risk of developing PD.
Increasing evidences have reported that the metabolism‐related indicators are associated with the occurrence and progression of PD. For example, disturbances in glucose metabolism in the SN can be a marker for the diagnosis of PD. 177 Xicoy et al. showed that the genetic overlap between the specific lipids in the blood and PD can be identified as a novel diagnostic biomarker in PD. 178 PD is related to the abnormal lipid metabolism according to integrated proteomics and metabolomics analysis which indicates that improving lipid metabolism is a promising method for the treatment of PD. 179 Also, abnormalities in amino acid metabolism have also been widely noted in PD such as glycine, glutamate and GABA. 1 , 180 Heilman et al. reported that the kynurenine metabolites may be a biomarker for PD and/or involved in the pathogenesis of PD. 181 Zhang et al. showed that modulation of glutamine metabolism has a potential therapeutic effect for the treatment of PD. 182 A recent study has shown that upregulation of serine levels is a biochemical signature of dopaminergic neuronal degeneration in patients with PD. 183 Therefore, it can be concluded that metabolism‐related indicators may be used as markers for monitoring PD progression. Drugs related to targeted metabolic pathways may have a potential role in the treatment of PD. Furthermore, since the role of PPP and cholesterol with PD is not well understood, it is worthwhile to investigate the potential role of PPP and cholesterol in the treatment of PD. In the future, we hope to improve the life of PD patients through research on energy metabolism.
AUTHOR CONTRIBUTIONS
The authors declare no competing financial interests. Xiang Li and Yan Chen were responsible for the study concept and design. Hangzhen Li, Fancai Zeng, Cancan Huang, and Qiqi Pu drafted the manuscript. Xiang Li and Elizabeth Rosalind Thomas provided a critical revision of the manuscript for important intellectual content. All authors read and approved the final version.
FUNDING INFORMATION
This study was financially supported by grants from the Science and Technology Strategic Cooperation Project of the Luzhou People's Government and Southwest Medical University (No. 2019LZXNYDJ34) and the undergraduate innovation and entrepreneurship training program (S202110632241). This study was financially supported by Sichuan Science and Technology Program (Grant No. 2022YFS0623 and 2023JDRC0109).
CONFLICT OF INTEREST STATEMENT
The authors declare no competing financial interests.
Li H, Zeng F, Huang C, et al. The potential role of glucose metabolism, lipid metabolism, and amino acid metabolism in the treatment of Parkinson's disease. CNS Neurosci Ther. 2024;30:e14411. doi: 10.1111/cns.14411
Hangzhen Li, Fancai Zeng, and Cancan Huang contributed equally to this work.
Contributor Information
Yan Chen, Email: chenyan0216@swmu.edu.cn.
Xiang Li, Email: lix2009@126.com.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in The National Center for Biotechnology Information at https://www.ncbi.nlm.nih.gov/.
REFERENCES
- 1. Emamzadeh FN, Surguchov A. Parkinson's disease: biomarkers, treatment, and risk factors. Front Neurosci. 2018;12:612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anis E, Xie A, Brundin L, Brundin P. Digesting recent findings: gut alpha‐synuclein, microbiome changes in Parkinson's disease. Trends Endocrinol Metab. 2022;33(2):147‐157. [DOI] [PubMed] [Google Scholar]
- 3. Tatton WG, Chalmers‐Redman R, Brown D, Tatton N. Apoptosis in Parkinson's disease: signals for neuronal degradation. Ann Neurol. 2003;53(Suppl 3):S61‐S70; discussion S70–S72. [DOI] [PubMed] [Google Scholar]
- 4. Janda E, Isidoro C, Carresi C, Mollace V. Defective autophagy in Parkinson's disease: role of oxidative stress. Mol Neurobiol. 2012;46(3):639‐661. [DOI] [PubMed] [Google Scholar]
- 5. Subramaniam SR, Chesselet MF. Mitochondrial dysfunction and oxidative stress in Parkinson's disease. Prog Neurobiol. 2013;106–107:17‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hirsch EC, Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382‐397. [DOI] [PubMed] [Google Scholar]
- 7. Quansah E, Peelaerts W, Langston JW, Simon DK, Colca J, Brundin P. Targeting energy metabolism via the mitochondrial pyruvate carrier as a novel approach to attenuate neurodegeneration. Mol Neurodegener. 2018;13(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu M, Jiao Q, Du X, Bi M, Chen X, Jiang H. Potential crosstalk between Parkinson's disease and energy metabolism. Aging Dis. 2021;12(8):2003‐2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Flores‐Dorantes MT, Diaz‐Lopez YE, Gutierrez‐Aguilar R. Environment and Gene Association with obesity and their impact on neurodegenerative and neurodevelopmental diseases. Front Neurosci. 2020;14:863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nam GE, Kim SM, Han K, et al. Metabolic syndrome and risk of Parkinson disease: a nationwide cohort study. PLoS Med. 2018;15, (8):e1002640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vikdahl M, Carlsson M, Linder J, Forsgren L, Haglin L. Weight gain and increased central obesity in the early phase of Parkinson's disease. Clin Nutr. 2014;33(6):1132‐1139. [DOI] [PubMed] [Google Scholar]
- 12. Roh JH, Lee S, Yoon JH. Metabolic syndrome and Parkinson's disease incidence: a nationwide study using propensity score matching. Metab Syndr Relat Disord. 2021;19(1):1‐7. [DOI] [PubMed] [Google Scholar]
- 13. Rossi S, Zanier ER, Mauri I, Columbo A, Stocchetti N. Brain temperature, body core temperature, and intracranial pressure in acute cerebral damage. J Neurol Neurosurg Psychiatry. 2001;71(4):448‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Camilleri A, Vassallo N. The centrality of mitochondria in the pathogenesis and treatment of Parkinson's disease. CNS Neurosci Ther. 2014;20(7):591‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gibson GE, Kingsbury AE, Xu H, et al. Deficits in a tricarboxylic acid cycle enzyme in brains from patients with Parkinson's disease. Neurochem Int. 2003;43(2):129‐135. [DOI] [PubMed] [Google Scholar]
- 16. Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983‐997. [DOI] [PubMed] [Google Scholar]
- 17. Graham SH, Liu H. Life and death in the trash heap: the ubiquitin proteasome pathway and UCHL1 in brain aging, neurodegenerative disease and cerebral ischemia. Ageing Res Rev. 2017;34:30‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xilouri M, Stefanis L. Chaperone mediated autophagy in aging: starve to prosper. Ageing Res Rev. 2016;32:13‐21. [DOI] [PubMed] [Google Scholar]
- 19. Reeve A, Simcox E, Turnbull D. Ageing and Parkinson's disease: why is advancing age the biggest risk factor? Ageing Res Rev. 2014;14(100):19‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reeg S, Grune T. Protein oxidation in aging: does it play a role in aging progression? Antioxid Redox Signal. 2015;23(3):239‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. da Silva Rosa SC, Martens MD, Field JT, et al. BNIP3L/nix‐induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy. 2021;17(9):2257‐2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salin Raj P, Swapna SUS, Raghu KG. High glucose induced calcium overload via impairment of SERCA/PLN pathway and mitochondrial dysfunction leads to oxidative stress in H9c2 cells and amelioration with ferulic acid. Fundam Clin Pharmacol. 2019;33(4):412‐425. [DOI] [PubMed] [Google Scholar]
- 23. Morris JK, Bomhoff GL, Stanford JA, Geiger PC. Neurodegeneration in an animal model of Parkinson's disease is exacerbated by a high‐fat diet. Am J Physiol Regul Integr Comp Physiol. 2010;299(4):R1082‐R1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Athauda D, Foltynie T. Insulin resistance and Parkinson's disease: a new target for disease modification? Prog Neurobiol. 2016;145–146:98‐120. [DOI] [PubMed] [Google Scholar]
- 25. Sinclair E, Trivedi DK, Sarkar D, et al. Metabolomics of sebum reveals lipid dysregulation in Parkinson's disease. Nat Commun. 2021;12(1):1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu Y, Jin X, Zhao P. Serum lipids and the pathogenesis of Parkinson's disease: a systematic review and meta‐analysis. Int J Clin Pract. 2021;75(4):e13865. [DOI] [PubMed] [Google Scholar]
- 27. Anandhan A, Jacome MS, Lei S, et al. Metabolic dysfunction in Parkinson's disease: bioenergetics, redox homeostasis and central carbon metabolism. Brain Res Bull. 2017;133:12‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pagano G, Polychronis S, Wilson H, et al. Diabetes mellitus and Parkinson disease. Neurology. 2018;90(19):e1654‐e1662. [DOI] [PubMed] [Google Scholar]
- 29. Ashraghi MR, Pagano G, Polychronis S, Niccolini F, Politis M. Parkinson's disease, diabetes and cognitive impairment. Recent Pat Endocr Metab Immune Drug Discov. 2016;10(1):11‐21. [DOI] [PubMed] [Google Scholar]
- 30. Shah K, Desilva S, Abbruscato T. The role of glucose transporters in brain disease: diabetes and Alzheimer's disease. Int J Mol Sci. 2012;13(10):12629‐12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Butterfield DA, Favia M, Spera I, Campanella A, Lanza M, Castegna A. Metabolic features of brain function with relevance to clinical features of Alzheimer and Parkinson diseases. Molecules. 2022;27(3):951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Puchades M, Sogn CJ, Maehlen J, Bergersen LH, Gundersen V. Unaltered lactate and glucose transporter levels in the MPTP mouse model of Parkinson's disease. J Parkinsons Dis. 2013;3(3):371‐385. [DOI] [PubMed] [Google Scholar]
- 33. Sarkar S, Chigurupati S, Raymick J, et al. Neuroprotective effect of the chemical chaperone, trehalose in a chronic MPTP‐induced Parkinson's disease mouse model. Neurotoxicology. 2014;44:250‐262. [DOI] [PubMed] [Google Scholar]
- 34. Burks S, Raymick J, Robinson B, Hanig J, Sarkar S. Neuroprotective effects of acetyl‐l‐carnitine (ALC) in a chronic MPTP‐induced Parkinson's disease mouse model: endothelial and microglial effects. Neurosci Lett. 2019;703:86‐95. [DOI] [PubMed] [Google Scholar]
- 35. Anandhan A, Lei S, Levytskyy R, et al. Glucose metabolism and AMPK signaling regulate dopaminergic cell death induced by gene (alpha‐synuclein)‐environment (Paraquat) interactions. Mol Neurobiol. 2017;54(5):3825‐3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Powers R, Lei S, Anandhan A, et al. Metabolic investigations of the molecular mechanisms associated with Parkinson's disease. Metabolites. 2017;7(2):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Foltynie T, Athauda D. Repurposing anti‐diabetic drugs for the treatment of Parkinson's disease: rationale and clinical experience. Prog Brain Res. 2020;252:493‐523. [DOI] [PubMed] [Google Scholar]
- 38. Cardoso S, Moreira PI. Antidiabetic drugs for Alzheimer's and Parkinson's diseases: repurposing insulin, metformin, and thiazolidinediones. Int Rev Neurobiol. 2020;155:37‐64. [DOI] [PubMed] [Google Scholar]
- 39. Saxena U. Bioenergetics failure in neurodegenerative diseases: back to the future. Expert Opin Ther Targets. 2012;16(4):351‐354. [DOI] [PubMed] [Google Scholar]
- 40. Edison P, Ahmed I, Fan Z, et al. Microglia, amyloid, and glucose metabolism in Parkinson's disease with and without dementia. Neuropsychopharmacology. 2013;38(6):938‐949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Borghammer P, Chakravarty M, Jonsdottir KY, et al. Cortical hypometabolism and hypoperfusion in Parkinson's disease is extensive: probably even at early disease stages. Brain Struct Funct. 2010;214(4):303‐317. [DOI] [PubMed] [Google Scholar]
- 42. Goyal MS, Hawrylycz M, Miller JA, Snyder AZ, Raichle ME. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014;19(1):49‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cai R, Zhang Y, Simmering JE, et al. Enhancing glycolysis attenuates Parkinson's disease progression in models and clinical databases. J Clin Invest. 2019;129(10):4539‐4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yellen G. Fueling thought: management of glycolysis and oxidative phosphorylation in neuronal metabolism. J Cell Biol. 2018;217(7):2235‐2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sakaue S, Kasai T, Mizuta I, et al. Early‐onset parkinsonism in a pedigree with phosphoglycerate kinase deficiency and a heterozygous carrier: do PGK‐1 mutations contribute to vulnerability to parkinsonism? NPJ Parkinsons Dis. 2017;3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sotiriou E, Greene P, Krishna S, Hirano M, DiMauro S. Myopathy and parkinsonism in phosphoglycerate kinase deficiency. Muscle Nerve. 2010;41(5):707‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Simmering JE, Welsh MJ, Schultz J, Narayanan NS. Use of glycolysis‐enhancing drugs and risk of Parkinson's disease. Mov Disord. 2022;37(11):2210‐2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang SQ, Tian Q, Li D, et al. Leptin mediates protection of hydrogen sulfide against 6‐hydroxydopamine‐induced Parkinson's disease: involving enhancement in Warburg effect. Neurochem Int. 2020;135:104692. [DOI] [PubMed] [Google Scholar]
- 49. Gimenez‐Cassina A, Lim F, Cerrato T, Palomo GM, Diaz‐Nido J. Mitochondrial hexokinase II promotes neuronal survival and acts downstream of glycogen synthase kinase‐3. J Biol Chem. 2009;284(5):3001‐3011. [DOI] [PubMed] [Google Scholar]
- 50. Corona JC, Gimenez‐Cassina A, Lim F, Diaz‐Nido J. Hexokinase II gene transfer protects against neurodegeneration in the rotenone and MPTP mouse models of Parkinson's disease. J Neurosci Res. 2010;88(9):1943‐1950. [DOI] [PubMed] [Google Scholar]
- 51. Hong CT, Chau KY, Schapira AH. Meclizine‐induced enhanced glycolysis is neuroprotective in Parkinson disease cell models. Sci Rep. 2016;6:25344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ali MZ, Dholaniya PS. Oxidative phosphorylation mediated pathogenesis of Parkinson's disease and its implication via Akt signaling. Neurochem Int. 2022;157:105344. [DOI] [PubMed] [Google Scholar]
- 53. Merlini E, Coleman MP, Loreto A. Mitochondrial dysfunction as a trigger of programmed axon death. Trends Neurosci. 2022;45(1):53‐63. [DOI] [PubMed] [Google Scholar]
- 54. Manini A, Abati E, Comi GP, Corti S, Ronchi D. Mitochondrial DNA homeostasis impairment and dopaminergic dysfunction: a trembling balance. Ageing Res Rev. 2022;76:101578. [DOI] [PubMed] [Google Scholar]
- 55. Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci. 2017;18(2):101‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bolam JP, Pissadaki EK. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord. 2012;27(12):1478‐1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gonzalez‐Rodriguez P, Zampese E, Stout KA, et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature. 2021;599(7886):650‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang W, Hao W, Meng Z, et al. Molecular regulatory mechanism and toxicology of neurodegenerative processes in MPTP/probenecid‐induced progressive Parkinson's disease mice model revealed by transcriptome. Mol Neurobiol. 2021;58(2):603‐616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Innos J, Hickey MA. Using rotenone to model Parkinson's disease in mice: a review of the role of pharmacokinetics. Chem Res Toxicol. 2021;34(5):1223‐1239. [DOI] [PubMed] [Google Scholar]
- 60. Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38(5):515‐517. [DOI] [PubMed] [Google Scholar]
- 61. Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38(5):518‐520. [DOI] [PubMed] [Google Scholar]
- 62. Ghavami S, Shojaei S, Yeganeh B, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24‐49. [DOI] [PubMed] [Google Scholar]
- 63. Kalani K, Yan SF, Yan SS. Mitochondrial permeability transition pore: a potential drug target for neurodegeneration. Drug Discov Today. 2018;23(12):1983‐1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zamzami N, Susin SA, Marchetti P, et al. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183(4):1533‐1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun. 2003;304(3):463‐470. [DOI] [PubMed] [Google Scholar]
- 66. Quintana DD, Garcia JA, Anantula Y, et al. Amyloid‐beta causes mitochondrial dysfunction via a Ca2+‐driven upregulation of oxidative phosphorylation and superoxide production in cerebrovascular endothelial cells. J Alzheimers Dis. 2020;75(1):119‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Surmeier DJ, Schumacker PT, Guzman JD, Ilijic E, Yang B, Zampese E. Calcium and Parkinson's disease. Biochem Biophys Res Commun. 2017;483(4):1013‐1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem. 2018;293(32):12649‐12652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Brace LE, Vose SC, Stanya K, et al. Increased oxidative phosphorylation in response to acute and chronic DNA damage. NPJ Aging Mech Dis. 2016;2:16022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pilchova I, Klacanova K, Tatarkova Z, Kaplan P, Racay P. The involvement of Mg(2+) in regulation of cellular and mitochondrial functions. Oxidative Med Cell Longev. 2017;2017:6797460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mazzio E, Soliman KF. The role of glycolysis and gluconeogenesis in the cytoprotection of neuroblastoma cells against 1‐methyl 4‐phenylpyridinium ion toxicity. Neurotoxicology. 2003;24(1):137‐147. [DOI] [PubMed] [Google Scholar]
- 72. Kim D, Jeon H, Ryu S, Koo S, Ha KT, Kim S. Proteomic analysis of the effect of Korean red ginseng in the striatum of a Parkinson's disease mouse model. PLoS One. 2016;11(10):e0164906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dunn L, Allen GF, Mamais A, et al. Dysregulation of glucose metabolism is an early event in sporadic Parkinson's disease. Neurobiol Aging. 2014;35(5):1111‐1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Perl A, Hanczko R, Telarico T, Oaks Z, Landas S. Oxidative stress, inflammation and carcinogenesis are controlled through the pentose phosphate pathway by transaldolase. Trends Mol Med. 2011;17(7):395‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mejias R, Villadiego J, Pintado CO, et al. Neuroprotection by transgenic expression of glucose‐6‐phosphate dehydrogenase in dopaminergic nigrostriatal neurons of mice. J Neurosci. 2006;26(17):4500‐4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Herrero‐Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolanos JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C‐Cdh1. Nat Cell Biol. 2009;11(6):747‐752. [DOI] [PubMed] [Google Scholar]
- 77. Tu D, Gao Y, Yang R, Guan T, Hong JS, Gao HM. The pentose phosphate pathway regulates chronic neuroinflammation and dopaminergic neurodegeneration. J Neuroinflammation. 2019;16(1):255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lei S, Zavala‐Flores L, Garcia‐Garcia A, et al. Alterations in energy/redox metabolism induced by mitochondrial and environmental toxins: a specific role for glucose‐6‐phosphate‐dehydrogenase and the pentose phosphate pathway in paraquat toxicity. ACS Chem Biol. 2014;9(9):2032‐2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Abraham S, Soundararajan CC, Vivekanandhan S, Behari M. Erythrocyte antioxidant enzymes in Parkinson's disease. Indian J Med Res. 2005;121(2):111‐115. [PubMed] [Google Scholar]
- 80. Gao L, Mir P, Diaz‐Corrales FJ, et al. Glucose‐6‐phosphate dehydrogenase activity in Parkinson's disease. J Neurol. 2008;255(11):1850‐1851. [DOI] [PubMed] [Google Scholar]
- 81. Takahashi S. Neuroprotective function of high glycolytic activity in astrocytes: common roles in stroke and neurodegenerative diseases. Int J Mol Sci. 2021;22(12):6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dringen R, Kussmaul L, Gutterer JM, Hirrlinger J, Hamprecht B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J Neurochem. 1999;72(6):2523‐2530. [DOI] [PubMed] [Google Scholar]
- 83. Mashima K, Takahashi S, Minami K, et al. Neuroprotective role of Astroglia in Parkinson disease by reducing oxidative stress through dopamine‐induced activation of pentose‐phosphate pathway. ASN Neuro. 2018;10:1759091418775562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ko CW, Qu J, Black DD, Tso P. Regulation of intestinal lipid metabolism: current concepts and relevance to disease. Nat Rev Gastroenterol Hepatol. 2020;17(3):169‐183. [DOI] [PubMed] [Google Scholar]
- 85. Yin F. Lipid metabolism and Alzheimer's disease: clinical evidence, mechanistic link and therapeutic promise. FEBS J. 2023;290(6):1420‐1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Alecu I, Bennett SAL. Dysregulated lipid metabolism and its role in alpha‐synucleinopathy in Parkinson's disease. Front Neurosci. 2019;13:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Outeiro TF, Lindquist S. Yeast cells provide insight into alpha‐synuclein biology and pathobiology. Science. 2003;302(5651):1772‐1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jana BA, Chintamaneni PK, Krishnamurthy PT, Wadhwani A, Mohankumar SK. Cytosolic lipid excess‐induced mitochondrial dysfunction is the cause or effect of high fat diet‐induced skeletal muscle insulin resistance: a molecular insight. Mol Biol Rep. 2019;46(1):957‐963. [DOI] [PubMed] [Google Scholar]
- 89. Emamzadeh FN, Allsop D. Alpha‐synuclein interacts with lipoproteins in plasma. J Mol Neurosci. 2017;63(2):165‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Fanning S, Selkoe D, Dettmer U. Vesicle trafficking and lipid metabolism in synucleinopathy. Acta Neuropathol. 2021;141(4):491‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Estes RE, Lin B, Khera A, Davis MY. Lipid metabolism influence on neurodegenerative disease progression: is the vehicle as important as the cargo? Front Mol Neurosci. 2021;14:788695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Scigliano G, Musicco M, Soliveri P, Piccolo I, Ronchetti G, Girotti F. Reduced risk factors for vascular disorders in Parkinson disease patients: a case‐control study. Stroke. 2006;37(5):1184‐1188. [DOI] [PubMed] [Google Scholar]
- 93. Guo X, Song W, Chen K, et al. The serum lipid profile of Parkinson's disease patients: a study from China. Int J Neurosci. 2015;125(11):838‐844. [DOI] [PubMed] [Google Scholar]
- 94. Saaksjarvi K, Knekt P, Mannisto S, Lyytinen J, Heliovaara M. Prospective study on the components of metabolic syndrome and the incidence of Parkinson's disease. Parkinsonism Relat Disord. 2015;21(10):1148‐1155. [DOI] [PubMed] [Google Scholar]
- 95. Vikdahl M, Backman L, Johansson I, Forsgren L, Haglin L. Cardiovascular risk factors and the risk of Parkinson's disease. Eur J Clin Nutr. 2015;69(6):729‐733. [DOI] [PubMed] [Google Scholar]
- 96. Guerreiro PS, Coelho JE, Sousa‐Lima I, et al. Mutant A53T alpha‐synuclein improves Rotarod performance before motor deficits and affects metabolic pathways. NeuroMolecular Med. 2017;19(1):113‐121. [DOI] [PubMed] [Google Scholar]
- 97. Meng X, Zheng R, Zhang Y, et al. An activated sympathetic nervous system affects white adipocyte differentiation and lipolysis in a rat model of Parkinson's disease. J Neurosci Res. 2015;93(2):350‐360. [DOI] [PubMed] [Google Scholar]
- 98. Rada P, Avena NM, Hoebel BG. Daily bingeing on sugar repeatedly releases dopamine in the accumbens shell. Neuroscience. 2005;134(3):737‐744. [DOI] [PubMed] [Google Scholar]
- 99. Chong MF, Fielding BA, Frayn KN. Metabolic interaction of dietary sugars and plasma lipids with a focus on mechanisms and de novo lipogenesis. Proc Nutr Soc. 2007;66(1):52‐59. [DOI] [PubMed] [Google Scholar]
- 100. Fanning S, Haque A, Imberdis T, et al. Lipidomic analysis of alpha‐synuclein neurotoxicity identifies stearoyl CoA desaturase as a target for Parkinson treatment. Mol Cell. 2019;73(5):1001‐1014 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ramirez A, Heimbach A, Grundemann J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P‐type ATPase. Nat Genet. 2006;38(10):1184‐1191. [DOI] [PubMed] [Google Scholar]
- 102. Marcos AL, Corradi GR, Mazzitelli LR, et al. The Parkinson‐associated human P5B‐ATPase ATP13A2 modifies lipid homeostasis. Biochim Biophys Acta Biomembr. 2019;1861(10):182993. [DOI] [PubMed] [Google Scholar]
- 103. Lucke C, Gantz DL, Klimtchuk E, Hamilton JA. Interactions between fatty acids and alpha‐synuclein. J Lipid Res. 2006;47(8):1714‐1724. [DOI] [PubMed] [Google Scholar]
- 104. Sharon R, Bar‐Joseph I, Frosch MP, Walsh DM, Hamilton JA, Selkoe DJ. The formation of highly soluble oligomers of alpha‐synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron. 2003;37(4):583‐595. [DOI] [PubMed] [Google Scholar]
- 105. Sharon R, Goldberg MS, Bar‐Josef I, Betensky RA, Shen J, Selkoe DJ. Alpha‐synuclein occurs in lipid‐rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid‐binding proteins. Proc Natl Acad Sci U S A. 2001;98(16):9110‐9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bousquet M, Calon F, Cicchetti F. Impact of omega‐3 fatty acids in Parkinson's disease. Ageing Res Rev. 2011;10(4):453‐463. [DOI] [PubMed] [Google Scholar]
- 107. Julien C, Berthiaume L, Hadj‐Tahar A, et al. Postmortem brain fatty acid profile of levodopa‐treated Parkinson disease patients and parkinsonian monkeys. Neurochem Int. 2006;48(5):404‐414. [DOI] [PubMed] [Google Scholar]
- 108. Meng Q, Luchtman DW, El Bahh B, Zidichouski JA, Yang J, Song C. Ethyl‐eicosapentaenoate modulates changes in neurochemistry and brain lipids induced by parkinsonian neurotoxin 1‐methyl‐4‐phenylpyridinium in mouse brain slices. Eur J Pharmacol. 2010;649(1–3):127‐134. [DOI] [PubMed] [Google Scholar]
- 109. Sanchez Campos S, Rodriguez Diez G, Oresti GM, Salvador GA. Dopaminergic neurons respond to iron‐induced oxidative stress by modulating lipid acylation and deacylation cycles. PLoS One. 2015;10(6):e0130726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sanchez Campos S, Alza NP, Salvador GA. Lipid metabolism alterations in the neuronal response to A53T alpha‐synuclein and Fe‐induced injury. Arch Biochem Biophys. 2018;655:43‐54. [DOI] [PubMed] [Google Scholar]
- 111. He Q, Wang M, Petucci C, Gardell SJ, Han X. Rotenone induces reductive stress and triacylglycerol deposition in C2C12 cells. Int J Biochem Cell Biol. 2013;45(12):2749‐2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Garcia‐Sanz P, M F G Aerts J, Moratalla R. The role of cholesterol in alpha‐synuclein and Lewy body pathology in GBA1 Parkinson's disease. Mov Disord. 2021;36(5):1070‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Cerqueira NM, Oliveira EF, Gesto DS, et al. Cholesterol biosynthesis: a mechanistic overview. Biochemistry. 2016;55(39):5483‐5506. [DOI] [PubMed] [Google Scholar]
- 114. Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12(2):105‐112. [DOI] [PubMed] [Google Scholar]
- 115. Korade Z, Kenworthy AK. Lipid rafts, cholesterol, and the brain. Neuropharmacology. 2008;55(8):1265‐1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Pingale TD, Gupta GL. Novel therapeutic approaches for Parkinson's disease by targeting brain cholesterol homeostasis. J Pharm Pharmacol. 2021;73(7):862‐873. [DOI] [PubMed] [Google Scholar]
- 117. Kubo S, Hatano T, Hattori N. Lipid rafts involvement in the pathogenesis of Parkinson's disease. Front Biosci (Landmark Ed). 2015;20(2):263‐279. [DOI] [PubMed] [Google Scholar]
- 118. Turri M, Marchi C, Adorni MP, Calabresi L, Zimetti F. Emerging role of HDL in brain cholesterol metabolism and neurodegenerative disorders. Biochim Biophys Acta Mol Cell Biol Lipids. 2022;1867(5):159123. [DOI] [PubMed] [Google Scholar]
- 119. Nunes VS, da Silva Ferreira G, Quintao ECR. Cholesterol metabolism in aging simultaneously altered in liver and nervous system. Aging (Albany NY). 2022;14(3):1549‐1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Orth M, Bellosta S. Cholesterol: its regulation and role in central nervous system disorders. Cholesterol. 2012;2012:292598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Magalhaes J, Gegg ME, Migdalska‐Richards A, Doherty MK, Whitfield PD, Schapira AH. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum Mol Genet. 2016;25(16):3432‐3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Bosco DA, Fowler DM, Zhang Q, et al. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha‐synuclein fibrilization. Nat Chem Biol. 2006;2(5):249‐253. [DOI] [PubMed] [Google Scholar]
- 123. Rantham Prabhakara JP, Feist G, Thomasson S, Thompson A, Schommer E, Ghribi O. Differential effects of 24‐hydroxycholesterol and 27‐hydroxycholesterol on tyrosine hydroxylase and alpha‐synuclein in human neuroblastoma SH‐SY5Y cells. J Neurochem. 2008;107(6):1722‐1729. [DOI] [PubMed] [Google Scholar]
- 124. Jin U, Park SJ, Park SM. Cholesterol metabolism in the brain and its association with Parkinson's disease. Exp Neurobiol. 2019;28(5):554‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Dai L, Zou L, Meng L, Qiang G, Yan M, Zhang Z. Cholesterol metabolism in neurodegenerative diseases: molecular mechanisms and therapeutic targets. Mol Neurobiol. 2021;58(5):2183‐2201. [DOI] [PubMed] [Google Scholar]
- 126. Eriksson I, Nath S, Bornefall P, Giraldo AM, Ollinger K. Impact of high cholesterol in a Parkinson's disease model: prevention of lysosomal leakage versus stimulation of alpha‐synuclein aggregation. Eur J Cell Biol. 2017;96(2):99‐109. [DOI] [PubMed] [Google Scholar]
- 127. Udayar V, Chen Y, Sidransky E, Jagasia R. Lysosomal dysfunction in neurodegeneration: emerging concepts and methods. Trends Neurosci. 2022;45(3):184‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Takahashi Y, Nada S, Mori S, Soma‐Nagae T, Oneyama C, Okada M. The late endosome/lysosome‐anchored p18‐mTORC1 pathway controls terminal maturation of lysosomes. Biochem Biophys Res Commun. 2012;417(4):1151‐1157. [DOI] [PubMed] [Google Scholar]
- 129. Garcia‐Sanz P, Orgaz L, Fuentes JM, Vicario C, Moratalla R. Cholesterol and multilamellar bodies: lysosomal dysfunction in GBA‐Parkinson disease. Autophagy. 2018;14(4):717‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Farooqui AA, Horrocks LA, Farooqui T. Interactions between neural membrane glycerophospholipid and sphingolipid mediators: a recipe for neural cell survival or suicide. J Neurosci Res. 2007;85(9):1834‐1850. [DOI] [PubMed] [Google Scholar]
- 131. Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164‐1178. [DOI] [PubMed] [Google Scholar]
- 132. Pan PY, Sheehan P, Wang Q, et al. Synj1 haploinsufficiency causes dopamine neuron vulnerability and alpha‐synuclein accumulation in mice. Hum Mol Genet. 2020;29(14):2300‐2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Paisan‐Ruiz C, Bhatia KP, Li A, et al. Characterization of PLA2G6 as a locus for dystonia‐parkinsonism. Ann Neurol. 2009;65(1):19‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Gregory A, Kurian MA, Maher ER, Hogarth P, Hayflick SJ. PLA2G6‐associated neurodegeneration. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews®. University of Washington, Seattle; 1993. https://www.ncbi.nlm.nih.gov/books/NBK1675/pdf/Bookshelf_NBK1675.pdf [PubMed] [Google Scholar]
- 135. Fais M, Dore A, Galioto M, Galleri G, Crosio C, Iaccarino C. Parkinson's disease‐related genes and lipid alteration. Int J Mol Sci. 2021;22(14):7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Mori A, Hatano T, Inoshita T, et al. Parkinson's disease‐associated iPLA2‐VIA/PLA2G6 regulates neuronal functions and alpha‐synuclein stability through membrane remodeling. Proc Natl Acad Sci U S A. 2019;116(41):20689‐20699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Sun WY, Tyurin VA, Mikulska‐Ruminska K, et al. Phospholipase iPLA(2)beta averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol. 2021;17(4):465‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kinghorn KJ, Castillo‐Quan JI, Bartolome F, et al. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain. 2015;138(Pt 7):1801‐1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Sanchez E, Azcona LJ, Paisan‐Ruiz C. Pla2g6 deficiency in zebrafish leads to dopaminergic cell death, axonal degeneration, increased beta‐synuclein expression, and defects in brain functions and pathways. Mol Neurobiol. 2018;55(8):6734‐6754. [DOI] [PubMed] [Google Scholar]
- 140. Ohman A, Forsgren L. NMR metabonomics of cerebrospinal fluid distinguishes between Parkinson's disease and controls. Neurosci Lett. 2015;594:36‐39. [DOI] [PubMed] [Google Scholar]
- 141. Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59(17):4383‐4391. [PubMed] [Google Scholar]
- 142. Iovino L, Tremblay ME, Civiero L. Glutamate‐induced excitotoxicity in Parkinson's disease: the role of glial cells. J Pharmacol Sci. 2020;144(3):151‐164. [DOI] [PubMed] [Google Scholar]
- 143. Huot P, Johnston TH, Koprich JB, Fox SH, Brotchie JM. The pharmacology of L‐DOPA‐induced dyskinesia in Parkinson's disease. Pharmacol Rev. 2013;65(1):171‐222. [DOI] [PubMed] [Google Scholar]
- 144. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Jiang T, Cheng H, Su J, et al. Gastrodin protects against glutamate‐induced ferroptosis in HT‐22 cells through Nrf2/HO‐1 signaling pathway. Toxicol In Vitro. 2020;62:104715. [DOI] [PubMed] [Google Scholar]
- 146. Mahoney‐Sanchez L, Bouchaoui H, Ayton S, Devos D, Duce JA, Devedjian JC. Ferroptosis and its potential role in the physiopathology of Parkinson's disease. Prog Neurobiol. 2021;196:101890. [DOI] [PubMed] [Google Scholar]
- 147. Morari M, Brugnoli A, Pisano CA, et al. Safinamide differentially modulates In vivo glutamate and GABA release in the rat hippocampus and basal ganglia. J Pharmacol Exp Ther. 2018;364(2):198‐206. [DOI] [PubMed] [Google Scholar]
- 148. Pisano CA, Brugnoli A, Novello S, et al. Safinamide inhibits in vivo glutamate release in a rat model of Parkinson's disease. Neuropharmacology. 2020;167:108006. [DOI] [PubMed] [Google Scholar]
- 149. Perry TL, Godin DV, Hansen S. Parkinson's disease: a disorder due to nigral glutathione deficiency? Neurosci Lett. 1982;33(3):305‐310. [DOI] [PubMed] [Google Scholar]
- 150. Perry TL, Yong VW. Idiopathic Parkinson's disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci Lett. 1986;67(3):269‐274. [DOI] [PubMed] [Google Scholar]
- 151. Sofic E, Lange KW, Jellinger K, Riederer P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson's disease. Neurosci Lett. 1992;142(2):128‐130. [DOI] [PubMed] [Google Scholar]
- 152. Holmay MJ, Terpstra M, Coles LD, et al. N‐acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin Neuropharmacol. 2013;36(4):103‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Sian J, Dexter DT, Lees AJ, et al. Alterations in glutathione levels in Parkinson's disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36(3):348‐355. [DOI] [PubMed] [Google Scholar]
- 154. Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson's disease. J Parkinsons Dis. 2013;3(4):461‐491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Hauser DN, Hastings TG. Mitochondrial dysfunction and oxidative stress in Parkinson's disease and monogenic parkinsonism. Neurobiol Dis. 2013;51:35‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. McNaught KS, Jenner P. Extracellular accumulation of nitric oxide, hydrogen peroxide, and glutamate in astrocytic cultures following glutathione depletion, complex I inhibition, and/or lipopolysaccharide‐induced activation. Biochem Pharmacol. 2000;60(7):979‐988. [DOI] [PubMed] [Google Scholar]
- 157. Pereira CM, Oliveira CR. Glutamate toxicity on a PC12 cell line involves glutathione (GSH) depletion and oxidative stress. Free Radic Biol Med. 1997;23(4):637‐647. [DOI] [PubMed] [Google Scholar]
- 158. Schubert D, Piasecki D. Oxidative glutamate toxicity can be a component of the excitotoxicity cascade. J Neurosci. 2001;21(19):7455‐7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Hu Y, Guo N, Yang T, Yan J, Wang W, Li X. The potential mechanisms by which artemisinin and its derivatives induce Ferroptosis in the treatment of cancer. Oxidative Med Cell Longev. 2022;2022:1458143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Jimenez‐Jimenez FJ, Alonso‐Navarro H, Garcia‐Martin E, Agundez JA. Cerebrospinal fluid biochemical studies in patients with Parkinson's disease: toward a potential search for biomarkers for this disease. Front Cell Neurosci. 2014;8:369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Li X, Wang W, Yan J, Zeng F. Glutamic acid transporters: targets for neuroprotective therapies in Parkinson's disease. Front Neurosci. 2021;15:678154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Haydar TF, Wang F, Schwartz ML, Rakic P. Differential modulation of proliferation in the neocortical ventricular and subventricular zones. J Neurosci. 2000;20(15):5764‐5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Kuruvilla KP, Nandhu MS, Paul J, Paulose CS. Oxidative stress mediated neuronal damage in the corpus striatum of 6‐hydroxydopamine lesioned Parkinson's rats: neuroprotection by serotonin, GABA and bone marrow cells supplementation. J Neurol Sci. 2013;331(1–2):31‐37. [DOI] [PubMed] [Google Scholar]
- 164. Marrs TC, Maynard RL. Neurotranmission systems as targets for toxicants: a review. Cell Biol Toxicol. 2013;29(6):381‐396. [DOI] [PubMed] [Google Scholar]
- 165. Schmitz Y, Castagna C, Mrejeru A, et al. Glycine transporter‐1 inhibition promotes striatal axon sprouting via NMDA receptors in dopamine neurons. J Neurosci. 2013;33(42):16778‐16789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Palavra NC, Lubomski M, Flood VM, Davis RL, Sue CM. Increased added sugar consumption is common in Parkinson's disease. Front Nutr. 2021;8:628845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Cassani E, Barichella M, Ferri V, et al. Dietary habits in Parkinson's disease: adherence to Mediterranean diet. Parkinsonism Relat Disord. 2017;42:40‐46. [DOI] [PubMed] [Google Scholar]
- 168. Aden E, Carlsson M, Poortvliet E, et al. Dietary intake and olfactory function in patients with newly diagnosed Parkinson's disease: a case‐control study. Nutr Neurosci. 2011;14(1):25‐31. [DOI] [PubMed] [Google Scholar]
- 169. Limbad C, Oron TR, Alimirah F, et al. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS One. 2020;15, (1):e0227887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Bonvento G, Bolanos JP. Astrocyte‐neuron metabolic cooperation shapes brain activity. Cell Metab. 2021;33(8):1546‐1564. [DOI] [PubMed] [Google Scholar]
- 171. Aubert A, Costalat R, Magistretti PJ, Pellerin L. Brain lactate kinetics: modeling evidence for neuronal lactate uptake upon activation. Proc Natl Acad Sci U S A. 2005;102(45):16448‐16453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Jia L, Liao M, Mou A, et al. Rheb‐regulated mitochondrial pyruvate metabolism of Schwann cells linked to axon stability. Dev Cell. 2021;56(21):2980‐2994 e6. [DOI] [PubMed] [Google Scholar]
- 173. Hantikainen E, Roos E, Bellocco R, et al. Dietary fat intake and risk of Parkinson disease: results from the Swedish National March Cohort. Eur J Epidemiol. 2022;37(6):603‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Galper J, Dean NJ, Pickford R, et al. Lipid pathway dysfunction is prevalent in patients with Parkinson's disease. Brain. 2022;145(10):3472‐3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Adibhatla RM, Hatcher JF. Altered lipid metabolism in brain injury and disorders. Subcell Biochem. 2008;49:241‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Dong MX, Wei YD, Hu L. The disturbance of lipid metabolism is correlated with neuropsychiatric symptoms in patients with Parkinson's disease. Chem Phys Lipids. 2021;239:105112. [DOI] [PubMed] [Google Scholar]
- 177. Schroter N, Blazhenets G, Frings L, et al. Nigral glucose metabolism as a diagnostic marker of neurodegenerative parkinsonian syndromes. NPJ Parkinsons Dis. 2022;8(1):123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Xicoy H, Klemann CJ, De Witte W, Martens MB, Martens GJ, Poelmans G. Shared genetic etiology between Parkinson's disease and blood levels of specific lipids. NPJ Parkinsons Dis. 2021;7(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Hu L, Dong MX, Huang YL, et al. Integrated metabolomics and proteomics analysis reveals plasma lipid metabolic disturbance in patients with Parkinson's disease. Front Mol Neurosci. 2020;13:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Havelund JF, Heegaard NHH, Faergeman NJK, Gramsbergen JB. Biomarker research in Parkinson's disease using metabolite profiling. Metabolites. 2017;7(3):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Heilman PL, Wang EW, Lewis MM, et al. Tryptophan metabolites are associated with symptoms and Nigral pathology in Parkinson's disease. Mov Disord. 2020;35(11):2028‐2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Zhang Q, Gao Y, Zhang J, et al. L‐Asparaginase exerts neuroprotective effects in an SH‐SY5Y‐A53T model of Parkinson's disease by regulating glutamine metabolism. Front Mol Neurosci. 2020;13:563054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Di Maio A, Nuzzo T, Gilio L, et al. Homeostasis of serine enantiomers is disrupted in the post‐mortem caudate putamen and cerebrospinal fluid of living Parkinson's disease patients. Neurobiol Dis. 2023;184:106203. [DOI] [PubMed] [Google Scholar]
- 184. Simmering JE, Welsh MJ, Liu L, Narayanan NS, Pottegard A. Association of glycolysis‐enhancing alpha‐1 blockers with risk of developing Parkinson disease. JAMA Neurol. 2021;78(4):407‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are openly available in The National Center for Biotechnology Information at https://www.ncbi.nlm.nih.gov/.