

Abstract
A missense mutation within the histone acetyltransferase (HAT) domain of the TATA binding protein-associated factor TAF1 induces ts13 cells to undergo a late G1 arrest and decreases cyclin D1 transcription. We have found that TAF1 mutants (Δ844-850 and Δ848-850, from which amino acids 844 through 850 and 848 through 850 have been deleted, respectively) deficient in HAT activity are unable to complement the ts13 defect in cell proliferation and cyclin D1 transcription. Chromatin immunoprecipitation assays revealed that histone H3 acetylation was reduced at the cyclin D1 promoter but not the c-fos promoter upon inactivation of TAF1 in ts13 cells. The hypoacetylation of H3 at the cyclin D1 promoter was reversed by treatment with trichostatin A (TSA), a histone deacetylase inhibitor, or by expression of TAF1 proteins that retain HAT activity. Transcription of a chimeric promoter containing the Sp1 sites of cyclin D1 and c-fos core remained TAF1 dependent in ts13 cells. Treatment with TSA restored full activity to the cyclin D1-c-fos chimera at 39.5°C. In vivo genomic footprinting experiments indicate that protein-DNA interactions at the Sp1 sites of the cyclin D1 promoter were compromised at 39.5°C in ts13 cells. These data have led us to hypothesize that TAF1-dependent histone acetylation facilitates transcription factor binding to the Sp1 sites, thereby activating cyclin D1 transcription and ultimately G1-to-S-phase progression.
Transcription of protein-encoding genes requires the action of a large complex of transcription factors. To date, the molecular actions of many of these protein components remain to be demonstrated. The formation of the basic transcription machinery involves the assembly of a functional preinitiation complex, which contains RNA polymerase II (pol II) and a set of general transcription factors (TFIIA, TFIIB, TFIID, TFIIE, TFIIF, and TFIIH) (6, 28). The first step in preinitiation complex formation is the binding of TFIID to promoter DNA and the subsequent recruitment of the remaining general factors and RNA pol II. Therefore, TFIID plays an essential role in transcriptional initiation.
TAF1 is the largest subunit of TFIID and is a histone acetyltransferase (HAT) that acetylates histones H3 and H4 in vitro (22). The importance of the HAT activity of TAF1 in the transcription process remains controversial and has yet to be clearly demonstrated. A single missense mutation (Gly to Asp) in the HAT domain of TAF1 induces the temperature-sensitive mutant ts13 cell line, derived from baby hamster kidney cells, to arrest in the late G1 phase of the cell cycle at the nonpermissive temperature of 39.5°C (5, 8). Transcription from a subset of protein-encoding genes including the cell cycle regulators cyclin A, cyclin D1, and cyclin E is also compromised (29, 30, 35, 40). We have demonstrated that the HAT activity of the ts13 allele of TAF1 is temperature sensitive in vitro such that the mutant protein is four to five times less effective than wild-type TAF1 in acetylating histones at higher temperatures (4). These data were the first indication that inactivation of TAF1 HAT activity is contributing to the ts13 mutant phenotype. Therefore, the ts13 cell line provides a portal to the action of TAF1 HAT activity in the transcription process.
Conditional mutations in the yeast homologue of TAF1 also produce G1 cell cycle arrest and defects in gene transcription (10, 40). Further characterization of yeast TAF1 mutants suggests that the transcriptional defect is due to a lack of TFIID recruitment (21). We have elected to further investigate the role of TAF1 in the transcription process by using cyclin D1 as our model gene. The decrease in cyclin D1 mRNA levels occurs relatively quickly after ts13 cells have been shifted to 39.5°C (35). This rapid response suggests that transcription of cyclin D1 is directly affected by the ts13 mutation in TAF1.
Consistent with studies described in yeast, we have reported that TAF1 is required for the efficient binding of TFIID to the initiator region of the cyclin D1 promoter in mammalian cells (9). Interestingly, the recruitment of TFIID, by insertion of a TATA binding protein (TBP) binding site upstream of the initiator, was sufficient to restore basal but not Sp1-activated cyclin D1 transcription at 39.5°C in ts13 cells. These findings suggest that TAF1 is serving dual functions at the cyclin D1 promoter, one essential for efficient binding of TFIID to the initiation site and the other to mediate Sp1 activation of cyclin D1 transcription.
Commitment to cell division is made in G1 in response to many stimuli, including growth factors and other mitogens. Cyclin D1, the first cyclin expressed in the cell cycle, is transcriptionally upregulated by many factors (1, 43). Analysis of the human cyclin D1 promoter has revealed the presence of binding sites for several transcription factors that are essential for promoter activity (1, 24). In the absence of growth factors, the cyclin D1 protein is rapidly degraded via a ubiquitin-dependent proteasome pathway (3). Therefore, cyclin D1 protein levels are controlled at both the transcriptional and posttranscriptional levels.
Chromatin remodeling by histone modification is an important aspect of transcriptional regulation. Acetylation of histones on N-terminal lysine residues is thought to alter the structure of nucleosomes and/or nucleosomal arrays, which in turn increases the accessibility of transcription factor binding sites in promoter DNA (14, 32, 34). We have carried out structure-function studies on the HAT domain of TAF1 to determine whether a causal relationship can be demonstrated between inactivation of TAF1 HAT activity and the ts13 defect in cyclin D1 transcription and cell proliferation. Here we report a positive correlation between the level of histone H3 acetylation and occupancy of the Sp1 sites at the cyclin D1 promoter with cyclin D1 promoter activity and TAF1 acetyltransferase activity. Our studies provide the first pieces of experimental data suggesting that histones are acetylated by a TAF1-dependent mechanism in vivo and that the acetylation of histone H3 within the proximal promoter region is required for Sp1 activation of cyclin D1 transcription.
MATERIALS AND METHODS
Tissue culture cell lines.
ts13 and ts13R3 cells were grown in Dulbecco's modified Eagle medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (JRH Biosciences), 2 mM l-glutamine, and penicillin/streptomycin in a humidified incubator containing 10% CO2 at 33.5°C and 39.5°C, respectively. To inactivate TAF1 and induce cell cycle arrest, ts13 cells were shifted to 39.5°C. ts13 cells that stably express hemagglutinin (HA)-tagged wild-type (WT) or mutant TAF1 were established as previously described (9) with the following modifications. Subclones stably expressing the mutant in which amino acids 574 through 595 have been deleted (Δ574-595) or the E742Q mutant were established by selection for growth at 39.5°C instead of resistance to 1 mg/ml G418. After 1 to 2 weeks of selection, expression of the HA-tagged TAF1 mutants in pools of clones was determined by anti-HA Western blot analysis. HA-positive pools were propagated in 200 μg/ml G418 at 33.5°C to maintain expression of the HA-TAF1 proteins. Sf9 insect cells were propagated in Hink's TNM-FH insect medium (JRH Biosciences) containing 10% FBS, l-glutamine, and penicillin/streptomycin. Cultures were grown in spinner flasks at 27°C in the absence of CO2.
Construction of hTAF1 mutants.
The EcoRI fragment of TAF1 was cloned into the EcoRI site of pBlueScript SK(+) to generate pSK-hTAF1EcoRI. To construct the deletion mutants, pSK-hTAF1EcoRI was linearized at the unique HindIII, ScaI, or NcoI site or an introduced StuI site (a change of ATA to GCC at positions 2548 through 2550) in the TAF1 coding region. The linearized DNA fragments were digested with Bal31 nuclease. The resulting products were blunt ended, gel purified, and circularized. DNA sequencing was carried out to determine the reading frame and the amino acids deleted. The E742Q, C746A, and G923/925D point mutations were generated by site-directed mutagenesis using the following primers: for E742Q, 5′-AATATGGGCAAACTGT-3′; for C746A, 5′-GTTTACGCCCATACATCT-3′; and for G923/925D, 5′-GATGCTGACTATGATGAGAAA-3′. The EcoRI fragment of in-frame deletions and point mutants was then subcloned into the full-length TAF1 coding sequence in pBlueScript SK(+). Each TAF1 mutant was then cloned into the cytomegalovirus-driven mammalian expression vector CS2+MT for synthesis of myc epitope-tagged versions of human TAF1 (hTAF1) in ts13 cells. Mammalian expression vectors for HA-tagged hTAF1 mutant proteins also were constructed. The unique EcoRV-PstI fragment from CS2+HA-WT-TAF1, a plasmid containing wild-type HA-TAF1 coding sequence downstream of the cytomegalovirus promoter, was replaced with the equivalent fragment derived from the CS2+MT-TAF1 mutant constructs described above.
ts13 complementation assays.
ts13 cells were seeded at ∼7.5 × 105 cells/10-cm dish and grown overnight at 33.5°C. Cells were transfected with 0.5 μg of the indicated hTAF1 expression construct by calcium phosphate precipitation. Approximately 6 h posttransfection, the DNA precipitate was removed, cells were washed with phosphate-buffered saline (PBS), and fresh medium (DMEM plus 10% FBS) was added. Half the plates were then shifted to the nonpermissive temperature of 39.5°C. The presence of viable proliferating cells was monitored after 1 to 2 weeks.
Reporter plasmid constructs.
Promoters for transient transfection assays were cloned into pGL2-basic (Promega), a reporter plasmid that contains the luciferase coding region. Human cyclin D1 reporter plasmid was constructed by subcloning the EcoRI to PvuII fragment of pD1-G065 (kindly provided by Yue Xiong) into the SmaI site of pGL2-basic. c-fos reporter contains sequence from −356 to +58 (−356/+58) of the mouse gene as previously described (41). −183/+21 cyclin D1 luciferase reporter construct (−183/+21 cycD1-luc) was generated by PCR amplification of −714/+109 cycD1-luc (9) with primers 5′-CGCTTTGGGCTTGTCCCCT-3′ and 5′-ATCTGCCGCTCTCTGCTACGCG-3′, followed by cloning of the PCR product into the SmaI site of pGL2-basic. The reporter construct containing only the c-fos core was constructed by introducing a SacII site at position −64 of −356/+58 c-fos-luc to generate SacII/−64 c-fos-luc. The SacII containing c-fos plasmid was then digested with SacII and DraIII, blunt ended with Klenow fragment, and the vector recircularized. Sp1/c-fos-luc chimera was constructed as follows. SacII/−64 c-fos-luc was digested with DraIII, incubated with Klenow fragment to create blunt ends, and then digested with SacII. The Sp1 sites from the cyclin D1 promoter were isolated by introducing a SacII site at position −53 of cyclin D1-pCRII TOPO using the QuikChange site-directed mutagenesis kit (Stratagene) and primers 5′-GTCACACGGACCCGCGGGGAGTTTTG-3′ and 5′-CAAAACTCCCCGCGGGTCCGTGTGAC-3′. A PCR product was amplified from the construct mentioned above using primers 5′-TTTGGGCTTGTCCCCTCGGCG-3′ and 5′-ATCTGCCGCTCTCTGCTACTG-3′, and it was then digested with SacII, and the resulting fragment was subcloned into the blunt DraIII/SacII-digested c-fos plasmid described above. The −183/+21 cycD1-luc plasmid containing upstream activation sites (UAS) was generated by introducing a SacII site at position −173 by using primers 5′-TTTGGGCTTGTCCCCGCGGCGCTCGCAG-3′ and 5′-CTGCGAGCGCCGCGGGGACAAGCCCAAA-3′, and the plasmid linearized at the newly engineered SacII site. UASs containing oligonucleotides 5′-CGGAAGACTCTCCTCCGGC-3′ and 5′-CGGAGGAGAGTCTTCCGGC-3′ were annealed to produce SacII overhangs, multimerized, and ligated into the SacII-linearized cyclin D1-luc vector described above. The resulting −183/+21 cycD1-luc plasmid contains three tandem UAS sites, as determined by DNA sequencing.
Transient transfection assays.
Before the introduction of DNA, ts13 cells, seeded into 24-well plates, were maintained at 33.5°C. Cells were then switched to DMEM containing no FBS and no antibiotics. In total, 0.2 μg of the indicated CS2+MT-hTAF1 expression vector, 0.05 μg simian virus 40 (SV40) beta-galactosidase expression plasmid, and 0.2 μg of the cyclin D1 or c-fos reporter construct in pGL2-basic were transfected into the cells using the Lipofectamine and Plus reagents as described by the manufacturer (Invitrogen) with the following modification; the amount of Plus reagent used was reduced by 50%. After 3 h, FBS to a 10% final concentration and antibiotics were added to the cell culture medium and half the cells were shifted to 39.5°C. Whole-cell lysates were prepared using Report lysis buffer (Promega) between 16 and 18 h post-serum addition. The amounts of luciferase and beta-galactosidase activity were determined as described previously (42). Beta-galactosidase activity was used to correct for differences in transfection efficiency. When cells were treated with histone deacetylase (HDAC) inhibitors, ts13 cells were maintained at 33.5°C and transfected with DNA as described above. Following the addition of antibiotics and 10% FBS (final concentration), sodium butyrate (SB) or trichostatin A (TSA) was added to the cell culture medium. The cells were maintained at 33.5°C for 15 min, after which, half the plates were shifted to 39.5°C. For the Sp1 recruitment experiments, 0.2 μg −183/+21 cycD1-luc or −183/+21 cycD1-luc containing UAS was cotransfected with 0.2 μg Sp1-Gal4 or Gal4-pcDNA3 (17). Promoter activity was determined 16 to 18 h after serum addition as described above.
Expression of recombinant hTAF1 proteins.
Recombinant wild-type and mutant hTAF1 proteins were expressed using a baculovirus expression system in Sf9 insect cells. Recombinant virus was generated using the BaculoGold transfection system (PharMingen). For protein production, Sf9 cells were seeded at ∼7.5 × 106 cells/10-cm dish and infected with recombinant baculovirus at a multiplicity of infection of 1 to 2. Forty-eight hours postinfection, whole-cell lysates were prepared by sonication in 0.4 M HEMG buffer (25 mM HEPES-KOH, pH 7.6, 400 1mM KCl, 12.5 mM MgCl2, 0.1 mM EDTA, 10% glycerol) containing 0.1% NP-40, 1 mM dithiothreitol (DTT), and 0.2 mM phenylmethylsulfonyl fluoride. Protein interaction assays were performed as follows. One hundred microliters of wild-type and mutant hTAF1 cell lysates were incubated with anti-HA ascites followed by protein A-Sepharose (PAS). Purified hTAF1 immobilized on anti-HA-PAS was incubated with crude Sf9 lysates containing FLAG-Drosophila TAF2 (dTAF2) or His-human TBP (hTBP) for 2 h at 4°C. After extensive washing, proteins retained on the resin were identified by Western blotting using anti-HA, anti-FLAG, and anti-TBP antibodies.
In vitro histone acetyltransferase assays.
Wild-type and mutant hTAF1 containing Sf9 cell lysates were incubated with either an anti-HA ascites or anti-hTAF1 monoclonal antibody (MAb) 6B3 followed by PAS. hTAF1 proteins immobilized on PAS were washed three times with 0.4 M HEMG buffer containing 0.1% NP-40 and 1 mM DTT, followed by two washes with assay buffer (50 mM Tris-HCl, pH 8.0, 10% glycerol, 0.1 mM EDTA, 1 mM DTT, 0.2 mM phenylmethylsulfonyl fluoride). The isolated complexes were resuspended in assay buffer, and one-third to one-quarter of the sample was used per reaction. Reaction mixtures containing 20 μg core histones (type II-AS from calf thymus; Sigma) and 0.25 μCi [3H]acetyl-coenzyme A (CoA) (Amersham) were incubated for 60 min at 30°C, filtered through P81 paper, and washed with 50 mM sodium carbonate (pH 9.2). The amount of acetylated histones retained on the filter was measured by liquid scintillation. A fraction of the purified hTAF1 proteins was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by silver staining to assess protein purity and levels.
Small-scale sucrose density gradient sedimentation.
Stable ts13 cell lines expressing WT, Δ844-850, or Δ848-850 TAF1 were cultured at 33.5°C and 39.5°C, and nuclear extracts were prepared as previously described (42). One-milliliter 10 to 30% linear sucrose gradients were generated as described previously (36), by layering 200 μl of 30%, 25%, 20%, 15%, and 10% sucrose solutions in a centrifuge tube. Diffusion was allowed to proceed for 1 h at room temperature, and the gradients subsequently were cooled at 4°C for a minimum of 30 min. Nuclear extracts (∼750 μg in a 100-μl volume) were loaded onto each gradient, and the gradients were centrifuged at 50,000 rpm for 16 h at 4°C in a Beckman TLS-55 rotor. Native protein molecular mass markers (carbonic anhydrase, 29 kDa; bovine serum albumin, 66 kDa; alcohol dehydrogenase, 150 kDa; and β-amylase, 200 kDa) were run in parallel as an indicator of the sedimentation profile. After centrifugation, fractions (100 μl each) were recovered, beginning at the top of the gradient as described previously (37), subjected to trichloroacetic acid precipitation, separated on 12% SDS-polyacrylamide gels, and analyzed by Western blotting using anti-HA (Santa Cruz), anti-TBP (rabbit polyclonal serum), anti-mouse TAF4 (32TA), and anti-human TAF5 (MAb 6B1) antibodies.
Chromatin immunoprecipitation.
ts13 cells, seeded on 10-cm plates, were incubated at 33.5°C or 39.5°C for 16 to 18 h and then cross-linked by the addition of 400 μl of 37% formaldehyde to 10 ml medium. After 15 min, cells were washed twice with PBS and lysed in 0.5 ml immunoprecipitation (IP) buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, and 0.5% Nonidet P-40) containing protease inhibitors. The nuclear pellet was washed and resuspended in 1.0 ml IP buffer and sonicated for 4 × 10 s (Branson Sonifier 250, output 4, duty cycle 80) to produce DNA fragments of 300 bp in average length. A volume of 0.5 ml of the nuclear extract was incubated with 1 μg anti-diacetylated histone H3 antibody (Upstate Biotechnology) for 2 h at 4°C, after which 25 μl PAS was added, and the samples incubated for an additional hour at 4°C. Immunoprecipitated complexes were washed five times with IP buffer, and bound DNA/protein complexes were eluted with 2× 250 μl elution buffer (1% SDS, 0.1 M NaHCO3). The cross-links were reversed by the addition of 20 μl 5 M NaCl and incubation overnight at 65°C. DNA/protein complexes were precipitated with ethanol and glycogen as carrier. The pellet was resuspended in 100 μl proteinase K buffer (10 mM Tris-HCl, pH 7.8, 5 mM EDTA, 0.5% SDS) and digested with 2 mg/ml proteinase K for 30 min at 50°C. DNA was purified by one phenol extraction, two phenol-chloroform extractions, and two chloroform-isoamyl alcohol extractions, ethanol precipitated, and resuspended in 25 μl Tris-EDTA. To control for the specificity of the immunoprecipitation, the anti-diacetylated histone H3 antibody was preincubated with 0.05 mM histone H3 (acetyl Lys9 and Lys14) blocking peptide before the antibody was added to the nuclear extract.
For PCR analysis, 0.5 to 2.0 μl of purified DNA was used as a template. PCRs were carried out in 65 mM Tris-HCl, pH 8.8, 40 mM NaCl, 10 mM β-mercaptoethanol, 3 mM MgCl2, 10% dimethyl sulfoxide with 0.4 mM deoxynucleoside triphosphates; and 1 U Vent Exo− (New England Biolabs). The cyclin D1 proximal promoter region was amplified with 30 to 35 cycles of 94°C for 30 s, 59°C for 30 s, and 72°C for 30 s, using primers 5′-CGCTTTGGGCTTGTCCCCT-3′ and 5′-ATCTGCCGCTCTCTGCTACGCG-3′. The c-fos promoter was amplified with 25 to 30 rounds of the cycle described above using primers 5′-AATCCTACATGCGGAGAGTCCAGG-3′ and 5′-GAGCGCGGTCACTGCTCGTT-3′. PCR products were subjected to 1.25% agarose gel electrophoresis and visualized by ethidium bromide staining.
In vivo genomic footprinting of the hamster cyclin D1 promoter.
ts13 and ts13R3 cells were arrested in G0 by serum starvation for 36 h at 33.5°C and then released at either 33.5°C or 39.5°C by the addition of 10% FBS. Approximately 8 h post-serum addition, cells were treated with 0.01% dimethyl sulfate (DMS) for 2 min. The methylated genomic DNA was isolated as described previously with the following minor modifications (23). For the preparation of genomic DNA, the ethyl ether extraction was eliminated from the protocol and the isopropanol precipitation was replaced with an ethanol precipitation. Methylated guanines and adenines were chemically cleaved, and the sites of cleavage in the hamster cyclin D1 promoter were mapped using the technique of ligation-mediated PCR (33). Briefly, 5 μg genomic DNA served as the template in a first-strand synthesis reaction with the following primers: for the upper strand, 5′-CTGCCTCGCCGTCTACTGCC-3′; and for the lower strand, 5′-TATCTACGAAGGTCGAGCTGG-3′. Linkers were then ligated onto the blunt-ended products. Next, a linker and nested gene-specific primer (for the upper strand, 5′-GTCTCCGAGCGCGCGAATCT-3′; for the lower strand, 5′-CGCTTTGGGCTTGTCCCCTCGG-3′) were used to amplify the cleavage products. The resulting PCR products were end labeled with the following 32P-labeled primers: for the upper strand, 5′-ATCTGCCGCTCTCTGCTACGCG-3′; and for the lower strand, 5′-TCGGCGCTCGCAGCTCCCGA-3′. Reaction products were separated on denaturing 6% polyacrylamide gels and visualized by autoradiography.
RESULTS
hTAF1 mutants defective in histone acetyltransferase activity.
Whether a primary function of TAF1 in RNA pol II-dependent transcription is to alter chromatin structure by histone acetylation is a question that has been somewhat controversial. Our work and that of others have demonstrated that TAF1 plays a role in the recruitment of TFIID to core promoters (9, 21). However, the observation that a mutation in TAF1 that disrupts HAT activity leads to defects in transcription is consistent with the idea that this enzymatic activity of TAF1 is also important for efficient mRNA synthesis.
Sequence alignment of HAT enzymes has revealed the presence of distinct classes, including the Gcn5/PCAF, MYST, and CBP superfamilies (19). Within each family, there is high sequence homology, similar substrate specificity, and similar functional context for the acetyltransferases. TAF1 shows very limited sequence similarity with other HATs and therefore has been categorized into its own distinct family (18). Structural analysis of the HAT domains of GCN5 and PCAF has identified four conserved motifs, termed A, B, C, and D, that form a central catalytic core capable of binding acetyl-CoA and the histone substrate (25). Alignment of the hTAF1 HAT domain with these conserved motifs revealed very limited sequence homology (Fig. 1A). Mutational analysis of HAT domains has identified residues in each motif necessary for enzymatic activity. In particular, the conserved glutamate residue at position 173 in yeast Gcn5 (yGcn5) is essential for HAT activity and is proposed to function as a general base for catalysis (37, 38). We used the alignment with yGcn5 and CBP, along with mutagenesis information, as a guideline for designing deletion and point mutations in hTAF1 that would potentially disrupt HAT activity (15, 20). A total of eight different mutations between amino acids 517 and 976 of hTAF1 were introduced into the coding region (Fig. 1B). The G923/925D double mutant has been previously described and was found to be functionally equivalent to wild-type TAF1 for HAT activity (4).
FIG. 1.

Mutations in HAT domain of human TAF1. (A) Alignment of the HAT domains of Gcn5, CBP, and hTAF1 in the conserved motifs is shown. The dashes indicate that the motif continues in that direction. Five independent alignments demonstrate that TAF1 exhibits very limited sequence similarity to other HATs. Amino acids that compromise yGcn5 HAT activity when mutated are indicated by the asterisks. An aspartic acid residue important for CBP HAT activity is indicated by the square in alignment 5. Based on these alignments, different regions spanning the TAF1 HAT domain were targeted via site-directed or deletion mutagenesis to identify residues essential for HAT activity. The amino acids deleted are underlined in black below the alignments. The positions of two single point mutants, E742Q and C746A, are shown in alignment 4. (B) The locations, within the TAF1 HAT domain, of five deletions, three point mutations, and the single missense mutation (G716D) of ts13 cells are indicated. The G923/925D double point mutant disrupts a putative acetyl-CoA binding site and has been previously described (3).
Next, we tested the ability of each hTAF1 mutant to acetylate histones in vitro. HA-tagged versions of wild-type and mutant hTAF1 were expressed, immunoaffinity purified (Fig. 2A), and added to reaction mixtures containing core histones and [3H]acetyl-CoA. A dramatic decrease in histone acetylation was detected with two overlapping hTAF1 deletion mutants, Δ844-850 and Δ848-850, from which 7 and 3 amino acids were removed, respectively (Fig. 2B). By contrast, the remaining deletion and point mutations had no significant effect on HAT activity. The inability to disrupt enzymatic activity by targeting regions of TAF1 that display limited amino acid homology to motifs important for catalysis by other well-characterized enzymes supports the idea that the HAT domain of TAF1 may be structurally distinct and will need to be more precisely defined for further investigation.
FIG. 2.

Histone acetyltransferase activity of hTAF1 mutants. (A) HA-tagged wild-type (WT) and mutant TAF1 proteins were immunopurified from baculovirus-infected Sf9 extracts using an anti-human TAF1 monoclonal antibody (6B3). An aliquot of each sample was run on SDS-polyacrylamide gels and visualized by silver staining. The positions of molecular weight standards (MW; on the left) and immunoglobulin heavy chain (IgH) are indicated. (B) hTAF1 proteins, immobilized on PAS, were added to reaction mixtures containing purified calf thymus histones and [3H]acetyl-coenzyme A. Assays were incubated for 60 min at 30°C, and the amounts of acetylated histones were measured by filter binding, followed by liquid scintillation. The data are averages from four independent experiments, each carried out in duplicate, with standard errors of the means (SEM) shown. Only the deletion of amino acids between 844 and 850 compromised TAF1 HAT activity.
Complementation of the ts13 mutant phenotype by hTAF1 mutants.
Although a complete loss of TAF1 protein is lethal, the single missense mutation found in ts13 and tsBN462 cells alters transcription of select genes and leads to G1 cell cycle arrest. The isolation of the identical glycine-to-aspartic acid missense mutation in two independent genetic screens points to the importance of this residue in TAF1 function (8). We have previously shown that the HAT activity of the ts13 allele of hTAF1 is decreased at higher temperatures in vitro suggesting that this enzymatic activity is impaired at nonpermissive temperature in ts13 cells (4). To further implicate the importance of TAF1 HAT activity in cell cycle progression, we assessed whether the mutations engineered into hTAF1 affected complementation of the ts13 proliferation defect. Expression constructs for the wild-type and mutant forms of hTAF1 and a control vector were transfected into ts13 cells maintained at the permissive (33.5°C) or nonpermissive (39.5°C) temperature. At 5 to 7 days posttransfection, expression of wild-type hTAF1 and all mutants that retained HAT activity resulted in viable proliferating cells at 39.5°C (data not shown). Complementation of the proliferation defect was not observed with the two HAT-defective mutants, Δ844-850 and Δ848-850. No visible change in cell growth was observed at 33.5°C in the presence of any of the hTAF1 mutant constructs. These data suggest that disruption of HAT activity compromises the ability of TAF1 to overcome the cell cycle arrest of ts13 cells.
Another hallmark of the ts13 mutant phenotype is the decrease in the transcription of a subset of protein-encoding genes, including cyclin D1 (29, 30, 35, 42). Expression of wild-type hTAF1 is sufficient to restore transcriptional activity to the cyclin D1 promoter (42). Therefore, we examined the ability of the hTAF1 mutants to support cyclin D1 transcription in transient transfection assays (Fig. 3). In the presence of the ts13 allele, the cyclin D1 promoter was 20 to 50 times more active at 33.5°C than at 39.5°C. This dramatic difference in transcriptional activity was quantified by dividing the amount of luciferase activity detected at 33.5°C by the amount of activity detected at 39.5°C, with larger ratios reflecting a greater dependence on TAF1 function (Fig. 3). Transcription from the c-fos reporter remained largely unaffected and exhibited a ratio between 1 and 3. When expressed in ts13 cells, only the TAF1 deletion mutants deficient in HAT activity, Δ844-850 and Δ848-850, were unable to restore transcription to the cyclin D1 promoter at 39.5°C (ratios greater than 30) while having little effect on c-fos promoter activity. This was in stark contrast to the remaining hTAF1 mutants that behaved similar to the wild-type protein in complementing the cyclin D1 transcriptional defect. We confirmed, by Western blotting using an anti-human TAF1 specific monoclonal antibody (6B3), that comparable amounts of the wild-type and mutant proteins were expressed in the transfected ts13 hamster cells (Fig. 3A).
FIG. 3.

Complementation of cyclin D1 transcriptional defect by hTAF1 mutants in ts13 cells. (A) ts13 cells, maintained at permissive (33.5°C) or nonpermissive (39.5°C) temperature, were transiently transfected with the control (Ctrl) or indicated myc-tagged hTAF1 expression vector, the human cyclin D1 or mouse c-fos luciferase reporter construct, and an SV40-driven beta-galactosidase plasmid. Approximately 24 h posttransfection, the transcriptional activity of the cyclin D1 or c-fos reporter was measured and corrected for transfection efficiency. (Lower panel) Dependence on hTAF1 function was calculated by dividing the amount of normalized luciferase activity for each reporter at 33.5°C by the amount of activity at 39.5°C. A larger ratio reflects a stronger requirement for TAF1 function. The data presented are from one representative experiment carried out in triplicate and have been reproducibly observed in three independent experiments. The error bars represent SEM. TAF1 mutants lacking HAT activity (Δ844-850 and Δ848-850) failed to complement the ts13 defect in cyclin D1 transcription. (Upper panel) The expression levels of the TAF1 mutants were determined by Western blotting using the anti-human TAF1-specific MAb 6B3 and found to be comparable for all proteins. The amount of protein loaded was corrected for differences in transfection efficiency by normalizing for beta-galactosidase activity expressed from the cotransfected SV40 beta-galactose construct. (B) HA-tagged wild-type, Δ669-685, and Δ844-850 hTAF1 proteins immobilized on anti-HA-PAS were incubated with Sf9 cell lysates containing FLAG-dTAF2 and His-hTBP. The retained proteins were analyzed by SDS-PAGE and Western blotting using anti-HA, anti-TBP, and anti-FLAG antibodies.
Integrity of TFIID complexes containing hTAF1 mutants.
TAF1 is an integral component of the TFIID complex and is thought to serve a critical scaffolding function (2, 31). The deletion of the residues from Δ844-850 and Δ848-850 could lead to gross protein misfolding and disruption of the TFIID complex. To eliminate this possibility, we investigated the binding of different TAF1 mutants to subunits of TFIID. Coimmunoprecipitation experiments were conducted in vitro with two hTAF1 binding partners: TAF2 and TBP. HA-tagged WT and mutant hTAF1 were immobilized on anti-HA-PAS and sequentially incubated with FLAG-TAF2 followed by His-hTBP. After extensive washing, bound proteins were analyzed by Western blotting. We observed that Δ844-850, a HAT-deficient mutant, and Δ669-685, a mutant that retains HAT and complementation function, both bound to FLAG-dTAF2 and His-hTBP as effectively as WT-TAF1 (Fig. 3B). Therefore, the deletion of residues 844 through 850 does not appear to cause gross changes in protein structure.
We also examined the abilities of Δ844-850 and Δ848-850 to assemble into TFIID by small-scale sucrose gradient sedimentation (36). ts13 subclones that constitutively express HA-tagged WT or mutant TAF1 were maintained at 33.5°C or 39.5°C for 24 h. Nuclear extracts were prepared and subjected to sedimentation analysis using 10 to 30% sucrose gradients (Fig. 4). As expected, WT-TAF1, from cells maintained at 33.5°C or 39.5°C, sedimented towards the bottom of the gradient, at a molecular mass significantly greater than 200 kDa. In parallel gradients, the sedimentation profiles for Δ844-850 and Δ848-850 showed that a fraction of the mutant proteins behaved similarly to WT-TAF1, indicating that the mutant proteins are able to associate with a large protein complex at 33.5°C and 39.5°C. Cosedimentation of WT and mutant TAF1 with TBP, TAF4, and TAF5 suggests that the TAF1 proteins are incorporated into TFIID, even under conditions in which the mutant TAF1s are unable to complement the ts13 proliferation and transcription defects. These data have led us to propose that the Δ844-850 and Δ848-850 mutations selectively disrupt the acetyltransferase activity of TAF1 and not the ability of TAF1 to assemble into the TFIID complex.
FIG. 4.

Sucrose density sedimentation of TFIID complexes from ts13 nuclear extracts containing WT or HAT-deficient TAF1 protein. Nuclear extracts prepared from ts13 cells, incubated at 33.5°C or 39.5°C, and expressing the indicated TAF1 protein, were layered onto 10 to 30% sucrose gradients and subjected to density sedimentation. The presences of the indicated TFIID subunits in collected fractions were determined by Western blotting using anti-HA (Santa Cruz) for HA-TAF1, anti-mouse TAF4 MAb, anti-human TAF5 MAb, and anti-TBP polyclonal antibodies. The sedimentation of native molecular weight standards is indicated by the arrows. Fraction 1 represents the top of the gradient, and fraction 11 represents the bottom.
HDAC inhibitors partially restore cyclin D1 transcription in ts13 cells.
To gather more evidence for the contribution of histone acetylation to cyclin D1 promoter activity, we tested the effects of HDAC inhibitors SB and TSA on cyclin D1 transcription in ts13 cells. We have previously cloned the hamster cyclin D1 promoter and found that a fragment between positions −183 and +21, conserved in rats, mice, and humans, was as active as the full-length promoter and highly dependent on TAF1 activity (9). Therefore, ts13 cells maintained at 33.5°C were transfected with either −183/+21 cycD1 or −356/+58 c-fos luciferase reporter construct, treated with 1 mM SB or 500 ng/ml TSA, and then either kept at 33.5°C or shifted to 39.5°C for 12 to 16 h. While both HDAC inhibitors slightly decreased c-fos transcription at 39.5°C (Fig. 5B), 6.4- and 5-fold increases in −183/+21 cycD1-luc transcription were observed in the presence of SB and TSA, respectively (Fig. 5A). This increase in promoter activity translated into a partial decrease in TAF1 dependence, as the ratios of transcription at 33.5°C and 39.5°C decreased from 58.4 to 14.2 and 5.8 after treatment with SB and TSA, respectively. Because the initial concentration of HDAC inhibitors used altered cyclin D1 transcription at 33.5°C, we treated ts13 cells with lower doses of drug. Under these conditions, no significant effect on cyclin D1 promoter activity was observed at 33.5°C (Fig. 6A), and both SB and TSA increased cyclin D1 transcription at 39.5°C in a dose-dependent manner (Fig. 6B). These findings support the conclusion that histone acetylation is a feature of the molecular mechanism regulating cyclin D1 transcription.
FIG. 5.

Effect of histone deacetylase inhibitors on cyclin D1 and c-fos promoter activity in ts13 cells. rlu, relative light units. (A) ts13 cells, incubated at 33.5°C, were transfected with −183/+21 cyclin D1 luciferase reporter construct and an SV40-driven beta-galactosidase gene, as a control for transfection efficiency. Cells were then treated with either 1 mM SB or 500 ng/ml TSA, and half the cells were shifted to 39.5°C. Approximately 24 h later, the transcriptional activities of the cyclin D1 reporter construct at the two temperatures were measured and corrected for variations in transfection efficiency. All experiments were performed in duplicate, and the data shown are the averages from three independent experiments, with SEM indicated. Treatment with the HDAC inhibitors significantly increased cyclin D1 promoter activity under conditions in which TAF1 function was compromised in ts13 cells (39.5°C). (B) The −356/+58 c-fos luciferase reporter was transfected into ts13 cells and analyzed as described for panel A. The results presented are averages from three separate experiments, each carried out in duplicate. Error bars represent SEM. The activity of the c-fos promoter was not significantly altered by the HDAC inhibitors at 39.5°C.
FIG. 6.

Dose response of cyclin D1 transcription to sodium butyrate and TSA in ts13 cells. ts13 cells were transfected with −183/+21 cyclin D1 luciferase reporter and an SV40-driven beta-galactosidase gene at 33.5°C. Cells were then treated with increasing concentrations of SB or TSA, and half the cells were shifted to 39.5°C. The amounts of luciferase activity, corrected for transfection efficiency, were determined at 33.5°C (A) and 39.5°C (B). The data presented are from one representative experiment carried out in triplicate, with standard errors provided (error bars). A dose-dependent increase in cyclin D1 transcription was detected but only at 39.5°C. rlu, relative light units.
Hypoacetylation of H3 at the cyclin D1 promoter upon inactivation of TAF1 in ts13 cells.
TAF1 was first identified as a histone acetyltransferase. It has been subsequently reported to acetylate nonhistone proteins, including the small subunit of the basal transcription factor TFIIE (11). Therefore, we used the technique of chromatin immunoprecipitation to directly assess the state of histone acetylation at the cyclin D1 and c-fos promoters in ts13 cells under conditions permissive and restrictive for TAF1 function. We focused on histone H3, as this protein has been reported to be the preferred histone substrate for TAF1 HAT activity in vitro (22). We found that the cyclin D1 promoter between positions −183 and +21 and the c-fos core promoter region were associated with acetylated histone H3 at 33.5°C in ts13 cells. However, the amount of acetylated H3 at the cyclin D1 promoter was reduced approximately fivefold when the cells were shifted to 39.5°C (Fig. 7B). No temperature-dependent reduction in H3 acetylation was observed at the c-fos promoter. The specificity of the chromatin immunoprecipitation reaction was demonstrated by the addition of a peptide that blocked the anti-acetylated H3 antibody. The presence of the blocking peptide prevented the precipitation of promoter fragments.
FIG. 7.

Hypoacetylation of histone H3 at the cyclin D1 promoter upon inactivation of TAF1 in ts13 cells. (A) ts13 cells and ts13 subclones that constitutively express the indicated HA-tagged TAF1 protein were maintained at 39.5°C for 24 h. Whole-cell lysates were prepared and subjected to 8% SDS-PAGE. The expression level of HA-tagged TAF1 proteins was determined by Western blotting using an anti-HA antibody. (B) Chromatin isolated from ts13 cells and ts13 subclones maintained at 33.5°C or 39.5°C was subjected to chromatin immunoprecipitation analysis using an antibody against diacetylated (acetyl Lys9 and Lys14) histone H3 (Upstate). The precipitated genomic DNA was purified and the presence of hamster cyclin D1 and c-fos promoter sequence was determined by PCR. Reaction products were analyzed by agarose gel electrophoresis and visualized by ethidium bromide staining. To control for antibody specificity, immunoprecipitation reactions were carried out in the presence (+) or absence (−) of a blocking peptide (acetyl [Ac] H3 peptide) that binds to the anti-diacetylated histone H3 antibody. PCRs carried out with the unbound supernatant or in the absence of DNA template are shown in lanes “unbound” and “no DNA,” respectively. In the absence of the AcH3 peptide, a decrease in histone H3 acetylation was observed at 39.5°C at the cyclin D1 promoter but not the c-fos promoter and only in cells expressing TAF1 proteins deficient in HAT activity (Δ844-850 and Δ848-850).
The state of histone acetylation was also examined in ts13 subclones that constitutively expressed either wild type or different mutants of hTAF1. In cells expressing WT TAF1, Δ574-595, or E742Q, comparable levels of H3 acetylation were associated with the cyclin D1 and c-fos promoters at 33.5°C and 39.5°C (Fig. 7B). By contrast, H3 remained hypoacetylated at 39.5°C in ts13 cells expressing the HAT-deficient TAF1 proteins Δ844-850 and Δ848-850 (Fig. 7B). The expression level of each TAF1 mutant was monitored at 39.5°C by anti-HA Western blotting and found to be similar for all subclones examined (Fig. 7A). These data collectively support the conclusions that loss of histone acetylation is a primary consequence of the ts13 mutation and that amino acids 848 to 850 of TAF1 are important for histone H3 acetylation at the cyclin D1 promoter.
Requirement for TAF1 activity in Sp1 site binding at the cyclin D1 promoter.
The acetylation of histones is a mechanism for increasing the accessibility of promoter DNA to transcription factors. Therefore, we examined the occupancy of the Sp1 sites of cyclin D1 at 33.5°C and 39.5°C by in vivo genomic footprinting. By monitoring protection from DMS methylation, we observed a significant increase in the accessibility of nucleotides within the three adjacent Sp1 sites at 39.5°C compared to at 33.5°C (Fig. 8A, compare lanes 2 and 3 and lanes 6 and 7). Under conditions of TAF1 inactivation (39.5°C), an increase in DNA methylation over the Sp1-B binding site and the guanine at position −148 of the Sp1-C binding site was detected on the upper strand. On the lower strand, all three Sp1 binding sites showed extensive increases in DNA methylation at 39.5°C. These footprinting studies enabled us to define a DNA element, which we have named TDE2, for TAF1-dependent DNA element 2, spanning nucleotides −109 to −159 (Fig. 8B). Within this region, the nucleotides that reproducibly displayed increases in DNA methylation mapped to the Sp1 binding sites. By contrast, temperature-dependent differences in the methylation protection patterns were not detected with genomic DNA isolated from ts13R3 cells, which constitutively express wild-type human TAF1 (Fig. 8A, compare lanes 4 and 5 and lanes 8 and 9). These findings suggest that inactivation of TAF1 HAT activity, and not heat shock effects, is responsible for the compromised interaction of Sp1 and/or other transcription factors with the Sp1 sites in the cyclin D1 promoter.
FIG. 8.

In vivo genomic footprinting reveals a reduction in Sp1 site occupancy at the cyclin D1 promoter when TAF1 is inactivated in ts13 cells. (A) In vivo DNA methylation pattern between nucleotides −180 and −65 of the hamster cyclin D1 promoter is shown. ts13 and ts13R3 cells were subjected to DMS treatment at the indicated temperature. The modified genomic DNA was chemically cleaved and the positions of methylated guanines and adenines were mapped by ligation-mediated PCR using nested cyclin D1 gene-specific primers. Both the upper and lower strands were examined. The cleavage pattern obtained from genomic DNA methylated in vitro in the absence of cellular protein is shown in lanes 1 and 10. The solid lines between the ts13 and ts13R3 samples indicate nucleotides that demonstrated increased methylation at 39.5°C in ts13 cells, but not in ts13R3 cells, in three independent experiments. The positions of putative binding sites for the transcription factors Sp1 and cyclic AMP responsive binding protein (CREB/ATF) are also shown. (B) The DNA sequence of the region analyzed is provided. The asterisks indicate nucleotides that repeatedly displayed increased methylation in ts13 but not in ts13R3 cells at 39.5°C. Protection of the Sp1 sites from DNA methylation decreased dramatically at 39.5°C.
Proximal Sp1 sites in the cyclin D1 promoter are dependent on TAF1 activity and histone acetylation.
We have previously proposed that TAF1 carries out two independent functions at the cyclin D1 promoter, one in the core region and the other at the upstream activating Sp1 sites (9). The role of TAF1 at the core appears to be in the recruitment of TFIID to the initiator, in agreement with studies carried out with yeasts (21). To determine if the Sp1 sites also can confer TAF1 dependence to the promoter, we eliminated the TAF1-dependent core region of cyclin D1 by constructing a hybrid promoter containing the Sp1 sites of cyclin D1 upstream of the TAF1-independent c-fos core. The c-fos core promoter was equally active at 33.5°C and 39.5°C in ts13 cells (Fig. 9A). The addition of the Sp1 sites onto the c-fos core (Sp1/c-fos) activated transcription at 33.5°C but not at 39.5°C (Fig. 9A). Therefore, the Sp1 sites of cyclin D1 are sufficient to confer TAF1 sensitivity to TAF1-independent promoter elements.
FIG. 9.

TAF1 function and H3 acetylation in Sp1 activation of cyclin D1 transcription. (A) Sp1 sites of the hamster cyclin D1 promoter confer TAF1 sensitivity in ts13 cells. The c-fos core promoter (c-fos) or a hybrid construct (Sp1/c-fos) consisting of Sp1 sites from the hamster cyclin D1 promoter upstream of the c-fos core was transfected into ts13 cells. The amount of luciferase activity, corrected for differences in transfection efficiency, was determined for cells incubated at 33.5°C or 39.5°C and treated with 0 (−) or 200 ng/ml (+) TSA. The data are the results from two independent experiments, each carried out in duplicate. Error bars indicate SEM. Treatment with TSA increased transcription from the cyclin D1 but not the c-fos promoter at 39.5°C in ts13 cells. rlu, relative light units. (B) Treatment with HDAC inhibitor TSA restores H3 acetylation at the cyclin D1 promoter in ts13 cells. The amount of histone H3 acetylation at the cyclin D1 (upper panels) and c-fos (lower panels) promoters in ts13 cells was determined at the indicated temperatures as described in Fig. 6B. For these studies, ts13 cells were treated with either 0 (−TSA) or 200 ng/ml TSA (+TSA) prior to chromatin immunoprecipitation and PCR analyses. Treatment with TSA was sufficient to reverse the decrease in H3 acetylation observed on the cyclin D1 promoter at 39.5°C while having no effect at the c-fos promoter. AcHe pep, blocking peptide for anti-diacetylated H3 antibody.
Next, we tested the effect of TSA on transcription from the Sp1/c-fos hybrid construct in ts13 cells. Treatment with 200 ng/ml TSA had no effect on c-fos core promoter activity but was able to restore full transcriptional activity to the Sp1/c-fos hybrid at 39.5°C (Fig. 9A). The observed stimulation of Sp1-dependent transcription upon the addition of the HDAC inhibitor suggests that acetylation of histones or other proteins within the cyclin D1 promoter is playing an important role in the activation of cyclin D1 transcription.
By chromatin immunoprecipitation, a significant increase in the amount of acetylated H3 bound to the cyclin D1 promoter was observed at 39.5°C in TSA-treated cells compared to untreated cells (Fig. 9B). By contrast, 200 ng/ml TSA had no effect on histone H3 acetylation at 33.5°C. The changes in H3 acetylation were specific to the cyclin D1 promoter, as no effect was observed at the c-fos promoter after TSA treatment (Fig. 9B). These findings lend further support to our hypothesis that histone H3 acetylation mediated by TAF1 is required for Sp1 activation of cyclin D1 transcription.
Recruitment of Sp1 partially bypasses the requirement for TAF1 activity at the cyclin D1 promoter.
The binding of Sp1 to the cyclin D1 promoter DNA appears to be disrupted at 39.5°C in ts13 cells. Therefore, would providing an alternative mechanism for recruiting Sp1 to the promoter overcome the requirement for TAF1 in activating cyclin D1 transcription? To address this question, three tandem DNA binding sites for Gal4 (UAS) were inserted immediately upstream of the Sp1 sites in the cyclin D1 promoter. The presence of the Gal4 binding sites allowed for the recruitment of Sp1 as a Gal4 DNA binding domain fusion in a manner that was independent of the endogenous Sp1 sites. We found that the cotransfection of the UAS-cyclin D1-luc reporter and a Gal4-Sp1 expression plasmid increased transcription levels at 39.5°C greater than eightfold but only from a reporter construct containing the UAS sites (Fig. 10, lower panel). A smaller, twofold increase in transcription was observed at 33.5°C (Fig. 10, upper panel). The combination of the two effects resulted in a significant decrease in the overall dependence on TAF1 activity. The inability to fully restore promoter activity at 39.5°C by artificial recruitment of Sp1 is consistent with our previous findings that the core promoter of cyclin D1 requires a function of TAF1 for recruitment of TFIID to the initiator (9).
FIG. 10.
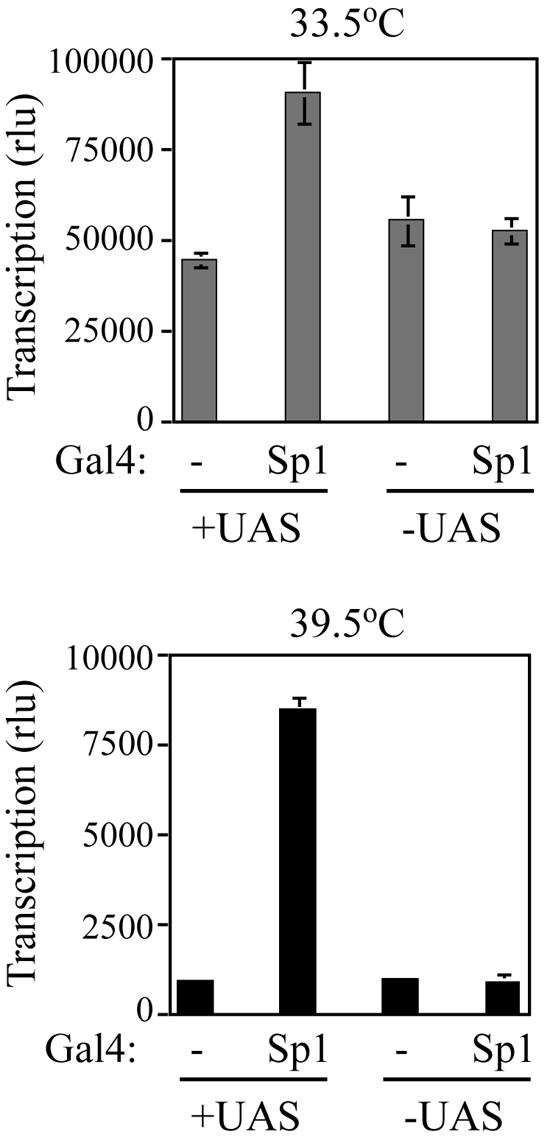
Artificial recruitment of Sp1 partially overcomes the requirement for TAF1 function at the cyclin D1 promoter. ts13 cells were transfected with −183/+21 cycD1-luc (−UAS) or −183/+21 cycD1-luc containing three tandem copies of the Gal4 DNA binding site upstream of the Sp1 sites (+UAS), the beta-galactosidase expression plasmid, and the Gal4 (−) or Gal4-Sp1 expression vector at 33.5°C. The transcriptional activity of cells maintained at 33.5°C or shifted to 39.5°C was determined and adjusted for differences in transfection efficiency. All experiments were performed in duplicate, and the averages of three independent experiments are given. The error bars represent SEM. The recruitment of Sp1 thru Gal4 binding sites increased cyclin D1 promoter activity more than eightfold at 39.5°C. A smaller twofold stimulation in transcription was detected at 33.5°C. The overall effect was a decrease in TAF1 dependence in ts13 cells. rlu, relative light units.
In summary, we provide the first piece of biochemical evidence suggesting that acetylation of histones, specifically histone H3, at the cyclin D1 promoter is TAF1 dependent in vivo and that this modification plays an important role in transcriptional regulation of cyclin D1 expression. To extend these findings we have gathered data suggesting that the binding of transcription factors to the Sp1 sites in the cyclin D1 promoter is mediated by TAF1 and histone H3 acetylation. Our current hypothesis is that at the cyclin D1 promoter, TAF1 is responsible for altering chromatin structure by histone acetylation, thereby facilitating Sp1 transcription factor binding and activating gene expression. The identification of signaling pathways that modulate the HAT activity of TAF1 will provide new insight into the regulation of cyclin D1 transcription and the control of cell cycle progression.
DISCUSSION
We have previously shown that the HAT activity of the ts13 conditional allele of TAF1 is severely compromised at elevated temperatures in vitro (4). Here, we identify additional HAT-defective hTAF1 mutants (Δ844-850 and Δ848-850) that fail to complement the G1 arrest and cyclin D1 transcription defect of ts13 cells. However, the molecular action of TAF1 HAT activity in mediating gene transcription remains unclear. Specifically, what are the physiological substrates of this enzymatic activity of TAF1 and is it important for transcriptional regulation? To tackle this problem, we have utilized the TAF1-dependent cyclin D1 promoter as our model system and provide data suggesting that TAF1 mediates acetylation of histone H3, an event required for transcription factor binding and activation of cyclin D1 transcription. We show that treatment with HDAC inhibitors restores Sp1-dependent transcriptional activation to the cyclin D1 promoter under conditions in which TAF1 HAT activity is compromised. Using the techniques of chromatin immunoprecipitation, in vivo genomic footprinting, and transient transfection, a direct correlation between the levels of histone H3 acetylation, Sp1 site occupancy, and cyclin D1 transcription can be demonstrated. These results provide evidence for a possible role of histone acetylation by TAF1 in the activation of gene transcription. Alternatively, TAF1, as a component of TFIID, may be directly or indirectly involved in the recruitment of a coactivator complex containing an enzyme that modifies the necessary histones to activate cyclin D1 transcription. Additional experiments will be needed to distinguish between these two possibilities.
There exists considerable evidence in support of a scaffolding function for TAF1in the TFIID complex. Mutations in TAF1 that disrupt the TFIID complex could lead to defects in mRNA synthesis and a cell cycle arrest. We do not favor this interpretation, because our protein-protein interaction and density gradient sedimentation studies indicate that the TAF1 mutants unable to complement the ts13 defects incorporate into TFIID. Characterization of yeast TAF1p has mapped the domain essential for TFIID assembly to a region distinct from the HAT domain (31). Deletions within the centrally conserved domain of yeast TAF1, which is homologous to the human TAF1 HAT domain, have little or no effect on the integrity of the yeast TFIID complex. Therefore, we conclude that the deletion of residues 844 to 850 selectively disrupts the HAT activity of TAF1. These amino acids display weak homology to a putative motif A in the histone acetyltransferase CBP. This region of CBP contains a potential acidic catalytic residue that, when mutated, compromises CBP HAT activity (20). Future studies will be needed to further characterize the contribution of each residue deleted in Δ848-850 to TAF1 HAT activity.
Expression of CIITA, a transcriptional activator and histone acetyltransferase, bypasses the promoter requirement for TAF1 in major histocompatibility complex class I transcription in ts13 cells (27). The restoration of promoter activity requires the acetyltransferase activity of CIITA and provides additional evidence that the primary defect in ts13 cells responsible for the mutant phenotype is a reduction in histone acetylation under nonpermissive conditions.
The promoter of cyclin D1 is an ideal model system to study TAF1 function in the transcription process. The proximal promoter of cyclin D1 contains the initiator region and binding sites for a limited number of transcription factors, including NF-κB, CREB, and Sp1. Changes in histone acetylation can influence the binding of one or more of these transcription factors. Sp1 sites are crucial for cyclin D1 transcription, as a deletion of this region in the hamster gene leads to a 10-fold decrease in promoter activity (9). Similar findings have been reported for the human and rat promoters (13, 24). Studies of epithelial cells have demonstrated that Sp1 is a G1 cell cycle-specific transcription factor and an important regulator of cyclin D1 expression (7). We have obtained evidence indicating that binding to the Sp1 sites of cyclin D1 is mediated by histone H3 acetylation. We propose that the acetylation event is TAF1 dependent and relieves the repressive chromatin environment thereby allowing for Sp1 binding, which is essential for growth factor responsiveness and recruitment of the transcriptional machinery. In support of this hypothesis, the binding of Sp1 to GC boxes in the SV40 promoter has been shown to be reduced 10- to 20-fold, although not completely abrogated, when assembled into nucleosomal DNA (16).
Cyclin D1 transcription occurs during the G1 phase of the cell cycle. Thus, it can be inferred that during other stages of the cell cycle, the cyclin D1 promoter is inactivated, possibly due to the presence of repressive nucleosomes that hinder the binding of transcription factors. The cyclin B1 promoter is regulated by its acetylation state across the cell cycle (12). Studies by Zhang et al. (44) demonstrate that the tumor suppressor INI1/hSNF5 represses cyclin D1 transcription by recruiting HDAC activity to the promoter region, thereby leading to the deacetylation of histones in the cyclin D1 promoter, transcriptional repression, and cell cycle arrest. Therefore, the robust activation of cyclin D1 transcription may be, in fact, a two-step process that first requires the removal or inactivation of an inhibitory deacetylase complex, followed by the recruitment and activation of HAT activity, which we propose is provided by TAF1, to stimulate gene transcription.
Other genes that appear to be affected by the ts13 mutation in TAF1 include EGFR (39), cyclin D3 (29, 30), and VDAC3 (26). A comparison of the promoter regions of these TAF1-dependent genes and cyclin D1 reveals two common characteristics: (i) the absence of a canonical TBP binding site or TATA box between positions −25 and −30 and (ii) the presence of GC-rich Sp1 binding sites in the proximal promoter region. Previously, we have demonstrated that TAF1 plays a role in the recruitment and binding of TFIID to core promoters that lack a strong TBP binding site. Here we present data describing a second function for TAF1 in Sp1 activation of cyclin D1 transcription. Further characterization of the ts13 transcription defect may allow us to uncover and classify promoters that are directly regulated by activities of TAF1. It appears that promoter architecture will determine which activities of the TAF1 protein are required for high levels of gene transcription.
Acknowledgments
We are indebted to K. Lewis for excellent technical support with tissue culture and molecular biology and Y. Kawata and K. Bomsztyk for assistance with the chromatin immunoprecipitation protocol. We thank R. Tjian for kindly providing the anti-HA ascites fluid, X. F. Wang for the Gal4 and Gal4-Sp1 fusion expression constructs, I. Davidson for the mouse TAF4 antibody, and S. Bajjalieh and K. Bomsztyk for critical reading of the manuscript. We especially thank past and present members of the Wang lab, including K. Lewis, R. Squillace, and K. White, for valuable discussions and support of this research.
T.L.H. was supported in part by National Research Service Award GM7750 from the National Institute of General Medical Sciences. E.L.D. was supported in part by National Research Service Award T32 GM07270 from the National Institute of General Medical Sciences. This work was supported by research project grants RPG-98-201-CCG and RSG-04-234-01-GMC from the American Cancer Society.
REFERENCES
- 1.Castro-Rivera, E., I. Samudio, and S. Safe. 2001. Estrogen regulation of cyclin D1 gene expression in ZR-75 breast cancer cells involves multiple enhancer elements. J. Biol. Chem. 276:30853-30861. [DOI] [PubMed] [Google Scholar]
- 2.Chen, J.-L., L. D. Attardi, C. P. Verrijzer, K. Yokomori, and R. Tjian. 1994. Assembly of recombinant TFIID reveals differential coactivator requirements for distinct transcriptional activators. Cell 79:93-105. [DOI] [PubMed] [Google Scholar]
- 3.Diehl, J. A., F. Zindy, and C. J. Sherr. 1997. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 11:957-972. [DOI] [PubMed] [Google Scholar]
- 4.Dunphy, E. L., T. Johnson, S. S. Auerbach, and E. H. Wang. 2000. Requirement for TAFII250 acetyltransferase activity in cell cycle progression. Mol. Cell. Biol. 20:1134-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Floros, J., T. Ashihara, and R. Baserga. 1978. Characterization of ts13 cells, a temperature-sensitive mutant of the G1 phase of the cell cycle. Cell Biol. Int. Rep. 2:259-269. [DOI] [PubMed] [Google Scholar]
- 6.Gill, G. 2001. Regulation of the initiation of eukaryotic transcription. Essays Biochem. 37:33-43. [DOI] [PubMed] [Google Scholar]
- 7.Grinstein, E., F. Jundt, I. Weinert, P. Wernet, and H.-D. Royer. 2002. Sp1 as G1 cell cycle phase specific transcription factor in epithelial cells. Oncogene 21:1485-1492. [DOI] [PubMed] [Google Scholar]
- 8.Hayashida, T., T. Sekiguchi, E. Noguchi, H. Sunamoto, T. Ohba, and T. Nishimoto. 1994. The CCG1/TAFII250 gene is mutated in thermosensitive G1 mutants of the BHK21 cell line derived from golden hamster. Gene 141:267-270. [DOI] [PubMed] [Google Scholar]
- 9.Hilton, T. L., and E. H. Wang. 2003. TFIID recruitment and Sp1 activation: dual function of TAF1 in cyclin D1 transcription. J. Biol. Chem. 15:12992-13002. [DOI] [PubMed] [Google Scholar]
- 10.Holstege, F. C. P., E. G. Jennings, J. J. Wyrick, T. I. Lee, C. J. Hengartner, M. R. Green, T. R. Golub, E. S. Lander, and R. A. Young. 1998. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95:717-728. [DOI] [PubMed] [Google Scholar]
- 11.Imhof, A., X.-J. Yang, V. V. Ogryzko, Y. Nakatani, A. P. Wolffe, and H. Ge. 1997. Acetylation of general transcription factors by histone acetyltransferases. Curr. Biol. 7:689-692. [DOI] [PubMed] [Google Scholar]
- 12.Katula, K., A. Fields, P. Apple, and T. Rotruck. 2002. Cell-cycle specific changes in the human cyclin B1 gene regulatory region as revealed by response to trichostatin A. Arch. Biochem. Biophys. 401:271-276. [DOI] [PubMed] [Google Scholar]
- 13.Kitazawa, S., R. Kitazawa, and S. Maeda. 1999. Transcriptional regulation of cyclin D1 gene by CpG methylation status in promoter region. J. Biol. Chem. 274:28787-28793. [DOI] [PubMed] [Google Scholar]
- 14.Kuo, M., and C. D. Allis. 1998. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 20:615-626. [DOI] [PubMed] [Google Scholar]
- 15.Kuo, M., J. Zhou, P. Jambeck, M. Churchill, and C. D. Allis. 1998. Histone acetyltransferase activity of yeast Gcn5p is required for the activation of target genes in vivo. Genes Dev. 12:627-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li, B., C. C. Adams, and J. L. Workman. 1994. Nucleosome binding by the constitutive transcription factor Sp1. J. Biol. Chem. 269:7756-7763. [PubMed] [Google Scholar]
- 17.Li, J. M., M. B. Datto, X. Shen, P. P. Hu, Y. Yu, and X. F. Wang. 1998. Sp1, but not Sp3, functions to mediate promoter activation by TGF-beta through canonical Sp1 binding sites. Nucleic Acids Res. 26:2449-2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marmorstein, R. 2001. Structure of histone acetyltransferases. J. Mol. Biol. 311:433-444. [DOI] [PubMed] [Google Scholar]
- 19.Marmorstein, R., and S. Roth. 2001. Histone acetyltransferases: function, structure, and catalysis. Curr. Opin. Genet. Dev. 2:155-161. [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Balbas, M., U. Bauer, S. Nielsen, A. Brehm, and T. Kouzarides. 2000. Regulation of E2F1 activity by acetylation. EMBO J. 19:662-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mencia, M., and K. Struhl. 2001. Region of yeast TAF130 required for TFIID to associate with promoter. Mol. Cell. Biol. 21:1145-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizzen, C. A., X. J. Yang, T. Kokubo, J. E. Brownell, A. J. Bannister, H.-T. Owen, J. Workman, L. Wang, S. L. Berger, T. Kouzarides, Y. Nakatani, and C. D. Allis. 1996. The TAF(II)250 subunit of TFIID has histone acetyltransferase activity. Cell 87:1261-1270. [DOI] [PubMed] [Google Scholar]
- 23.Mueller, P. R., B. Wold, and P. A. Garrity. 1992. Preparation of genomic DNA from monolayers cells for DMS footprinting, vol. 2. John Wiley and Sons, Inc., Boston, Mass.
- 24.Nagata, D., E. Suzuki, H. Nishimatsu, H. Satonaka, A. Goto, M. Omata, and Y. Hirata. 2001. Transcriptional activation of the cyclin D1 gene is mediated by multiple cis-elements, including Sp1 sites and a cAMP-responsive element in vascular endothelial cells. J. Biol. Chem. 276:662-669. [DOI] [PubMed] [Google Scholar]
- 25.Neuwald, A., and D. Landsman. 1997. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem. Sci. 22:154-155. [DOI] [PubMed] [Google Scholar]
- 26.O'Brien, T., and R. Tjian. 2000. Different functional domains of TAFII250 modulate expression of distinct subsets of mammalian genes. Proc. Natl. Acad. Sci. USA 97:2456-2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raval, A., T. K. Howcroft, J. Weissman, S. Kirshner, X. Shu, K. Yokoyama, J. Ting, and D. Singer. 2001. Transcriptional coactivator, CTIIA, is an acetyltransferase that bypasses the promoter requirement for TAFII250. Mol. Cell 7:105-115. [DOI] [PubMed] [Google Scholar]
- 28.Roeder, R. 1996. The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem. Sci. 21:327-335. [PubMed] [Google Scholar]
- 29.Rushton, J. J., R. A. Steinman, and P. A. Robbins. 1997. Differential regulation of transcription of p21 and cyclin D1 conferred by TAFII250. Cell Growth Differ. 8:1099-1104. [PubMed] [Google Scholar]
- 30.Sekiguchi, T., E. Noguchi, T. Hayashida, T. Nakashima, H. Toyoshima, T. Nishimoto, and T. Hunter. 1996. D-type cyclin expression is decreased and p21 and p27 CDK inhibitor expression is increased when tsBN462 CCG1/TAFII250 mutant cells arrest in G1 at the restrictive temperature. Genes Cells 1:687-705. [DOI] [PubMed] [Google Scholar]
- 31.Singh, M. V., C. E. Bland, and A. Weil. 2004. Molecular and genetic characterization of a Taf1p domain essential for yeast TFIID assembly. Mol. Cell. Biol. 24:4929-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sterner, D., and S. L. Berger. 2000. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64:435-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strauss, E. C., and S. H. Orkin. 1997. Guanine-adenine ligation-mediated PCR in vivo footprinting. Methods 11:164-170. [DOI] [PubMed] [Google Scholar]
- 34.Struhl, K. 1998. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 12:599-606. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki-Yagawa, Y., M. Guermah, and R. G. Roeder. 1997. The ts13 mutation in the TAFII250 subunit (CCG1) of TFIID directly affects transcription of D-type cyclin genes in cells arrested in G1 at the nonpermissive temperature. Mol. Cell. Biol. 17:3284-3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanese, N. 1997. Small-scale density gradient sedimentaion to separate and analyze multiprotein complexes. Methods 12:224-234. [DOI] [PubMed] [Google Scholar]
- 37.Tanner, K. R., M. Trievel, M. Kuo, R. Howard, S. Berger, C. D. Allis, R. Marmorstein, and J. Denu. 1999. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J. Biol. Chem. 274:18157-18160. [DOI] [PubMed] [Google Scholar]
- 38.Trievel, R. C., J. R. Rojas, D. E. Sterner, R. N. Venkataramani, L. Wang, J. Zhou, C. D. Allis, S. L. Berger, and R. Marmorstein. 1999. Crystal structure and mechanism of histone acetylation of the yeast GCN5 transcriptional coactivator. Proc. Natl. Acad. Sci. USA 96:8931-8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vargas, G. A., J. M. Isas, E. Fantino, J. J. Gargus, and H. T. Haigler. 1998. CCG1/TAF(II)250 regulates epidermal growth factor receptor gene transcription in cell cycle mutant ts13. J. Cell. Physiol. 176:642-647. [DOI] [PubMed] [Google Scholar]
- 40.Walker, S. S., W.-C. Shen, J. C. Reese, L. M. Apone, and M. R. Green. 1997. Yeast TAFII145 required for transcription of G1/S cyclin genes and regulated by the cellular growth state. Cell 90:607-614. [DOI] [PubMed] [Google Scholar]
- 41.Wang, E. H., and R. Tjian. 1994. Promoter selective transcriptional defect in cell cycle mutant ts13 rescued by hTAFII250 in vitro. Science 263:811-814. [DOI] [PubMed] [Google Scholar]
- 42.Wang, E. H., S. Zhou, and R. Tjian. 1997. TAFII250 dependent transcription of cyclin A is directed by ATF activator proteins. Genes Dev. 11:2658-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan, G.-Z., and E. B. Ziff. 1997. Nerve growth factor induces transcription of the p21 WAF1/CIP1 and cyclin D1 genes in PC12 cells by activating the Sp1 transcription factor. J. Neurosci. 17:6122-6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang, Z.-K., K. P. Davies, J. Allen, L. Zhu, R. G. Pestell, D. Zagzag, and G. V. Kalpana. 2002. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol. Cell. Biol. 22:5975-5988. [DOI] [PMC free article] [PubMed] [Google Scholar]