

Abstract

Understanding enzyme mechanisms is essential for unraveling the complex molecular machinery of life. In this review, we survey the field of computational enzymology, highlighting key principles governing enzyme mechanisms and discussing ongoing challenges and promising advances. Over the years, computer simulations have become indispensable in the study of enzyme mechanisms, with the integration of experimental and computational exploration now established as a holistic approach to gain deep insights into enzymatic catalysis. Numerous studies have demonstrated the power of computer simulations in characterizing reaction pathways, transition states, substrate selectivity, product distribution, and dynamic conformational changes for various enzymes. Nevertheless, significant challenges remain in investigating the mechanisms of complex multistep reactions, large-scale conformational changes, and allosteric regulation. Beyond mechanistic studies, computational enzyme modeling has emerged as an essential tool for computer-aided enzyme design and the rational discovery of covalent drugs for targeted therapies. Overall, enzyme design/engineering and covalent drug development can greatly benefit from our understanding of the detailed mechanisms of enzymes, such as protein dynamics, entropy contributions, and allostery, as revealed by computational studies. Such a convergence of different research approaches is expected to continue, creating synergies in enzyme research. This review, by outlining the ever-expanding field of enzyme research, aims to provide guidance for future research directions and facilitate new developments in this important and evolving field.
Introduction
Enzymes differ from conventional catalysts in their ability to accelerate chemical reactions, often exceeding a million-fold.1−11 This unparalleled catalytic power makes them indispensable for carrying out a vast array of biological functions that would not otherwise be possible.12 However, enzymes are not ordinary catalysts; they exhibit exceptional selectivity in recognizing and binding specific substrates, ensuring efficient catalysis while preventing wasteful or undesirable side reactions.13−17 What further distinguishes enzymes is that they are regulated by multiple and customized mechanisms so that each chemical process occurs with exquisite timing within the relevant cellular context.18−21 Throughout evolution, these unique features of enzymes have exerted evolutionary pressure, resulting in the optimization of their functions and even the development of new capabilities.22−29 In addition, the design of robust enzymes has received considerable attention in recent years.30 Therefore, a comprehensive understanding of the mechanisms of enzyme action has become essential to maximize their potential in a wide range of applications, from biotechnology31−34 to medicine.35−42 This understanding has also contributed significantly to our broader understanding of chemical processes in biology and life.
Extensive computational studies have been devoted to elucidating the role of enzymes in catalyzing reactions.3,43−48 Initially, much of this research focused on understanding how enzymes stabilize the high-energy transition state (TS) of a reaction—a concept famously theorized by Pauling.49−51 This stabilization effectively reduces the activation free energy required for reactions to proceed, resulting in a significant increase in the reaction rates. However, as scientific investigation has progressed, several alternative mechanisms have been proposed, including reactant-state destabilization,45,52 coupled dynamics,53−56 chemical control,57 chemical dynamics,58,59 and others.60,61 Recently, many investigations have focused on understanding protein allostery in the context of regulation of enzyme function.25,56,62−67 In principle, allosteric regulation involves the binding of effector molecules at sites other than the enzyme’s active site, thereby affecting its catalytic activity. Understanding these regulatory mechanisms has not only provided valuable insights into how enzymes respond to specific cellular demands, such as in cellular signaling,19,68,69 but has also shed light on the underlying factors contributing to the development of various human diseases and therapeutic strategies to target them.38,39,41,70
The mechanisms of ligand binding have also been extensively studied, leading to an ongoing debate between the induced-fit13 and conformational selection mechanisms.15,27,62,71−74 In the induced-fit model, the enzyme undergoes a conformational change upon binding of its substrate, rearranging its active site to accommodate the ligand. In contrast, the conformational selection model, also known as the population-shift model, assumes that the enzyme has multiple pre-existing conformations, one of which is a favorable fit to the ligand. While computational studies have played a central role in the investigation of these mechanisms,75−77 the search for the definitive mechanism behind ligand binding remains an active area of research. Moreover, molecular recognition extends beyond substrates and cofactors to include allosteric regulators, target proteins, and even drug binding. Therefore, developing the ability to theoretically model these processes will allow a more detailed exploration of how enzymes recognize and bind ligands and ultimately help to better understand these essential biological interactions.
Within the context of the topics outlined above, this review discusses the current state of several important areas within the field of computational enzymology (Figure 1). The discussion begins with a brief overview of recent advances, with particular emphasis on the enzyme systems that have been the focus of recent research from our laboratories. We then discuss the challenges inherent in studying complex enzymes, engineering enzymes for non-natural reactions, and developing innovative drugs targeting enzymes. Throughout this discussion, our goal is to highlight recent advances while recognizing the ongoing challenges that continue to drive research in the field. It is anticipated that this review will provide guidance for future directions and developments in the field.
Figure 1.
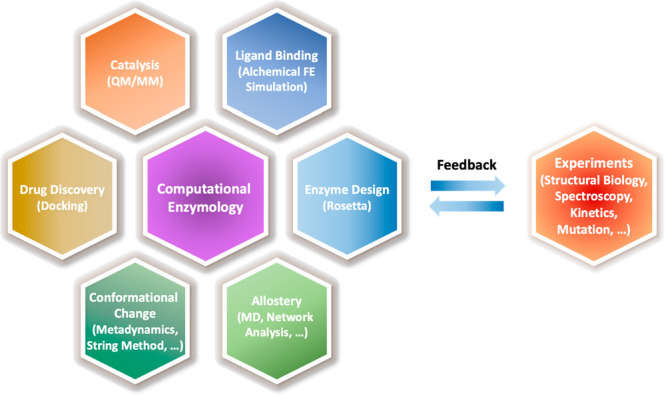
Topics in computational enzymology covered in this review and representative computational approaches. The synergistic connection with experiments is also indicated. Abbreviations: MD: molecular dynamics; QM/MM: quantum mechanical and molecular mechanical method; FE: free energy.
Modeling of Complex Enzyme Catalytic Mechanisms
While transition-state (TS) stabilization remains a fundamental principle,12,50,51,78 it represents only the tip of the iceberg underlying the full spectrum of catalytic strategies employed by enzymes.45,61,79,80 These strategies include, but are not limited to, acid–base catalysis,11,81 covalent catalysis,11,61,82 solvation/desolvation,83−85 reactant-state (RS) destabilization,45,52 strain distortion,86,87 near-attack conformation,61,88 chemical control,89,90 quantum tunneling,91−93 promoting vibration,60,94,95 and coupled dynamics.3,96−98 Some of these strategies, such as acid–base catalysis and covalent catalysis, are enzyme-specific, while others contribute more generally to catalytic acceleration. However, their impact on catalytic efficiency (i.e., kcat) varies and depends on the context (e.g., reactions and enzymes), with TS stabilization often being the most significant contributor. Furthermore, recognizing that many enzymes involve small- to large-scale conformational motions, recent studies,53,99−104 including our own,98,105 have investigated the interplay between protein dynamics and enzymatic catalysis, revealing how these motions contribute to enzyme catalysis and regulation (see the discussion in the section “Conformational Substrates and Their Role in Enzyme Activity”). Taken together, these mechanisms unraveled by computational studies have greatly enriched our understanding of how life orchestrates chemical reactions and evolves new functionalities.23,27,106 From the perspective of enzyme design and evolution, this mechanistic understanding also has important implications for further enhancing enzyme activity.
Computational studies in the area typically begin by determining the catalytic barriers associated with different chemical reaction steps (Figure 2).44,45,107 These barriers can be compared to experimental reaction rates through transition state theory (TST),44,108 as described by the equation:
| 1 |
In this equation, γ represents the generalized transmission coefficient and β = (kBT)−1, where kB is the Boltzmann constant and T is the temperature. The term ν‡, according to gas-phase reaction theory, is equal to (βh)−1, where h is Planck’s constant, and characterizes the inverse time for transitioning from the transition state to the product state (PS). Since the classical (recrossing) contribution to γ is typically in the range of 0.5 to 1.0,109−112 its contribution to the reaction rate is often ignored in most studies. Enzymes, which play a significant role, are primarily limited to reactions involving proton and/or hydride transfers with pronounced quantum mechanical effects, such as tunneling.8,91−93,113
Figure 2.

(A) Free energy landscapes visualizing the hierarchy of protein motions on different time scales. State A represents the reactant state in which multiple substates exist in equilibrium. The rate constants for exchange between substates occur on time scales ranging from picoseconds (ps) to nanoseconds (ns). The catalytic reaction takes place on microsecond (μs) ∼ millisecond (ms) time scales, forming products represented as state B. (B) Enzyme catalysis from the perspective of conformational substates, where the free energy landscape is plotted as a function of the catalytic reaction coordinate (ξ) and conformational substate (z). In each substate, the catalytic barrier and reaction free energy can be calculated by applying the QM/MM free energy simulation methods. Then, the overall reaction rate can be expressed as the sum of the population-weighted rates (eq 3).
The activation free energy, denoted as ΔG‡ in eq 1, is defined as the free energy difference between the transition state and the reactant state. In practical applications, it is often approximated as the difference between the potentials of mean force (PMFs) for TS and RS, as
| 2 |
where W(ξX) represents the PMF value at a given reaction coordinate value ξ = ξX, where X refers to either TS or RS.45 The calculation of PMF is commonly performed using enhanced sampling techniques, such as umbrella sampling114 and variants thereof,48,115 with stratification into narrow simulation windows to facilitate configuration sampling. Data-reweighting can be accomplished with the weighted histogram analysis method (WHAM),116 the multistate Bennett acceptance ratio (MBAR),117,118 the variational free energy profile (vFEP) method,119,120 and the dynamic histogram analysis method (DHAM).121 In recent years, methods such as metadynamics,122−124 string methods,125,126 and their variants127−132 have emerged as valuable tools for determining the minimum free energy paths (MFEP) and the corresponding PMF values along these paths. Nonetheless, these simulation techniques must be combined with hybrid quantum mechanical and molecular mechanical (QM/MM) potentials, using both semiempirical (SE) and ab initio/density functional theory (AI/DFT) methods as QM methods, to enable bond breaking and formation.133−140 Recent studies have also explored machine learning potentials (MLPs) employing deep learning techniques, which show great promise in speeding up calculations by using potentials that are surrogates for time-consuming QM methods,141−148 as well as approaches that use low-level QM theories to drive the QM/MM molecular dynamics (MD) simulations while high-level QM theories are used to provide corrections to the energy on the fly138,149 or as a posterior correction.150−163 Alternatively, the low-level QM potentials are reparameterized to reproduce results at high-level QM theories.135,164−168 While each approach has its own strengths and weaknesses—e.g., speed versus transferability in MLPs and reparameterized SE-QM models—the ultimate goal may be to incorporate high-level QM energies and forces directly into the simulations, e.g., by applying multiple time step approaches, to overcome the limitations of MLPs and SE-QM/MM methods without significantly increasing the overall computational cost.138,149,169,170
Despite substantial progress in free energy barrier calculations, elucidating the complete mechanism of enzymes remains a formidable theoretical challenge.45,171−173 To begin with, experimentally determined rates, such as kcat, derived by fitting the Michaelis–Menten equation, provide only apparent values that are collective reporters of the overall reaction without providing insights into the microscopic steps in catalysis. This limitation can lead to misleading interpretations of the specifics of reaction steps and underlying mechanisms, especially when catalytic turnover involves multiple reaction steps. To address this problem, integrated approaches combining various methods such as kinetics,174,175 high-throughput microfluidic kinetics,176 kinetic isotope effects,87,113,177 single-molecule spectroscopy,73,100,178−180 vibrational spectroscopy,9,181 X-ray crystallography,182−185 and nuclear magnetic resonance (NMR) spectroscopy7,97,186−192 have been used in conjunction with simulations to characterize individual steps and associated transient intermediates. NMR spectroscopy, in particular, has the advantage of providing both quantitative and qualitative insights into protein dynamics across time-scales ranging from picoseconds (ps) to hours, and at atomic resolution.7,189 An example demonstrating this is a study of the dynamics of a transition state mimic bound to the nucleoside monophosphate kinase UmpK.193 Based on the results, the authors proposed that the transition state is a rigid, low entropy state that can facilitate the chemical reaction while suppressing unproductive structural fluctuations. NMR-derived order parameters have also been used to estimate residue-specific contributions to entropy for the binding of substrate molecules to proteins;194−196 the estimated binding entropy can serve as a benchmark for computationally derived order parameters, such as those from MD simulations.71,197 Despite these advantages, NMR spectroscopy has limitations. For example, NMR spin relaxation rates include contributions from all of the parallel processes occurring during enzymatic reactions, which complicates their interpretation. Under these circumstances, biochemical tuning, such as the use of nonhydrolyzable ATP analogues198 or amino acid substitutions,199,200 can be used to study each process separately, which can also be combined with molecular simulations to provide more detailed insights into each process. Another widely used experimental approach is isothermal titration calorimetry (ITC), which can quantify the enthalpic and entropic contributions to the binding step in catalysis. However, this methodology provides only total contributions and can separate neither the contributions of protein versus water solvent in molecular recognition nor the contributions of individual residues.
From a computational point of view, the computational cost increases rapidly as the complexity of a reaction mechanism increases.120,201 For example, many enzymes catalyze their reactions through complex multistep processes, requiring the evaluation of the catalytic barrier for each step to identify the rate-determining step (RDS) and the underlying mechanisms.57,201,202 When alternative mechanisms are possible, each mechanism must be thoroughly investigated and compared with experimental rates or product distributions.57,90,201−203 This situation highlights the importance of being able to simulate multiple reaction steps within a single simulation framework to expedite the exploration of these mechanisms. Therefore, traditional methods such as umbrella sampling are less applicable, and new, more efficient approaches are needed. In addition, improving the efficiency of filtering out unrealistic mechanisms at low computational cost, as exemplified by EnzyDock’s consensus docking of reaction mechanisms,204 can significantly reduce the number of mechanisms and reaction steps that require computationally intensive investigation. Together, these innovations will serve to bridge the gap between experimental and computational approaches in enzyme mechanism studies.
Having identified the reaction mechanisms, the logical next step in exploring the catalytic power of an enzyme is to dissect its individual contributions to both the catalytic barrier and the overall free energy of the reaction.205,206 This can be done by decomposing the free energy values, including enthalpy–entropy decomposition,48,207,208 specifying the contributions of individual residues,203,209−220 and taking into account the electrostatic solvation effects of the enzyme and the surrounding solvent.164,221 While computational methods, some of which are reviewed in ref (222), have been instrumental in providing detailed insights into these aspects, they require experimental validation. To this end, a widely used experimental approach is to introduce amino acid substitutions into the enzyme and measure their effects on catalytic activity.105,223−225 These perturbations then provide feedback on computational predictions and accuracy of the simulation models. Likewise, simulations complement these efforts by providing additional perspectives on enzyme mechanisms. A notable example of this synergy between computation and experiment is our recent study of adenylate kinase.98,105 In this study, we investigated the role of catalytic residues in facilitating the key catalytic step of the enzyme using both QM/MM simulations and amino acid replacements to characterize the effects of individual residues on the kinetic parameters of the enzyme. Interestingly, this investigation revealed that these catalytic residues also have a significant effect on the global dynamics of the enzyme. This finding demonstrates the interplay between enzyme activity and protein dynamics that underlies the robust catalytic activity of this enzyme and the importance of combining simulations and experiments.
Hierarchy of Functional Protein Motions
Enzymes are inherently dynamic, involving a variety of motions on different time and length scales to coordinate their biological functions (Figure 2A).5,6,8,51,226−228 Among these motions, fast time-scale motions, such as bond vibrations and bending motions, manifest as fluctuations around the average enzyme structures during reactions and contribute to the overall entropy of these processes. MD simulations have proven powerful in exploring these fast time-scale motions and have provided valuable insights into their role in enzymatic function.43,226,229 For example, numerous QM/MM MD simulation studies have shown that active site residues of enzymes rearrange rapidly following catalytic reactions, providing transition state stabilization and thereby facilitating the catalytic transformation from reactant to product.3,44,98,174,203,220,230−234 Consequently, mutations in these residues directly impact their catalytic processes, which can be readily explored using both experiments and simulations.95,105,174,181,217,234,235
In contrast, large-scale collective protein motions, which occur on time scales ranging from microseconds (μs) to milliseconds (ms), play distinctive roles in enzyme function.53,186,190,198,227,236 Perhaps, their most prominent role is to bridge different conformational states of an enzyme, achieved by driving interconversion between them during processes such as ligand binding,180,198,237−240 allosteric activation,241−244 and even the chemical reaction.98,101,102,105 While their role in modulating enzymes’ conformational states is easy to conceptualize, understanding how they influence catalytic mechanisms remains elusive. For example, functional links between large-scale protein motions and key enzymatic processes may exist in enzymes with active sites located at flexible hinges.245 Particularly, in these enzymes, active sites undergo open–closed conformational transitions that accompany ligand binding and release,179,186,198,246 as well as catalysis.100 Numerous simulation studies, including long MD simulations using all atom potentials,229,247−250 have explored their role in enzyme function and provided valuable insights into protein dynamics relevant to conformational changes. Nevertheless, studying these large-scale motions and their involvement in the catalytic process remains a theoretical challenge and requires further development of advanced, enhanced sampling techniques to efficiently characterize their complex free energy landscapes.229,248,251−254 In addition, in recent years, the proposed involvement of protonation state changes during protein conformational change and function255−259 has added complexity to simulation studies. Such studies require the application of constant pH MD (CpMD) simulation techniques260 and/or the simulations must be performed at different protonation states.
The mechanisms governing large-scale conformational changes in enzymes have been studied extensively, particularly in the context of substrate binding and product release.15,27,71,73,74,76,237,240 This has led to the development of two widely discussed models: the induced-fit model13 and the conformational selection model.62,72,73,261 In the induced-fit model, enzymes are postulated to adopt an open conformation in their apo state. Upon ligand binding, the molecules undergo a conformational transition to a closed state. Following the reaction and subsequent release of the reaction products, the enzymes return to their open conformation, completing the catalytic cycle. In contrast, the conformational selection model assumes that enzymes populate multiple conformations including the conformation with optimal geometry for ligand binding even in the absence of ligands (e.g., substrates) and that ligand binding occurs predominantly with the closed conformation. In theory, these two models can be distinguished by examining the accessibility of the closed conformation in the apo state and/or by investigating the time scales of conformational changes between the enzyme in the apo versus holo states.240,262,263 For real enzymes, however, complications arise because most experimentally measured opening and closing rates reflect the residence times of the enzyme in its respective closed and open states rather than the actual time scales of conformational motions.73,180,187,198 Furthermore, as we have recently pointed out, caution must be exercised when comparing protein opening/closing motions in the presence of natural substrates versus non-natural substrates or inhibitors.105,240 For example, using adenylate kinase as a model enzyme, we demonstrated differences in the opening rates of the enzyme between the two ligand-binding subdomains, and these rates were significantly affected by the nature of the ligands (i.e., reactants, products, or inhibitors).98,240 These studies were carried out by applying the string free energy simulation method126 to simulate the opening of the enzyme and the concurrent release of ligands, which clearly showed a three-step process in enzyme opening and ligand release and differences in the free energy barriers of enzyme opening between the reactant and product states. These results suggest a measurable influence of active site residues on modulating the free energy of enzyme opening. In addition, the sequence of opening events differed between the apo- and holo-state enzymes.240 These results suggest that the conformational changes of the enzyme are more complex than those captured by a simple two-state model. Therefore, interpreting experimental data by fitting them to a two-state model may potentially lead to a misleading understanding of the underlying mechanism of enzyme opening and closing.240,264 Furthermore, for enzymes to exhibit robust activity, they must have evolved to minimize the closed-to-open conformational transitions during catalytic reactions. Ideally, they should remain in the closed conformation and transition to the open state only when products are released.105 In such scenarios, even if the chemical step is the actual rate-limiting step, experimentally measured rates may overlap with enzyme opening rates if they represent residence times. In addition, if these enzymes are small, the two events, i.e., conformational change and enzymatic catalysis, may not be completely separable, as the enzyme must share the same functional/structural units to perform both tasks as we have shown in adenylate kinase.98,105 In this case, therefore, it is possible that there exists a connection between large-scale protein motions and enzymatic processes, at least at the mechanistic level.
Conformational Substrates and Their Role in Enzyme Activity
In recent years, the existence of conformational substates within enzyme–substrate complexes and their role in enzymatic activity has been increasingly recognized.14,21,265−274 In essence, this concept proposes that enzymes do not function exclusively in a single, well-defined conformation in the holo-state. Instead, they exist in a dynamic equilibrium of multiple similar conformations, each associated with different degrees of catalytic activity. Early evidence supporting this idea came from a single-molecule experiment performed by English et al. in 2006.178 In their study, the authors observed a wide range of waiting times between catalytic events of the enzyme β-galactosidase. These variations, however, could not be explained by simple diffusion of the substrate to and from the enzyme alone. Instead, the authors proposed fluctuations in the catalytically relevant conformations of the enzyme, characterized by conformational transition time scales slower than those of substrate binding and catalysis.178,275
This observation can also be understood as a mechanistic coupling between large-scale conformational fluctuations and enzymatic catalysis (Figure 2B). However, due to the disparate time scales between the two, this coupling does not necessarily occur dynamically during the catalytic process. Instead, the enzyme continuously explores its conformational landscapes and establishes an equilibrium distribution along these landscapes. During this exploration, catalytic events proceed as expected, but with different catalytic barriers depending on the conformational state of the enzyme.99,273,276,277 In this scenario, the overall catalytic rate can be expressed as the sum of the population-weighted rates, represented as
| 3 |
where ρ represents the population at the conformational coordinate z along which conformational fluctuations occur, and kmicro denotes the (microscopic) rate constant of each conformational state (Figure 2B). If the substates are discrete, as implied in the case of β-galactosidase mentioned above,178 the integral can be replaced by a summation over the accessible substates. A pertinent example of such dynamic motions involves low-frequency hinge motions observed in enzymes with active sites located at their hinges.98,103,278 In these cases, motions along the hinges can modulate the relative orientations of the reacting species, transitioning between optimal and suboptimal orientations for catalysis or bringing them close together. These hinge motions can also contribute to the establishment of (discrete) conformational substates in the catalytic conformations of the enzyme. To computationally study this scenario, it is necessary to incorporate a conformational coordinate z into the reaction coordinate alongside the chemical events. This approach has recently been exploited by our group98 and others.104 In these studies, while the conformational coordinate z was restrained to discrete values, QM/MM simulations were performed to determine the catalytic free energy profiles and to compare the effects of restraints on the catalytic barrier. These simulations clearly show impacts on the catalytic barrier and/or reaction free energy in response to the restrained slow protein motions, supporting the proposed mechanistic coupling between the two events. Alternatively, QM/MM simulations can be performed for multiple protein conformations, which are sampled prior to the QM/MM simulations using enhanced sampling techniques, as demonstrated for dihydrofolate reductases.277 In addition, conformational substates and their role in catalysis can be studied by designing amino acid substitutions that selectively perturb either the catalytic barrier or the population distribution; in some cases, both aspects can be affected simultaneously, as recently demonstrated by Yabukarski et al. using ketosteroid isomerase as a model enzyme.273 However, despite these valuable studies, gaining a concrete understanding of the full spectrum of these conformational substates and their associated time scales remains an important subject for further investigation.
Protein Allostery for Enzyme Activity Regulation
Since the introduction of the term “allostery” in 1961,261 the concept has evolved to encompass long-range communication within proteins that affects activities at the active sites, as well as the influence of effector molecule binding.38,63,67,68,279 In the case of multimeric enzymes, this mode of communication is often manifested as positive or negative cooperativity.21,56,64,280 For instance, consider F1-ATPase, whose activity responds cyclically to the rotation of a central γ-subunit.281,282 Despite the substantial separation of more than 50 Å between the three active sites of the enzyme, they exhibit negative cooperativity, ensuring that only one active site functions at any given time to achieve high energy conversion efficiency.239 This phenomenon highlights the communication between spatially separated active sites within the enzyme. Another important example is found in protein kinases, where allosteric regulation plays a central role in cell signaling.19,23,283 Because of its importance, protein allostery has been studied extensively.23,64,243,249,284−287 While more comprehensive reviews are available elsewhere,21,65,66,279 our discussion here focuses on protein allostery within the context of protein dynamics and conformational landscapes.
Allosteric communication can occur through two primary mechanisms: conformationally driven and entropically driven.21,286,288,289 The conformationally driven mechanism, which is often associated with changes in protein structure, is relatively straightforward to study using structural biology techniques such as X-ray crystallography. This type of allostery, similar to ligand binding, can result from conformational changes resembling the induced-fit model, or it can involve transitions between different conformational states, such as population shift.103,290 Consequently, the principles established to explain ligand binding mechanisms can be readily applied to understand conformational allostery, while the investigation of the conformational change mechanism is largely left for computational studies. Notable examples of such studies include the Markov state modeling (MSM) studies of cSrc protein kinase,241 the metadynamics simulations of cyclin-dependent protein kinase 5291 and adenylyl cyclase,274 the string method simulation study of insulin receptor kinase (IRK),244 and the alchemical free energy simulation study of insulin-like growth factor 1 kinase (IGF-1RK).243 However, in contrast to the structure prediction problem, which can be approached using pairwise interactions, allosteric networks are generally orders of magnitude more complex and rely on clusters of interacting networks on multiple levels.10 In addition, the energetic separation between microscopic structural states in allosteric mechanisms can be minute, on the order of a few “kBT”,97 and therefore difficult to accurately quantify with existing computational approaches and force fields.
The entropically driven mechanism is characterized by minimal or no discernible changes in protein structure.286,288,290,292 Instead, it operates by modifying the dynamic behavior of enzymes, such as correlated protein dynamics (that can be characterized by covariance analysis293,294), to induce entropic effects and resultant changes in the free energy landscapes of the enzyme as discussed below. Notably, since no conformational changes are involved, these changes occur through the adjustment of protein–ligand and intraprotein interaction networks in response to ligand binding/release. Theoretical studies of this mechanism, however, have been challenging, which has led to the development of various approaches,295,296 such as principal component and covariance analyses,293,294,297 network models,298,299 normal mode/elastic network models,300−302 statistical coupling analysis,303 rigid residue scan304,305 and machine learning approaches,306,307 as well as through the determination of free energy profiles along the allosteric conformational change pathways.241,244,252 These methods have allowed the investigation of protein allostery in protein activation,238,244,249,308 ligand binding/recognition,187,238,285,309,310 and even catalysis,187,243,311 which revealed the complex role of protein dynamic motions in these functions and allosteric hotspots.305,310 In parallel, experimental techniques, such as amino acid substitutions223,312 and NMR spectroscopy,191,284,292,313 have been used to quantify changes in protein dynamics attributable to allosteric effects. This collaborative effort between experimental and theoretical methods serves as a powerful tool for understanding the intricate mechanisms underlying protein allostery and its role in biological processes.314 One particular example of the synergy between simulation and experiment can be found in our study of F1-ATPase.239 In this study, we used single-molecule spectroscopy data measured for the enzyme315,316 and previous computational results317,318 to construct free energy profiles of the enzyme along the entire 360° rotation cycle. The determined free energy profiles, together with covariance analyses of the enzyme,319 can explain the molecular events associated with the enzyme’s power-stroke process and the overall energy conversion efficiency achieved by modulating the free energy of substrate vs product binding during the rotational cycle of the enzyme.
Interestingly, these two types of allostery need not be mutually exclusive. Our recent computer simulation studies examining the activation mechanism of IRK and IGF-1RK have shown that they can indeed coexist within a single protein fold, each operating in distinct functional steps (Figure 3).243,244 For example, the conformational activation of these kinases is modulated by conformational allostery. In particular, phosphorylation of their activation loop (A-loop) restricts access to their inactive conformation, while the kinase predominantly adopts this inactive conformation in the inactive state. In contrast, allostery in the catalytic and ligand-binding steps is driven by entropic effects resulting from altered coordinated protein dynamics upon activation loop phosphorylation.243,311 Similarly, in the Src kinase, binding of its regulatory domain to the C-terminal tail of the kinase induces changes in the dynamics of the enzyme as demonstrated by using MD simulations.320 These changes then propagate to modulate the coupled dynamics between the two regulatory subdomains of the enzyme, ultimately leading to the association or dissociation of these regulatory subdomains from the kinase domain. Likewise, the catalytic activity of spleen tyrosine kinase (Syk) is regulated via modulation of the conformational dynamics (i.e., entropy) of the linker connecting the two regulatory domains of the kinase. This effect is mediated by phosphorylation in the linker which is located remotely from the regulatory domains binding surfaces and solvent exposed,321 and was probed by a combined application of MD simulations, isothermal calorimetry and NMR spectroscopy. These findings suggest that due to the structural similarities among protein kinases, these principles of allostery may be applicable to other protein kinases. However, further studies are needed to thoroughly examine and confirm this idea.
Figure 3.
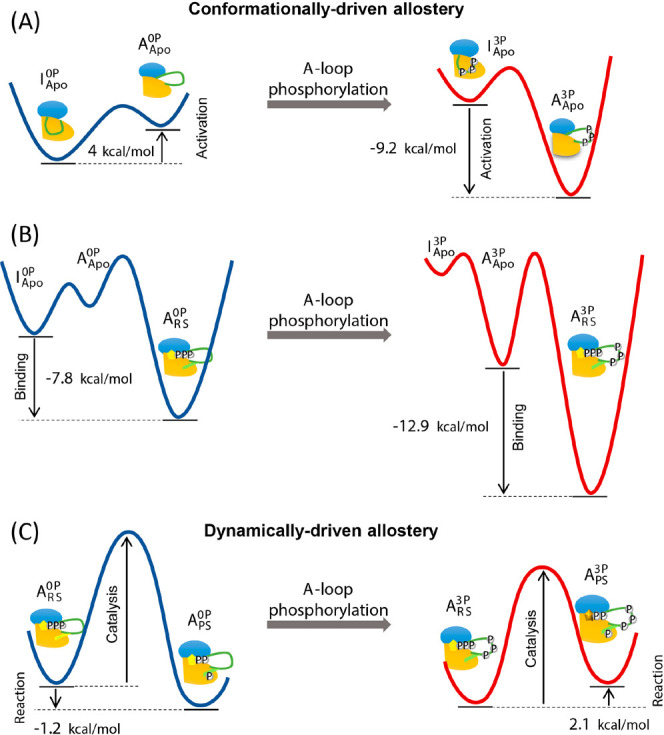
Schematic representation of the free energy changes and thus, the allosteric mechanisms of (A) the IGF-1RK conformational change that is shifted toward the activated state following phosphorylation, (B) increased substrate binding affinity following phosphorylation, and (C) increased phosphoryl transfer reaction partly caused by changes in underlying protein dynamics after phosphorylation. In each diagram, the changes of the free energy are indicated with the arrow, and the red and blue lines represent the free energy profiles of fully activated and inactive form kinases, respectively. Adapted with permission from ref (243). Copyright 2017 Royal Society of Chemistry.
De Novo Enzyme Design and Evolution
The ultimate goal in enzyme design is to engineer entirely new enzymes equipped with catalytic capabilities that match or even surpass those of their natural counterparts while catalyzing novel chemical reactions. Substantial progress has been made toward this goal over the past few decades, largely driven by the development of computational enzyme design strategies.31,322−334 Typically, de novo enzyme design begins with the creation and fine-tuning of “theozymes”, which comprise essential side chains positioned to stabilize reaction transition states.335 This initial design is followed by the selection of enzyme scaffolds capable of accommodating the theozyme configurations, forming the active site of the designed enzyme; recent development even allows the design of the scaffold proteins through deep learning approaches.336−339 Subsequently, sequence optimizations are performed to maximize the compatibility of the active site with the reaction mechanism of the theozyme while preserving the overall stability of the designed enzyme. Sequences produced through these processes are then subjected to experimental characterization to evaluate their catalytic efficiencies, often measured as the specificity constant kcat/KM. When necessary, the selected sequences, once their catalytic efficiency has been measured, undergo further refinement to enhance their catalytic performance and improve properties like thermostability. These strategies have been successfully employed to generate de novo enzymes with the capacity to catalyze a range of organic reactions, including Kemp eliminase,323 retro-aldolase,340,341 Diels–Alderase,342 esterase,327,343,344 and luciferase.345 Alternatively, existing enzymes or sequences are repurposed to catalyze increasingly complex chemical reactions,329,332,346−348 including poly(ethylene terephthalate) (PET) hydrolysis.349−352
Despite these successes, the designed enzymes have shown relatively modest catalytic activities, with kcat/KM values being several orders of magnitude lower than those of natural enzymes.326,353,354 To further enhance the catalytic potential of these designed enzymes, experimental approaches, e.g., directed evolution, have been employed.324,330,355−357 This combined approach, starting with computational design followed by experimentally directed evolution, has resulted in designed enzymes with catalytic efficiencies approaching those of their natural counterparts. It also provided valuable insights into the structural determinants of efficient catalysis. In particular, directed evolution introduces variations of active site residues, including the catalytic residues that were originally designed. Amino acid replacements from directed evolution can improve catalysis, for example, by introducing new catalytic groups, optimizing catalytic contacts and ligand-binding modes and enhancing transition-state complementarity within the binding pocket.348 In some cases, replacements are introduced at positions remote from the active site, affecting ligand-binding properties and/or populations of conformational substates.323,354,358 These observations, together with relatively poor selectivity of substrates in the designed enzymes, point to potential limitations of current computational methods and highlight the need for further development of computational enzyme design algorithms.272,341,354 Such improvements should aim to improve the accuracy of modeling catalytic and ligand-binding interactions while allowing the efficient exploration of the vast sequence space. In addition, the full spectrum of mechanisms contributing to enzyme catalysis, beyond the simple transition state stabilization, needs to be considered in the design of new enzymes to reach the catalytic activity of natural enzymes.
In recent years, the application of machine learning techniques has revolutionized the field of protein structure prediction,359,360 protein design,334,338,339,345,361 and enzyme engineering.334,361−366 These techniques focus primarily on either structure prediction or activity prediction. On the structure side, achievements such as AlphaFold2359 and RoseTTAFold2360 have significantly improved the accuracy of protein structure prediction. These structure prediction techniques have the potential to greatly improve our ability to design robust enzymes with high success rates.367 Nevertheless, challenges remain, including the need to incorporate aspects of protein dynamics,272,368 allostery and entropy-enthalpy compensation into enzyme design principles,333,369 as well as the design of enzymes capable of catalyzing multistep reactions.330,367 From the perspective of computational enzymology, the physics-based computational methods discussed above have proven instrumental in exploring these complexities of enzymes and understanding their mechanisms.329 Consequently, these methods need to be fully integrated with machine learning techniques into the enzyme design framework to streamline the effective refinement of the catalytic properties of the designed enzymes.30,272,329,330,333,367,370,371 On the activity prediction side, regression models, including linear regression and neural networks, have long been used to decipher the sequence–activity relationship of enzymes. Such relationships have then been used to guide (a) the optimization of enzyme variants372,373 and (b) the directed evolution of enzyme activity362,374,375 and product enantioselectivity.376,377
A compelling question related to enzyme design principles is how nature evolved enzymes to achieve the remarkable catalytic capabilities found in modern enzymes.28,67,106,272,332,353,358,368,371,378 To address this question, ancestral sequence reconstruction (ASR) algorithms379,380 can be combined with protein structure prediction algorithms to test evolutionary design principles, such as those governing their catalytic activities and thermal adaptation, and to rapidly evaluate hypotheses.331,332,381,382 This idea has already been adopted, where the high accuracy of proteins structure predictions such as AlphaFold2 and RoseTTAFold2 have been used to evaluate the structures of the ASR-generated sequences, followed by biochemical/biophysical characterization and structural elucidation.383−385 Notable examples of such studies include the study of monooxygenases’ functional diversification, which revealed relatively few mutations could be sufficient to induce diversification of the catalytic function of the enzyme,384 and the development of multiple xylulose isomerases by combining big data, ASR, and adaptive laboratory evolution techniques for biotechnology.385 We expect that this combined approach will be used more frequently to provide new opportunities for researchers. Furthermore, these approaches can also be applied to further evolve enzymes for biomedical and biotechnological applications. In these applications and in the development of new enzyme-design approaches, advanced physics-based computational methods, such as those discussed in this review, can provide a fundamental understanding of their working mechanisms and thereby help advance the field of enzyme design and its biotechnological applications.329
Emergence of Covalent Drugs and In Silico Docking Strategies
While drugs that form covalent bonds with protein targets have been known for more than a century, such as aspirin and penicillin, recent successes have renewed efforts to develop covalent drugs.386−389 Notable examples of these drugs include remdesivir390,391 and nirmatrelvir392,393 for COVID-19 and the G12C K-Ras inhibitor sotorasib,394,395 where K-Ras has long been considered undruggable. Compared to the conventional noncovalent drugs,396,397 the main advantage of covalent drugs is their ability to produce long-lasting pharmacological effects even at low doses, and they can even irreversibly inactivate target proteins, reducing the frequency of administration. For this advantage, numerous computational studies have been conducted to elucidate the chemical mechanism underlying the covalent bond formation (Figure 4).41,398−410 However, despite this recent interest in the development of covalent drugs, the lack of effective strategies remains a current technical challenge to the widespread development of covalent inhibitors.387,388,397,408,411−419
Figure 4.

(A) Proposed two-step binding of a covalent ligand. The first step is noncovalent binding with barrier ΔG⧧b, and the second step is covalent bond formation with ΔG⧧c as the associated barrier. The effectiveness of the covalent binders can be determined by the free energy barrier (ΔG⧧c) of the second step relative to the noncovalent binding free energy (ΔGnc) and/or the unbinding free energy barrier (ΔG⧧ub). Too high a barrier can result in premature release of the binder prior to covalent bond formation, while too low a barrier renders the binder reversible or ineffective as a covalent inhibitor. In the latter case, the warhead may be too reactive, leading to nonspecific binding. Therefore, modulation of both ΔGnc and ΔG⧧c is critical for the design of effective covalent binders, where the binding pocket of the target protein/enzyme provides an environment for nonspecific binding and functional modifications of the warhead control the reactivity of the binders. (B) SARS-CoV-2 Mpro with bound ligand, N3 inhibitor (PDB ID: 7BQY). The bound ligand is colored yellow, and water molecules are pink. The protein surface appears in gray. The Cys 145-His41 dyad is shown in deep teal. Adapted with permission from ref (404). Copyright 2021 American Chemical Society.
From the point of view of enzymatic catalysis, covalent drugs follow the general mechanism analogous to the Michaelis–Menten mechanism (Figure 4A).389,413 In essence, they first bind noncovalently and reversibly to the target protein, followed by the formation of a covalent bond between the ligand and the target residue. In this context, the first step corresponds to the substrate binding step, and the second step is analogous to the enzymatic catalysis step, where the enzyme environment and protein dynamics play a critical role in achieving high catalytic efficiency and specificity in substrate recognition. Therefore, in addition to achieving high binding affinity of the drug (in a noncovalent form), efforts in covalent drug design must take into account: (a) the reactivity of the “warheads”, i.e., the reactive functional groups of the drug, and (b) the presence of susceptible residues on the target proteins, such as cysteine (Cys) residues, among others.387,420,421 In particular, the reactivity of the warheads should be sufficient to react with the target residues, but not overly reactive to minimize off-target reactivity.415,420,422 For this reason, most known warheads are weak electrophiles. Upon binding, the warheads must also be properly oriented to react effectively with the nucleophile of the protein.411 While the first consideration requires quantum mechanical treatment,409,421 the second can be partially addressed using conventional, noncovalent drug docking approaches. For example, docking can be performed with restraints that position the warheads in proximity to the target residue and predict the docked poses driven by noncovalent interactions.411,413,417,423 The approach can then be switched to a QM/MM potential to optimize the docked poses in the presence of a covalent bond between the warhead and the protein.408,413,424,425 However, the actual binding of covalent drugs involves reaction transition states, whose barriers influence the time scale of the reaction. Understanding this process is therefore necessary to gain insight into the reactivity of the warheads and the reversibility of the covalently bound drugs.405,406,426−428 In this regard, knowledge gained from the study of enzyme catalysis can be applied to identify potential binding poses and subsequent reactions with target proteins.70,204,426,428 Furthermore, given the shared principles between enzyme catalysis and covalent drug binding, the multiscale computational approaches developed to study enzyme mechanisms (Figure 1) can be effectively applied to understand both covalent and noncovalent drug binding. For a similar reason, rational covalent drug design shares the same challenges as enzyme design. However, unlike enzyme design, where the goal is to maximize the catalytic activity of the designed enzymes, the challenge in covalent drug design lies in the modulation of the reactivity of the warheads specifically for the target residues while ensuring that they react within a reasonable time frame of noncovalent binding/unbinding.
Finally, similar to the case of enzyme design, machine learning algorithms have shown potential to improve the accuracy and efficiency of molecular docking.429−433 These machine learning, particularly deep learning, techniques include multilayer perceptrons, convolutional neural networks, and graph neural networks. Many of these methods have focused on virtual screening of ligand libraries,433,434 which can be helpful in identifying new lead molecules as inhibitors. Other methods have focused on improving the accuracy or efficiency of scoring functions. Deep learning methods for binding site identification have also been proposed. However, these methods have mostly focused on noncovalent compounds. Therefore, although they can be helpful for computational enzymologists to quickly generate initial structures, they have not been specifically designed toward generating high-quality guesses for Michaelis complex needed for enzyme modeling. Hence, as with any general docking tool, computational enzymologists should use these methods with caution and consider the docking results as initial guesses that should be refined using multiscale methods as defined in this review (Figure 1).
Future Perspectives
In this review, we have discussed several topics within the field of computational enzymology while highlighting several emerging frontiers that deserve future exploration. These include:
Computational approaches for complex enzymatic processes: Enzymatic catalysis involves interrelated events spanning multiple temporal and spatial scales, encompassing substrate binding, multistep catalysis, and product release. Exploring this complexity requires the development of advanced computational methods capable of integrating multiple levels of theory within a unified free energy simulation framework. While such methods have yet to reach full development, they remain a compelling theoretical challenge.
Real-time enzyme observations: The field of structural biology is advancing toward the real-time unraveling of dynamic molecular processes. Techniques, such as room-temperature X-ray free-electron laser (XFEL), time-resolved X-ray crystallography, cryo-electron microscopy, and single-molecule approaches, combined with molecular simulations, have the potential to transform the field by enabling real-time observation of enzymatic processes.
Conformational substates in enzymes: The study of conformational substates within enzyme–substrate complexes is an evolving field. The full spectrum of these substates, their temporal dynamics and their impact on enzyme mechanisms remain to be characterized.
Advanced enzyme design strategies: The development of robust enzyme design algorithms is an important frontier. These algorithms should improve the accuracy of modeling catalytic and ligand-binding interactions, while rapidly exploring the vast sequence and protein conformational space. To this end, the incorporation of machine learning techniques has shown the potential to significantly accelerate enzyme prediction and design. In addition, the development of enzyme design principles that incorporate our knowledge of protein dynamics, entropy and allostery is essential for improving the mechanistic properties of designed enzymes, such as catalytic activity, substrate specificity, and thermal stability.
Covalent drug design strategies: Developing effective strategies for the widespread development of covalent inhibitors is an ongoing technical challenge. Advancing our understanding of covalent drug interactions, warhead reactivity, and potential binding poses represents an emerging frontier where the integration of QM/MM methodologies can have an impact by providing more accurate descriptions of drug–protein interactions and the reaction transition states that govern the binding and reactivity of covalent drugs.
Together, these frontiers represent areas where ongoing research and innovation promise significant advances in our understanding of enzymes, protein dynamics, and drug and enzyme design. They also offer exciting opportunities to push the boundaries of what is currently achievable in these fields. Finally, we note that the synergistic integration of experimental and computational methods will continue to play a central role in providing valuable insights into enzymatic mechanisms and protein–ligand interactions.
Acknowledgments
The authors acknowledge the financial support from the National Institute of General Medical Sciences of the National Institute of Health: R01GM132481 (K.N. and M.W.W.), R44GM133270 (K.N. and Y.S.), R21GM148895 (K.N. and D.T.M.), and R01GM135392 (Y.S.); and the Swedish Research Council: VR 2021-04513 (M.W.W).
The authors declare no competing financial interest.
References
- Radzicka A.; Wolfenden R. A Proficient Enzyme. Science 1995, 267, 90–93. 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- Alberts B. The Cell as a Collection of Protein Machines: Preparing the Next Generation of Molecular Biologists. Cell 1998, 92, 291–294. 10.1016/S0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- Benkovic S. J.; Hammes-Schiffer S. A Perspective on Enzyme Catalysis. Science 2003, 301 (5637), 1196–1202. 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Houk K. N. Why Enzymes Are Proficient Catalysts: Byeond the Pauling Paradigm. Acc. Chem. Res. 2005, 38, 379–440. 10.1021/ar040257s. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K.; Kern D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Callender R.; Dyer R. B. The Dynamic Nature of Enzymatic Catalysis. Acc. Chem. Res. 2015, 48, 407–413. 10.1021/ar5002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovermann M.; Rogne P.; Wolf-Watz M. Protein dynamics and function from solution state NMR spectroscopy. Q. Rev. Biophys. 2016, 49, e6 10.1017/S0033583516000019. [DOI] [PubMed] [Google Scholar]
- Cheatum C. M. Low-Frequency Protein Motions Coupled to Catalytic Sites. Annu. Rev. Phys. Chem. 2020, 71, 267–288. 10.1146/annurev-physchem-050317-014308. [DOI] [PubMed] [Google Scholar]
- Zheng C.; Mao Y.; Kozuch J.; Atsango A. O.; Ji Z.; Markland T. E.; Boxer S. G. A two-directional vibrational probe reveals different electric field orientations in solution and an enzyme active site. Nat. Chem. 2022, 14, 891–897. 10.1038/s41557-022-00937-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam K.; Wolf-Watz M. Protein dynamics: The future is bright and complicated!. Struct. Dyn. 2023, 10, 014301. 10.1063/4.0000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A.Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 3rd ed.; John Wiley & Sons, Inc., 2023. [Google Scholar]
- Wolfenden R.; Snider M. J. The Depth of Chemical Time and the Power of Enzymes as Catalysts. Acc. Chem. Res. 2001, 34, 938–945. 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- Koshland D. E. Jr.; Némethy G.; Filmer D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry 1966, 5, 365–385. 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; Nussinov R.; Wright P. E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B.; Nussinov R. Enzyme dynamics point to stepwise conformational selection in catalysis. Curr. Opin. Chem. Biol. 2010, 14 (5), 652–659. 10.1016/j.cbpa.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitchagno G. T. M.; Nchiozem-Ngnitedem V.-A.; Melchert D.; Fobofou S. A. Demystifying racemic natural products in the homochiral world. Nat. Rev. Chem. 2022, 6, 806–822. 10.1038/s41570-022-00431-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogne P.; Rosselin M.; Grundström C.; Hedberg C.; Sauer U. H.; Wolf-Watz M. Molecular mechanism of ATP versus GTP selectivity of adenylate kinase. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 3012–3017. 10.1073/pnas.1721508115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes G. G.Enzyme Catalysis and Regulation; Elsevier Inc., 1982. [Google Scholar]
- Nolen B.; Taylor S.; Ghosh G. Regulation of Protein Kinases: Controlling Activity through Activation Segment Conformation. Mol. Cell 2004, 15, 661–675. 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Endres N. F.; Barros T.; Cantor A. J.; Kuriyan J. Emerging concepts in the regulation of the EGF receptor and other receptor tyrosine kinases. Trends Biochem. Sci. 2014, 39, 437–446. 10.1016/j.tibs.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Goodey N. M.; Benkovic S. J. Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 2008, 4, 474–482. 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- Kannan N.; Neuwald A. F. Did Protein Kinase Regulatory Mechanisms Evolve Through Elaboration of a Simple Structural Component?. J. Mol. Biol. 2005, 351, 956–972. 10.1016/j.jmb.2005.06.057. [DOI] [PubMed] [Google Scholar]
- Taylor S. S.; Kornev A. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrović D.; Risso V. A.; Kamerlin S. C. L.; Sanchez-Ruiz J. M. Conformational dynamics and enzyme evolution. J. R. Soc. Interface 2018, 15, 20180330. 10.1098/rsif.2018.0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campitelli P.; Modi T.; Kumar S.; Ozkan S. B. The Role of Conformational Dynamics and Allostery in Modulating Protein Evolution. Annu. Rev. Biophys. 2020, 49, 267–288. 10.1146/annurev-biophys-052118-115517. [DOI] [PubMed] [Google Scholar]
- Verma A.; Åberg-Zingmark E.; Sparrman T.; Mushtaq A. U.; Rogne P.; Grundström C.; Berntsson R.; Sauer U. H.; Backman L.; Sauer-Eriksson E.; Wolf-Watz M.; Nam K. Insights into the evolution of enzymatic specificity and catalysis: From Asgard archaea to human adenylate kinases. Sci. Adv. 2022, 8, eabm4089 10.1126/sciadv.abm4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P. Enabling Role of Ligand-Driven Conformational Changes in Enzyme Evolution. Biochemistry 2022, 61, 1533–1542. 10.1021/acs.biochem.2c00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbella M.; Pinto G. P.; Kamerlin S. C. L. Loop dynamics and the evolution of enzyme activity. Nat. Rev. Chem. 2023, 7, 536–547. 10.1038/s41570-023-00495-w. [DOI] [PubMed] [Google Scholar]
- Arcus V. L.; van der Kamp M. W.; Pudney C. R.; Mulholland A. Enzyme evolution and the temperature dependence of enzyme catalysis. Curr. Opin. Struct. Biol. 2020, 65, 96–101. 10.1016/j.sbi.2020.06.001. [DOI] [PubMed] [Google Scholar]
- Ferreira P.; Fernandes P. A.; Ramos M. J. Modern computational methods for rational enzyme engineering. Chem. Catal. 2022, 2, 2481–2498. 10.1016/j.checat.2022.09.036. [DOI] [Google Scholar]
- Li S.; Yang X.; Yang S.; Zhu M.; Wang X. TECHNOLOGY PROSPECTING ON ENZYMES: APPLICATION, MARKETING AND ENGINEERING. Comput. Struct. Biotechnol. J. 2012, 2, e201209017 10.5936/csbj.201209017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Singh S. Directed evolution: tailoring biocatalysts for industrial applications. Crit. Rev. Biotechnol. 2013, 33, 365–378. 10.3109/07388551.2012.716810. [DOI] [PubMed] [Google Scholar]
- Adrio J. L.; Demain A. L. Microbial Enzymes: Tools for Biotechnological Processes. Biomolecules 2014, 4, 117–139. 10.3390/biom4010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otte K. B.; Hauer B. Enzyme engineering in the context of novel pathways and products. Curr. Opin. Biotechnol. 2015, 35, 16–22. 10.1016/j.copbio.2014.12.011. [DOI] [PubMed] [Google Scholar]
- Chène P. ATPase AS DRUG TARGETS: LEARNING FROM THEIR STRUCTURE. Nat. Rev. Drug Discovery 2002, 1, 665–673. 10.1038/nrd894. [DOI] [PubMed] [Google Scholar]
- Capdeville R.; Buchdunger E.; Zimmermann J.; Matter A. GLIVEC (STI571, IMATINIB), A RATIONALLY DEVELOPED, TARGETED ANTICANCER DRUG. Nat. Rev. Drug Discovery 2002, 1, 493–502. 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- De Benedetti P. G.; Fanelli F. Computational quantum chemistry and adaptive ligand modeling in mechanistic QSAR. Drug Discovery Today 2010, 15, 859–866. 10.1016/j.drudis.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Nussinov R.; Tsai C.-J. Allostery in Disease and in Drug Discovery. Cell 2013, 153, 293–305. 10.1016/j.cell.2013.03.034. [DOI] [PubMed] [Google Scholar]
- Nussinov R.; Jang H.; Tsai C.-J.; Cheng F. Review: Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers. PLoS Comput. Biol. 2019, 15, e1006658 10.1371/journal.pcbi.1006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarnera E.; Berezovsky I. N. Allosteric drugs and mutations: changes, challenges, and necessity. Curr. Opin. Struct. Biol. 2020, 62, 149–157. 10.1016/j.sbi.2020.01.010. [DOI] [PubMed] [Google Scholar]
- Attwood M. M.; Fabbro D.; Sokolov A. V.; Knapp S.; Schiöth H. B. Trends in kinase drug discovery: targets, indicators and inhibitor design. Nat. Rev. Drug Discovery 2021, 20, 839–861. 10.1038/s41573-021-00252-y. [DOI] [PubMed] [Google Scholar]
- Wu K.; Karapetyan E.; Schloss J.; Vadgama J.; Wu Y. Advancements in small molecule drug design: A structural perspective. Drug Discovery Today 2023, 28, 103730. 10.1016/j.drudis.2023.103730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus M.; McCammon J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- Garcia-Viloca M.; Gao J.; Karplus M.; Truhlar D. G. How Enzymes Work: Analysis by Modern Rate Theory and Computer Simulations. Science 2004, 303 (5655), 186–195. 10.1126/science.1088172. [DOI] [PubMed] [Google Scholar]
- Gao J.; Ma S.; Major D. T.; Nam K.; Pu J.; Truhlar D. G. Mechanisms and Free Energies of Enzymatic Reactions. Chem. Rev. 2006, 106, 3188–3209. 10.1021/cr050293k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senn H. M.; Thiel W. QM/MM studies of enzymes. Curr. Opin. Chem. Biol. 2007, 11, 182–187. 10.1016/j.cbpa.2007.01.684. [DOI] [PubMed] [Google Scholar]
- McGeagh J. D.; Ranaghan K. E.; Mulholland A. J. Protein dynamics and enzyme catalysis: Insights from simulations. Biochim. Biophys. Acta 2011, 1814, 1077–1092. 10.1016/j.bbapap.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Kazemi M.; Himo F.; Åqvist J. Enzyme catalysis by entropy without Circe effect. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 2406–2411. 10.1073/pnas.1521020113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L. C. Molecular architecture and biological reactions. Chem. Eng. News 1946, 24, 1375–1377. 10.1021/cen-v024n010.p1375. [DOI] [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Xiang Y.; Liu H.; Olsson M. H. M. Electrostatic Basis for Enzyme Catalysis. Chem. Rev. 2006, 106, 3210–3235. 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- Schwartz S. D.; Schramm V. L. Enzymatic transition states and dynamic motion in barrier crossing. Nat. Chem. Biol. 2009, 5, 551–558. 10.1038/nchembio.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N.; Mo Y.; Gao J.; Pai E. F. Electrostatic stress in catalysis: Structure and mechanism of the enzyme orotidine monophosphate decarboxylase. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 2017–2022. 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman K. A.; Thai V.; Lei M.; Ott M.; Wolf-Watz M.; Fenn T.; Pozharski E.; Wilson M. A.; Petsko G. A.; Karplus M.; Hübner C. G.; Kern D. Intrinsic motions along an enzymatic reaction trajectory. Nature 2007, 450, 838–844. 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- Tuñón I.; Laage D.; Hynes J. T. Are there dynamical effects in enzyme catalysis? Some thoughts concerning the enzymatic chemical step. Arch. Biochem. Biophys. 2015, 582, 42–55. 10.1016/j.abb.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohen A. Role of Dynamics in Enzyme Catalysis: Substantial versus Semantic Controversies. Acc. Chem. Res. 2015, 48, 466–473. 10.1021/ar500322s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T. H.; Mehrabi P.; Ren Z.; Sljoka A.; Ing C.; Bezginov A.; Ye L.; Pomès R.; Prosser R. S.; Pai E. F. The role of dimer asymmetry and protomer dynamics in enzyme catalysis. Science 2017, 355, eaag2355 10.1126/science.aag2355. [DOI] [PubMed] [Google Scholar]
- Raz K.; Levi S.; Gupta P. K.; Major D. T. Enzymatic control of product distribution in terpene synthases: insights from multiscale simulations. Curr. Opin. Biotechnol. 2020, 65, 248–258. 10.1016/j.copbio.2020.06.002. [DOI] [PubMed] [Google Scholar]
- Weitman M.; Major D. T. Challenges Posed to Bornyl Diphosphate Synthase: Diverging Reaction Mechanisms in Monoterpenes. J. Am. Chem. Soc. 2010, 132, 6349–6360. 10.1021/ja910134x. [DOI] [PubMed] [Google Scholar]
- Major D. T.; Weitman M. Electrostatically Guided Dynamics—The Root of Fidelity in a Promiscuous Terpene Synthase?. J. Am. Chem. Soc. 2012, 134, 19454–19462. 10.1021/ja308295p. [DOI] [PubMed] [Google Scholar]
- Antoniou D.; Schwartz S. D. Internal enzyme motions as a source of catalytic activity: Rate-promoting vibrations and hydrogen tunneling. J. Phys. Chem. B 2001, 105, 5553. 10.1021/jp004547b. [DOI] [Google Scholar]
- Bruice T. C. Computational Approaches: Reaction Trajectories, Structures, and Atomic Motions. Enzyme Reactions and Profiency. Chem. Rev. 2006, 106, 3119–3139. 10.1021/cr050283j. [DOI] [PubMed] [Google Scholar]
- Monod J.; Wyman J.; Changeux J.-P. On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol. 1965, 12, 88–118. 10.1016/S0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Gunasekaran K.; Ma B.; Nussinov R. Is allostery an intrinsic property of all dynamic proteins?. Proteins 2004, 57, 433–443. 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- Cui Q.; Karplus M. Allostery and cooperativity revisited. Protein Sci. 2008, 17, 1295–1307. 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilser V. J.; Wrabl J. O.; Motlagh H. N. Structural and Energetic Basis of Allostery. Annu. Rev. Biophys. 2012, 41, 585–609. 10.1146/annurev-biophys-050511-102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.; Zhou H.-X. Protein Allostery and Conformational Dynamics. Chem. Rev. 2016, 116, 6503–6515. 10.1021/acs.chemrev.5b00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra H. G.; Wrabl J. O.; Anderson J. A.; Li J.; Hilser V. J. Dynamic allostery can drive cold adaptation in enzymes. Nature 2018, 558, 324–328. 10.1038/s41586-018-0183-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicena P.; Kuriyan J. Protein-protein interactions in the allosteric regulation of protein kinases. Curr. Opin. Struct. Biol. 2006, 16, 702–709. 10.1016/j.sbi.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Lengyel E.; Sawada K.; Salgia R. Tyrosine Kinase Mutations in Human Cancer. Curr. Mol. Med. 2007, 7, 77–84. 10.2174/156652407779940486. [DOI] [PubMed] [Google Scholar]
- Amaro R. E.; Mulholland A. Multiscale methods in drug design bridge chemical and biological complexity in the search for cures. Nat. Rev. Chem. 2018, 2, 0148. 10.1038/s41570-018-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange O. F.; Lakomek N.-A.; Farès C.; Schröder G. F.; Walter K. F. A.; Becker S.; Meiler J.; Grubmüller H.; Griesinger C.; de Groot B. L. Recognition Dynamics Up to Microseconds Revealed from an RDC-Derived Ubiquitin Ensemble in Solution. Science 2008, 320, 1471–1475. 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- Changeux J.-P.; Edelstein S. Conformational selection or induced fit? 50 years of debate unsolved. F1000 Biol. Rep. 2011, 3, 19. 10.3410/B3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzakis N. S. Single molecule insights on conformational selection and induced fit mechanism. Biophys. Chem. 2014, 186, 46–54. 10.1016/j.bpc.2013.11.003. [DOI] [PubMed] [Google Scholar]
- Chakrabarti K. S.; Agafonov R. V.; Pontiggia F.; Otten R.; Higgins M. K.; Schertler G. F. X.; Oprian D. D.; Kern D. Conformational Selection in a Protein-Protein Interaction Revealed by Dynamic Pathway Analysis. Cell Rep. 2016, 14, 32–42. 10.1016/j.celrep.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora K.; Brooks III C. L. Large-scale allosteric conformational transitions of adenylate kinase appear to involve a population-shift mechanism. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 18496–18501. 10.1073/pnas.0706443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki K.-i.; Takada S. Dynamic energy landscape view of coupled binding and protein conformational change: Induced-fit versus population-shift mechanisms. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 11182–11187. 10.1073/pnas.0802524105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer S.; Olsen K. W.; Nam K.; Karplus M. Unsuspected pathway of the allosteric transition in hemoglobin. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 5608–5613. 10.1073/pnas.1011995108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A. Electrostatic Origin of the Catalytic Power of Enzymes and the Role of Preorganized Active Sites. J. Biol. Chem. 1998, 273, 27035–27038. 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- Frey P. A.; Hegeman A. D.. Enzymatic Reaction Mechanisms; Oxford University Press: Oxford, 2007. [Google Scholar]
- Hanoian P.; Liu C. T.; Hammes-Schiffer S.; Benkovic S. Perspectives on Electrostatics and Conformational Motions in Enzyme Catalysis. Acc. Chem. Res. 2015, 48, 482–489. 10.1021/ar500390e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby A. J.Acid-Base Catalysis by Enzymes. In Encyclopedia of Life Sciences; John Wiley & Sons, 2010. [Google Scholar]
- Segel I. H.Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems; Wiley, 1993. [Google Scholar]
- Dewar M. J. S.; Storch D. M. Alternative view of enzyme reactions. Proc. Natl. Acad. Sci. U.S.A. 1985, 82, 2225–2229. 10.1073/pnas.82.8.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P.Catalysis in Chemistry and Enzymology; Dover Publication, Inc., 1987. [Google Scholar]
- Warshel A.; Åqvist J.; Creighton S. Enzymes work by solvation substitution rather than by desolvation. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 5820–5824. 10.1073/pnas.86.15.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokenson M. J.; Cope G. A.; Lewis E. R.; Oberg K. A.; Fink A. L. Enzyme-Induced Strain/Distortion in the Ground-State ES Complex in β-Lactamase Catalysis Revealed by FTIR. Biochemistry 2000, 39, 6538–6545. 10.1021/bi9928041. [DOI] [PubMed] [Google Scholar]
- Schramm V. L. Enzymatic Transition States, Transition-State Analogs, Dynamics, Thermodynamics, and Lifetimes. Annu. Rev. Biochem. 2011, 80, 703–732. 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightstone F. C.; Bruice T. C. Ground State Conformations and Entropic and Enthalpic Factors in the Efficiency of Intramolecular and Enzymatic Reactions. 1. Cyclic Anhydride Formation by Substituted Glutarates, Succinate, and 3,6-Endoxo-Δ4-tetrahydrophthalate Monophenyl Esters. J. Am. Chem. Soc. 1996, 118, 2595–2605. 10.1021/ja952589l. [DOI] [Google Scholar]
- Major D. T. Electrostatic Control of Chemistry in Terpene Cyclases. ACS Catal. 2017, 7, 5461–5465. 10.1021/acscatal.7b01328. [DOI] [Google Scholar]
- Dixit M.; Weitman M.; Gao J.; Major D. T. Chemical Control in the Battle against Fidelity in Promiscuous Natural Product Biosynthesis: The Case of Trichodiene Synthase. ACS Catal. 2017, 7, 812–818. 10.1021/acscatal.6b02584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel Z. D.; Klinman J. P. Tunneling and Dynamics in Enzymatic Hydride Transfer. Chem. Rev. 2006, 106, 3095–3118. 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- Major D. T.; Heroux A.; Orville A. M.; Valley M. P.; Fitzpatrick P. F.; Gao J. Differential quantum tunneling contributions innitroalkane oxidase catalyzed and the uncatalyzedproton transfer reaction. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 20734–20739. 10.1073/pnas.0911416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman J. P.; Kohen A. Hydrogen Tunneling Links Protein Dynamics to Enzyme Catalysis. Annu. Rev. Biochem. 2013, 82, 471–496. 10.1146/annurev-biochem-051710-133623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou D.; Basner J.; Núñez S.; Schwartz S. D. Computational and Theoretical Methods to Explore the Relation between Enzyme Dynamics and Catalysis. Chem. Rev. 2006, 106, 3170–3187. 10.1021/cr0503052. [DOI] [PubMed] [Google Scholar]
- Hay S.; Scrutton N. S. Good vibrations in enzyme-catalyzed reactions. Nat. Chem. 2012, 4, 161–168. 10.1038/nchem.1223. [DOI] [PubMed] [Google Scholar]
- Agarwal P. K.; Billeter S. R.; Rajagopalan P. T. R.; Benkovic S. J.; Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 2794–2799. 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr D. D.; McElheny D.; Dyson H. J.; Wright P. E. The dynamic energy landscape of dihydrofolate reductase catalysis. Science 2006, 313, 1638. 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- Ojeda-May P.; Mushtaq A. U.; Rogne P.; Verma A.; Ovchinnikov V.; Grundström C.; Dulko-Smith B.; Sauer U. H.; Wolf-Watz M.; Nam K. Dynamic Coupling between Enzymatic Catalysis and Collective Protein Motions. Biochemistry 2021, 60, 2246–2258. 10.1021/acs.biochem.1c00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisliakov A. V.; Cao J.; Kamerlin S. C. L.; Warshel A. Enzyme millisecond conformational dynamics do not catalyze the chemical step. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 17359–17364. 10.1073/pnas.0909150106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y.; Moody I. S.; Sims P. C.; Hunt S. R.; Corso B. L.; Perez I.; Weiss G. A.; Collins P. G. Single-Molecule Lysozyme Dynamics Monitored by an Electronic Circuit. Science 2012, 335, 319–324. 10.1126/science.1214824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi U.; McGowan L. C.; Ladani S. T.; Hamelberg D. Resolving the complex role of enzyme conformational dynamics in catalytic function. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 5699–5704. 10.1073/pnas.1117060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T.; Saito S. Dissecting the Dynamics during Enzyme Catalysis: A Case Study of Pin1 Peptidyl-Prolyl Isomerase. J. Chem. Inf. Model 2020, 16, 3396–3407. 10.1021/acs.jctc.9b01279. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Doruker P.; Kaynak B.; Zhang S.; Krieger J.; Li H.; Bahar I. Intrinsic dynamics is evolutionarily optimized to enable allosteric behavior. Curr. Opin. Struct. Biol. 2020, 62, 14–21. 10.1016/j.sbi.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost C. F.; Balasubramani S. G.; Antoniou D.; Schwartz S. D. Connecting Conformational Motions to Rapid Dynamics in Human Purine Nucleoside Phosphorylase. J. Phys. Chem. B 2023, 127, 144–150. 10.1021/acs.jpcb.2c07243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulko-Smith B.; Ojeda-May P.; Ådén J.; Wolf-Watz M.; Nam K. Mechanistic Basis for a Connection between the Catalytic Step and Slow Opening Dynamics of Adenylate Kinase. J. Chem. Inf. Model. 2023, 63, 1556–1569. 10.1021/acs.jcim.2c01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen V.; Wilson C.; Hoemberger M.; Stiller J. B.; Agafonov R. V.; Kutter S.; English J.; Theobald D. L.; Kern D. Evolutionary drivers of thermoadaptation in enzyme catalysis. Science 2017, 355, 289–294. 10.1126/science.aah3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi K.; Ito S.; Sugita Y. Exploring the Minimum-Energy Pathways and Free-Energy Profiles of Enzymatic Reactions with QM/MM Calculations. J. Phys. Chem. B 2021, 125, 4701–4713. 10.1021/acs.jpcb.1c01862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyring H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107. 10.1063/1.1749604. [DOI] [Google Scholar]
- Neria E.; Kuplus M. Molecular dynamics of an enzyme reaction: proton transfer in TIM. J. Chem. Phys. 1997, 267, 23–30. 10.1016/S0009-2614(97)00068-7. [DOI] [Google Scholar]
- Villà J.; Warshel A. Energetics and Dynamics of Enzymatic Reactions. J. Phys. Chem. B 2001, 105, 7887–7907. 10.1021/jp011048h. [DOI] [Google Scholar]
- Nam K.; Prat-Resina X.; Garcia-Viloca M.; Devi-Kesavan L. S.; Gao J. Dynamics of an Enzymatic Substition Reaction in Haloalkane Dehalogenase. J. Am. Chem. Soc. 2004, 126, 1369–1376. 10.1021/ja039093l. [DOI] [PubMed] [Google Scholar]
- Masgrau L.; Truhlar D. G. The Importance of Ensemble Averaging in Enzyme Kinetics. Acc. Chem. Res. 2015, 48, 431–438. 10.1021/ar500319e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M. E.; Piccirilli J. A.; York D. M. Integration of kinetic isotope effect analyses to elucidate ribonuclease mechanism. Biochim. Biophys. Acta 2015, 1854, 1801–1808. 10.1016/j.bbapap.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrie G. M.; Valleau J. P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187. 10.1016/0021-9991(77)90121-8. [DOI] [Google Scholar]
- Spiriti J.; Kamberaj H.; Van Der Vaart A. Development and application of enhanced sampling techniques to simulate the long-time scale dynamics of biomolecular systems. Int. J. Quantum Chem. 2012, 112, 33–43. 10.1002/qua.23139. [DOI] [Google Scholar]
- Kumar S.; Rosenberg J. M.; Bouzida D.; Swendsen R. H.; Kollman P. A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. 10.1002/jcc.540130812. [DOI] [Google Scholar]
- Shirts M.; Bair E.; Hooker G.; Pande V. S. Equilibrium Free Energies from Nonequilibrium Measurements Using Maximum-Likelihood Methods. Phys. Rev. Lett. 2003, 91, 140601. 10.1103/PhysRevLett.91.140601. [DOI] [PubMed] [Google Scholar]
- Shirts M.; Chodera J. D. Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys. 2008, 129, 124105. 10.1063/1.2978177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.-S.; Radak B. K.; Pabis A.; York D. M. A New Maximum Likelihood Approch for Free Energy Profile Construction from Molecular Simulations. J. Chem. Theory Comput. 2013, 9, 153–164. 10.1021/ct300703z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese T. J.; Ekesan Ş.; York D. M. Extension of the Variational Free Energy Profile and Multistate Bennett Acceptance Ratio Methods for High-Dimensional Potential of Mean Force Profile Analysis. J. Phys. Chem. A 2021, 125, 4216–4232. 10.1021/acs.jpca.1c00736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosta E.; Hummer G. Free Energies from Dynamic Weighted Histogram Analysis UsingUnbiased Markov State Model. J. Chem. Theory Comput. 2015, 11, 276–285. 10.1021/ct500719p. [DOI] [PubMed] [Google Scholar]
- Laio A.; Parrinello M. Escaping free-energy minima. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12562. 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G.; Laio A.; Parrinello M. Equilibrium Free Energies from Nonequilibrium Metadynamics. Phys. Rev. Lett. 2006, 96, 090601. 10.1103/PhysRevLett.96.090601. [DOI] [PubMed] [Google Scholar]
- Barducci A.; Bonomi M.; Parrinello M. Metadynamics. WIREs Comput. Mol. Sci. 2011, 1, 826–843. 10.1002/wcms.31. [DOI] [Google Scholar]
- Vanden-Eijnden E.; Venturoli M. Revisiting the finite temperature string method for the calculation of reaction tubes and free energies. J. Chem. Phys. 2009, 130, 194103. 10.1063/1.3130083. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov V.; Karplus M.; Vanden-Eijnden E. Free energy of conformational transition paths in biomolecules: The string method and its application to myosin VI. J. Chem. Phys. 2011, 134, 085103. 10.1063/1.3544209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock H. L.; Hodošček M.; Brooks B. R. Exploring SCC-DFTB Paths for Mapping QM/MM Reaction Mechanisms. J. Phys. Chem. A 2007, 111, 5720–5728. 10.1021/jp0714217. [DOI] [PubMed] [Google Scholar]
- Barducci A.; Bussi G.; Parrinello M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- Leines G. D.; Ensing B. Path Finding on High-Dimensional Free Energy Landscapes. Phys. Rev. Lett. 2012, 109, 020601. 10.1103/PhysRevLett.109.020601. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov V.; Nam K.; Karplus M. A Simple and Accurate Method to Calculate Free Energy Profiles and Reaction Rates from Restrained Molecular Simulatoins of Diffusive Processes. J. Phys. Chem. B 2016, 120, 8457–8472. 10.1021/acs.jpcb.6b02139. [DOI] [PubMed] [Google Scholar]
- Fu H.; Shao X.; Cai W.; Chipot C. Taming Rugged Free Energy Landscapes Using an Averaging Force. Acc. Chem. Res. 2019, 52, 3254–3264. 10.1021/acs.accounts.9b00473. [DOI] [PubMed] [Google Scholar]
- Xu J.; Cao X.-M.; Hu P. Accelerating Metadynamics-Based Free-Energy Calculations with Adaptive Machine Learning Potentials. J. Chem. Theory Comput. 2021, 17, 4465–4476. 10.1021/acs.jctc.1c00261. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- Field M. J.; Bash P. A.; Karplus M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 1990, 11, 700–733. 10.1002/jcc.540110605. [DOI] [Google Scholar]
- Warshel A.Computer Modeling of Chemical Reactions in Enzymes and Solutions; J. Wiley & Sons, Inc., 1991. [Google Scholar]
- Gao J.Methods and Applications of Combined Quantum Mechanical and Molecular Mechanical Potentials. In Rev. Comput. Chem., Lipkowitz K. B.; Boyd D. B., Eds.; John Wiley & Sons, Inc., 1996; pp 119–185. [Google Scholar]
- Cui Q.; Elstner M.; Kaxiras E.; Frauenheim T.; Karplus M. A QM/MM implementation of the self-consistent charge density functional tight binding (SCC-DFTB) method. J. Phys. Chem. B 2001, 105, 569–585. 10.1021/jp0029109. [DOI] [Google Scholar]
- Nam K. Acceleration of ab initio QM/MM calculations under periodic boundary conditions by multiscale and multiple time step approaches. J. Chem. Theory Comput. 2014, 10, 4175–4183. 10.1021/ct5005643. [DOI] [PubMed] [Google Scholar]
- Ojeda-May P.; Nam K. Acceleration of Semiempirical QM/MM Methods through Message Passing Interface (MPI), Hybrid MPI/Open Multiprocessing, and Self-Consisten Field Accelerator Implementations. J. Chem. Theory Comput. 2017, 13, 3525–3536. 10.1021/acs.jctc.7b00322. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Liu H.; Yang W. Free energy calculation on enzyme reactions with an efficient iterative procedure to determine minimum energy paths on a combined ab initio QM/MM potential energy surface. J. Chem. Phys. 2000, 112, 3483–3492. 10.1063/1.480503. [DOI] [Google Scholar]
- Shen L.; Wu J.; Yang W. Multiscale Quantum Mechanics/Molecular Mechanics Simulations with Neural Networks. J. Chem. Theory Comput. 2016, 12, 4934–4946. 10.1021/acs.jctc.6b00663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Shen L.; Yang W. Solvation Free Energy Calculations with Quantum Mechanics/Molecular Mechanics and Machine Learning Models. J. Phys. Chem. B 2019, 123, 901–908. 10.1021/acs.jpcb.8b11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.; Yang J.; Van R.; Epifanovsky E.; Ho J.; Huang J.; Pu J.; Mei Y.; Nam K.; Shao Y. Machine-Learning-Assisted Free Energy Simulation of Solution-Phase and Enzyme Reactions. J. Chem. Theory Comput. 2021, 17, 5745–5758. 10.1021/acs.jctc.1c00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J.; Giese T. J.; Ekesan Ş.; York D. M. Development of Range-Corrected Deep Learning Potentials for Fast, Accurate Quantum Mechanical/Molecular Mechanical Simulations of Chemical Reactions in Solution. J. Chem. Theory Comput. 2021, 17, 6993–7009. 10.1021/acs.jctc.1c00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böselt L.; Thürlemann M.; Riniker S. Machine Learning in QM/MM Molecular Dynamics Simulations of Condensed-Phase Systems. J. Chem. Theory Comput. 2021, 17, 2641–2658. 10.1021/acs.jctc.0c01112. [DOI] [PubMed] [Google Scholar]
- Giese T. J.; Zeng J.; Ekesan Ş.; York D. M. Combined QM/MM, Machine Learning Path Integral Approach to Compute Free Energy Profiles and Kinetic Isotope Effects in RNA Cleavage Reactions. J. Chem. Theory Comput. 2022, 18, 4304–4317. 10.1021/acs.jctc.2c00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W.; Zeng J.; York D. M.; Zhang L.; Wang H., Chapter 6: Learning DeePMD-Kit: A Guide to Building Deep Potential Models. In A Practical Guide to Recent Advances in Multiscale Modeling and Simulation of Biomolecules, Wang Y.; Zhou R., Eds.; AIP Publishing LLC, 2023. [Google Scholar]
- Snyder R.; Kim B.; Pan X.; Shao Y.; Pu J. Bridging semiempirical and ab initio QM/MM potentials by Gaussian process regression and its sparse variants for free energy simulation. J. Chem. Phys. 2023, 159, 054107. 10.1063/5.0156327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.; Van R.; Epifanovsky E.; Liu J.; Pu J.; Nam K.; Shao Y. Accelerating Ab Initio Quantum Mechanical and Molecular Mechanical (QM/MM) Molecular Dynamics Simulations with Multiple Time Step Integration and a Recalibrated Semiempirical QM/MM Hamiltonian. J. Phys. Chem. B 2022, 126, 4226–4235. 10.1021/acs.jpcb.2c02262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J. Absolute Free Energy of Solvation From Monte Carlo Simulations Using Combined Quantum and Molecular Mechanical Potentials. J. Phys. Chem. 1992, 96, 537–540. 10.1021/j100181a009. [DOI] [Google Scholar]
- Muller R. P.; Warshel A. Ab Initio Calculations of Free Energy Barriers for Chemical Reactions in Solution. J. Phys. Chem. 1995, 99, 17516–17524. 10.1021/j100049a009. [DOI] [PubMed] [Google Scholar]
- Martí S.; Andrés J.; Moliner V.; Silla E.; Tuñón I.; Bertrán J. Transition structure selectivity in enzyme catalysis: a QM/MM study of chorismate mutase. Theor. Chem. Acc. 2001, 105, 207–212. 10.1007/s002140000203. [DOI] [Google Scholar]
- Rod T. H.; Ryde U. Quantum Mechanical Free Energy Barrier for an Enzymatic Reaction. Phys. Rev. Lett. 2005, 94, 138302. 10.1103/PhysRevLett.94.138302. [DOI] [PubMed] [Google Scholar]
- Polyak I.; Benighaus T.; Boulanger E.; Thiel W. Quantum mechanics/molecular mechanics dual Hamiltonian free energy perturbation. J. Chem. Phys. 2013, 139, 064105. 10.1063/1.4817402. [DOI] [PubMed] [Google Scholar]
- König G.; Hudson P. S.; Boresch S.; Woodcock H. L. Multiscale Free Energy Simulations: An Efficient Method for Connecting Classical MD Simulations to QM or QM/MM Free Energies Using Non-Boltzmann Bennett Reweighting Schemes. J. Chem. Theory Comput. 2014, 10, 1406–1419. 10.1021/ct401118k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson P. S.; White J. K.; Kearns F. L.; Hodošček M.; Boresch S.; Woodcock H. L. Efficiently computing pathway free energies: New approaches based on chain-of-replica and Non-Boltzmann Bennett reweighting schemes. Biochim. Biophys. Acta 2015, 1850, 944–953. 10.1016/j.bbagen.2014.09.016. [DOI] [PubMed] [Google Scholar]
- Li P.; Jia X.; Pan X.; Shao Y.; Mei Y. Accelerated Computation of Free Energy Profile at ab Initio Quantum Mechanical/Molecular Mechanics Accuracy via a Semi-Empirical Reference Potential. I. Weighted Thermodynamics Perturbation. J. Chem. Theory Comput. 2018, 14, 5583–5596. 10.1021/acs.jctc.8b00571. [DOI] [PubMed] [Google Scholar]
- König G.; Brooks B. R.; Thiel W.; York D. M. On the convergence of multi-scale free energy simulations. Mol. Simul. 2018, 44, 1062–1081. 10.1080/08927022.2018.1475741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese T. J.; York D. M. Development of a Robust Indirect Approach for MM → QM Free Energy Calculations That Combines Force-Matched Reference Potential and Bennett’s Acceptance Ratio Methods. J. Chem. Theory Comput. 2019, 15, 5543–5562. 10.1021/acs.jctc.9b00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese T. J.; Zeng J.; York D. M. Multireference Generalization of the Weighted Thermodynamic Perturbation Method. J. Phys. Chem. A 2022, 126, 8519–8533. 10.1021/acs.jpca.2c06201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.-N.; Xue Y.; Li P.; Pan X.; Wang M.; Shao Y.; Mo Y.; Mei Y. Perspective: Reference-Potential Methods for the Study of Thermodynamic Properties in Chemical Processes: Theory, Applications, and Pitfalls. J. Phys. Chem. Lett. 2023, 14, 4866–4875. 10.1021/acs.jpclett.3c00671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.; Zhou Y.; Xie D. Accelerated Quantum Mechanics/Molecular Mechanics Simulations via Neural Networks Incorporated with Mechanical Embedding Scheme. J. Chem. Theory Comput. 2023, 19, 1157–1169. 10.1021/acs.jctc.2c01131. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Cui Q. Accurate and Efficient Multilevel Free Energy Simulations with Neural Network-Assisted Enhanced Sampling. J. Chem. Theory Comput. 2023, 19, 5394–5406. 10.1021/acs.jctc.3c00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Xia X. A Priori Evaluation of Aqueous Polarization Effects Through Monte Carlo QM-MM Simulations. Science 1992, 258, 631–635. 10.1126/science.1411573. [DOI] [PubMed] [Google Scholar]
- Rossi I.; Truhlar D. G. Parameterization of NDDO wavefunctions using genetic algorithms. An evolutionary approach to parameterizing potential energy surfaces and direct dynamics calculations for organic reactions. Chem. Phys. Lett. 1995, 233, 231–236. 10.1016/0009-2614(94)01450-A. [DOI] [Google Scholar]
- Nam K.; Cui Q.; Gao J.; York D. M. A specific reaction parameterization for the AM1/d Hamiltonian for transphosphorylation reactions. J. Chem. Theory Comput. 2007, 3, 486–504. 10.1021/ct6002466. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Pu J. Reaction path force matching: A new strategy of fitting specific reaction parameters for semiempirical methods in combined QM/MM simulations. J. Chem. Theory Comput. 2014, 10, 3038. 10.1021/ct4009624. [DOI] [PubMed] [Google Scholar]
- Kim B.; Snyder R.; Nagaraju M.; Zhou Y.; Ojeda-May P.; Keeton S.; Hege M.; Shao Y.; Pu J. Reaction Path-Force Matching in Collective Variables: Determining Ab Initio QM/MM Free Energy Profiles by Fitting Mean Force. J. Chem. Theory Comput. 2021, 17, 4961–4980. 10.1021/acs.jctc.1c00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Kale S.; Weare J.; Dinner A. R.; Roux B. Multiple Time-Step Dual-Hamiltonian Hybrid Molecular Dynamics - Monte Carlon Canonical Propagation Algorithm. J. Chem. Theory Comput. 2016, 12, 1449–1458. 10.1021/acs.jctc.5b00706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q.; Pal T.; Xie L. Biomolecular QM/MM Simulations: What Are Some of the ″Burning Issues″?. J. Phys. Chem. B 2021, 125, 689–702. 10.1021/acs.jpcb.0c09898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H.; Crespo A.; Marti M.; Estrin D.; Roitberg A. E. Free energy calculations with non-equilibrium methods: applications of the Jarzynski relationship. Theor. Chem. Acc. 2006, 116, 338–346. 10.1007/s00214-005-0072-2. [DOI] [Google Scholar]
- Carvalho A. T. P.; Barrozo A.; Doron D.; Kilshtain A. V.; Major D. T.; Kamerlin S. C. L. Challenges in computational studies of enzyme structure, function and dynamics. J. Mol. Graph. Model. 2014, 54, 62–79. 10.1016/j.jmgm.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Dietschreit J. C. B.; Diestler D. J.; Hulm A.; Ochsenfeld C.; Gomez-Bombarelli R. From free-energy profiles to activation free energies. J. Chem. Phys. 2022, 157, 084113. 10.1063/5.0102075. [DOI] [PubMed] [Google Scholar]
- Ganguly A.; Weissman B. P.; Giese T. J.; Li N.-S.; Hoshika S.; Rao S.; Benner S. A.; Piccirilli J. A.; York D. M. Confluence of theory and experiment reveals the catalytic mechanism of the Varkud satellite ribozyme. Nat. Chem. 2020, 12, 193–201. 10.1038/s41557-019-0391-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S.; Ollie E.; York D. M.; Piccirilli J. A.; Harris M. E. Rapid Kinetics of Pistol Ribozyme: Insights into Limits to RNA Catalysis. Biochemistry 2023, 62, 2079–2092. 10.1021/acs.biochem.3c00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markin C. J.; Mokhtari D. A.; Sunden F.; Appel M. J.; Akiva E.; Longwell S. A.; Sabatti C.; Herschlag D.; Fordyce P. M. Revealing enzyme functional architecture via high-throughput microfluidic enzyme kinetics. Science 2021, 373, eabf8761 10.1126/science.abf8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohen A.; Limbach H.-H.. Isotope Effects In Chemistry and Biology; Taylor & Francis, 2005. [Google Scholar]
- English B. P; Min W.; van Oijen A. M; Lee K. T.; Luo G.; Sun H.; Cherayil B. J; Kou S C; Xie X S. Ever-fluctuating single enzyme molecules: Michaelis-Menten equation revisited. Nat. Chem. Biol. 2006, 2, 87–94. 10.1038/nchembio759. [DOI] [PubMed] [Google Scholar]
- Seo M.-H.; Park J.; Kim E.; Hohng S.; Kim H.-S. Protein conformational dynamics dictate the binding affinity for a ligand. Nat. Commun. 2014, 5, 3724. 10.1038/ncomms4724. [DOI] [PubMed] [Google Scholar]
- Aviram H. Y.; Pirchi M.; Mazal H.; Barak Y.; Riven I.; Haran G. Direct observation of ultrafast large-scale dynamics of an enzyme under turnover conditions. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 3243–3248. 10.1073/pnas.1720448115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano P.; Guo Q.; Kohen A.; Cheatum C. M. Oscillatory Enzyme Dynamics Revealed by Two-Dimensional Infrared Spectroscopy. J. Phys. Chem. Lett. 2016, 7, 2507–2511. 10.1021/acs.jpclett.6b01154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z. Q.; Jacobsen G. M.; Young M. A. Briefly Bound to Activate: Transient Binding of a Second Catalytic Magnesium Activates the Structure and Dynamics of CDK2 Kinase for Catalysis. Structure 2011, 19, 675–690. 10.1016/j.str.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick R. B.; van den Bedem H.; Fraser J. S.; Wright P. E. Integrated description of protein dynamics from room-temperature X-ray crystallography and NMR. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, E445-E454 10.1073/pnas.1323440111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Yang W. Capture of a third Mg2+ is essential for catalyzing DNA synthesis. Science 2016, 352, 1334–1337. 10.1126/science.aad9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.; Meisburger S. P.; Ando N. Correlated Motions in Structural Biology. Biochemistry 2021, 60, 2331–2340. 10.1021/acs.biochem.1c00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach H.; Cole R.; Gill M. L.; Loria J. P. Conservation of μs-ms Enzyme Motions in the Apo- and Substrate-Mimicked State. J. Am. Chem. Soc. 2005, 127, 9167–9176. 10.1021/ja0514949. [DOI] [PubMed] [Google Scholar]
- Masterson L. R.; Cheng C.; Yu T.; Tonelli M.; Kornev A.; Taylor S. S.; Veglia G. Dynamics connect substrate recognition to catalysis in protein kinase A. Nature Chem. Biol. 2010, 6, 821–828. 10.1038/nchembio.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittier S. K.; Hengge A. C.; Loria J. P. Conformational Motions Regulate Phosphoryl Transfer in Related Protein Tyrosine Phosphatases. Science 2013, 341, 899–903. 10.1126/science.1241735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay L. E. New Views of Functionally Dynamic Proteins by Solution NMR Spectroscop. J. Mol. Biol. 2016, 428, 323–331. 10.1016/j.jmb.2015.11.028. [DOI] [PubMed] [Google Scholar]
- Kovermann M.; Grundstrom C.; Sauer-Eriksson E.; Sauer U. H.; Wolf-Watz M. Structural basis for ligand binding to an enzyme by a conformational selection pathway. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 6298. 10.1073/pnas.1700919114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi G. P.; East K. W.; Batista V. S.; Loria J. P. Altering the allosteric pathway in IGPS suppresses millisecond motions and catalytic activity. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E3414-E3423 10.1073/pnas.1700448114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiller J. B.; Jordan Kerns S.; Hoemberger M.; Cho Y.-J.; Otten R.; Hagan M. F.; Kern D. Probing the transition state in enzyme catalysis by high-pressure NMR dynamics. Nat. Catal. 2019, 2, 726–734. 10.1038/s41929-019-0307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeymer C.; Werbeck N. D.; Zimmermann S.; Reinstein J.; Hansen D. F. Characterizing Active Site Conformational Heterogeneity along the Trajectory of an Enzymatic Phosphoryl Transfer Reaction. Angew. Chem., Int. Ed. 2016, 55, 11533–11537. 10.1002/anie.201606238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akke M.; Brueschweiler R.; Palmer A. G. III, NMR order parameters and free energy: an analytical approach and its application to cooperative calcium(2+) binding by calbindin D9k. J. Am. Chem. Soc. 1993, 115, 9832–9833. 10.1021/ja00074a073. [DOI] [Google Scholar]
- Stone M. J. NMR Relaxation Studies of the Role of Conformational Entropy in Protein Stability and Ligand Binding. Acc. Chem. Res. 2001, 34, 379–388. 10.1021/ar000079c. [DOI] [PubMed] [Google Scholar]
- Diehl C.; Engström O.; Delaine T.; Håkansson M.; Genheden S.; Modig K.; Leffler H.; Ryde U.; Nilsson U. J.; Akke M. Protein Flexibility and Conformational Entropy in Ligand Design Targeting the Carbohydrate Recognition Domain of Galectin-3. J. Am. Chem. Soc. 2010, 132, 14577–14589. 10.1021/ja105852y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl C.; Genheden S.; Modig K.; Ryde U.; Akke M. Conformational entropy changes upon lactose binding to the carbohydrate recognition domain of galectin-3. J. Biomol. NMR 2009, 45, 157–169. 10.1007/s10858-009-9356-5. [DOI] [PubMed] [Google Scholar]
- Wolf-Watz M.; Thai V.; Henzler-Wildman K.; Hadjipavlou G.; Eisenmesser E. Z.; Kern D. Linkage between dynamics and catalysis in a thermophilic-mesophilic enzyme pair. Nat. Struct. Mol. Biol. 2004, 11, 945–949. 10.1038/nsmb821. [DOI] [PubMed] [Google Scholar]
- Ådén J.; Verma A.; Schug A.; Wolf-Watz M. Modulation of a Pre-existing Conformational Equilibrium Tunes Adenylate Kinase Activity. J. Am. Chem. Soc. 2012, 134, 16562–16570. 10.1021/ja3032482. [DOI] [PubMed] [Google Scholar]
- Kovermann M.; Ådén J.; Grundström C.; Sauer-Eriksson A. E.; Sauer U. H.; Wolf-Watz M. Structural basis for catalytically restrictive dynamics of a high-energy enzyme state. Nat. Commun. 2015, 6, 7644. 10.1038/ncomms8644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz K.; Driller R.; Dimos N.; Ringel M.; Brück T.; Loll B.; Major D. T. The Impression of a Nonexisting Catalytic Effect: The Role of CotB2 in Guiding the Complex Biosynthesis of Cyclooctat-9-en-7-ol. J. Am. Chem. Soc. 2020, 142, 21562–21574. 10.1021/jacs.0c11348. [DOI] [PubMed] [Google Scholar]
- Mauser H.; King W. A.; Gready J. E.; Andrews T. J. CO2 Fixation by Rubisco: Computational Dissection of the Key Steps of Carboxylation, Hydration, and C-C Bond Cleavage. J. Am. Chem. Soc. 2001, 123, 10821–10829. 10.1021/ja011362p. [DOI] [PubMed] [Google Scholar]
- Nam K.; Gao J.; York D. M. Electrostatic interactions in the hairpin ribozyme account for the majority of the rate acceleration without chemical participation by nucleobases. RNA 2008, 14, 1501–1507. 10.1261/rna.863108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S.; Shimshi M.; Raz K.; Nitoker Eliaz N.; Mhashal A. R.; Ansbacher T.; Major D. T. EnzyDock: Protein-Ligand Docking of Multiple Reactive States Along a Reaction Coordinate in Enzymes. J. Chem. Theory Comput. 2019, 15, 5116–5134. 10.1021/acs.jctc.9b00366. [DOI] [PubMed] [Google Scholar]
- Boresch S.; Archontis G.; Karplus M. Free energy simulations: The meaning of the individual contributions from a component analysis. Proteins 1994, 20, 25–33. 10.1002/prot.340200105. [DOI] [PubMed] [Google Scholar]
- Boresch S.; Karplus M. The Meaning of Component Analysis: Decomposition of the Free Energy in Terms of Specific Interactions. J. Mol. Biol. 1995, 254, 801–807. 10.1006/jmbi.1995.0656. [DOI] [PubMed] [Google Scholar]
- Kazemi M.; Åqvist J. Chemical reaction mechanisms in solution from brute force computational Arrhenius plots. Nat. Commun. 2015, 6, 7293. 10.1038/ncomms8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åqvist J.; Kazemi M.; Villy I. G.; Brandsdal B. O. Entropy and Enzyme Catalysis. Acc. Chem. Res. 2017, 50, 199–207. 10.1021/acs.accounts.6b00321. [DOI] [PubMed] [Google Scholar]
- Karplus M.; Evanseck J. D.; Joseph D.; Bash P. A.; Field M. J. Simulation analysis of triose phosphate isomerase: conformational transition and catalysis. Faraday Discuss. 1992, 93, 239–248. 10.1039/fd9929300239. [DOI] [PubMed] [Google Scholar]
- Liu H.; Zhang Y.; Yang W. How Is the Active Site of Enolase Organized To Catalyze Two Different Reaction Steps?. J. Am. Chem. Soc. 2000, 122, 6560–6570. 10.1021/ja9936619. [DOI] [Google Scholar]
- Cui Q.; Karplus M. Triosephosphate Isomerase: A Theoretical Comparison of Alternative Pathways. J. Am. Chem. Soc. 2001, 123, 2284–2290. 10.1021/ja002886c. [DOI] [PubMed] [Google Scholar]
- Garcia-Viloca M.; Truhlar D. G.; Gao J. Reaction-Path Energetics and Kinetics of the Hydride Transfer Reaction Catalyzed by Dihydrofolate Reductase. Biochemistry 2003, 42, 13558–13575. 10.1021/bi034824f. [DOI] [PubMed] [Google Scholar]
- Ranaghan K. E.; Ridder L.; Szefczyk B.; Hermann W. A. S.; Mulholland A.; Sokalski W. A. Insights into enzyme catalysis from QM/MM modelling: transition state stabilization in chorismate mutase. Mol. Phys. 2003, 101, 2695–2714. 10.1080/00268970310001593286. [DOI] [Google Scholar]
- Szefczyk B.; Mulholland A. J.; Ranaghan K. E.; Sokalski W. A. Differential Transition-State Stabilization in Enzyme Catalysis: Quantum Chemical Analysis of Interactions in the Chorismate Mutase Reaction and Prediction of the Optimal Catalytic Field. J. Am. Chem. Soc. 2004, 126, 16148–16159. 10.1021/ja049376t. [DOI] [PubMed] [Google Scholar]
- Graham S.; Syeda F.; Cisneros G. A. Computational Prediction of Residues Involved in Fidelity Checking for DNA Synthesis in DNA Polymerase I. Biochemistry 2012, 51, 2569–2578. 10.1021/bi201856m. [DOI] [PubMed] [Google Scholar]
- Jaña G. A.; Delgado E. J.; Medina F. E. How Important Is the Synclinal Conformation of Sulfonylureas To Explain the Inhibition of AHAS: A Theoretical Study. J. Chem. Inf. Model 2014, 54, 926–932. 10.1021/ci400721y. [DOI] [PubMed] [Google Scholar]
- Liu C. T.; Layfield J. P.; Stewart R. J. III; French J. B.; Hanoian P.; Asbury J. B.; Hammes-Schiffer S.; Benkovic S. J. Probing the Electrostatics of Active Site Microenvironments along the Catalytic Cycle for Escherichia coli Dihydrofolate Reductase. J. Am. Chem. Soc. 2014, 136, 10349–10360. 10.1021/ja5038947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kędzierski P.; Moskal M.; Sokalski W. A. Catalytic Fields as a Tool to Analyze Enzyme Reaction Mechanism Variants and Reaction Steps. J. Phys. Chem. B 2021, 125, 11606–11616. 10.1021/acs.jpcb.1c05256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galmés M. À.; Nödling A. R.; He K.; Luk L. Y. P.; Świderek K.; Moliner V. Computational design of an amidase by combining the best electrostatic features of two promiscuous hydrolases. Chem. Sci. 2022, 13, 4779–4787. 10.1039/D2SC00778A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghsoud Y.; Jayasinghe-Arachchige V. M.; Kumari P.; Cisneros G. A.; Liu J. Leveraging QM/MM and Molecular Dynamics Simulations to Decipher the Reaction Mechanism of the Cas9 HNH Domain to Investigate Off-Target Effects. J. Chem. Inf. Model 2023, 63, 6834–6850. 10.1021/acs.jcim.3c01284. [DOI] [PubMed] [Google Scholar]
- Bash P. A.; Field M. J.; Davenport R. C.; Petsko G. A.; Ringe D.; Karplus M. Computer simulation and analysis of the reaction pathway of triosephosphate isomerase. Biochemistry 1991, 30, 5826–5832. 10.1021/bi00238a003. [DOI] [PubMed] [Google Scholar]
- Pan X.; Van R.; Pu J.; Nam K.; Mao Y.; Shao Y. Free Energy Profile Decomposition Analysis for QM/MM Simulations of Enzymatic Reactions. J. Chem. Theory Comput. 2023, 19, 8234–8244. 10.1021/acs.jctc.3c00973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht A.Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; W. H. Freeman, 1999. [Google Scholar]
- Kulkarni Y. S.; Amyes T. L.; Richard J. P.; Kamerlin S. C. L. Uncovering the Role of Key Active-Site Side Chains in Catalysis: An Extended Brønsted Relationship for Substrate Deprotonation Catalyzed by Wild-Type and Variants of Triosephosphate Isomerase. J. Am. Chem. Soc. 2019, 141, 16139–16150. 10.1021/jacs.9b08713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher G.; Corbella M.; Alphey M. S.; Nicholson J.; Read B. J.; Kamerlin S. C. L.; da Silva R. G. Allosteric rescue of catalytically impaired ATP phosphoribosyltransferase variants links protein dynamics to active-site electrostatic preorganisation. Nat. Commun. 2022, 13, 7607. 10.1038/s41467-022-34960-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurplus M.; McCammon J. A. DYNAMICS OF PROTEINS: ELEMENTS AND FUNCTION. Annu. Rev. Biochem. 1983, 52, 263–300. 10.1146/annurev.bi.52.070183.001403. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K. A.; Lei M.; Thai V.; Kerns S. J.; Karplus M.; Kern D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450 (7171), 913–916. 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- Cheatum C. M.; Kohen A. Relationship of Femtosecond-Picosecond Dynamics to Enzyme-Catalyzed H-Transfer. Top. Curr. Chem. 2013, 337, 1–40. 10.1007/128_2012_407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror R. O.; Dirks R. M.; Grossman J. P.; Xu H.; Shaw D. E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. 10.1146/annurev-biophys-042910-155245. [DOI] [PubMed] [Google Scholar]
- Yang J.; Lior-Hoffmann L.; Wang S.; Zhang Y.; Broyde S. DNA Cytosine Methylation: Structural and Thermodynamic Characterization of the Epigenetic Marking Mechanism. Biochemistry 2013, 52, 2828–2838. 10.1021/bi400163k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recabarren R.; Zinovjev K.; Tuñón I.; Alzate-Morales J. How a Second Mg2+ Ion Affects the Phosphoryl-Transfer Mechanism in a Protein Kinase: A Computational Study. ACS Catal. 2021, 11, 169–183. 10.1021/acscatal.0c03304. [DOI] [Google Scholar]
- Dürr S. L.; Bohuszewicz O.; Berta D.; Suardiaz R.; Jambrina P. G.; Peter C.; Shao Y.; Rosta E. The Role of Conserved Residues in the DEDDh Motif: the Proton-Transfer Mechanism of HIV-1 RNase H. ACS Catal. 2021, 11, 7915–7927. 10.1021/acscatal.1c01493. [DOI] [Google Scholar]
- Berger M. B.; Cisneros G. A. Distal Mutations in the β-Clamp of DNA Polymerase III* Disrupt DNA Orientation and Affect Exonuclease Activity. J. Am. Chem. Soc. 2023, 145, 3478–3490. 10.1021/jacs.2c11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekesan Ş.; McCarthy E.; Case D. A.; York D. M. RNA Electrostatics: How Ribozymes Engineer Active Sites to Enable Catalysis. J. Phys. Chem. B 2022, 126, 5982–5990. 10.1021/acs.jpcb.2c03727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhashal A. R.; Pshetitsky Y.; Eitan R.; Cheatum C. M.; Kohen A.; Major D. T. Effect of Asp122 Mutation on the Hydride Transfer in E. coli DHFR Demonstrates the Goldilocks of Enzyme Flexibility. J. Phys. Chem. B 2018, 122, 8006–8017. 10.1021/acs.jpcb.8b05556. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; McElheny D.; Dyson H. J.; Wright P. E. Millisecond timescale fluctuations in dihydrofolate reductase are exquisitely sensitive to the bound ligands. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 1373–1378. 10.1073/pnas.0914163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga Y.; Fujisaki H.; Terada T.; Furuta T.; Moritsugu K.; Kidera A. Minimum Free Energy Path of Ligand-Induced Transition in Adenylate Kinase. PLoS Comput. Biol. 2012, 8, e1002555 10.1371/journal.pcbi.1002555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmstrom R. D.; Kornev A. P.; Taylor S. S.; Amaro R. E. Allostery through the computational microscope: cAMP activation of a canonical signalling domain. Nat. Commun. 2015, 6, 7588. 10.1038/ncomms8588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam K.; Karplus M. Insights into the origin of the high energy-conversion efficiency of F1-ATPase. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 15924–15929. 10.1073/pnas.1906816116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam K.; Arattu Thodika A. R.; Grundström C.; Sauer U. H.; Wolf-Watz M. Elucidating Dynamics of Adenylate Kinase from Enzyme Opening to Ligand Release. J. Chem. Inf. Model 2024, 64 (1), 150–163. 10.1021/acs.jcim.3c01618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla D.; Meng Y.; Roux B.; Pande V. S. Activation pathway of Src kinase reveals intermediate states as targets for drug designs. Nat. Commun. 2014, 5, 3397. 10.1038/ncomms4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y.; Lin Y.-l.; Roux B. Computational Study of the ″DFG-Flip″ Conformational Transition in c-Abl and c-Src Tyrosine Kinases. J. Phys. Chem. B 2015, 119, 1443–1456. 10.1021/jp511792a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Nam K. Dynamic, structural, and thermodynamic basis of insulin-like growth factor 1 kinase allostery mediated by activation loop phosphorylation. Chem. Sci. 2017, 8, 3453–3464. 10.1039/C7SC00055C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam K.; Tao Y.; Ovchinnikov V. Molecular Simulations of Conformational Transitions within the Insulin Receptor Kinase Reveal Consensus Features in a Multistep Activation Pathway. J. Phys. Chem. B 2023, 127, 5789–5798. 10.1021/acs.jpcb.3c01804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gora A.; Brezovsky J.; damborsky J. Gates of Enzymes. Chem. Rev. 2013, 113, 5871–5923. 10.1021/cr300384w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerns S. J.; Agafonov R. V.; Cho Y.-J.; Pontiggia F.; Otten R.; Pachov D. V.; Kutter S.; Phung L. A.; Murphy P. N.; Thai V.; Alber T.; Hagan M. F.; Kern D. The energy landscape of adenylate kinase during catalysis. Nat. Struct. Mol. Biol. 2015, 22, 124–131. 10.1038/nsmb.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y.; Seeliger M. A.; Eastwood M. P.; Frank F.; Xu H.; Jensen M. Ø.; Dror R. O.; Kuriyan J.; Shaw D. E. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 139–144. 10.1073/pnas.0811223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elber R. Perspective: Computer simulations of long time dynamics. J. Chem. Phys. 2016, 144, 060901. 10.1063/1.4940794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertig S.; Latorraca N. R.; Dror R. O. Revealing Atomic-Level Mechanisms of Protein Allostery with Molecular Dynamics Simulations. PLoS Comput. Biol. 2016, 12, e1004746 10.1371/journal.pcbi.1004746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita Y.; Feig M.. All-atom Molecular Dynamics Simulation of Proteins in Crowded Environments. In In-cell NMR Spectroscopy: From Molecular Sciences to Cell Biology, Ito Y.; Dötsch V.; Shirakawa M., Eds.; The Royal Society of Chemistry, 2019; pp 228–248. [Google Scholar]
- Schlick T.; Collepardo-Guevara R.; Halvorsen L. A.; Jung S.; Xiao X. Biomoleular modeling and simulation: a field coming of age. Q. Rev. Biophys. 2011, 44, 191–228. 10.1017/S0033583510000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elber R. Simulations of allosteric transitions. Curr. Opin. Struct. Biol. 2011, 21, 167–172. 10.1016/j.sbi.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Dokainish H. M.; Sugita Y. Exploring Large Domain Motions in Proteins Using Atomistic Molecular Dynamics with Enhanced Conformational Sampling. Int. J. Mol. Sci. 2021, 22, 270. 10.3390/ijms22010270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima H.; Re S.; Sugita Y. Replica-Exchange Umbrella Sampling Combined with Gaussian Accelerated Molecular Dynamics for Free-Energy Calculation of Biomolecules. J. Chem. Theory Comput. 2019, 15, 5199–5208. 10.1021/acs.jctc.9b00761. [DOI] [PubMed] [Google Scholar]
- Hansen A. L.; Kay L. E. Measurement of histidine pKa values and tautomer populations in invisible protein states. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, E1705-E1712 10.1073/pnas.1400577111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z.; CHen W.; Zgurskaya H. I.; Shen J. Constant pH Molecular Dynamics Reveals How Proton Release Drives the Conformational Transition of a Transmembrane Efflux Pump. J. Chem. Theory Comput. 2017, 13, 6405–6414. 10.1021/acs.jctc.7b00874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson J. A.; Harris R. C.; Tsai C.-J.; Shen J. How Ligand Protonation State Controls Water in Protein-Ligand Binding. J. Phys. Chem. Lett. 2018, 9, 5440–5444. 10.1021/acs.jpclett.8b02440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C.-J.; Yue Z.; Shen J. How Electrostatic Coupling Enables Conformational Plasticity in a Tyrosine Kinase. J. Am. Chem. Soc. 2019, 141, 15092–15101. 10.1021/jacs.9b06064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A. Y.; Brooks B. R.; Damjanovic A. Ion channel selectivity through ion-modulated changes of selectivity filter pKa values. Proc. Natl. Acad. Sci. U.S.A. 2023, 120, e2220343120 10.1073/pnas.2220343120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins de Oliveira V.; Liu R.; Shen J. Constant pH molecular dynamics simulations: Current status and recent applications. Curr. Opin. Struct. Biol. 2022, 77, 102498. 10.1016/j.sbi.2022.102498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J.; Jacob F. Teleonomic Mechanisms in Cellular Metabolism, Growth, and Differentiation. Cold Spring Harb. Symp. Quant. Biol. 1961, 26, 389. 10.1101/SQB.1961.026.01.048. [DOI] [PubMed] [Google Scholar]
- Vogt A. D.; Di Cera E. Conformational Selection or Induced Fit? A Critical Appraisal of the Kinetic Mechanism. Biochemistry 2012, 51, 5894–8902. 10.1021/bi3006913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul F.; Weikl T. R. How to Distinguish Conformational Selection and Induced Fit Based on Chemical Relaxation Rates. PLoS Comput. Biol. 2016, 12, e1005067 10.1371/journal.pcbi.1005067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orädd F.; Ravishankar H.; Goodman J.; Rogne P.; Backman L.; Duelli A.; Pedersen M. N.; Levantino M.; Wulff M.; Wolf-Watz M.; Andersson M. Tracking the ATP-binding response in adenylate kinase in real time. Sci. Adv. 2021, 7, eabi5514 10.1126/sciadv.abi5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedy D. A.; Kenner L. R.; Warkentin M.; Woldeyes R. A.; Hopkins J. B.; Thompson M. C.; Brewster A. S.; Van Benschoten A. H.; Baxter E. L.; Uervirojnangkoorn M.; McPhillips S. E.; Song J.; Alonso-Mori R.; Holton J. M.; Weis W. I.; Brunger A. T.; Soltis S. M.; Lemke H.; Gonzalez A.; Sauter N. K.; Cohen A. E.; van den Bedem H.; Thorne R. E.; Fraser J. S. Mapping the conformational landscape of a dynamic enzyme by multitemperature and XFEL crystallography. eLife 2015, 4, e07574 10.7554/eLife.07574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman J. P.; Hammes-Schiffer S.. Dynamics in Enzyme Catalysis; Springer, 2013. [Google Scholar]
- Klinman J. P. Dynamically Achieved Active Site Precision in Enzyme Catalysis. Acc. Chem. Res. 2015, 48, 449–456. 10.1021/ar5003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro A. J.; Santos-Martins D.; Russo N.; Ramos M. J.; Fernandes P. A. Enzymatic Flexibility and Reaction Rate: A QM/MM Study of HIV-1 Protease. ACS Catal. 2015, 5, 5617–5626. 10.1021/acscatal.5b00759. [DOI] [Google Scholar]
- Agarwal P. K.; Doucet N.; Chennubhotla C.; Ramanathan A.; Narayanan C. Conformational Sub-states and Populations in Enzyme Catalysis. Meth. Enzymol. 2016, 578, 273–297. 10.1016/bs.mie.2016.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P. K.; Bernard D. N.; Bafna K.; Doucet N. Enzyme Dynamics: Looking Beyond a Single Structure. ChemCatChem. 2020, 12, 4704–4720. 10.1002/cctc.202000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabukarski F.; Biel J. T.; Pinney M. M.; Doukov T.; Powers A. S.; Fraser J. S.; Herschlag D. Assessment of enzyme active site positioning and tests of catalytic mechanisms through X-ray-derived conformational ensembles. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 33204–33215. 10.1073/pnas.2011350117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crean R. M.; Gardner J. M.; Kamerlin S. C. L. Harnessing Conformational Plasticity to Generate Designer Enzymes. J. Am. Chem. Soc. 2020, 142, 11324–11342. 10.1021/jacs.0c04924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabukarski F.; Doukov T.; Pinney M. M.; Biel J. T.; Fraser J. S.; Herschlag D. Ensemble-function relationships to dissect mechanisms of enzyme catalysis. Sci. Adv. 2022, 8, eabn7738 10.1126/sciadv.abn7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coimbra J. T. S.; Neves R. P. P.; Cunha A. V.; Ramos M. J.; Fernandes P. A. Different Enzyme Conformations Induce Different Mechanistic Traits in HIV-1 Protease. Chem.—Eur. J. 2022, 28, e202201066 10.1002/chem.202201066. [DOI] [PubMed] [Google Scholar]
- Min W.; Xie X. S.; Bagchi B. Two-Dimensional Reaction Free Energy Surfaces of Catalytic Reaction: Effects of Protein Conformational Dynamics on Enzyme Catalysis. J. Phys. Chem. B 2008, 112, 454–466. 10.1021/jp076533c. [DOI] [PubMed] [Google Scholar]
- Mokhtari D. A.; Appel M. J.; Fordyce P. M.; Herschlag D. High throughput and quantitative enzymology in the genomic era. Curr. Opin. Struct. Biol. 2021, 71, 259–273. 10.1016/j.sbi.2021.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffucci I.; Laage D.; Sterpone F.; Stirnemann G. Thermal Adaptation of Enzymes: Impacts of Conformational Shifts on Catalytic Activation Energy and Optimum Temperature. Chem.—Eur. J. 2020, 26, 10045–10056. 10.1002/chem.202001973. [DOI] [PubMed] [Google Scholar]
- Yang L.-W.; Bahar I. Coupling between Catalytic Site and Collective Dynamics: A Requirement for Mechanochemical Activity of Enzymes. Structure 2005, 13, 893–904. 10.1016/j.str.2005.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C.-J.; Nussinov R. A Unified View of ″How Allostery Works″. PLoS Comput. Biol. 2014, 10, e1003394 10.1371/journal.pcbi.1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph E.; Le C. Q.; Nguyen T.; Oyugi M.; Hossain M. S.; Foss F. W. Jr.; Johnson-Winters K. Evidence of Negative Cooperativity and Half-Site Reactivity within an F420-Dependent Enzyme: Kinetic Analysis of F420H2:NADP+ Oxidoreductase. Biochemistry 2016, 55, 1082–1090. 10.1021/acs.biochem.5b00762. [DOI] [PubMed] [Google Scholar]
- Abrahams J. P.; Leslie A. G. W.; Lutter R.; Walker J. E. Structure at 2.8 A Resolution of F1-ATPase from Bovine Heart Mitochondria. Nature 1994, 370, 621–628. 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- Karplus M.; Gao Y. Biomolecular Motors: the F1-ATPase Paradigm. Curr. Opin. Struct. Biol. 2004, 14, 250–259. 10.1016/j.sbi.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Hubbard S. R.; Till J. H. Protein Tyrosine Kinase Structure and Function. Annu. Rev. Biochem. 2000, 69, 373–398. 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- Tzeng S.-R.; Kalodimos C. G. Protein dynamics and allostery: an NMR view. Curr. Opin. Struct. Biol. 2011, 21, 62–67. 10.1016/j.sbi.2010.10.007. [DOI] [PubMed] [Google Scholar]
- Masterson L. R.; Cembran A.; Shi L.; Veglia G. ALLOSTERY AND BINDING COOPERATIVITY OF THE CATALYTIC SUBUNIT OF PROTEIN KINASE A BY NMR SPECTROSCOPY AND MOLECULAR DYNAMICS SIMULATIONS. Adv. Protein Chem. Struct. Biol. 2012, 87, 363–389. 10.1016/B978-0-12-398312-1.00012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motlagh H. N.; Wrabl J. O.; Li J.; Hilser V. J. The ensemble nature of allostery. Nature 2014, 508, 331–339. 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besch A.; Marsiglia W. M.; Mohammadi M.; Zhang Y.; Traaseth N. J. Gatekeepr mutations activate FGF receptor tyrosine kinases by destablizing the autoinhibited state. Proc. Natl. Acad. Sci. U.S.A. 2023, 120, e2213090120 10.1073/pnas.2213090120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A.; Dryden D. T. F. Allostery without conformational change. Eur. Biophys. J. 1984, 11, 103–109. 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- Kornev A. P.; Taylor S. S. Dynamics-driven allostery in protein kinases. Trends Biochem. Sci. 2015, 40, 628. 10.1016/j.tibs.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern D.; Zuiderweg E. R. P. The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol. 2003, 13, 748–757. 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Berteotti A.; Cavalli A.; Branduardi D.; Gervasio F. L.; Recanatini M.; Parrinello M. Protein Conformational Transitions: The Closure Mechanism of a Kinase Explored by Atomistic Simulations. J. Am. Chem. Soc. 2009, 131, 244–250. 10.1021/ja806846q. [DOI] [PubMed] [Google Scholar]
- Popovych N.; Sun S.; Ebright R. H.; Kalodimos C. G. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 2006, 13, 831–838. 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Janezic D.; Karplus M. Harmonic analysis of large systems. I. Methodology. J. Comput. Chem. 1995, 16, 1522. 10.1002/jcc.540161209. [DOI] [Google Scholar]
- Roy A.; Post C. B. Detection of Long-Range Concerted Motions in Protein by a Distance Covariance. J. Chem. Theory Comput. 2012, 8, 3009–3014. 10.1021/ct300565f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier G.; Ortiz V. Emerging computational approaches for the study of protein allostery. Arch. Biochem. Biophys. 2013, 538, 6–15. 10.1016/j.abb.2013.07.025. [DOI] [PubMed] [Google Scholar]
- Schueler-Furman O.; Wodak S. J. Computational approaches to investigating allostery. Curr. Opin. Struct. Biol. 2016, 41, 159–171. 10.1016/j.sbi.2016.06.017. [DOI] [PubMed] [Google Scholar]
- Balsera M. A.; Wriggers W.; Oono Y.; Schulten K. Principal component analysis and long time protein dynamics. J. Phys. Chem. 1996, 100, 2567. 10.1021/jp9536920. [DOI] [Google Scholar]
- del Sol A.; Fujihashi H.; Amoros D.; Nussinov R. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol. Syst. Biol. 2006, 2, 1–12. 10.1038/msb4100063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi A.; Eargle J.; Black A. A.; Luthey-Schulten Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 6620–6625. 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C.; Tobi D.; Bahar I. Allosteric Changes in Protein Structure Computed by a Simple Mechanical Model: Hemoglobin T↔R2 Transition. J. Mol. Biol. 2003, 333, 153–168. 10.1016/j.jmb.2003.08.027. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Brooks B. R.; Thirumalai D. Low-frequency normal modes that describe allosteric transitions in biological nanomachines are robust to sequence variations. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 7664–7669. 10.1073/pnas.0510426103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flechsig H. Design of Elastic Networks with Evolutionary Optimized Long-Range Communication as Mechanical Models of Allosteric Proteins. Biophys. J. 2017, 113, 558–571. 10.1016/j.bpj.2017.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockless S. W.; Ranganathan R. Evolutionarily Conserved Pathways of Energetic Connectivity in Protein Families. Science 1999, 286, 295–299. 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- Kalescky R.; Liu J.; Tao P. Identifying Key Residues for Protein Allostery through Rigid Residue Scan. J. Phys. Chem. A 2015, 119, 1689–1700. 10.1021/jp5083455. [DOI] [PubMed] [Google Scholar]
- Kalescky R.; Zhou H.; Liu J.; Tao P. Rigid Residue Scan Simulations Systematically Reveal Residue Entropic Roles in Protein Allostery. PLoS Comput. Biol. 2016, 12, e1004893 10.1371/journal.pcbi.1004893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhivker G. M.; Agajanian S.; Hu G.; Tao P. Allosteric Regulation at the Crossroads of New Technologies: Multiscale Modeling, Networks, and Machine Learning. Front. Mol. Biosci. 2020, 7, 136. 10.3389/fmolb.2020.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agajanian S.; Alshahrani M.; Bai F.; Tao P.; Verkhivker G. M. Exploring and Learning the Universe of Protein Allostery Using Artificial Intelligence Augmented Biophysical and Computational Approaches. J. Chem. Inf. Model 2023, 63, 1413–1428. 10.1021/acs.jcim.2c01634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Cui Q. Multiple Pathways and Time Scales for Conformational Transitions in apo-Adenylate Kinase. J. Chem. Theory Comput. 2018, 14, 1716–1726. 10.1021/acs.jctc.7b01064. [DOI] [PubMed] [Google Scholar]
- Buchenberg S.; Sittel F.; Stock G. Time-resolved observation of protein allosteric communication. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E6804-E6811 10.1073/pnas.1707694114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y.; Deng J.; Cui Q. Molecular Dynamics Simulations Establish the Molecular Basis for the Broad Allostery Hotspot Distributions in the Tetracycline Repressor. J. Am. Chem. Soc. 2022, 144, 10870–10887. 10.1021/jacs.2c03275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda-May P.; Li Y.; Ovchinnikov V.; Nam K. Role of Protein Synamics in Allosteric Control of the Catalytic Phosphoryl Tranfer of Insulin Receptor Kinase. J. Am. Chem. Soc. 2015, 137, 12454–12457. 10.1021/jacs.5b07996. [DOI] [PubMed] [Google Scholar]
- Faure A. J.; Domingo J.; Schmiedel J. M.; Hidalgo-Carcedo C.; Diss G.; Lehner B. Mapping the energetic and allosteric landscapes of protein binding domains. Nature 2022, 604, 175–183. 10.1038/s41586-022-04586-4. [DOI] [PubMed] [Google Scholar]
- Aoto P. C.; Martin B. T.; Wright P. E. NMR Characterization of Information Flow and Allosteric Communities in the MAP Kinase p38γ. Sci. Rep. 2016, 6, 28655. 10.1038/srep28655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday M. J.; Camilloni C.; Armstrong G. S.; Vendruscolo M.; Eisenmesser E. Z. Networks of Dynamic Allostery Regulate Enzyme Function. Struct 2017, 25, 276–286. 10.1016/j.str.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R.; Okuno D.; Sakakihara S.; Shimabukuro K.; Iino R.; Yoshida M.; Noji H. Mechanical modulation of catalytic power on F1-ATPase. Nat. Chem. Biol. 2012, 8, 86–92. 10.1038/nchembio.715. [DOI] [PubMed] [Google Scholar]
- Adachi K.; Oiwa K.; Yoshida M.; Nishizaka T.; Kinosita K. Jr, Controlled rotation of the F1-ATPase reveals differential and continuous binding changes for ATP synthesis. Nat. Commun. 2012, 3, 1022. 10.1038/ncomms2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y. Q.; Yang W.; Marcus R. A.; Karplus M. A model for the cooperative free energy transduction and kinetics of ATP hydrolysis by F1-ATPase. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 11339–11344. 10.1073/pnas.1334188100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y. Q.; Yang W.; Karplus M. A Structure-based Model for the Synthesis and Hydrolysis of ATP by F1-ATPase. Cell 2005, 123, 195–205. 10.1016/j.cell.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Nam K.; Pu J.; Karplus M. Trapping the ATP binding state leads to a detailed understanding of the F1-ATPase mechanism. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 17851–17586. 10.1073/pnas.1419486111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M. A.; Gonfloni S.; Superti-Furga G.; Roux B.; Kuriyan J. Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell 2001, 105, 115–126. 10.1016/S0092-8674(01)00301-4. [DOI] [PubMed] [Google Scholar]
- Feng C.; Roy A.; Post C. B. Entropic allostery dominates the phosphorylation-dependent regulation of Syk tyrosine kinase release from immunoreceptor tyrosine-based activation motifs. Protein Sci. 2018, 27, 1780–1796. 10.1002/pro.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolon D. N.; Mayo S. L. Enzyme-like proteins by computational design. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 14274–14279. 10.1073/pnas.251555398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röthlisberger D.; Khersonsky O.; Wollacott A. M.; Jiang L.; DeChancie J.; Betker J.; Gallaher J. L.; Althoff E. A.; Zanghellini A.; Dym O.; Albeck S.; Houk K. N.; Tawfik D. S.; Baker D. Kemp elimination catalysts by computational enzyme design. Nature 2008, 453, 190–195. 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- Jäckel C.; Kast P.; Hilvert D. Protein Design by Directed Evolution. Annu. Rev. Biophys. 2008, 37, 153–173. 10.1146/annurev.biophys.37.032807.125832. [DOI] [PubMed] [Google Scholar]
- Richter F.; Leaver-Fay A.; Khare S. D.; Bjelic S.; Baker D. De Novo Enzyme Design Using Rosetta3. PLoS One 2011, 6, e19230 10.1371/journal.pone.0019230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kries H.; Blomberg R.; Hilvert D. De novo enzymes by computational design. Curr. Opin. Chem. Biol. 2013, 17, 221–228. 10.1016/j.cbpa.2013.02.012. [DOI] [PubMed] [Google Scholar]
- Khersonsky O.; Lipsh R.; Avizemer Z.; Ashani Y.; Goldsmith M.; Leader H.; Dym O.; Rogotner S.; Trudeau D. L.; Prilusky J.; Amengual-Rigo P.; Guallar V.; Tawfik D. S.; Fleishman S. J. Automated Design of Efficient and Functionally Diverse Enzyme Repertoires. Mol. Cell 2018, 72, 178–186. 10.1016/j.molcel.2018.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques S.; Planas-Iglesias J.; Damborsky J. Web-based tools for computational enzyme design. Curr. Opin. Struct. Biol. 2021, 69, 19–34. 10.1016/j.sbi.2021.01.010. [DOI] [PubMed] [Google Scholar]
- Lin H.-Y.; Chen X.; Dong J.; Yang J.-F.; Xiao H.; Ye Y.; Li L.-H.; Zhan C.-G.; Yang W.-C.; Yang G.-F. Rational Redesign of Enzyme via the Combination of Quantum Mechanics/Molecular Mechanics, Molecular Dynamics, and Structural Biology Study. J. Am. Chem. Soc. 2021, 143, 15674–15687. 10.1021/jacs.1c06227. [DOI] [PubMed] [Google Scholar]
- Lovelock S. L.; Crawshaw R.; Basler S.; Levy C.; Baker D.; Hilvert D.; Green A. P. The road to fully programmable protein catalysis. Nature 2022, 606, 49–58. 10.1038/s41586-022-04456-z. [DOI] [PubMed] [Google Scholar]
- Wittmund M.; Cadet F.; Davari M. D. Learning Epistasis and Residue Coevolution Patterns: Current Trends and Future Perspectives for Advancing Enzyme Engineering. ACS Catal. 2022, 12, 14243–14263. 10.1021/acscatal.2c01426. [DOI] [Google Scholar]
- Pinto G. P.; Corbella M.; Demkiv A. O.; Kamerlin S. C. L. Exploiting enzyme evolution for computational protein design. Trends Biochem. Sci. 2022, 47, 375–389. 10.1016/j.tibs.2021.08.008. [DOI] [PubMed] [Google Scholar]
- Vaissier Welborn V. Structural dynamics and computational design of synthetic enzymes. Chem. Catal. 2022, 2, 19–28. 10.1016/j.checat.2021.10.009. [DOI] [PubMed] [Google Scholar]
- Markus B.; Christian C G.; Andreas K.; Arkadij K.; Stefan L.; Gustav O.; Elina S.; Radka S. Accelerating Biocatalysis Discovery with Machine Learning: A Paradigm Shift in Enzyme Engineering, Discovery, and Design. ACS Catal. 2023, 13, 14454–14469. 10.1021/acscatal.3c03417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantillo D. J; Jiangang C.; Houk K. N Theozymes and compuzymes: theoretical models for biological catalysis. Curr. Opin. Chem. Biol. 1998, 2, 743–750. 10.1016/S1367-5931(98)80112-9. [DOI] [PubMed] [Google Scholar]
- Anishchenko I.; Pellock S. J.; Chidyausiku T. M.; Ramelot T. A.; Ovchinnikov S.; Hao J.; Bafna K.; Norn C.; Kang A.; Bera A. K.; DiMaio F.; Carter L.; Chow C. M.; Montelione G. T.; Baker D. De novo protein design by deep network hallucination. Nature 2021, 600, 547–552. 10.1038/s41586-021-04184-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Lisanza S.; Juergens D.; Tischer D.; Watson J. L.; Castro K. M.; Ragotte R.; Saragovi A.; Milles L. F.; Baek M.; Anishchenko I.; Yang W.; Hicks D. R.; Expòsit M.; Schlichthaerle T.; Chun J.-H.; Dauparas J.; Bennett N.; Wicky B. I. M.; Muenks A.; DiMaio F.; Correia B.; Ovchinnikov S.; Baker D. Scaffolding protein functional sites using deep learning. Science 2022, 377, 387–394. 10.1126/science.abn2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauparas J.; Anishchenko I.; Bennett N.; Bai H.; Ragotte R. J.; Milles L. F.; Wicky B. I. M.; Courbet A.; de Haas R. J.; Bethel N.; Leung P. J. Y.; Huddy T. F.; Pellock S.; Tischer D.; Chan F.; Koepnick B.; Nguyen H.; Kang A.; Sankaran B.; Bera A. K.; King N. P.; Baker D. Robust deep learning-based protein sequence design using ProteinMPNN. Science 2022, 378, 49–56. 10.1126/science.add2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J. L.; Juergens D.; Bennett N.; Trippe B. L.; Yim J.; Eisenach H. E.; Ahern W.; Borst A. J.; Ragotte R.; Milles L. F.; Wicky B. I. M.; Hanikel N.; Pellock S. J.; Courbet A.; Sheffler W.; Wang J.; Venkatesh P.; Sappington I.; Torres S. V.; Lauko A.; De Bortoli V.; Mathieu E.; Ovchinnikov S.; Barzilay R.; Jaakkola T. S.; DiMaio F.; Baek M.; Baker D. De novo design of protein structure and function with RFdiffusion. Nature 2023, 620, 1089–1100. 10.1038/s41586-023-06415-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L.; Althoff E. A.; Clemente F. R.; Doyle L.; Röthlisberger D.; Zanghellini A.; Gallaher J. L.; Betker J.; Tanaka F.; Barbas C. F. III; Hilvert D.; Houk K. N.; Stoddard B. L.; Baker D. De Novo Computational Design of Retro-Aldol Enzymes. Science 2008, 319, 1387–1391. 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Rivera A.; Garcia-Borràs M.; Osuna S. Role of Conformational Dynamics in the Evolution of Retro-Aldolase Activity. ACS Catal. 2017, 7, 8524–8532. 10.1021/acscatal.7b02954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel J. B.; Zanghellini A.; Lovick H. M.; Kiss G.; Lambert A. R.; St.Clair J. L.; Gallaher J. L.; Hilvert D.; Gelb M. H.; Stoddard B. L.; Houk K. N.; Michael F. E.; Baker D. Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science 2010, 329, 309–313. 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter F.; Blomberg R.; Khare S. D.; Kiss G.; Kuzin A. P.; Smith A. J. T.; Gallaher J.; Pianowski Z.; Helgeson R. C.; Grjasnow A.; Xiao R.; Seetharaman J.; Su M.; Vorobiev S.; Lew S.; Forouhar F.; Kornhaber G. J.; Hunt J. F.; Montelione G. T.; Tong L.; Houk K. N.; Hilvert D.; Baker D. Computational Design of Catalytic Dyads and Oxyanion Holes for Ester Hydrolysis. J. Am. Chem. Soc. 2012, 134, 16197–16206. 10.1021/ja3037367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Xu L.; ZHang H.; Liu J.; Yan J.; Yan Y. A De Novo Designed Esterase with p-Nitrophenyl Acetate Hydrolysis Activity. Molecules 2020, 25, 4658. 10.3390/molecules25204658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh A. H.-W.; Norn C.; Kipnis Y.; Tischer D.; Pellock S. J.; Evans D.; Ma P.; Lee G. R.; Zhang J. Z.; Anishchenko I.; Coventry B.; Cao L.; Dauparas J.; Halabiya S.; DeWitt M.; Carter L.; Houk K. N.; Baker D. De novo design of luciferases using deep learning. Nature 2023, 614, 774–780. 10.1038/s41586-023-05696-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth J.; Havranek J. J.; Duarte C. M.; Sussman D.; Monnat R. J. Jr; Stoddard B. L.; Baker D. Computational redesign of endonuclease DNA binding and cleavage specificity. Nature 2006, 441, 656–659. 10.1038/nature04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz S.; Iamurri S. M. Protein Engineering: Past, Present, and Future. Methods Mol. Biol. 2018, 1685, 1–12. 10.1007/978-1-4939-7366-8_1. [DOI] [PubMed] [Google Scholar]
- Gardner J. M.; Biler M.; Risso V. A.; Sanchez-Ruiz J. M.; Kamerlin S. C. L. Manipulating Conformational Dynamics To Repurpose Ancient Proteins for Modern Catalytic Functions. ACS Catal. 2020, 10, 4863–4870. 10.1021/acscatal.0c00722. [DOI] [Google Scholar]
- Han X.; Liu W.; Huang J.-W.; Ma J.; Zheng Y.; Ko T.-P.; Xu L.; Cheng Y.-S.; Chen C.-C.; Guo R.-T. Structural insight into catalytic mechanism of PET hydrolase. Nat. Commun. 2017, 8, 2106. 10.1038/s41467-017-02255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi I.; Yoshida S.; Hiraga K.; Miyamoto K.; Kimura Y.; Oda K. Biodegration of PET: Current Status and Application Aspects. ACS Catal. 2019, 9, 4089–4105. 10.1021/acscatal.8b05171. [DOI] [Google Scholar]
- Knott B. C.; Erickson E.; Allen M. D.; Gado J. E.; Graham R.; Kearns F. L.; Pardo I.; Topuzlu E.; Anderson J. J.; Austin H. P.; Dominick G.; Johnson C. W.; Rorrer N. A.; Szostkiewicz C. J.; Copie V.; Payne C. M.; Woodcock H. L.; Donohoe B. S.; Beckham G. T.; McGeehan J. E. Characterization and engineering of a two-enzyme system for plastics depolymerization. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 25476–25485. 10.1073/pnas.2006753117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier V.; Topham C. M.; Gilles A.; David B.; Folgoas C.; Moya-Leclair E.; Kamionka E.; Desrousseaux M.-L.; Texier H.; Gavalda S.; Cot M.; Guémard E.; Dalibey M.; Nomme J.; Cioci G.; Barbe S.; Chateau M.; André I.; Duquesne S.; Marty A. An engineered PET depolymerase to break down and recycle plastic bottles. Nature 2020, 580, 216–219. 10.1038/s41586-020-2149-4. [DOI] [PubMed] [Google Scholar]
- Goldsmith M.; Tawfik D. S. Enzyme engineering: reaching the maximal catalytic efficiency peak. Curr. Opin. Struct. Biol. 2017, 47, 140–150. 10.1016/j.sbi.2017.09.002. [DOI] [PubMed] [Google Scholar]
- Broom A.; Rakotoharisoa R. V.; Thompson M. C.; Zarifi N.; Nguyen E.; Mukhametzhanov N.; Liu L.; Fraser J. S.; Chica R. A. Ensemble-based enzyme design can recapitulate the effects of laboratory directed evolution in silico. Nat. Commun. 2020, 11, 4808. 10.1038/s41467-020-18619-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chica R. A.; Doucet N.; Pelletier J. N. Semi-rational approaches to engineering enzyme activity: combining the benefits of directed evolution and rational design. Curr. Opin. Biotechnol. 2005, 16, 378–384. 10.1016/j.copbio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Khersonsky O.; Röthlisberger D.; Dym O.; Albeck S.; Jackson C. J.; Baker D.; Tawfik D. S. Evolutionary Optimization of Computationally Designed Enzymes: Kemp Eliminases of the KE07 Series. J. Mol. Biol. 2010, 396, 1025–1042. 10.1016/j.jmb.2009.12.031. [DOI] [PubMed] [Google Scholar]
- Khersonsky O.; Röthlisberger D.; Wollacott A. M.; Murphy P.; Dym O.; Albeck S.; Kiss G.; Houk K. N.; Baker D.; Tawfik D. S. Optimization of the In-Silico-Designed Kemp Eliminase KE70 by Computational Design and Directed Evolution. J. Mol. Biol. 2011, 407, 391–412. 10.1016/j.jmb.2011.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong N.-S.; Petrović D.; Lee R.; Gryn’ova G.; Purg M.; Saunders J.; Bauer P.; Carr P. D.; Lin C.-Y.; Mabbitt P. D.; Zhang W.; Altamore T.; Easton C.; Coote M. L.; Kamerlin S. C. L.; Jackson C. J. The evolution of multiple active site configurations in a designed enzyme. Nat. Commun. 2018, 9, 3900. 10.1038/s41467-018-06305-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; Bridgland A.; Meyer C.; Kohl S. A. A.; Ballard A. J.; Cowie A.; Romera-Paredes B.; Nikolov S.; Jain R.; Adler J.; Back T.; Petersen S.; Reiman D.; Clancy E.; Zielinski M.; Steinegger M.; Pacholska M.; Berghammer T.; Bodenstein S.; Silver D.; Vinyals O.; Senior A. W.; Kavukcuoglu K.; Kohli P.; Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek M.; DiMaio F.; Anishchenko I.; Dauparas J.; Ovchinnikov S.; Lee G. R.; Wang J.; Cong Q.; Kinch L. N.; Schaeffer R. D.; Millán C.; Park H.; Adams C.; Glassman C. R.; Degiovanni A.; Pereira J. H.; Rodrigues A. V.; van Dijk A. A.; Ebrecht A. C.; Opperman D. J.; Sagmeister T.; Buhlheller C.; Pavkov-Keller T.; Rathinaswamy M. K.; Dalwadi U.; Yip C. K.; Burke J. E.; Garcia K. C.; Grishin N. V.; Adams P. D.; Read R. J.; Baker D. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouba P.; Kohout P.; Haddadi F.; Bushuiev A.; Samusevich R.; Sedlar J.; Damborsky J.; Pluskal T.; Sivic J.; Mazurenko S. Machine Learning-Guided Protein Engineering. ACS Catal. 2023, 13, 13863–13895. 10.1021/acscatal.3c02743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. K.; Wu Z.; Arnold F. H. Machine-learning-guided directed evolution for protein engineering. Nat. Methods 2019, 16, 687–694. 10.1038/s41592-019-0496-6. [DOI] [PubMed] [Google Scholar]
- Mazurenko S.; Prokop Z.; Damborsky J. Machine Learning in Enzyme Engineering. ACS Catal. 2020, 10, 1210–1223. 10.1021/acscatal.9b04321. [DOI] [Google Scholar]
- Russ W. P.; Figliuzzi M.; Stocker C.; Barrat-Charlaix P.; Socolich M.; Kast P.; Hilvert D.; Monasson R.; Cocco S.; Weigt M.; Ranganathan R. An evolution-based model for designing chorismate mutase enzymes. Science 2020, 369, 440–445. 10.1126/science.aba3304. [DOI] [PubMed] [Google Scholar]
- Wittmann B. J.; Johnston K. E.; Wu Z.; Arnold F. H. Advances in machine learning for directed evolution. Curr. Opin. Struct. Biol. 2021, 69, 11–18. 10.1016/j.sbi.2021.01.008. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; Ran X.; Yang Z. Data-driven enzyme engineering to identify function-enhancing enzymes. Protein Eng. Des. Sel. 2023, 36, gzac009. 10.1093/protein/gzac009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall G.; Duran C.; Osuna S. AlphaFold2 and Deep Learning for Elucidating Enzyme Conformational Flexibility and Its Application for Design. JACS Au 2023, 3, 1554–1562. 10.1021/jacsau.3c00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson K. E.; Lindorff-Larsen K. Structural heterogeneity and dynamics in protein evolution and design. Curr. Opin. Struct. Biol. 2018, 48, 157–163. 10.1016/j.sbi.2018.01.010. [DOI] [PubMed] [Google Scholar]
- Wayment-Steele H. K.; Ojoawo A.; Otten R.; Apitz J. M.; Pitsawong W.; Hömberger M.; Ovchinnikov S.; Colwell L.; Kern D. Predicting multiple conformations via sequence clustering and AlphaFold2. Nature 2023, 625, 832–839. 10.1038/s41586-023-06832-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr D. D.; D’Amico R. N.; O’Rourke K. F. Engineered control of enzyme structural dynamics and function. Protein Sci. 2018, 27 (4), 825–838. 10.1002/pro.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrović D.; Kamerlin S. C. L. Molecular modeling of conformational dynamics and its role in enzyme evolution. Curr. Opin. Struct. Biol. 2018, 52, 50–57. 10.1016/j.sbi.2018.08.004. [DOI] [PubMed] [Google Scholar]
- Liao J.; Warmuth M. K.; Govindarajan S.; Ness J. E.; Wang R. P.; Gustafsson C.; Minshull J. Engineering proteinase K using machine learning and synthetic genes. BMC Biotechnol. 2007, 7, 16. 10.1186/1472-6750-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berta D.; Gehrke S.; Nyíri K.; Vértessy B. G.; Rosta E. Mechanism-Based Redesign of GAP to Activate Oncogenic Ras. J. Am. Chem. Soc. 2023, 145, 20302–20310. 10.1021/jacs.3c04330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox R. J.; Davis S. C.; Mundorff E. C.; Newman L. M.; Gavrilovic V.; Ma S. K.; Chung L. M.; Ching C.; Tam S.; Muley S.; Grate J.; Gruber J.; Whitman J. C.; Sheldon R. A.; Huisman G. W. Improving catalytic function by ProSAR-driven enzyme evolution. Nat. Biotechnol. 2007, 25, 338–344. 10.1038/nbt1286. [DOI] [PubMed] [Google Scholar]
- Saito Y.; Oikawa M.; Sato T.; Nakazawa H.; Ito T.; Kameda T.; Tsuda K.; Umetsu M. Machine-Learning-Guided Library Design Cycle for Directed Evolution of Enzymes: The Effects of Training Data Composition on Sequence Space Exploration. ACS Catal. 2021, 11, 14615–14624. 10.1021/acscatal.1c03753. [DOI] [Google Scholar]
- Cadet F.; Fontaine N.; Li G.; Sanchis J.; Ng Fuk Chong M.; Pandjaitan R.; Vetrivel I.; Offmann B.; Reetz M. T. A machine learning approach for reliable prediction of amino acid interactions and its application in the directed evolution of enantioselective enzymes. Sci. Rep. 2018, 8, 16757. 10.1038/s41598-018-35033-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Kan S. B. J.; Lewis R. D.; Wittmann B. J.; Arnold F. H. Machine learning-assisted directed protein evolution with combinatorial libraries. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 8852–8858. 10.1073/pnas.1901979116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten R.; Pádua R. A. P.; Bunzel H. A.; Nguyen V.; Pitsawong W.; Patterson M.; Sui S.; Perry S. L.; Cohen A. E.; Hilvert D.; Kern D. How directed evolution reshapes the energy landscape in an enzyme to boost catalysis. Science 2020, 370, 1442–1446. 10.1126/science.abd3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton J. W.; Need E.; Crews D. Resurrecting the Ancestral Steroid Receptor: Ancient Origin of Estrogen Signaling. Science 2003, 301, 1714–1717. 10.1126/science.1086185. [DOI] [PubMed] [Google Scholar]
- Merkl R.; Sterner R. Ancestral protein reconstruction: techniques and applications. Biol. Chem. 2016, 397, 1–21. 10.1515/hsz-2015-0158. [DOI] [PubMed] [Google Scholar]
- Lim S. A.; Hart K. M.; Harms M. J.; Marqusee S. Evolutionary trend toward kinetic stability in the folding trajectory of RNases H. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 13045–13050. 10.1073/pnas.1611781113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll C. R.; Massari M.; Fraaije M. W.; Mascotti M. L.; Mattevi A. Impact of ancestral sequence reconstruction on mechanistic and structural enzymology. Curr. Opin. Struct. Biol. 2023, 82, 102669. 10.1016/j.sbi.2023.102669. [DOI] [PubMed] [Google Scholar]
- Nocedal I.; Laub M. T. Ancestral reconstruction of duplicated signaling proteins reveals the evolution of signaling specificity. eLife 2022, 11, e77346 10.7554/eLife.77346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailleul G.; Yang G.; Nicoll C. R.; Mattevi A.; Fraaije M. W.; Mascotti M. L. Evolution of enzyme functionality in the flavin-containing monooxygenases. Nat. Commun. 2023, 14, 1042. 10.1038/s41467-023-36756-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Xu Z.; Ding B.; Zhang Y.; Liu S.; Cai C.; Li M.; Dale B. E.; Jin M. Big data mining, rational modification, and ancestral sequence reconstruction inferred multiple xylose isomerases for biorefinery. Sci. Adv. 2023, 9, eadd8835 10.1126/sciadv.add8835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10, 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Lonsdale R.; Ward R. A. Structure-based design of targeted covalent inhibitors. Chem. Sci. 2018, 47, 3816–3830. 10.1039/C7CS00220C. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Hatcher J. M.; Teng M.; Gray N. S.; Kostic M. Recent Advances in Selective and Irreversible Covalent Ligand Development and Validatation. Cell Chem. Biol. 2019, 26, 1486–1500. 10.1016/j.chembiol.2019.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdeldayem A.; Raouf Y. S.; Constantinescu S. N.; Moriggl R.; Gunning P. T. Advances in covalent kinase inhibitors. Chem. Soc. Rev. 2020, 49, 2617–2687. 10.1039/C9CS00720B. [DOI] [PubMed] [Google Scholar]
- Beigel J. H.; Tomashek K. M.; Dodd L. E.; Mehta A. K.; Zingman B. S.; Kalil A. C.; Hohmann E.; Chu H. Y.; Luetkemeyer A.; Kline S.; Lopez de Castilla D.; Finberg R. W.; Dierberg K.; Tapson V.; Hsieh L.; Patterson T. F.; Paredes R.; Sweeney D. A.; Short W. R.; Touloumi G.; Lye D. C.; Ohmagari N.; Oh M.-d.; Ruiz-Palacios G. M.; Benfield T.; Fatkenheuer G.; Kortepeter M. G.; Atmar R. L.; Creech C. B.; Lundgren J.; Babiker A. G.; Pett S.; Neaton J. D.; Burgess T. H.; Bonnett T.; Green M.; Makowski M.; Osinusi A.; Nayak S.; Lane H. C. Remdesivir for the Treatment of Covid-19 — Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W.; Mao C.; Luan X.; Shen D.-D.; Shen Q.; Su H.; Wang X.; Zhou F.; Zhao W.; Gao M.; Chang S.; Xie Y.-C.; Tian G.; Jiang H.-W.; Tao S.-C.; Shen J.; Jiang Y.; Jiang H.; Xu Y.; Zhang S.; Zhang Y.; Xu H. E. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R.; Allerton C. M. N.; Anderson A. S.; Aschenbrenner L.; Avery M.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K. J.; Dantonio A.; Di L.; Eng H.; Ferre R.; Gajiwala K. S.; Gibson S. A.; Greasley S. E.; Hurst B. L.; Kadar E. P.; Kalgutkar A. S.; Lee J. C.; Lee J.; Liu W.; Mason S. W.; Noell S.; Novak J. J.; Obach R. S.; Ogilvie K.; Patel N. C.; Pettersson M.; Rai D. K.; Reese M. R.; Sammons M. F.; Sathish J. G.; Singh R. S. P.; Steppan C. M.; Stewart A. E.; Tuttle J. B.; Updyke L.; Verhoest P. R.; Wei L.; Yang Q.; Zhu Y. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Cully M. A tale of two antiviral targets - and the COVID-19 drugs that bind them. Nat. Rev. Drug Discovery 2022, 21, 3–5. 10.1038/d41573-021-00202-8. [DOI] [PubMed] [Google Scholar]
- Ostrem J. M.; Peters U.; Sos M. L.; Wells J. A.; Shokat K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanman B. A.; Allen J. R.; Allen J. G.; Amegadzie A. K.; Ashton K. S.; Booker S. K.; Chen J. J.; Chen N.; Frohn M. J.; Goodman G.; Kopecky D. J.; Liu L.; Lopez P.; Low J. D.; Ma V.; Minatti A. E.; Nguyen T. T.; Nishimura N.; Pickrell A. J.; Reed A. B.; Shin Y.; Siegmund A. C.; Tamayo N. A.; Tegley C. M.; Walton M. C.; Wang H.-L.; Wurz R. P.; Xue M.; Yang K. C.; Achanta P.; Bartberger M. D.; Canon J.; Hollis L. S.; McCarter J. D.; Mohr C.; Rex K.; Saiki A. Y.; San Miguel T.; Volak L. P.; Wang K. H.; Whittington D. A.; Zech S. G.; Lipford J. R.; Cee V. J. Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. 10.1021/acs.jmedchem.9b01180. [DOI] [PubMed] [Google Scholar]
- Bender B. J.; Gahbauer S.; Luttens A.; Lyu J.; Webb C. M.; Stein R. M.; Fink E. A.; Balius T. E.; Carlsson J.; Irwin J. J.; Shoichet B. K. A practical guide to large-scale docking. Nat. Protoc. 2021, 16, 4799–4832. 10.1038/s41596-021-00597-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttens A.; Gullberg H.; Abdurakhmanov E.; Vo D. D.; Akaberi D.; Talibov V. O.; Nekhotiaeva N.; Vangeel L.; De Jonghe S.; Jochmans D.; Krambrich J.; Tas A.; Lundgren B.; Gravenfors Y.; Craig A. J.; Atilaw Y.; Sandström A.; Moodie L. W. K.; Lundkvist A.; van Hemert M. J.; Neyts J.; Lennerstrand J.; Kihlberg J.; Sandberg K.; Danielson H.; Carlsson J. Ultralarge Virtual Screening Identifies SARS-CoV-2 Main Protease Inhibitors with Broad-Spectrum Activity against Coronaviruses. J. Am. Chem. Soc. 2022, 144, 2905–2920. 10.1021/jacs.1c08402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awoonor-Williams E.; Isley W. C. III; Dale S. G.; Johnson E. R.; Yu H.; Becke A. D.; Roux B.; Rowley C. N. Quantum Chemical Methods for Modeling Covalent Modification of Biological Thiols. J. Comput. Chem. 2020, 41, 427–438. 10.1002/jcc.26064. [DOI] [PubMed] [Google Scholar]
- Mondal D.; Warshel A. Exploring the Mechanism of Covalent Inhibition: Simulating the Binding Free Energy of α-Ketoamide Inhibitors of the Main Protease of SARS-CoV-2. Biochemistry 2020, 59, 4601–4608. 10.1021/acs.biochem.0c00782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Świderek K.; Moliner V. Revealing the molecular mechanisms of proteolysis of SARS-CoV-2 Mpro by QM/MM computational methods. Chem. Sci. 2020, 11, 10626–10630. 10.1039/D0SC02823A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Guzmán C. A.; Ruiz-Pernía J. J.; Tuñón I. A microscopic description of SARS-CoV-2 main protease inhibition with Michael acceptors. Strategies for improving inhibitor design. Chem. Sci. 2021, 12, 3489–3496. 10.1039/D0SC04978F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Guzmán C. A.; Ruiz-Pernía J. J.; Tuñón I. Inhibition Mechanism of SARS-CoV-2 Main Protease with Ketone-Based Inhibitors Unveiled by Multiscale Simulations: Insights for Improved Designs. Angew. Chem., Int. Ed. Engl. 2021, 60, 25933–25941. 10.1002/anie.202110027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arafet K.; Serrano-Aparicio N.; Lodola A.; Mulholland A. J.; González F. V.; Świderek K.; Moliner V. Mechanism of inhibition of SARS-CoV-2 Mpro by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chem. Sci. 2021, 12, 1433–1444. 10.1039/D0SC06195F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zev S.; Raz K.; Schwartz R.; Tarabeh R.; Gupta P. K.; Major D. T. Benchmarking the Ability of Common Docking Programs to Correctly Reproduce and Score Binding Modes in SARS-CoV-2 Protease Mpro. J. Chem. Inf. Model 2021, 61, 2957–2966. 10.1021/acs.jcim.1c00263. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Saha A.; Huang Z.; Warshel A. Fast and Effective Prediction of the Absolute Binding Free Energies of Covalent Inhibitors of SARS-CoV-2 Main Protease and 20S Proteasome. J. Am. Chem. Soc. 2022, 144, 7568–7572. 10.1021/jacs.2c00853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Hauner D.; Laureanti J. A.; Agustin K.; Raugei S.; Kumar N. Mechanistic investigation of SARS-CoV-2 main protease to accelerate design of covalent inhibitors. Sci. Rep. 2022, 12, 21037. 10.1038/s41598-022-23570-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos A. M.; Oliveira A. R. S.; da Costa C. H. S.; Kenny P. W.; Montanari C. A.; Varela J. d. J. G.; Lameira J. Assessment of Reversibility for Covalent Cysteine Protease Inhibitors Using Quantum Mechanics/Molecular Mechanics Free Energy Surfaces. J. Chem. Inf. Model. 2022, 62, 4083–4094. 10.1021/acs.jcim.2c00466. [DOI] [PubMed] [Google Scholar]
- Nochebuena J.; Cisneros G. A. Polarizable MD and QM/MM investigation of acrylamide-based leads to target the main protease of SARS-CoV-2. J. Chem. Phys. 2022, 157, 185101. 10.1063/5.0123698. [DOI] [PubMed] [Google Scholar]
- Martí S.; Arafet K.; Lodola A.; Mulholland A. J.; Świderek K.; Moliner V. Impact of Warhead Modulations on the Covalent Inhibition of SARS-CoV-2 Mpro Explored by QM/MM Simulations. ACS Catal. 2022, 12, 698–708. 10.1021/acscatal.1c04661. [DOI] [PubMed] [Google Scholar]
- Ramos-Guzmán C. A.; Andjelkovic M.; Zinovjev K.; Ruiz-Pernía J. J.; Tuñón I. The impact of SARS-CoV-2 3CL protease mutations on nirmatrelvir inhibitory efficiency. Computational insights into potential resistance mechanisms. Chem. Sci. 2023, 14, 2686–2697. 10.1039/D2SC06584C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N.; Miller R. M.; Krishnan S.; Uchida K.; Irwin J. J.; Eidam O.; Gibold L.; Cimermančič P.; Bonnet R.; Shoichet B. K.; Taunton J. Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol. 2014, 10, 1066–1072. 10.1038/nchembio.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco G.; Forli S.; Goodsell D. S.; Olson A. J. Covalent docking using autodock: Two-point attractor and flexible side chain methods. Protein Sci. 2016, 25, 295–301. 10.1002/pro.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotriffer C. Docking of Covalent Ligands: Challenges and Approaches. Mol. Inf. 2018, 37, 1800062. 10.1002/minf.201800062. [DOI] [PubMed] [Google Scholar]
- Scarpino A.; Ferenczy G. G.; Keserű G. M. Comparative Evaluation of Covalent Docking Tools. J. Chem. Inf. Model 2018, 58, 1441–1458. 10.1021/acs.jcim.8b00228. [DOI] [PubMed] [Google Scholar]
- Dalton S. E.; Campos S. Covalent Small Molecules as Enabling Platforms for Drug Discovery. Chem. Bio. Chem. 2020, 21, 1080–1100. 10.1002/cbic.201900674. [DOI] [PubMed] [Google Scholar]
- Mihalovits L. M.; Ferenczy G. G.; Keserű G. M. Affinity and Selectivity Assessment of Covalent Inhibitors by Free Energy Calculations. J. Chem. Inf. Model. 2020, 60, 6579–6594. 10.1021/acs.jcim.0c00834. [DOI] [PubMed] [Google Scholar]
- Scarpino A.; Petri L.; Knez D.; Imre T.; Ábrányi-Balogh P.; Ferenczy G. G.; Gobec S.; Keserű G. M. WIDOCK: a reactive docking protocol for virtual screening of covalent inhibitors. J. Comput. Aided Mol. Des. 2021, 35, 223–244. 10.1007/s10822-020-00371-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Guzmán C. A.; Ruiz-Pernía J. J.; Tuñón I. Multiscale Simulations of SARS-CoV-2 3CL Protease Inhibition with Aldehyde Derivatives. Role of Protein and Inhibitor Conformational Changes in the Reaction Mechanism. ACS Catal. 2021, 11, 4157–4168. 10.1021/acscatal.0c05522. [DOI] [PubMed] [Google Scholar]
- Oyedele A.-Q. K.; Ogunlana A. T.; Boyenle I. D.; Adeyemi A. O.; Rita T. O.; Adelusi T. I.; Abdul-Hammed M.; Elegbeleye O. E.; Odunitan T. T. Docking covalent targets for drug discovery: stimulating the computer-aided drug design community of possible pitfalls and erroneous practices. Mol. Divers. 2023, 27, 1879–1903. 10.1007/s11030-022-10523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon D. A.; Weerapana E. Covalent protein modification: the current landscape of residue-specific electrophiles. Curr. Opin. Chem. Biol. 2015, 24, 18–26. 10.1016/j.cbpa.2014.10.021. [DOI] [PubMed] [Google Scholar]
- Mazmanian K.; Chen T.; Sargsyan K.; Lim C. From quantum-derived principles underlying cysteine reactivity to combating the COVID-19 pandemic. WIREs Comput. Mol. Sci. 2022, 12, e1607 10.1002/wcms.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ábrányi-Balogh P.; Petri L.; Imre T.; Szijj P.; Scarpino A.; Hrast M.; Mitrović A.; Fonovič U. P.; Németh K.; Barreteau H.; Roper D. I.; Horváti K.; Ferenczy G. G.; Kos J.; Ilaš J.; Gobec S.; Keserű G. M. A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur. J. Med. Chem. 2018, 160, 94–107. 10.1016/j.ejmech.2018.10.010. [DOI] [PubMed] [Google Scholar]
- Bianco G.; Goodsell D. S.; Forli S. Selective and Effective: Current Progress in Computational Structure-Based Drug Discovery of Targeted Covalent Inhibitors. Trends Pharmacol. Sci. 2020, 41, 1038–1049. 10.1016/j.tips.2020.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmeister T.; Kesselring J.; Jung S.; Schneider T. H.; Weickert A.; Becker J.; Lee W.; Bamberger D.; Wich P. R.; Distler U.; Tenzer S.; Johé P.; Hellmich U. A.; Engels B. Quantum Chemical-Based Protocol for the Rational Design of Covalent Inhibitors. J. Am. Chem. Soc. 2016, 138, 8332–8335. 10.1021/jacs.6b03052. [DOI] [PubMed] [Google Scholar]
- Mihalovits L. M.; Ferenczy G. G.; Keserű G. M. The role of quantum chemistry in covalent inhibitor design. Int. J. Quantum Chem. 2022, 122, e26768 10.1002/qua.26768. [DOI] [Google Scholar]
- Yu H. S.; Gao C.; Lupyan D.; Wu Y.; Kimura T.; Wu C.; Jacobson L.; Harder E.; Abel R.; Wang L. Toward Atomistic Modeling of Irreversible Covalent Inhibitor Binding Kinetics. J. Chem. Inf. Model. 2019, 59, 3955–3967. 10.1021/acs.jcim.9b00268. [DOI] [PubMed] [Google Scholar]
- Voice A.; Tresadern G.; van Vlijmen H.; Mulholland A. Limitations of Ligand-Only Approaches for Predicting the Reactivity of Covalent Inhibitors. J. Chem. Inf. Model. 2019, 59, 4220–4227. 10.1021/acs.jcim.9b00404. [DOI] [PubMed] [Google Scholar]
- Ramos-Guzmán C. A.; Velázquez-Libera J. L.; Ruiz-Pernía J. J.; Tuñón I. Testing Affordable Strategies for the Computational Study of Reactivity in Cysteine Proteases: The Case of SARS-CoV-2 3CL Protease Inhibition. J. Chem. Theory Comput. 2022, 18, 4005–4013. 10.1021/acs.jctc.2c00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile F.; Agrawal V.; Hsing M.; Ton A.-T.; Ban F.; Norinder U.; Gleave M. E.; Cherkasov A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6, 939–949. 10.1021/acscentsci.0c00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Park S.; Min D.; Kim W. Comprehensive Survey of Recent Drug Discovery Using Deep Learning. Int. J. Mol. Sci. 2021, 22, 9983. 10.3390/ijms22189983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff D. E.; Shakhnovich E. I.; Coley C. W. Accelerating high-throughput virtual screening through molecular pool-based active learning. Chem. Sci. 2021, 12, 7866–7881. 10.1039/D0SC06805E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crampon K.; Giorkallos A.; Deldossi M.; Baud S.; Steffenel L. A. Machine-learning methods for ligand-protein molecular docking. Drug Discovery Today 2022, 27, 151–164. 10.1016/j.drudis.2021.09.007. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Yao K.; Repasky M. P.; Leswing K.; Abel R.; Shoichet B. K.; Jerome S. V. Efficient Exploration of Chemical Space with Docking and Deep Learning. J. Chem. Theory Comput. 2021, 17, 7106–7119. 10.1021/acs.jctc.1c00810. [DOI] [PubMed] [Google Scholar]
- Gentile F.; Yaacoub J. C.; Gleave J.; Fernandez M.; Ton A.-T.; Ban F.; Stern A.; Cherkasov A. Artificial intelligence-enabled virtual screening of ultra-large chemical libraries with deep docking. Nat. Protoc. 2022, 17, 672–697. 10.1038/s41596-021-00659-2. [DOI] [PubMed] [Google Scholar]