

Abstract
Here we present remarkable epoxyketone-based proteasome inhibitors with low nanomolar in vitro potency for blood-stage Plasmodium falciparum and low cytotoxicity for human cells. Our best compound has more than 2,000-fold greater selectivity for erythrocytic-stage P. falciparum over HepG2 and H460 cells, which is largely driven by the accommodation of the parasite proteasome for a D-amino acid in the P3 position and the preference for a difluorobenzyl group in the P1 position. We isolated the proteasome from P. falciparum cell extracts and determined that the best compound is 171-fold more potent at inhibiting the β5 subunit of P. falciparum proteasome when compared to the same subunit of the human constitutive proteasome. These compounds also significantly reduce parasitemia in a P. berghei mouse infection model and prolong survival of animals by an average of 6 days. The current epoxyketone inhibitors are ideal starting compounds for orally bioavailable anti-malarial drugs.
Keywords: Epoxyketone, Inhibition, Plasmodium, Proteasome, Malaria
Introduction
Malaria remains a worldwide public health problem, and Plasmodium falciparum is the protozoan parasite responsible for the most deaths by this disease.[1] The emergence of resistance to artemisinin combination therapies[2] has led to a need for new medications with novel mechanisms of action.[3,4] The P. falciparum proteasome is a multi-subunit protease involved in turnover of cellular proteins[5,6] and is essential for both protozoal replication and for invasion of host erythrocytes.[7] Additionally, proteasome inhibitors FDA approved to treat multiple myeloma have been used to validate the P. falciparum proteasome as a drug target. However, these compounds are too toxic to be used for treatment of malaria.[8,9] As a result, structural differences between the P. falciparum and human proteasomes have been exploited to develop potent inhibitors that are selective for P. falciparum.[10-13] These include covalent tripeptide-vinyl sulfone inhibitors,[11,14] amino-amide boronates[15] and noncovalent macrocyclic peptide inhibitors[11,14,16] with nanomolar potency against the parasite and micromolar potency against human cells. Most importantly, proteasome inhibitors strongly synergize artemisinin-mediated killing of Plasmodium in vitro and in vivo.[17]
We previously discovered a nanomolar potency covalent peptide-epoxyketone proteasome inhibitor, carmaphycin B, from a marine cyanobacterium, but it was only 3-fold selective for asexual blood stage P. falciparum cells over human HepG2 cells. We thus modified this scaffold to yield P. falciparum proteasome inhibitor J-18 that showed a 379-fold selectivity over HepG2 cells.[13] This compound contains a d-amino acid at the P3 position that enables a favorable interaction with the binding pocket of the parasite proteasome but not with the host proteasome. The epoxyketone (EK) group of J-18 compounds binds irreversibly to the catalytic threonine residue in the proteasome active site.
Results and Discussion
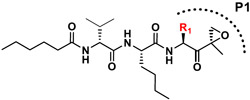
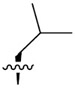
Building on our previous hit compound J-18, with selectivity index (SI) of 379,[13] we constructed 31 new compounds with modifications at P1, P2, P3, and P4 as summarized in Figure 1. We first replaced Leu at P1 with Phe (J-50), as we, as well as others, have shown that phenyl groups in this position increase selectivity.[13,15] This change reduced host cell cytotoxicity by 3.8-fold while only reducing potency for P falciparum by 2-fold, therefore increasing selectivity to 649 (Table 1).
Figure 1.

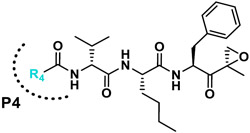
Summary of the amino acid modifications in the synthesized analogues based on published lead compound J-18.
Table 1.
Analogues with changes in the P1, P4 and P3 moieties (R1, R4, and R3, respectively). IC50 data presented as mean ± SEM for 3 biological replicates. Selectivity Index (SI) is the ratio of the compound’s IC50 for HepG2 to it’s IC50 for P. falciparum.
| No. | R-group | P. falciparum IC50 (nM) | HepG2 IC50 (nM) | SI |
|---|---|---|---|---|
| R1 |
|
|||
| J-18 |
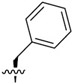
|
3.3 ± 0.23 | 1,240 ± 274 | 379.1 |
| J-50 |
|
7.1 ± 3.2 | 4,608 ± 721 | 649.0 |
| R4 |
|
|||
| J-50 |
|
7.1 ± 3.2 | 4,608 ± 721 | 645.6 |
| J-54 |
|
166 ± 10.1 | 1,546 ± 132 | 9.3 |
| J-55 |
|
20.0 ± 1.1 | 1,529 ± 15.9 | 76.5 |
| J-56 |
|
240 ± 19.2 | 1,587 ± 213 | 6.6 |
| J-75 |
|
36.2 ± 1.6 | 12,535 ± 122 | 346.2 |
| R3 |
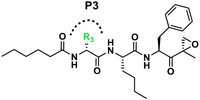
|
|||
| J-51 |
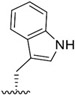
|
9.10 ± 1.7 | 308 ± 77.3 | 33.9 |
| J-53 |
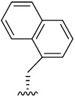
|
99.7 ± 0.23 | 1,068 ± 330 | 10.7 |
| J-64 |
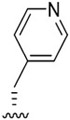
|
20.3 ± 4.4 | 5,882 ± 46.5 | 289.8 |
| J-66 |
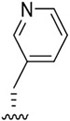
|
30.8 ± 4.5 | 2,303 ± 107 | 74.8 |
| J-71 |
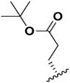
|
15.9 ± 0.19 | 15,680 ± 1,860 | 986.2 |
| J-74 |
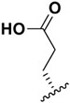
|
94.2 ± 5.1 | >100,000 | >1000 |
| J-76 |
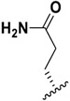
|
1006 ± 48.7 | 12,600 ± 3,870 | 12.5 |
Next, we explored the potency of different electrophilic warheads using the peptide backbone of J-18 and J-50. The warheads included aldehyde (J-69), ketone (J-72, J-73), enone (J-60, J-61), boronic acid ester (J-62) and vinyl sulfone with both cis (J-52, J-57) and trans groups (J-63).[11,14] All of these alternative warheads reduced both potency and selectivity relative to the EK moiety (Table S1). Because the most selective peptide vinyl sulfone inhibitors reported to date have an l-amino acid at P3,[11] we also evaluated this configuration at P3 (J-58, J-59), but found that the selectivity was significantly reduced when compared to J-50 (Table S1). In summary, inhibitors with a Phe-EK moiety had greater P. falciparum selectivity over all vinyl sulfone analogues and over Leu-EK. Therefore, the Phe-EK moiety was fixed for subsequent compounds in this series.
We next investigated analogues of J-50 by varying only in the N-terminal cap at P4. Increasing the alkyl chain length (J-54), and cyclization without (J-55) or with aromatization (J-56), lowered the potency against P. falciparum and increased the cytotoxicity against HepG2 cells (Table 1). Although 2-(methoxymethoxy)acetyl at P4 (J-75) improved solubility and reduced host cell cytotoxicity, selectivity was still lower than J-50. Therefore, we fixed the P4 hexanoyl group in subsequent compounds and explored various d-amino acids at P3 (Table 2). Placing d-Trp (J-51), d-naphthyl-Ala (J-53), d-4-pyridyl-Ala (J-64), d-3-pyridyl-Ala (J-66) at P3, resulted in lower selectivity while t-butyl d-Glu (J-71) or d-Glu (J-74) greatly reduced host cell cytotoxicity yielding higher selectivity indices (SI) values near or above 1000. Interestingly, the change from d-Glu to d-Gln (J-76) in P3 decreased potency against the parasite by 10-fold while also increased cytotoxicity against HepG2 by 10-fold, thereby reducing the parasite selectivity to 12.5. As a secondary screen for cell cytotoxicity, the same compounds were tested against H460 cells and IC50 values were found to be highly correlative (Pearson correlation coefficient 0.954) to those of HepG2 (Table S1, Fig S1). These data reveal that the side chain of the d-amino acid at P3 is particularly important for high potency and selectivity of this compound class.
Table 2.
ML9 analogues with EK warhead, N,N-diethyl Asn as the P3 moiety and different P4 N-terminal capping groups. IC50 data presented as mean ± SEM for 3 biological replicates
| No. | P4 | P3 |
P. falciparum IC50 [nM] |
HepG2 IC50 [nM] |
SI |
|---|---|---|---|---|---|
| J-77 |
|
l-N,N-Diethyl - Asn | 17.0 ± 2.4 | 2,580 ± 362 | 152 |
| J-78 |
|
d-N,N-Diethyl - Asn | 17.0 ± 0.71 | 30,300 ± 11,400 | 1,782 |
| J-79 |
|
l-N,N-Diethyl - Asn | 13.0 ± 3.2 | 1,417 ± 44.1 | 109 |
| J-80 |
|
d-N,N-Diethyl - Asn | 9.2 ± 1.8 | 24,300 ± 2,130 | 2,641 |
Next, insights were gained from previously described proteasome inhibitors that have amino acids in the P3 position with overlapping features to d-Glu or t-butyl protected d-Glu. In a study by Lin and colleagues, 1,600 N,C-capped dipeptide inhibitors were screened for potency against the mycobacterial proteasome and activity was compared to the β5 subunit of the human constitutive proteasome.[18] Many of the active compounds had Asp and Asn derivatives at P3 and 2,4-difluorobenzyl as the P1 substituent. Instead of modifying our compound series with these features, we added an EK reactive group to the Lin compound ‘ML9’, the most selective anti-mycobacterial compound from this series (Figure 2). This yielded compound J-77, which possesses a moderate SI of 155 (Table 2). Using compound J-77 as a new starting point, we subsequently modified the P3 N,N-diethyl Asn to the d-isomer. This resulted in a 2.4-fold increase in potency and a 5.4-fold decrease in HepG2 cytotoxicity. Substitution of the N-terminal cap with the preferred hexanoyl group yielded J-80, our most selective compound to date. J-80 inhibits P. falciparium replication with an IC50 of 9.2 nM and IC50 to HepG2 cells of 24.3 μM. This exceptional 2,641-fold selectivity for P. falciparum replication over human cells was achieved with a non-natural d-amino acid at P3, the fluorophenyl-containing Phe residues at P1 and P2, and an irreversible EK warhead.
Figure 2.

ML9, a reversible inhibitor of the mycobacterium 20S proteasome was modified with a C-terminal EK group, an N-terminal hexanoyl chain and d-N,N-Diethyl Asn at P3
To assess the potency and subunit selectivity of our most promising analogues, we isolated the P. falciparum 20S (Pf20S) proteasome and evaluated potency at the β1, β2 and β5 subunits using subunit specific fluorogenic reporter substrates as a read-out for activity. In parallel, we tested the same compounds with the purified human constitutive 20S (c20S) proteasome. None of the compounds showed appreciable inhibition of the β1 subunit of either Pf20S or c20S, and potency was largely driven by binding at β5, with some compounds also targeting β2 (Figure S2). We calculated the rate constant for inactivation, kinact, and potency, Kapp (Table 3). There was a strong correlation of both potency and selectivity between our cellular data and our enzyme inhibition data. Notably, the most selective analogues from our cellular studies, J-50, J-71, J-74, J-78 and J-80, all showed greater potency for Pf20S β5 than for c20S β5. Compounds J-74 and J-78 did not inhibit c20S β5 at concentrations up to 8.3 μM, so enzyme selectivity could not be calculated. Compound J-80 exhibited a 177-fold selectivity for Pf20S β5 over c20S β5 while compound J-71 was the most potent at inhibiting Pf20S β5 and β2. Dual inhibition of β5 and β2 has been shown to be important for killing at all stages of the P. falciparum life cycle, particularly when an inhibitor is administered for only a short period.[11,14] Therefore, the high potency of compound J-71 for two subunits and the high selectivity of J-80 for Pf20S β5 over c20S β5 make these two compounds ideal candidates for further studies.
Table 3.
Selectivity of epoxyketone analogues for Pf20S β5, c20S β5 and Pf20S β2 and c20S β2. All assays were performed in triplicate wells using the same preparation of Pf20S and c20S.
| No. | Compound Structure | Pf20S β5 | c20S β5 | SI (β5) | Pf20S β2 | c20S β2 | SI (β2) |
|---|---|---|---|---|---|---|---|
| kinact/Kapp [M−1.s−1] |
kinact/Kapp [M−1.s−1] |
kinact/Kapp [M−1.s−1] |
kinact/Kapp [M−1.s−1] |
||||
| J-18 |
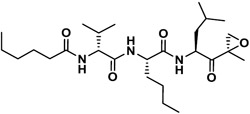
|
429 | 8.1 | 53 | NI | NI | - |
| J-50 |
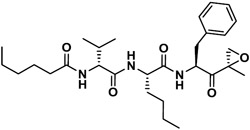
|
4,030 | 53.8 | 75 | 9 | NI | - |
| J-71 |
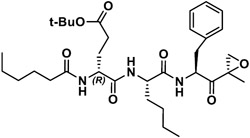
|
28,880 | 2,125 | 14 | 765 | 1,589 | 0.48 |
| J-74 |
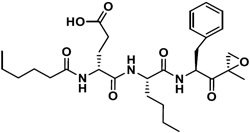
|
3,808 | NI | ∞ | 1 | NI | - |
| J-75 |
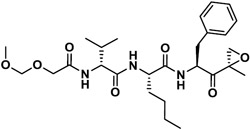
|
2,170 | 852 | 3 | 133 | 647 | 0.21 |
| J-76 |
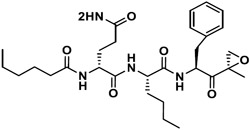
|
213.8 | 30.3 | 7 | 5 | NI | - |
| J-77 |
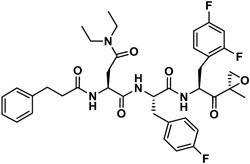
|
1,906 | 7,790 | 0.24 | 85 | 80 | 1.06 |
| J-78 |
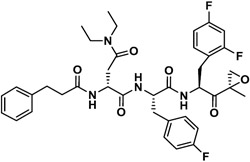
|
159 | NI | ∞ | NI | NI | - |
| J-79 |
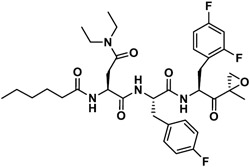
|
2,279 | 963 | 2 | 180 | 158 | 1.14 |
| J-80 |
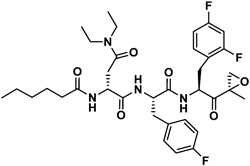
|
5,160 | 29.1 | 177 | NI | NI | - |
Molecular docking calculations suggest why compounds with a d-amino acid at P3 (J-50, J-75, J-64, J-71, J-78, J-80) achieve the best selectivity.[13] When these compounds are docked to Pf20S β5, most of the high-scoring (i.e., high probability) poses adopt an inverted binding mode, where the P3 group occupies the S4 pocket and the P4 group occupies the S3 pocket (Figure 3). The plausibility of this inverted binding mode is supported by a crystal structure of the yeast proteasome (4QLV), bound with a structurally similar ligand with a d configuration at P3 and where P3 is located in the S4 pocket and P4 in the S3 pocket.[19] For the highly selective compounds reported here, this inverted arrangement enables the ligand’s P4 carbonyl to accept a stabilizing hydrogen bond from the hydroxyl group of Ser27. This stabilizing ligand-protein interaction is not possible with human β5, because it has Ala instead of Ser at position 27 (Figure 3). In addition, the bulky aromatic P1 groups of these most selective ligands are predicted to occupy the S1 pocket, where they may form stabilizing hydrophobic interactions with Leu53 of the Pf20S β5; again, this stabilizing ligand-protein interaction is unavailable in the human enzyme where a polar Ser residue is present instead of the nonpolar Leu. This latter observation may explain the selectivity gain resulting from changing the P1 Leu to either a Phe or fluorinated phenylalanine.
Figure 3.

Visualization of compound J-80 docked to Pf20S in an inverted binding mode, where residues P3/P4 occupy sites S4/S3, respectively. Protein surface of Pf20S near the ligand is drawn semi-transparently. Ligand is drawn in ball and stick, while Ser27 of Pf20S β5 and Ala27 of structurally aligned human β5 are drawn in thick and thin sticks, respectively. The EK part of the compound and nonpolar hydrogens are hidden for clarity. Arrow and dashed line indicate the hydrogen bond from Pf20S Ser27 to P4 carbonyl.
We evaluated the toxicity of analogues J-71 and J-80 in a mouse toxicity model, and no acute toxicity effects were observed with up to 50 mg/kg administered intravenously (IV). In a pharmacokinetic study in rats, the elimination half-lives of compounds J-71 and J-80 were 5.1 and 2.4 hours, respectively following IV dosing. We evaluated J-71 and J-80 in the P. berghei luciferase (Pb-Luc) infection model by treating mice every 12 h with 50 mg/kg (IV) for 4 doses total starting five days post infection. We observed a significant reduction in parasitemia on days 7 and 8, but parasitemia rebounded at the end of the treatment period (Figure 4). These compounds led to prolongation of survival by 6 to 10 days but were not curative to mice at the doses and schedules given. Treatment with J-71 resulted in a greater reduction in parasitemia when compared to J-80, however, animals generally survived for longer following J-80 treatment. We postulate that the superior reduction in parasitaemia with J-71 is due to dual inhibition of β5 and β2, while the increased survival in mice treated with J-80 is due to this compound being unable to target the human proteasome and therefore it’s concentration in red blood cells is not reduced due to off-target binding to the human proteasome. In addition, metabolic profiling of J-71 and J-80 was performed with mouse liver microsomes. The main metabolic pathway of J-80 is the oxidation of the hexanoyl chain in P4, while J-71 was subject to some minor epoxide hydrolysis in addition to P4 oxidation (Figure S3-S8). These results indicate that P4 changes are required to extend the half-lives of these analogues.
Figure 4.

A. Therapeutic efficacy of J-71 and J-80 compared to chloroquine in mice infected with P. berghei luciferase (Pb-Luc). Dose 1 was administered intravenously when parasitemia reached 0.5% on Day 5 and 3 subsequent doses were given in 12 h intervals. Parasitemia was estimated from May Grunwald-Giemsa-stained blood smears (x1000 magnification) and a paired T-test was performed to determine the significance in parasitemia reduction on Day 8, 9 and 10 (*<0.05, **<0.001, ***<0.0005) compared to control. B. Survival curve of mice infected with Pb-Luc parasites for 25 days after infection. Comparison of infected mouse survival between the untreated (control) and treated groups (J-71, J-80 and chloroquine). Colors used in the survival curves match panel A. Four mice were used in each treatment group.
Conclusion
In summary, we have identified a series of EK-based proteasome inhibitors that are highly selective for the malaria proteasome over the human constitutive proteasome and have >2,600-fold greater selectivity for erythrocytic-stage P. falciparum over HepG2 cells. These analogues contain a hexanoyl group at P4, a d-amino acid at P3 and a fluorinated aromatic l-amino acid at P1. These analogues represent novel, potent and selective antimalarial drug leads with favorable pharmacokinetics properties and efficacy in animal studies, and therefore warrant further investigation and development for the potential treatment of malaria, possibly in co-administration with an artemisinin-based therapeutic.
Experimental Section
Material and Instrumentation.
All chemicals and solvents were purchased from commercial suppliers and used without further purification, unless stated otherwise. Anhydrous tetrahydrofuran and ether were freshly distilled from sodium and benzophenone before use. A Jasco P-2000 polarimeter 314 was used to measure optical rotations. NMR spectra were recorded on a Varian 500 MHz spectrometer (500 and 125 MHz for the 1H and 13C nuclei, respectively) using CDCl3 or CD3OD as solvents from Cambridge Isotope Laboratories, Inc. Spectra were referenced to residual CDCl3 or CD3OD solvent as the internal standard (for CDCl3 δH 7.26 and δC 77.1; and for CD3OD δH 4.78 and δC 49.2). LC-HRMS data for analysis of all compounds were obtained on an Agilent 6239 HR-ESI-TOFMS equipped with a Phenomenex Luna 5 μm C18 100 Å column (4.6 × 250 mm). LCMS data for purity analysis of the synthesized compounds were obtained with a Thermo Finnigan Surveyor Autosampler-Plus/LC-PumpPlus/PDA-Plus system and a Thermo Finnigan LCQ Advantage Max mass spectrometer (monitoring 200–600 nm and m/z 150–2000 in positive ion mode) using a linear gradient of 20 - 100% H2O/acetonitrile over 15 - 20 min; flow rate of 1 mL/min. Semipreparative HPLC purification was carried out using a Waters 515 with a Waters 996 photodiode array detector using Empower Pro software. Structural integrity and purity of the test compounds were determined from the composite of 1H and 13C NMR, HRMS and HPLC (see Supporting information), and all compounds were found to be > 90% pure. Chemical shifts (δ) are given in parts per million (ppm) and coupling constants (J) are reported in Hertz (Hz). The compounds are named in accordance with IUPAC rules as applied by ChemBioDraw Ultra (version 16.0). Compounds are stable for up to 3 years when stored as dry powders at room temperature and for up to 12-months as 10 mM DMSO stocks stored at −20°C.
Culturing of P. falciparum
P. falciparum Dd2 strain parasites were cultured under standard conditions,[20] using RPMI medium supplemented with 0.05 mg/mL gentamycin, 0.014 mg/mL hypoxanthine (prepared fresh), 38.4 mM HEPES, 0.2% sodium bicarbonate, 3.4 mM sodium hydroxide, 0.05% O+ human serum (denatured at 56°C for 40 min and from Interstate Blood Bank, Memphis, TN) and 0.0025% Albumax. Human O+ whole blood was obtained from The Scripps Research Institute blood bank (La Jolla, CA) and incubated at 37°C in an atmosphere of 1% O2, 5% CO2 and 94% N2. Cultures were monitored by Giemsa staining of methanol-fixed thin blood smears. The culture media were replaced every 48 h and parasitemia was maintained below 5% to ensure the health of the cultures.[13]
Potency of compounds in P. falciparum, HepG2 and H-460 cell cultures
Parasite susceptibility to the indicated compounds was measured using the malaria SYBR green I-based fluorescence assay.[21] Asynchronous P. falciparum parasites (Dd2 strain) were cultured in standard conditions prior to each assay. Compounds were tested after 72 h incubation in technical duplicates performed on three different days at 12-point distinct concentrations, as prepared by 3-fold dilution from 6.7 μM to 0.11 nM, with artemisinin used as a positive control. Compounds inactive within this range were retested in 3-fold dilution from 67 μM to 1.1 nM. IC50 values were obtained using background subtracted fluorescence intensity and analyzed in Prism 6 (GraphPad Software Inc.) via a nonlinear, variable slope, four-parameter regression curve-fit. HepG2 toxicity was assessed as previously reported.[22] Briefly, HepG2-A16-CD81EGFP cells were maintained at 37°C and 5% CO2 in DMEM media (Life Technologies, CA) containing 10% FBS, 0.29 mg/mL glutamine, 100 units of penicillin, and 100 μg/mL streptomycin. For the assays themselves, 3 × 103 cells per well in 5 μL of DMEM without phenol red (Life Technologies, CA) (containing 5% FBS and 5× Pen–Strep glutamine (Life Technologies, CA)) were added to 1536-well plates. Six hours later, 55 nL of compound was transferred via acoustic transfer system (ATS) (Biosera) into the assay plates. Compounds were tested in technical duplicates at 12 distinct concentrations, which were prepared by 3-fold dilution from 50 μM to 0.75 nM and cells were incubated in the presence of compound for 48 hours. Compound J-74 was further retested in 3-fold dilution beginning at 100 μM. Puromycin was used as a positive control. HepG2-A16-CD81EGFP cell viability were quantified by a bioluminescence measurement using an Envision multilabel reader (PerkinElmer), with IC50 values determined using Prism 6.
Cytotoxicity was also evaluated using the H-460 human lung carcinoma cell line with the MTT-staining method on a range of 10 concentrations prepared by serial half-logarithmic dilutions. The highest concentration for compounds was 20 μM. Each final concentration was tested in duplicate wells on each of three 96 well plates. IC50 values were calculated as an average of three IC50 dose-response curves, and their errors calculated as the standard deviation.
Preparation of Pf20S
Pf20S proteasome was purified from infected red blood cells using a modification of a published procedure.[13,23] Briefly, pellets of sorbitol-synchronized mature stage parasites were prepared from 550 mL of P. falciparum culture (5% haematocrit and 9.8% parasitaemia). Parasites were resuspended in 12 ml of lysis buffer containing 20 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 1 mM DTT, 5% glycerol and 10 μM E-64, and sonicated 3 times on ice. The lysate was clarified by centrifugation at 30,000 g for 20 min and filtered through a 0.22 μm syringe filter. The supernatant was applied to a 5 mL HiTrap DEAE FF column (GE Healthcare) in 25 mM Tris-HCl, pH 7.5, 5% glycerol and proteins were eluted using a 0 to 1 M NaCl gradient in 25 mM Tris-HCl, pH 7.5, 5% glycerol (100% in 100 min). Fractions were assayed with Ac-WLA-AMC (AdipoGen Life Sciences; SBB-PS0008) in 25 mM Tris, pH 7.5, 0.02% SDS at excitation 360 nm and emission 460 nm at 24°C on a Synergy HTX multimode reader (Biotek). Activity was evaluated in the presence and absence of 10 μM carfilzomib (SelleckChem S2853). Fractions that had catalytic activity in the absence of carfilzomib (0.001% DMSO) but were inhibited by carfilzomib were pooled and further purified by gel filtration using a Superose 6 column (GE Healthcare).
Activity and inhibition assays
2 nM human 20S constitutive proteasome (R&D Systems; E-360) was mixed with 28 nM of PA28α (R&D Systems; E-381) to activate the enzyme complex. Activities of the β1, β2 and β5 subunits of c20S were monitored using Z-LLE-AMC (R&D Systems; S-230), Z-VLR-AMC (AdipoGen Life Sciences; AG-CP3-0027) and Suc-LLVY-AMC (R&D Systems; S-280) respectively. 1 nM of Plasmodium 20S proteasome was activated with 150 nM of PA28α and β1, β2 and β5 subunits were measured using Ac-nPnD-AMC (Cayman Chemical; 21639), Bz-FVR-AMC (Bachem; 4003131) and Ac-WLA-AMC, respectively. β2 activity assays were performed in 50 mM HEPES pH 7.5, 1 mM DTT while β1 and β5 assays were performed in 50 mM HEPES pH 7.5, 1 mM DTT, 2.5 mM MgCl2. For inhibition assays, inhibitor (8.3 μM to 0.14 nM) and substrate were added simultaneously to the enzyme and the rate of AMC release was detected for 4 hours. The relative potency of each compound was compared to a DMSO vehicle control. All reactions were performed in triplicate wells on 384-well black plates at 37°C in a final volume of 30 μL. Fluorescence was measured at excitation of 360 nm and emission of 460 nm on a Biotek Synergy HTX plate reader. Kinetic datasets were analyzed by fitting the time-course data to a single exponential t0 derived kobs for each inhibitor concentration. A secondary plot of kobs vs [I] was used to determine kinact and Kapp. All analyses were carried out in GraphPad Prism 9 software.
Efficacy study in P. berghei infected mice
J-71 was dissolved in 29% DMSO, 29% Ethanol, 20% PEG400, and 22% PBS while J-80 was dissolved in 24% DMSO, 24% Ethanol, 20% PEG400 and 32% PBS. Chloroquine was resuspended in ultrapure water. In vivo efficacy in Swiss Webster mice against P. berghei Luciferase (Pb-Luc) was tested in 4 groups of animals with 4 mice per group). 50 mg/kg of each compound was administered intravenously on days five (one dose), six (two doses) and seven (one dose) and parasitemia was determined using standard May Grunwald-Giemsa-stained blood smears. Antimalarial activity was measured as a percent reduction in parasitaemia from day one until day ten post-infection. Two-tailed statistical tests were performed using data from day 8, 9 and 10. Animals were considered cured if there were no detectable parasites on day 25 post-infection. A comparison of infected mouse survival between the negative control and the treated groups was performed for Day 8, 9 and 10 p<0.0001, Log-rank (Mantel- Cox) test, p<0.0001 Gehan-Breslow-Wilcoxon tests, (n=4 at each group treated). The results are expressed as the mean ± SEM. The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC), UC San Diego, Protocol Number: S13013 with Elizabeth A. Winzeler as the principal investigator.
Methods for Docking study
The programs MOE and Docktite[24] were used for structure preparation, homology modeling, and covalent docking. 4QLV from RCSB PDB, a structure of yeast constitutive 20S proteasome in complex with an epoxyketone inhibitor containing a P3 D-amino acid, was used as the starting point for structure-based modeling, because its binding mode to β5 is likely shared by our highly-selective ligands containing P3 D-amino acids. The structure was prepared using MOE Quickprep default options, removing solvent atoms and all protein chains except β5 and β6 near the binding site, and using force field Amber14:EHT for energy minimization. Then, using MOE’s Homology Model module, protein-ligand complex structures for Pf20S/human c20S were created by mutating the prepared 4QLV structure to Pf/human sequence based on alignments with PDB 6MUW/4R67, respectively. We also prepared the 4R67 structure in the same way as 4QLV and found no discernable difference in subsequent docking and visualization between using the non-homology 4R67 structure and using the 4R67-4QLV homology structure. Then, following the Docktite workflow, our top ligands were covalently docked to Pf20S and human c20S structures, using pharmacophore constraints to ensure that epoxyketone part of the ligand is in place to form a 6-member ring with Thr1 residue. To identify and visualize the preferential hydrogen bond to Pf20S Ser27 drawn in the main text, the top pose of compound J-80 docked to the Pf20S homology structure was used, and the prepared human 4R67 structure was then aligned to the Pf20S structure.
In-vivo toxicity study procedure
Mice were injected IV via retro-orbital injection; each mouse is initially anesthetized in an isoflurane induction chamber. A 27-gauge needle, 1 mL syringe is inserted in the venous pool of the ventral eye socket of the mouse and administer study compound at 50 mg/kg. After IV injection the mouse was placed back in cage to recover from the anesthesia. The mouse was visually observed for toxicity issues that included ruffled fur, lethargy, closed eyes, and hindered mobility.
Microsomal stability study and metabolite identification study
Mouse microsome incubations were prepared in 24-well polystyrene tissue culture plates to a final volume of 2 mL per well. To quantify parent compound degradation, 2 μM of test compound was mixed with 0.5 mg/mL mouse liver microsomal protein (Xenogen), 3 mM MgCl2, 100 mM potassium phosphate buffer, pH 7.4 in a final volume of 2.0 mL. All incubations were conducted in triplicate. The reaction was initiated through the addition of NADPH to a final concentration of 1 mM and incubated at 37°C at 300 rpm. At each time point (0, 5, 30, 45, 60, 90, and 120 mins), 200 μL of the reaction was added to 600 μL of cold (4°C) acetonitrile/0.1% formic acid, vigorously vortexed and centrifuged at 10,000 rpm for 5 mins. The supernatant was removed and analyzed by LC-MS/MS. To monitor metabolite formation, the procedure outlined above was used except that 20 μM of test compound was mixed with mouse live microsomal protein and 950 μL of the mixture was added to 2,850 μL of the quenching solution after 0, 10 and 20 min incubation.
Supplementary Material
Acknowledgments
JA would like to acknowledge the deanship of scientific research at the University of Jordan for the scientific leave. PF received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 846688, ProTeCT. The research was supported by the Bill and Melinda Gates Foundation (to EAW, WHG and AJO), in addition to R01AI158612, R21AI133393, R21AI146387 and R21AI171824 (to AJO). JA would like to acknowledge the St. Baldrick’s Foundation for the International Scholar award 2022-2025.
Footnotes
Supporting Information
The Supporting Information contains analytical data and structures for all compounds used in this study. In addition, cellular IC50 data, enzyme inhibition dose-response curves, cytotoxicity data, microsomal stability and metabolite identification data are also present.
M.K.G. has an equity interest in and is a cofounder and scientific advisor of VeraChem LLC
References
- [1].Malaria World Report, World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- [2].Blasco B, Leroy Di., Fidock DA, Nat. Med 2017, 23, 917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han K-T, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, Rahman MR, Hasan MM, Islam A, Miotto O, Amato R, MacInnis B, Stalker J, Kwiatkowski DP, Bozdech Z, Jeeyapant A, Cheah PY, Sakulthaew T, Chalk J, Intharabut B, Silamut K, Lee SJ, Vihokhern B, Kunasol C, Imwong M, Tarning J, Taylor WJ, Yeung S, Woodrow CJ, Flegg JA, Das D, Smith J, Venkatesan M, Plowe CV, Stepniewska K, Guerin PJ, Dondorp AM, Day NP, White NJ, N. Engl. J. Med 2014, 371, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Straimer J, Gnädig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, Gregory PD, Urnov FD, Mercereau-Puijalon O, Benoit-Vical F, Fairhurst RM, Ménard D, Fidock DA, Science (80-. ) 2015, 347, 428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kirkman LA, Zhan W, Visone J, Dziedziech A, Singh PK, Fan H, Tong X, Bruzual I, Hara R, Kawasaki M, Imaeda T, Okamoto R, Sato K, Michino M, Alvaro EF, Guiang LF, Sanz L, Mota DJ, Govindasamy K, Wang R, Ling Y, Tumwebaze PK, Sukenick G, Shi L, Vendome J, Bhanot P, Rosenthal PJ, Aso K, Foley MA, Cooper RA, Kafsack B, Doggett JS, Nathan CF, Lin G, Proc. Natl. Acad. Sci 2018, 115, E6863–E6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Krishnan KM, Williamson KC, Transl. Res 2018, 198, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dekel E, Yaffe D, Rosenhek-Goldian I, Ben-Nissan G, Ofir-Birin Y, Morandi MI, Ziv T, Sisquella X, Pimentel MA, Nebl T, Kapp E, Ohana Daniel Y, Karam PA, Alfandari D, Rotkopf R, Malihi S, Temin TB, Mullick D, Revach OY, Rudik A, Gov NS, Azuri I, Porat Z, Bergamaschi G, Sorkin R, Wuite GJL, Avinoam O, Carvalho TG, Cohen SR, Sharon M, Regev-Rudzki N, Nat. Commun. 2021 121 2021, 12, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Manasanch EE, Orlowski RZ, Nat Rev Clin Oncol 2017, 14, 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Stokes BH, Yoo E, Murithi JM, Luth MR, Afanasyev P, da Fonseca PCA, Winzeler EA, Ng CL, Bogyo M, Fidock DA, PLoS Pathog. 2019, 15, DOI 10.1371/journal.ppat.1007722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li H, Tsu C, Blackburn C, Li G, Hales P, Dick L, Bogyo M, J. Am. Chem. Soc 2014, 136, 13562–13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yoo E, Stokes BH, De Jong H, Vanaerschot M, Kumar TRS, Lawrence N, Njoroge M, Garcia A, Van Der Westhuyzen R, Momper JD, Ng CL, Fidock DA, Bogyo M, J. Am. Chem. Soc 2018, 140, 11424–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Le Chapelain C, Groll M, Angew. Chemie Int. Ed 2016, 55, 6370–6372. [DOI] [PubMed] [Google Scholar]
- [13].LaMonte GM, Almaliti J, Bibo-Verdugo B, Keller L, Zou BY, Yang J, Antonova-Koch Y, Orjuela-Sanchez P, Boyle CA, Vigil E, Wang L, Goldgof GM, Gerwick L, O’Donoghue AJ, Winzeler EA, Gerwick WH, Ottilie S, J. Med. Chem 2017, 60, 6721–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li H, O’Donoghue AJ, Van Der Linden WA, Xie SC, Yoo E, Foe IT, Tilley L, Craik CS, Da Fonseca PCA, Bogyo M, Nature 2016, 530, 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xie SC, Metcalfe RD, Mizutani H, Puhalovich T, Hanssen E, Morton CJ, Du Y, Dogovski C, Huang SC, Ciavarri J, Hales P, Griffin RJ, Cohen LH, Chuang BC, Wittlin S, Deni I, Yeo T, Ward KE, Barry DC, Liu B, Gillett DL, Crespo-Fernandez BF, Ottilie S, Mittal N, Churchyard A, Ferguson D, Aguiar ACC, Guido RVC, Baum J, Hanson KK, Winzeler EA, Gamo FJ, Fidock DA, Baud D, Parker MW, Brand S, Dick LR, Griffin MDW, Gould AE, Tilley L, Proc. Natl. Acad. Sci. U. S. A 2021, 118, DOI 10.1073/PNAS.2107213118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhan W, Zhang H, Ginn J, Leung A, Liu YJ, Michino M, Toita A, Okamoto R, Wong TT, Imaeda T, Hara R, Yukawa T, Chelebieva S, Tumwebaze PK, Lafuente-Monasterio MJ, Martinez-Martinez MS, Vendome J, Beuming T, Sato K, Aso K, Rosenthal PJ, Cooper RA, Meinke PT, Nathan CF, Kirkman LA, Lin G, Angew. Chemie - Int. Ed 2021, 60, 9279–9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang H, Ginn J, Zhan W, Liu YJ, Leung A, Toita A, Okamoto R, Wong T-T, Imaeda T, Hara R, Yukawa T, Michino M, Vendome J, Beuming T, Sato K, Aso K, Meinke PT, Nathan CF, Kirkman LA, Lin G, J. Med. Chem 2022, DOI 10.1021/ACS.JMEDCHEM.2C00611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lin G, Chidawanyika T, Tsu C, Warrier T, Vaubourgeix J, Blackburn C, Gigstad K, Sintchak M, Dick L, Nathan C, J. Am. Chem. Soc 2013, 135, 9968–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].De Bruin G, Huber EM, Xin BT, Van Rooden EJ, Al-Ayed K, Kim KB, Kisselev AF, Driessen C, Van Der Stelt M, Van Der Marel GA, Groll M, Overkleeft HS, J. Med. Chem 2014, 57, 6197–6209. [DOI] [PubMed] [Google Scholar]
- [20].Trager W, Jensen JB, Science (80-. ) 1976, 193, 673–675. [DOI] [PubMed] [Google Scholar]
- [21].Swann J, Corey V, Scherer CA, Kato N, Comer E, Maetani M, Antonova-Koch Y, Reimer C, Gagaring K, Ibanez M, Plouffe D, Zeeman AM, Kocken CHM, McNamara CW, Schreiber SL, Campo B, Winzeler EA, Meister S, ACS Infect. Dis 2016, 2, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M, Antimicrob. Agents Chemother 2004, 48, 1803–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Xie SC, Metcalfe RD, Hanssen E, Yang T, Gillett DL, Leis AP, Morton CJ, Kuiper MJ, Parker MW, Spillman NJ, Wong W, Tsu C, Dick LR, Griffin MDW, Tilley L, Nat. Microbiol. 2019 411 2019, 4, 1990–2000. [DOI] [PubMed] [Google Scholar]
- [24].Scholz C, Knorr S, Hamacher K, Schmidt B, J. Chem. Inf. Model 2015, 55, 398–406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.