

Abstract
Autosomal recessive polycystic kidney disease (ARPKD) is caused primarily by mutations in PKHD1, encoding fibrocystin (FPC), but Pkhd1 mutant mice failed to reproduce the human phenotype. In contrast, the renal lesion in congenital polycystic kidney (cpk) mice, with a mutation in Cys1 and cystin protein loss, closely phenocopies ARPKD. Although the nonhomologous mutation diminished the translational relevance of the cpk model, recent identification of patients with CYS1 mutations and ARPKD prompted the investigations described herein. We examined cystin and FPC expression in mouse models (cpk, rescued‐cpk (r‐cpk), Pkhd1 mutants) and mouse cortical collecting duct (CCD) cell lines (wild type (wt), cpk). We found that cystin deficiency caused FPC loss in both cpk kidneys and CCD cells. FPC levels increased in r‐cpk kidneys and siRNA of Cys1 in wt cells reduced FPC. However, FPC deficiency in Pkhd1 mutants did not affect cystin levels. Cystin deficiency and associated FPC loss impacted the architecture of the primary cilium, but not ciliogenesis. No reduction in Pkhd1 mRNA levels in cpk kidneys and CCD cells suggested posttranslational FPC loss. Studies of cellular protein degradation systems suggested selective autophagy as a mechanism. In support of the previously described function of FPC in E3 ubiquitin ligase complexes, we demonstrated reduced polyubiquitination and elevated levels of functional epithelial sodium channel in cpk cells. Therefore, our studies expand the function of cystin in mice to include inhibition of Myc expression via interaction with necdin and maintenance of FPC as functional component of the NEDD4 E3 ligase complexes. Loss of FPC from E3 ligases may alter the cellular proteome, contributing to cystogenesis through multiple, yet to be defined, mechanisms.
Keywords: ARPKD, cpk, cystin, FPC, proteome, selective autophagy
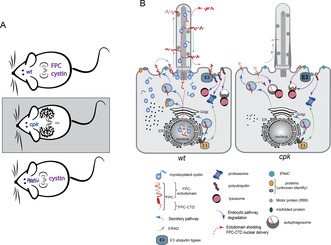
(A) In healthy mice (wt), cystin and FPC are both expressed in the kidneys. In cpk mice, cystin loss leads to FPC reduction and renal cystic disease. Pkhd1 mutant mice with loss of FPC, but without reduction in cystin, do not develop cystic kidneys. (B) In cells, cystin stabilizes FPC, regulates its ciliary traffic and delivery to E3 ubiquitin ligase complexes. In the absence of cystin, FPC is quickly degraded, leading to reduced polyubiquitination and cellular proteome dysregulation.
Abbreviations
- AQP2
aquaporin‐2
- ARPKD
autosomal recessive polycystickidney disease
- AT
acetylated α tubulin
- BF
bafilomycin
- CCD
cortical collecting duct
- CFTR
cystic fibrosis transmembraneconductance regulator
- CTD
C‐terminal domain
- cpk
C57BL/6JCys1 cpk/cpk mouse
- CQ
chloroquine
- CTD
C‐terminaldomain
- E‐cad
e‐cadherin
- ENaC
epithelialsodium channel
- ERAD
endoplasmicreticulum‐associated degradation
- FPC
fibrocystinpolyductin complex
- GFP
greenfluorescent protein
- HECT
homologousto E6AP C‐terminus
- I‐V
Current‐voltage
- LAMP1
lysosome‐associatedmembrane protein 1
- MG
MG‐132
- mTERT
mouse telomerase reversetranscriptase
- Pc2
polycystin‐2
- PKD
polycystickidney disease
- Ppia
peptidylpropylisomerase A
- r‐cpk
rescued‐cpk
- scr
scrambled
- siRNA
small‐interferingRNA
- UPS
ubiquitin‐proteasomesystem
- wt
wildtype
1. INTRODUCTION
The C57BL/6JCys1 cpkcpk (cpk) mouse is the first described experimental model of polycystic kidney disease (PKD), 1 in which a spontaneous mutation in Cys1 (encoding the protein cystin) causes renal cystic disease that phenocopies human ARPKD. 2 In contrast, causative mutations in the majority of ARPKD patients occur in PKHD1, encoding fibrocystin (FPC). 3 Recently, patients with ARPKD linked to mutations in CYS1 have been identified, 4 prompting interest in possible functional relationships between cystin and FPC. We and others have used various methodologies (co‐immunoprecipitation, tandem affinity purification, mass spectroscopy) to identify protein–protein interactions, but to our best knowledge, these efforts were unsuccessful. In the current study, we sought to examine potential functional linkages between cystin and FPC in cpk 4 and Pkhd1 mutant mouse kidneys 5 , 6 , 7 as well as cortical collecting duct (CCD) cell lines (wt, cpk). 8 , 9
The cpk phenotype is caused by a frameshift mutation within Cys1 exon 1, leading to reduced Cys1 mRNA levels and subsequent loss of cystin, a hydrophobic, 145‐amino acid, N‐terminally myristoylated protein. 10 In mouse renal epithelial cells, cystin is found in the primary cilium and co‐localizes with other PKD‐associated proteins, including polaris/Ift88. 11 N‐myristoylation modulates protein's association with plasma membrane and impacts protein function via subcellular trafficking and localization. 12 , 13 , 14 , 15 , 16 Cystin is myristoylated at amino acid residue G2 and associates with lipid raft membrane microdomains. 17 Myristoylation and a short peptide sequence between amino acids 28 and 35 (AxEGG) are both required for cystin trafficking to, and retention in, the primary cilium. 18 Cystin also localizes to the nucleus in a regulatory complex with necdin. 18 In the absence of cystin, necdin activates Myc transcription. Importantly, MYC levels are significantly elevated in both PKHD1‐deficient human ARPKD kidneys and mouse cpk cystic kidneys. 18 Dysregulated Myc/MYC expression has been implicated as a central driver of PKD. 19 , 20
In contrast to cpk mice, Pkhd1 mutants fail to develop an ARPKD‐like renal phenotype, 7 , 21 but reasons for the discordant phenotype have not been determined. FPC, encoded by PKHD1/Pkhd1, is a large transmembrane protein with an extensive extracellular N‐terminal domain, a single pass transmembrane domain, and a short cytoplasmic C‐terminal domain (CTD). 21 , 22 Although FPC function is largely unknown, FPC is required for the function of the NEDD4 family of HECT domain containing E3 ubiquitin ligases and co‐localizes in vesicles with the NEDD4 ubiquitin ligase interacting protein, NDFIP2. 23 FPC deficiency is associated with enhanced activity of ENaC, 23 an NEDD4 target protein, 24 which may contribute to the systemic hypertension that is characteristic of ARPKD. 25 , 26 However, the influence of FPC on the cellular proteome and the mechanisms through which FPC loss, or reduction alters cellular homeostasis remains unclear. Recent reports have provided new insights into the formation of E3 ligase complexes, determinants of substrate specificity, and regulation of the cellular proteome through ubiquitination. 27 , 28 , 29 Based on these developments, we speculate that FPC, through its function in E3 ligase complexes, may play a role in maintaining the cellular proteome and disruption of proteome homeostasis may contribute to ARPKD renal pathogenesis through multiple mechanisms depending upon the proteins which are affected by the proteome dysregulation.
We examined cystin and FPC levels in the kidneys of wt, Pkhd1 mutant 7 , 21 cpk, and rescued cpk (r‐cpk) mice with collecting duct‐specific expression of a wild‐type, green fluorescent protein (GFP)‐tagged Cys1 transgene, which reduced cyst formation in our previous studies. 4 We found that FPC levels were markedly reduced in cpk compared to wt kidneys and FPC abundance increased in r‐cpk kidneys. Conversely, FPC loss did not alter cystin levels in Pkhd1 mutants. Using previously developed and characterized renal CCD cell lines generated from wt and cpk mouse kidney CCD cells, 8 , 9 we observed that FPC levels were reduced in cystin‐deficient cells as well. Furthermore, siRNA depletion of cystin in wt CCD cells led to reduced FPC as well. FPC loss led to a significant, generalized reduction of polyubiquitination that likely causes alterations in the cellular proteome. As would be expected based on previous reports, 23 ENaC levels were elevated and sodium transport was increased in cpk cells, consistent with inhibited degradation of ENaC, an NEDD4 target protein. 30
Based on our observations, we propose that cystin is necessary to stabilize FPC that contributes to the regulation of the cellular proteome. Our results imply that the ARPKD‐like renal cystic phenotype in cpk mice results from the combined loss of cystin and FPC, leading to proteome alterations that disrupt cellular homeostasis. In addition, the loss of cystin‐dependent negative regulation of Myc transcription results in overexpression of Myc that can drive cyst formation as we have reported previously. 18 While the current studies were not designed to directly address the discordant renal phenotypes observed in PKHD1 mutation‐associated ARPKD and mouse Pkhd1 mutants, identification of a functional connection between cystin and FPC and demonstration that FPC loss in Pkhd1 mutant mice does not reduce cystin levels, leads us to speculate that in Pkhd1 mutant mice, cystin may contribute to mitigating an ARPKD‐like renal cystic phenotype by constraining Myc transcription. 18 , 31
2. MATERIALS AND METHODS
2.1. Mice
Kidney tissues were collected from 14‐day‐old, male animals. Animal procedures were approved by the Institutional Animal Care Committee of Children's National Research Institute.
2.2. Analysis of kidney tissue lysates
Kidney tissues were collected, homogenized, and processed for immunoblotting as previously described. 18 Immunoreactive protein bands were visualized using SuperSignal West Dura chemiluminescent substrate (Thermo Fisher Scientific #34076) and images were obtained using a ChemiDoc Imaging System (Bio‐Rad laboratory, Inc.). Densitometry data were collected using Image Lab (Bio‐Rad laboratory, Inc., Version 6.0) and statistical analysis was performed using GraphPad Prism software (RRID:SCR_002798).
2.3. Cell lines
mTERT‐immortalized mouse CCD cells were cultured as described previously 8 , 9 in 1:1 DMEM/F‐12 (Gibco GlutaMAX™; Fisher #10565018) with 5% FBS (Gibco One Shot™; Fisher #A3160602) and Penicillin/Streptomycin (Mediatech, Inc., Corning #30–002). Complete cell culture medium was supplemented with dexamethasone (0.2 μg/mL; Sigma #D8893‐1 mg), triiodothyronine (10 nM; Sigma #T5616‐1 mg), 1× insulin‐transferrin‐sodium selenite (Sigma #I1884‐1VL), and L‐Glutamine (200 mM; Corning Cellgro #25‐005‐CI).
2.4. Treatment of cells with inhibitors
MG‐132 (Thermo Fisher Scientific, #t: AAJ63250MCR) was used at 5 μM for 4–8 h. Chloroquine (Thermo Fisher Scientific, #: AC455240250) was used at 10 μM for 4 h.
2.5. Real‐time PCR analysis
Relative quantification of Pkhd1 mRNA in cells was performed using the delta‐CT method, 32 relative to Gapdh mRNA using the following primer sets: Pkhd1‐T forward 5’‐CAGTTCTTGCCAGAGCATTTAC‐3′, Pkhd1‐T reverse 5’‐CAGAATCTCACCTCCTGCTATG‐3′; Pkhd1‐M forward 5’‐CTGATAATGCACAGGGACCTAC‐3′, Pkhd1‐M reverse 5’‐GGCAAAGGATGAAATGGAAGTG‐3′; Pkhd1‐B forward 5′‐CCA CCAGAAACCATCCAGTAA‐3′, Pkhd1‐B reverse 5’‐AGACCTCTCCTCTCCCATTT‐3′ (Integrated DNA Technologies); mGapdh, forward 5’‐CATCACTGCCACCCAGAAGACTG‐3′, reverse 5′‐ATGCCAGTGAGCTTCCCGTTCAG‐3′ (Origene). The efficiency of siRNA‐mediated Cys1 mRNA reduction was tested by comparing the relative levels of Cys1 mRNA in untreated, scrambled (scr) siRNA transfected, and Cys1 siRNA transfected wt cells using the following primer sets: forward: 5’‐CTACGCCTGCTGGATCAGTTGC‐3′; reverse: 5’‐CAGCTGTCTTCAGGGTTGCCTT‐3′ (Integrated DNA Technologies), relative to Gapdh (Origene). Data are expressed as mean ± S.E; n = 3 per group. Relative Pkhd1 mRNA levels from kidneys were determined by measuring Pkhd1 ex66‐67 expression normalized to Ppia (peptidylpropyl isomerase A); data are expressed as mean ± S.E; n = 3 per group, using primers Pkhd1 forward 5’‐CCAGAAGACATATCTGAATCCCAGGC‐3′ (mPkhd1 e66F), reverse 5’‐AGCAAGAGATCCTGGAACACAGGT‐3′ (mPkhd1 e67R); Ppia forward 5’‐AGCACTGGAGAGAAAGGATT, reverse 5′‐ ATTATGGCGTGTAAAGTCACCA.
2.6. Antibodies
Primary antibodies used for WB analysis were produced against FPC (rat monoclonal (mAb), Baltimore Center of the PKD Research Resource Consortium; PKD‐RRC; PD1E1; 1:1000), cystin (rabbit, polyclonal (pAb), 70 053; 1:1000), 17 , 18 Pc2 (rabbit mAb, Baltimore Center of the PKD Research Resource Consortium; PKD‐RRC; 3374; 1:1000), β‐actin (mouse, mAb, Santa Cruz; sc‐47 778; 1:3000), Ift88 (rabbit pAb, GN593; 1:500), 33 GAPDH (rabbit pAb, Abcam; ab9485; 1:1000), e‐cadherin (rabbit polyclonal, Invitrogen #PA5‐85088), AQP2 (rabbit polyclonal, Invitrogen, #PA5‐38004), p62/SQSTM1 (mouse mAb, R&D Systems; MAB8028; 1:1000), pan‐polyubiquitin (rabbit pAb, Biomol: UG9510; 1:1000), polyubiquitin K63 (rabbit mAb, Millipore; 05‐1308; 1:1000), polyubiquitin K48 (rabbit pAb, Cell Signaling; 4289; 1:1000), ENaCα (rabbit pAb, StressMarq; SPC‐403; 1:1000), ENaCα (goat pAb, Santa Cruz; sc‐22 239; 1:200), and LAMP1 (mouse mAb: Sztul Lab; 1:20 34 ). The anti‐FPC (rat monoclonal antibody, PD1E1) recognizes the C‐terminal tail of the mouse FPC. This segment is localized intracellularly, or inside the axoneme of the primary cilium (Figure S2C).
2.7. Western blotting
Cells were lysed in RIPA buffer (Pierce; Thermo #89901) containing protease and phosphatase inhibitors (Halt; Thermo #1861281), sonicated, and centrifuged at 12000 rpm for 30 min. The supernatant was collected, and protein concentration measured using a BCA Protein Assay (Pierce; Thermo #23225). Equal amounts of proteins were denatured using NuPAGE reducing agent (Invitrogen; Thermo #NP0009), separated by SDS‐PAGE using NuPAGE 4–12% bis‐tris gel (Invitrogen; Thermo #WG1402BOX) or 3–8% tris‐acetate gel (Invitrogen; Thermo #WG1602BOX), and electrotransferred to a nitrocellulose membrane (Odyssey; Li‐Cor #926–31 092). Membranes were incubated with primary antibodies overnight at 4°C and secondary antibodies (Li‐Cor; 1:20000) for 1 h at room temperature. Results were imaged using a Li‐Cor Odyssey CLx imaging system.
2.8. Cell count
DAPI/Hoechst containing layer was selected and processed. Histogram equalization was performed to enhance the contrast between nuclei and the background. 35 After creating a binary mask of the nuclei, the periphery of the objects in the binary mask was dilated, 36 and small holes inside the objects were filled. 37 The edges of the nuclei were detected and traced using the bwperim function. 38 Lastly, using the bwboundaries function, the number of objects in the binary mask was counted and recorded as the number of cells in the image. 36
2.9. Cilia count
Cilia development was induced by serum starvation for 8 h as described previously. 39 Acetylated α tubulin (AT) staining was used to identify cilia and the percentage of cells with AT‐stained primary cilia was plotted relative to total cell number in a viewing area. Cilia with FPC expression were counted following single labeling of FPC and counting the cells with FPC‐positive cilia relative to total number of cells (Figure S2D).
2.10. Cilia length and thickness
These parameters were determined using ImageJ (RRID:SCR_003070) software. Results of measurements were plotted and analyzed in GraphPad Prism (RRID:SCR_002798). Cilia were traced with “straight line” or “segmented line” tool and then measured with “Analyze ‐> Measure” tool. Separate measurements were made to determine the length and width of each cilium. The results (in pixels) were converted into nanometer and then micrometer, with the scale bar serving as a point of reference (1pixel = 0.2066 nm). 61 cpk and 59 wt cilia were measured following AT labeling that has been shown to yield reproducible results for cilia measurements. 40 These measurements allowed statistical analysis to determine the length, width, and length/width ratio of the cilia. Images of cells used for cilia measurements are provided in Figure S3A,B.
2.11. Densitometry and statistical analysis
Image processing and densitometry were performed using Image Studio Lite software. Results were transferred to GraphPad Prism (RRID:SCR_002798) and data subjected to Student's t‐test or ANOVA analysis. “N” represents the number of experiments performed, while “n” is the total number of biological (mice, individual lysates) repeats.
2.12. Immunocytochemistry and microscopy
Cells were seeded on glass coverslips pretreated with HistoGrip (Fisher #008050) to enhance cell adhesion. After reaching 40–60% confluence, the cells were washed with PBS, fixed with 4% paraformaldehyde, quenched with ammonium chloride, permeabilized with 0.1% Triton X‐100, blocked with 2.5% goat serum, incubated with primary antibodies (1:200) and Alexa Fluor‐conjugated secondary antibodies (1:500), stained with Hoechst 33258 or DAPI, and mounted onto glass microscope slides. Following each step, the cells were washed with PBS+ 0.1% Tween20. Images were captured using Olympus BX51 microscope with a Q‐Color5 camera and CellSens Standard v1.13 software. Specificity testing of antibodies where the primary antibody was replaced with non‐immune IgG was performed for anti‐cystin and anti‐FPC antibodies (Figure S1A,B). Confocal microscopy was performed using Nikon A1R Confocal Microscope and Advanced NIS‐Elements software at the UAB, high‐resolution imaging facility (Figure S2A–C).
2.13. Patch clamp
Whole‐cell voltage‐clamp recordings were performed on wt and cpk cells at 24–32 h after seeding on plastic coverslips in regular growth medium as previously described. 41 , 42 Briefly, cells were bathed in an external solution containing (in mM) 145 NaCl, 2.7 KCl, 1.8 CaCl2, 2 MgCl2, 5.5 glucose, and 10 HEPES, pH 7.4, and continuously perfused (~3 mL/min). Patch pipette resistance was 2.5–3 MΩ when filled with an internal solution containing (in mM): 135 potassium glutamic acid, 10 KCl, 6 NaCl, 1 Mg2ATP, 5.5 glucose, 10 HEPES, and 0.5 EGTA, pH 7.3. Junction potentials were considered and corrected before breaking the cell membrane and the formation of a whole‐cell patch. To acquire individual cell capacitance, a transient current was induced by applying a short (15 ms) depolarization pulse from −30 to −20 mV before whole‐cell current recording. To measure amiloride‐sensitive currents in voltage clamp, 10 μM amiloride was applied after recording control current. Whole‐cell currents were evoked using a voltage step protocol from −90 to 30 mV in 20 mV increments of 450 ms duration at 5 s intervals. The cell membrane potential was held at −30 mV during recording. Signals from whole‐cell recording were filtered and sampled at 2 kHz. Current–voltage (I–V) relationships were constructed by measuring the steady‐state current values at the end of each voltage step. Data acquisition and analysis were performed using Axonpatch 200B amplifier, Digidata 1322A analog‐to‐digital converter, pClamp 9.2 software (RRID:SCR_011323, Axon Instruments/Molecular Devices, USA), and Origin Software (Microcal Software, Northampton, MA). All experiments were performed at room temperature.
3. RESULTS
3.1. Lack of cystin leads to reduced FPC levels in cpk kidneys and CCD cells
3.1.1. Cystin deficiency is associated with marked reduction of FPC in cpk mouse kidneys
We hypothesized that similar CYS1/Cys1 and PKHD1 renal disease phenotypes reflect disruption of an essential functional interplay between cystin and FPC. In the current study, we compared FPC and cystin levels in the kidneys of wt, cpk, and r‐cpk mice, a partially rescued cystic phenotype with collecting duct‐specific expression of the Cys1‐GFP transgene 4 (Figure 1A). Our results showed a marked (~90%) reduction (Figure 1B) of FPC in cpk kidneys compared to wt. We observed a three to fourfold increase in FPC levels in r‐cpk kidneys. Partial rescue of FPC expression in r‐cpk kidneys is consistent with diminished, but not completely eliminated cyst formation in r‐cpk mice, which is largely confined to proximal tubular segments. 4 In addition, the residual renal cystic disease may also result from diminished function of the cystin‐GFP fusion protein. 43 , 44 Endogenous cystin was present in wt but not in cpk and r‐cpk kidneys (Figure 1A, lower panel). Measurement of Pkhd1 mRNA levels indicated ~1.5‐fold increase in cpk kidney lysates compared to wt. This increase rather than reduction in Pkhd1 mRNA implies that FPC downregulation in cpk mice was not the result of reduced transcription and/or mRNA stability, but rather occurred at the protein level (Figure 1C).
FIGURE 1.
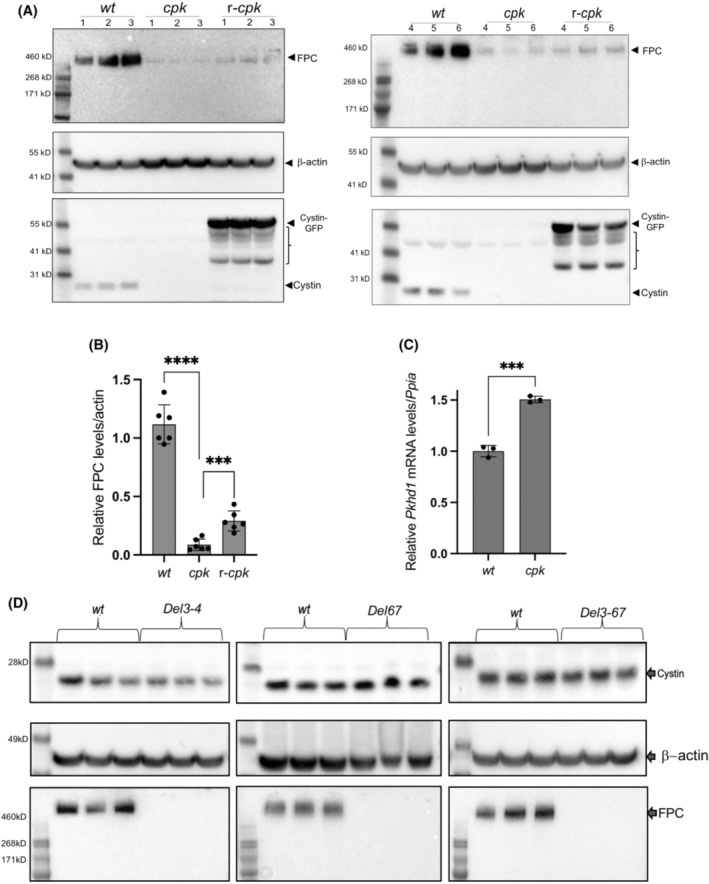
Cystin deficiency in cpk kidneys leads to FPC loss, with partial rescue of FPC following cystin–GFP expression (r‐cpk), but FPC loss (Del34, Del67 and Del367) does not alter cystin expression in mouse kidneys. (A) WB experiments demonstrate significantly lower FPC levels in cpk, compared to wt cells, with partial rescue of FPC levels in r‐cpk kidneys. Whole kidney lysates (N = 2, n = 6, male mice) from wt (C57BL/6), cpk and r‐cpk mouse kidneys were analyzed for FPC (anti‐FPC, C‐terminal, rat monoclonal antibody, top), β‐actin (loading control, middle) and cystin (anti‐cystin polyclonal, rabbit antibody, bottom). In wt samples, cystin is present (arrow). In r‐cpk samples, a cystin–GFP fusion protein (cystin‐GFP, arrow) and multiple unspecific bands are also present (brackets). No endogenous cystin is present in cpk and r‐cpk kidneys. (B) Densitometry confirms higher FPC levels in r‐cpk kidneys compared to cpk. Wt, cpk, and r‐cpk kidney lysates were tested for FPC levels are plotted relative to β‐actin (****: n = 6, p < .0001; ***p < .0005, unpaired t‐test). (C) No reduction in relative Pkhd1 mRNA levels in cpk kidneys compared to wt suggest FPC loss at the protein level. Relative Pkhd1 mRNA levels in wt and cpk kidneys were measured by real‐time RT‐PCR using Ppia mRNA as internal reference. Pkhd1 mRNA levels were higher, not lower in cpk kidneys compared to wt (n = 6, ***p < .001, unpaired t‐test). (D) Cystin levels are not reduced in Pkhd1 mutant (Del3‐4, Del67 and Del3‐67) kidneys. Whole kidney lysates from Pkhd1 mutant kidneys (Del3‐4, Del67, and Del3‐67) were tested for cystin and FPC expression and compared to age and gender matched wt kidney lysates (n = 3, male mice). Cystin expression was similar in wt and mutant (Del3‐4, Del67 and Del3‐67) kidney lysates (top). β‐actin was analyzed as loading control (middle). FPC levels were compared in the same lysates (bottom). We observed the loss of full‐length FPC in all Pkhd1 mutant kidneys tested (Del3‐4, Del67, and Del3‐67).
Because FPC was severely reduced in cystin‐deficient kidneys, we tested whether cystin levels were dependent on FPC expression. We analyzed Pkhd1 mutant (Del3‐4, Del67, Del3‐67) whole kidney lysates for cystin and FPC expression and found that in these Pkhd1 mutants, loss of full‐length FPC did not affect cystin expression (Figure 1D). Taken together, our results demonstrate significant FPC reduction in cystin‐deficient mouse kidneys and partial restoration of FPC levels following expression of a cystin–GFP fusion protein in r‐cpk kidneys. Conversely, loss of full‐length FPC in Pkhd1 mutant kidneys did not cause reduction in cystin levels.
3.1.2. FPC levels are linked to cystin expression in cortical collecting duct (CCD) cell lines
In subsequent experiments, we examined cell lines generated from wt and cpk mouse CCD cells 8 , 9 to better define the functional connections between cystin and FPC. We validated the nephron origin of these cell lines by assessing expression of the epithelial cell marker, e‐cadherin, and the CCD marker, AQP2 45 in multiple samples obtained from cell cultures of different passage numbers. We found similar levels of e‐cadherin and AQP2 in wt and cpk cells (Figure 2A,B). As expected, cystin was present in wt, but absent from cpk cells (Figure 2C). Similar to our observation in whole kidneys, FPC levels were greatly reduced (by 83 ± 6%) in cpk cells (Figure 2D). To determine whether the impact of cystin deficiency was FPC‐specific or extended to other ciliary and PKD‐associated proteins, we examined levels of polycystin‐2 (Pc2) and Ift88 proteins previously shown to co‐localize with cystin in the primary cilium. 10 Reduction of Pc2 abundance (30 ± 6%) in cpk cells was less severe than FPC (Figure 2E), consistent with previous reports that demonstrated Pc2 reduction in Pkhd1 knockout kidneys. 46 However, lack of cystin did not affect Ift88 levels (Figure 2F). These results are consistent with our observations in cpk kidneys (Figure 1) and support a specific relationship between cystin and FPC expression in CCD cells. Pkhd1 mRNA levels in wt and cpk CCD cells were similar (Figure 2G) and support the hypothesis that loss of cystin affects FPC abundance posttranslationally.
FIGURE 2.
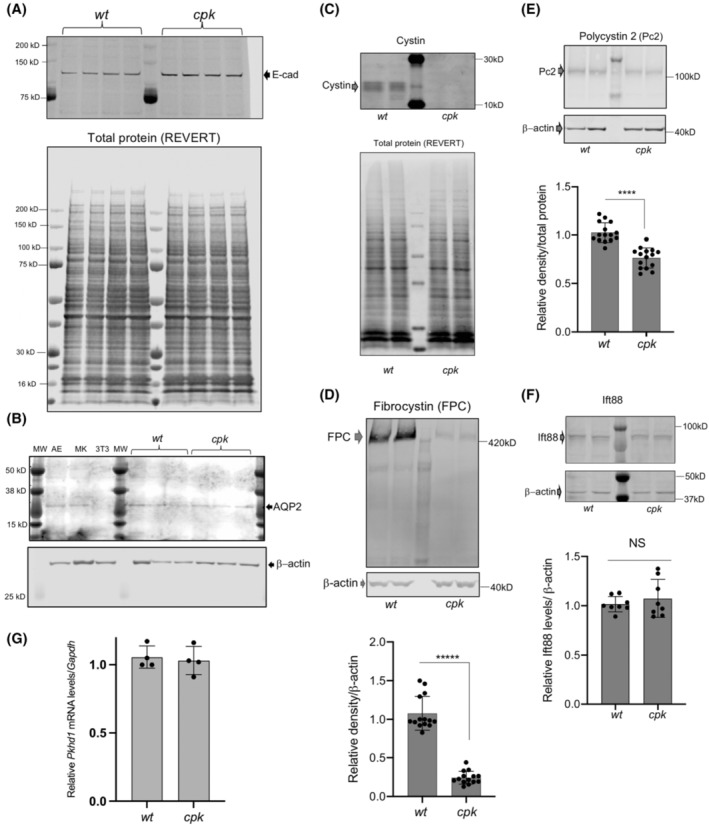
Reduced FPC levels in cpk, compared to wt CCD cells. (A,B) Characterization of wt and cpk CCD cell lines (n = 4). (A) E‐cadherin (E‐cad) expression was tested as epithelial marker. Cell lysates were randomly selected (wt, cpk) from experiments performed in a 6‐month period (n = 4). (B) Aquaporin‐2 (AQP2) expression (CCD marker, top) in randomly selected from wt and cpk cell lysates. AE: airway epithelial cells (positive control), MK: mixed mouse kidney epithelial cells, 3 T3: mouse fibroblast cell line (negative control). β‐actin as loading control (bottom). (C) Cpk cells lack cystin. Cystin was only present in wt cells. Cystin was labeled with rabbit polyclonal anti‐cystin antibody (top). Total protein stain (REVERT) is presented in the lower panel to demonstrate equal loading. (D) Reduced FPC levels in cpk cells compared to wt. 30 μg of total cellular proteins was analyzed by WB using an anti‐FPC C‐terminal, rat monoclonal antibody (representative gel on top). Relative FPC abundance was quantitatively assessed by densitometry and expressed relative to β‐actin (bottom). Experiments were performed using cell culture lysates from different passage numbers (N = 7, n = 14, unpaired t‐test). (E) Pc2 reduction in cpk cells. Pc2 WB using a rabbit polyclonal Pc2 antiserum (top). Pc2 abundance was quantitatively assessed by densitometry and expressed relative to β‐actin. Same cell lysates as for FPC were tested for Pc2 (N = 7, n = 14, ****p < .0001, unpaired t‐test). (F) Ift88 levels are similar in wt and cpk cells. Ift88 was labeled with anti‐Ift88 rabbit polyclonal antiserum. No significant differences were observed based on quantitative assessment of densitometry expressed relative to β‐actin (N = 4, n = 8, NS, unpaired t‐test). Same lysates were tested as for FPC and Pc2. (G) No reduction in relative Pkhd1 mRNA levels in cpk cells compared to wt. Plotted values indicate mRNA levels in four independently derived samples relative to Gapdh1 mRNA (from cell cultures at different passage number, n = 4, NS, unpaired t‐test).
We showed that overexpression of cystin–GFP in r‐cpk kidneys increased FPC levels (Figure 1A,B). To confirm a role for cystin in maintaining FPC levels and to eliminate possible off‐target effects of transient cystin overexpression on cellular functions, we used siRNA to reduce Cys1 expression in wt cells and evaluated both cystin and FPC levels. First, we tested the efficiency of the selected siRNA oligos (40 ng/mL) on reducing Cys1 mRNA levels 72 h post‐transfection (Figure 3A). Considering the high efficiency of siRNA oligos on mRNA levels, the following experiments concentrated on cystin protein levels. We achieved siRNA dose‐dependent (20–80 ng/mL) reductions in cystin levels 72 h after siRNA transfection. Reduction of FPC correlated with the efficiency of Cys1 knockdown (Figure 3B,C). Taken together, our in vivo and in vitro observations strongly support a role for cystin in maintaining FPC levels in both mouse kidneys and renal epithelial cells.
FIGURE 3.
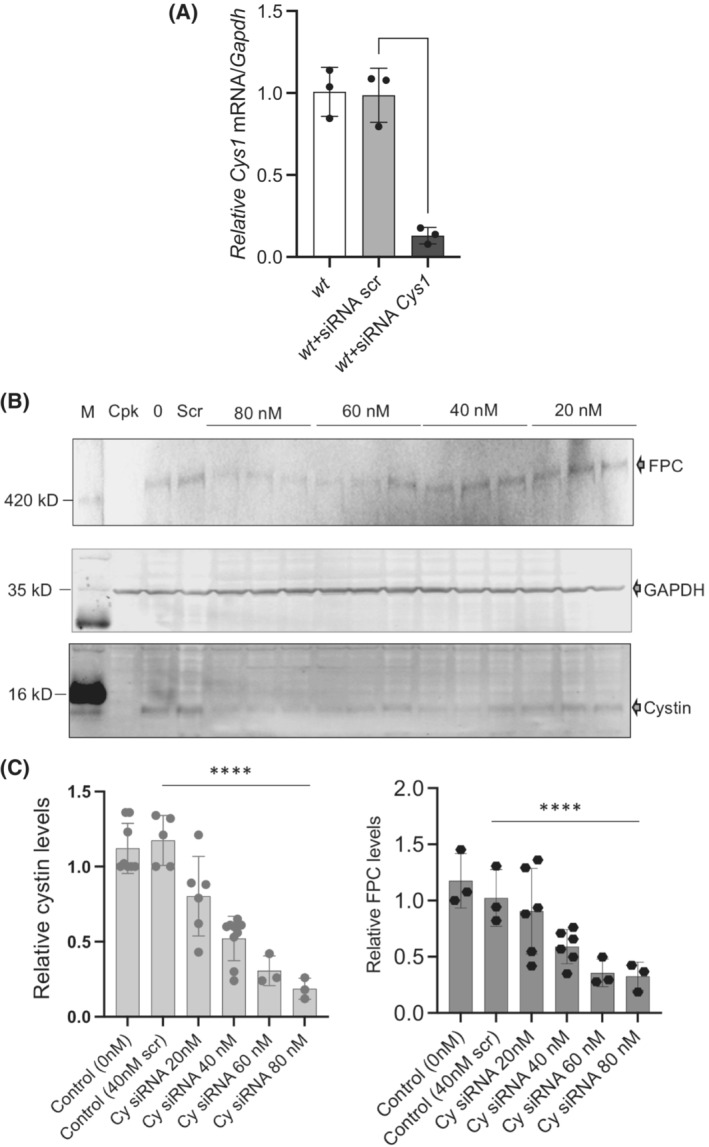
siRNA‐based cystin depletion leads to FPC reduction. (A) siRNA depletion of Cys1 mRNA. The efficiency of siRNA oligos (60 nM) to reduce Cys1 mRNA levels was measured relative to Gapdh1 mRNA in wt, wt + siRNA scrambled, and wt + siRNA Cys1 samples. Significant (**p < .0001, unpaired t‐test) reduction in Cys1 mRNA levels confirmed the specificity of the selected oligos. (B) siRNA depletion of Cys1 mRNA leads to reduction in FPC and cystin protein levels. Representative WB of FPC (top) and cystin (bottom) following cystin siRNA treatment for 72 h (siRNA concentrations indicated above image). GAPDH levels (middle) were used to normalize cystin and FPC expression. FPC expression was tested using anti‐FPC C‐terminal, rat monoclonal antibody, cystin expression was measured using anti‐cystin rabbit polyclonal antibody (bottom). (C) Densitometric analysis of cystin protein depletion (left) and corresponding FPC levels in the same samples (right). Values are plotted relative to GAPDH protein from N = 1–3 separate experiments, with n = 3–6 under each siRNA concentration (****p < .0001, unpaired t‐test, significance was determined between scrambled and cystin‐specific siRNA (80 nM)).
3.1.3. Loss of cystin affects ciliary architecture
Because both cystin and FPC are ciliary proteins, we examined how loss of cystin and FPC impacted cilia formation in cpk cells. For these experiments, we induced primary cilium development both in wt and cpk cells using serum starvation 39 and quantitated the fraction of cells with cilia (AT staining) relative to total cell number. We observed that the level of ciliation was very similar in wt and cpk cells with 77.35% of total wt and 75.82% of cpk cells exhibiting primary cilia (Figure 4A).
FIGURE 4.
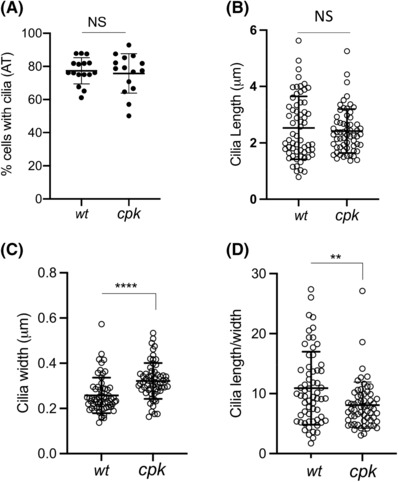
Loss of cystin in cpk cells does not affect cilia formation but leads to altered ciliary architecture. Acetylated tubulin (AT) staining was used to determine cilia number, thickness, and length. (A) Similar percentage of wt and cpk cells develop primary cilia. Cilia development was induced by serum starvation. Each data point represents the percentage of cells with cilia in a field with 22–44 cells in two experiments (wt: n = 492, cpk: n = 533). (B) Cilia length; (C) width (thickness, at the base of the cilia) was measured using ImageJ (see Materials and Methods). Each data point represents measurements of one cilium (n = 61, ****p < .0001). (D) Cilia length/width ratios are plotted from data points presented in (B) and (C) (n = 61, **: p = .0027).
Although there was no significant difference between the subsets of wt and cpk cells expressing cilia, the cilia of cpk cells structurally differed from those of wt cells (Figure 4A–C). Morphometric analyses (length, width, and width‐to‐length ratios) of cilia revealed no significant difference in the length of wt and cpk primary cilia (Figure 4B), but the cpk cilia were thicker at the base than wt (0.32 vs. 0.26 μm, p < .0001) (Figure 4C), and the cilia length/width ratio was reduced in cpk cells (10.9 (wt), 8.1 (cpk), p = .003) (Figure 4D). Therefore, the absence of cystin and significantly reduced FPC did not affect primary cilia formation but was associated with altered ciliary architecture in cpk cells (Figures 4 and S2).
3.2. The mechanism of FPC loss in cpk cells
3.2.1. Selective autophagy as a pathway for FPC removal
Relative Pkhd1 mRNA levels were not lower in cpk kidneys and CCD cells compared to wt (Figures 1 and 2). These results implied that FPC loss occurred at the protein level. If cystin plays an essential role in maintaining FPC levels, we reasoned that in the absence of cystin, FPC should undergo degradation through one of the intracellular protein degradation pathways (i.e., proteasomes, lysosomes, or autophagy).
Alternatively, enhanced FPC ectodomain shedding following Notch‐like processing could be a mechanism of FPC loss in cpk cells. 47 , 48 If FPC ectodomain shedding occurred in cpk cells, we would expect release of the FPC‐CTD into the cytoplasm and/or to the nucleus. However, immunocytochemistry using antibodies specifically directed against the mouse FPC‐CTD did not detect enhanced levels in the nuclei or the cytoplasm of cpk cells (Figure S2A). Neither did we observe increased FPC‐CTD staining in the nucleus or cytoplasm when cells were treated with proteasome and lysosomes inhibitors. These results suggest that intracellular release of FPC‐CTD was not increased.
Another possible mechanism by which cystin could modulate FPC levels is through vesicular trafficking by binding to the cytosolic side of FPC‐containing vesicles. In this scenario, we would have expected co‐localization of cystin and FPC, which was not observed (Figure S2A). The absence of vesicle‐associated cystin‐FPC co‐localization argues against cystin functioning as an escort protein of FPC‐containing vesicles.
Loss of FPC might also occur during translation in the endoplasmic reticulum, for example, endoplasmic reticulum‐associated degradation (ERAD). However, proteasome inhibition with MG‐132 (5 μM) for 4 h did not increase levels of FPC in cpk cells, suggesting that FPC reduction in cpk cells does not involve ERAD (Figure 5A). However, increased levels of polyubiquitinated proteins in MG‐132‐treated samples of both wt and cpk cells indicated efficient proteasome inhibition, as shown later in Figure 7B. Interestingly, prolonged (8 h) treatment with MG‐132 (5 μM) caused a severe reduction of FPC in both cpk and wt cells (Figure 5B), but without affecting levels of other cystoproteins Pc2 and Ift88 (Figure 5C). This prolonged MG‐132 effect is likely the result of selective autophagy activation as an alternative pathway for protein degradation when the proteasomes are inhibited. 49 , 50 , 51 We also found that inhibition of lysosomal fusion by chloroquine (CQ) did not alter the soluble fraction of FPC in either wt or cpk cells (Figure 5B). These observations argue against direct lysosomal FPC degradation as mechanism for removal. In contrast to non‐selective autophagy that is activated during conditions such as starvation, selective autophagy is responsible for the degradation of protein aggregates and damaged cellular organelles. 52 Therefore, if FPC was directed to this pathway physiologically, as it has been shown for other ciliary proteins, 53 , 54 selective autophagy activation could be responsible for the significant reduction in FPC levels following 8 h MG‐132 treatment.
FIGURE 5.
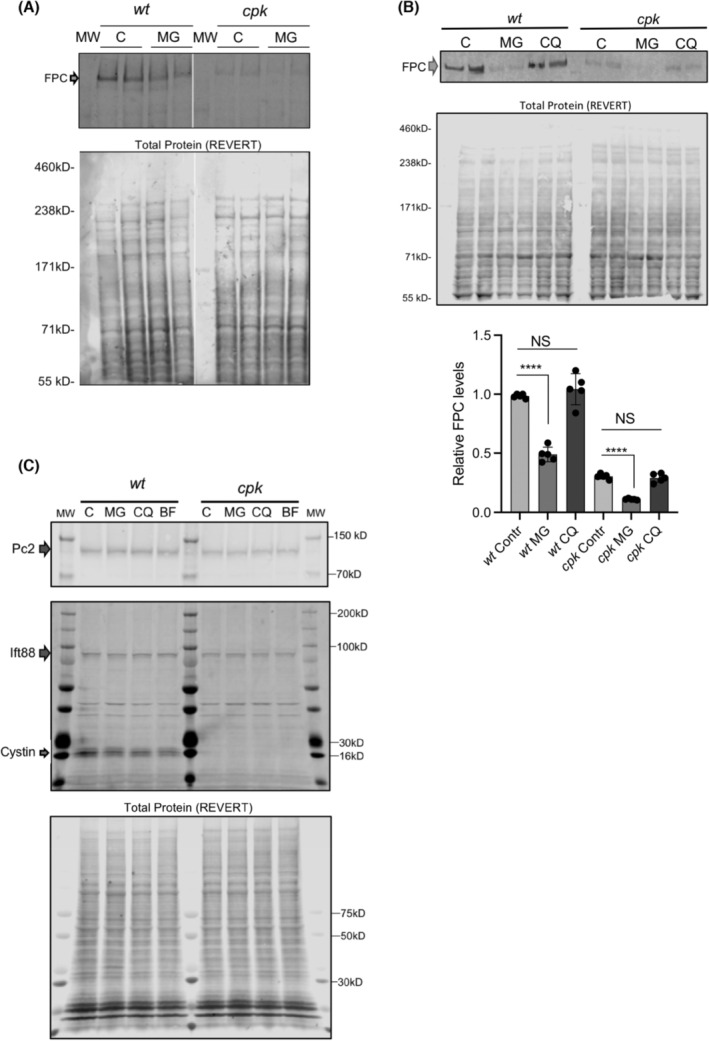
Proteasome or lysosome inhibition does not lead to increase in soluble FPC levels. (A) Representative gel is shown on top with biological duplicates for control (untreated) and proteasome inhibitor (MG, 4 h) treated wt and cpk cells (top). Total protein stain (REVERT) is shown to demonstrate equal loading. FPC and arrow demonstrate full‐length FPC. (B) Prolonged (8 h) proteasome inhibition (MG) leads to reduced FPC in wt and cpk cells. Top shows a representative gel image (n = 2). Middle shows a total protein gel image (Total Protein, REVERT). Densitometric measurement of relative FPC levels/total protein shown at bottom. (N = 3, n = 6, ****p < .0001, unpaired t‐test). Lysosome inhibition (CQ) for 8 h did not change FPC levels. (C) Levels of Pc2 and Ift88 did not change in the presence of proteasome (MG), or lysosome inhibitors (CQ, BF). MG, CQ, and bafilomycin (BF) were applied for 4 h, total protein (REVERT) staining is shown for reference (bottom).
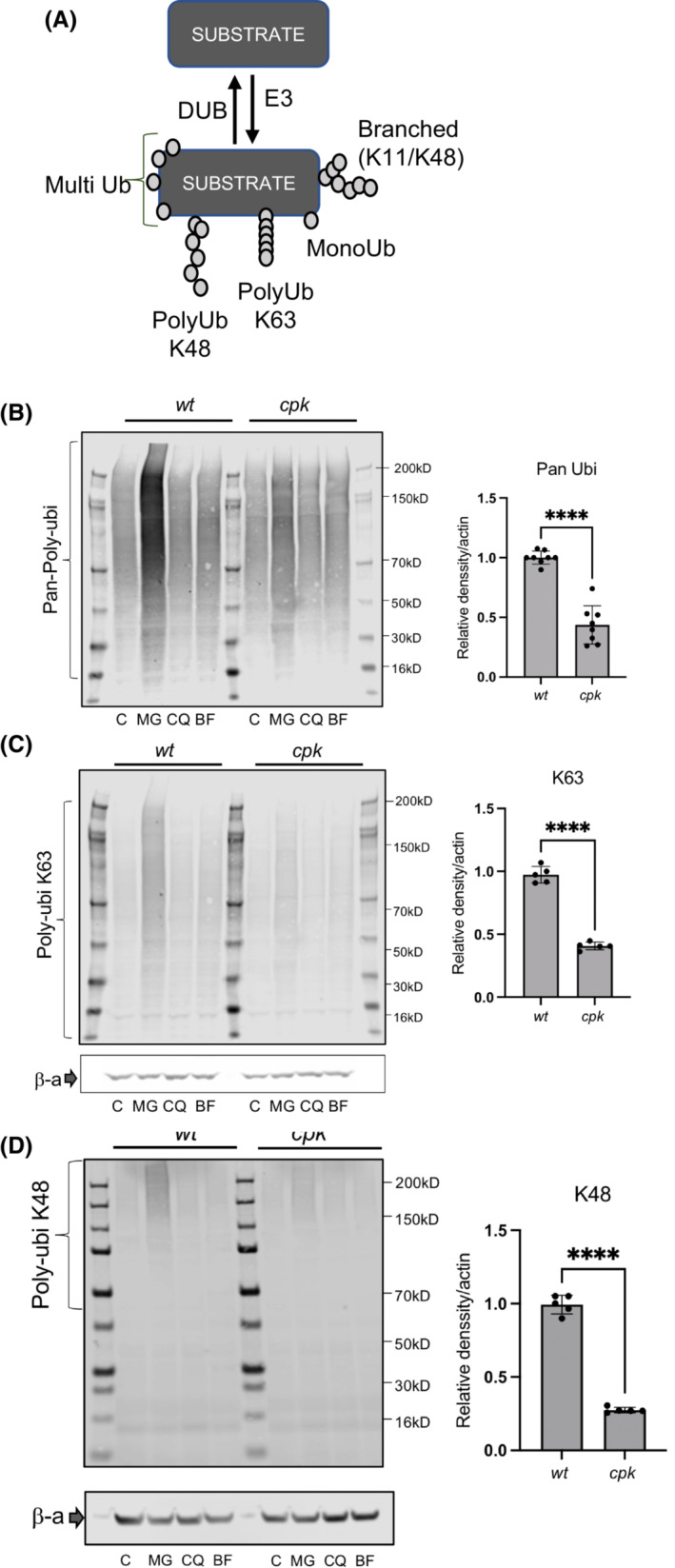
FIGURE 7.
Reduced polyubiquitination in cpk cells. (A) Scheme depicting types of ubiquitination. Proteins are polyubiquitinated prior to ERAD, lysosomal degradation, or autophagy. The length of the poly‐ubiquitin chain varies. (B–D) Reduced polyubiquitination in cpk cells. Representative gels (left) are shown for polyubiquitination of cellular proteins in control (C), MG, CQ, and BF‐treated cells (4 h). (B) Pan‐poly‐ubiquitination (N = 3, n = 8); (C) K63‐linked (N = 2, n = 5); and (D) K48‐linked polyubiquitination (N = 2, n = 5). Results of densitometric analysis (right) are plotted relative to β‐actin levels (****p < .00001, unpaired t‐test).
To test this possibility, we first determined if selective autophagy was activated following prolonged treatment with MG‐132. We used SQSTM1/p62 as reporter. SQSTM1/p62 links the autophagy receptor LC3 with ubiquitinated proteins 55 , 56 , 57 to regulate cytoplasmic segregation and autophagic degradation of ubiquitinated proteins. 58 , 59 , 60 In MG‐132‐treated (8 h) wt and cpk cells, we observed increased SQSTM1/p62 levels, consistent with activated autophagy (Figure 6A). However, cpk cells appeared to have higher baseline levels of SQSTM1/p62 and also exhibited p62‐oligomers, indicative of autophagy hyperactivation upon prolonged proteasome inhibition (Figure 6A). 61 Testing multiple samples of untreated wt and cpk lysates confirmed the higher abundance of SQSTM1/p62 in cpk compared to wt cells, suggestive of defective protein degradation and autophagy induction in cystin‐deficient cpk cells (Figure 6B).
FIGURE 6.
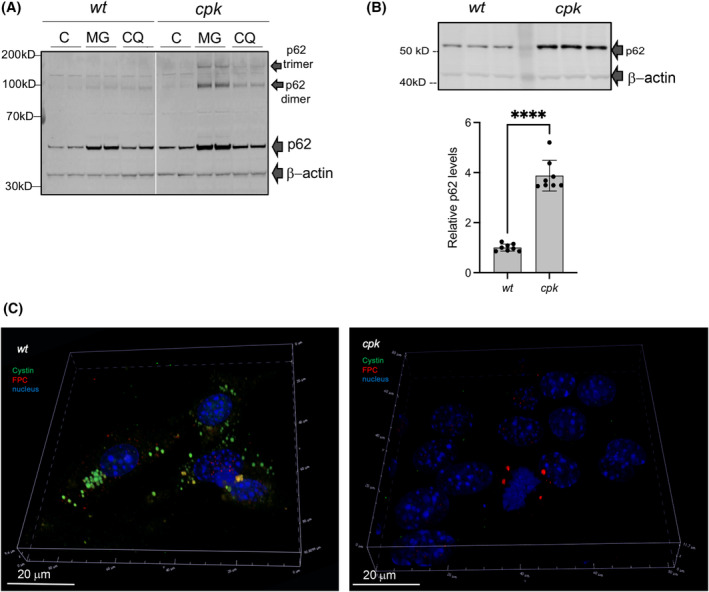
Autophagy as a pathway for FPC removal. (A) Hyperactivation of selective autophagy by proteasome inhibition, SQSTM1/p62 as reporter. Representative gel image (n = 2). (C) control, MG: MG‐132, CQ: chloroquine. (B) Higher baseline SQSTM1/p62 levels in cpk compared to wt cells. Representative gel image (top. Relative p62 levels/β‐actin plotted on bottom (N = 2, n = 8), ****p < .000, unpaired t‐test). (C) Co‐localization of cystin and FPC in cytoplasmic particles of wt cells following proteosome inhibition and lysosome inhibition. Wt cells were stained with anti‐cystin (green) and anti‐FPC (red) antibodies following treatment with MG‐132 (8 h) and CQ (4 h), to facilitate formation of autophagic particles. Cystin co‐localized with FPC in some particles (yellow staining). In cystin‐negative cpk cells, smaller aggregates of FPC were detected in some cells (right).
Intracellular protein degradation that maintains the cellular proteome involves three interactive systems, the proteosome, lysosomes, and autophagy, the latter involving lysosomal fusion of different types of autophagic particles. Therefore, we expected that when both the proteasomes and the lysosomes were inhibited, FPC would be seen sequestered in autophagic particles. To test this hypothesis, we treated the cells with MG‐132 for 8 h to induce autophagy by building up cytosolic protein aggregates that would be degraded by the proteasome in the absence of MG‐132. Such aggregates could contain FPC, but to detect it, we needed to inhibit the fusion of aggregates with the lysosomes (CQ, 4 h), thereby preventing degradation of sequestered proteins. We co‐labeled cystin and FPC and observed large cystin‐containing particles in wt cells, some of which also co‐stained with FPC (Figure 6C, left), suggesting that both cystin and FPC were aggregated and directed to autophagy. In cpk cells, we observed only few FPC‐containing particles and no cystin (Figure 6C, right). We also labeled SQSTM1/p62 in control and MG + CQ‐treated wt and cpk cells. Some of the cpk cells expressed higher levels of SQSTM1/p62 and we observed more particles than in wt (Figure S4A, top, white arrows). Treatment with MG‐132 (8 h, 5 mM) and CQ (4 h, 10 mM) resulted in the formation of multiple autophagic particles (autophagosomes) (Figure S4A, bottom, yellow arrows). Confocal microscopy shows more detailed structure of the SQSTM1/p62 particles (green) following MG + CQ treatment (Figure S4B, top, yellow arrows) in wt cells and co‐localization of p62 with FPC (red) in MG + CQ‐treated cells (Figure S4B, bottom, white arrows).
3.3. The consequences of FPC loss in cpk cells
As described above, we have shown that cystin deficiency results in the loss of FPC (~80%) and Pc2 (~30%) and that re‐expression of a GFP‐tagged cystin can reduce cyst formation in r‐cpk mice, 4 but results only in partial recovery of FPC levels (Figure 1). Therefore, we next sought to assess the functional consequences of FPC reduction in cpk cells. In previous collaborative work, we have shown that FPC is a functional component of the NEDD4‐family of E3 ubiquitin ligase complexes, which facilitate the ubiquitination of multiple proteins, and that lack of FPC compromised the function of intracellular protein degradation systems. 23 Therefore, ubiquitination may be compromised in cpk cells with minimal FPC levels, a condition which can itself lead to the activation of SQSTM1/p62‐mediated selective autophagy. 49 The data in the current study demonstrating higher SQSTM1/p62 levels in cpk cells compared to wt (Figure 6B) are consistent with compromised protein degradation in cpk cells. Therefore, we compared the efficiency of polyubiquitination in wt and cpk cells.
3.3.1. Reduced ubiquitination in cpk cells
The NEDD4 family of E3 ligases belong to the homologous to E6AP C‐terminus (HECT) domain E3 ligase group, which regulate multiple cellular activities through protein ubiquitination (for review: [ 62 ]). E3 ligases facilitate multiple types of ubiquitination 63 (Figure 7A). FPC deficiency‐associated E3 ligase dysfunction in cpk cells should, therefore, result in altered polyubiquitination of numerous proteins, impair the ubiquitin‐proteasome system (UPS) as well as lysosomal/autophagosome degradative systems, leading to altered proteome homeostasis. We inhibited the proteasomes with MG‐132 (5 μM) and the lysosomes with CQ (10 μM) or BF (10 μM) for 4 h and evaluated polyubiquitination, using linkage‐specific anti‐ubiquitin antibodies 64 to compare levels of polyubiquitination in wt and cpk cells. While proteasome inhibition led to increased ubiquitination in both wt and cpk cells, polyubiquitination was attenuated in cpk cells compared to wt (Figure 7B). Inhibition of the lysosomes with CQ or BF did not affect polyubiquitination and were excluded from further analysis.
To determine which type of ubiquitination was affected, we used anti‐ubiquitin antibodies recognizing: (1) all types of polyubiquitin links (Pan‐ubi), (2) K48‐linked ubiquitin, which is the primary linkage type for proteasomal degradation, and (3) K63‐linked ubiquitin, which is the primary linkage type degradation of cell surface receptors. 65 Cpk cells exhibited lower levels of polyubiquitination in general (Figure 7B), as well as reduced K63‐linked (Figure 7C), and K48‐linked polyubiquitin chains (Figure 7D). These results are consistent with reduced E3 ligase activity in cpk cells that may impede protein quality control by both the lysosomes and proteasomes and affect protein quality control at the endoplasmic reticulum and at the cell surface. Reduced polyubiquitination likely influences multiple cellular signaling pathways, as well as overall proteome homeostasis, 62 making the consequences of cystin and FPC loss more complicated and severe than expected.
3.3.2. Enhanced expression of the epithelial sodium channel (ENaCα) in cpk cells
Proteomic analyses were beyond the scope of the current study. Therefore, as a proof of concept, we selected a functionally relevant NEDD4 E3 substrate, the epithelial sodium channel (ENaC) 24 to analyze the functional consequences of FPC loss in cpk cells. Of note, enhanced renal ENaC activity is associated with high blood pressure, 66 , 67 and systemic hypertension is a hallmark of ARPKD. 25 , 68 Previous studies have shown that expression of the functional alpha subunit of ENaC (ENaCα) is regulated via ubiquitination and degradation by both the UPS and lysosomes. 30 , 69 , 70 Consistent with these results, we observed enhanced levels of ENaCα following both MG‐132 and CQ treatment for 4 h (Figure 8A). Interestingly, we also observed increased abundance of ENaCα in cpk compared to wt cells under control conditions (Figure 8A,B). Confocal microscopy confirmed higher membrane ENaCα levels in cpk cells (Figure 8C), with strong vesicular ENaCα staining and significant co‐localization with lysosome‐associated membrane protein 1 (LAMP1), which delivers proteins to the lysosome (Figure 8D). In wt cells, ENaCα was mostly observed in vesicular compartments and was not colocalized with LAMP1. These data suggest that ENaC turnover is reduced in cpk compared to wt cells, consistent with previous findings indicating that FPC deficiency is linked to reduced NEDD4 E3 ligase activity, leading to defective degradation of cell surface transporters including ENaC. 23
FIGURE 8.
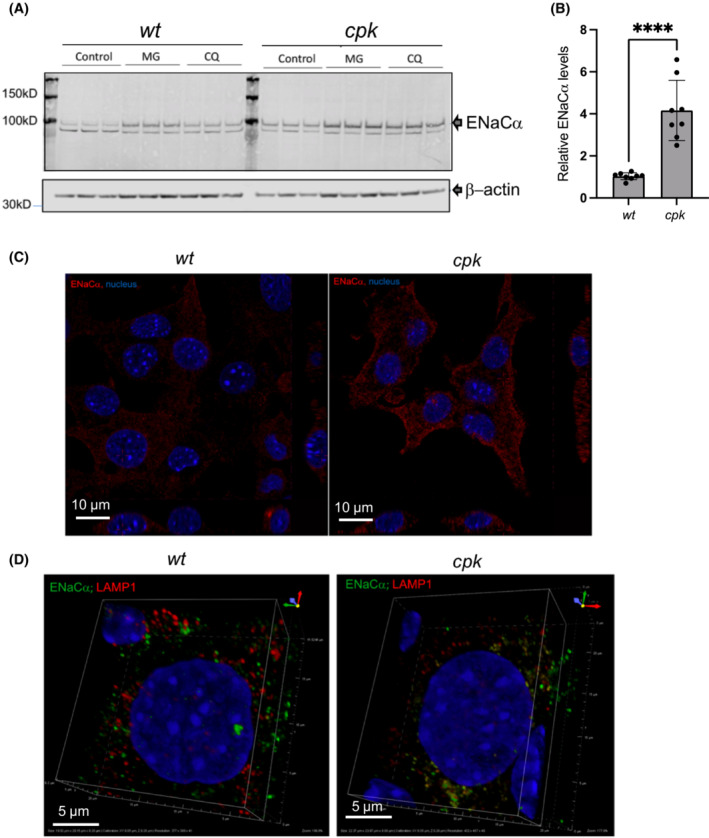
Higher ENaC levels in cpk cells. (A) Representative WB of ENaCα in wt and cpk cells. β‐actin was used as loading control. Both proteasome and lysosome inhibition (CQ) enhanced ENaCα levels, consistent with previously reported results. (B) Relative ENaCα levels. (N = 3, n = 7; ***p < .0001). (C) Increased ENaC staining (red) in cpk cells. Confocal microscopy images of wt (left) and cpk cells (right). (D) Co‐localization of ENaC and LAMP1 (lysosomal marker). Minimal co‐localization of ENaC (green) with LAMP1(red) in wt cells (left). ENaC (green) highly co‐localized with LAMP1 (red) in cpk cells (right).
3.3.3. Higher ENaCα levels correspond to enhanced sodium transport
To determine whether increased ENaCα membrane abundance correlated with increased sodium transport, we performed whole‐cell patch analysis on wt and cpk cells using previously published protocols. 41 , 42 Both cell types exhibited currents sensitive to the ENaC inhibitor amiloride (Figure 9A–F), supporting the biochemical evidence for ENaC expression in both cell types (Figure 8). We measured net amiloride‐sensitive currents by subtracting the currents in the presence of amiloride from control currents (Figure 9C,F) and plotted the mean I–V relation of amiloride‐sensitive currents from wt and cpk cells, respectively (Figure 9G,H). Mean I–V curves of absolute amiloride‐sensitive currents from cpk cells showed higher amiloride‐sensitive currents at each membrane potential compared to wt. At positive membrane potentials (from −10 to 30 mV) when the I–V relation was expressed by current density (pA/pF, Figure 9H), we observed that cpk cells showed significantly larger amiloride‐sensitive currents, suggesting that there are more fluxes of amiloride‐sensitive currents in cpk cells, representing more channels in the membrane. In addition, amiloride‐sensitive currents from cpk cells are outward‐rectified, in contrast to wt cells with inward rectified currents, contributing to high amiloride‐sensitive currents from cpk cells at positive membrane potentials. Our findings indicate a positive correlation between higher ENaCα membrane abundance and enhanced amiloride‐sensitive currents in cpk cells, a result consistent with the anticipated consequences of FPC deficiency‐linked changes in proteome homeostasis.
FIGURE 9.
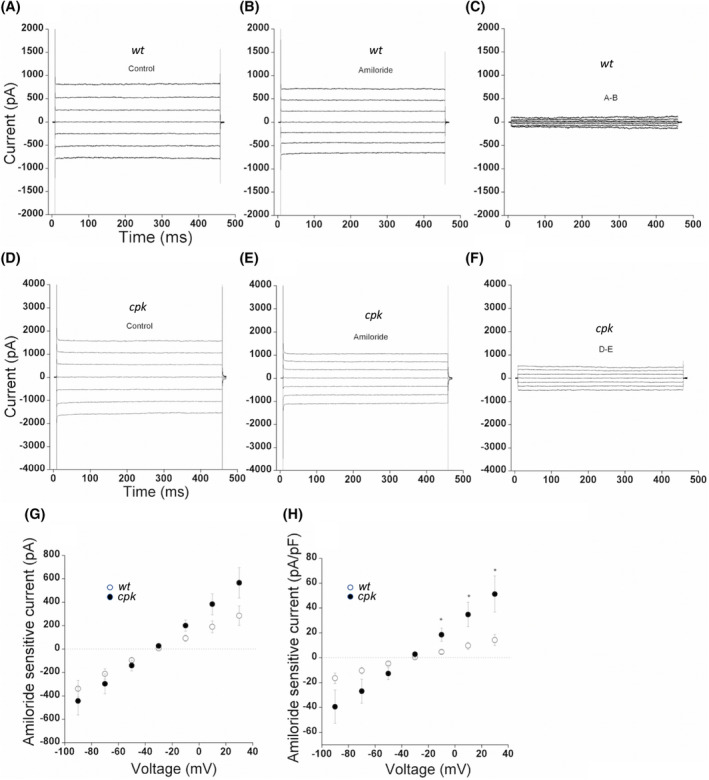
Higher relative amiloride‐sensitive (ENaC) currents in cpk compared to wt cells. Whole‐cell currents were evoked by applying voltage steps from −90 to 30 mV in 20 mV increments. Cells were held at −30 mV. (A–C) Representative whole‐cell current recordings from wt cells; (D–F) Representative whole‐cell current recordings from cpk cells; (A and D) Representative whole‐cell current recordings in the absence of amiloride. (B and E) Representative whole‐cell current recordings in the presence of amiloride (10 μM). (G) Amiloride‐sensitive currents (C and F) were measured by subtracting (B) from (A) and (E) from (D). Mean I–V relationship of amiloride‐sensitive currents expressed by total currents for wt (open circle, n = 15) and cpk (dark circle, n = 13) cells. (H) Mean I–V relationship of amiloride‐sensitive currents was expressed by current density for wt (open circle, n = 15) and cpk (dark circle, n = 13) cells. Values are means ±SE, *p < .05.
4. DISCUSSION
The current study demonstrates the cystin‐dependent regulation of FPC levels in mouse kidneys and CCD cell lines, providing the first evidence of a mechanistic connection between the phenotypes of cpk mice and patients with ARPKD caused by PKHD1 (encoding FPC) as well as CYS1 mutations (encoding cystin). Our studies demonstrate that in mouse kidneys and CCD cell lines, loss of cystin led to an 80–90% reduction in FPC levels, which occurred at the protein level. Both the natural mutation‐mediated loss of cystin in cpk kidneys and CCD cells, as well as siRNA removal of Cys1 mRNA in wt CCD cells led to FPC reduction. These results (in vivo, in vitro in cpk and siRNA of Cys1 in wt cells) argue against the possibility that reduced FPC in cpk cells is the result of a cpk CCD cell line‐specific phenomenon. We note that collecting duct‐specific expression of the cystin–GFP fusion protein in r‐cpk mice 4 partially rescued FPC levels (Figure 1A,B). It is possible that, despite its high level of expression, the cystin–GFP fusion protein does not have comparable function as endogenous cystin, as has been demonstrated for multiple GFP‐tagged proteins. 43 , 44 , 71 , 72 However, our observation that depleting cystin through siRNA in wt cells, which express endogenous cystin and FPC, caused a cystin depletion‐dependent FPC reduction, supports our thesis that cystin is necessary to stabilize FPC and when cystin is removed, FPC levels quickly decrease. These data and the observation that FPC deficiency did not reduce cystin expression in any of the tested Pkhd1 mutant mouse kidneys suggest that cystin‐mediated FPC expression regulation is unidirectional.
The level of FPC rescue that is necessary to mitigate renal cystic disease is not known. In comparison, we and others have examined the trafficking and regulated expression of the cystic fibrosis transmembrane conductance regulator (CFTR) and proposed that 10–35% of CFTR rescue is sufficient to prevent symptoms of cystic fibrosis pulmonary disease. 73 , 74 , 75 , 76 Subsequent development and application of pharmacological compounds to rescue different CFTR mutants corroborated this estimate. 76 , 77 , 78 , 79 , 80 Our knowledge of FPC biogenesis regulation, trafficking, and function is substantially more limited than what we know about CFT. Defining the level of FPC rescue that is necessary to prevent renal cystic phenotype will require additional studies, but our results presented in Figure 1, indicating partial rescue of FPC in r‐cpk mouse kidneys and mitigation of the cystic phenotype suggest that even low level of FPC rescue can be beneficial.
To our knowledge, no prior studies have analyzed primary cilia development in cpk cells. We demonstrate that loss of cystin and FPC did not disrupt ciliogenesis, but rather the architecture of the cilia was different in cpk versus wt cells (Figure 4). Currently, we do not know whether at low‐level FPC traffics to the cilia in cpk cells, or in the absence of cystin, the protein never reaches the cilial compartment. This very important question will be the subject of future studies and will require the development of additional antibodies as well as cell lines with inducible cystin expression and endogenous FPC expression. Importantly, our results provide the basis for future studies to investigate and define mechanisms that underpin this novel observation and whether ciliary structural alterations contribute to the cpk renal cystic phenotype.
In order to understand the mechanisms of FPC loss in the absence of cystin, we examined the role of intracellular protein degradation systems. We could not recover FPC by inhibiting the proteasomes or lysosomes in cpk cells (Figure 5). These data implicate autophagy as the primary mechanism responsible for FPC removal in the absence of cystin. However, the very low baseline levels of FPC in cpk cells confounded our ability to experimentally analyze this mechanism in cystin‐deficient cells. Moreover, we note that if FPC is a functional component of the cellular protein quality control, as proposed by Kaimori et al 23 and supported by our results (Figures 7, 8, 9), it may not be possible to recover FPC by simply inhibiting individual protein degradation systems, or with simultaneous pan inhibition, because these systems are functionally linked at multiple levels and their integration is required for cellular homeostasis [for reviews 81 , 82 , 83 ]. Inhibition of one protein degradation pathway activates alternative degradation pathways 49 and stress responses, leading to alterations in cellular functions, including translation inhibition. 73 , 74 , 84 In addition, once a protein is sequestered in autophagic particles, it may not be resolved by standard cell lysis conditions. 85 Analysis of protein aggregates would require FPC domain‐specific antibodies and those are not available for endogenously expressed FPC. Therefore, it was not feasible to further investigate the nature and localization of such sequestered complexes at this time.
We observed that SQSTM1/p62 protein levels, a reporter of selective autophagy, were higher in cystin‐deficient cpk cells at baseline (Figure 6B). This is likely the consequence of in E3 ligase function deficiency (Figure 7A), which is necessary for proper protein quality control and when this system is disturbed, alternative pathways are activated [for review 62 , 82 ]. The formation of protein aggregates when we hyperactivated autophagy and inhibited the fusion of the particles with lysosomes (Figure 6C) also suggests that autophagy is responsible for FPC removal. Moreover, we found that autophagy hyperactivation led to the degradation of FPC not only in cpk but also in wt cells (Figure 5B). Our results are also consistent with previous reports indicating that degradation of ciliary proteins occurs through selective autophagy. 53 , 54 Further studies will be necessary to define the mechanisms by which cystin shelters FPC from degradation and contributes to the maintenance of the cellular proteome. Although we do not understand all facets of the cystin–FPC interaction, the current studies, performed in mouse kidneys and CCD cell lines expressing endogenous FPC, clearly demonstrate that loss of cystin leads to significant FPC reduction. Conversely, FPC loss does not alter cystin expression in Pkhd1 mutant mice (Figure 1D) which lack classic ARPKD phenotype. 6 , 7
As an important consequence of cystin and FPC loss, we demonstrated significant reduction in polyubiquitination (Figure 7), supporting the findings of Kaimori and colleagues 23 that FPC plays role in protein quality control and proteome management. Our data are also consistent with the interactive feedback among intracellular protein degradation systems (proteasome, lysosomes, and different types of autophagy 86 ) in maintaining proteostasis. 87 When one of these systems is rendered dysfunctional, other protein degradation pathways undergo dynamic adjustments to maintain proteome integrity. 82 We propose that in the absence of cystin, FPC undergoes targeted degradation that renders at least the NEDD4 family of E3 ligase complexes dysfunctional. The downstream consequences involve dismantling of the integrated protein quality control mechanisms that maintain cellular proteome integrity. This hypothesis is supported by previous studies reporting attenuated expression of epithelial adhesion molecules, 88 , 89 altered basement membrane deposition, 90 and metabolic abnormalities 8 in cpk cells.
Proteomic analysis, combined with genomics, will be necessary to fully elucidate the extent and severity of consequences caused by cystin deficiency and the consequent reduction of FPC deficiency on the cellular proteome. Such studies will not only improve our understanding of the complexity of mechanisms leading to cyst formation but will also provide important new insights about the function of FPC and the FPC‐containing E3 ligase complexes. As an initial proof of concept, we examined a functionally relevant NEDD4 E3 substrate, the epithelial sodium channel (ENaC). 24 We observed elevated ENaC protein levels and increased epithelial sodium transport in cpk cells, which supports our hypothesis and reveals a possible mechanism underlying the systemic hypertension that is a hallmark symptom of ARPKD. 25 , 68
Our studies support the proposition that cystin maintains FPC levels, and FPC is required for the physiological function of certain HECT‐domain E3 ubiquitin ligase complexes, especially in renal epithelial cells. We have extended the previously proposed role of FPC in maintaining the integrity of signaling pathways, protein quality control, and turnover, 23 now to include cystin. We have shown that cystin affects FPC abundance in a unidirectional manner and that loss of FPC in Pkhd1 mutant mouse kidneys was not associated with reduced cystin levels. This observation may be critically important for clarifying an otherwise apparent paradox. That is, why does the loss of FPC cause renal cysts in cpk but not Pkhd1 mutant mice? It is possible that, in mice, cystin has at least two essential roles: maintenance of FPC as a required functional component of the NEDD4 E3 ligase complexes, 23 and inhibition of Myc expression via interaction with necdin, as we have demonstrated previously. 18 In mouse kidneys, more specifically in CCD epithelial cells, both FPC loss and Myc overexpression are required for cystogenesis. By contrast, Pkhd1 mutant mice lacking FPC retain normal cystin levels (Figure 1), which can keep Myc expression low through the cystin‐necdin interaction. 18 , 31 Our results imply that disruption of proteome homeostasis is a significant pathophysiological consequence of FPC loss, yet not sufficient for renal cyst formation in mice when cystin is present. Identification of all E3 ubiquitin ligases, components of the complexes, and the mechanisms by which the cellular proteome is affected by the loss of FPC in mouse and human kidneys will require further investigations. Those studies will likely shed light on factors that are responsible for the differences observed between human and mouse kidney phenotypes following FPC loss.
Finally, our data establishing a functional link between cystin and FPC expression in mouse kidneys and CCD cells, as well as the recent identification of patients with CYS1‐related ARPKD, support the use of the cpk mouse as an important experimental model to study ARPKD pathogenesis and experimental therapeutics. Our studies also provide novel perspectives on the renal phenotype in mouse models of ARPKD, suggesting that future mechanistic studies should evaluate the interactome that underpins FPC and cystin expression, Myc transcriptional regulation, and maintenance of the cellular proteome, as well as how disruption of these interacting pathways drives ARPKD pathogenesis.
AUTHOR CONTRIBUTIONS
Yiming J. Zhang contributed to experimental design, performed most of the experimental procedures, contributed to data analysis and manuscript writing. Chaozhe Yang designed experiments, prepared samples, and performed western blotting analysis of cystin and FPC expression in mouse kidneys. Wei Wang designed, performed, and analyzed the patch clamp studies. Naoe Harafuji designed the experiments and performed and analyzed mRNA expression levels in cells and kidney tissues. P. Darwin Bell developed the immortalized cell lines and helped to interpret initial results. Elizabeth Sztul's laboratory (Piotr Stasiak) performed the cilia morphology studies. Ljubica Caldovic contributed to the interpretation of data and manuscript writing. Lisa M. Guay‐Woodford supervised studies in wt and cpk mice, contributed to the interpretation of results and manuscript writing. Zsuzsanna Bebok conceived the study, co‐designed the majority of the experiments, supervised the analysis and presentation of experimental results, and wrote the manuscript.
FUNDING INFORMATION
UAB HRFD Core Center and O'Brien Center for Acute Kidney Injury Center Pilot Program (ZsB) P30DK072482, UAB CF Research and Translation Core Center and CCF‐ROWE19RO, UAB Research and Development Program (ZsB and WW); R01GM122802 (ES). R01DK121530, PKD Foundation, and The Moran Family Foundation (LGW).
DISCLOSURES
The authors have stated explicitly that there are no conflicts of interest in connection with this article.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
ACKNOWLEDGMENTS
We thank Dr. Adam Richman for critical reading and help with editing of the manuscript.
Zhang YJ, Yang C, Wang W, et al. Cystin is required for maintaining fibrocystin (FPC) levels and safeguarding proteome integrity in mouse renal epithelial cells. The FASEB Journal. 2023;37:e23008. doi: 10.1096/fj.202300100R
Contributor Information
Lisa M. Guay‐Woodford, Email: lguaywoo@childrensnational.org, Email: guaywoodfl@chop.edu, Email: guaywoodfl@chop.edu.
Zsuzsanna Bebok, Email: bebizs@me.com.
DATA AVAILABILITY STATEMENT
The data (western blot images, densitometry results, RT‐PCR readings, raw confocal microscopy images) that support the findings of this study are available from the corresponding author, [ZsB], upon reasonable request. Data acquisition details are described in the Materials and Methods section.
REFERENCES
- 1. Preminger GM, Koch WE, Fried FA, McFarland E, Murphy ED, Mandell J. Murine congenital polycystic kidney disease: a model for studying development of cystic disease. J Urol. 1982;127:556‐560. [DOI] [PubMed] [Google Scholar]
- 2. Schieren G, Pey R, Bach J, Hafner M, Gretz N. Murine models of polycystic kidney disease. Nephrol Dial Transplant. 1996;11(Suppl 6):38‐45. [DOI] [PubMed] [Google Scholar]
- 3. Guay‐Woodford LM. Murine models of polycystic kidney disease: molecular and therapeutic insights. Am J Physiol Renal Physiol. 2003;285:F1034‐F1049. [DOI] [PubMed] [Google Scholar]
- 4. Yang C, Harafuji N, O'Connor A, et al. Cystin genetic variants cause autosomal recessive polycystic kidney disease associated with altered Myc expression. Sci Rep. 2021;11:18274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Garcia‐Gonzalez MA, Menezes LF, Piontek KB, et al. Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum Mol Genet. 2007;16:1940‐1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Menezes LF, Zhou F, Kurashige MK, Outeda P, Watnick T, Germino G. Mice Lacking Pkhd1 Exons 3‐67 Develop Biliary but not Renal Abnormalities. ASN Kidney Week; 2018. Accessed May 12, 2023. https://www.asn‐online.org/education/kidneyweek/2018/program‐abstract.aspx?controlId=3019761 [asn‐online.org] [Google Scholar]
- 7. Outeda P, Menezes L, Hartung EA, et al. A novel model of autosomal recessive polycystic kidney questions the role of the fibrocystin C‐terminus in disease mechanism. Kidney Int. 2017;92:1130‐1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hwang VJ, Kim J, Rand A, et al. The cpk model of recessive PKD shows glutamine dependence associated with the production of the oncometabolite 2‐hydroxyglutarate. Am J Physiol Renal Physiol. 2015;309:F492‐F498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Steele SL, Wu Y, Kolb RJ, et al. Telomerase immortalization of principal cells from mouse collecting duct. Am J Physiol Renal Physiol. 2010;299:F1507‐F1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hou X, Mrug M, Yoder BK, et al. Cystin, a novel cilia‐associated protein, is disrupted in the cpk mouse model of polycystic kidney disease. J Clin Invest. 2002;109:533‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoder BK, Hou X, Guay‐Woodford LM. The polycystic kidney disease proteins, polycystin‐1, polycystin‐2, polaris, and cystin, are co‐localized in renal cilia. J Am Soc Nephrol. 2002;13:2508‐2516. [DOI] [PubMed] [Google Scholar]
- 12. Udenwobele DI, Su RC, Good SV, Ball TB, Varma Shrivastav S, Shrivastav A. Myristoylation: an important protein modification in the immune response. Front Immunol. 2017;8:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wright MH, Heal WP, Mann DJ, Tate EW. Protein myristoylation in health and disease. J Chem Biol. 2010;3:19‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kahn RA, Goddard C, Newkirk M. Chemical and immunological characterization of the 21‐kDa ADP‐ribosylation factor of adenylate cyclase. J Biol Chem. 1988;263:8282‐8287. [PubMed] [Google Scholar]
- 15. Franco M, Chardin P, Chabre M, Paris S. Myristoylation‐facilitated binding of the G protein ARF1GDP to membrane phospholipids is required for its activation by a soluble nucleotide exchange factor. J Biol Chem. 1996;271:1573‐1578. [DOI] [PubMed] [Google Scholar]
- 16. Kaczmarek B, Verbavatz JM, Jackson CL. GBF1 and Arf1 function in vesicular trafficking, lipid homoeostasis and organelle dynamics. Biol Cell. 2017;109:391‐399. [DOI] [PubMed] [Google Scholar]
- 17. Tao B, Bu S, Yang Z, et al. Cystin localizes to primary cilia via membrane microdomains and a targeting motif. J Am Soc Nephrol. 2009;20:2570‐2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu M, Yang C, Tao B, Bu S, Guay‐Woodford LM. The ciliary protein cystin forms a regulatory complex with necdin to modulate Myc expression. PLoS One. 2013;8:e83062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Parrot C, Kurbegovic A, Yao G, Couillard M, Cote O, Trudel M. c‐Myc is a regulator of the PKD1 gene and PC1‐induced pathogenesis. Hum Mol Genet. 2019;28:751‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trudel M. c‐Myc Signalling in the Genetic Mechanism of Polycystic Kidney Disease. In: Li X, ed. Polycystic Kidney Disease [Internet]. Brisbane: Codon Publications; 2015. [PubMed] [Google Scholar]
- 21. O'Connnor AK, Guay‐Woodford LM. The Polycystic Kidney Diseases and Other Hepato‐Renal Fibrocystic Diseases: Clinical Phenotypes, Molecular Pathobiology, and Variation between Mouse and Man. Elsevier; 2016. [Google Scholar]
- 22. Goggolidou P, Richards T. The genetics of autosomal recessive polycystic kidney disease (ARPKD). Biochim Biophys Acta Mol Basis Dis. 2022;1868:166348. [DOI] [PubMed] [Google Scholar]
- 23. Kaimori JY, Lin CC, Outeda P, et al. NEDD4‐family E3 ligase dysfunction due to PKHD1/Pkhd1 defects suggests a mechanistic model for ARPKD pathobiology. Sci Rep. 2017;7:7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eaton DC, Malik B, Bao HF, Yu L, Jain L. Regulation of epithelial sodium channel trafficking by ubiquitination. Proc Am Thorac Soc. 2010;7:54‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dorval G, Boyer O, Couderc A, et al. Long‐term kidney and liver outcome in 50 children with autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2021;36:1165‐1173. [DOI] [PubMed] [Google Scholar]
- 26. Guay‐Woodford LM. Autosomal recessive polycystic kidney disease: the prototype of the hepato‐renal fibrocystic diseases. J Pediatr Genet. 2014;3:89‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scott DC, Rhee DY, Duda DM, et al. Two distinct types of E3 ligases work in unison to regulate substrate ubiquitylation. Cell. 2016;166:1198‐1214.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morreale FE, Walden H. Types of ubiquitin ligases. Cell. 2016;165:248‐248.e1. [DOI] [PubMed] [Google Scholar]
- 29. Horn‐Ghetko D, Schulman BA. New classes of E3 ligases illuminated by chemical probes. Curr Opin Struct Biol. 2022;73:102341. [DOI] [PubMed] [Google Scholar]
- 30. Rotin D, Staub O. Nedd4‐2 and the regulation of epithelial sodium transport. Front Physiol. 2012;3:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang C, Harafuji N, Cuevas S, et al. Human and Mouse FPC‐CTD Activate the MYC/Myc P1 Promoter: Implications for Renal Cystogenesis in ARPKD. Vol 2019. ASN Kidney Week; 2019. Accessed May 12, 2023. https://www.asn‐online.org/education/kidneyweek/2019/program‐abstract.aspx?controlId=3233770 [asn‐online.org] [Google Scholar]
- 32. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402‐408. [DOI] [PubMed] [Google Scholar]
- 33. Taulman PD, Haycraft CJ, Balkovetz DF, Yoder BK. Polaris, a protein involved in left‐right axis patterning, localizes to basal bodies and cilia. Mol Biol Cell. 2001;12:589‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nelson DS, Alvarez C, Gao YS, Garcia‐Mata R, Fialkowski E, Sztul E. The membrane transport factor TAP/p115 cycles between the Golgi and earlier secretory compartments and contains distinct domains required for its localization and function. J Cell Biol. 1998;143:319‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zuiderveld K. Contrast limited adaptive Histograph equalization. In Heckbert PS, ed. Graphic Gems IV. Academic Press Professional; 1994:474‐485. [Google Scholar]
- 36. Gonzalez RC, Woods RE, Eddins SL. Digital Image Processing Using MATLAB. Gatesmark Publishing; 2009. [Google Scholar]
- 37. Soille P. Morphological Image Analysis: Principles and Applications. Springer‐Verlag; 1999. [Google Scholar]
- 38. MathWorks T. MATLAB Function Reference Version 9.6. The MathWorks; 2019. [Google Scholar]
- 39. Kukic I, Rivera‐Molina F, Toomre D. The IN/OUT assay: a new tool to study ciliogenesis. Cilia. 2016;5:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dummer A, Poelma C, DeRuiter MC, Goumans MJ, Hierck BP. Measuring the primary cilium length: improved method for unbiased high‐throughput analysis. Cilia. 2016;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lazrak A, Jurkuvenaite A, Chen L, et al. Enhancement of alveolar epithelial sodium channel activity with decreased cystic fibrosis transmembrane conductance regulator expression in mouse lung. Am J Physiol Lung Cell Mol Physiol. 2011;301:L557‐L567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lazrak A, Samanta A, Matalon S. Biophysical properties and molecular characterization of amiloride‐sensitive sodium channels in A549 cells. Am J Physiol Lung Cell Mol Physiol. 2000;278:L848‐L857. [DOI] [PubMed] [Google Scholar]
- 43. Garcia‐Mata R, Bebok Z, Sorscher EJ, Sztul ES. Characterization and dynamics of aggresome formation by a cytosolic GFP‐chimera. J Cell Biol. 1999;146:1239‐1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rappoport JZ, Simon SM. A functional GFP fusion for imaging clathrin‐mediated endocytosis. Traffic. 2008;9:1250‐1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nesterov V, Dahlmann A, Krueger B, Bertog M, Loffing J, Korbmacher C. Aldosterone‐dependent and ‐independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. Am J Physiol Renal Physiol. 2012;303:F1289‐F1299. [DOI] [PubMed] [Google Scholar]
- 46. Wang S, Zhang J, Nauli SM, et al. Fibrocystin/polyductin, found in the same protein complex with polycystin‐2, regulates calcium responses in kidney epithelia. Mol Cell Biol. 2007;27:3241‐3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hiesberger T, Gourley E, Erickson A, et al. Proteolytic cleavage and nuclear translocation of fibrocystin is regulated by intracellular Ca2+ and activation of protein kinase C. J Biol Chem. 2006;281:34357‐34364. [DOI] [PubMed] [Google Scholar]
- 48. Kaimori JY, Nagasawa Y, Menezes LF, et al. Polyductin undergoes notch‐like processing and regulated release from primary cilia. Hum Mol Genet. 2007;16:942‐956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Demishtein A, Fraiberg M, Berko D, Tirosh B, Elazar Z, Navon A. SQSTM1/p62‐mediated autophagy compensates for loss of proteasome polyubiquitin recruiting capacity. Autophagy. 2017;13:1697‐1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pan B, Li J, Parajuli N, et al. The calcineurin‐TFEB‐p62 pathway mediates the activation of cardiac macroautophagy by proteasomal malfunction. Circ Res. 2020;127:502‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Su H, Wang X. Proteasome malfunction activates the PPP3/calcineurin‐TFEB‐SQSTM1/p62 pathway to induce macroautophagy in the heart. Autophagy. 2020;16:2114‐2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2023;24:167‐185. [DOI] [PubMed] [Google Scholar]
- 53. Pampliega O, Cuervo AM. Autophagy and primary cilia: dual interplay. Curr Opin Cell Biol. 2016;39:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pampliega O, Orhon I, Patel B, et al. Functional interaction between autophagy and ciliogenesis. Nature. 2013;502:194‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol. 2005;171:603‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bjorkoy G, Lamark T, Johansen T. p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy. 2006;2:138‐139. [DOI] [PubMed] [Google Scholar]
- 57. Lamark T, Svenning S, Johansen T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017;61:609‐624. [DOI] [PubMed] [Google Scholar]
- 58. Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279‐289. [DOI] [PubMed] [Google Scholar]
- 59. Mori Y, Mori T, Wakabayashi M, et al. Involvement of selective autophagy mediated by p62/SQSTM1 in KLHL3‐dependent WNK4 degradation. Biochem J. 2015;472:33‐41. [DOI] [PubMed] [Google Scholar]
- 60. Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131‐24145. [DOI] [PubMed] [Google Scholar]
- 61. Wurzer B, Zaffagnini G, Fracchiolla D, et al. Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. Elife. 2015;4:e08941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang Y, Argiles‐Castillo D, Kane EI, Zhou A, Spratt DE. HECT E3 ubiquitin ligases – emerging insights into their biological roles and disease relevance. J Cell Sci. 2020;133:jcs228072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dikic I. Proteasomal and autophagic degradation systems. Annu Rev Biochem. 2017;86:193‐224. [DOI] [PubMed] [Google Scholar]
- 64. Newton K, Matsumoto ML, Wertz IE, et al. Ubiquitin chain editing revealed by polyubiquitin linkage‐specific antibodies. Cell. 2008;134:668‐678. [DOI] [PubMed] [Google Scholar]
- 65. Dikic I, Schulman BA. An expanded lexicon for the ubiquitin code. Nat Rev Mol Cell Biol. 2023;24:273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Butterworth MB. Regulation of the epithelial sodium channel (ENaC) by membrane trafficking. Biochim Biophys Acta. 2010;1802:1166‐1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pratt JH. Central role for ENaC in development of hypertension. J Am Soc Nephrol. 2005;16:3154‐3159. [DOI] [PubMed] [Google Scholar]
- 68. Goto M, Hoxha N, Osman R, Dell KM. The renin‐angiotensin system and hypertension in autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2010;25:2449‐2457. [DOI] [PubMed] [Google Scholar]
- 69. Liu L, Duke BJ, Malik B, Yue Q, Eaton DC. Biphasic regulation of ENaC by TGF‐{alpha} and EGF in renal epithelial cells. Am J Physiol Renal Physiol. 2009;296:F1417‐F1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Malik B, Price SR, Mitch WE, Yue Q, Eaton DC. Regulation of epithelial sodium channels by the ubiquitin‐proteasome proteolytic pathway. Am J Physiol Renal Physiol. 2006;290:F1285‐F1294. [DOI] [PubMed] [Google Scholar]
- 71. Chan KW, Csanady L, Seto‐Young D, Nairn AC, Gadsby DC. Severed molecules functionally define the boundaries of the cystic fibrosis transmembrane conductance regulator's NH(2)‐terminal nucleotide binding domain. J Gen Physiol. 2000;116:163‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Han W, Rhee JS, Maximov A, et al. C‐terminal ECFP fusion impairs synaptotagmin 1 function: crowding out synaptotagmin 1. J Biol Chem. 2005;280:5089‐5100. [DOI] [PubMed] [Google Scholar]
- 73. Bartoszewski R, Rab A, Fu L, Bartoszewska S, Collawn J, Bebok Z. CFTR expression regulation by the unfolded protein response. Methods Enzymol. 2011;491:3‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bartoszewski R, Rab A, Twitty G, et al. The mechanism of cystic fibrosis transmembrane conductance regulator transcriptional repression during the unfolded protein response. J Biol Chem. 2008;283:12154‐12165. [DOI] [PubMed] [Google Scholar]
- 75. Bebok Z, Mazzochi C, King SA, Hong JS, Sorscher EJ. The mechanism underlying cystic fibrosis transmembrane conductance regulator transport from the endoplasmic reticulum to the proteasome includes Sec61beta and a cytosolic, deglycosylated intermediary. J Biol Chem. 1998;273:29873‐29878. [DOI] [PubMed] [Google Scholar]
- 76. Kerem E. Pharmacologic therapy for stop mutations: how much CFTR activity is enough? Curr Opin Pulm Med. 2004;10:547‐552. [DOI] [PubMed] [Google Scholar]
- 77. Mall MA, Mayer‐Hamblett N, Rowe SM. Cystic fibrosis: emergence of highly effective targeted therapeutics and potential clinical implications. Am J Respir Crit Care Med. 2020;201:1193‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Matos AM, Gomes‐Duarte A, Faria M, et al. Prolonged co‐treatment with HGF sustains epithelial integrity and improves pharmacological rescue of Phe508del‐CFTR. Sci Rep. 2018;8:13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Solomon GM, Marshall SG, Ramsey BW, Rowe SM. Breakthrough therapies: cystic fibrosis (CF) potentiators and correctors. Pediatr Pulmonol. 2015;50(Suppl 40):S3‐S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhang L, Button B, Gabriel SE, et al. CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS Biol. 2009;7:e1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mesgarzadeh JS, Buxbaum JN, Wiseman RL. Stress‐responsive regulation of extracellular proteostasis. J Cell Biol. 2022;221:e202112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pohl C, Dikic I. Cellular quality control by the ubiquitin‐proteasome system and autophagy. Science. 2019;366:818‐822. [DOI] [PubMed] [Google Scholar]
- 83. Zhao L, Zhao J, Zhong K, Tong A, Jia D. Targeted protein degradation: mechanisms, strategies and application. Signal Transduct Target Ther. 2022;7:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rab A, Bartoszewski R, Jurkuvenaite A, Wakefield J, Collawn JF, Bebok Z. Endoplasmic reticulum stress and the unfolded protein response regulate genomic cystic fibrosis transmembrane conductance regulator expression. Am J Physiol Cell Physiol. 2007;292:C756‐C766. [DOI] [PubMed] [Google Scholar]
- 85. Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524‐530. [DOI] [PubMed] [Google Scholar]
- 86. Li Y, Li S, Wu H. Ubiquitination‐proteasome system (UPS) and autophagy two main protein degradation machineries in response to cell stress. Cell. 2022;11:851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916‐919. [DOI] [PubMed] [Google Scholar]
- 88. Rocco MV, Neilson EG, Hoyer JR, Ziyadeh FN. Attenuated expression of epithelial cell adhesion molecules in murine polycystic kidney disease. Am J Physiol. 1992;262:F679‐F686. [DOI] [PubMed] [Google Scholar]
- 89. Ziegler WH, Soetje B, Marten LP, Wiese J, Burute M, Haffner D. Fibrocystin is essential to cellular control of adhesion and epithelial morphogenesis. Int J Mol Sci. 2020;21:5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Taub M, Laurie GW, Martin GR, Kleinman HK. Altered basement membrane protein biosynthesis by primary cultures of cpk/cpk mouse kidney. Kidney Int. 1990;37:1090‐1097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Data Availability Statement
The data (western blot images, densitometry results, RT‐PCR readings, raw confocal microscopy images) that support the findings of this study are available from the corresponding author, [ZsB], upon reasonable request. Data acquisition details are described in the Materials and Methods section.