

Abstract
Despite decades of research on new diffuse intrinsic pontine glioma (DIPG) treatments, little or no progress has been made on improving patient outcomes. In this work, we explored novel scaffold modifications of M4K2009, a 3,5-diphenylpyridine ALK2 inhibitor previously reported by our group. Here we disclose the design, synthesis, and evaluation of a first-in-class set of 5- to 7-membered ether-linked and 7-membered amine-linked constrained inhibitors of ALK2. This rigidification strategy led us to the discovery of the ether-linked inhibitors M4K2308 and M4K2281 and the amine-linked inhibitors M4K2304 and M4K2306, each with superior potency against ALK2. Notably, M4K2304 and M4K2306 exhibit exceptional selectivity for ALK2 over ALK5, surpassing the reference compound. Preliminary studies on their in vivo pharmacokinetics, including blood–brain barrier penetration, revealed that these constrained scaffolds have favorable exposure and do open a novel chemical space for further optimization and future evaluation in orthotopic models of DIPG.
Introduction
Pediatric central nervous system (CNS) tumors constitute a rare and aggressive group of heterogeneous diseases.1,2 Diffuse intrinsic pontine glioma (DIPG) is a grade IV tumor located in the pons region of the brainstem and is the most frequent brainstem cancer in children between 5 and 10 years of age, accounting for nearly 80% of pediatric brainstem tumors.2,3 While accounting for only 10% of all pediatric brain tumors, the prognosis for patients with DIPG is poor, presenting a median overall survival of 9–12 months from diagnosis and a median progression survival of 7 months.3,4
Despite decades of research on new DIPG treatments, little or no progress has been made on improving patient outcomes.3,5 Focal radiotherapy is the only treatment which offers temporary tumor growth control, although >90% of patients suffer a relapse and die from the disease within 3–7 months.3,4 Moreover, numerous clinical trials of chemotherapeutic agents have failed to show an improved survival benefit beyond the one achieved with standard radiation therapy.2−5
Novel improvements in the safety of brainstem biopsy procedures, in addition to detailed molecular sequencing profiles of the tumors, have allowed researchers to define the genomic landscape of DIPG and to identify potential biological targets for therapeutic intervention.4,6,7 Over the past decade, several research groups have identified gain-of-function mutations (R206H, R258G, G328 E/V/W, and G356D) in the ACVR1 gene in ∼33% of DIPG patients.4,6−9
ACVR1 encodes the activin receptor-like kinase-2 (ALK2), a bone-morphogenic protein (BMP) type I receptor, which is involved in the BMP/transforming growth factor β (TGFβ) signaling pathway. ACVR1 mutations confer an aberrant sensitivity to the ligand activin A, leading to increased pathway activation via the phosphorylation of downstream transcription factors SMAD1/5/8 by ALK2.4,10 This results in a significantly increased expression of target genes ID1–3, which have been associated with tumor growth, chemoresistance, and metastasis.4,11
The mechanism by which ALK2 contributes to DIPG pathogenesis has not yet been fully elucidated, and ACVR1 mutants alone have failed to explain the initiation of tumors in DIPG patients.9,12,13 However, a recent study by Carvalho and co-workers demonstrates that the treatment of immunocompromised mice bearing orthotopic xenografts of H3.3K27M, ACVR1R206H mutant HSJD-DIPG-007 cells with 25 mg/kg of LDN-214117 for 28 days results in an extended survival of the mutant murine strains.4 Their work suggests that DIPG cells are dependent on enhanced signaling by the mutant receptor. Consequently, the development of ALK2 inhibitors with enhanced potency, selectivity, and favorable pharmacokinetic (PK) properties may play a major role in the development of effective chemotherapies for DIPG patients.
In previous studies, we expanded the structure–activity relationship (SAR) of LDN-214117, leading to the discovery of M4K2009. This compound was found to be a highly potent and selective ALK2 inhibitor, presenting a suitable blood–brain barrier (BBB) penetration profile and robust in vivo PK properties.10
With the intent to further expand the chemical space around the 3,5-diphenylpyridine chemotype ALK2 inhibitors and potentially realize increased levels of selectivity over other ALK isoforms, we began exploring novel scaffold modifications of M4K2009, and here we report the design, synthesis, and evaluation of conformationally constrained derivatives. Our interest in pursuing cyclic analogues emerged from reports highlighting the rigidification of small molecules as a promising drug design strategy for conferring high levels of target affinity14−19 and selectivity,16−19 as well as optimal in vivo PK properties.16−19
To evaluate the impact of conformationally constrained ALK2 inhibitors, a small set of 5- to 7-membered ether-linked and 7-membered amine-linked analogues was prepared based on the cocrystal structure of M4K2009 with ALK2 (PDB code 6SZM).10 As in our previous studies, we conducted the primary assessment of compound potency and selectivity against ALK2, ALK4, and ALK5 using a radiometric in vitro kinase assay performed by Reaction Biology Corporation (RBC). Activity assays in HEK-293 cells were performed by the Structural Genomics Consortium (SGC) with a nanoBRET assay used to test the compounds against ALK2 and a dual luciferase assay (DLA) used to determine activity against ALK5. It has been documented that ALK5 inhibition includes adverse effects such as cardiotoxicity and gastrointestinal inflammation. Therefore, achieving selectivity for ALK2 over ALK5 in biochemical and cell-based assays was considered crucial to create a safety profile and reduce the risk of toxicity.10,20
Results and Discussion
Synthesis of Analogues
From our previous work on the SAR expansion of LDN-214117, we learned that translocation of the C-2 methyl group to the C-4 position of the pyridyl core resulted in enhanced potency and selectivity for ALK2 over ALK5 in both the biochemical and cell-based assays. Crystallographic studies of the inhibitor M4K2009 bound to ALK2 suggest that the presence of the sterically bulky C-4 position methyl substituent induces a conformational lock on the bioactive conformation of the ligand.10 The steric strain between the methyl group and the hydrogens of the adjacent phenyl rings causes the ligand to adopt a favorable nonplanar conformation, with dihedral angles of 90° between the pyridyl ring and the C-3 position 3,4,5-trimethoxyphenyl ring and 47° between the pyridyl ring and the C-5 position 4-piperazinophenyl ring.
Based on this structural information, we were confident that the conformational lock induced by the methyl group was responsible for the improved activity and selectivity of M4K2009.10,21 In order to reinforce this conformational bias, we decided to bridge the C-4 position of the pyridyl ring and the C-5 position 4-piperazinophenyl moiety with 5-, 6-, and 7-membered ring systems. It was anticipated that the reduced rotational freedom conferred to the ligands by this bridging would induce a tighter conformational lock of their bioactive conformations while preserving the same binding mode as M4K2009 and therefore conserving the main interactions with the residues of the ALK2 pocket. This would result in highly specific binding modes22 to the hinge region of ALK2. Moreover, this would also lead to expanded ligand occupancy within the binding pocket. We decided to use oxygen and nitrogen heteroatoms to synthesize the ring systems and modify their position within the linker to evaluate their effect on potency and selectivity for ALK2. By choosing to link these two moieties instead of the pyridyl ring and the 3,4,5-trimethoxyphenyl ring, we were able to develop more accessible synthetic approaches to generate the final compounds and avoid the synthetic challenges inherent to the generation of a penta-substituted phenyl ring. However, even with a focus on synthetic efficiency, the resulting structural complexity of the proposed analogues precluded the use of a common route for their synthesis.
The route to the furanyl-bridged compound M4K2286 (8) is shown in Scheme 1. Commercially available 3,5-dibromo-4-hydroxypyridine (1) and 3,4,5-trimethoxyphenylboronic acid (2) were coupled under Suzuki–Miyaura conditions to afford intermediate 3, followed by a second coupling with 4-chloro-2-fluorophenylboronic acid (4) to obtain intermediate 5. Step a produced a significant amount of the undesired dicoupled product (∼40%), contributing to a low reaction yield. The subsequent ring closure to obtain intermediate 6 was achieved by an intramolecular nucleophilic aromatic substitution (SNAr).23 Treatment of 6 with 1-methylpiperazine (7) under Buchwald–Hartwig amination reaction conditions afforded the final compound, M4K2286 (8).
Scheme 1. Synthesis of Compound M4K2286 (8).
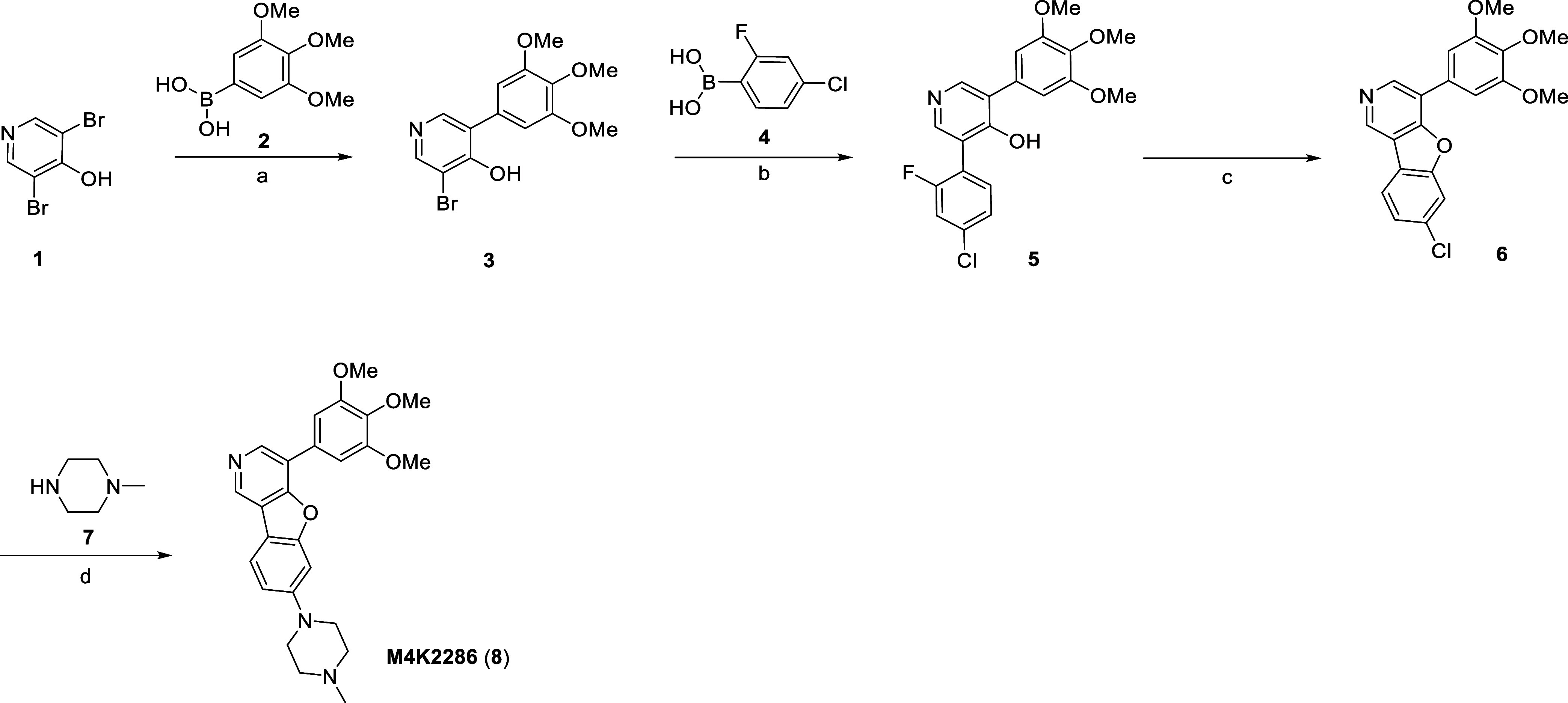
Reagents and conditions: (a) Pd(dppf)Cl2, K2CO3, 1,4-dioxane/H2O, 100 °C, 3 h, 23%; (b) Pd(dppf)Cl2, K2CO3, 1,4-dioxane/H2O, 100 °C, 3 h, 49%; (c) K2CO3, 100 °C, NMP, 3 h, 49%; (d) RuPhos Pd G3, NaOtBu, 1,4-dioxane, 100 °C, 16 h, 51%.
A slightly modified route, illustrated in Scheme 2, was employed for the preparation of the pyranyl-bridged inhibitor M4K2277 (14). The sequence of the cross-coupling reactions had to be modified to avoid a low reaction yield on the cyclization step. Commercially available 3,5-dibromoisonicotinaldehyde (9) and 4 were coupled under Suzuki–Miyaura conditions to yield intermediate 10, followed by NaBH4 reduction of the aldehyde to afford intermediate 11. Ring closure to access intermediate 12 was again achieved using an intramolecular SNAr reaction. Transformation to the final product M4K2277 (14) was carried out by coupling 12 and 2 under Suzuki–Miyaura conditions, followed by a Buchwald–Hartwig amination reaction with 7 to afford the final compound.
Scheme 2. Synthesis of Compound M4K2277 (14).
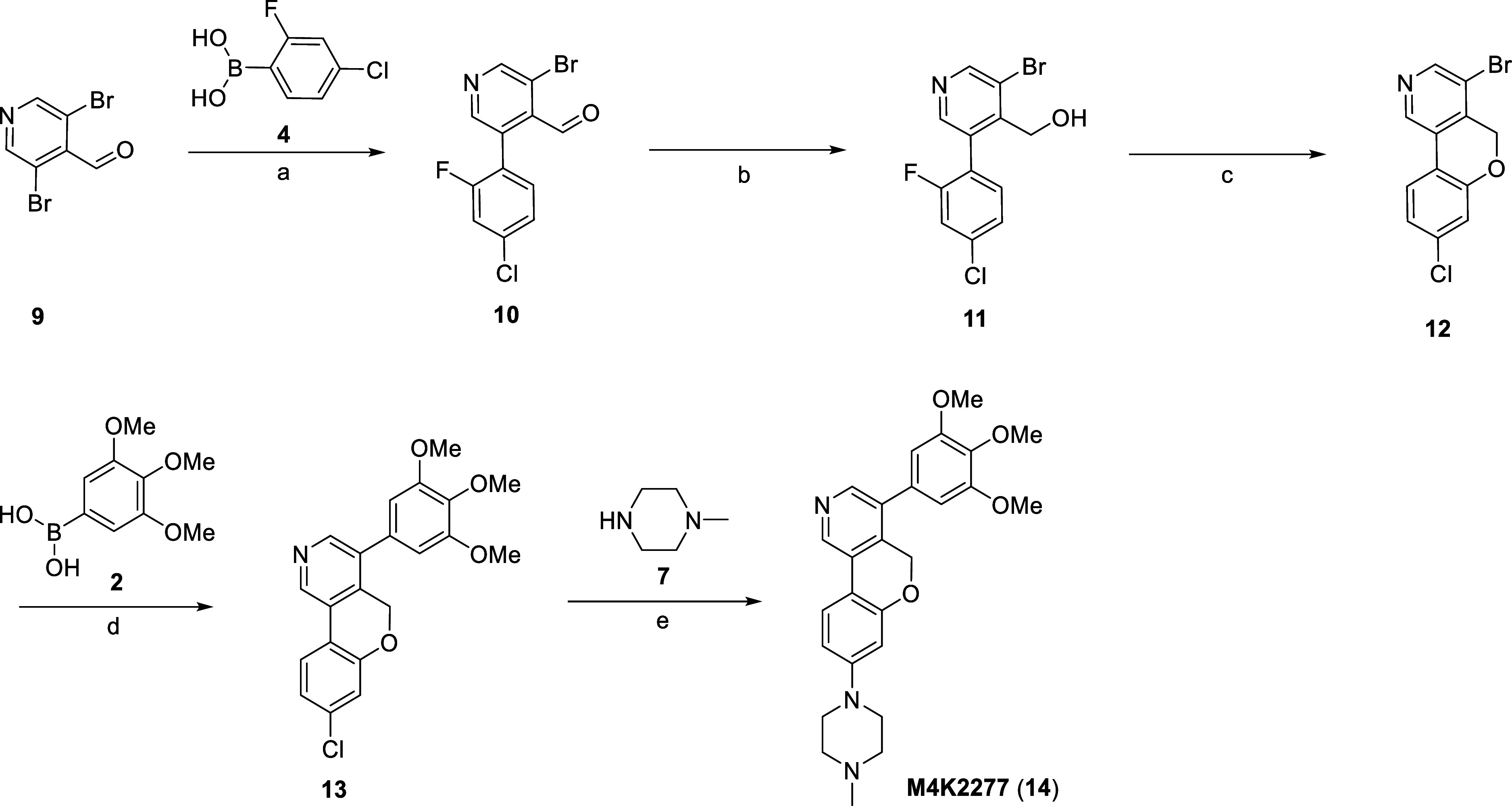
Reagents and conditions: (a) Pd(dppf)Cl2, Na2CO3, 1,4-dioxane/H2O, 100 °C, 16 h, 36%; (b) NaBH4, THF, 0 °C to rt, 10 min, 62%; (c) NaH, THF, 0 °C to rt, 10 min, 99%; (d) Pd(dppf)Cl2, K2CO3, 1,4-dioxane/H2O, 100 °C, 3 h, 97%; (e) RuPhos Pd G3, NaOtBu, 1,4-dioxane, 100 °C, 16 h, 33%.
Formation of 7-member bridged compounds M4K2207 (21) and M4K2303 (22) necessitated a further departure from the previously used synthetic routes, ultimately exploiting an alternate intramolecular cyclization process as shown in Scheme 3. Commercially available 9 and 2 were coupled under Suzuki–Miyaura conditions to afford intermediate 15, followed by NaBH4 reduction of the aldehyde to afford intermediate 16. Benzyl alcohol 16 was then treated with 2-bromomethyl-4-chlorophenylboronic acid pinacol ester (17) under Williamson conditions to access the cyclization precursor 18. Intermediate 18 was subsequently cyclized under Suzuki–Miyaura conditions to obtain penultimate intermediate 19. High dilution conditions were employed in step d to minimize any intermolecular coupling. Intermediate 19 was treated with 1-Boc-piperazine (20) under Buchwald–Hartwig amination conditions, followed by acid-mediated Boc deprotection to provide M4K2207 (21). The final product M4K2303 (22) was similarly prepared via Buchwald–Hartwig amination with 7.
Scheme 3. Synthesis of Compounds M4K2207 (21) and M4K2303 (22).
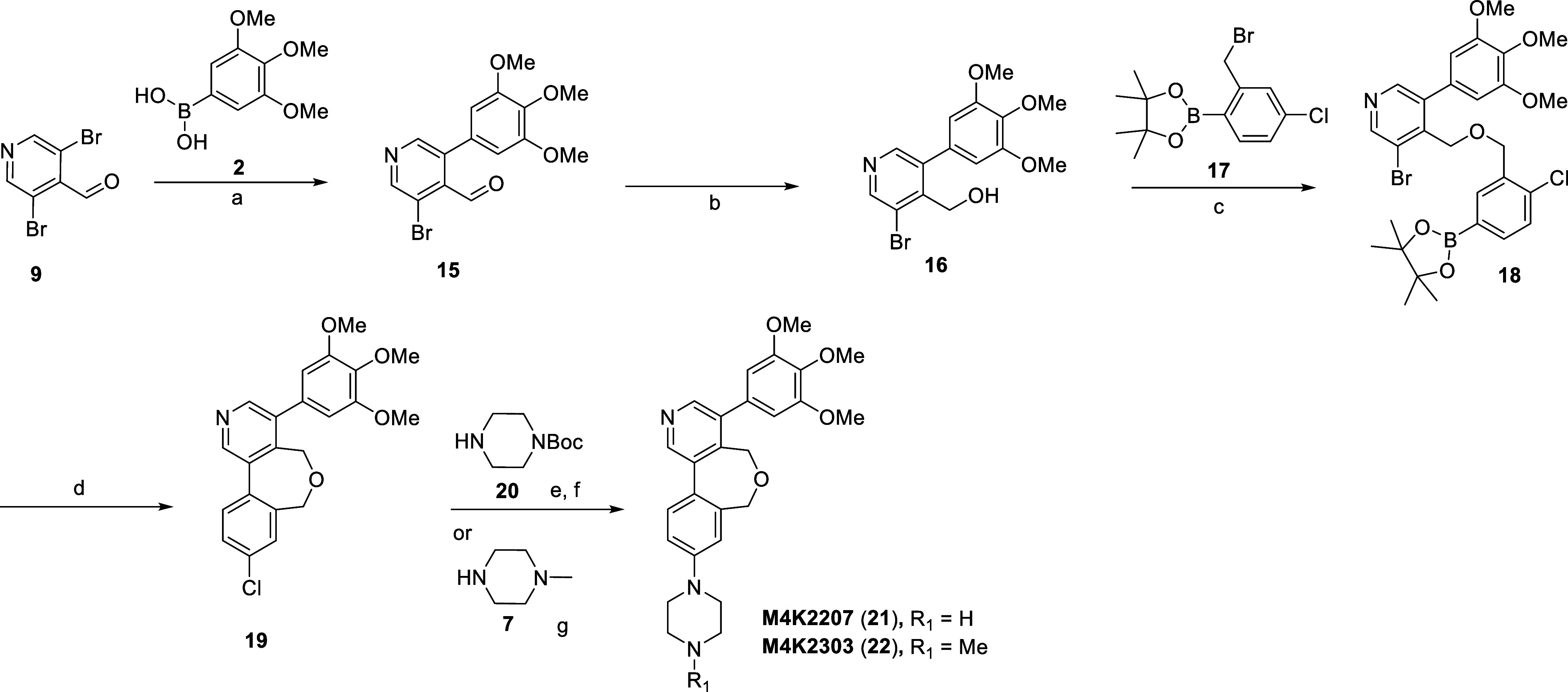
Reagents and conditions: (a) Pd(dppf)Cl2, K2CO3, 1,4-dioxane/H2O, 100 °C, 24 h, 29%; (b) NaBH4, THF/MeOH, 0 °C to rt, 10 min, 62%; (c) NaH, DMF, 0 °C to rt, 1 h, 49%; (d) Pd(dppf)Cl2, K2CO3, 1,4-dioxane/H2O, 100 °C, 2 h, 82%; (e) RuPhos Pd G3, NaOtBu, 1,4-dioxane, 100 °C, 16 h; (f) TFA, DCM, rt, 1 h, 51% over two steps; (g) RuPhos Pd G3, NaOtBu, 1,4-dioxane, 100 °C, 16 h, 52%.
An alternative route, illustrated in Scheme 4 and featuring an additional homologation step, was employed for the preparation of M4K2308 (32) and M4K2281 (33). The ring closure approach used for the preparation of 32 and 33 had to be completely reformulated from previous synthetic strategies due to the formation of a styrene when using SNAr reaction conditions. Commercially available 9 and 4-chloro-2-methoxyphenylboronic acid (23) were coupled under Suzuki–Miyaura conditions to afford intermediate 24. Aldehyde 24 was treated with (methoxymethyl)triphenylphosphonium chloride (25) under Wittig conditions to generate intermediate 26 as a mixture of the (E)- and (Z)-isomers. Vinyl ether hydrolysis gave rise to intermediate 27, followed by NaBH4 reduction of the aldehyde to afford alcohol 28. Intermediate 28 underwent BBr3-facilitated demethylation to provide the cyclization precursor 29. Ring closure to form tricycle 30 was readily achieved by an intramolecular Mitsunobu reaction, with subsequent coupling of 2 under Suzuki–Miyaura conditions producing intermediate 31. Treatment of intermediate 31 with 20 under Buchwald–Hartwig amination conditions was followed by acid-mediated Boc deprotection to provide M4K2308 (32). The final product M4K2281 (33) was also prepared via Buchwald–Hartwig amination with 7.
Scheme 4. Synthesis of Compounds M4K2308 (32) and M4K2281 (33).
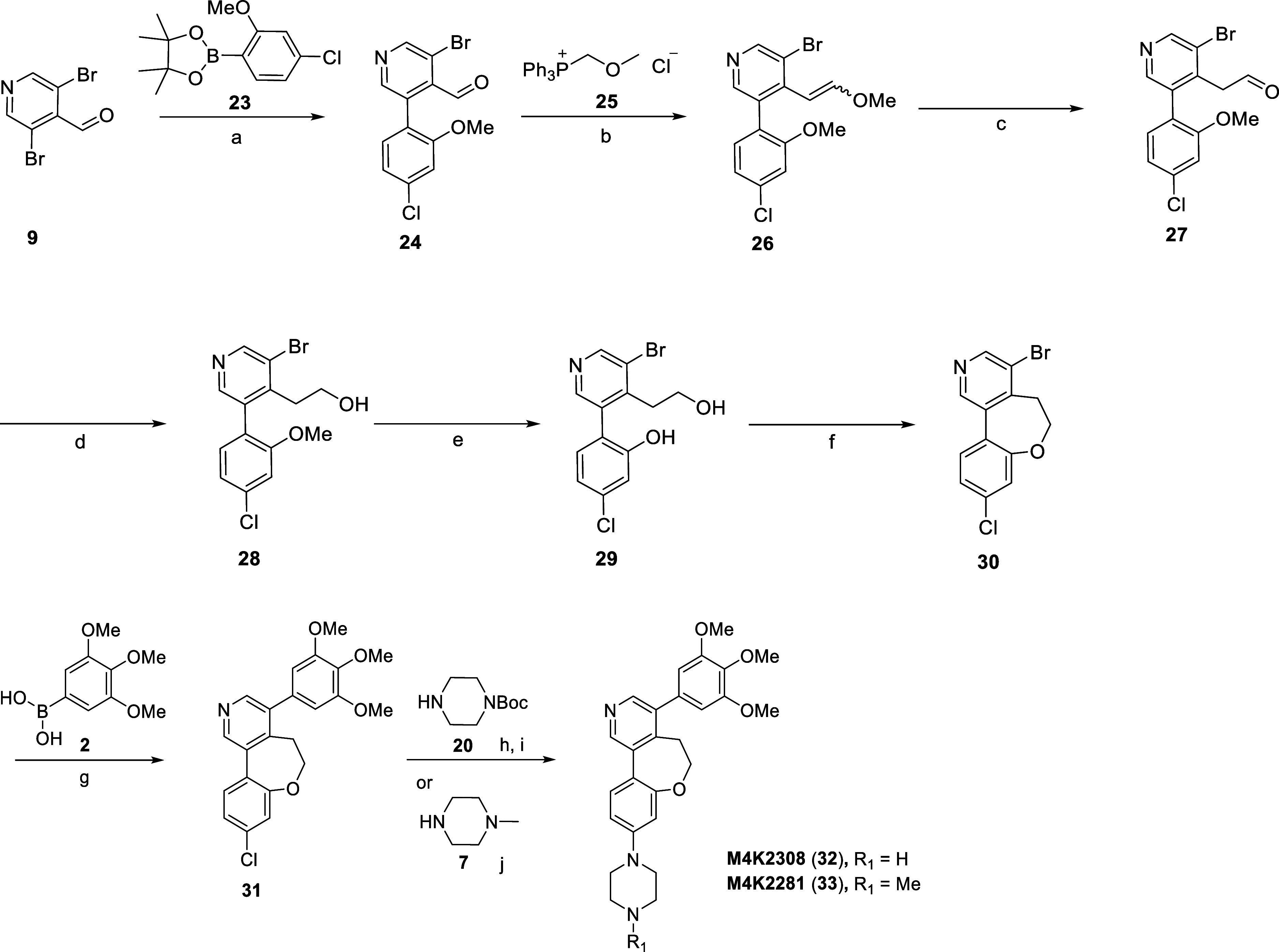
Reagents and conditions: (a) Pd(dppf)Cl2, K2CO3, 1,4-dioxane/H2O, 100 °C, 16 h, 35%; (b) LiHDMS, THF, 0 °C to rt, 1 h, 96%; (c) HCl, rt, 1 h, 85%; (d) NaBH4, THF/MeOH, 0 °C, 2 h, 77%; (e) BBr3, DCM, −78 °C to rt, 24 h, 62%; (f) triphenylphosphine, diisopropyl azodicarboxylate, THF, 0 °C to rt, 1.5 h, 68%; (g) Pd(dppf)Cl2, K2CO3, 1,4-dioxane/H2O, 100 °C, 2 h, 73%; (h) RuPhos Pd G3, NaOtBu, 1,4-dioxane, 100 °C, 16 h; (i) TFA, DCM, rt, 1 h, 29% over two steps; (j) RuPhos Pd G3, NaOtBu, 1,4-dioxane, 100 °C, 16 h, 49%.
The route to compounds M4K2304 (41) and M4K2306 (42), both featuring a 7-membered amine-containing bridge, is shown in Scheme 5. The initial reaction of 2-bromomethyl-4-chlorophenylboronic acid pinacol ester (34) and methylamine (35) through an SN2 reaction generated intermediate 36. Secondary amine 36 was treated with 9 under reductive amination conditions to provide a mixture of tertiary amine intermediates 37 and 38. A low yield is reported for step b, a function of the boronic ester hydrolysis complicating product elution during the chromatographic purification process. The mixture of these two intermediates was subjected to Suzuki–Miyaura conditions to obtain cyclized intermediate 39. A subsequent Suzuki–Miyaura coupling between intermediate 39 and 2 provided tetracycle 40. Intermediate 40 was treated with 20 under Buchwald–Hartwig amination conditions, followed by acid-mediated Boc deprotection to provide M4K2304 (41). The final product M4K2306 (42) was again prepared via Buchwald–Hartwig amination with 7.
Scheme 5. Synthesis of Compounds M4K2304 (41) and M4K2306 (42).
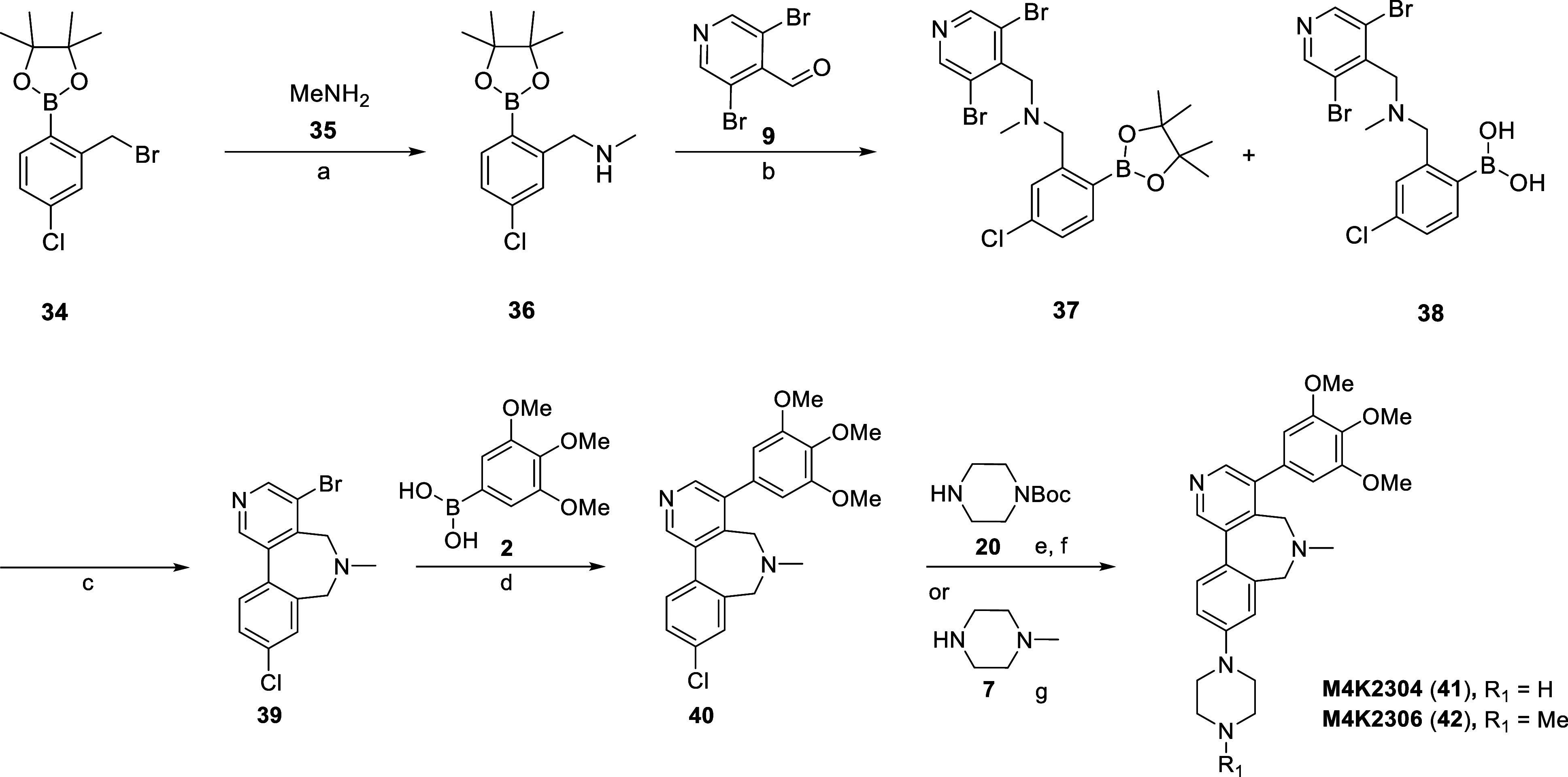
Reagents and conditions: (a) Pd(dppf)Cl2, K2CO3, 1,4-dioxane/H2O, 100 °C, 3 h, 23%; (b) Pd(dppf)Cl2, K2CO3, 1,4-dioxane/H2O, 100 °C, 3 h, 49%; (c) K2CO3, 100 °C, NMP, 3 h, 49%; (d) RuPhos Pd G3, NaOtBu, 1,4-dioxane, 100 °C, 16 h, 51%.
Structure Activity Relationship Studies
The 7-membered ring inhibitors M4K2207, M4K2303, M4K2308, M4K2281, M4K2304, and M4K2306, whether containing an ether or amine linkage, revealed impressive levels of potency against ALK2, with values below 10 nM and 20 nM in the biochemical and cellular assays, respectively (Table 1). Gratifyingly, all six compounds exhibited higher potencies than the lead compound M4K2009, suggesting that the 7-membered ring system induces a specific and favorable conformational lock on the bioactive conformation of these ligands. Moreover, compounds M4K2207, M4K2303, M4K2308, M4K2281, and M4K2304 are the most potent inhibitors of ALK2 reported to date (IC50 ≤ 3 nM).
Table 1. Inhibitory and Off-Target Activities of Novel M4K Series Compounds.
| Compound | ALK2a IC50 (nM) | ALK5a IC50 (nM) | Fold Selectivity | nanoBRET ALK2a IC50 (nM) | DLA ALK5a IC50 (nM) | Cell-based Fold Selectivity |
|---|---|---|---|---|---|---|
| M4K2009 | 13a | 2427a | 187a | 26 | 3297 | 126 |
| M4K2286 (8) | 609 | >10,000 | 7691 | |||
| M4K2277 (14) | 13 | 1541 | 119 | 45 | 1738 | 39 |
| M4K2207 (21) | 2 | 104 | 52 | 5 | 224 | 45 |
| M4K2303 (22) | 3 | 187 | 64 | 7 | 347 | 50 |
| M4K2308 (32) | 2 | 224 | 112 | 6 | 471 | 79 |
| M4K2281 (33) | 2 | 350 | 175 | 15 | 1193 | 80 |
| M4K2304 (41) | 3 | 2500 | 883 | 11 | 1690 | 154 |
| M4K2306 (42) | 7 | 3915 | 559 | 18 | 3964 | 220 |
Literature values.10
Average of triplicate measurements.
Inhibitor M4K2286 proved to be the least potent compound in the series, likely a consequence of the molecule’s planarity and attendant, unfavorable binding mode. Compound M4K2277, the 6-membered analogue, was found to be a less potent inhibitor of ALK2 in the cellular assay but comparable in biochemical activity to M4K2009.
We were particularly pleased to find that the 7-membered amine-linked inhibitors M4K2304 and M4K2306 also showed high levels of selectivity for ALK2 over ALK5, exceeding levels exhibited by M4K2009 in both the biochemical and cell-based assays.
Unsurprisingly, methylating the piperazine of the inhibitors M4K2207, M4K2308, and M4K2304 resulted in compounds with similar activity and selectivity profiles. This was encouraging, suggesting that this solvent-exposed position could be exploited for the potential introduction of moieties capable of influencing aggregate PK properties.
Structural Basis for ALK2 Inhibition and Selectivity
Crystallographic efforts yielded the cocrystal structures of the amine-linked inhibitor M4K2304 (PDB 8C7W) and the ether-linked inhibitor M4K2308 (PDB 8C7Z) in the ATP pocket of the ALK2 Ser/Thr kinase domain.
As expected, the X-ray structures showed that both 7-membered analogues adopt the same binding mode as the reference ligand M4K2009 (Figures 1 and 2). Consequently, the key ATP-mimetic hydrogen bond between the pyridine ring and the backbone amide of H286 in the hinge region is preserved with both constrained analogues. The water-mediated hydrogen bond between the meta-methoxy group of the 3,4,5-trimethoxyphenyl moiety and the K235 residue is also conserved with both structures. As shown in Figure 2, all exit vectors and moieties from M4K2009 and M4K2304 are completely superimposed in the 3D space.
Figure 1.

2D ligand interaction plots of (A) M4K2009 (PDB 6SZM), (B) M4K2304 (PDB 8C7W), and (C) M4K2308 (PDB 8C7Z) in complex with the ATP pocket of the ALK2 Ser/Thr kinase domain. The main protein–ligand interactions are represented. All protein residues within 5.0 Å from the ligands are displayed.
Figure 2.
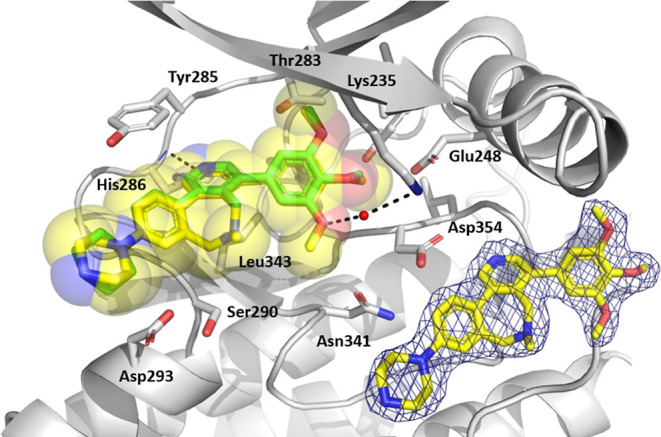
Cocrystal structure of the amine-linked inhibitor M4K2304 with ALK2 (PDB 8C7W). The ATP pocket of the ALK2 is shown as cartoon representation in gray. Compound M4K2304 is shown as yellow sticks. Hydrogen bonds (black dashed lines) are established with H286 and K235. Reference compound M4K2009 (PDB 6SZM) is shown as green sticks. The electron density of the ligand is displayed as a blue mesh (countered at 1.0 sigma).
The fact that both constrained inhibitors, whether containing an ether or amine linkage, show the same binding mode as the reference ligand suggests that their gain in potency is indeed the result of the 7-membered ring system, which induces a highly specific and favorable conformational lock of their bioactive conformations when bound to the ALK2 pocket (Figure 2). Moreover, the reduction of the number of degrees of freedom through this bridging could also lead to favorable entropic contributions to the ligands, which can have a positive impact on their potency values.
Selectivity among highly homologous proteins is often difficult to rationalize, even with high-resolution structural data and subsequent molecular modeling studies. In this work, the potency values against ALK2 of all 7-membered analogues (M4K2207, M4K2303, M4K2308, M4K2281, M4K2304, and M4K2306) are almost identical. However, they show significant differences in their potency values against ALK5 (Table 1). As we do not have a cocrystal structure of ALK5, we proceeded to perform molecular modeling studies on the differential residues within the binding pockets of ALK2 and ALK5. This allowed us to rationalize some general selectivity features of our compound set. Most of the first-shell residues around the ligands are identical in both kinases, except for T283S, L297R, and V214I in ALK5. With the T283S modification in ALK5, a slightly larger and deeper cavity is available. Our studies suggest that the 7-membered amine-linked inhibitors M4K2304 and M4K2306 would not completely occupy the ALK5 pocket, leading to fewer van der Waals interactions (e.g., the optimal distance between the carbon of the meta-methoxy group of the 3,4,5-trimethoxyphenyl moiety and the terminal methyl of the T283 in ALK2 would be lost in ALK5). With the L297R modification, the solvent-exposed basic amine of our ligands would be located near the basic side chain of the arginine residue in ALK5, resulting in suboptimal interactions. Finally, according to Maestro and SeeSAR evaluations, the V214I modification is predicted to have a minor impact on the selectivity of the compounds. The isoleucine rotamer of the cocrystal structure of ALK5 in complex with staurosporine (PDB 5E8W) does not create unfavorable clashes with our series in the in silico engineered structure of M4K2304 in complex with ALK2 (PDB 8C7W). It is important to mention, however, that none of these arguments (either from structural data or molecular modeling studies) are sufficient to explain the differences in selectivity of the ether-linked inhibitors (M4K2207, M4K2303, M4K2308, M4K2281).
Kinome Selectivity
We decided to further investigate the off–target activity of M4K2304 in the Eurofins in vitro Full Kinase Panel (422-member kinase set) KinaseProfiler, finding that at 1 μM only 3% of the kinases were inhibited by more than 50% (see Supporting Information, Table S1). Compared to M4K2009, inhibitor M4K2304 exhibited enhanced inhibition of ALK2 and a slight improvement in selectivity over ALK1 and ALK6. Although M4K2304 showed higher inhibition of ALK4 in comparison to M4K2009, biochemical data revealed a substantial 393-fold selectivity over this kinase (ALK4 IC50 = 1180 nM), easily meeting our desired selectivity criteria (a 30-fold selectivity window over subfamily proteins).
Outside of the ALK family, M4K2304 was found to have reasonable kinome-wide selectivity. The kinases RIPK2, DDR1, BRK, EGFR-L858R, EGFR-L861Q, TNIK, Lyn, and CK1 were the most sensitive to inhibition. DDR1 and TNIK showed almost complete inhibition when treated with M4K2009 at 1 μM but showed significant residual activity when treated with M4K2304. The favorable kinome-wide selectivity suggests that the rigidification of the ligand prevents any conformational rearrangement within the binding site of other protein kinases.
In our previous study, we screened M4K2009 against available ALK2 mutants (G328 V, R206H, and R258G) in both the biochemical and cell-based assays. The potency values against the mutants were comparable to ones obtained when screening the compound against wild-type (WT) ALK2.10 This was expected because the mutations in both the GS and Ser/Thr kinase domains of ALK2 have no significant effects on the way ATP and type I kinase inhibitors interact with the hinge region of the kinase.24 The lack of mutant protein availability and assay expense, in addition to our previous experience with M4K2009, led us to screen our set of inhibitors exclusively against WT ALK2.
In Vitro ADME
Phase I (CYP enzymes only) in vitro metabolic stability of our inhibitor set was determined in both mouse and human liver microsomes (MLM and HLM), while permeability was evaluated using a Caco-2 assay (Table 2). We were pleased to find that our most potent and selective inhibitors, M4K2304 and M4K2306, exhibited moderate (20–70% remaining) to high stability (>70% remaining) in both the MLM and HLM assays, with compound M4K2304 demonstrating the highest overall degree of stability (>70% remaining) after 60 min incubation at 37 °C. While M4K2207 and M4K2308 also exhibited high MLM and HLM stability, their 4-methylpiperazine counterparts, M4K2303 and M4K2281, as well as M4K2286 and M4K2277, were considerably less stable. Taken together, these results suggest that the 4-methylpiperazine moiety is labile under the prevailing conditions, generating the demethylated metabolite.
Table 2. In Vitro ADME Parameters for Novel M4K Series Compounds.
| Compound | Microsomal Stability Assayb |
Caco-2 Permeability
Assayc,d |
c Log D @ pH 7 | |||
|---|---|---|---|---|---|---|
| MLM (%) | HLM (%) | Papp_AB (10–6 cm/s) | Papp_BA (10–6 cm/s) | Efflux Ratio | ||
| M4K2009 | 65a | 71a | 5.4a | 1.5a | 0.3a | 2.9 |
| M4K2286 (8) | 9.5 | 37.4 | 4.0 | 2.5 | 0.6 | 3.7 |
| M4K2277 (14) | 0.9 | 34.8 | 8.3 | 9.0 | 1.1 | 3.1 |
| M4K2207 (21) | 75.7 | 62.3 | 4.2 | 9.4 | 2.2 | 2.0 |
| M4K2303 (22) | 4.3 | 29.7 | 16.5 | 13.6 | 0.8 | 2.9 |
| M4K2308 (32) | 70.9 | 66.5 | 5.3 | 11.6 | 2.2 | 2.5 |
| M4K2281 (33) | 10.3 | 21.2 | 13.9 | 11.5 | 0.8 | 3.4 |
| M4K2304 (41) | 98.6 | 73.2 | 0.8e,f | 4.2e | 5.0 | 2.3 |
| M4K2306 (42) | 65.0 | 58.3 | 1.5e,f | 2.5e | 1.6 | 3.2 |
Literature values.10
Stability classification: stable (>70%), moderately stable (20–70%), unstable (<20%) compound remaining.
Permeability classification: low (Papp < 1), medium (Papp 1–3) or high (Papp > 3) × 10–6 cm/s.
Efflux ratio (BA/AB) classification: negative (ratio <2), minor/moderate (ratio 2–7) or high (ratio >7).
Average of duplicate measurements.
Poor post-assay recovery in A to B chambers possibly due to low aqueous solubility, high lipophilicity, nonspecific binding to plastics or cells, and/or poor intracellular stability of the test article.
Caco-2 permeability values were high for all compounds (Papp_AB ≥ 3 × 10–6 cm/s) except M4K2304 and M4K2306, which showed only low and moderate permeability, respectively. Interestingly, both compounds also exhibited poor post-assay recovery (<15%). This suggests that adding a second basic amine moiety to the compounds could result in significantly lower permeability. Compounds M4K2286, M4K2277, M4K2303, and M4K2281 appeared to not be recognized by efflux proteins, while all other compounds suffered from only minor to moderate efflux. As expected, the 4-methylpiperazine moiety of M4K2303 and M4K2281 served to increase the Papp_AB value and decrease the efflux ratio of the inhibitors significantly. Although not as permeable, unmethylated analogues demonstrated permeability levels suitable for in vivo PK studies.
In Vivo PK Exposure in Mouse Model
Given the expense of obtaining a full PK profile, we decided to first evaluate the exposure levels of the 7-membered ether-linked inhibitors M4K2308 and M4K2281 and the 7-membered amine-linked inhibitors M4K2304 and M4K2306 in our internal rapid PK mouse model that was run by evaluating plasma concentrations of drugs at a limited number of time points (Table 3). The inhibitors were administered orally to NOD-SCID male mice as a 25 mg/kg dose formulation of 5% dimethyl sulfoxide (DMSO), 47.5% poly(ethylene glycol) (PEG), and 47.5% deionized water with 10% Tween80. Afterward, plasma concentration of the ligands was determined at five time points. We also proceeded to measure the plasma concentration of the demethylated metabolites of M4K2281 and M4K2306 to gain a better understanding of their ADME (absorption, distribution, metabolism, and excretion) profiles.
Table 3. In Vivo PK Exposure of M4K2304, M4K2306, M4K2308, and M4K2281.
| Compound | C @ 0.5 h (nM) | C @ 1 h (nM) | C @ 2 h (nM) | C @ 6 h (nM) | C @ 24 h (nM) |
|---|---|---|---|---|---|
| M4K2009 | 3021 | 4276 | 7489 | 13,432 | 1542 |
| M4K2308 (32) | 729 | 1069 | 1117 | 1890 | 63 |
| M4K2281 (33) | 3647 | 5053 | 3533 | 2233 | 0 |
| M4K2304 (41) | 323 | 466 | 523 | 1260 | 336 |
| M4K2306 (42) | 134 | 254 | 277 | 300 | 17 |
As expected, M4K2304 and M4K2306 exhibited significantly lower exposure compared to M4K2009 across the five time points. M4K2304 showed an apparently 2-fold higher exposure in the first three time points in contrast with its methylated counterpart, M4K2306. This is most likely the result of M4K2306 being metabolized into its demethylated metabolite (M4K2306 to M4K2304). M4K2304 was observed to be present at half (C2h = 241 nM) of the concentration of the inhibitor M4K2306, showing comparable exposure levels to M4K2304.
Although the ether-linked analogue M4K2308 showed comparable permeability values (Papp_AB) to M4K2009, it exhibited significantly lower exposure than the reference compound, possibly a function of M4K2009’s minimal efflux (Table 2). Nonetheless, M4K2308 showed a 2-fold higher exposure at the first three time points when compared to the amine-linked analogues. Finally, we were pleased to find that compound M4K2281 showed comparable exposure levels to M4K2009 at 30 and 60 min. This was expected because of its high in vitro permeability values and its negative efflux ratio (Table 2). The concentration of the ligand likely begins to decrease after 2 h of administration because of the rapid formation of its demethylated counterpart (M4K2281 to M4K2308). At 6 h after administration, we found that the plasma concentration of the metabolite M4K2308 was 4177 nM. This is encouraging from the standpoint of predicting potential in vivo efficacy, since both M4K2308 and M4K2281 show impressive levels of potency against ALK2 in both the biochemical and cellular assays (Table 1).
These results, in addition to the in vitro ADME profiles of these compounds (Table 2), suggest that the basic amine in the linker decreases the permeability of the inhibitors, a function of having two charged amines (protonated at physiological pH) as part of their structure. Additionally, we were able to confirm that the solvent-exposed 4-methylpiperazine moiety is labile under the prevailing conditions, generating the demethylated metabolites. This explains the significant differences in in vitro metabolic stability between the methylated inhibitors (M4K2303, M4K2281, and M4K2306) and their demethylated counterparts (M4K2207, M4K2308, and M4K2304). These results confirm that the solvent-exposed position is capable of influencing aggregate PK properties.
In Vivo BBB Penetration in Mouse Model
We decided to further investigate the BBB penetration of compounds M4K2281 and M4K2306 in the same strain of mice (Table 4). Both compounds were administered orally in a 10 mg/kg dose formulation. Subsequently, we measured the plasma and brain concentrations of the ligands and their corresponding demethylated metabolites (M4K2308 and M4K2304, respectively) at 2 and 4 h intervals.
Table 4. In Vivo BBB Penetration of M4K2281 and M4K2306.
| Compound | Cplasma @ 2 h (nM) | Cbrain @ 2 h (nM) | Cplasma @ 4 h (nM) | Cbrain @ 4 h (nM) | Cbrain/Cplasma @ 4 h |
|---|---|---|---|---|---|
| M4K2009 | 5250 | 2321 | 2683 | 888 | 0.3 |
| M4K2281 (33) | 980 | 2409 | 393 | 1473 | 3.7 |
| M4K2306 (42) | 75a | 4133a | 43a | 3252a | 75.6a |
Average of duplicate measurements (see Supporting Information, Table S4).
Compared to M4K2009, inhibitors M4K2281 and M4K2306 exhibited moderate to high brain-to-plasma ratios (Kp = Cbrain/Cplasma, or B/P) of 3.7 and 75.6, respectively, at 4 h. This indicates a significant increase in the concentration of the compounds crossing the BBB into the brain.
The differences in the physicochemical properties between these constrained analogues and the lead compound provide insights into a possible rationale for their enhanced B/P ratios (Table 5). M4K2281 and M4K2306 show the complete absence of H-bond donors, a feature achieved by the methylation of the solvent-exposed piperazine moiety. This modification was previously demonstrated in our in vitro ADME and in vivo PK experiments to increase the compounds’ permeability. This suggests that the absence of an H-bond donor may also significantly enhance BBB penetration. Although both analogues possess one additional H-bond acceptor due to the presence of oxygen and nitrogen heteroatoms within the linker, their total number of H-bonds remains below 8. This aligns with the criteria set for the target product profile.
Table 5. Physicochemical Properties of M4K2009, M4K2306, and M4K2281.

Another potential contributing factor to the enhanced BBB penetration of the constrained analogues is molecular flexibility, measured by the number of rotatable bonds. Drugs targeting the CNS typically have fewer rotatable bonds compared to other drugs, with most commercial CNS drugs possessing five or fewer rotatable bonds.25,26 By connecting the C-4 position of the pyridyl ring to the C-5 position 4-piperazinophenyl moiety via the 7-membered ring systems in both analogues, the number of rotatable bonds is reduced to five. Additionally, this structural modification may also hinder the free rotation of the C-3 position 3,4,5-trimethoxyphenyl ring due to steric strains between the hydrogens of the CH2 of the 7-membered ring systems and those of the trimethoxyphenyl ring moiety. This suggests that making the 3,5-diarylpyridine chemotype more rigid could enhance BBB permeability.
While the points mentioned above provide insight into the improved brain-to-plasma ratios of the constrained analogues compared to M4K2009, the stark B/P ratio disparity between M4K2281 and M4K2306 cannot be fully explained by only comparing their physicochemical properties. This is because the molecular weight, cLogP, pKa, and tPSA of both ligands do not display any significant variation that would explain the notable difference in their BBB permeability. Additional studies are required to determine whether the exceptional B/P ratio of M4K2306 is a consequence of the compound being actively transported into the brain by influx transporters.27
We also identified the demethylated counterparts of both M4K2281 and M4K2306 (M4K2308 and M4K2304, respectively) in the brain. At 4 h after administration, the concentration of M4K2308 in the brain was 857 nM, while that of M4K2304 was 728 nM. It remains unclear whether this is a result of direct brain metabolism, the permeation of the demethylated compounds into the brain, or both. However, this finding is promising, as both demethylated compounds have demonstrated significant potency against ALK2 in both biochemical and cellular assays.
Although the observed brain-to-plasma ratios of M4K2281 and M4K2306 show promising results and offer an initial insight into the concentration of these analogues entering the brain, it will be important to determine their unbound brain-to-plasma ratio (Kp,uu) to better understand their CNS exposure and to establish robust PK/PD correlations.27,28
Conclusions
The pathogenesis of DIPG has not yet been fully elucidated, making it one of the most challenging tumors to treat. Although the ACVR1 mutants alone have failed to explain the initiation of tumors in DIPG patients, the development of ALK2 inhibitors remains the most potentially relevant clinical strategy for developing effective treatments for this aggressive glioma.
In this work, we reported the design, synthesis, and evaluation of conformationally constrained inhibitors of ALK2. We explored novel scaffold modifications using M4K2009 as the lead compound to optimize potency against ALK2 and increase selectivity over important off-targets such as ALK5. A strategy of rigidification of the 3,5-diarylpyridine chemotype through a 7-membered ring system led us to generate the compounds M4K2207, M4K2303, M4K2308, M4K2281, and M4K2304, which are the most potent inhibitors of ALK2 reported to date (IC50 ≤ 3 nM). Analysis of the cocrystal structures of M4K2308 and M4K2304 with ALK2 showed that the rigidification of the ligands using a 7-membered ring system induces a favorable conformational lock of their bioactive conformations while preserving the main protein–ligand interactions evident with M4K2009. This structural analysis helped us rationalize the increase in potency of the 7-membered ring inhibitors. In addition, the 7-membered amine-linked analogues M4K2304 and M4K2306 exhibited high levels of selectivity for ALK2 over ALK5, exceeding levels exhibited by the reference compound (Table 6). M4K2304 also demonstrated excellent kinome-wide selectivity, inhibiting only 3% of 422 kinases by more than 50% at 1 μM.
Table 6. Alignment of M4K2009, M4K2281, M4K2304, and M4K2306 with the TPP.
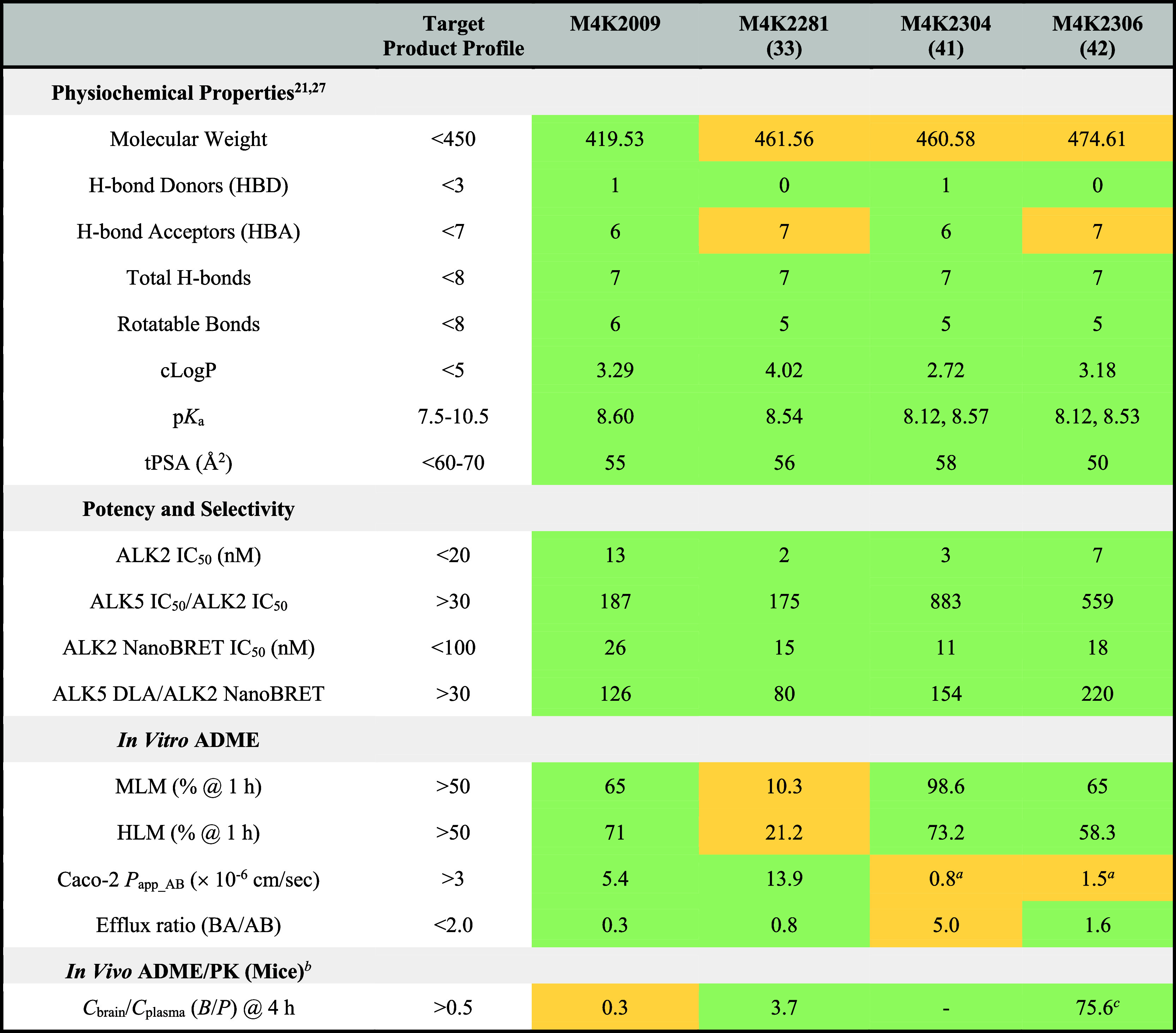
Poor post-assay recovery.
10 mg/kg PO dosing.
Average of duplicate measurements.
Although the in vivo PK properties of our compound set have not yet been fully studied, the initial in vivo mouse exposure assay of M4K2308, M4K2281, M4K2304, and M4K2306 (in addition to their in vitro ADME profiles) revealed that the solvent-exposed position can be explored to improve the physical and PK properties of the compounds, while preserving the key structural elements required for protein–ligand interaction. Additionally, the in vivo BBB penetration assay demonstrated that M4K2281 and M4K2306 exhibit notably higher brain-to-plasma ratios than M4K2009 (Table 6). This provides a promising initial insight into the CNS exposure of these constrained analogues. Therefore, M4K2308 and M4K2304 can be utilized to further optimize the in vivo PK profile of these compounds and test them in orthotopic mouse models of DIPG. It is important to mention that these cyclic scaffolds are structurally unique among the type I kinase inhibitors reported in the literature. It is therefore reasonable to suggest that this rigidification strategy can be extended to the design of other kinase inhibitors.
Experimental Section
Chemistry
All reagents were purchased from commercial vendors and used without further purification. Volatiles were removed under reduced pressure by rotary evaporation or by using the V-10 solvent evaporator system from Biotage. Very high boiling point (6000 rpm, 0 mbar, 56 °C), mixed volatile (7000 rpm, 30 mbar, 36 °C), and volatile (6000 rpm, 30 mbar, 36 °C) methods were used to evaporate solvents. The yields given refer to chromatographically purified and spectroscopically pure compounds, unless stated otherwise. Compounds were purified using a Biotage Isolera One system by normal-phase chromatography using Biotage SNAP KP-Sil or Sfär Silica D columns (part no. FSKO-1107/FSRD-0445) or by reverse-phase chromatography using Biotage SNAP KP-C18-HS or Sfär C18 D columns (part no. FSLO-1118/FSUD-040). If additional purification was required, compounds were purified by solid-phase extraction (SPE) using Biotage Isolute Flash SCX-2 cation exchange cartridges (part nos. 532-0050-C and 456-0200-D). Products were washed with two cartridge volumes of MeOH and eluted with a solution of MeOH and NH4OH (9:1 v/v). The final compounds were dried using the Labconco Benchtop FreeZoneTM Freeze-Dry System (4.5 L model). 1H NMRs were recorded on a Bruker Avance-III 500 MHz spectrometer at ambient temperature. Residual protons of CDCl3, DMSO-d6, and CD3OD solvents were used as internal references. Spectral data are reported as follows: chemical shift (δ in ppm), multiplicity (br = broad, s = singlet, d = doublet, dd = doublet of doublets, m = multiplet), coupling constants (J in Hz), and proton integration. Compound purity was determined by UV absorbance at 254 nm during tandem liquid chromatography/mass spectrometry (LCMS) using a Waters Acquity separations module. All final compounds had a purity of ≥95% as determined using this method. Low-resolution mass spectrometry was conducted in positive ion mode using a Waters Acquity SQD mass spectrometer (electrospray ionization source) fitted with a PDA detector. Mobile phase A consisted of 0.1% formic acid in water, while mobile phase B consisted of 0.1% formic acid in acetonitrile. One column was used: Acquity UPLC HSS T3 (2.1 × 50 mm, 100 Å, 1.8 μm. part no. 186003538). Method 1: the gradient went from 98% to 5% mobile phase A over 1.8 min, maintained at 5% for 0.5 min, then increased to 98% over 0.2 min for a total run time of 3 min. Method 2: the gradient went from 98% to 5% mobile phase A over 8.8 min, maintained at 5% for 0.5 min, then increased to 98% over 0.2 min for a total run time of 10 min. The flow rate was 0.4 mL/min throughout both runs. All columns were used with the temperature maintained at 25 °C. High-resolution mass spectrometry was conducted using a Waters Synapt G2-S quadrupole-time-of-flight (QTOF) hybrid mass spectrometer system coupled with an Acquity ultraperformance liquid chromatography (UPLC) I-Class system. Chromatographic separations were carried out on an Acquity UPLC HSS T3 (2.1 × 50 mm, 100 Å, 1.8 μm, part no. 186003538). The mobile phases were 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). Leucine Enkephalin was used as the lock mass. MassLynx 4.1 was used for data analysis.
3-Bromo-5-(3,4,5-trimethoxyphenyl)pyridin-4-ol (3)
A solution of 3,5-dibromo-4-hydroxypyridine (1) (1000 mg, 3.95 mmol), 3,4,5-trimethoxyphenylboronic acid (2) (838 mg, 3.95 mmol), [1,12-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (145 mg, 0.20 mmol), and potassium carbonate (1639 mg, 11.86 mmol) in 1,4-dioxane (12 mL) and water (2 mL) was heated to 100 °C and stirred for 3 h. Afterward, the reaction mixture was cooled to room temperature, diluted with water (40 mL), and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. This crude material was purified by reverse-phase chromatography [2–95% ACN (0.1% formic acid) in water (0.1% formic acid)] to afford intermediate 3 as a powder (310 mg, 23% yield). LC: method 1, tR = 1.27 min, MS (ESI): m/z = 340.07 [M + H]+, 342.13 [M + H]+ + 2.
3-(4-Chloro-2-fluorophenyl)-5-(3,4,5-trimethoxyphenyl)pyridin-4-ol (5)
A solution of 3 (290 mg, 0.85 mmol), 4-chloro-2-fluorophenylboronic acid (4) (297 mg, 1.70 mmol), [1,12-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (31 mg, 0.04 mmol), and potassium carbonate (353 mg, 2.56 mmol) in 1,4-dioxane (12 mL) and water (2 mL) was heated to 100 °C and stirred for 3 h. Afterward, the reaction mixture was cooled to room temperature, diluted with water (20 mL), and the aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. This crude material was purified by reverse-phase chromatography [2–95% ACN (0.1% formic acid) in water (0.1% formic acid)] to afford intermediate 5 as a powder (175 mg, 49% yield). LC: method 1, tR = 1.63 min, MS (ESI): m/z = 390.15 [M + H]+, 392.47 [M + H]+ + 2.
7-Chloro-4-(3,4,5-trimethoxyphenyl)benzofuro[3,2-c]pyridine (6)
A solution of 5 (175 mg, 0.45 mmol) and potassium carbonate (248 g, 1.79 mmol) in N-methyl-2-pyrrolidinone (NMP) was heated to 100 °C and stirred for 3 h. Afterward, the reaction mixture was cooled to room temperature, diluted with water (30 mL), and the aqueous layer was extracted with EtOAc (3 × 30 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford intermediate 6 as a brown powder (86 mg, 49% yield). No further purification was required. LC: method 1, tR = 1.95 min, MS (ESI): m/z = 370.22 [M + H]+, 372.35 [M + H]+ + 2.
7-(4-Methylpiperazin-1-yl)-4-(3,4,5-trimethoxyphenyl)benzofuro[3,2-c]pyridine (8) (M4K2286)
To a solution of 6 (60 mg, 0.16 mmol), RuPhos Pd G3 (37 mg, 0.05 mmol), and sodium tert-butoxide (47 mg, 0.49 mmol) in 1,4-dioxane (2 mL) at room temperature was added methylpiperazine (7) (33 mg, 0.32 mmol). After heating the resulting mixture at 100 °C for 16 h, it was cooled to room temperature. Volatiles were removed under reduced pressure, and the crude product was purified by reverse-phase chromatography [2–95% ACN (0.1% formic acid) in water (0.1% formic acid)]. Then, the product was purified by SPE. Freeze-drying for 2 days afforded 8 (M4K2286) as a white powder (36 mg, 51% yield). 1H NMR (500 MHz, MeOD-d4): δ 9.02 (s, 1H), 8.62 (s, 1H), 7.97 (d, J = 8.68 Hz, 1H), 7.25 (d, J = 1.83 Hz, 1H), 7.18 (s, 2H), 7.15 (dd, J = 8.74, 1.90 Hz, 1H), 3.95 (s, 6H), 3.86 (s, 3H), 3.40–3.37 (m, 4H), 2.71–2.68 (m, 4H), and 2.40 (s, 3H). HRMS (ESI) for C25H28N3O4 [M + H]+m/z: calcd, 434.2080; found, 434.2081.
3-Bromo-5-(4-chloro-2-fluorophenyl)isonicotinaldehyde (10)
A solution of 3,5-dibromoisonicotinaldehyde (9) (1854 mg, 7.00 mmol), 4-chloro-2-fluorophenylboronic acid (4) (1221 mg, 7.00 mmol), [1,12-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (180 mg, 0.25 mmol), and sodium carbonate (2600 mg, 21.00 mmol) in 1,4-dioxane (20 mL) and water (2 mL) was heated to 100 °C and stirred for 16 h. Afterward, the reaction mixture was cooled to room temperature, diluted with water (50 mL), and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. This crude material was purified by normal-phase chromatography (0–30% EtOAc/hexanes) to afford intermediate 10 as a powder (848 mg, 36% yield). LC: method 1, tR = 2.07 min, MS (ESI): m/z = 314.18 [M + H]+, 316.24 [M + H]+ + 2.
(3-Bromo-5-(4-chloro-2-fluorophenyl)pyridin-4-yl)methanol (11)
To a solution of 10 (600 mg, 1.91 mmol) in tetrahydrofuran (THF) (10 mL), cooled to 0 °C, was added sodium borohydride (72 mg, 1.91 mmol) in one portion. Upon completion of the addition, the mixture was warmed to room temperature with stirring for 10 min. The reaction mixture was then diluted with water (30 mL), and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. This crude material was purified by normal-phase chromatography (0–50% EtOAc/hexanes) to afford intermediate 11 as a powder (408 mg, 62% yield). LC: method 1, tR = 1.79 min, MS (ESI): m/z = 316.09 [M + H]+, 318.03 [M + H]+ + 2.
4-Bromo-8-chloro-5H-chromeno[4,3-c]pyridine (12)
To a solution of 11 (250 mg, 0.79 mmol) in THF (8 mL) at 0 °C, was added sodium hydride (60%, 126 mg, 3.16 mmol) in one portion. Upon completion of the addition, the mixture was warmed to room temperature with stirring for 10 min. The reaction mixture was then diluted with water (50 mL), and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude material 12 was used without further purification in the subsequent reaction (265 mg, 99% yield). LC: method 1, tR = 2.22 min, MS (ESI): m/z = 296.10 [M + H]+, 298.04 [M + H]+ + 2.
8-Chloro-4-(3,4,5-trimethoxyphenyl)-5H-chromeno[4,3-c]pyridine (13)
A solution of 12 (140 mg, 0.47 mmol), 2 (120 mg, 0.57 mmol), [1,12-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (17 mg, 0.02 mmol), and potassium carbonate (196 mg, 1.42 mmol) in 1,4-dioxane (5 mL) and water (1 mL) was heated to 100 °C and stirred for 3 h. Afterward, the reaction mixture was cooled to room temperature, diluted with water (30 mL), and the aqueous layer was extracted with EtOAc (3 × 30 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude material 13 was used without further purification in the subsequent reaction (199 mg, 97% yield). LC: method 1, tR = 2.13 min, MS (ESI): m/z = 384.20 [M + H]+, 386.45 [M + H]+ + 2.
8-(4-Methylpiperazin-1-yl)-4-(3,4,5-trimethoxyphenyl)-5H-chromeno[4,3-c]pyridine (14) (M4K2277)
To a solution of 13 (80 mg, 0.21 mmol), RuPhos Pd G3 (48 mg, 0.06 mmol), and sodium tert-butoxide (60 mg, 0.63 mmol) in 1,4-dioxane (2 mL) at room temperature was added 7 (33 mg, 0.32 mmol). After heating the resulting mixture at 100 °C for 16 h, it was cooled to room temperature. Volatiles were removed under reduced pressure, and the crude product was purified by reverse-phase chromatography [2–95% ACN (0.1% formic acid) in water (0.1% formic acid)]. Then, the product was purified by SPE. Freeze-drying for 2 days afforded 14 (M4K2277) as a white powder (31 mg, 33% yield). 1H NMR (500 MHz, DMSO-d6): δ 8.93 (s, 1H), 8.44 (s, 1H), 7.84 (d, J = 8.68 Hz, 1H), 6.74 (dd, J = 8.68, 2.08 Hz, 1H), 6.68 (s, 2H), 6.52 (d, J = 1.96 Hz, 1H), 5.14 (s, 2H), 3.82 (s, 6H), 3.72 (s, 3H), 3.23–3.20 (m, 4H), 2.45–2.42 (m, 4H), and 2.22 (s, 3H). HRMS (ESI) for C26H30N3O4 [M + H]+m/z: calcd, 448.2236; found, 448.2245.
3-Bromo-5-(3,4,5-trimethoxyphenyl)isonicotinaldehyde (15)
A solution of 9 (4000 mg, 15.10 mmol), 2 (3200 mg, 15.10 mmol), [1,12-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (552 mg, 0.75 mmol), and potassium carbonate (6260 mg, 45.30 mmol) in 1,4-dioxane (26 mL) and water (4 mL) was heated to 100 °C and stirred for 24 h. Afterward, the reaction mixture was cooled to room temperature, diluted with water (40 mL), and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. This crude material was purified by normal-phase chromatography (0–40% EtOAc/hexanes) to afford intermediate 15 as a powder (1627 mg, 29% yield). LC: method 2, tR = 4.73 min, MS (ESI): m/z = 352.11 [M + H]+, 354.22 [M + H]+ + 2.
(3-Bromo-5-(3,4,5-trimethoxyphenyl)pyridin-4-yl)methanol (16)
To a solution of 15 (1610 mg, 4.57 mmol) in THF (10 mL) and MeOH (10 mL), cooled to 0 °C, was added sodium borohydride (173 mg, 4.57 mmol) in one portion. Upon completion of the addition, the mixture was warmed to room temperature with stirring for 10 min. The reaction mixture was then diluted with water (50 mL), and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified by normal-phase chromatography (0–40% EtOAc/hexanes) to afford intermediate 16 as a powder (1023 mg, 62% yield). LC: method 1, tR = 1.69 min, MS (ESI): m/z = 353.82 [M + H]+, 355.82 [M + H]+ + 2.
3-Bromo-4-(((5-chloro-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl)oxy)methyl)-5-(3,4,5-trimethoxyphenyl)pyridine (18)
To a solution of 16 (500 mg, 1.41 mmol) in N,N-dimethylformamide (DMF) (5 mL) at 0 °C, was added sodium hydride (60%, 113 mg, 2.82 mmol) in one portion. The mixture was stirred for 10 min with the reaction temperature being maintained at 0 °C. To the mixture was added a solution of 2-bromomethyl-4-chlorophenylboronic acid, pinacol ester (17) (468 mg, 1.41 mmol) in DMF (2 mL). Upon completion of the addition, the mixture was warmed to room temperature with stirring for 1 h. The reaction mixture was then diluted with EtOAc (50 mL), washed with brine (3 × 50 mL), dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified by normal-phase chromatography (0–20% EtOAc/hexanes) to afford intermediate 18 as a powder (415 mg, 49% yield). LC: method 2, tR = 8.23 min, MS (ESI): m/z = 604.07 [M + H]+, 605.95 [M + H]+ + 2.
9-Chloro-4-(3,4,5-trimethoxyphenyl)-5,7-dihydrobenzo[5,6]oxepino[4,3-c]pyridine (19)
A solution of 18 (395 mg, 0.65 mmol), [1,12-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (48 mg, 0.06 mmol), and potassium carbonate (6260 mg, 45.30 mmol) in 1,4-dioxane (8 mL) and water (2 mL) was heated to 100 °C and stirred for 2 h. The reaction mixture was cooled to room temperature, diluted with water (20 mL), and the aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. This crude material was purified by normal-phase chromatography (0–40% EtOAc/hexanes) to afford intermediate 19 as a powder (212 mg, 82% yield). LC: method 1, tR = 2.11 min, MS (ESI): m/z = 398.01 [M + H]+, 400.33 [M + H]+ + 2.
9-(Piperazin-1-yl)-4-(3,4,5-trimethoxyphenyl)-5,7-dihydrobenzo[5,6]oxepino[4,3-c]pyridine (21) (M4K2207)
A solution of 19 (50 mg, 0.13 mmol), RuPhos Pd G3 (29 mg, 0.04 mmol), sodium tert-butoxide (36 mg, 0.38 mmol), and 1-boc-piperazine (20) (47 mg, 0.25 mmol) in 1,4-dioxane (2 mL) was heated to 100 °C for 16 h. Volatiles were removed under reduced pressure, and the crude product was purified by normal-phase chromatography (0–70% EtOAc/hexanes). Afterward, the purified product was dissolved in dichloromethane (DCM) (1 mL), treated with trifluoroacetic acid (TFA) (0.5 mL, 0.13 mmol), and stirred for 1 h. The solution was concentrated under reduced pressure prior to purification by SPE. Freeze-drying for 2 days afforded 21 (M4K2207) as a white powder (29 mg, 51% yield). 1H NMR (500 MHz, DMSO-d6): δ 8.74 (s, 1H), 8.65 (s, 1H), 7.59 (d, J = 8.56 Hz, 1H), 7.16 (d, J = 2.08 Hz, 1H), 7.11 (dd, J = 8.56, 2.20 Hz, 1H), 6.84 (s, 2H), 4.40 (s, 2H), 4.17 (s, 2H), 3.83 (s, 6H), 3.73 (s, 3H), 3.20–3.17 (m, 4H), and 2.88–2.85 (m, 4H). HRMS (ESI) for C26H30N3O4 [M + H]+m/z: calcd, 448.2236; found, 448.2236.
9-(4-Methylpiperazin-1-yl)-4-(3,4,5-trimethoxyphenyl)-5,7-dihydrobenzo[5,6]oxepino[4,3-c]pyridine (22) (M4K2303)
To a solution of 19 (50 mg, 0.13 mmol), RuPhos Pd G3 (29 mg, 0.04 mmol), and sodium tert-butoxide (36 mg, 0.38 mmol) in 1,4-dioxane (2 mL) at room temperature was added 7 (25 mg, 0.25 mmol). After heating the resulting mixture at 100 °C for 16 h, it was cooled to room temperature. Volatiles were removed under reduced pressure, and the crude product was purified by reverse-phase chromatography [2–95% ACN (0.1% formic acid) in water (0.1% formic acid)]. Then, the product was purified by SPE. Freeze-drying for 2 days afforded 22 (M4K2303) as a white powder (30 mg, 52% yield). 1H NMR (500 MHz, DMSO-d6): δ 8.74 (s, 1H), 8.65 (s, 1H), 7.59 (d, J = 8.44 Hz, 1H), 7.18 (d, J = 2.08 Hz, 1H), 7.13 (dd, J = 8.50, 2.14 Hz, 1H), 6.84 (s, 2H), 4.40 (s, 2H), 4.17 (s, 2H), 3.83 (s, 6H), 3.73 (s, 3H), 3.28–3.26 (m, 4H), 2.48–2.47 (m, 4H), and 2.24 (s, 3H). HRMS (ESI) for C27H32N3O4 [M + H]+m/z: calcd, 462.2393; found, 462.2401.
3-Bromo-5-(4-chloro-2-methoxyphenyl)isonicotinaldehyde (24)
A solution of 9 (8000 mg, 30.2 mmol), 4-chloro-2-methoxyphenylboronic acid (23) (4500 mg, 24.16 mmol), tetrakis(triphenylphosphine)palladium(0) (1745 mg, 1.51 mmol), and potassium carbonate (10,430 mg, 75 mmol) in 1,4-dioxane (336 mL) and water (56 mL) was heated to 100 °C and stirred for 16 h. Afterward, the reaction mixture was cooled to room temperature, diluted with water (150 mL), and the aqueous layer was extracted with EtOAc (3 × 150 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. This crude material was purified by normal-phase chromatography (0–10% EtOAc/hexanes) to afford intermediate 24 as a powder (3520 mg, 35% yield). LC: method 1, tR = 2.06 min, MS (ESI): m/z = 326.05 [M + H]+, 327.99 [M + H]+ + 2.
3-Bromo-5-(4-chloro-2-methoxyphenyl)-4-(2-methoxyvinyl)pyridine (26)
To a solution of (methoxymethyl)triphenylphosphonium chloride (25) (7390 mg, 21.56 mmol) in THF (50 mL), cooled to 0 °C, was added lithium bis(trimethylsilyl)amide (1 M in THF, 22 mL, 21.56 mmol). After 10 min of stirring at 0 °C, 24 (3518 g, 10.78 mmol) dissolved in 20 mL of THF was added in gradually, and the solution was stirred for 1 h at this temperature. The solution was then stirred for 1 h at room temperature. The reaction mixture was diluted with 100 mL of water, and the aqueous layer was extracted with EtOAc (3 × 100 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. This crude material was purified by normal-phase chromatography (0–20% EtOAc/hexanes) to afford intermediate 26 as a yellow liquid (3806 mg, 96% yield). LC: method 1, tR = 2.19 min, MS (ESI): m/z = 354.19 [M + H]+, 356.14 [M + H]+ + 2.
2-(3-Bromo-5-(4-chloro-2-methoxyphenyl)pyridin-4-yl)acetaldehyde (27)
Concentrated hydrochloric acid (4.46 mL, 53.6 mmol) was directly added to 26 (3800 mg, 10.72 mmol). Immediately, a red solution was formed. After stirring the reaction mixture for 1 h at room temperature, it was basified with aqueous 10% sodium hydroxide. After reaching a pH of 5, a yellow solid was formed. The mixture was extracted with EtOAc (3 × 100 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford intermediate 27 as a yellow powder (3194 mg, 85% yield). The crude material was used without further purification in the subsequent reaction. LC: method 1, tR = 2.02 min, MS (ESI): m/z = 339.9 [M + H]+, 341.28 [M + H]+ + 2.
2-(3-Bromo-5-(4-chloro-2-methoxyphenyl)pyridin-4-yl)ethan-1-ol (28)
To a solution of 27 (3190 mg, 9.37 mmol) in THF (50 mL) and MeOH (50 mL), cooled to 0 °C, was added sodium borohydride (1417 g, 37.5 mmol) in five portions over 2 h with stirring. The reaction mixture was then warmed to room temperature, diluted with water (30 mL), and the aqueous layer was extracted with EtOAc (3 × 30 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified by normal-phase chromatography (0–20% EtOAc/hexanes) to afford intermediate (28) as a white powder (2458 mg, 77% yield). LC: method 1, tR = 1.76 min, MS (ESI): m/z = 342.10 [M + H]+, 344.04 [M + H]+ + 2.
2-(5-Bromo-4-(2-hydroxyethyl)pyridin-3-yl)-5-chlorophenol (29)
To a solution of 28 (400 mg, 1.17 mmol) in DCM (12 mL) at −78 °C was added boron tribromide solution (1 M in CH2Cl2, 11.60 mL, 11.67 mmol). The reaction mixture was warmed to room temperature and vigorously stirred for 24 h. Then, the reaction mixture was diluted with cold water (50 mL), and the aqueous layer was extracted with DCM (3 × 50 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Afterward, the crude material was dissolved in water, treated with aqueous 2.5 M sodium hydroxide, and the solution was stirred for 30 min at room temperature. Then, the solution was acidified to pH 5 with aqueous 1 M HCl. A white precipitate was formed and extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified by normal-phase chromatography (0–50% EtOAc/hexanes) to afford intermediate 29 as a white powder (246 mg, 62% yield). LC: method 1, tR = 1.58 min, MS (ESI): m/z = 328.30 [M + H]+, 330.25 [M + H]+ + 2.
4-Bromo-9-chloro-5,6-dihydrobenzo[2,3]oxepino[4,5-c]pyridine (30)
To a solution 29 (244 mg, 0.74 mmol) and triphenylphosphine (390 mg, 1.48 mmol) in THF (3 mL) at 0 °C, was added diisopropyl azodicarboxylate (0.3 mL, 1.48 mmol). The mixture was warmed to room temperature and stirred for 1.5 h. Then, the reaction mixture was diluted with 20 mL of cold water, and the aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified by normal-phase chromatography (0–10% EtOAc/hexanes) to afford intermediate 30 as a powder (157 mg, 68% yield). LC: method 1, tR = 2.14 min, MS (ESI): m/z = 310.12 [M + H]+, 312.12 [M + H]+ + 2.
9-Chloro-4-(3,4,5-trimethoxyphenyl)-5,6-dihydrobenzo[2,3]oxepino[4,5-c]pyridine (31)
A solution of 30 (154 mg, 0.49 mmol), 2 (105 mg, 0.49 mmol), [1,12-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (36 mg, 0.049 mmol), and potassium carbonate (206 mg, 1.49 mmol) in 1,4-dioxane (4 mL) and water (0.7 mL) was heated to 100 °C and stirred for 2 h. Afterward, the reaction mixture was cooled to room temperature, diluted with water (20 mL), and the aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. This crude material was purified by normal-phase chromatography (0–40% EtOAc/hexanes) to afford intermediate 31 as a powder (144 mg, 73% yield). LC: method 1, tR = 1.99 min, MS (ESI): m/z = 398.39 [M + H]+, 400.45 [M + H]+ + 2.
9-(Piperazin-1-yl)-4-(3,4,5-trimethoxyphenyl)-5,6-dihydrobenzo[2,3]oxepino[4,5-c]pyridine (32) (M4K2308)
A solution of 31 (40 mg, 0.10 mmol), RuPhos Pd G3 (23 mg, 0.03 mmol), sodium tert-butoxide (29 mg, 0.30 mmol), and 20 (38 mg, 0.20 mmol) in 1,4-dioxane (2 mL) was heated to 100 °C for 16 h. Volatiles were removed under reduced pressure, and the crude product was purified by normal-phase chromatography (0–70% EtOAc/hexanes). Afterward, the purified product was dissolved in DCM (1 mL), treated with TFA (0.5 mL, 0.10 mmol), and stirred for 1 h. The solution was concentrated under reduced pressure prior to purification by SPE. Freeze-drying for 2 days afforded 32 (M4K2308) as a white powder (13 mg, 29% yield). 1H NMR (500 MHz, MeOD-d4): δ 8.48 (s, 1H), 8.37 (s, 1H), 7.41 (d, J = 8.56 Hz, 1H), 6.94 (dd, J = 8.56, 2.45 Hz, 1H), 6.78 (d, J = 2.45 Hz,1H), 6.67 (s, 2H), 4.57 (t, J = 6.36 Hz, 2H), 3.88 (s, 6H), 3.82 (s, 3H), 3.30–3.28 (m, 4H), 3.09–3.06 (m, 4H), and 2.81 (t, J = 6.30 Hz, 2H). HRMS (ESI) for C26H30N3O4 [M + H]+m/z = calcd, 448.2236; found, 448.2238.
9-(4-Methylpiperazin-1-yl)-4-(3,4,5-trimethoxyphenyl)-5,6-dihydrobenzo[2,3]oxepino[4,5-c]pyridine (33) (M4K2281)
To a solution of 31 (40 mg, 0.10 mmol), RuPhos Pd G3 (23 mg, 0.03 mmol), and sodium tert-butoxide (29 mg, 0.30 mmol) in 1,4-dioxane (2 mL) at room temperature was added 7 (20 mg, 0.20 mmol). After heating the resulting mixture at 100 °C for 16 h, it was cooled to room temperature. Volatiles were removed under reduced pressure, and the crude product was purified by reverse-phase chromatography [2–95% ACN (0.1% formic acid) in water (0.1% formic acid)]. Then, the product was purified by SPE. Freeze-drying for 2 days afforded 33 (M4K2281) as a white powder (23 mg, 49% yield). 1H NMR (500 MHz, MeOD-d4): δ 8.48 (s, 1H), 8.37 (s, 1H), 7.41 (d, J = 8.56 Hz, 1H), 6.95 (dd, J = 8.56, 2.57 Hz, 1H), 6.79 (d, J = 2.45 Hz, 1H), 6.67 (s, 2H), 4.57 (t, J = 6.36 Hz, 2H), 3.87 (s, 6H), 3.82 (s, 3H), 3.38–3.35 (m, 4H), 2.84–2.80 (m, 6H), and 2.51 (s, 3H). HRMS (ESI) for C27H32N3O4 [M + H]+m/z = calcd, 462.2393; found, 462.2395.
1-(5-Chloro-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-N-methylmethanamine (36)
To a solution of 2-bromomethyl-4-chlorophenylboronic acid, pinacol ester (34) (1000 mg, 3.02 mmol) in THF (15 mL) at 0 °C, methylamine (35) (2.0 M in THF, 5.5 mL, 151 mmol) was added. The reaction mixture was warmed to room temperature and stirred for 24 h. Volatiles were removed under reduced pressure. The reaction crude was then diluted with water (50 mL), and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Upon removal of the solvent, a white solid was obtained. The solid was washed with hexanes (3 × 100 mL) and dried under high vacuum. The crude product (36) (365 mg, 42%) was used directly in the next step without further purification.
N-(5-chloro-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl)-1-(3,5-dibromopyridin-4-yl)-N-methylmethanamine and (4-Chloro-2-((((3,5-dibromopyridin-4-yl)methyl)(methyl)amino)methyl)phenyl)boronic Acid (37 and 38)
To a solution of 9 (341 mg, 1.28 mmol) in DCM (5.5 mL) at room temperature, 36 (362 mg, 1.28 mmol) and acetic acid (0.1 mL, 1.75 mmol) were added, and the mixture was stirred for 1 h. The reaction mixture was then cooled to 0 °C, and sodium triacetoxyborohydride (545 mg, 2.57 mmol) was added in one portion. The mixture was warmed to room temperature and stirred for 2.5 h. Volatiles were evaporated under reduced pressure. The reaction crude was then diluted with water (40 mL), and the aqueous layer was extracted with EtOAc (3 × 40 mL). The combined organic extracts were dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified by normal-phase chromatography (0–50% MeOH/EtOAc) to afford a mixture of intermediates 37 and 38 as a powder (170 mg, 27% yield). LC (intermediate 37): method 1, tR = 2.40 min, MS (ESI): m/z = 528.31 [M + H]+, 529.32 [M + H]+ + 2. LC (intermediate 38): method 1, tR = 1.59 min, MS (ESI): m/z = 446.21 [M + H]+, 447.28 [M + H]+ + 2.
4-Bromo-9-chloro-6-methyl-6,7-dihydro-5H-benzo[c]pyrido[3,4-e]azepine (39)
A solution of a mixture of 37 and 38 (160 mg, 0.36 mmol), [1,12-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (26 mg, 0.04 mmol), and potassium carbonate (148 mg, 1.07 mmol) in 1,4-dioxane (4 mL) and water (0.7 mL) was heated to 100 °C and stirred for 2 h. Volatiles were evaporated under reduced pressure. The crude material was purified by normal-phase chromatography (0–60% EtOAc/hexanes) to afford intermediate 39 as a powder (115 mg, 50% yield). LC: method 1, tR = 1.22 min, MS (ESI): m/z = 323.29 [M + H]+, 325.17 [M + H]+ + 2.
9-Chloro-6-methyl-4-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[c]pyrido[3,4-e]azepine (40)
A solution of 39 (56 mg, 0.17 mmol), 2 (37 mg, 0.17 mmol), [1,12-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (13 mg, 0.02 mmol), and potassium carbonate (72 mg, 0.52 mmol) in 1,4-dioxane (2 mL) and water (0.3 mL) was heated to 100 °C and stirred for 2 h. Volatiles were evaporated under reduced pressure. The crude material was purified by normal-phase chromatography (0–60% EtOAc/hexanes) to afford intermediate 40 as a powder (40 mg, 57% yield). LC: method 1, tR = 1.41 min, MS (ESI): m/z = 411.24 [M + H]+, 413.43 [M + H]+ + 2.
6-Methyl-9-(piperazin-1-yl)-4-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[c]pyrido[3,4-e]azepine (41) (M4K2304)
A solution of 40 (38 mg, 0.09 mmol), RuPhos Pd G3 (21 mg, 0.03 mmol), sodium tert-butoxide (27 mg, 0.28 mmol), and 20 (34 mg, 0.18 mmol) in 1,4-dioxane (2 mL) was heated to 100 °C for 16 h. The reaction mixture was then cooled to room temperature, and volatiles were removed under reduced pressure. The crude product was purified by reverse-phase chromatography [2–95% ACN (0.1% formic acid) in water (0.1% formic acid)]. Afterward, the purified product was dissolved in DCM (1 mL), treated with HCl (4 M in dioxane), and stirred for 1 h. The solution was concentrated under reduced pressure prior to purification by SPE. Freeze-drying for 2 days afforded 41 (M4K2304) as a white powder (21 mg, 48% yield). 1H NMR (500 MHz, MeOD-d4): δ 8.58 (s, 1H), 8.49 (s, 1H), 7.51 (d, J = 8.44 Hz, 1H), 7.12 (dd, J = 8.50, 2.38 Hz, 1H), 7.04 (d, J = 2.32 Hz, 1H), 6.89 (s, 2H), 3.89 (s, 6H), 3.83 (s, 3H), 3.51 (s, 2H), 3.30–3.29 (m, 4H), 3.06–3.04 (m, 4H), 2.33 (s, 3H), and 1.29 (s, 2H). HRMS (ESI) for C27H33N4O3 [M + H]+m/z: calcd, 461.2553; found, 461.2554.
6-Methyl-9-(4-methylpiperazin-1-yl)-4-(3,4,5-trimethoxyphenyl)-6,7-dihydro-5H-benzo[c]pyrido[3,4-e]azepine (42) (M4K2306)
A solution of 40 (30 mg, 0.07 mmol), RuPhos Pd G3 (17 mg, 0.02 mmol), sodium tert-butoxide (21 mg, 0.21 mmol), and 7 (15 mg, 0.14 mmol) in 1,4-dioxane (2 mL) was heated to 100 °C for 16 h. The reaction mixture was then cooled to room temperature, and volatiles were removed under reduced pressure. The crude product was purified by normal-phase chromatography (0–50% MeOH/EtOAc). Freeze-drying for 2 days afforded 42 (M4K2306) as a yellow-white powder (28 mg, 80% yield). 1H NMR (500 MHz, MeOD-d4): δ 8.58 (s, 1H), 8.49 (s, 1H), 7.51 (d, J = 8.44 Hz, 1H), 7.13 (dd, J = 8.44, 2.45 Hz, 1H), 7.05 (d, J = 2.45 Hz, 1H), 6.89 (s, 2H), 3.89 (s, 6H), 3.83 (s, 3H), 3.52 (s, 2H), 3.37–3.35 (m, 4H), 2.73–2.70 (m, 4H), 2.42 (s, 3H), 2.34 (s, 3H), and 1.93 (s, 2H). HRMS (ESI) for C28H35N4O3 [M + H]+m/z: calcd, 475.2709; found, 475.2698.
Kinase Assay
The biochemical potencies of all compounds were measured by Reaction Biology Corporation (Malvern, Pennsylvania, United States). Compounds were tested against ALK2/ACVR1, ALK4/ACVR1β, and ALK5/TGFβ-R1 in 10-dose IC50 mode with a 3-fold serial dilution starting at 1 or 5 μM. Reactions were conducted at an ATP concentration of 10 μM. LDN-193189 was tested as a control in 10-dose IC50 mode with a 3-fold serial dilution starting at 10 μM. Reductions in enzymatic activity were determined relative to DMSO controls.
Cell Culture and Transfection
HEK-293 cells were maintained in Dulbecco’s modified Eagle medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher) and penicillin/streptomycin (Thermo Fisher). HEK-293 cells were transfected with the protein expression or reporter constructs using FuGENE HD (Promega) according to the manufacturer’s instructions. Briefly, DNA was diluted into phenol red-free Opti-MEM (Gibco) at a concentration of 10 μg/mL. Without coming in contact with the sides of the container, 3 μL of FuGENE HD was added for each μg of DNA used. After thorough mixing by inversion, FuGENE HD/DNA complexes were allowed to form by incubation at room temperature for 20 min. Transfection mixture (1 part) was added to 20 parts of HEK-293 cell suspension with a density of 200,000 cells per mL (volume/volume). HEK-293 cells were incubated in a humidified, 37 °C incubator with 5% CO2 for 24 h before they were used in the NanoBRET target engagement assay or the dual luciferase reporter assay.
NanoBRET Target Engagement Assay
ALK2-C-terminal nanoluciferase fusion with a GSSG linker was encoded by the pFC32K vector (Promega). ALK2-nanoluciferase fusion construct (1 part) was mixed with 9 parts of Transfection Carrier DNA (mass/mass) (Promega). Transfected cells were trypsinized and resuspended in Opti-MEM at a density of 200,000 cells per mL. Cells (17 μL) were dispensed into each well of a 384-well flat-bottom polypropylene plate (Greiner). A working solution (20 ×) of target engagement tracer PBI-6908 (Promega) was prepared by diluting DMSO stock in tracer dilution buffer (12.5 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES) pH 7.5, 31.25% PEG-400). Stocks (1000 ×) of test compounds in DMSO (Cell Signaling Technology) were diluted further in Opti-MEM for 10× working solutions. After the addition of 1 μL of 20× target engagement tracer and 2 μL of 10× working solutions, the contents of the wells were thoroughly mixed by agitating the plate at 500 rpm for 1 min. Cells were incubated in a humidified, 37 °C incubator with 5% CO2 for 2 h prior to bioluminescence resonance energy transfer (BRET) measurement. For BRET measurement, NanoBRET NanoGlo Substrate and Extracellular NanoLuc Inhibitor (Promega) were diluted 166× and 500 ×, respectively, in Opti-MEM to produce 3× working stock. A PHERAstar FSX microplate reader (BMG Labtech) with the LUM 610-LP 460–80 optical module was used to measure the intensity of dual emission. A measurement interval of 1 s and gain settings of 3600 and 1879 for 610 and 460 nm, respectively, were used. Milli-BRET units (mBU) were calculated by dividing the signal measured at 610 nm with the signal measured at 460 nm and multiplying by 1000. The apparent EC50 values of test compounds were estimated using the [Inhibitor] versus response (three parameter) nonlinear regression curve fitting function of GraphPad Prism 7.
Dual Luciferase Reporter Assay
CAGA-Luc and Renilla-luciferase constructs (a gift of Dr Petra Knaus, Free University of Berlin) were used as the reporters for ALK5 signaling and loading control, respectively. CAGA-Luc construct (4 parts) was mixed with 1 part of Renilla-luciferase construct (mass/mass). Ten thousand transfected cells were seeded into each well of a 96-well plate (Corning). Some 24 h after transfection, the cells were incubated with 10 ng/mL TGFβ1 (Peprotech, 100-21-10) and test compounds simultaneously at the concentrations indicated in a humidified, 37 °C incubator with 5% CO2. Another 24 h later, the cells were harvested, lysed, and processed for the measurement of luciferase activity using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. Briefly, the culture medium was aspirated completely, and cells were lysed in 50 μL of 1× PLB with 300 rpm agitation for 30 min. Cell lysate (10 μL) was dispensed into each well of a 384-well flat-bottom polypropylene plate (Greiner). The luminescent signal of firefly and Renilla luciferase activity was measured sequentially using a PHERAstar FS microplate reader (BMG Labtech) after the addition of 25 μL of LARII and Stop & Glo, respectively. A measurement interval of 2 s and gain setting of 3600 were used. The firefly luciferase signal was normalized to the cell number by division with Renilla luciferase signal. The relative luciferase unit (RLU) was obtained by further division with the signal from cells without TGFβ stimulation. The apparent EC50 values of test compounds were estimated using the [Inhibitor] versus response (three parameter) nonlinear regression curve fitting function of GraphPad Prism 7.
Caco-2 Permeability Assay
Caco-2 cells (C2BBe1) were purchased from the American Type Culture Collection (ATCC). Caco-2 cell cultures were routinely maintained in T-75 tissue culture flasks in DMEM containing 20% FBS, 0.1 mg/mL normocin, and 0.05 mg/mL gentamicin. These cells were seeded at a density of 40,000 cells per well on the 24-well polyethylene terephthalate membrane (1.0 μm pore size, 0.3 cm2 surface area) insert plates. Cell monolayers were grown for 21 or 22 days at 37 °C with 5% CO2 in a humidified incubator. The cell culture medium was replaced twice weekly during the cell growth period. Prior to beginning the permeability assay, cell monolayers were rinsed with Hank’s balanced salt solution (HBSS) twice to remove the residual cell culture medium. The assay buffer comprised HBSS containing 10 mM HEPES and 15 mM glucose at pH 7.4. The dosing buffer contained 5 μM metoprolol (positive control), 5 μM atenolol (negative control), and 100 μM Lucifer yellow in the assay buffer. The receiving buffer contained 1% bovine serum albumin (BSA) in the assay buffer. The concentration of the test compound was 5 μM in the dosing buffer (the final DMSO concentration was 0.1%). Digoxin at 10 μM was utilized as a P-gp substrate control. For the apical to basolateral (A to B) permeability experiment, 0.25 mL of the dosing buffer was added to the apical chambers, and 1.0 mL of the receiving buffer was added to the basolateral chambers of the assay plate. For the basolateral to apical (B to A) permeability experiment, 0.25 mL of the receiving buffer was added to the apical chambers, and 1.0 mL of dosing buffer was added to the basolateral chambers of the assay plates. The assay plates were then incubated at 37 °C for 2 h on an orbital shaker at 65 rpm. Sample solutions were taken from the donor chambers (10 μL) and receiver chambers (100 μL) after the incubation period. For each sample, there were two technical replicates. The sample solutions from donor chambers were diluted 10× with the receiving buffer. In order to extract test compounds and precipitate BSA from sample solutions, three volumes of acetonitrile (containing 0.5% formic acid and an internal standard) were added, and the plate was vigorously mixed. Sample solutions were then centrifuged at 4000 rpm for 10 min to remove debris and precipitated BSA. Approximately 150 μL of the supernatant was subsequently transferred to a new 96-well microplate for LC/MS analysis. Narrow-window mass extraction LC/MS analysis was performed for all samples using a Waters Acquity UPLC system with a Waters Xevo quadrupole time-of-flight mass spectrometer to determine the relative peak areas of the parent compounds. Chromatographic separations were performed on a Waters Acquity UPLC HSS T3 column (2.1 × 100 mm, 100 Å, 1.8 μm, part no. 186003539) at 30 °C. Mobile phases A and B were 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. The sample temperature was kept at 10 °C. The typical injection volume was 0.3 μL. Chromatographic gradients used are shown in the table below.
| time (min) | flow rate (mL/min) | % A | % B |
| 0.0 | 0.400 | 100 | 0 |
| 0.5 | 0.400 | 100 | 0 |
| 4.5 | 0.400 | 5 | 95 |
| 5.0 | 0.400 | 5 | 95 |
| 5.5 | 0.400 | 100 | 0 |
| 6.5 | 0.400 | 100 | 0 |
Data acquisition was performed in electrospray (ES) positive ion mode in the mass range of 100–1000 m/z. The following source parameters were used: capillary voltage, 0.8 kV; cone voltage, 25 V; source temperature, 150 °C; desolvation temperature, 500 °C; cone gas flow, 150 L/h; desolvation gas flow, 600 L/h. Accurate masses were measured in lock-spray automated exact mass measurement mode. The fragment ions of leucine-enkephalin were used as reference substances (lock-mass) in acquisition mode. The following lock-spray configuration was used: frequency, 10 s; cone voltage, 25 V; collision energy, 22 V. Data processing was performed using MassLynx 4.1 software (Waters). The codosed positive and negative controls were also measured for each well to monitor integrity of cell monolayers and well-to-well variability. The apparent permeability coefficient (Papp) and post-assay recovery are calculated using the following equations
where dC/dt is the slope of cumulative concentration in the receiver compartment versus time, Vr is the volume of the receiver compartment, Vd is the volume of the donor compartment, A is the membrane surface area, C0 is the initial compound concentration in the donor chamber, Crfinal is the cumulative receiver concentration at the end of the incubation period, and Cdfinal is the concentration of the donor at the end of the incubation period. Efflux ratio (ER) is defined as Papp (B-to-A)/Papp (A-to-B).
Liver Microsomal Metabolic Stability Assay
For this assay, stock solutions of test compounds in DMSO (1 mM) were initially diluted to a concentration of 40.0 μM using 0.1 M potassium phosphate buffer (pH 7.4). Test compounds were then added to reaction wells at a final concentration of 1 μM, which was assumed to be well below Km values to ensure linear reaction conditions (i.e., avoid saturation). The final DMSO concentration was kept constant at 0.1%. Each compound was tested in duplicate for both time points (0 and 60 min). CD-1 mouse (male) or pooled human liver microsomes (Corning Gentest) were added to the reaction wells at a final concentration of 0.5 mg/mL (protein). The final volume for each reaction was 100 μL, which included the NADPH-regeneration solution (NRS) mix (Corning Gentest). This NRS mix comprised glucose 6-phosphate dehydrogenase, NADP+, MgCl2, and glucose 6-phosphate. Reactions were carried out at 37 °C in an orbital shaker at 175 rpm. Upon completion of the 60 min time point, reactions were terminated by the addition of two volumes (200 μL) of ice-cold acetonitrile containing 0.5% formic acid and an internal standard. Samples were then centrifuged at 4000 rpm for 10 min to remove debris and precipitated proteins. Approximately 150 μL of supernatant was subsequently transferred to a new 96-well microplate for LC/MS analysis. Narrow-window mass extraction LC/MS analysis was performed for all samples using a Waters Acquity UPLC system with a Waters Xevo quadrupole time-of-flight (Q-TOF) mass spectrometer to determine relative peak areas of the parent compounds. Chromatographic separations were performed on a Waters Acquity UPLC HSS T3 column (2.1 × 100 mm, 100 Å, 1.8 μm, part no. 186003539) at 30 °C. Mobile phases A and B were 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. The sample temperature was kept at 10 °C. The typical injection volume was 0.3 μL. Chromatographic gradients used are shown in the table below.
| time (min) | flow rate (mL/min) | % A | % B |
| 0.0 | 0.450 | 100 | 0 |
| 2.5 | 0.450 | 5 | 95 |
| 3.0 | 0.450 | 5 | 95 |
| 3.5 | 0.450 | 100 | 0 |
| 4.5 | 0.450 | 100 | 0 |
Data acquisition was performed in ES positive ion mode in the mass range of 100–1000 m/Z. The following source parameters were used: capillary voltage, 0.8 kV; cone voltage, 25 V; source temperature, 150 °C; desolvation temperature, 500 °C; cone gas flow, 150 L/h; desolvation gas flow, 600 L/h. Accurate masses were measured in lock-spray automated exact mass measurement mode. The fragment ions of leucine-enkephalin were used as reference substances (lock-mass) in acquisition mode. The following lock-spray configuration was used: frequency, 10 s; cone voltage, 25 V; collision energy, 22 V. Data processing was performed using MassLynx 4.1 software (Waters). The percentage remaining values were calculated using the following equation
where A is area response after incubation, and A0 is area response at initial time point.
In Vivo Pharmacokinetic Exposure in Mouse Model
All animal-related procedures were in accordance with the regulations of the Canadian Council on Animal Care (CCAC) and the University Health Network (UHN). NOD-SCID male mice were orally dosed with 25 mg/kg of M4K2009, M4K2304, M4K2306, M4K2308, and M4K2281 in the OICR (Ontario Institute for Cancer Research) formulation–5% DMSO, 47.5% PEG, 47.5% Aq. (10% Tween80 in dH2O). For each compound, there were three biological replicates. Blood samples were drawn from the saphenous veins of mice at 0.5, 1, 2, 6, and 24 h. Samples were immediately centrifuged, and 20 μL of plasma was collected and stored at 80 °C until analysis. In addition, blank plasma was obtained from treatment-naïve mice for the negative control. For M4K2009, 40 μL of ACN was added to 20 μL of each thawed plasma sample. Samples were vortexed, then centrifuged at 14,000 rpm for 6.5 min. About 50 μL of supernatant was added to an high-performance liquid chromatography (HPLC) vial. A calibration curve with ten different concentrations of M4K2009 starting from 10 ng/mL up to 10,000 ng/mL was prepared by spiking blank plasma from treatment-naïve mice. One μL of each sample was injected for LC–MS/MS analysis. Chromatographic separations were carried out on a Waters Acquity UPLC BEH C18 (2.1 × 50 mm, 1.7 μm) column using the Waters Acquity UPLC I-Class system. Mobile phases A and B were 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. Chromatographic gradients used are shown in the table below.
| time (min) | flow rate (mL/min) | % A | % B |
| 0.0 | 0.450 | 95 | 5 |
| 4.5 | 0.450 | 5 | 95 |
| 5.0 | 0.450 | 5 | 95 |
| 5.5 | 0.450 | 95 | 5 |
| 6.0 | 0.450 | 95 | 5 |
A Waters Synapt G2S QTof mass spectrometer equipped with an atmospheric pressure ionization source was used for mass spectrometric analysis. MassLynx 4.1 was used for data analysis.
For M4K2304, M4K2306, M4K2308, and M4K2281, 50 μL of ACN was added to 20 μL of each thawed plasma sample. The samples were vortexed and centrifuged at 14,000 rpm for 6.5 min. Approximately 60 μL of the supernatant was transferred to an HPLC vial. A calibration curve with ten different concentrations of the test compounds from 10 nM to 10,000 nM was generated by spiking blank plasma from treatment-naïve mice. 0.5 μL of each sample was injected for LC–MS/MS analysis. Chromatographic separations were performed on a Waters Xbridge BEH C8 column (2.1 × 50 mm, 3.5 μm) using an Agilent 1200 series HPLC system. The mobile phases A and B were 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. The chromatographic gradients used are listed in the following table.
| time (min) | flow rate (mL/min) | % A | % B |
| 0.0 | 0.500 | 90 | 10 |
| 1.5 | 0.500 | 10 | 90 |
| 2.0 | 0.500 | 10 | 90 |
| 2.5 | 0.500 | 90 | 10 |
| 5.5 | 0.500 | 90 | 10 |
An AB Sciex QTRAP 5500 mass analyzer equipped with an atmospheric pressure ionization source was used for mass spectrometric analysis. Analyst 1.7 was used for data acquisition and analysis.
In Vivo BBB Penetration in Mouse Model
Male NOD-SCID mice were orally dosed with 10 mg/kg M4K2281 and M4K2306 in the OICR formulation. There were three biological replicates for each compound. Brain and plasma samples were collected at 2 and 4 h after dosing. Blood samples were collected by cardiac puncture, immediately centrifuged, and about 100 μL of plasma was collected. Brains were perfused with PBS, collected surgically, and immediately snap frozen on dry ice. All samples were stored at −80 °C until analysis. For brain samples, each brain was cut in half, and one part was used to quantify drug levels. Brain samples were weighed and transferred to plastic vials containing 1:2 (brain: water, e.g., 100 mg of brain with 200 μL of water) volume of water and 2 glass Pyrex beads (4 mm in size). Brains were then homogenized by bead beating (three 30-s pulses at 5000 rpm) using a Precellys 24 homogenizer and allowed to settle on ice for 5 min. The homogenized samples were centrifuged at 2000 rpm for 5 min to separate the suspension layer from the foam layer. 50 μL of the suspension layers were carefully transferred to microcentrifuge tubes. 100 μL of acetonitrile containing an internal standard was added to the suspensions and mixed vigorously. The samples were then centrifuged at 14,000 rpm for 6.5 min. Approximately 100 μL of the supernatant was transferred to an LCMS vial. For plasma samples, 20 μL of each thawed plasma was transferred to a microcentrifuge tube, and 40 μL of acetonitrile containing an internal standard was added. Then, samples were vortexed and centrifuged at 14,000 rpm for 6.5 min. Approximately 50 μL of the supernatant was transferred to an LCMS vial. A calibration curve with ten different concentrations of the test compounds from 10 nM to 10,000 nM was generated by spiking blank plasma from treatment-naïve mice. To quantify the demethylated metabolites in plasma and brain, M4K2308 and M4K2304 were included in the calibration curve. 0.5 μL of each sample was injected for LC–MS/MS analysis. Chromatographic separations were performed on a Waters Xbridge BEH C8 column (2.1 × 50 mm, 3.5 μm) using an Agilent 1200 series HPLC system. The mobile phases A and B were 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. The chromatographic gradients used are listed in the following table.
| time (min) | flow rate (mL/min) | % A | % B |
| 0.0 | 0.500 | 90 | 10 |
| 1.5 | 0.500 | 10 | 90 |
| 2.0 | 0.500 | 10 | 90 |
| 2.5 | 0.500 | 90 | 10 |
| 5.5 | 0.500 | 90 | 10 |
An AB Sciex QTRAP 5500 mass analyzer equipped with an atmospheric pressure ionization source was used for mass spectrometric analysis. Analyst 1.7 was used for data acquisition and analysis.
Co-crystallization of ALK2 with M4K2304 and M4K2308
Protein Expression and Purification
For protein expression, the kinase domain of ALK2 (residues 201-499 and Q207D activating mutation) was cloned into the pFastBac-derived vector pFB-LIC-Bse, which provides an N-terminal hexahistidine tag to enable purification, as previously described.29 Baculoviral expression was performed in Sf9 insect cells at 27 °C, shaking at 110 rpm. Cells were harvested at 72 h after infection, pelleted with centrifugation, and stored at −20 °C. For purification, cells pellets were resuspended in lysis buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 5 mM imidazole, 5% glycerol) supplemented with 1 mM tris(2-carboxyethyl)phosphine (TCEP) and protease inhibitor. Lysis was performed using sonication, and then 0.125% polyethylenimine was added to precipitate DNA. Insoluble material was excluded by centrifugation, and the clear supernatant was loaded onto Ni-affinity resin. Bound proteins were eluted with lysis buffer supplemented with increasing concentrations of imidazole (50–100–150–250 mM). Subsequently, relevant eluted proteins were cleaved with Tobacco Etch Virus protease and loaded for size-exclusion chromatography (S200 HiLoad 16/60 Superdex), equilibrated in 50 mM HEPES pH 7.5, 300 mM NaCl, and 0.5 mM TCEP. Aliquots containing ALK2 were combined, concentrated to 4 mg/mL, flash frozen, and stored at −80 °C. Purity was assessed by SDS-PAGE and the molecular weight verified by intact mass spectrometry analysis.
Crystallization
Frozen ALK2 protein was thawed and purified on a S200 Hiload 16/60 Superdex column, following the same protocol and buffer as described above. ALK2 was preincubated with 1 mM M4K compounds at a protein concentration of 10 and 8 mg/mL for M4K2304 and M4K2308, respectively. Crystallization was achieved using the sitting drop vapor diffusion method at 4 °C with a reservoir solution containing 1.6 ammonium sulfate, 0.1 M tris pH 8.5, 4% glycerol or 1.7 ammonium sulfate, 0.1 M tris pH 8, 8% glycerol for M4K2304 and M4K2308, respectively. Initial microcrystals were used to seed the protein solution and improve crystal growth. Viable crystals were obtained when the seeded protein solution was mixed with the reservoir solution at 1:2 and 2:1 ratios, respectively. Crystals were cryoprotected with mother liquor solution supplemented with 25% ethylene glycol, prior to vitrification in liquid nitrogen.
Data Collection, Phasing, and Refinement
Diffraction data were collected at the Diamond Light Source, beamline i03, at a temperature of 100 K using monochromatic radiation at a wavelength of 0.976254 Å. Data were processed with Xia2 dials and subsequently scaled using the program AIMLESS from the CCP4 suite.30,31 The structures were solved by molecular replacement using PHASER32 and the coordinate of an in-house model for ALK2 as a search model. Atomic models were refined using Phenix Refine and manually improved using COOT.33,34 Stereochemical validation of the final models was performed with MolProbity.35 Data collection and refinement statistics are summarized in Supporting Information, Tables S2 and S3.
Structural Analysis and Molecular Modeling
Analysis of the X-ray structures was performed using both PyMOL and Maestro software from Schrödinger. Protein structures were prepared using the Protein Preparation Workflow to add hydrogens and predict the orientation of water molecules. A final constrained minimization protocol (max RMSD deviation of 0.2 Å) was performed before generating the 2D ligand interaction plots with LID tool.
Acknowledgments
This work was funded by the Cancer Therapeutics Innovation Pipeline program at the Ontario Institute for Cancer Research (OICR). The OICR receives financial support from the Government of Ontario through the Ministry of Training, Colleges, and Universities. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute [OGI-1523 055], Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA, Darmstadt, Germany, MSD, Novartis Pharma AG, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z]. The authors thank the Diamond Light Source for beamtime (proposal mx28172) as well as the staff of beamline i03 for their guidance. J.C. and A.N.B. acknowledge funding from FOP Friends. A.N.B. also acknowledges funding from the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement No 875510 (EUbOPEN). The JU receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA and Ontario Institute for Cancer Research, Royal Institution for the Advancement of Learning McGill University, Kungliga Tekniska Hoegskolan, Diamond Light Source Limited. We thank Reaction Biology Corporation for their in-kind contributions in testing the biological activity of our compounds. We thank Lisa Rooney for discussions at the early stage of the project. H.G-A acknowledges The Mexican National Council of Science and Technology (CONACyT) for financial support of his work.
Glossary
Abbreviations
- ALK2
activin receptor-like kinase-2
- ALK5
activin receptor-like kinase-5
- BBB
blood–brain barrier
- BMP
bone morphogenetic protein
- BRET
bioluminescence resonance energy transfer
- CNS
central nervous system
- DIPG
diffuse intrinsic pontine glioma
- DLA
dual luciferase assay
- GS
glycine–serine
- HLM
human liver microsome
- MLM
mouse liver microsome
- TGFβ
transforming growth factor beta
- TPP
target product profile
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c02308.
Accession Codes
Author Contributions
The manuscript was written by H.G.-A. Compounds were synthesized by H.G.-A. and D.E. Compounds were designed, and their syntheses devised by H.G.-A., D.E., T.X., B.W., C.A.Z.-V., M.B–I., D.S., and R.A. NanoBRET and DLA data were generated by J.F.W. and supervised by A.N.B. Crystallography experiments were performed by J.C. and supervised by A.N.-B. M.N.S. purified the ALK2 protein and started the crystallization screening that yielded the co-crystal structure of M4K2304, supervised by A.N.-B. Structural analysis and molecular modeling were performed by L.H. HRMS, Caco-2, microsomal stability, and PK studies were conducted by T.K. and supervised by A.A., O.R. was CEO of M4K Pharm Inc. D.S. initiated the project, was involved in experimental design and interpretation of data, monitored project progress, and edited the paper. R.A. was involved in experimental design, monitored project progress, and revised the manuscript.
The authors declare no competing financial interest.
After this article was published ASAP March 18, 2024, a change was made to the TOC and abstract graphics, and Melissa N. Sweeney was added to the author list. The corrected version was reposted March 28, 2024.
Supplementary Material
References
- Girardi F.; Allemani C.; Coleman M. P. Worldwide Trends in Survival From Common Childhood Brain Tumors: A Systematic Review. J. Glob. Oncol. 2019, 5, 1–25. 10.1200/JGO.19.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren K. E. Diffuse Intrinsic Pontine Glioma: Poised for Progress. Front. Oncol. 2012, 2, 205. 10.3389/fonc.2012.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J. R.; Young C. C.; Vitanza N. A.; McGrath M.; Feroze A. H.; Browd S. R.; Hauptman J. S. Progress In Diffuse Intrinsic Pontine Glioma: Advocating for Stereotactic Biopsy in the Standard of Care. Neurosurg. Focus 2020, 48 (1), E4 10.3171/2019.9.FOCUS19745. [DOI] [PubMed] [Google Scholar]
- Carvalho D.; Taylor K. R.; Olaciregui N. G.; Molinari V.; Clarke M.; Mackay A.; Ruddle R.; Henley A.; Valenti M.; Hayes A.; Brandon A. D. H.; Eccles S. A.; Raynaud F.; Boudhar A.; Monje M.; Popov S.; Moore A. S.; Mora J.; Cruz O.; Vinci M.; Brennan P. E.; Bullock A. N.; Carcaboso A. M.; Jones C. ALK2 Inhibitors Display Beneficial Effects in Preclinical Models of ACVR1Mutant Diffuse Intrinsic Pontine Glioma. Commun. Biol. 2019, 2, 156. 10.1038/s42003-019-0420-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R. K.; Rutka J. T. Diffuse Intrinsic Pontine Glioma: Clinical Features, Molecular Genetics, and Novel Targeted Therapeutics. J. Korean Neurosurg. Soc. 2018, 61 (3), 343–351. 10.3340/jkns.2018.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay A.; Burford A.; Carvalho D.; Izquierdo E.; Fazal-Salom J.; Taylor K. R.; Bjerke L.; Clarke M.; Vinci M.; Nandhabalan M.; Temelso S.; Popov S.; Molinari V.; Raman P.; Waanders A. J.; Han H. J.; Gupta S.; Marshall L.; Zacharoulis S.; Vaidya S.; Mandeville H. C.; Bridges L. R.; Martin A. J.; Al-Sarraj S.; Chandler C.; Ng H. K.; Li X.; Mu K.; Trabelsi S.; Brahim D. H.; Kisljakov A. N.; Konovalov D. M.; Moore A. S.; Carcaboso A. M.; Sunol M.; de Torres C.; Cruz O.; Mora J.; Shats L. I.; Stavale J. N.; Bidinotto L. T.; Reis R. M.; Entz-Werle N.; Farrell M.; Cryan J.; Crimmins D.; Caird J.; Pears J.; Monje M.; Debily M. A.; Castel D.; Grill J.; Hawkins C.; Nikbakht H.; Jabado N.; Baker S. J.; Pfister S. M.; Jones D. T. W.; Fouladi M.; von Bueren A. O.; Baudis M.; Resnick A.; Jones C. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32 (4), 520–537.e5. 10.1016/j.ccell.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczkowicz P.; Hoeman C.; Rakopoulos P.; Pajovic S.; Letourneau L.; Dzamba M.; Morrison A.; Lewis P.; Bouffet E.; Bartels U.; Zuccaro J.; Agnihotri S.; Ryall S.; Barszczyk M.; Chornenkyy Y.; Bourgey M.; Bourque G.; Montpetit A.; Cordero F.; Castelo-Branco P.; Mangerel J.; Tabori U.; Ho K. C.; Huang A.; Taylor K. R.; Mackay A.; Bendel A. E.; Nazarian J.; Fangusaro J. R.; Karajannis M. A.; Zagzag D.; Foreman N. K.; Donson A.; Hegert J. V.; Smith A.; Chan J.; Lafay-Cousin L.; Dunn S.; Hukin J.; Dunham C.; Scheinemann K.; Michaud J.; Zelcer S.; Ramsay D.; Cain J.; Brennan C.; Souweidane M. M.; Jones C.; Allis C. D.; Brudno M.; Becher O.; Hawkins C. Genomic Analysis of Diffuse Intrinsic Pontine Gliomas Identifies Three Molecular Subgroups and Recurrent Activating ACVR1Mutations. Nat. Genet. 2014, 46 (5), 451–456. 10.1038/ng.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso A. M.; Papillon-Cavanagh S.; Schwartzentruber J.; Nikbakht H.; Gerges N.; Fiset P. O.; Bechet D.; Faury D.; De Jay N.; Ramkissoon L. A.; Corcoran A.; Jones D. T.; Sturm D.; Johann P.; Tomita T.; Goldman S.; Nagib M.; Bendel A.; Goumnerova L.; Bowers D. C.; Leonard J. R.; Rubin J. B.; Alden T.; Browd S.; Geyer J. R.; Leary S.; Jallo G.; Cohen K.; Gupta N.; Prados M. D.; Carret A. S.; Ellezam B.; Crevier L.; Klekner A.; Bognar L.; Hauser P.; Garami M.; Myseros J.; Dong Z.; Siegel P. M.; Malkin H.; Ligon A. H.; Albrecht S.; Pfister S. M.; Ligon K. L.; Majewski J.; Jabado N.; Kieran M. W. Recurrent Somatic Mutations in ACVR1 in Pediatric Midline High-grade Astrocytoma. Nat. Genet. 2014, 46 (5), 462–466. 10.1038/ng.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor K. R.; Mackay A.; Truffaux N.; Butterfield Y.; Morozova O.; Philippe C.; Castel D.; Grasso C. S.; Vinci M.; Carvalho D.; Carcaboso A. M.; de Torres C.; Cruz O.; Mora J.; Entz-Werle N.; Ingram W. J.; Monje M.; Hargrave D.; Bullock A. N.; Puget S.; Yip S.; Jones C.; Grill J. Recurrent Activating ACVR1Mutations in Diffuse Intrinsic Pontine Glioma. Nat. Genet. 2014, 46 (5), 457–461. 10.1038/ng.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smil D.; Wong J. F.; Williams E. P.; Adamson R. J.; Howarth A.; McLeod D. A.; Mamai A.; Kim S.; Wilson B. J.; Kiyota T.; Aman A.; Owen J.; Poda G.; Horiuchi K. Y.; Kuznetsova E.; Ma H.; Hamblin J. N.; Cramp S.; Roberts O. G.; Edwards A. M.; Uehling D.; Al-Awar R.; Bullock A. N.; O’Meara J. A.; Isaac M. B. Leveraging an Open Science Drug Discovery Model to Develop CNS-Penetrant ALK2 Inhibitors for the Treatment of Diffuse Intrinsic Pontine Glioma. J. Med. Chem. 2020, 63 (17), 10061–10085. 10.1021/acs.jmedchem.0c01199. [DOI] [PubMed] [Google Scholar]
- Roschger C.; Cabrele C. The Id-Protein Family in Developmental and Cancer-Associated Pathways. Cell Commun. Signaling 2017, 15 (1), 7. 10.1186/s12964-016-0161-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz A. K.; Paugh B. S.; Rankin S. L.; Ju B.; Li Y.; Zhu X.; Qu C.; Chen X.; Zhang J.; Easton J.; Edmonson M.; Ma X.; Lu C.; Nagahawatte P.; Hedlund E.; Rusch M.; Pounds S.; Lin T.; Onar-Thomas A.; Huether R.; Kriwacki R.; Parker M.; Gupta P.; Becksfort J.; Wei L.; Mulder H. L.; Boggs K.; Vadodaria B.; Yergeau D.; Russell J. C.; Ochoa K.; Fulton R. S.; Fulton L. L.; Jones C.; Boop F. A.; Broniscer A.; Wetmore C.; Gajjar A.; Ding L.; Mardis E. R.; Wilson R. K.; Taylor M. R.; Downing J. R.; Ellison D. W.; Zhang J.; Baker S. J.; The Genomic Landscape of Diffuse Intrinsic Pontine Glioma and Pediatric Non-Brainstem High-Grade Glioma. Nat. Genet. 2014, 46 (5), 444–450. 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacifici M.; Shore E. M. Common Mutations in ALK2/ACVR1, a Multi-Faceted Receptor, Have Roles in Distinct Pediatric Musculoskeletal and Neural Orphan Disorders. Cytokine Growth Factor Rev. 2016, 27, 93–104. 10.1016/j.cytogfr.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Tapia F.; Lou Y.; Brotherton-Pleiss C.; Kuglstatter A.; So S. S.; Kondru R. A Potent Seven-Membered Cyclic BTK (Bruton’s tyrosine Kinase) Chiral Inhibitor Conceived by Structure-Based Drug Design to Lock its Bioactive Conformation. Bioorg. Med. Chem. Lett. 2019, 29 (9), 1074–1078. 10.1016/j.bmcl.2019.03.001. [DOI] [PubMed] [Google Scholar]
- Li K.; McGee L. R.; Fisher B.; Sudom A.; Liu J.; Rubenstein S. M.; Anwer M. K.; Cushing T. D.; Shin Y.; Ayres M.; Lee F.; Eksterowicz J.; Faulder P.; Waszkowycz B.; Plotnikova O.; Farrelly E.; Xiao S. H.; Chen G.; Wang Z. Inhibiting NF-κB-Inducing Kinase (NIK): Discovery, Structure-Based Design, Synthesis, Structure-Activity Relationship, and Co-Crystal Structures. Bioorg. Med. Chem. Lett. 2013, 23 (5), 1238–1244. 10.1016/j.bmcl.2013.01.012. [DOI] [PubMed] [Google Scholar]
- Johnson T. W.; Richardson P. F.; Bailey S.; Brooun A.; Burke B. J.; Collins M. R.; Cui J. J.; Deal J. G.; Deng Y.-L.; Dinh D.; Engstrom L. D.; He M.; Hoffman J.; Hoffman R. L.; Huang Q.; Kania R. S.; Kath J. C.; Lam H.; Lam J. L.; Le P. T.; Lingardo L.; Liu W.; McTigue M.; Palmer C. L.; Sach N. W.; Smeal T.; Smith G. L.; Stewart A. E.; Timofeevski S.; Zhu H.; Zhu J.; Zou H. Y.; Edwards M. P. Discovery of (10R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h] [2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a Macrocyclic Inhibitor of Anaplastic Lymphoma Kinase (ALK) and c-ros Oncogene 1 (ROS1) with Preclinical Brain Exposure and Broad-Spectrum Potency against ALK-Resistant Mutations. J. Med. Chem. 2014, 57 (11), 4720–4744. 10.1021/jm500261q. [DOI] [PubMed] [Google Scholar]
- Gerasyuto A. I.; Arnold M. A.; Wang J.; Chen G.; Zhang X.; Smith S.; Woll M. G.; Baird J.; Zhang N.; Almstead N. G.; Narasimhan J.; Peddi S.; Dumble M.; Sheedy J.; Weetall M.; Branstrom A. A.; Prasad J. V. N.; Karp G. M. Discovery and Optimization of Indolyl-Containing 4-Hydroxy-2-Pyridone Type II DNA Topoisomerase Inhibitors Active against Multidrug Resistant Gram-negative Bacteria. J. Med. Chem. 2018, 61 (10), 4456–4475. 10.1021/acs.jmedchem.8b00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S.; Zhang L.; Xu S.; Luo J.; Lu X.; Zhang Z.; Xu T.; Liu Y.; Tu Z.; Xu Y.; Ren X.; Geng M.; Ding J.; Pei D.; Ding K. Design, Synthesis, and Biological Evaluation of Novel Conformationally Constrained Inhibitors Targeting Epidermal Growth Factor Receptor Threonine790 → Methionine790 Mutant. J. Med. Chem. 2012, 55 (6), 2711–2723. 10.1021/jm201591k. [DOI] [PubMed] [Google Scholar]
- Wu J.; Chen W.; Xia G.; Zhang J.; Shao J.; Tan B.; Zhang C.; Yu W.; Weng Q.; Liu H.; Hu M.; Deng H.; Hao Y.; Shen J.; Yu Y. Design, Synthesis, and Biological Evaluation of Novel Conformationally Constrained Inhibitors Targeting EGFR. ACS Med. Chem. Lett. 2013, 4 (10), 974–978. 10.1021/ml4002437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahn M.; Herbertz S.; Sawyer J. S.; Stauber A. J.; Gueorguieva I.; Driscoll K. E.; Estrem S. T.; Cleverly A. L.; Desaiah D.; Guba S. C.; Benhadji K. A.; Slapak C. A. Clinical Development of Galunisertib (LY2157299 Monohydrate), a Small Molecule Inhibitor of Transforming Growth Factor-Beta Signaling Pathway. Drug Des., Dev. Ther. 2015, 9, 4479–4499. 10.2147/DDDT.S86621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensan D.. Master of Science Thesis; University of Toronto, 2020.Developing Small Molecule Inhibitors of ALK2: a Serine/Threonine Kinase Implicated in Diffuse Intrinsic Pontine Glioma [Google Scholar]
- Bissantz C.; Kuhn B.; Stahl M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53 (14), 5061–5084. 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; Kim H. S.; Shin C. J.; Ryu D.; Yoon J.; Lee S.; Lim S.; Jung S.-H.. Triazine- and Carbazole-Based Compound and composition for Organic Optoelectronic and Display Devices. US Patent. 20200377489A1, 2020.
- Jiang J. K.; Huang X.; Shamim K.; Patel P. R.; Lee A.; Wang A. Q.; Nguyen K.; Tawa G.; Cuny G. D.; Yu P. B.; Zheng W.; Xu X.; Sanderson P.; Huang W. Discovery of 3-(4-sulfamoylnaphthyl)pyrazolo[1,5-a]pyrimidines as Potent and Selective ALK2 inhibitors. Bioorg. Med. Chem. Lett. 2018, 28 (20), 3356–3362. 10.1016/j.bmcl.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson P. D.; Davis A. M. Time-Related Differences in the Physical Property Profiles of Oral Drugs. J. Med. Chem. 2004, 47 (25), 6338–6348. 10.1021/jm049717d. [DOI] [PubMed] [Google Scholar]
- Pajouhesh H.; Lenz G. R. Medicinal Chemical Properties of Successful Central Nervous System Drugs. NeuroRx 2005, 2 (4), 541–553. 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankovic Z. CNS Drug Design: Balancing Physicochemical Properties for Optimal Brain Exposure. J. Med. Chem. 2015, 58 (6), 2584–2608. 10.1021/jm501535r. [DOI] [PubMed] [Google Scholar]
- Smith D. A.; Di L.; Kerns E. H. The Effect of Plasma Protein Binding on In Vivo Efficacy: Misconceptions in Drug Discovery. Nat. Rev. Drug Discovery 2010, 9 (12), 929–939. 10.1038/nrd3287. [DOI] [PubMed] [Google Scholar]
- Sanvitale C. E.; Kerr G.; Chaikuad A.; Ramel M.-C.; Mohedas A. H.; Reichert S.; Wang Y.; Triffitt J. T.; Cuny G. D.; Yu P. B.; et al. A New Class of Small Molecule Inhibitor of BMP Signaling. PLoS One 2013, 8 (4), e62721 10.1371/journal.pone.0062721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. Xia2: An Expert System for Macromolecular Crystallography Data Reduction. J. Appl. Crystallogr. 2010, 43 (1), 186–190. 10.1107/S0021889809045701. [DOI] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67 (4), 235–242. 10.1107/s0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J. Solving Structures of Protein Complexes by Molecular Replacement With Phaser. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63 (1), 32–41. 10.1107/s0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (2), 213–221. 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60 (12), 2126–2132. 10.1107/s0907444904019158. [DOI] [PubMed] [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B. III; Snoeyink J.; Richardson J. S.; Richardson D. C. MolProbity: All-atom Contacts and Structure Validation for Proteins and Nucleic Acids. Nucleic Acids Res. 2007, 35 (Web Server), W375–W383. 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.