

Abstract
The increasing application of meta‐omics approaches to investigate the structure, function, and intercellular interactions of microbial communities has led to a surge in available data. However, this abundance of human and environmental microbiome data has exposed new scalability challenges for existing bioinformatics tools. In response, we introduce Wekemo Bioincloud—a specialized platform for ‐omics studies. This platform offers a comprehensive analysis solution, specifically designed to alleviate the challenges of tool selection for users in the face of expanding data sets. As of now, Wekemo Bioincloud has been regularly equipped with 22 workflows and 65 visualization tools, establishing itself as a user‐friendly and widely embraced platform for studying diverse data sets. Additionally, the platform enables the online modification of vector outputs, and the registration‐independent personalized dashboard system ensures privacy and traceability. Wekemo Bioincloud is freely available at https://www.bioincloud.tech/.
Keywords: bioinformatics, meta‐omics, microbiome, user‐friendly platform, Wekemo Bioincloud
Wekemo Bioincloud is a specialized platform for meta‐omics studies that addresses scalability challenges in handling vast microbial community data with 22 workflows and 65 visualization tools. It provides comprehensive analysis, online vector output modification, and ensures privacy with a registration‐independent dashboard system.
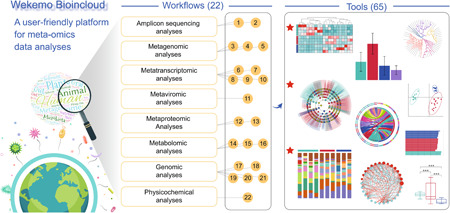
Highlights
Wekemo Bioincloud offers not just workflows for diverse meta‐omics data analysis but also a rich array of tools for effective data visualization.
Enables modifying vector outputs online, achieving publication‐ready figures.
Displays tools' popularity/usage trends, enhancing the platform's flexibility and user‐friendly experience.
INTRODUCTION
Recent development of meta‐omics approaches, spanning metagenomics, metatranscriptomics, metaproteomics, metaviromics, metabolomics, and physicochemical data, mark a transformative era in the comprehensive understanding of intricate biological systems [1, 2, 3]. The resulting multi‐omics data sets, encompassing the richness of microbial communities, present a profound need for robust bioinformatics tools that are both robust and user‐friendly [4], to unveil the profile of microbial communities and elucidate the nuanced interactions between environmental conditions and microbiome. Advancements in analytic platforms tailored for high‐throughput omics data further underscore the evolution in bioinformatics capabilities. Noteworthy examples include the application of QIIME 2 [5] and EasyAmplicon [6] for amplicon data analyses, Trimmomatic [7] or fastp [8] for stringent quality control, Kraken 2 [9] for precise taxonomic classification, HUMAnN3 pipeline [10] for comprehensive functional profiling, MultiPrime [11] for efficient minimal primer design, imageGP [12] for data visualization, and more, which collectively contribute to a more profound exploration of diverse omics data sets.
Conceptually, the standard omics data analysis workflow contains raw data processing, taxonomy annotation, functional profiling, and statistical analysis. Several workflows have been developed to streamline these processes [4, 13, 14, 15]. However, in diverse research contexts, personalized analysis approaches are crucial, emphasizing the necessity for customized analyses. Nowadays, various tools, pipelines, and online web services have been developed for ‐omics analyses. For instance, QIIME 2 [5] is a software primarily designed for amplicon sequencing analyses, which has expanded its capabilities to include metagenomic analyses. EasyAmplicon [6] is a pipeline specialized in amplicon sequencing analyses on the local server. MicrobiomeAnalyst [16] operates as a web server, mainly for amplicon sequencing analyses, metagenomic analyses, and metabolomic profiling. Notame [17] presents a dedicated workflow for metabolomic profiling. MetaProteomeAnalyzer [18] is a workflow for metaproteomic data analyses. Meanwhile, numerous innovative approaches have emerged for identifying reliable and stable biomarkers from ‐omics data [19, 20, 21, 22], and several research have diligently summarized and compared various R packages or software tools designed for ‐omics data [23, 24, 25, 26, 27, 28, 29, 30, 31]. However, the majority of these tools are oriented toward one or two specific types of ‐omics data analyses. Currently, integrative analysis across multiple ‐omics has become crucial for addressing scientific questions [1, 2]. Nevertheless, the diversity and complexity of analytical approaches mean that researchers not only need to install various tools or R packages for data analysis but also invest significant time in adapting to different tools or platforms. This highlights a considerable gap in the scientific community, emphasizing the need for an easily accessible web service specifically designed for the analysis and visualization of meta‐omics data [32].
Here, we present Wekemo Bioincloud, tailored for specific ‐omics studies, offering a comprehensive analysis solution that addresses the challenge of tool selection for users. The Wekemo Bioinclud platform securely stores users' raw sequencing data in the cloud and performs preprocessing tasks for various analysis tools in advance. Comprising two key modules, workflows and tools, the Wekemo Bioinclud platform satisfies a spectrum of requirements. Notably, this platform empowers users to oversee critical steps with dependencies in the workflow, facilitating systematic exploration of data and unveiling biological significance. This platform is openly accessible at https://www.bioincloud.tech/. For an in‐depth understanding of the platform, its usage, and result interpretation, comprehensive details can be found on the website.
RESULT
Overview of Wekemo Bioincloud
The Wekemo Bioincloud comprises two main components: the workflows and the tools. In the workflow module, users can analyze omics data step by step, generating reports that detail the software used and their respective parameters for each analysis (Figure 1). The tool module allows users to easily showcase their data, referencing our demo files. The platform is designed to be as convenient as possible for researchers to access analyses, which can modify all groups with one click in all analyses, and also run all analyses with one click. Notably, we offer more than just scalable vector graphics (SVG) editors for refining output images; users can also set up email reminders for each step, saving them valuable time. Additionally, we provide instructional videos covering tool usage, various workflows, and result analyses, enhancing user comprehension of their data. Our registration‐independent personalized dashboard system ensures privacy, traceability, and collaboration. There are two ways for researchers to use our platform. First, they have the option to analyze their data by providing tables through our tool module, referring to our online demo table. Alternatively, they can choose to submit their raw data to us. In return, we furnish them with standard analysis tables, then users have the flexibility to conduct personalized analyses either through our workflows or by using our tools online. All raw data and result reports for workflow analyses will be retained for 2 years, offering ample time for users to mine the data in depth, while the data used for tools will be deleted every day.
Figure 1.
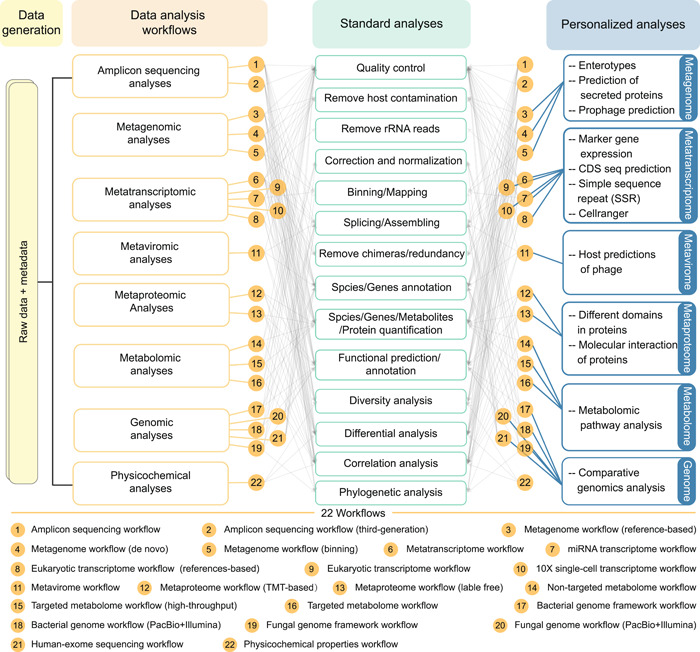
The framework of 22 analysis workflows for Wekemo Bioincloud. The platform now provides diverse workflows for meta‐omics data and accommodates both standardized and personalized analyses for various research. CDS, coding sequences; mRNA, messenger RNA; rRNA, ribosomal RNA; TMT, tandem mass tag.
After a thorough examination of all publications referencing Wekemo Bioincloud on Google Scholar until December 6, 2023, we observed that 157 publications have cited our platform. Following the removal of 17 publications due to unknown or repeated entries, a total of 140 distinct publications were identified. Notably, 42.14% of these publications opted for our workflows module, while the remaining 57.86% chose the tools module for visualizing their results. Common techniques employed for visualization include correlation tests, orthogonal partial least‐squares discriminant analysis (OPLS‐DA), principal coordinate analysis (PCoA), and linear discriminant analysis effect size (LEfSe) analysis (see Table S1).
Cloud workflows for meta‐omics data
The workflows module has currently been updated with 22 data analysis workflows, encompassing one‐step analyses of various types of data, including metagenome, metatranscriptome, metaproteome, metavirome, metabolome, genome, and physicochemical data (Figure 1). Each workflow comes with a comprehensive demo report, example processes, and interpretation of results, facilitating a quick start for new users. For routine ‐omics analysis, users only need to prepare raw sequencing data and metadata information by referring to our demo pipeline. Faced with plenty of meta‐omics analysis software, our workflows also include various software options. Users can easily choose different analysis algorithms/software based on the specific characteristics of their data, and all processing methods will be showcased in the output report.
Furthermore, we also provide some flexible choices for users to gain their personalized analysis. For example, the metagenomic pipeline can categorize the enterotypes of microbiome samples [33], and predict the prophage or secreted proteins of metagenomic binning. For profiling the transcriptional activity of individual cells, the 10× single‐cell transcriptome workflow empowers CellRanger (http://10xgenomics.com) to handle output results, incorporating processes such as alignment, quantification, clustering, and gene expression analysis. In addition, we have integrated different technologies for analyzing bacterial and fungal genomes using only Illumina short‐read sequencing, or Illumina short‐read sequencing with PacBio long‐read sequencing, facilitating the systematic exploration of data and unveiling biological significance.
Graphical tools for different purposes
To facilitate the intuitive presentation of scientific discoveries, the Bioincloud platform offers a diverse array of tools for visualizing, analyzing, and comparing the ‐omics data (Figure 2). Currently, it has launched 65 subfunctions, covering the analyses of (1) contribution, richness, composition of features or genes, (2) group comparison, (3) differences in data structure, (4) statistical analyses, (5) functional or metabolic pathways, (6) differentially expressed genes, (7) phylogenetic relationship, (8) correlation tests, (9) visualization pipelines, and (10) others. All subfunctions with popularity and difficulty scores help users gauge the usage frequency and complexity of each tool. The grouped cluster heatmap, LEfSe plots, and grouped percentage stacked bar plots are currently the three most popular tools, with usage counts reaching 36,098, 34,261, and 33,341, respectively, by December 10, 2023.
Figure 2.
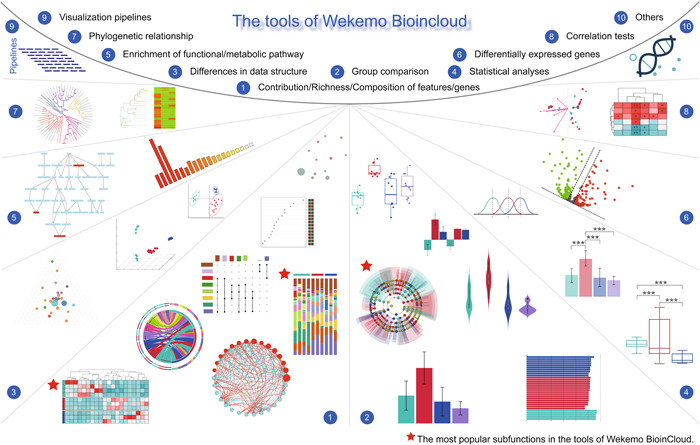
Example outputs generated by Wekemo Bioincloud Tools. The tools now contain 65 subfunctions, serving various purposes, including (1) the presentation of contribution, richness, composition of features or genes; (2) group comparison; (3) analysis of differences in data structure; (4) statistical analyses, such as analysis of variance tests, Kruskal–Wallis tests, and so forth; (5) enrichment of functional or metabolic pathways; (6) identification of differentially expressed genes; (7) construction of phylogenetic relationship; (8) correlation tests; (9) visualization pipelines, such as amplicon sequencing pipeline, metagenome taxonomy annotation pipeline, and so forth; (10) others, such as primer design, and so forth.
The tools module covers the majority of daily needs for ‐omics researchers through its 65 visualization and analysis functions, involving scatter plots, bar plots, bubble plots, violin plots, network plots, ternary plots, volcano plots, petal plots, heatmaps, pathway diagrams, and more (Figure 2). Additionally, basic significance tests such as analysis of variance (ANOVA), Kruskal–Wallis, and Dunn tests are available, along with common molecular tools like 16S ribosomal RNA (rRNA) gene blast and primer design. Moreover, it provides features such as the conversion of SVG to various formats, including PDF, JPG, PNG, and others. All detailed explanations and demo data sets are available on the website to address any potential user misunderstandings.
Case 1: Metagenomic data analysis workflows
The platform offers three workflows for analyzing metagenomic data, the reference‐based workflow, the de novo workflow, and the binning workflow (Figure 3). All raw reads are processed using KneadData (https://github.com/biobakery/kneaddata) to obtain clean reads, with Trimmomatic [7] used for the trimming of adapter sequences and low‐quality reads, and Bowtie employed for the removal of host genome contamination. Following this, all clean reads undergo further processing for various purposes.
Figure 3.
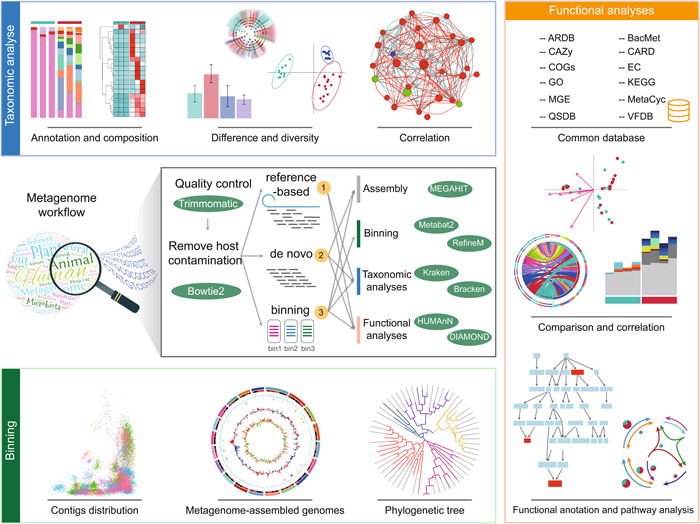
Metagenomic data analyses using Wekemo Bioincloud platform. The clean data could be analyzed with three different workflows, the reference‐based, the de novo, and the binning. The main software and visualization during assembly, binning, taxonomic analyses, and functional analyses were presented, making it suitable for various purposes or scenarios. The functional analyses include 12 diversity databases. ARDB, Antibiotic Resistance Genes Database; BacMet, Antibacterial Biocide and Metal Resistance Genes; CARD, Comprehensive Antibiotic Resistance Database; CAZy, Carbohydrate‐Active EnZymes; COGs, Clusters of Orthologous Groups of proteins; EC, Enzyme Commission; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; MetaCyc, Metabolic Pathway; MGE, Mobile Genetic Element; QS, Quorum Sensing; VFDB, Virulence Factors Database.
Notably, we leverage 12 commonly used bioinformatics databases in metagenomic analyses to predict the functions.
ARDB [34]: Antibiotic Resistance Genes Database is used to track antibiotic resistance genes.
BacMet [35]: Antibacterial Biocide and Metal Resistance Genes Database is used to confer the resistance to metals or antibacterial biocides.
CAZy [36]: Carbohydrate‐Active EnZyme is used to describe enzyme families responsible for cleaving or building complex carbohydrates.
CARD [37]: Comprehensive Antibiotic Resistance Database is employed to identify antibiotic resistance and virulence factors.
COGs [38]: Clusters of Orthologous Groups of proteins are attempted on a phylogenetic classification of the proteins.
EC [39]: Enzyme Commission, the numbers represent enzymes and enzyme genes. Evolutionary gene genealogy.
GO [40]: Gene Ontology, annotations report connections between gene products and the biological types.
KEGG [41]: Kyoto Encyclopedia of Genes and Genomes is utilized to identify functions within the biological system.
MGE [42]: Mobile Genetic Elements is used to carry various kinds of genes endowing their hosts with resistance to antibiotics and/or metals, pathogenicity, symbiosis, and metabolism of new substrates.
MetaCyc [43]: It contains pathways involved in both primary and secondary metabolism.
QSDB [44]: Quorum Sensing Database, a phenomenon in which the accumulation of signaling molecules allows a single cell to perceive the number of bacteria, enabling coordinated responses and behaviors among bacterial cells.
VFDB [45]: Virulence Factors Database is accessed to get bacterial virulence factors.
The reference‐based workflow involves mapping raw reads to different databases, including gene, nucleotide, or protein sequences. In this approach, Kraken [9] is utilized for taxonomic analyses with clean reads, and Bracken [46] is employed for estimating the species‐ or genus‐level abundance. To present the annotation or composition of species, bar plots, heatmaps, and Venn plots can be used. Comparisons between groups or samples can be demonstrated using ANOVA [47], DESeq2 [48], Kruskal–Wallis [49], and LEfSe [50]. The diversity of groups can be visualized through Bray–Curtis nonmetric multidimensional scaling, Bray–Curtis PCoA, and α‐diversity analyses. Correlations between groups or different factors can be explored using heatmaps, networks, and redundancy analysis/canonical correspondence analysis. Furthermore, clean reads are assigned to microbial metabolic pathways and functions using HUMAnN and the UniRef90 diamond annotated full reference database. CARD, COG, KEGG, MetaCyc, GO, EC, and CAZy are employed for function analyses. Then, the annotation or composition, comparison, or correlation of functional analyses can be shown like taxonomic analyses. In addition, using DiTing [51] enables the analysis of elemental cycles (carbon, nitrogen, phosphorus, sulfur) and the creation of cycle pathway diagrams. Besides, users can also select differential genes, comparing their differences or functional pathways.
The de novo workflow revolves around generating assembled contigs without relying on existing reference sequences, which allows for the discovery of more poorly described taxonomic groups. In this process, the classification and analyses of taxonomy follow a similar approach to the reference‐based workflow, which was mentioned earlier. However, the function analyses are based on the assembled contigs, and assembly is conducted using MEGAHIT [52], followed by gene prediction using Prodigal [53]. The CARD, COG, KEGG, GO, CAZy, MEG, ARDB, BacMet, and VFDB databases are used for functional annotation. Gene counting, which refers to the number of genes within a particular functional category, can also be calculated.
The binning workflow takes advantage of multiple features, such as the co‐abundance and coverage of contigs across samples, as well as the grouping of contigs based on similar Kmer frequencies and GC content. In this pipeline, the MEGAHIT [52] is applied to assemble clean reads and yields contigs. Then, MetaBAT [54] is employed to bin the contigs, RefineM [55] is used to eliminate contigs for removing the high contamination contigs, CheckM [56] is utilized to evaluate the completion and contamination of each bin, and dRep [57] is used to obtain a nonredundant bin. Then, the analyses and visualization for composition and function of bins can be shown as described in reference‐based workflow. The CAZy, COG, GO, KEGG, VFDB, MGE, and CARD databases are used for functional annotation. In addition, the metagenomic assembled genes of bins can be created to display the information about chromosome orientation GC content or GC skew of different contigs.
Case 2: Metabolomics data analysis workflow
Metabolomics can be broadly categorized into nontargeted and targeted metabolomics. Here, we offer three workflows, the nontargeted workflow, the targeted workflow, and the high‐throughput targeted workflow (Figure 4). Nontargeted metabolomics, characterized by its unbiased approach, facilitates a comprehensive analysis of the metabolites derived from the organisms, helping us to find some novel biomarkers. Targeted metabolomics employs standards, providing the absolute quantification of targeted metabolites, and it is reproducible. The high‐throughput targeted metabolomics allows the rapid and efficient analysis of a large number of metabolites in a sample, contributing to a more thorough understanding of the targeted metabolic profile. However, only targeted metabolomics can achieve absolute quantification of metabolites, the nontargeted and the high‐throughput targeted metabolomics are generally considered quantitative rather than absolute. The data analyses of the three different methods to gain metabolomics are mostly similar, containing compound detection, data preprocessing, statistical analyses, feature selection, and functional analyses.
Figure 4.
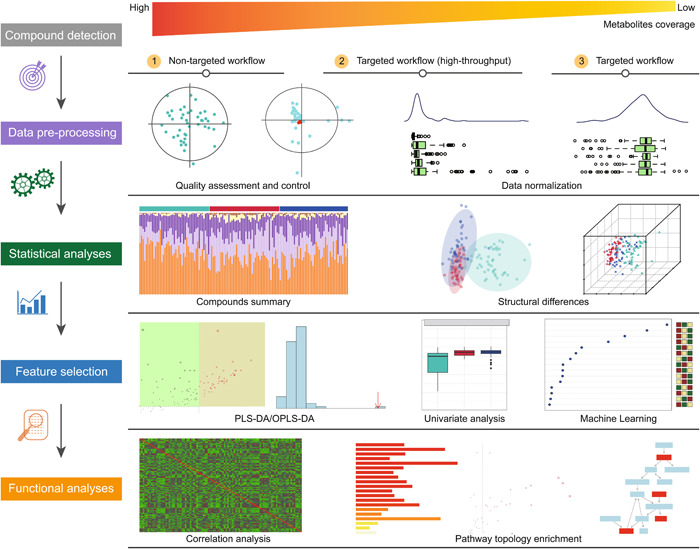
Metabolomic data analyses using the Wekemo Bioincloud platform. After employing three ways (nontargeted screening, targeted screening with high throughput and targeted screening) for compound detection, data preprocessing, statistical analyses, feature selection, and functional analyses are implemented in three workflows. Nontargeted screening provides the highest metabolite coverage, whereas targeted screening yields the lowest coverage. OPLS‐DA, orthogonal partial least‐squares discriminant analysis; PLS‐DA, partial least‐squares discriminant analysis.
MetaboAnalystR [26] is used to perform the nontargeted and targeted metabolomics data for potential detection of metabolite compounds in our platform.
For data preprocessing, we implement quality assessment and quality control to detect the outliers, and to remove metabolites or samples beyond threefold standard error. Then, users have the option to decide whether to perform data normalization, eliminating potential systematic biases during sample collection or metabolite detection.
After that, all metabolites are compared with the KEGG database br08001 [41] to determine the percentage content of each biological role. Standard statistical analyses, including compound summaries and the identification of structural differences, are then conducted.
For feature selection, we offer partial least‐squares discriminant analysis (PLS‐DA) [58] or OPLS‐DA [59] to underlying metabolite patterns discriminating between sample groups. Then, the univariate analysis and machine learning techniques, such as random forest and support vector machine, are also employed for the selection of differential metabolites.
For functional analyses, we offer the correlation analyses, over‐representation analysis (ORA) of pathways [60], topology analysis of pathways, and metabolic pathway maps, for providing intuitive insights into the relationships, functional patterns, and topological structure of metabolites within pathways.
Additionally, we apply maSigPro [61] package to enhance the analysis of metabolism time‐series data for nontargeted workflow and targeted workflow. This is achieved by implementing a generalized linear model, allowing users to identify significant regression relationships between various elements (such as genes, metabolites, or features) and temporal factors (like time, time squared, or specified groups). As part of our efforts, we have constructed a comprehensive high‐throughput targeted metabolism database, with more than 2500 plant metabolites and approximately 1800 animal metabolites.
DISCUSSION
Wekemo Bioincloud platform offers a robust and user‐friendly service, contributing to global collaborative initiatives in the field of multi‐omics research. In comparison to existing online servers for ‐omics data analyses, such as MicrobiomeAnalyst [16], which focused mainly on metagenomic analyses, MetaboAnalystR [26] was specifically tailored for metabolomic analyses, and GeNets [62] was dedicated to genomic analyses. Our platform stands out by providing 22 workflows (Figure 1), encompassing the analysis of amplicon sequencing data, metagenomic data, metatranscriptomic data, metaviromic data, metaproteomic data, genomic data, and physicochemical data. This extensive coverage facilitates the integrated analyses of multiple ‐omics data types, offering researchers a comprehensive and user‐friendly solution.
sMeanwhile, Wekemo Bioincloud platform also provides some personalized analyses tailored to researchers, aligning with their diverse research scopes. For instance, we provide comprehensive bioinformatics databases (ARDB, BacMet, CAZy, CARD, COGs, EC, GO, KEGG, MGE, MetaCyc, QS, VFDB) for in‐depth functional analyses of metagenomic data (Figure 3). The platform supports comparison of enterotypes in different groups, and the prediction secreted of proteins or prophage in samples (Figure 1). To our knowledge, while Majorbio Cloud is also a bioinformatic platform for multi‐omics analyses [13], our platform additionally offers 65 tools (Figure 2) and online SVG editors, empowering researchers to freely adjust their plots for publication.
In the future, we intend to integrate EasyMicrobiome [23] and EasyMetagenome [29] pipelines in our platform, and constantly update the platform within half a year. Additionally, we will enhance the platform with English video tutorials. While the website is currently accessible through Google Translate, our future plans include updating the English version to enhance accessibility for international researchers.
CONCLUSION
In summary, the Wekemo Bioincloud platform emerges as a valuable solution to the escalating challenges presented by the growing volume of meta‐omics data in microbial community studies. This specialized platform encompasses 22 workflows and 65 graphical tools, allowing for the modification of vector outputs. By addressing scalability issues inherent in existing bioinformatics tools, Wekemo Bioincloud enhances the platform's flexibility and user‐friendly experience.
METHODS
The Wekemo Bioincloud is designed as a web application, employing Javascript, HTML, Vue, and Bootstrap for front‐end development. For back‐end data preprocessing and analysis, it incorporates various widely used ‐omics analysis software/tools. The steps include, but are not limited to, quality control, removal of host contamination, filtering rRNA reads and chimeras, addressing redundancy, binning or mapping, splicing or assembling, species or gene annotation, quantification of species, genes, metabolites, proteins, and functional prediction or annotation, as well as the analysis of diversity, differences, correlations, and phylogeny.
All detailed information on all software/tools for each workflow is available on our website. Here, we briefly outline the steps of metagenome workflow (reference‐based). In summary, Trimmomatic [7] is employed for quality control, read filtering, and base correction for FASTQ data. The remaining reads were aligned to the host genome reference by Bowtie [63] to remove host DNA contamination. Kraken [9] is utilized to sequence abundance, MetaPhlAn and mOTUs3 for taxonomic abundance [64], and HUMAnN [10] is employed to identify microbial functions. The output file with .qzv file can be viewed using QIIME 2 [5], and most statistical analyses and plots are generated based on R scripts.
AUTHOR CONTRIBUTIONS
Shunyao Jiang and Guoxing Zhang developed the platform. Yong‐Xin Liu and Shunyao Jiang conceived and coordinated the study. Yunyun Gao drafted the manuscript. Yong‐Xin Liu, Guoxing Zhang, and Shunyao Jiang revised the manuscript. All authors have read the final manuscript and approved it for publication.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
Supporting information
Table S1: Overview of publications citing Wekemo Bioincloud on Google Scholar until December 6, 2023.
ACKNOWLEDGMENTS
The work was financially supported by the Agricultural Science and Technology Innovation Program (CAAS‐ZDRW202308) and the National Natural Science Foundation of China (U21A20182, U23A20148).
Gao, Yunyun , Zhang Guoxing, Jiang Shunyao, and Liu Yong‐Xin. 2024. “Wekemo Bioincloud: A User‐friendly Platform for Meta‐omics Data Analyses.” iMeta 3, e175. 10.1002/imt2.175
DATA AVAILABILITY STATEMENT
Data sharing does not applicable to this article as no data sets were generated or analyzed during the current study. All demonstration data for visualization purposes are available on the Wekemo Bioincloud website (https://www.bioincloud.tech/). Supplementary materials (tables, scripts, graphical abstract, slides, videos, Chinese translated version and update materials) may be found in the online DOI or iMeta Science http://www.imeta.science/.
REFERENCES
- 1. Gao, Yunyun , Li Danyi, and Liu Yong‐Xin. 2023. “Microbiome Research Outlook: Past, Present, and Future.” Protein & Cell 14: 709–712. 10.1093/procel/pwad031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang, Xu , Li Leyuan, Butcher James, Stintzi Alain, and Figeys Daniel. 2019. “Advancing Functional and Translational Microbiome Research Using Meta‐Omics Approaches.” Microbiome 7: 154. 10.1186/s40168-019-0767-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu, Yong‐Xin , Chen Tong, Li Danyi, Fu Jingyuan, and Liu Shuang‐Jiang. 2022. “iMeta: Integrated Meta‐Omics for Biology and Environments.” iMeta 1: e15. 10.1002/imt2.15 [DOI] [Google Scholar]
- 4. Lu, Yao , Zhou Guangyan, Ewald Jessica, Pang Zhiqiang, Shiri Tanisha, and Xia Jianguo. 2023. “MicrobiomeAnalyst 2.0: Comprehensive Statistical, Functional and Integrative Analysis Of Microbiome Data.” Nucleic Acids Research 51: W310–W318. 10.1093/nar/gkad407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bolyen, Evan , Rideout Jai Ram, Dillon Matthew R., Bokulich Nicholas A., Abnet Christian C., Al‐Ghalith Gabriel A., Alexander Harriet, et al. 2019. “Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2.” Nature Biotechnology 37: 852–857. 10.1038/s41587-019-0209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu, Yong‐Xin , Chen Lei, Ma Tengfei, Li Xiaofang, Zheng Maosheng, Zhou Xin, Chen Liang, et al. 2023. “EasyAmplicon: An Easy‐to‐Use, Open‐Source, Reproducible, and Community‐Based Pipeline for Amplicon Data Analysis In Microbiome Research.” iMeta 2: e83. 10.1002/imt2.83 [DOI] [Google Scholar]
- 7. Bolger, Anthony M. , Lohse Marc, and Usadel Bjoern. 2014. “Trimmomatic: A Flexible Trimmer for Illumina Sequence Data.” Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen, Shifu . 2023. “Ultrafast One‐Pass FASTQ Data Preprocessing, Quality Control, and Deduplication Using Fastp.” iMeta 2: e107. 10.1002/imt2.107 [DOI] [Google Scholar]
- 9. Wood, Derrick E. , Lu Jennifer, and Langmead Ben. 2019. “Improved Metagenomic Analysis with Kraken 2.” Genome Biology 20: 257. 10.1186/s13059-019-1891-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beghini, Francesco , McIver Lauren J., Blanco‐Míguez Aitor, Dubois Leonard, Asnicar Francesco, Maharjan Sagun, Mailyan Ana, et al. 2021. “Integrating Taxonomic, Functional, and Strain‐Level Profiling of Diverse Microbial Communities with Biobakery 3.” elife 10: e65088. 10.7554/eLife.65088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xia, Han , Zhang Zhe, Luo Chen, Wei Kangfei, Li Xuming, Mu Xiyu, Duan Meilin, et al. 2023. “MultiPrime: A Reliable and Efficient Tool for Targeted Next‐Generation Sequencing.” iMeta 2: e143. 10.1002/imt2.143 [DOI] [Google Scholar]
- 12. Chen, Tong , Liu Yong‐Xin, and Huang Luqi. 2022. “ImageGP: an Easy‐To‐Use Data Visualization Web Server for Scientific Researchers.” iMeta 1: e5. 10.1002/imt2.5 [DOI] [Google Scholar]
- 13. Ren, Yi , Yu Guo, Shi Caiping, Liu Linmeng, Guo Quan, Han Chang, Zhang Dan, et al. 2022. “Majorbio Cloud: A One‐Stop, Comprehensive Bioinformatic Platform for Multiomics Analyses.” iMeta 1: e12. 10.1002/imt2.12 [DOI] [Google Scholar]
- 14. Zhai, Peng , Yang Longshu, Guo Xiao, Wang Zhe, Guo Jiangtao, Wang Xiaoqi, and Zhu Huaiqiu. 2017. “MetaComp: Comprehensive Analysis Software for Comparative Meta‐Omics Including Comparative Metagenomics.” BMC Bioinformatics 18: 434. 10.1186/s12859-017-1849-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shen, Weitao , Song Ziguang, Zhong Xiao, Huang Mei, Shen Danting, Gao Pingping, Qian Xiaoqian, et al. 2022. “Sangerbox: A Comprehensive, Interaction‐Friendly Clinical Bioinformatics Analysis Platform.” iMeta 1: e36. 10.1002/imt2.36 [DOI] [Google Scholar]
- 16. Chong, Jasmine , Liu Peng, Zhou Guangyan, and Xia Jianguo. 2020. “Using MicrobiomeAnalyst for Comprehensive Statistical, Functional, and Meta‐Analysis of Microbiome Data.” Nature Protocols 15: 799–821. 10.1038/s41596-019-0264-1 [DOI] [PubMed] [Google Scholar]
- 17. Klåvus, Anton , Kokla Marietta, Noerman Stefania, Koistinen Ville M., Tuomainen Marjo, Zarei Iman, Meuronen Topi, et al. 2020. “‘Notame’: Workflow for Non‐Targeted LC–MS Metabolic Profiling.” Metabolites 10: 135. 10.3390/metabo10040135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schiebenhoefer, Henning , Schallert Kay, Renard Bernhard Y., Trappe Kathrin, Schmid Emanuel, Benndorf Dirk, Riedel Katharina, Muth Thilo, and Fuchs Stephan. 2020. “A Complete and Flexible Workflow for Metaproteomics Data Analysis Based on MetaProteomeAnalyzer and Prophane.” Nature Protocols 15: 3212–3239. 10.1038/s41596-020-0368-7 [DOI] [PubMed] [Google Scholar]
- 19. Li, Fengcheng , Zhou Ying, Zhang Ying, Yin Jiayi, Qiu Yunqing, Gao Jianqing, and Zhu Feng. 2022. “POSREG: Proteomic Signature Discovered by Simultaneously Optimizing its Reproducibility and Generalizability.” Briefings in Bioinformatics 23: bbac040. 10.1093/bib/bbac040 [DOI] [PubMed] [Google Scholar]
- 20. Li, Fengcheng , Yin Jiayi, Lu Mingkun, Yang Qingxia, Zeng Zhenyu, Zhang Bing, Li Zhaorong, et al. 2022. “ConSIG: Consistent Discovery of Molecular Signature from OMIC Data.” Briefings in Bioinformatics 23: bbac253. 10.1093/bib/bbac253 [DOI] [PubMed] [Google Scholar]
- 21. Yang, Qingxia , Gong Yaguo, and Zhu Feng. 2023. “Critical Assessment of the Biomarker Discovery and Classification Methods for Multiclass Metabolomics.” Analytical Chemistry 95: 5542–5552. 10.1021/acs.analchem.2c04402 [DOI] [PubMed] [Google Scholar]
- 22. Yang, Qingxia , Li Yi, Li Bo, and Gong Yaguo. 2022. “A Novel Multi‐Class Classification Model for Schizophrenia, Bipolar Disorder and Healthy Controls Using Comprehensive Transcriptomic Data.” Computers in Biology and Medicine 148: 105956. 10.1016/j.compbiomed.2022.105956 [DOI] [PubMed] [Google Scholar]
- 23. Wen, Tao , Niu Guoqing, Chen Tong, Shen Qingrong, Yuan Jun, and Liu Yong‐Xin. 2023. “The Best Practice for Microbiome Analysis Using R.” Protein & Cell 14: 713–725. 10.1093/procel/pwad024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li, Wenjun , Wang Likun, Li Xiaofang, Zheng Xin, Cohen Michael F., and Liu Yong‐Xin. 2022. “Sequence‐Based Functional Metagenomics Reveals Novel Natural Diversity of Functioning CopA in Environmental Microbiomes.” Genomics, Proteomics & Bioinformatics 20: 1–12. 10.1016/j.gpb.2022.08.006 [DOI] [PubMed] [Google Scholar]
- 25. Cambiaghi, Alice , Ferrario Manuela, and Masseroli Marco. 2017. “Analysis of Metabolomic Data: Tools, Current Strategies and Future Challenges for Omics Data Integration.” Briefings in Bioinformatics 18: 498–510. 10.1093/bib/bbw031 [DOI] [PubMed] [Google Scholar]
- 26. Pang, Zhiqiang , Chong Jasmine, Zhou Guangyan, de Lima Morais David Anderson, Chang Le, Barrette Michel, Gauthier Carol, et al. 2021. “MetaboAnalyst 5.0: Narrowing the Gap Between Raw Spectra and Functional Insights.” Nucleic Acids Research 49: W388–W396. 10.1093/nar/gkab382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen, Tong , Zhang Haiyan, Liu Yu, Liu Yong‐Xin, and Huang Luqi. 2021. “EVenn: Easy to Create Repeatable and Editable Venn Diagrams and Venn Networks Online.” Journal of Genetics and Genomics 48: 863–866. 10.1016/j.jgg.2021.07.007 [DOI] [PubMed] [Google Scholar]
- 28. Wen, Tao , Xie Penghao, Yang Shengdie, Niu Guoqing, Liu Xiaoyu, Ding Zhexu, Xue Chao, et al. 2022. “ggClusterNet: An R Package for Microbiome Network Analysis and Modularity‐Based Multiple Network Layouts.” iMeta 1: e32. 10.1002/imt2.32 [DOI] [Google Scholar]
- 29. Liu, Yong‐Xin , Qin Yuan, Chen Tong, Lu Meiping, Qian Xubo, Guo Xiaoxuan, and Bai Yang. 2021. “A Practical Guide to Amplicon and Metagenomic Analysis of Microbiome Data.” Protein & Cell 12: 315–330. 10.1007/s13238-020-00724-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu, Chi , Cui Yaoming, Li Xiangzhen, and Yao Minjie. 2021. “Microeco: An R Package for Data Mining in Microbial Community Ecology.” FEMS Microbiology Ecology 97: fiaa255. 10.1093/femsec/fiaa255 [DOI] [PubMed] [Google Scholar]
- 31. Gao, Chun‐Hui , Yu Guangchuang, and Cai Peng. 2021. “ggVennDiagram: An Intuitive, Easy‐to‐Use, and Highly Customizable R Package to Generate Venn Diagram.” Frontiers in Genetics 12: 706907. 10.3389/fgene.2021.706907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li, Jianfeng , Miao Benben, Wang Shixiang, Dong Wei, Xu Houshi, Si Chenchen, and Wang Wei, et al. 2022. “Hiplot: A Comprehensive and Easy‐to‐Use Web Service for Boosting Publication‐Ready Biomedical Data Visualization.” Briefings in Bioinformatics 23: bbac261. 10.1093/bib/bbac261 [DOI] [PubMed] [Google Scholar]
- 33. Beck, Lauren C. , Masi Andrea C., Young Gregory R., Vatanen Tommi, Lamb Christopher A., Smith Rachel, Coxhead Jonathan, et al. 2022. “Strain‐Specific Impacts of Probiotics are a Significant Driver of Gut Microbiome Development in Very Preterm Infants.” Nature Microbiology 7: 1525–1535. 10.1038/s41564-022-01213-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu, B. , and Pop M.. 2009. “ARDB—Antibiotic Resistance Genes Database.” Nucleic Acids Research 37: D443–D447. 10.1093/nar/gkn656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pal, Chandan , Bengtsson‐Palme Johan, Rensing Christopher, Kristiansson Erik, and Larsson DG Joakim. 2014. “BacMet: Antibacterial Biocide and Metal Resistance Genes Database.” Nucleic Acids Research 42: D737–D743. 10.1093/nar/gkt1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cantarel, B. L. , Coutinho P. M., Rancurel C., Bernard T., Lombard V., and Henrissat B.. 2009. “The Carbohydrate‐Active EnZymes Database (CAZy): An Expert Resource for Glycogenomics.” Nucleic Acids Research 37: D233–D238. 10.1093/nar/gkn663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alcock, Brian P. , Huynh William, Chalil Romeo, Smith Keaton W., Raphenya Amogelang R., Wlodarski Mateusz A., Edalatmand Arman, et al. 2023. “CARD 2023: Expanded Curation, Support for Machine Learning, and Resistome Prediction at the Comprehensive Antibiotic Resistance Database.” Nucleic Acids Research 51: D690–D699. 10.1093/nar/gkac920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tatusov, R. L. 2001. “The COG Database: New Developments in Phylogenetic Classification of Proteins from Complete Genomes.” Nucleic Acids Research 29: 22–28. 10.1093/nar/29.1.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bairoch, A. 2000. “The ENZYME Database in 2000.” Nucleic Acids Research 28: 304–305. 10.1093/nar/28.1.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gene Ontology Consortium . 2004. “The Gene Ontology (GO) Database and Informatics Resource.” Nucleic Acids Research 32: 258D–261D. 10.1093/nar/gkh036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kanehisa, M. 2000. “KEGG: Kyoto Encyclopedia of Genes and Genomes.” Nucleic Acids Research 28: 27–30. 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frost, Laura S. , Leplae Raphael, Summers Anne O., and Toussaint Ariane. 2005. “Mobile Genetic Elements: The Agents of Open Source Evolution.” Nature Reviews Microbiology 3: 722–732. 10.1038/nrmicro1235 [DOI] [PubMed] [Google Scholar]
- 43. Caspi, Ron , Billington Richard, Keseler Ingrid M., Kothari Anamika, Krummenacker Markus, Midford Peter E., Ong Wai Kit, et al. 2020. “The MetaCyc Database of Metabolic Pathways and Enzymes—A 2019 Update.” Nucleic Acids Research 48: D445–D453. 10.1093/nar/gkz862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Klein, Karsten , Garkov Dimitar, Rütschlin Sina, Böttcher Thomas, and Schreiber Falk. 2021. “QSDB—A Graphical Quorum Sensing Database.” Database 2021: baab058. 10.1093/database/baab058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu, Bo , Zheng Dandan, Zhou Siyu, Chen Lihong, and Yang Jian. 2022. “VFDB 2022: A General Classification Scheme for Bacterial Virulence Factors.” Nucleic Acids Research 50: D912–D917. 10.1093/nar/gkab1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu, Jennifer , Breitwieser Florian P., Thielen Peter, and Salzberg Steven L.. 2017. “Bracken: Estimating Species Abundance in Metagenomics Data.” PeerJ Computer Science 3: e104. 10.7717/peerj-cs.104 [DOI] [Google Scholar]
- 47. St»hle, Lars , and Wold Svante. 1989. “Analysis of Variance (ANOVA).” Chemometrics and Intelligent Laboratory Systems 6: 259–272. 10.1016/0169-7439(89)80095-4 [DOI] [Google Scholar]
- 48. Love, Michael , Anders Simon, and Huber Wolfgang. 2014. “Differential Analysis of Count Data—The DESeq. 2 Package.” Genome Biology 15: 10–1186. 10.1186/s13059-014-0550-8 [DOI] [Google Scholar]
- 49. McKight, Patrick E. , and Najab Julius. 2010. “Kruskal‐Wallis Test.” The Corsini Encyclopedia of Psychology 1: 1–10. 10.1002/9780470479216.corpsy0491 [DOI] [Google Scholar]
- 50. Segata, Nicola , Izard Jacques, Waldron Levi, Gevers Dirk, Miropolsky Larisa, Garrett Wendy S., and Huttenhower Curtis. 2011. “Metagenomic Biomarker Discovery and Explanation.” Genome Biology 12: R60. 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xue, Chun‐Xu , Lin Heyu, Zhu Xiao‐Yu, Liu Jiwen, Zhang Yunhui, Rowley Gary, Todd Jonathan D., Li Meng, and Zhang Xiao‐Hua. 2021. “DiTing: A Pipeline to Infer and Compare Biogeochemical Pathways from Metagenomic and Metatranscriptomic Data.” Frontiers in Microbiology 12: 698286. 10.3389/fmicb.2021.698286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li, Dinghua , Liu Chi‐Man, Luo Ruibang, Sadakane Kunihiko, and Lam Tak‐Wah. 2015. “MEGAHIT: An Ultra‐Fast Single‐Node Solution for Large and Complex Metagenomics Assembly via Succinct De Bruijn Graph.” Bioinformatics 31: 1674–1676. 10.1093/bioinformatics/btv033 [DOI] [PubMed] [Google Scholar]
- 53. Hyatt, Doug , Chen Gwo‐Liang, LoCascio Philip F., Land Miriam L., Larimer Frank W., and Hauser Loren J.. 2010. “Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification.” BMC Bioinformatics 11: 119. 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kang, Dongwan D. , Li Feng, Kirton Edward, Thomas Ashleigh, Egan Rob, An Hong, and Wang Zhong. 2019. “MetaBAT 2: An Adaptive Binning Algorithm for Robust and Efficient Genome Reconstruction from Metagenome Assemblies.” PeerJ 7: e7359. 10.7717/peerj.7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Parks, Donovan H. , Rinke Christian, Chuvochina Maria, Chaumeil Pierre‐Alain, Woodcroft Ben J., Evans Paul N., Hugenholtz Philip, and Tyson Gene W.. 2017. “Recovery of Nearly 8,000 Metagenome‐Assembled Genomes Substantially Expands the Tree of Life.” Nature Microbiology 2: 1533–1542. 10.1038/s41564-017-0012-7 [DOI] [PubMed] [Google Scholar]
- 56. Parks, Donovan H. , Imelfort Michael, Skennerton Connor T., Hugenholtz Philip, and Tyson Gene W.. 2015. “CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes.” Genome Research 25: 1043–1055. 10.1101/gr.186072.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Olm, Matthew R. , Brown Christopher T., Brooks Brandon, and Banfield Jillian F.. 2017. “Drep: A Tool for Fast and Accurate Genomic Comparisons that Enables Improved Genome Recovery from Metagenomes through De‐Replication.” The ISME Journal 11: 2864–2868. 10.1038/ismej.2017.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee, Loong Chuen , Liong Choong‐Yeun, and Jemain Abdul Aziz. 2018. “Partial Least Squares‐Discriminant Analysis (PLS‐DA) for Classification of High‐Dimensional (HD) Data: A Review of Contemporary Practice Strategies and Knowledge Gaps.” Analyst 143: 3526–3539. 10.1039/C8AN00599K [DOI] [PubMed] [Google Scholar]
- 59. Boccard, Julien , and Rutledge Douglas N.. 2013. “A Consensus Orthogonal Partial Least Squares Discriminant Analysis (OPLS‐DA) Strategy for Multiblock Omics Data Fusion.” Analytica Chimica Acta 769: 30–39. 10.1016/j.aca.2013.01.022 [DOI] [PubMed] [Google Scholar]
- 60. Wieder, Cecilia , Frainay Clément, Poupin Nathalie, Rodríguez‐Mier Pablo, Vinson Florence, Cooke Juliette, PJPJ Lai Rachel, et al. 2021. “Pathway Analysis in Metabolomics: Recommendations for the Use of Over‐Representation Analysis.” PLoS Computational Biology 17: e1009105. 10.1371/journal.pcbi.1009105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Conesa, Ana , Nueda María José, Ferrer Alberto, and Talón Manuel. 2006. “maSigPro: A Method to Identify Significantly Differential Expression Profiles in Time‐Course Microarray Experiments.” Bioinformatics 22: 1096–1102. 10.1093/bioinformatics/btl056 [DOI] [PubMed] [Google Scholar]
- 62. Li, Taibo , Kim April, Rosenbluh Joseph, Horn Heiko, Greenfeld Liraz, An David, Zimmer Andrew, et al. 2018. “GeNets: A Unified Web Platform for Network‐Based Genomic Analyses.” Nature Methods 15: 543–546. 10.1038/s41592-018-0039-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Langmead, Ben , and Salzberg Steven L.. 2012. “Fast Gapped‐Read Alignment with Bowtie 2.” Nature Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sun, Zheng , Huang Shi, Zhang Meng, Zhu Qiyun, Haiminen Niina, Carrieri Anna Paola, Vázquez‐Baeza Yoshiki, et al. 2021. “Challenges in Benchmarking Metagenomic Profilers.” Nature Methods 18: 618–626. 10.1038/s41592-021-01141-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Overview of publications citing Wekemo Bioincloud on Google Scholar until December 6, 2023.
Data Availability Statement
Data sharing does not applicable to this article as no data sets were generated or analyzed during the current study. All demonstration data for visualization purposes are available on the Wekemo Bioincloud website (https://www.bioincloud.tech/). Supplementary materials (tables, scripts, graphical abstract, slides, videos, Chinese translated version and update materials) may be found in the online DOI or iMeta Science http://www.imeta.science/.