

Abstract
We analyzed >700,000 single-nucleus RNA sequencing profiles from 106 donors during prenatal and postnatal developmental stages and identified lineage-specific programs that underlie the development of specific subtypes of excitatory cortical neurons, interneurons, glial cell types, and brain vasculature. By leveraging single-nucleus chromatin accessibility data, we delineated enhancer gene regulatory networks and transcription factors that control commitment of specific cortical lineages. By intersecting our results with genetic risk factors for human brain diseases, we identified the cortical cell types and lineages most vulnerable to genetic insults of different brain disorders, especially autism. We find that lineage-specific gene expression programs up-regulated in female cells are especially enriched for the genetic risk factors of autism. Our study captures the molecular progression of cortical lineages across human development.
Development of the human cerebral cortex spans months during prenatal stages and years after birth, generating tens to hundreds of cell types across multiple cortical areas. This complex process is orchestrated by lineage-specific gene expression programs that guide the production, migration, differentiation, and maturation of neuronal and glial cell types, as well as the formation of projections and neuronal circuits. Alterations in these regulatory gene programs during development lead to the pathogenesis of neurodevelopmental and psychiatric disorders, including autism spectrum disorder (ASD) and schizophrenia (SCZ). Most previous studies have focused on investigating the molecular processes that underly human cortical development during the second trimester of gestation (1–5), which is the peak of cortical neurogenesis and neuronal migration. These studies have revealed molecular signatures of progenitor cells and neuronal and glial cell types, as well as the early specification of neurons into broad subtypes and their arealization across the cortex. However, later stages of human cortical development—including the third trimester of gestation, birth, and neonatal and early postnatal development—have been largely studied using bulk genomic approaches.
Single-nucleus RNA sequencing analysis of prenatal and postnatal human cortical development
To gain a comprehensive view of human cortical development across prenatal and postnatal stages, we used single-nucleus RNA sequencing (snRNA-seq) (6) to profile 413,682 nuclei from 108 tissue samples derived from 60 neurotypical individuals. We sampled nuclei from ages spanning from the second trimester of gestation to adulthood, including samples from the third trimester and early postnatal stages that are often excluded or underrepresented in genomic studies of the human brain. We acquired data from the ganglionic eminences—the major source of cortical interneurons (7, 8)—and from the cortex. We used Seurat (9) to perform unbiased clustering and uniform manifold approximation and projection (UMAP) embedding. After removing a cluster of cell debris (fig. S1A), we retained 358,663 nuclei. To extend our analyses to more brain samples and nuclei, we integrated our data with published datasets of prenatal and postnatal human cortical development (10–12). After data integration (fig. S1B), our final dataset included 709,372 nuclei and 169 brain tissue samples from 106 individuals (Fig. 1A and data S1). We identified clusters corresponding to neural progenitors and to the major subtypes of excitatory and inhibitory neurons, glia, and vascular cells (Fig. 1, B and C), indicating that we were able to capture transcriptomic changes underlying differentiation and maturation of cortical cell types across development. We detected similar numbers of genes, transcripts, and mitochondrial RNA ratios across different samples (fig. S1C), with a median of 1106 genes and 1609 transcripts per nucleus and some variability from sample to sample. These relative numbers are comparable with published single-cell genomics data collected from the human brain (13), with mature neuron cell types expressing higher numbers of genes and transcripts than other cell types (fig. S1D). We observed neither batch effects (nuclei from different samples were well intermixed) nor clusters composed of nuclei from a single sample (fig. S1E). Nuclei were captured from the prefrontal, cingulate, temporal, insular, and motor cortices (fig. S1F). For prenatal samples that were not sex-identified, we determined their sex using sex-specific gene expression (fig. S1G). Our dataset included 45 female and 61 male subjects. We observed that nuclei clustered according to developmental age (Fig. 1D), suggesting that transcriptomic changes associated with development are a major driver of cell identity.
Fig. 1. Brain tissue samples used for data collection and initial clustering of snRNA-seq data.
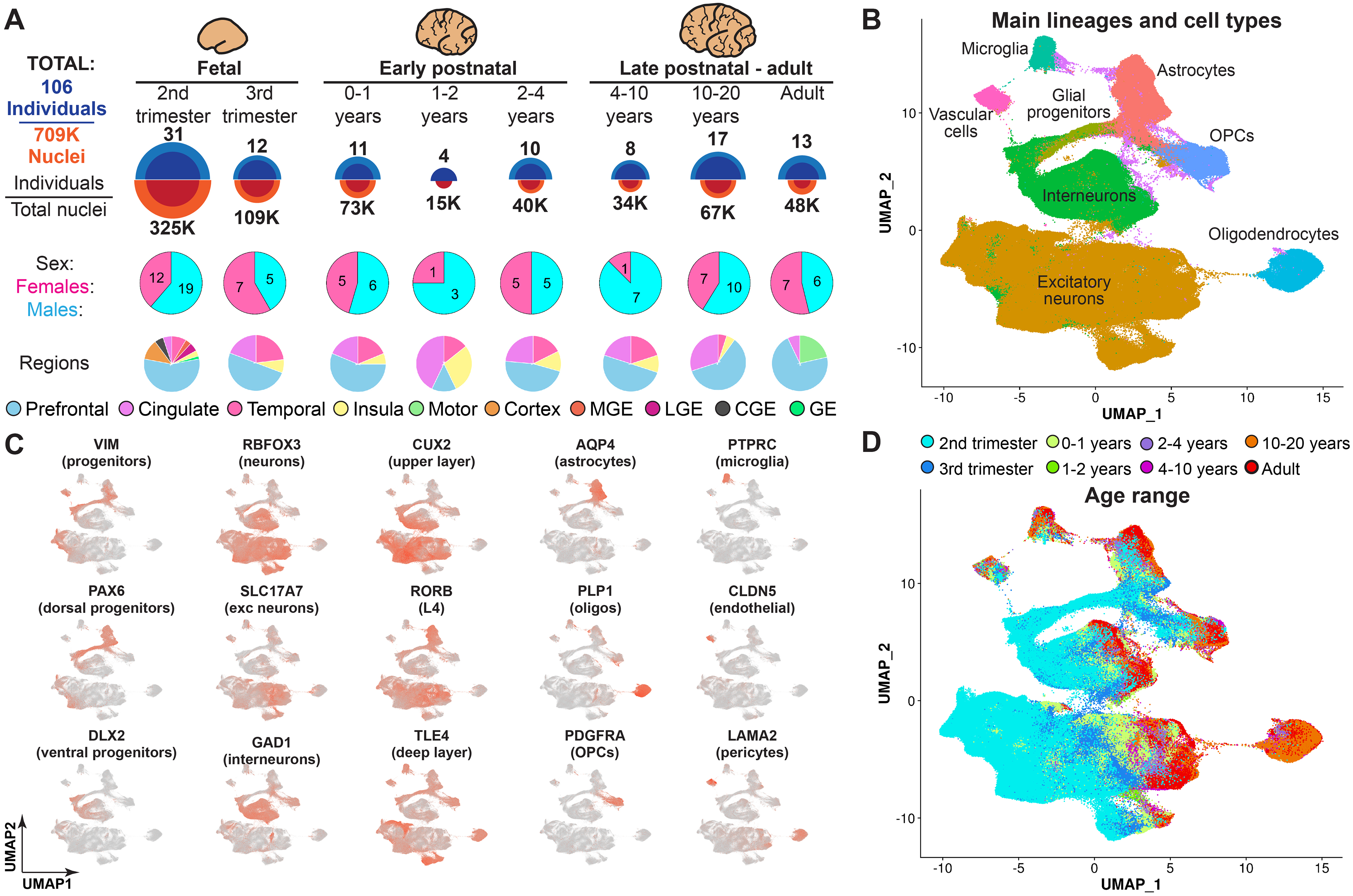
(A) Overview of the tissue samples used in this study, including the number of individuals and the ages and brain regions captured in the snRNA-seq dataset. MGE, medial ganglionic eminence; LGE, lateral ganglionic eminence; CGE, caudal ganglionic eminence; GE, ganglionic eminence. (B) Clustering of the entire dataset, with the major lineages labeled. (C) Expression of cell type–specific markers used to determine cardinal lineages. exc neurons, excitatory neurons. (D) Nuclei labeled by their developmental age.
Analysis of specific excitatory neuron and interneuron lineages
We next examined the developmental trajectories of excitatory and inhibitory neurons. First, we selected clusters corresponding to dorsal forebrain progenitors (including radial glia and intermediate precursor cells) as well as clusters containing excitatory neurons. By re-clustering this data and referencing molecularly defined cell types annotated in the Allen Brain Atlas (14), we identified clusters corresponding to known subtypes of excitatory neurons, including upper (L2–3) and deep-layer intratelencephalic (L5–6-IT) projection neurons, layer 4 neurons (L4), and layer 5 (L5) and layer 6 (L6) extratelencephalic projection neurons, as well as subplate neurons (SP) that were present transiently during the second trimester (Fig. 2A and fig. S2A). We next used the analysis toolkit Monocle 3 (15) along with custom scripts (see methods in the supplementary materials) to construct cellular trajectories on the basis of snRNA-seq data (Fig. 2A and fig. S2B), select trajectory branches corresponding to specific lineages, and calculate pseudotime for each nucleus. Pseudotime corresponded well to the developmental age of nuclei in each lineage (Fig. 2A). We identified several branching points in the trajectory: between two major groups of excitatory neurons L2–3, L4, and L5–6-IT (Ex1) and L5 and L6 (Ex2) and between L4 and L2–3 or L5–6-IT (Ex3). Next, we aimed to investigate developmental gene expression changes during differentiation and maturation of γ-aminobutyric acid–mediated interneuron (IN) lineages. We selected nuclei from both ventral forebrain progenitors and cortical interneurons, re-clustered the data, and identified known classes of cortical interneurons (Fig. 2B and fig. S2C), including interneurons expressing vasoactive intestinal polypeptide (VIP), calretinin (CALB2), reelin (RELN), and nitric acid synthase (NOS); chandelier (PV-CH) and basket (PV-BSK) interneurons expressing parvalbumin (PV), membrane metalloendopeptidase (MME), and tachykinin precursor 1 (TAC1); and interneurons expressing somatostatin (SST) and co-expressing SST and reelin (SST-RELN). We then reconstructed lineage trajectories corresponding to each interneuron subtype (Fig. 2B and fig. S2D), as well as point of trajectory divergence, such as trajectory branches including medial ganglionic eminence (MGE)–derived (IN1) and caudal ganglionic eminence (CGE)–derived (IN2) interneurons. We calculated pseudotime for each nucleus, which correlated well with the developmental age of the interneurons. Next, we asked whether different neuronal lineages in the human cortex mature at different rates. We correlated pseudotime with the developmental age in each neuronal lineage and observed that neuronal types fell into two main groups: those that mostly matured by the end of the second trimester, and those whose transcriptome profiles continued to change through the third trimester and after birth (Fig. 2C). The first group included L5, L5–6-IT, and all interneuron subtypes, whereas the second group contained L2–3, L4, and L6 excitatory neurons. This result suggests that certain types of human cortical neurons have a protracted maturation timeline.
Fig. 2. Analysis of excitatory and inhibitory neuron lineages.

(A) Cell types, reconstructed single-cell trajectories, and age distribution for subtypes of excitatory neurons. L2–3, upper-layer intratelencephalic projection neurons; L4, layer 4 neurons; L5–6-IT, deep-layer intratelencephalic projection neurons; L6, layer 6 neurons; L5, layer 5 neurons; SP, subplate neurons. (B) Identification of interneuron trajectories. (C) Rates of maturation of subtypes of excitatory neurons and interneurons. (D) GO analysis of genes with different age of onset of expression. FDR, false discovery rate; Reg., regulation. (E) Examples of top lineage- and branch-specific genes with transient and burst expression patterns. (F) Number of transient and burst genes in specific lineages and branches. (G) Spatial transcriptomic analysis of 140 lineage-specific genes, showing the spatial map of annotated cell types across development. GW22, 22 weeks of gestation; 2wk, 2 weeks postnatal; 25yo, 25-year-old; PFC, prefrontal cortex; Ex, excitatory; radial glial. (H) Examples of deep-layer neuronal markers with early patterned layer-specific expression (putative layer location is noted in parentheses).
Once we isolated trajectory branches corresponding to each neuronal lineage, we sought to identify lineage-specific gene expression programs. We used an approach that allows identification of lineage-specific programs by comparing dynamic expression profiles of each gene in a lineage of interest to all other neuronal, glial, and non-neural lineages (see methods). In addition, we applied this approach to identify genes specific to related lineages in the excitatory neuron and interneuron trajectory branches. In total, we identified 1062 lineage-specific genes and 405 branch-specific genes (data S2). We classified these genes according to the age of onset of gene expression (50% of the maximum expression) and performed Gene Ontology (GO) analysis for the genes up-regulated at each developmental time point (Fig. 2D). During the second trimester of gestation, we saw enrichment in pathways related to neurogenesis, differentiation, and process growth. Up-regulation of synaptogenesis and ion transport pathways could be observed during the third trimester but was most profound between birth and 1 year of age. Enrichment in synaptic pathways could be observed until adulthood.
In addition to classifying genes according to their age of appearance, we also characterized dynamic expression patterns of lineage-specific genes. The two most common patterns we observed were transient expression and burst expression where up-regulation would start at a certain age and continue into adulthood (Fig. 2E). Our analysis identified several putative regulators of neuronal lineage commitment, such as transcriptional regulator MN1 specific to L2–3, L5–6-IT, and L4 neurons; noncoding RNAs CYP1B1-AS1 and LINC00507 enriched in L2–3 neurons; and HS3ST4 specific to L5 neurons. We saw that genes enriched in more broad lineage branches tended to be transiently expressed, whereas genes specific to mature neuronal cell types mostly followed burst expression patterns (Fig. 2F). This suggests gradual commitment and specification of neuronal cell types through a series of transient and burst transcriptional events. We also classified additional less common expression patterns, such as biphasic expression (fig. S2E), and identified different biological processes enriched for genes with burst and transient expression patterns (fig. S2F). Finally, we identified genes dynamically expressed during the specification of subplate neurons by comparing lineages during the second and third trimester of gestation when these cells are present (fig. S2G). Using spatial transcriptomic analysis of 140 genes across three developmental time points, we were able to identify and visualize the spatial location of cell-specific clusters overlaid on the tissue cytoarchitecture. Focusing on early emerging lineage-specific genes, we validated the spatiotemporal expression of excitatory layer-specific markers (Fig. 2, G and H, and fig. S3). We observed that broad classes of excitatory neurons in the Ex1, Ex2, and Ex3 trajectory branches are restricted to specific cortical layers during the second trimester of gestation. Moreover, several markers of L4 neurons, such as hippocalcin (HPCA) and gremlin 2 (GREM2), are expressed in a layer-restricted manner during the second trimester of gestation, suggesting that L4 neuronal identity starts to be specified early in development. The layer identity of most excitatory neurons emerges by birth (fig. S3) based on the lineage-specific signatures that we find specify human cortical neurons and their segregation to cortical layers.
Dissection of glial and non-neural lineages
We further focused on the analysis of glial lineages, including astrocytes and oligodendrocytes. We re-clustered glial progenitors, oligodendrocyte precursor cells (OPCs), oligodendrocytes, and astrocytes and performed trajectory analysis (Fig. 3A). We identified two types of astrocytes: fibrous astrocytes with high expression of glial fibrillary acidic protein (GFAP) and protoplasmic astrocytes with low expression of GFAP and high expression of glutamate transporter GLAST (SLA1A3) (fig. S4A). Next, we performed identification of lineage-specific genes in the manner described for neuronal lineages (data S2). We first focused on genes that were expressed at the divergence of astrocyte and oligo trajectory branches (Fig. 3B). We observed well-known transcription factors guiding commitment to the oligo and astrocyte lineages, including OLIG1, OLIG2, ID4, and SOX9, as well as other putative regulators, such as the zinc finger protein ZCCHC24 specific to the oligo lineage and a DNA binding protein, STOX1, enriched in astrocytes. When comparing fibrous and protoplasmic astrocytes, we identified gene programs specific to these cell types (Fig. 3C). Genes up-regulated in protoplasmic astrocytes after birth and during the first year of life were mostly associated with the transport of glutamate and its metabolites (Fig. 3D), suggesting a maturation program to support neuronal firing during the early postnatal period. For oligodendrocytes, we observed that genes up-regulated during the second and third trimesters were associated with glial cell differentiation, whereas myelination genes were up-regulated after birth and continued to be expressed into adulthood (Fig. 3E). Analysis of microglia development (Fig. 3F) identified three cell trajectories (MG-1, −2, and −3), one of which (MG-3) was associated with highly activated microglia and was present in a small number of samples. These trajectories were confirmed by an alternative analysis using Slingshot (fig. S4B) (16). We focused on the nonactivated microglia trajectories (MG-1 and MG-2), which were differentiated from each other by expression of a proinflammatory microglia marker, IKZF1, expressed in MG-2. IKZF1 was the only gene differentiating MG-1 and MG-2, suggesting that these trajectories may represent two different states of the same microglia cell type rather than different subtypes; therefore, we focused on genes developmentally expressed in both of these microglia cell clusters. By performing GO analysis of microglia-specific genes up-regulated at different developmental stages, we observed complement genes associated with synaptic pruning up-regulated in microglia after birth and during the first year of life (Fig. 3G and fig. S4C). These findings suggest that the developmental period between birth and the first year of life is a critical period of synaptic formation and plasticity that involves not only neuronal lineages but also protoplasmic astrocytes and microglia. Finally, we identified gene programs associated with the maturation of brain endothelial cells and pericytes (fig. S4, D to F). Our data suggest a coordinated maturation of neuronal and glial cell functions that ensures proper formation and maintenance of neuronal circuits.
Fig. 3. Analysis of cortical glial lineages.
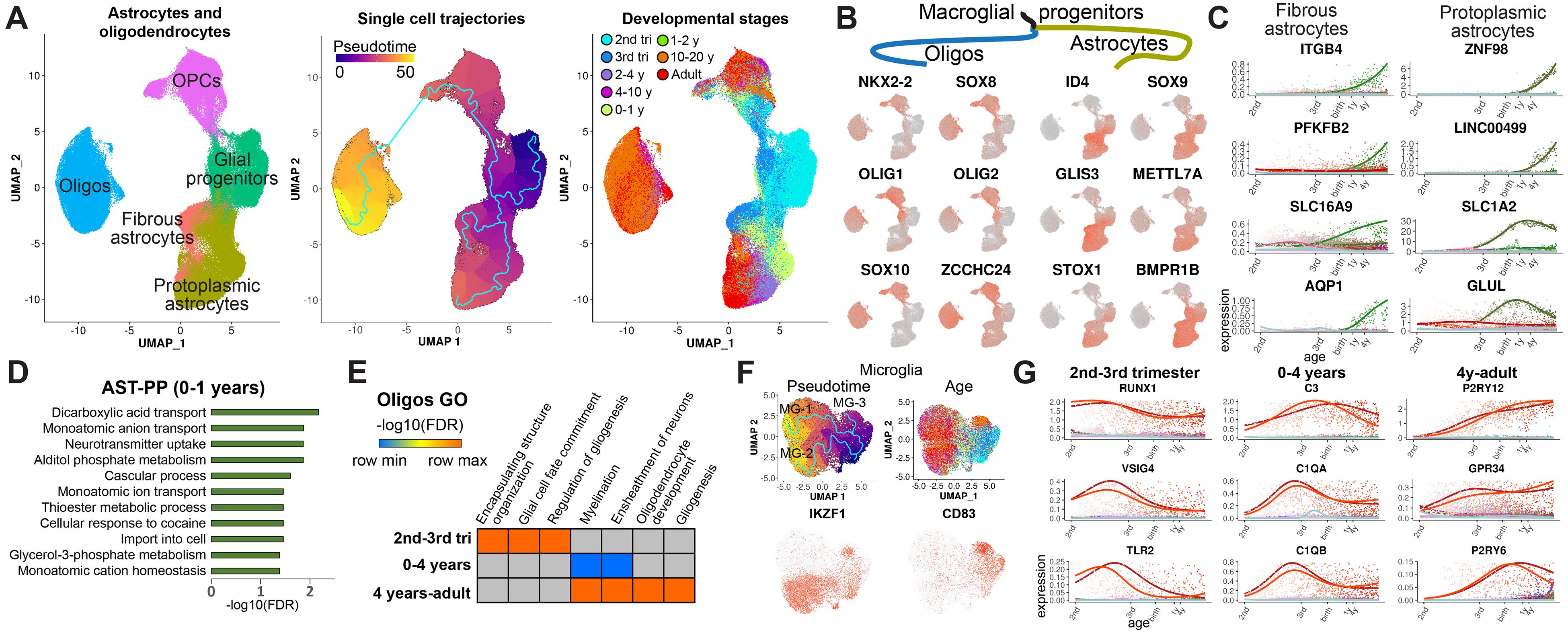
(A) Clusters and trajectories of glial progenitors, astrocytes, and oligodendrocytes. (B) Example genes specific to oligodendrocyte and astrocyte lineage branches. (C) Examples of top dynamically expressed genes specific to fibrous and protoplasmic astrocytes. (D) GO analysis of protoplasmic astrocyte-specific genes expressed during the first year of life. (E) Pathways enriched for oligo lineage-specific genes expressed at different developmental stages. (F) Analysis of microglia lineages. (G) Temporal patterns of developmental microglia genes.
Integration with single-cell open chromatin data and identification of lineage-specific gene regulatory networks
Epigenetic regulation plays a crucial role in cortical neuron lineage commitment and specification. To identify lineage-specific transcriptional and epigenetic regulators of the cortical lineages identified in the snRNA-seq data, we leveraged the recently published single-nucleus assay for transposase-accessible chromatin using sequencing (snATAC-seq) data from the developing human cortex during prenatal and postnatal stages (4, 10, 11, 17). First, we combined snATAC-seq data from four datasets, obtaining 284,907 snATAC-seq profiles from 57 tissue samples and 42 individuals across the second trimester and early postnatal stages of development, as well as adulthood. We then used Seurat to integrate the resulting snATAC-seq data with our snRNA-seq data and mapped the integrated snATAC-seq data to the snRNA-seq clusters, UMAP space, and cell types (Fig. 4A; see methods). We observed that the developmental ages for the snATAC-seq and snRNA-seq profiles were well aligned (Figs. 4A and 1D). Gene activity (open chromatin in the promoter and gene body) of cell type marker genes suggested that snATAC-seq profiles mapped to corresponding transcriptionally defined neuronal and glial cell types (fig. S5A). Next, we repeated the integration and mapping procedure for three major lineage classes: excitatory neurons, interneurons, and glia (astrocytes and oligodendrocytes) (Fig. 4, B to D, and fig. S5, B to D). We omitted microglia and vascular cells because of a low number of snATAC-seq profiles in these lineages. After mapping snATAC-seq data to the transcriptionally defined lineages, we selected snATAC-seq cells along each lineage branch (fig. S5, B to D). Not all lineages could be reliably recovered owing to the smaller size of the snATAC-seq dataset and the lack of key developmental stages, such as the third trimester. We therefore focused on lineages that had ATAC cells along the entire span of the trajectory, including four excitatory neuron lineages, five interneuron lineages, and both types of astrocytes and oligodendrocytes, as indicated in Fig. 4, B to D. Plots of lineage-specific gene activity over pseudotime demonstrated that we accurately mapped and selected lineage-specific snATAC-seq profiles (Fig. 4, B to D). Finally, we leveraged SCENIC+ (18), a recently developed algorithm that uses paired single-cell transcriptomic and open chromatin data to identify enhancer gene regulatory networks (eGRNs) and candidate transcription factors that regulate expression of target genes in these networks. We applied SCENIC+ to the snRNA-seq and snATAC-seq profiles in each lineage to identify open chromatin regions correlated with pseudotime, putative enhancers, candidate transcription factors (TFs) that bind them, and their association with lineage-specific dynamically expressed genes (data S3). In total, we identified 51 transcription factors regulating 1373 lineage-specific genes through predicted binding of 4846 regulatory chromatin regions. We observed networks regulated by previously known lineage-specific transcriptional regulators, such as SOX5 in deep-layer projection neurons (Fig. 4B), LHX6 in MGE-derived PV and SST interneurons (Fig. 4C and data S3), OLIG2 in oligodendrocytes, and SOX9 in astrocytes (Fig. 4D). Additionally, we identified previously unrecognized (to the best of our knowledge) putative lineage-specific transcriptional regulators, such as BACH2, predicted to regulate several key deep-layer transcription factors in L5 neurons, including FOXP2 and FEZF2, as well as NFIX and ZNF184 specific to L2–3 neurons and regulating expression of the upper-layer master transcription factor CUX2 (Fig. 4B). Our results also suggest the role of the transcription factor MAFB in parvalbumin interneuron specification (Fig. 4C), as well as of FOXN2 and RFX4 in determining the fate of oligodendrocytes and protoplasmic astrocytes, respectively (Fig. 4D). Our data shed new light on epigenetic control of neural lineage commitment and identify putative transcription factors and regulatory networks that define the fate of specific human cortical neuronal and glial cell types.
Fig. 4. Identification of lineage-specific epigenetic and transcriptional regulators.
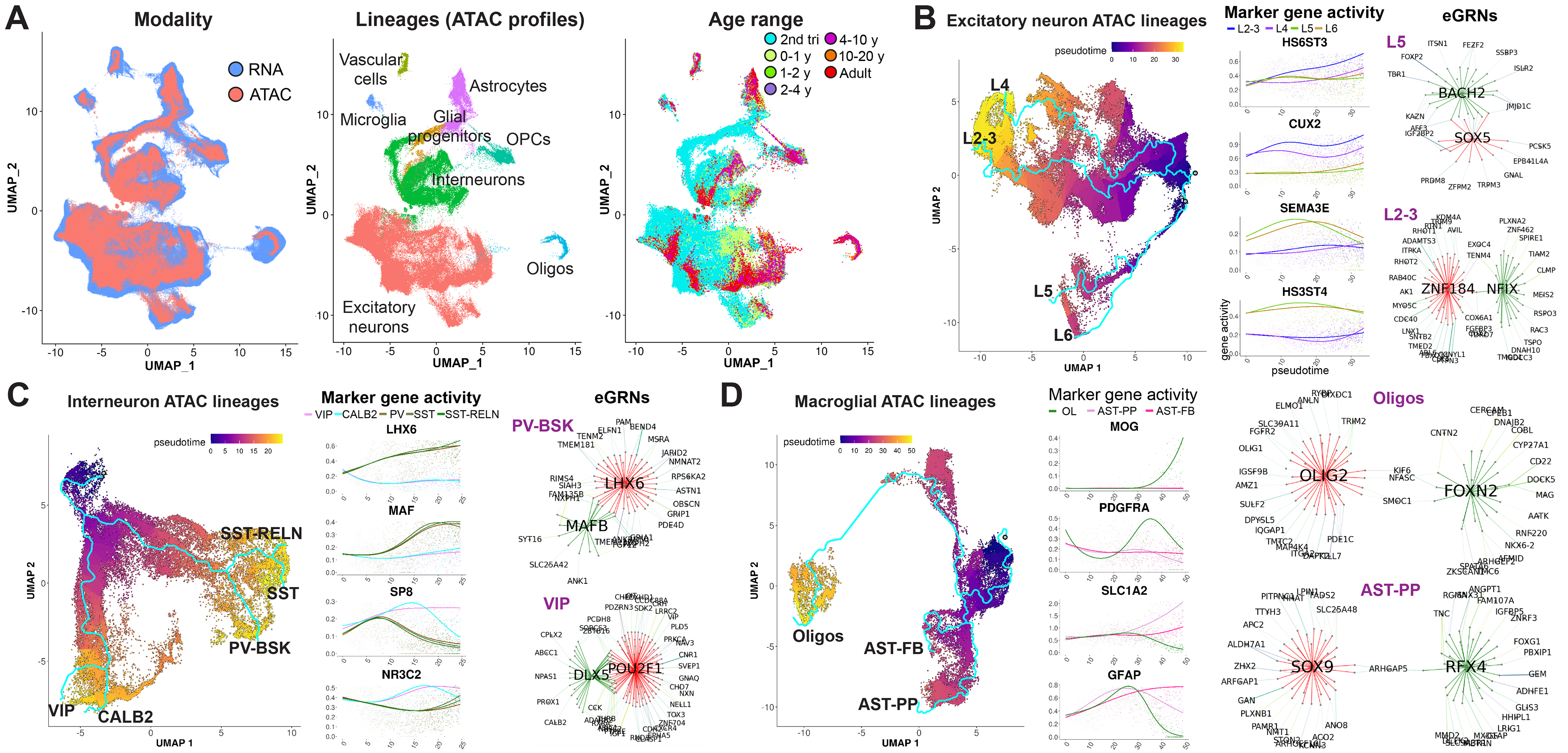
(A) Integration of snRNA-seq and snATAC-seq data. snATAC-seq data was mapped on the snRNA-seq coordinates, clusters, and cell types. (B to D) Analysis of eGRNs in excitatory neuron lineages (B), interneurons (C), and glial lineages (D). Network plots (eGRNs) display transcription factors predicted to bind enhancer regions to regulate lineage-specific transcriptional programs. Edge colors indicate regulation by different transcription factors. Top 20 genes on the basis of the predicted confidence of interaction are shown for each transcription factor network.
Identification of region- and sex-enriched lineage-specific gene programs
Given that we sampled our transcriptomic data from different cortical regions, we asked whether lineage-specific developmental gene expression profiles might be spatially defined and vary depending on cortical area. We focused on the frontal and prefrontal cortex (PFC) because we had the most complete sampling of these cortical areas across developmental stages (fig. S6A). We compared each neuronal and glial lineage trajectory in the PFC with the trajectories in all other cortical areas and identified PFC-enriched developmentally regulated genes in each lineage (data S4). We observed more PFC-specific genes in excitatory neuron lineages, especially in intratelencephalic upper (L2–3) and deep-layer (L5–6-IT) neurons, as well as in astrocytes and oligodendrocytes, whereas most interneuron lineages and microglia expressed fewer PFC-specific genes (fig. S6B). After performing GO analysis for PFC genes specific to neuronal lineages, we observed enrichment in cell adhesion and synaptic transmission pathways (fig. S6C). Analysis of glia-specific PFC genes demonstrated enrichment in different categories of biological pathways associated with cell division and cell migration (fig. S6D). Examples of neuronal PFC genes included synaptojanin 2 binding protein (SYNJ2BP) regulating receptor localization and signal transduction at the synapse and the cation channel TRPC7 (fig. S6E). PFC fibrous astrocytes up-regulated R-spondin 2 (RSPO2) and frizzled class receptor 8 (FZD8), which both participate in Wnt signaling and cell migration. Our results suggest cortical areal differences in lineage-specific transcriptomic programs, with synaptic genes up-regulated in neuronal cell types and cell division and cell migration programs activated in glial cells in the developing PFC. PFC-specific expression of synaptic genes in neuronal cell types suggests regional specification of neuronal circuits during development.
We next asked whether the development of specific cellular lineages is modulated in a sex-dependent manner. For each lineage analyzed, we selected female and male nuclei (Fig. 5A and fig. S7, A and B) and identified dynamically expressed genes enriched during either female or male development. In total, we identified 740 female-enriched genes and 312 male-enriched genes (data S5). A smaller fraction of male genes showed sex enrichment in a lineage-specific manner (181/312, 58%) compared with female-enriched genes (510/740, 69%). Despite several top sex-enriched genes located on X and Y chromosomes [including X inactive specific transcript (XIST) and protocadherin 11 Y-linked (PCDH11Y)], sex-enriched genes were evenly distributed across all chromosomes (fig. S7C), suggesting that sex-dependent developmental modulation of gene expression is not directly dependent on transcription from the sex chromosomes. We next performed GO analysis of female- and male-enriched genes, focusing on the neuronal, astrocyte, and oligodendrocyte lineages for which we had a large number of samples and nuclei from both sexes. We observed substantial differences between the biological processes associated with female-enriched genes and male-enriched genes: female genes were involved in developmental processes, including cell adhesion, central nervous system (CNS) development, synaptic transmission, and membrane potential regulation (Fig. 5B), whereas male genes were associated with RNA metabolism and translation (Fig. 5C). Only a small number of male-specific genes, such as Y-box binding protein 1 (YBX1) and leucine rich repeat and Ig domain containing 1 (LINGO1), were associated with developmental processes; however, these genes were enriched across multiple male lineages (fig. S7D). We classified sex-enriched genes according to their dynamic expression pattern and saw that the majority were expressed transiently (Fig. 5D), with >90% reaching medium expression during the second trimester (data S5). This suggests early and transient sex-dependent developmental modulation of cortical lineages. Sex-enriched genes were more abundant in excitatory neuron lineages than in interneurons (Fig. 5E) and were also abundant in female fibrous astrocytes. Several top lineage-specific female-enriched genes were associated with neuronal, glial, and endothelial development (Fig. 5F and fig. S7E). These included nuclear hormone receptor/transcription factor RORA in L2–3 neurons, synaptic protein neurexophilin 3 (NXPH3) in L6 neurons, transcription factor HES4 in fibrous astrocytes, and an actin filament depolymerization enzyme, MICAL3, in oligodendrocytes. Overall, our results point to modulation of neuronal and glial developmental programs during second trimester female brain development.
Fig. 5. Analysis of sex-specific developmental programs in human cortex.
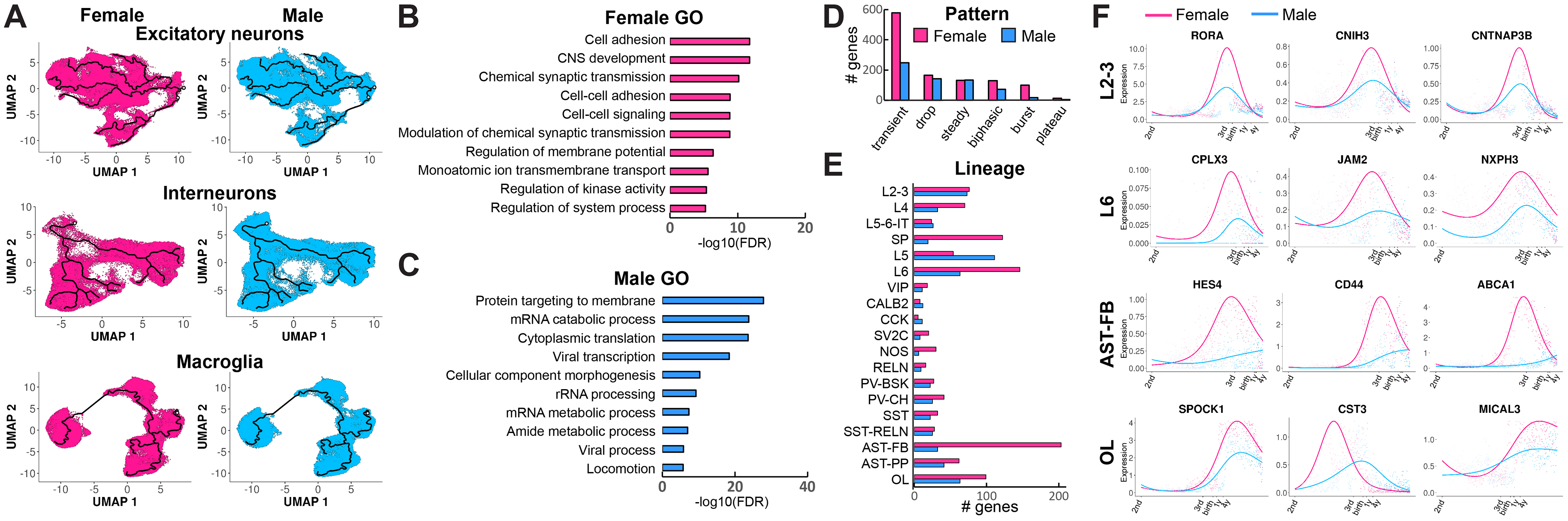
(A) Female and male developmental trajectories of excitatory neurons, interneurons, astrocytes, and oligodendrocytes. (B and C) GO analysis of female-enriched and male-enriched genes. (D) Dynamic expression patterns of sex-enriched genes. (E) Sex enrichment of developmental gene expression across neuronal and glial lineages. (F) Examples of top female-enriched genes in specific lineages.
Enrichment of lineage-specific developmental gene programs for risk factors of brain disorders
Once we defined lineage- and sex-specific developmental gene programs in human cortical cell types, we sought to investigate how these transcriptional programs may be affected in neurodevelopmental, psychiatric, and neurodegenerative disorders. We compiled all lineage-specific gene signatures for excitatory neurons, astrocytes, oligodendrocytes, interneurons, microglia, endothelial cells, and pericytes, in total obtaining 2796 distinct genes, and divided them into five groups according to their age of expression onset (50% of max expression). We then overlapped this gene list with lists of rare gene variants associated with the risk of ASD from the Simons Foundation Autism Research Initiative (SFARI) Gene database (19), as well as genome-wide association study genes for the risk of SCZ (20), bipolar disorder (BPD) (21), and Alzheimer’s disease (AD) (22) (Fig. 6A and data S6). We observed a large enrichment for genes associated with risk for ASD, SCZ, and BPD in the second trimester, with expression of ASD and BPD risk genes extending to the third trimester. The risk of neurodevelopmental disorders dropped during later stages of development. Expression of AD risk genes remained mostly flat and only slightly above the significance level, demonstrating a pattern different from neurodevelopmental and psychiatric disorders. We next analyzed enrichment of disease risk genes across cortical lineages (Fig. 6B). We were able to detect significant enrichment for ASD risk genes in L5–6-IT and L5 neurons, whereas AD risk genes were enriched in microglia. We focused on ASD because we observed the strongest enrichment for the risk of this disorder among developmentally regulated genes and because a large amount of genetic risk data are available for this disorder. We observed developmental enrichment of ASD risk genes with a SFARI score of 2 or 3 but not a score of 1 and did not find enrichment in syndromic ASD genes (Fig. 6C). We observed a significant enrichment among high-confidence ASD risk genes (ASD-HC) based on the TADA (transmitted and de novo association) analysis (23). We conclude that the genetic burden of ASD has the potential to affect the development of specific neuronal cell types, especially deep-layer intratelencephalic projection neurons and L5 neurons. We next explored enrichment of ASD risk genes in sex-specific developmental programs. We observed strong enrichment of female-specific developmental genes in both SFARI and ASD-HC gene lists (Fig. 6D). Male-specific genes were less frequently found among SFARI genes, and we did not find a meaningful overlap between male-enriched and ASD-HC genes. This finding points to a strong enrichment of the genetic risk of ASD among developmental genes that are more highly expressed in female cells. SFARI genes were enriched in female cells across multiple neuronal cell types, especially the subplate and L6 excitatory neurons, as well as oligodendrocytes and fibrous astrocytes, but not in microglia or vascular cell types (Fig. 6E). This suggests a role for the subplate in the pathogenesis of ASD. Examples of female-specific ASD-HC genes included the subplate-specific transcription factor NR4A2 and the neuronal transcription factor MEF2C that were up-regulated in female subplate cells, as well as a regulator of axon guidance and synaptogenesis, neurexin 2 (NRXN2), and PCDH15 encoding a cell adhesion molecule in female L6 neurons (Fig. 6F). Our findings provide strong evidence supporting the ASD female protective effect hypothesis (24) and suggest that fine-tuning of cortical cell lineages by sex-specific developmental programs can contribute to the male bias in the pathogenesis of ASD.
Fig. 6. Lineage enrichment of brain-disorder risk genes.
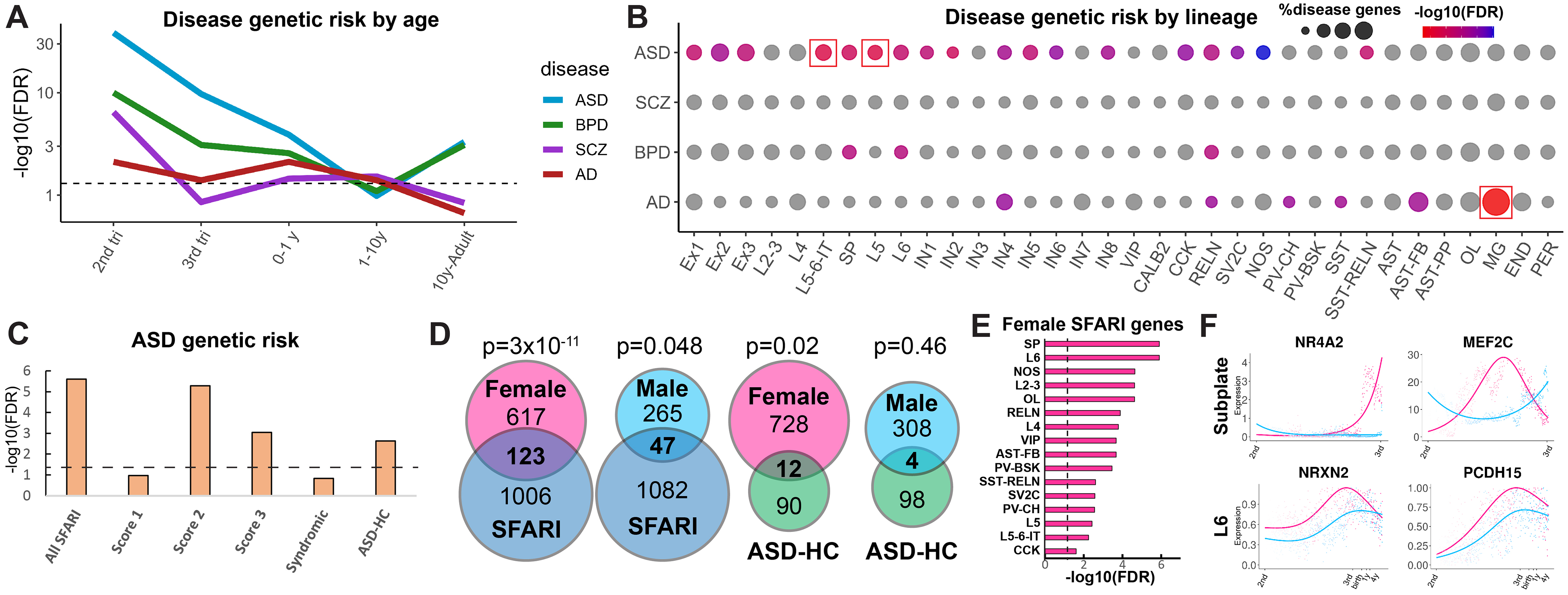
(A) Enrichment of disease risk genes across developmental stages. (B) Disease risk gene enrichment across lineages and lineage branches of neuronal, glial, and vascular cell types. Red squares indicate statistical significance. (C) Enrichment of lineage-specific developmentally regulated ASD risk genes of different categories and evidence scores. (D) Overlap between ASD risk genes and female and male-enriched developmental gene programs. (E) Enrichment of sex-specific genes across cortical lineages. (F) Temporal patterns of female-enriched genes that are known risk factors for ASD.
Discussion
By generating snRNA-seq data from the developing human cortex and integrating the findings with previously published datasets, we performed a large-scale unbiased transcriptomic analysis of human cortical development throughout the life span. By reconstructing single-cell trajectories and identifying genes that are expressed in a lineage-specific manner, we created a compendium of developmental programs for all the major cortical cell types. By integrating our data with published single-cell chromatin accessibility datasets, we identified enhancer gene regulatory networks and transcription factors that are predicted to control the commitment and differentiation of specific cortical neural lineages. In addition, we characterized sex- and brain region–specific gene programs that are used by particular lineages of cortical cell types. We find that female-enriched genes are associated with neurodevelopmental processes, whereas male-enriched genes are involved in protein translation control, suggesting sex-specific variation of developmental trajectories. We also find that developmental gene programs used by cortical excitatory neurons, astrocytes, and oligodendrocytes are the most region-specific. Interneurons, in contrast, express few region-specific genes during development, consistent with data on regional signatures of cortical cell types in the mature human brain (25).
We investigated the enrichment of genetic risk factors for brain disorders, focusing on ASD, and found that the developmental programs of both deep-layer intratelencephalic and extratelencephalic projection neurons are enriched for ASD risk genes. These data are in agreement with previous reports of enrichment of ASD genes in deep-layer cortical neurons during mid-gestation (26, 27) but also suggest that both deep-layer neurons projecting to other cortical areas and to subcortical locations could be affected. We previously reported that upper-layer cortical excitatory neurons are most dysregulated in the cortex of idiopathic ASD patients (28). It would be an important future direction to elucidate how changes in pan-excitatory neuron programs during development can culminate in dysfunction of specific cortical neuronal populations, such as L2–3 neurons. It would also be valuable to explore whether the molecular pathology of upper-layer neurons is specific to idiopathic ASD, and whether it is driven by common gene variants rather than by rare variants with strong effect sizes (29). In addition, we observed a strong enrichment of ASD genetic risk factors among female-specific developmental genes. As these female-enriched ASD risk genes have higher expression in females during cortical development, it is possible that this higher baseline expression renders the female brain more resistant to genetic insults causing autism, especially to haploinsufficiency that can reduce transcript or protein expression by affecting one of the two alleles. This finding might explain the 4:1 male-to-female ratio of individuals affected by ASD and suggests the importance of sexual dimorphism in human brain development. However, the role of sex hormones in the increased male-to-female ratio in ASD is not to be discounted, and additional studies are needed to reconcile the role of early development and later sex-specific processes in the pathogenesis of autism. Our preliminary findings indicate the cell type–specific risk of BPD and SCZ, but more detailed genetic studies are needed to further dissect cell type and developmental stage vulnerability. The data generated here may help enable fine-grained understanding of human brain development and provide insight into mechanisms of neurodevelopmental disorders. Interactive visualization of our data is available on the University of California Santa Cruz (UCSC) Cell Browser (https://pre-postnatal-cortex.cells.ucsc.edu).
Our study, however, is limited by the technical difficulty of integrating snRNA-seq and snATAC-seq data as well as by the lack of inclusion of earlier developmental stages, such as the first trimester, owing to challenges of integrating single-cell RNA sequencing (scRNA-seq) and snRNA-seq datasets. Overcoming these obstacles will allow for even more comprehensive future understanding of how specific human cortical lineages develop. Moreover, single-cell epigenetic analyses of human brain development would be necessary to determine whether imprinting plays a role in regulating sex enrichment of developmentally expressed genes.
Materials and methods summary
Brain tissue samples were sectioned using a cryostat to collect coronal cortical sections, lysed, and ultracentrifuged to isolate nuclei. Nuclei were captured using 10x Genomics Single Cell 3′ v.2 kits.
Raw sequencing data were processed using 10x Genomics CellRanger and aligning reads to unspliced human transcriptome to capture reads from pre-mRNAs. Dataset integration was performed using Harmony based on 10x chemistry, and clustering and UMAP embedding was carried out with Seurat. Monocle 3 was used to reconstruct lineage trajectories, and custom scripts were used to identify lineage-specific dynamically expressed genes (see supplementary materials).
snATAC-seq data were integrated with snRNA-seq data using canonical correlation analysis in Seurat, after which different snATAC-seq chemistries were integrated using Harmony. Enhancer gene regulatory networks were identified using SCENIC+.
Data and materials availability:
Raw data can be accessed at the Neuroscience Multi-omic (NeMo) Archive, under accession number nemo:dat-3ah9h9x (https://assets.nemoarchive.org/dat-3ah9h9x). Analyzed data (cell-count matrix and metadata) and data visualization can be accessed through the UCSC Cell Browser, collection “Human Cortical Development” (https://pre-postnatal-cortex.cells.ucsc.edu) and at CELLxGENE (https://cellxgene.cziscience.com/collections/bacccb91-066d-4453-b70e-59de0b4598cd). Code is available at https://github.com/velmeshevlab/dev_hum_cortex and in Zenodo (30).
Supplementary Material
Acknowledgments
We thank the NIH’s Brain Research through Advancing Innovative Neurotechnologies Initiative - Cell Census Network (BICCN) and the Brain Cell Data Center (BCDC) at the Allen Institute for Brain Science, as well as all of its members, for support of this work and helpful discussions. We also thank Dr. Maximilian Haeussler for his help with UCSC Cell Browser visualization.
Funding:
This study was funded by NIH BRAIN Initiative U01 grant U01MH114825 awarded to A.R.K.; NINDS R35 grant 5R35NS097305 awarded to A.R.K.; NINDS P01 NS083513 Neuropathology Core grant to E.J.H.; a Quantitative Biosciences Institute BOLD & BASIC Fellowship and NIMH K99/R00 Award to D.V.; a New York Stem Cell Foundation Robertson Neuroscience Investigator Award to T.J.N.; Deutsche Forschungsgemeinschaft grant MA 7374/1-1 to S.M.; and NHGRI grant 2U24HG002371–23 to B.W.
Footnotes
Competing interests: The authors have no competing interests.
References and notes
- 1.Bhaduri A et al. , An atlas of cortical arealization identifies dynamic molecular signatures. Nature 598, 200–204 (2021). doi: 10.1038/s41586-021-03910-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eze UC, Bhaduri A, Haeussler M, Nowakowski TJ, Kriegstein AR, Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat. Neurosci. 24, 584–594 (2021). doi: 10.1038/s41593-020-00794-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nowakowski TJ et al. , Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323 (2017). doi: 10.1126/science.aap8809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ziffra RS et al. , Single-cell epigenomics reveals mechanisms of human cortical development. Nature 598, 205–213 (2021). doi: 10.1038/s41586-021-03209-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li M et al. , Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, eaat7615 (2018). doi: 10.1126/science.aat7615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lake BB et al. , Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590 (2016). doi: 10.1126/science.aaf1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansen DV et al. , Non-epithelial stem cells and cortical interneuron production in the human ganglionic eminences. Nat. Neurosci. 16, 1576–1587 (2013). doi: 10.1038/nn.3541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma T et al. , Subcortical origins of human and monkey neocortical interneurons. Nat. Neurosci. 16, 1588–1597 (2013). doi: 10.1038/nn.3536 [DOI] [PubMed] [Google Scholar]
- 9.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). doi: 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herring CA et al. , Human prefrontal cortex gene regulatory dynamics from gestation to adulthood at single-cell resolution. Cell 185, 4428–4447.e28 (2022). doi: 10.1016/j.cell.2022.09.039 [DOI] [PubMed] [Google Scholar]
- 11.Trevino AE et al. , Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell 184, 5053–5069.e23 (2021). doi: 10.1016/j.cell.2021.07.039 [DOI] [PubMed] [Google Scholar]
- 12.Ramos SI et al. , An atlas of late prenatal human neurodevelopment resolved by single-nucleus transcriptomics. Nat. Commun. 13, 7671 (2022). doi: 10.1038/s41467-022-34975-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Habib N et al. , Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017). doi: 10.1038/nmeth.4407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodge RD et al. , Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). doi: 10.1038/s41586-019-1506-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao J et al. , The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). doi: 10.1038/s41586-019-0969-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Street K et al. , Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018). doi: 10.1186/s12864-018-4772-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Domcke S et al. , A human cell atlas of fetal chromatin accessibility. Science 370, eaba7612 (2020). doi: 10.1126/science.aba7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bravo González-Blas C et al. , SCENIC+: Single-cell multiomic inference of enhancers and gene regulatory networks. Nat. Methods 20, 1355–1367 (2023). doi: 10.1038/s41592-023-01938-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abrahams BS et al. , SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 4, 36 (2013). doi: 10.1186/2040-2392-4-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Y, Li X, Liu J, Luo XJ, Yao YG, SZDB2.0: An updated comprehensive resource for schizophrenia research. Hum. Genet. 139, 1285–1297 (2020). doi: 10.1007/s00439-020-02171-1 [DOI] [PubMed] [Google Scholar]
- 21.Li X et al. , dbBIP: A comprehensive bipolar disorder database for genetic research. Database 2022, baac049 (2022). doi: 10.1093/database/baac049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bellenguez C et al. , New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022). doi: 10.1038/s41588-022-01024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Satterstrom FK et al. , Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23 (2020). doi: 10.1016/j.cell.2019.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson EB, Lichtenstein P, Anckarsäter H, Happé F, Ronald A, Examining and interpreting the female protective effect against autistic behavior. Proc. Natl. Acad. Sci. U.S.A. 110, 5258–5262 (2013). doi: 10.1073/pnas.1211070110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tasic B et al. , Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78 (2018). doi: 10.1038/s41586-018-0654-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parikshak NN et al. , Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021 (2013). doi: 10.1016/j.cell.2013.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willsey AJ et al. , Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007 (2013). doi: 10.1016/j.cell.2013.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Velmeshev D et al. , Single-cell genomics identifies cell type–specific molecular changes in autism. Science 364, 685–689 (2019). doi: 10.1126/science.aav8130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaugler T et al. , Most genetic risk for autism resides with common variation. Nat. Genet. 46, 881–885 (2014). doi: 10.1038/ng.3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.velmeshevlab, velmeshevlab/dev_hum_cortex: beta 1.0, version 1, Zenodo (2022); 10.5281/zenodo.7245297. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.