

Summary
Hispanic/Latino children have the highest risk of acute lymphoblastic leukemia (ALL) in the US compared to other racial/ethnic groups, yet the basis of this remains incompletely understood. Through genetic fine-mapping analyses, we identified a new independent childhood ALL risk signal near IKZF1 in self-reported Hispanic/Latino individuals, but not in non-Hispanic White individuals, with an effect size of ∼1.44 (95% confidence interval = 1.33–1.55) and a risk allele frequency of ∼18% in Hispanic/Latino populations and <0.5% in European populations. This risk allele was positively associated with Indigenous American ancestry, showed evidence of selection in human history, and was associated with reduced IKZF1 expression. We identified a putative causal variant in a downstream enhancer that is most active in pro-B cells and interacts with the IKZF1 promoter. This variant disrupts IKZF1 autoregulation at this enhancer and results in reduced enhancer activity in B cell progenitors. Our study reveals a genetic basis for the increased ALL risk in Hispanic/Latino children.
Keywords: acute lymphoblastic leukemia, childhood leukemia, B-ALL, IKZF1, GWAS, fine-mapping, Indigenous American, cancer disparity, cancer predisposition, hematopoiesis
Graphical abstract
Highlights
-
•
IKZF1 variants contribute to the increased risk of ALL in Hispanic/Latino children
-
•
Risk allele is associated with Indigenous American ancestry and underwent selection
-
•
Risk variant impacts IKZF1 enhancer that is selectively active in B cell development
-
•
Risk allele reduces enhancer activity, chromatin accessibility, and IKZF1 expression
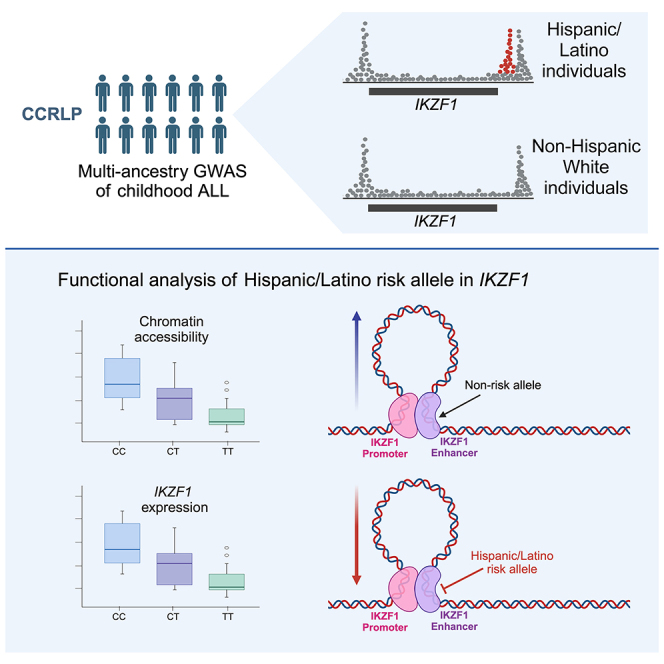
Genetic fine-mapping across the IKZF1 gene revealed a variant associated with childhood ALL that contributes to the increased risk of this disease in Hispanic/Latino individuals. The ALL risk allele reduces enhancer activity and IKZF1 expression specifically in B cell progenitors, likely resulting in stalled B cell development and an increased risk of ALL.
Introduction
The incidence of certain cancer types varies among specific racial/ethnic groups. Understanding the causes of this variation will be essential for attempts to alleviate health disparities and reveal etiologic insights.1 One notable example of such variation is acute lymphoblastic leukemia (ALL), the most common malignancy in children, for which self-reported Hispanic/Latino individuals have the greatest risk in the United States—an approximately 1.3-fold increased incidence compared to non-Hispanic White children.2 This difference rises to >2-fold in adolescents and young adults (AYAs).1,3 Moreover, Hispanic/Latino patients with ALL have lower overall survival compared to non-Hispanic White patients, even after accounting for social determinants of health.4,5,6 Intriguingly, the disparity in ALL incidence appears to be principally attributable to the B cell ALL immunophenotype,7,8 suggesting the possibility of specific underlying biological mechanisms.
Genome-wide association studies (GWASs) have identified several well-replicated single-nucleotide polymorphisms (SNPs) associated with childhood ALL risk, including variants near genes involved in B cell development and hematopoiesis such as IKZF1, ARID5B, CEBPE, and GATA3.9,10,11,12 Several ALL risk alleles have a higher frequency in Hispanic/Latino populations than in individuals of predominantly European ancestry, in particular at ARID5B and GATA313,14; the risk allele frequencies (RAFs) of these SNPs have also been positively correlated with Indigenous American (IAM) ancestry proportions,13,15,16 as have the RAF and effect size of SNPs at ERG, another key regulator of hematopoiesis.16,17 However, the three aforementioned risk loci fail to explain most of the disparity in ALL risk in Hispanic/Latino individuals. Moreover, the effect sizes of other known ALL GWAS loci do not differ substantially between Hispanic/Latino and non-Hispanic White individuals, and it is likely that additional genetic loci or variants within known loci contribute to the increased ALL risk in Hispanic/Latino individuals.
In a recent trans-ancestry GWAS of childhood ALL, we identified multiple SNP associations at the IKZF1 gene locus at chromosome 7p12.2.18,19 However, the total number of risk variants present at IKZF1, the causal variants underlying their association, the relevant cell types in which they operate, and their underlying mechanisms have not been examined. To address this, we carried out fine-mapping of the IKZF1 region in self-reported (see STAR Methods) Hispanic/Latino, non-Hispanic White, and East Asian childhood ALL cases and controls, followed by functional analysis of putative causal variants.
Results
Novel IKZF1 association in Hispanic/Latino children
In a GWAS of childhood ALL in self-reported Hispanic/Latino individuals (1,878 cases, 8,441 controls) from the California Cancer Records Linkage Project (CCRLP) (Table S1), we identified 109 genome-wide significant SNPs at IKZF1, with the top SNP, rs4917017 (odds ratio [OR] = 1.41, p = 2.05 × 10−17), located in the promoter region (“signal 1”) (Figure 1A; Table S2). There was a second distinct association peak (“signal 2”) spanning the IKZF1 3′ end. Adjusting for the signal 1 lead SNP, only signal 2 remained genome-wide significant with the top SNP, rs10272724 (OR = 1.29, p = 4.39 × 10−9), which is not in linkage disequilibrium (LD) with rs4917017 (r2 = 0.002) in admixed American (AMR) individuals in the 1000 Genomes Project (1KG) (Figure 1B). These two ALL risk loci were reported previously.9,10,20
Figure 1.

Novel childhood ALL risk locus at IKZF1 is associated with Indigenous American ancestry and demonstrates ancient origins and positive selection in Hispanic/Latino populations
(A–C) LocusZoom plots showing an approximately 600 Mb region at chromosome 7p12.2 centered on the IKZF1 gene region (±250 kb) from (A) signal 1, the unconditional GWAS of childhood ALL in Hispanic/Latino individuals; (B) signal 2, results conditioned on the lead SNP (rs4917017) in signal 1; and (C) signal 3, results conditioned on lead SNPs in signals 1 (rs4917017) and 2 (rs10272724), identifying the novel ALL risk locus tagged by lead SNP rs76880433. Diamond symbols in purple indicate the lead SNP in each locus. Color of remaining SNPs is based on linkage disequilibrium (LD) in the admixed American superpopulation in the 1KG Project (which includes Mexican ancestry in Los Angeles [MXL], Peruvian in Lima, Peru [PEL], Colombian, and Puerto Rican populations) as measured by r2 with the lead SNP in each signal. All coordinates are in genome build hg38.
(D) The rs76880433 risk allele T is positively associated with Indigenous American ancestry among Hispanic/Latino cases and controls in the California Cancer Record Linkage Project ALL GWAS. Error bars correspond to standard errors.
(E) Frequency of the haplotype derived from Indigenous American ancestry at IKZF1 signal 3 is significantly higher in childhood ALL cases than controls among Hispanic/Latino individuals.
(F) Mapping the origins of the putative causal rs1451367 SNP with shotgun sequencing data of ancient DNA samples suggests the mutation arose at least 12,700 years ago.
(G) Reconstructed genealogies in 1KG populations provide evidence for positive selection at the haplotype containing signal 3 lead SNP rs76880433 only in Hispanic/Latino populations (MXL and PEL) but not in European (IBS) or East Asian (CHS) populations.
Adjusting for signals 1 and 2 revealed a third, independent genome-wide significant peak (“signal 3”) spanning the 3′ end of IKZF1 in Hispanic/Latino individuals (Figure 1C). The signal 3 lead SNP rs76880433 (OR = 1.44, p = 4.67 × 10−11) was not in LD with the signal 1 (rs4917017, r2 = 0.09) or signal 2 (rs10272724, r2 = 0.07) lead SNP in AMR individuals; indeed, signal 3 SNP rs76880433 risk allele T and signal 2 SNP rs10272724 risk allele C lie on opposite haplotypes. The effect size of rs76880433 was increased when adjusting for both signals 1 and 2 (Table 1). We explored potential epistatic effects between the three lead SNPs in Hispanic/Latino individuals but found no evidence of any significant SNP-SNP interactions, although our power to detect an effect was likely limited.
Table 1.
Marginal and conditional association test results for the lead IKZF1 SNPs at three independent childhood ALL association signals in Hispanic/Latino individuals in the California Cancer Records Linkage Project
| IKZF1 SNP rsID (GWAS signal) | Chromosome 7 location (bp, hg38) | Risk allele | RAF |
Marginal test |
Conditional test 1a |
Conditional test 2b |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Overall | Cases | Controls | OR (95% CI) | p value | OR (95% CI) | p value | OR (95% CI) | p value | |||
| rs4917017 (signal 1) | 50295636 | A | 0.437 | 0.569 | 0.407 | 1.41 (1.33–1.49) | 2.05 × 10−17 | – | – | – | – |
| rs10272724 (signal 2) | 50409515 | C | 0.268 | 0.300 | 0.260 | 1.30 (1.22–1.39) | 7.49 × 10−10 | 1.29 (1.20–1.37) | 4.39 × 10−9 | – | – |
| rs76880433 (signal 3) | 50384350 | T | 0.153 | 0.267 | 0.127 | 1.37 (1.27–1.46) | 1.62 × 10−10 | 1.22 (1.12–1.32) | 8.43 × 10−5 | 1.44 (1.33–1.55) | 4.67 × 10−11 |
RAF, risk allele frequency; CI, confidence interval.
In conditional test 1, the association tests were repeated adjusting for the genotype of SNP rs4917017 (lead SNP in signal 1).
In conditional test 2, the association tests were repeated adjusting for the genotype of both SNP rs4917017 (lead SNP in signal 1) and SNP rs10272724 (lead SNP in signal 2).
In non-Hispanic White individuals (1,162 cases, 57,341 controls), there was a single genome-wide significant peak (Figure S1); top SNP rs17133805 is in near-perfect LD with the Hispanic/Latino signal 2 lead SNP (rs10272724, r2 = 0.995) in European individuals. Adjusting for rs17133805 revealed a second peak at the IKZF1 promoter region with top SNP rs9886239 (OR = 1.20, p = 4.01 × 10−5), which is in LD with the Hispanic/Latino signal 1 lead SNP (rs4917017, r2 = 0.88) in European individuals. Adjusting for rs17133805 and rs9886239 revealed no further significant loci. In East Asian individuals (318 cases, 5,017 controls), there were no genome-wide significant SNPs, but there appeared to be two independent risk loci at the 3′ end of IKZF1 (Figure S2).
Thus, Hispanic/Latino children are unique in having three independent IKZF1 risk loci for ALL, as non-Hispanic White individuals lacked the association signal 3 identified by rs76880433 in Hispanic/Latino individuals that confers >1.4-fold increased odds for developing ALL. SNP rs76880433 is relatively common in AMR (RAF = 17.7%) and East Asian (18.9%) individuals in the Genome Aggregation Database (gnomAD v.4.0) but rare in non-Finnish European individuals (RAF = 0.2%), with the highest reported RAF (50.0%) in Indigenous American individuals in the Human Genome Diversity Project (HGDP). Similarly, the signal 1 lead SNP rs4917017 has a higher RAF in AMR individuals (43.2%) than in non-Finnish European individuals (29.5%) and an even higher frequency (75.0%) in Indigenous American individuals in the HGDP. The signal 2 lead SNP rs10272724, on the other hand, has similar RAFs across AMR (23.3%), non-Finnish European (27.1%), and Indigenous American (24.0%) individuals. Given the large RAF differences for two of the IKZF1 signal lead SNPs, genetic variation at IKZF1 likely contributes to the increased incidence of ALL in Hispanic/Latino children, particularly in the case of the signal tagged by rs76880433, which is almost completely absent in European populations.
To explore the extent to which all of these variants in the IKZF1 locus contribute to risk disparities, we calculated an IKZF1 genetic risk score (GRS) comprising the lead independent risk variants in Hispanic/Latino (n = 3) and non-Hispanic White individuals (n = 2) weighted by their corresponding marginal or conditional effect estimates (Figure S3). Remarkably, the proportion of the variance in ALL risk explained on the liability scale by GRSIKZF1 was >3-fold higher in Hispanic/Latino (pseudo-R2 = 0.194) than in non-Hispanic White children (pseudo-R2 = 0.062), although further analysis in independent datasets will be needed to determine more accurate estimates of the variance in ALL risk explained.
Next, we assessed the RAF of the three independent Hispanic/Latino ALL risk variants among 335 Hispanic/Latino childhood ALL cases from the Children’s Oncology Group/St. Jude Children’s Research Hospital studies with available leukemia molecular subtype information.17 For signal 3 lead SNP rs76880433, there was a higher RAF in BCR::ABL1 (RAF = 0.40), Ph-like (0.43), and TCF3::PBX1 (0.53) subtypes compared to ETV6::RUNX1 (0.19), high hyperdiploid (0.26), hypodiploid (0.31), and KMT2A-rearranged (0.18) subtypes, a pattern that was significantly different from the expected distribution (chi-squared goodness of fit, p = 0.0001) and was not seen for signal 1 (rs4917017) or signal 2 (rs10272724) lead SNPs (p = 0.072 and 0.34, respectively) (Figure S4). It is interesting to note that these patterns of acquired driver mutations do differ compared to what has been reported with germline coding variation in IKZF1 that predisposes to ALL.21
Association of IKZF1 RAF and effect size with IAM ancestry
Given that the third independent ALL risk allele at IKZF1 was largely absent in European populations, we suspected that it may have derived from Indigenous American haplotypes among Hispanic/Latino individuals, who harbor varying extents of admixture from European, African, and IAM ancestries.22 Among Hispanic/Latino individuals in our study, admixture analysis revealed, on average, estimated 28.0% IAM, 67.6% European, and 4.4% African global genetic ancestry proportions (Figure S5). Both signal 1 (rs4917017) and signal 3 (rs76880433) lead SNP risk alleles were significantly positively correlated with global IAM ancestry proportions, whereas the signal 2 lead SNP (rs10272724) showed no correlation (Table S3; Figure 1D). Signal 3 SNP rs76880433 showed the greatest correlation with IAM ancestry, with similar estimates in Hispanic/Latino cases (estimate = 0.296, p = 3.28 × 10−39) and controls (estimate = 0.283, p = 1.76 × 10−38). The highest RAF for rs76880433 was in individuals with ≥50% IAM ancestry (Figure S6).
Analysis of local ancestry across IKZF1 revealed a significantly higher frequency of the Indigenous American haplotype at signal 3 in cases than in controls (p = 8.78 × 10−7) (Figure 1E). Furthermore, we found an increasing effect size of the signal 3 lead SNP with increasing numbers of the Indigenous American haplotype at this locus, a pattern not seen for the lead SNPs in signals 1 or 2 (Table S4).
Fine-mapping causal variants
Statistical fine-mapping identified three credible sets of causal variants in Hispanic/Latino individuals, with 44 putative causal variants underlying the three distinct IKZF1 risk signals (Table S5; Figure S7). Credible set 1 contained 10 variants in LD with the GWAS signal 3 lead SNP (rs76880433), one of which, rs1451367, overlapped a putative functional regulatory element. This variant is in high LD with rs76880433 (r2 > 0.96 in AMR individuals) and is located downstream of IKZF1 in a regulatory region defined by histone modification peaks. Credible set 2, anchored by the signal 2 lead SNP (rs10272724), contained 23 SNPs, of which 4 overlapped the same functional element, including rs17133807 that is located only 26 bp away from rs1451367 (Figure S8). As with tagging SNPs rs76880433 and rs10272724, the rs1451367 and rs17133807 risk alleles reside on opposing haplotypes. Credible set 3 contained 11 variants in LD with the signal 1 lead SNP rs4917017, with 3 variants mapping to within 10 kb of the IKZF1 promoter.
All 44 SNPs across the three credible sets identified in Hispanic/Latino individuals plus SNP rs4917017 had statistically significant effects on whole-blood IKZF1 expression (Figure S9; Table S6). In Mexican American children and in children with >50% IAM ancestry (IAMHigh), ALL risk-increasing allele rs4917017-A and lead SNPs from credible set 1 rs76880433-T and rs1451367-T were associated with lower IKZF1 expression, whereas credible set 2/signal 2 lead SNP rs10272724-C conferred an increase in whole-blood transcript levels (Figure S9; Table S6). Similarly, the ATT haplotype (rs4917017-A, rs76880433-T, rs10272724 non-risk allele T), which was common in Mexican American children (22.6%) and IAMHigh children (26.2%) but rare in those with <10% IAM ancestry (1.3%), was associated with decreased IKZF1 expression, whereas the GCC haplotype was positively associated with IKZF1 levels (Table S7). Results did not change when constructing haplotypes with rs1451367 instead of rs76880433. Few variants had significant effects on the expression of other nearby genes. In contrast to our results in Mexican American children, credible set 1 SNPs rs76880433 and rs1451367 were not identified as expression quantitative trait loci (eQTLs) either in the Genotype-Tissue Expression (GTEx) project23 or in the eQTLGen Consortium,24 as these include predominantly individuals with European ancestry. However, in an immune-cell-specific eQTL dataset in Japanese individuals, ImmuNexUT,25 rs76880433 and rs1451367 were both associated with reduced IKZF1 expression, most significantly in B cells (Table S8). SNP rs4917017 was associated with reduced whole-blood expression of IKZF1 in both the GTEx and eQTLGen, whereas in contrast with results in Mexican American children, rs10272724 was associated with reduced whole-blood expression of IKZF1 in eQTLGen (likely due to the lack of impact of the Indigenous American haplotype on IKZF1 expression in this dataset) (Table S8). Therefore, it is likely that all three ALL risk alleles identified reduce IKZF1 expression, but in Mexican American children, the impact of different haplotypes harboring rs76880433 and rs10272724 might confound the impact on expression of the latter allele.
In non-Hispanic White individuals, genetic fine-mapping identified two credible sets (Figure S7). Credible set 2 included 23 variants spanning the IKZF1 promoter, including rs4917017 and all 11 of the credible set 3 variants in Hispanic/Latino individuals. Two of the variants, rs11765436 and rs11761922, reside within the IKZF1 promoter region and are in strong LD (r2 > 0.92) with the tagging SNP rs4917017 (Table S5). In East Asian individuals, there was one credible set of 13 variants, including all 10 variants in Hispanic/Latino credible set 1.
Selection analyses
We next sought to understand more about when the childhood ALL risk locus (signal 3) tagged by the putative causal variant rs1451367 and enriched in Hispanic/Latino and East Asian populations may have arisen in human history. The oldest previously sequenced Indigenous American individual (Anzick, 12,700 years old) was heterozygous for the rs1451367 risk allele, supporting the idea that the SNP was present in the first migrants who entered the Americas ∼13,000 years ago. Importantly, many ancient individuals in a variety of locations in the Americas appeared to carry the risk allele (Figure 1F; Table S9).26
We examined signatures of positive selection at IKZF1 and found evidence for selection at the signal 3 lead SNP rs76880433 in Hispanic/Latino populations (MXL [Mexican ancestry in Los Angeles], p = 2.9 × 10−3; PEL [Peruvian in Lima, Peru], p = 5.3 × 10−3) but not in European (IBS [Iberian populations in Spain], p = 0.97) or East Asian (CHS [Southern Han Chinese], p = 0.31) populations (Figure 1G; Table S10). The estimated ages for this variant range from 28 to 52 ka based on the genealogies. The putative functional variant rs1451367, in high LD with rs76880433, had much older age estimates (130–260 ka) and, consequently, more modest evidence of selection (p = 0.03 in PEL, 0.12 in MXL). The lead SNPs at signals 1 and 2, rs4917017 and rs10272724, showed no evidence of selection (p > 0.05).
Functional assessment of Hispanic/Latino ALL risk variant
Having identified the signal 3 causal variant rs1451367 as an important contributor to the increased risk for Hispanic/Latino children to acquire ALL, we sought to define the mechanisms through which this risk arises. At the IKZF1 regulatory element harboring putative causal variants rs17133807 and rs1451367, at Hispanic/Latino GWAS signals 2 and 3, we found strong chromatin accessibility in B cells and their precursors, with the greatest accessibility at the pro-B stage but minimal or no accessibility in other lineages (Figures 2 and S10).27 These SNPs might, therefore, influence ALL predisposition during early B cell development, particularly during the transition from pro- to pre-B cells. This finding is consistent with the observation that IKZF1 SNPs appear to confer risk for B-precursor ALL and not for T cell ALL.28 In promoter capture Hi-C data from human B lymphoblastoid cells, this regulatory region showed a strong interaction with the promoter of IKZF1, which itself harbored the ALL risk variants tagging the signal 1 lead SNP rs4917017 (Figure 2A). This finding suggests that multiple risk-variant-harboring regulatory elements might interact in three-dimensional space to effectively alter gene regulation at the IKZF1 locus.
Figure 2.

Two independent childhood ALL risk alleles in Hispanic/Latino individuals reside in an enhancer for IKZF1 that is most active in pro-B cells
(A) Top: putative causal variants underlying Hispanic/Latino ALL GWAS signal 2 and signal 3, rs17133807 and rs1451367, respectively, lie 26 bp apart in a region with strong interaction with IKZF1 promoter in promoter capture Hi-C data. Bottom: single-cell assay for transposase-accessible chromatin using sequencing (ATAC sequencing) data showed strong accessibility in B cells and precursors, especially pro-B cells, but minimal or no accessibility in T cells or myeloid cells obtained from healthy donors (see Figure S10 for details).
(B) ATAC sequencing coverage in 156 patients with B cell ALL displayed by genotype for putative causal variants underlying GWAS signals 1–3. Genotypes are ordered by non-risk allele homozygotes, non-risk/risk allele heterozygotes, and risk allele homozygotes (rs11765436: T/T, T/A, A/A; rs1451367: C/C, C/T, T/T; rs17133807: G/G, G/A, A/A). For each SNP, the patients with B cell ALL were categorized into three groups based on a read coverage cutoff of 5: non-risk/non-risk, where non-risk allele reads are >5 and risk allele reads are <5; non-risk/risk, where both risk and non-risk allele reads are >5; and risk/risk, where risk allele reads are >5 and non-risk allele reads are <5. ATAC sequencing read counts normalized by reads per million are shown (see Table S11 for details), and the p values were calculated using one-tailed Wilcoxon tests.
We next examined a large collection of accessible chromatin data from 156 patients with B-precursor ALL across a range of subtypes29 and found that the rs1451367 and rs17133807 risk alleles both significantly reduced chromatin accessibility (as did the putative causal variant rs11765436 in the IKZF1 promoter region at signal 1) (Figure 2B; Table S11). We additionally observed that there was a significant allelic bias with reduced representation of the risk allele in individuals heterozygous for the variants (Figure S11).
Introduction of the regulatory region harboring variants rs1451367 and rs17133807 upstream of a minimal promoter solely exerted enhancer activity in REH cells, which resemble human pro-B cells, but did not have an impact on other human cell lines, including HEK293T and HepG2 cells (Figure 3A). Importantly, both the rs1451367 and rs17133807 risk alleles individually reduced enhancer activity in REH cells but not in the other cell models. When both variants were present, we noted even further reduced activity, although this is of unclear genetic relevance, as the risk variants are found on different haplotypes. Collectively, we show that the Hispanic/Latino-population-enriched ALL risk variant in the IKZF1 locus appears to reduce expression, which is likely to have the biggest impact on IKZF1 during the pro- to pre-B cell transition, a developmental stage at which transformation to ALL is thought to arise and a key stage at which IKZF1 has a critical role.30,31
Figure 3.

Functional impact of IKZF1 variants on enhancer activity, IKZF1 regulation, and implications for B cell development
(A) IKZF1 putative causal variants were found to reduce enhancer activity in lentiviral reporter assays. Lentiviral reporters were introduced at the same multiplicity of infection as assessed by quantitative PCR of the WPRE element into (top) REH, (middle) HepG2, and (bottom) HEK293T cells. Geometric mean fluorescence intensity of GFP was measured by flow cytometry. Two-sided p values of 0.05 or less were considered to indicate statistical significance. All data are presented as the mean ± the standard deviation (n = 3; ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
(B) rs1451367 overlaps IKZF1 binding peak in human B lymphoblastoid cell chromatin immunoprecipitation sequencing (ChIP-seq) data.
(C) rs1451367 risk allele disrupts an IKZF1 binding motif based on global ChIP-seq data analysis.
(D) IKZF1 expression changes along the B cell developmental trajectory. Single-cell RNA sequencing revealing increasing IKZF1 expression from early hematopoietic progenitors to mature B cells is shown as a pseudo-time trajectory.
(E) Reduced IKZF1 mRNA expression in B cell progenitors derived from differentiation of human HSPCs by qPCR after ribonucleoprotein-mediated introduction of microdeletions involving the IKZF1 transcription factor binding motif within the enhancer region. All data are presented as the mean ± the standard deviation (n = 3; ∗p < 0.05).
(F) Genome editing of IKZF1 results in an accumulation of CD10+CD19− B cell progenitors and impaired differentiation of CD19+CD10+ maturing B precursors after 3 weeks of coculture of human hematopoietic stem and progenitor cells on MS-5 stromal cells.
Next, we examined canonical motifs in the regulatory element and found that rs1451367 was near an IKZF1 binding motif.32,33 Intriguingly, we found a strong peak of IKZF1 binding to this precise region, with a peak around rs1451367 in human B lymphoblastoid cells suggesting direct chromatin occupancy at this region (Figure 3B). Employing a sequence-based method for defining how nucleotide variants may impact function that was trained on IKZF1 chromatin occupancy data, we observed that the risk allele at rs1451367 was predicted to be less likely to bind IKZF1 as well (Figure 3C). Given that IKZF1 increases in expression during the transition from early lymphoid progenitors to lineage-committed B cell precursors (Figure 3D), this regulatory element that harbors rs1451367 might have a critical role in enabling positive feedback of IKZF1 on its own expression during these transitions.
To test the concept that this variant-harboring motif was necessary for IKZF1 autoregulation during primary human B cell development, we employed CRISPR-Cas9 genome editing using dual single-guide RNAs (sgRNAs) to excise the IKZF1 binding motif and surrounding nucleotides (Figure S12A) in primary human hematopoietic stem and progenitor cells (HSPCs)34,35 that were then subject to coculture on MS-5 stromal cells to enable B cell differentiation.36 At 3 weeks of differentiation, when the population largely consists of pro- and pre-B cells, we found that IKZF1 expression was reduced by ∼20% in comparison with the AAVS1 control in the CD19+ fraction of cells (Figure 3E), with nearly 100% of alleles showing editing (Figure S12A). To assess the consequences of perturbing IKZF1 directly and to a greater extent than can be achieved with more modest expression alterations observed when perturbing individual regulatory elements, we employed two independent sgRNAs that significantly reduced IKZF1 protein expression (Figures S12B–S12D) and resulted in reduced B cell production, as noted by the presence of CD19+CD10+ cells, and an accumulation of more immature progenitor cells that solely expressed CD10 after 3 weeks of coculture of edited HSPCs on MS-5 stromal cells (Figure 3F).
Discussion
The increased incidence of ALL in Hispanic/Latino individuals is a well-established cancer disparity that impacts children and AYAs across the US and Latin America,3,37 and understanding its etiologies remains a priority in childhood health research. We characterize a novel association locus at the established risk gene IKZF1 that appears to explain a considerable portion of the increased ALL risk in Hispanic/Latino individuals compared to non-Hispanic White individuals, supported by its large effect on ALL risk (OR = 1.44, 95% confidence interval = 1.33–1.55), and the relatively high RAF (18%) in Hispanic/Latino populations but its near absence (<0.5%) in European populations.
Hispanic/Latino populations across the Americas are highly heterogeneous, comprising individuals who are culturally, phenotypically, and genetically diverse. Furthermore, the incidence of childhood ALL varies across different Hispanic/Latino populations and appears to correlate with genetic ancestry, with reported incidences being higher in countries where the populations have relatively high IAM ancestry proportions, such as Mexico and Ecuador, but lower in countries such as Argentina where the population has a larger European ancestral component.37,38,39,40 Similarly, the incidence of childhood ALL in Puerto Rico, where the population has relatively high proportions of European and African ancestry but much less IAM ancestry, is lower than in Hispanic/Latino children in the contiguous US.41,42 The majority of Hispanic/Latino patients with ALL in our California-based study would have origins in Mexico and other Central American countries (Table S1) and thus, on average, have higher proportions of IAM ancestry compared to Hispanic/Latino patients of, for example, Puerto Rican or Cuban origin. It is important to note, however, that individuals of Central American origin comprise the majority of Hispanic/Latino individuals both in California and in the US overall. Thus, our newly described IKZF1 risk variant, as well as other ALL risk alleles that correlate with IAM ancestry, are important ALL risk factors to consider for Hispanic/Latino children across the Americas and, in particular, those with relatively high levels of IAM ancestry.
Hispanic/Latino individuals appear to be unique in harboring three independent ALL risk loci at IKZF1, and results from our study suggest, for the first time, that variants in IKZF1 contribute to the increased risk of ALL in Hispanic/Latino children relative to children of predominantly European ancestry. This is supported by the considerable difference in RAFs between the two populations for both the IKZF1 signal 1 and 3 lead SNPs and the fact that, for both variants, the RAF reported in Indigenous American individuals is higher than in any other population.
The IKZF1 variants likely contribute significantly to the increased ALL risk in Hispanic/Latino individuals both in the US and in Latin America, with some of the highest global rates of childhood ALL reported in Mexico City.37,43 Efforts are underway to understand the genetic diversity that exists across Indigenous American subpopulations,39,40 and it will be important to elucidate how different Indigenous American ancestries influence allele frequencies and effect sizes at IKZF1 and other ALL risk loci for future precision prevention efforts to alleviate the disparity in ALL incidence in Hispanic/Latino individuals.
The strong correlation between the rs76880433 risk allele and IAM ancestry motivated our analysis of signatures of selection at this locus, which revealed significant selection in Hispanic/Latino populations but not in East Asian or European populations. While multiple aspects of this model require further confirmation, we speculate an evolutionary model in which the haplotype containing the rs76880433 risk allele (1) was introduced after the split of Asian and European populations, (2) drifted neutrally to a frequency of ∼20% prior to the split between East Asian and Indigenous American populations, and (3) was subsequently positively selected in Indigenous American individuals, resulting in a dramatically increased frequency in both ancient and modern American samples over at least the last 13,000 years (Figure 1G). We speculate that the risk allele arose after the split of Asian and European populations to explain its extremely low frequency (0.2%) among European populations; the few haplotypes found in IBS could be due to back-migrations from Indigenous American individuals. However, the estimated allele age (28–52 ka) spans the likely time frame of population split between European and Asian populations, and thus the allele may have predated the split but remained low in frequency among European populations due to other demographic events. Similarly, we suspected that the allele drifted neutrally before the split between East Asian and Indigenous American populations to explain the lack of selection signals in genealogies from CHS, but the relatively low frequency in East Asian individuals and the low sample size in our study posed a challenge in mapping, fine-mapping, and estimating selective effects in East Asian populations. Despite these uncertainties, today, the RAF is highest among Hispanic/Latino individuals with >50% estimated IAM ancestry (RAF > 30%; Figure S6) as well as in the Indigenous American samples from the HGDP (RAF = 0.50 from the gnomAD), possibly as a result of positive selection specific to the Americas. The precise events in human history that underlie this selection remain unknown, although it is interesting to note how many infectious diseases in human history have shaped selection in alleles that altered human immune responses,44 considering the role IKZF1 plays in immune function and development.45,46,47
In addition to investigating the evolutionary origins of the IKZF1 risk loci, we performed several analyses to shed light on their functional mechanisms. Fine-mapping pinpointed likely causal variants underlying the three association signals in Hispanic/Latino individuals. We focused our attention on the association signals 2 and 3 that spanned the IKZF1 3′ region and harbor putative causal variants that reside only 26 bp apart within the same regulatory element but on opposing haplotypes. We demonstrate that this region is an enhancer for IKZF1 that is most active in developing B cell progenitors.48 The Hispanic/Latino risk allele in this regulatory region appears to reduce enhancer activity, chromatin accessibility, and IKZF1 expression. Furthermore, we find that this regulatory element is occupied by IKZF1 itself, and the putative causal variant for the Hispanic/Latino-population-enriched association signal, rs1451367, alters an IKZF1 binding motif, which when deleted results in reduced IKZF1 expression, showing that this variant likely disrupts a positive autoregulatory mechanism. The precise mechanisms through which IKZF1 variants increase ALL risk are yet to be determined, although we note that a reduction of IKZF1 expression stalls development at a stage of differentiation when B cell progenitors have high proliferative potential and ongoing expression of RAG recombinases that may mediate the formation of ALL-causing deletions and translocations via illegitimate V(D)J recombination,49,50 thus increasing the vulnerability to malignant transformation.
In conclusion, our study demonstrates the molecular and evolutionary mechanisms through which a single genetic variant that impacts gene regulation explains a considerable portion of the disparity in ALL risk in Hispanic/Latino children while also uncovering mechanisms through which such risk can arise. Further research is warranted to fully understand the contribution of genetic variation to this disparity in ALL incidence and how this can impact other clinical endpoints beyond the risk of developing ALL that demonstrate racial/ethnic disparities, such as overall survival, risk of relapse, and cytokine release syndrome with chimeric antigen receptor T cell therapy.4,51,52
Limitations of the study
We note that although our results support that genetic variation at the IKZF1 locus contributes to the increased risk of ALL in Hispanic/Latino children, further analysis using GWASs with larger sample sizes will be required to obtain a more accurate estimate of the effects of IKZF1 risk alleles on childhood ALL risk. The novel IKZF1 risk allele is also common in East Asian populations (RAF = 19%), and our East Asian GWAS results suggest that this locus contributes to ALL risk in this population; however, East Asian populations appear to lack the IKZF1 promoter region association signal. Small sample size may have impaired our ability to detect independent signals at IKZF1 in East Asian individuals, although it is important to note that this population has a similar incidence of childhood ALL to non-Hispanic White individuals.53 We were also limited in our ability to investigate association signals in African American individuals, who have the lowest risk of ALL in the US, due to small sample size, with only 128 cases in the CCRLP.18 Future studies to understand the contribution of genetic variation to ALL risk in African American children will be important as well. Finally, while we have conducted a number of functional studies of this variant-harboring regulatory element using exogenous reporter assays, functional assessments from genomic data, and genome editing in human HSPCs, future studies will employ emerging tools, such as base editors,54 to more precisely define the functional impact of the single-nucleotide variant on B cell development and IKZF1 expression.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Association results | This study | Table S2 |
| Resource for Genetic Epidemiology Research on Aging (GERA) genotype data | Kvale et al.55 | dbGaP accession number: phs000788.v1.p2 |
| Children’s Oncology Group (COG)/St. Jude Children’s Research Hospital study genotype data | Qian et al.17 | dbGaP accession number: phs000638.v1.p1, phs000637.v1.p1 |
| Genes-environments and Admixture in Latino Asthmatics (GALA II) whole-genome sequencing and RNA sequencing data | Kachuri et al.41 | dbGaP accession number: phs000920 |
| Study of African Americans, Asthma, Genes, and Environments (SAGE) whole-genome sequencing and RNA sequencing data | Kachuri et al.41 | dbGaP accession number: phs000921 |
| 1000 Genomes Project Phase 3 data | 1000 Genomes Project Consortium56 | https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502 |
| Genome Aggregation Database (gnomAD) v3.1 | Karczewski et al.57 | https://gnomad.broadinstitute.org/downloads#v3 |
| Hematopoiesis single-cell ATAC-sequencing data | Yu et al.,27 Granja et al.,58 Satpathy et al.59 | https://github.com/GreenleafLab/MPAL-Single-Cell-2019, https://github.com/GreenleafLab/10x-scATAC-2019 |
| Hematopoiesis single-cell RNA-sequencing data | Granja et al.58 | https://github.com/GreenleafLab/MPAL-Single-Cell-2019 |
| ATAC-sequencing data from 156 B cell ALL patients | Barnett et al.29 | GEO: GSE226400 |
| IKZF1 ChIP-seq data for GM12878 | ENCODE Project Consortium60 | GEO: GSE105587 |
| Promoter capture Hi-C data for GM12878 | Bhattacharyya et al.61 | https://doi.org/10.5281/zenodo.3255048 |
| eQTL summary statistics, ancestry-specific eQTLs, and TWAS models developed using data from GALA II and SAGE participants | Kachuri et al.41 | https://doi.org/10.5281/zenodo.7735723 |
| GTEx Consortium eQTL data | GTEx Consortium23 | https://www.gtexportal.org/home/ |
| eQTLGen Consortium eQTL data | Võsa et al.24 | https://eqtlgen.org/cis-eqtls.html |
| ImmuNexUT eQTL data | Ota et al.25 | https://www.immunexut.org/ |
| Pre-calculated trees for each 1KG population | Speidel et al.62 | https://zenodo.org/record/3234689#.Y2VdouzML0p/ |
| Ancient genome shotgun sequencing data | Posth et al.,63 Mallick et al.,64 Moreno-Mayar et al.,65 Scheib et al.,66 Lindo et al.,67 de la Fuente et al.,68 Capodiferro et al.,69 Rasmussen et al.,70 Rasmussen et al.,71 Raghavan et al.,72 Skoglund et al.73 | ENA: PRJEB28961, PRJEB9586, ERP010710, PRJEB29074, PRJEB25445, PRJNA470966, PRJEB24629, PRJEB42372, PRJEB9733 NCBI SRA: SRX381032, SRS937952 |
| Human genome reference sequence hg38 | UCSC Genome Browser | https://hgdownload.soe.ucsc.edu/downloads.html |
| Software and algorithms | ||
| PLINK v2.0 | Chang et al.74 | https://www.cog-genomics.org/plink/2.0/ |
| LDLink | Machiela et al.75 | https://ldlink.nih.gov/ |
| TOPMed Imputation Server | Taliun et al.76 | https://imputation.biodatacatalyst.nhlbi.nih.gov/ |
| susieR | Zou et al.,77 Wang et al.78 | https://github.com/stephenslab/susieR |
| RFMix | Maples et al.79 | https://github.com/slowkoni/rfmix |
| ADMIXTURE | Alexander et al.80 | https://dalexander.github.io/admixture/download.html |
| Relate | Speidel et al.62 | https://myersgroup.github.io/relate/ |
| samtools | Li et al.81 | https://www.htslib.org/ |
| SRA Toolkit | Github | https://github.com/ncbi/sra-tools |
| cutadapt | Martin82 | https://cutadapt.readthedocs.io/en/stable/ |
| FastQC | Github | https://github.com/s-andrews/FastQC |
| Bowtie2 | Langmead et al.83 | https://bowtie-bio.sourceforge.net/bowtie2 |
| CHiCAGO | Cairns et al.84 | https://www.bioconductor.org/packages/release/bioc/html/Chicago.html |
| LI-COR Image Studio Lite | LICOR Biosciences | RRID: SCR_013715 |
| FlowJo | FlowJo LLC | RRID: SCR_008520 |
| Chemicals, peptides, and recombinant proteins | ||
| Roche EDTA-free complete protease inhibitor cocktail | Roche | Cat: 11697498001 |
| RIPA Buffer | Thermo Scientific | Cat: 89900 |
| Dulbecco’s Modified Eagle Medium-High Glucose (DMEM) | Life Technologies | Cat: 11965-118 |
| Iscove’s Modified Dulbecco’s Medium (IMDM) | StemCell Technologies | Cat: 36150 |
| Fetal Bovine Serum (FBS) | BioTechne | Cat: S11550 |
| Penicillin-Streptomycin | GIBCO | Cat: 15140-122 |
| StemSpan SFEM II medium | StemCell Technologies | Cat: 02690 |
| StemSpan CC100 | StemCell Technologies | Cat: 02690 |
| 2-mercaptoethanol | Gibco | Cat: 21985023 |
| PBS | GIBCO | Cat: 10010-023 |
| L-Glutamine | Thermo Fisher Scientific | Cat: 25-030-081 |
| Penicillin/Streptomycin | Life Technologies | Cat: 15140-122 |
| Electroporation enhancer | IDT | Cat: 1075915 |
| P3 Lonza buffer with supplement | Lonza | Cat: V4XP-3032 |
| HiFi Cas9 Nuclease V3 | IDT | Cat: 1081061 |
| RPMI | Life Technologies | Cat: 11875085 |
| EasySep Human CD34 Positive Selection Kit II | StemCell Technologies | Cat: 17856 |
| RosetteSep Human Hematopoietic Progenitor Cell Enrichment Cocktail | StemCell Technologies | Cat: 15066 |
| Ficoll-Paque | GE Healthcare | Cat: 45-001-751 |
| TPO | Peprotech | Cat: 300-18 |
| Recombinant IL7 | StemCell Technologies | Cat: 78053 |
| AllPrep DNA/RNA Mini kit | QIAGEN | Cat: 80204 |
| OneShot TOP10 Chemically Competent Cells | Invitrogen | Cat: C404006 |
| 4–12% Criterion XT Bis-Tris Protein Gel | Bio-Rad | Cat: 3450124 |
| Blocking buffer | LICOR Biosciences | Cat: 927-70001 |
| 7-AAD | BD Biosciences | Cat: 51-68981E |
| Recombinant Anti-Ikaros antibody | Abcam | Cat: ab191394; RRID: AB_3073859 |
| Monoclonal Beta Actin Antibody | Santa Cruz Biotechnology | Cat: sc-47778; RRID: AB_626632 |
| Platinum II Hot-Start PCR Master Mix | Invitrogen | Cat: 14000012 |
| Tween 20 | Sigma Aldrich | Cat: P9416 |
| IRDye Secondary Antibodies | LI-COR Biosciences | Cat: 926–68072; RRID: AB_10953628Cat: 926-32212; RRID: AB_621847 |
| FcR Blocking Reagent | Miltenyi | Cat: 130-059-901 |
| CD34-Alexa488 | BioLegend | Cat: 343517; RRID: AB_1937204 |
| CD10-PE | Beckman-Coulter | Cat: IM1915U; RRID: AB_131294 |
| CD19-BV421 | BD Biosciences | Cat: 562440; RRID: AB_11153299 |
| CD20-Pe-Cy7 | BD Biosciences | Cat: 560735; RRID: AB_1727450 |
| CD33-APC | BioLegend | Cat: 303407; RRID: AB_314351 |
| Lenti-X Integration Site Analysis Kit | Takara | Cat: 631263 |
| NucleoBond Xtra Maxi | Macherey-Nagel | Cat: 740424.50 |
| Oligonucleotides | ||
| Primers and oligonucleotides | IDT | See STAR Methods |
| sgRNAs | Synthego | See STAR Methods |
| Recombinant DNA | ||
| pLKO lentiviral construct | Genetic Perturbation Platform, Broad Institute | Cat: TRC046 |
| Biological samples | ||
| Human CD34+ hematopoietic stem and progenitor cells, adult | Fred Hutchinson Cancer Research Center | N/A |
| Cord Blood Unit for umbilical cord-derived CD34+ hematopoietic stem and progenitor cells | Dana Farber Pasquarello Tissue Bank | N/A |
| Experimental models: Cell lines | ||
| 293T cells | ATCC | Cat: CRL-3216 |
| Reh cells | ATCC | Cat: CRL-8286 |
| MS-5 | DSMZ-German Collection of Microorganisms and Cell Cultures | Cat: ACC 441 |
| HepG2 | ATCC | Cat: HB-8065 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vijay G. Sankaran (sankaran@broadinstitute.org).
Materials availability
All unique materials will be available upon request to the lead contact.
Data and code availability
This study included the analysis of data derived from biospecimens from the California Biobank Program (CCRLP study). Any uploading of genomic data and/or sharing of these biospecimens or individual data derived from these biospecimens has been determined to violate the statutory scheme of the California Health and Safety Code Sections 124980(j); 124991(b), (g), and (h); and 103850 (a) and (d), which protect the confidential nature of biospecimens and individual data derived from biospecimens. Should we be contacted by other investigators who would like to use the data, we will direct them to the California Department of Public Health Institutional Review Board to establish their own approved protocol to utilize the data, which can then be shared peer-to-peer.
The GWAS summary statistics at the IKZF1 locus are included in Table S2. Genotype data for the Genetic Epidemiology Research on Aging (GERA) study controls are available on dbGAP under accession number phs000788.v1.p2. Genotype data for the Children’s Oncology Group (COG)/St. Jude Children’s Research Hospital study ALL patients are available on dbGaP under accession number phs000638.v1.p1 and phs000637.v1.p1.
Whole genome sequencing and whole blood RNA sequencing data from the Genes-environments and Admixture in Latino Asthmatics (GALA II) study and the Study of African Americans, Asthma, Genes, and Environments (SAGE), generated as part of the NHLBI TOPMed program, are available dbGaP under accession numbers phs000920 (GALA II) and phs000921 (SAGE). Summary statistics for cis- and trans-eQTLs, a catalog of ancestry-specific eQTLs, and TWAS models developed using data from GALA II and SAGE participants have been posted in a public repository at https://doi.org/10.5281/zenodo.7735723.
All other data are available in the cited references.
Experimental model and study participant details
Cell lines
MS-5, REH, 293T, and HepG2 cells were cultured and handled as discussed below. Primary human hematopoietic stem and progenitor cells were isolated from deidentified and discarded cord blood samples made available through the Pasquarello Tissue Bank at the Dana-Farber Cancer Institute.
Study subjects
The study protocol was approved by the Institutional Review Boards at the California Health and Human Services Agency, University of Southern California, University of California San Francisco, and Yale University. This study includes childhood ALL cases and controls identified in the California Cancer Records Linkage Project (CCRLP).18 The acquisition of newborn dried bloodspot (DBS) samples has been described in detail previously.12 Briefly, DBS for cases and controls were obtained from the California Biobank Program (California Biobank Program SIS request # 1380), California Department of Public Health (CDPH), Genetic Disease Screening Program, with a waiver of consent from the Committee for the Protection of Human Subjects of the State of California. Childhood ALL cases (≤14 years of age) were identified through linkage between the CDPH statewide birth records (years 1982–2009) and the California Cancer Registry (CCR, diagnosis years 1988–2011), and controls were randomly selected and matched on year and month of birth, sex, and race and ethnicity (categorized as non-Hispanic White, non-Hispanic Black, Hispanic/Latino [any race], non-Hispanic Asian/Pacific Islander, other) as reported in the birth records. Additional controls were included from the Genetic Epidemiology Research on Aging (GERA) study (dbGaP accession: phs000788.v1.p2). ALL molecular subtype analysis was conducted using patients included in Children’s Oncology Group (COG)/St. Jude Children’s Research Hospital studies.17
Race and ethnicity are considered social constructs but they are also correlated with genetic ancestry.85 Since recruitment of participants in studies can only be practically done based on self-described race and ethnicity, we used those variables to select participants. In this study, analyses were limited to participants who were self-reported as Hispanic/Latino, non-Hispanic White, or East Asian, as described below. These participant groupings were retained for genetic association analyses to align the findings of this study with the well-documented disparity in ALL in the United States, with significantly higher incidence in the Hispanic/Latino population.
Method details
Genome-wide SNP data processing, quality control, and imputation
Genome-wide SNP genotyping was performed for all individuals in CCRLP and GERA using the Affymetrix Axiom World LAT array.12 Quality control (QC) of SNP array data and samples were carried out in each population group, as previously described in our recent trans-ancestry GWAS meta-analysis of ALL.18 In brief, for pre-imputation QC, we excluded the sex chromosomes and filtered out SNPs based on call rates <98%, minor allele frequency (MAF) < 0.01, and Hardy-Weinberg equilibrium in controls (p < 10−5), and removed samples based on genome-wide relatedness (PI_HAT>0.20), genome heterozygosity rate (mean heterozygosity± 6Std), and call rates <95%. In general, we included individuals in each of the three population groups based on their self-reported race/ethnicity and did not reassign individuals based on estimated genetic ancestry.18 Principal components analysis (PCA) was performed for each population group along with reference data from 1000 Genomes Project (1KG)56 to identify extreme outlier individuals that, consistent with incurring excessive technical errors during the data generation process, clustered separately from other individuals in their self-reported race-ethnicity groups. Among self-reported Asian individuals, we identified a small subset (29 cases and 51 controls from CCRLP, 31 individuals from GERA) that clustered with South Asian reference individuals and were subsequently removed from our analyses, with remaining individuals classified as East Asian individuals. In a global PCA plot (Figure S13), a small number of Hispanic/Latino individuals clustered with other populations, but these were retained in the GWAS as our inclusion criteria were based on self-reported race/ethnicity. Pre-imputation GWAS was performed between population-matched CCRLP controls and GERA individuals to protect against between-cohort batch effects.18
Whole-genome imputation was performed for each population group, separately for CCRLP and GERA datasets, using the TOPMed Imputation Server,76 with additional QC filtering performed post-imputation. We removed variants in each population group based on imputation quality (r2 < 0.3), MAF (<0.01), and allele frequency difference between non-Finnish Europeans in the Genome Aggregation Database (gnomAD)57 and CCRLP non-Hispanic White controls (>0.1). We next performed another GWAS between CCRLP controls and GERA individuals and removed variants with P < 1x10−5. CCRLP and GERA datasets for Hispanic/Latino, non-Hispanic White, and East Asian individuals were then merged to perform the GWAS of childhood ALL. After quality control filtering, the Hispanic/Latino GWAS included 1,878 cases and 8,441 controls, the non-Hispanic White GWAS included 1,162 cases and 57,341 controls, and the East Asian GWAS included 318 cases and 5,017 controls.
Cell lines
MS-5 cells (DSMZ) were cultured at 37°C in IMDM (StemCell) supplemented with 10% FBS, 50 μM 2-Mercaptoethanol (Gibco) and 1% penicillin/streptomycin. 24–48 h prior to establishment of co-culture with HSPCs, cells were pre-plated in a 24 well plate at 3 × 10ˆ4 per well. REH cells (ATCC) were cultured at 37°C in RPMI (Life Technologies) supplemented with 10% FBS and 1% penicillin/streptomycin. Both 293T cells and HepG2 cells (ATCC) were cultured at 37°C in DMEM (Life Technologies) supplemented with 10% FBS and 1% penicillin/streptomycin.
Primary cell culture
B-cell progenitors were derived from differentiation of human hematopoietic stem and progenitor cells (HSPCs). HSPCs were purified from discarded umbilical cord blood samples of healthy newborns using the EasySep Human CD34 Positive Selection Kit II following pre-enrichment with RosetteSep Pre-enrichment cocktail (Stem Cell Technologies) and mononuclear cell isolation on a Ficoll-Paque (GE Healthcare) density gradient. Cells were initially cultured and expanded at 37°C and 5% O2 in serum-free medium consisting of StemSpan II medium (Stem Cell Technologies) supplemented with CC100 cytokine cocktail (Stem Cell Technologies) and 50 ng/mL TPO (Peprotech).35 Confluency was maintained between 5 × 10ˆ5 and 1 × 10ˆ6 cells per mL. After 5 days of expansion culture, HSPCs were co-cultured with the MS-5 stromal layer to facilitate B-cell differentiation. Cells were maintained for 3–5 weeks with bi-weekly feedings with 5% FBS, IMDM (StemCell), and 1% penicillin/streptomycin (Gibco) supplemented with 10 ng/mL recombinant IL7 (StemCell).36
CRISPR/Cas9-genome editing and analysis
CRISPR/Cas9 genome editing of HSPCs was performed while cells were in progenitor maintenance media.35 Electroporation was performed 48 h after thawing of cord-blood derived CD34+ HSPCs using the Lonza 4D Nucleofector kit. The RNP complex was made by combining 100 pmol Cas9 (IDT) and 100 pmol modified sgRNA (Synthego) targeting IKZF1 or AAVS1 (using single synthetic guide RNAs to introduce indels: IKZF1 sg1: 5′-CCUGUAAGCGAUACUCCAGA-3′; sg2: 5′-CCCUGUAAGCGAUACUCCAG-3′ or AAVS1 5′-GGGGCCACUAGGGACAGGAU-3′) or the IKZF1 binding motif within the enhancer sequence (using pairs of synthetic guide RNAs to introduce microdeletions: IKZF1 Enh sg1: 5′-GCAGATGGGCCCTGGCAACT-3′; sg2: 5′-GCACAACTGGAACCTGCTGG-3′ or AAVS1 sg1: 5′-GGGGCCACUAGGGACAGGAU-3′; sg2: 5′-GGGACCACCUUAUAUUCCCA-3′) and incubating at room temperature for 30 min. Between 1 × 10ˆ5 and 5 × 10ˆ5 HSPCs were resuspended in 20 μL P3 solution with 1 μL electroporation enhancer (IDT) and mixed with the RNP to undergo nucleofection with program DZ-100. Cells were returned to HSC medium and editing efficiency was measured at 48 h after electroporation, unless otherwise indicated. For this, genomic DNA and RNA was extracted using the AllPrep DNA/RNA Mini kit (QIAGEN) according to the manufacturer’s instructions. Genomic DNA PCR was performed using Platinum II Hotstart Mastermix (Thermo Fisher). Edited allele frequency was detected by Sanger sequencing and analyzed by Inference of CRISPR Edits (ICE) (ice.syngthego.com). The following primer pairs were used: IKZF1-exon 3 (forward: 5′-AGTGTCTGGGATTATAGGTGATTG-3′; reverse: 5′-CCCATCCTGCTGATCTTTGT-3′); IKZF1 enhancer region (forward: 5′-CACGTCTGGGATCTGGGCTTCT-3′; reverse: 5′-GGGATCACATGTGGTCGCAACC-3′) and AAVS1 (forward: 5′-GGCTCTGGTTCTGGGTACTT-3′; reverse: 5′-TCTCTCCTTGCCAGAACC -3′). The experiment was performed in triplicate, representative editing efficiencies are displayed in Figure S12.
Western blotting
The effect of genome editing of HSPCs on IKZF1 protein expression was detected by western blotting. Cells were harvested, washed with ice-cold PBS and resuspended in radioimmunoassay lysis buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate) (Thermo Scientific) supplemented with 1× Complete Protease Inhibitor Cocktail (Roche). After centrifugation at 14,000 rpm for 15 min at 4°C to remove cellular debris, whole cell lysates were denatured at 90°C for 10 min. Equal amounts of protein were separated by SDS gel electrophoresis using 4–12% polyacrylamide gels (Bio-Rad). Subsequently, proteins were transferred onto a polyvinylidene fluoride membrane. Membranes were incubated with blocking buffer (LICOR Biosciences) and probed with rabbit monoclonal antibody to IKZF1 (ab191394; Abcam), and mouse monoclonal antibody to actin (sc-47778; Santa Cruz Biotechnology) all at a 1:1,000 dilution overnight at 4°C. Membranes were then washed with PBS with 0.1% Tween, incubated with secondary antibodies (IRDye Secondary Antibodies; LI-COR Biosciences), and analyzed using the LI-COR Odyssey imaging system.
Flow cytometry
Cells were harvested, filtered, washed with PBS supplemented with 2% FBS, incubated with FcR Blocking Reagent (Miltenyi) and stained with the following panel of antibodies: anti-CD34-Alexa488 (BioLegend, 343517), anti-CD10-PE (Beckman-Coulter, IM1915U), anti-CD19-BV421 (BD, 562440), anti-CD20-Pe-Cy7 (BD, 560735) and anti-CD33-APC (BioLegend, 303407). Flow cytometric analyses were conducted on a BD Fortessa analyzer and all data were analyzed using FlowJo software.
Lentiviral reporter assays
The lentiviral reporter constructs were designed to deliver enhancer elements containing risk and non-risk alleles of rs1451367 and/or rs17133807 positioned upstream of a minimal promoter (TATA box) driving a reporter eGFP.86 The constructs were generated after modification of a pLKO lentiviral construct (TRC046, Genetic Perturbation Platform, Broad Institute). Lentiviral supernatants produced by transient transfection in 293Ts were concentrated and titered by qPCR. Provirus copy number per cell were quantified using the Lenti Integration Site Analysis Kit (Takara). Titered lentivirus was then used to transduce REH, 293T, and HepG2 cells. Cell lines were cultured for 72 h and reporter GFP expression was measured by flow cytometry.
Quantification and statistical analysis
Chromosome 7p12 association testing and conditional analysis
SNP association testing was limited to a ∼600 kb region centered +/−250 kb around the IKZF1 gene at chromosome 7p12.2-p12.1 (chr7: 50,054,716–50,655,101, hg38). In each population group, we used PLINK v2.074 to test the association between imputed genotype dosage and case-control status in logistic regression, after adjusting for the top 20 principal components (PCs). We did not include sex as a covariate, and we found sex was not correlated with genotype dosage of any of the putatively associated SNPs (p > 0.05). Next, we repeated the association testing in each population group conditioning on the top SNP in that group, by including it as an additional covariate in the logistic regression model (conditional test 1). Then, we repeated the association testing conditioning on the top SNP in the main GWAS plus the top SNP in conditional test 1 (conditional test 2). In Hispanic/Latino individuals, where 3 independent loci were identified, we repeated analysis conditioning on top SNPs from the main marginal GWAS and conditional tests 1 and 2. A genome-wide threshold of p < 5 x 10−8 was used for significance in each test. Linkage disequilibrium (LD) between SNPs in 1KG populations was assessed using the LDlink tool.75
The IKZF1 GRS was calculated in Hispanic/Latino individuals including the three lead independent risk variants weighted by their corresponding marginal (signal 1 lead SNP rs4917017) or conditional (signal 2 lead SNP rs10272724 adjusted for signal 1, and signal 3 lead SNP rs76880433 adjusted for signals 1 and 2) effect estimates obtained from the Hispanic/Latino GWAS of ALL. The IKZF1 GRS in non-Hispanic White individuals included the two lead independent risk variants weighted by their marginal (signal 1 lead SNP rs17133805) or conditional (signal 2 lead SNP rs9886239 adjusted for signal 1) effect estimates obtained from the non-Hispanic White ALL GWAS.
Statistical fine-mapping
To prioritize potentially causal variants underlying the observed risk in GWAS summary data, we performed summary-based statistical fine-mapping using SuSiE77 focusing on ∼2.1Mb region centered around risk variants rs4917017 and rs10272724, which included 2191, 2152, and 2067 variants in Hispanic/Latino, non-Hispanic White, and East Asian individuals, respectively. To account for LD among test statistics, we estimated in-sample LD for each population group after regressing out GWAS covariates from genotype dosages, an approach similar to that described in ref. 87. To ensure our GWAS test statistics were robust to residualized LD estimates and reduce false positives due to misassigned ref/alt alleles (i.e., allele flips), we performed a kriging analysis using SuSiE. Using the SuSiE-recommended LRT thresholds, we found little support for inconsistencies between LD estimates and GWAS results. Lastly, we set the maximum number of causal variants as 10 and reported 90% credible sets for each population.
Genetic ancestry analysis
We assessed the association between IKZF1 risk alleles and Indigenous American ancestry among CCRLP Hispanic/Latino individuals. Global and local ancestry inference were performed on CCRLP Hispanic/Latino cases and controls using RFMix,79 using a reference panel consisting of 671 non-Finnish European individuals for European ancestry, 708 African individuals for African ancestry, and 94 Latino/Admixed American (AMR) individuals (7 Colombian, 12 Karitianan, 14 Mayan, 4 Mexican ancestry in Los Angeles [MXL], 37 Peruvian in Lima Peru [PEL], 12 Pima, and 8 Surui) for Indigenous American ancestry from gnomAD v3.1 release.57 The 94 AMR individuals were selected based on having more than 85% Indigenous American ancestry in an unsupervised analysis of global ancestry of AMR individuals from the 1KG,56 using ADMIXTURE.80 After estimating local ancestry for each individual, we stratified Hispanic/Latino individuals into whether they carried zero copies or at least 1 copy of the haplotype derived from Indigenous American ancestry, and in each group estimated the odds ratio (OR) for association of the three independent IKZF1 SNPs with ALL risk in a logistic regression model for a series of conditional analyses on the top SNPs, adjusting for age, sex, and 20 principal components using PLINK v2.0.74 For each individual, we calculated their proportions of global Indigenous American, European, and African ancestry by summing local ancestry estimates across the genome.
Selection analysis
We retrieved pre-compiled estimates of the allelic ages and p values for positive selection for each of the IKZF1 risk alleles for IBS (Iberian populations in Spain), CHS (Southern Han Chinese), PEL, and MXL populations from the 1KG. The estimates of allelic ages and evidence of selection were based on an inferred genealogical tree spanning the IKZF1 locus. The genealogical trees were inferred using Relate,62 and pre-calculated trees for each 1KG population are available for download at: https://zenodo.org/record/3234689#.Y2VdouzML0p/. The allelic age is estimated as the mid-branch time in the number of generations for the branch of the genealogy where the derived allele of each risk-associated SNPs arose. The age in generations was converted to years assuming 28 years per generation. The evidence of positive selection is based on the over-representation of lineages carrying the derived allele at the tip of the genealogy, given the distribution of carrier and non-carrier haplotypes when the branch carrying the allele of interest first branched into two sublineages.
Expression quantitative trait loci (eQTL) analysis
To examine the effects of the ALL risk SNPs on the expression of IKZF1 and nearby genes, we analyzed whole genome and whole blood RNA sequencing data from the Genes-environments and Admixture in Latino Asthmatics (GALA II) study and the Study of African Americans, Asthma, Genes, and Environments (SAGE), which recruited asthma cases and controls between 8 and 21 years of age.41 Effects on gene expression were estimated in 784 self-identified Mexican Americans and in participants grouped based on Indigenous American (IAM) genetic ancestry. Associations in individuals with >50% IAM ancestry (IAMHigh; n = 610) were compared to those with <10% (IAMLow; n = 1257), as described.41 Single-variant and haplotype analyses were performed using linear regression with adjustment for age, sex, case-control status, top 5 genetic ancestry PC’s, and 80 PEER factors to account for hidden confounding factors in the gene expression data. Cis-eQTL effects were considered statistically significant for variants with p values less than the beta-approximated FastQTL p value at the false discovery rate <0.05 for a given gene (approximately PeQTL<6 × 10−4). For the lead SNPs at signal 1, signal 2, and signal 3, we also looked for significant eQTL associations in publicly available datasets including GTEx,23 eQTLGen,24 and the Immune Cell Gene Expression Atlas from the University of Tokyo (ImmuNexUT).25
Analysis of ancient genomes
To assess the allelic age of a putative causal variant at IKZF1, analysis of shotgun sequencing data from ancient American genomes was performed, as has been described previously.63 Briefly, we analyzed the number of reads at the IKZF1 gene using samtools version 1.3.181,88 mpileup with the settings -d 8000 -B -q30 -Q30 to obtain information about each read from the bam files of our samples (Table S9). We used the fasta file from human genome GRCh37 (hg19) for the pileup. We counted the number of derived and ancestral variants at each analyzed position using a custom Python script. In order to reduce the effect of damage characteristic of ancient DNA (we note that this allele is a C to T change and thus similar to changes that are seen in ancient DNA), we used primarily samples treated with UDG, which substantially reduces the effect of these transitions. We also clip the last 3 bp for UDG-treated samples and 10 bp for non-UDG-treated samples to further decrease the effect of ancient damage. Lastly, we note that the most important samples showing the finding of a T allele in the ancient Americas have a substantial fraction of their alleles being T, which would not be possible if they only resulted from DNA damage characteristic of ancient DNA. The results were plotted based on coordinates available for all ancient individuals using ggplot2.
Epigenomic and long-range chromatin interaction data analysis
The colocalization analysis was performed between functional signals, including chromatin accessibility and IKZF1 chromatin occupancy, and each SNP individually. Accessible chromatin data spanning the full spectrum of human hematopoiesis were analyzed, as previously described.27 The processed IKZF1 ChIP-seq (GSE105587) and promoter capture Hi-C datasets61 from human B-lymphoblastoid cell line GM12878 were used to infer functional effects of SNPs. The IKZF1 binding motif was determined using IKZF1 ChIP-seq data based on relative occupancy and the similarity to the determined motif was then compared in the presence or absence with different variants at rs1451367. The long range chromatin interactions of IKZF1 promoter with CHiCAGO score greater than 5 were kept for further analysis.84 A single cell RNA-seq dataset of hematopoiesis was analyzed58 and the gene expression of IKZF1 of cells belonging to B-cell developmental trajectory, ranging from early hematopoietic progenitors to mature B cells, was examined.
Analysis of chromatin accessibility at IKZF1 risk variants in B-cell ALL patients
Raw ATAC-sequencing data from 156 B-cell ALL patients29 in SRA format were retrieved from the NCBI database (BioProject PRJNA940132) and converted into fastq format using the fastq-dump-orig (v3.0.7) tool in SRA Toolkit. The adapter sequences were sequentially removed with cutadapt (v 2.5),82 employing a minimum length cutoff of 5 (-m 5) and an adapter error rate of 0.2. FastQC (v 0.12.1) was used to assess the quality of sequencing data. The reference genome of assembly hg38, chromosome 7, where IKZF1 variants of interest are located, was obtained from UCSC. Bowtie2 (v 2.3.4.3)83 was utilized to map the reads to the reference genome, with parameters set to multimapping = 2, -X 2000, and others kept as default. Samtools (v 1.9)81 mpileup was employed to extract genotype information from genomic reads that overlap with inspected SNPs from the aligned BAM files for each variant. A custom parser was developed and used for allele frequency calculation and result interpretation. Normalized ATAC-sequencing read counts were compared between genotype groups for each SNP using the Wilcoxon rank-sum test (one-tailed). Read counts for non-risk allele versus risk allele among SNP heterozygotes were compared using the paired Wilcoxon signed rank test (one-tailed).
Acknowledgments
We thank Drs. Stephen Sallan, David Nathan, Melissa Burns, and Steve Smale, as well as members of our laboratories, for valuable discussions and suggestions. This work was supported by grants from the National Institutes of Health (R01CA262263 to A.J.d.S., C.W.K.C., J.L.W., C.M., X.M., J.J.Y., and N.M.; R01CA155461 to J.L.W. and X.M.; R00CA246076 to L.K.; R35GM142783 to C.W.K.C.; and R01DK103794 and R01CA265726 to V.G.S.), the New York Stem Cell Foundation (to V.G.S.), and the Dana-Farber Cancer Institute Presidential Priorities Initiative (to L.W. and V.G.S.). A.J.d.S. is a Scholar of the Leukemia & Lymphoma Society. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The collection of cancer incidence data used in this study was supported by the California Department of Public Health as part of the statewide cancer reporting program mandated by California Health and Safety Code Section 103885; the National Cancer Institute’s Surveillance, Epidemiology, and End Results Program under contract HHSN261201000140C awarded to the Cancer Prevention Institute of California, contract HHSN261201000035C awarded to the University of Southern California, and contract HHSN261201000034C awarded to the Public Health Institute; and the Centers for Disease Control and Prevention’s National Program of Cancer Registries under agreement U58DP003862-01 awarded to the California Department of Public Health. Data for control individuals partially came from a grant, the Resource for Genetic Epidemiology Research in Adult Health and Aging (RC2 AG033067; Schaefer and Risch, PIs), awarded to the Kaiser Permanente Research Program on Genes, Environment, and Health (RPGEH) and the UCSF Institute for Human Genetics. The RPGEH was supported by grants from the Robert Wood Johnson Foundation, the Wayne and Gladys Valley Foundation, the Ellison Medical Foundation, Kaiser Permanente Northern California, and the Kaiser Permanente National and Northern California Community Benefit Programs. The RPGEH and the Resource for Genetic Epidemiology Research in Adult Health and Aging are described here: https://divisionofresearch.kaiserpermanente.org/research/research-program-on-genes-environment-and-health/. The biospecimens and/or data used in this study were obtained from the California Biobank Program (SIS request #1380), in accordance with Section 6555(b), 17 CCR. The California Department of Public Health is not responsible for the results or conclusions drawn by the authors of this publication. Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program were supported by the National Heart, Lung, and Blood Institute (NHLBI). Genome and RNA sequencing for NHLBI TOPMed Genes-Environments and Admixture in Latino Asthmatics (GALA II) Study (phs000920) and NHLBI TOPMed Study of African Americans, Asthma, Genes, and Environments (SAGE) (phs000921) was performed at NYGC Genomics (3R01HL117004-02S3). Core support, including phenotype harmonization, data management, sample-identity quality control, and general program coordination, was provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). Additionally, E.G.B. is supported by the Sandler Family Foundation, the American Asthma Foundation, the RWJF Amos Medical Faculty Development Program, the Harry W. and Diana V. Hind Distinguished Professor in Pharmaceutical Sciences II, the NHLBI (R01HL117004, R01HL135156, X01HL134589, and U01HL138626), and the National Institutes of Health and Environmental Health Sciences (R01ES015794). The authors acknowledge the Center for Advanced Research Computing (https://carc.usc.edu/) at the University of Southern California for providing computing resources that have contributed to the research results reported within this publication.
Author contributions
A.J.d.S., L.W., C.W.K.C., and V.G.S. conceived and designed the study. A.J.d.S., L.W., S.J., L.K., S.B., J.L., L.D.C., N.N., T.-F.C., G.X., S.M., W.Y., S.G., C.E., D.H., E.G.B., E.Z., C.M., N.M., J.J.Y., X.M., J.L.W., F.Y., C.W.K.C., and V.G.S. performed analyses and experiments and/or contributed key resources. A.J.d.S., L.W., and V.G.S. wrote and edited the manuscript with input from all authors.
Declaration of interests
V.G.S. serves as an advisor to and/or has equity in Branch Biosciences, Ensoma, and Cellarity, all unrelated to the present work.
Published: March 26, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2024.100526.
Contributor Information
Adam J. de Smith, Email: desmith@usc.edu.
Vijay G. Sankaran, Email: sankaran@broadinstitute.org.
Supplemental information
References
- 1.Zavala V.A., Bracci P.M., Carethers J.M., Carvajal-Carmona L., Coggins N.B., Cruz-Correa M.R., Davis M., de Smith A.J., Dutil J., Figueiredo J.C., et al. Cancer health disparities in racial/ethnic minorities in the United States. Br. J. Cancer. 2021;124:315–332. doi: 10.1038/s41416-020-01038-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ward E., DeSantis C., Robbins A., Kohler B., Jemal A. Childhood and adolescent cancer statistics. CA Cancer J. Clin. 2014;64:83–103. doi: 10.3322/caac.21219. [DOI] [PubMed] [Google Scholar]
- 3.Feng Q., de Smith A.J., Vergara-Lluri M., Muskens I.S., McKean-Cowdin R., Kogan S., Brynes R., Wiemels J.L. Trends in Acute Lymphoblastic Leukemia Incidence in the United States by Race/Ethnicity From 2000 to 2016. Am. J. Epidemiol. 2021;190:519–527. doi: 10.1093/aje/kwaa215. [DOI] [PubMed] [Google Scholar]
- 4.Linabery A.M., Ross J.A. Childhood and adolescent cancer survival in the US by race and ethnicity for the diagnostic period 1975-1999. Cancer. 2008;113:2575–2596. doi: 10.1002/cncr.23866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rivera-Luna R., Perez-Vera P., Galvan-Diaz C., Velasco-Hidalgo L., Olaya-Vargas A., Cardenas-Cardos R., Aguilar-Ortiz M., Ponce-Cruz J. Triple-hit explanation for the worse prognosis of pediatric acute lymphoblastic leukemia among Mexican and Hispanic children. Front. Oncol. 2022;12:1072811. doi: 10.3389/fonc.2022.1072811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatia S., Sather H.N., Heerema N.A., Trigg M.E., Gaynon P.S., Robison L.L. Racial and ethnic differences in survival of children with acute lymphoblastic leukemia. Blood. 2002;100:1957–1964. doi: 10.1182/blood-2002-02-0395. [DOI] [PubMed] [Google Scholar]
- 7.Quiroz E., Venkateswaran A.R., Nelson R., Aldoss I., Pullarkat V., Rego E., Marcucci G., Douer D. Immunophenotype of acute lymphoblastic leukemia in minorities- analysis from the SEER database. Hematol. Oncol. 2022;40:105–110. doi: 10.1002/hon.2945. [DOI] [PubMed] [Google Scholar]
- 8.Dores G.M., Devesa S.S., Curtis R.E., Linet M.S., Morton L.M. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood. 2012;119:34–43. doi: 10.1182/blood-2011-04-347872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papaemmanuil E., Hosking F.J., Vijayakrishnan J., Price A., Olver B., Sheridan E., Kinsey S.E., Lightfoot T., Roman E., Irving J.A.E., et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat. Genet. 2009;41:1006–1010. doi: 10.1038/ng.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Treviño L.R., Yang W., French D., Hunger S.P., Carroll W.L., Devidas M., Willman C., Neale G., Downing J., Raimondi S.C., et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat. Genet. 2009;41:1001–1005. doi: 10.1038/ng.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Migliorini G., Fiege B., Hosking F.J., Ma Y., Kumar R., Sherborne A.L., da Silva Filho M.I., Vijayakrishnan J., Koehler R., Thomsen H., et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood. 2013;122:3298–3307. doi: 10.1182/blood-2013-03-491316. [DOI] [PubMed] [Google Scholar]
- 12.Wiemels J.L., Walsh K.M., de Smith A.J., Metayer C., Gonseth S., Hansen H.M., Francis S.S., Ojha J., Smirnov I., Barcellos L., et al. GWAS in childhood acute lymphoblastic leukemia reveals novel genetic associations at chromosomes 17q12 and 8q24.21. Nat. Commun. 2018;9:286. doi: 10.1038/s41467-017-02596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu H., Cheng C., Devidas M., Pei D., Fan Y., Yang W., Neale G., Scheet P., Burchard E.G., Torgerson D.G., et al. ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2012;30:751–757. doi: 10.1200/JCO.2011.38.0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perez-Andreu V., Roberts K.G., Harvey R.C., Yang W., Cheng C., Pei D., Xu H., Gastier-Foster J., E S., Lim J.Y.-S., et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat. Genet. 2013;45:1494–1498. doi: 10.1038/ng.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walsh K.M., de Smith A.J., Chokkalingam A.P., Metayer C., Roberts W., Barcellos L.F., Wiemels J.L., Buffler P.A. GATA3 risk alleles are associated with ancestral components in Hispanic children with ALL. Blood. 2013;122:3385–3387. doi: 10.1182/blood-2013-08-524124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Smith A.J., Walsh K.M., Morimoto L.M., Francis S.S., Hansen H.M., Jeon S., Gonseth S., Chen M., Sun H., Luna-Fineman S., et al. Heritable variation at the chromosome 21 gene ERG is associated with acute lymphoblastic leukemia risk in children with and without Down syndrome. Leukemia. 2019;33:2746–2751. doi: 10.1038/s41375-019-0514-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qian M., Xu H., Perez-Andreu V., Roberts K.G., Zhang H., Yang W., Zhang S., Zhao X., Smith C., Devidas M., et al. Novel susceptibility variants at the ERG locus for childhood acute lymphoblastic leukemia in Hispanics. Blood. 2019;133:724–729. doi: 10.1182/blood-2018-07-862946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeon S., de Smith A.J., Li S., Chen M., Chan T.F., Muskens I.S., Morimoto L.M., DeWan A.T., Mancuso N., Metayer C., et al. Genome-wide trans-ethnic meta-analysis identifies novel susceptibility loci for childhood acute lymphoblastic leukemia. Leukemia. 2022;36:865–868. doi: 10.1038/s41375-021-01465-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu K., Li S., Pandey P., Kang A.Y., Morimoto L.M., Mancuso N., Ma X., Metayer C., Wiemels J.L., de Smith A.J. Investigating DNA methylation as a mediator of genetic risk in childhood acute lymphoblastic leukemia. Hum. Mol. Genet. 2022;31:3741–3756. doi: 10.1093/hmg/ddac137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vijayakrishnan J., Qian M., Studd J.B., Yang W., Kinnersley B., Law P.J., Broderick P., Raetz E.A., Allan J., Pui C.-H., et al. Identification of four novel associations for B-cell acute lymphoblastic leukaemia risk. Nat. Commun. 2019;10:5348. doi: 10.1038/s41467-019-13069-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Churchman M.L., Qian M., Te Kronnie G., Zhang R., Yang W., Zhang H., Lana T., Tedrick P., Baskin R., Verbist K., et al. Germline Genetic IKZF1 Variation and Predisposition to Childhood Acute Lymphoblastic Leukemia. Cancer Cell. 2018;33:937–948.e8. doi: 10.1016/j.ccell.2018.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price A.L., Patterson N., Yu F., Cox D.R., Waliszewska A., McDonald G.J., Tandon A., Schirmer C., Neubauer J., Bedoya G., et al. A genomewide admixture map for Latino populations. Am. J. Hum. Genet. 2007;80:1024–1036. doi: 10.1086/518313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Võsa U., Claringbould A., Westra H.-J., Bonder M.J., Deelen P., Zeng B., Kirsten H., Saha A., Kreuzhuber R., Yazar S., et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021;53:1300–1310. doi: 10.1038/s41588-021-00913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ota M., Nagafuchi Y., Hatano H., Ishigaki K., Terao C., Takeshima Y., Yanaoka H., Kobayashi S., Okubo M., Shirai H., et al. Dynamic landscape of immune cell-specific gene regulation in immune-mediated diseases. Cell. 2021;184:3006–3021.e17. doi: 10.1016/j.cell.2021.03.056. [DOI] [PubMed] [Google Scholar]
- 26.Fu Q., Li H., Moorjani P., Jay F., Slepchenko S.M., Bondarev A.A., Johnson P.L.F., Aximu-Petri A., Prüfer K., de Filippo C., et al. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature. 2014;514:445–449. doi: 10.1038/nature13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu F., Cato L.D., Weng C., Liggett L.A., Jeon S., Xu K., Chiang C.W.K., Wiemels J.L., Weissman J.S., de Smith A.J., Sankaran V.G. Variant to function mapping at single-cell resolution through network propagation. Nat. Biotechnol. 2022;40:1644–1653. doi: 10.1038/s41587-022-01341-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian M., Zhao X., Devidas M., Yang W., Gocho Y., Smith C., Gastier-Foster J.M., Li Y., Xu H., Zhang S., et al. Genome-Wide Association Study of Susceptibility Loci for T-Cell Acute Lymphoblastic Leukemia in Children. J. Natl. Cancer Inst. 2019;111:1350–1357. doi: 10.1093/jnci/djz043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barnett K.R., Mobley R.J., Diedrich J.D., Bergeron B.P., Bhattarai K.R., Monovich A.C., Narina S., Yang W., Crews K.R., Manring C.S., et al. Epigenomic mapping reveals distinct B cell acute lymphoblastic leukemia chromatin architectures and regulators. Cell Genom. 2023;3:100442. doi: 10.1016/j.xgen.2023.100442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joshi I., Yoshida T., Jena N., Qi X., Zhang J., Van Etten R.A., Georgopoulos K. Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat. Immunol. 2014;15:294–304. doi: 10.1038/ni.2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwickert T.A., Tagoh H., Gültekin S., Dakic A., Axelsson E., Minnich M., Ebert A., Werner B., Roth M., Cimmino L., et al. Stage-specific control of early B cell development by the transcription factor Ikaros. Nat. Immunol. 2014;15:283–293. doi: 10.1038/ni.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lo K., Landau N.R., Smale S.T. LyF-1, a transcriptional regulator that interacts with a novel class of promoters for lymphocyte-specific genes. Mol. Cell Biol. 1991;11:5229–5243. doi: 10.1128/mcb.11.10.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gounari F., Kee B.L. Fingerprinting ikaros. Nat. Immunol. 2013;14:1034–1035. doi: 10.1038/ni.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao J., Jia Y., Mahmut D., Deik A.A., Jeanfavre S., Clish C.B., Sankaran V.G. Human hematopoietic stem cell vulnerability to ferroptosis. Cell. 2023;186:732–747.e16. doi: 10.1016/j.cell.2023.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voit R.A., Tao L., Yu F., Cato L.D., Cohen B., Fleming T.J., Antoszewski M., Liao X., Fiorini C., Nandakumar S.K., et al. A genetic disorder reveals a hematopoietic stem cell regulatory network co-opted in leukemia. Nat. Immunol. 2023;24:69–83. doi: 10.1038/s41590-022-01370-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo X.M., Maarschalk E., O’Connell R.M., Wang P., Yang L., Baltimore D. Engineering human hematopoietic stem/progenitor cells to produce a broadly neutralizing anti-HIV antibody after in vitro maturation to human B lymphocytes. Blood. 2009;113:1422–1431. doi: 10.1182/blood-2008-09-177139. [DOI] [PubMed] [Google Scholar]
- 37.Quiroz E., Aldoss I., Pullarkat V., Rego E., Marcucci G., Douer D. The emerging story of acute lymphoblastic leukemia among the Latin American population - biological and clinical implications. Blood Rev. 2019;33:98–105. doi: 10.1016/j.blre.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 38.Homburger J.R., Moreno-Estrada A., Gignoux C.R., Nelson D., Sanchez E., Ortiz-Tello P., Pons-Estel B.A., Acevedo-Vasquez E., Miranda P., Langefeld C.D., et al. Genomic Insights into the Ancestry and Demographic History of South America. PLoS Genet. 2015;11:e1005602. doi: 10.1371/journal.pgen.1005602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ziyatdinov A., Torres J., Alegre-Díaz J., Backman J., Mbatchou J., Turner M., Gaynor S.M., Joseph T., Zou Y., Liu D., et al. Genotyping, sequencing and analysis of 140,000 adults from Mexico City. Nature. 2023;622:784–793. doi: 10.1038/s41586-023-06595-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sohail M., Palma-Martínez M.J., Chong A.Y., Quinto-Cortés C.D., Barberena-Jonas C., Medina-Muñoz S.G., Ragsdale A., Delgado-Sánchez G., Cruz-Hervert L.P., Ferreyra-Reyes L., et al. Mexican Biobank advances population and medical genomics of diverse ancestries. Nature. 2023;622:775–783. doi: 10.1038/s41586-023-06560-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kachuri L., Mak A.C.Y., Hu D., Eng C., Huntsman S., Elhawary J.R., Gupta N., Gabriel S., Xiao S., Keys K.L., et al. Gene expression in African Americans, Puerto Ricans and Mexican Americans reveals ancestry-specific patterns of genetic architecture. Nat. Genet. 2023;55:952–963. doi: 10.1038/s41588-023-01377-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montes-Rodríguez I.M., Soto-Salgado M., Torres-Cintrón C.R., Tomassini-Fernandini J.C., Suárez E., Clavell L.A., Cadilla C.L. Incidence and Mortality Rates for Childhood Acute Lymphoblastic Leukemia in Puerto Rican Hispanics, 2012-2016. Cancer Epidemiol. Biomarkers Prev. 2023;32:1030–1037. doi: 10.1158/1055-9965.EPI-22-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pérez-Saldivar M.L., Fajardo-Gutiérrez A., Bernáldez-Ríos R., Martínez-Avalos A., Medina-Sanson A., Espinosa-Hernández L., Flores-Chapa J.d.D., Amador-Sánchez R., Peñaloza-González J.G., Alvarez-Rodríguez F.J., et al. Childhood acute leukemias are frequent in Mexico City: descriptive epidemiology. BMC Cancer. 2011;11:355. doi: 10.1186/1471-2407-11-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quintana-Murci L. Human Immunology through the Lens of Evolutionary Genetics. Cell. 2019;177:184–199. doi: 10.1016/j.cell.2019.02.033. [DOI] [PubMed] [Google Scholar]
- 45.Cytlak U., Resteu A., Bogaert D., Kuehn H.S., Altmann T., Gennery A., Jackson G., Kumanovics A., Voelkerding K.V., Prader S., et al. Ikaros family zinc finger 1 regulates dendritic cell development and function in humans. Nat. Commun. 2018;9:1239. doi: 10.1038/s41467-018-02977-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuehn H.S., Nunes-Santos C.J., Rosenzweig S.D. IKAROS-Associated Diseases in 2020: Genotypes, Phenotypes, and Outcomes in Primary Immune Deficiency/Inborn Errors of Immunity. J. Clin. Immunol. 2021;41:1–10. doi: 10.1007/s10875-020-00936-x. [DOI] [PubMed] [Google Scholar]
- 47.Kuehn H.S., Boisson B., Cunningham-Rundles C., Reichenbach J., Stray-Pedersen A., Gelfand E.W., Maffucci P., Pierce K.R., Abbott J.K., Voelkerding K.V., et al. Loss of B Cells in Patients with Heterozygous Mutations in IKAROS. N. Engl. J. Med. 2016;374:1032–1043. doi: 10.1056/NEJMoa1512234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown A.L., de Smith A.J., Gant V.U., Yang W., Scheurer M.E., Walsh K.M., Chernus J.M., Kallsen N.A., Peyton S.A., Davies G.E., et al. Inherited genetic susceptibility to acute lymphoblastic leukemia in Down syndrome. Blood. 2019;134:1227–1237. doi: 10.1182/blood.2018890764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Papaemmanuil E., Rapado I., Li Y., Potter N.E., Wedge D.C., Tubio J., Alexandrov L.B., Van Loo P., Cooke S.L., Marshall J., et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet. 2014;46:116–125. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mullighan C.G., Miller C.B., Radtke I., Phillips L.A., Dalton J., Ma J., White D., Hughes T.P., Le Beau M.M., Pui C.-H., et al. BCR–ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–114. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 51.Gupta S., Dai Y., Chen Z., Winestone L.E., Teachey D.T., Bona K., Aplenc R., Rabin K.R., Zweidler-McKay P., Carroll A.J., et al. Racial and ethnic disparities in childhood and young adult acute lymphocytic leukaemia: secondary analyses of eight Children’s Oncology Group cohort trials. Lancet Haematol. 2023;10:e129–e141. doi: 10.1016/S2352-3026(22)00371-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Faruqi A.J., Ligon J.A., Borgman P., Steinberg S.M., Foley T., Little L., Mackall C.L., Lee D.W., Fry T.J., Shalabi H., et al. The impact of race, ethnicity, and obesity on CAR T-cell therapy outcomes. Blood Adv. 2022;6:6040–6050. doi: 10.1182/bloodadvances.2022007676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moore K.J., Hubbard A.K., Williams L.A., Spector L.G. Childhood cancer incidence among specific Asian and Pacific Islander populations in the United States. Int. J. Cancer. 2020;147:3339–3348. doi: 10.1002/ijc.33153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin-Rufino J.D., Castano N., Pang M., Grody E.I., Joubran S., Caulier A., Wahlster L., Li T., Qiu X., Riera-Escandell A.M., et al. Massively parallel base editing to map variant effects in human hematopoiesis. Cell. 2023;186:2456–2474.e24. doi: 10.1016/j.cell.2023.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kvale M.N., Hesselson S., Hoffmann T.J., Cao Y., Chan D., Connell S., Croen L.A., Dispensa B.P., Eshragh J., Finn A., et al. Genotyping Informatics and Quality Control for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics. 2015;200:1051–1060. doi: 10.1534/genetics.115.178905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.1000 Genomes Project Consortium A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Granja J.M., Klemm S., McGinnis L.M., Kathiria A.S., Mezger A., Corces M.R., Parks B., Gars E., Liedtke M., Zheng G.X.Y., et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol. 2019;37:1458–1465. doi: 10.1038/s41587-019-0332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Satpathy A.T., Granja J.M., Yost K.E., Qi Y., Meschi F., McDermott G.P., Olsen B.N., Mumbach M.R., Pierce S.E., Corces M.R., et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 2019;37:925–936. doi: 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhattacharyya S., Chandra V., Vijayanand P., Ay F. Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nat. Commun. 2019;10:4221. doi: 10.1038/s41467-019-11950-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Speidel L., Forest M., Shi S., Myers S.R. A method for genome-wide genealogy estimation for thousands of samples. Nat. Genet. 2019;51:1321–1329. doi: 10.1038/s41588-019-0484-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Posth C., Nakatsuka N., Lazaridis I., Skoglund P., Mallick S., Lamnidis T.C., Rohland N., Nägele K., Adamski N., Bertolini E., et al. Reconstructing the Deep Population History of Central and South America. Cell. 2018;175:1185–1197.e22. doi: 10.1016/j.cell.2018.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mallick S., Li H., Lipson M., Mathieson I., Gymrek M., Racimo F., Zhao M., Chennagiri N., Nordenfelt S., Tandon A., et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature. 2016;538:201–206. doi: 10.1038/nature18964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moreno-Mayar J.V., Vinner L., de Barros Damgaard P., de la Fuente C., Chan J., Spence J.P., Allentoft M.E., Vimala T., Racimo F., Pinotti T., et al. Early human dispersals within the Americas. Science. 2018;362:eaav2621. doi: 10.1126/science.aav2621. [DOI] [PubMed] [Google Scholar]
- 66.Scheib C.L., Li H., Desai T., Link V., Kendall C., Dewar G., Griffith P.W., Mörseburg A., Johnson J.R., Potter A., et al. Ancient human parallel lineages within North America contributed to a coastal expansion. Science. 2018;360:1024–1027. doi: 10.1126/science.aar6851. [DOI] [PubMed] [Google Scholar]
- 67.Lindo J., Haas R., Hofman C., Apata M., Moraga M., Verdugo R.A., Watson J.T., Viviano Llave C., Witonsky D., Beall C., et al. The genetic prehistory of the Andean highlands 7000 years BP though European contact. Sci. Adv. 2018;4:eaau4921. doi: 10.1126/sciadv.aau4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.de la Fuente C., Ávila-Arcos M.C., Galimany J., Carpenter M.L., Homburger J.R., Blanco A., Contreras P., Cruz Dávalos D., Reyes O., San Roman M., et al. Genomic insights into the origin and diversification of late maritime hunter-gatherers from the Chilean Patagonia. Proc. Natl. Acad. Sci. USA. 2018;115:E4006–E4012. doi: 10.1073/pnas.1715688115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Capodiferro M.R., Aram B., Raveane A., Rambaldi Migliore N., Colombo G., Ongaro L., Rivera J., Mendizábal T., Hernández-Mora I., Tribaldos M., et al. Archaeogenomic distinctiveness of the Isthmo-Colombian area. Cell. 2021;184:1706–1723.e24. doi: 10.1016/j.cell.2021.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rasmussen M., Anzick S.L., Waters M.R., Skoglund P., DeGiorgio M., Stafford T.W., Jr., Rasmussen S., Moltke I., Albrechtsen A., Doyle S.M., et al. The genome of a Late Pleistocene human from a Clovis burial site in western Montana. Nature. 2014;506:225–229. doi: 10.1038/nature13025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rasmussen M., Sikora M., Albrechtsen A., Korneliussen T.S., Moreno-Mayar J.V., Poznik G.D., Zollikofer C.P.E., de León M.P., Allentoft M.E., Moltke I., et al. The ancestry and affiliations of Kennewick Man. Nature. 2015;523:455–458. doi: 10.1038/nature14625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raghavan M., Steinrücken M., Harris K., Schiffels S., Rasmussen S., DeGiorgio M., Albrechtsen A., Valdiosera C., Ávila-Arcos M.C., Malaspinas A.S., et al. POPULATION GENETICS. Genomic evidence for the Pleistocene and recent population history of Native Americans. Science. 2015;349:aab3884. doi: 10.1126/science.aab3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Skoglund P., Mallick S., Bortolini M.C., Chennagiri N., Hünemeier T., Petzl-Erler M.L., Salzano F.M., Patterson N., Reich D. Genetic evidence for two founding populations of the Americas. Nature. 2015;525:104–108. doi: 10.1038/nature14895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Machiela M.J., Chanock S.J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–3557. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taliun D., Harris D.N., Kessler M.D., Carlson J., Szpiech Z.A., Torres R., Taliun S.A.G., Corvelo A., Gogarten S.M., Kang H.M., et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–299. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zou Y., Carbonetto P., Wang G., Stephens M. Fine-mapping from summary data with the “Sum of Single Effects” model. PLoS Genet. 2022;18:e1010299. doi: 10.1371/journal.pgen.1010299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang G., Sarkar A., Carbonetto P., Stephens M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Series B Stat. Methodol. 2020;82:1273–1300. doi: 10.1111/rssb.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maples B.K., Gravel S., Kenny E.E., Bustamante C.D. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet. 2013;93:278–288. doi: 10.1016/j.ajhg.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alexander D.H., Novembre J., Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j. 2011;17:10–12. [Google Scholar]
- 83.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cairns J., Freire-Pritchett P., Wingett S.W., Várnai C., Dimond A., Plagnol V., Zerbino D., Schoenfelder S., Javierre B.-M., Osborne C., et al. CHiCAGO: robust detection of DNA looping interactions in Capture Hi-C data. Genome Biol. 2016;17:127. doi: 10.1186/s13059-016-0992-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Borrell L.N., Elhawary J.R., Fuentes-Afflick E., Witonsky J., Bhakta N., Wu A.H.B., Bibbins-Domingo K., Rodríguez-Santana J.R., Lenoir M.A., Gavin J.R., 3rd, et al. Race and Genetic Ancestry in Medicine - A Time for Reckoning with Racism. N. Engl. J. Med. 2021;384:474–480. doi: 10.1056/NEJMms2029562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bao E.L., Nandakumar S.K., Liao X., Bick A.G., Karjalainen J., Tabaka M., Gan O.I., Havulinna A.S., Kiiskinen T.T.J., Lareau C.A., et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature. 2020;586:769–775. doi: 10.1038/s41586-020-2786-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Luo Y., Li X., Wang X., Gazal S., Mercader J.M., 23 and Me Research Team. SIGMA Type 2 Diabetes Consortium. Neale B.M., Florez J.C., Auton A., et al. Estimating heritability and its enrichment in tissue-specific gene sets in admixed populations. Hum. Mol. Genet. 2021;30:1521–1534. doi: 10.1093/hmg/ddab130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study included the analysis of data derived from biospecimens from the California Biobank Program (CCRLP study). Any uploading of genomic data and/or sharing of these biospecimens or individual data derived from these biospecimens has been determined to violate the statutory scheme of the California Health and Safety Code Sections 124980(j); 124991(b), (g), and (h); and 103850 (a) and (d), which protect the confidential nature of biospecimens and individual data derived from biospecimens. Should we be contacted by other investigators who would like to use the data, we will direct them to the California Department of Public Health Institutional Review Board to establish their own approved protocol to utilize the data, which can then be shared peer-to-peer.
The GWAS summary statistics at the IKZF1 locus are included in Table S2. Genotype data for the Genetic Epidemiology Research on Aging (GERA) study controls are available on dbGAP under accession number phs000788.v1.p2. Genotype data for the Children’s Oncology Group (COG)/St. Jude Children’s Research Hospital study ALL patients are available on dbGaP under accession number phs000638.v1.p1 and phs000637.v1.p1.
Whole genome sequencing and whole blood RNA sequencing data from the Genes-environments and Admixture in Latino Asthmatics (GALA II) study and the Study of African Americans, Asthma, Genes, and Environments (SAGE), generated as part of the NHLBI TOPMed program, are available dbGaP under accession numbers phs000920 (GALA II) and phs000921 (SAGE). Summary statistics for cis- and trans-eQTLs, a catalog of ancestry-specific eQTLs, and TWAS models developed using data from GALA II and SAGE participants have been posted in a public repository at https://doi.org/10.5281/zenodo.7735723.
All other data are available in the cited references.