

Abstract
Delivery of mRNA-based therapeutics to the perinatal brain holds great potential in treating congenital brain diseases. However, non-viral delivery platforms that facilitate nucleic acid delivery in this environment have yet to be rigorously studied. Here, we screen a diverse library of ionizable lipid nanoparticles (LNPs) via intracerebroventricular (ICV) injection in both fetal and neonatal mice and identify an LNP formulation with greater functional mRNA delivery in the perinatal brain than an FDA-approved industry standard LNP. Following in vitro optimization of the top-performing LNP (C3 LNP) for co-delivery of an adenine base editing platform, we improve the biochemical phenotype of a lysosomal storage disease in the neonatal mouse brain, exhibit proof-of-principle mRNA brain transfection in vivo in a fetal nonhuman primate model, and demonstrate the translational potential of C3 LNPs ex vivo in human patient-derived brain tissues. These LNPs may provide a clinically translatable platform for in utero and postnatal mRNA therapies, including gene editing, in the brain.
Keywords: ionizable lipid nanoparticles, congenital brain disease, mRNA delivery, gene editing, fetal gene therapy
Graphical Abstract
INTRODUCTION:
Approximately 7,000–10,000 diseases result from mutations in single genes, of which 17% have a neurologic component including developmental delay, motor and cognitive dysfunction, and progressive neuronal degeneration (1–4). Although some genetic diseases, including inborn errors of metabolism, that affect the CNS can be treated with enzyme replacement therapies, few curative therapies exist and current clinical management often focuses on symptom reduction (1, 2). Furthermore, the pathology of many genetic CNS diseases begins before birth and is difficult to reverse after onset resulting in significant morbidity by the time of or shortly after birth (3). Progress in prenatal care and DNA sequencing technology now allows for the prenatal diagnosis of many genetic diseases, including the identification of disease-causing mutations, highlighting the potential to treat disease before birth and the onset of irreversible pathology (4).
Advances in mRNA-based gene editing tools, including CRISPR-Cas9 and base editing platforms, provide an opportunity for “one-and-done” treatments for monogenic diseases (5, 6). In utero gene editing takes advantage of normal fetal ontogeny to deliver therapies in a potentially more efficient manner, mitigating genetic disease before pathologic insult. Specifically, small fetal size maximizes dose of a therapy per recipient weight, fetal stem/progenitor cells are more accessible and abundant supporting the persistence of the therapeutic edit, and a tolerant fetal immune system minimizes an immune barrier to gene editing tools (4). Previously, we demonstrated the therapeutic potential of in utero gene editing in mouse models of human diseases, rescuing the lethal phenotype of hereditary tyrosinemia type 1, improving the pulmonary phenotype of surfactant protein C deficiency, and ameliorating metabolic, musculoskeletal, and cardiac disease in mucopolysaccharidosis type I (MPS-IH, Hurler syndrome) (7–9). These proof-of-concept studies demonstrated the ability to efficiently target the fetal liver and heart following systemic delivery of viral vectors and pulmonary epithelial cells following intraamniotic viral vector delivery. Although encouraging, CNS targeting via these approaches was not efficient, highlighting the need to evaluate alternative delivery approaches to treat genetic CNS diseases.
Although viral vectors are a promising approach to deliver gene editing technology to the CNS, viral delivery platforms can be limited by pre-existing viral immunity, transgene size constraints, undesired vector integration, and adverse events related to the viral vector (10–12). Ionizable lipid nanoparticles (LNPs) are a promising non-viral delivery platform for mRNA nanomedicines (13, 14) that have been used to deliver siRNA and CRISPR-based gene editing therapies to the liver in postnatal mouse, non-human primate, and clinical studies with encouraging results (15–18). A distinct advantage of using LNPs is their modularity, but this necessitates designing LNPs specifically for the target delivery location, intended cargo, and intended patient population including the developmental stage of the patient. We previously identified LNPs for mRNA delivery to the fetal mouse liver following in utero intravascular injection (19). These studies demonstrate the potential of LNPs as a delivery platform for nucleic acid-based therapies, including gene editing technologies, especially for diseases amenable to liver targeting. However, the applicability of LNPs for genetic CNS diseases – as delivery vehicles for gene editing platforms in the perinatal brain – remains to be determined.
In the current study, we engineer LNPs for delivery of mRNA base editing platforms to the perinatal brain. We first screen a diverse library of LNPs in vivo to identify LNPs that strongly transfect the fetal and neonatal mouse brain following intracerebroventricular (ICV) injection. After determining the cellular tropism of our top performing LNP in the perinatal mouse brain, we optimize the LNP formulation in vitro for three formulation parameters relevant to base editing applications. Optimized LNPs were then used to deliver adenine base editor (ABE) mRNA and synthetic guide RNA (sgRNA) by ICV injection in the neonatal mouse model of MPS-IH to correct the Idua G→A (W392X) disease-causing mutation. We demonstrate successful LNP-mediated on-target base editing, improved biochemical parameters of the disease, and minimal immunologic response to delivery vector. The same LNP formulation was then used to deliver mRNA to the brain in a fetal cynomolgus macaque following ICV injection. Finally, we demonstrate the translational potential of this delivery platform by confirming the stability of the optimized LNP in human cerebrospinal fluid (CSF), exhibiting LNP-mediated mRNA transfection in patient-derived neurons, and displaying LNP-mediated base editing of the human IDUA gene at the site of the common MPS-IH disease-causing mutation in human precision cut brain slices.
RESULTS:
Synthesis and characterization of an ionizable LNP library
A library of 12 LNPs was prepared as previously described (20). First, ionizable lipids were synthesized using Michael addition chemistry, whereby alkyl tails (denoted by tail length: A = C12, B = C14, C = C16) were reacted with polyamine molecules (labeled numerically 1 through 4) to form polyamine-lipid cores. These ionizable lipids were combined with cholesterol, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and lipid-anchored PEG at ratios determined by previous optimization studies in adult mice (21) and then mixed with firefly luciferase mRNA through a herringbone-style microfluidic device (Fig. 1A).
Fig. 1 |. Design and evaluation of LNP library for perinatal brain mRNA delivery.
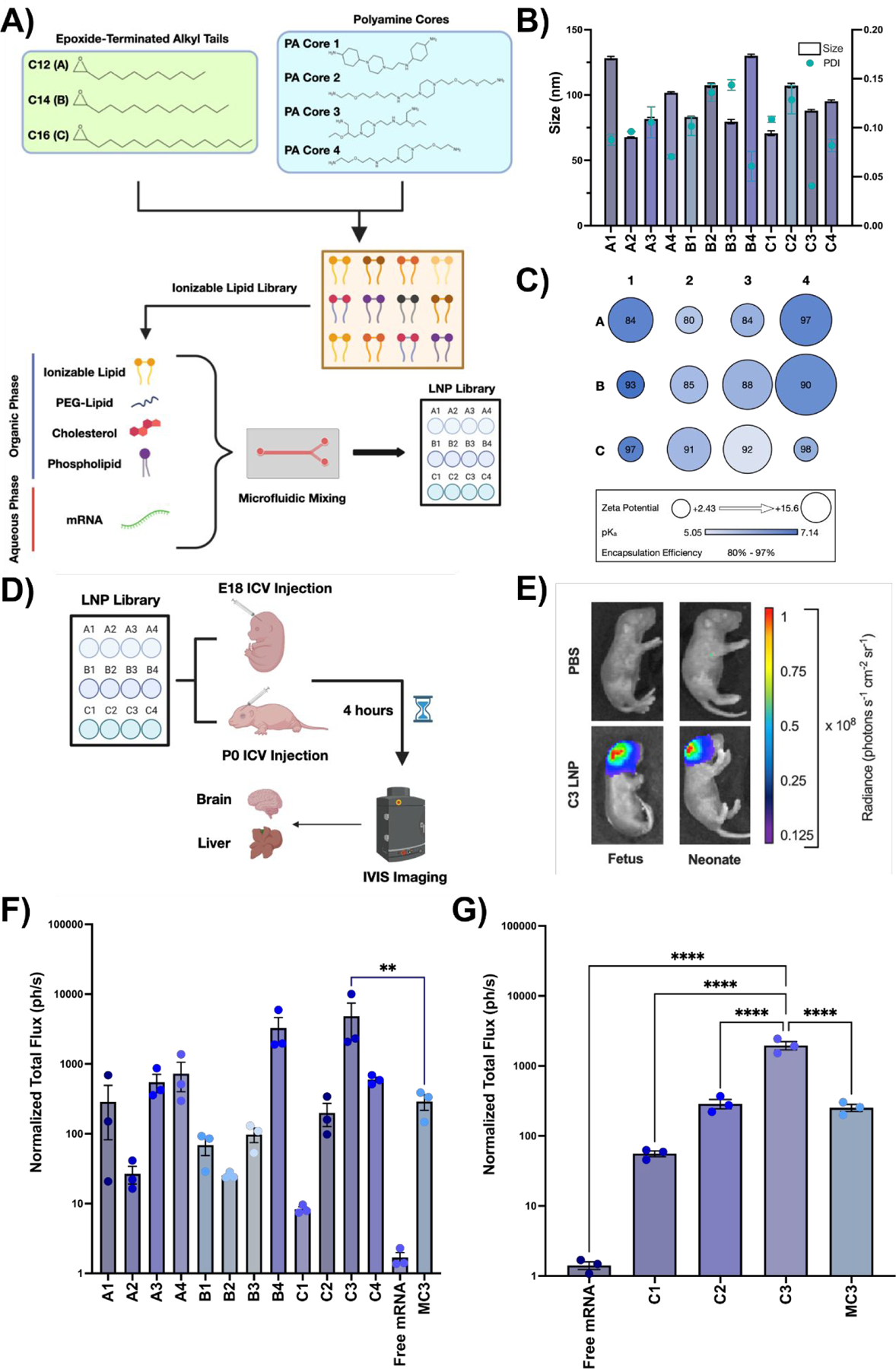
(A) Chemical structures of epoxide-terminated alkyl tails (green box) and polyamine cores (blue box) combined to generate an ionizable lipid library. Formulation of LNPs via microfluidic mixing with an ethanol phase containing ionizable lipid, PEG-lipid, cholesterol, and DOPE and an aqueous phase containing luciferase mRNA is also visualized. LNPs were named based on the alkyl tail length (A-C) and polyamine core (1–4) of the ionizable lipid incorporated into the formulation. (B) Size and PDI of each LNP formulation in the LNP library. (C) Zeta potential (radius of circle), pKa (gradient color), and encapsulation efficiency (centered number) for each LNP formulation. (D) Scheme demonstrating LNP screening in E18 BALB/c fetuses or P0 BALB/c neonates via ICV injection. (E) IVIS imaging showing luciferase expression in a representative C3 LNP-treated fetus (left) and neonate (right) relative to PBS-treated controls. (F) Quantification of luciferase signal from fetal brains treated with each LNP. (G) Quantification of luciferase signal from the neonatal brains treated with a subset of LNPs. All luminescence readings are represented as normalized total flux. ** p < 0.01, **** p < 0.0001 by one-way analysis of variance (ANOVA) with post-hoc Dunnett’s test compared to MC3 (fetus) and C3 (neonate). Outliers were detected using Grubbs’ test and removed from analysis; minimum n = 3 per treatment group; error bars represent SEM.
The resultant library of LNPs was characterized by size, encapsulation efficiency, pKa, and zeta potential (Table S1). Hydrodynamic diameter for all LNP formulations – measured via dynamic light scattering intensity – ranged from 67.8 to 128.3 nm with a maximum polydispersity value of 0.154 (Fig. 1B), indicating small and relatively monodisperse LNPs (PDI < 0.3) (19). Encapsulation efficiencies were also high, ranging from 79.9% to 97.6% (Fig. 1C). Next, LNPs were assessed for their pKa, which reflects their ability to undergo endosomal escape and release nucleic acid cargo. All LNP formulations had a pKa value between 5.05 and 7.14 (Fig. 1C), which is within an acceptable range for in vivo nucleic acid delivery (22). Finally, the zeta potential, a function of surface charge, was measured via a Zetasizer Nano (23). In this library, LNP zeta potentials ranged from 15.6 mV to 2.43 mV (Fig. 1C).
LNP-mediated mRNA delivery to the perinatal mouse brain
After characterizing the LNP library, we evaluated it for mRNA delivery to the brain in fetal and neonatal BALB/c mice (Fig. 1D). LNPs encapsulating luciferase mRNA at an mRNA dose of 1 mg/kg were injected ICV into the lateral ventricles of gestational day (E) 18 BALB/c fetuses, a neuro-developmental stage akin to a mid-gestation human fetus and a timepoint where therapeutic intervention is technically feasible (24). ICV injection was selected as the mode of introduction since it has been used clinically to safely and effectively deliver therapeutic agents for a broad range of neurological diseases (25). Fetuses were assessed 4 hours after ICV injection for luciferase expression using an in vivo imaging system (IVIS). This timepoint was selected based on previous studies (19, 26) and in vitro verification of LNP-mediated luciferase expression by 4 hours (Fig. S1). For each injected dam, phosphate-buffered saline (PBS) injected fetuses served as negative controls. Naked luciferase mRNA was included as a treatment group to demonstrate baseline nucleic acid uptake in the brain without an LNP carrier. Finally, DLin-MC3-DMA ionizable lipid – an FDA approved industry standard (18) – was used to encapsulate luciferase mRNA as a positive control.
IVIS imaging of both the whole fetus (Fig. 1E) as well as individually dissected fetal brains (Fig. 1F) demonstrated a range of mRNA delivery and luciferase expression in the brain with all LNPs in the library. All LNP formulations yielded greater mRNA delivery than injection of naked mRNA, confirming that LNP encapsulation protects mRNA cargo from systemic degradation and facilitates intracellular delivery (Fig. 1F). Each ionizable lipid produced LNPs with variable performance, demonstrating the crucial role of this component for mRNA delivery (Fig. 1F). Interestingly, we observed trends for in vivo transfection efficacy in the fetal brain with decreasing pKa, increasing size, and increasing zeta potential (Fig. S2). The top-performing LNP (C3 LNP) resulted in 17-fold greater mRNA expression in the fetal brain than the industry standard (p < 0.05). Of note, IVIS imaging of individually dissected fetal livers demonstrated minimal luciferase expression after treatment with any of the LNPs in this library (Fig. S3). Finally, we assessed the in vivo biodistribution of C3 LNPs administered ICV to E18 Balb/c fetuses. We observed strong luciferase expression in the fetal brain but no significant expression in the heart, lung, intestine, liver, spleen, or kidney (Fig. S4).
In this in vivo screen, long-term survival post-injection was not assessed. However, fetal mouse ICV injection has previously been associated with intrauterine mortality due to the technical challenge of this procedure in small animals (27). In contrast, postnatal day 0 (P0) mouse ICV injections are technically easier, associated with good long-term survival, and still approximate the CNS development of a mid-to-late gestation human fetus. Therefore, additional studies were performed to determine if the results of LNP-mediated mRNA delivery to the fetal brain were maintained following delivery in the neonatal mouse model. LNPs identified as high- (C3), mid-(C2), and low-performing (C1) in the fetal ICV screening studies, the industry standard LNP (MC3), or free luciferase mRNA were injected ICV at an mRNA dose of 1 mg/kg in P0 BALB/c neonates. Survival at 4 hours after neonatal ICV injection, the terminal analysis time point, was 100% in Balb/c neonates. IVIS imaging of neonatal brains confirmed the performance trends observed in the fetal brain (Fig. 1F and 1G), with C3 LNPs facilitating 8-fold greater mRNA expression in the neonatal brain than the industry standard LNP (p < 0.0001). These results, in combination with improved experimental feasibility, supported the use of neonatal mouse models in subsequent studies.
C3 LNPs facilitate gene modulation in periventricular cells of the murine brain
To gain insight into the cellular tropism of LNP-mediated mRNA delivery in the neonatal brain, we studied the brain biodistribution of high- (C3), mid- (C2), and low-performing (C1) LNPs containing Cre recombinase mRNA (1 mg/kg) after ICV administration to P0 R26mT/mG mice. R26mT/mG mice contain a membrane-targeted tdTomato (mT) cassette flanked by loxP sites and constitutively express red fluorescence in all cells (Fig. 2A). Upon expression of translated Cre protein, the mT cassette and transcriptional stop cassette are deleted, resulting in expression of a downstream mG-enhanced green fluorescent protein (mG-EGFP) cassette. Thus, successful LNP-mediated delivery of Cre mRNA leads to expression of green fluorescence in the transfected cell.
Fig. 2 |. Cellular tropism of C3 LNPs in the neonatal mouse brain.
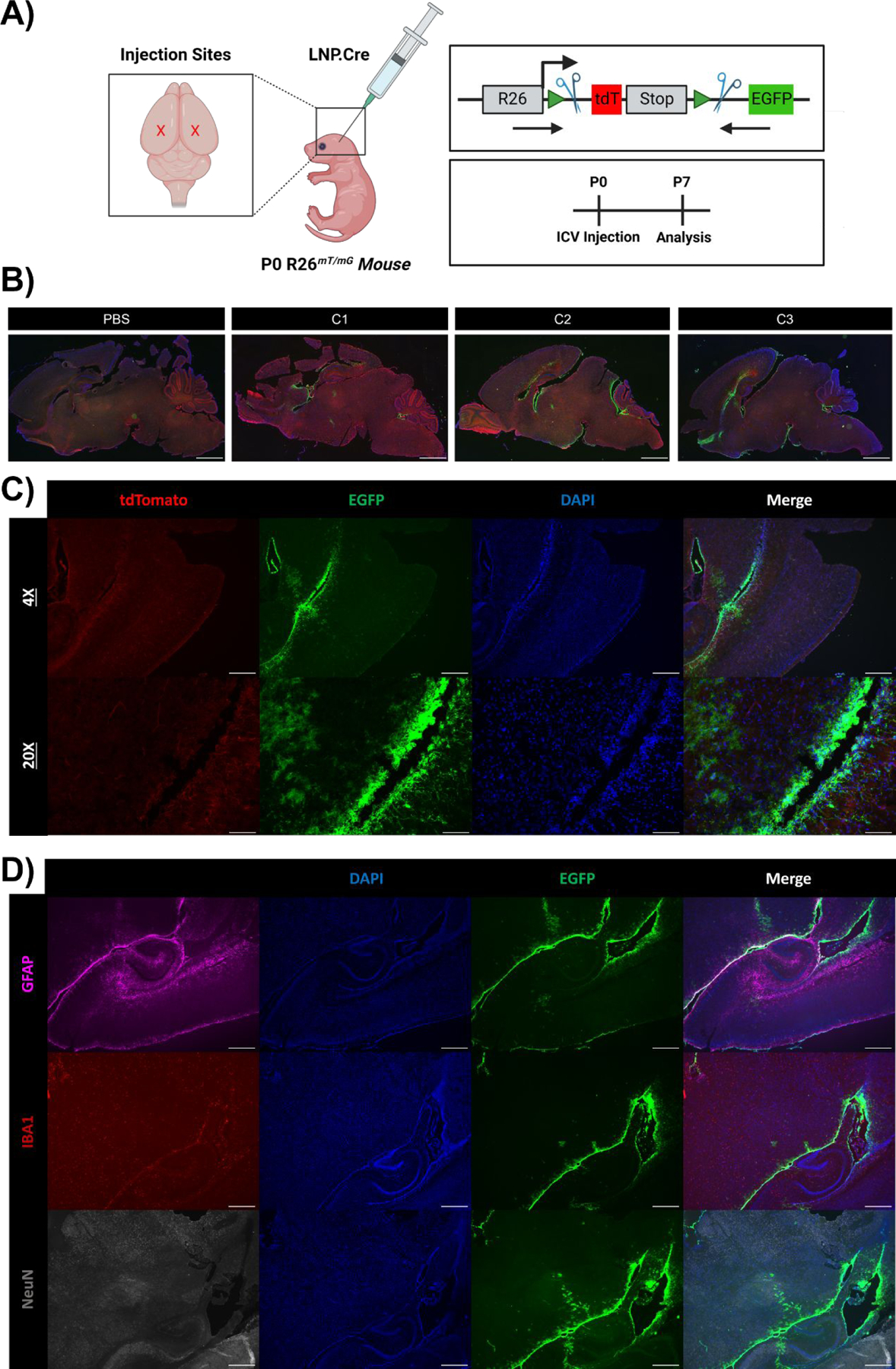
(A) Scheme demonstrating application of LNPs encapsulating Cre mRNA to P0 R26mT/mG neonates via bilateral ICV injection. Genome modulation in the R26mT/mG mouse model and experimental scheme are also visualized in the right panel. (B) Whole brain histology of neonatal brains 7 days after PBS, C1, C2, or C3 LNP ICV injection, displaying unedited (tdTomato+) and edited (GFP+) regions, imaged at 1X. (C) Histology focused on the brain ventricular lining 7 days after C3 LNP ICV injection, imaged at 4X and 20X. (D) Histology focused on the brain ventricular lining with intracellular staining to capture successful C3 LNP-mediated delivery (GFP+) to astrocytes (GFAP+), microglia (IBA1+), and neurons (NeuN+). Scale bars: 1 mm (1X), 200 μm (4x) and 50 μm (20x).
Survival after neonatal ICV injection was 100% in R26mT/mG neonates. One week after injection, harvested neonatal brains were analyzed for gene modulation via tissue histology. Although we observed similarities in whole brain distribution across LNPs, the magnitude of GFP expression and was proportional to LNP performance in our initial screening studies (C3 > C2 > C1) (Fig. 2B). None of the LNPs demonstrated significant diffusion deep into the brain parenchyma beyond the periventricular space. C3 LNP-treated neonates had strong GFP expression in periventricular areas and adjacent cortical structures (Fig. 2B and 2C). Flow cytometry (Fig. S5) and cell-specific brain histology (Fig. 2D) confirmed GFP expression in microglia lining the ventricles and vicinal astrocytes and neurons with limited penetration to internal brain structures. Despite low whole brain gene modulation and delivery to specific brain cells or internal brain structures, efficient targeting of periventricular cells is a potential target to produce therapeutic proteins for secretion into CSF circulation and utilize a paracrine effect for treatment of disease.
In vitro optimization and validation of C3 LNPs for co-delivery of base editing technology
The molar ratios at which the components of LNPs are combined can be optimized for specific applications to better encapsulate nucleic acid cargo, enhance uptake by target cells, or alter biodistribution and protein corona formation (28). In our initial library screen, LNPs were formulated with standard parameters known to facilitate mRNA delivery. However, the optimal parameters to produce C3 LNPs specifically for base editing applications in the brain have not previously been determined. Thus, we used a multi-stage optimization protocol for C3 LNPs to determine the effects of excipient molar ratio, N:P ratio, and ratio of co-delivered mRNA on delivery performance in neural-origin cell lines (Fig. 3A).
Fig. 3 |. In vitro optimization of C3 LNPs for co-delivery of base editing platforms.
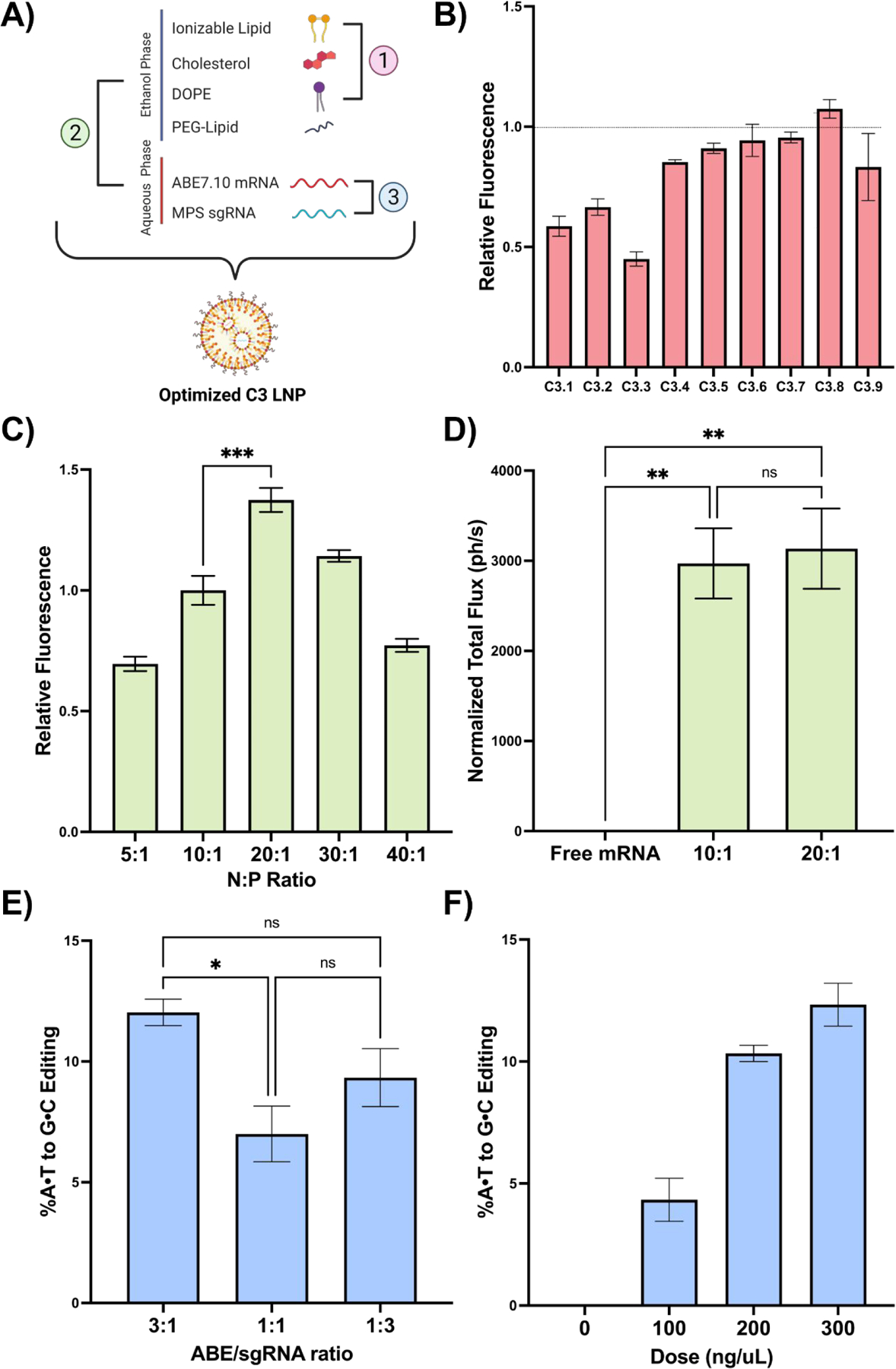
(A) Scheme demonstrating the three LNP formulation parameters evaluated: 1) excipient molar ratios, 2) N:P ratio, and 3) ABE to sgRNA mass ratio. (B) Normalized mean fluorescence intensity (MFI) in Neuro-2a cells after treatment with the C3 LNP DOE library relative to the original C3 LNP formulation (dotted line). (C) Normalized MFI in Neuro-2a cells after treatment with C3 LNPs formulated at a range of N:P ratios. (D) Quantification of luminescence signal (normalized total flux) from brains of neonates treated with C3 LNPs prepared at the top-performing N:P ratios in vitro. (E) Sequencing results at the expected site of base editing following C3 LNP-mediated base editing in primary MPS-I murine fibroblasts at three ABE/sgRNA mass ratios. (F) Sequencing results in primary MPS-I murine neurons treated with C3.MPS LNPs at a range of doses. * p < 0.05, ** p < 0.01, *** p < 0.001 by one-way ANOVA with post-hoc Dunnett’s test compared positive controls described in the text. Outliers were detected using Grubbs’ test and removed from analysis; minimum n = 3 replicates per treatment group; error bars represent SEM.
While the ionizable lipid – in this case C3 – is a major determinant of intracellular delivery by both directly complexing with mRNA and facilitating endosomal escape, the remaining excipients also play critical roles: cholesterol provides stability and enables membrane fusion, DOPE supplies structural support, and PEG reduces aggregation and non-specific endocytosis (29). In prior work, we demonstrated that modulating excipient molar ratios improves in vitro delivery to fetal lung cells and corresponds to enhanced in vivo performance following intra-amniotic LNP injection (28). Here, we used an orthogonal design of experiments (DOE) methodology to investigate a design space of 27 LNP formulations, generated using three molar ratios each of C3 ionizable lipid, cholesterol, and DOPE with a fixed PEG-lipid ratio (Fig. S6). A representative library of 9 LNPs encapsulating GFP mRNA (C3.1-C3.9) was screened against the original C3 LNP (C3.0) for delivery and cytotoxicity in Neuro-2a cells – immortalized mouse neuroblasts isolated from brain tissue. Although none of these formulations significantly enhanced mRNA delivery compared to the original C3 LNP formulation (Fig. 3B), we did identify relationships between the molar ratio of each excipient and mRNA delivery (Fig. S7). Of note, these trends are distinct from those observed in our prior studies that utilized different ionizable lipids (28, 30), demonstrating the importance of delineating these parameters in an application-specific manner. For all future studies, C3 LNPs were formulated at a molar ratio of 35:16:46.5:2.5 of C3 ionizable lipid, DOPE, cholesterol, and PEG, respectively.
The N:P ratio is the quantitative relationship between nitrogen on the ionizable lipid to phosphate on mRNA. Previous studies have demonstrated that the N:P ratio impacts both nucleic acid delivery and toxicity, with higher ratios leading to diminishing returns in cellular transfection due to increased cell death (31). The ideal N:P ratio is known to be distinct for each ionizable lipid (18). As such, C3 LNPs were formulated at a range of N:P ratios to encapsulate GFP mRNA and screened for delivery and cytotoxicity in Neuro-2a cells. In comparison to the original N:P ratio of 10:1, LNPs formulated at an N:P ratio of 20:1 had a significant improvement in mRNA delivery (Fig. 3C, p < 0.001) albeit with enhanced toxicity (Fig. S8B). To explore if this improvement was maintained in vivo, C3 LNPs were formulated with luciferase mRNA at both 10:1 and 20:1 N:P ratios and injected ICV in P0 BALB/c mice. IVIS imaging revealed that while both LNP formulations had similar performance in the neonatal brain to the initial screening study, there was no significant difference between the LNPs formulated at different N:P ratios (Fig. 3D). Given the increased in vitro toxicity seen with the 20:1 NP ratio and to minimize reactive excess ionizable lipid in solution, the C3 LNP formulation at a 10:1 N:P ratio was selected for further study.
While reporter mRNA was used in the first two phases of in vitro optimization to improve throughput, our goal was to deliver CRISPR-base editing mRNA and sgRNA to perinatal brain cells. Thus, we evaluated the feasibility of C3 LNPs to co-encapsulate both ABE7.10 mRNA and an sgRNA specific for the Idua G→A (W392X) mutation present in the mouse model of MPS-IH (C3.MPS LNPs) (9). C3 LNPs encapsulating ABE7.10 mRNA and sgRNA at three different mass ratios were formulated and assessed for on-target editing in MPS-IH mouse fibroblasts. A 3:1 ratio of ABE7.10 mRNA:sgRNA within C3 LNPs resulted in the highest level of editing (Fig. 3E) and was subsequently used in a dose-response study for on-target editing in MPS-IH mouse neurons, as well as for toxicity studies in MPS-IH mouse fibroblasts and neurons. These studies demonstrated dose-dependent A→G editing of the target adenine in the mouse Idua gene (Fig. 3F) and no toxicity in either cell type (Fig. S8C). Based on these in vitro optimization studies, future experimentation was conducted with the C3 LNP at the original formulation parameters, an N:P ratio of 10:1, and an mRNA:sgRNA ratio of 3:1.
C3.MPS LNPs mediate biochemical rescue of disease in the neonatal MPS-IH mouse brain
We next tested the safety and efficacy of the optimized C3 LNP formulation in vivo in a murine model of congenital brain disease (Fig. 4A). P0 Idua-W392X neonates were injected ICV with C3.MPS LNPs containing 1mg/kg of RNA and assessed at 1 month of age for on-target editing and amelioration of biochemical manifestations of MPS-IH. PBS injected age- and sex-matched Idua-W392X mice and C57Bl/6 (B6) mice served as controls. Next-generation sequencing (NGS) demonstrated low-level on-target base editing at the disease-causing locus in several regions of harvested brain tissue (Fig. 4B). Overall editing efficiency in the brain was comparable to that observed previously in the R26mT/mG mouse model. Notably, no editing above baseline was noted in genomic DNA isolated from the liver and gonads (Fig. S9), demonstrating similar brain specific delivery observed in our initial LNP library screen. Survival after neonatal ICV injection was 100% in IDUA-W392X neonates.
Fig. 4 |. Efficacy and safety of C3.MPS LNPs in Idua-W392X neonates.
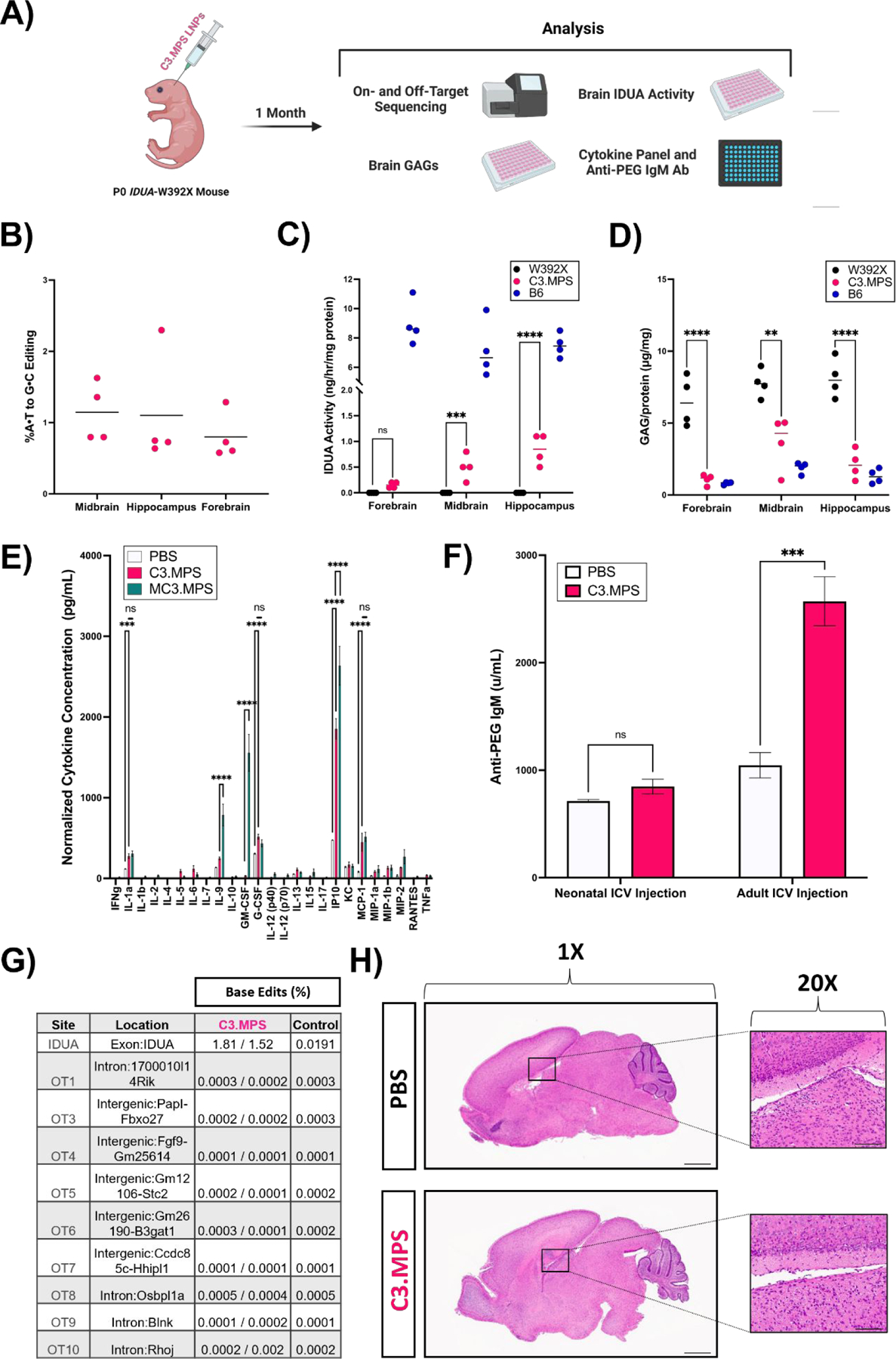
(A) Scheme for genetic, biochemical, and safety analysis of P0 Idua-W392X neonates injected ICV with C3.MPS LNPs. (B) NGS results at the expected site of base editing in three different sections of harvested brain tissue normalized to negative control (PBS); mean represented by horizontal line for each group (C) IDUA activity measured after harvest in three different sections of brain tissue from B6 mice (positive control), C3.MPS LNP-treated Idua-W392X mice (experimental group), and untreated Idua-W392X mice (negative control). (D) GAG amounts in three sections of brain tissue from B6, C3.MPS LNP-treated Idua-W392X, and untreated Idua-W392X mice. (E) Cytokine analysis in serum of Idua-W392X neonates treated 24 hours prior with C3.MPS LNPs. ** p < 0.01, *** p < 0.001, **** p < 0.0001 by two-way analysis of variance (ANOVA) with post-hoc Šídák’s multiple comparisons test; minimum n = 3 per treatment group; error bars represent SEM. (F) Serum anti-PEG IgM antibody levels in neonatal or adult mice one week following C3.MPS LNP treatment. *** p < 0.001 by Student’s t test with α = 0.05; minimum n = 3 per treatment group; error bars represent SEM. (G) NGS results at the Idua on-target site and the top computationally predicted off-target sites in brain genomic DNA of two C3.MPS LNP-treated Idua-W392X mice and one PBS-treated negative control. (H) Hematoxylin and eosin (H&E) stained whole brain tissue sections of PBS or C3.MPS treated Balb/c neonates with focus on the lateral ventricle. Scale bars: 1 mm (1X) and 50 μm (20x).
Next, we explored if durable biochemical corrections existed in the brains of Idua-W392X mice treated with C3.MPS LNPs. Similar to patients with MPS-IH, Idua-W392X mice have undetectable α-L- iduronidase (IDUA) enzyme activity and elevated tissue glycosaminoglycans (GAGs) (32). At one month, mice treated with C3.MPS LNPs demonstrated increased IDUA activity in the midbrain (p < 0.001, 6.97% of normal) and hippocampus (p < 0.0001, 11.3% of normal) relative to untreated Idua-W392X mice that was associated with a reduction in GAG levels in the respective brain regions (Figs. 4C and 4D). These data suggest that C3.MPS LNPs facilitate partial biochemical correction of disease in the neonatal Idua-W392X mouse brain, demonstrating potential for mitigation of brain disease in MPS-IH.
C3.MPS LNPs exhibit genomic and immunologic safety in the neonatal MPS-IH mouse
To further study the safety of C3.MPS LNPs, we investigated the acute, systemic immune response following LNP treatment. A cohort of Idua-W392X mice was injected ICV at P0 with C3.MPS LNPs containing 1mg/kg of RNA, and serum cytokines were assessed 24 hours later. Serum from PBS-injected mice and mice injected with LNPs formulated with MC3 ionizable lipid (MC3.MPS) served as controls. Four pro-inflammatory cytokines, G-CSF (regulator of neutrophil production), IL-1a (driver of inflammation), IP10 (immune cell chemotaxis), and MCP-1 (monocyte migration) were elevated in C3.MPS LNP and MC3.MPS treated neonates compared to PBS-treated controls (Fig. 4E). Furthermore, MC3.MPS LNP treated mice demonstrated a significant increase in serum IP10, IL-9 (pleiotropic cytokine with both pro- and anti-inflammatory function), and GM-CSF (regulator of macrophage production) levels relative to mice treated with C3.MPS LNPs. Taken together, these data suggest that while C3.MPS LNPs do produce an acute serum cytokine response that is generally pro-inflammatory, this reaction is at a lower level than that produced by FDA-approved MC3 LNPs.
Recent studies have revealed that PEGylated therapies give rise to anti-PEG antibodies in animal models and patients (33). While PEGylation of LNPs attracts a water shell, reducing adsorption of opsonins and evading recognition of LNPs by the mononuclear phagocyte system, antibodies to PEG-lipid may cause adverse immune effects, reduce therapeutic efficacy, and limit the potential for repeat dosing due to the accelerated blood clearance phenomenon (34). To characterize the anti-PEG antibody response elicited by C3.MPS LNPs, P0 Idua-W392X mice were injected ICV, and serum was assessed one week later for anti-PEG IgM antibodies. No significant difference in anti-PEG IgM levels was observed between C3.MPS LNP-treated and PBS-treated neonates. In contrast, a 2.5-fold increase in serum anti-PEG IgM antibodies in C3.MPS LNP-treated adult mice compared to PBS-treated adult mice (p < 0.001) was observed (Fig. 4F). These data are consistent with our previous study demonstrating that the immune system of a P0 mouse is immature (35) – similar to that of a developing human fetus – and imply that a mature immune system, as is present in an adult mouse, is capable of producing an anti-PEG IgM response to C3.MPS LNPs.
Next, we investigated the genomic safety of our LNP-mediated base editing strategy. Next-generation sequencing of brain genomic DNA from experimental mice did not demonstrate off-target base editing above background at nine previously identified possible off-target sites for the sgRNA targeting the mouse Idua G→A mutation were assessed by NGS (9). No editing above background at these sites in the brain was noted (Fig. 4G). Finally, we assessed the potential toxicity of C3.MPS LNPs in P0 Balb/c neonates via brain hemotoxin and eosin (H&E) (Fig. 4H). Relative to PBS-treated brain tissue sections, there was no evidence of gross structural damage or inflammatory cell infiltration near the injection site in LNP-treated brains.
C3 LNPs enable mRNA delivery to cerebral ventricular cells in a fetal macaque
We next evaluated the feasibility of C3 LNP-mediated mRNA delivery to the brain in a fetal large animal model. Relative to fetal and neonatal mouse models, non-human primate models have more structurally similar brain anatomy to humans and allow for the evaluation of the delivery approach, a minimally invasive, ultrasound-guided ICV, that would be used clinically. Thus, we evaluated the translational potential of our delivery platform in a 0.61G Macaca fascicularis, the gestational age that approximates the developmental stage of a mid-gestation human fetus when therapeutic intervention is clinically feasible.
C3 LNPs encapsulating GFP mRNA (C3.GFP) were administered ICV via transabdominal ultrasound guidance at a dose of 1 mg/kg into the lateral ventricle of a 0.61G macaque (Fig. 5A). Successful delivery was confirmed by observation of transient ventricular swelling (Fig. 5B). Forty-eight hours after injection, the fetus was delivered by caesarian section and GFP expression in the brain was assessed by immunohistochemistry. Tissue sections of harvested fetal macaque brain demonstrated strong GFP expression in the cells lining the lateral ventricles, similar to the region transfected in our previous R26mT/mG mouse studies (Fig. 5C). This result demonstrates that the mRNA delivery performance of C3 LNPs in the perinatal brain translates to a larger scale animal model more developmentally similar to a human, while highlighting the need for future optimization to enhance targeting deeper regions of the non-human primate brain.
Fig. 5 |. C3 LNP-mediated in utero mRNA delivery to the NHP brain and ex vivo performance in pediatric biological fluids and brain tissue.
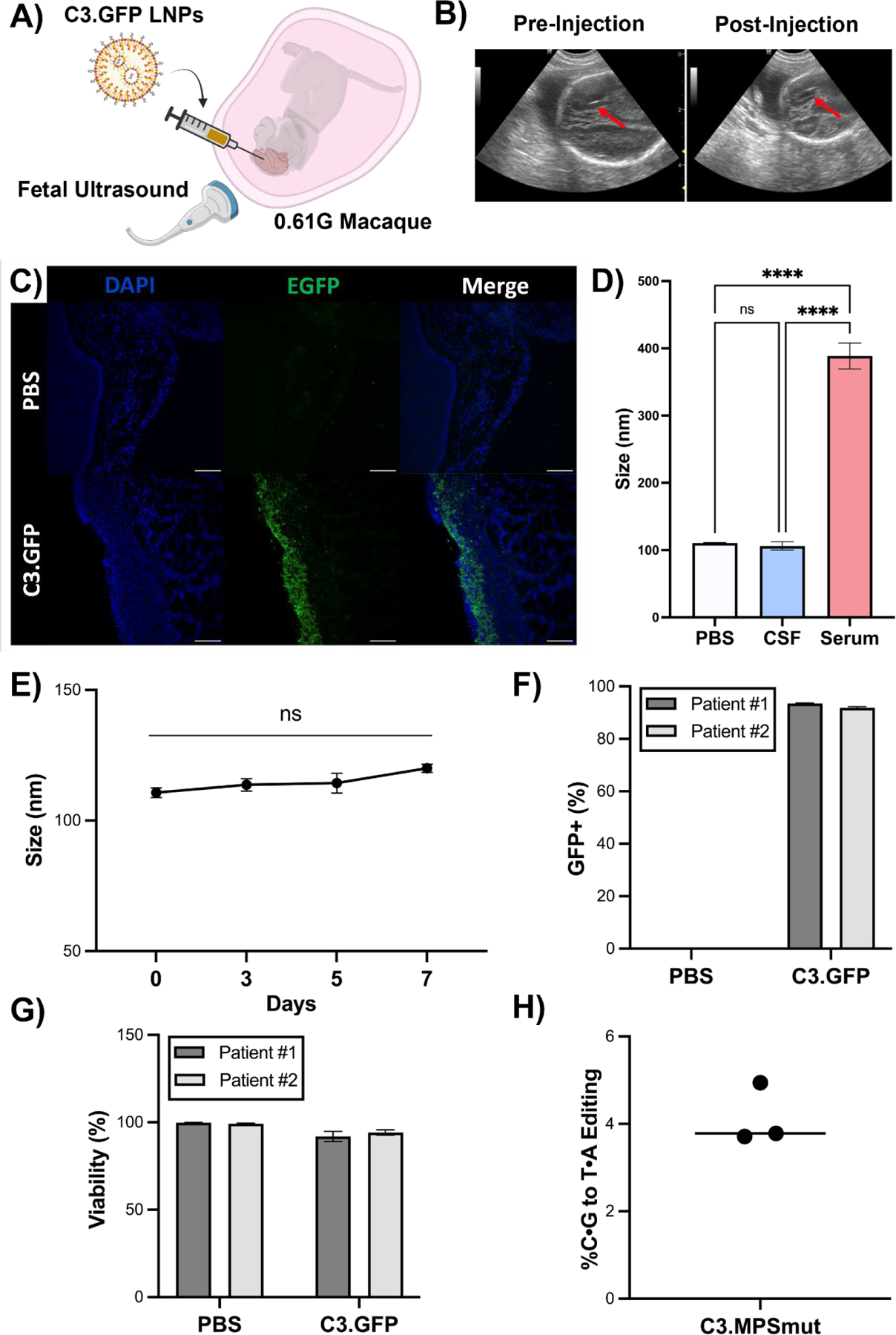
(A) Experimental scheme depicting ultrasound guided ICV injection of C3 LNP.GFP in a 0.61G Macaca fascicularis. (B) Ultrasound images (red arrow points to needle) pre- and post-ICV injection of C3.GFP LNPs in fetal macaque. (C) GFP immunohistochemistry on brain sections from C3 LNP.GFP-treated and negative control animals. Scale bars: 100 μm (10X). (D) Size and PDI measurements of C3.MPS LNPs incubated in PBS, human CSF, or human serum. **** p < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. (E) Size measurements of C3.MPS LNPs incubated in human CSF over a 7-day time course. (F) GFP positivity in two patient-derived primary cell lines enriched for neurons after C3.GFP LNP treatment. (G) Viability after C3.GFP LNP treatment in both patient-derived cell lines. (H) NGS results in patient-derived precision cut brain slices treated with C3.MPS LNPs normalized to control (PBS). Outliers were detected using Grubbs’ test and removed from analysis; minimum n = 3 per treatment group; error bars represent SEM.
LNPs demonstrate translational potential in patient-derived brain tissues
To further evaluate the potential clinical relevance of C3 LNPs for mRNA delivery to the human brain, we tested their performance in three patient-derived samples – cerebrospinal fluid (CSF), neurons derived from brain tissue, and precision-cut three-dimensional brain slices. We previously developed an assay to evaluate LNP stability in fetal fluids and demonstrated that ex vivo stability predicts in vivo mRNA delivery (28). As such, C3.MPS LNPs were incubated in pediatric cerebrospinal fluid (CSF), the fluid into which they would be clinically delivered, and assessed for stability via size and charge measurements (Fig. 5D and Fig. S10). No significant change in size or polydispersity of C3.MPS LNPs incubated in human CSF was observed (Fig. 5D and Fig. 5E), and this result was confirmed over a one-week time course, supporting the stability of our LNP. In contrast, incubation of C3.MPS LNPs in human serum resulted in significant aggregation and clustering (Fig. 5D). Previous studies have reported that LNP zeta potential measurements become more negative at increasing concentrations of protein-rich fluid (28). However, the zeta potential of C3.MPS LNPs did not change at increasing concentrations of human CSF (Fig. S10C), implying that these LNPs avoid significant protein adhesion that can reduce cellular uptake.
Next, neurons were cultured from cerebral cortex tissue of two pediatric patients undergoing neurosurgical procedures. C3.GFP LNPs resulted in a high transfection efficiency (Fig. 5F) with no significant effect on viability (Fig. 5G) of these cells. We then sought to demonstrate C3 LNP-mediated base editing in human brain tissue. Using an established protocol (36), precision cut slices of pediatric brain tissue were generated and cultured ex vivo in a 50:50 mixture of human CSF and serum-free media. C3 LNPs (C3.MPSmut LNPs) encapsulating cytosine base editor (BE3) mRNA and an sgRNA to introduce a C→T mutation (W402X) in the IDUA gene – a common mutation in MPS-IH patients – were applied to precision cut brain slices. NGS demonstrated ~4% on-target editing (Fig. 5H) supporting the ability of C3 LNPs to co-deliver a functional base editor mRNA and sgRNA in an ex vivo human brain tissue model.
DISCUSSION:
Most genetic brain diseases lack reasonably effective therapies due to the complexity of the CNS, the limited regenerative capacity of the tissue, and the difficulty of therapeutics crossing the blood-brain barrier (37). Since congenital brain pathology often begins before birth, fetal intervention provides a therapeutic opportunity to mitigate irreversible disease burden (3). Through correction of underlying pathogenic mutations, gene editing technologies offer a potential curative strategy for genetic diseases with CNS features after a single treatment. However, the applicability of gene editing therapies in the perinatal CNS is currently limited by the lack of a safe and effective nucleic acid delivery platform. Here, we screen a diverse ionizable LNP library for delivery of mRNA to the murine fetal and neonatal CNS and demonstrate LNP-mediated base editing and partial biochemical correction of disease after in vitro optimization of the lead LNP platform. In addition, we achieve LNP-mediated mRNA delivery to the CNS of a fetal NHP in vivo and characterize the stability and base editing performance of our optimized LNP ex vivo in patient-derived human brain tissues. This proof-of-concept study supports the safety and efficacy of LNPs for delivery of mRNA-based gene editing therapies to the CNS.
In this work, a mouse model of MPS-IH was chosen to study LNP-mediated perinatal delivery of a gene editing therapy since the neurodevelopmental pathology in patients with this disease starts prior to or shortly after birth, is irreversible, and is resistant to currently available treatments, including enzyme replacement therapy and hematopoietic stem cell transplant (9). MPS-IH is caused by genetic mutations in the IDUA gene resulting in IDUA enzyme deficiency. One of the most common IDUA disease-causing mutations is amenable to correction by adenine base editing resulting in restoration of IDUA enzyme activity and improvement in disease pathology including the reduction in lysosomal accumulation of GAGs (9). Despite low-level LNP-mediated adenine base editing at the pathogenic locus in the neonatal MPS-IH mouse brain, we observed partial restoration of IDUA enzyme activity (7–11% of normal) and reduction in the accumulation of GAGs in multiple brain regions. We previously demonstrated stable editing in multiple organs from 1 month to 6 months after in utero AAV-mediated adenine base editing in the MPS-IH mouse model (9). Similarly, future studies investigating the long-term stability of both C3 LNP-mediated base edits and biochemical correction as treated animals grow from neonates to adults are important. However, since studies suggest that 1% of normal IDUA enzyme activity may be sufficient to transition from severe Hurler’s syndrome to the milder Scheie syndrome (38), the low-level editing observed via the current approach may beneficially affect the neurobehavioral phenotype in MPS-IH, although assessment of this outcome is limited in the current study due to the lack of a strong neurobehavioral phenotype in this mouse model at the short time points assessed (9). The improvement in IDUA and GAG levels is likely due to secretion of IDUA enzyme into the CNS by a small number of corrected periventricular cells, which is supported by our studies in neonatal reporter mice demonstrating the predominance of editing confined to periventricular cells after intraventricular injection of LNPs. Thus, while the lead LNP engineered in this study holds promise for CNS diseases in which a paracrine effect of restoring normal protein function following periventricular cell targeting is adequate, further LNP optimization is required to apply this delivery modality to a broader range of diseases involving non-periventricular structures or specific neural cell types.
Engineering of LNPs is an iterative process, and characterization of our lead LNP has established critical parameters for LNP-mediated nucleic acid delivery to the perinatal CNS. LNPs containing the C3 ionizable lipid were small (<100 nm), monodisperse (<0.20 PDI), well-encapsulated (>90%), and on the lower end of the known pKa range (~5) for efficient mRNA delivery. Given the more acidic pH of CSF and size of intercellular junctions adjoining brain cell types (39), we hypothesize that these parameters convey better in vivo efficacy in the perinatal brain. More broadly, the trends observed for in vivo transfection efficiency of our LNP library in the fetal mouse brain matched those seen in the neonatal mouse brain, validating that the neonatal mouse model can be used to approximate LNP performance in the fetal mouse CNS. Importantly, the top-performing LNP following in utero ICV mRNA delivery was in the bottom quartile of LNPs for targeting the fetal liver following in utero intravascular mRNA delivery (19). These findings highlight the variability of cell-type specific in vivo LNP targeting dependent on LNP characteristics. In addition to the identity of the ionizable lipid, a range of variables during LNP formulation are known to influence in vivo delivery of gene editing platforms. We assessed several of these variables in vitro in neuronal origin cells. Modulation of LNP lipid ratio parameters suggested that our original LNP formulation mediated efficient mRNA delivery. An optimal weight ratio of base editor mRNA to sgRNA within the lead LNP formulation (3:1) was also identified, differing from previous reports of LNPs co-encapsulating base editor mRNA and sgRNA (16, 40). Further strategies to improve LNP-mediated gene editing include in vivo screening of a large DOE library of LNPs via mRNA barcoding, optimization of mRNA cargo modifications, or the addition of cell-specific targeting moieties to the LNP surface.
Highly effective “traditional” replacement gene therapy to the perinatal CNS has previously been achieved in several animal studies via viral vectors. For instance, Waddington and colleagues utilized AAV9 to rescue a mouse model of Gaucher’s disease and demonstrated brain transduction following ICV administration of AAV9 in the fetal NHP model (41). While AAVs were shown to be effective for these applications, recent concern over AAV toxicity at high doses motivates the development of non-viral alternatives (42). This is especially important when considering fetal gene therapy or gene editing in which the fetus may be more prone to toxicities of the delivery vehicle and the mother, who, in most cases, is unaffected by the disease, must not be harmed. In this study, C3 LNPs did not result in morphological changes to the fetal or neonatal murine brain parenchyma on gross examination. In addition, ICV delivery of C3 LNPs encapsulating a base editing platform to treat the mouse model of MPS-IH did not result in demonstrable editing in the liver and gonads. Thus, while this delivery modality holds promise for the CNS pathology of MPS-IH and other genetic diseases limited to CNS pathology, diseases with multi-organ pathology would require additional systemic delivery approaches for complete treatment.
Assessment of the acute serum cytokine response following C3 LNP treatment of MPS-IH neonates exhibited elevated levels of a small subset of cytokines; however, this response was lower than that which was produced by FDA-approved LNPs encapsulating the same base editing platform. ICV administration of C3 LNPs in the mouse neonate, which immunologically mimics a mid- to late-gestation human fetus, also did not elicit an anti-PEG IgM antibody response, in contrast to that seen following injection of the same LNPs in the adult mouse brain. These results highlight the opportunity for repeat LNP injections to boost longitudinal therapeutic efficacy (18). While these results coupled with our survival data in neonatal mice, brain tissue histology, and off-target genomic analysis are encouraging, additional safety analyses of our LNP base editing platform are required prior to translation, including unbiased genomic off-target analyses, long-term anti-PEG IgG response, immune response to repeat doses of an LNP-based therapeutic, and evaluation of long-term systemic organ toxicity.
Although valuable insights can be gained from in vitro and in vivo small animal studies, testing delivery carriers in models that better account for the size, complexity, and species variability of the human CNS is critical for consideration of clinical translation (42). To this end, we evaluated the ability of C3 LNPs to deliver mRNA to the fetal brain in the NHP via ICV administration at a gestational age akin to a mid-gestation human fetus and demonstrated transfection of the brain ventricular lining akin to our studies in the mouse model. In addition, given the ultimate goal of developing an LNP platform for delivery into the CSF-filled ventricles of patients, we assessed the stability of C3 LNPs in human CSF and demonstrated that LNPs retained their physiochemical properties upon incubation in CSF but not human serum ex vivo. Prior studies have demonstrated that the molecular composition of an LNP modulates the endogenous proteins that bind the nanoparticle and govern its biological fate (43). Both the abundance and identity of proteins in the CSF are distinct relative to the serum (44), which may contribute to the difference in stability, and subsequently performance, of LNPs in these two biological fluids. Proteomics-based identification of specific proteins that bind C3 LNPs in the CSF in future studies may allow for enhanced LNP-mediated cellular uptake in the perinatal brain through receptor targeting. Finally, we generated precision-cut slices of human cortical brain tissue and demonstrated C3 LNP-mediated base editing of the gene implicated in MPS-IH. Taken together, these experiments provide the foundation for additional translational studies and demonstrate base editing facilitated by a non-viral delivery carrier in the NHP fetal brain and primary human brain tissue.
CONCLUSIONS:
Delivery of mRNA-based therapeutics to the perinatal brain holds great potential in treating congenital brain disorders with limited therapeutic options. Although previous work has evaluated virally-mediated gene delivery to the CNS, non-viral mediated delivery of mRNA therapeutics to the CNS has been less robustly studied and offers potential safety advantages over viral vector delivery approaches. Here, we engineer an LNP platform to begin to address this need, demonstrating its efficacy and safety in a mouse model of a human disease, in the fetal NHP model, and in patient-derived brain tissues. With future optimization, these LNPs may offer a translatable delivery platform for in utero and neonatal CNS-directed gene editing.
MATERIALS AND METHODS:
Ionizable lipid synthesis
Ionizable lipids were prepared via Michael addition chemistry as previously described (20). Epoxide-terminated alkyl chains (Avanti Polar Lipids) were reacted with polyamine cores (Enamine Incorporated). The components were combined at an excess of lipid epoxides in a 4mL amber vial with magnetic stir bar. The crude product was transferred to a Rotavapor R-300 (BUCHI) for solvent evaporation, and the lipids were suspended in ethanol for use.
mRNA synthesis and production
mRNA was produced using standard in vitro transcription methods. Luciferase, GFP, ABE7.10, and BE3 gene sequences were codon optimized and cloned into proprietary mRNA production plasmids. The m1Ψ UTP nucleoside modified mRNA was co-transcriptionally capped with a trinucleotide cap1 analogue (TriLink) and engineered to contain a 101 nucleotide-long poly(A) tail. Transcription was performed using MegaScript T7 RNA polymerase (Invitrogen), and mRNA was precipitated before purification via cellulose chromatography. mRNAs were analyzed by agarose gel electrophoresis, sequenced, subjected to a standard J2 dot blot, assayed for INF induction in human monocyte derived dendritic cells, and stored at −80°C.
Cre recombinase mRNA fully substituted with 5-methoxyuridine was sourced from TriLink Biotechnologies via the CleanCap® platform. sgRNAs were sourced from Integrated DNA Technologies via the Alt-R™ platform. The mouse Idua gene targeting protospacer and PAM was 5’-ACTCTAGGCAGAGGTCTCAA | AGG-3’ as described in our previous work (9). The protospacer and PAM to introduce the most common mutation in the W402X human Idua gene was 5’-CCAGAGCTGCTCCTCATCTG | CGG-3’.
LNP formulation
Ionizable lipids, prepared as described above, or DLin-MC3-DMA (MedChem Express) were combined in an ethanol phase with cholesterol (Sigma-Aldrich), DOPE (Avanti), and 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (C14-PEG2000, Avanti) at a molar ratio of 35:46.5:16:2.5 or at molar ratios specified in Fig. S6. An aqueous phase was prepared consisting of 25μg of luciferase mRNA, 25μg of GFP mRNA, or 25μg of total mRNA at a variable mass ratio of ABE to sgRNA in 10 mM citrate buffer (pH 3). The ethanol and aqueous phases were then combined at a 3:1 ratio through channels in a microfluidic device facilitated by a Pump33DS syringe pump (Harvard Apparatus) or rapidly mixed and incubated. LNPs were dialyzed against 1X PBS for 2 hours before sterile filtration via 0.22μM filters. All materials were prepared and handled ribonuclease-free.
LNP characterization
Zetasizer Nano (Malvern Instruments) was used to measure the diameter (z-average) and polydispersity index (PDI) of LNPs suspended in 1X PBS at a dilution factor of 1 to 100. Zeta potential measurements were collected for LNPs suspended in deionized water at a dilution factor of 1 to 100 using the Zetasizer Nano and DTS1070 zeta potential cuvettes (Malvern Panalytical).
pKa measurements were conducted as previously described (22). Buffered solutions of 150mM sodium chloride, 20mM sodium phosphate, 20mM ammonium acetate, and 25mM ammonium citrate were each adjusted to pH 2 to 12 in increments of 0.5. 200μL of each pH-adjusted solution was combined with 5μL of each LNP formulation. TNS [6-(p-toluidinyl)naphthalene-2-sulfonic acid] was added to each well at a final TNS concentration of 6μM. Fluorescence intensity was read on an Infinite 200 Pro plate reader (Tecan), and pKa was calculated as the pH at which the fluorescence intensity was 50% of its maximum value, reflective of 50% protonation.
Encapsulation efficiencies were calculated using Quant-iT RiboGreen (Thermo Fisher Scientific) assay as previously described (19). Equal concentrations of LNPs were treated with either Triton X-100 (Sigma-Aldrich) or left untreated, and after incubation, the groups were plated alongside RNA standards. RiboGreen reagent was added, and fluorescence was measured on a plate reader at an absorbance range of 480 nm / 520 nm. A standard curve was generated using RNA standards and used to quantify RNA content and calculate encapsulation efficiency.
Animal experiments
All mouse animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at Children’s Hospital of Philadelphia (IACUC 21-001417) and followed guidelines in the NIH’s Guide for the Care and Use of Laboratory Animals.
BALB/c, B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J (R26mTmG), and B6.126S-Iduatm1.1Kmke/J (Idua-W392X) were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were housed in the Laboratory Animal Facility at the Children’s Hospital of Philadelphia.
Macaque procedures were performed in accordance with the IACUC at the National University of Singapore and Singapore Health Services (IACUC 2009-SHS-512). The macaque experiments were conducted at the SingHealth Experimental Medicine Centre, accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Mouse ICV injection
In utero, neonatal, and adult bilateral ICV injection were performed as described previously (41, 45, 46). Fetus: 18-day gestation BALB/c fetuses were used to screen the LNP library in vivo. Briefly, under isoflurane anesthesia, a maternal midline laparotomy was performed to expose the uterine horns. Either 4μL of PBS or LNPs containing 1mg/kg mRNA was injected into the lateral ventricles using an 80μm beveled glass micropipette and a Narishige IM-400 Electric Microinjector (Narishige International). After successful injection, confirmed by visualizing clearance of the injectate and transient swelling of the ventricle, the uterus was returned to the peritoneal cavity, the abdomen was closed, and mice recovered in a warm cage. Neonate: P0 BALB/c or Idua-W392X pups were anesthetized on ice and 4μL of PBS or LNPs containing 1mg/kg mRNA was injected into the lateral ventricles under a dissecting microscope using an 80-μm beveled glass micropipette and automated microinjector. After successful injection, pups were recovered on a warming pad and returned to a nursing foster mother. Adult: After appropriate analgesia and isoflurane anesthesia, a 31-guage needle was used to penetrate the skull and access the lateral ventricles in 12-week-old BALB/c mice. 10 μL of either PBS or LNPs containing 1mg/kg mRNA was injected. Following injection, skin was closed with running 5-0 absorbable suture, and animals were recovered.
Luciferase imaging
Mice were imaged 4 hours after injection with LNPs on an in vivo imaging system (IVIS, PerkinElmer). Prior to imaging, animals were injected intraperitoneally with D-luciferin (150mg/kg) and potassium salt (Biotium). After sacrifice, whole specimens or dissected organs were imaged via IVIS, and luminescence signal was detected with a standardized exposure time. Image analysis was conducted in the Living Image software (PerkinElmer). To quantify luminescent flux, a rectangular ROI was placed over each sample, and an ROI of the same size was placed in an area without any luminescent signal in the same image. Normalized flux was calculated by dividing total flux from the sample area by the total flux from the background area.
R26mT/mG and Balb/c mouse studies and brain immunohistochemistry
P0 R26mT/mG neonates were injected ICV with either PBS or C3 LNP. One week after injection, animals were harvested by standard transcardiac perfusion using 4% PFA followed by overnight fixation. After 24 hours, samples were transferred to 30% sucrose for cryoprotection. Tissues were embedded in OCT compound and sectioned on a cryostat. Sections were mounted on Superfrost Plus slides (Thermo Fisher Scientific) and were frozen at −80°C.
For immunostaining, slides were dried at 60°C via a slide warmer and rehydrated in 1X PBS. Tissues were blocked with 10% donkey serum (Sigma-Aldrich) followed by overnight incubation with 1:500 dilutions of the following primary antibodies: NeuN (Rabbit mAb #12943, Cell Signaling Technology), IBA1 (Rabbit mAb 019–19741, FUJIFILM Wako Pure Chemical Corporation), and GFAP (Rabbit mAb GA524, Agilent Technologies). After 24 hours, slides were washed with 1X PBS and incubated with Alexa Fluor-conjugated secondary antibodies for 2 hours at room temperature. Slides were mounted on Fluoroshield Mounting Medium with DAPI (Abcam) and imaged on a fluorescence microscope (BZ-X, Keyence). A similar protocol was executed for immunostaining of sectioned Balb/c neonatal brain tissue for H&E via the Hematoxylin and Eosin Staining Kit (ab245880).
In vitro LNP optimization studies
Neuro-2a cells (ATCC no. CCL-131) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with l-glutamine (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% penicillin-streptomycin (Gibco). Cells were plated at a density of 1000 cells/uL, incubated overnight, and dosed with LNPs encapsulating 100 ng of GFP mRNA in serum-free media. After 24 hours, cells were detached via trypsin, washed with 1X PBS, and resuspended in flow cytometry (FACS) buffer (Ca2+/Mg2+ Free PBS, 0.5% BSA, 0.5 mM EDTA). Samples were analyzed for fluorescence via flow cytometry (BD FACSAria™ Cell Sorter) for GFP+. Viability was assessed via Live/Dead™ Cytotoxicity Kit (Thermo Fisher Scientific).
Primary fibroblasts or primary neurons were harvested from an adult Idua-W392X mouse. The mouse was euthanized, and organ tissue was isolated prior to mechanical digestion and filtering through 100μm cell strainers. Cells were washed with 1X PBS and subsequently cultured in DMEM supplemented with 15% FBS and 1% penicillin-streptomycin. Either primary fibroblasts or primary neurons were plated at a cellular density of 1000 cells/uL, incubated overnight, and treated with LNPs at a range of ABE to sgRNA ratios and doses in serum-free media. After 5 days, cells were detached via trypsin, washed in 1X PBS, and resuspended in QuickExtract™ DNA Extraction Solution (Lucigen Corporation). Q5 polymerase was used to amplify the genomic region encompassing the W392X mutation with the following primers (F: 5’-TGCTAGGTATGAGAGAGCCA-3’, R: 5’-AGTGTAGATGAGGACTGTGGT-3’) at an annealing temperature of 66°C. PCR products were evaluated using an agarose gel, purified using the Qiagen PCR Purification Kit, and analyzed by Sanger sequencing (Azenta Life Sciences).
Idua-W392X mouse studies (on- and off-target editing, IDUA activity, tissue GAG assay)
P0 Idua-W392X neonates were injected ICV with C3.MPS LNPs containing 1mg/kg total mRNA. Untreated Idua-W392X mice and wild-type B6 mice served as positive and negative controls. After one month, animals were sacrificed, and brain tissue (forebrain, midbrain, hippocampus), liver, and gonads was harvested. DNA was extracted using the Qiagen DNEasy Blood and Tissue Kit (Qiagen), PCR amplified and purified, and analyzed via NGS (Azenta Life Sciences).
The top ten-off target sites were predicted using CRISPOR and ranked using the Cutting Frequency Determination off-target score. These sites were PCR amplified using Platinum SuperFi II Hi-Fidelity DNA Polymerase (Thermo Fisher Scientific), PCR amplified and purified, and analyzed via NGS (Azenta Life Sciences).
Tissue IDUA activity was assayed as described previously (9). Tissue samples were homogenized with 0.1% Triton X-100 lysis buffer using a TissueLyser LT (Qiagen). IDUA enzyme activity was induced using 4-methyl-umbelliferyl-α-L-iduronide (Glycosynth). After incubation, reactions were arrested using glycine carbonate buffer, and fluorescence was measured via plate reader. Enzyme activity was normalized to sample protein content measured via Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). After obtaining IDUA enzyme activity, 0.5mL of papain solution (Sigma-Aldrich) was added to homogenized tissue lysates. Samples were incubated at 65°C for 3 hours and centrifuged to clarify supernatant. Total sulfated GAG content in each sample was assessed using the Blyscan Glycosaminoglycan Assay (Biocolour). Tissue GAG content was normalized to sample protein content.
Idua-W392X mouse studies (cytokine analysis, anti-PEG IgM antibody)
P0 Idua-W392X were injected ICV with either 1X PBS or C3.MPS/MC3.MPS LNPs containing 1mg/kg total mRNA. Animals were bled after 24 hours prior to analysis via 25-proinflammatory cytokine panel (MilliporeSigma). A standard curve for each plate was prepared by serial dilution of the provided standards in the appropriate dilutant. Multiplex plates were run on a MAGPIX® system (Luminex Corporation). Each cytokine was assessed using a five-parameter regression algorithm and normalized to sample protein concentration determined by microBCA assay.
P0 Idua-W392X neonates and adults were injected ICV with either PBS or C3.MPS containing 1mg/kg total mRNA. Animals were bled after 7 days, and the concentration of circulating IgM anti-PEG antibodies was assessed by Mouse Anti-PEG IgM ELISA (Life Diagnostics Incorporated). Concentration of IgM anti-PEG antibody was calculated based on a standard curve.
LNP administration to fetal macaque and brain immunohistochemistry
A cynomolgus macaque (Macaca fasicularis) was used for a proof-of-principle non-human primate (NHP) study. Macaques underwent timed mating with pregnancy confirmed through ultrasound evidence of a fetal pole and fetal heart activity. Dating was performed as previously reported (41), and in utero ICV injection was performed at 0.61G. In brief, anesthesia was induced via sevoflurane. A 25G Quincke needle (Becton-Dickenson) was used to target the lateral ventricle closest to the anterior maternal abdomen under continuous ultrasound imaging. C3 LNP containing 1mg/kg total mRNA was injected as a slow bolus, and transient ventricular swelling validated successful delivery. The needle was removed, and both mother and fetus were monitored post-operatively. Animals were sacrificed after 48 hours, and macaque brains were harvested using isoflurane and pentobarbitone, cardiac puncture, and perfusion of 1% PFA.
After 24 hours, samples were cryoprotected, embedded in OCT compound, sectioned, and mounted on Superfrost Plus slides (Thermo Fisher Scientific). For immunostaining, slides were dried at 60°C via a slide warmer and rehydrated in 1X PBS. Tissues were blocked with 10% donkey serum (Sigma-Aldrich) and incubated overnight with 1:100 dilution of GFP antibody (Rabbit mAb ab290). After 24 hours, slides were washed with 1X PBS, incubated with an Alexa Fluor-conjugated secondary antibody for 2 hours at room temperature, and mounted on Fluoroshield Mounting Medium with DAPI prior to imaging.
LNP stability in human fluids
All human specimens were collected following Institutional Review Board (IRB) approval at the Children’s Hospital of Philadelphia (IRB 21-018470). LNP stability in patient-derived fluids was assessed via our previously published protocol (28). In brief, C3.MPS LNPs were incubated in a range of human serum or CSF percentages – 0%, 25%, 50%, 75%, 90% and 100% (volume biological fluid/total sample volume) for 30 minutes at 37°C under gentle agitation. After 30 minutes, each sample was diluted 1 to 100 in 1X PBS and transferred to a standard cuvette for size, PDI, or zeta potential measurement via Zetasizer Nano.
Isolation of patient-derived precision cut brain slices and brain cells
Brain tissue was dissected and transferred into ice-cold patient cerebrospinal fluid equilibrated with carbogen (95% O2, 5% CO2). Tissue was trimmed and 300μm slices were prepared using a vibratome (Leica Biosystems). Slices were transferred to tissue culture plates and cultured in media modified from prior protocols (36): 50% DMEM, 48% Neurobasal™ Media (Thermo Fisher Scientific), 1X GlutaMAX™ (Gibco), and 20mM HEPES (Thermo Fisher Scientific).
Primary human brain cells were isolated via an established protocol (47). Patient-derived brain tissue was mechanically dissociated before enzymatic digestion in papain and DNase I (Invitrogen Corporation) for 30 minutes at 37°C. Cell suspension was passed through a 100μm cell strainer, centrifuged, and resuspended in the media described above. Cells were plated onto poly-d-lysine-coated cell culture surfaces, and 50% of culture media was exchanged every 48 hours.
LNP studies in patient-derived cells and tissue
Patient-derived primary cells were seeded at a cellular density of 1000 cells/uL in a poly-d-lysine coated 96-well plate and allowed to incubate overnight. Cells were treated with either PBS or C3 LNPs containing 100 ng of GFP mRNA. After 24 hours, cells were detached, washed with 1X PBS, and resuspended in FACS buffer. Samples were analyzed for fluorescence (GFP+) via flow cytometry and viability via Live/Dead™ Cytotoxicity Kit (Thermo Fisher Scientific).
Prior to treatment, precision cut brain slices were cultured in 50% primary brain media and 50% human CSF. Slices were treated with either PBS or C3.MPSmut LNPs containing 500 ng of total mRNA in serum-free media. After 5 days, slices were mechanically digested, passed through a 100 μm cell strainer, and resuspended in QuickExtract™ DNA extraction solution. The genomic region encompassing the human W402X mutation was amplified with KAPA HiFi DNA Polymerase (Roche, Basel, Switzerland) using the following primers (F: 5’-CAATGCCTTCCTGAGCTACCAC-3’, R: 5’-AGGTAGCGCGTGACGTAGAC-3’) at an annealing temperature of 57°C. PCR products were evaluated using an agarose gel, purified using the Qiagen PCR Purification Kit, and analyzed by NGS (Azenta Life Sciences).
Statistical analysis
All quantifications represent the mean and standard error of the mean (SEM) acquired from at least three biological replicates (cells or animals) per treatment group. Means were compared via one-way ANOVA with post-hoc Dunnett’s test, two-way ANOVA with post-hoc Šídák’s multiple comparisons test, or Student’s t test. For all experiments, outliers were detected using Grubbs’ test and removed from analysis.
Supplementary Material
ACKNOWELDGEMENTS:
We acknowledge the Children’s Hospital of Philadelphia Small Animal Imaging Facility for maintenance of the IVIS. Figure schematics were created with BioRender.
Funding:
This study was supported by the U.S. National Institutes of Health (NIH) Director’s New Innovator Awards (DP2TR002776 to M.J.M and DP2HL152427 to W.H.P), NIH R01DK123049 to W.H.P. and M.J.M, and a Burroughs Wellcome Fund Career Award at the Scientific Interface (CASI) to M.J.M. R.P. was supported by an NIH Medical Scientist Training Program grant (T32GM07170) and an NIH NHLBI F30 fellowship (F30HL162465-01A1).
Footnotes
SUPPORTING INFORMATION:
The supporting information is available free of charge at [insert link here]. This includes a table with the physiochemical properties of the LNP library; LNP-mediated luciferase expression and N2A cell viability after treatment at three different time points; the relationship between LNP library physiochemical properties and LNP fetal mouse brain transfection efficacy in vivo; LNP-mediated luciferase expression in the fetal mouse liver; biodistribution of lead LNP in the fetal mouse after ICV injection; flow cytometry data showing sub-populations of gene modulated brain cells; table displaying range of excipient molar ratios tested in DOE library; impact trend curves from in vitro DOE screen; viability after treatment of LNPs from in vitro optimization scheme; absence of LNP-mediated base editing in the gonads and liver; characterization of LNP stability in human CSF and human serum (PDF).
Competing interests: R.P., W.H.P., and M.J.M. have filed a patent application based on this work. D.W. is an inventor on several patents related to this work filed by the Trustees of the University of Pennsylvania (11/990,646; 13/585,517, 13/839,023; 13/839,155; 14/456,302; 15/339,363; and 16/299,202). M.J.M. is an inventor on a patent related to this work filed by the Trustees of the University of Pennsylvania (PCT/US20/56252). The authors declare that they have no other competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present within the main text and/or the Supplementary Information. All data and materials are stored at the University of Pennsylvania and Children’s Hospital of Philadelphia facilities in the Mitchell and Peranteau laboratories. Additional data may be requested from the co-corresponding authors: M.J.M. (mjmitch@seas.upenn.edu) and/or W.H.P. (peranteauw@chop.edu).
REFERENCES:
- 1.Eisengart JB, Pierpont EI, Kaizer AM, Rudser KD, King KE, Pasquali M, Polgreen LE, Dickson PI, Le SQ, Miller WP, Tolar J, Orchard PJ, Lund TC, Intrathecal enzyme replacement for Hurler syndrome: biomarker association with neurocognitive outcomes. Genet. Med 21, 2552–2560 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freeze H, Eklund EA, Ng B, Patterson MC, Neurology of inherited glycosylation disorders. Lancet Neurol. 11, 453–466 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peranteau WH, Flake AW, The Future of In Utero Gene Therapy. Mol. Diagn. Ther 24, 135–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palanki R, Peranteau WH, Mitchell MJ, Delivery Technologies for In Utero Gene Therapy. Adv. Drug Deliv. Rev 169, 51 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cox DBT, Platt RJ, Zhang F, Therapeutic Genome Editing: Prospects and Challenges. Nat. Med 21, 121–131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anzalone AV, Koblan LW, Liu DR, Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol 38, 824–844 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Rossidis AC, Stratigis JD, Chadwick AC, Hartman HA, Ahn NJ, Li H, Singh K, Coons BE, Li L, Lv W, Zoltick PW, Alapati D, Zacharias W, Jain R, Morrisey EE, Musunuru K, Peranteau WH, In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat. Med 24, 1513–1518 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alapati D, Zacharias WJ, Hartman HA, Rossidis AC, Stratigis JD, Ahn NJ, Coons B, Zhou S, Li H, Singh K, Katzen J, Tomer Y, Chadwick AC, Musunuru K, Beers MF, Morrisey EE, Peranteau WH, In utero gene editing for monogenic lung disease. Sci. Transl. Med 11, eaav8375 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bose SK, White BM, Kashyap MV, Dave A, De Bie FR, Li H, Singh K, Menon P, Wang T, Teerdhala S, Swaminathan V, Hartman HA, Jayachandran S, Chandrasekaran P, Musunuru K, Jain R, Frank DB, Zoltick P, Peranteau WH, In utero adenine base editing corrects multi-organ pathology in a lethal lysosomal storage disease. Nat. Commun 12, 4291 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paunovska K, Loughrey D, Dahlman JE, Drug delivery systems for RNA therapeutics. Nat. Rev. Genet 23, 265–280 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bulcha JT, Wang Y, Ma H, Tai PWL, Gao G, Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther 6, 1–24 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.High-dose AAV gene therapy deaths. Nat. Biotechnol 38, 910–910 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Huang X, Kong N, Zhang X, Cao Y, Langer R, Tao W, The landscape of mRNA nanomedicine. Nat. Med 28, 2273–2287 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Xiao Y, Tang Z, Huang X, Chen W, Zhou J, Liu H, Liu C, Kong N, Tao W, Emerging mRNA technologies: delivery strategies and biomedical applications. Chem. Soc. Rev 51, 3828–3845 (2022). [DOI] [PubMed] [Google Scholar]
- 15.Song C-Q, Jiang T, Richter M, Rhym LH, Koblan LW, Zafra MP, Schatoff EM, Doman JL, Cao Y, Dow LE, Zhu LJ, Anderson DG, Liu DR, Yin H, Xue W, Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed. Eng 4, 125–130 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musunuru K, Chadwick AC, Mizoguchi T, Garcia SP, DeNizio JE, Reiss CW, Wang K, Iyer S, Dutta C, Clendaniel V, Amaonye M, Beach A, Berth K, Biswas S, Braun MC, Chen H-M, Colace TV, Ganey JD, Gangopadhyay SA, Garrity R, Kasiewicz LN, Lavoie J, Madsen JA, Matsumoto Y, Mazzola AM, Nasrullah YS, Nneji J, Ren H, Sanjeev A, Shay M, Stahley MR, Fan SHY, Tam YK, Gaudelli NM, Ciaramella G, Stolz LE, Malyala P, Cheng CJ, Rajeev KG, Rohde E, Bellinger AM, Kathiresan S, In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 593, 429–434 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, Seitzer J, O’Connell D, Walsh KR, Wood K, Phillips J, Xu Y, Amaral A, Boyd AP, Cehelsky JE, McKee MD, Schiermeier A, Harari O, Murphy A, Kyratsous CA, Zambrowicz B, Soltys R, Gutstein DE, Leonard J, Sepp-Lorenzino L, Lebwohl D, CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med 385, 493–502 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Han X, Zhang H, Butowska K, Swingle KL, Alameh M-G, Weissman D, Mitchell MJ, An ionizable lipid toolbox for RNA delivery. Nat. Commun 12, 7233 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riley RS, Kashyap MV, Billingsley MM, White B, Alameh M-G, Bose SK, Zoltick PW, Li H, Zhang R, Cheng AY, Weissman D, Peranteau WH, Mitchell MJ, Ionizable lipid nanoparticles for in utero mRNA delivery. Sci. Adv 7, eaba1028 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kauffman KJ, Dorkin JR, Yang JH, Heartlein MW, DeRosa F, Mir FF, Fenton OS, Anderson DG, Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 15, 7300–7306 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Oberli MA, Reichmuth AM, Dorkin JR, Mitchell MJ, Fenton OS, Jaklenec A, Anderson DG, Langer R, Blankschtein D, Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 17, 1326–1335 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hajj KA, Ball RL, Deluty SB, Singh SR, Strelkova D, Knapp CM, Whitehead KA, Branched-Tail Lipid Nanoparticles Potently Deliver mRNA In Vivo due to Enhanced Ionization at Endosomal pH. Small 15, 1805097 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Basha G, Novobrantseva TI, Rosin N, Tam YYC, Hafez IM, Wong MK, Sugo T, Ruda VM, Qin J, Klebanov B, Ciufolini M, Akinc A, Tam YK, Hope MJ, Cullis PR, Influence of Cationic Lipid Composition on Gene Silencing Properties of Lipid Nanoparticle Formulations of siRNA in Antigen-Presenting Cells. Mol. Ther 19, 2186–2200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ratajczak CK, Fay JC, Muglia LJ, Preventing preterm birth: the past limitations and new potential of animal models. Dis. Model. Mech 3, 407–414 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Cohen-Pfeffer JL, Gururangan S, Lester T, Lim DA, Shaywitz AJ, Westphal M, Slavc I, Intracerebroventricular Delivery as a Safe, Long-Term Route of Drug Administration. Pediatr. Neurol 67, 23–35 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, Madden TD, Hope MJ, Weissman D, Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Controlled Release 217, 345–351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mattar CN, Wong AMS, Hoefer K, Alonso-Ferrero ME, Buckley SMK, Howe SJ, Cooper JD, Waddington SN, Chan JKY, Rahim AA, Systemic gene delivery following intravenous administration of AAV9 to fetal and neonatal mice and late-gestation nonhuman primates. FASEB J. 29, 3876–3888 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swingle KL, Billingsley MM, Bose SK, White B, Palanki R, Dave A, Patel SK, Gong N, Hamilton AG, Alameh M-G, Weissman D, Peranteau WH, Mitchell MJ, Amniotic fluid stabilized lipid nanoparticles for in utero intra-amniotic mRNA delivery. J. Control. Release Off. J. Control. Release Soc 341, 616–633 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou X, Zaks T, Langer R, Dong Y, Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater 6, 1078–1094 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Billingsley MM, Hamilton AG, Mai D, Patel SK, Swingle KL, Sheppard NC, June CH, Mitchell MJ, Orthogonal Design of Experiments for Optimization of Lipid Nanoparticles for mRNA Engineering of CAR T Cells. Nano Lett. 22, 533–542 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gary DJ, Min JB, Kim Y, Park K, Won Y-Y, The Effect of N/P Ratio on the In Vitro and In Vivo Interaction Properties of PEGylated Poly(2-(dimethylamino)ethyl methacrylate)-Based siRNA Complexes. Macromol. Biosci 13, 1059–1071 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D, Shukla C, Liu X, Schoeb TR, Clarke LA, Bedwell DM, Keeling KM, Characterization of an MPS I-H knock-in mouse that carries a nonsense mutation analogous to the human IDUA-W402X mutation. Mol. Genet. Metab 99, 62–71 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Estapé Senti M, de Jongh CA, Dijkxhoorn K, Verhoef JJF, Szebeni J, Storm G, Hack CE, Schiffelers RM, Fens MH, Boross P, Anti-PEG antibodies compromise the integrity of PEGylated lipid-based nanoparticles via complement. J. Controlled Release 341, 475–486 (2022). [DOI] [PubMed] [Google Scholar]
- 34.Szebeni J, Storm G, Ljubimova JY, Castells M, Phillips EJ, Turjeman K, Barenholz Y, Crommelin DJA, Dobrovolskaia MA, Applying lessons learned from nanomedicines to understand rare hypersensitivity reactions to mRNA-based SARS-CoV-2 vaccines. Nat. Nanotechnol 17, 337–346 (2022). [DOI] [PubMed] [Google Scholar]
- 35.Riley JS, McClain LE, Stratigis JD, Coons BE, Ahn NJ, Li H, Loukogeorgakis SP, Fachin CG, Dias AIBS, Flake AW, Peranteau WH, Regulatory T cells promote alloengraftment in a model of late-gestation in utero hematopoietic cell transplantation. Blood Adv. 4, 1102–1114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwarz N, Uysal B, Welzer M, Bahr JC, Layer N, Löffler H, Stanaitis K, PA H, Weber YG, Hedrich UB, Honegger JB, Skodras A, Becker AJ, Wuttke TV, Koch H, Slutsky I, Marder E, Mansvelder HD, Eds. Long-term adult human brain slice cultures as a model system to study human CNS circuitry and disease. eLife 8, e48417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ingusci S, Verlengia G, Soukupova M, Zucchini S, Simonato M, Gene Therapy Tools for Brain Diseases. Front. Pharmacol 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clarke LA, Mucopolysaccharidosis Type I (University of Washington, Seattle, 2021). [PubMed] [Google Scholar]
- 39.Akaishi T, Onishi E, Abe M, Toyama H, Ishizawa K, Kumagai M, Kubo R, Nakashima I, Aoki M, Yamauchi M, Ishii T, The human central nervous system discharges carbon dioxide and lactic acid into the cerebrospinal fluid. Fluids Barriers CNS 16, 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Villiger L, Rothgangl T, Witzigmann D, Oka R, Lin PJC, Qi W, Janjuha S, Berk C, Ringnalda F, Beattie MB, Stoffel M, Thöny B, Hall J, Rehrauer H, van Boxtel R, Tam YK, Schwank G, In vivo cytidine base editing of hepatocytes without detectable off-target mutations in RNA and DNA. Nat. Biomed. Eng 5, 179–189 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Massaro G, Mattar CNZ, Wong AMS, Sirka E, Buckley SMK, Herbert BR, Karlsson S, Perocheau DP, Burke D, Heales S, Richard-Londt A, Brandner S, Huebecker M, Priestman DA, Platt FM, Mills K, Biswas A, Cooper JD, Chan JKY, Cheng SH, Waddington SN, Rahim AA, Fetal gene therapy for neurodegenerative disease of infants. Nat. Med 24, 1317–1323 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benatti HR, Gray-Edwards HL, Adeno-Associated Virus Delivery Limitations for Neurological Indications. Hum. Gene Ther 33, 1–7 (2022). [DOI] [PubMed] [Google Scholar]
- 43.Dilliard SA, Cheng Q, Siegwart DJ, On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci 118, e2109256118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dayon L, Cominetti O, Wojcik J, Galindo AN, Oikonomidi A, Henry H, Migliavacca E, Kussmann M, Bowman GL, Popp J, Proteomes of Paired Human Cerebrospinal Fluid and Plasma: Relation to Blood–Brain Barrier Permeability in Older Adults. J. Proteome Res 18, 1162–1174 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Kim J-Y, Grunke SD, Levites Y, Golde TE, Jankowsky JL, Intracerebroventricular Viral Injection of the Neonatal Mouse Brain for Persistent and Widespread Neuronal Transduction. JoVE J. Vis. Exp, e51863 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cetin A, Komai S, Eliava M, Seeburg PH, Osten P, Stereotaxic gene delivery in the rodent brain. Nat. Protoc 1, 3166–3173 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Park TI-H, Schweder P, Lee K, Dieriks BV, Jung Y, Smyth L, Rustenhoven J, Mee E, Heppner P, Turner C, Curtis MA, Faull RLM, Montgomery JM, Dragunow M, Isolation and culture of functional adult human neurons from neurosurgical brain specimens. Brain Commun. 2, fcaa171 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present within the main text and/or the Supplementary Information. All data and materials are stored at the University of Pennsylvania and Children’s Hospital of Philadelphia facilities in the Mitchell and Peranteau laboratories. Additional data may be requested from the co-corresponding authors: M.J.M. (mjmitch@seas.upenn.edu) and/or W.H.P. (peranteauw@chop.edu).