

Summary
Lung cancer mortality is exacerbated by late-stage diagnosis. Emerging evidence indicates the potential clinical significance of distinct microbial signatures as diagnostic and prognostic biomarkers across various cancers. However, circulating microbiome DNA (cmDNA) profiles are underexplored in lung cancer (LC). Here, whole-genome sequencing is performed on plasma of LC patients and healthy controls (HCs). Differentially enriched microbial species are identified between LC and HC. A diagnostic model is developed, which has a high sensitivity of 87.7% and achieves an AUC of 93.2% in the independent validation dataset. Crucially, this model demonstrates the capability to detect early-stage LC, achieving a sensitivity of 86.5% for stage I and 87.1% for tumors <1 cm. In addition, we construct a cmDNA model for recurrence, which precisely predicts LC recurrence after surgery. Overall, this study highlights the significant alterations of cmDNA profiles in LC, indicating its potential as biomarkers for early diagnosis and recurrence.
Keywords: early detection, recurrence, circulating microbiome DNA, biomarker
Graphical abstract
Highlights
-
•
The cmNDA profiles of lung cancer patients exhibit unique characteristics
-
•
A cmDNA-based diagnostic model of lung cancer exhibits high accuracy
-
•
cmDNA can predict lung cancer recurrence after surgery
Chen et al. reveal that circulating microbiome DNA can be a biomarker for lung cancer diagnosis and recurrence, establishing a paradigm of cancer liquid biopsy.
Introduction
Lung cancer (LC) is the second most common cancer and the leading cause of cancer mortality worldwide.1 The prognosis of LC is significantly associated with the stages at which cancer patients are diagnosed.2 If diagnosed early, the 5-year survival rate is >90%, but when diagnosed at the late metastatic stage, it drops to <5%.2 Therefore, early detection is a critical strategy in reducing LC mortality rates. Unfortunately, >80% of LC cases are detected at an advanced stage, which contributes to the high mortality rates.3 Chest low-dose computed tomography screening has been demonstrated to reduce cancer-related deaths by 20% in large randomized trials4,5. However, given its unsatisfactory high rate of false positive imaging results, radiation exposure, and cost,4,5 its use as an early-screening method is limited. Surgery is the treatment of choice for early-stage LC patients. Although the surgery cures most cancer patients with stages I–IIIA, 10%–50% of them experience recurrence after surgery, particularly within 3 years, which exacerbates LC mortality.2 Therefore, postoperative recurrence prediction is essential for obtaining a favorable prognosis and deciding on the appropriate adjuvant therapy. Hence, it is crucial to develop approaches with a high degree of accuracy to improve early LC detection and identify patients with high postoperative recurrence risk.
The lungs are a complex and unique organ system, which harbor one of the largest surface areas in the human body.6,7 The mucosal surfaces of the lungs are exposed to the external environment, facilitating the colonization of a vast number of microbiota communities.8 These microbial communities play an important role in the development, diagnosis, and prognosis of lung cancer.7 Previous studies have shown that commensal bacteria from LC promote cancer cell proliferation through cross-talk with myeloid cells and γδ T cells.9 In addition, a study revealed that the bacterial burden in tumor cells is significantly higher than that in immune cells and stroma in LC.10 Our previous report indicated that LC manifesting as solid nodules and subsolid nodules have distinct microbiome compositions, and alpha diversity is greater in the subsolid nodules subtype, which has more indolent clinical behavior.11 Importantly, reducing the pulmonary bacterial load in LC is associated with fewer regulatory T cells and enhanced T cell activation and leads to a significant reduction in cancer metastases.12 These findings demonstrate that the microbiome community is not only closely associated with LC development but also modulates cancer metastasis and recurrence.13
In recent years, various cancer tissues, including lung, breast, colorectal, and prostate cancers, have been found to harbor the tumor microbiome.7 With advancements in molecular biology and sequencing techniques, studies have highlighted the causal implications of commensal microbiota for tumorigenesis and its potential as diagnostic biomarkers of cancer.7,14,15 Recently, Poore et al.16 revealed that circulating microbiome DNA (cmDNA), nonhuman, microorganism-derived cell-free DNA isolated from peripheral blood, could discriminate between multiple cancers and healthy controls (HCs), opening up a new paradigm for cancer liquid biopsy and suggesting cmDNA as a valuable tool in cancer detection.17,18,19 In addition, several studies have reported that cmDNA has high discriminatory performance as a promising biomarker for noninvasive cancer detection, including colorectal cancer,17 esophageal adenocarcinoma,18 and hepatocellular carcinoma.20 Xiao et al.17 found reduced bacterial diversity in the cmDNA profiles of colorectal cancer and built a classifier that could differentiate it from HCs. However, circulating microbial profiles in LC, particularly their role as diagnostic markers of early-stage cancer detection and postoperative recurrence, have not been systematically characterized.
Therefore, this study used whole-genome sequencing (WGS) of plasma samples for a systematic investigation of cmDNA profiles in 416 participants with LC and HCs. We have identified the distinct profiles of cmDNA and evaluated their potential as noninvasive diagnostic biomarkers for early-stage detection of lung cancer and postoperative recurrence.
Results
Distinct cmDNA profiles in LC
The overall design of this study is shown in Figure 1. The 315 participants from the LC detection model study made up the training cohort (LC: 69; HC: 97), and two independent validation cohorts, including the validation I (LC: 48; HC: 48) and the validation II (LC: 33; HC: 20) (Tables S1, S2, and S3). Circulating free (cfDNA) was extracted from plasma samples of the training cohort and underwent WGS, with an average sequence depth of 5× (Figure 1). Human reads were removed, and the remaining reads were classified using Kraken2 and estimated using Braken to acquire cmDNA profiles. The sequencing results showed that the mean percentages of human reads were 98.04% in HC and 97.25% in LC. The mean percentages of microbial reads were 0.012% in HC and 0.009% in LC (Table S4). These results were consistent with a circulating bacterial DNA study of colorectal cancer.17 Next, the biodiversity and composition of cmDNA in LC patients were compared to HCs. Within the healthy group, the total number of species was significantly higher than that in the LC group (Figure S1A). In addition, alpha diversity analysis indicated that both the Shannon diversity and Simpson diversity of the healthy group were significantly higher compared to that of LC patients (Figures S1B and S1C). Intriguingly, the reduced bacterial diversity in cmDNA was consistent with the lower diversity of gut microbiome in LC, as previously reported by Liu et al.21
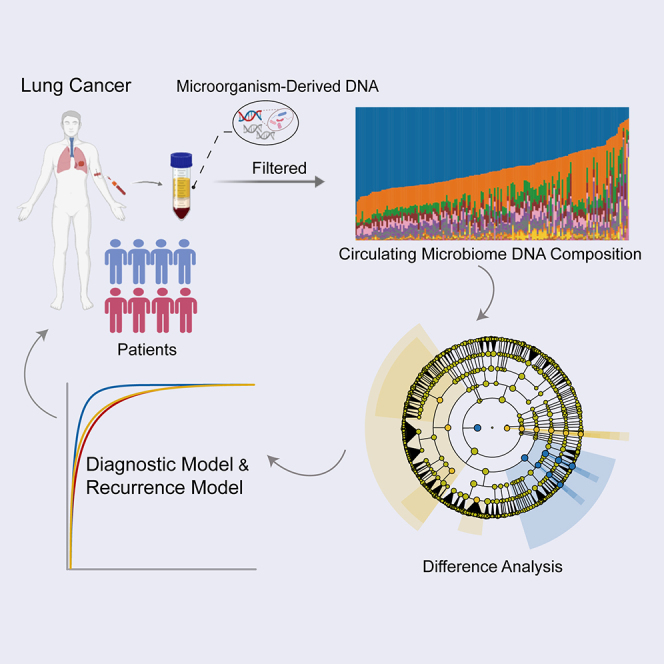
Figure 1.

Study workflow
A total of 416 participants were included in this study. WGS of plasma cfDNA was performed, and the cmDNA features of each subject were profiled. LC, lung cancer; R, recurrence; NR, non-recurrence.
To investigate the association between cmDNA and lung cancer, the relative abundances were compared between the LC and HC groups at different phylogenetic levels. At the phylum level, Proteobacteria, Actinobacteria, and Firmicutes dominated the circulating microbial communities in both groups, followed by Bacteroidetes and Ascomycota (Figure S2A). The abundance of Proteobacteria was higher in the LC group than that in the HC group, although there was no statistical significance (Figure S2A). It was noteworthy that an increase of the Proteobacteria phylum has previously been observed in the lung tissue of LC patients.22 We observed a similar pattern of composition at the order level (Figure 2A). Specifically, the relative abundance of Pseudomonadales from the Proteobacteria phylum was significantly higher in the LC group (p = 0.006), whereas the relative abundance of Corynebacteriales from the Actinobacteria phylum tended to be higher in the HC group (p = 0.012; Figure 2B). Intriguingly, we performed intratumor microbiome analysis of lung tissue from a subgroup of 15 patients’ with cmDNA, including tumor tissue and paired normal tissue, and found that the Pseudomonadales order was also enriched in the tumor group, although there was no significant difference (Table S5; Figures S2B and S2C). The higher abundance of Pseudomonadales order in lung tissue was reported to be associated with a worse disease-free survival among LC patients,23 whereas a higher abundance of Corynebacteriales was associated with a reduced risk of several cancers.24 Furthermore, the top 20 enriched species belonged mainly to the Proteobacteria phylum, of which Pseudomonas azotoformans, Pseudomonas sp. SXM-1, and Pseudomonas fluorescens were enriched in the LC group (p = 0.004, p = 0.014, p = 0.005, respectively) (Figures 2C and S2D). Among the statistically different genera or species, most bacterial taxa enriched in the LC group belonged to the Proteobacteria phyla. In particular, Acinetobacter, a well-known microbe enriched in bronchoalveolar lavage fluid of LC,25 showed significantly higher abundance in the LC group (p < 0.001; Figure 2C). In addition, the microbiome analysis among different subgroups of LC separated according to tumor size and tumor stage revealed that the cmDNA composition remained relative stable at the order level (Figure 2D). Taken together, these findings suggested that baseline signatures of cmDNA microbiome diversity and composition were correlated with LC patients, indicating that cmDNA may serve as a possible biomarker for distinguishing LC patients from HCs.
Figure 2.

cmDNA microbiome composition of all participants in the training cohort
(A) cmDNA microbiome composition at the order level (ordered by the most abundant taxa, Pseudomonadales order).
(B) Relative abundance comparisons of Pseudomonadales, Moraxellales, Hyphomicrobiales, and Corynebacteriales in the LC and HC groups at the order level (Wilcoxon test).
(C) Relative abundance comparison of significant taxa in the LC and HC groups at the genus level (left) and the species level (right) (Wilcoxon test).
(D) Dynamic microbial composition of different tumor size subgroups (left) and different tumor stage subgroups (right) in the LC and HC groups at the order level.
cmDNA microbial panel as a novel diagnostic biomarker for early-stage lung cancer
In addition to the significant disparity observed in alpha diversity, the beta diversity, measured via the Bray-Curtis distance, exhibited a clear separation between LC patients and HCs by nonmetric multidimensional scaling (NMDS; Adonis R2 = 0.027, p = 0.001; Figure 3A). Next, we performed linear discriminant analysis effect size (LEfSe) analysis to further compare the cmDNA microbial features of each group. Initially, we conducted pairwise analyses at all taxonomic levels and created a cladogram to visualize the differences in relative abundance of each taxa when comparing the LC and HC groups (linear discriminant analysis [LDA] >2.0 and p < 0.05) (Figure 3B; Table S6). Subsequently, we identified 46 species enriched in LC patients and 130 species enriched in HCs that potentially correlated with lung cancer (Figure 3C; Table S7). Notably, Pseudomonasa zotoformans, Acinetobacter guillouiae, Acinetobacter johnsonii, Pseudomonas fluorescens, and Pseudomonas sp. SXM-1 were the most enriched species in the LC group. In contrast, Fusarium oxysporum and Delftia sp. WY8 were significantly enriched in the HC group (Figure 3C).
Figure 3.

Differential taxa identified and lung cancer detection model development
(A) The NMDS plot showing beta diversity based on the Bray-Curtis distance between HC and LC groups. Significant differences were observed between LC patients and HCs with Adonis test (R2 = 0.027, p = 0.001).
(B) Taxonomic cladogram from LEfSe showed significantly different taxa enriched in the HC and LC groups (the top 30 according to LDA).
(C) LEfSe identified significantly differentially abundant species in the HC and LC groups (the top 30 according to LDA).
(D) The receiver operating characteristic curve showed an AUC value of 95.6% in the training cohort.
(E) The receiver operating characteristic curve of 5-fold cross-validation in predicting LC with different tumor size subgroups, the subgroup with smaller tumors (<1 cm, red line), and the subgroup with larger tumors (≥1 cm, green line).
Next, we investigated the possibility of identifying LC patients from HCs based on the significant species. We built a random forest machine learning model based on differentially expressed species data. The 119 important features were chosen for model development that had mean decrease accuracy scores >1 with random forest analysis. This model achieved an area under the receiver operating characteristic curve (AUC) of 95.6%, along with a sensitivity of 81.2%, a specificity of 90.7%, and an accuracy of 86.8% (Figure 3D; Table S8). Furthermore, we depicted cancer scores for each participant in the training cohort in Figure S3. The predicted scores of cancer patients in the early stage (stage I) and late stages (stages II–IV) were significantly higher than those for HCs. Moreover, we evaluated the predictive ability of the detection classifier on two subgroups based on tumor diameter: one with smaller tumors (<1 cm) and the other with larger tumors (≥1 cm). Fivefold cross-validation was used to test the discriminate efficacy in the subgroups. We obtained AUCs of 91.5% and 94.0% for the subgroup with smaller tumors (<1 cm) and the subgroup with larger tumors (≥1 cm), respectively (Figure 3E).
Before we developed the model, a decontamination pipeline was implemented to eliminate potential contaminants. Specifically, a threshold analysis was conducted on three negative controls (see STAR Methods) and a list of potential contaminants was created based on previous studies.16 We found that the selected important species genomes included in our model had no overlap with the possible microbial contamination (Table S9).
Independent validation of the early detection model
Next, we evaluated the performance of the detection model in two independent validation datasets separately. The age and gender distribution of LC patients and HCs were similar in both datasets (Tables S1, S2, and S3). The LC group was highlighted by the majority of early-stage diseases in the independent validation I (stage I, 45/48, 93.8%), whereas LC patients in the independent validation II were at the late stage (stages III–IV, 33/33, 100.0%). Intriguingly, the model showed high AUC values in external validations, with 92.1% (95% confidence interval [CI]: 86.7%–97.5%) and 97.2% (95% CI: 93.7%–100.0%) in validation I and validation II, respectively (Figure 4A). The resultant sensitivities are 87.5% (95% CI: 74.1%–94.8%) in validation I and 87.9% (95% CI: 70.9%–96.0%) in validation II (Figure 4B; Table S10). Based on the 75.0% specificity in the validation I cohort, we chose a cancer score cutoff of 0.511 as optimal. This model also exhibited a specificity of 90.0% in the validation II cohort. When combining the two validation cohorts, the model achieved a sensitivity of 87.7% (95% CI: 78.0%–93.6%) and a specificity of 79.4% (95% CI: 67.5%–87.9%), with an AUC of 93.2% (95% CI: 89.2%–97.2%) (Figures 4A and 4B). Furthermore, in both validation datasets, patients with LC had significantly higher predicted cancer scores than HCs (Figure 4C; Table S11). Importantly, our model exhibited sensitivities of 86.5% and 87.1% for tumors at very early-stage (stage I) and small-size (<1cm), respectively, pointing to its powerful detection capabilities for identifying early-stage traits. To further evaluate the stability and robustness of this model, we applied WGS data with a reduced coverage depth in an independent, small-sized validation dataset (Table S12). Upon reducing WGS coverages to 1×, we found that the predicted cancer scores of these cancer patients were higher than the cutoff value of 0.511 (Figures 4D and S4; Table S12). In conclusion, our findings suggested the superior and reliable performance of this LC detection model based on cmDNA.
Figure 4.

Independent validation of the lung cancer detection model
(A) The receiver operating characteristic curve evaluating the performance of the LC detection model in the combined validation cohort and its validation I and II cohorts separately.
(B) Sensitivities of the LC detection model are 87.7% for the combined validation cohort, 87.5% for the validation I, and 87.9% for the validation II, respectively.
(C) The boxplots showing the distribution of cancer scores in the LC and HC groups of the independent validation cohorts. The cutoff score for the independent validation I set is 0.511, and a Wilcoxon test was performed for the comparison between LC and HC subsets.
(D) The boxplots showing the distribution of cancer scores in additional shallow-coverage validation dataset with the coverage depth of 1×, compared to WGS data of 5×. Error bars represent each group’s mean ± SD.
Distinct circulating microbial profiles in postoperative recurrence patients
In view of the potential clinical application of cmDNA in LC detection, we next investigated the association between the cmDNA microbial features and recurrence in resected LC patients with clinical stage T1. It has been observed that LC patients generally experience postoperative recurrence within 3 years.26 We first established an early-stage LC recurrence cohort, including 36 patients who suffered recurrence within 3 years after surgery (R group) and 65 long-term survivors who survived >3 years without recurrence (NR group) (Figure 1). The patients in the R and NR groups were matched with respect to age, gender, cigarette smoking history, tumor diameter, and pathology (Table S13). Subsequently, we randomly split the recurrence cohort into training and test sets at a ratio of 6:4. In the training set, cmDNA was derived from plasma samples of 61 LC patients (22 R and 39 NR) (Table S13). All of the samples of this recurrence cohort were preoperative samples, acquired on the morning of patients’ surgeries.
Next, we investigated the changes of cmDNA microbial taxa at the genus and species levels that corresponded to distinct groups. At the genus level, Acinetobacter, Comamonas, Cutibacterium, and Escherichia were the top enriched genera in the training set (Figure 5A). Specifically, Comamonas and Cutibacterium were lower in the R group, whereas Acinetobacter and Escherichia were significantly more abundant (Figure S5A). Furthermore, at the species level, we identified the five most abundant species, namely Acinetobacter bereziniae, Acinetobacter johnsonii, Acinetobacter lwoffii, Comamonas testosterone, and Cutibacterium acnes, among the microbial compositions between the R and NR groups (Figure 5B). We noted that Acinetobacter johnsonii and Acinetobacter lwoffii were more prominent in the R group’s plasma specimens as compared to the NR group. In contrast, Acinetobacter bereziniae, Comamonas testosterone, and Cutibacterium acnes were significantly more abundant in the NR group (Figure S5B).
Figure 5.

Differential taxa were enriched in the R group and NR group of the 61 patients with LC
(A and B) Descriptive visual representation of top 10 microbial taxa showing the distinctive profile of microbiota between patients in the R and NR groups, at the genus level (A) and the species level (B), respectively.
(C) LEfSe identified significantly differentially abundant taxa in the R and NR groups (the top 30 according to LDA).
cmDNA signatures as a novel biomarker for recurrence of LC
We identified 23 taxa enriched in the R group and 39 taxa enriched in the NR group, which may relate to postoperative recurrence through LEfSe analysis (LDA >2, p < 0.05) (Figure 5C; Table S14). Of note, the Candidatus Nanosynbacteraceae family, the Staphylococcus genus, and the Candidatus Nanopelagicales order were the most enriched taxa in the R group, whereas the Propionibacteriales order, the Pseudomonasa zotoformans species, and the Ralstonia mannitolilytica species were the most enriched taxa in the NR group (LDA >2, p < 0.05) (Figure 5C). We then assessed whether these microbial features could discriminate between the R and NR groups. Based on taxa DNA relative abundance of each sample, the principal-component analysis (PCA) revealed that the R group could be distinguished from the NR group in the training set (Figure S5C). In addition, we performed PCA on the test set and found that the R group was still separated from the NR group (Figure S5D). No statistically different taxa were found in the possible contaminants list (Table S9).
Subsequently, we explored the feasibility of differentiating patients belonging to the R group from those in the NR group by using the significant microbial characteristics. A machine learning model was implemented and 5-fold cross-validation was performed in the training set. The 5-fold cross-validation yields a high accuracy of 85.3%, with a mean AUC value of 87.3% (Figure S5E). This model, based on cmDNA markers, exhibited a sensitivity of 72.7% and a specificity of 84.6% in the training set, with an AUC of 88.1% (95% CI, 79.7%–96.6%) (Figure 6A). In addition, in the test set, the model achieved a sensitivity of 71.3% and specificity of 84.6%, with an AUC of 80.9% (Figure 6B). We observed that the predicted recurrence scores of the R group were significantly higher than those of the NR group in the training set (p < 0.001) and the test set (p = 0.002) (Figure 6C). Furthermore, patients were categorized into high- and low-risk groups according to the median of predicted recurrence scores. The Kaplan-Meier survival curve revealed that the recurrence-free survival (RFS) in the high-risk group was significantly shorter (p < 0.001) than that of the low-risk group (Figure 6D). In addition, we displayed the top 20 crucial taxa resulting from random forest analysis, such as the Methylophilaceae family, in Figure 6E. Importantly, we found that Methylophilaceae, the top-most significant feature, was significantly correlated with RFS (p < 0.001; Figure 6F).
Figure 6.

The postoperative recurrence predicting model
(A and B) Receiver operating characteristic curve delineating the association between predictive probability and the R group in the training (A) and test (B) sets.
(C) The boxplots showing the distribution of recurrence scores in the R and NR groups of the training and test sets. The cutoff score set is 0.347, and a Wilcoxon test was performed for the comparison between the R group and the NR group.
(D) The Kaplan-Meier method with log rank test estimates the RFS for patients with higher or lower levels of recurrence model-predicting scores.
(E) Top 20 circulating microbial features prioritized by random forest analysis ranked by the mean decrease in accuracy.
(F) The Kaplan-Meier method with log rank test estimates the median RFS for patients with higher or lower abundance of the Methylophilaceae family.
As expected, RFS in the high-risk group proved to be significantly shorter in the test set (Figure S5F). Furthermore, Kaplan-Meier survival curve analysis demonstrated a significant association between Methylophilaceae and RFS between the R and NR groups (Figure S5G). Remarkably, the test set had a larger number of patients with TNM stage II or III (p = 0.002; Table S13). Therefore, we conducted univariable and multivariable analyses (Table S15), which demonstrated that the model predicting score remained an independent predictor of RFS in the multivariate Cox regression model (hazard ratio = 27.8, 95%CI: 3.6–216.5, p = 0.001) even after adjusting for TNM stage.
Discussion
Previous researchers have highlighted the significance of the tumor microbiome in both tumor development and diagnosis,7,9,16,27 providing opportunities for biomarker identification in many fields of cancer. Most studies of microbial markers, however, tend to focus either on cancer detection27,28 or treatment.13 Specifically, early-stage cancer detection methods, as previously reported by Zheng et al.27 have limited diagnostic accuracy. In our study, we aimed to improve the diagnostic sensitivity of early-stage LC and postoperative recurrence. Unlike conventional microbial investigations derived from fecal samples,27 we established a machine learning model based on cmDNA in the LC detection study, which achieved high sensitivity in discriminating between early-stage LC and noncancer subjects (86.5% sensitivity for stage I and 87.1% sensitivity for tumors <1 cm in independent validation datasets). Furthermore, a random forest classifier in the LC recurrence study exhibited high discriminatory performance between patients with or without recurrence, with AUC values of 88.1% in the training set and 80.9% in the test set, respectively.
A recent study by Poore et al. has proposed a new cancer diagnostic approach based on cmDNA through microbiome analyses of blood, demonstrating high accuracy.16 Furthermore, cmDNA is a promising tool in the diagnosis and prognosis of esophageal adenocarcinoma.18 In the present study, our results demonstrated distinct cmDNA profiles between LC patients and HCs. Consistent with previous studies,13,21,29,30 LC patients displayed lower microbiota diversity than did HCs. We additionally identified that cmDNA levels of 315 taxa were significantly altered in LC. Notably, some of these taxa, including Actinomyces31,32 and Acinetobacter,33 have been previously identified as being significantly correlated with LC development. In addition, the top significant feature, Gammaproteobacteria, enriched in the LC group, is associated with poor response to checkpoint-based immunotherapy in non-small cell lung cancer.29 Using 119 significant species, we developed a robust classifier model predicting early-stage LC. An important feature, Granulicatella adiacens,25 is observed to be significantly more abundant in LC, further substantiating our diagnostic approach. Importantly, our model outperformed the previous models27 of gut microbiome signature in distinguishing LC and noncancer subjects. Fivefold cross-validation demonstrated high accuracy, with mean AUC values of 91.5% for tumor sizes <1 cm and 94.0% for tumor sizes ≥1 cm, respectively. Notably, our model sensitively identified early pathological features in independent datasets.
Several studies have suggested that lower airway microbiota is associated with the recurrence of LC and could facilitate tumor progression.34,35 In this study, we performed a comprehensive analysis of cmDNA signatures between the R group and the NR group. Using a classifier based on cmDNA microbial features, we achieved high performance levels in discriminating between the R and NR groups. Moreover, the predictive scores of the recurrence model were significantly associated with RFS, further demonstrating its potential as a noninvasive biomarker. Notably, our cmDNA-based model provides a convenient and noninvasive detection method for LC. Although some liquid biopsy approaches, such as cell-free DNA methylation, could improve LC detection performance, their application is limited due to their high cost.36 To ensure adequate performance while reducing expenses, we used WGS data with a coverage depth of 5× for model construction. The use of the low-coverage WGS method significantly reduces the cost of our model when compared to other liquid biopsy techniques. Moreover, the performance of the model remains robust even with shallow WGS data of 1× coverage depth.
The origin of cmDNA is still a mystery, however. In this study, we found that the distribution of order-level phylotypes was similar between cmDNA and paired intratumor bacterial. Although some studies have confirmed that tumors contain intracellular bacteria,37 the relative contribution of intratumor microbes at nontumor sites is not clear, which needs to be further characterized. In addition, the composition of the microbiome of a participant is affected by factors including lifestyle and diet, as well as treatments of antibiotics. In our study, we carefully matched patient and control groups with respect to age, gender, and smoking status, particularly using larger sample sizes from multicenters of the south and the north of China to enable us to mitigate interindividual variation. Given the complexity of the microbiome, there are likely further unknown biological confounders, which should be considered in further study.
This study has some limitations that need to be addressed. First, although our model based on cmDNA showed high accuracy in LC detection, the specificity of the predictive model in validation cohorts was not sufficient for the large population screening. To mitigate this LC detection challenge, multi-omics approaches that combine multiple cfDNA signatures, including microbiome, methylation, and fragment markers, may boost sensitivity and specificity for early cancer detection. The multi-omic liquid biopsy approaches need larger sample sizes and more robust integrated analyses, which may be investigated in the future. Second, we were unable to conduct independent validations of our LC recurrence model due to our limited number of samples. Thus, further follow-up validations using larger sample sizes are necessary. Third, although our model demonstrated stability and robustness with shallow WGS data, the additional shallow-coverage dataset had a small sample size of late-stage patients. Therefore, it is necessary to validate our model further with a larger dataset that includes early-stage patients processed in the shallow-coverage test.
In summary, our study provides valuable insights into the significant alterations in cmDNA profiles in LC and HC patients, as well as the microbial signatures of patients with recurrence. We developed a highly sensitive cmDNA-based model that effectively distinguished early-stage LC patients from noncancer controls. Our findings also suggest the potential use of cmDNA as a promising biomarker for postoperative recurrence, which could significantly improve patient outcomes.
Limitations of the study
First, our study has revealed the consistent composition of cmDNA and intratumor microbiome using a small sample size. Further research necessitates a larger sample size of tumor tissues. Second, the recurrence cohort predominantly comprised lung adenocarcinoma patients. It warrants additional investigation as to whether the findings could expand to different populations. Future researchers should be aware of this factor when incorporating our data into their analyses.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| Life-MAGMAX Cell-free DNA Kit | Thermo Fisher | Cat#A29319 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher | Q32851 |
| Hieff NGS® Ultima Pro DNA Library Prep Kit | Yeasen | Cat#12201ES24 |
| FastDNA™ Spin Kit | MP Biomedicals | Cat#6560-200 |
| Deposited data | ||
| Raw data | This paper | BIG: HRA005896 |
| Custom data processing scripts | This paper | https://zenodo.org/records/10605234 |
| Software and algorithms | ||
| R version 4.2.0 | R Development Core Team | https://www.r-project.org |
| Bowtie2 (v2.3.5.1) | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml | N/A |
| SPSS Version 22 | IBM Corporation | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mantang Qiu (qiumantang@163.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive of the Beijing Institute of Genomics (BIG) Data Center, BIG, Chinese Academy of Sciences, under accession code HRA005896 and are publicly accessible at http://bigd.big.ac.cn/gsa-human. The codes to process and analyze data are publicly available at https://zenodo.org/records/10605234.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
Patients and sample information
A total of 416 participants took part in the study (Figure 1). For the LC Detection Model study, we recruited 166 participants in the training cohort, which comprised healthy control (HC, n = 97) from Aerospace 731 Hospital and previously untreated LC patients (n = 69) from Peking University People’s Hospital, China. The LC patients included adenocarcinoma (n = 51), squamous cell carcinoma (n = 12), small cell carcinoma (n = 5), and large-cell neuroendocrine carcinoma (n = 1), as detailed in Tables S1, S2, and S3. After constructing the model, we conducted two prospective independent validation cohorts using plasma samples of 149 participants. Validation cohort I had 48 HC and 48 LC participants, whereas Validation cohort II had 20 healthy participants and 33 LC participants. The healthy participants in the validation cohorts were from Jiangsu Province Geriatric Hospital, while LC patients in Validation cohort I were from The Second People’s Hospital of Shenzhen, and LC patients in Validation cohort II were from Jiangsu Province Geriatric Hospital. The LC and HC cohorts were gender and age-matched as shown in Table S3. For the LC Recurrence Model study, we enrolled 101 participants. Patients and related samples were selected from specimen repository in Peking University People’s Hospital between 2013 and 2018 according to follow criteria: 1. Clinical stage T1 lung cancer; 2. Received radical surgery; 3. Recurrence or death in 3 years. Thirty-six patients were included as recurrence group (R group) and after matching for various clinicopathologic variables, The sixty-five patients without known recurrence was chosen to the non-recurrence group (NR group). This study was approved by the ethics committee at Peking University People’s Hospital (Approval No. 2022PHB454). All participants provided written informed consent.
Method details
DNA extraction and library preparation
Whole blood samples were collected in EDTA tubes after skin surfaces were sterilized twice and processed immediately. The separation of plasma and cellular components was achieved through centrifugation at 1600g for 10 min at 4°C. Subsequently, plasma was centrifuged again at 16,000 g at 4°C to eliminate any residual cellular debris and stored at −80°C until the point of DNA extraction. Cell-free DNA was extracted from plasma using Life-MAGMAX Cell-free DNA Kit. The NGS cfDNA libraries were established for whole genome sequencing using 10 to 250 ng of cfDNA. In brief, the concentration of cfDNA was measured using the Qubit dsDNA HS Assay Kit in compliance with the manufacturer’s recommendations. Then, genomic libraries were produced using the Hieff NGS Ultima Pro DNA Library Prep Kit for Illumina. Whole genome libraries were sequenced using 100-bp paired-end runs on the DNBSEQ-T7 (Geneplus-Beijing Institute, Beijing, China).
To avoid contamination, we conducted three negative controls, wherein the complete DNA extraction and sequencing procedure were repeated with blank tubes instead of participants’ plasma samples.
Sequence data processing
FastQC was used for FASTQ file quality control. All reads from the samples were initially mapped to the hg19 reference sequence of human genome using Bowtie2 software (v2.3.5.1) with default parameters.38 To get microbial data of all plasma samples and ensure human-derived reads were removed, we applied three bioinformatic analysis steps to the aligned results. First of all, reads that aligned to human genome were removed using Samtools software with SAM-flags of “-f 12” and “-F 256”. The SAM flag of “-f 12” could extract only alignments with both paired reads unmapped to human genome and the SAM flag of “-F 256” required primary alignment to be exracted.39 Secondly, the filtered reads were then aligned to the microbial reference genome databases available in the NCBI using a k-mer-based algorithm through Kraken2.40 We used the complete genomes from NCBI, including the standard Kraken2 database of archaea, bacteria, human, Univec_Core and viral, and supplement genome database of fungi, to avoid potential contamination from draft genomes. Kraken2 hits accumulating less than 10% of K-mers matching the reference sequence were discarded and a hit was considered true positive only if at least 50 reads were aligned to the reference database. Thirdly, the taxonomy labels assigned by Kraken2 were analyzed by Bracken with a parameter of 32 k-mer distribution to estimate the species-level read abundance.41 Bracken re-estimated species abundances from the Kraken2 output results by probabilistically re-distributing reads in the taxonomic tree.
To assess the reproducibility of this workflow, we compared circulating microbial DNA profiles both the day before and the day of surgery. We processed sequencing data analysis of two patients and found that genus-level taxa were similar in the adjacent two days (Figure S6). Specifically, compared to the samples of the first day, there were 15 identical taxa in the sample of the second day in the patient LC01 (15/20) and 14 identical taxa in the patient LC02 (14/20) (Tables S16), which confirmed the robustness of our cmDNA analysis.
Differentially abundant taxa identification
For phylogenetic diversity between clinical groups, alpha diversity was computed using the R package vegan to evaluate the richness and evenness of each sample, and then compared with the Wilcoxon test. Beta diversity based on Bray-Curtis metrics was applied to compare the dissimilarities between different groups with non-metric multidimensional scaling (NMDS). In order to identify significantly differential taxa between clinical groups, we used the Wilcoxon test based on their relative abundance. Linear discriminant analysis effect size (LEfSe) was further applied to identify significantly differentially enriched taxa between clinical groups,42 with Linear Discriminant Analysis (LDA) threshold set at 2.0 and p < 0.05.
Quality filtering
To mitigate the potential contamination effect, we applied two filtering steps to the significant taxa obtained from LEfSe analysis. Firstly, we utilized the negative control samples to identify contaminant species. Specifically, we identified microbial reads and computed the relative abundance of three negative control (NC) samples. Then, we performed a threshold analysis, similar to a previous study,22 where any significant species detected in the NC samples, with a relative abundance higher than 5% in any NC sample, was considered contamination and flagged. Secondly, we curated a list of genera and species that were reported as contaminants in previous studies,16,43 especially in the circulating microbiome research on multiple cancers from Poore et al.16 We removed any significant taxa detected in the curated list.
Machine learning model
The LEfSe results identified significant features, which were utilized as inputs for the random forest analysis. The caret package (https://cran.r-project.org/web/packages/caret/) and the randomForest R package (https://cran.r-project.org/web/packages/randomForest/index.html) were employed for this purpose. In the analysis, 1000 trees were constructed using the randomForest R package (version 4.7–1.1) with 5-fold cross-validation, and the process was repeated 100 times. The pROC R package was used to generate class predictions and the receiver operating characteristics (ROC) curve.
DNA extraction and 16S rRNA gene sequencing
Microbial community genomic DNA was extracted from lung tissue samples using the FastDNA Spin Kit for Soil (MP Biomedicals, Southern California, U.S.) according to manufacturer’s instructions. The DNA extraction was checked on 1% agarose gel, and DNA concentration and purity were determined with NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, USA). The bacterial 16S rRNA genes were amplified using the universal bacterial primers 27F (5′-AGRGTTYGATYMTGGCTCAG-3′) and 1492R (5′-RGYTACCTTGTTACGACTT-3′). PCR reactions were performed in triplicate condition. After electrophoresis, PCR products were purified using AMPure PB beads (Pacifc Biosciences, CA, USA) and quantified with Quantus Fluorometer (Promega, WI, USA). All purified products were pooled in equimolar and the DNA library was constructed using the SMRTbell prep kit 3.0 (Pacifc Biosciences, CA, USA) according to PacBio’s instructions. The purified SMRTbell libraries were sequenced on the Pacbio Sequel IIe System (Pacifc Biosciences, CA, USA) by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China).
Analysis of 16S rRNA sequencing data
All PacBio raw reads were processed using the SMRTLink analysis software (version 11.0) to obtain high-quality Hifi reads with a minimum of three full passes and 99% sequence accuracy. The Hifi reads were barcode-identified and length-filtered. The sequencing reads with a length <1,000 or >1,800 bp were removed. The Hifi reads were denoised using DADA244 plugin in the Qiime2,45 with recommended parameters. Taxonomic assignment of DADA2 denoised sequences, known as amplicon sequence variants (ASVs) was performed using the Naive bayes consensus taxonomy classifier implemented in Qiime2 and the Nucleotide Sequence Database.
Quantification and statistical analysis
The Wilcoxon test was employed to compare the relative abundance at different phylogenetic levels between different clinical groups. The Kaplan-Meier method and log rank test within the R package, survminer, were used to perform univariate survival analysis of RFS between the groups. The two-sided P-values <0.05 were considered statistically significant. All statistical analyses were executed in R (version 4.2.0) and SPSS software (version 22.0; IBM Corporation Armonk, NY, USA).
Acknowledgments
We thank Hao Li and Siao Jiang from the Beijing Institute of Radiation Medicine for assistance in the data analysis. This work was supported by the National Natural Science Foundation of China (92059203, 32270714 and 82173386), the Beijing Nova Program (20230484314), the Research Unit of Intelligence Diagnosis and Treatment in Early Non-small Cell Lung Cancer (Chinese Academy of Medical Sciences, 2021RU002), the CAMS Innovation Fund for Medical Sciences (2022-I2M-C&T-B-120), the Beijing Natural Science Foundation (L222021), the Peking University People’s Hospital Research and Development Founds (RZ2022-04 and RDH2020-10), the Peking University Medicine Sailing Program for Young Scholars’ Scientific & Technological Innovation (BMU2023YFJHMX010), the Shenzhen Second People’s Hospital Clinical Research Fund of Guangdong Province High-level Hospital Construction Project (20213357024), the Science and Technology Development Fund Project of Shenzhen (JCYJ20220530150813029), and the Open Project of Jiangsu Provincial Science and Technology Resources (Clinical Resources) Coordination Service Platform (TC2022B010). The cell-free DNA WGS sequencing was performed by Geneplus-Beijing Institute (Beijing, China).
Author contributions
M.Q., Y.L., and H.W. conceived and designed this study. H.C. and Y.L. performed the WGS data analysis. H.C. performed machine learning and interpreted the results. H.C., Y.M., J.X., W.W., and F.Y. collected samples and made clinical diagnoses. C.Q. and H.L. assisted with the data analysis. H.C., Y.L., and M.Q. wrote the manuscript, with all of the authors contributing to writing and providing feedback.
Declaration of interests
The authors declare no competing interests.
Published: April 5, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101499.
Contributor Information
Yiming Lu, Email: ylu.phd@gmail.com.
Hao Wu, Email: haowudr@hotmail.com.
Mantang Qiu, Email: qiumantang@163.com.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer Statistics, 2021. CA. Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Chansky K., Detterbeck F.C., Nicholson A.G., Rusch V.W., Vallières E., Groome P., Kennedy C., Krasnik M., Peake M., Shemanski L., et al. The IASLC Lung Cancer Staging Project: External Validation of the Revision of the TNM Stage Groupings in the Eighth Edition of the TNM Classification of Lung Cancer. J. Thorac. Oncol. 2017;12:1109–1121. doi: 10.1016/j.jtho.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 3.Blandin Knight S., Crosbie P.A., Balata H., Chudziak J., Hussell T., Dive C. Progress and prospects of early detection in lung cancer. Open Biol. 2017;7 doi: 10.1098/rsob.170070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.National Lung Screening Trial Research Team. Church T.R., Black W.C., Aberle D.R., Berg C.D., Clingan K.L., Duan F., Fagerstrom R.M., Gareen I.F., Gierada D.S., et al. Results of initial low-dose computed tomographic screening for lung cancer. N. Engl. J. Med. 2013;368:1980–1991. doi: 10.1056/NEJMoa1209120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.National Lung Screening Trial Research Team. Aberle D.R., Adams A.M., Berg C.D., Black W.C., Clapp J.D., Fagerstrom R.M., Gareen I.F., Gatsonis C., Marcus P.M., Sicks J.D. Reduced lung-cancer mortality with low-dose computed tomographic screening. N. Engl. J. Med. 2011;365:395–409. doi: 10.1056/NEJMoa1102873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong Q., Chen E.S., Zhao C., Jin C. Host-Microbiome Interaction in Lung Cancer. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.679829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y., Wu F.H., Wu P.Q., Xing H.Y., Ma T. The Role of The Tumor Microbiome in Tumor Development and Its Treatment. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.935846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Binnewies M., Roberts E.W., Kersten K., Chan V., Fearon D.F., Merad M., Coussens L.M., Gabrilovich D.I., Ostrand-Rosenberg S., Hedrick C.C., et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018;24:541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin C., Lagoudas G.K., Zhao C., Bullman S., Bhutkar A., Hu B., Ameh S., Sandel D., Liang X.S., Mazzilli S., et al. Commensal Microbiota Promote Lung Cancer Development via gammadelta T Cells. Cell. 2019;176:998–1013.e16. doi: 10.1016/j.cell.2018.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong-Rolle A., Dong Q., Zhu Y., Divakar P., Hor J.L., Kedei N., Wong M., Tillo D., Conner E.A., Rajan A., et al. Spatial meta-transcriptomics reveal associations of intratumor bacteria burden with lung cancer cells showing a distinct oncogenic signature. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2022-004698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma Y., Qiu M., Wang S., Meng S., Yang F., Jiang G. Distinct tumor bacterial microbiome in lung adenocarcinomas manifested as radiological subsolid nodules. Transl. Oncol. 2021;14 doi: 10.1016/j.tranon.2021.101050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Noci V., Guglielmetti S., Arioli S., Camisaschi C., Bianchi F., Sommariva M., Storti C., Triulzi T., Castelli C., Balsari A., et al. Modulation of Pulmonary Microbiota by Antibiotic or Probiotic Aerosol Therapy: A Strategy to Promote Immunosurveillance against Lung Metastases. Cell Rep. 2018;24:3528–3538. doi: 10.1016/j.celrep.2018.08.090. [DOI] [PubMed] [Google Scholar]
- 13.Riquelme E., Zhang Y., Zhang L., Montiel M., Zoltan M., Dong W., Quesada P., Sahin I., Chandra V., San Lucas A., et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell. 2019;178:795–806.e12. doi: 10.1016/j.cell.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pandey H., Tang D.W.T., Wong S.H., Lal D. Gut Microbiota in Colorectal Cancer: Biological Role and Therapeutic Opportunities. Cancers. 2023;15 doi: 10.3390/cancers15030866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee S.H., Sung J.Y., Yong D., Chun J., Kim S.Y., Song J.H., Chung K.S., Kim E.Y., Jung J.Y., Kang Y.A., et al. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung Cancer. 2016;102:89–95. doi: 10.1016/j.lungcan.2016.10.016. [DOI] [PubMed] [Google Scholar]
- 16.Poore G.D., Kopylova E., Zhu Q., Carpenter C., Fraraccio S., Wandro S., Kosciolek T., Janssen S., Metcalf J., Song S.J., et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. 2020;579:567–574. doi: 10.1038/s41586-020-2095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao Q., Lu W., Kong X., Shao Y.W., Hu Y., Wang A., Bao H., Cao R., Liu K., Wang X., et al. Alterations of circulating bacterial DNA in colorectal cancer and adenoma: A proof-of-concept study. Cancer Lett. 2021;499:201–208. doi: 10.1016/j.canlet.2020.11.030. [DOI] [PubMed] [Google Scholar]
- 18.Zaidi A.H., Pratama M.Y., Omstead A.N., Gorbonova A., Mansoor R., Melton-Kreft R., Jobe B.A., Wagner P.L., Kelly R.J., Goel A. A blood-based circulating microbial metagenomic panel for early diagnosis and prognosis of oesophageal adenocarcinoma. Br. J. Cancer. 2022;127:2016–2024. doi: 10.1038/s41416-022-01974-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H., Ma Y., Liu Z., Li J., Li X., Yang F., Qiu M. Circulating microbiome DNA: An emerging paradigm for cancer liquid biopsy. Cancer Lett. 2021;521:82–87. doi: 10.1016/j.canlet.2021.08.036. [DOI] [PubMed] [Google Scholar]
- 20.Cho E.J., Leem S., Kim S.A., Yang J., Lee Y.B., Kim S.S., Cheong J.Y., Cho S.W., Kim J.W., Kim S.M., et al. Circulating Microbiota-Based Metagenomic Signature for Detection of Hepatocellular Carcinoma. Sci. Rep. 2019;9:7536. doi: 10.1038/s41598-019-44012-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu F., Li J., Guan Y., Lou Y., Chen H., Xu M., Deng D., Chen J., Ni B., Zhao L., et al. Dysbiosis of the Gut Microbiome is associated with Tumor Biomarkers in Lung Cancer. Int. J. Biol. Sci. 2019;15:2381–2392. doi: 10.7150/ijbs.35980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greathouse K.L., White J.R., Vargas A.J., Bliskovsky V.V., Beck J.A., von Muhlinen N., Polley E.C., Bowman E.D., Khan M.A., Robles A.I., et al. Interaction between the microbiome and TP53 in human lung cancer. Genome Biol. 2018;19:123. doi: 10.1186/s13059-018-1501-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peters B.A., Pass H.I., Burk R.D., Xue X., Goparaju C., Sollecito C.C., Grassi E., Segal L.N., Tsay J.C.J., Hayes R.B., Ahn J. The lung microbiome, peripheral gene expression, and recurrence-free survival after resection of stage II non-small cell lung cancer. Genome Med. 2022;14:121. doi: 10.1186/s13073-022-01126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayes R.B., Ahn J., Fan X., Peters B.A., Ma Y., Yang L., Agalliu I., Burk R.D., Ganly I., Purdue M.P., et al. Association of Oral Microbiome With Risk for Incident Head and Neck Squamous Cell Cancer. JAMA Oncol. 2018;4:358–365. doi: 10.1001/jamaoncol.2017.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cameron S.J.S., Lewis K.E., Huws S.A., Hegarty M.J., Lewis P.D., Pachebat J.A., Mur L.A.J. A pilot study using metagenomic sequencing of the sputum microbiome suggests potential bacterial biomarkers for lung cancer. PLoS One. 2017;12 doi: 10.1371/journal.pone.0177062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van den Berg L.L., Klinkenberg T.J., Groen H.J.M., Widder J. Patterns of Recurrence and Survival after Surgery or Stereotactic Radiotherapy for Early Stage NSCLC. J. Thorac. Oncol. 2015;10:826–831. doi: 10.1097/JTO.0000000000000483. [DOI] [PubMed] [Google Scholar]
- 27.Zheng Y., Fang Z., Xue Y., Zhang J., Zhu J., Gao R., Yao S., Ye Y., Wang S., Lin C., et al. Specific gut microbiome signature predicts the early-stage lung cancer. Gut Microb. 2020;11:1030–1042. doi: 10.1080/19490976.2020.1737487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Q., Hou K., Tang M., Ying S., Zhao X., Li G., Pan J., He X., Xia H., Li Y., et al. Screening of potential microbial markers for lung cancer using metagenomic sequencing. Cancer Med. 2023;12:7127–7139. doi: 10.1002/cam4.5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boesch M., Baty F., Albrich W.C., Flatz L., Rodriguez R., Rothschild S.I., Joerger M., Früh M., Brutsche M.H. Local tumor microbial signatures and response to checkpoint blockade in non-small cell lung cancer. OncoImmunology. 2021;10 doi: 10.1080/2162402X.2021.1988403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hosgood H.D., Cai Q., Hua X., Long J., Shi J., Wan Y., Yang Y., Abnet C., Bassig B.A., Hu W., et al. Variation in oral microbiome is associated with future risk of lung cancer among never-smokers. Thorax. 2021;76:256–263. doi: 10.1136/thoraxjnl-2020-215542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kovaleva O., Podlesnaya P., Rashidova M., Samoilova D., Petrenko A., Zborovskaya I., Mochalnikova V., Kataev V., Khlopko Y., Plotnikov A., Gratchev A. Lung Microbiome Differentially Impacts Survival of Patients with Non-Small Cell Lung Cancer Depending on Tumor Stroma Phenotype. Biomedicines. 2020;8 doi: 10.3390/biomedicines8090349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dumont-Leblond N., Veillette M., Racine C., Joubert P., Duchaine C. Development of a robust protocol for the characterization of the pulmonary microbiota. Commun. Biol. 2021;4:164. doi: 10.1038/s42003-021-01690-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomes S., Cavadas B., Ferreira J.C., Marques P.I., Monteiro C., Sucena M., Sousa C., Vaz Rodrigues L., Teixeira G., Pinto P., et al. Profiling of lung microbiota discloses differences in adenocarcinoma and squamous cell carcinoma. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-49195-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patnaik S.K., Cortes E.G., Kannisto E.D., Punnanitinont A., Dhillon S.S., Liu S., Yendamuri S. Lower airway bacterial microbiome may influence recurrence after resection of early-stage non-small cell lung cancer. J. Thorac. Cardiovasc. Surg. 2021;161:419–429.e16. doi: 10.1016/j.jtcvs.2020.01.104. [DOI] [PubMed] [Google Scholar]
- 35.Tsay J.C.J., Wu B.G., Sulaiman I., Gershner K., Schluger R., Li Y., Yie T.A., Meyn P., Olsen E., Perez L., et al. Lower Airway Dysbiosis Affects Lung Cancer Progression. Cancer Discov. 2021;11:293–307. doi: 10.1158/2159-8290.CD-20-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lo Y.M.D., Han D.S.C., Jiang P., Chiu R.W.K. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science. 2021;372 doi: 10.1126/science.aaw3616. [DOI] [PubMed] [Google Scholar]
- 37.Nejman D., Livyatan I., Fuks G., Gavert N., Zwang Y., Geller L.T., Rotter-Maskowitz A., Weiser R., Mallel G., Gigi E., et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science. 2020;368:973–980. doi: 10.1126/science.aay9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wood D.E., Lu J., Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu J., Rincon N., Wood D.E., Breitwieser F.P., Pockrandt C., Langmead B., Salzberg S.L., Steinegger M. Metagenome analysis using the Kraken software suite. Nat. Protoc. 2022;17:2815–2839. doi: 10.1038/s41596-022-00738-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woerner J., Huang Y., Hutter S., Gurnari C., Sánchez J.M.H., Wang J., Huang Y., Schnabel D., Aaby M., Xu W., et al. Circulating microbial content in myeloid malignancy patients is associated with disease subtypes and patient outcomes. Nat. Commun. 2022;13:1038. doi: 10.1038/s41467-022-28678-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Callahan B.J., McMurdie P.J., Rosen M.J., Han A.W., Johnson A.J.A., Holmes S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolyen E., Rideout J.R., Dillon M.R., Bokulich N.A., Abnet C.C., Al-Ghalith G.A., Alexander H., Alm E.J., Arumugam M., Asnicar F., et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive of the Beijing Institute of Genomics (BIG) Data Center, BIG, Chinese Academy of Sciences, under accession code HRA005896 and are publicly accessible at http://bigd.big.ac.cn/gsa-human. The codes to process and analyze data are publicly available at https://zenodo.org/records/10605234.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.