

Abstract
BACKGROUND
Impairment of the ubiquitin‐proteasome system (UPS) has been implicated in abnormal protein accumulation in Alzheimer's disease. It remains unclear if genetic variation affects the intrinsic properties of neurons that render some individuals more vulnerable to UPS impairment.
METHODS
Induced pluripotent stem cell (iPSC)‐derived neurons were generated from over 50 genetically variant and highly characterized participants of cohorts of aging. Proteomic profiling, proteasome activity assays, and Western blotting were employed to examine neurons at baseline and in response to UPS perturbation.
RESULTS
Neurons with lower basal UPS activity were more vulnerable to tau accumulation following mild UPS inhibition. Chronic reduction in proteasome activity in human neurons induced compensatory elevation of regulatory proteins involved in proteostasis and several proteasome subunits.
DISCUSSION
These findings reveal that genetic variation influences basal UPS activity in human neurons and differentially sensitizes them to external factors perturbing the UPS, leading to the accumulation of aggregation‐prone proteins such as tau.
Highlights
Polygenic risk score for AD is associated with the ubiquitin‐proteasome system (UPS) in neurons.
Basal proteasome activity correlates with aggregation‐prone protein levels in neurons.
Genetic variation affects the response to proteasome inhibition in neurons.
Neuronal proteasome perturbation induces an elevation in specific proteins involved in proteostasis.
Low basal proteasome activity leads to enhanced tau accumulation with UPS challenge.
Keywords: Aβ, Alzheimer's, autophagy, BAG3, bortezomib, HSPA6, immunoproteasome, lysosome, MAPT, polygenic risk, proteasome, proteostasis, tau, ubiquitin
1. BACKGROUND
Regulated protein homeostasis is particularly essential for postmitotic, long‐lived cells such as neurons. Human brain cells accumulate oxidative injury during aging, which can lead to widespread protein aggregation, increasing the demand for cellular protein degradation systems such as the ubiquitin‐proteasome system (UPS) and the autophagy‐lysosome pathway (ALP). 1 With more than 800 proteins involved, the UPS degrades most water‐soluble proteins in cells. 2 The proteasome comprises a multiprotein catalytic core particle (20S) and multiprotein regulatory components (19S or 11S). The process of UPS starts with an enzymatic cascade that marks proteasome substrates with ubiquitin chains. Once recognized by the proteasome, substrates are deubiquitylated by deubiquitinases, unfolded by ATPases, and translocated into the 20S chamber for hydrolysis.
Impaired UPS activity has been reported in neurodegenerative diseases, including Alzheimer's disease (AD) 3 , 4 and Parkinson's disease (PD). 5 Abnormal protein accumulation is a common pathological feature of several neurodegenerative diseases, and reductions in normal protein turnover can contribute to the aggregation and accumulation of amyloid‐β (Aβ), tau, and α‐synuclein. 6 , 7 Therefore, the accumulation of abnormal protein aggregates, including excessively phosphorylated microtubule‐associated protein tau (p‐tau) 8 , 9 , 10 and α‐synuclein, 11 , 12 is likely mediated at least in part by the UPS. Aging is the leading risk factor for neurodegeneration, 13 and studies show reduced UPS activity in aged cells 14 , 15 , 16 and organs. 17 , 18 , 19 , 20 , 21 Thus, disruption of the UPS may be an important link between aging, neurodegenerative disorders, and protein aggregation. 22 However, the molecular mechanisms underlying why some individuals develop neurodegeneration while others of the same age do not are poorly understood. There is also evidence that accumulating toxic aggregates of p‐tau, 23 Aβ, 24 , 25 , 26 and α‐synuclein 6 , 7 , 26 can disrupt proteasome function in transgenic animals and cells. Still, whether reduced UPS activity in neurons causes abnormal protein accumulation is unclear. This study provides evidence that in neurons, basal UPS activity is, in part, encoded within the genetic landscape of an individual and can influence the propensity for protein aggregation.
Human induced pluripotent stem cell (iPSC) ‐derived neurons (iNs) provide a highly controlled and reproducible model system for the study of disease biology. This system is particularly well‐suited to analyze the initiating events in the disease that are directly downstream of genetic risk variants. 27 We have generated iPSC lines from over 50 individuals in the Religious Order Study or Memory and Aging Project (ROSMAP) cohorts. 28 These cohorts are composed of Catholic nuns, priests, and monks across the United States (ROS) and older persons recruited from communities around the Chicago metropolitan area (MAP). Upon enrollment, ROSMAP participants are cognitively unaffected and are primarily over the age of 65. Participants agree to take annual physiological examinations and cognitive function tests and consent to donate their organs, including their brain, after death. 29 Brain tissue is assessed to quantify neuropathology, including Aβ plaques and tau tangles, and several layers of ‐omic profiles are acquired. 30 , 31
Studying iNs derived from these well‐characterized ROSMAP participants that span the clinical and neuropathological spectrum of aging allows for examining the biological processes affected by genetic risk for the disease and elucidating the molecular pathways contributing to risk and resilience for disease. In this study, unbiased proteomic profiling of ROSMAP iNs led us to further interrogate basal UPS activity and tau accumulation in iNs across different genetic backgrounds. These studies revealed person‐specific differences in UPS‐mediated protein homeostasis in human neurons that were accompanied by differences in the accumulation of aggregation‐prone proteins. We also confirm previous reports that accumulation of tau reduces proteasome activity 23 and additionally report that modeling chronic reduced proteasome capacity with mild UPS inhibition induces accumulation of tau, 32 , 33 suggesting a positive feedback loop in excitatory neurons that supports tau accumulation following an initiation event. Last, we show that neurons from different genetic backgrounds are differentially vulnerable to mild UPS insult, resulting in differences in tau accumulation. Taken together, we provide evidence for a genetically encoded risk of tau accumulation that impacts UPS activity in human neurons.
Research in context
Systematic review: A systematic review was performed to examine the state of knowledge regarding genetic background effects on neuronal proteostasis.
Interpretation: Our findings suggest that genetic variation influences basal ubiquitin‐proteasome system (UPS) activity in human neurons and that this genetic variation differentially sensitizes neurons to external factors that can perturb the UPS resulting in the accumulation of aggregation‐prone proteins such as tau.
Future directions: Future directions will involve identifying the genetic variants affecting UPS vulnerability which will allow for the determination of a “UPS vulnerability score.” These studies would provide a foothold for identifying subtypes of neurodegenerative disease that may be especially responsive to interventions that boost proteostasis.
2. METHODS
2.1. iPSC‐derived neurons
The iPSC from the ROSMAP cohort and the MAPTP301L carrier were generated as described previously. 28 , 34 Induced neurons were generated and cultured to Day 21 as described previously. 28 An Institutional Review Board approved ROS and MAP studies of Rush University Medical Center. All participants signed informed consent, an Anatomical Gift Act, and a repository consent to allow their data and biospecimens to be repurposed. The iPSC lines were generated following Institutional Review Board (IRB) review and approval through Partners/BWH IRB (#2015P001676), and we certify that the study was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments. See Supplemental Table S1 for details on human subjects' cognition and pathology.
2.2. Liquid chromatography‐tandem mass spectrometry proteomic analysis
For each ROSMAP iN line, 1.2 million Day 4 iNs were plated per well of a six‐well plate and were cultured for 21 days. On Day 21, iNs were washed twice with ice‐cold Dulbecco's phosphate‐buffered saline (DPBS) and flash‐frozen in dry ice before being lysed in 100 μL lysis buffer. Liquid chromatography coupled to tandem mass spectrometry (MS) and the analyses were performed as described previously. 35 Briefly, protein concentrations were determined by the bicinchoninic acid method. Tandem mass tag (TMT) labeling and high pH fractionation were performed. MS was conducted with a high‐field asymmetric waveform ion mobility spectrometry Pro equipped with Orbitrap Eclipse (Thermo Scientific). Raw data were analyzed using the Proteome Discoverer Suite (version 2.3, Thermo Scientific). MS/MS spectra were searched against the UniProtKB human proteome database. Following spectral assignment, peptides were assembled into proteins. Post‐quantification quality control procedures included normalization to a Global Internal Standard, removal of peptides with excessive missing, and imputation with the k‐nearest neighbor algorithm. A ComBat algorithm was used to remove variance induced by the cell harvest batch. The final iN proteomics data file included 94,162 peptides with 11,508 unique proteins.
2.3. Polygenic risk score calculation
Genotype data were available for 2,067 ROS/MAP subjects between two batches: nbatch1 = 1,686 genotyped using the Affymetrix GeneChip 6.0 and nbatch2 = 381 genotyped using the Illumina OmniQuad Express platform. Details of raw genotype quality control (QC) have been previously described. 36 Briefly, genotypes were first preprocessed using the TOPMed Imputation Server‐recommended data preparation pipeline (https://topmedimpute.readthedocs.io/en/latest/prepare‐your‐data.html). Following data preparation, each batch was imputed separately using the TOPMed Imputation Server (TOPMed reference r2), 37 including Eagle (v2.4) for allelic phasing and Minimac4 (v1.5.7) for imputation. The resulting imputed data were then filtered for imputation quality (removing SNPs with r2 < 0.8) and minor allele frequency (removing single nucleotide polymorphisms [SNPs] with MAF < 2.5e‐3). All tri‐allelic variants were removed before the imputed genotype data were merged and all overlapping SNPs mapped to rsIDs (dbSNP build 155). This resulted in a set of 9,329,439 high‐quality, bi‐allelic autosomal SNPs.
PRSice (v2.3.3) 38 was used to calculate an Alzheimer's disease (AD) polygenic risk score (PRS) using the largest available genome‐wide association study (GWAS) on AD and related dementias. 39 To account for patterns of linkage disequilibrium (LD), we applied a 500Kb sliding window and r2 threshold of 0.1 during variant clumping. We then set the SNP inclusion p‐value threshold to 5e‐8, including only genome‐wide significant SNPs during variant thresholding. This resulted in 126 independent SNPs that were carried forward to the PRS calculation. An individual's PRS is a weighted sum of their allelic dosage at each of the 126 selected SNPs, weighted by the corresponding effect size estimate of that SNP estimated by the AD GWAS.
2.4. Gene set enrichment analyses
Gene set enrichment analyses (GSEA) were performed using the desktop GSEA application (https://www.gsea‐msigdb.org/gsea/index.jsp). 40 For GSEA of correlations, we generated rank files using the ‐log10(p‐value)∗(r‐value) to factor both the significance as well as the magnitude and directionality of the effect. KEGG subset of canonical pathways (cp.kegg.v2022.1.Hs.symbol.gmt) was used. One thousand permutations were performed. Normalized Enrichment Scores (NES) reflect the amount of enrichment of each gene set in the top (+) or bottom (‐) of the gene rank files that were normalized to account for gene set sizes.
2.5. Proteasome activity assay in iN protein lysates
Induced neurons vehicle‐treated or treated with 5 nM bortezomib (2‐, 8‐, 24‐, or 72 h) or 50 nM ONX‐0914 (72 h) were washed with ice‐cold DPBS on Day 21, followed by homogenization in 25 mM Hepes‐KOH (pH 7.5), 5 mM MgCl2, 10% glycerol, 0.1% NP‐40, 1 mM dithiothreitol (DTT), 1 mM adenosine triphosphate (ATP), 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM NaF, followed by sonication in a water bath sonicator (B2500A‐MT, VWR) for 20 times for 10 s each, with 30 s pause in between pulses. Protein lysates were centrifuged at 10,000 × g for 10 min at 4°C. Samples normalized for protein concentration (5∼10 μg total protein) were loaded into a black‐walled 96‐well plate. Proteasome activity was monitored using Infinite 200 PRO plate reader (Tecan) with the i‐control microplate reader software (Tecan). The reaction was started by adding 20 μM Suc‐LLVY‐amc (for chymotrypsin‐like activity), or 20 μM Boc‐LRR‐amc (for trypsin‐like activity), or 40 μM Ac‐nLPnLD‐amc (for caspase‐like activity) in 50 mM Tris, 40 mM KCl, 5 mM MgCl2, 0.05 mg/mL Bovine Serum Albumin (BSA), 1 mM ATP, 1 mM DTT, 1 mM NaF at the zero‐time point. For background detection, 2 μM epoxomicin was added to the reactions with Suc‐LLVY‐amc; 100 μM MG132 was added to reactions with Boc‐LRR‐amc or Ac‐nLPnLD‐amc. The activity was assayed by measuring the fluorescence intensity for 1 h at 37°C in 1‐min intervals (excitation 380 nm; emission 460 nm).
2.6. Western blot analysis
The cell lysates were separated by 4‐12% Bis‐Tris gels (Thermo Fisher), followed by transfer to nitrocellulose membrane using Criterion blotter system (Bio‐Rad). Revert 700 total protein stain (LI‐COR) was used to quantify total protein levels. Nitrocellulose membranes were incubated with Intercept blocking buffer (LI‐COR) for 1 h at room temperature, followed by overnight incubation with mouse anti‐Ubiquitin (Ubi‐1) (1:500, Millipore), rabbit anti‐PSMB5 (1:2000, Bethyl), rabbit anti‐synaptophysin (1:10,000, Abcam), rabbit anti‐HOMER1 (1:1,000, Synaptic Systems), chicken anti‐MAP2 (1:20,000, Abcam), chicken anti‐β‐III tubulin (1:10,000, Novus Biologicals), rabbit anti‐tau (K9JA, 1:1000, Dako), mouse anti‐tau (Tau‐5, 1:1000, Invitrogen), mouse anti‐pT181‐tau (1:1000, Invitrogen), mouse anti‐pS202/T205‐tau (AT8, 1:250, Invitrogen), rabbit anti‐pS214‐tau (1:1,000, Cell Signaling), rabbit anti‐pT217‐tau (1:1000, GeneTex), mouse anti‐pS396‐tau (1:1000, Cell Signaling), rabbit anti‐pS404‐tau (1:1000, Invitrogen), mouse anti‐glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH; 1:10,000, Proteintech). Membranes were washed three times with TBST and incubated with fluorescent dye‐conjugated secondary antibodies (anti‐mouse or anti‐rabbit, 1:10,000, LI‐COR) for 1 h at room temperature. Membranes were washed three times with TBST and then two times with TBS, followed by scanning using an Odyssey Infrared Imaging System (LI‐COR).
2.7. Amyloid‐β enzyme‐linked immunosorbent assay
Quantification of Aβ in the conditioned media collected from the ROSMAP iN cultures was performed using a multiplex enzyme‐linked immunosorbent assay (ELISA) for Aβ38, Aβ40, and Aβ42 (V‐Plex Plus Aβ Panel, Mesoscale Discovery). Aβ37 was quantified as previously described. 41
2.8. Immunocytochemistry
Day 21 iPSC‐derived neurons were washed with ice‐cold PBS and fixed with 4% paraformaldehyde in PBS. Following fixation, the cells were permeabilized with 1% Triton X‐100 in donkey serum (Jackson ImmunoResearch). The cells were probed with primary antibodies: chicken anti‐β‐tubulin III (1:500, Novus Biologicals), rabbit anti‐Brn2 (1:100, Cell Signaling Technology), followed by secondary antibodies: anti‐chicken Cy3 (1:2000, Jackson ImmunoResearch), anti‐rabbit Cy5 (1:2000, Jackson ImmunoResearch). DAPI (1 μg/mL, Thermo Analysis) was used for nuclear staining. Images were acquired using an LSM710 confocal microscope (Zeiss).
2.9. Statistical analysis
Pearson correlation coefficients were used for examining the relationships between ROSMAP iN proteomics and (i) LOAD PRS, (ii) iN basal proteasome activity, and (iii) p‐tau levels. Z‐Scores were used to compare basal proteasome activity, PSMB5 (measured by WB), and pan‐ubiquitinated protein across ROSMAP iNs. Unpaired t‐tests were used to make the comparison between (i) NCI and AD, (ii) MAPT +/+ and MAPTP301L /+, (iii) DMSO and Bortezomib‐treated, and (iv) DMSO and ONX‐0914‐treated.
3. RESULTS
3.1. Analyses of neurons from over 50 individuals implicate genetically encoded proteasome vulnerability in AD
To determine the pathways and molecular processes downstream of genetic risk for late‐onset AD (LOAD) in human neurons, we performed unbiased proteomic profiling by TMT‐MS of iNs from 53 ROSMAP participants (Figure 1A, Supplemental Tables S1, S2). IPSC models capture genetic risk and resilience factors from the person from whom they were derived, and in this cohort a range of PRS for LOAD is represented (Figure 1B, Supplemental Table S1). We performed correlation analyses between proteomic profiles of iNs and LOAD PRS, followed by GSEA. Strikingly, the only KEGG (Kyoto Encyclopedia of Genes and Genomes) gene set negatively associated with LOAD PRS was “proteasome” (FDR = 0.017) (Figure 1C, Supplemental Table S3). Differential protein analysis comparing iNs from non‐cognitively impaired (NCI) individuals (n = 35) and those from AD individuals (n = 18) revealed 760 differentially expressed proteins (DEPs) defined by an adjusted p‐value < 0.05 and a logFC of > 0.25 or < −0.25 (Supplemental Table S4). Pathway analysis of DEPs revealed associations with several processes, including the downregulation of proteins involved in proteostasis (Figure 1D). Examples of proteins driving these associations include proteasome components, ubiquitin ligases, and deubiquitinases (Figure 1E,F). In line with these findings, pseudobulk RNAseq data 42 , 43 from human brain comparing AD and NCI reveals that proteasome components are downregulated in excitatory neurons in the AD brain (Figure 1G).
FIGURE 1.
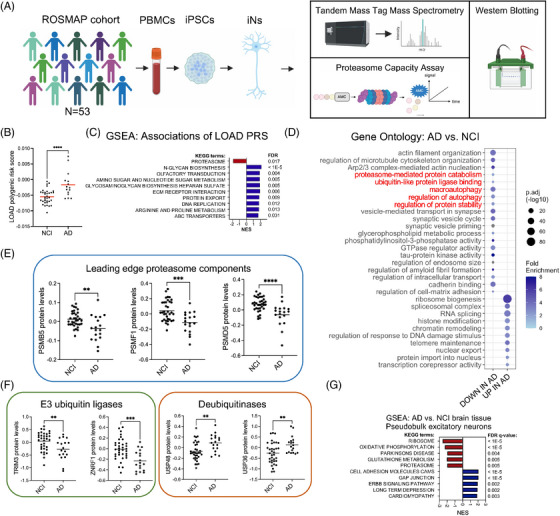
Genetically encoded proteasome vulnerability in Alzheimer's disease (AD) implicated in the analyses of induced pluripotent stem cell (iPSC) ‐derived neurons from over 50 individuals. (A) Overview of the study. (B) Polygenic risk scores (PRS) for late‐onset AD (LOAD) (n = 18) and not cognitively impaired (NCI) (n = 35) individuals in the ROSMAP (Religious Order Study or Memory and Aging Project) cohort examined in this study. Red bars represent the mean. Statistical analysis used unpaired t‐tests. (C) Gene set enrichment analysis (GSEA) using the KEGG (Kyoto Encyclopedia of Genes and Genomes) database of proteins correlated with LOAD PRS (by Pearson correlation analysis) in the ROSMAP iPSC‐derived neurons (iNs). (D) Gene ontology analysis of differentially expressed proteins in AD versus not cognitively impaired (NCI) individuals; adjusted p‐value < 0.05. Terms relating to protein turnover are highlighted in red. (E‐F) Protein expression of PSMB5, PSMF1, PSMD5, TRIM3, ZNRF1, USP48, and USP36 in NCI (n = 35) or AD (n = 18) individuals. Black bars represent the mean. Statistical analysis used unpaired t‐tests. (G) Single nucleus (sn)RNAseq data available on the AMP‐AD Knowledge Portal from human brain tissue (DLPFC [dorsolateral prefrontal cortex], BA9) from 131 AD and 162 NCI ROSMAP participants was used to compare pseudobulk RNAseq data between AD and NCI excitatory neurons. Shown are the results of GSEA of genes up and downregulated in AD versus NCI excitatory neurons, graphing only the top 5 terms in each direction. For comparisons in B, E, F: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
3.2. Genetic variation influences basal proteasome activity in human neurons
A key advantage of the iPSC system is that it captures human genetic variation in a well‐controlled reductionist system, allowing for the study of biological domains impacted by known and unknown genetic risk and resilience variants. 44 We, therefore, sought to determine whether neurons from these varied genetic backgrounds display differences in proteasome activity as suggested by our unbiased‐omics analysis (Figure 2A). To determine if changes in protein levels of UPS components result in differential proteasome activity, we performed in vitro proteasome activity assays following cell lysis using the fluorogenic substrate Suc‐LLVY‐amc, which fluoresces when cleaved and detects chymotrypsin‐like activity in protein lysates. To assess the proportion of cleavage due to the proteasome, parallel protein lysates receiving no treatment were compared to those receiving epoxomicin, a proteasome inhibitor targeting PSMB5 (the subunit responsible for chymotrypsin‐like activity in the 20S proteasome). Of note, chymotrypsin‐like activity is the rate‐limiting catalytic site in proteasomal proteolysis. 45 Across ROSMAP iNs, a spectrum of basal proteasome activity was observed (Figure 2B, Supplemental Table S5). Chymotrypsin‐like activity significantly correlated to PSMB5 protein levels, supporting the validity of the LLVY cleavage assay (Figure 2C). The NGN2 direct induction protocol generates cultures with > 95% of cells expressing neuronal markers. 28 Immunocytochemical staining of neurons (Figure 2D), proteomic profiles, and Western blotting of neuronal protein lysates (Supplemental Figure 1) provide evidence that the differences in proteasome activity observed are not likely to be due to differences in neuronal differentiation efficiency. Rather, analyses of the correlation between TMT‐MS profiles and proteasome activity revealed that the top three associated pathways were “oxidative phosphorylation,” “Parkinson's disease,” and “Alzheimer's disease,” each of which was elevated with lower proteasome activity (Figure 2E, Supplemental Tables S6, S7). Each of the leading edge proteins driving the negative associations in Figure 2E is shown in the Venn diagram in Figure 2F. This Venn diagram reveals the highly interconnected nature of these KEGG terms with regard to the specific proteins elevated with low proteasome activity (Figure 2F).
FIGURE 2.
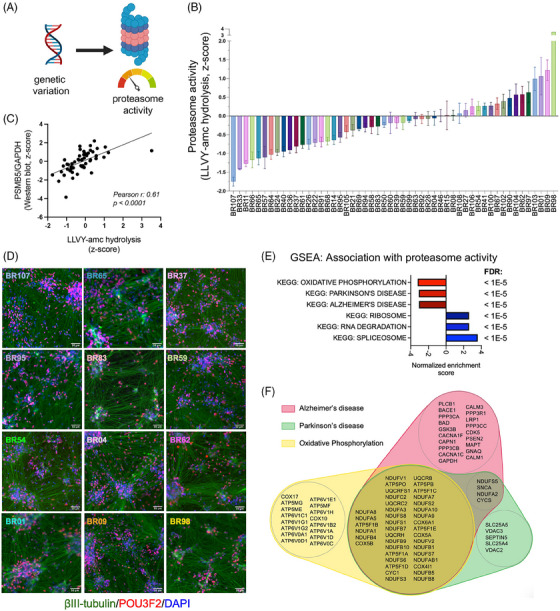
Genetic variation influences basal proteasome activity in human neurons. (A) A schematic illustrates varied genetic backgrounds mediating differences in proteasome activity. (B) Waterfall plot of a spectrum of basal proteasome activity measured by LLVY‐amc hydrolysis assay in protein lysates from the ROSMAP (Religious Order Study or Memory and Aging Project) induced pluripotent stem cell‐derived neurons (iNs). Error bars denote mean ± SEM. (C) Scatter plot of basal proteasome activity and PSMB5 protein expression, as measured by Western blot, in iN protein lysates. (D) Representative immunofluorescence staining of β‐tubulin III (green), POU3F2 (POU domain, class3, transcription factor 2) (red), and DAPI (blue) in the ROSMAP iNs. Lines are ordered from lowest to highest proteasome activity. Scale bar = 50 μm. (E) Gene set enrichment analysis (GSEA) of proteins correlated with proteasome activity (by Pearson correlation analysis). Top associations that are positively and negatively associated are shown. (F) A Venn diagram of the leading‐edge genes from GSEA in (E) driving the associations with Alzheimer's disease (red), Parkinson's disease (green), and oxidative phosphorylation (yellow).
3.3. Basal proteasome activity correlates with the level of aggregation‐prone proteins in iNs
The accumulation of aggregation‐prone proteins is a common feature of several neurodegenerative disorders, including AD, PD, frontotemporal dementia (FTD), other tauopathies, amyotrophic lateral sclerosis (ALS), and Huntington's disease (HD). Here, we examined the relationship between basal proteasome activity and expression levels of aggregation‐prone proteins relevant to these diseases, including amyloid‐beta (Aβ), microtubule‐associated protein tau (MAPT), α‐synuclein (SNCA), superoxide dismutase [Cu‐Zn] (SOD1), huntingtin (HTT), prion protein (PRNP), RNA‐binding protein FUS (FUS), TAR DNA‐binding protein 43 (TARDBP), and progranulin (GRN) in iNs. Proteasome activity was significantly associated with protein levels of several of these genes across ROSMAP iNs (Figure 3A,B). As proteasome activity increases, APP and Aβ, MAPT, SNCA, PRNP, and SOD1 are reduced (Figures 3B–E, Supplemental Figure 2). Intriguingly, the opposite is true for the RNA‐binding proteins TDP‐43 and FUS, which are elevated with higher proteasome activity (Figure 3B, Supplemental Figure 2). Examination of a correlation matrix of these proteins reveals strong associations among these different aggregation‐prone proteins (Supplemental Figure 2). While separate from the focus of this study, future experiments are warranted to determine whether there is a causal relationship between levels of these proteins in human neurons (for example, is there a causal link between levels of tau and TDP‐43 in this set of iNs similar to what is observed in experimental models of TARDBP mutations).
FIGURE 3.
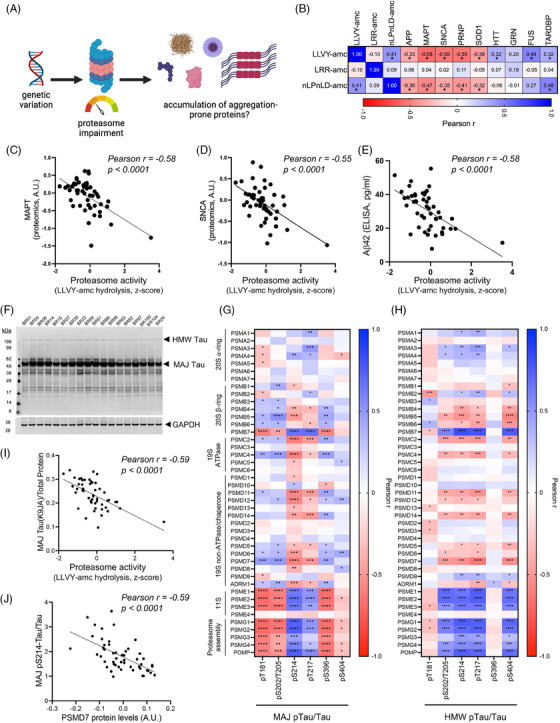
Correlations between basal proteasome activity and the level of aggregation‐prone proteins in the ROSMAP (Religious Order Study or Memory and Aging Project) induced pluripotent stem cell‐derived neurons (iNs). (A) A schematic illustrates the proposed relationship between genetic variation, basal proteasome activity, and expression levels of the aggregation‐prone proteins. (B) A heatmap of Pearson correlation coefficients with proteasome activity and levels of aggregation‐prone proteins, including APP (amyloid precursor protein), MAPT (microtubule‐associated protein tau, all isoforms), SNCA (α‐synuclein), SOD1 (superoxide dismutase [Cu‐Zn]), HTT (huntingtin), PRNP (prion protein), FUS (RNA‐binding protein FUS), TARDBP (TAR DNA‐binding protein 43), and GRN (progranulin) across ROSMAP iNs. (C‐D) Scatter plots of basal proteasome activities (LLVY‐amc hydrolysis assay) and expressions of (C) MAPT and (D) SNCA in iN protein lysates. (E) Scatter plot of basal proteasome activities (LLVY‐amc hydrolysis assay) in iN protein lysate and Aβ42 level in conditioned media. (F) Example Western blot for the major forms of 50‐60 kDa (MAJ) tau and the high molecular weight (HMW) form of aggregated tau recognized by the K9JA antibody. GAPDH (glyceraldehyde‐3‐phosphate dehydrogenase) serves as loading control. See also Supplemental Figure 3). (G‐H) Heatmaps of correlations between proteasome components and different phospho‐tau epitopes in the major forms (G) or the HMW form (H). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (I) Scatter plot of proteasome activities (LLVY‐amc hydrolysis assay) and the major forms of tau in iN protein lysate. (J) Scatter plot of PSMD7 protein level and the major form of pS214‐tau in iN protein lysate.
Tau levels showed the strongest association with proteasome activity compared to each of the other aggregation‐prone proteins analyzed by TMT‐MS (Figure 3B). Posttranslational regulation of tau is complex, and different modifications of tau affect its function and aggregation state. Thus, we next performed a deeper analysis of tau to look at both the major forms of 50‐60 kDa (MAJ) tau, as well as the high molecular weight (HMW) form of aggregated tau previously reported by us 28 and others. 46 In these Western blot analyses, we also examined different phospho‐epitopes to determine whether distinct proteoforms of tau are differentially associated with particular proteasome components (Figures 3F–J, see Supplemental Figure 3 for representative Western blots for each epitope, Supplemental Table S8 for all quantifications). Consistent with TMT‐MS data, levels of the major form of total tau (measured with antibody K9JA) are reduced with increasing proteasome activity (Figure 3I). However, distinct associations are observed when specific phospho‐epitopes are quantified, and differences are also observed when examining MAJ versus HMW forms of tau (Figure 3G,H). Intriguingly, levels of different phospho‐epitopes of tau MAJ are differentially associated with proteasome subunit levels (Figure 3G). For example, levels of pS214 tau are strongly and inversely associated with several subunits of the 19S ATPases and chaperones (Figure 3G,J) but positively associated with 11S immunoproteasome subunits and proteasome assembly proteins (Figure 3G). Other phospho‐tau epitopes, such as pT181 and pS202/T205, show positive correlations with various core proteasome components and negative associations with 11S and proteasome assembly proteins (Figure 3G).
Phosphorylated HMW‐tau forms that associate with cognition 28 also significantly and negatively correlate with the expression of multiple proteasome subunits, including the core catalytic subunits in the 20S proteasome (PSMB5 and PSMB6), the 19S ATPases (PSMC2 and PSMC4), and the deubiquitinases (PSMD7 and PSMD14) in the 19S proteasome (Figure 3H). The expression of proteasome assembly chaperone proteins (PSMG1, PSMG2, PSMG3, PSMG4, and POMP) positively correlates to the level of several phosphor‐epitopes on HMW‐tau (Figure 3H). Additionally, expressions of subunits in the 11S immunoproteasome (PSME1, PSME2, and PSME3) strongly positively correlate to phosphorylated HMW‐tau (Figure 3H).
3.4. Accumulation of phosphorylated tau can induce a reduction in proteasome activity
Prior studies have demonstrated the causal role of p‐tau accumulation in proteasome impairment in tau transgenic animals 23 , 47 (Figure 4A). Studies of iPSC‐derived neurons harboring FTD‐associated MAPT mutations previously showed an accumulation of p‐tau and dysregulation of the proteostasis network, including proteasome activity 48 and lysosomal function. 49 , 50 Here, we validated these findings using our iN system. In human iNs with mutant MAPT (MAPTP301L/+), 34 we observed increased p‐tau (Figure 4B–D). In addition, we found that MAPT mutant neurons had reduced proteasome activity and a reduction in PSMB5 levels (Figure 4E,F). Interestingly, TMT‐MS analyses revealed reduced levels of additional proteasome subunits (Figure 4G, Supplemental Tables S9, S10), supporting prior findings that MAPT mutations lead to tau accumulation and proteasome disruption. Interestingly, gene sets involved in neuronal activity (KEGG long‐term potentiation, KEGG long‐term depression) are positively associated with MAPT mutation (Supplemental Table S11), supporting the prior finding that proteasome inhibition prevents the turnover of synaptic proteins that mediate synaptic activity. 51
FIGURE 4.
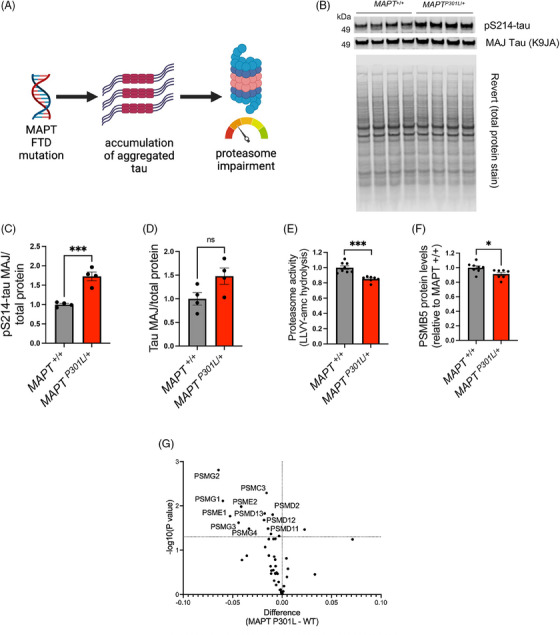
Reduced proteasome activity in MAPT (microtubule‐associated protein tau, all isoforms) mutant induced pluripotent stem cell‐derived neurons (iNs). (A) A schematic illustrates the proposed causal role of p‐tau accumulation in proteasome impairment that has been proposed based on tau transgenic animal studies. (B‐D) Western blot and quantification of pS214‐tau and the major (MAJ) form of tau in MAPTP301L /+ iNs and isogenic control iNs (MAPT +/+). The total protein level was measured by REVERT stain. Error bars denote mean ± SEM. Statistical analysis used unpaired t‐tests; N = 4. (E‐F) Basal proteasome activities (Suc‐LLVY‐amc hydrolysis) and PSMB5 levels in MAPTP301L /+ and the isogenic control (MAPT +/+) iNs. Error bars denote mean ± SEM. Statistical analysis used unpaired t‐tests; N = 7 ∼ 8. (G) Volcano plot of the expression of proteasome subunits in MAPTP301L /+ and the isogenic control (WT) iNs. Statistical analysis used unpaired t‐tests. N = 3 ∼ 4. For all quantifications: *p < 0.05, ***p < 0.001. ns, not significant.
3.5. Establishing a neuronal system to model chronic, mild reductions in proteasome activity
Having observed that MAPT mutations can induce accumulation of tau which in turn leads to proteasome dysfunction, we next interrogated whether reductions in proteasome activity in human neurons would induce accumulation of tau. To this end, we aimed to identify conditions that could model a chronic, partial reduction in proteasome activity. We chose two proteasome inhibitors to examine: bortezomib, an Food and Drug Administration (FDA)‐approved cancer treatment that binds reversibly to PSMB5, and ONX‐0914, a selective inhibitor of the chymotrypsin‐like subunit of the immunoproteasome (PSMB8) (Figure 5A). To explore all protein‐level changes in human neurons with mild proteasome inhibition, we first examined the proteomic profile of iNs treated with 5 nM bortezomib for 72 h (Supplemental Table S12). 5 nM was chosen based on findings that bortezomib inhibits proteasome‐mediated intracellular proteolysis of long‐lived proteins with a concentration of ∼0.1 μM that inhibits 50% of the proteolysis and has been shown to kill cells at 24 and 48 h with IC50s of 100 and 20 nM, respectively. 52 At 72 h, neuronal morphology remained grossly intact (Supplemental Figure 4A), and the level of apoptosis regulators Bax and p53 were unaltered (Supplemental Figure 4B), while proteasome activity was reduced by 75% with an accumulation of ubiquitinated proteins (Figure 5B,C, Supplemental Figure 4C). Using a cutoff of qval < 0.01 and absolute value of logFC > 0.2, 1,361 proteins were differentially expressed with bortezomib treatment for 72 h (Supplemental Table S13). Multiple proteasome subunits, including PSMC4, PSMC5, and PSMD1, are significantly increased with bortezomib, suggesting a compensatory response to reduced proteasome activity in neurons. Interestingly, however, the expression of nuclear factor erythroid‐derived 2‐related factor 1 (NRF1), a well‐established master transcription factor regulating the expression of proteasome subunits, 53 was decreased by 10% in bortezomib‐treated iNs compared to control after 72 h of low‐dose proteasome inhibition (Supplemental Table S13). Several deubiquitinases (DUBs), including PSMD7 and PSMD14 that form the minimal DUB‐competent complex in the proteasome, 54 , 55 are elevated in bortezomib‐treated iNs (Figure 5D). Additionally, multiple ubiquitin‐conjugating enzymes, including UBE2J1, whose expression in iNs inversely correlates to LOAD PRS (Supplemental Table S3), are upregulated in bortezomib‐treated iNs (Supplemental Table S13). The increase of proteins that regulate ubiquitination indicates a neuronal response to the global accumulation of ubiquitinated proteins during proteasome inhibition. Intriguingly, HSP70 co‐chaperone BCL‐2‐associated athanogene 3 (BAG3), a necessary component of chaperone‐assisted selective macroautophagy, 56 is among the top 5 proteins significantly upregulated in bortezomib‐treated neurons compared to control (Figures 5D), supporting prior findings in other cellular models. 57 , 58
FIGURE 5.
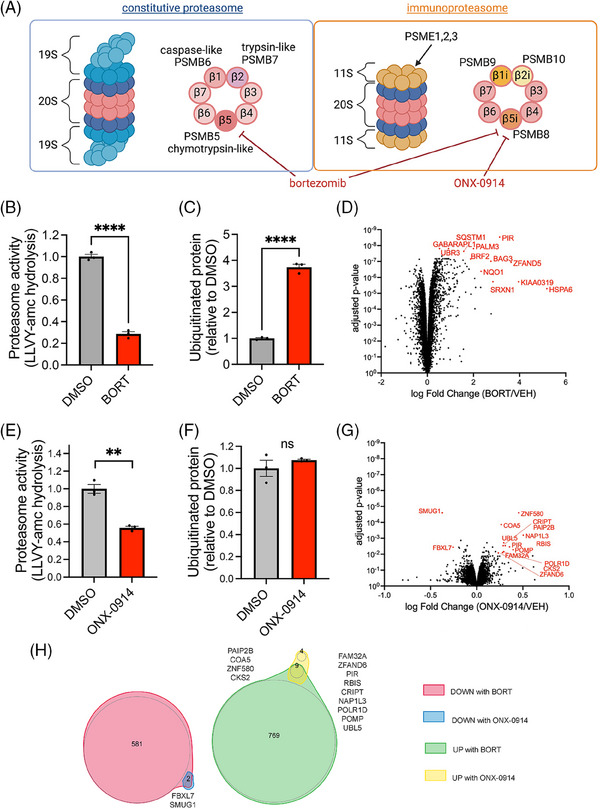
A neuronal model for extended, partial reduction in proteasome activity. (A) Schematics illustrate the targets of bortezomib and ONX‐0914 in constitutive proteasomes and immunoproteasomes. (B‐C) Proteasome activities (Suc‐LLVY‐amc hydrolysis) and ubiquitinated protein levels in the ROSMAP (Religious Order Study or Memory and Aging Project) induced pluripotent stem cell‐derived neurons (iNs) treated with 5 nM bortezomib or vehicle control (DMSO). Statistical analysis used unpaired t‐tests, N = 4. (D) Volcano plot of the fold change of proteins in the ROSMAP iNs treated with 5 nM bortezomib or vehicle control (DMSO). Statistical analysis used unpaired t‐tests. N = 4. (E‐F) Proteasome activities (Suc‐LLVY‐amc hydrolysis) and ubiquitinated protein levels in the ROSMAP iNs treated with 50 nM ONX‐0914 or vehicle control (DMSO). Statistical analysis used unpaired t‐tests, N = 3. (G) Volcano plot of the fold change of proteins in iNs treated with 50 nM ONX‐0914 or vehicle control (DMSO). Statistical analysis used unpaired t‐tests. N = 3. (H) Venn diagrams of DEPs in iNs treated with 5 nM bortezomib or 50 nM ONX‐0914. For quantifications in B, C, E, F: ns, not significant. **p < 0.01, ****p < 0.0001.
PSME1, PSME2, and PSME3 in the 11S immunoproteasome strongly correlate to the levels of p‐tau in iNs (Figure 3G,H). To understand the impact of immunoproteasome inhibition on proteomic profile in human neurons, we treated iNs with 50 nM ONX‐0914, an immunoproteasome‐specific inhibitor, 59 for 72 h. ONX‐0914 reduced LLVY‐amc hydrolysis in iN protein lysates by 50% (Figure 5E). Surprisingly, levels of pan‐ubiquitinated protein were not significantly increased (Figure 5F), and only 15 proteins were significantly differentially expressed in ONX‐0914‐treated iNs (Figure 4G, qval < 0.01 and absolute value of logFC > 0.2). Several of these 15 DEPs overlapped with DEPs from the bortezomib analysis, including POMP, PIR, and UBL5 (Figure 5H).
3.6. Genetic variation affects differential vulnerability to proteasome perturbation
To understand the consequence of the extended reduction of proteasome activity on p‐tau homeostasis, we treated a set of iNs from eight genetic backgrounds that span the spectrum of proteasome activity with bortezomib at a low dose (5 nM) (Figure 6A,B). Proteasome activity in iNs was significantly reduced by 40% after 2 h of the treatment, regardless of basal proteasome activity, and it continued to decrease to approximately 25% of basal levels by 72 h (Figure 6C). Ubiquitinated protein began to accumulate after only 2 h of the treatment (Figure 6D, Supplemental Figure 4C). HMW‐pS214‐tau began to accumulate between 8 and 24 h of proteasome inhibition (Figure 6E), while HMW‐pS202/T205‐tau began to accumulate after 24 h of proteasome inhibition (Figure 6F). Accumulation of HMW‐pS214‐tau before HMW‐pS202/T205‐tau supports the previously proposed hypothesis that tau phosphorylation at S214 is a priming event for further phosphorylation of tau at S202/T205 and other phosphorylation sites commonly found in aggregated tau. 60 Intriguingly, iNs with lower basal proteasome activity (from individuals BR107 and BR33) showed a greater accumulation of HMW‐p‐tau than those with higher basal proteasome activity (from individuals BR09 and BR98) (Figure 6E,F). These results indicate that iNs with lower basal proteasome activity are more vulnerable to HMW‐p‐tau accumulation mediated by proteasome perturbation.
FIGURE 6.
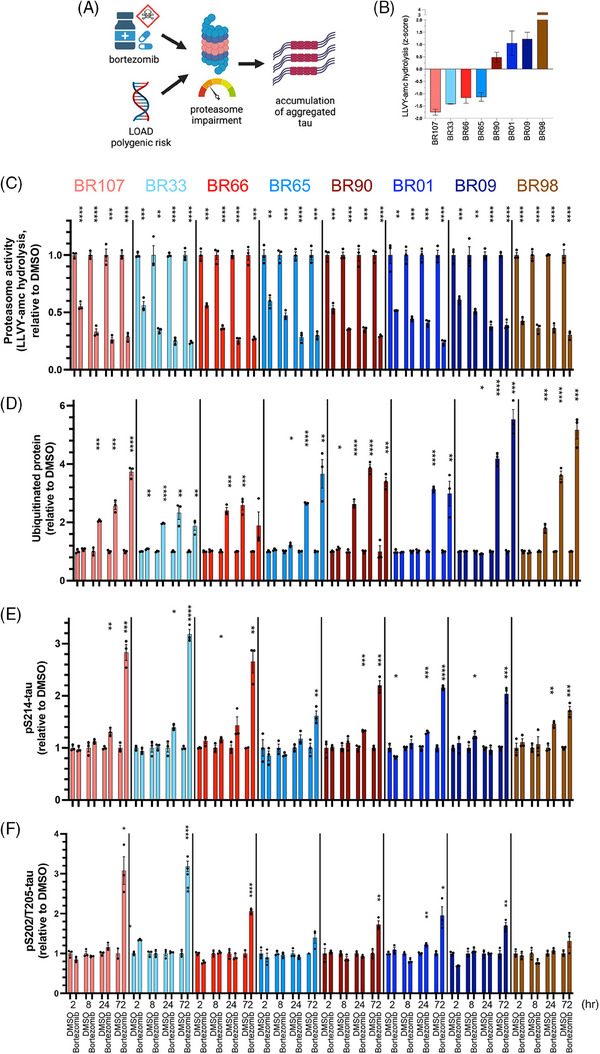
Differential responsiveness of neurons in different genetic backgrounds to the proteasome inhibitor bortezomib. (A) A schematic of the causality between genetically or pharmacologically induced proteasome impairment and p‐tau accumulation. (B) Bar graph of relative basal proteasome activities (Suc‐LLVY‐amc hydrolysis) in ROSMAP (Religious Order Study or Memory and Aging Project) induced pluripotent stem cell‐derived neurons (iNs) from eight individuals. (C‐F) Bar graph of (C) proteasome activities (Suc‐LLVY‐amc hydrolysis), (D) pan‐ubiquitinated protein, (E) HMW (high molecular weight)‐pS214‐tau, and (F) HMW‐pS202/T205‐tau in eight ROSMAP iNs treated with 5 nM bortezomib or vehicle control (DMSO) for 2‐, 8‐, 24‐, or 72 h. Error bars denote mean ± SEM. Statistical analysis used unpaired t‐tests. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. N = 3.
Treatment with immunoproteasome‐specific ONX‐0914 had only minor or no effects on tau accumulation, even though proteasome activity was reduced by approximately 50% (Supplemental Figure 5). Thus, while p‐tau levels are highly associated with immunoproteasome components (Figure 3G,H), the reduction of immunoproteasome activity does not dramatically affect p‐tau levels.
4. DISCUSSION
This study reveals person‐specific differences in UPS‐mediated proteostasis in human neurons by integrating proteomic profiles, proteasome activity assay, and p‐tau characterization in ROSMAP iNs. First, we identify the ubiquitin‐proteasome system as a molecular pathway in AD. In ROSMAP iNs with a spectrum of basal proteasome activity, reduced proteasome components are associated with high LOAD PRS and tau accumulation. Second, we find that basal proteasome activity correlates with the level of aggregation‐prone proteins in iNs. Third, we reveal the differential responsiveness of neurons in different genetic backgrounds to the proteasome inhibitor bortezomib. Last, we show that proteasome perturbation significantly increases specific proteins involved in proteostasis in iNs. Overall, our data support the hypothesis that individuals with low basal proteasome activity are more susceptible to proteasome perturbation and prone to protein accumulation (schematized in Figure 7). Our datasets also implicate the crucial components involved in the UPS that may mediate differential susceptibility to abnormal protein accumulation in human neurons.
FIGURE 7.
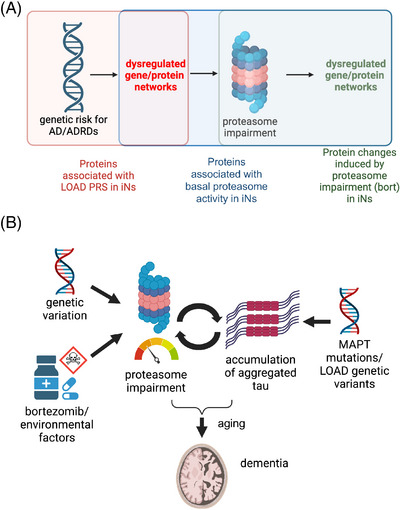
Candidate proteins that may mediate the susceptibility of neurons to abnormal protein accumulation. (A) A schematic outlining the datasets used to identify candidate proteins that may mediate the susceptibility of neurons to abnormal protein accumulation. See also Supplemental Tables S3, S6, and S13. (B) A model outlining the hypotheses that individuals with low basal proteasome activity are more susceptible to proteasome perturbation and prone to protein accumulation.
The data presented herein can be utilized to identify candidate proteins that may mediate the vulnerability of neurons to abnormal protein accumulation by comparing proteins in iNs that are (i) associated with LOAD PRS, (ii) correlated with basal proteasome activity, and (iii) differentially expressed under mild proteasome inhibition (Figure 7A,B, Supplemental Tables). Multiple proteins in the UPS, including RNF145, ING3, and NBEAL1, are associated with both LOAD PRS and basal proteasome activity. Numerous proteins in the UPS that correlate with basal proteasome activity in iNs are also differentially expressed in iNs with proteasome perturbation (Supplemental Table S14), including multiple proteasome subunits and DUBs. AMFR (an E3 ligase), DNAJB14 (a heat shock protein), SEC24D (involved in vesicle trafficking), SRPRB (a transmembrane GTPase on the ER membrane), and VSTM2L (involved in negative regulation of neuron apoptosis) each are differentially expressed in iNs with proteasome inhibition. Interestingly, their protein expression also correlates with both LOAD PRS and basal proteasome activity, suggesting their critical roles in mediating the susceptibility of neurons to abnormal protein accumulation.
4.1. Proteostasis in human neurons in the early stage of the disease
Proteostasis is especially crucial in post‐mitotic, long‐lived neurons, and the function of UPS and ALP, the two major pathways maintaining proteostasis in cells, plays a vital role in neuronal health. Much younger than neurons in aged brains, ROSMAP iNs studied here provide an experimental system for understanding cellular pathways mediating neuropathogenesis directly downstream of genetic risk factors. Here we show an association of LOAD genetic risk with reduced expression of proteins involved in proteostasis in human iNs (Figure 1), indicating the genetic contribution to dysfunctional proteostasis pathways in LOAD. Several top hits involved in proteostasis pathways are linked to neurodegeneration. NBR1 (“neighbor of BRCA1 gene”) is an autophagy receptor that accumulates in Lewy bodies. 61 UBE2J1, a ubiquitin‐conjugating enzyme, is differentially expressed in AD. 62 Additionally, GSEA suggests that the UPS is a significantly altered pathway in AD iNs, emphasizing the essential role of efficient proteasome‐mediated protein turnover in AD. These results indicate that at least a subset of individuals who developed LOAD carried genetic risk factors associated with lower expression of proteins involved in proteostasis. More studies are warranted to identify the genetic drivers for the downregulation of proteins involved in UPS.
4.2. The impact of proteasome inhibition on neuronal proteomic profiles
In iNs treated with low‐dose bortezomib for 72 h, we observe a dramatic increase in levels of multiple proteins associated with the cellular stress response, including HSPA6 and BAG3, whose elevated expression was also reported in aging human tissues. 63 , 64 HSPA6, a not well‐studied member of the heat shock 70‐kDa proteins, is robustly induced and colocalized with cytoplasmic protein aggregates in human neurons under thermal stress or treated with MG132, a proteasome inhibitor. 65 , 66 HSPA6 is the most highly elevated protein in bortezomib‐treated neurons (Figure 5D) and is not present in the mouse or rat genome, underscoring the advantage of a controlled human model system for studying processes of complex neurobiological regulation. BAG3 regulates multiple cellular pathways and can mediate an increase in autophagic flux. 56 Further, BAG3 has been identified as a master regulator in tau homeostasis. 67 Its upregulation has been shown to reduce tau accumulation in primary neurons, while its downregulation exacerbated this effect. 67 , 68 Highly elevated BAG3 levels in bortezomib‐treated neurons suggest that neurons are responding to partial proteasome inhibition by enhancing the ALP, supporting cooperative crosstalk between the UPS and the ALP in human neurons. 69 Additionally, in the bortezomib‐treated neurons, highly elevated ZFAND5, a proteasome activator that binds ubiquitin conjugates and the proteasome, 70 , 71 indicates a compensatory effect mediated by the components within the UPS. Interestingly, in both bortezomib‐ and ONX‐0914‐treated neurons, an elevation in PIR (pirin, a nuclear redox sensor 72 ) protein levels was observed, suggesting an elevation of oxidative stress induced by proteasome inhibition. The increased level of POMP (proteasome maturation protein), an essential factor facilitating the formation of the 20S proteasome, 73 indicates a neuronal response to offset proteasome inhibition. In summary, this study aids in the identification of crucial elements in the proteostasis network (Figure 7B) and also enhances our understanding of the cellular response to the perturbation of proteostasis in human neurons.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
CONSENT STATEMENT
Informed consent was obtained by all human participants with proper IRB approvals.
DIVERSITY, EQUITY, AND INCLUSION STATEMENT
This study analyzed cells derived from deceased human subjects in the ROS and MAP cohorts. In these cohorts, race is self‐declared and ethnicity is determined using genetic data. These data are provided in supplemental tables. Ethnicity and race distribution in this study match the overall statistics for the cohort. Ongoing studies are expanding the iPSC cohort to purposefully expand diversity in the iPSC lines available for study.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We thank the NeuroTechonology Studio at Brigham and Women's Hospital for providing Zeiss LSM710 Confocal instrument access and consultation on data acquisition and data analysis; iPSC NeuroHub at Brigham and Women's Hospital for technical assistance with iN differentiations; Alfred Goldberg and his lab members at Harvard Medical School for sharing techniques and insights on characterizations of the UPS; members of the Young‐Pearse lab and Dennis Selkoe for their constructive input. The results published here are in whole or in part based on data obtained from the AD Knowledge Portal https://adknowledgeportal.synapse.org/. This work was supported by an Alzheimer's Association Research Fellowship AARF‐21‐720471 and NIH grants R01AG055909, U01AG072572, U01AG061356, RF1NS117446.
Hsieh Y‐C, Augur ZM, Arbery M, et al. Person‐specific differences in ubiquitin‐proteasome mediated proteostasis in human neurons. Alzheimer's Dement. 2024;20:2952–2967. 10.1002/alz.13680
REFERENCES
- 1. Reichmann D, Voth W, Jakob U. Maintaining a healthy proteome during oxidative stress. Mol Cell. 2018;69(2):203‐213. doi: 10.1016/j.molcel.2017.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Collins GA, Goldberg AL. The logic of the 26s proteasome. Cell. 2017;169(5):792‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Keck S, Nitsch R, Grune T, Ullrich O. Proteasome inhibition by paired helical filament‐tau in brains of patients with Alzheimer's disease. J Neurochem. 2003;85(1):115‐122. doi: 10.1046/j.1471-4159.2003.01642.x [DOI] [PubMed] [Google Scholar]
- 4. Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem. 2001;75(1):436‐439. doi: 10.1046/j.1471-4159.2000.0750436.x [DOI] [PubMed] [Google Scholar]
- 5. McNaught KSP, Jenner P. Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci Lett. 2001;297(3):191‐194. doi: 10.1016/S0304-3940(00)01701-8 [DOI] [PubMed] [Google Scholar]
- 6. Lindersson E, Beedholm R, Højrup P, et al. Proteasomal inhibition by α‐synuclein filaments and oligomers. J Biol Chem. 2004;279(13):12924‐12934. doi: 10.1074/jbc.M306390200 [DOI] [PubMed] [Google Scholar]
- 7. Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. Aggregated and monomeric α‐synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278(14):11753‐11759. doi: 10.1074/jbc.M208641200 [DOI] [PubMed] [Google Scholar]
- 8. Petrucelli L, Dickson D, Kehoe K, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13(7):703‐714. doi: 10.1093/hmg/ddh083 [DOI] [PubMed] [Google Scholar]
- 9. Shimura H, Schwartz D, Gygi SP, Kosik KS. CHIP‐Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem. 2004;279(6):4869‐4876. doi: 10.1074/jbc.M305838200 [DOI] [PubMed] [Google Scholar]
- 10. Hartl FU, Bracher A, Hayer‐Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475(7356):324‐332. doi: 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- 11. Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. α‐synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278(27):25009‐25013. doi: 10.1074/jbc.M300227200 [DOI] [PubMed] [Google Scholar]
- 12. Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM. Degradation of α‐synuclein by proteasome. J Biol Chem. 1999;274(48):33855‐33858. doi: 10.1074/jbc.274.48.33855 [DOI] [PubMed] [Google Scholar]
- 13. Hou Y, Dan X, Babbar M, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15(10):565‐581. doi: 10.1038/s41582-019-0244-7 [DOI] [PubMed] [Google Scholar]
- 14. Bulteau AL, Petropoulos I, Friguet B. Age‐related alterations of proteasome structure and function in aging epidermis. Exp Gerontol. 2000;35:767‐777. doi: 10.1016/S0531-5565(00)00136-4 [DOI] [PubMed] [Google Scholar]
- 15. Carrard G, Dieu M, Raes M, Toussaint O, Friguet B. Impact of ageing on proteasome structure and function in human lymphocytes. Int J Biochem Cell Biol . 2003;35(5):728‐739. doi: 10.1016/S1357-2725(02)00356-4 [DOI] [PubMed] [Google Scholar]
- 16. Petropoulos I, Conconi M, Wang X, et al. Increase of oxidatively modified protein is associated with a decrease of proteasome activity and content in aging epidermal cells. J Gerontol A Biol Sci Med Sci . 2000;55(5):B220‐B227. doi: 10.1093/gerona/55.5.B220 [DOI] [PubMed] [Google Scholar]
- 17. Bardag‐Gorce F, Farout L, Veyrat‐Durebex C, Briand Y, Briand M. Changes in 20S proteasome activity during ageing of the LOU rat. Mol Biol Rep. 1999;26(1‐2):89‐93. doi: 10.1023/a:1006968208077 [DOI] [PubMed] [Google Scholar]
- 18. Bulteau AL, Szweda LI, Friguet B. Age‐dependent declines in proteasome activity in the heart. Arch Biochem Biophys. 2002;397(2):298‐304. doi: 10.1006/abbi.2001.2663 [DOI] [PubMed] [Google Scholar]
- 19. Conconi M, Szweda LI, Levine RL, Stadtman ER, Friguet B. Age‐related decline of rat liver multicatalytic proteinase activity and protection from oxidative inactivation by heat‐shock protein 90. Arch Biochem Biophys. 1996;331(2):232‐240. doi: 10.1006/abbi.1996.0303 [DOI] [PubMed] [Google Scholar]
- 20. Ferrington DA, Husom AD, Thompson LV. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J . 2005;19(6):1‐24. doi: 10.1096/fj.04-2578fje [DOI] [PubMed] [Google Scholar]
- 21. Shibatani T, Nazir M, Ward WF. Alteration of rat liver 20S proteasome activities by age and food restriction. J Gerontol A Biol Sci Med Sci . 1996;51(5):B316‐B322. doi: 10.1093/gerona/51A.5.B316 [DOI] [PubMed] [Google Scholar]
- 22. Zabel C, Nguyen HP, Hin SC, Hartl D, Mao L, Klose J. Proteasome and oxidative phoshorylation changes may explain why aging is a risk factor for neurodegenerative disorders. J Proteomics. 2010;73(11):2230‐2238. doi: 10.1016/j.jprot.2010.08.008 [DOI] [PubMed] [Google Scholar]
- 23. Myeku N, Clelland CL, Emrani S, et al. Tau‐driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP‐PKA signaling. Nat Med. 2016;22(1):46‐53. doi: 10.1038/nm.4011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tseng BP, Green KN, Chan JL, Blurton‐Jones M, LaFerla FM. Aβ inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008;29(11):1607‐1618. doi: 10.1016/j.neurobiolaging.2007.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Almeida CG, Takahashi RH, Gouras GK. β‐amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin‐proteasome system. J Neurosci . 2006;26(16):4277‐4288. doi: 10.1523/JNEUROSCI.5078-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thibaudeau TA, Anderson RT, Smith DM. A common mechanism of proteasome impairment by neurodegenerative disease‐associated oligomers. Nat Commun. 2018;9(1):1097. doi: 10.1038/s41467-018-03509-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klimmt J, Dannert A, Paquet D. Neurodegeneration in a dish: advancing human stem‐cell‐based models of Alzheimer's disease. Curr Opin Neurobiol. 2020;61:96‐104. doi: 10.1016/j.conb.2020.01.008 [DOI] [PubMed] [Google Scholar]
- 28. Lagomarsino VN, Pearse RV, Liu L, et al. Stem cell‐derived neurons reflect features of protein networks, neuropathology, and cognitive outcome of their aged human donors. Neuron. 2021;109(21):3402‐3420. doi: 10.1016/j.neuron.2021.08.003. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious orders study and rush memory and aging project. J Alzheimer's Dis . 2018;64(s1):S161‐S189. doi: 10.3233/JAD-179939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. De Jager PL, Ma Y, McCabe C, et al. A multi‐omic atlas of the human frontal cortex for aging and Alzheimer's disease research. Sci Data. 2018;5:180142. doi: 10.1038/sdata.2018.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Green GS, Fujita M, Yang HS, et al. Cellular dynamics across aged human brains uncover a multicellular cascade leading to Alzheimer's disease. bioRxiv. 2023. doi: 10.1101/2023.03.07.531493. Published online January 1. :2023.03.07.531493. bioRxiv. [DOI] [Google Scholar]
- 32. Liu YH, Wei W, Yin J, et al. Proteasome inhibition increases tau accumulation independent of phosphorylation. Neurobiol Aging. 2009;30(12):1949‐1961. doi: 10.1016/j.neurobiolaging.2008.02.012 [DOI] [PubMed] [Google Scholar]
- 33. Goldbaum O, Oppermann M, Handschuh M, et al. Proteasome inhibition stabilizes tau inclusions in oligodendroglial cells that occur after treatment with okadaic acid. J Neurosci . 2003;23(26):8872‐8880. doi: 10.1523/jneurosci.23-26-08872.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Karch CM, Kao AW, Karydas A, et al. A comprehensive resource for induced pluripotent stem cells from patients with primary tauopathies. Stem Cell Reports. 2019;13(5):939‐955. doi: 10.1016/j.stemcr.2019.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu L, Hsieh YC, Pearse RV, et al. Association of AK4 protein from stem cell–derived neurons with cognitive reserve: an autopsy study. Neurology. 2022. Published online August 10. doi: 10.1212/wnl.0000000000201120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. De Jager PL, Ma Y, McCabe C, et al. Data descriptor: a multi‐omic atlas of the human frontal cortex for aging and Alzheimer's disease research. Sci Data. 2018;5. doi: 10.1038/sdata.2018.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature. 2021;590(7845):290‐299. doi: 10.1038/s41586-021-03205-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Choi SW, O'Reilly PF. PRSice‐2: polygenic risk score software for biobank‐scale data. Gigascience. 2019;8(7):giz082. doi: 10.1093/gigascience/giz082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54(4). doi: 10.1038/s41588-022-01024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545‐15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu L, Lauro BM, He A, et al. Identification of the Aβ37/42 peptide ratio in CSF as an improved Aβ biomarker for Alzheimer's disease. Alzheimers Dement. 2022. doi: 10.1002/alz.12646. Published online March 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fujita M, Gao Z, Zeng L, et al. Cell‐subtype specific effects of genetic variation in the aging and Alzheimer cortex. bioRxiv. 2022. doi: 10.1101/2022.11.07.515446. Published online January 1. :2022.11.07.515446. bioRxiv. [DOI] [Google Scholar]
- 43. Green GS, Fujita M, Yang HS, et al. Cellular dynamics across aged human brains uncover a multicellular cascade leading to Alzheimer's disease. bioRxiv. 2023. doi: 10.1101/2023.03.07.531493. Published online March 9. bioRxiv. [DOI] [Google Scholar]
- 44. Young‐Pearse TL, Lee H, Hsieh YC, Chou V, Selkoe DJ. Moving beyond amyloid and tau to capture the biological heterogeneity of Alzheimer's disease. Trends Neurosci. 2023;46(6):426‐444. doi: 10.1016/j.tins.2023.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kisselev AF, Akopian TN, Castillo V, Goldberg AL. Proteasome active sites allosterically regulate each other, suggesting a cyclical bite‐chew mechanism for protein breakdown. Mol Cell. 1999;4(3):395‐402. doi: 10.1016/S1097-2765(00)80341-X [DOI] [PubMed] [Google Scholar]
- 46. Dujardin S, Commins C, Lathuiliere A, et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer's disease. Nat Med. 2020:1‐8. doi: 10.1038/s41591-020-0938-9. Published online June 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. VerPlank JJS, Tyrkalska SD, Fleming A, Rubinsztein DC, Goldberg AL. cGMP via PKG activates 26S proteasomes and enhances degradation of proteins, including ones that cause neurodegenerative diseases. Proc Natl Acad Sci U S A. 2020;117(25):14220‐14230. doi: 10.1073/pnas.2003277117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lines G, Arber C, Preza E, et al. Investigating changes in the proteostasis capabilities of iPSC‐neurons during development and in FTD using iPSC‐neurons with MAPT mutations. Alzheimers Dement. 2021;17:e058308. doi: 10.1002/alz.058308 [DOI] [Google Scholar]
- 49. Mahali S, Martinez R, King M, et al. Defective proteostasis in induced pluripotent stem cell models of frontotemporal lobar degeneration. Transl Psychiatry. 2022;12(1):508. doi: 10.1038/s41398-022-02274-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Verheyen A, Diels A, Reumers J, et al. Genetically engineered iPSC‐Derived FTDP‐17 MAPT neurons display mutation‐specific neurodegenerative and neurodevelopmental phenotypes. Stem Cell Reports. 2018;11(2):363‐379. doi: 10.1016/j.stemcr.2018.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Türker F, Cook EK, Margolis SS. The proteasome and its role in the nervous system. Cell Chem Biol. 2021;28(7):903‐917. doi: 10.1016/j.chembiol.2021.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Adams J, Palombella VJ, Sausville EA, et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999;59(11):2615‐2622. [PubMed] [Google Scholar]
- 53. Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, Deshaies RJ. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell. 2010;38(1):17‐28. doi: 10.1016/j.molcel.2010.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pathare GR, Nagy I, Śledź P, et al. Crystal structure of the proteasomal deubiquitylation module Rpn8‐Rpn11. Proc Natl Acad Sci U S A. 2014;111(8):2984‐2989. doi: 10.1073/pnas.1400546111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Worden EJ, Padovani C, Martin A. Structure of the Rpn11‐Rpn8 dimer reveals mechanisms of substrate deubiquitination during proteasomal degradation. Nat Struct Mol Biol. 2014;21(3):220‐227. doi: 10.1038/nsmb.2771 [DOI] [PubMed] [Google Scholar]
- 56. Rosati A, Graziano V, de Laurenzi V, Pascale M, Turco MC. BAG3: a multifaceted protein that regulates major cell pathways. Cell Death Dis. 2011;2(4):e141. doi: 10.1038/cddis.2011.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Du ZX, Zhang HY, Meng X, et al. Proteasome inhibitor MG132 induces BAG3 expression through activation of heat shock factor 1. J Cell Physiol. 2009;218(3):631‐637. doi: 10.1002/jcp.21634 [DOI] [PubMed] [Google Scholar]
- 58. Wang HQ, Liu HM, Zhang HY, Guan Y, Du ZX. Transcriptional upregulation of BAG3 upon proteasome inhibition. Biochem Biophys Res Commun. 2008;365(2):381‐385. doi: 10.1016/j.bbrc.2007.11.001 [DOI] [PubMed] [Google Scholar]
- 59. Muchamuel T, Basler M, Aujay MA, et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med. 2009;15(7):781‐787. doi: 10.1038/nm.1978 [DOI] [PubMed] [Google Scholar]
- 60. Arnsten AFT, Datta D, Del Tredici K, Braak H. Hypothesis: tau pathology is an initiating factor in sporadic Alzheimer's disease. Alzheimer's and Dementia. 2021;17(1):115‐124. doi: 10.1002/alz.12192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Odagiri S, Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K. Autophagic adapter protein NBR1 is localized in Lewy bodies and glial cytoplasmic inclusions and is involved in aggregate formation in α‐synucleinopathy. Acta Neuropathol. 2012;124(2):173‐186. doi: 10.1007/s00401-012-0975-7 [DOI] [PubMed] [Google Scholar]
- 62. Liang WS, Dunckley T, Beach TG, et al. Neuronal gene expression in non‐demented individuals with intermediate Alzheimer's Disease neuropathology. Neurobiol Aging. 2010;31(4):549‐566. doi: 10.1016/j.neurobiolaging.2008.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO Journal. 2009;28(7):889‐901. doi: 10.1038/emboj.2009.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vo TKD, Godard P, de Saint‐Hubert M, et al. Transcriptomic biomarkers of human ageing in peripheral blood mononuclear cell total RNA. Exp Gerontol. 2010;45(3):188‐194. doi: 10.1016/j.exger.2009.12.001 [DOI] [PubMed] [Google Scholar]
- 65. Deane CAS, Brown IR. Intracellular targeting of heat shock proteins in differentiated human neuronal cells following proteotoxic stress. J Alzheimer's Dis . 2018;66(3):1295‐1308. doi: 10.3233/JAD-180536 [DOI] [PubMed] [Google Scholar]
- 66. Khalouei S, Chow AM, Brown IR. Stress‐induced localization of HSPA6 (HSP70B) and HSPA1A (HSP70‐1) proteins to centrioles in human neuronal cells. Cell Stress Chaperones. 2014;19(3):321‐327. doi: 10.1007/s12192-013-0459-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fu H, Possenti A, Freer R, et al. A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat Neurosci. 2019;22(1):47‐56. doi: 10.1038/s41593-018-0298-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lei Z, Brizzee C, Johnson GVW. BAG3 facilitates the clearance of endogenous tau in primary neurons. Neurobiol Aging. 2015;36(1):241‐248. doi: 10.1016/j.neurobiolaging.2014.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kocaturk NM, Gozuacik D. Crosstalk between mammalian autophagy and the ubiquitin‐proteasome system. Front Cell Dev Biol. 2018;6:128. doi: 10.3389/fcell.2018.00128. OCT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lee D, Takayama S, Goldberg AL. ZFAND5/ZNF216 is an activator of the 26S proteasome that stimulates overall protein degradation. Proc Natl Acad Sci U S A. 2018;115(41):E9550‐E9559. doi: 10.1073/pnas.1809934115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Goldberg AL, Kim HT, Lee D, Collins GA. Mechanisms that activate 26s proteasomes and enhance protein degradation. Biomolecules. 2021;11(6):779. doi: 10.3390/biom11060779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu F, Rehmani I, Esaki S, et al. Pirin is an iron‐dependent redox regulator of NF‐κB. Proc Natl Acad Sci U S A. 2013;110(24):9722‐9727. doi: 10.1073/pnas.1221743110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fricke B, Heink S, Steffen J, Kloetzel PM, Krüger E. The proteasome maturation protein POMP facilitates major steps of 20S proteasome formation at the endoplasmic reticulum. EMBO Rep. 2007;8(12):1170‐1175. doi: 10.1038/sj.embor.7401091 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information