

ABSTRACT
Macroautophagy/autophagy is the intracellular degradation process of cytoplasmic content and damaged organelles. Autophagy is strongly associated with the progression of Alzheimer disease (AD). Microglia are brain-resident macrophages, and recent studies indicate that autophagy in microglia protects neurons from neurodegeneration. Postnatal neurogenesis, the generation of new neurons from adult neural stem cells (NSCs), is impaired in AD patients as well as in AD animal models. However, the extent to which microglial autophagy influences adult NSCs and neurogenesis in AD animal models has not been studied. Here, we showed that conditional knock out (cKO) of Atg5 (autophagy related 5) in microglia inhibited postnatal neurogenesis in the dentate gyrus (DG) of the hippocampus, but not in the subventricular zone (SVZ) of a 5×FAD mouse model. Interestingly, the protection of neurogenesis by Atg5 in microglia was only observed in female AD mice. To confirm the roles of autophagy in microglia for postnatal hippocampal neurogenesis, we generated additional cKO mice to delete autophagy essential genes Rb1cc1 or Atg14 in microglia. However, these rb1cc1 cKO and atg14 cKO mice did not exhibit neurogenesis defects in the context of a female AD mouse model. Last, we used the CSF1R antagonist to deplete ATG5-deficient microglia and this intervention restored neurogenesis in the hippocampus of 5×FAD mice. These results indicate that microglial ATG5 is essential to maintain postnatal hippocampal neurogenesis in a mouse model of AD. Our findings further support the notion that ATG5 in microglia supports NSC health and may prevent neurodegeneration.
Abbreviations: 5×FAD: familial Alzheimer disease; Aβ: β-amyloid; AD: Alzheimer disease; AIF1: allograft inflammatory factor 1; ATG: autophagy related; BrdU: 5-bromo-2ʹ-deoxyuridine; CA: Cornu Ammonis; cKO: conditional knock out; CSF1R: colony stimulating factor 1 receptor; Ctrl: control; DCX: doublecortin; DG: dentate gyrus; GFAP: glial fibrillary acidic protein; GZ: granular zone; H&E: hematoxylin and eosin; IF: immunofluorescence; LD: lipid droplet; LDAM: lipid droplets accumulated microglia; LPS: lipopolysaccharides; MAP1LC3B/LC3: microtubule-associated protein 1 light chain 3 beta; NSCs: neural stem cells; RB1CC1: RB1-inducible coiled-coil 1; SOX2: SRY (sex determining region Y)-box 2; SGZ: subgranular zone; SVZ: subventricular zone; WT: wild type.
KEYWORDS: Alzheimer disease, animal model, autophagy, microglia, neural stem cell, neurogenesis
Introduction
Alzheimer disease (AD) is the most common form of dementia with hallmarks of β-amyloid (Aβ) deposits, neurofilament tangles, synaptic and neuronal loss in the patient’s brain. AD results in loss of memory and cognitive functions in people of an aging society. There are 6 million Americans living with AD and this number is projected to rise to 14 million by 2050 [1]. There are no effective drugs or therapies in treating the causes or slowing down AD progression [2]. One underlying reason for this is that we understand little about the early pathological changes of AD that lead to the deterioration of brain functions.
Neurogenesis, the generation of new neurons from neural stem cells (NSCs) to replace dead or damaged neurons, occurs throughout life in the adult mammalian brain [3–5]. There is strong evidence that continuous postnatal neurogenesis happens in subventricular zone (SVZ) of the lateral ventricle and subgranular zone (SGZ) of dentate gyrus (DG) in the hippocampus. The DG is integrated in brain circuits that mediate learning and memory, and it is one of the first regions to suffer at the early onset of AD. Moreover, several studies suggest that there is progressive failure of neurogenesis in the brain of AD patients and that this decline is evident before overt AD-related pathologies [6–8]. In different AD mouse models, NSC functions also decrease in the SVZ and DG [9–12]. It is reported that blocking adult hippocampal neurogenesis exacerbates neuronal loss and cognitive deficits in 5×FAD (familial Alzheimer disease) mouse model [13]. Altogether these results suggest that postnatal neurogenesis plays critical functions in maintaining the existing neural populations in AD hippocampus. These previous studies also imply that deficient neurogenesis in AD brains might precipitate disease progression; thereby making neurogenesis a potential early phase intervention target.
Microglia support neurogenesis through release of neurotrophic factors and phagocytosis of debris during development [14–17]. They could dampen NSCs’ maintenance or maturation of new neurons by neuroinflammations [18–20]. These brain-resident macrophages are well-established as key regulators of AD initiation and progression [21,22]. Microglia are activated by Aβ accumulation and they form a protective barrier around amyloid deposits to prevent plaque spreading [23] and neuronal damage during the early stages of AD [24]. More recent studies show that aberrantly activated microglia also eliminate functioning synapses and exacerbate spine loss in AD. AD patients exhibit impaired neurogenesis and activated microglia in the hippocampus [7], but whether there is a causative correlation between these two phenomena is still unknown. In PSEN1/PS1M146V/+ AD mice housed in an environmental enrichment situation, depleting activated microglia restores neurogenesis deficits of the hippocampus [25], suggesting that uncontrolled microglia activation contributes to declined NSC function in AD. Although more research on this topic is needed, the molecular mechanisms of microglia’s impact on AD neurogenesis remain largely unexplored.
Autophagy is a self-degradation process of cytoplasmic content or organelles to maintain homeostasis and it has been implicated in aging and neurodegenerative diseases [26,27]. Autophagy activity in neural cells, including neurons, NSCs, and microglia, decreased with aging and in late stages of neurodegenerative diseases [28–30]. Interestingly, acute Aβ treatment increases autophagy level in cultured microglia [31], which is different from the consistently reduced autophagy in neurons after exposure to Aβ [32]. These studies imply the specific response of autophagy in microglia during the progression of neurodegenerative diseases. Recently, studies from Heckmann et al. suggest that ATG5 (autophagy related 5) and RUBCN/Rubicon in microglia protect 5×FAD mice from Aβ accumulation, neuroinflammation, and neurodegeneration. Other autophagy genes, Rb1cc1 (RB1-inducible coiled-coil 1) and Atg14 (autophagy related 14) have no beneficial functions in AD microglia [33]. Mechanistically, canonical autophagy-independent functions of MAP1LC3B/LC3 (microtubule-associated protein 1 light chain 3 beta)-associated endocytosis enables removal of Aβ and ameliorates pathology in these murine AD models. However, the functions of canonical autophagy in microglia to regulate AD neurogenesis are still not clear.
In this study, we demonstrated that conditional knockout of Atg5, but not Atg14 and Rb1cc1, in microglia was associated with defective NSCs maintenance and neurogenesis in AD mouse models. We also observed sex differences in these effects as deficits in NSCs and neurogenesis were only prominent in female mice lacking Atg5 expression in microglia. Importantly, elimination of ATG5-deficient microglia restored the pool of NSCs and the neurogenesis in hippocampus. These preclinical studies provided significant insights into the role and mechanisms of microglia autophagy in maintenance of NSCs and neurogenesis in a neurodegenerative disease. Moreover, our results indicated that modulation of microglial autophagy might provide therapeutic benefits in neurodegenerative diseases and improve human healthspan.
Results
Atg5 deficiency in microglia exacerbated NSC loss and defective neurogenesis in SGZ of female 5×FAD atg5 [CX3CR1] cKO mice
To study the functions of microglia autophagy in AD neurogenesis, we crossed Atg5 flox/flox mice (designated as Atg5 WT) with 5×FAD [34] mice to get 5×FAD;Atg5 flox/flox mice (FAD;Atg5fl/fl, designated as FAD Ctrl). We mated a FAD;Atg5fl/fl mouse with a CX3CR1-CreERT2 transgenic mouse line [35] to get FAD atg5[CX3CR1] cKO (designated as FAD atg5 cKO) mice. We used female mice because AD is more prevalent in female adults [36]. After intraperitoneal injection of tamoxifen (1 mg each injection for 4 times) into female FAD Ctrl and FAD atg5 cKO mice at 1 month of age, we collected brain samples 3 months later for examination. We isolated the hippocampus from FAD Ctrl and FAD atg5 cKO mice to label dissociated cells with ITGAM/CD11b, P2RY12, and TREM2 for microglia sorting by FACS [37]. Our qPCR result showed a significant decrease of Atg5 mRNA level in microglia isolated from FAD atg5 cKO mice compared to that in microglia isolated from FAD Ctrl mice (Figure 1A). Next, we stained Aβ in the DG but we did not find differences in the total area of Aβ plaque between FAD Ctrl mice and FAD atg5 cKO mice at 4 months of age (Figure 1B,C). We used AIF1 (allograft inflammatory factor 1) to label microglia and we found a comparable number of AIF1+ cells in the DG of FAD Ctrl mice and FAD atg5 cKO mice (Figure 1D,E). Nevertheless, atg5 cKO microglia in the AD brain exhibited hypertrophic processes and the loss of ramified branches as compared to FAD Ctrl microglia (arrows in Figure 1D). These results suggested that even though atg5 deletion did not affect the survival and/or proliferation of microglia, it altered microglia’s morphology and activation in 5×FAD mice at 4 months of age. We used GFAP (glial fibrillary acidic protein) and SOX2 (SRY (sex determining region Y)-box 2) to label postnatal NSCs in the SGZ with a radial glia morphology [38]. We observed a significantly reduced number of GFAP+ SOX2+ NSCs in the DG of FAD atg5 cKO hippocampus compared to those in FAD Ctrl mice (Figure 1F,G). Atg5 cKO mice without FAD background had no defects in NSC maintenance (Figures 1F,G). These results suggested that ATG5 in microglia protected NSCs pools in the female AD hippocampus.
Figure 1.
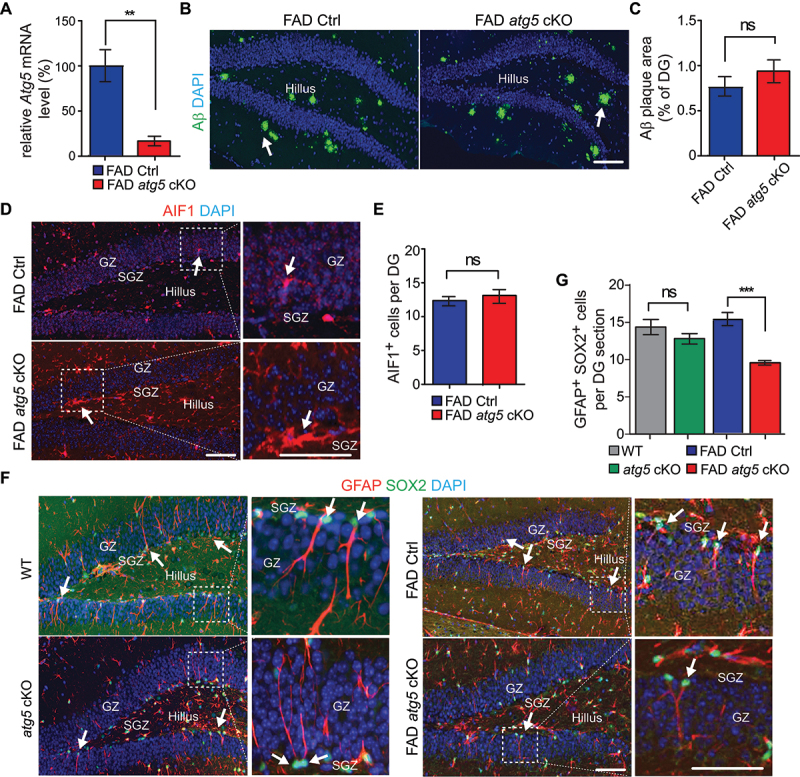
NSC degeneration in DG of female FAD atg5[CX3CR1] cKO mice at 4 months old. (A) mean ± SE of the relative mRNA level of Atg5 in sorted hippocampal microglia from female FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old were shown. n = 4 for each genotype. (B) if of Aβ and DAPI in DG of female FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old. Arrows indicated Aβ plaque. (C) mean ± SE of the percentile of Aβ plaque area of total DG area in female FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old were shown. (D) if of AIF1 and DAPI in DG of female FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old. Boxed areas were shown in detail on the right. Arrows indicated AIF1+ cells. (E) mean ± SE of the number of microglia in DG of female FAD Ctrl and FAD atg5[CX3CR1] cKO mice were shown. (F) if of GFAP, SOX2, and DAPI in SGZ of female WT, atg5[CX3CR1] cKO, FAD Ctrl, and FAD atg5[CX3CR1] cKO mice at 4 months old. Boxed areas were shown in detail on the right. Arrows indicated GFAP+ SOX2+ NSCs. (G) mean ± SE of GFAP+ SOX2+ cell number of female WT, atg5[CX3CR1] cKO, FAD Ctrl, and FAD atg5[CX3CR1] cKO mice were shown. n = 8 for female FAD Ctrl mice and 9 for female FAD atg5[CX3CR1] cKO mice in C, E, and G; n = 6 for female WT mice and 6 for female atg5[CX3CR1] cKO mice in G. GZ: granular zone, SGZ: subgranular zone. Bar: 100 µm. ns: no significance, **: p < 0.01, ***: p < 0.001. Student’s t test and two-way Anova were used for statistical analysis.
We examined the DCX (doublecortin)-positive (DCX+) immature neurons in DG and we found a significantly reduced number of DCX+ cells in female FAD atg5 cKO mice compared to FAD Ctrl mice (Figure 2A,B). To confirm the neurogenesis defects in NSCs from FAD atg5 cKO hippocampus, we performed long-term BrdU retention experiments [38] to label proliferative NSCs and neuronal progenitors in the hippocampus at 3 months old. One month after BrdU incorporation, we found a significantly lower percentage of BrdU+ cells in the granular zone (GZ) and a significantly higher percentage of BrdU+ cells in the SGZ of the DG from FAD atg5 cKO mice (Figure 2C,D), which suggested impaired neuronal generation in the FAD atg5 cKO hippocampus. We did not find a difference in BrdU+ cell number in the hillus from both FAD Ctrl and FAD atg5 cKO mice (Figure 2C,D). Last, we quantified the area of the DG and compared the Cornu Ammonis region 1 (CA1) in WT, atg5 cKO, FAD Ctrl, and FAD atg5 cKO hippocampus, however, we did not find obvious differences in DG area and thickness of CA1 between these animals at 4 months old (Figure 2E,F). It is reported that 5×FAD mice did not show significant neuron loss in hippocampus at 4 months old [39], which indicates that ATG5 deficiency in microglia specifically impaired NSC and neurogenesis but not mature neurons in AD hippocampus before overt neurodegeneration.
Figure 2.
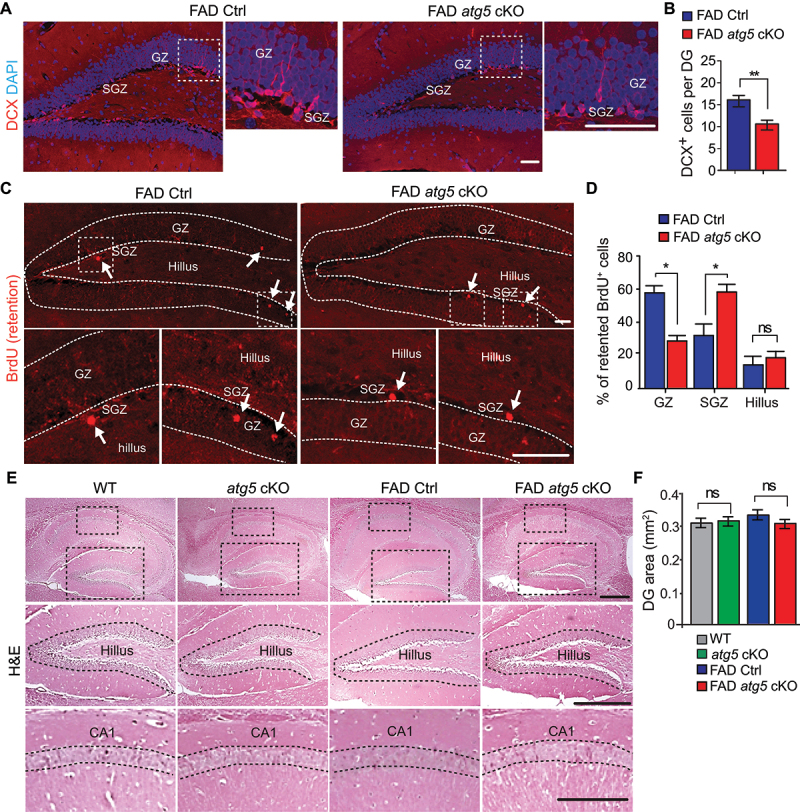
Impaired neurogenesis in DG of female FAD atg5[CX3CR1] cKO mice at 4 months old. (A) if of DCX and DAPI in DG of female FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old. Boxed areas were shown in detail on right. (B) mean ± SE of DCX+ cell number in DG of female FAD Ctrl and FAD atg5[CX3CR1] cKO mice were shown. n = 9 for female FAD Ctrl and 9 for FAD atg5[CX3CR1] cKO. (C) if of retentioned BrdU in DG of female FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old. Arrows indicated BrdU+ cells. Boxed areas were shown in detail at the bottom. Dashed lines indicated the borders of DG. (D) mean ± SE of the percentage of BrdU+ cells in GZ, SGZ, and hillus of DG from female FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old were shown. n = 5 for FAD Ctrl mice and 5 for FAD atg5[CX3CR1] cKO mice. (E) H&E staining of hippocampus in female FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old. (F) mean ± SE of DG area in female FAD Ctrl and FAD atg5[CX3CR1] cKO mice were shown. n = 8 for female FAD Ctrl mice, 8 for female FAD atg5[CX3CR1] cKO mice, 6 for female WT mice, and 6 for female atg5[CX3CR1] cKO mice. CA1: cornu Ammonis 1, GZ: granular zone, SGZ: subgranular zone. Bar: 100 µm. ns: no significance, *: p < 0.05, **: p < 0.01. Student’s t test and two-way Anova were used for statistical analysis.
Deletion of Atg5 in myeloid lineage by Lyz2-cre also impaired NSCs for neurogenesis in AD hippocampus
We also crossed Atg5 flox/flox;5×FAD mice with a Lyz2/LysM-Cre transgenic mouse line to get atg5[Lyz2] cKO;5×FAD mice (designated as FAD atg5[Lyz2] cKO mice). The Lyz2-Cre mouse is used to delete autophagy genes in peripheral myeloid cells [40–42] as well as in microglia [43]. We isolated cortical cells from neonatal WT (Atg5 flox/flox) and atg5[Lyz2] cKO (Atg5 flox/flox;Lyz2-Cre+) mice for primary microglia and we examined the deletion of ATG5 in atg5[Lyz2] cKO microglia. Our data showed that ATG5 was completely depleted in microglia from atg5[Lyz2] cKO mice. The atg5[Lyz2] cKO microglia showed a lower level of LC3-II and a significantly increased amount of SQSTM1/p62, suggesting a decline in autophagy activity (Figure S1A). We examined the female FAD Ctrl and FAD atg5[Lyz2] cKO mice at 4 months old. Female FAD atg5[Lyz2] cKO mice displayed no differences in Aβ deposition and microglia number when compared to their FAD Ctrl littermates (Figure S1B-S1E). Nevertheless, we found a significantly reduced number of GFAP+ SOX2+ NSCs as well as GFAP+ NES/nestin+ NSCs in DG of 4-months-old FAD atg5[Lyz2] cKO hippocampus (Figure S1F-S1H). Moreover, the labeling of proliferative NSCs (GFAP+ SOX2+ MKI67+ cells) indicated a reduced portion of dividing NSCs in SGZ of FAD atg5[Lyz2] cKO mice (3.3 ± 1.2% in FAD Ctrl vs 1.3 ± 0.5% in FAD atg5[Lyz2] cKO, p < 0.01). Next, we prepared SGZ cells to examine their neurosphere formation ability. We found significantly fewer and smaller primary neurospheres from DG of FAD atg5[Lyz2] cKO mice compared with neurospheres from WT, atg5[Lyz2] cKO, and FAD Ctrl mice (Figure S1I and S1J). We also found significantly reduced number of DCX+ cells and MKI67+ cells (Figure S1K-S1M) in DG of female FAD atg5[Lyz2] cKO mice. We noticed a modest reduction of DG area in FAD atg5[Lyz2] cKO mice compared to that in FAD Ctrl mice (Figure S1N and S1O). The number of NSCs, neurospheres, immature neurons, proliferative cells in SGZ, as well as DG area were comparable between female WT and atg5[Lyz2] cKO mice (Figure S1F-S1M, and data not shown).
In summary, using two independent Cre lines (CX3CR1-CreERT2 and Lyz2-Cre), our data indicated that ATG5 deficiency in microglia, but not likely in other myeloid lineages, caused defects in NSC maintenance and their generation of new neurons in hippocampus of female AD mice at early disease stage.
Atg5 deficiency in microglia had no effect on SVZ NSCs in female 5×FAD mice
We also examined the other postnatal neurogenic niche of SVZ, but we did not find changes in SVZ cell number or GFAP+ SOX2+ NSCs between female FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old (Figure S2A-S2D). Moreover, we examined the SVZ of WT, atg5[Lyz2] cKO, FAD Ctrl, and FAD atg5[Lyz2] cKO mice at 4 months old. We did not find differences in SVZ cell number or GFAP+ SOX2+ NSCs between these mice (Figure S2E-S2H). We prepared SVZ cells from 4-months-old WT, atg5[Lyz2] cKO, FAD Ctrl, and FAD atg5[Lyz2] cKO mice to examine neurosphere formation. We did not notice difference in the number and size of primary neurospheres from the SVZ of these mice (Figure S2I and S2J). These data suggested a niche specific function of ATG5 in microglia to protect the maintenance and neurogenesis of NSCs in female AD mice.
Sex-dependent effects of atg5 deletion in microglia on NSC and neurogenesis in AD mice
To examine whether microglia ATG5 performed the same functions in protecting NSCs and neurogenesis in male AD mice, we collected samples from male FAD Ctrl mice and male FAD atg5[CX3CR1] cKO mice at 4 months old. We did not find any difference in Aβ accumulation in the DG between male FAD Ctrl and FAD atg5 cKO mice (Figure 3A,B), but we noticed fewer Aβ plaques in male FAD mice than those in female FAD mice at 4 months old. We did not find a difference in the number of AIF1+ microglia between male FAD atg5 cKO mice and male FAD Ctrl mice (Figure 3C,D). We did not observe any difference in the number of GFAP+ SOX2+ NSCs between male FAD atg5 cKO mice and male FAD Ctrl mice (Figure 3E,F). Similarly, the generation of DCX+ immature neurons was comparable between male FAD atg5 cKO mice and FAD Ctrl mice (Figure 3G,H). Therefore, we did not notice any difference in DG area and CA thickness between these mice at 4 months old by H&E staining (Figure 3I,J). Taken together, our results suggested that ATG5 in male microglia had different functions from female microglia to protect hippocampal neurogenesis in AD mouse model.
Figure 3.
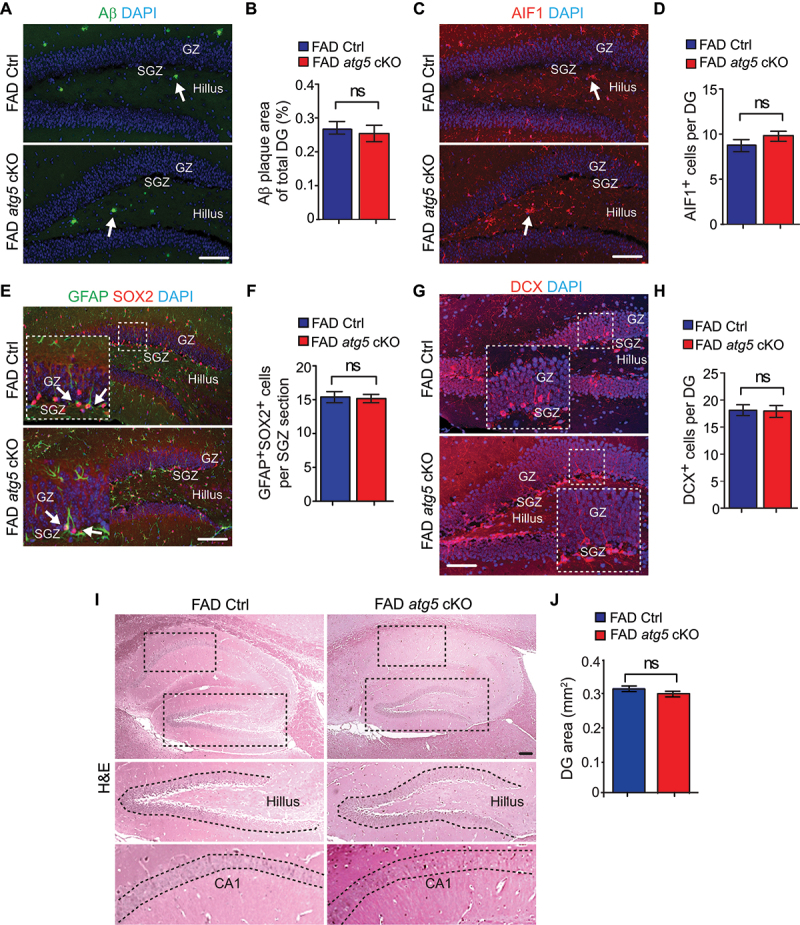
Intact NSC pool and neurogenesis in male FAD atg5[CX3CR1] cKO mice at 4 months old. (A) if of Aβ and DAPI in DG of male FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old. Arrows indicated Aβ plaques. (B) mean ± SE of the percentile of Aβ plaque area of total DG area in male FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old were shown. (C) if of AIF1 and DAPI in DG of male FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old. Arrows indicated AIF1+ microglia. (D) mean ± SE of the number of microglia in DG of male FAD Ctrl and FAD atg5[CX3CR1] cKO mice were shown. (E) if of GFAP, SOX2, and DAPI in DG of male FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old. Arrows indicated GFAP+ SOX2+ NSCs. Boxed areas were shown in detail as insets. (F) mean ± SE of the GFAP+ SOX2+ cell number in DG of male FAD Ctrl and FAD atg5[CX3CR1] cKO mice were shown. (G) if of DCX and DAPI in DG of male FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old. Boxed areas were shown in detail as insets. (H) mean ± SE of the DCX+ cell number in DG of male FAD Ctrl and FAD atg5[CX3CR1] cKO mice were shown. (I) H&E staining of hippocampus in male FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old. (J) mean ± SE of DG area of male FAD Ctrl and FAD atg5[CX3CR1] cKO mice were shown. For B, D, F, H, and J, n = 6 for male FAD Ctrl mice and 6 for male FAD atg5[CX3CR1] cKO mice. GZ: granular zone, SGZ: subgranular zone. Bar: 100 µm. ns: no significance. Student’s t test was used for statistical analysis.
We also used male FAD atg5[Lyz2] cKO mice and FAD Ctrl mice at 4 months old for another comparison. We did not find differences in Aβ accumulation and microglia number between male FAD atg5[Lyz2] cKO mice and male FAD Ctrl mice (Figure S3A-S3D). Most microglia in male FAD atg5[Lyz2] cKO DG and male FAD Ctrl DG kept ramified morphology (Figure S3C). We did not observe any differences in the number of hippocampal GFAP+ SOX2+ NSCs and DCX+ immature neurons as well as the DG areas between male FAD atg5[Lyz2] cKO mice and male FAD Ctrl mice (Figure S3E-S3J). In summary, this data confirmed that ATG5 in microglia had no protective functions on hippocampal neurogenesis in male AD mouse models.
Deletion of Rb1cc1 and Atg14 in microglia had no effects on NSC maintenance and neurogenesis in AD hippocampus
The canonic functions of Atg5 participate in autophagosome elongation and maturation, so we expanded our study to examine other autophagy genes that play distinct roles for autophagy induction (e.g., Rb1cc1) and autophagy initiation (e.g., Atg14) in microglia for postnatal neurogenesis in AD mice. To achieve this, we generated FAD rb1cc1[CX3CR1] cKO (designated as FAD rb1cc1 cKO) mice and FAD atg14[CX3CR1] cKO (designated as FAD atg14 cKO) mice along with their respective FAD controls (5×FAD;Rb1cc1 flox/flox mice and 5×FAD;Atg14 flox/flox mice). We only used female FAD rb1cc1 cKO mice and female FAD atg14 cKO mice with their respective FAD Ctrl at 4 months old. We found that deletion of Atg14 and Rb1cc1 in female microglia modestly decreased the formation of Aβ plaques in DG compared to that in FAD Ctrl mice (Figure 4A,B). We did not find changes in microglia number in the DG of female FAD rb1cc1 cKO mice or FAD atg14 cKO mice when compared with that in FAD Ctrl mice (Figure 4C,D). Along with the data for atg5 cKO in microglia, these results indicated that classic autophagy genes were not essential for the proliferation and/or survival of microglia in AD brains. To clarify the impact of rb1cc1 cKO and atg14 cKO in microglia on AD neurogenesis, we examined NSCs and immature neurons in DG of 4-months-old mice. We did not find a significant difference in the number of hippocampal GFAP+ SOX2+ NSC from FAD atg14 cKO and FAD rb1cc1 cKO mice when compared with their respective FAD Ctrl, although atg14 deletion led to a trend for NSC reduction (Figure 4E,F). The generation of DCX+ immature neurons was not affected in the DG of FAD atg14 cKO and FAD rb1cc1 cKO mice (Figure 4G,H), and there was no difference in DG areas and CA thickness of these mice when compared to FAD Ctrl mice (Figure 4I,J). These results suggested a mechanism for specific autophagy genes in microglia to regulate AD neurogenesis.
Figure 4.
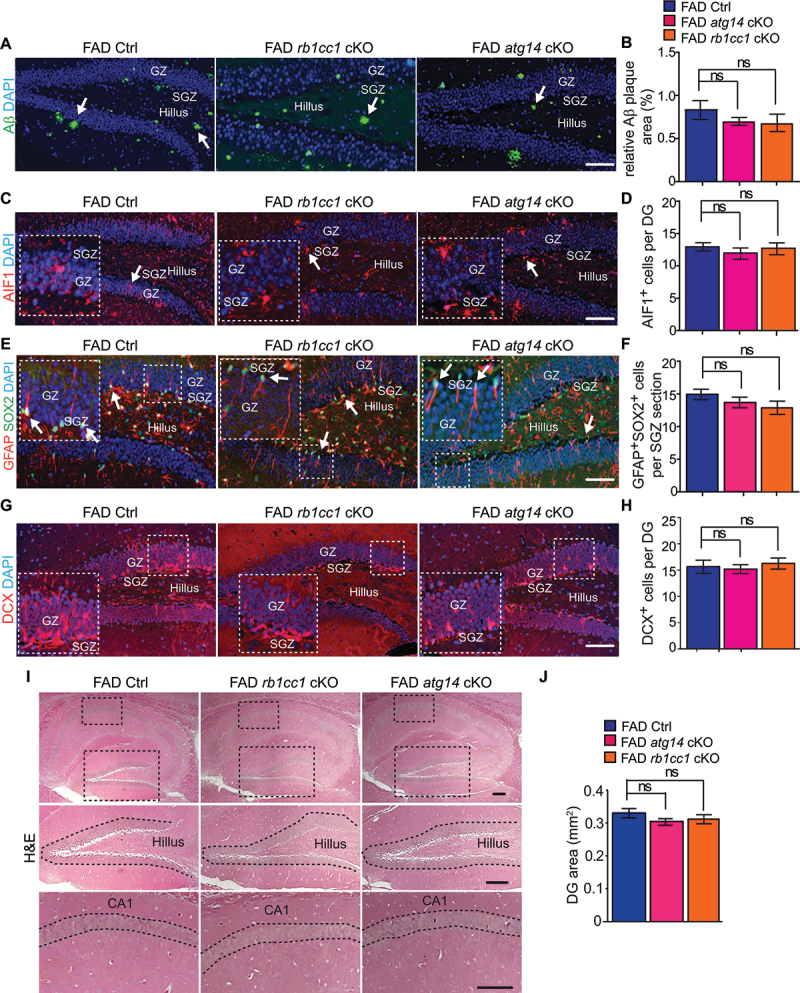
Normal NSC maintenance and functions in hippocampus of female FAD atg14[CX3CR1] cKO mice and FAD rb1cc1[CX3CR1] cKO mice at 4 months old. (A) if of Aβ and DAPI in DG of female FAD Ctrl, FAD rb1cc1[CX3CR1] cKO, and FAD atg14[CX3CR1] cKO mice at 4 months old. Arrows indicated Aβ plaques. (B) mean ± SE of the relative percentile of Aβ plaque area of total DG area in FAD Ctrl, FAD atg14[CX3CR1] cKO, and FAD rb1cc1[CX3CR1] cKO mice were shown. (C) if of AIF1 and DAPI in DG of female FAD Ctrl, FAD atg14[CX3CR1] cKO, and FAD rb1cc1[CX3CR1] cKO mice at 4 months old. Arrows indicated AIF1+ microglia. Boxed areas were shown in detail as insets. (D) mean ± SE of GFAP+ SOX2+ cell number of FAD Ctrl, FAD atg14[CX3CR1] cKO, and FAD rb1cc1[CX3CR1] cKO mice at 4 months old were shown. (E) if of GFAP, SOX2, and DAPI in DG of female FAD Ctrl, FAD atg14[CX3CR1] cKO, and FAD rb1cc1[CX3CR1] cKO mice at 4 months old. Arrows indicated GFAP+ SOX2+ NSCs. Boxed areas were shown in detail as insets. (F) mean ± SE of GFAP+ SOX2+ cell number of FAD Ctrl, FAD atg14[CX3CR1] cKO, and FAD rb1cc1[CX3CR1] cKO mice at 4 months old were shown. (G) if of DCX and DAPI in SGZ of female FAD Ctrl, FAD atg14[CX3CR1] cKO, and FAD rb1cc1[CX3CR1] cKO mice at 4 months old. Boxed areas were shown in detail as insets. (H) mean ± SE of DCX+ cell number in DG of FAD Ctrl, FAD atg14[CX3CR1] cKO, and FAD rb1cc1[CX3CR1] cKO mice at 4 months old were shown. (I) H&E staining of hippocampus of female FAD Ctrl, FAD atg14[CX3CR1] cKO, and FAD rb1cc1[CX3CR1] cKO mice. The dashed lines indicated the borders of DG. (J) mean ± SE of the DG area of FAD Ctrl, FAD atg14[CX3CR1] cKO, and FAD rb1cc1[CX3CR1] cKO mice were shown. For B, D, F, H, and J, n = 8 for female FAD Ctrl mice, 6 for female FAD rb1cc1 cKO mice, 6 for female FAD atg14 cKO mice. GZ: granular zone, SGZ: subgranular zone. Bar: 100 µm. ns: no significance. Student’s t test and One-way Anova was used for statistical analysis.
We also generated FAD rb1cc1[Lyz2] cKO mice and FAD atg14[Lyz2] cKO mice by crossing 5×FAD;Rb1cc1 flox/flox mice and 5×FAD;Atg14 flox/flox mice with a Lyz2-Cre mouse. We found that deletion of Atg14 in female microglia increased the area of Aβ plaques in DG, while deletion of Rb1cc1 had no effect on Aβ plaque areas in DG (Figure S4A and S4B). We did not find changes in microglia number in DG of FAD atg14[Lyz2] cKO mice and FAD rb1cc1[Lyz2] cKO mice when compared with FAD Ctrl (Figure S4C-S4F). Like CX3CR1-CreERT2 mediated gene deletion, we did not find a significant difference in the number of hippocampal GFAP+ SOX2+ NSC from FAD atg14[Lyz2] cKO and FAD rb1cc1[Lyz2] cKO mice, except that rb1cc1 deletion led to a trend for NSC reduction (Figure S5A and S5B). The DCX+ immature neurons and DG areas were not affected in FAD atg14[Lyz2] cKO and FAD rb1cc1[Lyz2] cKO mice (Figure S5C-S5F). Together, these studies from multiple FAD rb1cc1 cKO and FAD atg14 cKO mouse lines indicated that ATG14 and RB1CC1 in microglia were not involved in the regulation of postnatal NSC maintenance and neurogenesis in AD hippocampus.
Lipid droplets accumulation in Atg5 deficient microglia after Aβ stimulation
To reveal the functions of ATG5 in microglia to protect NSCs in SGZ of female AD mice, we examined the morphology of microglia in DG of FAD Ctrl mice and FAD atg5[CX3CR1] cKO mice for both sexes at 4 months old. We classified the morphology of microglia [18] and we found more round or amoeboid shapes in FAD mice compared to WT mice as well as atg5 cKO mice (data not shown), suggesting the activation of microglia in early AD brains. Interestingly, ATG5 deficiency significantly increased the ratio of microglia with amoeboid shape and round microglia in DG of female (please see Figure 1D for representative morphologies), but not male FAD atg5[CX3CR1] cKO mice (please see Figure 3C for representative morphologies)(Figure 5A). We also characterized the microglia morphology in FAD atg5[Lyz2] cKO mice for both sexes (please see Figure S1D and S3C for representative morphologies) and we got comparable results to what we found in FAD atg5[CX3CR1] cKO mice (Figure 5B). The results from two independent Cre lines indicated that ATG5 differentially regulated microglia activation in the hippocampus of female and male FAD mice at the initial stage of AD.
Figure 5.
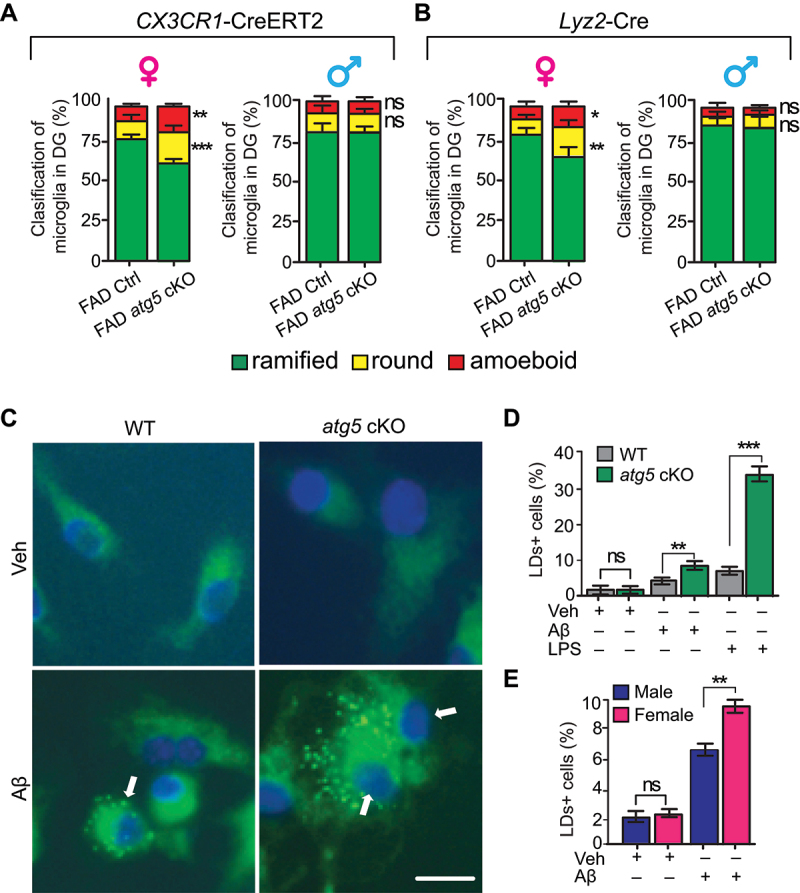
Female atg5 cKO microglia showed activated morphology in vivo and abnormal lipid droplets accumulation upon Aβ treatment in vitro. (A) mean ± SE of the percentage of resting microglia, round microglia, and amoeboid-shape microglia in DG of female (left) and male (right) FAD Ctrl and FAD atg5[CX3CR1] cKO mice at 4 months old were shown. n = 8 female mice for FAD Ctrl and 9 for FAD atg5[CX3CR1] cKO mice, n = 6 for male FAD Ctrl mice and 4 for male FAD atg5[CX3CR1] cKO mice. (B) mean ± SE of the percentage of resting microglia, round microglia, and amoeboid-shape microglia in DG of female (left) or male (right) FAD Ctrl and FAD atg5[Lyz2] cKO mice at 4 months old were shown. n = 5 for female FAD Ctrl and FAD atg5[Lyz2] cKO mice, n = 3 for male FAD Ctrl and FAD atg5[Lyz2] cKO mice. (C) BODIPY staining of LDs in WT and atg5[Lyz2] cKO microglia after vehicle and oAβ treatment for 24 h. Arrows indicated LDs+ microglia. (D and E) mean ± SE of the percentage of LDs+ microglia after vehicle, oAβ, or LPS treatment (D) and the percentage of LDs+ male or LDs+ female atg5-null microglia after oAβ treatment (E) was shown. For D, 305 primary microglia from 3 WT mice and 320 primary microglia from 3 atg5[Lyz2] cKO mice were counted; for E, 335 primary microglia from 3 female atg5[Lyz2] cKO mice and 352 primary microglia from 3 male atg5[Lyz2] cKO mice were counted. Bar: 20 µm. ns: no significance, *: p < 0.05, **: p < 0.01, ***: p < 0.001. Student’s t test and two-way Anova was used for statistical analysis.
Alterations of lipid composition have been reported in postmortem AD brain and in AD mice [9,44]. Recently, Marschallinger et al. report a new proinflammatory phenotype of lipid droplets accumulated microglia (LDAM) in neuroinflammation and in neurodegenerative diseases [45]. To investigate the regulatory functions of ATG5 for lipid droplets (LDs) in microglia, we used vehicle (PBS), oligomer Aβ (oAβ), or 5 ng/ml lipopolysaccharides (LPS, as a positive control) to treat WT and atg5-null microglia for 24 h before staining LDs with BODIPY493/503. Under resting condition, we found that only 1–2% of WT and atg5-null microglia contained LDs. After oAβ treatment, the number of LDs+ cells in both WT and atg5 cKO microglia significantly increased and we observed more LDs+ atg5-null microglia (Figure 5C–E). As a positive control, LPS generated most LDs in atg5-null microglia from our experimental settings (Figure 5D). Interestingly, after we separated the data, we found more LDAM from female atg5 cKO mice than from male atg5 cKO mice after oAβ treatment (Figure 5E). These results suggested that Aβ induced more LDAM in female atg5 cKO microglia, which might be an underling mechanism for their activation and reduced neurogenesis in AD hippocampus.
Depletion of microglia restored NSC in SGZ of FAD atg5 cKO mice
We wanted to know when the neurogenesis in FAD atg5 cKO mice started to decline compared to FAD Ctrl mice, so we checked the NSCs’ content and their functions in female FAD Ctrl and FAD atg5[Lyz2] cKO mice at 2 months old. We found that the number of GFAP+ SOX2+ NSCs, DCX+ immature neurons, and DCX+ MKI67+ proliferative neuroblasts in the DG were all comparable between female FAD Ctrl and FAD atg5[Lyz2] cKO mice (Figure S6A-S6E). These results indicated a therapeutic window to target ATG5 deficient microglia in young adult AD mice.
To determine the role of ATG5-deficient microglia in defective hippocampal NSCs and their functions in female AD mice, we first fed 1 months old female WT mice with CSF1R (colony stimulating factor 1 receptor) antagonist diet (PLX5622 at 1200 ppm) [23] for 10 days. We found that PLX5622 efficiently depleted microglia (>90%) in DG, cortex (Figure S6F-S6H), and other brain regions (data not shown) without affecting the whole-body weight and activities of the WT mice. Next, we provided continued supplement of CSF1R antagonist in diet to deplete microglia in FAD Ctrl, FAD atg5[CX3CR1] cKO, and FAD atg5[Lyz2] cKO mice from 2 months old to 4 months old. We did not notice obvious changes in body weight and activity in PLX5622 fed animals compared with mice fed with normal chow. As expected, we found a significant reduction of AIF1+ microglia in the DG from FAD Ctrl, FAD atg5[CX3CR1] cKO, and FAD atg5[Lyz2] cKO mice (Figure 6A,B). The residual individual microglia in PLX5622-treated mice surrounded Aβ plaques within DG and we did not find a notable change of Aβ plaque depositions in DG after the treatment, even though the trends for Aβ accumulation in FAD atg5[CX3CR1] cKO and FAD atg5[Lyz2] cKO mice after PLX5622 treatment were different (Figure 6A–C). After the majority of atg5 cKO microglia were depleted by PLX5622, we found the restoration of NSCs and DCX+ immature neurons in DG of FAD atg5[CX3CR1] cKO and FAD atg5[Lyz2] cKO mice (Figure 6D–G). Depletion of microglia by CSF1R antagonist in FAD Ctrl mice had no effect on NSC maintenance and differentiation (Figure 6D–G). The results from the depleting experiments further confirmed an Aβ independent protective function of microglial ATG5 in the maintenance and differentiation of postnatal hippocampal NSCs in AD mouse models.
Figure 6.
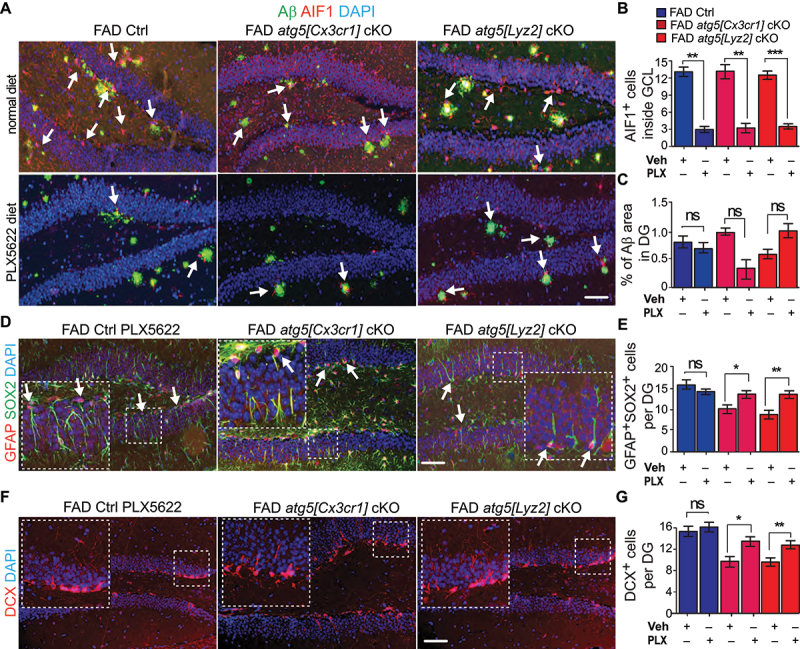
Depletion of atg5 cKO macroglia restored NSC pool and neurogenesis in DG of AD mice. (A) if of Aβ, AIF1, and DAPI in DG of female FAD Ctrl, FAD atg5[CX3CR1] cKO, and FAD atg5[Lyz2] cKO mice after receiving control chow and PLX5622 chow for 2 months. Arrows indicated AIF1+ cells in adjacent to Aβ plaques. (B) mean ± SE of number of microglia in DG of FAD Ctrl, FAD atg5[CX3CR1] cKO, and FAD atg5[Lyz2] cKO mice after receiving control chow and PLX5622 chow were shown. (C) mean ± SE of percentile of Aβ plaque area of total DG area in FAD Ctrl, FAD atg5[CX3CR1] cKO, and FAD atg5[Lyz2] cKO mice after receiving control chow and PLX5622 chow were shown. (D) if of GFAP, SOX2, and DAPI in DG of female FAD Ctrl, FAD atg5[CX3CR1] cKO, and FAD atg5[Lyz2] cKO mice after receiving PLX5622 chow for 2 months. Arrows indicated GFAP+ SOX2+ NSCs. Boxed areas were shown in detail as insets. (E) mean ± SE of number of GFAP+ SOX2+ cells in DG of FAD Ctrl and FAD atg5 cKO mice after receiving control chow and PLX5622 chow were shown. (F) if of DCX and DAPI in DG of female FAD Ctrl, FAD atg5[CX3CR1] cKO, and FAD atg5[Lyz2] cKO mice after receiving PLX5622 chow for 2 months. Boxed areas were shown in detail as insets. (G) mean ± SE of the DCX+ cells number in DG of FAD Ctrl, FAD atg5[CX3CR1] cKO, and FAD atg5[Lyz2] cKO mice after receiving control chow and PLX5622 chow for 2 months were shown. n = 3 for FAD Ctrl mice, 3 for FAD atg5[CX3CR1] cKO mice, and 5 for FAD atg5[Lyz2] cKO mice. Bar: 100 µm. ns: no significance, **: p < 0.01, ***: p < 0.001. Student’s t test and two-way Anova was used for statistical analysis.
Discussion
Aging is a main risk factor for AD and a hallmark of aging is the exhaustion of tissue stem cells [46]. In the brain, ongoing neurogenesis in hippocampus is susceptible to age-related declines and the hippocampus is one of the primary regions affected in AD patients. Indeed the presence of Aβ plaques and neurofilament tangles in the hippocampus is strongly correlated with deterioration of cognitive functions [47]. Impaired neurogenesis also compromises hippocampal function and plays a role in cognitive deficits in AD models [13,48]. Multiple longitudinal studies demonstrate that neuroinflammation mediated by activated microglia begins early in AD [49]. Nevertheless, the molecular mechanisms of microglia in hippocampal neurogenesis in AD are not clear. Using conditional microglia-specific depletion of autophagy genes in mice, we demonstrate that Atg5, particularly in female microglia, limits loss of NSC pools and neurogenesis in the hippocampus of an AD mouse model.
The reduction of GFAP+ SOX2+ radial glia in hippocampus of 4-months-old FAD atg5 cKO mice indicated an interruption of NSC maintenance by dysfunctional microglia at early onset of AD (Figure 1F,G, S1F-S1H). The ability of NSCs to generate DCX+ new neurons was also impaired in FAD atg5 cKO mice (Figure 2A,B, S3C and S3D). After depletion of microglia in FAD atg5 cKO mice, we restored the functions of NSCs and the number of newborn neurons in the hippocampus (Figure 6). For the first time, these results indicated a detrimental function of atg5 cKO microglia on NSCs’ maintenance and differentiation in AD. Since the levels of autophagy and autophagy protein BECN1/Beclin1 decrease in the aging brain and more drastically in AD brains [50–53], previous and current studies suggest that decreased autophagy negatively affects NSCs and neurogenesis through both cell-autonomous and non-cell-autonomous mechanisms. We noticed a trend of smaller DG area in FAD atg5 cKO mice at 4 months old (Figure 2E,F, S1N and S1O). This modest neuronal degeneration was consistent with previous reports that neuronal loss is not obvious in hippocampus of 4.5-months-old and 9-months-old 5×FAD mice [34,39]. Since we did not detect difference of apoptosis in DG between FAD Ctrl and FAD atg5 cKO mice (data not shown), the trend of reduced DG area was likely to be a consequence of impaired neurogenesis to supply new neurons in FAD atg5 cKO animals. Together, our finding raises a novel intercellular communication mechanism mediated by autophagy in niche cells, such as microglia, to protect NSC and neurogenesis in AD hippocampus.
Compared to studies of autophagy in neurons, far less is known about autophagy and autophagy related genes in microglia. Only recently, studies of autophagy are performed in microglia during development and in neurodegenerative disease models [54,55]. Cheng et al. report that atg5 cKO in ITGAM/CD11b+ cells lead to the development of Parkinson disease (PD)-like symptoms [56]. Nevertheless, another study shows that atg5 cKO microglia in Sall1-CreER mice is dispensable for microglia development and CNS homeostasis [57], which is consistent with our results that without 5×FAD transgene, autophagy-deficient microglia were largely normal and they did not affect maintenance of NSC and integrity of the hippocampus in mice (Figures 1, 2, S1, S2). In neurodegenerative disease models, previous studies reveal that atg5 cKO microglia accelerate neurodegeneration in 5×FAD mice [33] and atg7 cKO microglia contribute to synaptic degeneration in both 5×FAD and PS19 AD mice [58,59]. Atg5- and atg7-deficient microglia aggravate dopaminergic neuron loss through impaired autophagy activity in PD mouse models [60,61]. Besides ATG5 and ATG7, BECN1 affect microglia activation by modulating neuroinflammation via NLRP3 (NLR family, pyrin domain containing 3) degradation in AD [62]. Interestingly, microglia deficiency of ATG7 but not ULK1 (another upstream autophagy-related protein in a complex with RB1CC1) prevent mice from recovery with experimental autoimmune encephalomyelitis to mimic multiple sclerosis [63]. These atg7-null microglia show notable transcriptional and functional similarities to disease-associated microglia found in mouse AD brains [64,65]. However, it should be noted that atg5 cKO microglia are not necessary for the progression of experimental autoimmune encephalomyelitis and atg5 cKO microglia do not change their inflammatory status compared to WT microglia [57]. These data suggest that different autophagy genes, even though their protein products participate in the same autophagy process, may still perform distinct functions under different disease contexts. One interpretation is that proteins in autophagosome elongation process (e.g., ATG5, ATG16L1, and ATG7), autophagy induction process (e.g., RB1CC1), or initiation process (e.g., ATG14) play distinct functions in AD microglia. Alternatively, ATG5-dependent non-canonical autophagic functions of LC3-associated endocytosis [33] might be crucial for protective roles of microglia in AD. To reconcile the discrepancies in these findings, additional ATG knockin mutations with loss of autophagy function while preserving other non-autophagy functions, e.g., RB1CC14A knockin mouse that we recently generate [66], are needed in future animal research.
In this study, we observed sex differences for Atg5 in microglia for protecting NSC and neurogenesis in female 5×FAD mice (Figures 1, 3, S1-S3). Autophagy is involved in numerous aspects of sex differences in diseases, including neurodegeneration [67]. Female sex is a known risk factor for both developing AD and more severe pathology [68,69]. Sex differences in gene expression and functions of microglia are observed, and these differences are more obvious in aged brains [70,71]. For example, both male and female microglia increase their phagocytosis of neural debris with aging and microglia from aged females performed better than microglia from aged males. However, aged female microglia could not adapt their phagocytosis to the inflammatory conditions. These findings suggest sex differences in aged microglia play a role in neurodegenerative diseases [72]. Using PS19 AD mice, Kodama et al. show that deletion of microRNAs in male microglia leads to transcriptome changes toward disease-associated microglia and increased MAPT/tau pathology [73], suggesting additional mechanisms for sex difference in AD. Female AD transgenic mouse models classically have earlier onset of Aβ pathology than male counterparts [34,74,75], possibly due to the estrogen response element on Thy1 promoter. We observed higher levels of Aβ deposition in female AD mice than in male mice (Figures 1, 3, S1, S3). Nevertheless, the sex difference in atg5 cKO microglia functions was not dependent on Aβ accumulation in hippocampus of 4-months-old male AD mice (Figure 3 and S3) as we found significantly more Aβ plaques in hippocampus of 8 months old male FAD Ctrl and FAD atg5[Lyz2] cKO mice with no significant difference in NSC maintenance and differentiation between these mice (data not show). Even though it is not our focus in the current study, the sex differences of atg5 cKO in microglia between female and male AD mice need further investigation.
We observed differences of atg5-deficient microglia in their functions for NSCs between SVZ and hippocampus (Figures 1, 2, S1, S2). This discrepancy might be attributed to the intrinsic difference between SVZ and SGZ neurogenesis, mainly in the niche organization, vascular structure, neuronal subtype differentiation, and migration of newborn neurons [5]. It was also possible that fewer Aβ accumulations in SVZ and striatum did not trigger an extensive reactivation response in atg5 cKO microglia at 4 months old. Currently, we do not know the exact mechanisms for this regional difference in FAD atg5 cKO mice. It should be noted that our study focused on microglia autophagy in neurogenesis at early stage of AD while other mechanisms involved in neuronal degeneration by microglia were not within the scope of current study. There is no obvious neuronal loss in hippocampus of 4.5-months-old and 9-months-old 5×FAD mice [34,39]. We did not observe neuronal loss, severe microgliosis, and Aβ aggregates in 4-months-old FAD atg5 cKO mice (Figures 1, 3, S1-S3). However, these neurodegenerative features are prominent in another study using similar microglia-specific FAD atg5 cKO models [33]. In a MAPT/tau model (PS19) for AD mice, there is significant synaptic degeneration but no overt neuronal loss in atg7 cKO AD mice at 12 months of age [58]. During the revision of this manuscript, Choi et al. publish their results from microglia specific 5×FAD atg7 cKO mice [59]. This study also describes abnormal synapses but does not provide information about neuronal loss. Even though atg7 deletion significantly alters the association of microglia with plaques, microglia number and Aβ plaques number are comparable to those in FAD Ctrl mice at 8 months old. More interestingly, Choi et al. use senolytic drugs (dasatinib and quercetin) to remove senescent atg7 cKO microglia and alleviate AD-related pathologies. They find that after senolytic drug treatment, the number of Aβ plaque associated microglia increases to benefit Aβ removal. Consistent with this observation, we found that the residual microglia after PLX5622 treatment in atg5 cKO mice were associated with Aβ plaques (Figure 6), indicating that these surviving atg5 cKO microglia might perform protective functions for NSCs in AD. Elmore et al. show that elimination of microglia has no effect on neurogenesis in aged (22 months old) mice and it only benefits young (3 months old) mice for BrdU incorporation but not DCX+ cells in hippocampus [76]. These studies suggest that removal of activate microglia in aged and/or degenerative mice is not generally beneficial for improving neurogenesis, which is consistent with our findings in FAD Ctrl mice after feeding with PLX5622 (Figure 6). In summary, additional studies are needed to gain a better understanding of the mechanisms of microglial autophagy in AD pathologies, which is required for the development of selective therapy to target AD microglia.
The Lyz2-Cre has microglial penetrance of 90% and we showed results that microglia isolated from atg5[Lyz2] cKO mice were deficient in ATG5 (Figure S1A). However, it is known and supported by evidence that a high percentage of Lyz2-Cre mediated recombination in the brain occurs in neurons rather than in microglia [77]. This raised a possibility for the impaired NSC in atg5[Lyz2] cKO mice being a consequence of atg5 deletion in granular neurons. Previously, we use hGFAP-Cre [78] to delete Rb1cc1, Atg5, and Atg16l1 in postnatal NSCs and we only observe premature degeneration of NSCs in rb1cc1 cKO mice [38,79,80]. hGFAP-Cre targets all astrocytes, oligodendrocytes, many cortex neurons, and all granular neurons in the DG of a mouse brain. These results suggest that neuronal leakage for atg5 deletion was not a major contribution to NSCs defects in FAD atg5[Lyz2] cKO mice (Figure S1-S3). Lyz2-Cre also depletes autophagy related genes in peripheral monocyte lineage cells including macrophages [42]. We could not evaluate the infiltration of peripheral monocytic cells into the brain of FAD atg5[Lyz2] cKO animals, however, similar findings from CX3CR1-CreERT2 transgenic mice confirmed the microglial specific functions of ATG5 on NSCs in AD. Our tremendous work also suggests that even though Lyz2-Cre transgenic mice are valuable to study gene functions in myeloid lineage, future studies should not use this tool for microglia to avoid complication and confusion.
In conclusion, our study revealed that ATG5 in female microglia has a role in regulating NSCs and neurogenesis in an AD mouse model. The results of this study provided a non-cell-autonomous mechanism of how impaired microglial autophagy could contribute to NSC exhaustion and dysfunction in AD. Moreover, our findings supported the idea that microglial autophagy played a protective role in AD, and promotion or enhancement of autophagy in microglia might have therapeutic potential for improving AD outcomes.
Materials and methods
Animals
WT, 5×FAD, Atg5 flox/flox, Atg14 flox/flox, Rb1cc1 flox/flox, Lyz2-Cre;Atg5 flox/flox;5×FAD and CX3CR1-CreERT2;Atg5 flox/flox;5×FAD, Lyz2-Cre;Atg14 flox/flox;5×FAD and CX3CR1-CreERT2;Atg14 flox/flox;5×FAD, Lyz2-Cre;Rb1cc1 flox/flox;5×FAD and CX3CR1-CreERT2;Rb1cc1 flox/flox;5×FAD mice with B6 background were described as before. Lyz2-Cre, Atg5 flox/flox, and Atg14 flox/flox mice were gifted from Dr. Herbert Virgin’s lab at the Washington University, Rb1cc1 flox/flox mice were generated in Dr. Jun-Lin Guan’s lab at the University of Cincinnati, CX3CR1-CreERT2 mouse was purchased from Jackson lab (JAX 021,160). FAD Ctrl, CX3CR1-CreERT2;Atg5 flox/flox;5×FAD, CX3CR1-CreERT2;Atg14 flox/flox;5×FAD, and CX3CR1-CreERT2;Rb1cc1 flox/flox;5×FAD mice were intraperitoneally injected with 1 mg tamoxifen (Sigma, T5648) at 1 month old for 4 times to activate Cre. Mice were housed and handled according to local, state, and federal regulations. All experimental procedures were conducted according to the guidelines of Institutional Animal Care and Use Committee (IACUC) at University of Cincinnati (21-06-21-02).
Neurosphere formation assay and microglia culture
Neurospheres were cultured in neurobasal medium (ThermoFisher 21,103,049) supplemented with B27 (Invitrogen 17,504,044), 10 ng/ml FGF2/bFGF (Invitrogen, RP-8626) and 20 ng/ml EGF (epidermal growth factor; Invitrogen, A42556) in Ultra-Low Attachment dishes (Corning, CLS3471), essentially as described in our previous reports. Neurospheres with diameter larger than 50 µm was counted 7–9 d after culturing [38,79].
Mixed cortical cultures were prepared essentially as described previously [81]. In brief, newborn pups were decapitated and the cortices were removed from meninges, hippocampi, basal ganglia, and midbrain and kept in cold DMEM (ThermoFisher 11,965,118). The cortical tissue was cut into ~1 mm3 cubes, digested in 0.2% trypsin-EDTA (ThermoFisher 25,200,114) at 37°C for 20 min, and mechanically triturated. After filtration with 70-µm mesh, the cell suspension was plated in mixed cortical medium (DMEM with 10% FBS (ThermoFisher, A3840002)) onto tissue culture dishes and cultured at 37°C and 95% humidity. Media was changed 3 d after plating. Microglia were harvested 10 d after plating by adding 15 mM lidocaine (Sigma 1,366,002) into the medium for 15 min at room temperature. The medium containing the floating microglia was collected and centrifuged at 400 g for 5 min. The cell pellet was then resuspended, and the cell number was counted. Dispersed microglia were seeded in DMEM with 1% FBS and the purity of the microglial cultures was > 98% as examined by AIF1 staining. The microglia were used for experiments after 24–48 h of culture.
Antibodies and reagents
Primary antibodies used were mouse anti-Aβ (Biosensis, M-1742-50-B), anti-NES/nestin (DSHB, Rat-401), anti-GFAP (Cell Signaling Technology, 3670), anti-BrdU (Invitrogen, MA3–071), anti-P2RY12 (BioLegend 848,001), anti-VCL/vinculin (Sigma, V9131); rabbit anti-AIF1 (WAKO, 019–19741), anti-MKI67/Ki67 (Spring Bioscience 151,213), anti-GFAP (DAKO, M0761), anti-ATG5 (Cell Signaling Technology 12,994), anti-MAP1LC3B/LC3 (Cell Signaling Technology, 2755), anti-SOX2 (Millipore, AB5603); rat anti-ITGAM/CD11b (BD Pharmingen 557,397), anti-MKI67 (BioLegend 151,213), anti-TREM2 (R&D Systems, MAB17291); and guinea pig anti-DCX (Millipore, AB2253). Secondary antibodies were goat anti-rabbit IgG-FITC (Jackson Immunology, 111-095-003), goat anti-rabbit IgG-Alexa Fluor (Jackson Immunology, 111-586-003), goat anti-mouse IgG-FITC (Jackson Immunology, 115-095-003), goat anti-mouse IgG-Texas Red (Jackson Immunology, 115-295-003), goat anti-mouse IgM-Rhodamine (Jackson Immunology, 115-025-020), goat anti-guinea pig IgG-Texas Red (Jackson Immunology, 106-585-003), goat anti-mouse IgG-HRP (Jackson Immunology,115-035-00.), and goat anti-rabbit IgG-HRP (Jackson Immunology, 111–035–144).
BODIPY493/503 was purchased from Invitrogen (Invitrogen, D3922). DAPI (Sigma 268,298) and LPS (Sigma, L2755) were purchased from Sigma. Synthetic Aβ (1–42) peptide was purchased from Bachem (Bachem 4,014,447) and dissolved in DMSO (ThermoFisher, J66650-AK) before oligomer preparation. Aβ aggregation was prepared according to previous report [82]. oAβ were incubated with microglia overnight before experiments. PLX5622 was purchased from Chemgood (C-1521). For long term dosing, the compounds were formulated in AIN-76A standard chow by Research Diets Inc. at 1200 ppm. The PLX5622-mixed chow and control diet were given to 2 months old FAD mice for 2 mon.
Histology and immunofluorescence (IF)
Mice were euthanized using CO2, and brain was harvested during necropsy. Fixation was conducted for 16 h at 4°C using 4% (w:v) freshly made, pre-chilled PBS (Invitrogen 10,010,023) buffered paraformaldehyde/PFA. The brain tissues were all sagittally separated into two hemispheres through midline and one hemisphere was embedded in paraffin, sectioned at 5 μm. Slides from histologically comparable positions (triangular lateral ventricle with intact rostral migratory stream) were stained with hematoxylin and eosin (H&E) for routine histological examination or left unstained for immunofluorescence (IF). H&E-stained sections were examined under a B×41light microscope (Olympus America, Inc., Center Valley, PA), and images were captured with an Olympus digital camera (model DP70) using DP Controller software (Version 1.2.1.10 8). For immunofluorescence, unstained tissues were first deparaffinized in 3 washes of xylene (3 min each) and then were rehydrated in graded ethanol solutions (100, 95, 70, 50, and 30%). After heat-activated antigen retrieval (Retriever 2000, PickCell Laboratories B.V., Amsterdam, Holland) according to the manufacturer’s specifications, sections were treated with Protein Block Serum Free (Agilent, X090930–2) at room temperature for 10 min. Slices were then incubated with the primary antibodies at 4°C for 16 h in a humidified chamber, washed in PBS for 3 times (5 min each) and incubated with the 1:200 secondary antibodies for 1 h at room temperature. After incubation with secondary antibodies and washed in PBS for 3 times (5 min each), nuclei were stained with DAPI and mounted with Vectashield mounting medium (Vector Laboratories, H-1200-10). Digital photography was carried out as described previously [83].
BrdU incorporation assay
BrdU (Sigma, B5002) was administrated intraperitoneally at 100 µg/gram for 3 times with 2 h interval every day for 3 consecutive days. Mice were euthanized 4 weeks later for long term BrdU retention, and tissues were processed as described above. For BrdU detection, the samples were treated with 2 M HCL at room temperature for 20 min to denature the nucleotides and then neutralized with 0.1 M sodium borate at room temperature for another 20 min. After 3 washes in PBS (5 min each), slides were incubated with mouse anti-BrdU antibody and secondary antibodies as described in IF staining. Nuclei were stained with DAPI and mounted with Vectashield mounting medium. Histological examination and digital photography were conducted as described above.
Percoll gradient isolation and FACS for enriched microglia
The method for isolation of microglia was as what we described before [37]. In brief, dissected cortex and hippocampus were passed through a 70-μm cell strainer. Homogenates were centrifuged at 600 g for 6 min. Supernatants were removed, and cell pellets were resuspended in 70% isotonic Percoll (GE Healthcare, 17-5445-01). A discontinuous Percoll density gradient was layered as follows: 50%, 35%, and 0% isotonic Percoll. The gradient was centrifuged for 20 min at 2000 g, and enriched microglia were collected from the interphase between the 70% and 50% Percoll layers. Enriched microglia were labeled with antibodies for flow cytometry and sorted based on ITGAM/CD11b, P2RY12, and TREM2 expression using a Bio-Rad S3e cytometer/cell sorter (Bio-Rad 12,007,058).
Real time PCR
Total RNAs were isolated from sorted microglia using the Single Cell RNA Purification Kit (Norgen Biotek Corp., 51800) according to the user manual. Reverse transcription complementary DNAs (cDNAs) were synthesized with iScript cDNA Synthesis Kit (Bio-Rad 1,708,891). Real-time PCR was performed with iQ SYBR Green Supermix Kit (Bio-Rad, 170–8880). Expression values were normalized to Actb/β-actin. The mouse Atg5 primer was obtained from PrimerBank (https://pga.mgh.harvard.edu/primerbank/) unless specific references were cited. The specificity of primer was validated with their dissociation curves.
Protein extraction, SDS-PAGE, and western blotting
Cells were used for protein extraction by homogenization in modified radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 7.4, 1% Triton X-100 (ThermoFisher 85,112), 0.2% sodium deoxycholate (ThermoFisher 89,905), 0.2% SDS, 1 mm sodium EDTA) supplemented with protease inhibitor (Sigma, P8340) and 1 mm phenylmethylsulfonyl fluoride (Sigma 52,332). After removing cell debris by centrifugation at 15,000 g for 10 min at 4°C, protein concentration was determined using Bio-Rad protein assay reagent (Bio-Rad 5,000,001). The lysates were boiled for 5 min in 1×SDS sample buffer (50 mm Tris-HCl, pH 6.8, 12.5% glycerol, 1% SDS, 0.01% bromophenol blue) containing 5% β-mercaptoethanol (Sigma, M6250). They were then analyzed by SDS-PAGE followed by western blotting using various antibodies, as described previously [18,38,79,84].
Statistical analysis
Lengths, areas, and the number of cells from comparable sections were quantified using the ImageJ software package. Statistical significance was evaluated by One-way ANOVA, Two-way ANOVA, and student’s t-test with p < 0.05 as indicative of statistical significance using Graph Pad Prism (Version 7.0). The number of animals used for quantification was indicated in the figure legends.
Supplementary Material
Acknowledgements
We thank Dr. Jun-Lin Guan for his suggestions and revision for the manuscript. We thank Dr. Aarti Nagayach, Ms. Gabrielle Angst, and Ms. Haley N Hurst in Dr. Chenran Wang’s lab for their work on the project.
Funding Statement
The work was supported by the Alzheimer's Association Research Grant [AARG-NTF-21-849207]; National Cancer Institute [CA273586]; National Heart, Lung, and Blood Institute [HL145176]; National Institute of Neurological Disorders and Stroke [R01NS103981].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2277634
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].Hebert LE, Weuve J, Scherr PA, et al. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013;80(19):1778–1783. doi: 10.1212/WNL.0b013e31828726f5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Velazquez R, Ferreira E, Winslow W, et al. Maternal choline supplementation ameliorates Alzheimer’s disease pathology by reducing brain homocysteine levels across multiple generations. Mol Psychiatry. 2020;25(10):2620–2629. doi: 10.1038/s41380-018-0322-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gage FH. Mammalian neural stem cells. Science. 2000;287(5457):1433–1438. doi: 10.1126/science.287.5457.1433 [DOI] [PubMed] [Google Scholar]
- [4].Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32(1):149–184. doi: 10.1146/annurev.neuro.051508.135600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70(4):687–702. doi: 10.1016/j.neuron.2011.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Moreno-Jimenez EP, Flor-García M, Terreros-Roncal J, et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med. 2019;25(4):554–560. doi: 10.1038/s41591-019-0375-9 [DOI] [PubMed] [Google Scholar]
- [7].Terreros-Roncal J, Moreno-Jiménez EP, Flor-García M, et al. Impact of neurodegenerative diseases on human adult hippocampal neurogenesis. Science. 2021;374(6571):1106–1113. doi: 10.1126/science.abl5163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhou Y, Su Y, Li S, et al. Molecular landscapes of human hippocampal immature neurons across lifespan. Nature. 2022;607(7919):527–533. doi: 10.1038/s41586-022-04912-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hamilton LK, Dufresne M, Joppé SE, et al. Aberrant lipid metabolism in the forebrain niche suppresses adult neural stem cell proliferation in an animal model of Alzheimer’s disease. Cell Stem Cell. 2015;17(4):397–411. doi: 10.1016/j.stem.2015.08.001 [DOI] [PubMed] [Google Scholar]
- [10].Moon M, Cha MY, Mook-Jung I. Impaired hippocampal neurogenesis and its enhancement with ghrelin in 5XFAD mice. J Alzheimers Dis. 2014;41(1):233–241. doi: 10.3233/JAD-132417 [DOI] [PubMed] [Google Scholar]
- [11].Zaletel I, Schwirtlich M, Perović M, et al. Early impairments of hippocampal neurogenesis in 5xFAD mouse model of Alzheimer’s disease are associated with altered expression of SOXB transcription factors. J Alzheimers Dis. 2018;65(3):963–976. doi: 10.3233/JAD-180277 [DOI] [PubMed] [Google Scholar]
- [12].Scopa C, Marrocco F, Latina V, et al. Impaired adult neurogenesis is an early event in Alzheimer’s disease neurodegeneration, mediated by intracellular Aβ oligomers. Cell Death Differ. 2020;27(3):934–948. doi: 10.1038/s41418-019-0409-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Choi SH, Bylykbashi E, Chatila ZK, et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science. 2018;361(6406). doi: 10.1126/science.aan8821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sato K. Effects of microglia on neurogenesis. Glia. 2015;63(8):1394–1405. doi: 10.1002/glia.22858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sierra A, Encinas JM, Deudero JJ, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7(4):483–495. doi: 10.1016/j.stem.2010.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gemma C, Bachstetter AD. The role of microglia in adult hippocampal neurogenesis. Front Cell Neurosci. 2013;7:229. doi: 10.3389/fncel.2013.00229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Walton NM, Sutter BM, Laywell ED, et al. Microglia instruct subventricular zone neurogenesis. Glia. 2006;54(8):815–825. doi: 10.1002/glia.20419 [DOI] [PubMed] [Google Scholar]
- [18].Wang C, Yeo S, Haas MA, et al. Autophagy gene FIP200 in neural progenitors non–cell autonomously controls differentiation by regulating microglia. Journal Of Cell Biology. 2017;216(8):2581–2596. doi: 10.1083/jcb.201609093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302(5651):1760–1765. doi: 10.1126/science.1088417 [DOI] [PubMed] [Google Scholar]
- [20].Appel JR, Ye S, Tang F, et al. Increased microglial activity, impaired adult hippocampal neurogenesis, and depressive-like behavior in microglial VPS35-depleted mice. J Neurosci. 2018;38(26):5949–5968. doi: 10.1523/JNEUROSCI.3621-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Bio. 2018;217(2):459–472. doi: 10.1083/jcb.201709069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17(3):157–172. doi: 10.1038/s41582-020-00435-y [DOI] [PubMed] [Google Scholar]
- [23].Spangenberg E, Severson PL, Hohsfield LA, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun. 2019;10(1):3758. doi: 10.1038/s41467-019-11674-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yuan P, Condello C, Keene C, et al. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. 2016;92(1):252–264. doi: 10.1016/j.neuron.2016.09.016 [DOI] [PubMed] [Google Scholar]
- [25].Ortega-Martinez S, Palla N, Zhang X, et al. Deficits in enrichment-dependent neurogenesis and enhanced anxiety behaviors mediated by expression of Alzheimer’s disease-linked Ps1 variants are rescued by microglial Depletion. J Neurosci. 2019;39(34):6766–6780. doi: 10.1523/JNEUROSCI.0884-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12(9):823–830. doi: 10.1038/ncb0910-823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Aman Y, Schmauck-Medina T, Hansen M, et al. Autophagy in healthy aging and disease. Nat Aging. 2021;1(8):634–650. doi: 10.1038/s43587-021-00098-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–695. doi: 10.1016/j.cell.2011.07.030 [DOI] [PubMed] [Google Scholar]
- [29].Wang C, Haas M, Yeo SK, et al. Enhanced autophagy in Becn1 F121A/F121A knockin mice counteracts aging-related neural stem cell exhaustion and dysfunction. Autophagy. 2022;18(2):409–422. doi: 10.1080/15548627.2021.1936358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lucin KM, O’Brien C, Bieri G, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron. 2013;79(5):873–886. doi: 10.1016/j.neuron.2013.06.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cho MH, Cho K, Kang H-J, et al. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10(10):1761–1775. doi: 10.4161/auto.29647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lin CL, Cheng Y-S, Li H-H, et al. Amyloid-β suppresses AMP-activated protein kinase (AMPK) signaling and contributes to α-synuclein-induced cytotoxicity. Exp Neurol. 2016;275(Pt 1):84–98. doi: 10.1016/j.expneurol.2015.10.009 [DOI] [PubMed] [Google Scholar]
- [33].Heckmann BL, Teubner BJW, Tummers B, et al. LC3-associated endocytosis facilitates β-amyloid clearance and mitigates neurodegeneration in murine Alzheimer’s disease. Cell. 2019;178(3):536–551 e14. doi: 10.1016/j.cell.2019.05.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Oakley H, Cole SL, Logan S, et al. Intraneuronal β-amyloid Aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Goldmann T, Wieghofer P, Müller PF, et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci. 2013;16(11):1618–1626. doi: 10.1038/nn.3531 [DOI] [PubMed] [Google Scholar]
- [36].Vina J, Lloret A. Why women have more Alzheimer’s disease than men: gender and mitochondrial toxicity of amyloid-β peptide. J Alzheimers Dis. 2010;20 Suppl 2(s2):S527–33. doi: 10.3233/JAD-2010-100501 [DOI] [PubMed] [Google Scholar]
- [37].Wohleb ES, Terwilliger R, Duman CH, et al. Stress-induced neuronal colony stimulating factor 1 provokes microglia-mediated neuronal remodeling and depressive-like behavior. Biol Psychiatry. 2018;83(1):38–49. doi: 10.1016/j.biopsych.2017.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang C, Liang C-C, Bian ZC, et al. RB1CC1 is required for maintenance and differentiation of postnatal neural stem cells. Nat Neurosci. 2013;16(5):532–542. doi: 10.1038/nn.3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kim S, Nam Y, Jeong Y-O, et al. Topographical visualization of the reciprocal projection between the medial septum and the hippocampus in the 5XFAD mouse model of Alzheimer’s disease. IJMS. 2019;20(16):20(16. doi: 10.3390/ijms20163992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kimmey JM, Huynh JP, Weiss LA, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015;528(7583):565–569. doi: 10.1038/nature16451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Martinez J, Cunha LD, Park S, et al. RETRACTED ARTICLE: noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533(7601):115–119. doi: 10.1038/nature17950 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [42].Wang YT, Zaitsev K, Lu Q, et al. Select autophagy genes maintain quiescence of tissue-resident macrophages and increase susceptibility to listeria monocytogenes. Nat Microbiol. 2020;5(2):272–281. doi: 10.1038/s41564-019-0633-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kim HJ, Cho M-H, Shim WH, et al. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry. 2017;22(11):1576–1584. doi: 10.1038/mp.2016.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12(5):284–296. doi: 10.1038/nrn3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Marschallinger J, Iram T, Zardeneta M, et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci. 2020;23(2):194–208. doi: 10.1038/s41593-019-0566-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lopez-Otin C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Näslund J, Haroutunian V, Mohs R, et al. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283(12):1571–1577. doi: 10.1001/jama.283.12.1571 [DOI] [PubMed] [Google Scholar]
- [48].Hollands C, Tobin MK, Hsu M, et al. Depletion of adult neurogenesis exacerbates cognitive deficits in Alzheimer’s disease by compromising hippocampal inhibition. Mol Neurodegener. 2017;12(1):64. doi: 10.1186/s13024-017-0207-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang Q, Chen G, Schindler SE, et al. Baseline microglial activation correlates with brain amyloidosis and longitudinal cognitive decline in Alzheimer disease. Neurol Neuroimmunol Neuroinflamm. 2022;9(3):9(3. doi: 10.1212/NXI.0000000000001152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest. 2008;118(6):2190–2199. doi: 10.1172/JCI33585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Shibata M, Lu T, Furuya T, et al. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem. 2006;281(20):14474–14485. doi: 10.1074/jbc.M600364200 [DOI] [PubMed] [Google Scholar]
- [52].Lipinski MM, Zheng B, Lu T, et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2010;107(32):14164–14169. doi: 10.1073/pnas.1009485107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tramutola A, Triplett JC, Di Domenico F, et al. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem. 2015;133(5):739–749. doi: 10.1111/jnc.13037 [DOI] [PubMed] [Google Scholar]
- [54].Plaza-Zabala A, Sierra-Torre V, Sierra A. Assessing autophagy in microglia: a two-step model to determine autophagosome formation, degradation, and net turnover. Front Immunol. 2020;11:620602. doi: 10.3389/fimmu.2020.620602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jülg J, Strohm L, Behrends C. Canonical and non-canonical autophagy pathways in microglia. Mol Cell Biol. 2020;41(3). doi: 10.1128/MCB.00389-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cheng J, Liao Y, Dong Y, et al. Microglial autophagy defect causes parkinso n disease-like symptoms by accelerating inflammasome activation in mice. Autophagy. 2020;16(12):2193–2205. doi: 10.1080/15548627.2020.1719723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Srimat Kandadai K, Kotur MB, Dokalis N, et al. ATG5 in microglia does not contribute vitally to autoimmune neuroinflammation in mice. Autophagy. 2021;17(11):3566–3576. doi: 10.1080/15548627.2021.1883880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Xu Y, Propson NE, Du S, et al. Autophagy deficiency modulates microglial lipid homeostasis and aggravates tau pathology and spreading. Proc Natl Acad Sci U S A. 2021;118(27). doi: 10.1073/pnas.2023418118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Choi I, Wang M, Yoo S, et al. Autophagy enables microglia to engage amyloid plaques and prevents microglial senescence. Nat Cell Biol. 2023;25(7):963–974. doi: 10.1038/s41556-023-01158-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Qin Y, Qiu J, Wang P, et al. Impaired autophagy in microglia aggravates dopaminergic neurodegeneration by regulating NLRP3 inflammasome activation in experimental models of Parkinson’s disease. Brain Behav Immun. 2021;91:324–338. doi: 10.1016/j.bbi.2020.10.010 [DOI] [PubMed] [Google Scholar]
- [61].Choi I, Zhang Y, Seegobin SP, et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 2020;11(1):1386. doi: 10.1038/s41467-020-15119-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Houtman J, Freitag K, Gimber N, et al. Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP3. The EMBO Journal. 2019;38(4):38(4. doi: 10.15252/embj.201899430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Berglund R, Guerreiro-Cacais AO, Adzemovic MZ, et al. Microglial autophagy–associated phagocytosis is essential for recovery from neuroinflammation. Sci Immunol. 2020;5(52):5(52. doi: 10.1126/sciimmunol.abb5077 [DOI] [PubMed] [Google Scholar]
- [64].Keren-Shaul H, Spinrad A, Weiner A, et al. A Unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276–1290 e17. doi: 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- [65].Deczkowska A, Keren-Shaul H, Weiner A, et al. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell. 2018;173(5):1073–1081. doi: 10.1016/j.cell.2018.05.003 [DOI] [PubMed] [Google Scholar]
- [66].Chen S, Wang C, Yeo S, et al. Distinct roles of autophagy-dependent and -independent functions of RB1CC1 revealed by generation and analysis of a mutant knock-in mouse model. Genes Dev. 2016;30(7):856–869. doi: 10.1101/gad.276428.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shang D, Wang L, Klionsky DJ, et al. Sex differences in autophagy-mediated diseases: toward precision medicine. Autophagy. 2021;17(5):1065–1076. doi: 10.1080/15548627.2020.1752511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ferretti MT, Iulita MF, Cavedo E, et al. Sex differences in Alzheimer disease — the gateway to precision medicine. Nat Rev Neurol. 2018;14(8):457–469. doi: 10.1038/s41582-018-0032-9 [DOI] [PubMed] [Google Scholar]
- [69].Mazure CM, Swendsen J. Sex differences in Alzheimer’s disease and other dementias. Lancet Neurol. 2016;15(5):451–452. doi: 10.1016/S1474-4422(16)00067-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Mangold CA, Wronowski B, Du M, et al. Sexually divergent induction of microglial-associated neuroinflammation with hippocampal aging. J Neuroinflammation. 2017;14(1):141. doi: 10.1186/s12974-017-0920-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kang SS, Ebbert MTW, Baker KE, et al. Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J Exp Med. 2018;215(9):2235–2245. doi: 10.1084/jem.20180653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Yanguas-Casás N, Crespo‐Castrillo A, Arevalo M-A, et al. Aging and sex: impact on microglia phagocytosis. Aging Cell. 2020;19(8):e13182. doi: 10.1111/acel.13182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kodama L, Guzman E, Etchegaray JI, et al. Microglial microRNAs mediate sex-specific responses to tau pathology. Nat Neurosci. 2020;23(2):167–171. doi: 10.1038/s41593-019-0560-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Gallagher JJ, Minogue AM, Lynch MA. Impaired performance of female APP/PS1 mice in the Morris water maze is coupled with increased Aβ accumulation and microglial activation. Neurodegener Dis. 2013;11(1):33–41. doi: 10.1159/000337458 [DOI] [PubMed] [Google Scholar]
- [75].Hirata-Fukae C, Li H-F, Hoe H-S, et al. Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 2008;1216:92–103. doi: 10.1016/j.brainres.2008.03.079 [DOI] [PubMed] [Google Scholar]
- [76].Elmore MRP, Hohsfield LA, Kramár EA, et al. Replacement of microglia in the aged brain reverses cognitive, synaptic, and neuronal deficits in mice. Aging Cell. 2018;17(6):e12832. doi: 10.1111/acel.12832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Orthgiess J, Gericke M, Immig K, et al. Neurons exhibit Lyz2 promoter activity in vivo: Implications for using LysM-Cre mice in myeloid cell research. Eur J Immunol. 2016;46(6):1529–1532. doi: 10.1002/eji.201546108 [DOI] [PubMed] [Google Scholar]
- [78].Zhuo L, Theis M, Alvarez‐Maya I, et al. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31(2):85–94. doi: 10.1002/gene.10008 [DOI] [PubMed] [Google Scholar]
- [79].Wang C, Chen S, Yeo S, et al. Elevated p62/SQSTM1 determines the fate of autophagy-deficient neural stem cells by increasing superoxide. J Cell Bio. 2016;212(5):545–560. doi: 10.1083/jcb.201507023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Liu H, Wang C, Yi F, et al. Non-canonical function of RB1CC1 is required for neural stem cell maintenance and differentiation by limiting TBK1 activation and p62 aggregate formation. Sci Rep. 2021;11(1):23907. doi: 10.1038/s41598-021-03404-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gao Z, Nissen JC, Ji K, et al. The experimental autoimmune encephalomyelitis disease course is modulated by nicotine and other cigarette smoke components. PLoS One. 2014;9(9):e107979. doi: 10.1371/journal.pone.0107979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Stine WB, Jungbauer L, Yu C, et al. Preparing synthetic Aβ in different aggregation states. Methods Mol Biol. 2011;670:13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Wei H, Wei S, Gan B, et al. Suppression of autophagy by RB1CC1 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25(14):1510–1527. doi: 10.1101/gad.2051011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wang C, Yoo Y, Fan H, et al. Regulation of Integrin β 1 recycling to lipid rafts by Rab1a to promote cell migration. J Biol Chem. 2010;285(38):29398–29405. doi: 10.1074/jbc.M110.141440 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.