

Abstract
Immune cells rely on transient physical interactions with other immune and non-immune populations to regulate their function1. To study these “kiss-and-run” interactions directly in vivo, we previously developed LIPSTIC (Labeling Immune Partnerships by SorTagging Intercellular Contacts)2, an approach that uses enzymatic transfer of a labeled substrate between the molecular partners CD40L and CD40 to label interacting cells. Reliance on this pathway limited the use of LIPSTIC to measuring interactions between CD4+ helper T cells and antigen presenting cells, however. Here, we report the development of a universal version of LIPSTIC (uLIPSTIC), which can record physical interactions both among immune cells and between immune and non-immune populations irrespective of the receptors and ligands involved. We show that uLIPSTIC can be used, among other things, to monitor the priming of CD8+ T cells by dendritic cells, reveal the steady-state cellular partners of regulatory T (Treg) cells, and identify germinal center (GC)-resident T follicular helper (Tfh) cells based on their ability to interact cognately with GC B cells. By coupling uLIPSTIC with single-cell transcriptomics, we build a catalog of the immune populations that physically interact with intestinal epithelial cells (IECs) at steady state and profile the evolution of the interactome of lymphocytic choriomeningitis virus (LCMV)-specific CD8+ T cells in multiple organs upon systemic infection. Thus, uLIPSTIC provides a broadly useful technology for measuring and understanding cell–cell interactions across multiple biological systems.
Physical interactions in which cells exchange signals through membrane-bound molecules at the core of multiple tissue functions3,4. In the immune system such interactions feature prominently, from the priming of T cells by dendritic cells (DCs) that initiates the adaptive immune response to the CD4+ T cell help that enables B cells to produce high-affinity antibodies1,5. More recent work has explored the role of interactions between immune and non-immune cells, such as those forming the epithelial barrier of the gut and skin, which are thought to drive transcriptional changes in immune cells that in turn enable them to support tissue function6,7. Despite their importance, direct observation of cell-cell interactions has traditionally been done by microscopy8, which has the key limitation that interacting cells cannot be retrieved for downstream analysis. Thus, the impact of the interaction on cell behavior and the cellular features that lead the interaction to occur in the first place cannot be inferred from traditional imaging alone. More recently, spatial transcriptomics and high-density imaging technologies have allowed for more in-depth characterization of the states of cells in the same neighborhood9. However, even when capable of high resolution, transcriptomic and imaging techniques still report on proximity between cells rather than on true physical interaction and signal exchange between membranes, requiring additional indirect methods and assumptions to infer functional interactions computationally (e.g.10). High throughput identification of cellular interactors and full deconvolution of the transcriptomic effects of physical interaction on cellular behavior and function are therefore yet to be achieved.
Many such limitations can be overcome by proximity-based labeling across cellular membranes2,11–16. These approaches rely on equipping “donor” cells with enzymes or other signals that act over short distances to identify “acceptor” cells in either close proximity or physical contact. An early example was our development of LIPSTIC, which uses enzymatic labeling across immune synapses to directly record cell-cell interactions in vivo2. In its first iteration, LIPSTIC labeled only interactions delivered through CD40 and CD40L, which restricted its utility to interactions involving effector CD4+ T cells. Here, we report the development of a universal (u)LIPSTIC tool, which enables us to record interactions between an extended array of cell types, regardless of the surface molecules involved. Coupling uLIPSTIC to standard single-cell mRNA sequencing (scRNA-seq) methods allows for atlas-type characterization of the “cellular interactome” of a population of interest and for the definition of the molecular pathways associated with such interactions. Thus, uLIPSTIC enables us to achieve truly quantitative interaction-based transcriptomics without need for computational inference of transcriptomes or interacting molecules.
Results
Universal recording of cell interactions
LIPSTIC uses the Staphylococcus aureus transpeptidase Sortase A (SrtA) to covalently transfer a peptide substrate containing the motif LPETG onto an N-terminal pentaglycine (G5) acceptor2. In its original version, catalysis by the very low-affinity (~1.8 mM) interaction between LPXTG-loaded SrtA and its G5 target17,18 was favored by genetically fusing each component to one of the members of a receptor–ligand pair2, thus raising local concentration of the reactants above the threshold required for substrate transfer (Fig. 1A). We reasoned that a similarly high local concentration of enzyme and target could also be achieved in a “universal,” receptor–ligand-independent manner by driving very high expression of SrtA and G5 on apposing cell membranes without direct fusion to the interacting molecules, potentially providing a readout for physical interactions between cells of any type (Fig. 1B).
Figure 1 |. The uLIPSTIC system.
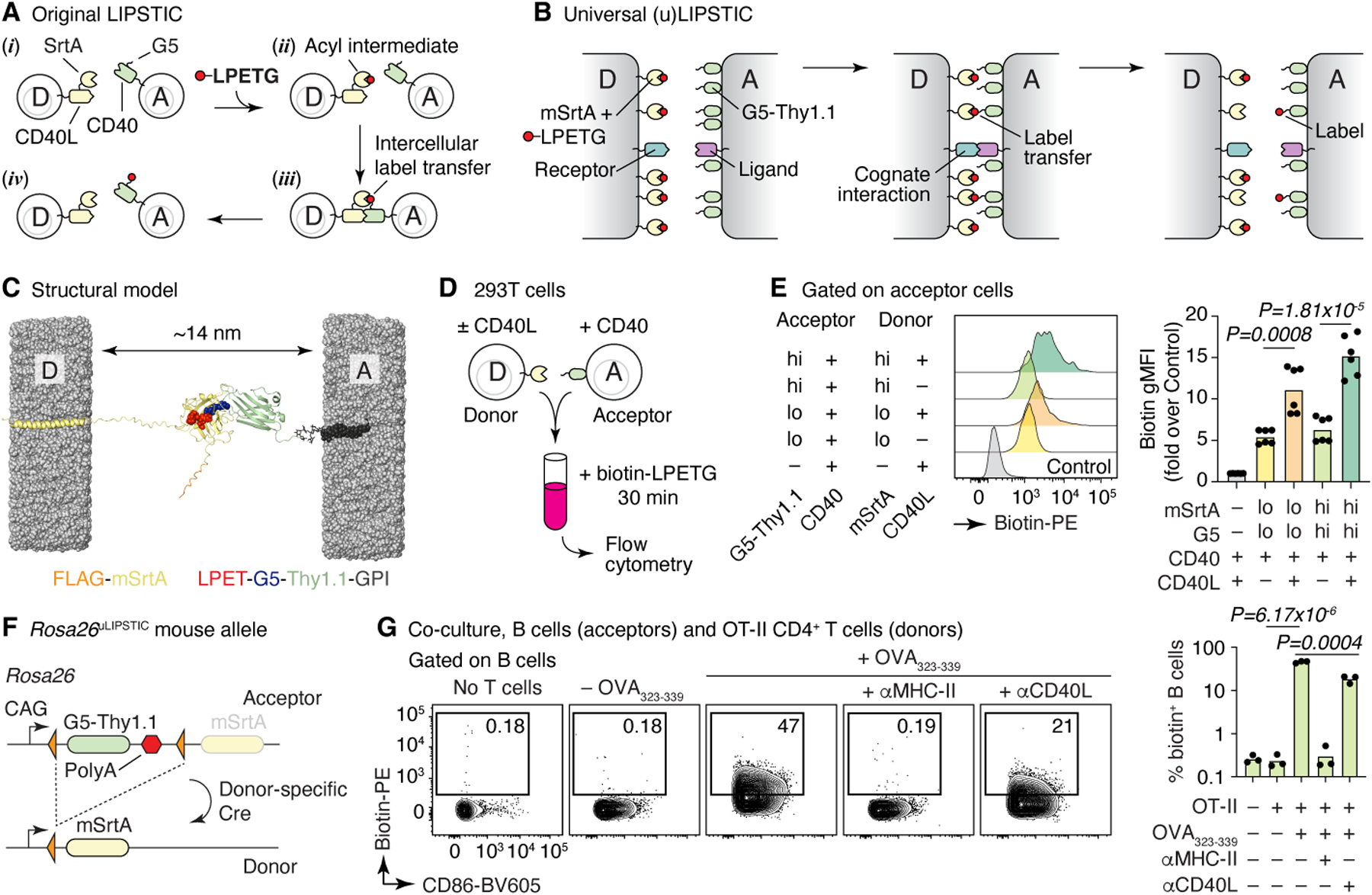
(A, B) Schematic comparison of the original2 and universal LIPSTIC systems. In the original system (A), SrtA and G5 were brought into proximity by fusion to a receptor–ligand pair involved in a cell–cell interaction, allowing intercellular transfer of labeled substrate (LPETG) from donor cell “D” to acceptor cell “A.” In uLIPSTIC (B), SrtA and G5 (fused to the irrelevant protein Thy1.1) are anchored non-specifically to the cell membrane at high density; the enzymatic reaction is allowed to proceed when apposing membranes come within a short distance (< 14 nm) of each other, which can be driven by interactions between any receptor–ligand pair of the appropriate dimensions. (C) Computational model depicting the inter-membrane span of fully extended mSrtA upon transfer of the LPETG substrate onto G5-Thy1.1. (D,E) Populations of 293T cells co-transfected with high or low levels of either mSrtA or G5-Thy1.1 were co-incubated in the presence of biotin-LPETG for 30 min and analyzed by flow cytometry. Histograms show the extent of labeling of acceptor cells. Each symbol on column plot represents one technical replicate, pooled from two independent experiments. (F) Rosa26uLIPSTIC allele. Using the Ai9 high-expression backbone20, a LoxP-flanked G5-Thy1.1 is followed by mSrtA. Cre-recombinase switches cells from “acceptor” (G5-Thy1.1+) to “donor” (mSrtA+) modes. (G) Rosa26uLIPSTIC/+.CD4-Cre OT-II donor T cells were co-cultured with Rosa26uLIPSTIC/+ acceptor B cells in the presence or absence of OVA323–339 peptide and blocking antibodies to CD40L and MHC-II. Flow cytometry plots show biotin-LPETG transfer from T to B cells. Each symbol in column plot represents a biological replicate from three independent experiments. For (E) and (G), P-values were calculated using two-tailed Student’s tests.
To test this, we generated a donor–acceptor pair consisting of the “PDK” version of SrtA17 targeted to the plasma membrane by fusion to the human PDGFRB transmembrane domain2 (mSrtA) and the G5 peptide fused to the N-terminus of the mouse Thy1.1 GPI-anchored protein. 3D modeling (Fig. 1C) predicted the maximal distance between membranes at which label transfer would occur to be approximately 14 nm, comparable to the inter-membrane span required, for example, for the TCR-MHC interaction (~15 nm), and narrower than the typical distance separating juxtaposed cell membranes in the absence of receptor-ligand interactions, set by glycocalyx repulsion19. Given the negligible affinity (~1.8 mM) between SrtA-LPETG and G517, such a design would in principle allow for label transfer only when cells were functionally interacting at a close intermembrane distance, without driving artificial interactions between its engineered components. We then transfected HEK293T cells with high or low concentrations of plasmids expressing either mSrtA or G5-Thy1.1, adding biotin-LPETG substrate to combined cell populations as described2 (Fig. 1D). Label transfer was detectable above background when donor and acceptor populations were forced to interact by co-transfection of constructs encoding CD40L and CD40, respectively, and further increased when the uLIPSTIC components were transfected at the highest concentration (Fig. 1E). Thus, high membrane expression of SrtA and G5 allows LIPSTIC labeling in the absence of fusion to specific receptor–ligand pairs.
We generated a Rosa26uLIPSTIC mouse allele in which high expression of mSrtA (preceded by a FLAG-tag) or G5-Thy1.1 were driven by the strong cytomegalovirus early enhancer/chicken beta-actin/rabbit beta-globin (CAG) promoter introduced into the ubiquitously expressed Rosa26 locus20 (Fig. 1F and Extended Data Fig. 1A). The G5-Thy1.1 is flanked by LoxP sites, so that Cre-mediated recombination leads to expression of a previously silent downstream mSrtA, switching Cre-expressing cells from uLIPSTIC acceptors into uLIPSTIC donors (Extended Data Fig. 1D). To test this system, we crossed Rosa26uLIPSTIC mice to the CD4-Cre and OT-II T cell receptor transgenes to generate mSrtA+ uLIPSTIC donor T cells specific for peptide 323–339 of the model antigen chicken ovalbumin (OVA). Efficient transfer of labeled substrate between co-cultured T and B cells in occurred only in the presence of OVA323–339 (Fig. 1G). Substrate transfer was abrogated by addition of a blocking antibody to MHC-II, necessary for the cognate B cells–T cell interaction, but not by an antibody to CD40L (Fig. 1G). Loading of mSrtA+ donor T cells and its transfer onto G5-Thy1.1 acceptor DCs increased progressively in the first two hours of labeling, after which it plateaued (Extended Data Fig. 2A–E). Gradually decreasing peptide-MHC concentration or the affinity of the complex towards the OT-II TCR using truncated altered peptide ligands (APLs) as described21,22 led to reduced labeling of DCs in vitro (Extended Data Fig. 2F,G). We conclude that uLIPSTIC enables trans-synaptic labeling of contacts between immune cells regardless which receptor(s) and ligand(s) drive these interactions.
uLIPSTIC labeling in vivo
To test uLIPSTIC labeling in vivo, we used a well-established in vivo T cell priming model8,23, where G5-Thy1.1+ DCs loaded with OVA323–339 are injected into the footpads of mice followed by adoptive transfer of mSrtA+ OT-II T cells. Lymphatic migration of DCs to the draining popliteal lymph node (pLN) allows DC–T cell interactions to take place at this site (Fig. 2A). Footpad injection of biotin-LPETG substrate 24 h after T cell transfer led to detectable labeling of on average 6.5% of transferred DCs (Fig. 2B). Comparable numbers were obtained when using the original CD40L–CD40 LIPSTIC system2 (Fig. 2C). Treatment with anti-MHC-II prior to substrate injection blocked labeling in both settings (whereas treatment with anti-CD40L blocked transfer only by the original LIPSTIC), indicating that the uLIPSTIC components alone are insufficient to artificially drive interactions between neighboring cells also in vivo. Thus, uLIPSTIC labeling is equivalent to receptor–ligand-specific LIPSTIC for recording the binding patterns of CD4+ T cells and DCs during in vivo priming. Pulsing DCs with OVA323–339 APLs showed that the fraction of labeled DCs decreased as peptide-MHC affinity for the OT-II TCR decreased (Extended Data Fig. 2H–J). Transferring decreasing numbers of mSrtA+ donor T cells also decreased the degree to which interacting DCs were labeled (Extended Data Fig. 3A–E). Lastly, increasing the time interval between substrate administration and tissue harvesting led to a gradual decrease in biotin detection on the surface of acceptor cells, so that little substrate was detectable 4–6 h after the last injection of substrate (Extended Data Fig. 3F–I). Therefore, uLIPSTIC signal detection is useful for acute but not long-term tracking of interacting cells.
Figure 2 |. uLIPSTIC labeling of cell-cell interactions in vivo.
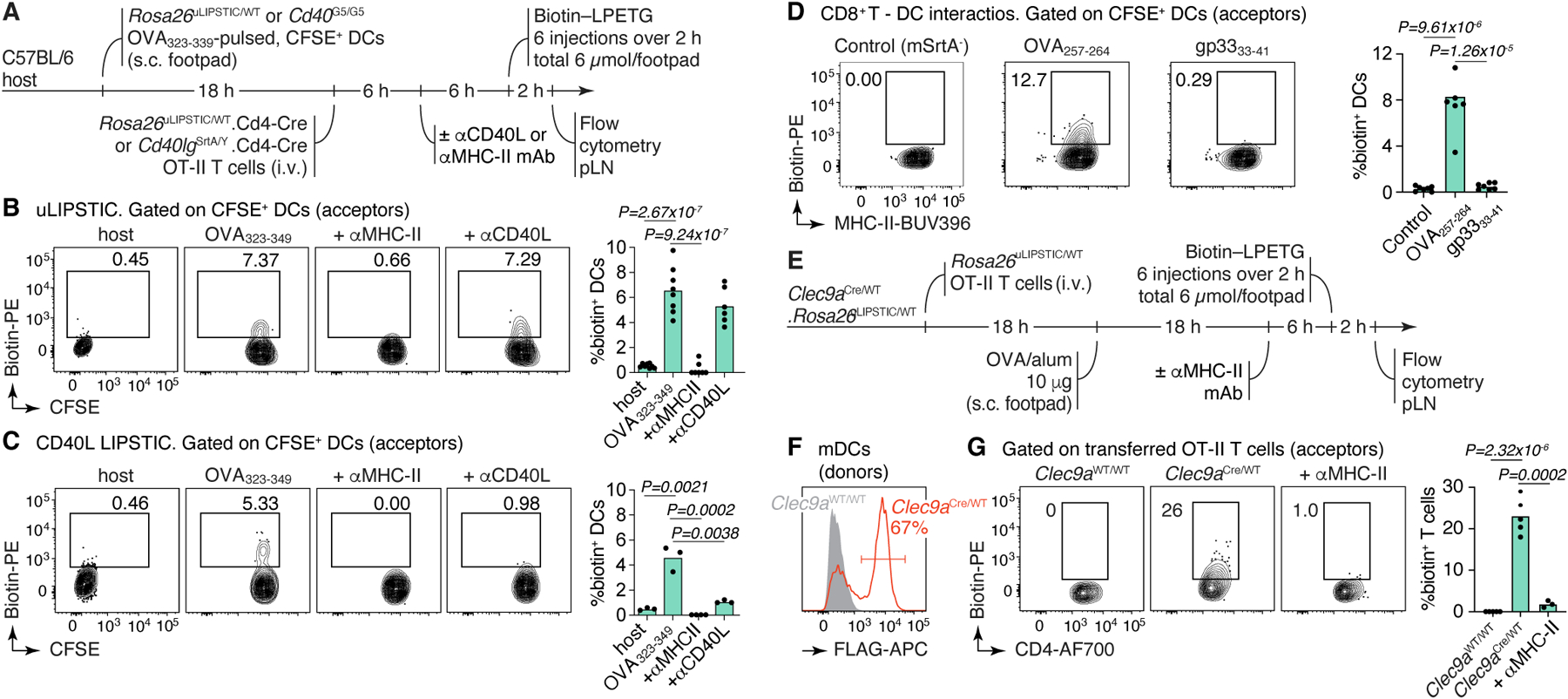
(A) Experimental layout for the experiments in panels (B,C). (B,C) uLIPSTIC (B) and CD40L LIPSTIC (C) labeling of adoptively-transferred DCs in an in vivo priming model. Flow cytometry plots are gated on transferred (CFSE-labeled) DCs. Column plot on the right summarize the extent of DC labeling. (D) uLIPSTIC labeling of DCs by CD8+ T cells. Experimental setup as in (A), but DCs were pulsed either with cognate (OVA257–264) or control (LCMV gp3333–41) peptides and transferred along with Rosa26uLIPSTIC/WT.CD4-Cre OT-I CD8+ donor T cells or control mSrtA– Rosa26uLIPSTIC/WT OT-I CD8+ T cells. Labeling of DCs is summarized in column plot. (E-G) Labeling of antigen-specific CD4+ T cells by Clec9a-expressing DCs. (E) Experimental layout. (F) efficiency of recombination of the uLIPSTIC allele in migratory (m)DCs by Clec9aCre. (G) Left, labeling of adoptively transferred OT-II T cells upon immunization with OVA/alum. Right, summary of data. All results shown in column plots are from two independent experiments, with each symbol representing one mouse. P-values were calculated using two-tailed Student’s tests.
We next used uLIPSTIC to record T cell–DC interactions that were inaccessible to the original LIPSTIC system, either because they do not involve the CD40L/CD40 interaction or because directionality is reversed. mSrtA+ OT-I CD8+ T cells labeled on average 8.3% of DCs pulsed with their cognate peptide (OVA257–264) but only background levels (0.5%) of DCs pulsed with the LCMV GP33–41 peptide (Fig. 2D). Inverting the uLIPSTIC reaction so that endogenous mSrtA+ DCs (in Rosa26uLIPSTIC/+.Clec9aCre/+ mice, in which most DCs are labeled due to Clec9a expression in common DC progenitors24) labeled adoptively transferred Rosa26uLIPSTIC/+ OT-II CD4+ T cells upon OVA immunization (Fig. 2E–G) led to detectable labeling of roughly 22% of transferred T cells (likely because of incomplete recombination of donor cDC2 cells by Clec9aCre; Extended Data Fig. 1E). Labeling was again fully abrogated by prior injection of a blocking antibody to MHC-II (Fig. 2F–G). Thus, uLIPSTIC can label interactions between T cells and DCs bidirectionally.
To test uLIPSTIC in settings other than naïve T cell priming, we first determined the identity of the cellular partners of regulatory T (Treg) cells in the steady state lymph node, using the Foxp3CreERT2 driver25 to achieve tamoxifen-dependent recombination of Rosa26uLIPSTIC specifically in Treg cells (Fig. 3A,B). Broad characterization of biotin+ acceptors showed that DCs are the primary population engaged by Treg cells at steady state, with a smaller contribution from macrophages (Extended Data Fig. 4A). Closer examination of the DC population showed pronounced labeling of most DCs with migratory (MHC-IIhiCD11cint) phenotype, whereas labeling of resident (MHC-IIintCD11chi) DCs was markedly lower (Fig. 3B,C). Importantly, labeling of CD8+ T cells and Foxp3– CD4+ T cells was negligible in this setting, confirming that simple colocalization of these populations with donor Treg cells within the same microenvironment is not sufficient to drive label transfer (Fig. 3B, left and Extended Data Fig. 4A). Expression of mSrtA+ in roughly equivalent numbers of Treg cells or total conventional CD4+ T cells (the latter achieved by low-dose tamoxifen administration to Rosa26uLIPSTIC/+.CD4-CreERT2 mice26) (Extended Data Fig. 4B,C) resulted in much less efficient labeling of migratory DCs by conventional T cells (Extended Data Fig. 4D). Thus, interaction with migratory DCs at steady state, although not a unique property of Treg cells, is more pronounced among this subset. Treg cell labeling of migratory DCs was decreased but not completely abrogated by administration of a blocking antibody to MHC-II, confirming that the strong interaction between Treg cells and migratory DCs is partly driven by the TCR-MHC-II axis but suggesting that other receptor-ligand pairs may also contribute to this process (Fig. 3B,C).
Figure 3 |. uLIPSTIC identifies cellular partners of Treg cells, Tfh cells, and IECs.
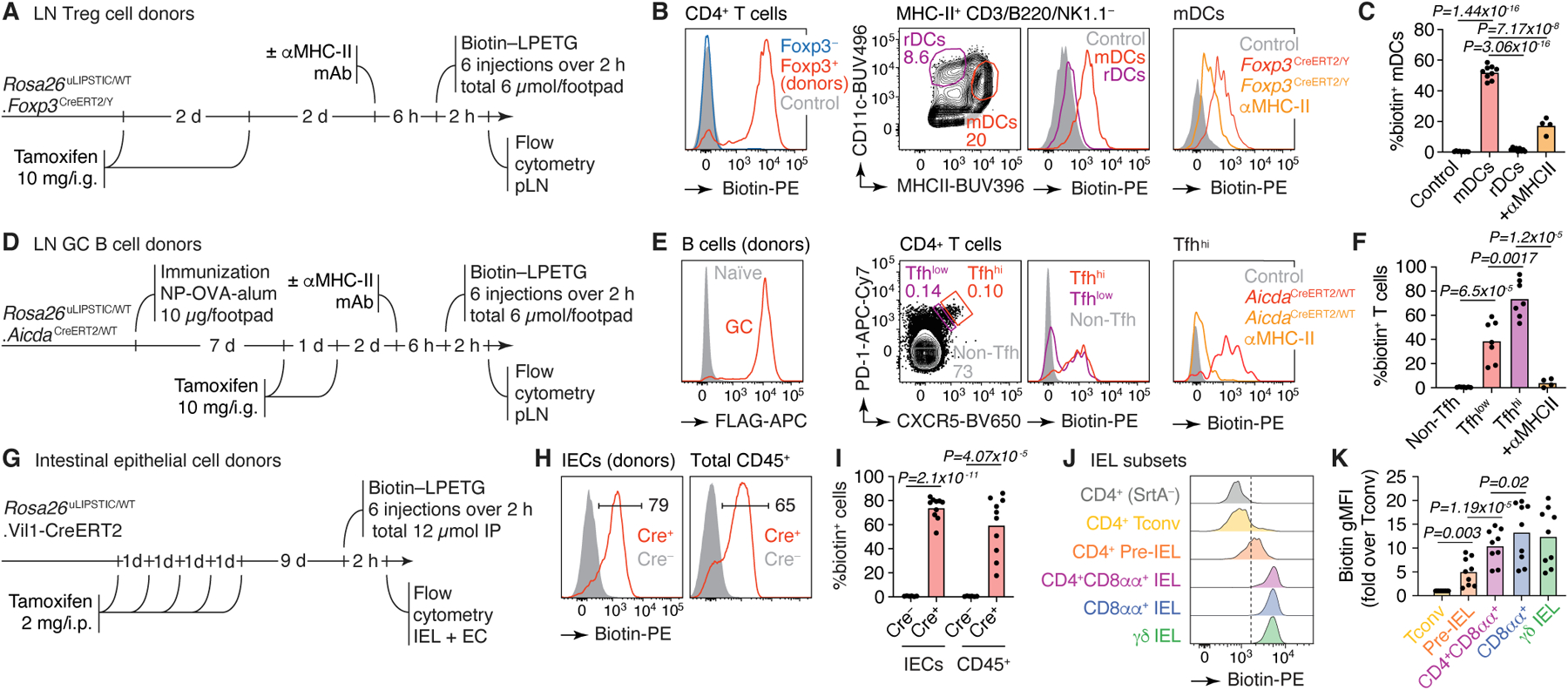
(A) Experimental layout for panels (B,C). (B) Left, efficiency of recombination of the uLIPSTIC allele in Treg cells by Foxp3CreERT2. Biotin signal represents the acquisition of substrate by Treg cells (the biotin-LPET-SrtA acyl intermediate) and also shows the absence of transfer of substrate to Foxp3– T cells. Center, labeling of migratory (m)DCs and resident (r)DCs by Treg cells at steady state. Right, labeling of mDCs upon injection of a blocking antibody to MHC-II. (C) Summary of data from 3 independent experiments. (D) Experimental layout for panels (E,F). (E) Labeling of Tfh cells by GC B cells. Left, efficiency of recombination of the uLIPSTIC allele in GC B cells by AicdaCreERT2 after 2 doses of tamoxifen, as in (B). Center, labeling of Tfh cells by GC B cells at 10 days after immunization with NP-OVA/alum. T cells are gated as high or low expressors of Tfh markers CXCR5 and PD-1 (Tfhhi and Tfhlo, respectively). Right, labeling of Tfhhi cells upon injection of a blocking antibody to MHC-II. (F) Summary of data from 2 independent experiments. (G) Experimental layout for panels (H-K). (H) Left, Efficiency of conversion of IECs into uLIPSTIC donors and substrate capture in Vil1-CreERT2 mice (as in (B)). Right, labeling of total CD45+ intraepithelial leukocytes. (I) Summary of data from three independent experiments. (J) Differential labeling of selected IEL populations by IEC donors. The dashed line is placed for reference. (K) Biotin geometric mean fluorescence intensity (gMFI) from three independent experiments is summarized. For all column plots, each symbol represents one mouse, bars represent the mean. P-values were calculated using two-tailed Student’s tests.
We next determined the phenotype of the T cells that provide help to B cells in germinal centers (GCs), which can be difficult to identify unambiguously using canonical Tfh markers CXCR5 and PD-127. We immunized Rosa26uLIPSTIC/+.AicdaCreERT2/+ mice28 in the footpads with the model antigen 4-hydroxi-3-nitro-phenylacetyl (NP)-OVA to generate GCs, then treated these mice with tamoxifen 7 and 8 days later to induce mSrtA expression in GC B cells (Fig. 3D). GC B cells replaced Thy1.1-G5 with mSrtA much faster than did resting CD4+ T cells, indicating that replacement kinetics vary across donor populations (Extended Data Fig. 4E–G). Biotin-LPETG injection 10 days post-immunization led to substantial labeling of CXCR5hiPD-1hi Tfh acceptor cells but not CXCR5–PD-1− non-Tfh cells in the pLN (Fig. 3E,F). Only a fraction of CXCR5intPD-1int T cells were labeled by GC B cells, indicating that relatively few of the cells in this population are indeed engaged with GC B cells. Again, blocking of MHC-II led to total loss of Tfh cell labeling, confirming the specificity of the reaction (Fig. 3E,F).
Lastly, we sought to test the ability of uLIPSTIC to record interactions between immune and non-immune cells outside of secondary lymphoid organs. Intraperitoneally (i.p.) injected LIPSTIC substrate reaches donor cells in multiple organs in mice (including brain, bone marrow, kidney, lungs, spleen, and thymus), and its use is therefore not limited to draining LNs (Extended Data Fig. 5). As a test case, we measured substrate transfer from intestinal epithelial cells (IECs) to the intraepithelial T lymphocytes (IELs) that reside within this compartment29. We crossed Rosa26uLIPSTIC mice to villin-1 (Vil1)-CreERT230 to generate IEC donors upon tamoxifen treatment (Fig. 3G). Intraperitoneally administered biotin-LPETG was transferred efficiently onto a large fraction (median 65%) of CD45+ IELs (Fig. 3H,I and Extended Data Fig. 6A,B). Labeling followed a gradient corresponding to the stage of differentiation of these cells: whereas “natural” TCRγδ+ and CD8αα+/TCRαβ+ IELs displayed uniformly high uLIPSTIC signal, labeling among induced CD4+ IELs31 followed closely their developmental trajectory6,32, from background levels in the CD4+CD8αα−CD103− “conventional” subset to intermediate labeling in CD4+CD8αα−CD103+ pre-IELs and levels comparable to those of natural IELs in the epithelium-adapted CD4+CD8αα+CD103+ population (Fig. 3J,K). Thus, uLIPSTIC is capable of recording interactions between epithelial and immune cells in the small intestine.
We conclude that uLIPSTIC can be used to label a wide variety of immune cell interactions in vivo across multiple organs, both in adoptive transfer and in fully endogenous models. In the latter, uLIPSTIC revealed the interaction preferences of steady-state LN Treg cells, identified populations of Tfh cells capable of providing help to B cells in the GC, and showed stepwise acquisition by intraepithelial CD4+ T cells of the ability to physically interact with IECs.
uLIPSTIC-based transcriptomics
A key feature of uLIPSTIC is its ability to identify the full cellular interactome of a given cell type in an unbiased manner. Reading out this interactome is best achieved by single-cell RNA sequencing (scRNA-seq), which is also unbiased in its ability to identify labeled cell populations. Because LIPSTIC labeling has a wide dynamic range2, coupling it to scRNA-seq also has the potential to identify genes and transcriptional programs quantitatively associated with the degree of interaction between two cell types, which can in principle reveal the molecular pathways that drive a given interaction (Fig. 4A). To explore these possibilities, we labeled Rosa26uLIPSTIC/WT.Vil1-CreERT2 mice as in Fig. 3G. Sorted CD45+ cells (enriched for rarer leukocyte populations as in Extended Data Fig. 6C) stained with a DNA-barcoded anti-biotin antibody were then profiled by droplet-based scRNA-seq using the 10X Genomics platform. Immune cell populations were identified by marker gene expression/TCR reconstruction and by comparison with publicly available gene signatures (Fig. 4B, Extended Data Figs. 6D–K and 7–8, and Supplementary Tables 1,2). uLIPSTIC revealed broad variation in the extent to which different populations interacted with IECs, which aligned with the data obtained by flow cytometry. Labeling was high among natural IELs (TCRγδ and TCRαβ+CD8αα+), low or negligible among B cell subsets, and intermediate in plasmacytoid DCs (Fig. 4B,C). uLIPSTIC also labeled two less clearly defined populations that interacted strongly with IECs, including a small cluster of likely myeloid cells and a larger cluster marked by high expression of genes such as Atxn1 and Btbd11 (Fig. 4C and Extended Data Figs. 6D,J,K and 7). CD4+ T cells again showed a gradient in their ability to interact with IECs, which became more apparent when these cells were clustered into subpopulations (Fig. 4D, Extended Data Fig. 8A–C, and Supplementary Table 3). The ability to acquire the biotin label largely followed a developmental trajectory (determined from gene expression alone) that began with a highly polyclonal naïve-like population with low uLIPSTIC signal and followed through a pre-IEL intermediate into a fully differentiated, oligoclonal CD4-IEL state6,32 (Fig. 4D–F) labeled to a similar extent as natural IELs (Fig. 4C).
Figure 4 |. Using uLIPSTIC for interaction-based transcriptomics.
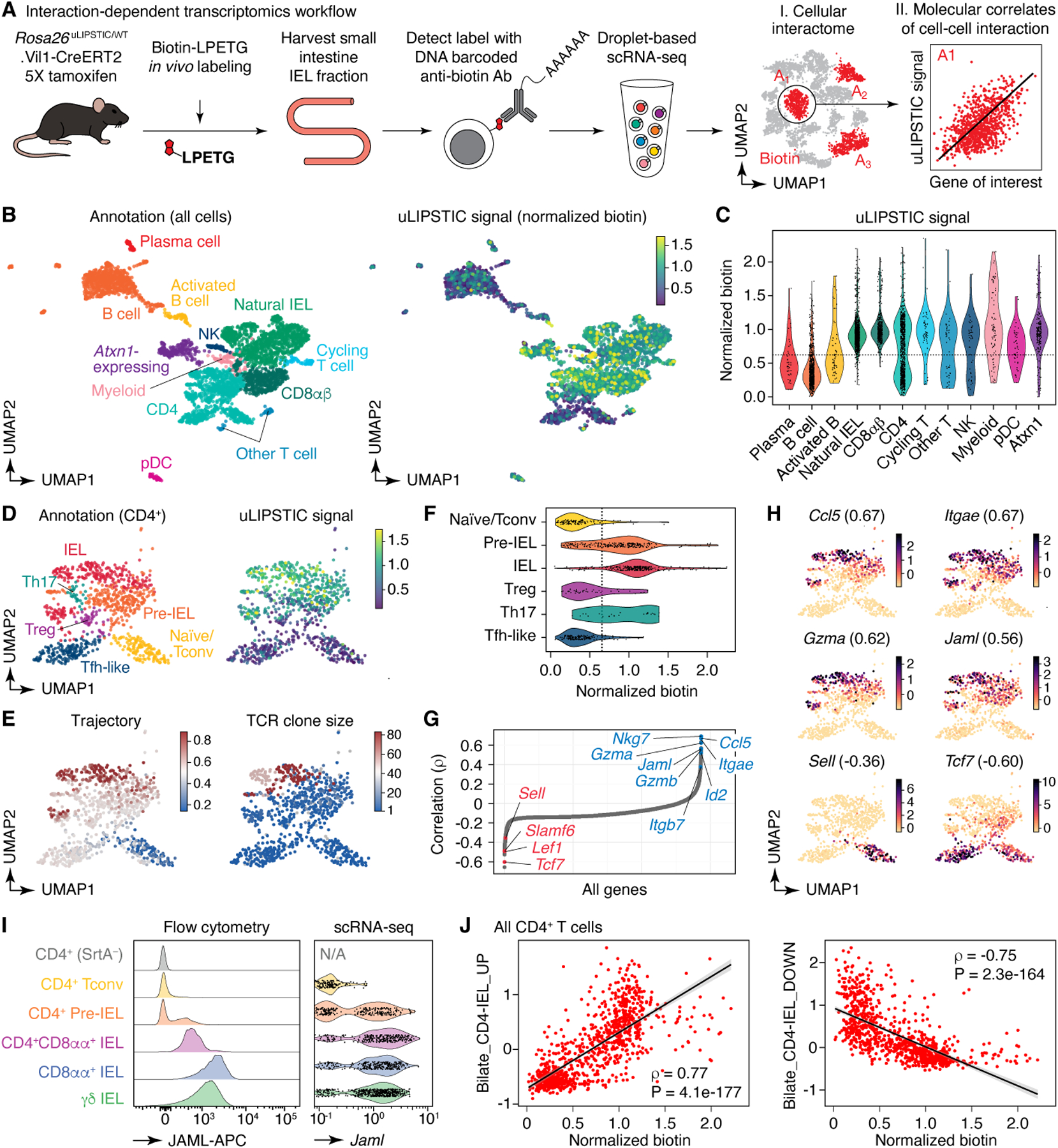
(A) Experimental workflow. (B) UMAP plots of the CD45+ intraepithelial immune cell fraction from a uLIPSTIC reaction as in Fig. 3G. Data pooled from three mice. Left, major cell populations (see Extended Data Figs. 6,7). Right, normalized uLIPSTIC signal in log-scaled arbitrary units. (C) Normalized uLIPSTIC signal among CD45+ cell populations. (D) UMAP plots of CD4+ T cells from (B), n=915 cells. Left, major cell subpopulations (see Extended Data Fig. 11). Right, normalized uLIPSTIC signal. (E) Left, inferred trajectory and right, αβTCR diversity (plotted as clone size) among CD4+ T cells. (F) Normalized uLIPSTIC signal among CD4+ T cell subpopulations. (G) Correlation (Spearman’s ρ) between normalized uLIPSTIC signal and normalized gene expression, calculated for each gene over all CD4+ T cells, shown in order of increasing correlation. Selected significantly correlated genes (FDR < 1e-23) are highlighted. (H) Normalized expression of selected genes. Correlation with normalized uLIPSTIC shown in parentheses. (I) Representative samples showing in vivo staining of JAML in IELs and scRNA-seq expression of Jaml in the equivalent populations. In the latter, CD8αα+ and γδ IEL were separated from within the “Natural IEL” cluster by the presence of rearranged αβ TCRs or expression of the Trdc gene. (J) Relationship between normalized uLIPSTIC signal among all CD4+ T cells and expression of gene signatures up and downregulated as epithelial T cells transition from Tconv (CD4+CD103−CD8αα−) to CD4-IEL (CD4+CD103+CD8αα+) phenotypes (signatures based on data from Bilate et al.6). Trend line and error are for linear regression with 95% confidence interval, Spearman’s ρ and two-sided P-value are listed.
Correlating the uLIPSTIC signal within CD4+ T cells with the expression of all detected genes in our dataset (Fig. 4G,H) revealed multiple significant correlations with markers of IEL differentiation. These included negative correlations with naïve T cell markers such as Sell (encoding for L-selectin) and Tcf7 and positive correlations with CD4-IEL associated genes such as Ccl5, Gzma, Itgae, Itgb7, and Jaml (Fig. 4G,H, Extended Data Fig. 8D,E, and Supplementary Table 4)6. The last three are of particular interest, given that CD103 (the αEβ7 integrin, encoded by Itgae and Itgb7) and JAML (junction adhesion molecule-like, encoded by Jaml) are interacting partners of E-cadherin and of the coxsackie and adenovirus receptor (CAR), respectively, both of which are expressed in the tight junctions of the intestinal epithelium33–36. Flow cytometry confirmed the correlation between biotin acquisition and expression of CD103 (Extended Data Fig. 8F), and in vivo staining with an anti-JAML antibody confirmed stepwise acquisition of this molecule during CD4-IEL development (Fig. 4I). Search for correlations among “canonical” (M2.CP) pathways in the MSigDB database37 revealed a significant positive correlation between biotin acquisition by CD4+ T cells and expression of genes in the Biocarta cytotoxic T lymphocyte (CTL) pathway, among others (Extended Data Fig. 8G). Targeted correlation analysis showed strong positive and negative correlations (|Spearman’s ρ| > 0.75) between biotin acquisition and expression of genes modulated as conventional T cells develop into CD4+ IELs6 (Fig. 4J, Extended Data Fig. 8H,I, and Supplementary Table 5). We conclude that uLIPSTIC allows for quantitative interaction-based transcriptomics, enabling us not only to define the cellular interactomes of populations of interest, but also to discern specific genes and signatures associated with acquisition of the ability to form specific cell-cell interactions.
Applying uLIPSTIC to LCMV infection
Finally, we investigated the interacting partners of virus-specific CD8+ T cells in a classic systemic infection model, using the Armstrong strain of LCMV38. We infected uLIPSTIC acceptor (Rosa26uLIPSTIC/WT) mice that had previously received CD8+ uLIPSTIC-donor T cells carrying the LCMV-specific P14 TCR (Rosa26uLIPSTIC/+.CD4-Cre.P14-tg) with LCMV by the i.p. route. We administered LIPSTIC substrate to these mice at different timepoints prior to harvesting the mediastinal (m)LN, a focal point of the early immune response in this model39 (Fig. 5A and Extended Data Fig. 9A). uLIPSTIC detected the expected engagement of P14 T cells with DCs as early as 36 hours post-infection (hpi), which peaked at 50 hpi and then declined by 96 hpi (Fig. 5B). However, DCs accounted for an average of only 5.4% of the full P14 cellular interactome at all timepoints analyzed (Fig. 5C and Extended Data Fig. 9B), suggesting that other populations in addition to DCs may contribute to the initial activation of LCMV-specific CD8+ T cells. uLIPSTIC-coupled single-cell transcriptomics identified the majority of P14-interacting cells at 36 hpi as monocytic lineage cells (potentially monocytes or macrophages) expressing high levels of Ly6c2 (“Mo/MF1” cluster; Fig. 5D–F, Extended Data Fig. 9D–J, and Supplementary Table 6). Following a phase of broader uLIPSTIC labeling spread evenly across most mLN populations at 50 hpi, the P14 interactome at 96 hpi became strongly enriched in a second cluster of monocytic cells expressing lower Ly6c2 and higher H2-Aa (the “Mo/MF2” cluster, comprising either a distinct population of macrophages or a further differentiation stage of Ly6c2-high monocytes40; Fig. 5D–F and Extended Data Fig. 9D–J). Flow cytometry of uLIPSTIC-labeled cells confirmed this transition: whereas P14 interactors in the monocyte/macrophage gate (F4/80+MHC-IIlo-int) consisted almost entirely of Ly-6ChiMHC-II– monocytes at 36 hpi, this population shifted markedly towards a Ly-6CintMHC-IIint phenotype at the 96 hpi timepoint (Fig. 5G). To determine whether these interactions were antigen-dependent, we compared labeling between mice infected with wild-type LCMV (LCMVWT) or with a mutant strain lacking the P14 epitope (LCMVΔP14)41. Whereas uLIPSTIC labeled a large fraction of monocytic lineage cells in mice infected with LCMVWT, such labeling was completely absent from LCMVΔP14-infected mice at all timepoints (Fig. 5H). Thus, the interactions between CD8+ T cells and monocyte lineage cells revealed by uLIPSITC are antigen-dependent, suggesting that the latter may acquire and present viral antigen in vivo at early timepoints after LCMV infection.
Figure 5 |. Using uLIPSTIC to dissect the early events in CD8+ T cell priming upon LCMV infection.
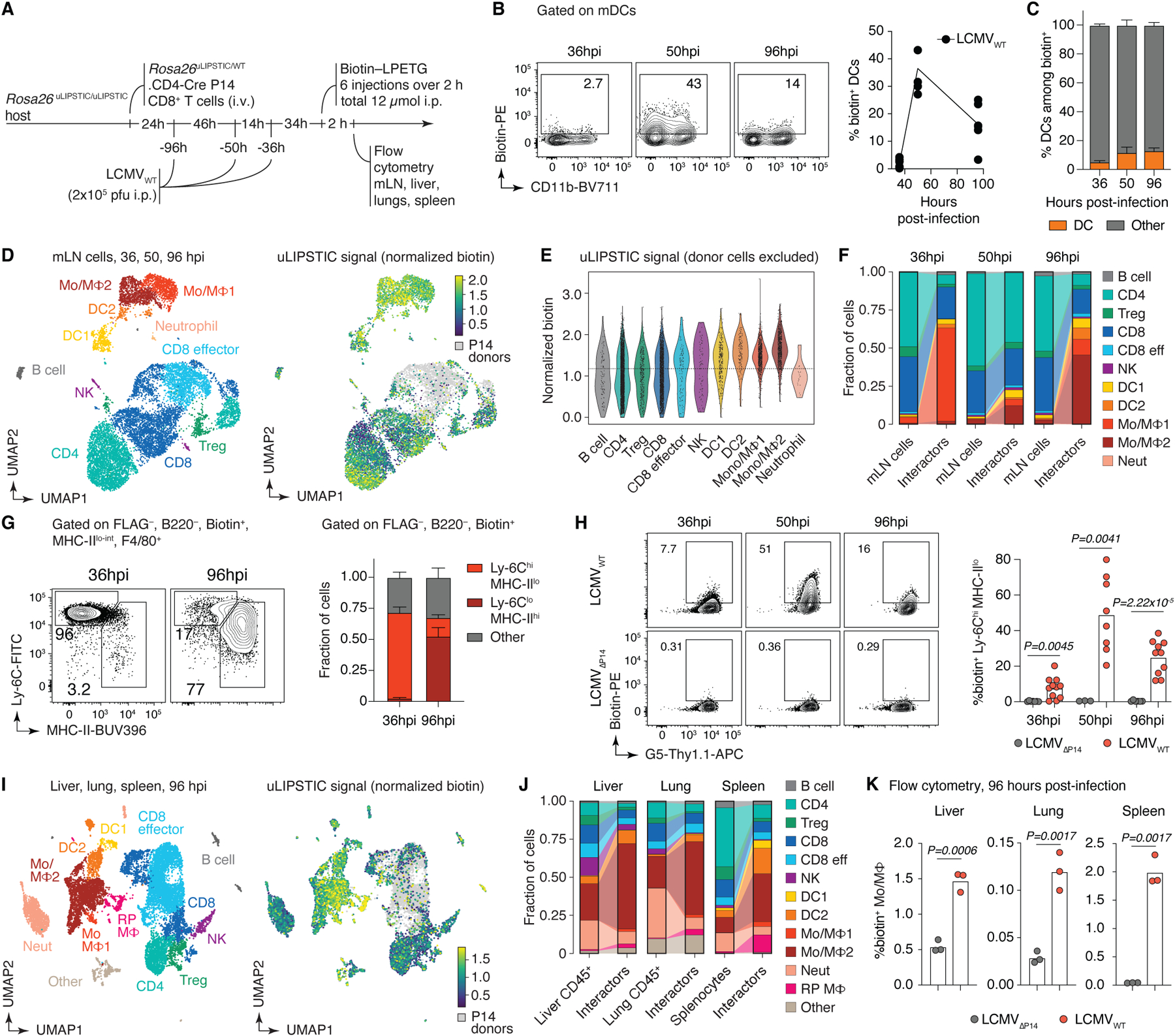
(A) Experimental layout. (B) Left, labeling of DCs by P14 cells at the indicated timepoint. Right, summary of data from three independent experiments. (C) Proportion of DCs among biotin+ acceptor cells, as determined by flow cytometry. Data for six mice per timepoint from three independent experiments. (D) UMAP plots of mLN cells sorted as in (A). Data pooled from 36, 50, and 96 hpi, with 2–3 mice per timepoint. Cells were enriched for uLIPSTIC acceptors and depleted of B cells as described in Extended Data Fig. 9C. Left, major cell type annotations (see Extended Data Fig. 9G–I). Right, normalized uLIPSTIC signal (biotin), excluding donor P14 cells. (E) Normalized uLIPSTIC signal among all cell populations, excluding donor P14 cells. (F) Distribution of cell types as in (D) in total mLN cells vs. in the biotin+ acceptor fraction (excluding P14 donors). (G) Left, distribution of uLIPSTIC labeled monocytic cells (Mo/MΦ) at indicated timepoints. Right, abundance of the indicated populations as a fraction of all uLIPSTIC-labeled acceptor cells. Data for four mice per timepoint from one experiment. (H) Left, uLIPSTIC labeling of Ly6Chi monocytes (Mo/MΦ1) at 36 hpi after infection with either LCMVWT or LCMVΔP14. Right, quantification of data from three independent experiments. (I) As in (D) but for pooled samples from liver, lung, and spleen at 96 hpi. (J) As in (F) but for pooled samples from liver, lung, and spleen at 96 hpi. (K) uLIPSTIC labeling of MHC-IIhi monocytes/macrophages (Mo/MΦ2) in organs of mice treated as in (A) but infected with either LCMVWT or LCMVΔP14, analyzed at 96 hpi. Data from one experiment. Bar plots in (C) and (G) show mean ± SEM. For (B), (H) and (K), each symbol represents one mouse and P-values were calculated using two-tailed Student’s test.
We also profiled the interactomes of P14 CD8+ T cells in the liver, lung, and spleen at 96 hpi, when accumulation of donor cells becomes evident by flow cytometry (not shown). As with mLNs, interactions between P14 T cells and monocytic lineage cells were observed at all three sites (Fig. 5I, Extended data Fig. 10A–G, and Supplementary Table 7). Labeled monocytic cells included a large cluster that closely matched the Mo/MF2 phenotype found in the mLN, as well as a smaller population of cells that resembled Mo/MF1 cells, in addition to a further cluster that comprised splenic red-pulp macrophages in the spleen and related populations in liver and lung (“Red pulp MF” cluster) (Fig. 5I and Extended data Fig. 10D–G). Infection with mutant (LCMVΔP14) virus confirmed that P14 interactions with monocytes/macrophages in all tissues was dependent on the presence of the P14 epitope (Fig. 5K and Extended Data Fig. 10H), again underscoring the ability of uLIPSTIC to detect antigen-dependent interactions. We conclude that the combination of uLIPSTIC and single-cell transcriptomics enables the identification of non-canonical cell-cell interactions even in well-characterized models, revealing a previously unappreciated predominance of monocytic cells as the primary interaction partners of CD8+ T cells in mLN during early systemic LCMV infection.
Discussion
This study describes a generalization of the LIPSTIC method2 that does not require cognate interaction between a pre-specified receptor-ligand pair for label transfer, allowing one to probe the full cellular interactome of a population of interest in an unbiased manner. Although most of the experiments we present involve interactions between immune cells, and particularly T cells, uLIPSTIC is in principle applicable to any population of cells that interact physically with each other. However, unlike our original cognate system2, where labeling reports on the engagement of a pre-defined pathway, the nature and function of interactions revealed by uLIPSTIC must be determined downstream, on a case-by-case basis. uLIPSTIC, especially when coupled to single-cell transcriptomics, is therefore best conceived of as a hypothesis-generating tool.
In the absence of a requirement for cognate interactions, the specificity of uLIPSTIC is ensured by the short intermembrane distance spanned by its components (~14 nm) and their low intrinsic affinity for each other (millimolar Km). Specificity is confirmed experimentally by the finding that (i) labeling is abrogated by antibodies that block known drivers of the cellular interaction; and (ii) not all cells that are physically juxtaposed label each other, as exemplified by the low degree of labeling of conventional T cells or resident DCs by Treg cell donors. uLIPSTIC thus complements methods such as synthetic Notch receptor variants, which, although they can be used to drive transcription of downstream reporter genes13,16, are based on molecular partners that have high (nanomolar) affinity for each other, are thus are thus themselves capable of driving cellular interactions42; as well as methods based on the spread of cell-permeable labels or barcoded virions between neighboring cells by extracellular diffusion12,15, which mark cellular microniches rather than physical interactions between cells. uLIPSTIC has advantages over cell doublet-based methods43, in that labeling is quantitative rather than binary, and it does not require computational deconvolution of single-cell transcriptional profiles from doublets; on the other hand, our system has the relative disadvantage of requiring genetic engineering of its components. Other limitations of uLIPSTIC include the need for relatively high numbers of donor cells to ensure that the true signal is detectable over the noise inherent to flow cytometry, especially when the target acceptor populations are prone to binding detection reagents, as is the case for B cells. Moreover, in acute inflammatory settings, such as in the 50h timepoint of LCMV infection (Fig. 5F), labeling appears to broaden to most LN-resident populations, suggesting that non-cognate labeling may occur in altered tissue environments. Again, downstream validation of interactions detected by uLIPSTIC will be critical in such cases.
A central feature of uLIPSTIC is that it can be coupled directly to droplet-based scRNA-seq to achieve quantitative interaction-based transcriptomics. This property can be used in both an “atlas” mode, where the objective is to identify which populations of acceptor cells interact with a given donor lineage, and in “mechanistic” mode, where correlations between uLIPSTIC signal intensity and expression of individual genes or gene signatures allow us to establish the molecular basis of an interaction of interest. Using this approach, we show that the ability of CD4+ T cells to interact physically with IECs in the small intestine is acquired developmentally as these cells adapt to the intestinal tissue environment and acquire the phenotypic and transcriptional features of CD4-IELs6,32,44. We also show that LCMV infection triggers CD8+ T cells to engage in interactions with monocytic cells that, greatly outnumber their interactions with DCs and may thus play a role in CTL priming. Our findings complement those of a previous study showing the expansion, following acute LCMV infection, of a monocytic population capable of priming CD8+ T cells in vitro45.
In conclusion, uLIPSTIC provides an unbiased platform for measurement of known cell-cell interactions as well as discovery of new ones. When coupled to scRNA-seq, uLIPSTIC interaction-based transcriptomics has the ability to quantify correlations between the intensity of cell-cell interactions and gene expression, allowing insight into the biology of the interaction itself. We expect this tool will be broadly useful for studying cellular interactions in immunology and beyond.
Methods
Plasmids
All constructs were cloned into the pMP71 vector46, which was modified to express a fluorescent reporter (eGFP or tdTomato) followed by the porcine teschovirus-1 self-cleavable 2A peptide47 and the protein of interest. The SrtA sequence, including an N-terminal Flag-tag, was attached by a single Gly-Gly-Gly-Gly-Ser linker48 to the human PDGFRB transmembrane domain to form mSrtA. The pentaglycine (G5) acceptor sequence was fused at the N terminus of the mouse Thy1.1 protein, downstream of the signal peptide. Sequences of all constructs are included in Supplementary Table 8.
Mice
CD45.2 (C57BL6/J), CD45.1 (B6.SJL Ptprca), CD4-Cre49, CD4-CreERT249, Foxp3eGFP-CreERT2 25, and Cx3cr1CreER mice were purchased from the Jackson Laboratories (strain numbers 000664, 002014, 022071, 022356, 016961, and 020940, respectively). Clec9aCre mice50 were a kind gift from C. Reis e Sousa (Francis Crick Institute, UK), S1pr2-CreERT2 BAC-transgenic mice51 were generated and kindly provided by T. Kurosaki and T. Okada (Osaka University and RIKEN-Yokohama), and AicdaCreERT2 mice28 were a kind gift from Claude-Agnès Reynaud and Jean-Claude Weill (Université Paris-Descartes). OT-II TCR transgenic (Y chromosome)52 mice were bred and maintained in our laboratory. The Rosa26uLIPSTIC mouse strain was generated by the Rockefeller University Gene Targeting and Transgenics facilities, as described below. All genetically modified strains are bred and maintained under specific pathogen-free conditions at the Rockefeller University’s Comparative Biosciences Center in accordance with institutional guidelines and ethical regulations. P14 TCR transgenic mice specific for LCMV-GP33–41 on a CD45.1. B6 background were originally provided by Dr. R. Ahmed (Emory), maintained at the Icahn School of Medicine at Mount Sinai vivarium, and bred with uLipstic mice. 6–12 week old adult male and female mice on the C57BL/6J background were used in all cases. Mice were housed at 72 °F (22.2 °C) and 30–70% humidity in a 12-h light/dark cycle with ad libitum access to food and water. All protocols were approved by the Rockefeller University and Mount Sinai School of Medicine Institutional Animal Care and Use Committees (protocol numbers 22058-H and IACUC-2018–0018/PROTO201900609, respectively).
Generation of the Rosa26uLIPSTIC allele
Rosa26uLIPSTIC mice were generated by gene targeting in C57BL/6 embryonic stem cells (ESCs). The Rosa26uLIPSTIC targeting vector is a modification of the Ai9 Rosa26 conditional expression vector20 (Addgene plasmid #22799). G5-Thy1.1 cDNA preceded by a mouse CD40 signal peptide was inserted into a NruI enzyme site in Ai9 immediately downstream of the first loxP site, whereas FLAG-mSrtA cDNA was introduced in place of the tdTomato gene using FseI enzyme sites. Expression of the cassette in ESCs was screened by standard Southern blotting analysis after EcoRI digestion and using a 32P probe targeting a sequence near the promoter region, shortly upstream of the left homology arm. Positive ESCs (7.3 kb band) were karyotyped, injected into blastocysts and chimeric founders were backcrossed to the C57BL6 background for at least six generations. The full sequence of the uLIPSTIC targeting vector and the Southern blot probe is reported in Supplementary Table 8. uLIPSTIC mice were deposited at Jackson Labs under strain number 038221.
Isolation of splenic dendritic cells (DCs), CD4+ T cells, and B cells
To isolate DCs, spleens were collected, cut into smaller pieces and incubated for 30 min at 37 °C in HBSS (Gibco) supplemented with CaCl2, MgCl2, and collagenase D at 400 U ml−1 (Roche). After digestion, tissue was forced 5 times through a 21-gauge needle and filtered through a 70 μm strainer into a 15 ml falcon tube with PBS supplemented with 0.5% BSA and 2 mM EDTA (PBE). Red-blood cells were lysed with ACK buffer (Gibco), and the resulting cell suspensions were filtered through a 70-μm mesh into PBE. DCs were obtained by magnetic cell separation (MACS) using anti-CD11c beads (Miltenyi Biotec), as per the manufacturer’s instructions. To isolate CD4+ T cells and CD8+ T cells spleens were forced through a 70 μm strainer, ACK-lysed and the resulting suspension was isolated by negative selection using a cocktail of biotinylated antibodies targeting Ter119, CD11c, CD11b, CD25, B220, NK1.1, and either CD8 (for CD4+ isolation) or CD4 (for CD8+ isolation), followed by anti-biotin beads (Miltenyi Biotec), as per the manufacturer’s instructions. B cells were were processed similarly as T cells from the spleens and isolated by negative selection using anti-CD43 beads (Miltenyi Biotec), as per the manufacturer’s instructions.
Adoptive cell transfers
For DC transfer experiments, splenic DCs were isolated as described above from mice subcutaneously injected with 1 × 106 B16 melanoma cells that constitutively secrete FMS-like tyrosine kinase 3 ligand (Flt3L)53 10 days prior to harvest. Cells were resuspended at 107 cells/ml and incubated with 10 μM OVA323–339, LCMV-GP61–80, OVA257–264, or LCMV276–286 peptides (Anaspec) in RPMI + 10% FBS, for 30 min at 37 °C. For cell labelling, CFSE or CTV (ThermoFisher Scientific) was added to a final concentration of 2 μM during the last 5 or 20 minutes of incubation, respectively. Cells were washed three times in RPMI + 10% FBS and resuspended at 2 × 107 cells/ml in PBS supplemented with 0.4 μg ml−1 LPS (Sigma-Aldrich). DCs were injected (5 × 105 cells in 25 μl) subcutaneously into the hind footpads. For CD4+ T cell and CD8+ T cell transfer experiments, 3×105 T cells isolated as described above were injected intravenously in 100 μl PBS per mouse. For LCMV infection experiments, 2×106 P14 CD8+ T cells were transferred intravenously 24 hours before infection.
Immunizations
Mice were immunized by subcutaneous injection into the hind footpad with 10 μg OVA or 10 μg NP-OVA (Biosearch Technologies) adsorbed in alum (Imject Alum, ThermoFisher Scientific) at 2:1 antigen:alum (v:v) ratio in 25 μl volume.
LCMV infections
For acute LCMV infections, mice were injected i.p. with 2×105 plaque forming units (PFU) of LCMV Armstrong (LCMVWT; originally provided by Dr. Michael Oldstone, The Scripps Research Institute) or a recombinant LCMV Armstrong strain (LCMVΔP14; a gift from Dr. Dirk Homann, Icahn School of Medicine at Mount Sinai) where the valine at position 35 of the LCMV glycoprotein is replaced by alanine thus precluding recognition by P14 or endogenous H2-Db-GP33–41-specific CD8 T cells as previously reported for an in vivo-selected LCMV variant41. LCMVΔP14 was originally generated by Dr. Juan-Carlos de la Torre (The Scripps Research Institute, La Jolla) using an established plasmid-based viral rescue strategy54–56 and will be described in an upcoming publication by van der Heide et al. elsewhere. Both LCMV strains were produced in BHK-21 cells (ATCC Cat# CCL-10) in DMEM with 2% FBS and infectious viral titers were assessed by plaque assays on Vero E6 (ATCC Cat# CRL-1586) monolayers.
Antibody treatments
For CD40L and MHC Class II blocking experiments in vivo, mice were injected intravenously with 200 μg of CD40L-blocking antibody (clone MR-1, BioXCell) or subcutaneously with 150 μg of MHC-II (I-A/I-E) blocking antibody (clone M5/114, BioXCell), four hours prior to the first injection of substrate.
Tamoxifen treatment
For induction of SrtA expression in regulatory T cells and conventional T cells, Foxp3eGFP-CreERT2/Y.Rosa26uLIPSTIC/WT mice and CD4-CreERT2.Rosa26uLIPSTIC/WT mice, respectively, were given two intragastric doses of 10 mg of tamoxifen (Sigma-Aldrich) dissolved in corn oil (Sigma-Aldrich) at 4 and 2 days prior to the end point (day 0). For SrtA expression in germinal center B cells, S1pr2-CreERT2.Rosa26uLIPSTIC/WT mice and AicdaCreERT2/+.Rosa26uLIPSTIC/WT mice, two doses of 10 mg of tamoxifen were administered intragastrically at 3 and 2 days prior to the end point. SrtA expression in gut epithelial cells was induced by daily intraperitoneal injections of tamoxifen (2 mg per injection) for 5 consecutive days, starting 14 days before the end point. For SrtA expression in microglia of Cx3cr1CreER.Rosa26uLIPSTIC/WT mice, two doses of 10 mg of tamoxifen each were administered intragastrically at 6 and 4 days prior to the end point.
In vivo substrate administration
Biotin-aminohexanoic acid-LPETGS, C-terminus amide at 95% purity (biotin-LPTEG) was purchased from LifeTein (custom synthesis) and stock solutions were prepared in PBS at 20 mM. For in vivo LIPSTIC and uLIPSTIC labeling experiments in popliteal lymph nodes (pLNs), biotin–LPETG was injected subcutaneously into the hind footpad (20 μl of 2.5 mM solution in PBS) six times 20 min apart, and pLNs were collected 20 min after the last injection, as described57. Mice were briefly anaesthetized with isoflurane at each injection. For in vivo labeling of gut IELs, DCs and/or microglia in various tissues, and for LCMV experiments, biotin-LPETG substrate was injected intraperitoneally (i.p.) (100 μl of 20 mM solution in PBS) six times, 20 min apart. Organs were collected 20 min after the last injection.
Isolation of lymphocytes from lymphoid organs
Spleen and popliteal, mediastinal and mesenteric lymph nodes were collected into microfuge tubes with 500 μl HBSS (Gibco) supplemented with CaCl2, MgCl2, and collagenase D at 400 U ml−1 (Roche). LNs were cut into small pieces and incubated for 30 min at 37 °C. After digestion, tissue was forced 5 times through a 21-gauge needle and filtered through a 70 μm strainer into a 15 ml falcon tube with PBE.
Isolation of cells from non-lymphoid organs
Intraepithelial leukocytes were isolated as previously described58. Briefly, small intestines were harvested and washed in PBS. Peyer’s patches were surgically removed and the intestine was segmented in ~1 cm pieces prior to incubation with 1 mM dithiothreitol for 10 min at room temperature followed by addition of 30 mM EDTA and incubation for 30 min at 37°C. Intraepithelial cells were recovered from the supernatant of dithiothreitol and EDTA washes and mononuclear cells were isolated by collecting the middle ring after 40% and 80% gradient Percoll centrifugation. Bone marrow cells were collected by centrifugation of punctured tibiae and femurs at up to 10,000 x G for 10 s, then treated with ACK red blood cell lysing buffer. Immune cells from the kidney, lungs, spleen, thymus and liver were isolated by incubating the fragmented tissue in 1.5 ml HBSS supplemented with collagenase D at 400 U ml−1, 0.1 mg ml-1 Dnase 1 (Sigma) and 0.8 mg ml-1 dispase 1 (Sigma) for 30 min at 37°C. After digestion, tissue was forced 5 times through a 21-gauge needle and filtered through a 70 μm strainer into a 15 ml falcon tube with PBE. Red-blood cells were lysed with ACK buffer and the resulting cell suspensions were filtered through a 70-μm mesh into PBE. To harvest immune cells from the brain, mice were anesthetized and perfused transcardially with 10 ml ice-cold HBSS without Ca2+ and Mg2+ (HBSS--, Gibco), and the brains were removed and kept in ice-cold HBSS before further process. To stain and discard CD45+ cells from blood vessels for downstream analysis, anti-CD45 antibodies were retro-orbitally injected 15 min before perfusion. The entire brain was minced by mashing through a 150 μm cell strainer and the strainer was washed thoroughly by ice-cold HBSS to collect as many cells as possible. Minced tissues were spun down at 290 g for 5 min at 4°C to discard the supernatant and digested in 2 ml of Digestion solution (2 mg ml−1 collagenase D, 25 mM HEPES, 1 mM sodium pyruvate, 14 μg ml−1 DNase 1 in HBSS) for 20 min at 37°C without shaking. Digestion was stopped by adding 2 ml ice-cold HBSS and the tissues were homogenized with syringes fitted with 21G, 25G, and 27G needles, sequentially. The homogenates were filtered through a 70 μm mesh and spun down at 420 g for 7 min at 4°C to discard the supernatant. The pellets were resuspended in 37% Percoll solution in HBSS and centrifuged at 500 g for 10 min at room temperature to discard supernatant with a myelin layer. The cells in pellets were washed and resuspended in HBSS for further analysis.
Flow Cytometry and cell sorting
Single-cell suspensions were washed with PBE, incubated with 1 μg ml−1 anti-CD16/32 (2.4G2, BioXCell) for 5 min at room temperature and then stained for cell surface markers at 4 °C for 20 min in PBS using the reagents listed in Supplementary Table 9. Cells were washed with PBE and stained with Zombie fixable viability dye (BioLegend) or fixable Aqua dead cell stain kit (Invitrogen) at room temperature for 15 min, then washed with PBE and filtered through a 40 μm strainer for acquisition. For in vivo JAML staining of IELs, mice were injected i.p. with 100 μg of anti-JAML AF646 antibody 12 or 6 h prior to the end point. For single-cell transcriptomic analysis, stained cells were further incubated with DNA-barcoded anti-biotin and sample hashtag (anti-MHC-I) antibodies (BioLegend) for 20 minutes in PBE, washed three times with PBE, and bulk-sorted. For substrate detection in vivo, an anti-biotin–PE antibody (Miltenyi Biotec) was exclusively used, as described previously57. Samples were acquired on FACSymphony A5 or Fortessa analyzers or sorted on FACSAriaII/III or FACSymphony S6 cell sorters (BD Biosciences). Data were analyzed using FlowJo v.10.6.2 software.
uLIPSTIC labeling in vitro
HEK293T cells (ATCC) were transfected by calcium phosphate transfection with the indicated expression vectors at high (1 μg μl−1) and low (0.1 μg μl−1) concentrations of Thy1.1-G5 and mSrtA constructs. Forty hours after transfection, cells were detached TrypLE Express cell dissociation solution (ThermoFisher Scientific), washed and resuspended at 106 cell per ml in PBS. Donor cell populations transfected with CD40L and/or mSrtA constructs and acceptor cell populations transfected with CD40 and/or Thy1.1-G5 were mixed at a 1:1 ratio (105 cells of each population) in a 1.5-ml conical tube, to which biotin–LPETG was added to a final concentration of 100 μM. Cells were incubated at room temperature for 30 min and washed three times with PBE to remove excess biotin–LPETG before FACS staining.
uLIPSTIC labeling ex vivo
B cells from Rosa26uLIPSTIC/WT mice and CD4+ T cells from OT-II CD4-Cre.Rosa26uLIPSTIC/WT mice were isolated from mouse spleens as described above. Isolated T cells were activated with CD3/CD28 dynabeads (ThermoFisher Scientific) for 24 h and then co-cultured with isolated B cells (2 × 105 cells per well, 1:1 ratio) in the presence or absence of OVA323–339 peptide in RPMI, 10% FBS supplemented with 0.1% 2 mercaptoethanol (Gibco) in U-bottom 96-well plates for 20 h. Blocking antibodies were added at the beginning of the co-culture at a final concentration of 150 μg ml−1. To label interactions ex-vivo, biotin-LPETG substrate was added 30 min before harvest at a final concentration of 100 μM.
Library preparation for single cell-RNA sequencing
In addition to fluorescent antibodies, cells were co-stained prior to sorting with hashtag oligonucleotide (HTO)-labeled antibodies to CD45 and MHC-I for sample separation (two hashtags per sample) and HTO-anti-biotin for detection of the uLIPSTIC signal. Sorted cells were collected into a microfuge tube with 300 μl PBS supplemented with 0.4% BSA. After the sort, tubes were topped with PBS 0.4% BSA, centrifuged and the buffer was carefully reduced by removing the volume with a pipette to a final volume of 40 μl. Cells were counted for viability and immediately submitted to library preparation. The scRNA-seq library was prepared using the 10X Single Cell Chromium system, according to the manufacturer’s instructions, at the Genomics Core of Rockefeller University and was sequenced on an Illumina NovaSeq SP flowcell to a minimum sequencing depth of 30,000 reads per cell using read lengths of 26 bp read 1, 8 bp i7 index, 98 bp read 2.
Computational analysis of uLIPSTIC + single-cell RNA sequencing data in intraepithelial immune cells
Gene expression unique molecular identifier (UMI) counts, along with sample and biotin (uLIPSTIC) HTO counts, were generated with CellRanger v6.0.1 “count” using “Feature Barcode” counts and otherwise default parameters, with mm10 reference. TCR data were preprocessed with CellRanger “vdj” with default parameters. Applying default cellranger filtering, this resulted in a filtered gene expression UMI count matrix including 4,607 cells and 32,285 genes.
We then performed a multi-step analysis of the data to annotate cells with cell types, including data preprocessing, normalization, clustering, and analysis of known marker genes from the literature as well as objective differential gene expression analysis. The scanpy package v1.9.1 was used for all analysis of the gene expression data59. Cell barcodes with unresolved sample HTOs, a low or extremely high number of expressed genes, a large fraction of expressed mitochondrial genes, or likely doublets were removed. Genes expressed in a low number of cells were removed. This resulted in a filtered gene expression matrix of 3,677 cells and 14,332 genes with a matching biotin HTO count in each cell representing uLIPSTIC signal.
Gene counts were normalized using Pearson residual normalization with theta = 1. Principal component analysis (PCA) was run with default parameters, and then k nearest neighbor (kNN) graph was constructed using 40 PCs, k = 30 and otherwise default parameters. Then Leiden clustering was performed with a resolution of 1, resulting in 26 clusters. Cluster 10 was further split into two subclusters containing cycling T and B cells (Extended Data Fig 6F,G).
uLIPSTIC normalized values were obtained for each cell by dividing the uLIPSTIC HTO counts by the number of sample-encoding HTO read counts in a cell. The 5th percentile of these normalized values was added as a pseudocount, and then log10 applied. These values were then shifted by the minimum log-scaled value, so the scale starts at 0. This resulted in arbitrary units of the normalized uLIPSTIC signal, subject to comparison between cells from a single dataset.
Known marker genes as well as TCR data were used to annotate the Leiden clusters. The scirpy package v0.10.1 was used for the TCR data preprocessing and analysis60. Cluster 10 was split into two subclusters that contained cycling T and B cells. Annotations were confirmed by scoring PanglaoDB immune cell marker gene sets61 using the score_genes() function in scanpy and by exploring significantly differentially expressed genes in each cluster as compared with all cells outside the cluster, obtained using a custom script. For differential expression analysis, log2 fold change (log2FC) of expression was calculated as the ratio of pseudobulk raw UMI counts summed over cells within and outside the cluster (then normalized by total amount of UMI counts inside and outside the cluster), p-values were calculated using Mann-Whitney U test applied to Pearson residual normalized expression values in single cells within and outside the cluster, and Benjamini-Hochberg correction for multiple hypothesis testing applied to all genes. This analysis resulted in final cell type annotations; some clusters received the same cell type annotations.
The analysis then focused on the CD4 T cell subset of 944 cells. A new kNN graph was generated for this subset, again using k = 30 neighbors and 40 PCs, and Leiden clustering performed with resolution = 1.3. Clusters were annotated using known marker genes and TCR clonality information, and one cluster was filtered out due to trouble annotating it, resulting in a dataset of 915 cells. Trajectory analysis and subsequent cell pseudotime calculation were performed using Wishbone v0.5.262 using default parameters as available in scanpy and using as the root the cell in the Naive/Tconv cluster with the highest value of Sell expression. uLIPSTIC signal (normalized biotin) data was not used to generate the trajectory.
To identify candidate genes involved in cell-cell interactions, for every gene the Spearman correlation was calculated between the Pearson residual normalized value of expression of that gene and the uLIPSTIC signal across all cells in the CD4 T cell subset. Bonferroni correction was used for multiple hypothesis testing on all genes. This calculation was separately performed when removing Tfh-like and naïve/memory cells, or when restricting to cells from each individual mouse, with consistent results (Extended Data Fig. 8D,E).
For violin plot of scRNA-seq expression of Jaml (Fig. 4I), Pearson residual normalized values were shifted so that the minimum value is zero, bottom 5th percentile of all values (across cell groups) omitted, and then plotted on log scale. The T cell subpopulations for the plot were defined as follows. The subpopulations of CD4 T cells, “Naïve/Tconv”, “Pre-IEL”, “IEL” (Fig. 4D–H, Extended Data Fig. 8A,B), were used as CD4+ Tconv, CD4+ Pre-IEL and CD4+ CD8αα+ IEL, respectively. The “Natural IEL” cells (Fig. 4B, Extended Data Fig. 6,7) were separated into three groups: CD8αα+ IEL if TCR ab chain was detected (301 cells), otherwise γδ IEL if normalized expression of Trdc was above 0 (517 cells), and other (163 cells) which were not included in the plot.
MSigDB canonical pathways were scored using scanpy’s score_genes() function over all CD4+ T cells. Spearman correlation with normalized biotin values was calculated for all pathways, and p-values were adjusted using q-value approach63 for pathways with positive correlation values. Top 5 pathway scores are shown by correlation value, for those with q < 0.05 (Extended Data Fig. 8G). CD4+CD103+CD8αα+ and CD4+CD103−CD8αα− gene signatures were generated from scRNA-seq (library 2) from Bilate et al6. tdTomato−CD4+CD8αα+ cells (Cluster 2) were compared to tdTomato– “recent epithelial immigrants” (REI, Cluster 5) using the Wilcoxon Rank Sum test. P-values were adjusted using Bonferroni correction. All genes with adjusted p-values < 0.05 were included in the signature. Genes with positive fold-change (enriched in Cluster 2) were included in “Bilate_CD40-IEL_UP” and with negative fold-change (enriched in Cluster 5) were included in “Bilate_CD40-IEL_DOWN”. Signatures were scored on the uLIPSTIC scRNA-seq data using scanpy’s score_genes() function, and Spearman correlation with normalized biotin values for both gene signatures was calculated over all CD4+ T cells, or over all CD4+ T cells excluding Tconv and Tfh-like cells. Linear regression fit with 95% confidence interval overlayed over scatter plots was calculated using geom_smooth() in ggplot2 using default parameters.
Computational analysis of uLIPSTIC + scRNA-seq data in LCMV infection
Gene expression UMI counts, along with sample, biotin (uLIPSTIC), and FLAG (to capture donor cells) HTO counts were generated for each of the three sequencing lanes with CellRanger 7.0.1 “count” using “Feature Barcode” counts and otherwise default parameters, with mm10 reference. All downstream analysis was performed using the scanpy package v1.9.1. Initial QC steps and normalization were performed separately for each of the three sequencing lanes. Cells were filtered based on high mitochondrial counts and total counts. Genes were filtered based on being present in at least 0.5% of cells in the sample, and cells were filtered to include at least 200 genes. Each cell was assigned to a sample if the fraction of all sample HTOs coming from that sample HTO was greater than 80%. For each cell, biotin read counts and donor (FLAG) read counts were normalized by dividing by the total number of HTO counts of the sample to which the cell was assigned. Gene expression counts were then normalized with analytical Pearson residual normalization from scanpy, using a theta of 1 for all 3 samples. After normalization, the three samples were concatenated. Non-protein coding genes were also filtered out based on the CellRanger mm10 GTF file. This resulted in a dataset of 27,043 cells and 11,558 genes.
LCMV lymph node cells from WT infection were selected for a separate analysis. This resulted in a dataset of 11,846 cells (and the same 11,558 genes). PCA was run with 100 components, KNN graph was built using 30 neighbors, 50 PCs and cosine metric, and Leiden clustering was performed with resolution of 1. Known marker genes were used to annotate the Leiden clusters.
As described above, sample-normalized biotin values were further adjusted by performing log10 transformation, using the 5th percentile as a pseudocount, and then shifted by the minimum log-scaled value, so the scale starts at 0. Differential gene expression analysis was performed as described above. Unless stated otherwise, in most plots, cells with high donor levels (using a threshold based on the distribution of these normalized FLAG counts) were filtered out.
Next, LCMV organ cells (from spleen, liver, and lung) in WT infection were selected for a separate analysis, yielding a dataset of 12,324 cells and 11,558 genes. PCA was run with 100 components, KNN graph was built using 30 neighbors, 50 PCs and cosine metric, and Leiden clustering was performed with resolution of 1. To annotate these clusters and compare them to the lymph node data, a dendrogram of transcriptional similarities between cells in clusters was created. For this, we combined the mean profile for each tissue Leiden cluster and each cell type annotation in the lymph node data over all genes. This enabled us to find tissue Leiden clusters that have similar expression profiles as the lymph node annotations. Known marker gene expression was used to confirm these lymph-node based annotations in the tissue data. Certain clusters did not relate to lymph node annotations, so most significant differentially expressed genes (following the same protocol as described above) for these clusters were used to assign annotations with the ImmGen My_Geneset tool (www.immgen.org). These annotations were again confirmed with marker gene expression.
As described above, biotin read counts and donor (FLAG) read counts were normalized by dividing by the total number of HTO counts of the sample to which the cell was assigned. The sample-normalized biotin values were further adjusted by performing log10, using the 5th percentile as a pseudocount, and then shifted by the minimum log-scaled value, so the scale starts at 0. For most plots, cells with high donor levels (using a threshold based on the distribution of the sample-normalized FLAG counts) were filtered out.
Modelling the SrtA-Thy1.1 complex on cells surfaces
First, structures of G5-Thy1.1 and FLAG-SrtA-PDGFRb were generated using Alphafold264. Next, in the FLAG-SrtA-PDGFRB model, the domain constructing peptide binding domain was substituted with the substrate bound Sortase A structure (PDB: 1T2W). Additionally, the flexible linker connecting SrtA domain to PDGFRB transmembrane helix was rebuilt to an extended conformation using COOT v. 0.8.9.265 to better estimate the maximum distance the protein is able to extend to. The Thy1.1 was aligned to SrtA using the substrate of 1T2W and 5G acceptor motif of G5-Thy1.1. Any resulting interprotein clashes were corrected using GalaxyRefineComplex from GalaxyWEB server (https://galaxy.seoklab.org/)66. To build the GPI anchor and the lipid bilayers we used CHARMM-GUI (https://www.charmm-gui.org/)67. The anchor glycolipid was generated based on the human prion protein (PrP) GPI68. Next theFLAG-SrtA-PDGFRB:G5-Thy1.1 complex was modelled in the POPC/cholesterol lipid bilayer using CHARMM-GUI. The GPI anchor was placed in the second bilayer using ChimeraX v. 1.469. Finally, both bilayers, with the protein complex and the GPI anchor were aligned in ChimeraX. The distance between two bilayers was measured in PyMOL v. 2.4.270.
Statistical analysis
Statistical tests were performed in GraphPad Prism 9.0 software and edited for appearance using Adobe Illustrator 27.1.1. Comparisons between two treatment conditions were analyzed using unpaired, two-tailed Student’s t-test and multivariate data were analyzed by one-way ANOVA with Tukey’s post-hoc tests to further examine pairwise differences.
Extended Data
Extended Data Figure 1 |. Design and characterization of Rosa26uLIPSTIC mice.
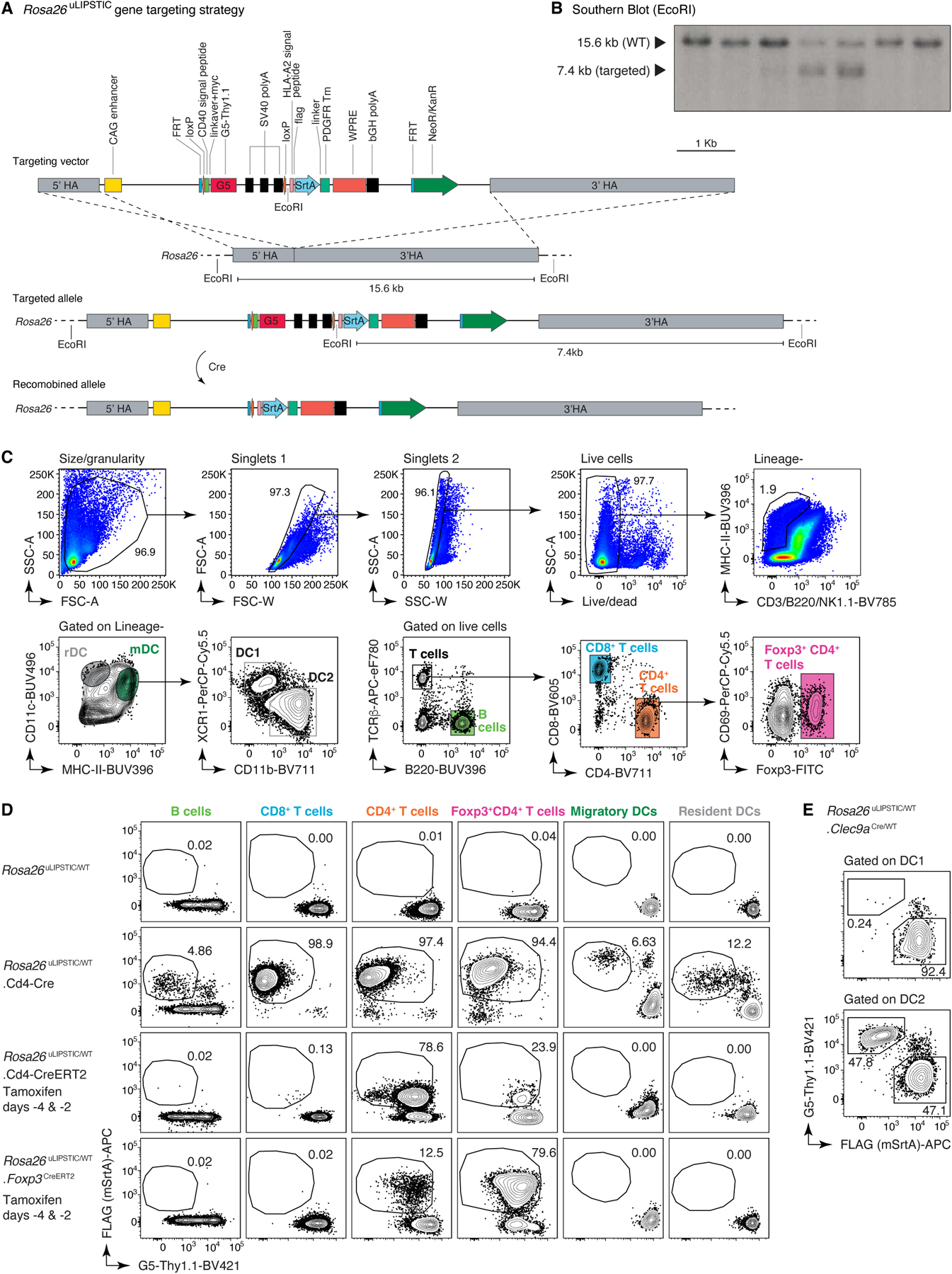
(A) The uLIPSTIC cassette carrying the lox-stop-lox G5Thy1.1 followed by mSrtA-PDGFRtm fused to FLAG tag was cloned into the Ai9 Rosa26 targeting plasmid. (B) Insertion of the uLIPSTIC cassette was assessed in embryonic stem (ES) cells by Southern blotting using a 32P-labeled probe (Supplementary Table 8 and Supplementary Figure 1 for gel source data) annealing upstream of the left arm after EcoRI digestion. ESCs carrying the insertion exhibit an extra EcoRI restriction site, resulting in a 7.4 kb fragment upon enzymatic digestion. The blot shows 2 heterozygous integrations out of 7 ES cell clones screened. (C-E) The specificity and efficiency of Rosa26uLIPSTIC recombination are determined by the Cre driver used. (C) Representative gating strategy for resident dendritic cells (rDCs; LIN–, MHC-IIint, CD11chi), migratory dendritic cells (mDCs; LIN–, MHC-IIhi, CD11c+), CD4+ T cells, CD8+ T cells, regulatory T (Treg) cells and B cells in lymph nodes. (D) SrtA expression (determined by FLAG detection) is induced by Cre recombination. Use of a constitutive Cre line (e.g., CD4-Cre) results in efficient but non-specific SrtA expression, generating T cells that can only be used in adoptive cell transfer experiments. The use of inducible Cre lines such as CD4-CreERT2 and Foxp3CreERT2 can often resolve specificity issues, enabling the implementation of uLIPSTIC in fully endogenous models. (E) SrtA expression in conventional DC subsets 1 (cDC1) and 2 (cDC2) in Rosa26uLIPSTIC/WT.Clec9aCre/WT mice.
Extended Data Figure 2 |. Kinetics and sensitivity of the uLIPSTIC reaction.
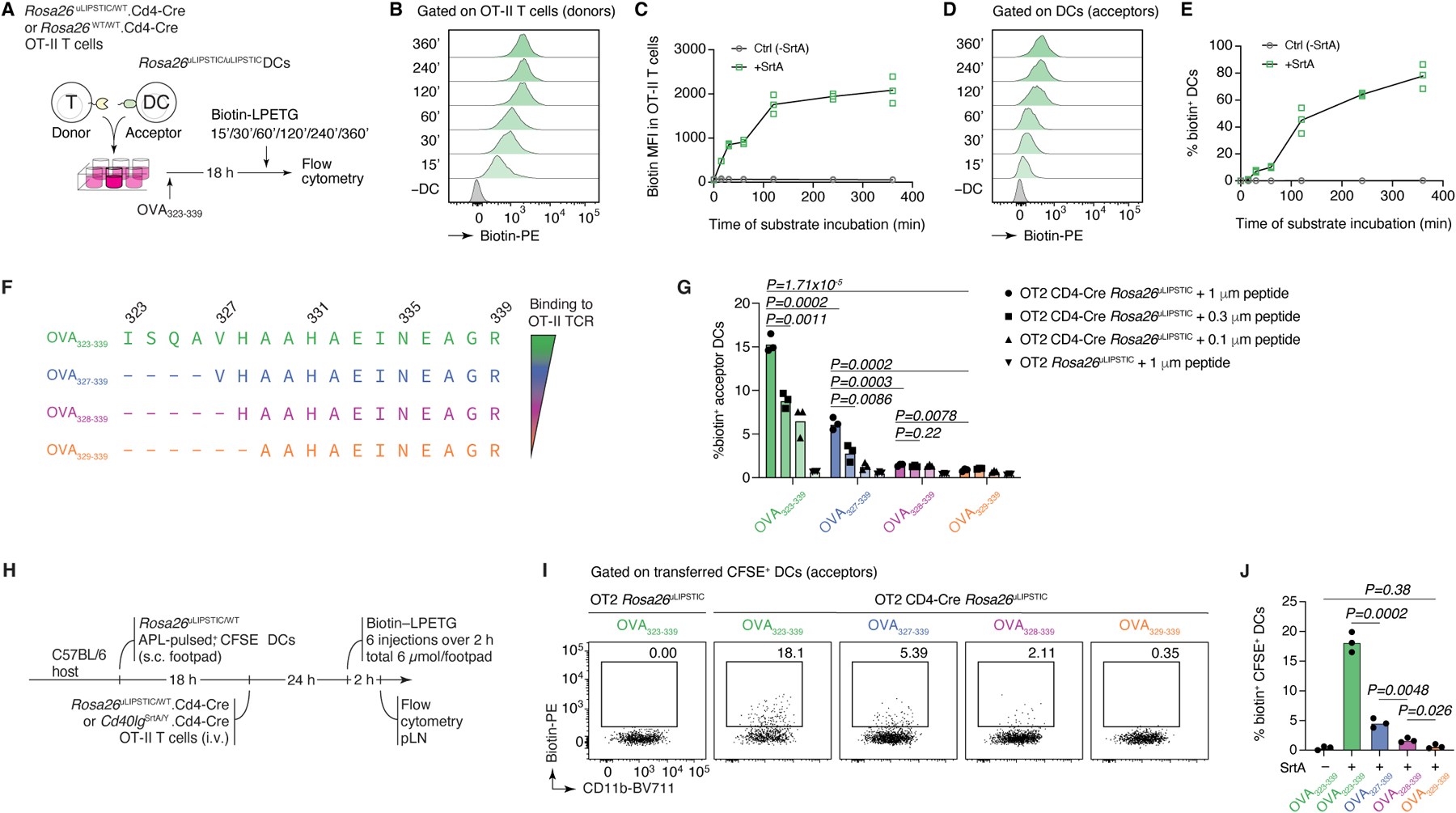
(A-E) Kinetics of the uLIPSTIC reaction. (A) Experimental setup for panels (B-E). OT-II CD4+ T cells from Rosa26uLIPSTIC/WT.CD4-Cre or Rosa26WT/WT.CD4-Cre control mice were co-incubated ex vivo with Rosa26uLIPSTIC/uLIPSTIC acceptor DCs in the presence of OVA323–339 cognate peptide. LIPSTIC substrate was added during the final minutes of incubation as indicated. (B,C) Efficiency of formation of the acyl intermediate (loading of LIPSTIC substrate onto SrtA) in OT-II SrtA+ donor T cells increases gradually with time. (D,E) Transfer of LIPSTIC substrate onto the surface of interacting acceptor DCs followed similar kinetics as acyl intermediate formation. (F-J) uLIPSTIC can resolve differences in peptide concentration and affinity both in vitro and in vivo. (F) Altered peptide ligands (APLs) of the OVA323–339 peptide, when complexed with MHC-II, display decreasing affinities for the OT-II TCR. (G) In vitro co-culture of Rosa26uLIPSTIC/WT.CD4-Cre OT-II T cells with Rosa26uLIPSTIC/uLIPSTIC DCs loaded with its APLs results in a reduction in LIPSTIC labeling that aligns with both the affinity of the peptide-MHCII complex to the OT-II TCR and the peptide concentration gradients. (H) Experimental layout for panels (I,J). (I) In vivo labeling of APL-pulsed DCs show decreased uLIPSTIC labeling in accordance with the affinity to the fixed OT-II TCR. Quantified in (J). Data for all plots are for three mice per condition from one experiment. P-values were calculated using two-tailed Student’s tests.
Extended Data Figure 3 |. uLIPSTIC labeling of T cell–DC interactions in adaptive transfer models.
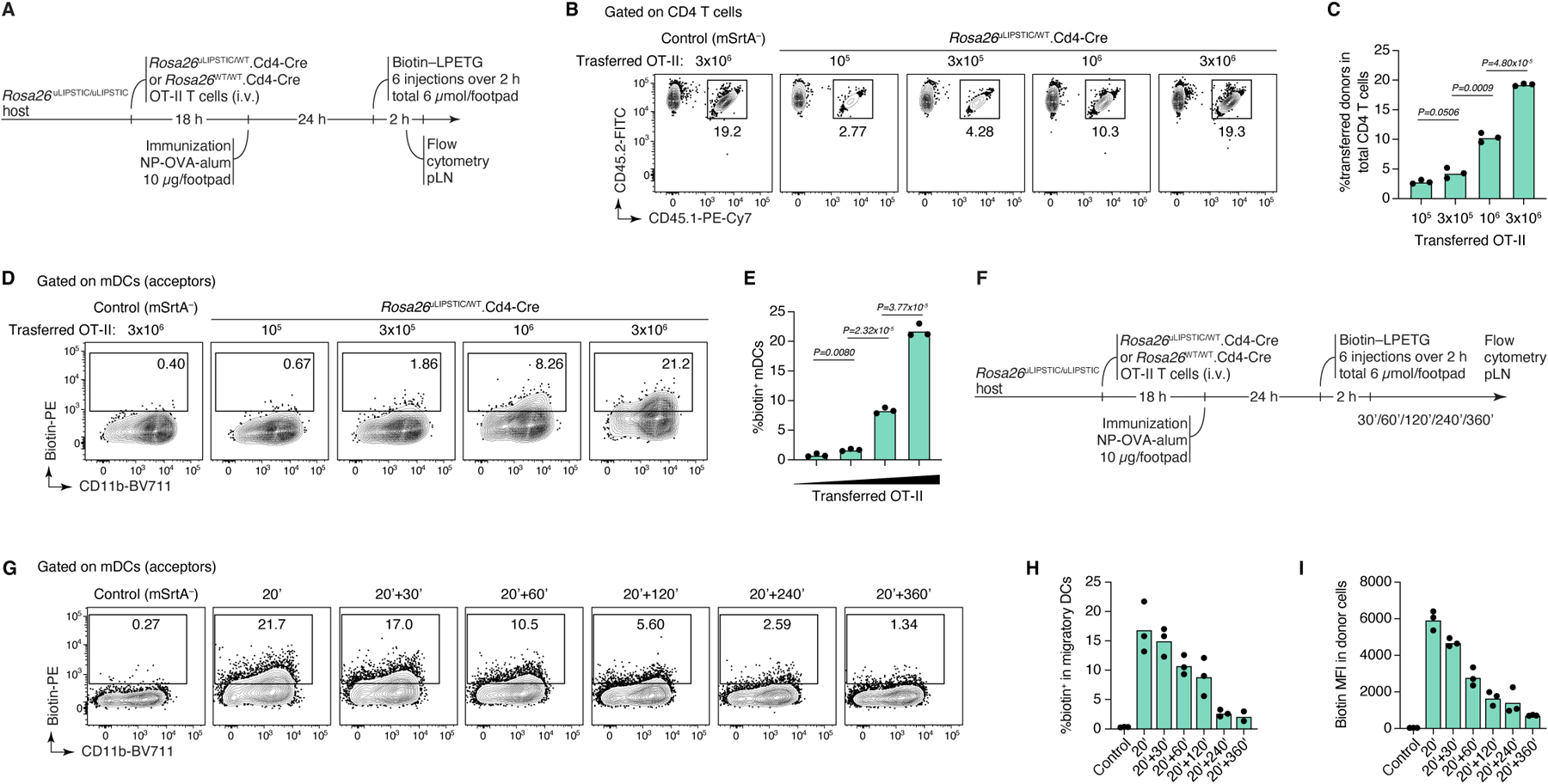
(A-E) mSrtA+ donor cell numbers determine the degree of uLIPSTIC labeling. (A) Experimental layout for panels (B-E). Increasing numbers (105, 3 × 105, 106, 3 × 106) of Rosa26uLIPSTIC/+.CD4-Cre OT-II CD4+ T cells were adoptively transferred into recipient Rosa26uLIPSTIC/ uLIPSTIC mice, followed by OVA/alum immunization 18 h post-transfer and LIPSTIC substrate injection one day later. The number of transferred cells (CD45.1/2) determined the proportion of donor cells in the CD4+ T cell compartment (B-C) and the corresponding percentage of labeled interacting cells in the mDC compartment (D-E). (F-I) Persistence of label on acceptor cells with time. (F) Experimental layout for panels (G-I). (G) uLIPSTIC labeling of mDCs after incremental delays between substrate injection and tissue harvest. Quantified in (H,I). Data for all plots are for three mice per condition from one experiment. P-values were calculated using two-tailed Student’s tests.
Extended Data Figure 4 |. uLIPSTIC labeling in inducible Cre lines in fully endogenous models.
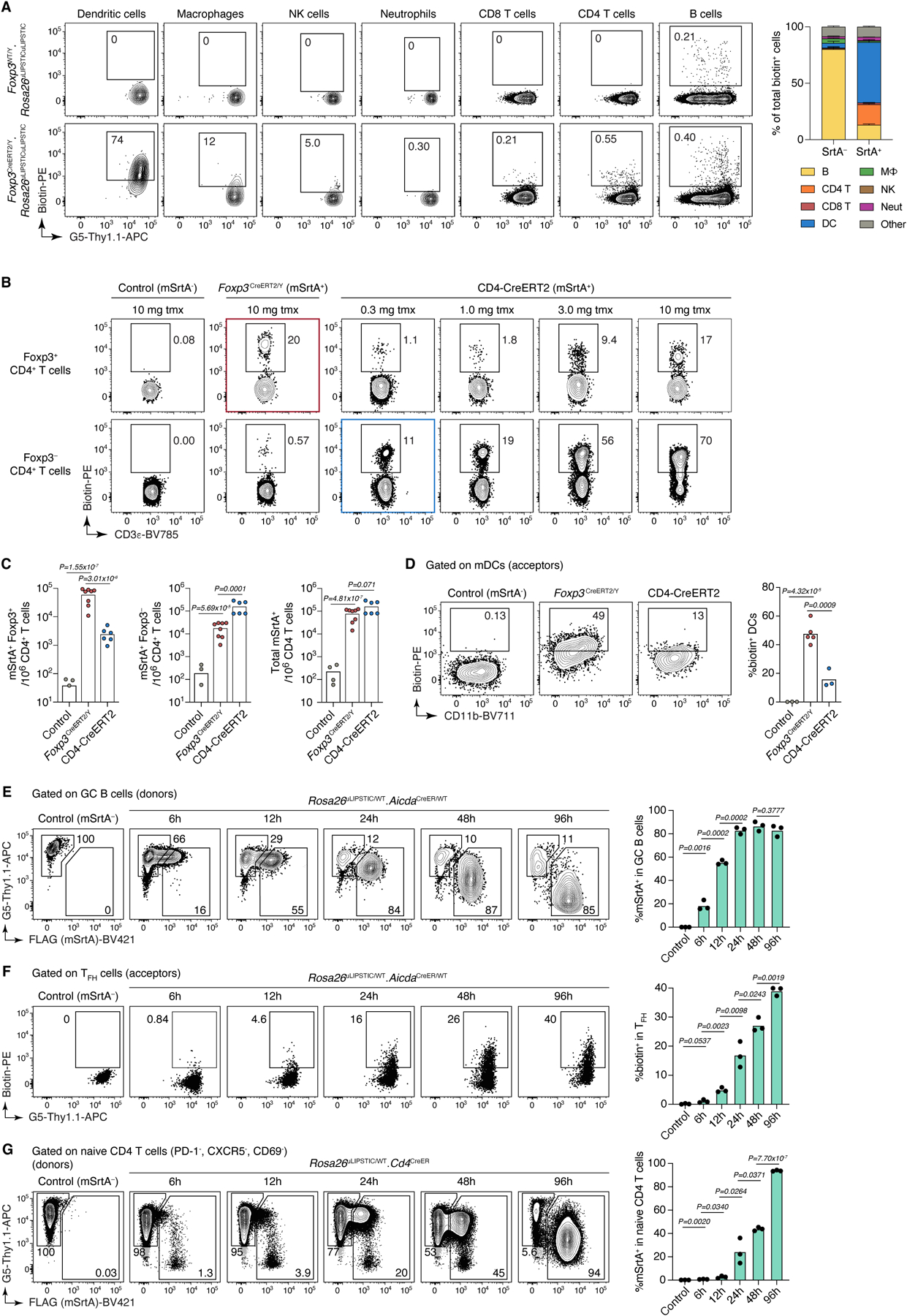
(A-D) uLIPSTIC labeling of Treg cell interactions in the steady-state pLN. (A) Cellular interactome of Tregs at steady-state. Rosa26uLIPSTIC/WT.Foxp3CreERT2/Y experimental mice and Rosa26WT/WT.Foxp3CreERT2/Y controls were given tamoxifen and administered LIPSTIC substrate in the footpad 2 days later. Left, flow cytometry plots show uLIPSTIC labeling in selected immune populations in control (top) and experimental (bottom) mice. The presence of residual labeling in B cells is an artifact common to uLIPSTIC and to other flow-cytometry based methods aimed at identifying rare B cell populations, likely due to B cell receptor-dependent binding of detection components by polyclonal B cells. Right, quantification of the proportion of all labeled cells belonging to each major immune population in control (SrtA–) or experimental (SrtA+) mice. Data for three mice per condition from one experiment, bar plots show mean ± SEM. (B-D) Treg cells interact with mDCs to a greater extent than conventional CD4+ T cells. (B) To test if enhanced interaction with mDCs is a specific feature of Treg cells or a general feature of all CD4+ T cells, we titrated the dose of tamoxifen in Rosa26uLIPSTIC/+.CD4-CreERT2 mice to achieve a similar percentage of SrtA-expression among total CD4+ T cells as in Rosa26uLIPSTIC/+.Foxp3CreERT2/Y mice. (C) At a dose of 0.3 mg of tamoxifen, Rosa26uLIPSTIC/+.CD4-CreERT2 mice showed SrtA expression in a small number of Treg cells (left), with most SrtA+ cells observed in CD4+ conventional T cells (center) and overall numbers of SrtA+ cells among total CD4+ T cells that were comparable with those of Rosa26uLIPSTIC/+.Foxp3CreERT2/Y mice treated with 10 mg tamoxifen (right). (D) When numbers of Treg and CD4+ conventional donor cells are equalized, acceptor mDCs show stronger interaction with Treg cell partners. For (C) and (D), data from two independent experiments with each symbol representing one mouse, P-values were calculated using two-tailed Student’s tests. (E-G) Kinetics of tamoxifen-driven recombination of the Rosa26uLIPSTIC allele according to cell type. (E) SrtA expression in the highly proliferative mesenteric lymph node GC B cells of Rosa26uLIPSTIC/WT.AicdaCreER/WT mice was assessed at different timepoints after tamoxifen administration. The fraction of recombined cells plateaus at 24 h post-tamoxifen administration (hpt), while SrtA protein expression is still increasing by 96 hpt. (F) Labeling of GC B cell interacting partners can be detected as early as 12 hpt, increasing thereafter according to SrtA expression levels. (G) In contrast, SrtA expression in quiescent naïve (PD-1−CXCR5−CD69−) CD4+ T cells in Rosa26uLIPSTIC/WT.Cd4-CreERT2 mice increased at a slower rate than, reaching >80% positive cells only at 96 hpt. For (E), (F) and (G), each plot used three mice per condition from one experiment, P-values were calculated using two-tailed Student’s tests
Extended Data Figure 5 |. Intraperitoneally-injected LIPSTIC substrate reaches cells in multiple tissues.
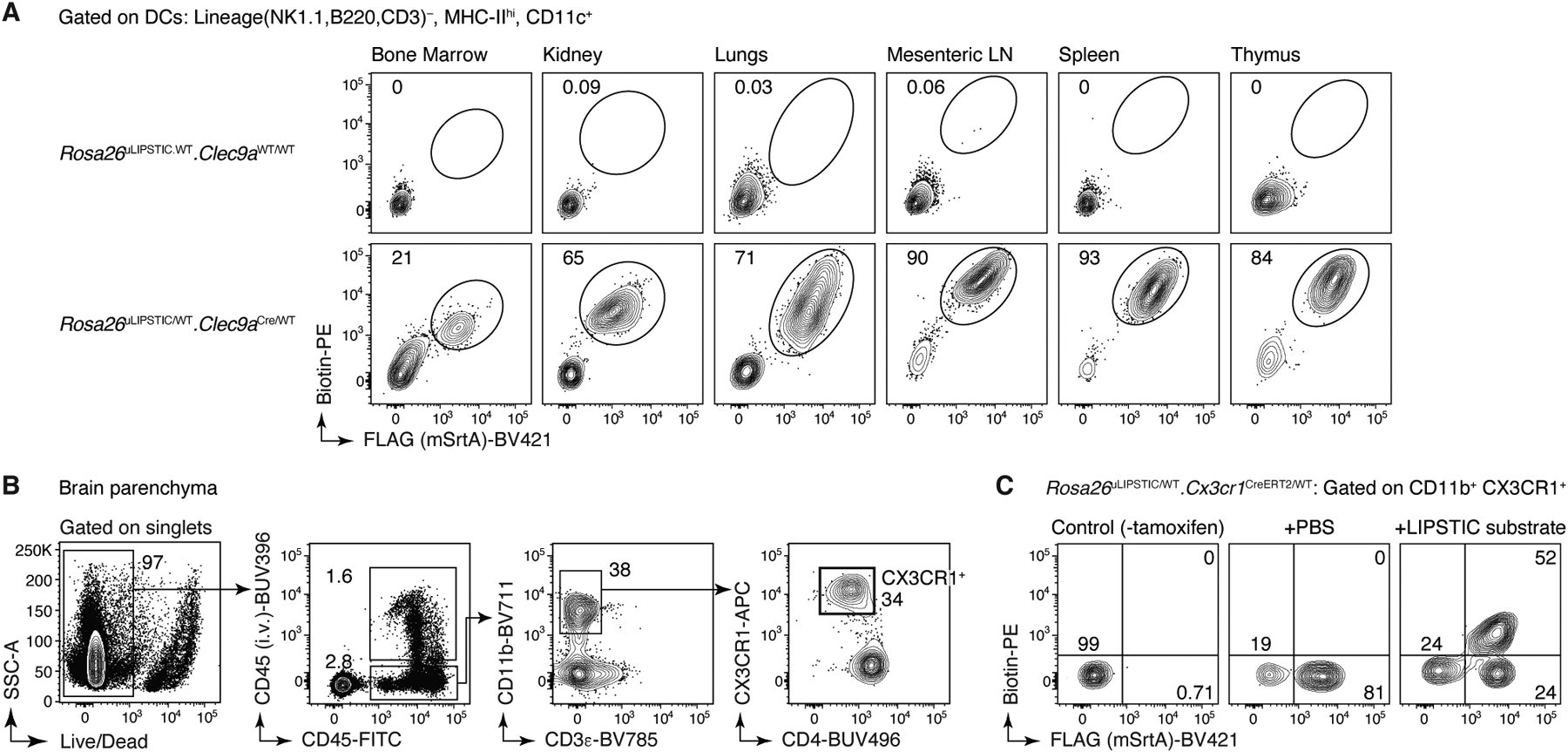
(A) Steady state Rosa26uLIPSTIC/WT.Clec9aCreER/WT or Rosa26uLIPSTIC/WT.Clec9aWT/WT control mice were injected i.p. with the LIPSTIC substrate and its loading onto DCs was achieved in all analyzed tissues. (B-C) I.p. injection of the LIPSTIC substrate reaches the brain in Rosa26uLIPSTIC/WT.Cx3xr1CreERT2/WT mice. (B) Flow cytometry gating strategy to analyze CX3CR1-expressing microglia in the brain. To discriminate resident from circulating immune cells in the brain, α-CD45 antibody was injected intravenously to mark the latter. (C) SrtA expression was detected in ~80% of CD11b+CX3CR1+ cells, 68% of which acquired i.p.-administered LIPSTIC substrate.
Extended Data Figure 6 |. uLIPSTIC to study epithelial cell – immune cell interactions in the gut.
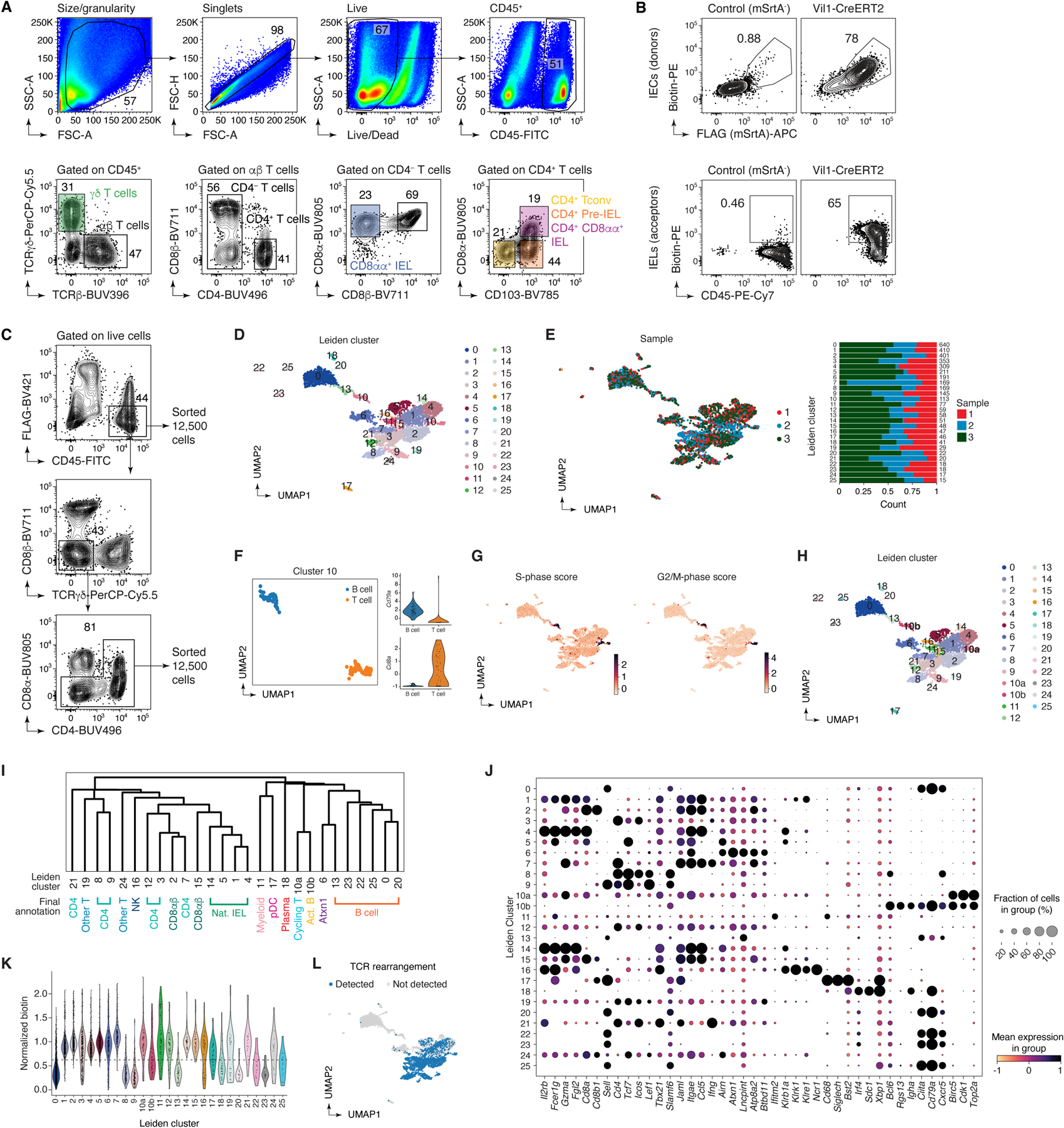
(A-C) Flow cytometry strategy for intraepithelial immune cells. (A) Representative gating strategy for γδ TCR and αβ TCR (Cd8αα+, CD8αβ+, and CD4+) IEL subsets. (B) Top, expression of SrtA (FLAG) and capture of LIPSTIC substrate by IEC donor cells and bottom, transfer of substrate onto CD45+ acceptor cells in SrtA-expressing and control mice. (C) Sorting strategy for the scRNA-seq experiment. Samples were enriched for rarer (e.g., B cell, CD4-IEL) populations by first sorting 12,500 total cells then an additional 12,500 cells depleted of the dominant γδ, CD8αα, and CD8αβ IEL populations. Three independent samples were sorted and stained with different hashtag oligos for downstream identification. (D-L) Clustering analysis of the immune interactome of IECs in the small intestine. (D) UMAP colored by Leiden clustering of the entire scRNA-seq/uLIPSTIC dataset (n=3,677 cells) used as an intermediate step in cell type annotations. (E) Left, UMAP colored by biological replicate. Right, bar plot indicating cluster composition by biological replicate, cluster size indicated at the right of each bar. (F-G) Further analysis of cluster 10 shows that is a composite comprising proliferating T and B cells. This co-clustering of B and T cells held true for varying number of PCs between 20 and 100 (not shown). (F) Left, Leiden cluster 10 was isolated and sub-clustered, yielding two separate clusters (UMAP). Right, normalized expression of Cd79a and Cd8a for these two sub-clusters of cluster 10 determines their annotation as either B or T cells. (G) UMAP showing the S and G2M phase cell cycle gene list scores (obtained using the `score_genes_cell_cycle()` function with lists from the Seurat package71), characterizing Leiden cluster 10 as proliferating cells, thus explaining their co-clustering. (H) UMAP showing final clustering of the entire data, with Leiden cluster 10 subdivided into clusters 10a and 10b. (I) Dendrogram representing transcriptional similarities among clusters. Differentially expressed genes were identified for each cluster (log2FC > 1, FDR < 0.05, see Methods), and normalized expression of all such genes (5,956 genes total), averaged per cluster, was used for the hierarchical clustering analysis that produced the dendrogram. Final annotation clusters shown in Fig. 4 are indicated below the Leiden cluster numbers. (J) Dot plot of marker genes indicating their level of expression in each cluster. Dot size indicates the fraction of cells in the cluster with Pearson residual normalized expression greater than 0, dot color represents level of expression. (K) Violin plot showing levels of normalized uLIPSTIC signal for each Leiden cluster. (L) UMAP showing presence of rearranged TCRα and β in each cell.
Extended Data Figure 7 |. Expression of marker genes and gene signatures in the annotated scRNA-seq data.
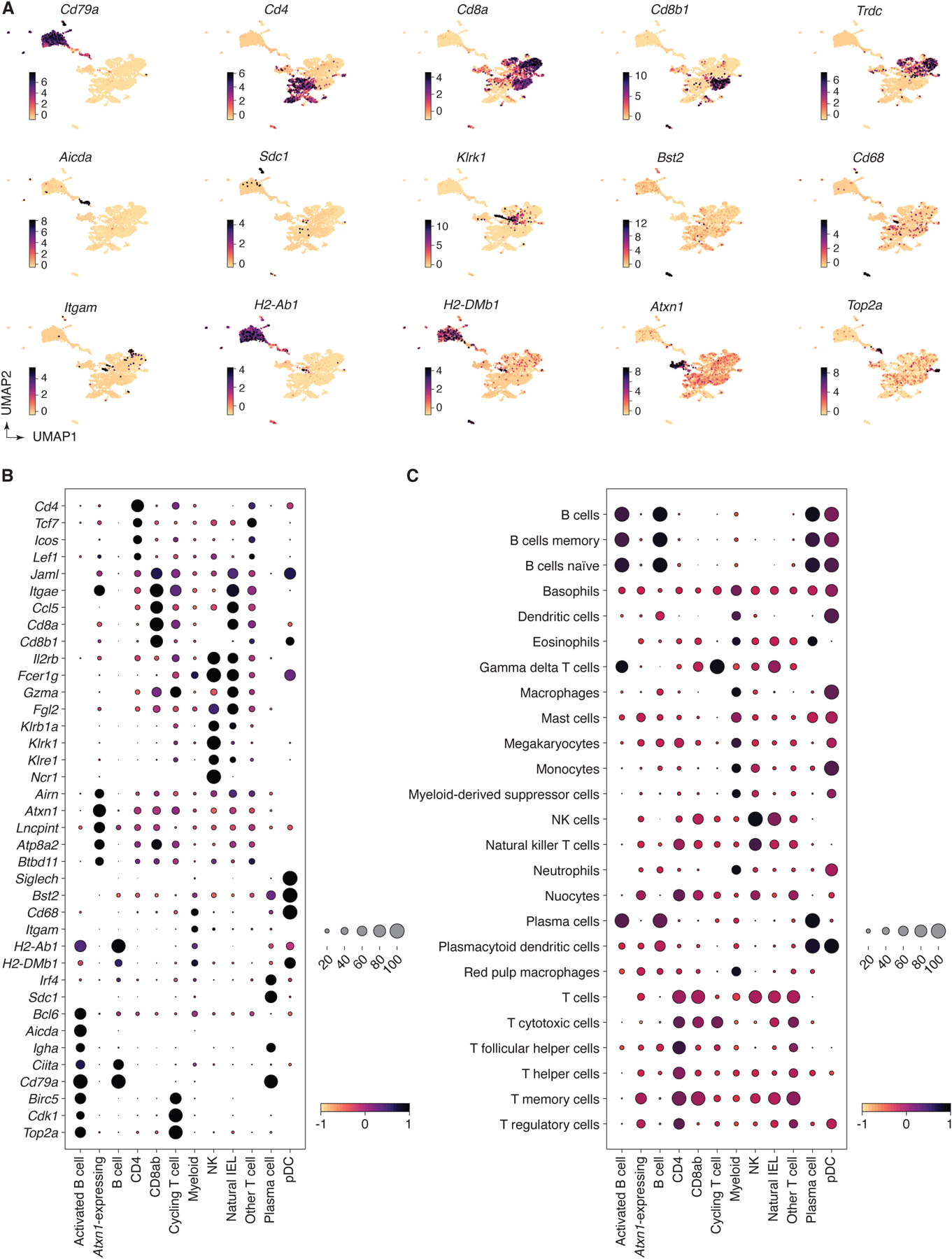
(A) UMAP plots showing normalized gene expression levels for selected marker genes characteristic of the final annotation clus. (B) Dot plot of marker genes indicating level of expression for each cell type annotation. (C) Dot plot of scores for gene signatures of immune cell types from PanglaoDB61. For both dot plots, dot size indicates the fraction of cells in the population with values greater than 0, dot color represents level of value (Pearson residual normalized expression or gene signature scores for B and C, respectively).
Extended Data Figure 8 |. Analysis of combined scRNA-seq + uLIPSTIC data for CD4+ T cells.
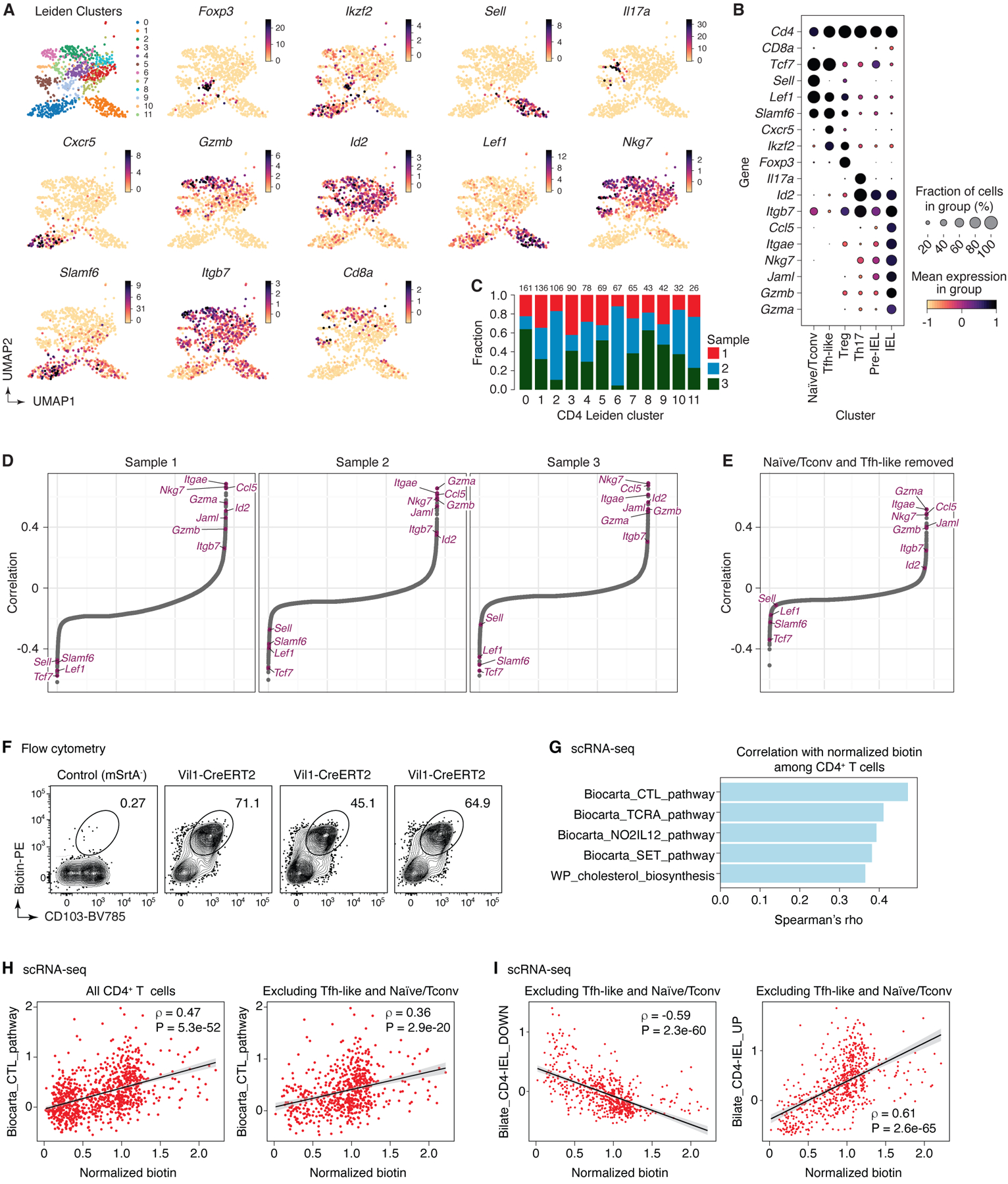
(A) UMAP for CD4+ T cells showing new Leiden sub-clusters and expression of selected marker genes in each cluster (n=915). (B) Dot plot of marker genes for each annotated subset of CD4+ T cells. Dot size indicates the fraction of cells in the cluster with Pearson residual normalized expression greater than 0, dot color represents level of expression. (C) Bar plot indicating CD4 Leiden cluster composition by biological replicate, cluster size indicated at the top of each bar. (D) Spearman correlation values, in increasing order, for uLIPSTIC signal and normalized expression of a gene, calculated separately for cells from each biological replicate, indicating consistency across mice. (E) Spearman correlation values, in increasing order, for uLIPSTIC signal and normalized expression of a gene, calculated when removing Tfh-like and naïve/conventional T cells (Leiden CD4 sub-clusters 0 and 1). (F-I) Correlation between acquisition of uLIPSTIC label and expression of CD103 and selected gene signatures by CD4-IELs. (F) Flow cytometry plots show uLIPSTIC signal and CD103 expression in one control Rosa26uLIPSTIC/WT and three Vil1-Cre.Rosa26uLIPSTIC/WT mice treated as in Fig. 3G. (G) Gene signatures from the MSigDB “canonical pathways” (M2.CP) database showing significant positive association with normalized biotin signal in scRNA-seq analysis over all CD4+ T cells. Plots show Spearman’s ρ value for each signature. (H) Correlation between acquisition of uLIPSTIC signal by CD4+ T cells (shown for all T cells and excluding Tfh-like and Naïve/Tconv clusters) and expression of the Biocarta CTL gene signature. Trend line and error are for linear regression with 95% confidence interval, Spearman’s ρ and two-sided P-value are listed. (I) Correlation between acquisition of uLIPSTIC signal by CD4+ T cells (shown for T cells excluding Tfh-like and Naïve/Tconv clusters) and expression of gene signatures up and downregulated as epithelial T cells transition from Tconv (CD4+CD103−CD8αα−) to CD4-IEL (CD4+CD103+CD8αα+) phenotypes (signatures based on data from Bilate et al.6). Trend line and error are for linear regression with 95% confidence interval, Spearman’s ρ and two-sided P-value are listed.
Extended Data Figure 9 |. Using uLIPSTIC to study CD8+ T cell priming during acute systemic LCMV infection.
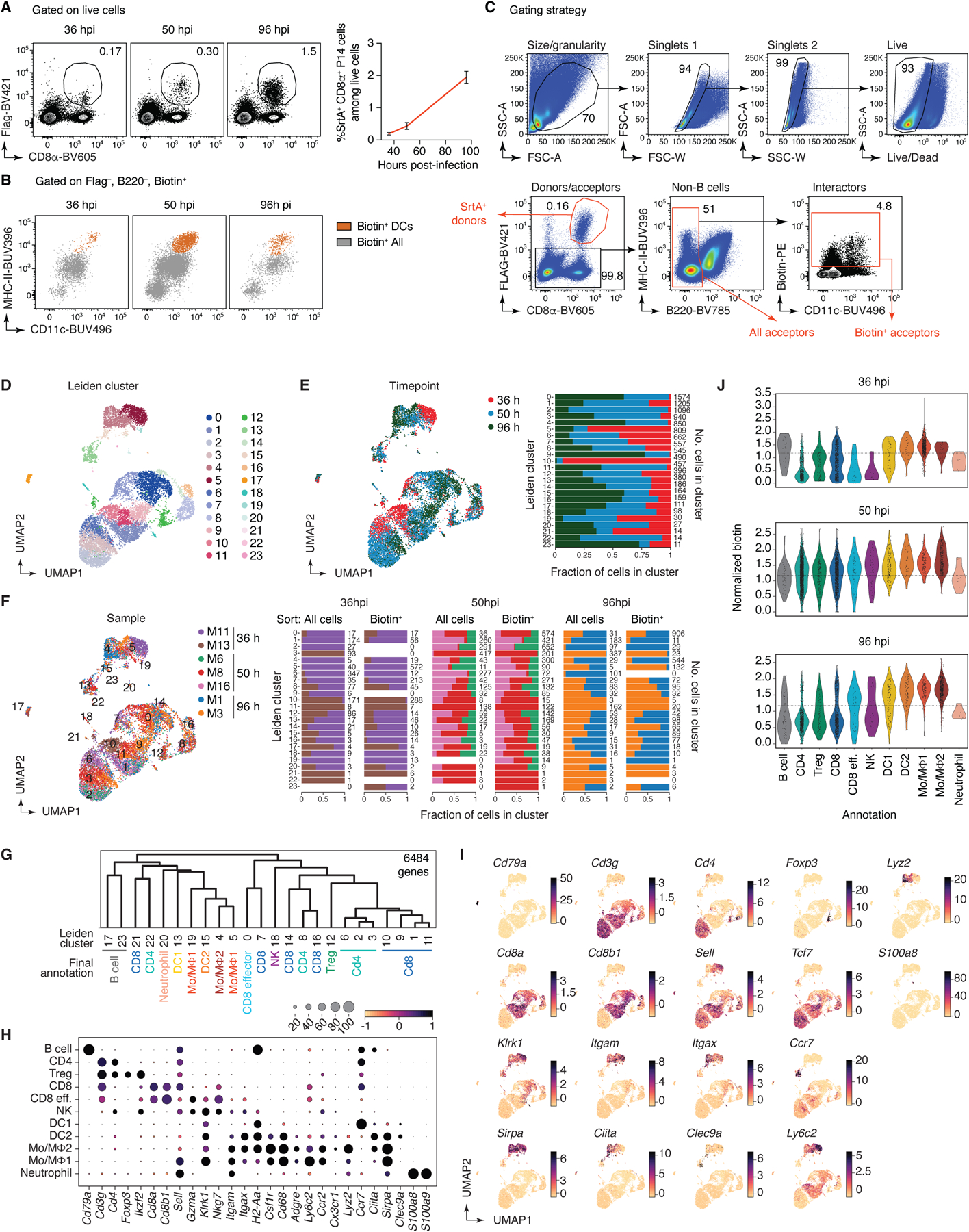
(A) Left, adoptively transferred LCMV-specific P14 CD8+ T cells infiltrated the mediastinal (m)LN of LCMV-infected Rosa26uLIPSTIC/uLIPSTIC hosts as early as 36 hpi. Right, fraction of P14 cells in total lymphocytes at the indicated timepoint. Data for ten mice per timepoint from three independent experiments, bar plots show mean ± SEM. (B) uLIPSTIC labeling of the P14-interactome (“Biotin+ All” in grey) showed that DCs (“Biotin+ DCs,” in orange) make up only a fraction of all interacting cells. (C) Sorting strategy for the scRNA-seq experiment. Immune cells—excluding B cells—were sorted both in an unbiased and biased manner, enriching for biotin+ acceptor cells and Flag+ donor cells using distinct hashtag oligos for downstream classification. Two-three independent samples per timepoint were sorted and stained with different hashtag oligos for downstream identification. (D-J) scRNA-seq nalysis of the immune interactome of P14 CD8+ T cells in the mLN during acute LCMV infection. (D) UMAP colored by Leiden clustering of the entire scRNA-seq/uLIPSTIC dataset (n=11,846 cells). (E) Left, UMAP colored by timepoint. Right, bar plot indicating cluster composition by timepoint, cluster size indicated at the right of each bar the right. (F) Left, UMAP colored by biological replicate. Right, bar plot indicating cluster composition by biological replicate, separated by whether the sample was sorted as total mLN cells or biotin-enriched mLN cells, as specified in (C). The cluster size is indicated at the right of each bar. (G) Dendrogram representing transcriptional similarities among clusters. Differentially expressed genes were identified for each cluster (log2FC > 1, FDR < 0.01, see Methods), and normalized expression of all such genes (6,484 genes total), averaged per cluster, was used for the hierarchical clustering analysis that produced the dendrogram. Final annotation clusters shown in Fig. 5 are indicated below the Leiden cluster numbers. (H) Dot plot of marker genes indicating their level of expression in each cell type annotation. Dot size indicates the fraction of cells in the cluster with Pearson residual normalized expression greater than 0, dot color represents level of expression. (I) UMAPs showing normalized gene expression levels for selected marker genes. (J) Violin plot showing levels of normalized uLIPSTIC signal for each cell type annotation, separated by timepoint and excluding P14 donor cells (high FLAG).
Extended Data Figure 10 |. Analysis of combined scRNA-seq + uLIPSTIC data for LCMV tissues (profiled at 96 hpi).
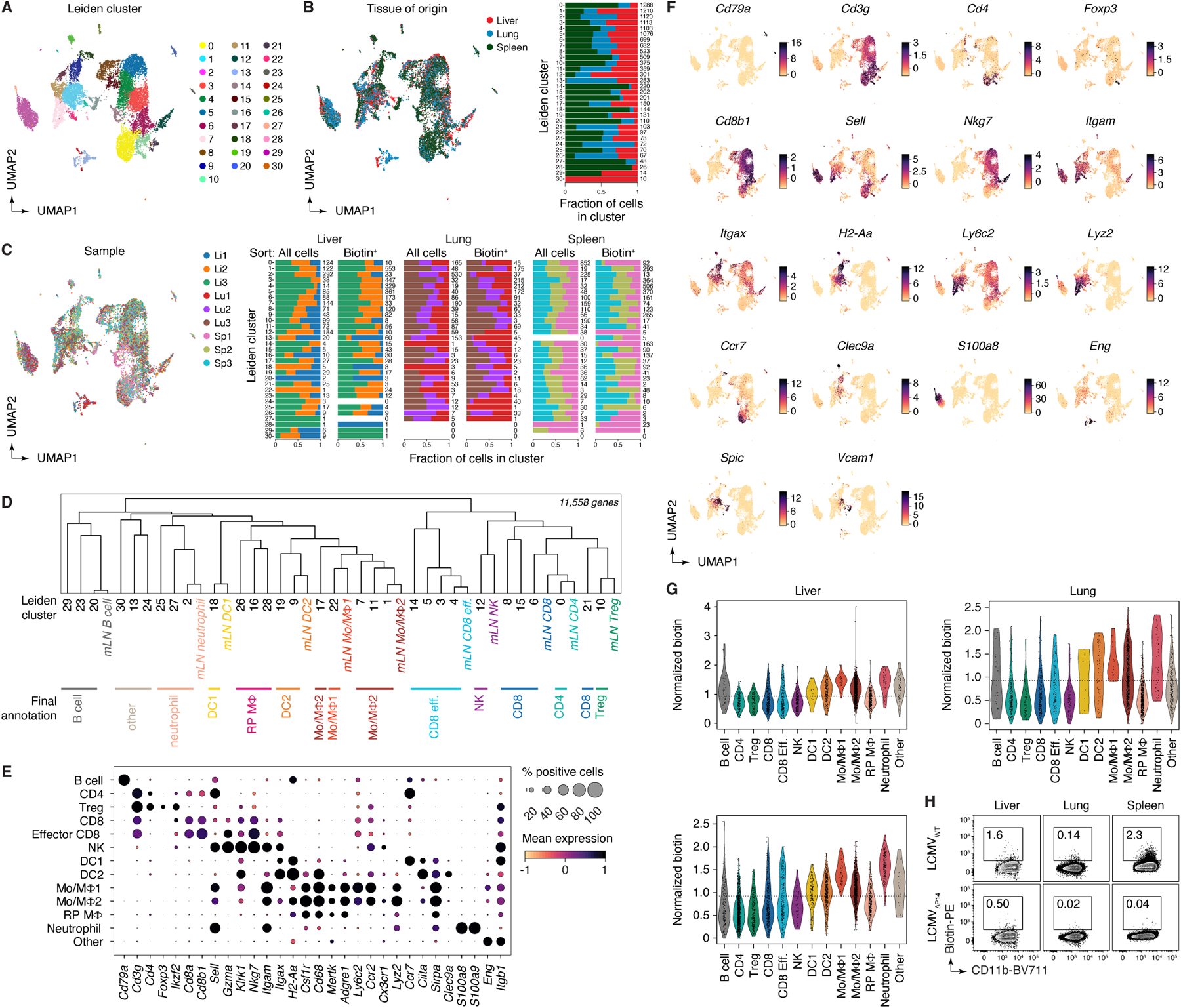
(A) UMAP colored by Leiden clustering of the entire scRNA-seq/uLIPSTIC dataset (n=12,324 cells). (B) Left, UMAP colored by tissue type. Right, bar plot indicating cluster composition by tissue, cluster size indicated at the right of each bar. (C) Left, UMAP colored by biological replicate. Right, bar plot indicating cluster composition by biological replicate, separated by whether the sample was unsorted cells or sorted as biotin-enriched cells. The cluster size is indicated at the right of each bar. (D) Dendrogram representing transcriptional similarities among tissue Leiden clusters with annotations from the mLN data. Normalized expression of all genes in the LCMV datasets (11,558 genes total), averaged per Leiden cluster for the tissue data and averaged per annotation for the mLN data, was used for the hierarchical clustering analysis that produced the dendrogram. Final annotation clusters shown in Fig. 5 are indicated below the Leiden cluster numbers. (E) Dot plot of marker genes indicating their level of expression in each cell type annotation. Dot size indicates the fraction of cells in the population with Pearson residual normalized expression greater than 0, dot color represents level of expression. (F) UMAP plots showing normalized gene expression levels for selected marker genes characteristic of the final annotation clusters. (G) Violin plot showing levels of normalized uLIPSTIC signal for each cell type annotation, separated by tissue type and excluding P14 donor cells (high FLAG). (H) uLIPSTIC labeling of MHC-IIhi monocytes/macrophages (Mo/MΦ2) in organs of mice treated as in Fig. 5A but infected with either LCMVWT or LCMVΔP14, analyzed at 96 hpi. Data from one experiment with each symbol representing one mouse, P-values were calculated using two-tailed Student’s test.
Supplementary Material
Acknowledgements:
We would like to thank the Rockefeller University Transgenics and Gene Targeting facilities for generating the uLIPSTIC mouse strain, the Comparative Biosciences Center for mouse housing, Tiago B.R. Castro for bioinformatics assistance, Cecilio Lemes Ferreira for mouse genotyping, Kristie Gordon and Jean-Philip Truman for cell sorting, and all Rockefeller University staff for their continuous support. We thank Caetano Reis e Sousa (Francis Crick Institute, UK), Tomohiro Kurosaki (U. Osaka, Japan), Takaharu Okada (RIKEN-Yokohama, Japan), and Claude-Agnès Reynaud and Jean-Claude Weill (Université Paris-Descartes, France) for mice; and Telmo Catarino (Instituto de Investigação e Inovação em Saúde, Portugal) and Henrique Veiga-Fernandes (Champalimaud Institute, Portugal) for their preliminary contributions to the development of the uLIPSTIC system. This study was funded by NIH grants DP1AI144248 (Pioneer award) and R01AI173086 to G.D.V., DP2AI171161 to Y.P., R01AR050452 and R01AR27883 to E.F., R01AI153363 to A.O.K. and V.D.H, and Starr Consortium grant I10-0044 to G.D.V. Work in the Victora laboratory is additionally supported by the Robertson Foundation, and work in the Pritykin lab by the Ludwig Institute for Cancer Research. S.N.-H. was supported by a Bulgari Women & Science Fellowship, S.W. by the NIH/NHGRI training grant 5T32HG003284, M.C.C.C. by the Pew Latin-American Fellows Program, A.C. by a Damon Runyon Postdoctoral Fellowship, and S.M.P. by a CRI/Carson Family Postdoctoral Fellowship (CRI4498). E.F. and D.M. are HHMI investigators. G.D.V. is a Burroughs-Wellcome Investigator in the Pathogenesis of Infectious Disease and Pew-Stewart Scholar.
Footnotes
Competing Interests: G.D.V. is an advisor for and owns stock futures in the Vaccine Company, Inc. E.F. recently served on the SABs of L’Oreal and Arsenal Biosciences and owns stock futures in the latter company.
Code availability: The full code used to analyze the scRNA-seq data is available at https://github.com/pritykinlab/ulipstic-analysis.
Data availability:
Final scRNA-seq datasets are available from GEO under accession number GSE253000. Processed scRNA-seq data is available at https://github.com/pritykinlab/ulipstic-analysis.
References
- 1.Dustin ML The immunological synapse. Cancer Immunol Res 2, 1023–1033 (2014). 10.1158/2326-6066.CIR-14-0161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pasqual G et al. Monitoring T cell-dendritic cell interactions in vivo by intercellular enzymatic labelling. Nature 553, 496–500 (2018). 10.1038/nature25442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenwald I & Rubin GM Making a difference: the role of cell-cell interactions in establishing separate identities for equivalent cells. Cell 68, 271–281 (1992). 10.1016/0092-8674(92)90470-w [DOI] [PubMed] [Google Scholar]
- 4.Sudhof TC & Malenka RC Understanding synapses: past, present, and future. Neuron 60, 469–476 (2008). 10.1016/j.neuron.2008.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Victora GD & Nussenzweig MC Germinal centers. Annu Rev Immunol 30, 429–457 (2012). 10.1146/annurev-immunol-020711-075032 [DOI] [PubMed] [Google Scholar]
- 6.Bilate AM et al. T Cell Receptor Is Required for Differentiation, but Not Maintenance, of Intestinal CD4(+) Intraepithelial Lymphocytes. Immunity 53, 1001–1014 e1020 (2020). 10.1016/j.immuni.2020.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niec RE, Rudensky AY & Fuchs E Inflammatory adaptation in barrier tissues. Cell 184, 3361–3375 (2021). 10.1016/j.cell.2021.05.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mempel TR, Henrickson SE & Von Andrian UH T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 427, 154–159 (2004). 10.1038/nature02238 [DOI] [PubMed] [Google Scholar]
- 9.Moses L & Pachter L Museum of spatial transcriptomics. Nat Methods 19, 534–546 (2022). 10.1038/s41592-022-01409-2 [DOI] [PubMed] [Google Scholar]
- 10.Efremova M, Vento-Tormo M, Teichmann SA & Vento-Tormo R CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nature protocols 15, 1484–1506 (2020). 10.1038/s41596-020-0292-x [DOI] [PubMed] [Google Scholar]
- 11.Liu DS, Loh KH, Lam SS, White KA & Ting AY Imaging trans-cellular neurexin-neuroligin interactions by enzymatic probe ligation. PLoS One 8, e52823 (2013). 10.1371/journal.pone.0052823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ombrato L et al. Metastatic-niche labelling reveals parenchymal cells with stem features. Nature 572, 603–608 (2019). 10.1038/s41586-019-1487-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang S et al. Monitoring of cell-cell communication and contact history in mammals. Science 378, eabo5503 (2022). 10.1126/science.abo5503 [DOI] [PubMed] [Google Scholar]
- 14.Bechtel TJ, Reyes-Robles T, Fadeyi OO & Oslund RC Strategies for monitoring cell-cell interactions. Nat Chem Biol 17, 641–652 (2021). 10.1038/s41589-021-00790-x [DOI] [PubMed] [Google Scholar]
- 15.Yassin M et al. Upregulation of PD-1 follows tumour development in the AOM/DSS model of inflammation-induced colorectal cancer in mice. Immunology 158, 35–46 (2019). 10.1111/imm.13093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weizman OE et al. Developing synthetic tools to decipher the tumor-immune interactome. Proc Natl Acad Sci U S A 120, e2306632120 (2023). 10.1073/pnas.2306632120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dorr BM, Ham HO, An C, Chaikof EL & Liu DR Reprogramming the specificity of sortase enzymes. Proc Natl Acad Sci U S A 111, 13343–13348 (2014). 10.1073/pnas.1411179111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guimaraes CP et al. Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions. Nature protocols 8, 1787–1799 (2013). 10.1038/nprot.2013.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dustin ML & Depoil D New insights into the T cell synapse from single molecule techniques. Nat Rev Immunol 11, 672–684 (2011). 10.1038/nri3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madisen L et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature neuroscience 13, 133–140 (2010). 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robertson JM, Jensen PE & Evavold BD DO11.10 and OT-II T cells recognize a C-terminal ovalbumin 323–339 epitope. J Immunol 164, 4706–4712 (2000). 10.4049/jimmunol.164.9.4706 [DOI] [PubMed] [Google Scholar]
- 22.Merkenschlager J et al. Dynamic regulation of TFH selection during the germinal centre reaction. Nature 591, 458–463 (2021). 10.1038/s41586-021-03187-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stoll S, Delon J, Brotz TM & Germain RN Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science 296, 1873–1876 (2002). 10.1126/science.1071065 [DOI] [PubMed] [Google Scholar]
- 24.Frederico B et al. DNGR-1-tracing marks an ependymal cell subset with damage-responsive neural stem cell potential. Dev Cell 57, 1957–1975 e1959 (2022). 10.1016/j.devcel.2022.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubtsov YP et al. Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671 (2010). 10.1126/science.1191996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aghajani K, Keerthivasan S, Yu Y & Gounari F Generation of CD4CreER(T(2)) transgenic mice to study development of peripheral CD4-T-cells. Genesis 50, 908–913 (2012). 10.1002/dvg.22052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shulman Z et al. T follicular helper cell dynamics in germinal centers. Science 341, 673–677 (2013). 10.1126/science.1241680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dogan I et al. Multiple layers of B cell memory with different effector functions. Nat Immunol 10, 1292–1299 (2009). 10.1038/ni.1814 [DOI] [PubMed] [Google Scholar]
- 29.McDonald BD, Jabri B & Bendelac A Diverse developmental pathways of intestinal intraepithelial lymphocytes. Nat Rev Immunol 18, 514–525 (2018). 10.1038/s41577-018-0013-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.el Marjou F et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193 (2004). 10.1002/gene.20042 [DOI] [PubMed] [Google Scholar]
- 31.Mucida D et al. Transcriptional reprogramming of mature CD4(+) helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat Immunol 14, 281–289 (2013). 10.1038/ni.2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.London M, Bilate AM, Castro TBR, Sujino T & Mucida D Stepwise chromatin and transcriptional acquisition of an intraepithelial lymphocyte program. Nat Immunol 22, 449–459 (2021). 10.1038/s41590-021-00883-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cepek KL et al. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature 372, 190–193 (1994). 10.1038/372190a0 [DOI] [PubMed] [Google Scholar]
- 34.Zen K et al. Neutrophil migration across tight junctions is mediated by adhesive interactions between epithelial coxsackie and adenovirus receptor and a junctional adhesion molecule-like protein on neutrophils. Mol Biol Cell 16, 2694–2703 (2005). 10.1091/mbc.e05-01-0036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen CJ et al. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc Natl Acad Sci U S A 98, 15191–15196 (2001). 10.1073/pnas.261452898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pazirandeh A et al. Multiple phenotypes in adult mice following inactivation of the Coxsackievirus and Adenovirus Receptor (Car) gene. PLoS One 6, e20203 (2011). 10.1371/journal.pone.0020203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oldstone MB et al. Virus and immune responses: lymphocytic choriomeningitis virus as a prototype model of viral pathogenesis. Br Med Bull 41, 70–74 (1985). 10.1093/oxfordjournals.bmb.a072029 [DOI] [PubMed] [Google Scholar]
- 39.Olson MR, McDermott DS & Varga SM The initial draining lymph node primes the bulk of the CD8 T cell response and influences memory T cell trafficking after a systemic viral infection. PLoS Pathog 8, e1003054 (2012). 10.1371/journal.ppat.1003054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jakubzick C et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 39, 599–610 (2013). 10.1016/j.immuni.2013.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puglielli MT et al. In vivo selection of a lymphocytic choriomeningitis virus variant that affects recognition of the GP33–43 epitope by H-2Db but not H-2Kb. J Virol 75, 5099–5107 (2001). 10.1128/JVI.75.11.5099-5107.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stevens AJ et al. Programming Multicellular Assembly with Synthetic Cell Adhesion Molecules. Nature (2022). 10.1038/s41586-022-05622-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giladi A et al. Dissecting cellular crosstalk by sequencing physically interacting cells. Nat Biotechnol 38, 629–637 (2020). 10.1038/s41587-020-0442-2 [DOI] [PubMed] [Google Scholar]
- 44.Sujino T et al. Tissue adaptation of regulatory and intraepithelial CD4(+) T cells controls gut inflammation. Science 352, 1581–1586 (2016). 10.1126/science.aaf3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shin KS et al. Monocyte-Derived Dendritic Cells Dictate the Memory Differentiation of CD8(+) T Cells During Acute Infection. Front Immunol 10, 1887 (2019). 10.3389/fimmu.2019.01887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engels B et al. Retroviral vectors for high-level transgene expression in T lymphocytes. Hum Gene Ther 14, 1155–1168 (2003). 10.1089/104303403322167993 [DOI] [PubMed] [Google Scholar]
- 47.Kim JH et al. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One 6, e18556 (2011). 10.1371/journal.pone.0018556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Argos P An investigation of oligopeptides linking domains in protein tertiary structures and possible candidates for general gene fusion. J Mol Biol 211, 943–958 (1990). 10.1016/0022-2836(90)90085-Z [DOI] [PubMed] [Google Scholar]
- 49.Lee PP et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15, 763–774 (2001). 10.1016/s1074-7613(01)00227-8 [DOI] [PubMed] [Google Scholar]
- 50.Schraml BU et al. Genetic tracing via DNGR-1 expression history defines dendritic cells as a hematopoietic lineage. Cell 154, 843–858 (2013). 10.1016/j.cell.2013.07.014 [DOI] [PubMed] [Google Scholar]
- 51.Shinnakasu R et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol 17, 861–869 (2016). 10.1038/ni.3460 [DOI] [PubMed] [Google Scholar]
- 52.Barnden MJ, Allison J, Heath WR & Carbone FR Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunology and cell biology 76, 34–40 (1998). 10.1046/j.1440-1711.1998.00709.x [DOI] [PubMed] [Google Scholar]
- 53.Danciu C et al. A characterization of four B16 murine melanoma cell sublines molecular fingerprint and proliferation behavior. Cancer Cell Int 13, 75 (2013). 10.1186/1475-2867-13-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanchez AB & de la Torre JC Rescue of the prototypic Arenavirus LCMV entirely from plasmid. Virology 350, 370–380 (2006). 10.1016/j.virol.2006.01.012 [DOI] [PubMed] [Google Scholar]
- 55.Emonet SF, Garidou L, McGavern DB & de la Torre JC Generation of recombinant lymphocytic choriomeningitis viruses with trisegmented genomes stably expressing two additional genes of interest. Proc Natl Acad Sci U S A 106, 3473–3478 (2009). 10.1073/pnas.0900088106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iwasaki M, Ngo N, Cubitt B, Teijaro JR & de la Torre JC General Molecular Strategy for Development of Arenavirus Live-Attenuated Vaccines. J Virol 89, 12166–12177 (2015). 10.1128/JVI.02075-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pasqual G, Angelini A & Victora GD Triggering positive selection of germinal center B cells by antigen targeting to DEC-205. Methods in molecular biology 1291, 125–134 (2015). 10.1007/978-1-4939-2498-1_10 [DOI] [PubMed] [Google Scholar]
- 58.Bilate AM et al. Tissue-specific emergence of regulatory and intraepithelial T cells from a clonal T cell precursor. Sci Immunol 1, eaaf7471 (2016). 10.1126/sciimmunol.aaf7471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolf FA, Angerer P & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome biology 19, 15 (2018). 10.1186/s13059-017-1382-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sturm G et al. Scirpy: a Scanpy extension for analyzing single-cell T-cell receptor-sequencing data. Bioinformatics 36, 4817–4818 (2020). 10.1093/bioinformatics/btaa611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Franzen O, Gan LM & Bjorkegren JLM PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford) 2019 (2019). 10.1093/database/baz046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Setty M et al. Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat Biotechnol 34, 637–645 (2016). 10.1038/nbt.3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Storey JD & Tibshirani R Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100, 9440–9445 (2003). 10.1073/pnas.1530509100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132 (2004). 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 66.Ko J, Park H, Heo L & Seok C GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res 40, W294–297 (2012). 10.1093/nar/gks493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jo S, Kim T, Iyer VG & Im W CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29, 1859–1865 (2008). 10.1002/jcc.20945 [DOI] [PubMed] [Google Scholar]
- 68.Paulick MG & Bertozzi CR The glycosylphosphatidylinositol anchor: a complex membrane-anchoring structure for proteins. Biochemistry 47, 6991–7000 (2008). 10.1021/bi8006324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goddard TD et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25 (2018). 10.1002/pro.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schrödinger LLC & DeLano W PyMOL v.2.4.0. (2020).
- 71.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Final scRNA-seq datasets are available from GEO under accession number GSE253000. Processed scRNA-seq data is available at https://github.com/pritykinlab/ulipstic-analysis.