

Abstract
Cystic fibrosis (CF), one of the most common fatal hereditary disorders, is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The CFTR gene product is a multidomain adenosine triphosphate-binding cassette (ABC) protein that functions as a chloride (Cl−) channel that is regulated by intracellular magnesium [Mg2+]i. The most common mutations in CFTR are a deletion of a phenylalanine residue at position 508 (ΔF508-CFTR, 70–80 % of CF phenotypes) and a Gly551Asp substitution (G551D-CFTR, 4–5 % of alleles), which lead to decreased or almost abolished Cl− channel function, respectively. Magnesium ions have to be finely regulated within cells for optimal expression and function of CFTR. Therefore, the melastatin-like transient receptor potential cation channel, subfamily M, member 7 (TRPM7), which is responsible for Mg2+ entry, was studies and [Mg2+]i measured in cells stably expressing wildtype CFTR, and two mutant proteins (ΔF508-CFTR and G551D-CFTR). This study shows for the first time that [Mg2+]i is decreased in cells expressing ΔF508-CFTR and G551D-CFTR mutated proteins. It was also observed that the expression of the TRPM7 protein is increased; however, membrane localization was altered for both ΔF508del-CFTR and G551D-CFTR. Furthermore, both the function and regulation of the TRPM7 channel regarding Mg2+ is decreased in the cells expressing the mutated CFTR. Ca2+ influx via TRPM7 were also modified in cells expressing a mutated CFTR. Therefore, there appears to be a direct involvement of TRPM7 in CF physiopathology. Finally, we propose that the TRPM7 activator Naltriben is a new potentiator for G551D-CFTR as the function of this mutant increases upon activation of TRPM7 by Naltriben.
Keywords: Cystic fibrosis, TRPM7, CFTR, G551D-CFTR, ΔF508-CFTR, Naltriben
Introduction
Cystic fibrosis (CF), one of the most common fatal hereditary disorders in Caucasians, is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene on chromosome 7 [1–3]. Translation of CFTR mRNA results is a 1480 residue with a predicted molecular mass of 168 kilodaltons (kDa) [2]. CFTR is a multidomain adenosine triphosphate-binding cassette (ABC) protein responsible for the regulation of chloride (Cl−) transmembrane transport, as well as many other ions such as halides (permeation sequence P I > P Br > P Cl > P F; [4]) and polyatomic anions [5], and also to some small uncharged molecules [6]. Also, CFTR function is critical in maintaining fluid homeostasis, airway fluid clearance and airway submucosal glands secretion [7, 8].
The most common mutation causing CF is a deletion of a phenylalanine residue at position 508 (ΔF508-CFTR), which is responsible for 70–80 % of CF phenotypes worldwide [8–10]. The ΔF508-CFTR protein is misfolded, retained in the endoplasmic reticulum (ER) and prematurely degraded. The limited amount of ΔF508-CFTR protein that reaches the plasma membrane is unstable, resulting in a rapid redirection from endosomal recycling towards lysosomal delivery and degradation [11–13]. Another mutation, Gly551Asp (G551D) also called “the Celtic mutation”, accounts for 4–5 % of alleles, and exhibits normal protein expression and cell surface localization. Nevertheless, it is associated with a severe form of CF pathology as Cl− channel function is lacking [14–17].
Under normal conditions, wildtype CFTR (WT-CFTR) is a Cl− channel activated by protein kinases A and C (PKA and PKC) and deactivated by dephosphorylation by a high number of protein phosphatases [15, 16]. WT-CFTR is also regulated by protein–protein interactions involving synaptosomal-associated protein 23 (SNAP23) and syntaxin 1A, Na+/H+ exchanger regulatory factor (NHERF), CFTR-associated protein of 70 kDa (CAP70), CFTR–associated ligand (CAL) and annexin A5 [20–28]. CFTR is also regulated by divalent cations, the most studied being calcium (Ca2+) ions. Indeed, it was clearly reported that Ca2+ homeostasis is altered in CF [29]. An abnormal spatial diffusion of Ca2+ in airway epithelial cells derived from CF patients with the ΔF508-CFTR mutation was observed, due to an accumulation of inositol 1,4,5-trisphosphate (IP3) in the ER [30] and to dysfunction Ca2+ uptake in mitochondria [31]. An increased Ca2+ uptake is a hallmark of CF [32], but when the function of CFTR is restored, for example using Miglustat, Ca2+ homeostasis is normalized [33]. Intracellular magnesium ([Mg2+]i) is also important for the regulation of CFTR. The use of ionophore A23187 or ionomycine in human bronchial cells increases [Mg2+]i and leads to a time- and dose-dependent decrease in the expression of CFTR mRNA [34]. By contrast, decreased [Mg2+]i leads to reduced opening and closing rates of the CFTR channel [34]. In fact, [Mg2+]i controls the closing rate of CFTR, likely in conjuction with ATP. It is well established that ATP hydrolysis by nucleotide binding domain 2 (NBD2) of CFTR is decreased when [Mg2+]i is also decreased [35], the role of Mg-ATP upon CFTR’s function being more efficient than that of ATP alone [36].
Due to the fact that Mg2+ has to be finely regulated within cells for optimal CFTR expression and function, our aim was to study the melastatin-like transient receptor potential cation channel, subfamily M, member 7 (TRPM7) in non-transfected Hela cells (NT) and in cells expressing WT-CFTR, ΔF508-CFTR and G551D-CFTR. TRPM7, which is a member of the melastatin-like transient receptor potential cation channel, subfamily M (TRPM) family, is ubiquitously expressed and possesses both an ion pore channel and a functional serine/threonine α-kinase domain to provide cellular influx for Ca2+, Mg2+ and divalent metals [37–40]. Being a channel and having an enzymatic activity, TRPM7 is often called chanzyme. The TRPM7 protein, which is the main cellular regulator of [Mg2+]i [41], is slightly more permeable to Mg2+ than to Ca2+ and is regulated by Mg2+ or Mg-ATP [38, 42–44]. Indeed, TRPM7 has the following selectivity profile for divalent cations: Zn2+ ≈ Ni2+ > > Ba2+ > Co2+ > Mg2+ ≥ Mn2+ ≥ Sr2+ ≥ Cd2+ ≥ Ca2+ [43]. A remarkable finding being that trace divalent metal ions are significantly more permeable through TRPM7 than the dominant physiological divalent metal ions Ca2+ and Mg2+ [43]. Therefore, we studied the expression, the function and the channel regulation of TRPM7 by [Mg2+]i in NT cells and in cells expressing WT-CFTR or mutated CFTR (ΔF508-CFTR and G551D-CFTR). The function of CFTR was further studied when TRPM7 function was modulated by specific drugs.
We show for the first time that [Mg2+]i is decreased in cells expressing ΔF508-CFTR and G551D-CFTR. The protein expression of TRPM7 was increased in these cells whereas its function and Mg2+ regulation were decreased. In order to highlight a possible functional link between TRPM7 and CFTR, TRPM7 activity was increased in cells expressing ΔF508-CFTR and G551D-CFTR by the use of Naltriben and the function of CFTR was studied. We found that the activation of TRPM7 resulted in a decreased CFTR function in ΔF508-CFTR cells and in a highly increased function in cells expressing G551D-CFTR. Mg2+ and Ca2+ flux through TRPM7 were found to be modulated in cells expressing mutated CFTR. We conclude that the link between TRPM7 and CFTR varies with the type of CFTR mutation. Finally, we propose that Naltriben is a new potentiator of G551D-CFTR with potential implications for CF therapy. Indeed, the TRPM7 activator Naltriben rescues the altered function associated with the G551D-CFTR mutation.
Materials and methods
Cell culture, quantitative PCR and western blotting
HeLa cells stably expressing WT-CFTR, ΔF508-CFTR and G551D-CFTR were obtained by lentiviral infection in which the cDNA encoding the human wildtype CFTR (WT-CFTR, Transgene SA, pTG5985, France, access Genbank No. M28668) was used, before and after site directed mutagenesis. Hela cells stably expressing WT-CFTR, ΔF508-CFTR or G551D-CFTR were characterized previously [45]. Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; E Healthcare, USA) plus 10 % calf serum in the presence of blasticidine (2 μg/ml).
Total RNA was extracted from cells using the RNeasy Plus Mini kit (Qiagen, France), according to the manufacturer’s instructions, and the quality was assessed using an Agilent 2100 Bioanalyzer (Agilent, France). RNA was further quantified using a nanophotometer for further use in Reverse Transcription Polymerase Chain Reaction (RT-PCR). RT-PCR was performed using Superscript II (InVitrogen, France). Quantitative PCR was performed using a Quantitect SYBR Green kit (Qiagen, France) and 3 μg of cDNA as tempate. TRPM7 primers were used as following: forward—CTTATGAAGAGGCAGGTCATGG and reverse—CATCTTGTCTGAAGGACTG. Actin primers were forward—GCATGGAGTCCTGTGGCATCC and reverse—GAGGCCAGGATGGAGCCG. Primers were tested for efficiency (E) using a standard curve analysis. E values were 107.83 and 105.32 % (R 2 > 0.98) for TRPM7 and actin, respectively. Optimal annealing temperature for the qPCR assay was determined by testing a range of temperatures above and below the calculated Tm (54 °C) of the primers. PCRs yielded 213 and 238 bp amplicons for TRPM7 and actin, respectively [46]. Reactions for TRPM7 were carried out with the following parameters: initial denaturation at 95 °C for 15 min followed by 40 cycles of denaturation at 95 °C for 30 s, annealing at 54 °C for 30 s and extension at 72 °C for 30 s. Relative quantification of the transcripts were assessed in a one-step format (300 ng total RNA) using a QuantiTect® SYBR® Green RT-PCR Kit (Qiagen) with an ABI PRISM® 7700 sequence detection system.
The cells were lyzed in 50 mM Tris–HCl, 100 mM NaCl, 0.1 % SDS, in the presence of 1.1 μM leupeptin, 0.7 μM aprotinin, 120 μM phenylmethanesulfonylfluoride (PMSF), 1 μM iodoacetamide, 0.7 μM pepstatin and 1 mM diisopropyl phosphorofluoridate (DIFP). Protein concentrations were determined using Lowry’s methodology with bovine serum albumin as a standard [47]. Equal amounts of total protein for each sample (50 µg) were subjected to 4–12 % gradient SDS/PAGE (InVitrogen, France). After blotting, the polyvinylidene difluoride (PVDF) membranes were incubated with anti-TRPM7 (1/1000; Abcam, France, [48]) or anti-Glycerol-3-Phosphate Dehydrogenase (G3PDH, 1/7000) antibodies and with secondary antibody (anti-mouse, abcam ab6820, 1/20,000). Visualization was performed using an ECL + detection kit (Amersham, France). Densitometric analysis of the signals was performed using a GelDoc 2000 apparatus (Bio-Rad, France) and each value was normalized to the total amount of loaded proteins per lane determined by Coomassie blue staining of the membrane or to the G3PDH, which was estimated by densitometric analysis.
Measurements of [Mg2+]i with Mag-Fura-2-AM
Cells were grown in glass bottomed, black wall 96-well plates (BD Falcon) for 1 day before experiments. They were washed three times with normal buffered salt solution (NBSS) containing: 135 mM NaCl, 5.4 mM KCl, 1.2 mM CaCl2, 1 mM MgCl2, 0.33 mM NaH2PO4, 5 mM Glucose and 10 mM HEPES (pH 7.4). Measurement of the background fluorescence was performed after the last wash. Cells were further incubated for 30 min at 37 °C in NBSS containing 5 µM Mag-Fura-2-AM (Molecular Probes, France) and 0.1 % Pluronic F-127. Cells were washed twice with NBSS containing 0.1 mM K2EGTA in place of 1.2 mM CaCl2 for 20 min at 37 °C and measurement of fluorescence was performed. All fluorescence measurements were performed on Varioskan® Flash (Thermo Scientific) at 37 °C. Excitation wavelengths were 335 nm and 375 nm and the emission wavelength was 505 nm (12 nm bandwidth). All measurements of background fluorescence were subtracted from fluorescence measurements for each wavelength. [Mg2+]i was calculated from the ratio of fluorescence at the two excitation wavelengths described above, using a dissociation constant (K d) of 1.9 mM, for the Mag-Fura-2-AM/Mg2+ complex. Fluorescence at 335 (F(335)) and 375 (F(335)) nm were used to determine the R value (R = F(335)/F(375)). All values of the measured R were normalized to the standard R value with identical optics and converted to [Mg2+]i using the standard equation [Mg2+]i = K d × (R − R min/R max − R). Cells were permeabilized with 20 µM digitonin and the Rmax value was obtained by the addition of 50 mM MgCl2 in absence of Ca2. Rmin was determined by chelating Mg2+ with addition of 100 mM EDTA (pH 7.2).
Measurements of Ca2+ influx through TRPM7
Cells were plated (30,000 cells/well) in glass bottomed, black wall 96-well plates (BD Falcon) in DMEM containing 10 % calf serum without blasticidine during 1 day before experiment. The culture medium was removed and replaced with loading buffer containing: 137 mM NaCl, 4.7 mM KCl, 1.8 mM CaCl2, 560 µM MgCl2, 1 mM Na2HPO4, 5.5 mM Glucose, 10 mM HEPES (pH 7.4) and the Fura-2 QBT Calcium dye (Molecular Devices) during 60 min at 37 °C in 5 % CO2 atmosphere. The Fura-2 QBT™ Calcium Kits contain Fura-2, AM, a cell permeable calcium indicator that is ratiometric and UV-light excitable. Loading buffer was removed and replaced by the same buffer without Fura-2 and Ca2+ (1.8 mM CaCl2 was replaced by 100 µM EDTA). Fluorescence quenching measurements (excitation wavelength: 360 nm; emission wavelength: 510 nm) were realized using Flextation3 plate reader (Molecular Devices; SoftMax Pro v5.4) at 37 °C [49]. Basal fluorescence at 360 nm excitation was read during 2 min before the addition of 2 mM MnCl2 to follow constitutive calcium entry for 13 min. In order to assess the implication of TRPM7 in Ca2+ entries, activator Naltriben and inhibitor Waixenicin A were added 15 min before starting the reading (50 and 15 µM, respectively). Slope values were first normalized to average initial fluorescence (F 0) measured between 0 and 2 min (F t = (ΔF/F 0; ΔF = F t(360) – F 0) and then normalized to control values (WT-CFTR = 100 %). The slope of the quenching which was estimating Ca2+ entry was calculated with GraphPad Prism 5 and presented as average ± SD. Different conditions were compared using t test.
Patch clamp analysis
Patch-clamp experiments were performed with an automatic electrophysiology workstation, Port-a-Patch (Nanion Technologies GmbH, Germany), coupled to an external amplifier unit HEKA EPC-10 [28, 50]. Whole-cell patch-clamp recordings were performed at room temperature on isolated cells. Before recording, the culture medium was replaced by the external solution contained the following: 140 mM NaCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES and 5 mM d-glucose monohydrate [41, 51]. The final pH value was adjusted to 7.4 with NaOH and the osmolarity was 298 mOsmol. The internal solution contained the following: 50 mM CsCl, 10 mM NaCl, 60 mM Cs-Fluoride, 20 mM EGTA and 10 mM HEPES/CsOH (pH 7.2; 285 mOsmol). Inhibition of TRPM7 was performed by increasing the free [Mg2+]i from 0 to 200 μM, using MaxChelator software available at http://www.stanford.edu/~cpatton/webmaxcS.htm, for Mg2+ calculation [41, 52]. In some experiments, seals were enhanced using the seal enhancer solution containing: 80 mM NaCl, 3 mM KCl, 10 mM MgCl2, 35 mM CaCl2, 10 mM HEPES (298 mOsmol). All voltages were corrected for a liquid junction potential of 10 mV between external and internal solutions. For TRPM7 measurements, currents were elicited by a ramp protocol from −100 to +100 mV over 50 ms acquired at 0.5 Hz and a holding potential of 0 mV, as previously described [41, 52]. Voltage-clamp pulses were generated and data were captured using the Patchmaster program (Nanion Technologies GmbH, Germany). Outward currents at +80 mV, normalized to cell size ranging from 20 to 35 pF, and plotted against time, were recorded. For CFTR measurements, voltage-dependent properties were assessed by applying voltage steps of 100 ms duration from a holding potential of −80 mV to test potentials ranging from −80 to +80 mV, with 10 mV increments [28]. Activation and inhibition of TRPM7 were obtained by Naltriben methanesulfonate (50 μM) and Carvacrol (500 μM), respectively, as previously described [53–56]. CFTR-inhibitor solution (10 μM CFTRinh172, Sigma) and CFTR-activator solution [10 μM forskolin (FSK) and 30 μM genistein (GST)], both from Sigma were added to the external solution to inhibit or activate CFTR, respectively. For all graphic representations of I–V relationships, currents were normalized by membrane capacitances (20–35 pF) to remove variability due to differences in cell sizes. For each patch-clamp analysis, measurements were made on three different cells from four different cultures.
Immunofluorescence
Cells were seeded on cover slips and incubated at 37 °C until 50–60 % confluence was obtained. After two washes with phosphate-buffered saline (PBS; 1X), cells were fixed in 4 % Paraformaldehyde (PFA) for 10 min. After fixation and three 5 min washes with PBS, cells were permeabilized with 0.2 % Triton X-100 in PBS (PBS-T) for 10 min. After 5 min washes with PBS, non-specific binding was prevented by blocking with 3 % Bovine Serum Albumin (BSA) in 1X PBS (2 h at room temperature). Primary antibody raised against TRPM7 (ab85016, Abcam) was applied overnight at room temperature with 3 % BSA in 1X PBS. After three 5 min washes with PBS-T, cells were incubated with Alexafluor 488 donkey anti-mouse IgG (Santa Cruz) secondary antibody (1:800 in 3 % BSA) for 1 h. Nuclei were labelled with 4′,6-diamidino-2-phenylindole (DAPI, Sigma; 1:1000 in PBS 1X). Slides were mounted with Fluorsave (Calbiochem) and examined under a Zeiss AxioStar-Plus microscope. Images were collected with a Zeiss 100× objective.
Cell surface biotinylation
Cell surface biotinylation was performed as previously described [28]. Cells were grown in serum-free medium overnight. Culture medium was removed and cells were washed three times with PBS (1X plus 0.1 mM CaCl2 and 1 mM MgCl2) and once with PBS (1X). Cells were then exposed to sulfo-NHS-SS-biotin for 30 min on ice, rinsed twice with BSA quenching buffer (1 % BSA in PBS 1X, pH 7.4) and incubated 10 min in the same buffer. Cells were scraped in PBS (pH 7.4) and centrifuged at 2400 rpm (6 min, 4 °C). Cell pellets were suspended in RadioImmunoPrecipitation Assay buffer (RIPA) plus anti-proteases and incubated for 30 min on ice. The resulting lysates were centrifuged at 16,000g for 15 min at 4 °C and total cellular protein content was determined using Lowry’s method [47]. The supernatants were incubated with streptavidin beads overnight at 4 °C. After a brief centrifugation, supernatants were removed and the beads were washed four times in RIPA. Biotinylated proteins were eluted from streptavidin beads using 5X sample buffer containing 2-mercaptoethanol to cleave the NHS-SS-biotin. Biotinylated proteins (membrane proteins) and eluated proteins (non biotinylated proteins) were probed to assess TRPM7 expression using western blot experiments (mouse monoclonal anti-TRPM7, 1:200, N-17, sc-19562; Santa Cruz Biotechnology, USA). For normalization and to assess the membrane protein enrichment, Na+/K+-ATPase (α1 isoform) expression was assessed on the same blots, probed with a mouse monoclonal antibody (1:200, clone 9-A5, Santa Cruz Biotechnology, USA). For the detection of TRPM7 and Na+/K+-ATPase, the secondary antibody was anti-mouse (1:20,000; abcam ab6820). Experiments were performed twice with two different cultures.
STRING query of TRPM7’s interaction network
The potential interactions of TRPM7 were evaluated using the online Search Tool for the Retrieval of Interacting Genes/Protein (STRING 9.0, http://string-db.org) to predict both direct and indirect interactions [57]. The confident scoring of the interactions were based on known and predicted protein interactions collected from direct (physical) and indirect (functional) associations. The score was set at 0.7 to represent a high confidence level. The database quantitatively integrates interaction data from four sources—genomic context, high-throughput experiments, co-expression and previous knowledge from research publications. The results are presented with the input protein (in this case TRPM7) in the context of a graphical network of interaction partners. Associations in STRING are provided with a probabilistic confidence score, which represents an estimate of how likely a given association describes a functional linkage between two proteins. The computed score indicates a high confidence when more than one type of information supports the association (direct physical interactions, text mining or experimental).
Statistics
Results were expressed as mean ± standard error of the mean (SEM). Differences between experimental groups were evaluated by a two-tailed unpaired Student’s t test. p < 0.05 was considered statistically significant (*), p < 0.001 was considered very statistically significant (**) and p < 0.0001 was considered highly statistically significant (***).
Results
Measurements of [Mg2+]i in cells expressing WT-CFTR, ΔF508-CFTR or G551D-CFTR
HeLa cells stably expressing WT-CFTR, ΔF508-CFTR and G551D-CFTR were previously described regarding CFTR expression and function [45, 57]. [Mg2+]i was measured using Mag-fura-2-AM, an intracellular Mg2+ indicator that is ratiometric and UV light-excitable [59–61]. Measurements were performed at 37 °C because at room temperature cells did not stick in the wells and because this temperature was physiologically relevant. As shown in Fig. 1, the statistical analysis indicates that [Mg2+]i is significantly lower in cells expressing either ΔF508-CFTR or G551D-CFTR compared to those expressing WT-CFTR. Furthermore, the levels of [Mg2+]i in the cells expressing the mutated CFTR were close to that observed in NT Hela cells. [Mg2+]i was also significantly lower in cells expressing G551D-CFTR compared to those expressing ΔF508-CFTR.
Fig. 1.

Ratiometric measurement of [Mg2+]i. [Mg2+]i was estimated using Mag-Fura-2-AM in NT cells and in WT-CFTR, ΔF508-CFTR or G551D-CFTR cells. The bar chart represents the result of the statistical analysis (n = 6) and shows that [Mg2+]i is significantly lower in F508del-CFTR and G551D-CFTR experiments compared to comparable levels in cells expressing WT-CFTR or NT cells. Furthermore, [Mg2+]i is significantly lower in cells expressing G551D-CFTR compared to those expressing ΔF508-CFTR. Statistical analysis was performed with results obtained from three different cell cultures
Detection and quantitation of TRPM7’s mRNA and protein
Basal TRPM7 mRNA expression was assessed in NT, WT-CFTR, ΔF508-CFTR and G551D-CFTR cells. As shown in Fig. 2a (left panel), a single PCR amplicon (200 bp) was observed indicating that endogenous TRPM7 was relatively abundant in all four cell-lines. No signal was observed in the negative control. A control, G3PDH, was also amplified by PCR (with an amplicon of 121 bp) in the same samples and showed no variation in signal intensity (not shown). TRPM7 mRNA expression was quantified by qPCR and no differences between the cells were observed (Fig. 2a, right panel).
Fig. 2.

Expression of TRPM7. a The left image shows (1) the molecular weight marker and an example of representative PCR amplicons for TRPM7 (200 bp) in Hela cells (2) not transfected, or transfected with (3) WT-CFTR, (4) ΔF508-CFTR or (5) G551D-CFTR. (6) is the negative control. The right bar chart is the quantitative analysis of TRPM7 gene expression, determined by quantitative PCR in each cell line. Data were analyzed by using ∆Ct (target—reference) methodologies [28] with no differences observed between cells lines. b The left image shows representative immunodetection experiments of the TRPM7 protein (212 kDa) in Hela cells (1) not transfected, or expressing (2) WT-CFTR, (3) ΔF508-CFTR or (4) G551D-CFTR. Statistical analysis (right) shows that the expression of TRPM7 is increased in both ΔF508-CFTR and G551D-CFTR experiments compared wildtype assays. Nevertheless, there is less TRPM7 protein in cells expressing G551D-CFTR compared to those expressing ΔF508-CFTR. Furthermore, TRPM7 expression is decreased in cells expressing WT-CFTR compared to NT cells (n = 6)
Western blots were performed using whole cell lysates to detect the basal expression of TRPM7 protein (212 kDa, Fig. 2b, left panel) in the cell-lines. TRPM7 protein was observed in all four cell-lines. Statistical analysis showed some differences between TRPM7 protein levels between the cells, after normalization with an internal control, G3PDH (37 kDa), or total protein (Fig. 2b, right panel). The levels of TRPM7 protein were significantly higher in cells expressing ΔF508-CFTR and G551D-CFTR compared to cells expressing WT-CFTR. No difference was seen between cells expressing mutated CFTR. Lower levels of TRPM7 protein were observed in cells expressing WT-CFTR or G551D-CFTR when compared to NT cells. This difference was not found in cells expressing ΔF508-CFTR.
Cellular localization of TRPM7
Cellular localization of TRPM7 using a specific primary antibody was performed in NT cells and in cells expressing WT-CFTR, ΔF508-CFTR and G551D-CFTR (Fig. 3). First, negative controls in which the anti-TRPM7 antibody was omitted were performed in order to show the specificity of the labelling (Fig. 3a–d). Whereas the membrane localization of TRPM7 was likely not observed in NT cells (Fig. 3e–g), the protein was observed at the plasma membrane in the cells expressing WT-CFTR (Fig. 3h–j) and ΔF508-CFTR (Fig. 3k–m) despite a less strong labelling in these latest cells. As in all cell types, the protein was observed throughout the entire cell with a vesicular pattern in G551D-CFTR cells, with no labelling at the membrane (Fig. 3n–p). The membrane localization of TRPM7 was thus increased in WT-CFTR, decreased in ΔF508-CFTR and absent in membranes of cells expressing G551D-CFTR. Nevertheless, the protein was observed throughout the entire cell with a vesicular pattern in all cell lines.
Fig. 3.
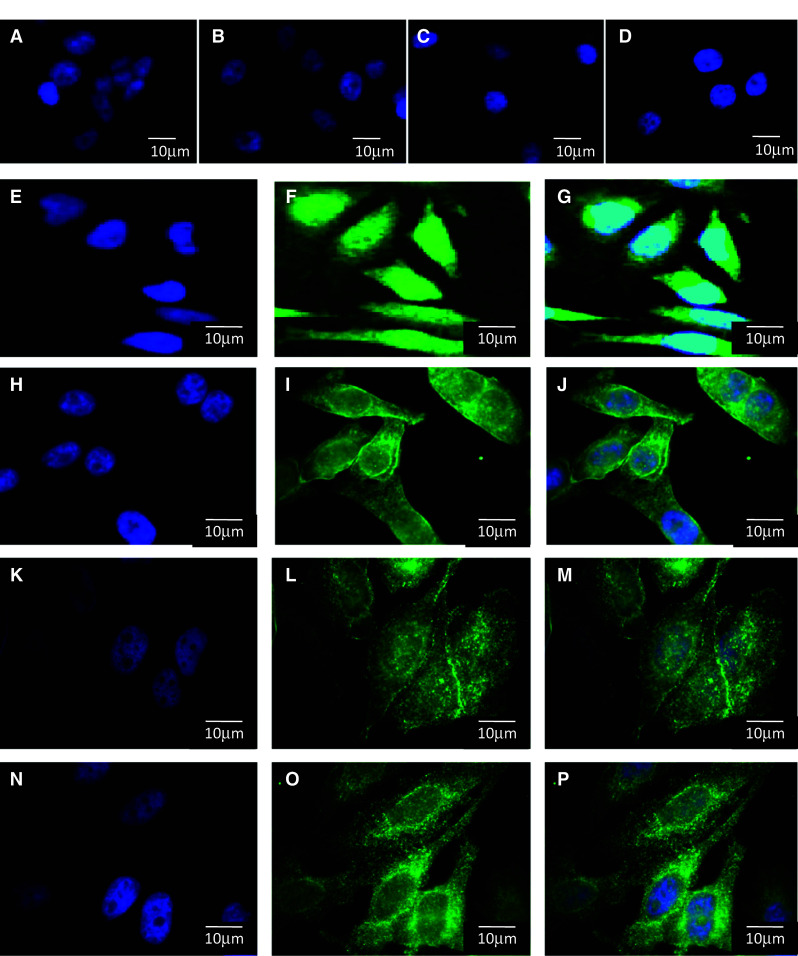
Cellular localization of TRPM7. Immunofluorescence was performed to localize TRPM7 in NT cells (a, e, f, g) and in cells expressing WT-CFTR (b, h, i, j), ΔF508-CFTR (c, k, l, m) and G551D-CFTR (d, n, o, p). First, negative controls in which first antibody is omitted shows the specificity of the labelling (a, b, c, d). Vesicular labelling in the cytoplasm is observed in all four cell types; however, membrane localization is mostly seen in cells expressing WT-CFTR (i, j) and ΔF508-CFTR (l, m). This membrane localization is likely not observed in NT cells (f, g). The labelling is less visible in ΔF508-CFTR (l, m) and lost in G551D-CFTR cells (o, p). Nuclei were DAPI stained (blue, a, b, c, d, e, h, i, j, k, l, m, n). Images were collected with a 100× objective. Bars represent 10 μm
To further confirm our findings regarding the localization of TRPM7, biotinylation experiments were performed (Fig. 4) [28]. Membrane proteins were biotinylated and TRPM7 expression was assessed by western blottings (Fig. 4a). In order to show the enrichment of the membrane proteins within the biotinylated fractions, Na+/K+-ATPase detection was performed. It was observed that the expression of TRPM7 is higher in cells expressing WT-CFTR and ΔF508-CFTR than in NT cells. The expression was found to be lower in ΔF508-CFTR than in WT-CFTR cells. The lowest expression of TRPM7 was observed in the membrane of cells expressing G551D-CFTR. Despite no statistical analysis was done because of the low number of experiments (n = 2), bar charts were drawn (Fig. 4b). After normalization by Na+/K+-ATPase expression, the densitometric analysis of the TRPM7 bands showed that the membrane localization of the protein was consistent with the results obtained by immunofluorescence.
Fig. 4.

Detection of TRPM7 within membranes by biotinylation experiments. a Representative immunoblots showing the detection of TRPM7 and Na+/K+-ATPase in NT (1), WT-CFTR (2), ΔF508-CFTR (3) and G551D-CFTR (4) cells, in biotinylated surface proteins and in non biotinylated proteins (Eluates). TRPM7 is present in the membrane fraction whereas it is likely not observed in non biotinylated proteins. The lower panel showing the presence of a specific membrane protein (Na+/K+-ATPase) in the biotinylated fractions indicates that membrane proteins were enriched in the biotinylated fractions. b Bar chart of the densitometric analysis of the cell surface expression of TRPM7 after normalization by Na+/K+-ATPase. The level of TRPM7 is higher in the membranes of WT-CFTR and ΔF508-CFTR cells than in NT and G551D-CFTR cells
Function and regulation of TRPM7
The native function of TRPM7 channels in the plasma membrane was assessed by recording of whole-cell currents in NT cells and in cells expressing WT-CFTR, ΔF508-CFTR and G551D-CFTR (Fig. 5). Activation of endogenous TRPM7-mediated currents was obtained by a voltage ramp (−100 to +100 mV in 50 ms intervals) and was used to determine current–voltage relationships. I–V relationships were determined at the time point when current magnitude reached maximum for each cell, with an internal Mg2+ free solution. I–V curves were drawn after normalization of the currents by cell capacitances. From the I–V curves (Fig. 5a, left panel) it was observed that TRPM7 channel function is drastically reduced in cells expressing mutant CFTR when compared to that of NT cells or to those expressing WT-CFTR. Currents in G551D-CFTR cells were weaker than those measured in cells expressing ΔF508-CFTR. Statistical analysis was performed and is presented as bar chart (Fig. 5a, right panel). Whereas currents are similar in NT and WT-CFTR cells, they were significantly decreased in cells expressing ΔF508-CFTR and G551D-CFTR. A difference was observed between cells expressing the mutated CFTR with lower currents in G551D-CFTR cells than in ΔF508-CFTR cells. TRPM7 is inhibited by Mg2+ [48]. Therefore, inhibition was induced by adding 200 μM Mg2+ in the intracellular solution, and I–V curves were recorded and compared to those obtained without divalent ions (Fig. 5b, left panel). It was observed that the presence of Mg2+ inhibited TRMP7 currents in NT cells as well as in cells expressing WT-CFTR. No effect was seen in ΔF508-CFTR or G551D-CFTR experiments. Statistical analysis (Fig. 5b, right panel) showed that 200 μM Mg2+ inhibited TRPM7 in NT and WT-CFTR cells and had no effect in the cells expressing either ΔF508-CFTR or G551D-CFTR. The latter results suggested that TRPM7 regulation by intracellular Mg2+ is altered in cells expressing mutant CFTR.
Fig. 5.

Function and regulation of TRPM7. Patch-clamp analysis was performed in NT Hela cells and in cells expressing, WT-CFTR, ΔF508-CFTR or and G551D-CFTR. a The left panel shows example current–voltage (I-V) relationships for TRPM7 channels obtained in the four cell lines with Mg2+-free intracellular solution. I–V relationships were taken at the time point when current magnitude reached maximum for each cell. Statistical analysis was performed using the pA/pF values taken at 100 mV and are presented as bar chart (right panel). Whereas currents are similar in NT and WT-CFTR cells, they were significantly decreased in ΔF508-CFTR and G551D-CFTR cells. A difference is also observed between cells expressing the mutated CFTR. The currents are significantly lower when in G551D-CFTR cells than in ΔF508-CFTR cells. b Comparison of the I–V relationships for TRPM7 channels in the four cell lines, with 200 μM Mg2+ and without Mg2+ in the intracellular solution (left panel). Bar chart (right panel) shows that whereas the function of TRPM7 is inhibited by Mg2+ in NT and WT-CFTR cells, no inhibition is observed in the cells expressing either ΔF508-CFTR or G551D-CFTR
Modulation of TRPM7 channel function by Mg-ATP, Naltriben and Carvacrol
Outward currents at +80 mV were recorded, and curves were drawn after normalization to cell sizes and plotted against time. Inhibition and activation of TRPM7 were achieved by using 500 μM Carvacrol and 50 μM Naltriben in cells expressing WT-CFTR, respectively. The drugs were applied 100 s after the start of the experiments (Fig. 6a). Activation was performed and observed in cells expressing either ΔF508-CFTR or G551D-CFTR, as currents were already very low when compared to those observed in WT-CFTR experiments (Fig. 6b, c, respectively). These results indicated that the recorded currents were due to TRPM7, which is consistent with previous reports [53–56]. Nevertheless, due to lower basal currents in ΔF508-CFTR and G551D-CFTR experiments, the effect of Naltriben was lower when compared to cells expressing WT-CFTR. The measurements presented in Fig. 5 also confirm that the function of TRPM7 is altered when mutant CFTR is expressed in cells.
Fig. 6.

Modulation of TRPM7 function in the absence of intracellular Mg2+. The temporal development of TRPM7 currents were extracted from individual ramp current recordings by measuring current amplitudes at a voltage of +80 mV. a (upper panel) Curve showing time-dependent currents measured in the absence of intracellular Mg2+ (black) and in the presence of Carvacrol (grey) in cells expressing WT-CFTR. The inhibitory effect of Carvacrol is observed. a (lower panel). Curve showing time-dependent currents measured in the absence of intracellular Mg2+ (black) and in the presence of Naltriben (grey) in cells expressing WT-CFTR. Activation of TRPM7 is observed. b Curve showing time-dependent currents measured in the absence of intracellular Mg2+ (black) and in the presence of Naltriben (grey) in cells expressing ΔF508-CFTR. Activation of TRPM7 is observed. c Curve showing time-dependent currents measured in the absence of intracellular Mg2+ (black) and in the presence of Naltriben (grey) in cells expressing G551D-CFTR. Activation of TRPM7 is observed. The curves presented in a, b and c are the mean curves of three cells from three different cell cultures
Mg-ATP is requested to measure the Cl− channel activity of CFTR [19, 36] and also inhibits TRPM7 [38]; therefore, the effects of Carvacrol and Natrilben in cells expressing WT-CFTR were assessed in the presence of intracellular Mg-ATP (Fig. 7). The same conditions were used as the previous set of experiments (see Fig. 6), except that 5 mM Mg-ATP was present in the intracellular solution. Figure 7a shows the time course of the currents with or without Mg-ATP in the intracellular buffer. It is observed that Mg-ATP inhibited TRPM7 activity in WT-CFTR experiments as expected. Furthermore, inhibition and activation of TRPM7 by Carvacrol and Natrilben, respectively, were obtained in cells expressing WT-CFTR in the presence of Mg-ATP, (Fig. 7b, c). However, the action of the drugs was lowered by decreased TRPM7 function due to the presence of 5 mM Mg-ATP in the intracellular solution. Figure 8 shows the activation and the inactivation of TRPM7 in cells expressing either ΔF508-CFTR or G551D-CFTR in the presence of 5 mM Mg-ATP. Figure 8a (upper panel) shows the time course of ΔF508-CFTR-induced currents, with or without Mg-ATP in the intracellular buffer, and it was deduced that Mg-ATP inhibited TRPM7 activity in the presence of mutant CFTR. Figure 8a (lower panel) shows that TRPM7 was activated and inactivated in the presence of Naltriben and Carvacrol, respectively, despite the presence of Mg-ATP in the intracellular solution. In Fig. 8b (upper panel), it was observed that TRPM7 currents in cells expressing G551D-CFTR were poorly inhibited by Mg-ATP in the intracellular buffer. Once again, TRPM7 was activated in this mutant cell-line in the presence of Naltriben, despite the presence of Mg-ATP in the intracellular solution (Fig. 8b, lower panel). Inactivation by Carvacrol was lower than in ΔF508-CFTR cells.
Fig. 7.

Modulation of TRPM7 function in WT-CFTR experiments, in the presence of 5 mM Mg-ATP. The same conditions as in Fig. 5 were used except that 5 mM Mg-ATP was present in the intracellular solution. a Time course of currents with (grey) or without (black) Mg-ATP in the intracellular buffer. Mg-ATP inhibited TRPM7 activity in cells expressing WT-CFTR. The inlet shows the initial decrease of currents due to Mg-ATP, whereas in the absence of Mg-ATP the initial currents increase. b Inhibition of TRPM7 by Carvacrol (grey). c Activation of TRPM7 by Natrilben
Fig. 8.

Activation of TRPM7 function in cells expressing either ΔF508-CFTR or G551D-CFTR, in the presence of 5 mM Mg-ATP. The same conditions as in Fig. 5 were used except that 5 mM Mg-ATP was present in the intracellular solution. a ( upper panel ) Time course of currents in cells expressing F508del-CFTR, with (grey) or without (black) Mg-ATP in the intracellular buffer. Mg-ATP inhibited TRPM7 activity in F508del-CFTR experiments. a (lower panel ) TRPM7 is activated and inhibited in the presence of Naltriben or Carvacrol, respectively, despite the presence of Mg-ATP in the intracellular solution, b (upper panel). Time course of currents in cells expressing G551D-CFTR, with (grey) or without (black) Mg-ATP in the intracellular buffer. Mg-ATP failed to inhibit TRPM7 activity in these cells. b (lower panel) TRPM7 is activated and poorly inhibited by Naltriben or Carvacrol, respectively in cells expressing G551D-CFTR, despite the presence of Mg-ATP in the intracellular solution
Measurement of CFTR channel function in the presence of Naltriben
CFTR function was measured by patch-clamping in cells expressing WT-CFTR, ΔF508-CFTR or G551D-CFTR (n = 3 cells from four different cell cultures for each cell type). Activators (FSK and GST) and inhibitor (Inhibitor 172) of CFTR were used to ensure the specificity of the recordings (Fig. 9a). Figure 9b shows the normalized I–V curve obtained in WT-CFTR experiments, in the presence of either Naltriben or Carvacrol. It was observed that TRPM7 inhibition had no effect on CFTR function, whereas TRPM7 activation by Naltriben led to decreased WT-CFTR channel function (Fig. 9b). A significant decrease in WT-CFTR function (−26 %) was observed in the presence of Naltriben, but no significant effect was detected by the addition of Carvacrol (Fig. 9c). For cells expressing ΔF508-CFTR (Fig. 10), I–V curves which were obtained in the presence of Naltriben and showed a significant decreased function of the mutated protein (Fig. 10a). This decreased function was more important than that of the specific inhibitor 172 (−39.8 %, Fig. 10b). I–V curves were also obtained in the presence of Naltriben in cells expressing G551D-CFTR (Fig. 11). I–V curves showed that Naltriben activated the function of G551D-CFTR and that this activation was higher than that of the addition of FSK plus GST. Indeed, a significantly increased chloride channel activity of G551D-CFTR (+824.2 %) was observed (Fig. 11b).
Fig. 9.

Inhibition of WT-CFTR currents by Naltriben. a I–V curves (normalized by cell capacitance pF, mean ± SEM) for CFTR currents in WT-CFTR expressing cells, with 5 mM Mg-ATP in the intracellular buffer. CFTR was activated by FSK and GST, and inhibited by CFTR inhibitor 172, indicating that the recorded currents were due to CFTR. b Curves obtained in the presence of Naltriben and Carvacrol were plotted. Naltriben and Carvacrol activates and inhibites CFTR, respectively. c Bar chart showing the statistical analysis of the currents noted at +80 mV indicating that WT-CFTR function is decreased in the presence of Naltriben
Fig. 10.

Inhibition of ΔF508-CFTR currents by Naltriben. a I–V normalized curves for CFTR currents in cells expressing ΔF508-CFTR (whole-cell configuration with 5 mM Mg-ATP in the intracellular buffer) in the presence or absence of Naltriben. CFTR was activated by FSK and GST and inhibited by CFTR inhibitor 172, indicating that the recorded currents were due to CFTR. The inhibition by Naltriben is more efficient than that of inhibitor 172. b Bar chart showing the statistical analysis of currents (+80 mV) indicating that ΔF508-CFTR function is decreased by Naltriben
Fig. 11.

Activation of G551D-CFTR currents by Naltriben. a I–V normalized curves for CFTR currents in cells expressing G551D-CFTR (with 5 mM Mg-ATP in the intracellular buffer) plus Naltriben. CFTR was activated by FSK and GST, and inhibited by CFTR inhibitor 172, indicating that the recorded currents were due to CFTR. The activation by Naltriben is more efficient than that of FSK plus GST. b Bar chart showing the statistical analysis of currents (+80 mV) indicating that G551D-CFTR function is very significantly increased by Naltriben
Measurements of Ca2+ influx through TRPM7
TRPM7 channel conductance was monitored using a FlexStation 3 scanning fluorometer to monitor the Ca2+-independent (360-nm excitation/510-nm emission) fura-2 quenching by Mn2+ in cells, as previously described [62]. As shown in Fig. 12a, the curves representing Ca2+ influx which were measured in all the cell types (n = 6 for each cell type), presented some differences among the cell types. Bar charts (Fig. 12b) were drawn using the average slope values of the quenching. Quenching rates were not different in WT-CFTR and NT cells. Ca2+ influx through TRPM7 was lower in cells expressing ΔF508-CFTR than in WT-CFTR cells. At the opposite, it was increased in the cells expressing G551D-CFTR. Whereas, Waixenicin A had no significant effect upon the quenching rates, Naltriben significantly increased the Ca2+ influx in cells expressing ΔF508-CFTR when compared to non-treated ΔF508-CFTR cells. The same recordings, using the same conditions as for Hela cells were performed in native HEK293 cells (n = 13). The quenching curves are presented in Fig. 12c (upper panel). The statistical analysis was performed and is presented as bar charts in Fig. 12c (lower panel). In these cells, a significant activation and inhibition of the Ca2+ influx through TRPM7 was obtained by the use of Naltriben (n = 10) and Waixenicin A (n = 6), respectively. This indicates that the absence of modulation observed in the Hela cells expressing WT-CFTR is likely due to the cell types and is not due to technical artifacts.
Fig. 12.
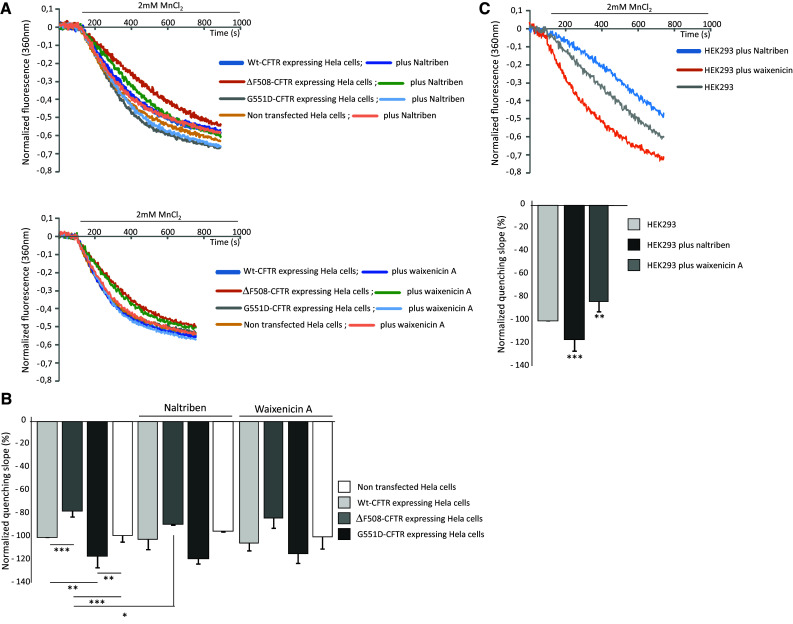
Measurements of Ca2+ influx through TRPM7. a Example of quenching curves showing the Ca2+ influx which were measured in all the cell types. The upper panel shows a set of curves obtained in the absence and in the presence of TRPM7 activator. The lower panel shows the curves obtained without and in the presence of TRPM7 inhibitor. b Bar charts show the average slope values of the quenching, taken at the 300 s time point. A significant difference was observed among the different cell types. Without any modulator of TRPM7, the quenching rate was significantly higher in G551D-CFTR than in WT-CFTR or NT cells. At the contrary, the quenching rate was lower in cells expressing ΔF508-CFTR than in WT-CFTR, G551D-CFTR and NT cells. Whereas, the TRPM7 inhibitor Waixenicin A had no significant effect upon the quenching rates, Naltriben significantly increased the quenching rate in cells expressing ΔF508-CFTR. c Ca2+ influx through TRPM7 was measured in HEK293 cells. The upper panel shows an example of quenching curves obtained without and with modulation of TRPM7. The lower panel shows the corresponding bar charts indicating that Naltriben and Waixenicin A significantly activates and inhibits TRPM7, respectively
Prediction of the protein–protein interaction network of TRPM7
The interaction network of TRPM7 was evaluated with the online Search Tool for the Retrieval of Interacting Genes/Protein to predict both direct and indirect interactions [54]. As shown in Fig. 13, based on bibliographic analysis, putative interacting partners of TRPM7 were found to be annexin A1 (ANXA1, score: 0.925); myosin heavy chain 9 (non-muscle, MYH9, score: 0.910); kininogen 1 (KNG1, score: 0.885); transient receptor potential cation channel 5, subfamily V, member 5 (TRPV5, score: 0.869); transient receptor potential cation channel 6, subfamily VI, member 6 (TRPV6, score: 0.859); myosin heavy chain 10 (non-muscle, MYH10, score: 0.853); transient receptor potential cation channel, subfamily V, member 1, (VR1, score: 0.834); transient receptor potential cation channel, subfamily V, member 1 (TRPV1, score: 0.832); transient receptor potential cation channel, subfamily V, member 4 (TRPV4, score: 0.832); and phospholipase C (beta 1, PLCB1, score: 0.836). Biochemical evidence was only depicted for annexinA1 (ANXA1), myosin heavy chain 9 (MYH9), myosin heavy chain 10 (MYH10) and phospholipase C beta 1 (PLCB1). ANXA1 is a Calcium/phospholipid-binding protein which promotes membrane fusion and is involved in exocytosis. It also regulates phospholipase A2 activity. MYH9 and MYH10 are non-muscle cellular myosins that play a role in cytokinesis, cell shape and secretion. PLCB1 which is phosphoinositide-specific, catalyzes the formation of inositol 1,4,5-trisphosphate and diacylglycerol from phosphatidylinositol 4,5-bisphosphate. This reaction uses calcium as a cofactor and plays an important role in the intracellular transduction of many extracellular signals.
Fig. 13.

Network of predicted interactions for TRPM7 obtained by STRING. Blue connecting lines indicate that the interaction/s are mentioned in PubMed abstracts. Interactions with Annexin A1 (ANXA1), myosin heavy chain 9 non-muscle (MYH9), myosin heavy chain 10 non-muscle (MYH10) and phospholipase C beta 1 (PLCB1) were given due to experimental data. Scores were set above 0.7 for high confidence
Discussion
Magnesium ions, being one of the most abundant intracellular cations present at a cellular molar concentration of 1 × 10−2 [59], are essential for a wide variety of biochemical reactions and physiological functions. Mg2+ is especially important for enzymes that require nucleotides (e.g. kinases, G-proteins and ATPases), as the biologically active form of ATP is the Mg-ATP complex. [Mg2+]i is known to regulate the expression of CFTR. Knowing that a decrease in [Mg2+]i leads to reduced opening and closing rates of the CFTR channel [34], we first estimated the Mg2+ content of cells expressing WT-CFTR, and ΔF508-CFTR and G551D-CFTR mutants. The ΔF508 and G551D mutations were chosen because once mutant proteins reach plasma membranes, they exhibit decreased and altered Cl− channel function, independently of their membrane localisation [14–16]. We observed a decreased [Mg2+]i in cells expressing ΔF508-CFTR or G551D-CFTR compared to that of cells expressing WT-CFTR. The effects of changes in the concentration of free Mg2+ on the activity of CFTR Cl− channels were previously investigated and it was shown that Mg2+ facilitates ATP binding that opens the CFTR channel [36]. Indeed, in an excised inside-out membrane patch experiment, removing Mg2+ from the cytosolic solution reduced, but did not abolish, CFTR activity and addition of Mg2+ restored the Cl− currents. Therefore, altered levels of [Mg2+]i in cells expressing mutant CFTR could explain, at least in part, the observed altered Cl− channel functions related to some mutations of CFTR. This hypothesis is reinforced by the fact that we saw less Mg2+ in cells expressing G551D-CFTR than in those expressing ΔF508-CFTR. This decreased [Mg2+]i in cells expressing ΔF508-CFTR and G551D-CFTR can be correlated to the described specific alterations in the channel function mediated by these two mutants [14–17, 36, 45, 58]. It was also observed that [Mg2+]i is higher in cells expressing WT-CFTR compared to NT cells. This latter point led us to postulate that CFTR itself is likely involved in the regulation of intracellular Mg2+, as it does for Ca2+ regulation [32]. Nevertheless, this hypothesis requires further evidence for its validation.
[Mg2+]i is finely regulated by precise mechanisms that balance Mg2+ uptake, intracellular Mg2+ storage and Mg2+efflux. Whereas extremely slow turn-over of Mg2+ across the cell plasma membrane under quiescent conditions is described, some efforts have been made to identify and characterize the complex network of Mg2+-specific transporters (for review [61]). The main vertebrate Mg2+ cellular transporters identified are Ancient Conserved Domain Protein (Acdp2) [63]; Magnesium Transporter 1 (MagT1) [64]; Mitochondrial RNA splicing 2 protein (Mrs2) [65]; Claudin 16 [66]; SoLute Carrier family (SLC41A1 and A2) [67, 68]; TRPM6 [69]; and TRPM7 [38, 42]. We focused our study on the ubiquitous TRPM7 chanzyme, which is presented as the main [Mg2+]i regulator of [41].
The quantitative mRNA analysis of TRPM7 showed that there was no difference in the presence or absence of CFTR within cells or when mutant CFTR is expressed. Therefore, the gene expression of TRPM7 itself is unlikely involved in the [Mg2+]i differences that were observed. The quantitation of TRPM7 protein levels in the cells was surprising when it was correlated to the measured [Mg2+]i. Indeed, low [Mg2+]i in cells expressing either ΔF508-CFTR or G551D-CFTR corresponded to high levels of TRPM7 protein expression. By contrast, high [Mg2+]i, which was observed in WT-CFTR experiments, correlated well to a low amount of protein. Thus, we hypothesized that the cellular localization, function, and regulation of the TRPM7 channel could be involved in the decreased [Mg2+]i in the cells expressing mutant CFTR.
Immunofluorescence and biotinylation experiments showed a vesicular labelling in the cytoplasm in all three cell types, with membrane expression being mostly seen in WT-CFTR experiments. Membrane localization was lower in cells expressing ΔF508-CFTR and likely absent when G551D-CFTR was expressed. These results are consistent with previously described studies [37, 41, 70, 71]. Furthermore, it was reported that TRPM7 can be translocated within cells in response to external stimulation. For example, TRPM7 accumulates within cells in response to an increased fluid flow and is localized at the plasma membrane in physiological condition, in <100 s [70]. In our experiments, the cellular modifications due to the CFTR mutations could explain the altered membrane localization of TRPM7. Furthermore, the altered membrane localization of TRPM7 in ΔF508-CFTR and mostly in G551D-CFTR cells could fully explain the decreased [Mg2+]i in these cells.
Although the decreased membrane localization of TRPM7 in cells expressing either ΔF508-CFTR or G551D-CFTR is sufficient to explain decreased [Mg2+]i, we studied its channel function. We observed that TRPM7 channel activity was decreased and almost abolished in ΔF508-CFTR and G551D-CFTR experiments, respectively. TRPM7 regulation by increasing Mg2+ was also evaluated according to previous studies [41–44] and we found that it did not respond in cells containing mutant CFTR. As in our cell model, amyotrophic lateral sclerosis and Parkinson’s disease are epidemiologically linked to a severe deficiency in Mg2+ [72]. In these disorders, a TRPM7 variant was found in some patients that produces a protein with a missense mutation (T1482I). Further analysis indicated that a recombinant T1482I-TRPM7 mutant exhibited the same kinase catalytic activity as wildtype TRPM7, with an increased sensitivity to inhibition by intracellular Mg2+ [69]. Therefore, in our study the kinase activity of the alpha-kinase domain fused to the ion channel moiety of TRPM7 (for review, [44]) is unlikely to be involved in altering channel function and regulation. The functional interaction between the TRPM7 channel and kinase activity is poorly understood, although it has been shown that TRPM7 kinase activity is not essential for channel activation [39]. Nevertheless, it is also reported that the kinase activity of TRPM7 plays a role in modulating channel activity and that it is itself affected by Mg2+ availability [41, 42, 44]. Thus, we propose that a vicious circle occurs in cells expressing mutant CFTR in which altered TRPM7 function leads to a decrease in [Mg2+]i, which in turn modifies the regulation of TRPM7 channel activity.
Mg2+ coordinates the γ phosphate of ATP and is required for hydrolysis by most enzymes [73]. Indeed, chelating Mg2+ abolishes ATP hydrolysis [32], which is necessary for CFTR channel function [19, 35]. Therefore, we hypothesized that the altered activity of TRPM7 presented here is linked to modifications in the function of ΔF508-CFTR and G551D-CFTR and we speculated that restoring TRPM7 function could play a positive role in the activity of CFTR. To answer this question, we performed experiments using Naltriben to activate TRPM7 [53, 54]. Whereas Mg-ATP inhibits TRPM7 [38], it has to be used in patch-clamp experiments to measure the function of CFTR [19, 36]. We performed these experiments in the presence of 5 mM Mg-ATP. This experimental condition permitting the electrophysiological recording of CFTR showed that TRPM7 was still active, despite a decreased function. CFTR Cl− channel function was thus recorded in the presence of a TRPM7 activator, according to previously described whole-cell methods [45, 58]. Naltriben led to a decrease in Cl− channel activity for WT-CFTR and ΔF508-CFTR. By contrast, we observed that TRPM7 activation leads to an increased G551D-CFTR channel function. CFTR is unique amongst ion channels as it is dependent not only on PKA-mediated phosphorylation, but also on ATP binding and hydrolysis [74, 75] by the nucleotide-binding domains (NBDs) which require Mg-ATP [35]. Mg-ATP was found to increase channel activity with a saturating effect above 1 mmol/l [76]; however, it was observed that whole-cell CFTR Cl− currents (activated by FSK) were diminished by increasing the cytosolic free Mg2+ level, even when maintaining Mg-ATP in the cytosol above millimolar concentrations [77]. This could explain why activation of TRPM7 leads to decreased CFTR function in cells expressing WT-CFTR and ΔF508-CFTR. In G551D-CFTR experiments, [Mg2+]i is much lower compared to cells expressing either WT-CFTR or ΔF508-CFTR and TRPM7 is poorly present in membranes were it exhibits very weak channel activity. Therefore, it can be postulated that the activation of TRPM7 by Naltriben leads to increased [Mg2+]i that remains below the inhibitory level for CFTR. Therefore, we show here for the first time that Naltriben is a new potentiator of the G551D-CFTR mutant protein. The presented negative effect of Naltriben on the function of ΔF508-CFTR was also reported for other potentiators such as VX-770 (Ivacaftor, [78]). Indeed, whereas VX-770 treatments rescue G551D CFTR function, it inhibits functional rescue of ΔF508 CFTR and reduces the membrane stability of WT-CFTR.
The role of TRPM7 upon CFTR function is unlikely due to [Mg2+]i modulation alone, but may also involve protein–protein interactions and/or the serine/threonine protein kinase domain at the distal C-terminus. Indeed, the C-terminus of TRPM7 contains a TRP box (~25 highly conserved residues) that may interact with phosphatidylinositol 4,5-bisphosphate (PIP2, for review [79]), which activates CFTR in the complete absence of Mg2+ [80]. Knowing that the kinase domain of TRPM7 also shows some similarity to cAMP-dependent PKA which also activates CFTR [18, 19], TRPM7 may act on CFTR in many ways involving a multitude of protein interactions. Amongst the proteins predicted to interact with TRPM7, biochemical evidence was only identified for ANXA1, MYH9, MYH10 and PLCB1. MYH and PLCB1 were shown to be involved in CFTR localization and/or function [81, 82]. Thus, it is clear that further experiments are required to ascertain whether altered TRPM7 function and regulation acts upon mutant CFTR or whether the converse interaction occurs.
Beside protein–protein interactions, Ca2+ is more likely involved in the altered function and regulation of TRPM7 in CF cells, for at least three reasons. First, it was previously described that TRPM7 channels are inhibited by intracellular Ca2+ [37–39, 44, 71, 83]. Second, a larger mobilization of Ca2+ ions in CF bronchial epithelial cells is well described [29–33, 84]. Third, the implication of other TRP channels (TRPV5 and TRPV6) [82] in the increase of Ca2+ influx in CF bronchial epithelial cells were studied and it was shown that this increase is mostly due to an up-regulation of TRPV6 activity. Therefore, we propose that the altered function and regulation of TRPM7 presented here are due to an increase in Ca2+ influx in ΔF508-CFTR cells. This is reinforced by our findings where decreased [Mg2+]i in cells expressing mutant CFTR is not involved in the altered function of TRPM7. In order to assess our hypothesis, we measured Ca2+ influx via TRPM7 in cells expressing WT-CFTR, ΔF508-CFTR and G551D-CFTR. The Ca2+ conductance of TRPM7 has been previously measured using fluorescent cation binding dyes [43], commonly using the ratiometric properties of the Ca2+ binding dye fura-2 [61]. Based on its TRPM7 permeability and high fura-2 binding affinity, we used Mn2+ as the quenching reagent in our assay. Measuring TRPM7-mediated Mn2+ entry, rather than Ca2+, affords several advantages. First, some competing cations entry pathways [e.g., calcium-release activated calcium (CRAC) channels] are less permeable than TRPM7 to Mn2+. Second, CRAC channel currents can be further disconnected by avoiding Ca2+-deficient assay conditions, which are needed for optimal measurement of Ca2+ influx. Third, it allowed us to realize our experiment in the presence of physiological levels of Ca2+ and Mg2+ [43]. Because Waixenicin A (a xenicane diterpenoid from the Hawaiian soft coral S. edmondsoni) [85] inhibits TRPM7-mediated Mn2+ quench in a dose-dependent manner and because its inhibitory effect is strongly dependent on the intracellular Mg2+ concentration [86], it was used, due to the observed variations in [Mg2+]i among our cell types. We observed a decreased and increased Ca2+ influx through TRPM7 in ΔF508-CFTR and G551D-CFTR, respectively. Regarding the cells expressing ΔF508-CFTR, this is in accordance with our results showing a decreased function of TRPM7 in these cells. Regarding the cells expressing G551D-CFTR, we found an increased Ca2+ influx via TRPM7 which was not in accordance with a decreased function of the channel. Therefore, we hypothesize that in this cell type the behaviour of TRPM7 may vary among ions. This point has to be further studied. We also noticed that the drug modulation of TRPM7 by Naltriben and Waixenicin A was difficult to detect in Hela cells. In order to show that this was not due to our experimental conditions, we performed the same experiments in HEK293 cells which have a high level of TRPM7 expression [38]. In native HEK293 cells, a modulation of TRPM7 was observed. This led us to conclude that the TRPM7 modulation was altered in Hela cells expressing a mutated CFTR and that the involvement of TRPM7 in constitutive Ca2+ influx is different among cell types. This cell-dependent contribution in constitutive Ca2+ currents due to TRPM7 was previously suggested [87]. Indeed, it was reported that it can be difficult to detect Ca2+ influx mediated by TRPM7 in some cells because its contribution can be small, relative to the large variety of constitutive currents [43, 88]. Our conclusion is that TRPM7 is likely not involved in the large Ca2+ influx described in cells expressing ΔF508-CFTR and that the described [Ca2+]i overload in these cells is likely responsible for the altered function of TRPM7. Second, we show here for the first time that Ca2+ influx via TRPM7 in cells expressing G551D-CFTR are increased.
We show here, for the first time, that the expression and function of TRPM7 are altered in cells expressing either ΔF508-CFTR or G551D-CFTR (Fig. 14). We also propose that there is a functional link between TRPM7 and CFTR via decreased [Mg2+]i and increased Ca2+ influx in CF cells. Hypomagnesaemia has been reported as a common feature in CF patients [89]. Mean serum magnesium range in CF patients was shown to be 0.46–1.03 mmol/L whereas the normal range is 0.74–1.1 [89]. This could explain the low levels of [Mg2+]i that we observed in cells expressing the mutated CFTR. Patients who receive oral magnesium present better clinical symptoms [90]. If we hypothesize that Mg2+ supplementation induces a decreased function of TRPM7, this could be in opposition with our results showing that its activation by Naltriben has a positive effect on the function of G551D-CFTR. Nevertheless, the mechanism of action of Mg2+ in pulmonology is not fully understood due to a small correlation between serum and intracellular concentrations [90]. For example, [Mg2+]i in erythrocytes, mononuclear cells and granulocytes do not reflect the body’s Mg2+ store because it is primarily concentrated in muscles and bones [91]. Therefore, we propose that the effect of supplementation may be different among the type of mutation of CFTR, which is linked to variations in the localization and function of TRPM7. Our results together with the improvement of the symptoms in CF by Mg2+ supplementation also reinforce the poor correlation between serum and intracellular concentrations of Mg2+.
Fig. 14.

Summary of results. The table presents previously published information regarding CFTR (grey) and the [Mg2+]i and TRPM7 (black) results of this study in order to provide a synthetic view of the correlation between CFTR modulation and our findings
From a pharmacological point of view, we show here that Naltriben modifies the phenotype of CFTR-expressing cells (Fig. 15). Interestingly, the activation of TRPM7 leads to increased G551D-CFTR function; thus, we propose that Naltriben is a new potentiator for this particular mutation.
Fig. 15.

Diagram presenting the results and the effect of Naltriben. The three cells represent WT-CFTR, ΔF508-CFTR and G551D-CFTR experiments with their status related to [Mg2+]i, CFTR and TRPM7. The effect of Naltriben is shown by arrows which indicate that the drug modifies TRPM7 activity and subsequently CFTR function
Acknowledgments
We thank the association Gaëtan Salaün and the French foundation “Vaincre la Mucoviscidose” for funding the present work. We also thank Dr Mohamed Keir (Nanion Technology, Germany) for his help. We acknowledge the grant that funds the ability to provide Waixenicin A: U.S. National Institute of Health Grant P20GM 103466.
Contributor Information
C. Férec, Phone: 33.(0)2.98.22.36.79, Email: claude.ferec@univ-brest.fr
P. Trouvé, Phone: 33.(0)2.98.22.36.79, Email: pascal.trouve@univ-brest.fr
References
- 1.Kerem BS, Rommens JM, Buchanan JA, Markiewicz D, Cox T, Chakravarti A, Buchwald M, Tsui LC. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- 2.Riordan JR, Rommens JM, Kerem BS, Alon N, Rozmahel R, Grzelczak Z, Zielinski J, Lok S, Plavsik N, Chou JL, Drumm ML, Ianuzzi MC, Collins FS, Tsui LC. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 3.Zielinski J, Rozmahel R, Bozon D, Kerem BS, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics. 1991;10:214–228. doi: 10.1016/0888-7543(91)90503-7. [DOI] [PubMed] [Google Scholar]
- 4.Tabcharani JA, Linsdell P, Hanrahan JW. Halide permeation in wild-type and mutant CFTR chloride channels. J Gen Physiol. 1997;110:341–354. doi: 10.1085/jgp.110.4.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linsdell P, Tabcharani JA, Rommens JM, Hou YX, Chang XB, Tsui LC, Riordan JR, Hanrahan JW. Permeability of wild-type and mutant CFTR chloride channels to polyatomic anions. J Gen Physiol. 1997;110:355–364. doi: 10.1085/jgp.110.4.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hasegawa H, Skach W, Baker O, Calayag MC, Lingappa V, Verkman AS. A multifunctional aqueous channel formed by CFTR. Science. 1992;258:1477–1479. doi: 10.1126/science.1279809. [DOI] [PubMed] [Google Scholar]
- 7.Ballard ST, Spadafora D. Fluid secretion by submucosal glands of the tracheobronchial airways. Respir Physiol Neurobiol. 2007;159:271–277. doi: 10.1016/j.resp.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–726. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- 9.Rosenstein BJ, Zeitlin PL. Cystic fibrosis. Lancet. 1998;351:277–282. doi: 10.1016/S0140-6736(97)09174-5. [DOI] [PubMed] [Google Scholar]
- 10.Ratjen F, Döring G. Cystic fibrosis. Lancet. 2003;361:681–689. doi: 10.1016/S0140-6736(03)12567-6. [DOI] [PubMed] [Google Scholar]
- 11.Amaral MD, Farinha CM. Rescuing mutant CFTR: a multi-task approach to a better outcome in treating cystic fibrosis. Curr Pharm Des. 2013;19:3497–3508. doi: 10.2174/13816128113199990318. [DOI] [PubMed] [Google Scholar]
- 12.Farinha CM, Matos P, Amaral MD. Control of cystic fibrosis transmembrane conductance regulator membrane trafficking: not just from the endoplasmic reticulum to the Golgi. FEBS J. 2013;280:4396–4406. doi: 10.1111/febs.12392. [DOI] [PubMed] [Google Scholar]
- 13.Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, Young JC, Lukacs GL. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science. 2010;329:805–810. doi: 10.1126/science.1191542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melin P, Norez C, Callebaut I, Becq F. The glycine residues G551 and G1349 within the ATP-binding cassette signature motifs play critical roles in the activation and inhibition of cystic fibrosis transmembrane conductance regulator channels by phloxine B. J Membr Biol. 2006;208(3):203–212. doi: 10.1007/s00232-005-7001-0. [DOI] [PubMed] [Google Scholar]
- 15.Bompadre SG, Sohma Y, Li M, Hwang TC. G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. J Gen Physiol. 2007;129:285–298. doi: 10.1085/jgp.200609667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bompadre SG, Li M, Hwang TC. Mechanism of G551D-CFTR potentiation by a high affinity ATP analog. J Biol Chem. 2008;283:5364–5369. doi: 10.1074/jbc.M709417200. [DOI] [PubMed] [Google Scholar]
- 17.Pasyk S, Li C, Ramjeesingh M, Bear CE. Direct interaction of a small-molecule modulator with G551D-CFTR, a cystic fibrosis-causing mutation associated with severe disease. Biochem J. 2009;418:185–190. doi: 10.1042/BJ20081424. [DOI] [PubMed] [Google Scholar]
- 18.Gadsby DC, Nairn AC. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev. 1999;79:S77–S107. doi: 10.1152/physrev.1999.79.1.S77. [DOI] [PubMed] [Google Scholar]
- 19.Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999;79:S23–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- 20.Cormet-Boyaka E, Di A, Chang SY, Naren AP, Tousson A, Nelson DJ, Kirk KL. CFTR chloride channels are regulated by a SNAP-23/syntaxin 1A complex. Proc Natl Acad Sci USA. 2002;99:12477–12482. doi: 10.1073/pnas.192203899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naren AP, Quick MW, Collawn JF, Nelson DJ, Kirk KL. Syntaxin 1A inhibits CFTR chloride channels by means of domain-specific protein–protein interactions. Proc Natl Acad Sci USA. 1998;95:10972–10977. doi: 10.1073/pnas.95.18.10972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raghuram V, Mak DD, Foskett JK. Regulation of cystic fibrosis transmembrane conductance regulator single-channel gating by bivalent PDZ-domain–mediated interaction. Proc Natl Acad Sci USA. 2001;98:1300–1305. doi: 10.1073/pnas.98.3.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Short DB, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, Stutts MJ, Milgram SL. An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton. J Biol Chem. 1998;273:19797–19801. doi: 10.1074/jbc.273.31.19797. [DOI] [PubMed] [Google Scholar]
- 24.Wang S, Yue H, Derin RB, Guggino WB, Li M. Accessory protein facilitated CFTR–CFTR interaction, a molecular mechanism to potentiate the chloride channel activity. Cell. 2000;103:169–179. doi: 10.1016/S0092-8674(00)00096-9. [DOI] [PubMed] [Google Scholar]
- 25.Cheng J, Moyer BD, Milewski M, Loffing J, Ikeda M, Mickle JE, Cutting GR, Li M, Stanton BA, Guggino WB. A Golgi-associated PDZ domain protein modulates cystic fibrosis transmembrane regulator plasma membrane expression. J Biol Chem. 2002;277:3520–3529. doi: 10.1074/jbc.M110177200. [DOI] [PubMed] [Google Scholar]
- 26.Trouvé P, Le Drévo MA, Kerbiriou M, Friocourt G, Fichou Y, Gillet D, Férec C. Annexin V is directly involved in cystic fibrosis transmembrane conductance regulator’s function. Biochim Biophys Acta. 2007;1772(10):1121–1133. doi: 10.1016/j.bbadis.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 27.Le Drévo MA, Benz N, Kerbiriou M, Gioux M, Pennec JP, Trouvé P, Férec C. Annexin A5 increases the cell surface expression and the chloride channel function of the ΔF508-cystic fibrosis transmembrane regulator. Biochim Biophys Acta. 2008;1782:605–614. doi: 10.1016/j.bbadis.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 28.Benz N, Le Hir S, Norez C, Kerbiriou M, Calvez M-L, Becq F, Trouvé P, Férec C. Improvement of chloride transport defect by gonadotropin-releasing hormone (GnRH) in cystic fibrosis epithelial cells. PLoS One. 2014;9(2):e88964. doi: 10.1371/journal.pone.0088964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antigny F, Norez C, Becq F, Vandebrouck C. CFTR and Ca2+ signaling in cystic fibrosis. Front Pharmacol. 2011;2:67. doi: 10.3389/fphar.2011.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antigny F, Norez C, Cantereau A, Becq F, Vandebrouck C. Abnormal spatial diffusion of Ca2+ in F508del-CFTR airway epithelial cells. Respir Res. 2008;9:70. doi: 10.1186/1465-9921-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Antigny F, Girardin N, Raveau D, Frieden M, Becq F, Vandebrouck C. Dysfunction of mitochondria Ca2+ uptake in cystic fibrosis airway epithelial cells. Mitochondrion. 2009;9(4):232–241. doi: 10.1016/j.mito.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 32.Antigny F, Norez C, Becq F, Vandebrouck C. Calcium homeostasis is abnormal in cystic fibrosis airway epithelial cells but is normalized after rescue of F508del-CFTR. Cell Calcium. 2008;43(2):175–183. doi: 10.1016/j.ceca.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 33.Norez C, Antigny F, Becq F, Vandebrouck C. Maintaining low Ca2+ level in the endoplasmic reticulum restores abnormal endogenous F508del-CFTR trafficking in airway epithelial cells. Traffic. 2006;7(5):562–573. doi: 10.1111/j.1600-0854.2006.00409.x. [DOI] [PubMed] [Google Scholar]
- 34.Bargon J, Trapnell BC, Chu CS, Rosenthal ER, Yoshimura K, Guggino WB, Dalemans W, Pavirani A, Lecocq JP, Crystal RG. Down-regulation of cystic fibrosis transmembrane conductance regulator gene expression by agents that modulate intracellular divalent cations. Mol Cell Biol. 1992;12(4):1872–1878. doi: 10.1128/MCB.12.4.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li C, Ramjeesingh M, Wang W, Garami E, Hewryk M, Lee D, Rommens JM, Galley K, Bear CE. ATPase activity of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1996;271(45):28463–28468. doi: 10.1074/jbc.271.45.28463. [DOI] [PubMed] [Google Scholar]
- 36.Ikuma M, Welsh MJ. Regulation of CFTR Cl2 channel gating by ATP binding and hydrolysis. PNAS. 2000;97(15):8675–8680. doi: 10.1073/pnas.140220597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vissera D, Middelbeekb J, van Leeuwenb FN, Jalink K. Function and regulation of the channel-kinase TRPM7 in health and disease. Eur J Cell Biol. 2014;93(10–12):455–465. doi: 10.1016/j.ejcb.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Nadler MJ, Hermosura M, Inabe K, Perraud AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg AM, Fleig A. LTRPC7 is a Mg. ATP-regulated divalent cation channel required for cell viability. Nature. 2001;411(6837):590–595. doi: 10.1038/35079092. [DOI] [PubMed] [Google Scholar]
- 39.Penner R, Fleig A. The Mg2+ and Mg2+-nucleotide-regulated channel-kinase TRPM7. Handb Exp Pharmacol. 2007;179:313–328. doi: 10.1007/978-3-540-34891-7_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Middelbeek J, Clark K, Venselaar H, Huynen MA, van Leeuwen FN. The alpha-kinase family: an exceptional branch on the protein kinase tree. Cell Mol Life Sci. 2010;67(6):875–890. doi: 10.1007/s00018-009-0215-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryazanova LV, Rondon LJ, Zierler S, Hu Z, Galli J, Yamaguchi TP, Mazur A, Fleig A, Ryazanov AG. TRPM7 is essential for Mg2+ homeostasis in mammals. Nat Commun. 2010;1:109. doi: 10.1038/ncomms1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmitz C, Perraud AL, Johnson CO, Inabe K, Smith MK, Penner R, Kurosaki T, Fleig A, Scharenberg AM. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell. 2003;114(2):191–200. doi: 10.1016/S0092-8674(03)00556-7. [DOI] [PubMed] [Google Scholar]
- 43.Monteilh-Zoller MK, Hermosura MC, Nadler MJ, Scharenberg AM, Penner R, Fleig A. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J Gen Physiol. 2003;121(1):49–60. doi: 10.1085/jgp.20028740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paravicinia TM, Chubanovb V, Gudermannb T. TRPM7: a unique channel involved in magnesium homeostasis. Int J Biochem Cell Biol. 2012;44(8):1381–1384. doi: 10.1016/j.biocel.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 45.Teng L, Kerbiriou M, Taiya M, Le Hir S, Benz N, Trouvé P, Férec C. Proteomic Identification of Calumenin as a G551D: CFTR associated protein. PLoS One. 2012;7(6):e40173. doi: 10.1371/journal.pone.0040173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baldoli E, Castiglioni S, Maier JA. Regulation and function of TRPM7 in human endothelial cells: TRPM7 as a potential novel regulator of endothelial function. PLoS One. 2013;8(3):e59891. doi: 10.1371/journal.pone.0059891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lowry OH, Rosebrough HJ, Farr A, Randall RJ. Protein measurement with the Pholin phenol reagent. J Biol Chem. 1951;183:263–275. [PubMed] [Google Scholar]
- 48.Yu M, Huang C, Huang Y, Wu X, Li X, Li J. Inhibition of TRPM7 channels prevents proliferation and differentiation of human lung fibroblasts. Inflamm Res. 2013;62(11):961–970. doi: 10.1007/s00011-013-0653-9. [DOI] [PubMed] [Google Scholar]
- 49.Luo J, Zhu Y, Zhu MX, Hu H. Cell-based calcium assay for medium to high throughput screening of TRP channel functions using FlexStation 3. J Vis Exp. 2011;54:e3149. doi: 10.3791/3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farre C, Stoelzle S, Haarmann C, George M, Brüggemann A, Fertig N. Automated ion channel screening: patch clamping made easy. Expert Opin Ther Targets. 2007;11(4):557–565. doi: 10.1517/14728222.11.4.557. [DOI] [PubMed] [Google Scholar]
- 51.Yang YM, Jung HH, Lee SJ, Choi HJ, Kim MS, Shin DM. TRPM7 is essential for RANKL-induced osteoclastogenesis. Korean J Physiol Pharmacol. 2013;17:65–71. doi: 10.4196/kjpp.2013.17.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chokshi R, Matsushita M, Kozak JA. Patch-clamp electrophysiology. Detailed examination of Mg2+ and pH sensitivity of human TRPM7 channels. Am J Physiol Cell Physiol. 2012;302(7):C1004–C1011. doi: 10.1152/ajpcell.00422.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chubanov V, Schäfer S, Ferioli S, Gudermann T. Natural and synthetic modulators of the TRPM7 channel. Cells. 2014;3:1089–1101. doi: 10.3390/cells3041089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hofmann T, Schäfer S, Linseisen M, Sytik L, Gudermann T, Chubanov V. Activation of TRPM7 channels by small molecules under physiological conditions. Pflug Arch: Eur J Physiol. 2014;466:2177–2189. doi: 10.1007/s00424-014-1488-0. [DOI] [PubMed] [Google Scholar]
- 55.Parnasa M, Petersa M, Dadona D, Leva S, Vertkinb I, Slutskyb I, Minkea B. Carvacrol is a novel inhibitor of Drosophila TRPL and mammalian TRPM7 channels. Cell Calcium. 2009;45:300–309. doi: 10.1016/j.ceca.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen W, Xu B, Xiao A, Liu L, Fang X, Liu R, Turlova E, Barszczyk A, Zhong X, Sun CLF, Britto LRG, Feng ZP, Sun HS. TRPM7 inhibitor carvacrol protects brain from neonatal hypoxic-ischemic injury. Molecular Brain. 2015;8:11. doi: 10.1186/s13041-015-0102-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szklarcyk D, Franchescini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, Von Mering C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucl Acids Res. 2011;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trouvé P, Kerbiriou M, Teng L, Benz N, Taiya M, Le Hir S, Férec C. G551D-CFTR needs more bound actin than wild-type CFTR to maintain its presence in plasma membranes. Cell Biol Int. 2015 doi: 10.1002/cbin.10456. [DOI] [PubMed] [Google Scholar]
- 59.Trapani V, Schweigel-Röntgen M, Cittadini A, Wolf FI. Chapter twenty-two: intracellular magnesium detection by fluorescent indicators. Methods Enzymol. 2012;505:421–444. doi: 10.1016/B978-0-12-388448-0.00030-9. [DOI] [PubMed] [Google Scholar]
- 60.Kim SJ, Kang HS, Jeong CW, Park SY, Kim IS, Kim NS, Kim SZ, Kwak YG, Kim JS, Quamme GA. Immunosuppressants inhibit hormone-stimulated Mg2+ uptake in mouse distal convoluted tubule cells. Biochem Biophys Res Commun. 2006;341(3):742–748. doi: 10.1016/j.bbrc.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 61.Schmitz C, Deason F, Perraud AL. Molecular components of vertebrate Mg2+-homeostasis regulation. Magnes Res. 2007;20(1):6–18. [PubMed] [Google Scholar]
- 62.Grynkiewicz G, Poenie M, Tsien Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;360:3440–3450. [PubMed] [Google Scholar]
- 63.Goytain A, Quamme GA. Functional characterization of ACDP2 (ancient conserved domain protein), a divalent metal transporter. Physiol Genomics. 2005;22:382–389. doi: 10.1152/physiolgenomics.00058.2005. [DOI] [PubMed] [Google Scholar]
- 64.Goytain A, Quamme GA. Identification and characterization of a novel mammalian Mg2+ transporter with channel-like properties. BMC Genom. 2005;6:48. doi: 10.1186/1471-2164-6-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kolisek M, Zsurka G, Samaj J, Weghuber J, Schweyen RJ, Schweigel M. Mrs2p is an essential component of the major electrophoretic Mg2+ influx system in mitochondria. EMBO J. 2003;22:1235–1244. doi: 10.1093/emboj/cdg122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, Casari G, Bettinelli A, Colussi G, Rodriguez-Soriano J, McCredie D, Milford D, Sanjad S, Lifton RP. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science. 1999;285:103–106. doi: 10.1126/science.285.5424.103. [DOI] [PubMed] [Google Scholar]
- 67.Wabakken T, Rian E, Kveine M, Aasheim HC. The human solute carrier SLC41A1 belongs to a novel eukaryotic subfamily with homology to prokaryotic MgtE Mg2+ transporters. Biochem Biophys Res Commun. 2003;306:718–724. doi: 10.1016/S0006-291X(03)01030-1. [DOI] [PubMed] [Google Scholar]
- 68.Sahni J, Nelson B, Scharenberg AM. SLC41A2 encodes a plasma-membrane Mg2+ transporter. Biochem J. 2007;401:505–513. doi: 10.1042/BJ20060673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Voets T, Nilius B, Hoefs S, van der Kemp AW, Droogmans G, Bindels RJ, Hoenderop JG. TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem. 2004;279:19–25. doi: 10.1074/jbc.M311201200. [DOI] [PubMed] [Google Scholar]
- 70.Oancea E, Wolfe JT, Clapham DE. Functional TRPM7 channels accumulate at the plasma membrane in response to fluid flow. Circ Res. 2006;98(2):245–253. doi: 10.1161/01.RES.0000200179.29375.cc. [DOI] [PubMed] [Google Scholar]
- 71.Simon F, Varela D, Cabello-Verrugio C. Oxidative stress-modulated TRPM ion channels in cell dysfunction and pathological conditions in humans. Cell Signal. 2013;25:1614–1624. doi: 10.1016/j.cellsig.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 72.Hermosura MC, Nayakanti H, Dorovkov MV, Calderon FR, Ryazanov AG, Haymer DS, Garruto RM. TRPM7 variant shows altered sensitivity to magnesium that may contribute to the pathogenesis of two Guamanian neurodegenerative disorders. Proc Natl Acad Sci USA. 2005;102(32):11510–11515. doi: 10.1073/pnas.0505149102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Rourke B. Ion channels as sensors of cellular energy. Mechanisms for modulation by magnesium and nucleotides. Biochem Pharmacol. 1993;46(7):1103–1112. doi: 10.1016/0006-2952(93)90456-7. [DOI] [PubMed] [Google Scholar]
- 74.Hennager DJ, Ikuma M, Hoshi T, Welsh MJ. A conditional probability analysis of cystic fibrosis transmembrane conductance regulator gating indicates that ATP has multiple effects during the gating cycle. Proc Natl Acad Sci USA. 2001;98:3594–3599. doi: 10.1073/pnas.051633298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hwang TC, Sheppard DN. Molecular pharmacology of the CFTR Cl− channel. Trend Pharmacol Sci. 1999;20:448–453. doi: 10.1016/S0165-6147(99)01386-3. [DOI] [PubMed] [Google Scholar]
- 76.Zeltwanger S, Wang F, Wang G-T, Gillis KD, Hwang TC. Gating of cystic fibrosis transmembrane conductance regulator chloride channels by adenosine triphosphate hydrolysis: quantitative analysis of a cyclic gating scheme. J Gen Physiol. 1999;113:541–554. doi: 10.1085/jgp.113.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhou SS, Takai A, Okada Y. Regulation of cardiac CFTR Cl− channel activity by a Mg2+-dependent protein phosphatase. Pflugers Arch: Eur J Physiol. 2002;444:327–334. doi: 10.1007/s00424-002-0822-0. [DOI] [PubMed] [Google Scholar]
- 78.Cholon DM, Quinney NL, Fulcher ML, Esther CR, Jr, Das J, Dokholyan NV, Randell SH, Boucher RC, Gentzsch M. Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci Transl Med. 2014;6(246):246ra96. doi: 10.1126/scitranslmed.3008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Park HS, Hong C, Kim BJ, So I. The pathophysiologic roles of TRPM7 channel. Korean J Physiol Pharmacol. 2014;18(1):15–23. doi: 10.4196/kjpp.2014.18.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Himmel B, Nagel G. Protein kinase-independent activation of CFTR by phosphatidylinositol phosphates. EMBO Rep. 2004;5(1):85–90. doi: 10.1038/sj.embor.7400034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kravtsov DV, Caputo C, Collaco A, Hoekstra N, Egan ME, Mooseker MS, Ameen NA. Myosin Ia is required for CFTR brush border membrane trafficking and ion transport in the mouse small intestine. Traffic. 2012;13(8):1072–1082. doi: 10.1111/j.1600-0854.2012.01368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vachel L, Norez C, Jayle C, Becq F, Vandebrouck C. The low PLC-δ1 expression in cystic fibrosis bronchial epithelial cells induces upregulation of TRPV6 channel activity. Cell Calcium. 2015;57(1):38–48. doi: 10.1016/j.ceca.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 83.Sun Y, Selvaraj S, Varma A, Derry S, Sahmoun AE, Singh BB. Increase in serum Ca2+/Mg2+ ratio promotes proliferation of prostate cancer cells by activating TRPM7 channels. J Biol Chem. 2013;288:255–263. doi: 10.1074/jbc.M112.393918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ribeiro CM, Paradiso AM, Carew MA, Shears SB, Boucher RC. Cystic fibrosis airway epithelial Ca2+ signaling: the mechanism for the larger agonist-mediated Ca2+ signals in human cystic fibrosis airway epithelia. J Biol Chem. 2005;280:10202–10209. doi: 10.1074/jbc.M410617200. [DOI] [PubMed] [Google Scholar]
- 85.Zierler S, Yao G, Zhang Z, Kuo WC, Pörzgen P, Penner R, Horgen FD, Fleig A. Waixenicin A inhibits cell proliferation through magnesiumdependent block of transient receptor potential melastatin 7 (TRPM7) channels. J Biol Chem. 2011;286(45):39328–39335. doi: 10.1074/jbc.M111.264341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Visser D, Langeslag M, Kedziora KM, Klarenbeek J, Kamermans A, Horgen FD, Fleig A, van Leeuwen FN, Jalink K. TRPM7 triggers Ca2+ sparks and invadosome formation in neuroblastoma cells. Cell Calcium. 2013;54:404–415. doi: 10.1016/j.ceca.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Oh HG, Chun YS, Park CS, Kim TW, Park MK, Chung S. Regulation of basal autophagy by transient receptor potential melastatin 7 (TRPM7) channel. Biochem Biophys Res Commun. 2015;463(1–2):7–12. doi: 10.1016/j.bbrc.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 88.Wei WL, Sun HS, Olah ME, Sun X, Czerwinska E, Czerwinski W, et al. TRPM7 channels in hippocampal neurons detect levels of extracellular divalent cations. Proc Natl Acad Sci USA. 2007;104:16323–16328. doi: 10.1073/pnas.0701149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gupta A, Eastham KM, Wrightson N, Spencer DA. Hypomagnesaemia in cystic fibrosis patients referred for lung transplant assessment. J Cyst Fibros. 2007;6:360–362. doi: 10.1016/j.jcf.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 90.Gontijo-Amaral C, Guimaraes EV, Camargos P. Oral magnesium supplementation in children with cystic fibrosis improves clinical and functional variables: a double-blind, randomized, placebo-controlled crossover trial. Am J Clin Nutr. 2012;96:50–56. doi: 10.3945/ajcn.112.034207. [DOI] [PubMed] [Google Scholar]
- 91.Nel S, Mervaala E, Karppanen H, Khawaja JÁ, Lewenstam A. Magnesium: an update on physiological, clinical and analytical aspects. Clin Chim Acta. 2000;294:1–26. doi: 10.1016/S0009-8981(99)00258-2. [DOI] [PubMed] [Google Scholar]