

Abstract
The exportin CRM1 binds nuclear export signals (NESs), and mediates active transport of NES-bearing proteins from the nucleus to the cytoplasm. Structural and biochemical analyses have uncovered the molecular mechanisms underlying CRM1/NES interaction. CRM1 binds NESs through a hydrophobic cleft, whose open or closed conformation facilitates NES binding and release. Several cofactors allosterically modulate the conformation of the NES-binding cleft through intramolecular interactions involving an acidic loop and a C-terminal helix in CRM1. This current model of CRM1-mediated nuclear export has not yet been evaluated in a cellular setting. Here, we describe SRV100, a cellular reporter to interrogate CRM1 nuclear export activity. Using this novel tool, we provide evidence further validating the model of NES binding and release by CRM1. Furthermore, using both SRV100-based cellular assays and in vitro biochemical analyses, we investigate the functional consequences of a recurrent cancer-related mutation, which targets a residue near CRM1 NES-binding cleft. Our data indicate that this mutation does not necessarily abrogate the nuclear export activity of CRM1, but may increase its affinity for NES sequences bearing a more negatively charged C-terminal end.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-016-2292-0) contains supplementary material, which is available to authorized users.
Keywords: NES, XPO1, Recurrent mutation, Chronic lymphocytic leukemia, Cellular assay
Introduction
The localization and function of many cellular proteins is regulated by active transport between the nucleus and the cytoplasm [1]. This transport is carried out by receptors belonging to the Karyopherin family that recognize and bind specific amino acid sequences (transport signals) in their cargo proteins. Nuclear import receptors, also known as importins, bind nuclear localization signals (NLSs) and mediate cargo entry into the nucleus, whereas nuclear export receptors, or exportins, recognize nuclear export signals (NESs) and mediate cargo exit to the cytoplasm (reviewed in [2]). The best characterized exportin is CRM1, which mediates the nuclear export of cargos bearing a type of NES termed leucine-rich NES (LR-NES). The directionality of nucleocytoplasmic transport is crucially regulated by the small GTPase Ran. A step concentration gradient of GTP- or GDP-bound Ran across the nuclear envelope controls receptor/cargo binding and dissociation. Thus, high levels of RanGTP inside the nucleus promote dissociation of importin/NLS complexes, facilitating release of the imported cargo. In contrast, as described below in more detail, RanGTP and cargo NESs bind CRM1 in a cooperative manner, forming a stable trimeric complex in the nucleus that is exported through the nuclear pore complex (NPC). Upon reaching the cytoplasmic side of the NPC, several factors, including Ran-binding proteins RanBP1 and RanBP2 and the RanGTPase activating protein RanGAP1, facilitate dissociation of the RanGTP/NES/CRM1 export complex to release the cargo.
The results of relatively recent structural analyses have led to a more detailed understanding of the molecular mechanisms that underlie CRM1 function as a nuclear export receptor [3–6]; reviewed in [7–9]. Human CRM1 is a 1071 amino acid long, ring-shaped protein, composed of 21 HEAT repeats (H1–H21). Each HEAT repeat is formed by two α-helices, termed A and B, connected by loops of different length. Three regions of CRM1, schematically illustrated in Fig. 1a, have been shown to play critical roles in the process of cargo binding and release. A hydrophobic cleft in the outer surface of CRM1 formed by HEAT repeats H11 and H12 serves as a binding site for LR-NESs [3, 4]. LR-NESs are short amino acid segments with a series of characteristically spaced hydrophobic residues (Φ0–Φ4) that fit a loose consensus pattern. NES peptides have been shown to adopt a helix-loop conformation that permits the docking of their hydrophobic residues in the pockets of CRM1 NES-binding cleft (Fig. 1b). The cycle of NES binding and release is mechanistically related to structural rearrangements of CRM1, which crucially involve the loop that connects H9A and H9B helices (H9 loop) and the carboxy-terminal helix H21B (C-helix). In cargo-free CRM1, the H9 loop and the C-helix contact the inner surface of H11 and H12 repeats, acting as allosteric auto inhibitors that stabilize a closed conformation of the NES-binding cleft [10]. Upon cooperative binding of RanGTP and NES, the H9 loop and the C-helix undergo major conformational changes that facilitate cleft opening. Rearrangements of the H9 loop and the C-helix are also important for dissociation of the export complex in the cytoplasm, which is greatly accelerated by Ran-binding proteins through an allosteric mechanism [11, 12].
Fig. 1.
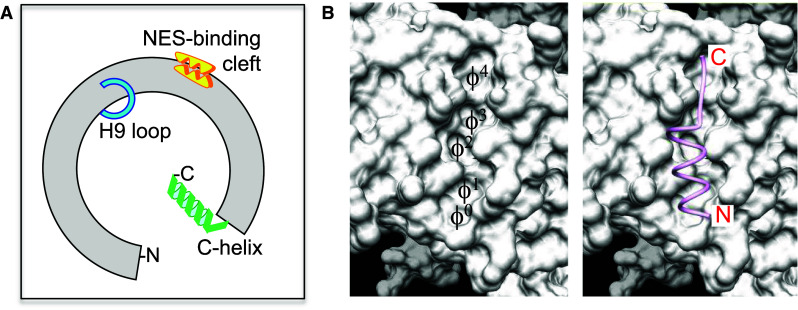
CRM1 domains involved in NES binding and release. a Schematic representation of CRM1 protein showing the position of the NES-binding cleft, the loop connecting H9A and H9B helices (H9 loop) and the carboxy-terminal helix (C-helix) domains. b Detailed view of the NES-binding cleft on the molecular surface of CRM1 (Structure 3GJX, [4]). The image was produced using the UCSF Chimera package [53]. The pockets that serve as docking sites for NES hydrophobic residues Φ0–Φ4 are indicated on the left panel. The right panel depicts a classical leucine-rich NES peptide (ribbon representation, pink) with helix-loop conformation bound to the cleft
CRM1 function appears to be frequently altered in human tumors. Overexpression of CRM1 has been reported in pancreatic, esophageal, ovarian and gastric tumors, as well as in gliomas and osteosarcomas [13–18], and is usually associated with poorer patient prognosis. CRM1 inhibition is emerging as a potential therapeutic strategy in cancer [19]. The rationale behind targeting CRM1 is to promote the nuclear accumulation of CRM1 cargoes, such as p53, APC, or BRCA1 that may carry out a tumor suppressive function in the nucleus [20]. A novel class of CRM1 inhibitors, such as KPT330 (Selinexor), which show a more favorable toxicity profile than the classical CRM1 inhibitor leptomycin B (LMB) [21, 22], have shown promising results in preclinical studies [23–28] and are currently undergoing clinical trials in patients with different tumor types.
The development of CRM1-targeted therapeutic drugs has been greatly facilitated by the structural analyses on CRM1-NES interaction described above [29]. The results of these analyses have been supported by in vitro assays using structure-guided CRM1 mutants. Thus, the affinity of CRM1 for the NESs of well-established cargos, such as snurportin (SPN1), the cAMP-dependent protein kinase inhibitor (PKI), and HIV Rev protein, is drastically reduced by site-directed mutagenesis of residues in the NES-binding cleft [3, 30]. Conversely, mutation of specific H9 loop residues and deletions in the C-helix increase the affinity of CRM1 for an NES-cargo in the absence of RanGTP [5, 12], and H9 loop mutations additionally reduce the rate of NES release [11].
Of note, the effect of these structure-guided experimental mutants of CRM1 has not yet been tested in a cellular setting. Evaluating CRM1 export function in an intact cell is challenging because of the many factors that modulate nucleocytoplasmic transport [31]. Nevertheless, these tests are crucial to further validate our current understanding of CRM1-mediated nuclear export.
Importantly, in 2011 somatic mutations in the XPO1 gene, encoding CRM1, were identified in patients with chronic lymphocytic leukemia (CLL) [32]. Numerous studies have subsequently confirmed that approximately 5 % of CLL patients bear XPO1 mutations (Online Resource 1). When interpreting the relevance of this finding, it must be taken into account that, unlike other types of leukemia, CLL is characterized by a high genetic heterogeneity, with low mutation recurrence and that several genes are recurrently found to be mutated at low frequency in this disease [33]. In this regard, a saturation analysis of cancer genes across different tumor types identified XPO1 as one of seven significantly mutated genes in CLL [34]. Of note, low-frequency mutations, such as those in XPO1, represent independent prognostic factors for CLL patients [35]. Furthermore, XPO1 mutations have been recently shown to be clonal before therapy, indicative of an early pathogenic role in CLL [36]. Thus, there is significant evidence supporting the view that the XPO1 mutation constitutes a “driver” alteration with a causative role in CLL. Strikingly, nearly 90 % of XPO1 mutations change CRM1 residue E571 in the H12A helix, near the NES-binding cleft, usually into a lysine (E571K). However, the molecular mechanism that underlies the pathogenic effect of the recurrent E571K amino acid change remains unexplored.
In this work, we describe a novel reporter, termed SRV100, to interrogate the nuclear export function of CRM1 in a cellular setting. We first use this reporter to evaluate how several experimental mutations previously characterized in vitro affect CRM1 export function in cells. Next, we investigate the functional consequences of the cancer-related E571K mutation using both SRV100-based cellular assays and in vitro biochemical assays. Our data show that the E571K mutation induces a subtle increase in CRM1 binding affinity for NESs with a more negatively charged C-terminal end.
Materials and methods
Molecular cloning and site-directed mutagenesis
Several cloning steps were performed to create the plasmid encoding the SRV100 reporter. First, the DNA sequence encoding GFP in pEGFP-N1 plasmid was replaced by a DNA sequence encoding a 3 × Flag epitope using NotI and AgeI restriction sites. Next, a DNA fragment encoding the NLS of SV40 large T antigen (SV40 NLS) was cloned upstream of 3 × Flag using BamHI and HindIII sites. Subsequently, a PCR-generated DNA fragment encoding survivin amino acid segment 1–100 (wild type or NES mutant) was cloned upstream of the SV40 NLS using BglII and HindIII sites. Finally, a DNA sequence encoding a second SV40 NLS was cloned downstream of the 3 × Flag epitope using the NotI restriction site. The SRV100 charge-modifying variants EDE and KKK were generated by PCR, using reverse primers with the desired mutations to amplify the DNA sequence encoding survivin 1–100. PCR products were purified and cloned as BglII/HindIII fragments.
The plasmid encoding YFP-CRM1 has been previously described [37]. Mutations were introduced into YFP-CRM1 using the Quick-Change Lightning Site-Directed Mutagenesis Kit (Stratagene), according to the manufacturer’s directions.
The cDNA sequence encoding wild type and E571K variants (bearing the H9 loop and C-terminal “activating” mutations [12]) was subcloned into a modified pTG-A20 vector [4] using BamHI and NotI sites, for expression of (His)10-ZZ tagged constructs in E. coli. TFP-NES constructs for fluorescence anisotropy binding assays were generated as in [38].
All the constructs generated were subjected to DNA sequencing (Stabvida), and the absence of any unwanted mutation was confirmed.
Cell culture, transfection and drug treatment
Human embryonic kidney 293T (HEK293T) cells, HeLa cells and U2OS cells were grown in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10 % fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin (all from Invitrogen). Twenty four hours before transfection, cells were seeded onto glass coverslips placed into 12-well tissue culture plates. Transfections were carried out using X-tremeGENE 9 transfection reagent (Roche Diagnostics) following manufacturer’s protocol. CRM1 inhibitors Leptomycin B (Apollo Scientific) and KPT330 (Selleckchem) were used at a final concentration of 6 ng/ml and 1 μM, respectively.
Immunofluorescence and microscopy analysis
Cells were fixed with 3.7 % formaldehyde in phosphate-buffered saline (PBS) for 30 min, permeabilized with 0.2 % Triton X-100 in PBS for 10 min and blocked in 3 % bovine serum albumin (BSA) in PBS for 1 h. Cells were incubated with anti-Flag M2 antibody (Sigma), diluted 1:300 in blocking solution for 1 h to detect SRV100. After washing with PBS, cells were incubated with an AlexaFluor594-conjugated anti-mouse secondary antibody (Invitrogen) diluted 1:400 for 1 h. Finally, samples were washed and mounted onto microscope slides using DAPI-containing Vectashield (Vector).
Semiquantitative assessment of SRV100 nucleocytoplasmic localization was carried out by determining the localization of the reporter in at least 200 co-transfected cells per slide using a Zeiss Axioskop fluorescence microscope. Slides were coded to ensure unbiased scoring. On the other hand, single-slice images were acquired using an Olympus Fluoview FV500 confocal microscope. Sequential acquisition of each fluorochrome was performed to avoid overlapping of fluorescent emission spectra.
Imaging flow cytometry
For imaging flow cytometry, 293T cells were detached from the plate using trypsin prior to anti-Flag immunofluorescence staining. The staining procedure was essentially as described above, but DAPI was added to the blocking solution containing the secondary antibody. Analysis was carried out using an ImageStream X-100 cytometer (Amnis-Millipore) as detailed previously [39]. Briefly, after gating single cells co-expressing the different SRV100 reporter variants with YFP, YFP-CRM1 or YFP-CRM1E571K, a mask to delineate the nucleus was created based on the DAPI signal. Based on this mask, the ratio of the amount of SRV100 in the entire cell to the amount of SRV100 in the nucleus was calculated. This ratio was log transformed to generate the “nuclear translocation score” for each cell.
Graphs representing the nuclear translocation score vs. the intensity of the YFP fluorescence were generated using GraphPad.
Bacterial protein expression, purification and fluorescence anisotropy measurements
CRM1 and TFP-NES proteins, all of them including poly-His tags, were produced from E. coli as previously described [38]. Purification was based on Ni–NTA affinity (the His-ZZ tag was removed from CRM1 proteins using proteolysis and a second Ni–NTA step) and size exclusion chromatography.
Binding assays were performed by adding increasing concentrations of CRM1 to 50 nM TFP-NES, as previously described [38], and fitting the relative increase in anisotropy of TFP to a quadratic function corresponding to a single site binding model.
Results and discussion
A novel reporter to interrogate CRM1 nuclear export activity in a cellular setting
Survivin is a nucleocytoplasmic shuttling protein whose nuclear export is mediated by CRM1 [40]. Survivin bears a LR-NES (residues 86–98), and a second C-terminal export (CTE) motif that does not fit the LR-NES consensus pattern (Fig. 2a) [41, 42]. The LR-NES partially overlaps a region that mediates the formation of survivin dimers and thus, homodimerization modulates CRM1-mediated export of survivin [42]. We have previously shown that Flag-tagged human survivin fused to a heterologous NLS, accumulates into the nucleus, but readily relocates to the cytoplasm upon co-expression with YFP-CRM1 [43]. Based on these previous observations, we set out to design a chimeric protein that could be used as a reporter to interrogate the export function of CRM1 in a cellular setting. Since the presence of two separate export motifs and the ability to dimerize might potentially complicate interpretation of results, we deleted the last 42 amino acids of survivin, thus removing the CTE and disrupting the dimerization motif. Two NLS sequences from the SV40 large T antigen and three tandem copies of the Flag epitope (3 × Flag) were fused to the survivin fragment 1–100, as illustrated in Fig. 2a, to create a chimeric protein that we named SRV100. We generated two versions of SRV100, one bearing a wild type NES sequence, and the other one with NES-inactivating mutations (SRV100NESm) to be used as a control (Fig. 2b).
Fig. 2.
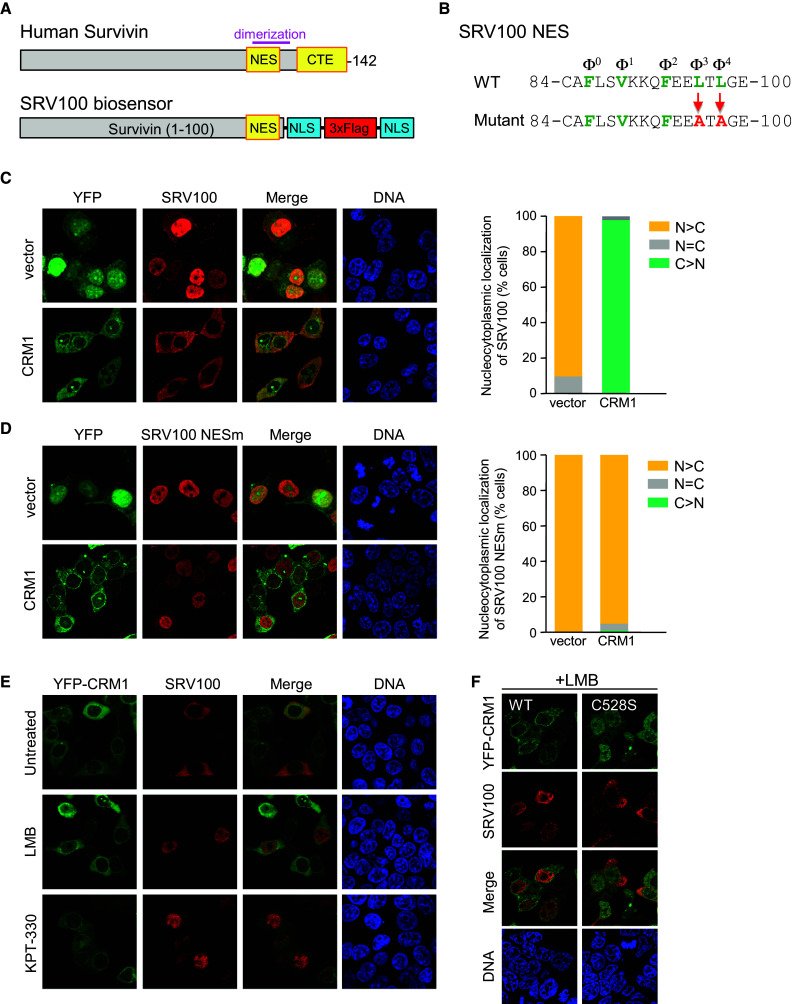
SRV100, a novel reporter to interrogate CRM1 nuclear export activity in a cellular setting. a Shematic representation of human survivin protein and the SRV100 reporter. Deletion of the last 42 amino acids eliminates a carboxy-terminal export motif (CTE) in survivin and disrupts its dimerization domain. Two SV40 NLSs (blue) and a 3 × Flag epitope tag (red) replace this segment in SRV100. b Amino acid sequence of survivin NES indicating the hydrophobic residues (green) that conform to the Φ0–Φ4 consensus. As a control, we generated a mutant version of the NES bearing alanine replacements of Φ3 and Φ4. c, d Images show representative examples of 293T cells co-expressing SRV100 and either YFP vector or YFP-CRM1. The graphs indicate the percentage of co-transfected cells showing predominantly nuclear (N > C), nuclear and cytoplasmic (N = C) or predominantly cytoplasmic (C > N) localization of SRV100. At least 200 cells were counted per sample. c The results with wild type SRV100 and d the results with the NES mutant (NESm) version of SRV100. e Images show representative examples of 293T cells co-expressing wild type SRV100 and YFP-CRM1. Cells were either left untreated or treated with CRM1 inhibitors LMB (6 ng/ml) or KPT330 (1 μM) for 3 h. f Images showing that, unlike wild-type YFP-CRM1, an LMB-insensitive mutant YFP-CRM1C528S induces SRV100 cytoplasmic relocation in LMB-treated cells
SRV100 accumulated in the nucleus of transfected 293T cells when co-expressed with YFP vector, but relocalized to the cytoplasm when co-expressed with YFP-CRM1 (Fig. 2c). Similar experiments were carried out in HeLa and U2OS cells (Online Resource 2). Surprisingly, in contrast to the clear effect observed in 293T cells, expression of YFP-CRM1 only marginally increased the cytoplasmic localization of SRV100 in HeLa and U2OS cells. We speculate that this observation could be due to yet-to-be characterized differences in the nuclear transport machinery between the different cell-lines. From these experiments, we concluded that the 293T cell line is a suitable cellular system to evaluate CRM1 export activity using the SRV100 reporter and, therefore, this cell line was used in all subsequent experiments.
Next, we carried out a series of control experiments to demonstrate that YFP-CRM1-mediated cytoplasmic relocalization of SRV100 is dependent on the presence of a functional NES in the reporter and on CRM1 export function. Indeed, an NES mutant version of the reporter (SRV100NESm) remained in the nucleus even when co-expressed with YFP-CRM1 (Fig. 2d), whereas cell treatment with CRM1 inhibitors LMB and KPT330 blocked SRV100 nuclear export (Fig. 2e). Finally, to investigate a possible effect of endogenous CRM1 on SRV100 export, we used a mutant version of YFP-CRM1 that is not inhibited by LMB (YFP-CRM1C528S). Cells transfected with this mutant were treated with LMB to inhibit endogenous CRM1. Under these conditions, SRV100 was located in the cytoplasm of YFP-CRM1C528S-expressing cells (Fig. 2f), thus showing that endogenous CRM1 activity does not play a crucial role on SRV100 localization.
Altogether, these results indicate that, when used in an appropriate cell line, the SRV100 constitutes a valid tool to interrogate CRM1 export function in a cellular setting. There are previously described reporters (termed translocation biosensors) that can be used to study CRM1-mediated export in a variety of cell lines [44]. The key difference between these reporters and SRV100 is their localization in basal conditions. In contrast to SRV100, these translocation biosensors are cytoplasmic proteins that relocate to the nucleus when CRM1 function is blocked. Thus, they are well-suited to search for novel CRM1 inhibitors [45]. SRV100, on the other hand, is better suited to investigate the functional consequences of experimental and naturally-occurring CRM1 mutations in co-transfection experiments.
Experimental mutations in the NES-binding cleft of CRM1 severely disrupt SRV100 export
The structural analyses that led to the identification of a hydrophobic cleft in the outer surface of CRM1 as the NES-binding site [3, 4] pinpointed several residues in this cleft that would engage in hydrophobic interactions with bound NES peptides. These residues included, as illustrated in Fig. 3a, I521 and L525 in helix H11A, and F561 and F572 in helix H12A. Supporting the structural data, site-directed mutagenesis of these amino acids was found to disrupt NES binding. Thus, a quadruple mutant I521A, L525A, F561A, F572A, termed 4X hereafter, was unable to bind three different GST-tagged NESs in pull-down assays [3]. In a subsequent study that shed further light on how CRM1 recognizes LR-NESs, Güttler et al. mutated a single residue (A541) to lysine on one of CRM1 NES-binding pockets (Fig. 3a). This bulkier residue is expected to interfere with NES docking. Indeed, the CRM1A541K mutant failed to bind to immobilized NES peptides [30].
Fig. 3.
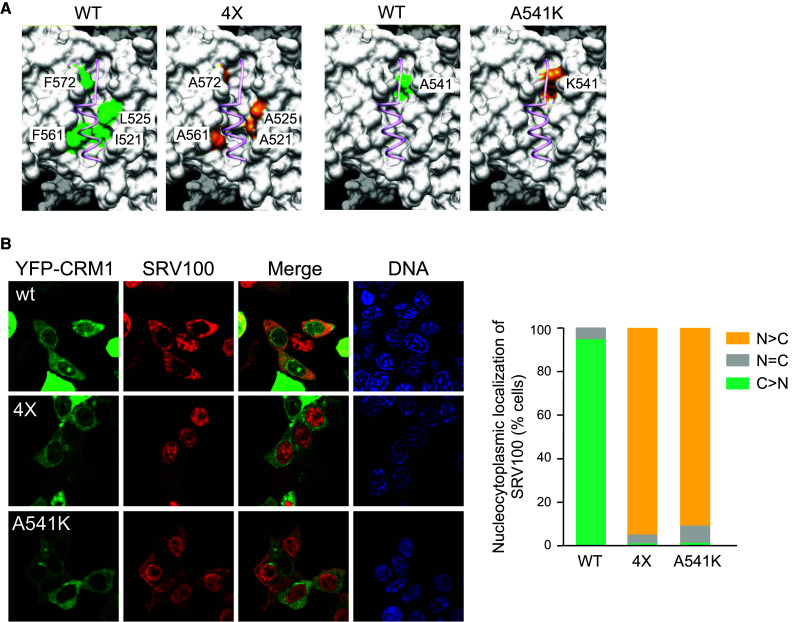
Experimental mutations in the NES-binding cleft of CRM1 severely disrupt SRV100 export. a View of CRM1 NES-binding cleft highlighting residues whose mutation has been reported to interfere with CRM1/NES peptide interaction in previous in vitro experiments. A quadruple mutant (×4) bearing alanine replacements of I521, L525, F561 and F572 [3] is shown on the left, whereas a single A541K mutant [30] is shown on the right. Wild type residues are highlighted in green and mutant residues are highlighted in orange. An NES peptide is depicted in pink using ribbon representation. b Images show representative examples of 293T cells co-expressing SRV100 with either wild type YFP-CRM1 or with the NES-binding cleft mutants YFP-CRM14X and YFP-CRM1A541K. The graph indicates the percentage of co-transfected cells showing predominantly nuclear (N > C), nuclear and cytoplasmic (N = C) or predominantly cytoplasmic (C > N) localization of SRV100. At least 200 cells were counted per sample
In an attempt to determine how the results of these in vitro assays translate to the more physiological setting of intact cells, we evaluated the ability of YFP-CRM14X and YFP-CRM1A541K mutants to promote nuclear export of SRV100 in 293T cells. As shown in Fig. 3b, SRV100 remained exclusively nuclear when co-expressed with YFP-CRM14X. On the other hand, a faint cytoplasmic SRV100 signal could be detected in some cells co-expressing YFP-CRM1A541K, but the reporter remained mostly located in the nucleus. In contrast, SRV100 was nearly exclusively cytoplasmic in cells co-expressing wild type YFP-CRM1.
These results show that mutations that alter the NES-binding cleft of CRM1 severely disrupt its nuclear export activity in cells. Our data, therefore, provide further support to the model that describes the molecular basis of NES recognition by CRM1 NES-binding cleft.
Experimental mutations in CRM1 H9 acidic loop and C-terminal helix partially impair SRV100 export
The H9 loop and the C-helix of CRM1 are key regulatory elements in the allosteric modulation of NES binding and release [10–12, 46], reviewed in [47]. In free CRM1, the H9 loop and the C-helix contact the inner surface of the receptor beneath the NES-binding cleft, stabilizing the cleft in a closed state that hinders NES access. Conformational rearrangements of these motifs upon RanGTP binding mediate the cooperative assembly of the trimeric export complex by switching the NES-binding cleft to an open state. Conversely, the contact of the H9 loop with the surface behind the NES-binding cleft is restored upon RanBP1 binding, which ultimately leads to closure of the cleft and extrusion of the NES, thus facilitating the disassembly of the export complex [11].
Supporting this model, in vitro analyses of structure-guided CRM1 variants have shown that deletions or mutations in the C-terminal domain or the H9 loop of CRM1 dramatically enhance the affinity of CRM1 for NES peptides in the absence of RanGTP, and that H9 loop mutations reduce the rate of RanBP1-accelerated NES release [5, 10–12, 38]. In particular, using fluorescence anisotropy, Fox et al. have reported that deletion of the last nine amino acids at the extreme distal tip of CRM1 C-terminal helix, a mutant hereafter called CRM11063X (schematically represented in Fig. 4a) or alanine substitution of three H9 loop residues (430VLV > AAA), hereafter termed CRM1AAA (Fig. 4a), increase affinity for PKI NES in the absence of RanGTP [12]. Importantly, CRM1 affinity is further enhanced by simultaneous combination of both changes (CRM1AAA/X), to reach a level comparable with that observed in the presence of RanGTP [12]. We introduced these mutations into YFP-CRM1 and tested their ability to promote SRV100 export. As shown in Fig. 4b, YFP-CRM11063X and YFP-CRM1AAA were only marginally impaired in their ability to export SRV100, as most cells expressing these CRM1 variants showed cytoplasmic localization of SRV100, similar to cells expressing wild type YFP-CRM1. In contrast, SRV100 export was clearly less efficient when co-expressed with the combined mutant YFP-CRM1AAA/X. In most cells expressing this mutant, the biomarker was evenly distributed between the nucleus and cytoplasm. These results indicate that interfering with the normal ability of the NES-binding cleft to switch between open and closed conformations, enhances CRM1/NES binding affinity [12], but impairs export cycle in a cellular context. Our data are in line with the previous observation that artificial supraphysiological NES peptides with extremely high affinity for CRM1 impair nuclear export complex disassembly [48]. Of note, although natural NESs with relatively high affinity, such as the PKI NES, can be efficiently exported, CRM1/NES interactions are usually weak.
Fig. 4.
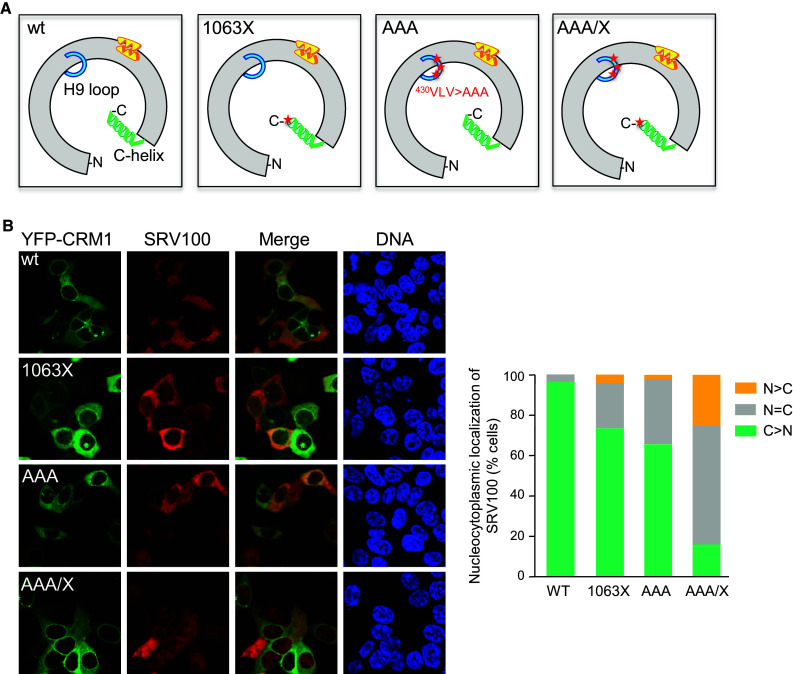
Experimental mutations in CRM1 H9 acidic loop and C-terminal helix partially impair SRV100 export. a Schematic representation of the CRM1 protein illustrating the position of mutations previously reported to increase NES-binding affinity [12]. CRM11063X lacks the last nine amino acids at the extreme distal tip of CRM1 C-terminal helix. CRM1AAA bears alanine substitution of three H9 loop residues (430VLV > AAA). The mutant CRM1AAA/X combines both alterations. b Images show representative examples of 293T cells co-expressing SRV100 with either wild type YFP-CRM1 or with the indicated mutants. The graph indicates the percentage of co-transfected cells showing predominantly nuclear (N > C), nuclear and cytoplasmic (N = C) or predominantly cytoplasmic (C > N) localization of SRV100. At least 200 cells were counted per sample
The cancer-associated CRM1 mutation E571K does not abrogate SRV100 export
The cancer-associated E571K mutation targets an amino acid near CRM1 NES-binding cleft (Fig. 5a). The recurrent nature of this mutation in CLL patients suggests that it may represent a “driver” alteration that contributes to leukemia development [32], but its potential impact on CRM1 function has not yet been investigated. Thus, we set out to test the effect of the E571K mutation on YFP-CRM1 export activity using the SRV100 reporter. For comparison, we introduced a single amino acid change targeting the next residue (F572A), which lies inside the cleft and engages in interactions with NES peptides (Fig. 5b). As shown in Fig. 5c, SRV100 was mostly nuclear in cells expressing YFP-CRM1F572A, indicating that this single amino acid mutation abrogates SRV100 export. In contrast, cells expressing YFP-CRM1E571K showed predominantly cytoplasmic localization of SRV100, very similar to cells expressing wild type YFP-CRM1. This observation indicates that the E571K mutation does not result in a general impairment of CRM1 export activity.
Fig. 5.
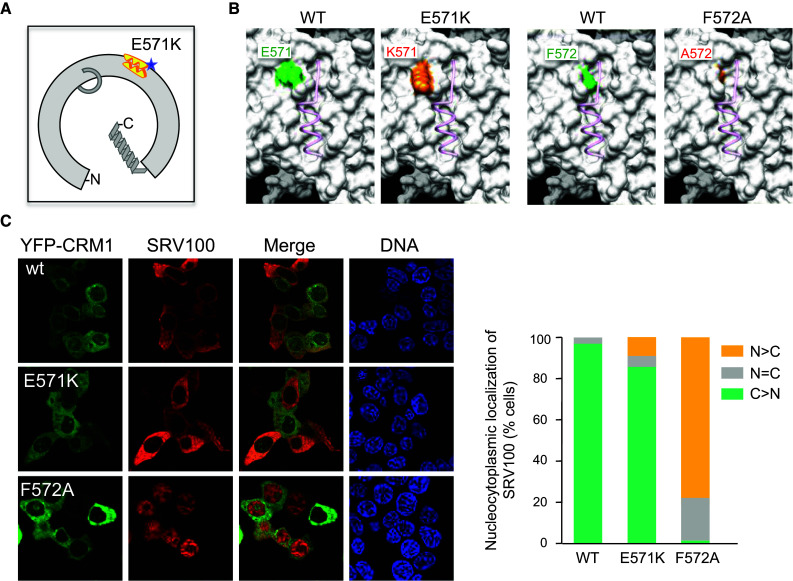
The cancer-associated CRM1 mutation E571K does not abrogate SRV100 export. a Schematic representation of the CRM1 protein illustrating the position of the recurrent cancer mutation E571K (blue star) proximal to the NES-binding cleft. b View of CRM1 NES-binding cleft highlighting wild type (green) and mutant (orange) versions of residues 571 (left panels) and 572 (right panels). An NES peptide is depicted in pink using ribbon representation. c Images show representative examples of 293T cells co-expressing SRV100 with either wild type YFP-CRM1 or with the indicated mutants. The graph indicates the percentage of co-transfected cells showing predominantly nuclear (N > C), nuclear and cytoplasmic (N = C) or predominantly cytoplasmic (C > N) localization of SRV100. At least 200 cells were counted per sample
The E571K mutation leads to subtle differences in the export of SRV100 variants with differently charged C-terminal end
As illustrated in Fig. 6a, the replacement of glutamic acid by a lysine residue in the E571K mutant increases the positive charge adjacent to the NES-binding cleft. An inspection of the 3D structure of the RanGTP/CRM1/NES complex (PDB entry 3GJX, [4]) revealed that the side chain of residue 571 locates closest to NES hydrophobic residues Φ3 and Φ4. We hypothesized that the negative-to-positive charge inversion might increase the affinity of CRM1E571K for NES sequences bearing a negatively charged C-terminal end, while decreasing the affinity for positively charged NESs. To test this hypothesis, we generated two variants of the SRV100 reporter introducing charge-modifying mutations into the C-terminal end of the NES (Fig. 6b). The wild type survivin NES includes two acidic residues between Φ2 and Φ3 and a further one in position Φ4 + 2. We introduced an additional acidic residue between Φ3 and Φ4 (T97D mutation), thus generating a more negatively charged NES version, hereafter termed “EDE”. On the other hand, we replaced NES residues E95, T97 and E100 with lysines to generate a more positively charged NES version termed “KKK”.
Fig. 6.
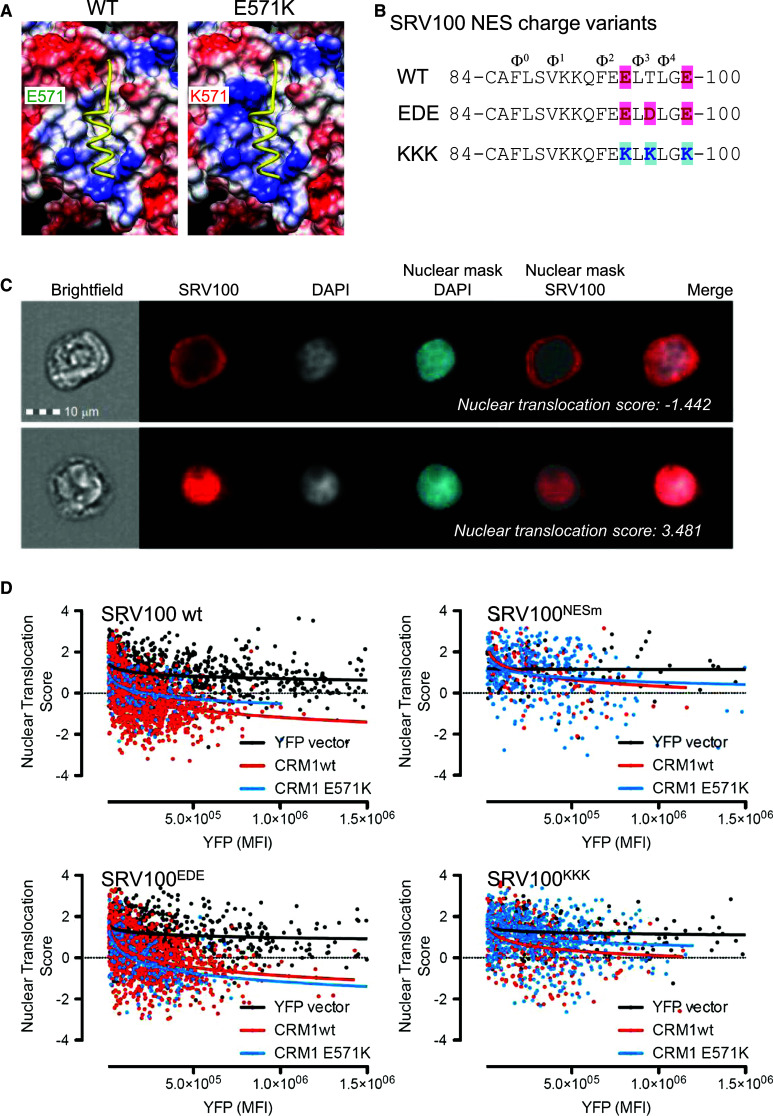
The E571K mutation leads to subtle differences in the export of SRV100 variants with differently charged C-terminal end. a Representation of the electrostatic surface of CRM1 NES-binding cleft (structure 3GJX, [4]) with blue and red colors corresponding to positive and negative charges, respectively. An NES peptide is depicted in yellow using ribbon representation. The E571K mutation increases the positive charge of the CRM1 surface near the cleft region that interacts with the C-terminal part of the NES. b Amino acid sequence of the NESs in the SRV100 charge variants (wild type, EDE and KKK), illustrating the substitutions in the region Φ3–Φ4. Acidic and basic residues mutated to create the charge variants are highlighted in pink and blue, respectively. c Images show examples of SRV100 nuclear translocation analysis in 293T cells using imaging flow cytometry. A mask delineating the nucleus is created based on the DAPI staining, and used to calculate a nuclear translocation score for SRV100 (see “Materials and methods” section for details). A low nuclear translocation score corresponds to cytoplasmic localization of the reporter (upper panels). A high score corresponds to nuclear localization of the reporter (lower panels). d Graphs representing the nuclear translocation score (y axis) vs. the intensity of the YFP fluorescence (x axis) for different SRV100 variants co-expressed with YFP vector (black) YFP-CRM1 wild type (red) or YFP-CRM1E571K (blue). Each dot represents a single cell. At least 100 cells per condition were analyzed. MFI median fluorescence intensity
293T cells were co-transfected with plasmids encoding these SRV100 variants and either wild type YFP-CRM1 or YFP-CRM1E571K. As negative controls, the empty YFP vector and the SRV100NESm mutant were used in these experiments. In an attempt to obtain a more detailed and quantitative view of the results, samples were analyzed using imaging flow cytometry. For each individual cell, the intensity of the YFP signal and the localization of the SRV100 reporter were determined. As illustrated in Fig. 6c, and described in detail in the “Materials and methods” section, a nuclear translocation score was automatically computed for each cell, reflecting the nuclear/cytoplasmic distribution of SRV100 (a lower score indicating a more pronounced cytoplasmic localization of the reporter). The nuclear translocation score was plotted against the intensity of the YFP signal (Fig. 6d). As expected, a progressively lower score for wild type SRV100 was observed in cells expressing increasing levels of wild type YFP-CRM1, indicating that higher expression levels of the receptor result in a more efficient SRV100 export. A similar but less pronounced effect was noted in cells expressing YFP-CRM1E571K. In contrast, expression of YFP vector did not reduce the score. As a control, the nuclear translocation score for SRV100NESm remained virtually unaltered even in cells expressing high levels of YFP-CRM1 (wt or E571K). Interestingly, the nuclear translocation score for SRV100EDE was slightly lower in cells expressing YFP-CRM1E571K. Conversely, the score for SRV100KKK was higher in cells expressing YFP-CRM1E571K. Of note, the nuclear translocation score for SRV100KKK remained relatively higher when co-expressed with either CRM1 variant, suggesting that the introduced mutations negatively affect NES activity. Three independent experiments that showed reproducible results were performed with SRV100EDE and two with SRV100KKK. The median nuclear translocation score for SRV100EDE was consistently lower in cells expressing YFP-CRM1E571K (Online Resource 3). These results suggest that the positive charge introduced by the E571K mutation might favor the export of a subset of NESs with a more negative charge in their C-terminal end.
The E571K mutation increases the affinity of CRM1 for NESs with a more negatively charged C-terminal end
The observed differences between wild type and E571K mutant CRM1 in the export of SRV100EDE and SRV100KKK reporter variants were reproducible, but small. Therefore, to further substantiate these observations, we used in vitro biochemical assays to compare the affinity of wild type and E571K mutant CRM1 for differently charged NESs.
We expressed and purified recombinant forms of human wild type CRM1 and CRM1E571K. It must be noted that, in both cases, the recombinant proteins also bear the H9 loop mutations (430VLV > AAA) and the C-terminal deletion described above, to increase their NES binding affinity. The use of these “high affinity mutants” allows formation of the CRM1/NES complex in the absence of RanGTP, and therefore facilitates in vitro characterization of the binding process [12, 38]. On the other hand, we expressed and purified the EDE and KKK var.iants, as well as the wild type (ETE) version of survivin NES fused to the C-terminal end of teal fluorescent protein (TFP-ETE, TFP-EDE and TFP-KKK). We estimated CRM1/NES binding affinities by monitoring the increase in fluorescence anisotropy of TFP, as previously described for mouse CRM1 [38].
The E571K mutant displayed a 2 fold higher affinity for TFP-EDE NES than wild type CRM1 (K D 413 vs. 844 nM) (Fig. 7a, b). Conversely, binding of the mutant to the TFP-KKK NES was disfavored with respect to wild type CRM1, (K D 3026 vs. 947 nM), whereas there was no significant difference in their affinity for the wild type version TFP-ETE (Fig. 7b). Thus, in line with the results from cell experiments using the SRV100 reporter, the results of these in vitro biochemical assays indicate that the E571K mutation increases the affinity of CRM1 for a NES bearing negative charges in the vicinity of Φ3–Φ4, and decreases the affinity for a NES with positive charges in that region.
Fig. 7.
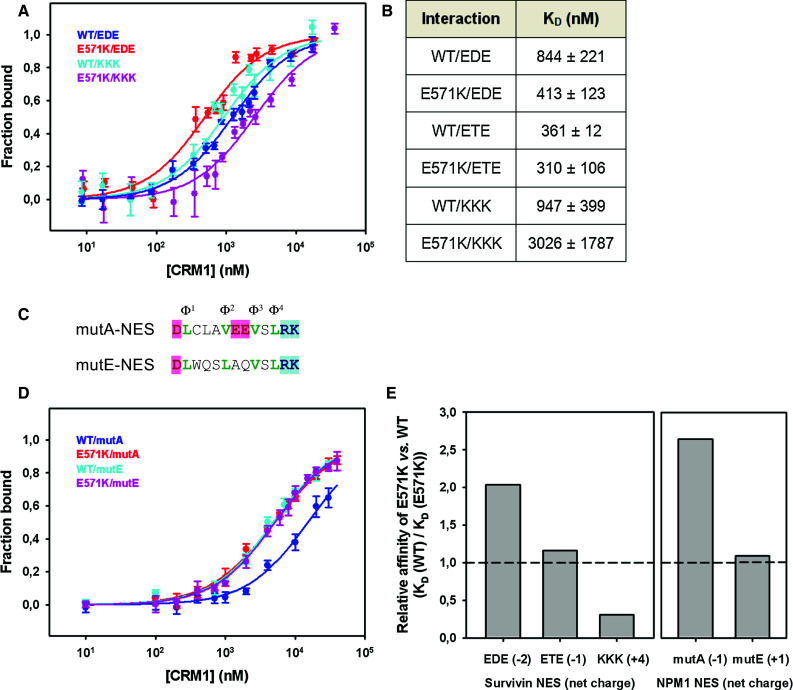
The E571K mutation increases the affinity of CRM1 for NESs with a more negatively charged C-terminal end. a Graph represents binding of WT and E571K CRM1 variants to TFP-EDE and TFP-KKK NESs, based on the increase in fluorescence anisotropy of TFP. Data points were fitted to a quadratic function (lines) corresponding to a single site binding model. b Estimated K D parameters (mean and standard deviation). 5–8 independent experiments were performed for each interaction pair, except for the WT/ETE and E571K/ETE combinations, where the number of independent experiments was 2. The differences between K D values of WT and E571K are statistically significant for EDE and KKK (two-tailed Student t test P = 0.001 and 0.02, respectively). c Amino acid sequences of the acquired NESs of AML-related nucleophosmin mutants A and E, highlighting acidic (pink) and basic (blue) residues. d Graph represents binding of WT and E571K CRM1 variants to TFP-mutA-NES and TFP-mutE-NES. Analysis was performed as in a. e Relative affinity (mean values) of CRM1E571K vs. wild type CRM1 for the five differently charged NESs tested
In an attempt to extend these findings to naturally occurring NESs, we investigated the effect of the E571K mutation on the affinity of CRM1 for two different NES sequences mutationally acquired by the oncoprotein nucleophosmin (NPM1) in a subtype of acute myeloid leukemia (AML) [49]. Two AML-associated NPM1 mutants, termed A and E, harbor frameshift mutations that create novel NESs, with different amino acid sequences, in the C-terminal end of the protein [50]. We and others have previously characterized the export activity of these NESs and their recognition by CRM1, showing that the NES of mutant E (mutE-NES) is more efficiently exported and is recognized in vitro with higher affinity by wild type CRM1 than the NES of mutant A (mutA-NES) [38, 51], probably due to a more optimal combination of hydrophobic residues [30]. Interestingly, mutA-NES and mutE-NES differ in the charge of the residues immediately preceding Φ3. As shown in Fig. 7c, mutA-NES has a net charge of −1, while mutE-NES has a charge of +1. These sequences may therefore represent an appropriate model system to further explore the effect of the E571K mutation on CRM1 affinity for differently charged NESs.
We used fluorescence anisotropy to determine the binding affinity of wild type and E571K mutant CRM1 to TFP-mutA-NES and TFP-mutE-NES. Consistent with our previous results using mouse CRM1 [38], we found that wild type human CRM1 binds with higher affinity to mutE-NES (K D 4.8 μM) than to mutA-NES (K D 15.1 μM) (Fig. 7d). In contrast, CRM1E571K bound to both mutants with similar affinity (K D 5.1 μM for mutE-NES and K D 5.9 μM for mutA-NES). This increase in the relative affinity of CRM1E571K for the negatively charged mutA-NES could be explained by the presence of acidic residues contiguous to Φ3 in this NES sequence, thus supporting the data with the artificial EDE and KKK SRV100 mutants described above.
In fact, although the number of NES peptides analyzed is limited, there appears to be a correlation between the net charge of the NES and the relative affinity for CRM1E571K (Fig. 7e), the mutant being a stronger binder for sequences with a negative charge.
Altogether, these biochemical data are in line with the results of cellular SRV100-based assays, and support our view that a phenotypic consequence of the E571K mutation is to increase the affinity of the receptor for NESs with a more negatively charged C-terminal end. It must be noted that a recent structural study has described that some NESs may bind CRM1 in a “reverse” orientation [6]. In these cases, it is possible that the electrostatic properties of the N-terminal part of the NES might also influence differential affinity for CRM1E571 mutant.
Conclusions
The SRV100 reporter described here represents a novel tool to interrogate the nuclear export function of CRM1 in a cellular setting. Using this tool to complement previous structural and biochemical data, we provide cellular evidence further supporting the model that describes the molecular basis of NES recognition by CRM1 NES-binding cleft and the regulation of NES binding and release by the H9 acidic loop and the C-terminal helix of the receptor.
Furthermore, we analyze for the first time the functional consequences of the recurrent cancer-related mutation CRM1E571K. Our results indicate that this mutation does not severely disrupt the nuclear export activity of CRM1, but may increase its affinity for NES sequences bearing a more negatively charged C-terminal end. Certainly, this may not be the only phenotypic consequence of the mutation. The E571K change might also disrupt interaction with other proteins that bind CRM1 independently of the NES, or it might affect other aspect of CRM1 function unrelated to export, such as mitotic regulation [52]. Nevertheless, it is tempting to speculate that these slight differences in NES binding affinity may alter the nucleocytoplasmic distribution of a subset of substrates that are preferentially exported by wt or E571K mutant CRM1, altering cell homeostasis and thus mediating a role of mutant CRM1E571K in tumorigenesis.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Dr. Fernando Moro for helping with the analysis of binding curves and Dr. René Medema for his support. We thank the staff from the High Resolution Microscopy Facility (SGIker-UPV/EHU) for technical support. This work is funded by the Spanish Ministry of Economy (Grant SAF2014-57743-R to SB and JAR), and by the University of the Basque Country (UFI 11/20). IG-S is a recipient of a postdoctoral fellowship from the Department of Education of the Basque Country Government.
Abbreviations
- AML
Acute myeloid leukemia
- CLL
Chronic lymphocytic leukemia
- CTE motif
C-terminal export motif
- LR-NES
Leucine rich NES
- NES
Nuclear export signal
- NLS
Nuclear localization signal
- NPC
Nuclear pore complex
- KD
Equilibrium dissociation constant
Footnotes
A patent application on the SRV100 biosensor has been submitted by the University of the Basque Country UPV/EHU.
Contributor Information
Sonia Bañuelos, Phone: +34 94 601 8050, Email: sonia.banuelos@ehu.es.
Jose A. Rodríguez, Phone: +34 94 601 8072, Email: josean.rodriguez@ehu.es
References
- 1.Yoneda Y. Nucleocytoplasmic protein traffic and its significance to cell function. Genes Cells. 2000;5:777–787. doi: 10.1046/j.1365-2443.2000.00366.x. [DOI] [PubMed] [Google Scholar]
- 2.Pemberton LF, Paschal BM. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic. 2005;6:187–198. doi: 10.1111/j.1600-0854.2005.00270.x. [DOI] [PubMed] [Google Scholar]
- 3.Dong X, Biswas A, Süel KE, et al. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009;458:1136–1141. doi: 10.1038/nature07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monecke T, Güttler T, Neumann P, et al. Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science. 2009;324:1087–1091. doi: 10.1126/science.1173388. [DOI] [PubMed] [Google Scholar]
- 5.Dong X, Biswas A, Chook YM. Structural basis for assembly and disassembly of the CRM1 nuclear export complex. Nat Struct Mol Biol. 2009;16:558–560. doi: 10.1038/nsmb.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fung HY, Fu SC, Brautigam CA, Chook YM. Structural determinants of nuclear export signal orientation in binding to exportin CRM1. Elife. 2015;4:e10034. doi: 10.7554/eLife.10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fung HY, Chook YM. Atomic basis of CRM1-cargo recognition, release and inhibition. Semin Cancer Biol. 2014;27:52–61. doi: 10.1016/j.semcancer.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuura Y. Mechanistic Insights from Structural Analyses of Ran-GTPase-Driven Nuclear Export of Proteins and RNAs. J Mol Biol. 2016;428:2025–2039. doi: 10.1016/j.jmb.2015.09.025. [DOI] [PubMed] [Google Scholar]
- 9.Dickmanns A, Monecke T, Ficner R. Structural basis of targeting the exportin CRM1 in cancer. Cells. 2015;4:538–568. doi: 10.3390/cells4030538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saito N, Matsuura Y. A 2.1-Å-resolution crystal structure of unliganded CRM1 reveals the mechanism of autoinhibition. J Mol Biol. 2013;425:350–364. doi: 10.1016/j.jmb.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 11.Koyama M, Matsuura Y. An allosteric mechanism to displace nuclear export cargo from CRM1 and RanGTP by RanBP1. EMBO J. 2010;29:2002–2013. doi: 10.1038/emboj.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fox AM, Ciziene D, McLaughlin SH, et al. Electrostatic interactions involving the extreme C terminus of nuclear export factor CRM1 modulate its affinity for cargo. J Biol Chem. 2011;286:29325–29335. doi: 10.1074/jbc.M111.245092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noske A, Weichert W, Niesporek S, et al. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer. 2008;112:1733–1743. doi: 10.1002/cncr.23354. [DOI] [PubMed] [Google Scholar]
- 14.Shen A, Wang Y, Zhao Y, et al. Expression of CRM1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery. 2009;65:153–159. doi: 10.1227/01.NEU.0000348550.47441.4B. [DOI] [PubMed] [Google Scholar]
- 15.Huang WY, Yue L, Qiu WS, et al. Prognostic value of CRM1 in pancreas cancer. Clin Invest Med. 2009;32:E315. [PubMed] [Google Scholar]
- 16.Yao Y, Dong Y, Lin F, et al. The expression of CRM1 is associated with prognosis in human osteosarcoma. Oncol Rep. 2009;21:229–235. [PubMed] [Google Scholar]
- 17.Zhou F, Qiu W, Yao R, et al. CRM1 is a novel independent prognostic factor for the poor prognosis of gastric carcinomas. Med Oncol. 2013;30:726. doi: 10.1007/s12032-013-0726-1. [DOI] [PubMed] [Google Scholar]
- 18.Lin DC, Hao JJ, Nagata Y, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014;46:467–473. doi: 10.1038/ng.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conforti F, Wang Y, Rodriguez JA, et al. Molecular pathways: anticancer activity by inhibition of nucleocytoplasmic shuttling. Clin Cancer Res. 2015;21:4508–4513. doi: 10.1158/1078-0432.CCR-15-0408. [DOI] [PubMed] [Google Scholar]
- 20.Azmi AS. The evolving role of nuclear transporters in cancer. Semin Cancer Biol. 2014;27:1–2. doi: 10.1016/j.semcancer.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 21.Kudo N, Wolff B, Sekimoto T, et al. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp Cell Res. 1998;242:540–547. doi: 10.1006/excr.1998.4136. [DOI] [PubMed] [Google Scholar]
- 22.Sun Q, Carrasco YP, Hu Y, et al. Nuclear export inhibition through covalent conjugation and hydrolysis of Leptomycin B by CRM1. Proc Natl Acad Sci USA. 2013;110:1303–1308. doi: 10.1073/pnas.1217203110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lapalombella R, Sun Q, Williams K, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012;120:4621–4634. doi: 10.1182/blood-2012-05-429506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ranganathan P, Yu X, Na C, et al. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood. 2012;120:1765–1773. doi: 10.1182/blood-2012-04-423160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pathria G, Wagner C, Wagner SN. Inhibition of CRM1-mediated nucleocytoplasmic transport: triggering human melanoma cell apoptosis by perturbing multiple cellular pathways. J Invest Dermatol. 2012;132:2780–2790. doi: 10.1038/jid.2012.233. [DOI] [PubMed] [Google Scholar]
- 26.Kojima K, Kornblau SM, Ruvolo V, et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood. 2013;121:4166–4174. doi: 10.1182/blood-2012-08-447581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Etchin J, Sanda T, Mansour MR, et al. KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br J Haematol. 2013;161:117–127. doi: 10.1111/bjh.12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inoue H, Kauffman M, Shacham S, et al. CRM1 blockade by selective inhibitors of nuclear export attenuates kidney cancer growth. J Urol. 2013;189:2317–2326. doi: 10.1016/j.juro.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalid O, Toledo Warshaviak D, Shechter S, et al. Consensus induced fit docking (cIFD): methodology, validation, and application to the discovery of novel Crm1 inhibitors. J Comput Aided Mol Des. 2012;26:1217–1228. doi: 10.1007/s10822-012-9611-9. [DOI] [PubMed] [Google Scholar]
- 30.Güttler T, Madl T, Neumann P, et al. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17:1367–1376. doi: 10.1038/nsmb.1931. [DOI] [PubMed] [Google Scholar]
- 31.Terry LJ, Shows EB, Wente SR. Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science. 2007;318:1412–1416. doi: 10.1126/science.1142204. [DOI] [PubMed] [Google Scholar]
- 32.Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodríguez D, Bretones G, Arango JR, et al. Molecular pathogenesis of CLL and its evolution. Int J Hematol. 2015;101:219–228. doi: 10.1007/s12185-015-1733-0. [DOI] [PubMed] [Google Scholar]
- 34.Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winkelmann N, Rose-Zerilli M, Forster J, et al. Low frequency mutations independently predict poor treatment-free survival in early stage chronic lymphocytic leukemia and monoclonal B-cell lymphocytosis. Haematologica. 2015;100:e237–e239. doi: 10.3324/haematol.2014.120238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amin N, Seymour EK, Saiya-Cork K, et al. A quantitative analysis of Subclonal and clonal gene mutations pre- and post-therapy in chronic lymphocytic leukemia. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodríguez JA, Henderson BR. Identification of a functional nuclear export sequence in BRCA1. J Biol Chem. 2000;275:38589–38596. doi: 10.1074/jbc.M003851200. [DOI] [PubMed] [Google Scholar]
- 38.Arregi I, Falces J, Olazabal-Herrero A, et al. Leukemia-associated mutations in nucleophosmin alter recognition by CRM1: molecular basis of aberrant transport. PLoS One. 2015;10:e0130610. doi: 10.1371/journal.pone.0130610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huijts CM, Schneiders FL, Garcia-Vallejo JJ, et al. mTOR inhibition per se induces nuclear localization of FOXP3 and conversion of invariant NKT (iNKT) cells into immunosuppressive regulatory iNKT cells. J Immunol. 2015;195:2038–2045. doi: 10.4049/jimmunol.1402710. [DOI] [PubMed] [Google Scholar]
- 40.Rodríguez JA, Span SW, Ferreira CG, et al. CRM1-mediated nuclear export determines the cytoplasmic localization of the antiapoptotic protein Survivin. Exp Cell Res. 2002;275:44–53. doi: 10.1006/excr.2002.5492. [DOI] [PubMed] [Google Scholar]
- 41.Stauber RH, Rabenhorst U, Rekik A, et al. Nucleocytoplasmic shuttling and the biological activity of mouse survivin are regulated by an active nuclear export signal. Traffic. 2006;7:1461–1472. doi: 10.1111/j.1600-0854.2006.00486.x. [DOI] [PubMed] [Google Scholar]
- 42.Engelsma D, Rodriguez JA, Fish A, et al. Homodimerization antagonizes nuclear export of survivin. Traffic. 2007;8:1495–1502. doi: 10.1111/j.1600-0854.2007.00629.x. [DOI] [PubMed] [Google Scholar]
- 43.Rodriguez JA, Lens SM, Span SW, et al. Subcellular localization and nucleocytoplasmic transport of the chromosomal passenger proteins before nuclear envelope breakdown. Oncogene. 2006;25:4867–4879. doi: 10.1038/sj.onc.1209499. [DOI] [PubMed] [Google Scholar]
- 44.Knauer SK, Moodt S, Berg T, et al. Translocation biosensors to study signal-specific nucleo-cytoplasmic transport, protease activity and protein-protein interactions. Traffic. 2005;6:594–606. doi: 10.1111/j.1600-0854.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- 45.Fetz V, Knauer SK, Bier C, et al. Translocation biosensors—cellular system integrators to dissect CRM1-dependent nuclear export by chemicogenomics. Sensors (Basel) 2009;9:5423–5445. doi: 10.3390/s90705423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dian C, Bernaudat F, Langer K, et al. Structure of a truncation mutant of the nuclear export factor CRM1 provides insights into the auto-inhibitory role of its C-terminal helix. Structure. 2013;21:1338–1349. doi: 10.1016/j.str.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 47.Monecke T, Dickmanns A, Ficner R. Allosteric control of the exportin CRM1 unraveled by crystal structure analysis. FEBS J. 2014;281:4179–4194. doi: 10.1111/febs.12842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Engelsma D, Bernad R, Calafat J. Supraphysiological nuclear export signals bind CRM1 independently of RanGTP and arrest at Nup358. EMBO J. 2004;23:3643–3652. doi: 10.1038/sj.emboj.7600370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 50.Falini B, Bolli N, Shan J, et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc + AML. Blood. 2006;107:4514–4523. doi: 10.1182/blood-2005-11-4745. [DOI] [PubMed] [Google Scholar]
- 51.Bolli N, Nicoletti I, De Marco MF, et al. Born to be exported: COOH-terminal nuclear export signals of different strength ensure cytoplasmic accumulation of nucleophosmin leukemic mutants. Cancer Res. 2007;67:6230–6237. doi: 10.1158/0008-5472.CAN-07-0273. [DOI] [PubMed] [Google Scholar]
- 52.Forbes DJ, Travesa A, Nord MS, et al. Nuclear transport factors: global regulation of mitosis. Curr Opin Cell Biol. 2015;35:78–90. doi: 10.1016/j.ceb.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.