

Abstract
Hemes (a, b, c, and o) and heme d 1 belong to the group of modified tetrapyrroles, which also includes chlorophylls, cobalamins, coenzyme F430, and siroheme. These compounds are found throughout all domains of life and are involved in a variety of essential biological processes ranging from photosynthesis to methanogenesis. The biosynthesis of heme b has been well studied in many organisms, but in sulfate-reducing bacteria and archaea, the pathway has remained a mystery, as many of the enzymes involved in these characterized steps are absent. The heme pathway in most organisms proceeds from the cyclic precursor of all modified tetrapyrroles uroporphyrinogen III, to coproporphyrinogen III, which is followed by oxidation of the ring and finally iron insertion. Sulfate-reducing bacteria and some archaea lack the genetic information necessary to convert uroporphyrinogen III to heme along the “classical” route and instead use an “alternative” pathway. Biosynthesis of the isobacteriochlorin heme d 1, a cofactor of the dissimilatory nitrite reductase cytochrome cd 1, has also been a subject of much research, although the biosynthetic pathway and its intermediates have evaded discovery for quite some time. This review focuses on the recent advances in the understanding of these two pathways and their surprisingly close relationship via the unlikely intermediate siroheme, which is also a cofactor of sulfite and nitrite reductases in many organisms. The evolutionary questions raised by this discovery will also be discussed along with the potential regulation required by organisms with overlapping tetrapyrrole biosynthesis pathways.
Keywords: Heme, Heme d1, Tetrapyrrole biosynthesis, Siroheme, Modified tetrapyrrole, Alternative heme biosynthesis
Tetrapyrroles and the pigments of life
The hemes belong to a family of metallo-prosthetic groups that are all based on the same structural framework assembled from four polymerized pyrrole rings (labeled A–D) that are arranged into a large macrocycle. A pyrrole is a five-membered ring containing one atom of nitrogen. Modified tetrapyrroles participate in a diverse range of fundamental biological processes and are indispensable components of the metabolism of practically all organisms on earth; they include molecules such as the chlorophylls, cobalamins, siroheme, heme d 1, and coenzyme F430. They play a central role in electron transfer-dependent energy-generating processes from photosynthesis to methanogenesis. The secret to the successful integration of tetrapyrrole-derivatives into so many biological systems reflects the photodynamic properties of the macrocycle and its ability to bind a range of metals. The structures of all modified tetrapyrroles are based on the molecular blueprint of uroporphyrinogen III (uro’gen III, Fig. 1), the first macrocyclic intermediate of the tetrapyrrole pathway, where four pyrrole rings are connected by four bridging carbon atoms to generate the overall structure. Variation in the oxidation state of the macrocycle, prototropic rearrangements assisted by regiospecific methylations and insertion of different metal ions accounts for the variation in the chemical properties and color of this family of compounds.
Fig. 1.
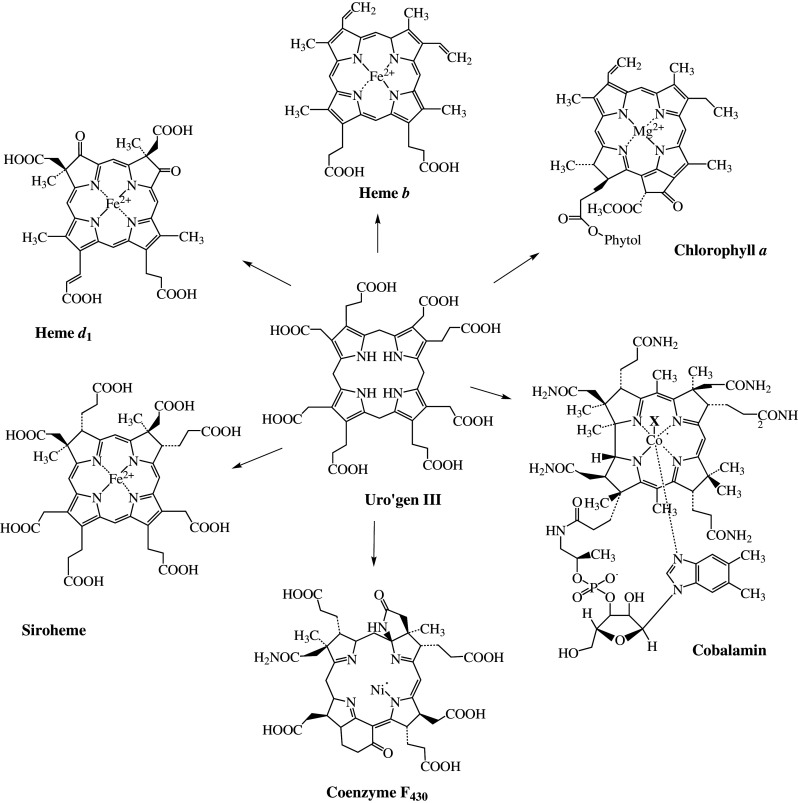
The structures of important representatives of naturally occurring tetrapyrroles and their common precursor uroporphyrinogen III
Heme b (also called protoheme or Fe-protoporphyrin-IX), for example, represents a fully oxidized derivative of uro’gen III, is an iron-containing porphyrin and, when bound to globin, is responsible for the red color of blood. The chlorophyll macrocycle contains one less double bond and is a magnesium-containing chlorin that is responsible for the green color of leaves. Because of their color and the essential role these molecules play in so many biological processes, they are often referred to as the “pigments of life” [1]. The essential functions of these molecules are briefly described below:
The most commonly found tetrapyrrole in nature is chlorophyll. The biosynthesis of this magnesium-containing pigment is the most notable process on earth, with an estimated 109 tons being produced (and broken down) each year with the change in the seasons [2]. Chlorophyll is responsible for harvesting and trapping light during photosynthesis and converting it into the chemical energy of the cell. This process produces oxygen as a byproduct, therefore giving rise to life on earth as it is known today [3]. The bacterial equivalent to chlorophyll is bacteriochlorophyll. It is solely employed in anoxic photosynthesis and probably displays the evolutionary ancestor of chlorophyll [4, 5]. The role of the Mg2+ has received relatively little attention, but a key point is that this cation is unable to undergo oxidation and reduction under any feasible physiological conditions and provides a rigidity to the macrocyle alongside conferring the ability to bind to axial ligands. There is evidence that Zn2+ can substitute for Mg2+ and it is possible that the greater abundancy of Mg is the reason for its ‘selection’ over Zn as the cation in chlorophyll.
Members of the cobalt-containing corrinoids such as cobalamin (vitamin B12) are characterized by a missing methine bridge in the tetrapyrrole structure, which means that rings C and D are linked directly [6]. Different forms of cobalamin facilitate different types of enzymatic transformations including rearrangement, methylation, and reductive dehalogenation processes. Coenzyme F430 incorporates nickel and serves as prosthetic group of methyl-coenzyme M reductase, an enzyme involved in archaeal methanogenesis [7, 8]. In fact, cobalamin and coenzyme F430 join forces to help mediate the final two stages in methanogenesis and are responsible for the production of over a billion tons of methane gas every year [8].
Heme b is the prosthetic group of hemoglobin and myoglobin that is involved in the transport of oxygen [9]. Different forms of heme are integral components of cytochromes and are involved in electron transport chains associated with energy recovery. These are found in almost all forms of life [10]. Heme b is also a cofactor of many enzymes including catalases, peroxidases, and cytochrome P450, which are involved in detoxification processes [11, 12]. The molecule is also part of gas sensing systems, can serve as a source of iron, act as transcriptional regulator or be involved in the control of pathogenicity [13].
The lesser-known modified tetrapyrroles include heme d 1 and siroheme. The heme-containing cytochrome cd 1 is the only known protein harboring the cofactor heme d 1 and one of the two enzymes responsible for the reduction of nitrite to nitric oxide in denitrifying bacteria [14]. Siroheme, an iron-containing isobacteriochlorin, is the cofactor of assimilatory sulfite and nitrite reductases for which the reaction products are sulfide and ammonium respectively [15].
Structure of heme
Hemes and heme d 1 are very different molecules. Hemes (which include heme a, b, c, and o) are iron-porphyrins, whereas heme d 1 is not actually a heme at all, but rather is an iron-containing dioxo-isobacteriochlorin. True hemes are derivatives of iron-protoporphyrin IX. The iron coordinated in the heme macrocycle can occur in two different oxidation states, FeII or FeIII, and fine tuning to the redox potential of the cofactor is achieved by changes to its environment, both axial ligands provided by the protein, and the general chemical properties of the heme pocket in the protein. This contributes greatly towards the functional diversity of the heme cofactor [16].
The heme macrocycle displays a system of fully conjugated double bonds causing heme to be photochemically active giving rise to characteristic absorption patterns in the visible spectrum and its strong coloration. The properties of light absorption are altered by relocation of the double bonds, which is achieved through substitutions on the ring and are also dependent on the protein the cofactor is embedded in [16, 17]. Further variations in functionality arise from the type of substituents on the pyrrole rings and interaction of the cofactor as heme can be covalently attached to the protein such is the case for cytochrome c. Hemes are classified according to their modifications relative to heme b. An overview of the substituents of hemes a, b, c, and o and their positions of modification on the macrocycle is shown in Fig. 2a; [17, 18]. Heme c differs from heme b through its covalent attachment to a protein through thioether bonds that are formed between the cysteines of the protein and the two vinyl groups of the heme [18]. The cysteine residues involved in heme binding follow a conserved CXXCH binding motif. Hemes a and o differ in that they both have farnesyl additions to ring A and heme a additionally a formyl group at ring D [19].
Fig. 2.
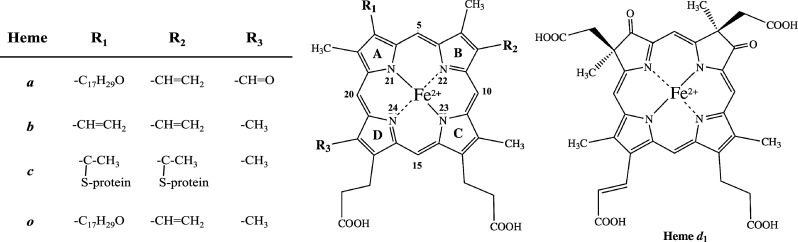
Structures of biological important variants of heme [17] and the dioxo-isobacteriochlorin heme d 1. a Ligands at different residues on the heme macrocycle are donated R 1, R 2, and R 3 and listed on the left. In archaea, the side chains at R1 of heme a and o can have longer groups [171]. The pyrrole rings are labeled A–D in a clockwise direction, the bridging carbon atoms are numbered 5, 10, 15 and 20, and the nitrogens 21–24 according to IUPAC nomenclature. b In comparison, the dioxo-isobacteriochlorin heme d 1
Heme not only plays an important role as a prosthetic group to many proteins processes but it also represents an important iron repository. Pathogens that do not synthesize this cofactor themselves have evolved intricate systems for heme scavenging from their host as this is essential for their survival [13, 20] and some of these processes represent important drug targets. The photodynamic properties of tetrapyrroles, especially the porphyrins, have been utilized in medicine through the use of photodynamic therapy. This application is based on the production of reactive oxygen species (ROS) from molecular oxygen after exposure of the porphyrin to light [21]. ROS are short-lived species that have a destructive effect on their close environment and for that reason the process has been used to treat certain cancers [22] and microbial infections [23], and has also been adapted for herbicidal and insecticidal activities [21].
Structure and function of heme d1
The structure of heme d 1 was solved in 1985 by NMR and UV–Vis spectroscopy [24] and further confirmed through a total synthesis [24–26]. From the experimental evidence it became clear that heme d 1 is a dioxo-isobacteriochlorin, with a number of structural features distinguishing it from other modified tetrapyrroles (Fig. 2b). Compared to heme b, the vinyl groups at positions C3 and C8 are replaced by electron withdrawing carbonyl groups and positions C2 and C7 carry acetate side chains as well as methyl groups leading to the saturation of pyrrole rings A and B. The final difference is the presence of an acrylate side chain at position C17 at ring D replacing the propionic acid side chain at this position in heme b.
Heme d 1 plays a key role in the function of cytochrome cd 1 nitrite reductase and thereby contributes to the global nitrogen cycle. Four sequential reactions are responsible for the stepwise reduction of nitrate (NO3 −) to dinitrogen gas (N2). Denitrification, i.e., complete reduction of nitrate to nitrogen via the gaseous intermediates nitric oxide and nitrous oxide, is a respiratory process used by many bacteria under the condition of low oxygen or complete anaerobiosis [27, 28]. The enzymes involved in the denitrification process are coupled to respiratory electron transport chains and the intermediate nitrogen-oxide(s) serve as the terminal electron acceptors. This is the only biological process by which the soil-fixed nitrogen is returned as gas to the atmosphere. Thus, denitrification is commonly used for removing excess nitrate/nitrite from sewage, groundwater, and wastewater [29].
During the denitrification process, the first committed step involves the conversion of nitrite to nitric oxide and water that is catalyzed by dissimilatory nitrite reductases. There are two main dissimilatory nitrite reductases, a heme-containing cytochrome cd 1 nitrite reductase (NirS) and a copper-containing nitrite reductase (NirK). Their presence in different bacterial species is mutually exclusive [30]. In fact, the biological function of heme d 1 is exclusive to cytochrome cd 1 nitrite reductase, NirS. Well-studied denitrifiers include Pseudomonas aeruginosa, Paracoccus denitrificans, Paracoccus pantotrophus, Roseobactor denitrificans, and Pseudomonas stutzeri, all of which contain NirS.
The structure of the periplasmic cytochrome cd 1 nitrite reductase from P. pantotrophus was first published in 1995 [31], revealing that each monomer consists of two domains. The smaller, N-terminal domain, binds a c-type heme and serves as the center for accepting electrons from donor protein(s) [32]. The larger C-terminal domain carries the non-covalently attached heme d 1 that acts as the site of nitrite reduction. Both the bacterial source and the oxidized/reduced state of NirS, together with axial heme ligands in cytochrome cd 1 nitrite reductase, affect its biochemical and spectroscopic properties [33].
Due to the presence of the carbonyl side chains, the ferric/ferrous redox couple potential of heme d 1 should be more positive than that of other iron-porphyrins, iron-chlorins, and isobacteriochlorins. A value of +175 mV has been estimated in a variant form of cytochrome cd 1 in which the heme c potential was perturbed by mutation to be well removed from the value for the heme d 1 and any cooperativity between the potentials had been removed [34]. This value is higher than that normally found for a heme b in proteins, but there are examples of similar or even greater positive values found for heme b in proteins. Thus, although the redox potential of the heme d 1 means that it is suitable to receive electrons that pass through the bc 1 complex and is also tuned to permit the reduction of nitrite to nitric oxide but not to ammonium, the redox potential alone cannot be the explanation for the occurrence of the structural features of heme d 1 in this enzyme. A variety of investigations over recent years have shown that the reduced form of the heme d 1 has both a higher affinity for the anionic substrate nitrite and a lower affinity for the product nitric oxide than would have been expected by extrapolation from the properties of ferric b-type heme which is the form, relative to the ferrous species, with a higher affinity for nitrite and lower affinity for nitric oxide. What this means for the cytochrome cd 1 mechanism is that the nitrite probably binds to the fully reduced form whereupon reduction of nitrite to give nitric oxide bound to ferric heme d 1 is followed by electron transfer from the c-type heme. Thus, the ferric heme d 1 NO adduct is reduced to the ferrous state and then NO is released. This description explains why the enzyme needs to be able to hold two electrons on each polypeptide chain for a one-electron reaction, but does not itself explain exactly why the unusual substituents are present on the heme d 1. Some insight into this has been provided by an advanced biophysical study [35] but the description is not complete.
Overview of heme biosyntheses
The biosynthesis of heme can be divided into three major sections: (1) The formation of the tetrapyrrole building block 5-aminolevulinic acid (ALA), (2) the synthesis of the first macrocyclic intermediate uro’gen III, and (3) the decoration of the ring. In most organisms, heme is synthesized via a highly conserved pathway that is initiated with the synthesis of the first common metabolite ALA. This small, five-carbon aminoketone is the precursor to all tetrapyrroles. There are two distinct and unrelated routes leading to ALA formation. The first pathway to be described is termed C-4 or ‘Shemin pathway’, which starts from glycine and succinyl coenzyme A and is found in animals, fungi, and the α-group of proteobacteria [36, 37]. The second route for ALA synthesis is referred to as the C-5 pathway, and is unique in that it utilizes glutamyl tRNA as a substrate for something other than protein synthesis. The C5 pathway is present in plants, most bacteria, and archaea [38, 39]. Organisms that make tetrapyrroles operate one or other of these routes although it has been shown in the photosynthetic organism Euglena gracilis that both pathways are functional [40, 41].
The middle section of heme synthesis involves the construction of the tetrapyrrole framework upon which all modified tetrapyrroles are derived. These three steps are found in all organisms that make tetrapyrrole-derivatives and involve the incorporation of eight molecules of ALA into the first macrocyclic intermediate, uro’gen III. In brief, this is achieved by the condensation of two molecules of ALA into the pyrrole porphobilinogen (PBG), four of which are oligomerized to the linear pre-uroporphyrinogen (also known as hydroxymethylbilane). Ring closure and inversion of the terminal D ring of the bilane results in the formation of the type III isomer of uro’gen III (Fig. 3). This is the only unsymmetrical isomer of uro’gen, which may have been selected as the template for tetrapyrrole synthesis since it provides a molecular spatial handle for further specific transformation of the molecule.
Fig. 3.
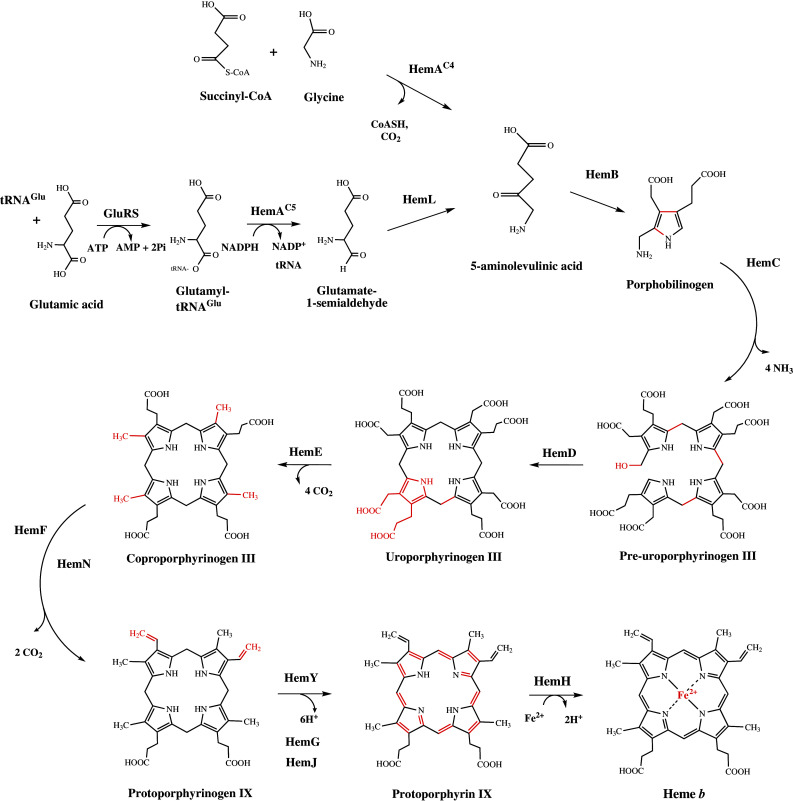
Overview of the classical heme biosynthesis pathway
The final stage of heme synthesis involves modifications to some of the side chains of the macrocycle, followed by aromatization and iron insertion to give protoheme or heme b. The net effect is to make the macrocycle more hydrophobic, presumably allowing the molecule to integrate more easily into proteinaceous environments. The initial steps of the macrocycle modification result in the decarboxylation of the acetate side chains (giving coproporphyrinogen III), which is followed by the oxidative decarboxylation of the ‘northern’ propionate side chains (on rings A and B) to give protoporphyrinogen IX. The removal of six electrons and six protons from the macrocycle introduces aromaticity generates planarity and color, and is a prerequisite to the chelation of iron that produces protoheme. It had been thought that although this part of the pathway proceeded via the same intermediates, there was some variation in the nature of the enzymes that catalyzed the various transformations. The reason for the variation in the nature of the enzymes reflects the fact that some organisms utilize specific aerobic or anaerobic pathways for heme synthesis.
Amongst Prokaryotes, there is a higher degree of variation along the later steps of the pathway [42] that imply an altogether different pathway for heme synthesis. For instance, in sulfate-reducing bacteria, as well as some archaea, orthologues to any of the enzymes responsible for the conversion of uro’gen III to heme are completely absent. Indeed, recently it has been shown that the pathway proceeds via the intermediate precorrin-2, a bis-methylated derivative of uro’gen III, which is also a precursor to cobalamin, cofactor F430, siroheme, and heme d 1. It has therefore been referred to as the alternative heme biosynthesis pathway and the enzymes involved have been given the abbreviation Ahb [43, 44]. In light of the discovery of an alternative heme biosynthetic pathway, which will be discussed later, the two routes for heme synthesis will be referred to as the ‘classic’ and ‘alternative’ pathways. Along the classical pathway, ALA represents the exclusive source of all the carbon and nitrogen atoms that are integrated into heme. An overview is given in Fig. 3.
In Eukaryotes, the enzymes involved in heme biosynthesis are highly conserved members of the classic pathway and are well characterized. In humans, enzyme dysfunction between ALA and heme gives rise to a series of metabolic disorders called the porphyrias. This is an umbrella name that covers one of seven separate disorders that have a range of symptoms that are dependent upon which biosynthetic step is affected. The symptoms include neurological problems, possibly due to the accumulation of ALA, and photosensitivity due to the photodynamic properties of accumulated porphyrins.
ALA syntheses
C-4 or Shemin pathway
In the C-4 pathway, the synthesis of ALA is achieved in a single step through the condensation of glycine and succinyl-CoA. The requirement of glycine for porphyrin synthesis was shown by Shemin through digestion of 15N-labeled glycine, which resulted in the isotopic labeling of his own heme [36]. The second source of carbon atoms, succinyl CoA, was identified through labeling studies using radioactive acetate [45]. The enzyme mediating this condensation reaction is the pyridoxal-5′-phosphate (PLP) dependent 5-aminolevulinic acid synthase (ALAS), encoded by hemA C4, which was first isolated from Rhodobacter sphaeroides [37]. The catalytic cycle and stereochemistry of the enzyme have been well studied. ALAS is highly specific for its substrate glycine and does not recognize any other amino acid [46]. After the binding of glycine to the enzyme, succinyl-CoA is incorporated with the release of coenzyme A and CO2 prior to the release of the product ALA [47]. The first and only crystal structure of a full-length ALAS has been solved from the bacterium Rhodobacter capsulatus [48]. The protein forms a homodimer with each monomer consisting of an N-terminal-, catalytic-, and C-terminal domain. The homodimer symmetrically binds two molecules of the cofactor PLP through a lysine residue that is conserved in all ALASs.
C-5 pathway
It took another 25 years before the ‘C5 pathway’ was discovered. In contrast to the C-4 pathway here, the initial substrate is not glycine but glutamyl tRNA. It was found that 14C labeled glutamate, glutamine, and α-ketoglutarate were incorporated into ALA in plants and a number of bacteria [38]. Subsequently, it was shown that it is the intact carbon skeleton of glutamate that is incorporated into ALA [49]. The C-5 route is a more complex process requiring the action of three separate enzymes.
During the first reaction, glutamyl-tRNA synthetase (GluRS), encoded by gltX, charges a glutamate-accepting tRNA (tRNAGlu) with glutamate in an ATP-dependent reaction [10, 50]. This reaction proceeds in the same manner as observed in protein synthesis. In the following step, glutamyl-tRNA reductase (GluTR), encoded by hemA C5, catalyzes the reduction of the glutamic acid residue of the tRNAGlu to glutamate-1-semialdehyde (GSA) [10, 51, 52]. This reaction is an NADPH-dependent reaction, which with the role of the tRNA, has no parallel in any other metabolic process. The third and final reaction is the transamination of GSA into ALA catalyzed by the PLP-dependent glutamate-1-semialdehyde-2,1-aminomutase (GSAM), encoded by hemL [10, 53, 54].
GSA is a highly reactive aldehyde. In order to ensure efficient synthesis of ALA without any loss of substrate, substrate channeling of GSA between GluTR and GSAM has been suggested. The structures of GluTR [55] and GSAM [54] have been solved and analysis revealed indeed structural complementarity leading to the model of a GluTR/GSAM complex [56]. Existence of this complex could further be confirmed by showing the interaction of the two enzymes in vivo [57, 58]. Substrate channeling has also been suggested to occur during the synthesis of cobalamin, where some of the intermediates are highly sensitive to oxygen [59]. It is further interesting to note that GSAM is structurally related to ALAS, suggesting that ALAS may have evolved from GSAM [56].
The biosynthesis of uroporphyrinogen III
The last common intermediate, and the first branch point in the biosynthesis of all modified tetrapyrroles, is uro’gen III (Fig. 1). The first macrocyclic intermediate of the pathway is made from eight molecules of ALA, which involves the action of three different enzymes.
The first enzyme ALA dehydratase (ALAD) or porphobilinogen synthase (PBGS), encoded by hemB, condenses two molecules of ALA asymmetrically to yield the pyrrole derivative PBG [60]. Most ALADs require metals to be fully functional though the existence of metal-independent enzymes has been reported [61]. ALADs share a high degree of sequence similarity although enzymes from different organisms display differential metal dependency. In yeast and mammals, the enzyme contains two zinc ions [62] while the enzyme from bacteria, such as Escherichia coli, contains one active site zinc ion but is further stimulated by magnesium [63]. Bacteria such as Pseudomonas aeruginosa require magnesium for their activity [64] and the same is the case for ALADs from plants [65]. Structural studies on the enzymes from yeast [66] and E. coli [67] have helped dissect the role of the two active site lysines that assist in the binding and catalysis of the ALA molecules through a Knorr type condensation. The quaternary structure reveals that the enzyme generally forms homo-octamers, although the reason for such a large macromolecular assembly is not clear.
In the proceeding reaction, four molecules of PBG are linked consecutively by porphobilinogen deaminase (PBGD or hydroxymethylbilane synthase) to produce the linear tetrapyrrole pre-uroporphyrinogen (or hydroxymethylbilane) [68]. This enzyme, which is encoded by hemC, uses a unique dipyrromethane cofactor that is itself derived from two molecules of PBG [69, 70]. The covalently linked cofactor serves as a primer for the oligomerization of four PBG molecules, which proceeds in a sequential fashion. Starting with ring A, the subsequent addition of the remaining units (B–C–D) to the growing polypyrrole chain results in the formation of a protein-bound linear hexapyrrole with four molecules of ammonia being released concomitantly [68]. After the addition of ring D, the tetrapyrrole is cleaved from the cofactor and pre-uroporphyrinogen is released leaving reactivated enzyme [71]. The structure of the enzyme reveals how the cofactor is tethered via a thio-ether linkage to a conserved cysteine residue within the active site that is formed between the three domains of this monomeric enzyme [72].
The final reaction is catalyzed by uro’gen III synthase (UROS), encoded by hemD. The enzyme mediates the inversion of ring D of the linear pre-uroporphyrinogen followed by closure of the macrocyclic ring through formation of a link between the first (ring A) and fourth (ring D) pyrrole units generating uro’gen III [73].
In the absence of UROS, pre-uroporphyrinogen spontaneously cyclizes to the isomer uroporphyrinogen I. Uro’gen I is the symmetrical isomer of uro’gen III as ring D is not inverted. Even though uro’gen I is not further involved in the pathway of tetrapyrrole biosynthesis [74], its oxidized form, uroporphyrin I, is found as a cofactor in the rubredoxin oxidoreductase of Desulfovibrio gigas, a terminal oxidase involved in reduction of oxygen into water [75]. The structure of the uro’gen III synthase has been solved, revealing a bi-domain structure. The enzyme has comparatively few conserved residues and the mechanism of ring inversion is still not fully understood, even though an enzyme–product complex has been determined by protein crystallography [76]. The most likely mechanism for this remarkable reaction involves a spiro intermediate, especially as a spiro-lactam analogue has been shown to act as a competitive inhibitor.
Completion of heme synthesis in the classical pathway
Uro’gen III represents the branchpoint at which the classic and alternative heme biosynthetic pathways diverge. Along the classical route, heme is produced through modifications of the side chains of uro’gen III, followed by oxidation of the macrocycle and is completed by the insertion of iron.
The first reaction starts with the successive decarboxylation of the four acetate side chains of uro’gen III in clockwise direction starting at ring D, followed by A, B, and C, forming coproporphyrinogen III [77]. This reaction is catalyzed by uro’gen decarboxylase (UROD), which is encoded by hemE, and releases four molecules of CO2 [78]. UROD is an unusual decarboxylase, as it does not require any cofactor for its activity but instead employs the pyrrole rings as electron sinks to help facilitate catalysis. The structure of this homodimeric enzyme shows how the substrate is accommodated within the active site and suggests that the substrate has to leave the site and re-orientate itself to allow for each subsequent decarboxylation event [79, 80].
In the next step, the two propionate side chains of coproporphyrinogen III on rings A and B are decarboxylated in an oxidative manner to yield vinyl groups under the release of two molecules of CO2, thereby generating protoporphyrinogen IX. This reaction is mediated by one of two structurally and mechanistically unrelated coproporphyrinogen oxidases (CPO) [10]. The distinction is made on whether molecular oxygen is required as terminal electron acceptor for the oxidative decarboxylation, which proceeds via the same stereochemistry in both cases. The oxygen-dependent enzyme HemF is mainly found in Eukaryotes [42], and the oxygen-independent HemN is present in most bacteria [78].
HemF is metal-independent [81], and through the formation of the reaction intermediate harderoporphyrinogen decarboxylates the propionate at ring A prior to that on ring B [82–84]. The structures of Saccharomyces cerevisiae [85] and human [86] HemF could only be obtained in the absence of tetrapyrrole. However, through mutagenesis study of the human CPO it has been suggested that one highly conserved aspartate and two arginine residues play an essential role in substrate binding and catalysis [87]. So far, only putative reaction mechanisms for HemF have been proposed [88, 89].
Due to similarities to enzymes involved in the alternative heme and heme d 1 synthesis, the enzyme HemN is discussed in greater detail. HemN is a radical S-adenosyl-l-methionine (SAM) enzyme and was the first of this class to have its structure determined [90]. Radical SAM enzymes catalyze a diverse range of biochemical transformations by activation and functionalization of unreactive C–H bonds [91, 92]. They require SAM and a reductant for their activity and are generally involved in anaerobic processes. Enzymes of this family contain an [4Fe–4S] center in which three iron atoms are coordinated by three conserved cysteine residues following a CX3CX2C motif [93]. The fourth iron atom is ligated by one molecule of SAM. During one reaction cycle, the iron sulfur cluster is reduced by a reductase. The electron is transferred onto SAM, which is then homolytically cleaved, generating a [4Fe–4S]2+-methionine intermediate and a 5′ deoxyadenosyl radical, which can subsequently abstract a hydrogen atom from the substrate. The mechanism of the reaction has been determined through isotopic labeling revealing that after formation of the highly reactive 5′ deoxyadenosyl radical, a hydrogen atom is abstracted from the propionate side chain of the substrate forming a 5′-deoxyadenosine and an allylic substrate radical initiating the elimination of a CO2 [94]. For HemN, the reductase and final electron acceptor have not been identified, although it was found that two molecules of SAM are cleaved during one reaction cycle.
It is worth noting that some annotations for HemN enzymes actually encode proteins not directly involved in heme biosynthesis. The protein present in bacteria, plants, and animals has therefore been named HemW [95]. For HemW from Lactococcus lactis, which has ~50 % similarity to E. coli HemN, it was demonstrated that the protein has no CPO activity but binds heme and was therefore proposed to be involved in heme transport [95].
The penultimate step of heme biosynthesis, the oxidation of protoporphyrinogen IX into protoporphyrin IX, gives rise to a fully conjugated, aromatic and planar macrocycle. During the reaction, which is catalyzed by protoporphyrinogen IX oxidase (PPO), six electrons and six protons are abstracted (from two of the four imino groups of the pyrrole and from the four bridging carbons C5, C10, C15, and C20) [78]. So far, three distinct enzymes have been identified to catalyze this reaction, including those encoded by hemY [96], hemG [97], and hemJ [98].
HemY is a flavo-dinucleotide-dependent enzyme utilizing molecular oxygen as terminal acceptor for the electrons abstracted from protoporphyrinogen IX resulting in the generation of H2O2. In most organisms, HemY is membrane-associated (bound to the inner mitochondrial membrane in Eukaryotes and to the cytoplasmic membrane in Prokaryotes) [96, 99] but was also found to be soluble in the cytoplasm as in Bacillus subtilis [100, 101]. The structures of several HemYs have been solved by protein crystallography, revealing how the flavin binds in proximity to the porphyrinogen binding site [102–104]. It is not clear if the mechanism involves reorientation of the substrate during the reaction to help facilitate the removal of the protons and electrons.
In enteric γ-proteobacteria such as E. coli and Salmonella enterica, HemY is replaced by the flavo-mononucleotide-dependent HemG [105]. The enzyme has been reported to couple heme biosynthesis to the electron transport chains for energy generation [106]. Here, the six-electron abstraction does not result in the production of H2O2 but is coupled to ATP generation, depending on availability, employing oxygen, fumarate, or nitrate as terminal electron acceptors.
The third PPO that has been reported is encoded by hemJ. The gene was identified through comparative genomic analysis and occurs in the genomes of almost all bacteria, which produce heme via the classical pathway but whose genomes lack a hemY or a hemG [98]. Its role as a PPO was verified through complementation of a hemJ mutant in Actinobacter baylyi. It has been suggested that the enzyme could employ oxygen as a final electron acceptor [107] though further experimental evidence is needed to support this view. So far, a functional enzyme has not been recombinantly produced and purified and there is evidence that the enzyme is dependent on an additional component for activity [98].
Heme biosynthesis along the classical pathway is completed by the insertion of Fe2+ into protoporphyrin IX generating heme b, the precursor to hemes a, c, and o. During the reaction catalyzed by ferrochelatase or HemH [108], the planar macrocycle has to be distorted by the enzyme to allow for metal chelation. In Eukaryotes, HemH is associated with the matrix side of the inner mitochondrial membrane and in many cases is found to contain a [2Fe–2S] center [109]. Membrane-association and the presence of an iron–sulfur cluster are also observed in some prokaryotic ferrochelatases [108]. Nevertheless, many ferrochelatases do not contain iron–sulfur clusters and can be soluble, such as the enzyme from B. subtilis [110]. The exact function of the Fe–S center is unknown. The structures of the B. subtilis, S. cerevisiae, and human enzymes have revealed that the enzymes promote chelation through a distortion of the porphyrin ring, although there is a debate as to the route the metal ion follows to the active site. The ferrochelatases appear to share a common structure with chelatases associated with some siroheme synthases and the cobalatochelatase of the anaerobic cobalamin pathway, indicating that they are all derived from a common ancestral source [111].
It has been reported that in bacteria employing the soluble form of HemY heme synthesis is dependent on the action of an additional protein, HemQ [112]. The exact role of HemQ remains unclear, though it appears to be essential in these organisms as it is required for HemY and HemH activity.
Biosynthesis of siroheme
Although this review focuses on the biosynthesis of heme and heme d 1, an understanding of the biosynthesis of siroheme is required, as this pathway and its intermediates are key to understanding the alternative heme biosynthesis pathway.
In comparison to other tetrapyrroles, the synthesis of siroheme is relatively simple and proceeds from uro’gen III in a three-step process (Fig. 4): (1) addition of two methyl groups from SAM at positions C2 and C7 forming precorrin-2, (2) oxidation of precorrin-2 to sirohydrochlorin, and (3) chelation of iron to form siroheme. Different organisms can use a variety of enzymes for the synthesis of siroheme. Bacillus megaterium contains three separate enzymes for the formation of siroheme, a SAM-dependent uro’gen III methyltransferase (SUMT) SirA, a precorrin-2 dehydrogenase SirC and a ferrochelatase SirB (an enzyme that shares structural similarity to HemH, as described above) [113]. Saccharomyces cerevisiae uses only two enzymes, the SUMT Met1p and the bi-functional precorrin-2 dehydrogenase and ferrochelatase Met8p [114]. Escherichia coli on the other hand has one multifunctional enzyme, CysG, that catalyzes all three steps from uro’gen III to siroheme [115]. CysG contains two domains termed CysGA and CysGB. CysGA is homologous to SirA and Met1p, and CysGB is responsible for precorrin-2 decarboxylation and iron chelation (Fig. 4).
Fig. 4.
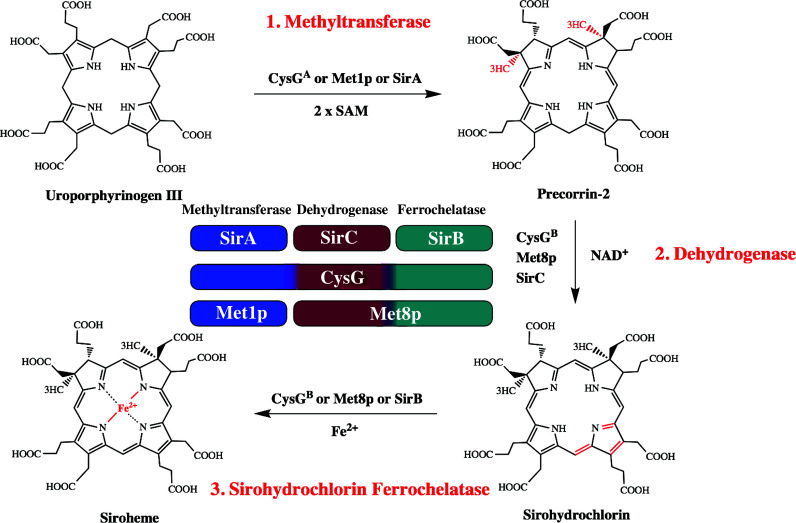
Biosynthesis of nature’s simplest tetrapyrrole, siroheme. Siroheme is synthesized from the common tetrapyrrole precursor uroporphyrinogen III in three consecutive enzymatic steps via the intermediates precorrin-2 and sirohydrochlorin. In E. coli, multifunctional siroheme synthase (CysG) catalyzes all the three reactions, which include the SAM-dependent methylation of uroporphyrinogen III, NAD+-dependent dehydrogenation of precorrin-2 and the iron insertion into sirohydrochlorin. In yeast, the methyltransferase Met1p and the bifunctional dehydrogenase/ferrochelatase Met8p are responsible for synthesis of siroheme and in B. megaterium three monofunctional enzymes exist for siroheme formation, SirA (methyltransferase), SirC (dehydrogenase), and SirB (chelatase). The center shows a functional alignment that does not represent the amino acid sequence homologies
Interestingly, the siroheme pathway shares intermediates of cobalamin synthesis with the aerobic and anaerobic pathways branching at different points. Precorrin-2 represents the branch point for the aerobic pathway where cobalamin synthesis proceeds with the production of precorrin-3 by CobI [116]. For the anaerobic pathway precorrin-2 is converted to sirohydrochlorin and chelation of cobalt by CbiK or CbiX directs the intermediate to cobalamin production instead of siroheme [113].
In some organisms capable of heme d 1 synthesis, production of siroheme is yet to be characterized. Almost all of the organisms that make heme via the alternative pathway contain siroheme-dependent sulfite and nitrite reductases and as such contain an intact siroheme synthesis pathway. The nir gene cluster contains a SUMT (NirE) but lacks genes encoding enzymes capable of converting precorrin-2 to siroheme. This may be due to the requirement of precorrin-2 in other pathways as when not in denitrifying conditions transcription of NirE would not occur, therefore making precorrin-2 unavailable. In fact, denitrifying bacteria appear to contain multiple SUMTs, suggesting that each tetrapyrrole pathway has its own dedicated SUMT with the other genes (i.e., homologues of sirB and sirC, cysG, or met8p) found elsewhere in the genome. Some denitrifiers contain a copy of cysG, which is indispensable for heme d 1 synthesis in Ps. aeruginosa [117]. All of the denitrifiers also have one or more copies of chelatase genes (e.g., sirB or cbiX), which may be involved in iron chelation in these organisms, although this is yet to be verified.
As with heme d 1-synthesizing organisms, complete synthesis of siroheme in most bacteria and archaea that synthesize heme via the alternative pathway is yet to be unraveled. Many archaeal genomes appear to have a SUMT, although annotations are varied between cobA, cysG-1, cysG-2, cysG, uroM, or hemX. Likewise, the gene encoding precorrin-2 dehydrogenase is referred to as sirC, hemX, cysG, or cysG1 [118]. A unique siroheme ferrochelatase appears to be absent from archaeal genomes but it is conceivable that the short cobaltochlatase CbiXS found in archaea may have a duel role in both iron and cobalt chelation into sirohydrochlorin. Indeed it was demonstrated that CbiXS from M. barkeri and Methanobacter thermoautotrophicum are capable of chelating iron into sirohydrochlorin, although at a lower rate to that of cobalt insertion [119]. Desulfovibrio vulgaris possesses a bi-functional uro’gen III synthase/methyltransferase (HemD-CobA) and a precorrin-2 dehydrogenase and is therefore capable of synthesizing sirohydrochlorin [120]. Again, a unique siroheme ferrochelatase is absent in the genome but the organism contains two putative CbiK cobaltochelatases (CbiKC and CbiKP), which are capable of chelating both cobalt and iron into sirohydrochlorin [121]. The genes encoding these proteins are not restricted to D. vulgaris but are in fact present in the genomes of other deltaproteobacteria including the groups Desulfovibrio, Desulfobulbus, Desulfatibacillum, and Desulfobacterium [122].
Biosynthesis of heme d1
The elucidation of the biosynthesis of heme d 1 has proven challenging due to difficulties in identifying the pathway intermediates and their enzymes. These proteins function under anaerobic conditions and both their potential substrates and the products can be unstable in laboratory conditions due to temperature, light, and oxygen sensitivity.
Heme d 1 is structurally closely related to siroheme (Fig. 1), as it carries the same combination of methyl and acetate side chains at positions C2 and C7. In siroheme, the methyl groups are derived from SAM [123]. Indeed, stable isotope labeling experiments in Ps. aeruginosa demonstrated that this is also the case in heme d 1 [124]. This result suggested that the synthesis of heme d 1 may proceed via precorrin-2 after which possibly five enzymatic steps may be required for the formation of heme d 1 including; (1) the conversion of propionate side chains at positions C3 and C8 to keto groups, (2) the introduction of a double bond into the propionate side chain attached to C17, (3) the oxidation of the tetrapyrrole macrocycle to yield an isobacteriochlorin, (4) the decarboxylation of acetate side chains at positions C12 and C18 in the southern half of the tetrapyrrole to give methyl groups, and finally, (5) the insertion of iron into the macrocycle. The order of these chemical transformations was difficult to predict, although it was thought that metal ion insertion would be the last step during biosynthesis, owing to the potential cyto-toxicity of the metal-chelated intermediates and the analogy to heme biosynthesis where the ferrochelatase is the terminal enzyme. After the biosynthesis of heme d 1 is completed, the molecule has to be transported across the membrane to the periplasm where it, along with heme c, is inserted into apo-cytochrome cd 1 [125].
Analysis of the of nirS loci from diverse species of denitrifying bacteria and some archaea revealed the presence of a set of contiguous genes downstream of nirS that were found to be essential for heme d 1 biogenesis [126, 127]. Depending on the organism, these loci consist of eight or nine open reading frames (ORFs). In Ps. stutzeri, the downstream region harbors the genes nirFDLGHJE. In Paracoccus pantotrophus and Paracoccus denitrificans, these genes are organized in the operon nirECFD-LGHJN and their transcription activated in the presence of nitric oxide [14].
Mutagenesis of the nirS downstream regions in Ps. stutzeri and Ps. aeruginosa resulted in the synthesis of semi apo cytochrome cd 1 (i.e., cytochrome cd 1, which lacks heme d 1) [128, 129]. In analogous studies of nirD, nirL, nirG, and nirH mutational analysis in Ps. stutzeri and Ps. aeruginosa, the indispensable nature of nirF, genes during heme d 1 biogenesis has been shown [28, 130]. Paracoccus denitrificans mutant strains were generated by disrupting the genes nirS, nirE, nirC, and nirF and all these mutants strains were reported to be unable to synthesize heme d 1 [131]. In addition, heterologous expression of nirS in organisms lacking cytochrome cd 1 yields an inactive form of NirS without heme d 1 [130]. However, it was reported that an active form of NirS, (i.e., a holo-cytochrome cd 1 with both c and heme d 1) was produced in E. coli when transformed with a plasmid including nirS from P. denitrificans and about 10 kbp of its downstream region [132]; it would be valuable if this work were to be confirmed and extended.
A comparison of the genetic organization of nir clusters from some of the well-studied denitrifiers is shown in Fig. 5. It is worth noting that Ps. stutzeri and Ps. aeruginosa harbor two separate genes nirD and nirL, which are fused encoding a single protein NirDL in Paracoccus species as well as in several other denitrifying bacteria. Many denitrifying bacteria additionally have a nirM gene encoding cytochrome c 551, the physiological electron donor for NirS within the nir cluster. However, in P. denitrificans, NirM is absent but it has been shown that NirS has two periplasmic physiological electron donors; cytochrome c 550 and copper-containing pseudoazurin [32]. In addition, nirC, which is present in the nir cluster of most of these organisms, also encodes a small periplasmic cytochrome c, which in some cases is able to transfer electrons to NirS. However, in the case of Paracoccus species, this appears not to apply in vivo, as a double mutant strain lacking both cytochrome c 550 and pseudoazurin was unable to reduce nitrite, an observation indicating another role for the product of the nirC gene. There was a report that nirC is required for biogenesis of the d 1 heme cofactor in Paracoccus (Van Spanning, pers. comm.) but further work, for example complementation studies, is needed to confirm this proposal.
Fig. 5.
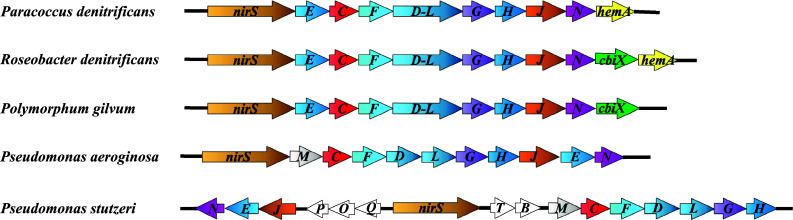
Genetic organization of the nirS loci in different denitrifiers. Clusters were generated from the “Microbial Genome Database for Comparative Analysis” (http://mbgd.genome.ad.jp)
Another interesting observation is the presence of cbiX in the nir gene clusters of Roseobacter denitrificans, Dinoroseobacter shibae, and Polymorphum gilvum. The gene cbiX is also present in P. denitrificans and some other denitrifiers, though elsewhere in the genome, but it is absent in the genomes of Ps. aeruginosa and Ps. stutzeri. This suggests that CbiX could act as a ferrochelatase for iron insertion during the heme d 1 biosynthesis in these organisms.
Biosynthesis of heme d1: structure and role of individual Nir proteins
Unfortunately, there are no reports on the accumulation of heme d 1 precursors in the aforementioned mutant strains. The classical approaches of solving a biosynthetic pathway by blocking the pathway at one stage in order to accumulate intermediates have failed in the case of heme d 1 biosynthesis. Furthermore, until recently, very little was known about the individual roles of the nine Nir proteins (i.e., NirE, NirC, NirF, NirD, NirL, NirG, NirH, NirJ, and NirN) in heme d 1 biogenesis.
Out of all Nir proteins, to date NirE is the most well characterized and shares a very high sequence homology to known SUMTs including the C-terminal region of CysG, CobA (methyltransferase of the cobalamin pathway), SirA, and Met1p [133]. NirE from P. pantotrophus has been shown to catalyze the conversion of uro’gen III to precorrin-2 in vitro; for Ps. aeruginosa, the same has been demonstrated both in vitro and in vivo [14, 134].
As with other SUMTs, NirE from Ps. aeruginosa exhibits substrate inhibition at high uro’gen III concentrations and product inhibition by S-adenosyl-l-homocysteine [134]. In organisms with overlapping tetrapyrrole biosynthesis pathways, such regulation could ensure direction of common intermediates towards the appropriate pathways. However, it is interesting to note that substrate inhibition by uro’gen III is not observed for NirE from P. pantotrophus [14].
Pseudomonas aeruginosa has both CysG and CobA, which carry out the same catalytic activity as NirE. However, the coordinated co-transcription of the nirE gene with the other heme d 1 biosynthesis genes, under denitrifying conditions, has been proposed to ensure that the cells can cope with the increased demands for heme d 1 intended for anaerobic respiration. Therefore, NirE is suggested to be specifically required for heme d 1 biosynthesis in organisms where precorrin-2 is precursor to various other tetrapyrroles. The crystal structure of NirE revealed that the protein is a dimer with a similar active site to that of CysG and CobA suggesting a conserved reaction mechanism [135].
Role of NirC, NirN, and NirF
NirC and NirN are c-type cytochromes that harbor N-terminal Sec signal sequences. Recombinant expression in E. coli demonstrated that both NirN and NirC are targeted to the periplasm via the Sec translocon [14]. Genomic deletion of either nirN or nirC have established that both proteins are not essential for the formation of heme d 1 [14, 136] and several proposals for their physiological role have been made as discussed above for NirC.
NirN can bind heme d 1 at neutral to low pH to produce a stoichiometric complex due to a C-terminal binding domain [14]. Therefore, it has been suggested that the protein could act as a chaperon for newly synthesized heme d 1 in the periplasm before its capture by the semi apo-cytochrome cd 1. This would prevent nonspecific transport out of the cell by porins located in the outer membrane. It has been shown in vitro that heme d 1 can be transferred from Paracoccus NirN to semi apo NirS (i.e., containing the c-type heme only) [14]. It was also proposed that NirN could serve as an electron donor to cytochrome cd 1, a signal transducer element or a catalyst for the degradation of excess heme d 1. NirC can act as an electron donor to both the NirN-heme d 1 complex and to cytochrome cd 1 in vitro [14, 136] though it is unlikely in vivo as the oxidation/reduction potential of NirC is not optimal for the transfer of electrons from the cytochrome bc 1 complex to NirS [14]. Therefore, the physiological roles of NirN and NirC remain unclear, as both are not essential for heme d 1 biogenesis in at least some organisms [14, 28]. There are nevertheless indications that at least under some circumstances the absence of NirN is deleterious. Thus, in a recent study with P. aeruginosa [137], there are indications for production of at least a fraction of a modified version of heme d 1 in the absence of NirN.
NirF is a protein of about 43 kDa and like NirN harbors a C-terminal heme d 1 binding domain though neither NirF nor NirN can function as nitrite reductases [28, 127]. NirF is essential for the production of heme d 1. Addition of purified periplasmic NirF to heme d 1 causes a large shift in the UV–Vis spectrum, confirming binding [125]. Mutagenesis identified an amino acid (His41, P. pantotrophus NirF) as essential for in vitro binding of heme d 1 and in vivo formation of holo-cytochrome cd 1 [125]. However, transfer of heme d 1 to either NirS or NirN could not be shown. Sequence analysis of all known nirF genes has revealed the presence of an N-terminal Sec targeting signal sequence with the exceptions for nirFs from Ps. aeruginosa and Magnetospirillum magneticum. NirF from P. pantotrophus has been confirmed to be periplasmic by both cell fractionation work and signal sequence deletion/replacement studies in vitro as well as in vivo [125]. As the location of NirF is in the periplasm, and given the non-essential nature of the two other known periplasmic proteins, NirC and NirN, it seems sensible to assume that the last step of heme d 1 synthesis is catalyzed in the periplasm. Interestingly, it was previously suggested that NirF may act as a dehydrogenase in the terminal steps of heme d 1 synthesis due to the presence of a glycine-rich region resembling a Rossman fold motif (GXGX2G) [125, 138]. This motif is also found in several dehydrogenases that are involved in the siroheme biosynthesis pathways [133]. However, mutagenesis studies established that the glycine residues are not essential for heme d 1 biosynthesis, so the role of NirF also still remains elusive.
The absence of a periplasmic targeting sequence in NirF from P. aeruginosa has had a disproportionate influence, leading to the widespread belief that NirF is a cytoplasmic protein. A dual location, periplasmic in some organisms and cytoplamsic in others, was rendered unlikely by the finding that a N-terminally truncated form, and thus cytoplasmic form, of P. aeruginosa NirF was unable to complement a Paracoccus mutant that lacked its endogenous NirF protein [139]. Furthermore, a recent study has shown that the globular domain of P. aeruginosa is exposed to the periplasm and that the protein is a lipoprotein, a feature that anchors it to the membrane [137]. Hence, it is now safe to say that NirF is involved in the last step of heme d 1 production, presumably through acting as an enzyme to catalyze a chemical transformation. There is, however, little clue as to what that reaction might be. For the moment, it can be considered that the synthesis of heme d 1 is completed in the periplasm and thus that contrary to expectation it is not heme d 1 itself but rather an immediate precursor that is transported from the cytoplasm to the periplasm.
It is clear that, at the time of writing, the roles of the NirF and NirN proteins are enigmatic, although both have been shown to bind specifically heme d 1 in vitro. A recent study, based mainly on the results of immunoprecipitation studies, has shown that NirS can interact not only with NirN but also with NirF, leading to the conclusion that the optimal final step in formation of holo NirS involves a complex of the three proteins [137].
Role of NirDL, NirG, and NirH
As stated earlier, in several denitrifying bacteria, the genes nirD and nirL are fused to give a single protein. In P. pantotrophus, NirDL (~37 kDa), NirG (~17 kDa), and NirH (~18 kDa) are all cytoplasmic proteins. They share very high sequence homology to each other (~40 % similarity for N-terminal part of NirDL and NirG, and ~42 % similarity for the C-terminal part of NirDL and NirH). Despite the extensive sequence homology, functional redundancy does not exist as all four proteins were shown to be essential for nitrite reductase function [127, 129]. There is also a charge complementarity between these proteins, which suggests that pairs of them might be involved in forming an αβ–α′β′ multimeric protein complex that may participate in symmetrical reactions of heme d 1 biosynthesis such as decarboxylation of acetate side chain at position C12 and C18 or conversion of propionate side chains at C3 and C8 to oxo side chains [129].
Due to the experimental evidence for precorrin-2 being an intermediate in both the siroheme and heme d 1 pathways it, was proposed that sirohydrochlorin might be a potential intermediate during heme d 1 biosynthesis, even though no gene encoding a precorrin-2 dehydrogenase is located in the nir cluster. When total cell lysates of E. coli over-expressing a different combination of Nir proteins (e.g., NirDL or NirDLGH from P. pantotrophus) were incubated with sirohydrochlorin, a new bis-decarboxylated intermediate, 12,18-didecarboxysiroheme (DDSH), was produced [44]. Further analysis revealed that the true substrate for NirDLGH is not sirohydrochlorin but the iron-containing siroheme. All four proteins were shown to be involved in catalyzing a symmetrical decarboxylation at C12 and C18 of siroheme. The details of these decarboxylation reactions were not reported in the study but a probable reaction mechanism for the chemical transformation was discussed (Fig. 6) [44]. This result furthermore explains the lack of a dedicated ferrochelatase encoding gene in the nir cluster.
Fig. 6.

Potential reaction mechanism of the NirDLGH-catalyzed double decarboxylation. The double decarboxylation of siroheme is catalyzed by the NirDLGH and occurs via the formation of an iminium ion. Here, ring D is shown to undergo decarboxylation at position C18 prior to the decarboxylation of the acetate side chain at ring C. The actual order of these transformations is not known
It has been reported that in Ps. stutzeri the maturation of active cytochrome cd 1 requires an intact Tat transport system because a deleterious mutation of tatC, encoding one component of the Tat transport system, leads to the formation of an inactive nitrite reductase that lacks heme d 1 [140]. The Tat (twin-arginine translocation) system is a bacterial protein transport pathway that transports folded proteins across the cytoplasmic membrane into the periplasm. This suggests that at least one protein involved in the biosynthesis/assembly of heme d 1 is translocated to the periplasm via the Tat system. It was also concluded that NirD from Ps. stutzeri is a periplasmic protein, since a twin arginine motif was found in the N-terminal region of NirD sequence [140]. However, homologues in other organisms do not have this sequence motif and now that a function has been assigned to NirD it seems very unlikely that just NirD, and not NirLGH, is periplasmic. Nevertheless, if variants of P. stutzeri lacking a functional Tat system cannot make holo NirS, this result still requires explanation. One possibility is that it may be a secondary consequence of the absence of a functional cytochrome bc 1 complex as export of the Rieske FeS protein depends on the Tat machinery. For example, it was shown that transcription of the nir genes for heme d 1 synthesis required nitric oxide, which presumably binds to the transcription factor nnr (or dnr in some organisms) [34]. Clearly, if electron transfer is blocked because of an inactive bc 1 complex, then the necessary nitric oxide cannot be produced, and thus heme d 1 biosynthesis is also blocked.
The sequences of NirD, NirL, NirG, and NirH reveal similarities to the AsnC/Lrp family of transcriptional regulators. Leucine Responsive Protein (Lrp) homologues are mostly involved in the regulation of genes relating to amino acid metabolism [141]. It was demonstrated that NirL from Heliophilum fasciatum is able to bind DNA, specifically the putative promoter of the nir operon in this organism [142], confirming that NirL is likely to be a transcriptional regulator as previously annotated. As these proteins also have enzymatic functions [44] it suggests they may play both a catalytic and regulatory role in heme d 1 synthesis.
Role of NirJ
NirJ (~45 kDa) is a monomeric cytoplasmic protein [129] harboring two conserved cysteine-based motifs. The N-terminal motif (CX3CX2C) corresponds exactly to the signature motif of the ‘radical SAM’ superfamily such as the aforementioned HemN that is involved in classic heme biosynthesis [143, 144]. The presence of an iron sulfur cluster was confirmed by electron paramagnetic resonance (EPR) spectroscopy [145]. NirJ also contains a C-terminal cysteine-rich motif (CX2CX5C) that is suspected to bind a second iron–sulfur cluster although this was not detected in the EPR analysis [28, 145]. However, it can be difficult to resolve and assign individual centers in proteins with more than one [4Fe–4S] cluster, and the obtained spectra could have been the sum of resonances emerging from two [4Fe–4S] clusters.
The NirJ protein shares a high sequence similarity with radical SAM enzymes, in particular PqqE involved in pyrroloquinoline quinone biosynthesis, MoaA involved in molybdopterin cofactor biosynthesis, and NifB participating in iron-molybdenum cofactor biosynthesis in nitrogenase [28]. All these proteins contain a second, C-terminal cysteine-rich region in addition to their [4Fe–4S] binding N-terminal CX3CX2C motif. The crystal structure of MoaA shows that [4Fe–4S]2+ clusters are bound, both at the N-terminal radical SAM motif and the C-terminal CX2CX12C motif, each with a non-cysteinylated site [146, 147], though the role of the latter cluster is unclear. Most importantly, there is a recent report on the true enzymatic activity of the second [4Fe–4S] cluster from AlbA protein, which is involved in generating the three thioether bonds by radical-based chemistry during subtilosin A maturation [148]. Based on this result and due to the conservation of this second potential [4Fe–4S] binding motif in NirJ sequences, it is possible that both these motifs may be important for the NirJ-catalyzed chemical transformation of the heme d 1 biosynthesis pathway.
It has been suggested that NirJ could catalyze the elimination of propionate side chains at position C3 and C8 of DDSH by activating the alkyl leaving groups in a radical mechanism [44]. However, the source of the oxygen atoms in the resulting carbonyl groups is not known. Interestingly, there are recent examples of radical SAM enzymes with multiple [4Fe–4S] clusters that are involved in two-electron oxidation/dehydrogenation reactions on the organic substrate or on cognate proteins [149–152]. In particular, BtrN, an enzyme involved in the biosynthesis of antibiotic butirosin B, catalyzes the two-electron oxidation of the C3 alcohol of 2-deoxy-scyllo-inosamine to give amino-2-scyllo-inosose [149]. Another interesting aspect of this conversion of an alcohol to a ketone is that the protein was not reported to utilize any of the usual cofactors such as flavin-, pyridine-, or quinone-containing nucleotide metabolites [149]. Therefore, it is possible that NirJ also catalyzes the two-electron oxidation of the propionate side chain at position C17 of DDSH to yield acrylate functionality of heme d 1 molecule in a similar fashion as that of BtrN. However, so far there is no evidence to support the presence of these activities.
Biosynthesis of heme d1: pathway intermediates and their transport to the periplasm
To transform DDSH into heme d 1, at least two chemical steps are required. These steps include (1) the removal of the propionate side chains at positions C3 and C8 of DDSH and their replacement by carbonyl groups and (2) the dehydrogenation of the propionate group at C17 to generate a double bond. The order in which these steps occur is not known, but it is speculated that NirJ carries out the transformation at C3 and C8 of DDSH to generate “pre-heme d 1″, which could then be converted into heme d 1 by the action of NirF [44]. NirF is the only essential periplasmic enzyme during heme d 1 biosynthesis and it has been shown to bind heme d 1. It is also possible that NirJ catalyzes both chemical transformations to convert DDSH to heme d 1 in a novel, yet unidentified radical-based chemistry.
It is still unclear how the intermediate pre-heme d 1 (if it is the di-oxo intermediate that acts as the substrate for NirF in the periplasm) and or heme d 1 is transported across the membrane to reach the site of cytochrome cd 1 assembly. Furthermore, it is not known which enzyme is responsible for the insertion of heme d 1 into semi apo cytochrome cd 1 or if insertion occurs spontaneously in the periplasm.
NirN has been shown to capture heme d 1 and this NirN-heme d 1 complex could transfer the heme d 1 to semi apo-cytochrome cd 1 in vitro [14]. However, the rate at which the NirN-heme d 1 complex is made is faster than the rate at which transfer of heme d 1 to semi apo-cytochrome cd 1 takes place and also NirN is not essential during heme d 1 biosynthesis as shown by mutagenesis study in P. pantotrophus [14]. This suggests that heme d 1 insertion might be a spontaneous process, unless the transfer is catalyzed by either NirF or some other yet unidentified protein under the physiological conditions. NirF has been shown to bind heme d 1 to give a stoichiometric complex, but this complex was unable to transfer the heme d 1 to semi apo-cytochrome cd 1 under the experimental conditions [44]. There are no obvious enzymes/transporters in the genomes of denitrifying bacteria that could be assigned the function of heme d 1 transport from cytoplasm to periplasm.
Discovery of the alternative heme biosynthesis pathway
As discussed earlier, some organisms contain heme but lack the later enzymes of the classical heme pathway that are required to convert uro’gen III into protoheme. The following sections outline the discovery of the alternative heme biosynthesis pathway and the enzymes involved.
Early biochemical evidence for an alternative heme biosynthesis pathway
Evidence for the existence of an alternative heme biosynthesis pathway was discovered some 20 years ago by serendipity [153]. During an experiment to determine redox potentials of cytochrome c3 from the sulfate-reducing bacterium D. vulgaris Miyazaki F, it was noted that labeled methionine was incorporated not only into the protein structure but also into the bound heme prosthetic group. It was found that the methyl groups at positions C2 and C7 in heme c were not derived from ALA, as would be the case for synthesis via the classical pathway, but were in fact from methionine, presumably via SAM. As previously mentioned, the two methyl groups at carbons C2 and C7 of precorrin-2 and sirohydrochlorin are derived from SAM [154] and the same has been demonstrated for the methyl groups at C2 and C7 of heme d 1 [124]. Combined with this knowledge and the fact that sulfate reducers contain siroheme and also sirohydrochlorin, it was considered likely that either precorrin-2 or sirohydrochlorin was an intermediate of the alternative heme biosynthesis pathway.
Further to these initial findings, cultures of D. vulgaris were supplemented with deuterated l-methionine-methyl-d 3 and porphyrins produced were extracted in an attempt to detect labeled intermediates of the proposed alternative pathway [43]. Several compounds containing deuterated methyl groups at positions C2 and C7 were discovered which included sirohydrochlorin, coproporphyrin III, and protoporphyrin IX as well as a previously unknown hexacarboxylic acid termed 12,18-didecarboxysirohydrochlorin. Based on these results, a new pathway was devised deviating from the classical pathway at uro’gen III to precorrin-2, but then later re-joining the classical pathway with the formation of coproporphyrinogen III, which then proceeds to heme. The steps of the alternative pathway starting at uro’gen III were suggested as follows: (1) methylation of C2 and C7 positions of uro’gen III to form precorrin-2, (2) decarboxylation of C12 and C18 positions to form 12,18-didecarboxyprecorrin-2, (3) elimination of acetate groups from C2 and C7 positions to form coproporphyrinogen III, (4) decarboxylation of propionate side chains on C3 and C8 positions to vinyl groups forming protoporphyrinogen IX, (5) oxidation of ring system to form protoporphyrin IX, (6) insertion of ferrous iron to form heme. As aforementioned, iron chelation was accepted to be the final stage of heme synthesis in due to the potential cyto-toxicity of the metal-chelated intermediates. Since the majority of these reactions are homologous to reactions of previously characterized pathways, the search for similar enzymes began.
Genetic evidence for an alternative heme biosynthesis pathway
The constantly growing number of sequenced genomes of bacteria and archaea allowed for detailed genetic analysis of the alternative heme biosynthesis pathway. Several independent studies have been able to show that this pathway is not restricted to sulfate-reducing bacteria as once believed [42, 118, 155].
It was discovered that some bacteria and archaea only contain the genes corresponding to early heme biosynthesis up to uro’gen III (i.e., hemA, hemL, hemB, hemC, and hemD) and lack the necessary genes to convert uro’gen III to heme via the classical pathway (hemE, hemF/N, hemY/G/J, and hemH, Fig. 3) [42, 155]. This information showed that completion of heme biosynthesis in these organisms must proceed via an alternative route which may occur via precorrin-2 and sirohydrochlorin. At the same time, this also meant that the latter stages of the proposed pathway, from coproporphyrinogen III, could not occur as they lacked homologues of the genes mediating these steps. Therefore a unique set of enzymes that catalyzed the conversion of precorrin-2 to heme must exist in these organisms.
The archeon Methanosarcina barkeri was shown to be one of the organisms lacking the latter half of the classical pathway and similar labeling studies were used as with D. vulgaris to analyze the incorporation pattern of deuterated methionine in heme [156]. These experiments revealed that, like D. vulgaris, two methyl groups in heme from M. barkeri are also derived from SAM and not ALA, therefore suggesting that they use the same pathway to synthesize heme.
Further genomic analysis of archaea showed that many species (47 out of 59 studied) only contained the hem genes for the production of uro’gen III and not the late hem genes needed for the completion of heme biosynthesis [118]. It was also noted that the early hem genes were often clustered with genes encoding SUMT and precorrin-2 dehydrogenase enzymes, required for the conversion of uro’gen III to sirohydrochlorin via precorrin-2, supporting the utilization of sirohydrochlorin as a precursor in the alternative heme pathway. Furthermore, it was found that homologues of the nir genes (nirD, nirH, and nirJ) were also present in many of these gene clusters, suggesting involvement in the alternative pathway as only three of 27 of the species containing these genes also contained a potential cytochrome cd 1.
The same observation was made in D. vulgaris, showing it too only contained the hem genes for the production of uro’gen III [120]. Furthermore, it was noted that it contained the necessary genes to convert uro’gen III to sirohydrochlorin and homologues of nirD and nirJ, despite the fact that it too does not produce cytochrome cd 1. As these gene products are not involved with heme d 1 synthesis, they will be subsequently referred to as the ahb genes (alternative heme biosynthesis) A, B, C, and D, which are homologous to nirD, H, and J, respectively (with nirJ being homologous to both ahbC and D) (Fig. 7a).
Fig. 7.

a Representation of genes involved in heme d 1 (nir) and alternative heme (ahb) biosyntheses and their homology. b The alternative heme and proposed heme d 1 biosynthesis pathways starting from siroheme (chemical modifications are highlighted in red)
Given the homology to the heme d 1 biosynthesis enzymes and their previously suggested functions [129], a new pathway was proposed. Decarboxylation of the acetate side chains at C12 and C18 positions of precorrin-2 were attributed to AhbA and AhbB, as was suggested for NirDLGH. Next, the removal of acetate side chains at carbons C2 and C7 was assigned to AhbC; this had been suggested previously in regard to a NirJ-like protein from D. vulgaris [120], which is now known to be AhbC in this organism [44]. Finally, a reaction not required for heme d 1 synthesis, the oxidative decarboxylation of propionate side chains on carbons C3 and C8 to vinyl groups, was suggested to be catalyzed by AhbD. This reaction occurs in the classical heme biosynthesis pathway and is catalyzed by HemF or HemN. AhbD is from the radical SAM enzyme family, as is HemN, and it was therefore a suitable candidate for the formation of the vinyl groups.
Although potential biosynthesis enzymes had been highlighted and their function suggested, no activity studies had been performed and no obvious ferrochelatase was known to be involved in the alternative pathway, which was believed to be the terminal step in analogy to the classical heme synthesis pathway [10].
Finding the missing link in the alternative heme biosynthesis pathway
As was the case for the Shemin/C5 ALA synthesis routes, the alternative heme biosynthesis pathway was discovered and characterized some 20 years after its counterpart. For many years it had been proposed that sirohydrochlorin was the substrate for the alternative pathway but recently the true substrate for the Ahb enzymes was discovered and a surprising link between the alternative heme and the heme d 1 biosynthesis pathways was found.
As mentioned earlier, the substrate for the first step in heme d 1 synthesis is in fact siroheme and not sirohydrochlorin as previously thought [44]. The heme d 1 pathway enzymes NirDLGH unexpectedly formed DDSH from siroheme. In the same study, incubations of AhbA and B from D. vulgaris and D. desulfuricans with purified siroheme yielded the same result, showing that this reaction was also a conserved step in the alternative heme biosynthesis pathway. Furthermore, the conversion of siroheme to heme was demonstrated using cell lysates of D. vulgaris incubated with purified siroheme. Heme was detected as well as intermediates of the pathway including monodecarboxysiroheme, DDSH, and monovinyl iron coproporphyrin.
By utilizing siroheme as a substrate, a chelatase unique to the alternative heme biosynthesis pathway is not required, and hence explains why one was not discovered in early genomic analysis. Previous work had found that D. vulgaris could chelate iron into sirohydrochlorin using homologues of CbiK, CbiKP, and CbiKC, which were capable of chelating both iron and cobalt into sirohydrochlorin [121]. As mentioned previously, siroheme is the prosthetic group of sulfite and nitrite reductases, making it an important molecule in sulfate-reducing bacteria. Siroheme is also found in sulfite reductases in archaea [157], again suggesting a prevalence of this molecule in these organisms and an existing biosynthetic pathway for its formation. In fact, in a previous study of archaeal genomes, only four organisms (Aeropyrum pernix K1, Ignicoccus hospitalis KIN4/I, Methanosphaerula palustris E1-9c, and Hyperthermus butylicus DSM 5456) were shown to contain ahbA, B, C, and D homologues but did not have evidence of a siroheme-containing sulfite or nitrite reductase or lacked cobalamin biosynthesis genes [118].
This finding revealed a new branch point in tetrapyrrole biosynthesis where the heme d 1 and alternative heme biosynthesis pathways diverge at the point of DDSH.
Roles of the Ahb enzymes
Since the discovery of the true substrate for the alternative heme biosynthesis pathway and the enzymes involved, the biosynthetic steps can be summarized as follows (Fig. 7b).
The first reaction in the alternative heme biosynthesis pathway involves decarboxylation of acetic acid side chains at positions C12 and C18 of siroheme to form DDSH [44]. This is catalyzed by AhbA (~20 kDa) and AhbB (~18 kDa), although it is unknown if these enzymes function simultaneously or if they sequentially decarboxylate siroheme. A monodecarboxylated form of siroheme has been detected, although the order of decarboxylation was not determined [44]. It is interesting to note that this same reaction is catalyzed by 3–4 indispensable proteins (NirDLGH) in heme d 1 biosynthesis as opposed to just two in the alternative heme pathway. The relevance of this is yet to be explained, although it could be linked to the regulatory roles of NirDLGH mentioned earlier.
In the next step, DDSH is converted to iron coproporphyrin III, catalyzed by AhbC (~44 kDa). This reaction involves oxidative loss of the northern acetic acid side chains at carbons C2 and C7. AhbC is a member of the radical SAM family, suggesting that the reaction proceeds via adenosyl radicals that activate the leaving groups on the substrate. AhbC contains two cysteine-rich motifs, which are characteristic of Fe–S centers, although it is currently unknown if both centers are required for formation of radicals and enzyme activity.
The final step of the alternative pathway is the conversion of iron coproporphyrin III to heme by the oxidative decarboxylation of the propionate groups at positions C3 and C8 to vinyl groups. This is catalyzed by AhbD (~41 kDa) most likely using a similar mechanism to HemN to transform coproporphyrinogen III to protoporphryinogen IX in the classical pathway [158].
Regulatory requirements of the Ahb pathway
Recent advances in the understanding of the alternative heme and heme d 1 biosynthesis pathways have revealed the increased importance of two intermediates, precorrin-2 and sirohydrochlorin, in tetrapyrrole biosynthesis as they stand as the branch points between cobalamin, coenzyme F430, siroheme, heme, and heme d 1 synthesis (Fig. 8). Therefore, regulation of these biosynthesis pathways must be highly coordinated to direct the common intermediate toward the appropriate pathway.
Fig. 8.

The known branch points of important tetrapyrrole biosynthesis pathways starting from uro’gen III, potential points of regulation between coenzyme F430 cobalamin, siroheme heme, and heme d 1 are displayed as dashed lines
For example, D. vulgaris produces siroheme, cobalamin (via the anaerobic pathway), and heme (via the alternative pathway). Cobalamin and siroheme formation may be regulated at the stage of cobalt or iron chelation into sirohydrochlorin. However, there must also be regulation of the Ahb enzymes to prevent use of siroheme as a substrate. To compound matters, it has been shown that in D. desulfuricans, iron coproporphyrin III is used as a unique cofactor in bacterioferritin, again requiring a further point of regulation on the alternative heme pathway in this organism [159].
Sequence alignments of CysGB from Salmonella enterica and Paenibacillus macerans as well as Met8p from S. cerevisiae and SirC from B. megaterium revealed a conserved hydroxyl group in the form of a serine or threonine [135]. This residue may serve as a phosphorylation site for the regulation of these enzymes. Phosphorylation potentially alters ferrochelatase activity allowing regulation of siroheme or cobalt-sirohydrochlorin production or may affect precorrin-2 dehydrogenation, allowing conservation of precorrin-2 for aerobic synthesis of cobalamin or coenzyme F430 synthesis, this is as yet unclear. Therefore, it can be envisaged that this regulatory mechanism may be present in homologues of these enzymes in organisms containing the heme d 1/alternative heme pathway and a coenzyme F430 and/or cobalamin synthesis pathway. It is also apparent that there is a trend in heme d 1/alternative heme-synthesizing organisms to have one or more cobaltochelatases, some of which have been shown to chelate iron into sirohydrochlorin [119, 121]. This may represent another level of regulation of the sirohydrochlorin pool in the cell, allowing direction to either cobalamin or siroheme synthesis. This being said, further regulation would be required to control the use of siroheme as a substrate for the production of heme/heme d 1 in organisms that require siroheme as a cofactor.
It was referred to earlier that NirD, L, G, and H may play a regulatory role in heme d 1 synthesis as transcription factors. Similar to NirD, L, G, and H, both AhbA and AhbB are annotated as transcription factors belonging to the Lrp/AsnC family. As mentioned before, NirL was shown to bind DNA containing the putative promoter of the nir operon in Heliophilum fasciatum, and as such it could be expected that AhbA and AhbB function in a similar manner. It was also observed that heme and iron induces transcription of nirL in P. aeruginosa [160]. This evidence points towards the possibility that AhbA and AhbB not only play a catalytic role in the alternative biosynthesis of heme but they may also serve as transcriptional regulators to the pathway. It is possible that a feedback mechanism exists allowing the reduction of transcription of ahb genes in the presence of high levels of heme, therefore allowing for the careful regulation of siroheme availability in the cell, while maintaining heme production when required, although this is yet to be experimentally verified.
Evolutionary questions
The recent advances in the research aimed at unraveling alternative heme and heme d 1 biosynthesis raise several interesting evolutionary questions. Which of the modified tetrapyrroles among heme, heme d 1, siroheme, cobalamin/B12, or cofactor F430 is evolutionarily the oldest? Did siroheme evolve first to be the intermediate on the biosynthetic pathways for making heme and heme d 1, and then found itself hijacked for use as a cofactor, in the nitrite and sulfite assimilatory pathways? Or could it have been the other way round? There is also an interesting aspect of gene regulation here. An organism such as P. denitrificans can assimilate nitrite under aerobic and anaerobic conditions provided ammonium is not available as the nitrogen source. Thus, the production of siroheme in such an organism is presumably inhibited under aerobic conditions in which nitrite assimilation is not required, but is activated when assimilation is required. However, under anaerobic conditions, siroheme production must be activated under all conditions, as siroheme production is obligatory in order to sustain the nitrite reductase step of the respiratory denitrification pathway.
In the past, it has been envisaged that cobalamin is possibly the oldest modified tetrapyrrole since it is involved in ribonucleotide reduction and contains a nucleotide loop that may represent a remnant of the RNA world [161, 162]. However, it has been suggested that the main function of cobalamin was to produce an adenosyl radical that could be used to initiate a number of rearrangement reactions, which involve degradation of ethanolamine, propanediol, and glycerol to name a few [163–165]. It has also been shown that cobalamin is required for the anaerobic formation of chlorophyll in some Gram-negative bacteria [166]. Another important function of cobalamin is in methylation reactions, as seen in case of the methionine synthase where methylcobalamin transfers a methyl group to homocysteine. However, both the methyl transfer and adenosyl radical involving chemical transformations are also carried out by “radical SAM” enzymes [91, 144] and therefore there was no evolutionary driving force that particularly required cobalamin formation in the earliest life forms.
Coenzyme F430 is the nickel-containing cofactor found in methyl coenzyme M reductase, an enzyme involved in methanogenesis. Due to its highly reduced ring structure (Fig. 1), which is a prerequisite in prebiotic life and its involvement in methanogenesis, it has also been suggested to be a candidate [167].
The argument that heme is an ancient tetrapyrrole is weakened by the fact that not all species of archaea, which are believed to be the evolutionary older organisms, possess heme [118]. The genetic information to make heme might have been acquired by lateral gene transfer from species of bacteria, such as the sulfate-reducing bacteria, that possess the alternate heme biosynthesis pathway involving siroheme. Interestingly, it has recently been found that archaea and sulfate-reducing bacteria also share a common system for attaching heme to proteins during maturation of c-type cytochromes, as this type of cytochrome is not common in archaea, this suggests that gene transfer could have occurred here, too [168]. There are indications that reduction of nitrite by cytochrome cd 1 may have preceded the appearance of oxygen, whereas assimilation of sulfite and nitrite may have occurred only after oxygen appeared on earth [169]. This might suggest that heme d 1 arose before siroheme and therefore, siroheme potentially has acted as a precursor to other tetrapyrroles, before being utilized as a prosthetic group in assimilatory sulfite and nitrite reductases.
However, in most archaea and sulfate-reducing bacteria, heme is made via the newly discovered alternative route involving siroheme. Since the biosynthesis of heme d 1 is also shown to proceed via siroheme, it is very tempting to conclude that siroheme may be the most ancient tetrapyrrole. It is structurally the simplest of all modified tetrapyrroles and enzymatically the easiest to make [133]. Furthermore, some obligate anaerobes lack cytochromes but can synthesize siroheme [170]. All these facts together promote the antiquity of siroheme in the early biotic life.
Given the close relationship between the syntheses of cobalamin, siroheme, coenzyme F430, and heme d 1, it is conceivable that the alternative heme biosynthesis pathway actually represents the original route for heme synthesis and the classical pathway may have emerged later. Conversion of uro’gen III to heme via the classical pathway utilizes only two molecules of SAM in comparison to the six required by the alternative. This lower-energy consumption is likely to be the driving force behind the successful integration of the classical pathway throughout all kingdoms of life.
Conclusions
In conclusion, as summarized in Fig. 7b, the chemical reactions involved in alternative heme and heme d 1 biosynthesis have been outlined. Siroheme has firmly been established as an intermediate in both pathways, as has DDSH, which represents a new branch point in tetrapyrrole biosynthesis.
The functions of the Ahb enzymes have been established and the intermediates of the pathway are known, although the order of decarboxylation of acetate groups of siroheme and the reaction mechanisms of AhbC and D are yet to be determined.
As with alternative heme synthesis, the mechanism of siroheme decarboxylation and exact roles of NirD, L, G, and H are yet to be established. Questions about NirC, N, and F also remain unanswered. The periplasmic location of NirC, NirN, and NirF suggests that their roles must be at the final stages of heme d 1 production. The exact enzymatic roles of NirF and NirJ still await experimental evidence. As there are more enzymes involved in the synthesis of heme d 1 than heme even though the same amount of chemical transformations are required from the point the two pathways diverge, those proteins are most likely to be responsible for the transport of heme d 1 to the periplasm as well as insertion into nitrite reductase. A summary of the proposed heme d 1 biosynthesis pathway is shown in Fig. 9.
Fig. 9.

Summary of the heme d 1 biosynthesis pathway. Schematic representation of all known proteins of the heme d 1 biosynthesis pathway and the reactions they catalyze. Please note that cytochrome cd 1 nitrite reductase is a homodimeric protein and requires two molecules of heme d 1 per molecule of protein
Integration of siroheme in alternate heme and heme d 1 biosynthesis pathways represents a beautiful example of patchwork model of pathway evolution. In this model, pathways evolve in the forward direction due to the promiscuity of the enzymes and undeveloped regulation mechanisms of the system producing multiple end products.
With the discovery of a new branch point in tetrapyrrole biosynthesis there are also many questions still to be answered relating to the regulation of overlapping metabolic pathways within some organisms (Fig. 8), with the potential of the early Ahb and Nir proteins playing a novel role as both catalytic enzymes and transcriptional regulators.
Acknowledgments
This work was supported by BBSRC grants BBE0229441 and BB/E024203 to S.J.F and M.J.W.
Footnotes
S. Bali, D. J. Palmer, and S. Schroeder contributed equally to this work.
References
- 1.Battersby AR. Tetrapyrroles: the pigments of life. Nat Prod Rep. 2000;17(6):507–526. doi: 10.1039/b002635m. [DOI] [PubMed] [Google Scholar]
- 2.Rudiger W. Chlorophyll metabolism: from outer space down to the molecular level. Phytochemistry. 1997;46(7):1151–1167. [Google Scholar]
- 3.Moulin M, Smith AG. Regulation of tetrapyrrole biosynthesis in higher plants. Paper presented at the Conference on coenzymology: biochemistry of vitamin biogenesis and cofactor-containing enzymes. Cambridge: King Coll; 2005. [Google Scholar]
- 4.Lockhart PJ, Larkum AWD, Steel MA, Waddell PJ, Penny D. Evolution of chlorophyll and bacteriochlorophyll: the problem of invariant sites in sequence analysis. Proc Natl Acad Sci USA. 1996;93(5):1930–1934. doi: 10.1073/pnas.93.5.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blankenship RE, Hartman H. The origin and evolution of oxygenic photosynthesis. Trends Biochem Sci. 1998;23(3):94–97. doi: 10.1016/s0968-0004(98)01186-4. [DOI] [PubMed] [Google Scholar]
- 6.Friedmann HC, Thauer RK. Macrocyclic tetrapyrrole biosynthesis in bacteria. Encyclopedia of microbiology. New York: Academic Press; 1992. pp. 1–19. [Google Scholar]
- 7.Thauer RK, Bonacker LG. Biosynthesis of coenzyme F430, a nickel porphinoid involved in methanogenesis. In: Chadwick DJA, editor. Biosynthesis of the tetrapyrrole pigments. Chichester: CIBA Foundation Symposia, John Wiley & Sons Ltd; 1994. pp. 210–222. [DOI] [PubMed] [Google Scholar]
- 8.Friedmann HC, Klein A, Thauer RK. Biochemistry of coenzyme F430 a nickel porphinoid involved in methanogenesis. In: Jordan PME, editor. New comprehensive biochemistry. New York: Elsevier Science Publishers B.V; 1991. pp. 139–154. [Google Scholar]
- 9.O’Brian MR, Thony-Meyer L. Biochemistry, regulation and genomics of haem biosynthesis in prokaryotes. Adv Microb Physiol. 2002;46:257–318. doi: 10.1016/s0065-2911(02)46006-7. [DOI] [PubMed] [Google Scholar]
- 10.Layer G, Reichelt J, Jahn D, Heinz DW. Structure and function of enzymes in heme biosynthesis. Protein Sci. 2010;19(6):1137–1161. doi: 10.1002/pro.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernhardt R. Cytochromes P450 as versatile biocatalysts. J Biotechnol. 2006;124(1):128–145. doi: 10.1016/j.jbiotec.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 12.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-containing oxygenases. Chem Rev. 1996;96(7):2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 13.Wilks A, Burkhard KA. Heme and virulence: how bacterial pathogens regulate, transport and utilize heme. Nat Prod Rep. 2007;24(3):511–522. doi: 10.1039/b604193k. [DOI] [PubMed] [Google Scholar]
- 14.Zajicek RS, Bali S, Arnold S, Brindley AA, Warren MJ, Ferguson SJ. d 1 haem biogenesis: assessing the roles of three nir gene products. FEBS J. 2009;276(21):6399–6411. doi: 10.1111/j.1742-4658.2009.07354.x. [DOI] [PubMed] [Google Scholar]
- 15.Simon J, Kroneck PM. Microbial sulfite respiration. Adv Microb Physiol. 2013;62:45–117. doi: 10.1016/B978-0-12-410515-7.00002-0. [DOI] [PubMed] [Google Scholar]
- 16.Wilks A. Analysis of heme and hemoproteins. In: Alison G, Smith MW, editors. Heme, chlorophyll, and bilines. Totova: Humana Press; 2002. pp. 157–184. [Google Scholar]
- 17.Sanders CTS, Onder O, Frawley ER, Kranz RG, Koch HG, Daldal F. Biogenesis of c-type cytochromes and cytochrome complexes. In: Hunter NCDF, Thurnauer MC, Beatty JT, editors. The purple phototrophic bacteria. Netherlands: Springer Science + Business Media B.V; 2009. pp. 407–423. [Google Scholar]
- 18.Kranz RG, Richard-Fogal C, Taylor JS, Frawley ER. Cytochrome c biogenesis: mechanisms for covalent modifications and trafficking of heme and for heme-iron redox control. Microbiol Mol Biol Rev. 2009;73(3):510–528. doi: 10.1128/MMBR.00001-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hederstedt L. Heme a biosynthesis. Biochim Biophys Acta. 2012;1817(6):920–927. doi: 10.1016/j.bbabio.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 20.Wandersman C, Delepelaire P. Bacterial iron sources: from siderophores to hemophores. Annu Rev Microbiol. 2004;58:611–647. doi: 10.1146/annurev.micro.58.030603.123811. [DOI] [PubMed] [Google Scholar]
- 21.DeRosa MC, Crutchley RJ. Photosensitized singlet oxygen and its applications. Coordin Chem Rev. 2002;233:351–371. [Google Scholar]
- 22.Dougherty TJ. Photosensitizers: therapy and detection of malignant tumors. Photochem Photobiol. 1987;45(6):879–889. doi: 10.1111/j.1751-1097.1987.tb07898.x. [DOI] [PubMed] [Google Scholar]
- 23.Demidova TN, Hamblin MR. Photodynamic therapy targeted to pathogens. Int J Immunopathol Pharmacol. 2004;17(3):245–254. doi: 10.1177/039463200401700304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang CK. On the structure of heme-d 1: an isobacteriochlorin derivative as the prosthetic group of dissimilatory nitrite reductase. J Biol Chem. 1985;260(17):9520–9522. [PubMed] [Google Scholar]
- 25.Chang CK, Timkovich R, Wu W. Evidence that heme d 1 is a 1,3-porphyrindione. Biochemistry. 1986;25(26):8447–8453. doi: 10.1021/bi00374a019. [DOI] [PubMed] [Google Scholar]
- 26.Chang CK, Wu W. The porphinedione structure of heme-d 1: synthesis and spectral properties of model compounds of the prosthetic group of dissimilatory nitrite reductase. J Biol Chem. 1986;261(19):8593–8596. [PubMed] [Google Scholar]
- 27.Carlson CA, Ingraham JL. Comparison of denitrification by Pseudomonas stutzeri, Pseudomonas aeruginosa, and Paracoccus denitrificans . Appl Environ Microbiol. 1983;45(4):1247–1253. doi: 10.1128/aem.45.4.1247-1253.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zumft WG. Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev. 1997;61(4):533–616. doi: 10.1128/mmbr.61.4.533-616.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones WL, Schroeder ED, Wilderer PA. Denitrification in a batch waste-water treatment system using sequestered organic-substances. Res J Water Pollut C. 1990;62(3):259–267. [Google Scholar]
- 30.Tavares P, Pereira AS, Moura JJG, Moura I. Metalloenzymes of the denitrification pathway. J Inorg Biochem. 2006;100(12):2087–2100. doi: 10.1016/j.jinorgbio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Fulop V, Moir JWB, Ferguson SJ, Hajdu J. The anatomy of a bifunctional enzyme: structural basis for reduction of oxygen to water and synthesis of nitric-oxide by cytochrome cd 1 . Cell. 1995;81(3):369–377. doi: 10.1016/0092-8674(95)90390-9. [DOI] [PubMed] [Google Scholar]
- 32.Pearson IV, Page MD, van Spanning RJM, Ferguson SJ. A mutant of Paracoccus denitrificans with disrupted genes coding for cytochrome c 550 and pseudoazurin establishes these two proteins as the in vivo electron donors to cytochrome cd 1 nitrite reductase. J Bacteriol. 2003;185(21):6308–6315. doi: 10.1128/JB.185.21.6308-6315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen JW, Ferguson SJ, Fülöp V. Cytochrome cd 1 nitrite reductase. Encyclop Inorgan Bioinorgan Chem. 2011 [Google Scholar]
- 34.Zajicek RS, Cartron ML, Ferguson SJ. Probing the unusual oxidation/reduction behavior of Paracoccus pantotrophus cytochrome cd 1 nitrite reductase by replacing a switchable methionine heme iron ligand with histidine. Biochemistry. 2006;45(37):11208–11216. doi: 10.1021/bi0604983. [DOI] [PubMed] [Google Scholar]
- 35.Radoul M, Bykov D, Rinaldo S, Cutruzzola F, Neese F, Goldfarb D. Dynamic hydrogen-bonding network in the distal pocket of the nitrosyl complex of P. aeruginosa cd 1 nitrite reductase. J Am Chem Soc. 2011;133(9):3043–3055. doi: 10.1021/ja109688w. [DOI] [PubMed] [Google Scholar]
- 36.Shemin D, Rittenberg D. The biological utilization of glycine for the synthesis of the protoporphyrin of hemoglobin. J Biol Chem. 1946;166(2):621–625. [PubMed] [Google Scholar]
- 37.Kikuchi G, Kumar A, Talmage P, Shemin D. The enzymatic synthesis of delta-aminolevulinic acid. J Biol Chem. 1958;233(5):1214–1219. [PubMed] [Google Scholar]
- 38.Beale SI, Castelfranco PA. 14 C incorporation from exogenous compounds into––aminolevulinic acid by greening cucumber cotyledons. Biochem Biophys Res Commun. 1973;52(1):143–149. doi: 10.1016/0006-291x(73)90966-2. [DOI] [PubMed] [Google Scholar]
- 39.Jahn D, Heinz DW. Biosynthesis of 5-aminolevulinic acid. In: Warren MJ, Smith AG, editors. Tetrapyrroles: birth, life, and death. Austin: Landes Bioscience; 2009. [Google Scholar]
- 40.Weinstein JD, Beale SI. Separate physiological roles and subcellular compartments for two tetrapyrrole biosynthetic pathways in Euglena gracilis . J Biol Chem. 1983;258(11):6799–6807. [PubMed] [Google Scholar]
- 41.Iida K, Mimura I, Kajiwara M. Evaluation of two biosynthetic pathways to delta-aminolevulinic acid in E. gracilis . Eur J Biochem. 2002;269(1):291–297. doi: 10.1046/j.0014-2956.2001.02651.x. [DOI] [PubMed] [Google Scholar]
- 42.Cavallaro G, Decaria L, Rosato A. Genome-based analysis of heme biosynthesis and uptake in prokaryotic systems. J Proteome Res. 2008;7(11):4946–4954. doi: 10.1021/pr8004309. [DOI] [PubMed] [Google Scholar]
- 43.Ishida T, Yu L, Akutsu H, Ozawa K, Kawanishi S, Seto A, Inubushi T, Sano S. A primitive pathway of porphyrin biosynthesis and enzymology in D. vulgaris . Proc Natl Acad Sci USA. 1998;95(9):4853–4858. doi: 10.1073/pnas.95.9.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bali S, Lawrence AD, Lobo SA, Saraiva LM, Golding BT, Palmer DJ, Howard MJ, Ferguson SJ, Warren MJ. Molecular hijacking of siroheme for the synthesis of heme and d 1 heme. Proc Natl Acad Sci USA. 2011;108(45):18260–18265. doi: 10.1073/pnas.1108228108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muir HM, Neuberger A. The biogenesis of porphyrins. 2. The origin of the methyne carbon atoms. Biochem J. 1950;47(1):97–104. doi: 10.1042/bj0470097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferreira GC, Gong J. 5-aminolevulinate synthase and the first step of heme-biosynthesis. J Bioenerg Biomembr. 1995;27(2):151–159. doi: 10.1007/BF02110030. [DOI] [PubMed] [Google Scholar]
- 47.Fanica-Gaignier M, Clement-Metral J. 5-aminolevulinic-acid synthetase of Rhodopseudomonas spheroides Y. kinetic mechanism and inhibition by ATP. Eur J Biochem. 1973;40(1):19–24. doi: 10.1111/j.1432-1033.1973.tb03164.x. [DOI] [PubMed] [Google Scholar]
- 48.Astner I, Schulze JO, van den Heuvel J, Jahn D, Schubert WD, Heinz DW. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 2005;24(18):3166–3177. doi: 10.1038/sj.emboj.7600792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beale SI, Gough SP, Granick S. Biosynthesis of delta-aminolevulinic acid from the intact carbon skeleton of glutamic acid in greening barley. Proc Natl Acad Sci USA. 1975;72(7):2719–2723. doi: 10.1073/pnas.72.7.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schon A, Krupp G, Gough S, Berry-Lowe S, Kannangara CG, Soll D. The RNA required in the first step of chlorophyll biosynthesis is a chloroplast glutamate tRNA. Nature. 1986;322(6076):281–284. doi: 10.1038/322281a0. [DOI] [PubMed] [Google Scholar]
- 51.Moser J, Lorenz S, Hubschwerlen C, Rompf A, Jahn D. Methanopyrus kandleri glutamyl-tRNA reductase. J Biol Chem. 1999;274(43):30679–30685. doi: 10.1074/jbc.274.43.30679. [DOI] [PubMed] [Google Scholar]
- 52.Chen MW, Jahn D, O’Neill GP, Soll D. Purification of the glutamyl-tRNA reductase from Chlamydomonas reinhardtii involved in delta-aminolevulinic acid formation during chlorophyll biosynthesis. J Biol Chem. 1990;265(7):4058–4063. [PubMed] [Google Scholar]
- 53.Hoober JK, Kahn A, Ash DE, Gough S, Kannangara CG. Biosynthesis of delta-aminolevulinate in greening barley leaves. IX. Structure of the substrate, mode of gabaculine inhibition, and the catalytic mechanism of glutamate 1-semialdehyde aminotransferase. Carlsberg Res Commun. 1988;53(1):11–25. doi: 10.1007/BF02908411. [DOI] [PubMed] [Google Scholar]
- 54.Hennig M, Grimm B, Contestabile R, John RA, Jansonius JN. Crystal structure of glutamate-1-semialdehyde aminomutase: an alpha2-dimeric vitamin B6-dependent enzyme with asymmetry in structure and active site reactivity. Proc Natl Acad Sci USA. 1997;94(10):4866–4871. doi: 10.1073/pnas.94.10.4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moser J, Schubert WD, Beier V, Bringemeier I, Jahn D, Heinz DW. V-shaped structure of glutamyl-tRNA reductase, the first enzyme of tRNA-dependent tetrapyrrole biosynthesis. EMBO J. 2001;20(23):6583–6590. doi: 10.1093/emboj/20.23.6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schulze JO, Schubert WD, Moser J, Jahn D, Heinz DW. Evolutionary relationship between initial enzymes of tetrapyrrole biosynthesis. J Mol Biol. 2006;358(5):1212–1220. doi: 10.1016/j.jmb.2006.02.064. [DOI] [PubMed] [Google Scholar]
- 57.Luer C, Schauer S, Mobius K, Schulze J, Schubert WD, Heinz DW, Jahn D, Moser J. Complex formation between glutamyl-tRNA reductase and glutamate-1-semialdehyde 2,1-aminomutase in E. coli during the initial reactions of porphyrin biosynthesis. J Biol Chem. 2005;280(19):18568–18572. doi: 10.1074/jbc.M500440200. [DOI] [PubMed] [Google Scholar]
- 58.Nogaj LA, Beale SI. Physical and kinetic interactions between glutamyl-tRNA reductase and glutamate-1-semialdehyde aminotransferase of Chlamydomonas reinhardtii . J Biol Chem. 2005;280(26):24301–24307. doi: 10.1074/jbc.M502483200. [DOI] [PubMed] [Google Scholar]
- 59.Deery E, Schroeder S, Lawrence AD, Taylor SL, Seyedarabi A, Waterman J, Wilson KS, Brown D, Geeves MA, Howard MJ, Pickersgill RW, Warren MJ. An enzyme-trap approach allows isolation of intermediates in cobalamin biosynthesis. Nat Chem Biol. 2012;8(11):933–940. doi: 10.1038/nchembio.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gibson KD, Neuberger A, Scott JJ. The enzymic conversion of delta-aminolaevulic acid to porphobilinogen. Biochem J. 1954;58(4):xli–xlii. [PubMed] [Google Scholar]
- 61.Bollivar DW, Clauson C, Lighthall R, Forbes S, Kokona B, Fairman R, Kundrat L, Jaffe EK. Rhodobacter capsulatus porphobilinogen synthase, a high activity metal ion independent hexamer. BMC Biochem. 2004;5:17. doi: 10.1186/1471-2091-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Senior NM, Brocklehurst K, Cooper JB, Wood SP, Erskine P, Shoolingin-Jordan PM, Thomas PG, Warren MJ. Comparative studies on the 5-aminolaevulinic acid dehydratases from Pisum sativum, E. coli and S. cerevisiae . Biochem J. 1996;320(2):401–412. [PMC free article] [PubMed] [Google Scholar]
- 63.Jaffe EK. The porphobilinogen synthase family of metalloenzymes. Acta Crystallogr D Biol Crystallogr. 2000;56(2):115–128. doi: 10.1107/s0907444999014894. [DOI] [PubMed] [Google Scholar]
- 64.Shoolingin-Jordan PM, Spencer P, Sarwar M, Erskine PE, Cheung KM, Cooper JB, Norton EB. 5-aminolaevulinic acid dehydratase: metals, mutants and mechanism. Edinburgh: Paper presented at the Colloquium on Tetrapyrroles: Their Life, Birth and Death Heriot-Watt; 2002. [DOI] [PubMed] [Google Scholar]
- 65.Frankenberg N, Erskine PT, Cooper JB, Shoolingin-Jordan PM, Jahn D, Heinz DW. High-resolution crystal structure of a Mg2+-dependent porphobilinogen synthase. J Mol Biol. 1999;289(3):591–602. doi: 10.1006/jmbi.1999.2808. [DOI] [PubMed] [Google Scholar]
- 66.Erskine PT, Newbold R, Brindley AA, Wood SP, Shoolingin-Jordan PM, Warren MJ, Cooper JB. The X-ray structure of yeast 5-aminolaevulinic acid dehydratase complexed with substrate and three inhibitors. J Mol Biol. 2001;312(1):133–141. doi: 10.1006/jmbi.2001.4947. [DOI] [PubMed] [Google Scholar]
- 67.Erskine PT, Norton E, Cooper JB, Lambert R, Coker A, Lewis G, Spencer P, Sarwar M, Wood SP, Warren MJ, Shoolingin-Jordan PM. X-ray structure of 5-aminolaevulinic acid dehydratase from E. coli complexed with the inhibitor levulinic acid at 2.0-A resolution. Biochemistry. 1999;38(14):4266–4276. doi: 10.1021/bi982137w. [DOI] [PubMed] [Google Scholar]
- 68.Warren MJ, Jordan PM. Investigation into the nature of substrate binding to the dipyrromethane cofactor of E. coli porphobilinogen deaminase. Biochemistry. 1988;27(25):9020–9030. doi: 10.1021/bi00425a021. [DOI] [PubMed] [Google Scholar]
- 69.Awan SJ, Siligardi G, Shoolingin-Jordan PM, Warren MJ. Reconstitution of the holoenzyme form of E. coli porphobilinogen deaminase from apoenzyme with porphobilinogen and pre-uroporphyrinogen: a study using circular dichroism spectroscopy. Biochemistry. 1997;36(30):9273–9282. doi: 10.1021/bi9702602. [DOI] [PubMed] [Google Scholar]
- 70.Hart GJ, Miller AD, Leeper FJ, Battersby AR. Biosynthesis of the natural porphyrins: proof that hydroxymethylbilane synthase (Porphobilinogen Deaminase) uses a novel binding group in its catalytic action. J Chem Soc Chem Comm. 1987;23:1762–1764. [Google Scholar]
- 71.Warren MJ, Scott AI. Tetrapyrrole assembly and modification into the ligands of biologically functional cofactors. Trends Biochem Sci. 1990;15(12):486–491. doi: 10.1016/0968-0004(90)90304-t. [DOI] [PubMed] [Google Scholar]
- 72.Louie GV, Brownlie PD, Lambert R, Cooper JB, Blundell TL, Wood SP, Warren MJ, Woodcock SC, Jordan PM. Structure of porphobilinogen deaminase reveals a flexible multidomain polymerase with a single catalytic site. Nature. 1992;359(6390):33–39. doi: 10.1038/359033a0. [DOI] [PubMed] [Google Scholar]
- 73.Shoolingin-Jordan PM. Porphobilinogen deaminase and uroporphyrinogen III synthase: structure, molecular biology, and mechanism. J Bioenerg Biomembr. 1995;27(2):181–195. doi: 10.1007/BF02110033. [DOI] [PubMed] [Google Scholar]
- 74.Burton G, Fagerness PE, Hosozawa S, Jordan PM, Scott AI. 13C NMR evidence for a new intermediate, pre-uroporphyrinogen, in the enzymic transformation of porphobilinogen into uroporphyrinogens I and III. J Chem Soc Chem Comm. 1979;5:202. [Google Scholar]
- 75.Timkovich R, Burkhalter RS, Xavier AV, Chen L, Legall J. Iron uroporphyrin-I and a heme c-derivative are prosthetic groups in D. gigas rubredoxin oxidase. Bioorg Chem. 1994;22(3):284–293. [Google Scholar]
- 76.Schubert HL, Raux E, Matthews MAA, Phillips JD, Wilson KS, Hill CP, Warren MJ (2002) Structural diversity in metal ion chelation and the structure of uroporphyrinogen III synthase. In: Colloquium on tetrapyrroles: their life, birth and death, Heriot-Watt, Edinburgh, pp 595–600 [DOI] [PubMed]
- 77.Luo J, Lim CK. Order of uroporphyrinogen III decarboxylation on incubation of porphobilinogen and uroporphyrinogen III with erythrocyte uroporphyrinogen decarboxylase. Biochem J. 1993;289(2):529–532. doi: 10.1042/bj2890529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dailey HA. Terminal steps of haem biosynthesis. Biochem Soc Trans. 2002;4:590–595. doi: 10.1042/bst0300590. [DOI] [PubMed] [Google Scholar]
- 79.Whitby FG. Crystal structure of human uroporphyrinogen decarboxylase. EMBO J. 1998;17(9):2463–2471. doi: 10.1093/emboj/17.9.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Phillips JD, Warby CA, Whitby FG, Kushner JP, Hill CP. Substrate shuttling between active sites of uroporphyrinogen decarboxylase is not required to generate coproporphyrinogen. J Mol Biol. 2009;389(2):306–314. doi: 10.1016/j.jmb.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Medlock AE, Dailey HA. Human coproporphyrinogen oxidase is not a metalloprotein. J Biol Chem. 1996;271(51):32507–32510. doi: 10.1074/jbc.271.51.32507. [DOI] [PubMed] [Google Scholar]
- 82.Cavaleiro JA, Kenner GW, Smith KM. Pyrroles and related compounds. XXXII. Biosynthesis of protoporphyrin-IX from coproporphyrinogen-III. J Chem Soc Perkin. 1974;110:1188–1194. doi: 10.1039/p19740001188. [DOI] [PubMed] [Google Scholar]
- 83.Elder GH, Evans JO, Jackson JR, Jackson AH. Factors determining sequence of oxidative decarboxylation of 2-propionate and 4-propionate substituents of coproporphyrinogen-Iii by coproporphyrinogen oxidase in rat-liver. Biochem J. 1978;169(1):215–223. doi: 10.1042/bj1690215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jackson AH, Jones DM, Philip G, Lash TD, Batlle AM, Smith SG. Synthetic and biosynthetic studies of porphyrins, Part IV. Further studies of the conversion of corporporhyrinogen-III to protoporphyrin-IX: mass spectrometric investigations of the incubation of specifically deuteriated corporporhyringen-III with chicken red cell haemolysates. Int J Biochem. 1980;12(5–6):681–688. doi: 10.1016/0020-711x(80)90144-5. [DOI] [PubMed] [Google Scholar]
- 85.Phillips JD, Whitby FG, Warby CA, Labbe P, Yang C, Pflugrath JW, Ferrara JD, Robinson H, Kushner JP, Hill CP. Crystal structure of the oxygen-dependant coproporphyrinogen oxidase (Hem13p) of S. cerevisiae . J Biol Chem. 2004;279(37):38960–38968. doi: 10.1074/jbc.M406050200. [DOI] [PubMed] [Google Scholar]
- 86.Lee DS, Flachsova E, Bodnarova M, Demeler B, Martasek P, Raman CS. Structural basis of hereditary coproporphyria. Natl Acad Sci USA. 2005;102(40):14232–14237. doi: 10.1073/pnas.0506557102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stephenson JR, Stacey JA, Morgenthaler JB, Friesen JA, Lash TD, Jones MA. Role of aspartate 400, arginine 262, and arginine 401 in the catalytic mechanism of human coproporphyrinogen oxidase. Protein Sci. 2007;16(3):401–410. doi: 10.1110/ps.062636907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Silva PJ, Ramos MJ. A comparative density-functional study of the reaction mechanism of the O2-dependent coproporphyrinogen III oxidase. Bioorg Med Chem Lett. 2008;16(6):2726–2733. doi: 10.1016/j.bmc.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 89.Lash TD. The enigma of coproporphyrinogen oxidase: how does this unusual enzyme carry out oxidative decarboxylations to afford vinyl groups? Bioorg Med Chem Lett. 2005;15(20):4506–4509. doi: 10.1016/j.bmcl.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 90.Layer G, Grage K, Teschner T, Schunemann V, Breckau D, Masoumi A, Jahn M, Heathcote P, Trautwein AX, Jahn D. Radical S-adenosylmethionine enzyme coproporphyrinogen III oxidase HemN: functional features of the [4Fe–4S] cluster and the two bound S-adenosyl-l-methionines. J Biol Chem. 2005;280(32):29038–29046. doi: 10.1074/jbc.M501275200. [DOI] [PubMed] [Google Scholar]
- 91.Atta M, Mulliez E, Arragain S, Forouhar F, Hunt JF, Fontecave M. S-Adenosylmethionine-dependent radical-based modification of biological macromolecules. Curr Opin Struc Biol. 2010;20(6):684–692. doi: 10.1016/j.sbi.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 92.Booker SJ. Anaerobic functionalization of unactivated C–H bonds. Curr Opin Chem Biol. 2009;13(1):58–73. doi: 10.1016/j.cbpa.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Layer G, Heinz DW, Jahn D, Schubert WD. Structure and function of radical SAM enzymes. Curr Opin Chem Biol. 2004;8(5):468–476. doi: 10.1016/j.cbpa.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 94.Layer G, Pierik AJ, Trost M, Rigby SE, Leech HK, Grage K, Breckau D, Astner I, Jansch L, Heathcote P, Warren MJ, Heinz DW, Jahn D. The substrate radical of E. coli oxygen-independent coproporphyrinogen III oxidase HemN. J Biol Chem. 2006;281(23):15727–15734. doi: 10.1074/jbc.M512628200. [DOI] [PubMed] [Google Scholar]
- 95.Abicht HK, Martinez J, Layer G, Jahn D, Solioz M. Lactococcus lactis HemW (HemN) is a haem-binding protein with a putative role in haem trafficking. Biochem J. 2012;442(2):335–343. doi: 10.1042/BJ20111618. [DOI] [PubMed] [Google Scholar]
- 96.Dailey HA, Dailey TA. Protoporphyrinogen oxidase of Myxococcus xanthus. Expression, purification, and characterization of the cloned enzyme. J Biol Chem. 1996;271(15):8714–8718. doi: 10.1074/jbc.271.15.8714. [DOI] [PubMed] [Google Scholar]
- 97.Sasarman A, Letowski J, Czaika G, Ramirez V, Nead MA, Jacobs JM, Morais R. Nucleotide-sequence of the hemG gene involved in the protoporphyrinogen oxidase activity of E. coli K12. Can J Microbiol. 1993;39(12):1155–1161. doi: 10.1139/m93-174. [DOI] [PubMed] [Google Scholar]
- 98.Boynton TO, Gerdes S, Craven SH, Neidle EL, Phillips JD, Dailey HA. Discovery of a gene involved in a third bacterial protoporphyrinogen oxidase activity through comparative genomic analysis and functional complementation. Appl Environ Microbiol. 2011;77(14):4795–4801. doi: 10.1128/AEM.00171-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dailey TA, Dailey HA (1997) Expression, purification, and characteristics of mammalian protoporphyrinogen oxidase. In: McCormick D, Suttie J, Wagner C (eds.) Methods in Enzymology, vol 281. pp 340–349 [DOI] [PubMed]
- 100.Hansson M, Hederstedt L. Bacillus subtilis HemY is a peripheral membrane-protein essential for protoheme-IX synthesis which can oxidize coproporphyrinogen-III and protoporphyrinogen-IX. J Bacteriol. 1994;176(19):5962–5970. doi: 10.1128/jb.176.19.5962-5970.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dailey TA, Meissner P, Dailey HA. Expression of a cloned protoporphyrinogen oxidase. J Biol Chem. 1994;269(2):813–815. [PubMed] [Google Scholar]
- 102.Koch M, Breithaupt C, Kiefersauer R, Freigang J, Huber R, Messerschmidt A. Crystal structure of protoporphyrinogen IX oxidase: a key enzyme in haem and chlorophyll biosynthesis. EMBO J. 2004;23(8):1720–1728. doi: 10.1038/sj.emboj.7600189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Corradi HR, Corrigall AV, Boix E, Mohan CG, Sturrock ED, Meissner PN, Acharya KR. Crystal structure of protoporphyrinogen oxidase from Myxococcus xanthus and its complex with the inhibitor acifluorfen. J Biol Chem. 2006;281(50):38625–38633. doi: 10.1074/jbc.M606640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Heinemann IU, Diekmann N, Masoumi A, Koch M, Messerschmidt A, Jahn M, Jahn D. Functional definition of the tobacco protoporphyrinogen IX oxidase substrate-binding site. Biochem J. 2007;402(3):575–580. doi: 10.1042/BJ20061321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boynton TO, Daugherty LE, Dailey TA, Dailey HA. Identification of E. coli HemG as a novel, menadione-dependent flavodoxin with protoporphyrinogen oxidase activity. Biochemistry. 2009;48(29):6705–6711. doi: 10.1021/bi900850y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mobius K, Arias-Cartin R, Breckau D, Hannig AL, Riedmann K, Biedendieck R, Schroder S, Becher D, Magalon A, Moser J, Jahn M, Jahn D. Heme biosynthesis is coupled to electron transport chains for energy generation. Proc Natl Acad Sci USA. 2010;107(23):10436–10441. doi: 10.1073/pnas.1000956107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kato K, Tanaka R, Sano S, Tanaka A, Hosaka H. Identification of a gene essential for protoporphyrinogen IX oxidase activity in the cyanobacterium Synechocystis sp. PCC6803. Proc Natl Acad Sci USA. 2010;107(38):16649–16654. doi: 10.1073/pnas.1000771107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dailey HA, Dailey TA, Wu CK, Medlock AE, Wang KF, Rose JP, Wang BC. Ferrochelatase at the millennium: structures, mechanisms and [2Fe–2S] clusters. Cell Mol Life Sci. 2000;57(13–14):1909–1926. doi: 10.1007/PL00000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Corrigall AV, Siziba KB, Maneli MH, Shephard EG, Ziman M, Dailey TA, Dailey HA, Kirsch RE, Meissner PN. Purification of and kinetic studies on a cloned protoporphyrinogen oxidase from the aerobic bacterium B. subtilis . Arch Biochem Biophys. 1998;358(2):251–256. doi: 10.1006/abbi.1998.0834. [DOI] [PubMed] [Google Scholar]
- 110.Hansson M, Hederstedt L. Purification and characterisation of a water-soluble ferrochelatase from B. subtilis . Eur J Biochem. 1994;220(1):201–208. doi: 10.1111/j.1432-1033.1994.tb18615.x. [DOI] [PubMed] [Google Scholar]
- 111.Romao CV, Ladakis D, Lobo SA, Carrondo MA, Brindley AA, Deery E, Matias PM, Pickersgill RW, Saraiva LM, Warren MJ. Evolution in a family of chelatases facilitated by the introduction of active site asymmetry and protein oligomerization. Proc Natl Acad Sci USA. 2011;108(1):97–102. doi: 10.1073/pnas.1014298108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dailey TA, Boynton TO, Albetel AN, Gerdes S, Johnson MK, Dailey HA. Discovery and characterization of HemQ: an essential heme biosynthetic pathway component. J Biol Chem. 2010;285(34):25978–25986. doi: 10.1074/jbc.M110.142604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Raux E, Leech HK, Beck R, Schubert HL, Santander PJ, Roessner CA, Scott AI, Martens JH, Jahn D, Thermes C, Rambach A, Warren MJ. Identification and functional analysis of enzymes required for precorrin-2 dehydrogenation and metal ion insertion in the biosynthesis of sirohaem and cobalamin in Bacillus megaterium . Biochem J. 2003;370(2):505–516. doi: 10.1042/BJ20021443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Raux E, McVeigh T, Peters SE, Leustek T, Warren MJ. The role of S. cerevisiae Met1p and Met8p in sirohaem and cobalamin biosynthesis. Biochem J. 1999;338:701–708. [PMC free article] [PubMed] [Google Scholar]
- 115.Warren MJ, Bolt EL, Roessner CA, Scott AI, Spencer JB, Woodcock SC. Gene dissection demonstrates that the E. coli cysG gene encodes a multifunctional protein. Biochem J. 1994;302(3):837–844. doi: 10.1042/bj3020837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Warren MJ, Raux E, Schubert HL, Escalante-Semerena JC. The biosynthesis of adenosylcobalamin (vitamin B12) Nat Prod Rep. 2002;19(4):390–412. doi: 10.1039/b108967f. [DOI] [PubMed] [Google Scholar]
- 117.Platt MD, Schurr MJ, Sauer K, Vazquez G, Kukavica-Ibrulj I, Potvin E, Levesque RC, Fedynak A, Brinkman FS, Schurr J, Hwang SH, Lau GW, Limbach PA, Rowe JJ, Lieberman MA, Barraud N, Webb J, Kjelleberg S, Hunt DF, Hassett DJ. Proteomic, microarray, and signature-tagged mutagenesis analyses of anaerobic P. aeruginosa at pH 6.5, likely representing chronic, late-stage cystic fibrosis airway conditions. J Bacteriol. 2008;190(8):2739–2758. doi: 10.1128/JB.01683-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Storbeck S, Rolfes S, Raux-Deery E, Warren MJ, Jahn D, Layer G. A novel pathway for the biosynthesis of heme in Archaea: genome-based bioinformatic predictions and experimental evidence. Archaea. 2010;2010:175050. doi: 10.1155/2010/175050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Brindley AA, Raux E, Leech HK, Schubert HL, Warren MJ. A story of chelatase evolution: identification and characterization of a small 13–15-kDa “ancestral” cobaltochelatase (CbiXS) in the archaea. J Biol Chem. 2003;278(25):22388–22395. doi: 10.1074/jbc.M302468200. [DOI] [PubMed] [Google Scholar]
- 120.Lobo SA, Brindley A, Warren MJ, Saraiva LM. Functional characterization of the early steps of tetrapyrrole biosynthesis and modification in D. vulgaris Hildenborough. Biochem J. 2009;420(2):317–325. doi: 10.1042/BJ20090151. [DOI] [PubMed] [Google Scholar]
- 121.Lobo SAL, Brindley AA, Romao CV, Leech HK, Warren MJ, Saraiva LM. Two distinct roles for two functional cobaltochelatases (CbiK) in D. vulgaris Hildenborough. Biochemistry. 2008;47(21):5851–5857. doi: 10.1021/bi800342c. [DOI] [PubMed] [Google Scholar]
- 122.Lobo SAL, Warren MJ, Saraiva LM. Chapter seven––sulfate-reducing bacteria reveal a new branch of tetrapyrrole metabolism. In: Robert KP, editor. Advance microbiol physiology. Netherlands: Amsterdam; 2012. pp. 267–295. [DOI] [PubMed] [Google Scholar]
- 123.Tripathy BC, Sherameti I, Oelmuller R. Siroheme: an essential component for life on earth. Plant Sign Behav. 2010;5(1):14–20. doi: 10.4161/psb.5.1.10173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yapbondoc F, Bondoc LL, Timkovich R, Baker DC, Hebbler A. C-methylation occurs during the biosynthesis of heme d 1 . J Biol Chem. 1990;265(23):13498–13500. [PubMed] [Google Scholar]
- 125.Bali S, Warren MJ, Ferguson SJ. NirF is a periplasmic protein that binds d 1 heme as part of its essential role in d 1 heme biogenesis. FEBS J. 2010;277(23):4944–4955. doi: 10.1111/j.1742-4658.2010.07899.x. [DOI] [PubMed] [Google Scholar]
- 126.Suzuki M, Hirai T, Arai H, Ishii M, Igarashi Y. Purification, characterization, and gene cloning of thermophilic cytochrome cd 1 nitrite reductase from Hydrogenobacter thermophilus TK-6. J Biosci Bioeng. 2006;101(5):391–397. doi: 10.1263/jbb.101.391. [DOI] [PubMed] [Google Scholar]
- 127.Palmedo G, Seither P, Korner H, Matthews JC, Burkhalter RS, Timkovich R, Zumft WG. Resolution of the nirD locus for heme d 1 synthesis of cytochrome cd 1 (respiratory nitrite reductase) from Pseudomonas stutzeri . Eur J Biochem. 1995;232(3):737–746. [PubMed] [Google Scholar]
- 128.Glockner AB, Zumft WG. Sequence analysis of an internal 9.72-kb segment from the 30-kb denitrification gene cluster of Pseudomonas stutzeri . Biochimica Et Biophysica Acta-Bioenergetics. 1996;1277((1-2)):6–12. doi: 10.1016/s0005-2728(96)00108-9. [DOI] [PubMed] [Google Scholar]
- 129.Kawasaki S, Arai H, Kodama T, Igarashi Y. Gene cluster for dissimilatory nitrite reductase (nir) from P. aeruginosa: sequencing and identification of a locus for heme d 1 biosynthesis. J Bacteriol. 1997;179(1):235–242. doi: 10.1128/jb.179.1.235-242.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Silvestrini MC, Cutruzzola F, D’Alessandro R, Brunori M, Fochesato N, Zennaro E. Expression of P. aeruginosa nitrite reductase in P. putida and characterization of the recombinant protein. Biochem J. 1992;285(2):661–666. doi: 10.1042/bj2850661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Deboer APN, Reijnders WNM, Kuenen JG, Stouthamer AH, Vanspanning RJM. Isolation, sequencing and mutational analysis of a gene-cluster involved in nitrite reduction in Paracoccus denitrificans . Anton Leeuw Int J G. 1994;66(1–3):111–127. doi: 10.1007/BF00871635. [DOI] [PubMed] [Google Scholar]
- 132.Ohshima T, Sugiyama M, Uozumi N, Iijima S, Kobayashi T. Cloning and sequencing of a gene encoding nitrite reductase from Paracoccus denitrificans and expression of the gene in E. coli . J Ferment Bioeng. 1993;76(2):82–88. [Google Scholar]
- 133.Warren MJ, Smith AG, Deery E, Rose RS (2009) Biosynthesis of Siroheme and Coenzyme F430. In: Tetrapyrroles: birth, life and death. Molecular biology intelligence unit. Springer, Berlin Heidelberg New York, pp 344–351, doi: 10.1007/978-0-387-78518-9_22
- 134.Storbeck S, Walther J, Mueller J, Parmar V, Schiebel HM, Kemken D, Duelcks T, Warren MJ, Layer G. The P. aeruginosa nirE gene encodes the S-adenosyl-l-methionine-dependent uroporphyrinogen III methyltransferase required for heme d 1 biosynthesis. FEBS J. 2009;276(20):5973–5982. doi: 10.1111/j.1742-4658.2009.07306.x. [DOI] [PubMed] [Google Scholar]
- 135.Stroupe ME, Leech HK, Daniels DS, Warren MJ, Getzoff ED. CysG structure reveals tetrapyrrole-binding features and novel regulation of siroheme biosynthesis. Nat Struct Biol. 2003;10(12):1064–1073. doi: 10.1038/nsb1007. [DOI] [PubMed] [Google Scholar]
- 136.Hasegawa N, Arai H, Igarashi Y. Two c-type cytochromes, NirM and NirC, encoded in the nir gene cluster of P. aeruginosa act as electron donors for nitrite reductase. Biochem Biophys Res Commun. 2001;288(5):1223–1230. doi: 10.1006/bbrc.2001.5919. [DOI] [PubMed] [Google Scholar]
- 137.Nicke T, Schnitzer T, Munch K, Adamczack J, Haufschildt K, Buchmeier S, Kucklick M, Felgentrager U, Jansch L, Riedel K, Layer G. Maturation of the cytochrome cd 1 nitrite reductase NirS from P. aeruginosa requires transient interactions between the three proteins NirS. Biosci Rep NirN NirF. 2013 doi: 10.1042/BSR20130043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rossmann MG, Moras D, Olsen KW. Chemical and biological evolution of a nucleotide-binding protein. Nature. 1974;250(5463):194–199. doi: 10.1038/250194a0. [DOI] [PubMed] [Google Scholar]
- 139.Bali S, Ferguson SJ (2011) Assembly of respiratory proteins of the nitrogen cycle In: Moir JWB (ed) Nitrogen cycling in bacteria: molecular analysis, pp 163–175
- 140.Heikkila MP, Honisch U, Wunsch P, Zumft WG. Role of the tat transport system in nitrous oxide reductase translocation and cytochrome cd 1 biosynthesis in Pseudomonas stutzeri . J Bacteriol. 2001;183(5):1663–1671. doi: 10.1128/JB.183.5.1663-1671.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Brinkman AB, Ettema TJ, de Vos WM, van der Oost J. The Lrp family of transcriptional regulators. Mol Microbiol. 2003;48(2):287–294. doi: 10.1046/j.1365-2958.2003.03442.x. [DOI] [PubMed] [Google Scholar]
- 142.Xiong J, Bauer CE, Pancholy A. Insight into the haem d 1 biosynthesis pathway in heliobacteria through bioinformatics analysis. Microbiol Sgm. 2007;153:3548–3562. doi: 10.1099/mic.0.2007/007930-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucl Acids Res. 2001;29(5):1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Frey PA, Hegeman AD, Ruzicka FJ. The radical SAM superfamily. Crit Rev Biochem Mol. 2008;43(1):63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 145.Brindley AA, Zajicek R, Warren MJ, Ferguson SJ, Rigby SE. NirJ, a radical SAM family member of the d 1 heme biogenesis cluster. FEBS Lett. 2010;584(11):2461–2466. doi: 10.1016/j.febslet.2010.04.053. [DOI] [PubMed] [Google Scholar]
- 146.Hanzelmann P, Schindelin H. Crystal structure of the S-adenosylmethionine-dependent enzyme MoaA and its implications for molybdenum cofactor deficiency in humans. Proc Natl Acad Sci USA. 2004;101(35):12870–12875. doi: 10.1073/pnas.0404624101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hanzelmann P, Hernandez HL, Menzel C, Garcia-Serres R, Huynh BH, Johnson MK, Mendel RR, Schindelin H. Characterization of MOCS1A, an oxygen-sensitive iron–sulfur protein involved in human molybdenum cofactor biosynthesis. J Biol Chem. 2004;279(33):34721–34732. doi: 10.1074/jbc.M313398200. [DOI] [PubMed] [Google Scholar]
- 148.Fluhe L, Knappe TA, Gattner MJ, Schafer A, Burghaus O, Linne U, Marahiel MA. The radical SAM enzyme AlbA catalyzes thioether bond formation in subtilosin A. Nat Chem Biol. 2012;8(4):350–357. doi: 10.1038/nchembio.798. [DOI] [PubMed] [Google Scholar]
- 149.Yokoyama K, Numakura M, Kudo F, Ohmori D, Eguchi T. Characterization and mechanistic study of a radical SAM dehydrogenase in the biosynthesis of butirosin. J Am Chem Soc. 2007;129(49):15147–15155. doi: 10.1021/ja072481t. [DOI] [PubMed] [Google Scholar]
- 150.Benjdia A, Subramanian S, Leprince J, Vaudry H, Johnson MK, Berteau O. Anaerobic sulfatase-maturating enzymes, first dual substrate radical S-adenosylmethionine enzymes. J Biol Chem. 2008;283(26):17815–17826. doi: 10.1074/jbc.M710074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Grove TL, Ahlum JH, Sharma P, Krebs C, Booker SJ. A consensus mechanism for radical SAM-dependent dehydrogenation? BtrN contains two 4Fe–4S clusters. Biochemistry. 2010;49(18):3783–3785. doi: 10.1021/bi9022126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Grove TL, Lee K-H, St. Clair J, Krebs C, Booker C. In vitro characterization of AtsB, a radical SAM formylglycine-generating enzyme that contains three 4Fe-4S clusters. Biochemistry. 2008;47(28):7523–7538. doi: 10.1021/bi8004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Akutsu H, Park JS, Sano S. l-methionine methyl is specifically incorporated into the C-2 and C-7 positions of the porphyrin of cytochrome-c3 in a strictly anaerobic bacterium, D. vulgaris . J Am Chem Soc. 1993;115(25):12185–12186. [Google Scholar]
- 154.Scott AI. Mechanistic and evolutionary aspects of vitamin-B12 biosynthesis. Acc Chem Res. 1990;23(9):308–317. [Google Scholar]
- 155.Panek H, O’Brian MR. A whole genome view of prokaryotic haem biosynthesis. Microbiol Sgm. 2002;148:2273–2282. doi: 10.1099/00221287-148-8-2273. [DOI] [PubMed] [Google Scholar]
- 156.Buchenau B, Kahnt J, Heinemann IU, Jahn D, Thauer RK. Heme biosynthesis in Methanosarcina barkeri via a pathway involving two methylation reactions. J Biol Chem. 2006;188(24):8666–8668. doi: 10.1128/JB.01349-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Susanti D, Mukhopadhyay B. An intertwined evolutionary history of methanogenic archaea and sulfate reduction. Plos One. 2012 doi: 10.1371/journal.pone.0045313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Layer G, Moser J, Heinz DW, Jahn D, Schubert WD. Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of Radical SAM enzymes. EMBO J. 2003;22(23):6214–6224. doi: 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Romao CV, Louro R, Timkovich R, Lubben M, Liu MY, LeGall J, Xavier AV, Teixeira M. Iron-coproporphyrin III is a natural cofactor in bacterioferritin from the anaerobic bacterium Desulfovibrio desulfuricans . FEBS Lett. 2000;480(2–3):213–216. doi: 10.1016/s0014-5793(00)01939-6. [DOI] [PubMed] [Google Scholar]
- 160.Oglesby-Sherrouse AG, Vasil ML. Characterization of a heme-regulated non-coding RNA encoded by the prrF locus of P. aeruginosa . PLoS ONE. 2010;5(4):e9930. doi: 10.1371/journal.pone.0009930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Benner SA, Ellington AD, Tauer A. Modern metabolism as a palimpsest of the RNA world. Proc Natl Acad Sci USA. 1989;86(18):7054–7058. doi: 10.1073/pnas.86.18.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dickman SR. Ribonucleotide reduction and possible role of cobalamin in evolution. J Mol Evol. 1977;10(3):251–260. doi: 10.1007/BF01764600. [DOI] [PubMed] [Google Scholar]
- 163.Banerjee R, Ragsdale SW. The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu Rev Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 164.Frey PA, Magnusson OT. S-Adenosylmethionine: a wolf in sheep’s clothing, or a rich man’s adenosylcobalamin? Chem Rev. 2003;103(6):2129–2148. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- 165.Nicolet Y, Drennan CL. AdoMet radical proteins: from structure to evolution––alignment of divergent protein sequences reveals strong secondary structure element conservation. Nucl Acids Res. 2004;32(13):4015–4025. doi: 10.1093/nar/gkh728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gough SP, Petersen BO, Duus JO. Anaerobic chlorophyll isocyclic ring formation in Rhodobacter capsulatus requires a cobalamin cofactor. Proc Natl Acad Sci USA. 2000;97(12):6908–6913. doi: 10.1073/pnas.97.12.6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Holliday GL, Thornton JM, Marquet A, Smith AG, Rebeille F, Mendel R, Schubert HL, Lawrence AD, Warren MJ. Evolution of enzymes and pathways for the biosynthesis of cofactors. Nat Prod Rep. 2007;24(5):972–987. doi: 10.1039/b703107f. [DOI] [PubMed] [Google Scholar]
- 168.Ferguson SJ. Remarkable diversity in biosynthesis of c-type cytochromes in eukaryotes and prokaryotes. FEBS J. 2011;278(22):4169. doi: 10.1111/j.1742-4658.2011.08378.x. [DOI] [PubMed] [Google Scholar]
- 169.Ducluzeau AL, van Lis R, Duval S, Schoepp-Cothenet B, Russell MJ, Nitschke W. Was nitric oxide the first deep electron sink? Trends Biochem Sci. 2009;34(1):9–15. doi: 10.1016/j.tibs.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 170.Crane BR, Getzoff ED. The relationship between structure and function for the sulfite reductases. Curr Opin Struc Biol. 1996;6(6):744–756. doi: 10.1016/s0959-440x(96)80003-0. [DOI] [PubMed] [Google Scholar]
- 171.Lubben M, Morand K. Novel prenylated hemes as cofactors of cytochrome oxidases: archaea have modified heme a and heme o . J Biol Chem. 1994;269(34):21473–21479. [PubMed] [Google Scholar]